Изобретение относится к фармацевтической композиции, в частности касается композиции для липофильных лекарственных препаратов, которая может увеличить биодоступность таких препаратов.
Многие липофильные лекарственные препараты, например, сердечно-сосудистые подвергаются экстенсивному и к тому же изменчивому метаболическому "эффекту первого прохождения". Это происходит потому, что вводимые внутрь препараты, всасывающиеся из желудочнокишечного тракта, транспортируются по портальной системе кровоснабжения непосредственно в печень. Поскольку печень является основным местом, в котором метаболизируются лекарственные вещества, а липофильные препараты более склонны к быстрому метаболизму, большая часть всасывающегося липофильного препарата может не достигнуть системой циркуляции. Это в особенности касается таких групп препаратов как липофильные бета-блокаторы и блокаторы кальциевых каналов. Однако и у других липофильных препаратов отмечается высокая степень метаболизма при первом прохождении через печень.
При назначении обычных доз препаратов, восприимчивых к метаболизму первого прохождения следует учитывать то, что метаболизм первого происхождения через печень проявляется по разному у различных больных или у одного и того же больного и в различное время, что приводит к непредсказуемости терапевтической реакции на дозу препарата, назначенного больному.
Факторами, определяющими величину метаболического воздействия при первом происхождении через печень, является:
(i) экспрессия генов, контролирующих уровни ферментов печени, метабилизирующих лекарственные вещества;
(ii) непредсказуемость или высокая вариабельность снижения метаболического воздействия печени на лекарственные вещества у взрослых;
(iii) изменчивость метаболического воздействия ферментов на лекарственные вещества у детей.
Липофильные сердечно-сосудистые препараты, в частности бета-блокаторы и блокаторы кальциевых каналов, относятся к тем препаратам, которые всасываются из желудочно-кишечного тракта и пассивно попадают в портальную систему кровоснабжения. Всасывание препаратов, как предполагают, практически завершается, но при движении они вариабельно и интенсивно метаболизируются до поступления в системную циркуляцию. Метаболизм может иметь место в полости желудочно-кишечного тракта, в стенке кишечника или в печени, но обычно считается, что в основном метаболизм происходит в печени вследствие практически полного попадания препарата в кровоток портальной системы. Метаболизирование лекарственного вещества во время его первого происхождения через печень получило название "эффекта первого прохождения", описанного выше.
Установлено, что эффект первого прохождения характерен для липофильных оснований (напр. пропранолола) и эфиров липофильных кислот (напр. ацетилсалициловой кислоты), но не характерен для липофильных кислот (напр. салициловой кислоты). В связи с интенсивностью процессов в печени, связанных с экстракцией и метаболизмом, биодоступность этих препаратов составляет менее 50% при значительной вариабельности данного показателя (более чем в два раза).
Непредсказуемость выведения из печени лекарственного вещества вызывает широкий спектр меж- и внутриклеточных субъектных изменений стабильных концентраций лекарственного вещества в плазме у проходящих курс лечения хронических больных и, тем самым, обеспечивает неуверенность при выборе дозы для данного лекарства. Очевидно также то, что лекарственные вещества, претерпевающие интенсивный метаболизм при первом прохождении, могут продуцировать различную концентрацию метаболитов в плазме в зависимости от временных показателей после приема внутрь и парентерального введения.
При применении сердечно-сосудистых лекарственных веществ обычно отмечается насыщение метаболических путей в печени и увеличение концентраций лекарственных веществ в портальной системе кровоснабжения. Такой эффект насыщения, который обычно отмечается в тех случаях, когда физиологические концентрации вступают в противоречие с нормальным дозированием, называется эффектом Микаэлиса-Ментэна, а лекарственный препарат, как полагают, обнаруживает свойства кинетики Микаэлиса-Ментэна.
Если концентрация препарата в портальной системе достаточно низкая и не вызывает насыщения метаболических путей, или если концентрация препарата настолько высокая, что вызывает полное насыщение метаболических частей, принято считать, что фармакокинетика является линейной. Это означает, что количество препарата, вошедшего в системную циркуляцию, пропорционально дозировке. Когда речь идет о насыщении метаболических путей, требуется пороговая доза, чтобы насыщение метаболических путей наступило до того момента, когда линейность станет очевидной.
Когда действует кинетика Микаэлиса-Ментэна, фармакокинетика является нелинейной, так как с увеличением дозы, увеличивается степень насыщения метаболических путей, что приводит к непропорциональному увеличению биодоступности. Это свойство создает трудности при титровании дозировки.
Биодоступность ряда препаратов, которые подвергаются интенсивному метаболизму при первом прохождении через печень после приема одной дозы, увеличивается при длительном введении лекарства. Такая нелинейность по всей вероятности объясняется насыщением метаболизма с учетом более высокой концентрации препаратов, достигаемой при повторных приемах.
Чем быстрее происходит высвобождение препарата в раствор в тонком кишечнике, тем больше вероятность насыщения и тем больше будет эффект насыщения, ведущий к увеличению биодоступности. И наоборот, чем медленнее происходит высвобождение, тем больше вероятность того, что насыщение не наступит и, следовательно, эффект первого происхождения будет максимальным. Таким образом, препараты с длительным высвобождением обычно имеют более низкую биодоступность, чем препараты с быстрым высвобождением.
Сердечно-сосудистые лекарственные препараты, который наиболее соответствуют целям доставки препарата к участку действия, включают, в соответствии с настоящим изобретением, бета-блокаторы пропранолол метопролол, лабетолол, окспренолол, тимолол и ацебутолол, применяемые в сочетании с антагонистами кальциевых каналов, такими как нифедипин, дилтиазем, никардипин, верапамил. Все эти препараты характеризуются обширным клиренсом и метаболизмом первого прохождения в отличие от гидрофильных бета-блокаторов атенолола и надолола, которые неинтенсивно метаболизируются печенью. В общих чертах, в настоящем изобретении дано описание соответствующих препаратов, используемых для доставки к участку действия, которые в большой степени подвергаются клиренсу и метаболизму при первом прохождении через печень, что приводит к низкой биодоступности в системной циркуляции после приема препарата внутрь, и физическое состояние которых совместно с компонентами системы доставки (напр. молекула препарата представлена как основание, а не хлоргидрат).
Верапамил и дилтиазем проявляют свойства линейной фармакокинетики при разовой дозе препарата, когда отсутствует насыщение метаболизма первого прохождения Микаэлиса-Ментэна, но присутствует насыщение и повышенная биодоступность при многоразовых дозах. Пропранолол и метопролол проявляют свойства линейной фармакокинетики при разовой и многоразовых дозах. Нифедипин проявляет свойства линейной фармакокинетики таким образом, что вопрос многоразового дозирования может быть решен в зависимости от результатов разового дозирования.
Нелинейность фармакокинетики препарата проявляется в том, что при назначении препаратов лекарственных форм длительного высвобождения фактически требуется увеличение суточных, доз чем при назначении препаратов обычного быстрого высвобождения. Предполагается, что это обстоятельство можно рассматривать как приемлемое, если учитывать преимущества, связанные со снижением побочных эффектов, возникающих сразу же после введения препарата, и с необходимостью уменьшения частоты дозирования.
Примером такого лекарственного вещества является нифедипин, у которого проявляется такие побочные явления как прилив крови и тахикардия у некоторых больных, причем большая часть этих явлений относится, как предполагают, к особенностям фармокинетики препарата. Обычные лекарственные формы должны дозироваться 2 - 3 раза в сутки, что может продуцировать значительную флуктуацию пиковых и нижних уровней концентраций. С целью снижения указанных побочных явлений, а также, что представляется более важным, с целью снижения числа суточных доз, разработаны таблетированные лекарственные формы нифедипина с медленным высвобождением.
В этой связи, возникает необходимость как для конкретно указанных выше препаратов, так и для других липофильных препаратов, в такой системе доставки лекарственных средств, которая улучшит биодоступность и позволит преодолеть трудности, связанные с нелинейной фармакокинетикой.
Заявка WO-A-9012583 предлагает лекарственные формы, которые безусловно улучшают биодоступность определенных липофильных фармацевтических активных веществ, в том числе сердечно-сосудистых средств. Эта заявка использует в качестве наполнителей естественную смесь составных частей желчи. Хотя эти формы и оказывают хорошее действие, предпочтительно более точно определить в силу ряда причин, их фармацевтическую рецептуру. Было бы также целесообразно усовершенствовать эти известные лекарственные формы.
Заявка WO-A-9003164 предлагает лекарственные формы двухфазного действия фармацевтически активных гидрофильных агентов, включающих инсулин, кальцитонин и соматотропин. Олеиновая кислота входит в число тех веществ, которые могут войти в состав предложенных относительно сложных форм, но учитывая то, что олеиновая кислота имеет липофильную природу, а действующие ингредиенты - гидрофильную, природу, олеиновую кислоту и действующие ингредиенты следует в значительной степени разделить по разным фазам.
В заявке EP-A-0255002, являющейся прототипом настоящего изобретения, описаны препараты для перорального введения, обладающие противовоспалительной, жаропонижающей и анальгетической активностью, например, арилуксусная и арилпропионовая кислоты. Препараты содержат гранулы, в которых активный компонент высвобождается немедленно, смешанные с гранулами, в которых активный компонент предназначен для замедленного высвобождения. Однако, в этой заявке не раскрывается препарат, включающий C12-C24 жирную кислоту.
Настоящее изобретение касается иного подхода к вопросу о разработке лекарственных форм с применением липофильных фармацевтически действующих веществ.
Первой особенностью настоящего изобретения является предложенная лекарственная форма, содержащая (а) жирную кислоту C12-C24 и (б) фармацевтически активное вещество, в которой часть жирной кислоты C12-C24 предназначена для недлительного высвобождения при непарантеральном введении, а часть жирной кислоты C12-C24 и по меньшей мере часть действующего вещества предназначены для длительного высвобождения при непарантеральном введении.
Предполагается, что такая лекарственная форма позволяет, путем отвода, абсорбироваться большей части фармацевтически активного вещества в лимфатических абсорбционных путях и тем самым значительно уменьшает количество препарата, входящего в портальную систему кровоснабжения а, следовательно, снижает эффект первого прохождения у больных с высокой степенью метаболизации препарата печенью.
Часть лимфатической системы с лимфой, оттекающей от желудочно-кишечного тракта, играет важную роль, выполняя функцию носителя, транспортирующего определенные липидорастворимые питательные вещества. Эти вещества, в число которых входят жирорастворимые витамины A, E, D, K, холестерин и длинноцепочечные жирные кислоты, переносятся в лимфе в основном в составе липопротеидов или связанные с ними. Липопротеиды, в том числе липопротеиды очень низкой плотности и хиломикроны, вырабатываются абсорбционными энтероцитами и переносятся в лимфу, откуда они, минуя на начальном отрезке своего пути печень, попадают непосредственно в системное кровоснабжение. Для сравнения отметим, что молекулы, будучи по своей природе относительно более гидрофильными и/или имеющими более низкий молекулярный вес, чем те, которые транспортируются в лимфе, проходят в капиллярное русло, окружающее желудочно-кишечный тракт, а затем попадают по кровотоку в печень через портальную систему кровоснабжения. Прохождение через печень, предваряющее попадание в системную циркуляцию, создает возможность для метаболической модификации.
Компонентом (а) композиции изобретения является жирная кислота C12-C24. Жирная кислота может быть насыщенной или ненасыщенной. К предпочтительной ненасыщенной кислоте относится стеариновая кислота (C18:0). Ненасыщенная кислота может быть моно-ненасыщенной или поли-ненасыщенной. К предпочтительной моно-ненасыщенной жирной кислоте относится олеиновая кислота (C18:1). К предпочтительным поли-ненасыщенным жирным кислотам относятся линолевая кислота (C18:2) в линоленовая кислота (C18:3).
Арахидоновая кислота (омега W6 жирная кислота) является важным предшествующим продуктом для 2 ряда простагландинов, которые оказывают мощное воздействие на сокращение гладкой мускулатуры и агрегацию кровяных пластинок. Докосогексановая кислота (C22:6W3) относится к омега 3 основным жирным кислотам, вырабатываемым из рыбьего жира, которые, как нашли, ингибируют агрегацию кровяных пластинок и оказывают благоприятное действие на сердечно-сосудистые факторы риска, увеличивая число липопротеидов высокой плотности (ЛВП) по отношению и липолпротеидам низкой плотности (ЛНП). При совместном введении этих жирных кислот и фармацевтических оснований можно получить ряд потенциальных благоприятных эффектов. Например, есть многократные подтверждения того, что терапевтическое действие бета-блокаторов вызывает неблагоприятные изменения липопротеидов крови, что может привести к увеличению риска сердечно-сосудистых заболеваний (Накамура, Х.А., журн. Кардиол, 1987, 60, 24E-28E; Робертс, В.С., журн. Кардиол, 1987, 60, 33E-35E). Поэтому помощь в снижении этих неблагоприятных действий смогут, по-видимому, сказать лекарственные формы бета-блокаторов с указанными жирными кислотами. При исследовании применения пропранолола в сочетании с терапией рыбьего жира отмечалось потенцирование снижения кровяного давления, вызванное действием пропранолола (П, Зингер, С. Мельчер, М., Гошель и С. Аугустин, журн. Хайпертэншн, 1990, 16, (6) 682-691). Однако, лекарственные формы двухфазного действия настоящего изобретения и преимущества, которые получают от их применения, не рассматривают и не предлагаются.
Жирные кислоты могут использоваться самостоятельно или в комбинации друг с другом. Они могут быть в виде свободной кислоты или в виде соли с фармацевтически приемлемым катионом, например кальцием или натрием.
Компонент (б) фармацевтической композиции в соответствии с изобретением представляет собой фармацевтически активное вещество, которое обычно является липофильным и предпочтительно растворимым в жирной кислоте. Для фармацевтически действующего вещества нет необходимости быть всегда и при всех условиях растворимым в жирной кислоте; наоборот, предпочтительно, чтобы действующее вещество обладало достаточной растворимостью в жирной кислоте для того, чтобы дать возможность легко приготовить лекарственные формы. Растворенное количество обычно связано с эффективностью дозы. Утверждая это, однако, следует считать предпочтительным, чтобы действующее вещество находилось в такой форме, которая позволяла бы ей легко растворяться в носителе жирной кислоты. Обычно препарат в форме основания, но не исключается применение солей. При определенных условиях можно смешивать фармацевтически активное вещество с солью жирной кислоты, эфиром, амидом или другим соединением. Пользуясь настоящим изобретением, можно составлять смеси различных действующих веществ.
Липофильность препарата можно определить по его коэффициенту распределения октанол/вода, который, как предполагают, дает величину сопоставления проницаемости его клеточной мембраны. Логарифмическая координата p равная по меньшей мере 2 обычно является показателем препарата, который обладает достаточной гидрофобностью или липофильностью для переноса, важного по своей значимости, в систему гидрофобной ЦНС организма.
Обычно лекарственные средства, которые в высокой степени подвержены клиренсу и метаболизму при первом прохождении через печень, могут в соответствии с настоящим изобретением входит в состав лекарственной формы при условии, что они физически совместимы. Однако лекарственная форма, включающая сердечно-сосудистые средства пропранолол, метопролол, тимолол, верапамил и дилтиазем в виде свободных оснований, является особо предпочтительной. Предпочтительными могут быть и другие препараты, например, нифедипин, нитрендипин, фелодипин и нимоджипин.
К другим препаратам, подверженным метаболизму первого прохождения и являющимся, в соответствии с настоящим изобретением, наиболее соответствующими кандидатурами для лекарственной формы, относятся, но ими не ограничиваются, лабетолол, никардепин, оксипентилфиллин, окспренолол, адреналин, допамин, фенотерол, иборамин (SKLF 100168), орципреналин, фенилэфрин, римитерол, ритодрин, салбутамол, тербуталин, фенолдопам (SKLF 82526), имипрамин и тримипрамин.
Антагонисты кальциевых каналов, бета-блокатора, бета2-агонисты (особенно салбутамол), трициклические антидепрессанты могут быть соответствующими кандидатурами, с помощью настоящего изобретения, для включения в лекарственную композицию.
Фармацевтически активное вещество может быть в форме пролекарства. Примерами таких пролекарств являются эфиры амиды, образованные в результате реакции (независимо от того проводится ли реакция до или после включения в лекарственную композицию между жирной кислотой и фармацевтически действующим соединением. Эфир может быть образован в том случае, если фармацевтически активное вещество имеет соответствующую гидроксильную группу, а амид может быть образован в том случае, когда фармацевтически активное вещество имеет соответствующие первичную или вторичную аминогруппы.
Как указано выше, часть жирной кислоты C12-C24 вводится в состав лекарственной композиции для недлительного (и обычного быстрого) высвобождения при непарантеральном (обычно оральном введении), а часть жирной кислоты C12-C24 и по меньшей мере часть (но в ряде случаев все количество) фармацевтически активного вещества вводится в лекарственную композицию для длительного высвобождения при парантеральном (тоже, как правило, оральном) введении. Поэтому, в соответствии с изобретением, композиции несут в себе характеристики двухфазного действия с высвобождением.
Предполагается, что фармацевтически активные вещества, которые вводятся или которые стремятся ввести в состав лекарственных форм с целью длительного высвобождения, будут использоваться с максимальными преимуществами с помощью настоящего изобретения, поскольку препараты в составе форм будут оптимально подвергаться метаболизму при прохождении через печень. Это связано с тем, что они доставляются в печень в малых концентрациях таким образом, что метаболические пути не насыщаются. Отведение абсорбционного процесса к путям лимфатической абсорбции должно поэтому привести к значительному увеличению биодоступности.
Выражение "длительное высвобождение" хорошо известно химикам-фармацевтам, разрабатывающим рецептуры лекарственных форм и не требует специального разъяснения. Тем не менее, когда обычно речь идет о длительности высвобождения действующего вещества, следует иметь в виде, что период его высвобождения длится свыше или по меньшей мере в течение 30 минут и предпочтительно по меньшей мере 1, 2, 5 и даже более часов. Лекарственная форма с длительным высвобождением может быть, но не обязательно должна быть формой с замедленным высвобождением.
Часть жирной кислоты C12-C24 с недлительным высвобождением может использоваться самостоятельно или в лекарственной композиции относительно простого состава. Если действующее вещество входит в состав рецептуры с недлительным высвобождением, оно может смешиваться с жирной кислотой C12-C24 или, что предпочтительно, растворяться в ней. Хотя в принципе нет оснований для того, чтобы эта часть общей формы не могла быть приготовлена более сложным способом, чтобы отвечать конкретным условиям (например, препарат с быстрым высвобождением может также быть составлен в форме гранул, содержащих жирную кислоту и действующее вещество), предпочтение часто отдается приготовлению самых простых лекарственных форм.
Соотношение жирных кислот и фармацевтически активных ингредиентов в фазе недлительного высвобождения меняется от формы к форме. Обычно, соотношение (вес:вес) жирных кислот и действующих ингредиентов имеет диапазон от 10:1 до 0,1:1, предпочтительное соотношение от 5:1 до 1:1.
Часть жирной кислоты C12-C24 и часть (или все количество, если отсутствует часть для недлительного высвобождения) действующего вещества используются для разработки формы с длительным высвобождением. Для приготовления формы такого свойства имеется несколько вариантов. Первый, когда компонент(ы) для длительного высвобождения могут гранулироваться, например, путем использования производных целлюлозы (например, оксипропиловая целлюлоза) или камедей; технология изготовления гранул хорошо известна. Гранулы затем могут быть покрыты оболочкой длительного высвобождения, например, оболочкой из этилцеллюлозы. После этого, гранулы в оболочке можно диспергировать в фазе с недлительным высвобождением лекарственной композиции.
Второй вариант, когда компонент(ы) для длительного высвобождения разрабатываются как размываемые и/или термопластические твердые вещества (при физиологических температурах). С этой целью, компонент(ы) для длительного высвобождения могут смешиваться с одним или более глицеридов или с другими соответствующими и физиологически совместимыми соединениями, имеющими переходную температуру (точку плавления) свыше 37oC. Соответствующими глицеридами являются ди- и три-глицериды, такие как, например, большая часть разнообразных соединений типа "Гелусир", являющихся эфирами гидрогенизированных жирных кислот фирмы Гаттефосс (олово "Гелусир" - товарный знак фирмы). Известны другие товарные значки соответствующих глицеридов, например, "Лабрафил" и "Пресирол". Предпочтительно использовать соединения "Гелусир" и другие соответствующие соединения, имеющие переходные температуры от 45oC до 70oC. Ниже приводятся конкретные примеры образцов соединений "Гелусир" и их эквивалентов:
- "Телусир" 50/02
- "Гелусир" 54/02 (также имеется в наличии "Пресирол")
- "Гелусир" 62/05
- "Гелусир" 64/02 (также имеется "Прессирол" WL 2155).
Две первые цифры цифровой части товарного знака "Гелусир" характеризуют температуру переходного периода в градусах Цельсия жидкой/твердой фазы, а вторые две цифры характеризуют величину гидрофильно-липофильного баланса (ГЛБ). Предпочтительны низкие значения величин ГЛБ (например, 6 или 5 или более низкие) для соединений "Гелусир", поскольку эти соединения, имеющие температуры, наиболее соответствующие переходному периоду фаз, имеют тенденцию к низким значениям величин ГЛБ, а также поскольку соединения "Гелусир" с низкими значениями ГЛБ имеют свойства водного диспергирования, более соответствующие параметрам настоящего изобретения. Однако, возможно применение других соединений "Гелусир", не имеющих предпочтительных характеристик, изложенных выше, дополнительно в качестве модификаторов скоростей длительного высвобождения.
Могут использоваться разнообразные вспомогательные средства для лекарственных композиций. Например, поверхностно-активное вещество (ПАВ). Одно или более ПАВ, рассматриваемые ниже более подробно, могут быть включены в фазу длительного высвобождения лекарственной формы. ПАВ используются при разработке рецептур форм как способствующие совместной растворимости компонентов и имеющие тенденцию к снижению барбортирования, если лекарственная форма заключается в капсулу. Кроме того, ПАВ может способствовать решению проблемы размываемости фазы длительного высвобождения формы in vivo. Другим вспомогательным средством, которое может использоваться при разработке форм, является флюидизатор и/или загущающий агент. Выполняющий две эти функции препарат двуокиси кремния имеет торговый знак "Аэросил" (например, "Аэросил 200"). Компонент двуокиси кремния может также благотворно влиять на характеристику размываемости лекарственной формы.
Третий вариант, когда при разработке компонента(ов) для длительного высвобождения формы используется тиксотропный материал. Такие материалы имеют свойства жидкостей при воздействии сдвигающих сил, которые могут быть вызваны перемешиванием или накачкой, но становятся нетекучими гелями при снятии сдвигающей нагрузки. Подробно термопластическим материалом, описанным выше, тиксотропные материалы применяются при изготовлении твердых желатиновых капсул как наилучшим образом отвечающие требованиям технологии. К тиксотропным материалам этого типа относятся коллоидные двуокиси кремния (например, упомянутый выше препарат "Аэросил 200") и этиловая целлюлоза (также описанная выше, как материал для покрытия фазы длительного высвобождения). В этом варианте осуществления изобретения предполагается смешивание тиксотропного материала с компонентом(ами) для длительного высвобождения. Другими компонентами могут быть промоторы геля и вспомогательные диспергирующие материалы. Гликоли, например, полиэтиленгликоль (ПЭГ) (например, ПЭГ 400) используются в качестве промоторов геля в тиксотропных лекарственных формах и, кроме того, способствуют диспергированию. Могут использоваться неоиногенные поверхностно-активные вещества (НПАВ), например, полиэтоксилированное произвольно гидрированное касторовое масло, имеющее значения величин ГЛБ в диапазоне от 12 до 14 или от 14 до 16.
Гелевая композиция может варьироваться в очень широком диапазоне при сохранении рабочих параметров. Ниже приводятся факторы, влияющие на изменение физических свойств геля.
(i) 1-2% гликоля, например, ПЭГ 400, достаточно для получения плотного геля с приемлемыми диспергирующими характеристиками. Исключением гликоля этой марки может быть получена лекарственная форма с повышенными показателями вязкости масла, и диспергирование будет недостаточно хорошим.
(ii) Увеличение препарата "Аэросил" или схожего с ним компонента на 10% при оптимальных 6% условиях приводит к получению очень плотного геля, но не приводит к ухудшению качества диспергирования. Однако, ригидность гелей, содержащих повышенное количество препарата "Аэросил" может создать трудности при производстве форм в промышленном масштабе.
(iii) Увеличение концентрации фармацевтически действующего вещества (например, пропранолола) свыше 20% может привести к размягчению геля (т.е. ослаблению структуры). Возможно также ухудшение диспергирующих характеристик. Предпочтительно поэтому удерживать концентрацию действующего вещества ниже 20% (вес./вес.).
Аналогично фазе с недлительным высвобождением, соотношение имеет диапазон от 10:1 до 0,1:1, предпочтительное соотношение от 5:1 до 1:1.
Лекарственные формы, в соотношении с изобретением, могут, как об этом упомянуто выше, быть заключены в твердые или мягкие желатиновые капсулы. Твердые желатиновые капсулы могут быть более предпочтительными; когда используются твердые желатиновые капсулы, и лекарственная форма, заключенная в них, содержит одно или более ПАВ, желательно включать в твердую желатиновую оболочку добавки, предупреждающие ломкость оболочек, как это предложено в заявке W-A-9102520.
Включение в лекарственную форму любого другого ингредиента не обязательно. Однако, в некоторых случаях может быть желательным добавление одного или более ингибиторов окисления для предохранения ненасыщенных двойных связей, входящих в состав жирных кислот. К соответствующим ингибиторам окисления относятся д-альфа-токоферол, дл-альфа-токоферол, бутилированный окситолуол (БОТ) и бутилированный оксианизол (БОА), ингибиторы окисления могут использоваться как самостоятельно, так и в комбинации.
Другим произвольным ингредиентом является поверхностно-активное вещество (ПАВ), о котором кратко говорилось выше. Соответствующие ПАВ бывают либо ионогенные (ИПАВ), либо неионогенные (НПАВ), но обычно не включают желчные кислоты или их соли. Предпочтительными являются НПАВ. Подходящим диапазоном ГЛБ для ПАВ, если оно входит в состав, будет широкий диапазон от 0 до 20, предпочтительный диапазон от 6 до 18 и обычный диапазон от 10 до 18. Примерами соответствующих ПАВ, которые могут использоваться как самостоятельно, так и в комбинации, являются эфиры полиоксиэтилен-сорбитановой жирной кислоты (напр. , полисорбат 80, полисорбат 60, полисорбат 40, полисорбат 20), полиоксиэтилен стеараты (напр. полиоксил-40 стеарат) и производные поликсиэтилированного произвольно гидрированного касторового масла, например, продукты Кремофор RH 40 и EL.
Обычно предпочтительно, чтобы лекарственные формы, в соответствии с изобретением, были в сущности не водными, в этом смысле, что вода в формы не добавляется. В состав используемых ингредиентов некоторое количество воды может входит. Однако, свободные от воды лекарственные формы не должны быть предпочтительными во всех случаях их применения.
Лекарственные композиции, в соответствии с изобретением, могут быть покрыты энтеросолюбильными оболочками или защищены каким-либо другим способом для обеспечения лучшего сохранения фармацевтически действующего соединения при транспорте через желудок. Можно использовать любой способ этерозащиты. Капсулы с лекарственной формой могут покрываться энтеросолюбильными оболочками, например, из гидроксипропил метилцеллюлоза фталата или способом промышленного покрытия фирмы Фарма-Винчи P/S.
Лекарственные формы настоящего изобретения являются стимуляторами абсорбционного перераспределения транспорта липофильных препаратов и лимфатическую систему, обусловленного рядом факторов. Во-первых, липофильные препараты в силу их большей растворяемости в липидных системах имеют предрасположенность к всасыванию при транспорте через лимфатическую систему. Во-вторых, подобным же образом происходит всасывание длинноцепочечных жирных кислот из желудочно-кишечного тракта в лимфатическую систему. Наконец, что особенно важно, ненасыщенные жирные кислоты, в том числе олеиновая кислота, имеют важное биохимическое свойство выполнять функции гормонов и переключаться на секрецию хиломикронов из энтероцитов и стимулировать образование лимфы. Таким образом, предполагается, что лекарственные формы действуют следующим образом: жирная кислота и липидорастворимый препарат высвобождаются из соответствующей формы и всасывается эритроцитами, окружающими желудочно-кишечный тракт. Благодаря липофильной природе лекарственного препарата, он имеет природную склонность быть подвергнутым обработке как липид и, следовательно, предрасположен к отбору для лимфатической секреции эритроцитами. Присутствие олеиновой кислоты или другой жирной кислоты обеспечивает наличие обильного материала для синтеза триглицеридов и другого материала, транспортируемых в лимфу в составе липопротеинов, в основном хиломикроны. Предполагается, что на этой стадии препарат связывается или входит в состав липопротеидов, транспортируемых в лимфу. Более того, жирная кислота, например, олеиновая кислота, выступает в роли биохимического диспетчера, регулируя транспорт хиломикронов в лимфатическую систему, Препарат таким образом транспортируется непосредственно в системную циркуляцию через лимфу, минуя печень.
Регуляция липопротеидного метаболизма на молекулярном уровне изучена недостаточно. В частности, неизвестна роль диетических жирных кислот в управлении синтезом и секрецией хиломикронов, липопротеидов с очень низкой плотностью, низкой плотностью и высокой плотностью. С целью попытки дать объяснение этим метаболическим процессам применялись модели для печени и кишечника с использованием клеточных линий HepG2 и Caco2, выращенных соответственно в культуре.
Исследования, проведенные Пуллингером и соавт. (журн. Липид Рисэрч, 1989, 3, 1065-77), Моберли и соавт. (журн. Биохим Биофиз. Акта, 1990, 1042 70-80) и Дашти и соавт. (журн. Липид Рисэрч, 1990, 31 113-123) показали, что олеиновая кислота повышает синтез аполипротеина B(апоB) и его секрецию как клетками HеpG2, так и клетками Caco2. Предполагается, что стимуляция происходит посттранскрипционно и здесь вероятно имеет место случай контрансляции или посттрансляции. Показано, что эффект олеиновой кислоты является дозозависимым до пороговой концентрации в ImM в культуре клетки (Моберли и соавт.).
В исследованиях Грина и Хэдграфта (журн. Интернэшнл Дж. Фармэсьютикс, 1987, 37 251-255) использовалась система искусственной клеточной мембраны для того, чтобы показать, что олеиновая кислота способствует всасыванию катионоактивных препаратов, включая метопролол, окспренолол пропранолол, через механизм ионных пар. Способность олеиновой кислоты ускорять усвоение везикул, содержащих мембраны из щеточной каемки эпителия кишечника продемонстрировали Симпсон и соавт. (журн.Биохим., Биофиз, Акта, 1988, 941 39-47).
Дальнейшие исследования показали, что олеиновая кислота может выступать в роли H+ ионоформы, стимулирующей окисление внутриклеточных структур Ригглзворт и соавт. , Биохим., журн., 1990, 270 109-118), процесс, связанный с прорастанием секреторных сосудов.
Предполагается, что жирные кислоты, например, олеиновая кислота, используемая в настоящем изобретении, выступает в роли гормонов, способных включиться в процесс синтеза и секреции хиломикронов и других липопротеидов с участием абсорбирующих клеток, окружающий желудочно-кишечный тракт. Кроме того, присутствие жирной кислоты в тесном взаимодействии с клеточными мембранами, окружающими желудочно-кишечный тракт, увеличивает абсорбцию фармацевтических оснований за счет эффекта ионовых пар. Этот эффект обычно потенцируется применением свободного основания препарата, поскольку это позволит осуществить совместное растворение лекарственных форм, тесную связь после высвобождения их формы и увеличить эффект пар ионов олеиновой кислоты в мембране. Следует в то же время отметить, что наблюдаемая эффективность лекарственных форм изобретения не зависит от корректности этих гипотез.
Фармацевтические основания легко растворяются в ненасыщенных жирных кислотах, например, в олеиновой кислоте, линолевой кислоте и докосогексановой кислоте.
Лекарственные композиции настоящего изобретения, в силу их способности избегать метаболизма при первом прохождении через печень, могут дать возможность значительно снизить (обычно на 50%) дозировку принимаемого лекарственного препарата. Такое снижение химической нагрузки может быть достигнуто при сохранении таких же терапевтических уровней препарата в системной циркуляции, какие имеются при использовании имеющихся в настоящее время стандартных лекарственных форм препарата. Кроме того, уровни лекарственного средства в системной циркуляции после введения лекарственных форм настоящего изобретения могут быть подвергнуты меньшей изменчивости по сравнению с обычными формами лекарственных средств. Предполагается, что увеличение предсказуемости ответной реакции у больных на дозировку препарата придаст еще больше уверенности клинической терапии. Поэтому появление у больных определенных симптомов может контролироваться быстрее и проще при уменьшении побочных эффектов и риске токсичности.
Для получения фармацевтических композиций можно использовать, в соответствии с изобретением, любой удобный способ. Как отмечалось выше, вторым объектом изобретения является предлагаемый способ получения фармацевтической композиции, включающий композицию фаз недлительного и длительного высвобождения, в котором фаза недлительного высвобождения включает часть жирной кислоты C12-C24, а фаза длительного высвобождения включает часть жирной кислоты C12-C24 и по меньшей мере часть фармацевтически активного вещества и совмещение фаз недлительного и длительного высвобождения.
В предпочтительных вариантах осуществления изобретения, фармацевтически активное вещество растворяется в жирной кислоте. В зависимости от природы в составе компонентов, входящих в препарат, возможно тепловое воздействие. На чистоту раствора указывает отсутствие любых нерастворившихся частиц.
В соответствии с третьим аспектом настоящего изобретения, предусматривается использование лекарственной композиции, описанной выше, как лекарства для лечения болезни или состояния с помощью фармацевтически действующего вещества. В некоторых воплощениях осуществления изобретения, заболевание или состояние относятся к области сердечно-сосудистых, и в этом случае действующим компонентом формы будет сердечно-сосудистый препарат. В этой связи можно рассматривать изобретение как способ лечения заболевания или состояния с помощью фармацевтически действующего вещества, в частности, случаев, относящихся к сердечно-сосудистой системе, рассмотренных выше.
В рассмотренные точки зрения и особенности настоящего изобретения, которым отдано предпочтение можно ввести необходимые изменения.
Изобретение иллюстрируется далее следующими примерами. Соотношения, приведенные в примерах, весовые, если они не оговариваются особо.
Пример 1. В рассматриваемом ниже примере показан процесс приготовления соединения, включающего компоненты быстрого и медленного высвобождения. В качестве медленного высвобождающегося компонента используется термопластический материал. Обычно эти пластификаты при нагревании плавятся, позволяя тем самым применять технологию смешивания и накачки для жидкостного наполнения.
А. Компонент медленного высвобождения (см. табл. А).
Олеиновую кислоту, Гелусир и Кремафор нагревают до 50-55oC до получения чистого раствора, затем добавляют при перемешивании основание пропранолола, поддерживая температуру смеси 50oC, продолжают перемешивать до полного растворения основания пропранолола и в конце добавляют Аэросил при перемешивании. Полученным количеством - 274 мг - смеси заполняют в нагретом состоянии твердожелатиновые капсулы большого размера и при охлаждении дают возможность смеси затвердеть.
Эквивалентная доза пропранолола в частично заполненных капсулах на данной стадии составляет 40 мг.
В. Компонент быстрого высвобождения (см. табл В).
Олеиновую кислоту и d-альфа-токоферол нагревают при перемешивании до 45-50oC, добавляют пропраналол и при перемешивании растворяют. Полученное количество - 154 мг - композиции добавляют желатиновые капсулы большого размера с уже заполненным затвердевшим компонентом медленного высвобождения. Суммарная доза пропранолола в двухкомпонентной капсуле составляет 80 мг. В результате капсулы содержат затвердевшую массу компонента длительного высвобождения, покрытую жидким компонентом быстрого высвобождения (допустим незначительный весовой излишек при производстве лекарственной формы, входящий в весовой показатель заполнителя).
C. Порядок покрытия капсул энтеросолюбильной оболочкой
Ниже приводится способ покрытия капсул длительного высвобождения энтеросолюбильной оболочкой.
Для защиты состава формы от окисляющего воздействия полости желудка и замедления высвобождения содержимого капсул до момента прохождения формы в двенадцативерстную кишку, твердожелатиновые капсулы покрывают энтеросолюбильной оболочкой.
В качестве материала для энтеросолюбильной оболочки использовали фталат гидроксипропилметилцеллюлозы (HP 55 фирмы Шин-Етсу), который наносили с использованием раствора (приведен ниже) после герметизации содержимого капсул, используя комплект (торговое название "Ликапс"), поставляемый фирмой капсугель.
Состав раствора для нанесения энтеросолюбильной оболочки:
HP55 - 6,0%
этанол - 84,5%
Очищенная вода - 9,5%
Указанный раствор наносили на капсулы, используя слой флюидизированного материала (торговое название "Униглат").
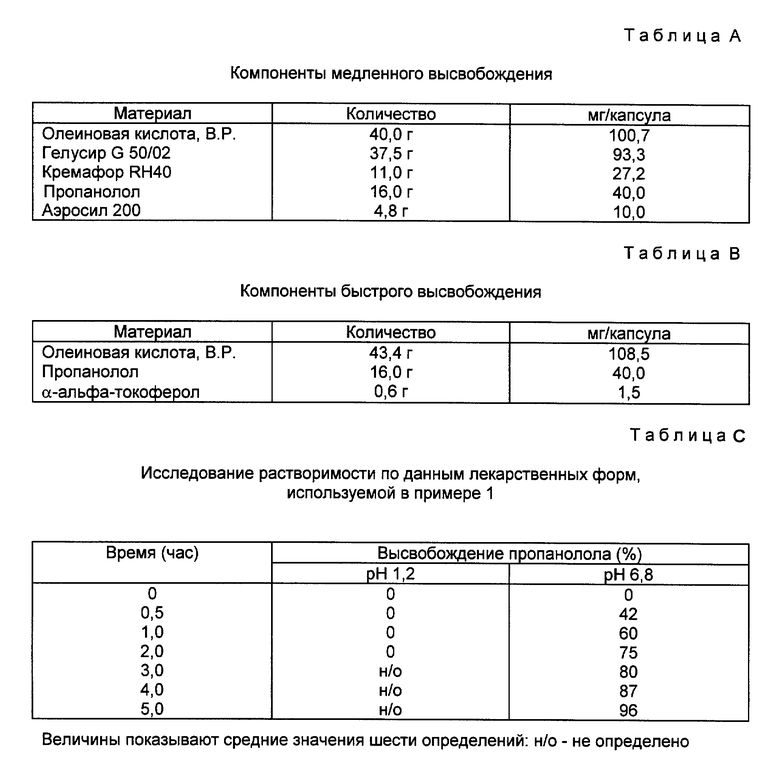
Пример 2. Исследование растворимости.
Для оценки характера диспергирования экспериментальных позиций был разработан метод исследования, за основу которого взято испытание растворимости, опубликованное в Фармакопее США XXII издания для таблеток и капсул. Цель метода - создание для образцов лекарственных проб условий подобных тем, которые имеются в среде кишечника. При определении продолжительности диспергирования остановились на 6-часовом процессе как достаточном для полного высвобождения экспериментальных лекарственных средств. При этом исходили из того, что процесс лимфатической абсорбции в основном происходит в тонком кишечнике.
Для исследования использовали аппарат, рекомендованный фармакопеей США XXII издания (аппарат 2) с фосфатным буферным раствором Соренсенса, pH 6,8, содержащим 0,2% холата натрия в 0,1% деоксихолата натрия, уравновешенного до 37oC. Общий объем буфера, добавленного в каждый из сосудов для растворения составлял 900 мл при скорости вращения мешалки 75 об/мин. Высоту мешалки отрегулировали таким образом, чтобы верхняя кромка лопастей была на уровне поверхности жидкости. Пробный образец опускали в среду для растворения и начинали вращение мешалки. В течение всего опыта образцу давали возможность свободно перемещаться на поверхности изделия. Выбиралось несколько контрольных временных точек, во время которых бралось 5 мл аликвоты растворяемой среды для пробы и заменялось 5 мл свежего буферного раствора. Взятую для пробы аликвоту разбавляли 5 мл метанола, полученный раствор пропускали через 0,8 mM мембранный фильтр (Сарториус, Минисарт NML) до начала определения спектральной поглощательной способности в области 290 нм по данным УФ-спектроскопии, используя УФ однолучевой спектрофотометр. Концентрацию пропранолола в растворимой среде рассчитывали, используя данные заданной калибровочной кривой для пропранолола.
Исследование растворимости по данным лекарственной формы, используемой в примере 1, приведено в табл. С.
В результате исследования растворимости композиции примера 1 установлено, что через пять часов произошло полное высвобождение лекарственного средства. Это достигнуто как за счет быстрого высвобождения масляного компонента лекарственной формы на начальной стадии опыта, так и за счет последующей постепенной эрозии парафиновой матрицы. Высвобождение пропранолола за счет эрозии является важным свидетельством того, что олеиновая кислота высвобождается в желудочно-кишечном тракте одновременно с пропранололом, а, следовательно, повышается поглощающая способность лимфатической системы. В результате проведенного пробного метода изучения растворяемости при pH 1,2 установлено, что энтеросолюбильная оболочка, нанесенная на капсулы, оставалась неповрежденной в течение двух часов.
Лекарственная форма двухфазного действия предназначена для орального введения. Форма состоит из двух фаз: фазы недлительного высвобождения и фазы длительного высвобождения компонентов. Форма включает жирную кислоту C12-C24 и фармацевтически активное липофильное вещество. Фаза недлительного высвобождения содержит часть жирной кислоты. Фаза длительного высвобождения содержит остальную часть жирной кислоты и по меньшей мере часть фармацевтически активного вещества. Массовое соотношение жирной кислоты и активного вещества составляет от 10: 1 до 1:1. Жирная кислота C12-C24 представлена олеиновой и/или линоленовой кислотой. Фармацевтически активное липофильное вещество является пропранололом, верапамилом, нифедипином, дилтиаземом, метопрололом, никардипином и/или лабетололом. Новая лекарственная форма способствует абсорбционному перераспределению активного вещества из портальной системы кровоснабжения в лимфатическую систему, исключается метаболизм лекарства при первом прохождении через печень. Увеличивается биодоступность лекарственных препаратов. 2 с. и 11 з.п.ф-лы.
| НОЖНИЦЫ ДЛЯ ОБРЫВА ПРОКАТА | 0 |
|
SU255002A1 |
