Изобретение относится к производным 2-циано-3-гидроксиенамидов, к способам их получения, а также к содержащим их фармацевтическим композициям.
Более конкретно, изобретение касается производных 2-циано-3-оксоенамидов общей формулы (I)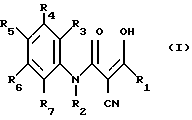 ,
,
в которой
R1 представляет собой группу формулы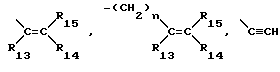 ,
,
где
R13 - R15 являются атомом водорода или C1-C3-алкилом;
n- принимает значения 1,2 или 3;
R2 - является атомом водорода;
R3 - R7 одинаковые или различные и являются атомом водорода, нитро- или цианогруппой, атомом галогена, C1-C3-алкилом, группой формулы - (CH2)mCF3 или -O-(CH2)mCF3, или -S-(CH2)mCF3, где m = 0, или R3 - R7 являются группой формулы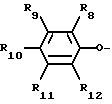 ,
,
где
R8 - R12 являются атомом водорода, галогена, группой CF3.
Среди соединений формулы I можно назвать соединения, в которых R3, R4, R5, R6, R7 одинаковые или различные, представляют собой атом водорода, атом фтора, хлора, брома или йода, радикал, такой как метильный, этильный, трет-бутильный, метоксильный, метилтионильный, трифторметильный, трифторметоксильный, трифторметилтионильный, пентафторэтильный, бром-дифторметоксильный, цианильный, нитро-, феноксильную, 4-хлор-феноксильную группу, или R4 и R5 совместно образуют группу -O-CH2-O-; R2 представляет собой атом водорода или метильный радикал; R1, R3, R6 и R7 имеют указанное выше значение.
Среди последних можно назвать производные формулы I, в которой R1 представляет собой группу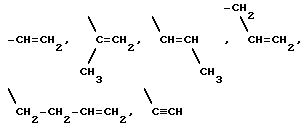 ,
,
R2 представляет собой атом водорода или метильный радикал; R3, R4, R5, R6, R7 одинаковые или различные, представляют собой атом водорода, атом фтора, хлора или брома, радикал, такой как метильный, трифторметильный, трифторметоксильный, цианильный, нитро- или феноксильную группу.
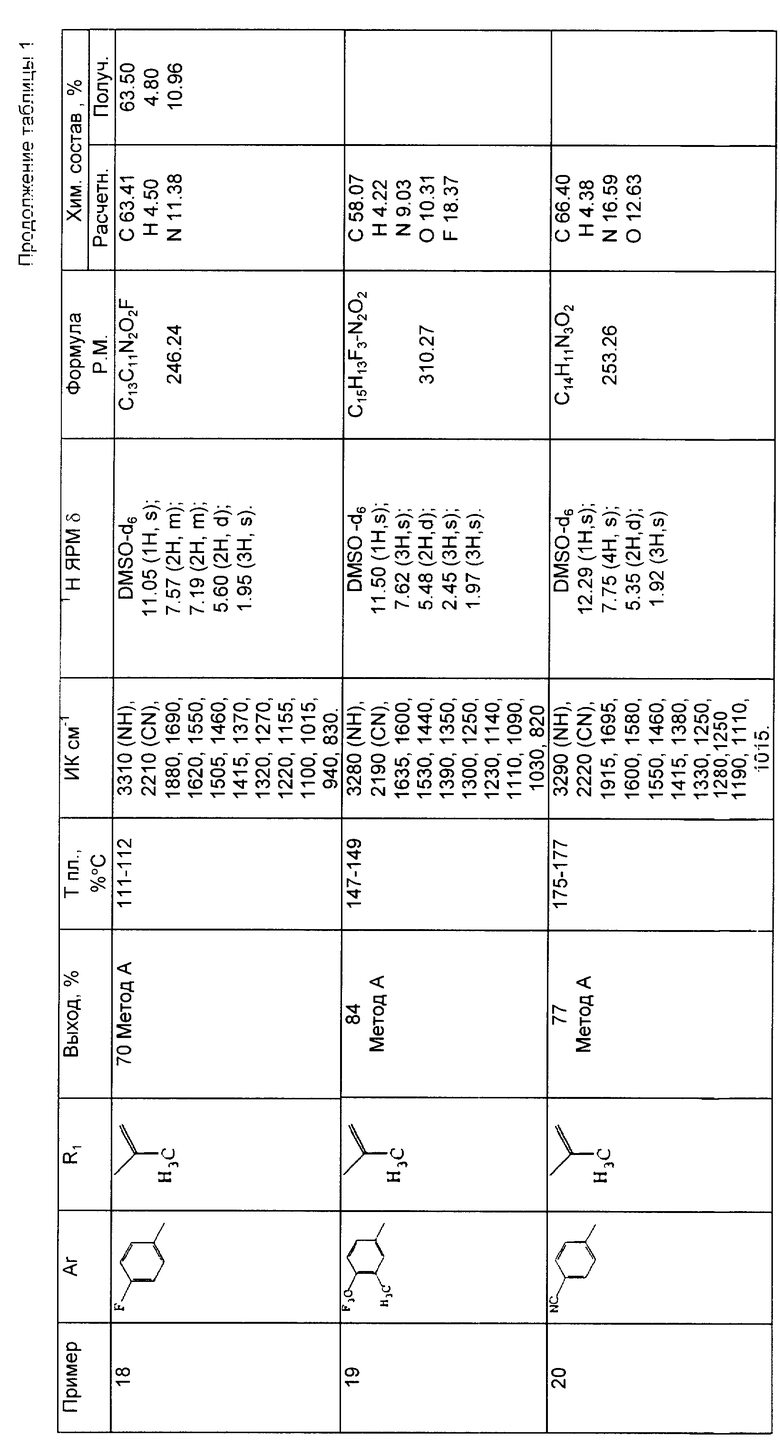
Из последних соединений наиболее интересными являются производные формулы I, которые представляют собой соединения:
2-циано-3-гидрокси-4-метил-N-(4-трифторметилфенил) пента-2,4-диенамид;
2-циано-3-гидрокси-N-(4-трифторметилфенил)-гекса-2,5-диенамид;
2-циано-3-гидрокси-4-метил-N-(4-хлор-3-трифторметилфенил)-пента- 2,4-диенамид;
2-циано-3-гидрокси-4-метил-N-(4-трифторметоксифенил)-пента-2,4-диенамид;
2-циано-3-гидрокси-4-метил-N-(4-бромфенил)-пента-2,4-диенамид;
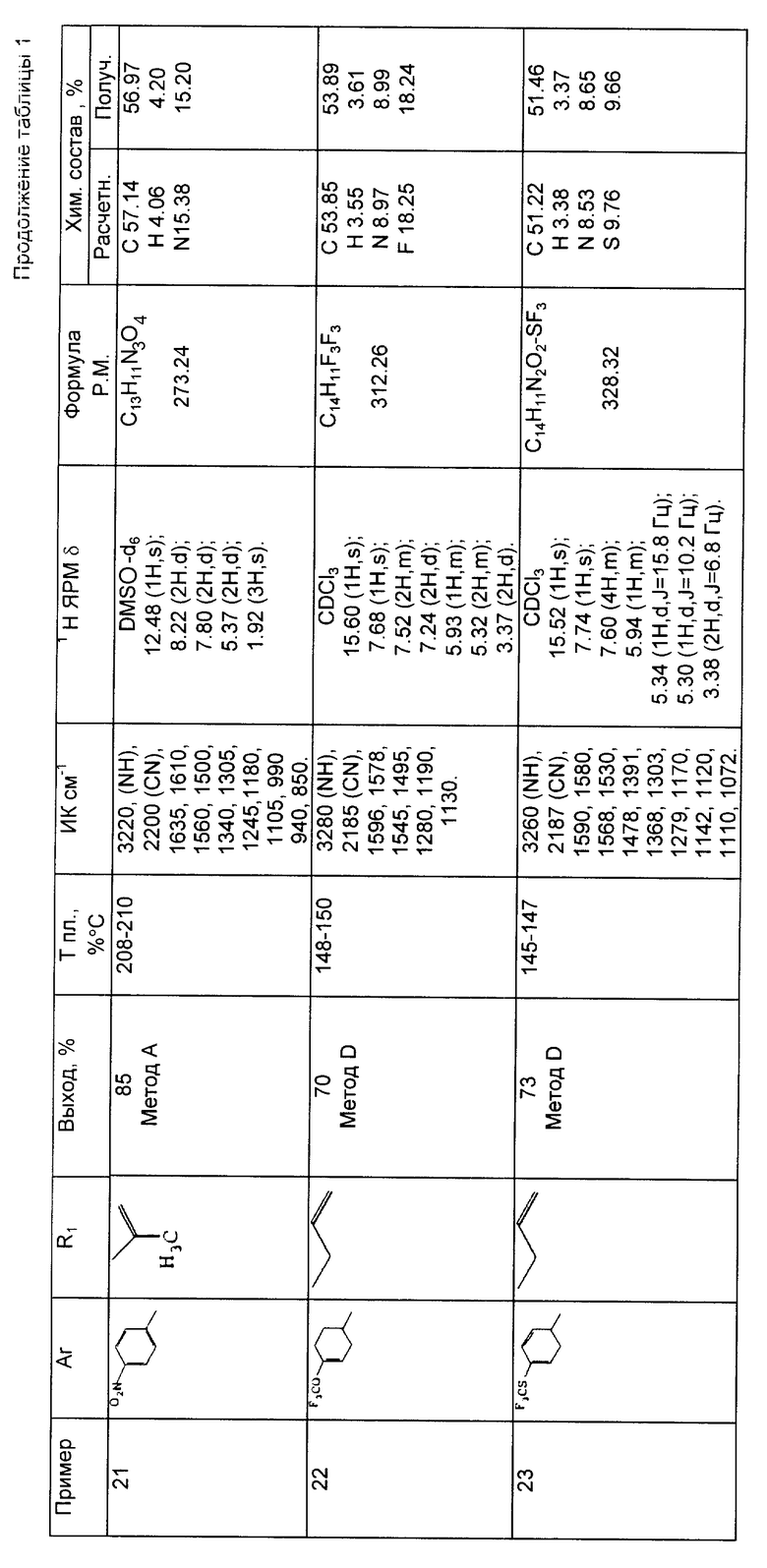
2-циано-3-гидрокси-N-(4-трифторметилфенил)-гепта-2-ен-6-инамид;
2-циано-3-гидрокси-N-(4-хлор-3-трифторметилфенил)-гекса-2,5-диенамид;
2-циано-3-гидрокси-N-(3-метил-4-трифторметилфенил)-гепта-2,6-диенамид;
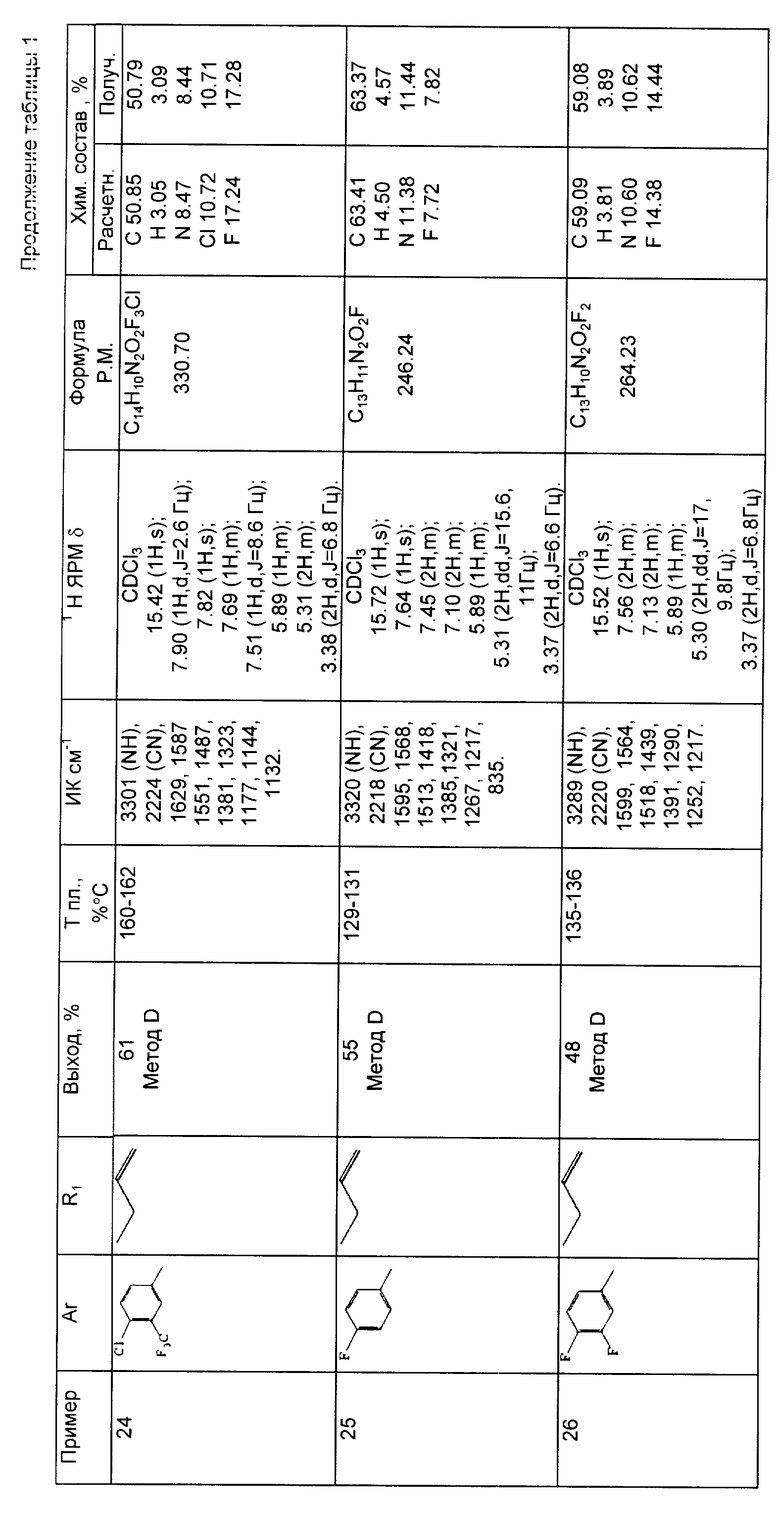
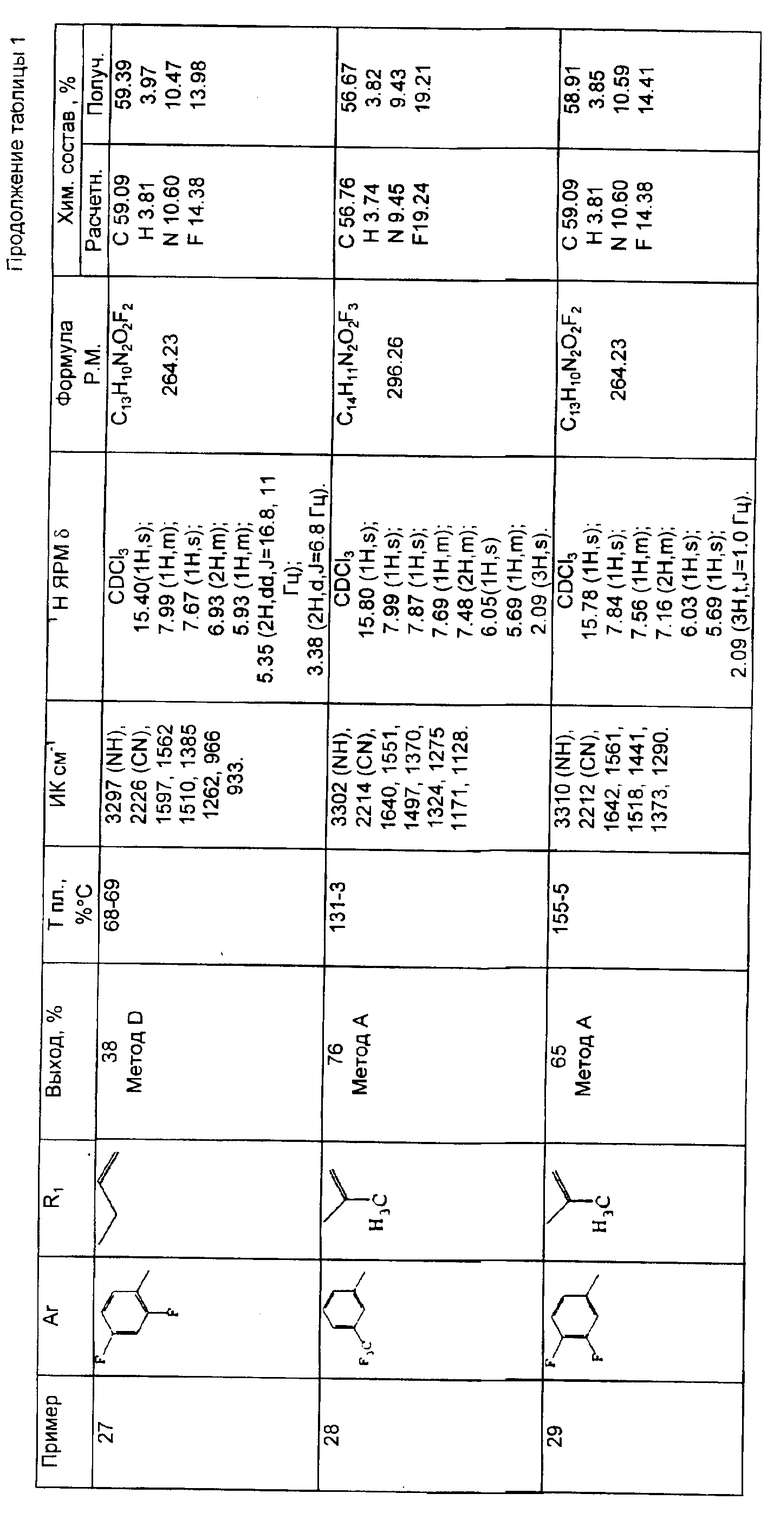
2-циано-3-гидрокси-N-(4-трифторметилфенил)-гепта-2,6-диенамид.
Изобретение относится также к способу получения 2-циано-3-гидроксиенамидов формулы (1), заключающемуся в том, что продукт формулы (II):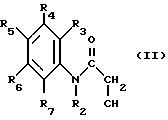 ,
,
в котором
R2, R3, R4, R5, R6 и R7 имеют значение, указанное выше, вводится последовательно в реакцию с гидридом натрия в присутствии катализатора, а затем:
a) либо с соединением формулы (III)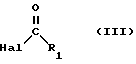 ,
,
в которой
Hal представляет собой атом галогена, а R1 имеет значение, указанное выше;
б) либо с соединением формулы (IIIA)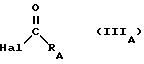 ,
,
в которой
Hal представляет собой атом галогена, а RA является радикалом R1, защищенным должным образом, для получения продукта формулы (IA)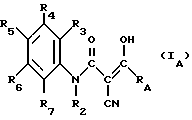 ,
,
в которой
RA, R2, R3, R4, R5, R6 и R7 имеют указанное выше значение; при необходимости, защиту полученного таким образом продукта формулы (IA) снимают для получения соответствующего продукта формулы (I), который выделяют.
В предпочтительных условиях осуществления указанного способа реакцию продукта формулы (II) с гидридом натрия производят в органическом растворителе, таком как тетрагидрофуран, в присутствии катализатора, такого как имидазол, или в отсутствии катализатора; реакцию с соединением формулы (III) или (IIIA) производят в органическом растворителе, таком как тетрагидрофуран, при низкой температуре; радикал R1 может быть защищен арилселеновой группой, такой как фенилселеновая; устранение защиты может производиться окислением с помощью перекисного соединения, такого как пероксид водорода в отсутствии растворителя или же в растворителе, или смеси органических растворителей, такой как смесь метанола и дихлорметана.
Изобретение относится также к способу получения 2-циано-3-гидроксиенамидов формулы (I), в которой R1 представляет собой группу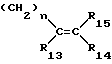 ,
,
в которой
R13, R14 и R15 имеют значение, указанное выше, а n имеет значение 2 или 3, заключающемуся в том, что соединения формулы (V):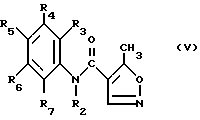 ,
,
в которой
R2, R3, R4, R5, R6, и R7 имеют значение, указанное выше, вводят в реакцию соответственно с соединением формулы (VI)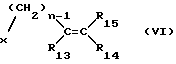 ,
,
в которой
X представляет собой легко удаляемую группу, предпочтительно йод, а n, R13, R14 и R15 имеют вышеприведенное значение, в присутствии сильного основания для получения соответствующего продукта формулы (I), который выделяют.
В предпочтительных условиях осуществления этого способа, когда используют соединение формулы (VI), где X представляет собой атом йода, реакцию взаимодействия соединения формулы (V) с соединением формулы (VI) производят в органическом растворителе, таком как тетрагидрофуран, при низкой температуре, и используемым сильным основанием может быть бутиллитий.
Некоторые продукты формулы (II) описаны в патентной заявке EP N 91402890.7, поданной фирмой-заявителем 29 октября 1991 г. Некоторые другие продукты формулы (II) являются новыми и отвечают формуле (II)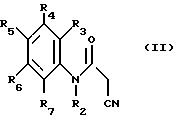 ,
,
в которой
R2 имеет вышеуказанное значение, а один из заместителей R3, R4, R5, R6 и R7 представляют собой группу ,
,
определенную выше, а остальные заместители имеют указанное выше значение.
Среди продуктов формулы (II) можно, в частности, назвать: [4-(4'-хлорфенокси)фенил] -цианоацетанилид; и [4- (4'-трифторметилфенокси)фенил]-цианоацетанилид; или же 4-трифторметоксицианоацетанилид.
Продукты формулы (II) могут быть получены, как указано в упомянутой заявке на патент N 91402890.7, из продуктов формулы (IV)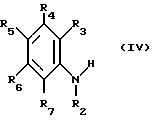 ,
,
в которой
R2, R3, R4, R5, R6 и R7 имеют указанное выше значение, по методу, аналогичному методу описанному А. Нохара, Т.Ишигуро и др. в J.Med.Chem (1985) 28 (5), 559-566, согласно следующей схеме: .
.
Продуктами формулы (IV), используемыми в упомянутом методе, являются, как правило, известные продукты, но они могут быть получены также методом диазотирования (реакция соли диазония с соответствующей солью щелочного металла или меди, например с хлористой медью, иодистым калием, цианистым натрием) с последующим восстановлением соответствующих нитроанилинов по методу, известному специалистам.
Используемые нитроанилины могут быть получены по методу, описанному в Т. Сура и др. в "Synthetic communications" (1988) 18 (16-17) 2161-5.
Некоторые анилины формулы (IV) могут быть получены по методу, указанному в патентной заявке EP N 206951, или путем восстановления соответствующих известных нитробензолов.
Некоторые нитробензолы являются новыми и могут быть получены, как описывается ниже в примерах.
Соединениями формулы (V), используемыми в вышеуказанном методе, как правило, являются известные продукты, но они могут также быть получены по методу, описанному в патентной заявке N WO 91/17748.
Соединения формулы (I) кислые. Их можно перевести в аддитивные соли в результате реакции с неорганическим или органическим основанием в соотношении, очень близком к стехиометрическому. Соли могут быть получены без выделения соответствующих кислот.
Соединения, являющиеся объектом настоящего изобретения, обладают ценной фармакологической активностью, в частности противовоспалительной.
Из патента Франции N 2334350 известны наиболее близкие к предложенным соединения - производные анилинов циануксусной кислоты, входящие в состав лекарственных противовоспалительных препаратов.
Предлагаемые соединения расширяют арсенал противовоспалительных средств. При этом отмечается, в частности, их исключительно высокая противовоспалительная активность. Они тормозят, с одной стороны, воспалительные явления, вызываемые раздражающими агентами, а с другой стороны, - аллергические реакции замедленного типа, препятствуя активации иммуноклеток специфическим антигеном.
Указанные свойства иллюстрированы ниже, в экспериментальной части.
Следовательно, объектом настоящего изобретения является также фармацевтическая композиция, обладающая противовоспалительной активностью, содержащая производное цианамида в качестве активного начала и фармацевтически приемлемые добавки, в качестве производного цианамида она содержит соединение формулы (I) по п. 1 в эффективном количестве.
Фармацевтические составы могут быть, например, твердыми или жидкими и иметь любую фармацевтическую форму, широко применяемую при лечении человека, например простые или дражевидные таблетки, капсулы, гранулы, свечи, препараты для инъекций; они производятся обычными методами. Действующее начало вводится в основы, обычно используемые при изготовлении фармацевтических составов, такие как тальк, аравийская камедь, лактоза, крахмал, стеарат магния, какао-масло, водные или безводные связующие, жиры животного или растительного происхождения, производные парафина, гликоли, различные увлажняющие, диспергирующие или эмульсионные агенты, консерванты.
Указанные композиции могут найти применение, например, при лечении ревматоидного артрита и хронических воспалительных болезней иммунного или неиммунного происхождения (при пересадках, трансплантациях органов, увеитах, раке и других болезнях, связанных с иммунологией).
Обычно применяемые дозы могут, в зависимости от используемого препарата, от особенностей больного и от заболевания, составлять от 0,1 до 200 мг в день при приеме внутрь.
Приводимые далее примеры показывают возможности осуществления изобретения, вместе с тем не ограничивая его.
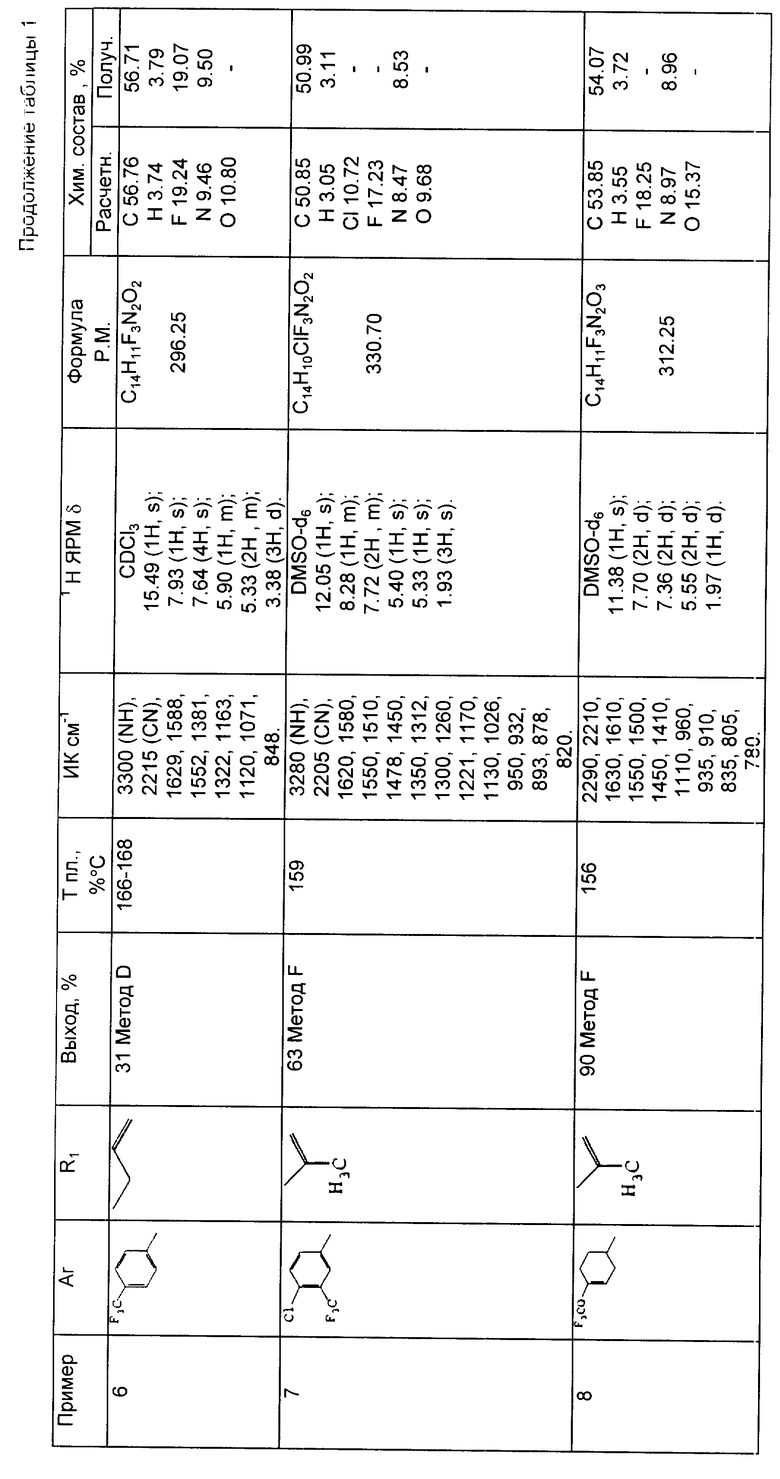
Пример 1. 2-Циано-3-гидрокси-4-метил-N-(4-трифторметилфенил)пента-2,4-диенамид (метод F).
Целевой продукт получен из соответствующих исходных продуктов с применением метода, описанного в примере 8.
Пример 2. 2-Циано-3-гидрокси-N-(4-трифторметилфенил)пента-2,4-диенамид (метод C).
Этап А: 2-циано-3-гидрокси-5-фенилселено-N-(4-трифторметилфенил) пента-2,4-диенамид.
В среде азота смешивают 7 г 4'-трифторметилцианоацетанилида в 200 см3 безводного тетрагидрофурана с добавлением 0,02 г имидазола и 2,3 г гидрида натрия. Затем взбалтывают в течение 2 часов при комнатной температуре, охлаждают до -78oC и добавляют 9,11 г 3-(фенилселено) пропионилхлорида, полученного по методу, описанному в J.Med.Chem. (1988), 31, 1190-6. После этого взбалтывают в течение 90 мин при температуре -78oC, вливают в смесь хлористоводородной кислоты и льда и фильтруют. Твердый остаток растворяют в метиленхлориде, промывают водой, высушивают и удаляют растворитель при пониженном давлении. После порошкования в этиловом эфире получают 13,4 г искомого продукта. Выход: 99%.
Этап Б: 2-циано-3-гидрокси-N-(4-трифторметилфенил)пента-2,4-диенамид (метод C).
8 г продукта, полученного на этапе А, в 200 см3 метиленхлорида охлаждают до температуры 0oC, добавляют 4 см3 30%-ной перекиси водорода и полученную суспензию селеноокиси взбалтывают в течение 30 мин. Затем разбавляют с использованием 40 см3 метанола и 200 см3 метиленхлорида, взбалтывают в течение 1 ч. при комнатной температуре и фильтруют на двуокиси кремния. Растворитель удаляют при пониженном давлении, остаток забирают в этиловом эфире и получают 2,8 г целевого продукта. Выход: 54%.
Пример 3. 2-Циано-3-гидрокси-4-метил-N-(4-бром-3-метилфенил)пента-2,4-диенамид (метод А).
В среде азота взбалтывают 6,3 г 4'-бром-3'-метилцианоацетанилида, растворенного в 200 см3 тетрагидрофурана с добавлением 0,02 г имидазола и 1,85 г гидрида натрия. Суспензию взбалтывают в течение 1 ч. при комнатной температуре, охлаждают до -78oC, добавляют 2,95 см3 свежедистиллированного метакрилоилхлорида и взбалтывают в течение 90 мин при температуре -20oC. Затем вливают в смесь хлористоводородной кислоты и льда и фильтруют. Твердый остаток растворяют в метиленхлориде, промывают водой, высушивают и удаляют растворитель при пониженном давлении. В результате получают 8 г целевого продукта. Выход: 99,7%.
Пример 4. 2-Циано-3-гидрокси-N-(4-трифторметилфенил)пента-2,4-диенамид (метод Е).
В среде азота смешивают 5 г 4'-трифторметилцианоацетанилида в 150 см3 тетрагидрофурана и добавляют 2 г гидрида натрия. Затем взбалтывают в течение 1 часа при комнатной температуре и охлаждают до -70oC. На колбе с реакционной средой устанавливают ацетоно-ледяной конденсатор, соединенный с дистилляционным аппаратом, заправленным 3,01 см3 пропиоловой кислоты и 15 г бензоилфторида, как описано в JACS (1974) 96 (18) 5855-9. Затем нагревают до 150oC и конденсируют выделяющийся пропионилфторид в растворе карбаниона. После этого взбалтывают в течение 1 ч при температуре -70oC, вливают в смесь хлористоводородной кислоты и льда, экстрагируют с использованием этилацетата, высушивают и удаляют растворитель при пониженном давлении. После порошкования в этиловом эфире и хроматографии маточных растворов на двуокиси кремния (элюант: метиленхлорид) получают 2,49 г целевого продукта. Выход: 40%.
Пример 5. (Е)-2-циано-3-гидрокси-N-(4-трифторметилфенил)-гекса-2,4-диенамид (метод B).
В среде азота смешивают 6 г 4'-трифторметилцианоацетанилида в 200 см3 тетрагидрофурана с добавлением 0,02 г имидазола и 1,95 г гидрида натрия. Затем взбалтывают в течение 2 ч при комнатной температуре, охлаждают до -78oC и добавляют 3,06 г свежедистиллированного кротоноилхлорида. После этого взбалтывают при температуре -78oC в течение 2 ч, вливают в смесь хлористоводородной кислоты и льда и фильтруют. Твердый остаток растворяют в метиленхлориде, промывают водой, высушивают и удаляют растворитель при пониженном давлении. В результате получают 7,65 г целевого продукта. Выход: 99%.
Пример 6. 2-Циано-3-гидрокси-N-(4-трифторметилфенил)гекса-2,5-диенамид (метод D).
В среде азота смешивают 6 г 4'-трифторметилцианоацетанилида в 200 см3 тетрагидрофурана и добавляют 1,95 г гидрида натрия. Затем взбалтывают в течение 30 мин при комнатной температуре, охлаждают до -50oC и добавляют 3,3 г 3-бутено-илхлорида, полученного по методу, описанному в J.Chem.Soc. (1948) 661. После этого взбалтывают при температуре -50oC в течение 2 ч, вливают в смесь хлористоводородной кислоты и льда и фильтруют. В результате хроматографии остатка на двуокиси кремния (элюант: метиленхлорид) получают 2,4 г целевого продукта в виде бесцветных кристаллов. Выход: 31%.
Пример 7. 2-Циано-3-гидрокси-4-метил-N-(4-хлор-3-трифторметилфенил)пента-2,4-диенамид.
Целевой продукт получен из соответствующих исходных продуктов с применением метода, описанного в примере 8.
Пример 8. 2-Циано-3-гидрокси-4-метил-N-(4-трифторметоксифенил)пента-2,4-диенамид (метод F).
В среде азота взбалтывают 0,5 г 4'-трифторметоксицианоацетанилида, растворенного в 22 см3 тетрагидрофурана с добавлением каталитического количества имидазола и 0,15 г гидрида натрия. Суспензию взбалтывают в течение 10 мин при комнатной температуре, охлаждают до -78oC, добавляют 0,24 см3 свежедистиллированного метакрилоилхлорида и взбалтывают в течение 30 мин. Затем добавляют 0,3 см3 ледяной уксусной кислоты и вновь взбалтывают в течение 30 мин. Затем вливают в ледяную хлористоводородную кислоту, фильтруют, трижды промывают в 5 см3 воды и 5 см3 эфира и после высушивания получают 575 мг целевого продукта. Выход: 90%.
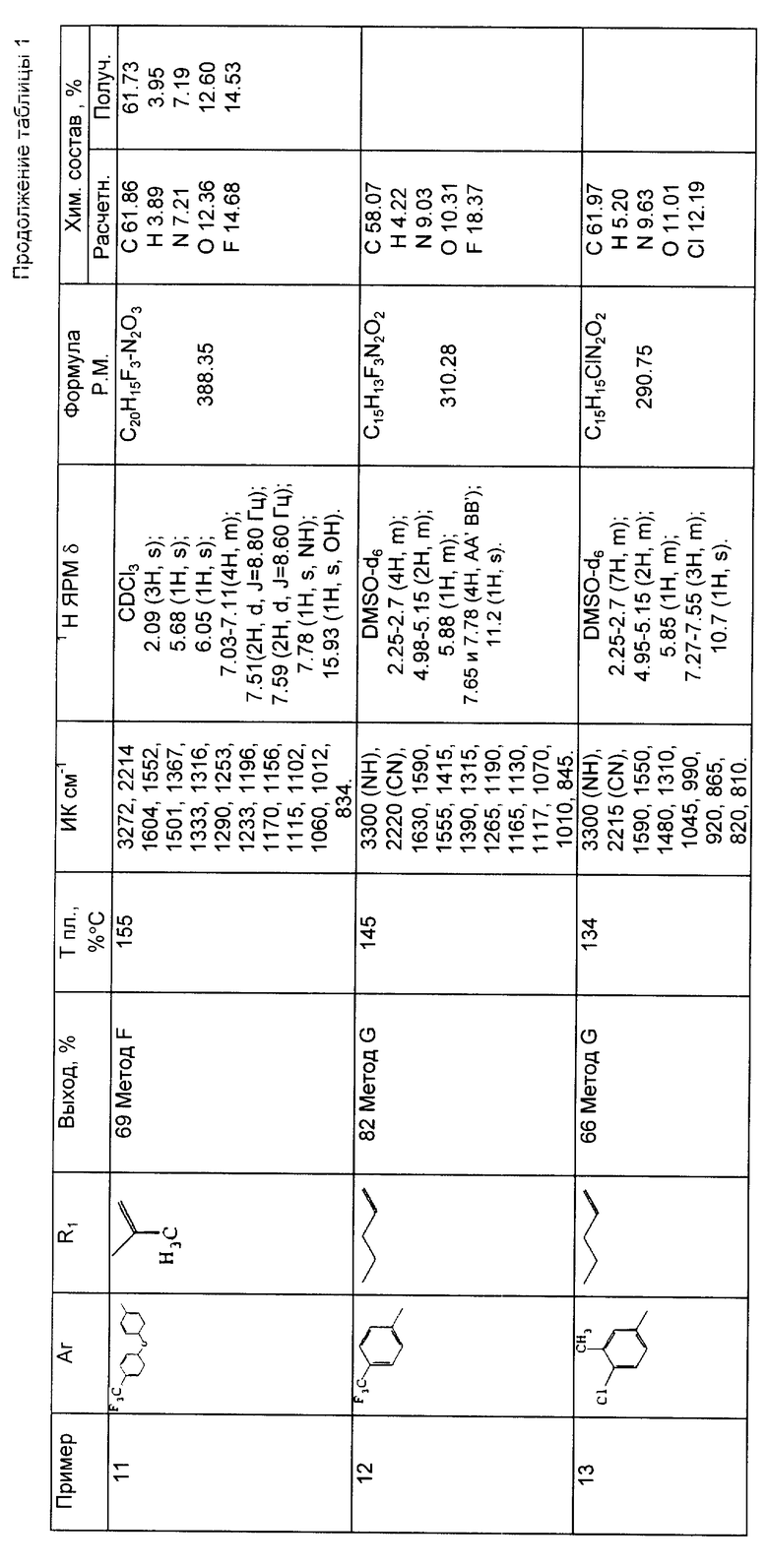
Пример 9. 2-Циано-3-гидрокси-4-метил-N-[4-(4'-хлорфенокси)-фенил]пента-2,4-диенамид.
Целевой продукт получен исходя из соответствующих исходных продуктов, без использования имидазола, с применением метода, описанного в примере 8.
Пример 10. 2-Циано-3-гидрокси-4-метил-N-[4-бромфенил]пента-2,4-диенамид (метод F).
Целевой продукт получен из соответствующих исходных продуктов с применением метода, описанного в примере 8.
Пример 11. 2-Циано-3-гидрокси-4-метил-N-[4-(4'трифторметилфенокси)фенил] пента- 2,4-диенамид.
Целевой продукт получен из соответствующих исходных продуктов с применением метода, описанного в примере 8.
Пример 12. 2-Циано-3-гидрокси-N-(4-трифторметилфенил)-гепта-2,6-диенамид (метод G).
6,75 г 5-метил-4-[N-(4-трифторметил)фенил]карбамоилизоксазола растворяют в 500 см3 абсолютного тетрагидрофурана в инертной среде аргона. Затем охлаждают до -78oC, добавляют 32 см3 2,5 н. раствора бутиллития в гексане, взбалтывают в течение 90 мин, добавляют 10,8 cм3 иодистого аллила и продолжают взбалтывать в течение еще 2 ч, затем добавляют 20 см3 воды и дают температуре подняться до 0oC. После этого добавляют 500 см5 этилацетата и 200 см3 1н. хлористоводородной кислоты, выделяют органическую фазу, промывают ее водой, высушивают и выпаривают растворитель. После перекристаллизации из смеси ацетон-вода-хлористоводородная кислота получают 6,35 г целевого продукта. Т.пл. 145oC.
Пример 13. 2-Циано-3-гидрокси-N-(4-хлор-3-метилфенил)-гепта-2,6-диенамид.
7,5 г целевого продукта получены из 5-метил-4-(N-(4-хлор-3-метил)фенил)карбамоилизоксазола с применением метода, описанного в примере 12 (метод G). Т.пл. 134oC.
Пример 14. 2-Циано-3-гидрокси-N-(4-трифторметилфенил)-гепта-2-ен-6-инамид.
4 г целевого продукта получены из 5-метил-4-(N-(4-трифторметил)фенил)карбамоилизоксазола с применением метода, описанного в примере 12 (метод G), и использованием в качестве алкилирующего средства иодистого пропаргила. Т.пл. 172oC.
Пример 15. 2-Циано-3-гидрокси-N-(4-хлор-3-метилфенил)гепта- 2-ен-6-инамид.
3,2 г целевого продукта получены из 5-метил-4-(N-(4-хлор-3-метил)фенил)карбамоилизоксазола с применением метода, описанного в примере 12 (метод G), и использованием в качестве алкилирующего средства иодистого пропаргила. Т.пл. 147oC.
По методу, описанному в примере 3 (метод A) или 6 (метод G), с применением соответствующих продуктов были получены следующие продукты:
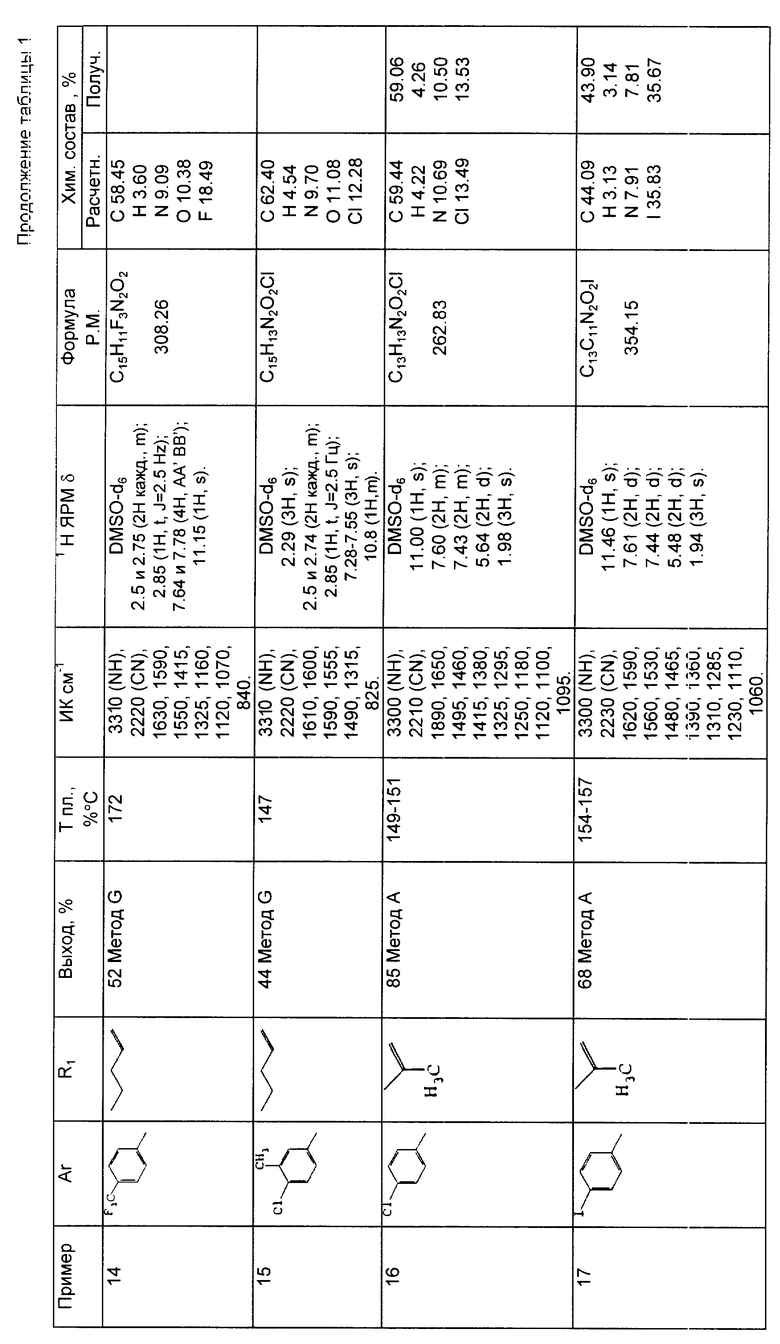
Пример 16. 2-Циано-3-гидрокси-4-метил-N-(4-хлорфенил)-пента-2,4-диенамид (метод A).
Пример 17. 2-Циано-3-гидрокси-4-метил-N-(4-иодофенил)-пента-2,4-диенамид (метод A).
Пример 18. 2-Циано-3-гидрокси-4-метил-N-(4-фторфенил)-пента-2,4-диенамид (метод A).
Пример 19. 2-Циано-3-гидркоси-4-метил-N-(3-метил-4-трифторметилфенил)-пента-2,4-диенамид (метод A).
Пример 20. 2-Циано-3- гидрокси-4-метил-N-(4-цианофенил)-пента-2,4-диенамид (метод A).
Пример 21. 2-Циано-3-гидрокси-4-метил-N-(4-нитрофенил)-пента-2,4-диенамид (метод A).
Пример 22. 2-Циано-3-гидрокси-N-(4-трифторметоксифенил)-гекса-2,5-диенамид (метод D).
Пример 23. 2-Циано-3-гидрокси-N-(4-трифторметилтиофенил)-гекса-2,5-диенамид (метод D).
Пример 24. 2-Циано-3-гидрокси-N-(4-хлор-3-трифторметилфенил)-гекса-2,5-диенамид (метод D).
Пример 25. 2-Циано-3-гидрокси-N-(4-фторфенил)-гекса-2,5-диенамид (метод D).
Пример 26. 2-Циано-3-гидрокси-N-(3,4-дифторфенил)-гекса-2,5-диенамид (метод D).
Пример 27. 2-Циано-3-гидрокси-N-(2, 4-дифторфенил)-гекса-2,5-диенамид (метод D).
Пример 28. 2-Циано-3-гидрокси-4-метил-N-(3-трифторметилфенил)-пента-2,4-диенамид (метод A).
Пример 29. 2-Циано-3-гидрокси-4-метил-N-(3,4-дифторфенил)-пента-2,4-диенамид (метод A).
Пример 30. 2-Циано-3-гидрокси-4-метил-N-(3-хлор-4-фторфенил)-пента-2,4-диенамид (метод A).
Пример 31. 2-Циано-3-гидрокси-4-метил-N-(3,4-дихлорфенил)-пента-2,4-диенамид (метод A).
Пример 32. 2-Циано-3-гидрокси-N-(4-хлорфенил)-гепта-2,6-диенамид.
4,3 г целевого продукта получены из 5-метил-4-(N-(4-хлорфенил)карбамоилизоксазола с применением метода, описанного в примере 12 (метод G). Т.пл. 138oC.
Пример 33. 2-Циано-3-гидрокси-N-(3-метил-4-трифторметилфенил)-гепта-2, 6-диенамид.
4,29 г целевого продукта получены из 5-метил-4-(N-(4-трифторметил-3-метил)фенил)карбамоилизоксазола с применением метода, описанного в примере 12 (метод G). Т.пл. 133oC.
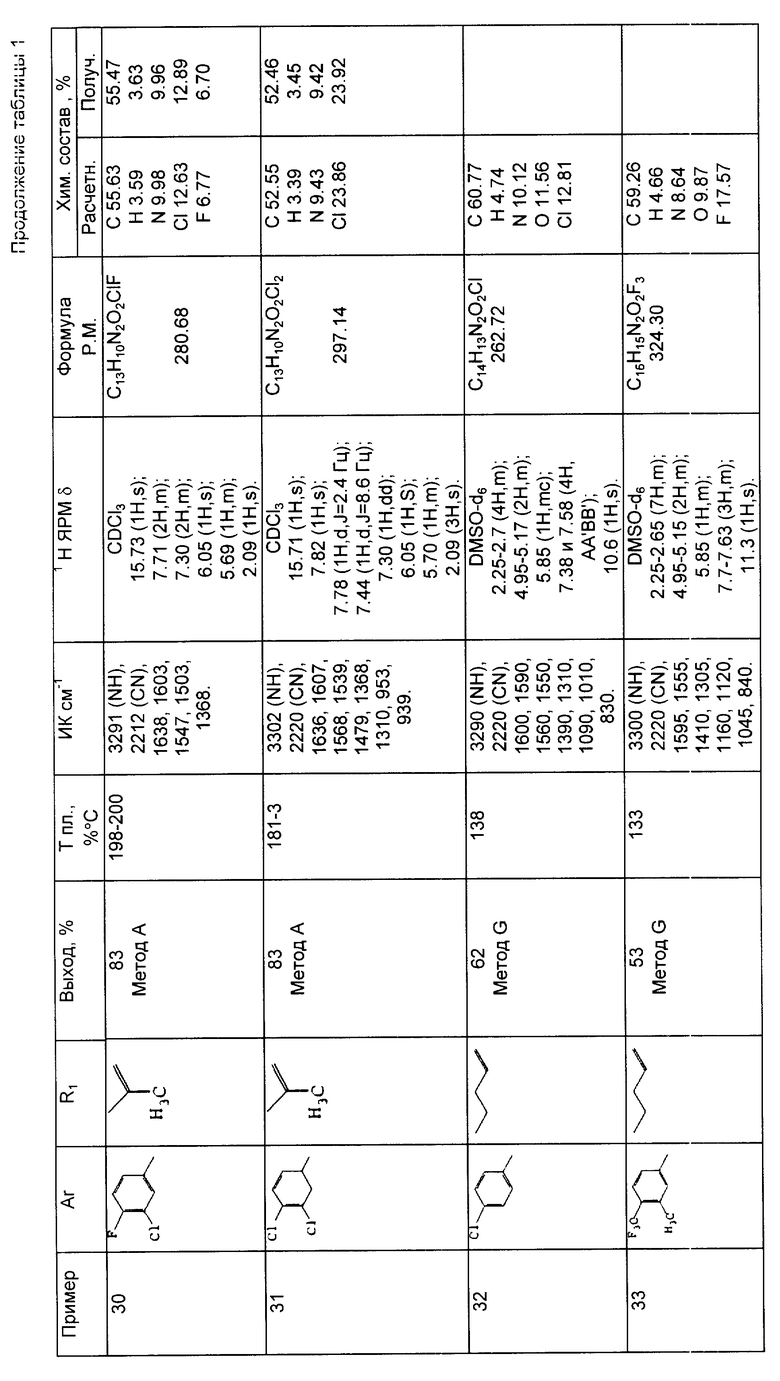
Результаты спектрометрического анализа, выход, температура плавления и результаты микроанализа вышеприведенных соединений приводятся в таблице 1.
Пример 34. Приготовлены были таблетки, отвечающие следующей формуле, мг: соединение примера 1 20; основа для готовой таблетки 150 (экципиенты основы: лактоза, крахмал, тальк, стеарат магния).
Пример 35. Приготовлены были таблетки, отвечающие следующей формуле, мг: соединение примера 2 20; основа для готовой таблетки 150 (экципиенты основы: лактоза, крахмал, тальк, стеарат магния).
Фармакологические результаты.
Методы биохимических тестов.
Тест 1. Отек ножки крысы (PO-R), вызванный каррагенином.
Через 1 ч после введения внутрь тестируемых соединений или контрольного эксципиента группам крыс (количество 6 - 12, самцы CFHB, вес: от 160 до 180 г) вводят в виде инъекции 1 мг каррагенина, растворенного в 0,2 мл рассола, в подушечки фаланг задней правой лапки. В противоположные лапки вводится контрольный солевой раствор. Реакции отека лапок оцениваются по истечении трех часов.
Тест 2. Отек ножки мыши (DTH-M) с аллергической реакцией замедленного типа.
Группы мышей (количество 8 - 10, самцы CD-1, вес: от 25 до 30 г) сенсибилизированы путем введения подкожно 1 мг метилированного сывороточного альбумина крупного рогатого скота (MBSA) в объеме 0,2 мл соляно-эмульсионного раствора комплексного стимулятора Фрэнда (FCA). Отрицательным контрольным группам вводится в виде инъекции соляно-эмульсионный раствор FCA.
Реакции DHT отека лапки оцениваются по истечении 24 ч после введения в подушечки фаланг задней правой лапки 0,1 мг MBSA в объеме 0,05 мл соляного раствора, которое производится на 7-е сутки после сенсибилизации. В противоположные лапки вводится контрольный солевой раствор. Тестируемые соединения или контрольные эксципиенты вводятся внутрь один раз в день на 4-е, 5-е и 6-е сутки и два раза в день на 7-е сутки, за 1 час до и через 6 часов после введения MBSA.
Тест 3-й. Отек ножки крысы (DTH-R) с аллергической реакцией замедленного типа.
Группы крыс (количество 8 - 12, самцы CFHB, вес: от 160 до 180 г) сенсибилизированы путем введения подкожно в основание хвоста объемов 0,1 мл FCA. Отрицательным контрольным группам вводится в виде инъекции некомплексный стимулятор Фрэнда. Реакции DHT отека лапки оцениваются по истечении 24 ч после введения в подушечки фаланг задней правой лапки 0,4 мг антигенного экстракта микобактерий Mycobacterium tuberculosis в объеме 0,2 мл соляного раствора, которое производится на 7-е сутки после сенсибилизации. В противоположные лапки вводится контрольный солевой раствор.
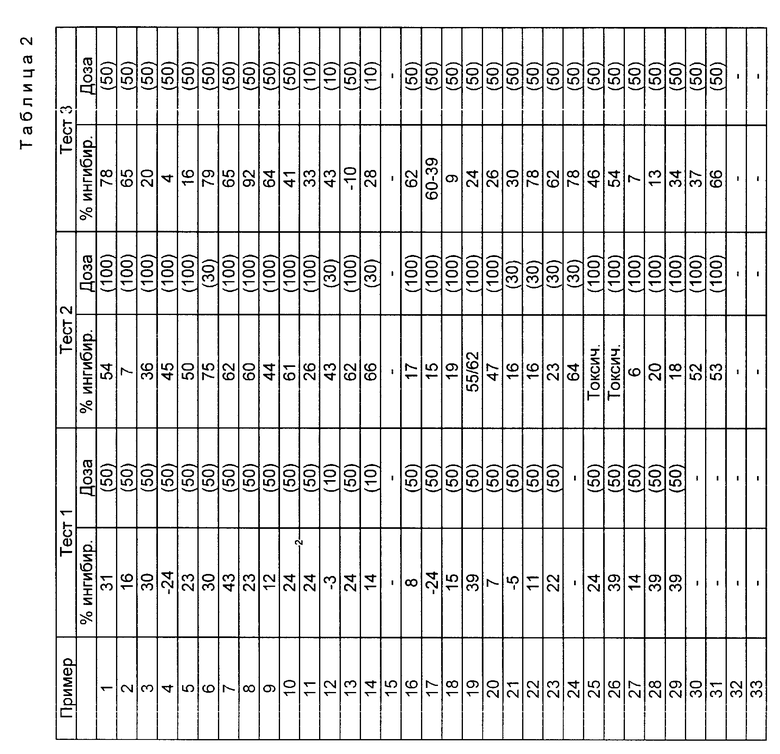
Тестируемые соединения вводятся внутрь один раз в день на 4-е, 5-е и 6-е сутки и два раза в день на 7-е сутки, за 1 ч до и через 6 ч после введения антигенного экстракта. Результаты вышеописанных тестов приводятся в табл.2. Дозировка приводится в единицах мг/кг веса.
Результаты, полученные при наблюдении за ингибированием реакции отека у крысы и мыши по трем описанным тестам, представлены в % ингибирования (значения без знака). Отрицательные значения соответствуют увеличению отека при исследуемой дозе.
Ни одно из соединений табл.2 не показывает отсутствия противовоспалительной активности в трех тестах.
Указание "токсичн." обозначает плохую переносимость соединения при вводимой дозе в данном конкретном тесте, поэтому процент ингибирования воспаления не может быть измерен. Однако, ни одно из соединений не показывает плохой переносимости во всех описанных трех тестах.



Изобретение относится к производным 2-циано-3-гидроксиенамидов формулы I  где R1 представляет группу -C(R13)= C(R14, R15), (-CH2)n, R13)C=C (R14, R15), -C ≡ CH, где R13 - R15 - атом водорода или C1-C3-алкил; n = 1, 2, 3; R2 - атом водорода; R3 - R7 - атом водорода, нитро- или цианогруппа, атом галогена, C1-C3-алкил, группа формулы -(CH2)m-CF3 или -O-(CH2)mCF3, или -S-(CH2)m-CF3, где m = 0 или R3-R7 - группа формулы
где R1 представляет группу -C(R13)= C(R14, R15), (-CH2)n, R13)C=C (R14, R15), -C ≡ CH, где R13 - R15 - атом водорода или C1-C3-алкил; n = 1, 2, 3; R2 - атом водорода; R3 - R7 - атом водорода, нитро- или цианогруппа, атом галогена, C1-C3-алкил, группа формулы -(CH2)m-CF3 или -O-(CH2)mCF3, или -S-(CH2)m-CF3, где m = 0 или R3-R7 - группа формулы 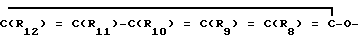 , где R8-R12 - атом водорода, галоген, CF3. Соединения I получают взаимодействием продукта II
, где R8-R12 - атом водорода, галоген, CF3. Соединения I получают взаимодействием продукта II 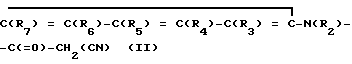 , с гидридом натрия в присутствии катализатора, а затем либо с соединением IIIA: Hal-C(=O)-RA (IIIA), где RA - радикал R1, который защищен. Соединение I, где R1 - группа ((CH2)n, R17)C=C(R14, R15), получают взаимодействием соединения V
, с гидридом натрия в присутствии катализатора, а затем либо с соединением IIIA: Hal-C(=O)-RA (IIIA), где RA - радикал R1, который защищен. Соединение I, где R1 - группа ((CH2)n, R17)C=C(R14, R15), получают взаимодействием соединения V 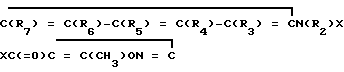 с соединением VI (CXH2)n-1(R13)C=C(R14, R15). Фармацевтическая композиция включает в качестве активного начала соединение I в эффективном количестве и фармацевтически приемлемые разбавители, носители и/или эксципиенты, обладает противовоспалительной активностью. 4 с. и 7 з.п.ф-лы, 2 табл.
с соединением VI (CXH2)n-1(R13)C=C(R14, R15). Фармацевтическая композиция включает в качестве активного начала соединение I в эффективном количестве и фармацевтически приемлемые разбавители, носители и/или эксципиенты, обладает противовоспалительной активностью. 4 с. и 7 з.п.ф-лы, 2 табл.
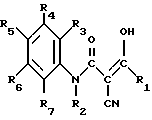
где R1 - группа общей формулы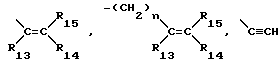
где R1 3 - R15 - водород или C1 - C3-алкил;
n = 1, 2 или 3;
R2 - водород;
R3 - R7, одинаковые или различные, - водород, нитро- или цианогруппа, галоген, C1 - C3-алкил, группа формулы - (CH2)m - CF3, или -O - (CH2)m - CF3, или -S -(CH2)m - CF3, где m = O, или R3 - R7 - группа общей формулы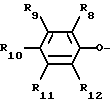
где R8 - R1 2 - водород, галоген, CF3.
R2 - водород или метильный радикал;
R1, R3, R6 и R7 имеют указанные значения.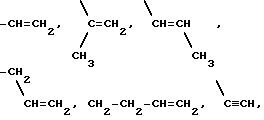
R2 - водород или метильный радикал, R3 - R7, одинаковые или различные, - водород, фтор, хлор или бром, радикал, такой, как метильной, трифторметильный, трифторметоксильный, цианильный, нитро- или феноксильная группа.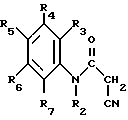
в котором R2 - R7 имеют значения, указанные в формуле I,
вводится последовательно в реакцию с гидридом натрия в присутствии катализатора, а затем а) либо с соединением общей формулы III
в которой Hal - галогенный атом;
R1 имеет значение, указанное в формуле I;
б) либо с соединением общей формулы IIIа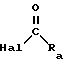
в которой Hal - галогенный атом;
Ra - является радикалом R1, защищенным должным образом для получения продуктов формулы Iа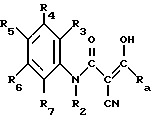
в которой Ra, R2 - R7 имеют указанные значения,
при необходимости защиту полученного таким образом продукта формулы Ia снимают для получения соответствующего продукта формулы I, который выделяют.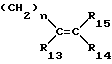
в которой R1 3, R1 4 и R1 5 имеют значения, указанные в формуле I;
n - 2 или 3,
отличающийся тем, что соединения формулы V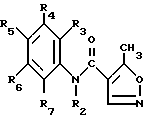
в которой R2 - R7 имеют значение, указанное в формуле I,
вводят в реакцию соответственно с соединением формулы VI
в которой X - легко удаляемая группа, предпочтительно йод;
n, R1 3 - R1 5 имеют указанные значения,
в присутствии сильного основания для получения соответствующего продукта формулы I, который выделяют.
