Предметом изобретения являются энантиомеры ароматических азотсодержащих производных и их солей, а также способ их получения.
Новые соединения могут найти применение в составах для терапевтического использования, особенно при патологических явлениях, которые связаны с системой нейрокининов: боли, аллергия и воспаление, сердечная недостаточность, желудочно-кишечные расстройства, расстройства органов дыхания.
Были описаны эндогенные лиганды с рецепторами нейрокининов, такие как вещество P (SP), нейрокинин A (NKA) (S. J. Bailey et al., Substance P, P. S. Кrabanck ed. Boole Press Dublin, 1983, 16 - 17) и нейрокинин (Б) NKB (S.P. Watson, Life Sciences, 1983, 25, 797 - 808).
Рецепторы нейрокининов в настоящее время классифицированы по трем типам: NK1, NK2 и NK3. Большинство изученных до настоящего времени препаратов обладают несколькими типами рецепторов, например подвздошная шишка морской свинки (NK1, NK2 и NK3), однако некоторые из них имеют только один тип, такие как сонная артерия собаки (NK1), легочная артерия кролика, лишенного эндотелия (NK2), и воротная вена крысы (NK3).
Более точные характеристики различных рецепторов стали известны благодаря синтезу селективных агонистов.
Так, [Sar9, Met - (O2)11SP, [Nle10]NKA4-10 и [Me Phe7] - NKB обладают, по-видимому, селективностью в отношении рецепторов NK1, NK2 и NK3 (D. Regoli, 1988, 1980, указанные выше).
Сейчас найдено, что некоторые ароматические аминосодержащие соединения обладают интересными фармакологическими свойствами, например являются антагонистами рецепторов нейрокининов, и особенно полезны при лечении любой патологии, обусловленной веществом P и нейрокинином.
Таким образом, настоящее изобретение относится к способу получения энантиомеров общей формулы: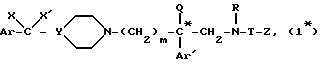
в которой
* - означает, что атом углерода, помещенный таким образом, имеет определенную абсолютную конфигурацию (+) или (-);
m - является целым числом от 2 до 3;
Ar и Ar' представляет собой независимо друг от друга тиенильную группу; фенильную группу, незамещенную, моно- или дизамещенную атомом галогена, алкилом C1 - C3, трифторметилом, алкоксигруппой, в которой алкил является радикалом C1 - C3, гидроксилом, метилендиоксигруппой; имидазолильную группу, причем Ar' может также означать бензотиенильную группу, незамещенную или замещенную галогеном, нафтильную группу, незамещенную или замещенную галогеном, бифенильную группу, индолил, незамещенный или замещенный на азоте бензильной группой;
X представляет собой водород,
X' представляет собой водород, гидроксильную группу или соединен с X'', определенным ниже, с образованием связи углерод-углерод;
или же X и X' образуют вместе группу оксо- или диалкиламиноалкилоксиимино- формулы =N-O-/CH2/p-Am, где p равно 2 или 3, а Am является диалкиламиногруппой, причем каждый алкил может содержать от 1 до 4 атомов углерода;
Y - представляет собой атом азота или группу C(X''),
где
X'' является водородом или, вместе с X' образует связь углерод-углерод;
Q представляет собой водород, алкильную группу C1 - C4 или аминоалкильную группу формулы -/CH2/q - Am',
где q равно 2 или 3, а Am' является пиперидино-, 4-бензилпиперидино- или диалкиламиногруппой, причем каждый алкил может содержать от 1 до 4 атомов углерода;
R представляет собой водород, метильную группу или группу /CH2/n - L, где n является целым числом от 2 до 6, а L является водородом или аминогруппой;
T представляет собой группу, выбранную среди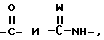
где
W является атомом кислорода или серы;
Z представляет собой либо водород, либо M или OM, когда T представляет собой группу 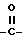 , либо M, когда T представляет собой группу
, либо M, когда T представляет собой группу 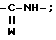
M представляет собой водород или алкил, прямой или разветвленный C1 - C6; фенилалкил, в котором алкильная группа содержит от 1 до 3 атомов углерода, незамещенный, моно- или полизамещенный в ароматическом цикле галогеном, гидроксилом, алкоксигруппой, содержащей от 1 до 4 атомов углерода, алкилом, содержащим от 1 до 4 атомов углерода; пиридилалкил, в котором алкильная группа содержит от 1 до 3 атомов углерода; нафтилалкил, в котором алкильная группа содержит от 1 до 3 атомов углерода, пиридилтиоалкил, в котором алкильная группа содержит от 1 до 3 атомов углерода, стирил, фенил, замещенный /1-метил/-2-имидазолил-тиоалкилом, в котором алкильная группа содержит от 1 до 3 атомов углерода; 1-оксо-3-фенилиндан-2-ил; ароматическую или гетероароматическую группу, незамещенную, моно или полизамещенную;
и их солей с неорганическими или органическими кислотами.
Соли соединений формулы (I) согласно настоящему изобретению включают как соли с неорганическими или органическими кислотами, которые позволяют проводить разделение или кристаллизацию соединений формулы (I), такими как пикриновая кислота, или щавелевая кислота, или оптически активная кислота, например миндальная или камфосульфоновая кислота, так и соли, которые являются фармацевтически допустимыми, такие как хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, малеат, фумарат, 2-нафталинсульфонат, гликолят, глюконат, цитрат, изатионат.
Этот способ заключается в том, что обрабатывают соединение формулы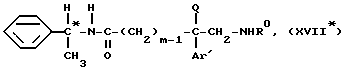
в таком растворителе, как, например, диоксан, в кислой среде, например, в присутствии хлороводородной кислоты, чтобы получить аминокислоту формулы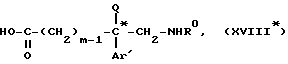
которую этерифицируют в алканоле AlkOH, где Alk является алкилом, содержащим от 1 до 4 атомов углерода, в кислой среде, затем обрабатывают соответствующий сложный эфир формулы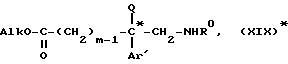
в которой
Alk, O, Ar', R0 и m таковы, как определено выше,
либо при помощи функционального производного кислоты формулы
HO - CO - Z, (III)
либо при помощи изо(тио)цианата формулы
W = C = N - Z, (III')
причем Z и W определены выше, чтобы получить сложный эфир формулы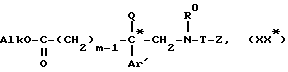
который затем восстанавливают в соответствующий спирт формулы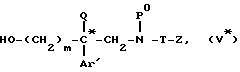
который подвергают взаимодействию с метансульфонилхлоридом с получением мезилата формулы (VI*)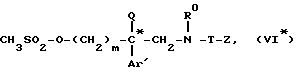
который подвергают взаимодействию с вторичным амином формулы (VII)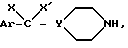
где
Ar, Y, X и X' имеют указанное выше значение, при необходимости удаляют O- и N-защитные группы и получают соединение формулы (I)*, которое при желании переводят в соль.
В качестве функционального производного кислоты (III) используют саму кислоту, активированную подходящим образом, например, при действии циклогексилкарбодиимида или гексафторфосфата бензотриазолил-N-окситрис-диметиламинофосфония (ВОФ), или же одно из функциональных производных, которые реагируют с аминами, например ангидрид, смешанный ангидрид, хлорангидрид или активированный сложный эфир. Когда Z является группой OМ, то соответствующая кислота является угольной кислотой, и в качестве функционального производного используют монохлорангидрид, а именно хлорформиат Cl-CO-OM.
Соединения с формулой (XVII*) известны или могут быть легко получены в соответствии с методом, описанным G. Helmchen et al., Angew. Chem. Int. Ed. Engl., 1979, 1, 18, 65, согласно следующей схеме 3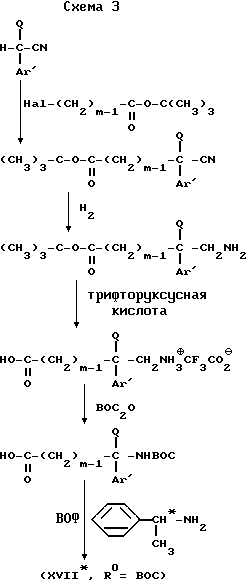 .
.
Полученные таким образом продукты формулы (I*) выделяются в виде свободного основания или соли в соответствии с упомянутыми методами.
Когда соединение с формулой (I*) получается в виде свободного основания, то образование соли осуществляется при действии выбранной кислоты в органическом растворителе. При обработке свободного основания, растворенного, например, в таком спирте, как изопропанол, при помощи раствора выбранной кислоты в том же растворителе, получают соответствующую соль, которая выделяется в соответствии с упомянутыми методами.
Таким образом, получают, например, хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, оксалат, малеат, фумарат, 2-нафталинсульфонат.
По окончании реакции соединения формулы (I*) могут быть выделены в виде одной из их солей, например хлоргидрата или оксалата, в этом случае, если это необходимо, свободное основание может быть получено при нейтрализации указанной соли неорганическим или органическим основанием.
Соединения согласно изобретению подвергались биохимическим и фармакологическим исследованиям.
Соединения формулы (I*) и их соли проявили свойства, противодействующие связыванию вещества P, в опытах, проведенных на мембранах из коры головного мозга крысы и лимфобластических клеток 1 М9, согласно M.A. Cascieri et al., J. Biol. Chem., 1983, 258, 5158-5164 и D.D. Paya et al., J. Immunol., 1984, 133, 3260-3265.
Те же самые соединения и их соли проявили свойства, противодействующие связыванию NKA, в опытах, проведенных на мембранах из двенадцатиперстной кишки крысы, согласно L. Bergstom et al., Mol. Pharmacol., 1987, 32, 764-771.
Те же самые соединения и их соли проявили свойства, противодействующие специфическим агонистам рецепторов NK1, NK2, NK3, в опытах, проведенных на различных выделенных органах, согласно D. Regoli et al., Trends Pharmacol. Sci., 1988, 9, 290-295.
Те же самые соединения и их соли проявили свойства глобальных антагонистов NK1, NK2, NK3 в опытах, проведенных на различных выделенных органах, согласно D. Regoli et al., Trends Pharmacol. Sci., 1988, 9, 290-295 и Pharmacology, 1989, 38, 1-15.
Те же самые соединения и их соли проявили свойства, противодействующие сверхподвижности, вызванной у крысы веществом P, в фармакологических опытах, проведенных Elliot et al., Brain Res., 1986, 381, 68-76.
Свойства, противодействующие слюнотечению, вызванному у крысы веществом P или специфическим агонистом NK1 (/Sar9Met (02) 11/SP), были выявлены в фармакологических опытах, проведенных согласно Takeda Y. and Krause J.E. Proc. Natl. Acad. Sci. USA, 1989, 86, 392-3963.
Обезболивающие свойства были выявлены в фармакологических опытах, проведенных над крысой, больной артритом, согласно U. Kayser et al.; Proceedings of the V-th World Congress on Pain, Dummer R. et al., ed. Elsevier Biomedical Division, 1988, 72-79.
Соединения по настоящему изобретению являются малотоксичными, как раз такая их токсичность совместима с их применением в качестве лекарства. Для такого использования вводят млекопитающим эффективное количество соединения формулы (I*) или одной из его солей, фармацевтически допустимых.
Соединения по настоящему изобретению обычно вводятся в единичных дозах. Указанные единицы дозировки преимущественно сформулированы в фармацевтических составах, в которых активный компонент смешан с фармацевтическими индифферентными веществами.
Соединения формулы (I*) и их соли, фармацевтически допустимые, могут быть использованы с дневными дозами от 0,01 до 100 мг на кг веса тела млекопитающего, подлежащего лечению, преимущественно с дневными дозами от 0,1 до 50 мг/кг. Для человека доза может варьироваться преимущественно от 0,56 до 4000 мг в день, предпочтительнее от 2,5 до 1000 мг, в соответствии с возрастом субъекта, подлежащего лечению, или с типом лечения: профилактическое или для выздоровления.
Фармацевтические составы могут выпускаться для орального, подъязычного, подкожного, внутримышечного, внутривенного, а также для нанесения через кожу, местного или ректального применения.
Соответственно, препаративными формами этих составов могут быть таблетки, капсулы, порошки, гранулы и растворы или суспензии для орального введения, формы подъязычного введения и через рот, формы для введения подкожного, внутримышечного, внутривенного, внутриносового или внутриглазного и формы ректального введения.
Когда готовят твердые составы в форме таблеток, то смешивают основной активный компонент с фармацевтическим носителем, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или их аналоги. Можно покрыть таблетки сахарозой или другими подходящими веществами или еще можно их обработать таким образом, чтобы они имели длительную или замедленную активность и чтобы они высвобождали непрерывно предопределенное количество активного компонента.
Препарат в капсулах получают при смешении активного компонента с разбавителем, приливая полученную смесь в мягкие или твердые капсулы.
Препарат в форме сиропа или эликсира может содержать активный компонент вместе с подслащивающим компонентом, преимущественно бескалорийным, с метилпарабеном и с пропилпарабеном в качестве антисептика, а также с агентом, придающим вкус и с подходящим красителем.
Порошки или гранулы, диспергируемые в воде, могут содержать активный компонент в смеси с диспергирующими агентами, или со смачивающими агентами, или с агентами для перевода в суспензию, например поливинилпирролидон, а также с подслащивающими компонентами или с корректорами вкуса.
Для ректального введения используют свечи, которые готовят со связующими веществами, плавящимися при ректальной температуре, например масло, какао или полиэтиленгликоли.
Для введения парентерального, внутриносового или внутриглазного применяют водные суспензии, солевые изотонические растворы или стерильные растворы, способные к впрыскиванию, которые содержат диспергирующие агенты и/или смачивающие агенты, фармакологически допустимые, например пропиленгликоль или бутиленгликоль.
Активный компонент может быть также использован в виде микрокапсул в случае необходимости с одним или несколькими носителями или добавками.
Следующие примеры иллюстрируют изобретение, не являясь в то же время ограничивающими.
Пример 1. Хоргидрат N-/4-(4-бензил-1-пиперидинил)-2-(3,4- дихлорфенил)-бутил/-2,4-дихлорбензамида /+/SR 47050 А.
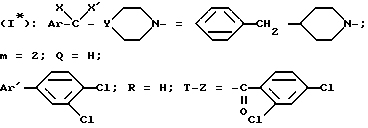
Вращательные способности соединений, приведенных ниже, были измерены при 25oC.
А) N-/1-фенилэтил/- β -/трет-бутоксикарбониламинометил/-3,4- дихлорбензолпропанамид.
Стадия 1
В трехгорлую колбу с объемом 2 л, продуваемую азотом, вносят 39,6 мл диизопропиламина, растворенных в 200 мл безводного ТГФ. Охлаждают до -60oC и прибавляют при этой температуре в следующем порядке:
176 мл 1,6 М расвтора бутиллития в гексане,
50 г 3,4-дихлорбензолацетонитрила в 300 мл ТГФ, затем
39,4 мл трет-бутилбромацетата в 100 мл ТГФ.
Температуру доводят до 0oC за 2 ч 30 мин. Реакционную смесь приливают к 3 л насыщенного водного раствора хлорида аммония. Экстрагируют 2 раза эфиром, сушат над сульфатом магния, выпаривают растворители. Полученную маслянистую жидкость хроматографируют на 1 кг диоксида кремния H. Элюирование осуществляют смесью циклогексан/этилацетат : 95/5. Таким образом получают 44,3 г β -циано-3,4-дихлор-третбутилбензолпропаноата. Т.пл. = 67oC.
Стадия 2
Смесь, содержащая 40 г полученного перед этим продукта (на стадии 1), 700 мл абсолютного этанола, 200 мл концентрированного гидрата окиси аммония (20%) и 3 шпателя с никелем Ренея, перемешивают в атмосфере водорода в течение 5 ч. После фильтрования от катализатора и выпаривания растворителей получают 38,8 г β - аминометил-3,4-дихлор-третбутилбензопропаноата в виде маслянистой жидкости,
Стадия 3
Раствор, содержащий 23,5 г полученного перед этим продукта (на стадии 2) в 150 мл дихлорметана, охлаждают до -10oC. Прибавляют 250 мл трифторуксусной кислоты, температуру доводят до 20oC за 1 ч 30 мин.
После удаления растворителей получают 27 г трифторацетата β - аминометил-3,4-дихлор-бензолпропановой кислоты в виде масляной жидкости.
Стадия 4
К 27 к полученного перед этим продукта (на стадии 3), растворенного в 150 мл воды, добавляют 150 мл диоксана, затем 30 мл триэтиламина, потом раствор, содержащий 23 г дитретбутилдикарбоната в 50 мл диоксана. Нагревают при 100oC в течение 1 ч. Диоксан удаляют под вакуумом и полученный раствор промывают изопропиловым эфиром. Водную фазу приливают к 1,5 л фосфатного буферного раствора (pH 2). После экстрагирования эфиром и сушки над сульфатом магния выпаривают растворители. Полученное жидкое масло кристаллизуется в изопропиловом эфире, что дает 20,3 г β -/трет-бутоксикарбониламинометил/-3,4-дихлорбензолпропановой кислоты.
Стадия 5
К 10 г полученного перед этим продукта (на стадии 4), растворенного в 150 мл дихлорметана, прибавляют в следующем порядке:
8 мл триэтиламина,
3,5 г S/-/- α -метилбензиламина,
14 г БОФ /гексафторфосфата бензотриазолил-N-окситрисдиметиламинофосфония/.
После перемешивания при комнатной температуре в течение 1 ч промывают водой, затем фосфатным буферным раствором (pH 2), потом насыщенным водным раствором бикарбоната натрия. Сушат на сульфате магния, удаляют растворители под вакуумом, в результате чего получают 12 г /N-(1-фенилэтил)/- β -трет-бутоксикрабониламинометил/ -3,4-дихлорбензолпропанамида.
B) Метиловый эфир β -/2,4-дихлорбензоиламинометил/-3,4-дихлорбензолпропановой /+/ кислоты.
Стадия 1
Разделение диастереоизомеров /N-(1-фенилэтил)/- β -/трет-бутоксикарбониламинометил/-3,4-дихлорбензолпропанамида.
Сырой продукт является смесью двух диастереоизомеров. Они могут быть разделены методом тонкослойной хроматографии. Их разделяют препаративным методом хроматографии на 400 г диоксида кремния H при элюировании смесью толуол/этилацетат : 80/20. Менее полярный изомер выходит первым и его собирают в количестве 5,8 г. Т.пл. = 146 - 147oC.
(α)D = -43,6o (c = 1 в хлороформе).
Стадия 2
Раствор, содержащий 5 г полученного перед этим продукта в 10 мл диоксана и 50 мл 6 н хлороводородной кислоты, нагревают при орошении флегмой в течение ночи. После охлаждения раствора его промывают эфиром, затем постепенно нейтрализуют водную фазу твердым бикарбонатом натрия до pH 7. В результате получают осадок, который фильтруют, промывают водой, изопропанолом, затем эфиром. После сушки получают 1,88 г β -аминометил-3,4-дихлорбензолпропановой кислоты. Т.пл. = 202 - 204oC.
Стадия 3
К суспензии, состоящей из 1,85 г полученного перед этим продукта (на стадии 2) в 20 мл метанола, которую охлаждают азотом до -20oC, прибавляют 1,10 мл тионилхлорида, затем дают температуре подняться до 230oC. Спустя 2 ч добавляют 200 мл эфира, фильтруют и промывают эфиром продукт, который затем перекристаллизовывают. После сушки получают 2,15 г β -аминометил-3,4-дихлорбензол-метилпропаноата /-/. Т.пл. = 184 - 186oC.
(α)D = -4,3o (c = 1 в метаноле).
Стадия 4
К охлажденному до 0oC раствору, содержащему 2,0 г полученного перед этим продукта (на стадии 3) и 1,5 г триэтиламина в 20 мл дихлорметана, прибавляют раствор, содержащий 1,54 г 2,4-дихлорбензоилхлорида в 5 мл дихлорметана. Спустя 5 мин раствор концентрируют досуха, добавляют воду и экстрагируют этилацетатом. Затем полученный остаток кристаллизуют в изопропиловом эфире. Получают в результате 2,72 г β -/2,4 дихлорбензоиламиноэтил/-бензолметилпропаноата /+/. Т.пл. = 105 - 107oC.
(α)D = +26,6o (c = 1 в хлороформе).
C) Метиловый эфир β -/2,4-бихлорбензоиламинометил/-3,4-дихлорбензолпропановой /-/ кислоты.
Стадия 1
Поступая, как это описано в примере -B на стадии 1, собирают более полярный изомер при элюировании смесью толуолэтилацетат : 80/20, потом 60/40. Концентрирование фракций приводит к получению 5,4 г /N-(1-фенилэтил)- β -/трет-бутоксикарбониламинометил/-3,4-дихлорбензолпропанамида. Т.пл. = 161 - 162oC.
(α)D = -18,4o (c = 1 в хлороформе).
Стадия 2
Поступая, как это описано в примере 1-B на стадии 2, получают β -аминометил-3,4-дихлорбензолпропановую кислоту. Т.пл.= 202 - 204oC.
Стадия 3
Поступая, как это описано в примере 1-B на стадии 3, получают β -аминометил-3,4-дихлорбензолметилпропаноат /+/. Т.пл.= 184 - 185oC.
(α)D = +3,9o (c = 1 в метаноле).
Стадия 4
Поступая, как это описано в примере 1-B на стадии 4, получают β -/2,4-дихлорбензоиламинометил/3,4-дихлорбензолметилпропаноат /-/. Т. пл. = 108 - 109oC.
(α)D = -27,7o (c = 1 в хлороформе).
D) Восстановление метиловых эфиров β -/2,4-дихлорбензоиламинометил/-3,4-дихлорбензолпропановой кислоты /+/ или /-/.
Приготавливают сначала 0,5 М раствор боргидрата кальция в ТГФ при перемешивании в течение 3 ч суспензии, состоящей из боргидрида натрия (0,1 моль) и хлорида кальция (0,05 моль) в 100 мл ТГФ. Затем прибавляют 13 мл этого раствора к раствору, содержащему 2,5 г метилового эфира β -/2,4-дихлорбензоиламинометил/-3,4-дихлорбензолпропановой кислоты /+/ или /-/ в 20 мл ТГФ. Перемешивают в течение ночи. На следующий день раствор охлаждают до 0oC, затем гидролизуют водой, а потом разбавленной хлороводородной кислотой. После экстрагирования эфиром получают спирт /+/ или /-/, практически чистый, в виде маслянистой жидкости.
E/ Получение мезилатных производных: метансульфоната γ -/2,4-дихлорбензоиламинометил/-3,4-дихлорбензолпропанола /+/ или /-/.
Растворяют 1,3 г полученного перед этим спирта в 30 мл дихлорметана, затем прибавляют к охлажденному до 0oC раствору 0,5 мл триэтиламина и 0,3 мл мезилхлорида. Реакционную смесь перемешивают при 0oC в течение 45 мин, промывают 3 раза ледяной водой, декантируют, сушат над MgSO4, и концентрируют под вакуумом.
Остаток хроматографируют на силикагеле, элюент - этилацетат/пентан: 60/40. Фракции, не содержащие примесей, концентрируют под вакуумом.
Таким образом, получают, исходя из сложного эфира /+/, остаток, который перекристаллизовывают в изопропиловом эфире, что дает 1,1 г метансульфоната γ - /2,4 - дихлорбензоиламинометил/-3,4-дихлорбензолпропанола - /+/. Т. пл. = 74 - 77oC.
(α)D = +21,2o (с = 1 в хлороформе).
Таким же образом получают, исходя из сложного эфира /-/, проводя реакцию, как описано выше, метансульфонат γ /-/ 2,4-дихлорбензоиламинометил/ - 3,4 - дихлорбензолпропанола /-/. Т.пл. = 72 - 76oC.
(α)D = - 22,5o (с = 1 в хлороформе).
F/ Получение хлоргидрата N-/4-(4-бензил-1-пиперидинил)-2-(3,4-дихлорфенил)- бутил/2,4-дихлорбензамида /+/ SR 47050 A.
Растворяют 0,6 г полученного перед этим мезилата /+/ и 0,54 г - 4-бензилпиперидина в 1 мл диметилформамида и реакционную смесь нагревают при 60oC в течение 30 мин. Прибавляют воду и экстрагируют этилацетатом. Органическую фазу концентрируют под вакуумом и остаток хроматографируют на силикагеле, элюент - дихлорметан/метанол: 97/3.
Фракции с чистым продуктом концентрируют под вакуумом, остаток разбавляют дихлорметаном, а добавление хлорристоводородного эфира позволяет получить хлоргидрат. m = 0,5 г.
(α)D = +14,0o (с = 1 в хлороформе).
Пример 2. Хлоргидрат N-/4-(4-бензил-1-пиперидинил)-2-(3,4-дихлорфенил)-бутил/-2,4- дихлорбензамида /-/. SR 47051 A.
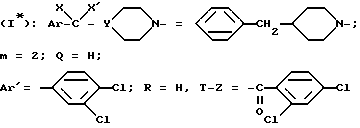
Поступая тем же способом, как указано выше (в соответствии с примером 1 F), но используя в качестве исходного продукта изомер /-/ мезилата, получают целевой продукт SR = 47051 A.
(α)D = -14,5o (с = 1 в хлороформе).
Пример 3. Поступая, как это описано в приведенном выше примере 1, получают
хлоргидрат N-/4-(4-бензил-1-пиперидинил)-2-(3,4-дифторфенил) - бутил/-2,4-дихлорбензамида /-/. SR 47243 A.
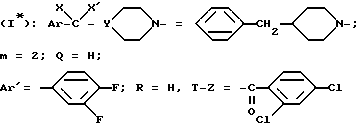
(α)D = -8,5o (с = 1 в хлороформе).
Пример 4. Поступая, как это описано в приведенном выше примере, получают
хлоргидрат N-/4-(4-бензил-1-пиперидинил)-2-(3,4-фторфенил)-бутил/- 2,4-дихлорбензамида /+/. SR 47238 A.
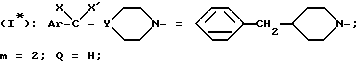
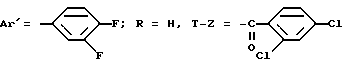
(α)D = + 7,3o (c = 1 в хлороформе).
Пример 5. Хлоргидрат N-/4-(4-бензил-1-пиперидинил)-2- (3,4-дихлорфенил)-бутил/-4-фтор-1-нафталинкарбоксамида /+/ и /-/.
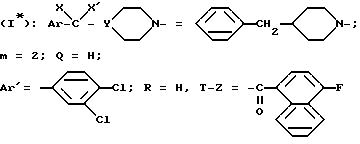
Стадия 1
1-амино-2-/3,4-дихлорфенил/-4-гидрокси бутан.
Прибавляют 150 мл насыщенного раствора хлористоводородного эфира к 149 г 4-/2-тетрагидропиранилокси/-2-/3,4-дихлорфенил/-1-амино бутана, растворенного в 700 мл метанола. Смесь перемешивают в течение получаса при комнатной температуре, концентрируют под вакуумом и остаток извлекают посредством 500 мл воды и промывают эфиром. Водную фазу подщелачивают раствором гидроксида натрия и экстрагируют 2 раза дихлорметаном. Органические фазы сушат над MgSO4, фильтруют и концентрируют под вакуумом. Остаток извлекают посредством 400 мл изопропилового эфира и смесь перемешивают в течение одного часа при комнатной температуре. Осадок фильтруют и промывают эфиром. m = 98,2 г. Т. пл. = 90 - 91oC.
Стадия 2
Энантиомер /+/ 1-амино-2-/3,4-дихлорфенил/-4-гидроксибутана.
К нагретому до образования флегмы раствору, содержащему 59, 65 г винной кислоты Д /-/ в 2 л метанола, прибавляют 93 г рацемической смеси, полученной перед этим в соответствии со стадией 1 и растворенной в 300 мл метанола. Температуру доводят до комнатной, фильтруют кристаллы, промывают метанолом и сушат под вакуумом при 50oC на P2O5. m= 64,8 г.
(α)D = -5,2 (c = 1 в воде).
Затем перекристаллизовывают из 2,96 л метанола, фильтруют кристаллы, промывают их в метаноле и сушат под вакуумом при 50oC на P2O5. m = 45,3 г.
(α)D = -4,5o (c= 1 в воде). Т.пл. = 201oC.
Тартрат Д /-/ извлекают посредством 250 мл воды, смесь подщелачивают концентрированным раствором гидроксида натрия и экстрагируют 3 раза 200 мл дихлорметана, промывают насыщенным раствором хлорида натрия, декантируют, сушат над MgSO4, фильтруют и концентрируют под вакуумом. Остаток извлекают изопропиловым эфиром, смесь перемешивают в течение одного часа при комнатной температуре, кристаллы фильтруют и промывают изопропиловым эфиром. m= 24,7 г
(α)D = +9,0o (c =1 в метаноле). Т.пл. = 79 - 80oC.
Энантиомер /-/ 1-амино-2-/3,4-дихлорфенил/-4-гидрокси-бутана.
Поступая, как указано выше, и используя L/ +/ винную кислоту, получают энантиомер /-/.
(α)D = -9,2o (c = 1 в метаноле). Т.пл. = 79-80oC.
Стадия 3
N-/2-(3,4- дихлорфенил)-4-мезилоксибутил/4-фтор-1- нафталинкарбоксамид/ энантиомер (-)/.
Раствор, содержащий 4,45 г хлорангидрида 4-фторнафтойной кислоты в 50 мл дихлорметана, прибавляют по каплям при -60oC к раствору, содержащему 5 г полученного перед этим продукта (энантиомера (+)) в 100 мл дихлорметана в присутствии 2,6 г триэтиламина. Перемешивают смесь в течение 15 мин при -60oC и доводят затем температуру до -30oC. Затем добавляют 2,5 г триэтиламина и 2,7 г мезилхлорида и температуру доводят до комнатной. Промывают водой, сушат органическую фазу над MgSO4 и концентрируют под вакуумом. Остаток хроматографируют на силикагеле, элюент : дихлорметан/метанол 99,5/ 0,5.
Фракции, не содержащие примесей, концентрируют под вакуумом. m = 8,4 г.
(α)D = -22,8 (c = 1 в метаноле).
N-/2-(3,4-дихлорфенил)-4-мезилоксибутил/-4-фтор-1- нафталинкарбоксамид/энантиомер (+)/.
Энантиомер /+/ получают согласно условиям, описанным на стадии 3, но используя энантиомер /-/, полученный на стадии 2.
(α)D = +22,7o (c = 1 в метаноле).
Стадия 4
Хлоргидрат N-/4-(4-бензил-1-пиперидинил)-2-(3,4-дихлорфенил) - бутил/-4-фтор-1-нафталинкарбоксамида. Энантиомер /-/ SR 48225 A.
Растворяют 7 г полученного на стадии 3 энантиомера /-/ и 5,02 г 4- бензилпиперидина в 15 мл диметилформамида и нагревают реакционную смесь при 70oC в течение 2 ч. Смесь приливают к воде экстрагируют этилацетатом, органические фазы промывают насыщенным раствором хлорида натрия, сушат над MgSO4 и концентрируют под вакуумом. Остаток хроматографируют на силикагеле, элюент - дихлорметан/ метанол : 97/3.
Фракции с чистым продуктом концентрируют под вакуумом, остаток разбавляют дихлорметаном, а добавление хлористоводородного эфира позволяет получить хлоргидрат. m= 6,2 г.
(α)D = -35,5o (c = 1 в метаноле).
Энантомер /+/. SR 48226 A.
Поступая тем же образом, как и для получения энантиомера /-/, но исходя из энантиомера /+/, полученного на стадии 3, получают энантиомер /+/. m = 7 г.
(α)D = + 36,0o (c = 1 в метаноле).
1.Фармакологические исследования in vitro
A. Сродство соединений к рецепторам нейрокининов различных мембранных препаратов.
Сродство соединений к рецепторам нейрокининов было выявлено в результате следующих опытов:
ингибирование связывания вещества P с его рецептором на мембранах коры головного мозга крысы и лимфобластических клеток человека (IM9) с использованием в качестве лиганда 1251 - вещество P (согласно Cascieri M.A., и Lang T., J.Biol. Chem., 258: 5158-5164 (1983); Payan D.G, Brewster D.R., и Gaetrl, F. J., J.Immunol., 133: 3260-3265 (1984));
ингибирование связывание нейрокинина - A с его рецептором в опытах на мембранах двенадцатиперстной кишки крысы с использованием в качестве лиганда 125I - нейрокинин- A, (согласно Berstrom и др. Molecular Pharmacol., 32: 764-771 (1987));
ингибирование связывания нейрокинина B с его рецептором на мембранах из коры головного мозга крысы с использованием в качестве лиганда 125I - эледуазина (согласно Cascieri M.A. и др. J.Biol.Chem., 260: 1501-1507 (1985)).
Активность соединений выражена в % ингибирования при концентрации 10 мкМ и в Ki(нМ), рассчитанном из IC50 (согласно Burt, D.R., in Receptor Binding in drug research, Clinical Pharmacology 5, O'Brien R.A., изд. Marcel Dekker, Inc., Нью-Йорк, 3-29 (1986)).
Результаты
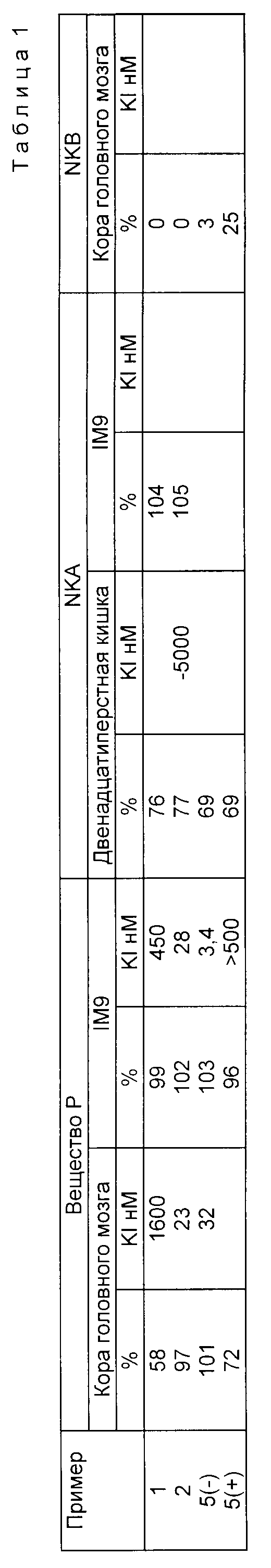
В табл.1 представлены значения % ингибирования при 10 мкМ и К; (нМ) для различных исследуемых соединений. Результаты показывают, что эти соединения обладают сродством к рецепторам нейрокининов, особенно к рецептору вещества - P, и в меньшей степени к рецептору нейрокинина - A (табл.1).
B. Антагонистические свойства соединений по отношению к рецепторам нейрокининов различных выделенных органов. Рецепторы нейрокининов были выявлены на многочисленных препаратах и классифицированы на три типа; NK1, NK2 и NK3.
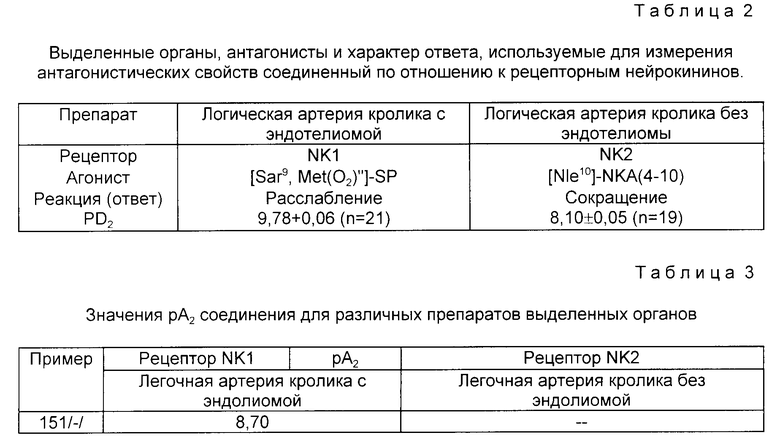
Антагонистические свойства соединений были исследованы в соответствии с методикой: Regoli, D. и др., Trends Pharmacol. Sci., 9:290-295 (1988). Regoli, D. и др. Pharmacology, 38:1-15 (1989) с использованием препаратов и агонистов, приведенных в табл.2. Антагонистические свойства соединений характеризуются величиной pA2. Значения pA2 были подсчитаны по методу: Van Rossum, J.M., Arch. Int. Pharmacodym. Ther., 143: 299-330 (1963).
Результаты.
В табл.3 представлены значения pA2 для различных исследуемых соединений. Результаты исследований показывают, что эти соединений являются антагонистами нейрокининов, в особенности на уровне рецептора NKI, и в меньшей степени на уровне рецепторов NK2.
Таблица 2
Выделенные органы, агонисты и характер ответа, используемые для измерения антагонистических свойств соединений по отношению к рецепторам нейрокининов.
II. Фармакологические исследования in vivo
A. Модель нейрогенного воспаления
Раздражающий препарат капсаицин вызывает выделение целого набора нейромедиаторов, среди которых находятся нейрокинины и, в частности, вещество P. Местное наложение капсаицина на ухо или лапу мыши провоцирует нейрогенное воспаление, что вызывает определенные поведенческие реакции: почесывание уха или облизывание лапы. На данной модели соединение примера 1(-) препятствует появлению этих поведенческих реакций при интраперитональном введении в дозе 5 мг/кг (почесывание) или при оральном введении в дозе 20 мг/кг (облизывание).
B. Модель сердечно-сосудистой системы
Внутривенное введение животному вещества P вызывает у него гипотонию (см. Regoli D. и др. TIPS, 9:290-295 (1988) и Maggi C.A. и др. J.Autton Pharmacol., 7:11-32 (1987)).
Моделируют гипотонию у кролика введением 5 нг/кг вещества P согласно методу Maggi C.A. и др. J.Autton Pharmacol., 1987. Соединение примеров I(-) ингибирует гипотонию при их внутривенном введении в дозе 5 мг/кг.
C. Модель центральной нервной системы
Крысе вводят интрацеребравентрикулярным путем (ИВВ) 10 мг вещества P, вызывающего у нее сверхподвижное поведение (см. Elliott, P.J. и Iversem, S. D., Brain Res, 381: 68-76 (1986)).
I. Интрацеребровентрикулярное введение крысе 2 мкг вещества P ингибирует позыв к утолению жажды, вызванной различными факторами, такими как подкожное вливание раствора NaCl, ИЦВ - вливание карбахолина или лишение питья (см. De Caro и др. J.Physiology, 279:133-140 (1978), De Caro и Massi Peptides, 6 : 181-185 (1985), Cautalamessa и др.J.Physiology, 79:524-530 (1984)).
В данной модели соединения примера 5(-), вводимое интерперитонально с дозировкой 10 мг/кг, ослабляет действие вещества P в случае, когда жажда вызвана лишением питья.
Использование: в медицине для лечения аллергии, сердечной недостаточности, желудочно-кишечных расстройств. Сущность изобретения: продукты: соединения ф-лы (1*), где * означает, что помеченный таким образом атом углерода имеет определенную (+) или (-) абсолютную конфигурацию, Ar - фенил, Х и Х' - водород, Y - группа СН, m -2 или 3, Ar'-фенил, замещенный одним или двумя атомами галогена, Q - водород, R - водород Т-С(О), Z - фенил или нафтил, замещенные одним или двумя атомами галогена или их соли. Реагент 1: соединение ф-лы (II), где Ro означает атом водорода или трет.-бутилкарбонильную N - защитную группу, которое подвергают кислотному гидролизу, образующийся продукт этерифицируют, образующийся эфир подвергают взаимодействию с функциональным производным кислоты для получения эфира, который подвергают восстановлению, образующийся спирт обрабатывают хлоридом метансульфонила, с последующим взаимодейст- вием образующегося продукта с амином ф-лы (III). 2 с. и 5 з. п. ф-лы, 3 табл.
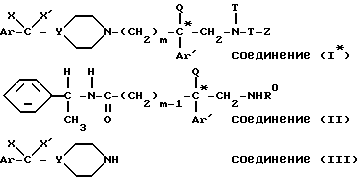 с
с
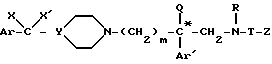
где * означает, что помеченный таким образом атом углерода имеет определенную (+) или (-) абсолютную конфигурацию,
Ar - фенил;
X X' - водород;
Y - группа CH;
m = 2 или 3;
Ar' - фенил, замещенный одним или двумя атомами галогена;
Q - водород;
R - водород;
T - группа C(O);
Z - фенил или нафтил, замещенные одним или двумя атомами галогена;
и их соли с неорганическими или органическими кислотами.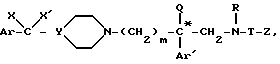
где * означает, что атом углерода, помеченный таким образом, имеет определенную (+) или (-) абсолютную конфигурацию;
Ar, X, X', Y, Q, Ar', R, T, Z и m имеют значения по п.1,
или одной из их солей с неорганическими или органическими кислотами, отличающийся тем, что соединение общей формулы XVII*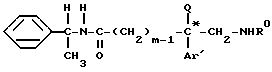
где Ro - водород или трет-бутилкарбонильная N-защитная группа,
подвергают кислому гидролизу в растворителе для получения аминокислоты формулы XVIII*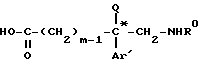
которую этерифицируют в алканоле AlkOH, где Alk-C1-C4-алкил, в кислой среде, образующийся эфир формулы XIX*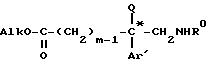
подвергают взаимодействию с функциональным производным кислоты формулы III
HO-CO-Z
для получения эфира общей формулы XX*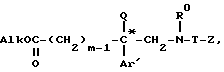
который подвергают восстановлению, образующийся спирт формулы V*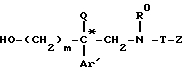
обрабатывают хлоридом метансульфонила с последующим взаимодействием образующегося продукта общей формулы VI*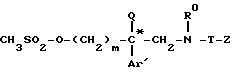
с амином общей формулы VII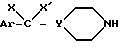
где Ar, X, X' и Y имеют значения по п.1,
снятием в случае необходимости защиты и выделением продукта формулы (I*) или в случае необходимости превращением его в одну из солей.
| EP, заявка, 0226516, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 0261842, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 0288352, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, з аявка, 0308328, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
