Изобретение относится к новым замещенным гетероциклическим соединениям, способу получения указанных соединений и фармацевтическим композициям, содержащим их в качестве активного вещества.
Настоящее изобретение, в частности, относится к новому классу замещенных гетероциклических соединений, предназначенных для лечения патологических состояний, вызываемых тахикининами, таких как болевые ощущения (D. Regoli et al. , Life Sciences, 1987, 40, 109-117), аллергия и воспаление (J.E. Morlay et al. , Life Sciences, 1987, 41, 527-544), недостаточность кровообращения (J. Losay et al., 1977, Substance P, Von Euler, I.S. and Pernow ed., 287-293, Raven Press, New York), нарушения пищеварительной системы (D. Regoli et al. , Trends Pharmacol. Sci., 1985, 6, 481-484), нарушения органов дыхания (J. Mizrahi et al., Pharmacology, 1982, 25, 39-50), неврологические нарушения и нейропсихиатрические расстройства (C. A. Maggi et al., J. Autonomic Pharmacol., 1993, 13, 23-93), причем эти примеры не ограничивают объем изобретения и не исключают другие заболевания.
За последние годы были проведены многочисленные исследования тахикининов и их рецепторов. Тахикинины широко присутствуют как в центральной нервной системе, так и в периферической нервной системе. Обнаруженные рецепторы тахикинина классифицированы на три типа: NK1, NK2, NK3. Вещество P (SP) является эндогенным лигандом рецепторов NK1, нейрокинин A (NKA) является эндогенным лигандом рецепторов NK2 и нейрокинин B (NKB) является эндогенным лигандом рецепторов NK3.
Рецепторы NK1, NK2 и NK3 обнаружены в разных видах животных. В обзоре C. A. Maggi et al. рассматриваются рецепторы тахикинина и их антагонисты, описываются фармакологические исследования и применение полученных препаратов для лечения людей (J. Autonomic Pharmacol., 1933, 13, 23-93).
В качестве антагонистов, специфичных для рецепторов NK1, можно указать следующие непептидные соединения: CP-96345 (J. Med. Chem., 1992, 35, 2591-2600), RP-68651 (Proc. Natl. Acad. Sci. USA, 1991, 88, 10208-10212), SR 140333 (Curr. J. Pharmacol., 1993, 250, 403-413).
Подробно описан непептидный селективный антагонист, SR 48968, для рецептора NK2 (Life Sci., 1992, 50 PL101-PL106).
Что касается рецептора NK3, то описаны определенные непептидные соединения, обладающие сродством с рецептором NK3 мозга крыс и морских свинок (FASEB J. , 1993, 7 (4), A710-4104); описан также пептидный антагонист, [Trp7, β-Ala8] NKA, который обладает слабой специфичностью для рецептора NK3 крыс (J. Autonomic Pharmacol., 1993, 13, 23-93).
В заявке на европейский патент N 336230 описываются производные пептида, которые представляют собой антагонисты вещества P и нейрокинина A, пригодные для лечения и профилактики астмы.
В заявках на международный патент N 90/05525, N 90/05729, N 91/09844 и N 91/18899 и в заявках на европейский патент N 0436334, N 0429466 и N 0430771 описываются антагонисты вещества P.
В заявках на европейский патент N 0474561, N 512901, N 515240, N 559538 и N 591040 также рассматриваются антагонисты рецепторов нейрокинина.
Обнаружены новые замещенные гетероциклические соединения, которые являются антагонистами рецепторов нейрокинина.
Таким образом, одним объектом настоящего изобретения являются соединения формулы
где A является двухвалентным радикалом, выбираемым из
A1) -O-CO-
A2) -CH2-O-CO
A3) -O-CH2-CO
A4) -O-CH2-CH2-
A5) -N(R1)-CO-
A6) -N(R1)-CO-CO-
A7) -N(R1)-CH2-CH2-
A8) -O-CH2-
где R1 обозначает водород или (C1-C4)-алкил;
m равен 2 или 3;
Ar1 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил, трифторметил и метилендиокси, причем указанные заместители являются одинаковыми или разными; тиенил, который не замещен или замещен атомом галогена; бензотиенил, который не замещен или замещен атомом галогена; нафтил, который не замещен или замещен атомом галогена; индолил, который не замещен или N-замещен (C1-C4)-алкилом или бензилом; имидазолил, который не замещен или замещен атомом галогена; пиридил, который не замещен или замещен атомом галогена; или бифенил;
T обозначает группу, выбираемую из CH2-Z, -CH(C6H5)2 и -C(C6H5)3; T может также быть группой -CO-B-Z, если A является двухвалентным радикалом, выбираемым из -O-CH2-CH2-, -N(R1)-CH2-CH2- и -O-CH2-;
B обозначает прямую связь или метилен;
Z обозначает возможно замещенную моно-, ди- или трициклическую ароматическую или гетероароматическую группу; и
Am обозначает:
i - либо группу Am1 формулы
где J1 является:
i1 - или группой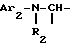
где Ar2 обозначает пиридил; фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил, трифторметил, нитро и метилендиокси, причем указанные заместители являются одинаковыми или разными; тиенил; пиримидил; или имидазолил, который не замещен или замещен (C1-C4)-алкилом; и
R2 обозначает водород; (C1-C7)-алкил; бензил; формил; или (C1-C7)-алкилкарбонил;
i2 - или группой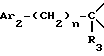
где Ar2 имеет указанные выше значения;
n = 0 или 1; и
R3 является группой, выбираемой из:
(1) водорода;
(2) (C1-C7)-алкила;
(3) формила;
(4) (C1-C7)-алкилкарбонила;
(5) циано;
(6) -(CH2)q-OH;
(7) -(CH2)q-O-(C1-C7)-алкила;
(8) -(CH2)q-OCHO;
(9) -(CH2)q-OCOR17;
(10) -(CH2)q-OCONH-(C1-C7)-алкила;
(11) -NR4R5;
(12) -(CH2)q-NR6C(=W1)R7;
(13) -(CH2)q-NR6COOR8;
(14) -(CH2)q-NR6SO2R9;
(15) -(CH2)q-NR6C(=W1)NR10R11;
(16) -CH2-NR12R13;
(17) -CH2-CH2-NR12-R13;
(18) -COOH;
(19) (C1-C7)-алкоксикарбонила;
(20) -C(=W1)NR10R11;
(21) -CH2-COOH;
(22) (C1-C7)-алкоксикарбонилметила;
(23) -CH2-C(=W1)NR10R11;
(24) -O-CH2CH2-OR18;
(25) -NR6COCOR19;
(26) -CO-NR20-NR21R22;
(27)
(28)
или R3 образует двойную связь между атомом углерода, к которому он присоединен, и смежным атомом углерода пиперидинового кольца;
q равен 0, 1 или 2;
W1 обозначает атом кислорода или атом серы;
R4 и R5 независимо друг от друга обозначают водород или (C1-C7)-алкил; R5 может также быть (C3-C7)-циклоалкилметилом, бензилом или фенилом; либо R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина, пергидроазепина и пиперазина, который не замещен или замещен в 4-положении (C1-C4)-алкилом;
R6 обозначает водород или (C1-C7)-алкил;
R7 обозначает водород; (C1-C7)-алкил; винил; фенил; бензил; пиридил; (C3-C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами; фурил; тиенил; пирролил или имидазолил;
либо R6 и R7 вместе обозначают группу -(CH2)p-;
p равен 3 или 4;
R8 обозначает (C1-C7)-алкил или фенил;
R9 обозначает (C1-C7)-алкил; аминогруппу, которая является свободной или замещена одним или двумя (C1-C7)-алкилами; или фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, (C1-C7)-алкил, трифторметил, гидроксил, (C1-C7)-алкокси, карбоксил, (C1-C7)-алкоксикарбонил, (C1-C7)-алкилкарбонилокси, циано, нитро и аминогруппу, которая является свободной или замещена одним или двумя (C1-C7)-алкилами, причем указанные заместители являются одинаковыми или разными;
R10 и R11 независимо друг от друга обозначают водород или (C1-C7)-алкил; R11 может также быть (C3-C7)-циклоалкилом, (C3-C7)-циклоалкилметилом, гидроксилом, (C1-C4)-алкокси, бензилом или фенилом; либо R10 и R11 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина и пергидроазепина;
R12 и R13 независимо друг от друга обозначают водород или (C1-C7)-алкил; R13 может также быть (C3-C7)-циклоалкилметилом или бензилом;
R17 обозначает (C1-C7)-алкил; (C3-C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами; фенил или пиридил;
R18 обозначает водород; (C1-C7)-алкил; формил или (C1-C7)-алкилкарбонил;
R19 обозначает (C1-C4)-алкокси;
R20 обозначает водород или (C1-C7)-алкил;
R21 и R22 независимо друг от друга обозначают водород или (C1-C7)-алкил;
либо R21 и R22 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из пирролидина, пиперидина и морфолина;
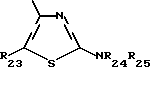
R23 обозначает водород или (C1-C7)-алкил; и
R24 и R25 независимо друг от друга обозначают водород или (C1-C7)-алкил; R25 может также быть формилом или (C1-C7)-алкилкарбонилом;
i3 - либо группой
где R3 имеет указанные выше значения;
R14 обозначает (C1-C7)-алкил или (C3-C7)-циклоалкил; R14 может также быть группой -CONR15R16, если R3 обозначает водород, или группой -NR15R16, если R3 обозначает водород, циано, карбоксил, (C1-C7)-алкоксикарбонил или группу -C(=W1)NR10R11; и
R15 и R16 независимо друг от друга обозначают (C1-C7)-алкил; либо R15 и R16 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина и пергидроазепина;
ii - либо группу Am2 формулы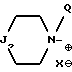
где J2 является:
ii2 - или группой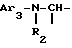
где Ar3 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными; и
R2 имеет значения, указанные выше для J1;
ii2 - или группой
где Ar3 имеет указанные выше значения;
n равен 0 или 1;
Q обозначает (C1-C6)-алкил или бензил, причем указанный заместитель занимает аксиальное положение или экваториальное положение; и
X⊖ является анионом;
iii - либо группу Am3 формулы
где Ar2 имеет указанные выше значения;
n равен 0 или 1; и
X⊖ является анионом;
iv - либо группу Am4 формулы
где Ar2 имеет указанные выше значения;
n равен 0 или 1; и
X⊖ является анионом;
и при желании их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Соединения формулы I по настоящему изобретению включают также рацематы, оптически чистые изомеры, а также аксиальные и экваториальные изомеры, если Am является Am2 в соединении формулы I.
Радикал Z, в частности, может представлять собой фенильную группу, которая может быть не замещена или может иметь один или несколько заместителей.
Если Z является фенильной группой, она может быть моно- или дизамещена, особенно в 2,4-положении, но также, например, в 2,3-, 4,5-, 3,4- или 3,5-положении; она может также быть трехзамещенной, особенно в 2,4,6-положении, но также, например, в 2,3,4-, 2,3,5-, 2,4,5- или 3,4,5-положении, тетразамещенной, например, в 2,3,4,5-положении, или пентазаместителей.
Радикал Z может также быть бициклической ароматической группой, такой как 1- или 2-нафтил либо 1-, 2-, 3-, 4-, 5-, 6- или 7-инденил, в котором одна или несколько связей могут быть гидрированы, причем указанные группы могут быть не замещены или могут иметь один или несколько заместителей, таких как алкил, фенил, циано, гидроксиалкил, гидроксил, оксо, алкилкарбониламино, алкоксикарбонил, тиоалкил, галоген, алкокси или трифторметил, где алкилами являются C1-C4-алкилы.
Радикал Z может также представлять собой пиридил, тиадиазолил, индолил, индазолил, имидазолил, бензимидазолил, бензотриазолил, бензофуранил, бензотиенил, бензотиазолил, бензизотиазолил, хинолил, изохинолил, бензоксазолил, бензизоксазолил, бензоксазинил, бензодиоксинил, изоксазолил, бензопиранил, тиазолил, тиенил, фурил, пиранил, хроменил, изобензофуранил, пирролил, пиразолил, пиразинил, пиримидинил, пиридазинил, индолизинил, фталазинил, хиназолинил, акридинил, изотиазолил, изохроманил или хроманил, в которых одна или несколько двойных связей могут быть гидрированы, причем указанные группы могут быть не замещены или могут иметь один или несколько заместителей, таких как алкил, фенил, циано, гидроксиалкил, гидроксил, алкилкарбониламино, алкоксикарбонил или тиоалкил, где алкилами являются C1-C4-алкилы.
Данное изобретение относится, в частности, к соединениям формулы I, в которой:
Z является Z' и обозначает:
- фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена; трифторметил; циано; гидроксил; нитро; фенил, который не замещен либо моно- или полизамещен галогеном, трифторметилом, (C1-C4)-алкилом, гидроксилом или (C1-C4)-алкокси, причем указанные заместители являются одинаковыми или разными; аминогруппу, которая не замещена, либо моно- или полизамещена (C1-C4)-алкилом; бензиламино; карбоксил; (C1-C10)-алкил; (C3-C8)-циклоалкил, который не замещен либо моно- или полизамещен метилом; (C1-C10)-алкокси; (C3-C8)-циклоалкокси, который не замещен либо моно- или полизамещен метилом; меркапто; (C1-C10)-алкилтио; формилокси; (C1-C6)-алкилкарбонилокси; формиламино; (C1-C6)-алкилкарбониламино; бензоиламино; (C1-C4)-алкоксикарбонил; (C3-C7)-циклоалкоксикарбонил; (C3-C7)-циклоалкилкарбонил, карбамоил, который не замещен либо моно- или дизамещен (C1-C4)-алкилом; уреидо, который не замещен либо моно- или дизамещен в 3-положении (C1-C4)-алкилом или (C3-C7)-циклоалкилом; и (пирролидин-1-ил)карбониламино, причем указанные заместители являются одинаковыми или разными;
- нафтил, который не замещен либо моно- или полизамещен галогеном, трифторметилом, (C1-C4)-алкилом, гидроксилом или (C1-C4)-алкокси; или
- пиридил, тиенил, индолил, хинолил, бензотиенил или имидазолил.
Можно получить соли соединений формулы I, которые не являются солями четвертичного аммония. Эти соли включают соли, получаемые при взаимодействии с минеральными и органическими кислотами, обеспечивающими необходимое отделение или кристаллизацию соединений формулы I, такими как пикриновая кислота, щавелевая кислота или оптически активная кислота, например миндальная или камфорсульфоновая кислота, а также с минеральными и органическими кислотами, образующими фармацевтически приемлемые соли, такие как гидрохлорид, гидробромид, сульфат, бисульфат, первичный кислый фосфат, метансульфонат, метилсульфат, малеат, фумарат, нафталин-2-сульфонат, бензолсульфонат, глюконат, цитрат, изэтионат или паратолуолсульфонат.
Анионами X- являются такие анионы, которые обычно используют для образования соли с ионами четвертичного аммония и предпочтительно представляют собой ионы хлорида, бромида, йодида, ацетата, бисульфата, метансульфоната, паратолуолсульфоната и бензолсульфоната.
Предпочтение отдается фармацевтически приемлемым анионам, например хлориду, метансульфонату или бензолсульфонату.
В соответствии с настоящим изобретением алкильные или алкоксильные группы являются группами с нормальными или разветвленными цепями; атомом галогена является атом хлора, брома, фтора или йода.
В заместителях группы Z = фенил заместитель (C1-C10)-алкил означает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил или н-пентил, гексил или н-гексил, гептил или н-гептил, октил или н-октил, нонил или н-нонил, децил или н-децил; (C3-C8)-циклоалкил, возможно замещенный метилом, означает, например, циклопропил, циклобутил, циклопентил, 1-, 2- или 3-метилциклопентил, циклогексил, 1-, 2-, 3- или 4-метилциклогексин, циклогептил или циклооктил; (C1-C10)-алкокси означает, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, гексилокси, гептилокси, нонилокси или децилокси; (C3-C8)-циклоалкокси, возможно замещенный метилом, означает, например, циклопропокси, циклогексилокси, 1-, 2-, 3- или 4-метилциклогексилокси, циклогептилокси или циклооктилокси; (C1-C10)-алкилтио означает, например, метилтио, этилтио, н-пропилтио, изопропилтио, н-бутилтио, изобутилтио, втор-бутилтио, трет-бутилтио, пентилтио, гексилтио, гептилтио, октилтио, нонилтио или децилтио; (C1-C6)-алкилкарбонилокси означает, например, ацетокси, пропионилокси, бутирилокси, валерилокси, капроилокси или гептаноилокси; (C1-C6)-алкилкарбониламино означает, например, ацетиламино, пропиониламино, бутириламино, изобутириламино, валериламино, капроиламино или гептаноиламино; (C1-C4)-алкоксикарбонил означает, например, метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил, н-бутоксикарбонил, изобутоксикарбонил, втор-бутоксикарбонил или трет-бутоксикарбонил; и (C3-C7)-циклоалкоксикарбонил означает, например, циклопропоксикарбонил, циклобутоксикарбонил, циклопентоксикарбонил, циклогексилоксикарбонил или циклогептилоксикарбонил.
Радикал Z предпочтительно является фенилом, который не замещен либо моно- или полизамещен атомом галогена, в частности атомом хлора, фтора или йода, трифторметилом, (C1-C4)-алкилом, гидроксилом или (C1-C4)-алкокси; нафтилом, который не замещен либо моно- или полизамещен галогеном, трифторметилом, (C1-C4)-алкилом, гидроксилом или (C1-C4)-алкокси; пиридилом; тиенилом; индолилом, хинолилом, бензотиенилом или имидазолилом.
В соответствии с настоящим изобретением предпочтительными соединениями являются соединения формулы I, в которых:
A является двухвалентным радикалом, выбираемым из:
A1) -O-CO-
A2) -CH2-O-CO
A3) -O-CH2-CO
A4) -O-CH2-CH2-
A5) -N(R1)-CO-
A6) -N(R1)-CO-CO-
A7) -N(R1)-CH2-CH2-
где R1 обозначает водород или (C1-C4)-алкил; и
Am обозначает:
i - либо группу Am1 формулы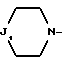
где J1 является:
i1 - либо группой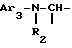
где Ar2 обозначает пиридил; фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил, трифторметил, причем указанные заместители являются одинаковыми или разными; и
R2 обозначает водород; (C1-C7)-алкил; бензил; формил; или (C1-C7)-алкилкарбонил;
i2 - либо группой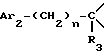
где Ar2 имеет указанные выше значения;
n равен 0 или 1; и
R3 обозначает водород; (C1-C7)-алкил; формил; (C1-C7)-алкилкарбонил; циано; группу -(CH2)q-OH; группу (C1-C7)-aлкил-O-(CH2)q-; группу HCOO-(CH2)q-; группу (C1-C7)-алктил-COO-(CH2)q-; группу (C1-C7)-алкил-NHCOO-(CH2)q-; группу -NR4R5; группу -(CH2)q-NR6COR7; группу -(CH2)q-NR6COOR8; группу -(CH2)q-NR6SO2R9; группу -(CH2)q-NR6CONR10R11; группу -CH2-NR12R13; группу -CH2-CH2-NR12R13; карбоксил; (C1-C7)-алкоксикарбонил; группу -CONR10R11; карбоксиметил; (C1-C7)-алкоксикарбонилметил; группу -CH2-CONR10R11; или 2-аминотиазол-4-ил, где аминогруппа является свободной или замещена одним или двумя (C1-C7)-алкилами;
или R3 образует двойную связь между атомом углерода, к которому он присоединен, и смежным атомом углерода пиперидинового кольца;
q равен 0, 1 или 2;
R4 и R5 независимо друг от друга обозначают водород или (C1-C7)-алкил; R5 может также быть (C3-C7)-циклоалкилметилом, бензилом или фенилом; либо R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина, пергидроазепина;
R6 обозначает водород или (C1-C7)-алкил;
R7 обозначает водород; (C1-C7)-алкил; винил; фенил; бензил; пиридил; (C3-C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами;
либо R6 и R7 вместе обозначают группу -(CH2)p-;
p равен 3 или 4;
R8 обозначает (C1-C7)-алкил или фенил;
R9 обозначает (C1-C7)-алкил; аминогруппу, которая является свободной или замещена одним или двумя (C1-C7)-алкилами; или фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, (C1-C7)-алкил, трифторметил, гидроксил, (C1-C7)-алкокси, карбоксил, (C1-C7)-алкоксикарбонил, (C1-C7)-алкилкарбонилокси, циано, нитро и аминогруппу, которая является свободной или замещена одним или двумя (C1-C7)-алкилами, причем указанные заместители являются одинаковыми или разными;
R10 и R11 независимо друг от друга обозначают водород или (C1-C7)-алкил; R11 может также быть (C3-C7)-циклоалкилом, (C3-C7)-циклоалкилметилом, гидроксилом, (C1-C4)-алкокси, бензилом или фенилом; либо R10 и R11 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина и пергидроазепина; и
R12 и R13 независимо друг от друга обозначают водород или (C1-C7)-алкил; R13 может также быть (C3-C7)-циклоалкилметилом или бензилом;
i3 - либо группой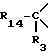
где R3 имеет указанные выше значения;
R14 обозначает (C1-C7)-алкил или (C3-C7)-циклоалкил; R14 может также быть группой -CONR15R16, если R3 является водородом, или группой -NR15R16, если R3 является циано, карбоксилом, (C1-C7)-алкоксикарбонилом или группой -CONR10R11; и
R15 и R16 независимо друг от друга обозначают (C1-C7)-алкил; либо R15 и R16 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина и пергидроазепина;
ii - либо группу Am2 формулы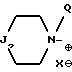
где J2 является:
ii2 - или группой
где Ar3 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными; и
R2 имеет значения, указанные выше для J1;
ii2 - или группой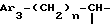
где Ar3 имеет указанные выше значения;
n равен 0 или 1;
Q обозначает (C1-C6)-алкил или бензил, причем указанный заместитель занимает аксиальное или экваториальное положение; и
X⊖ является анионом;
iii - либо группу Am3 формулы
где Ar2 имеет указанные выше значения;
n равен 0 или 1;
X⊖ является анионом;
iv - либо группу Am4 формулы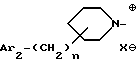
где Ar2 имеет указанные выше значения;
n равен 0 или 1; и
X⊖ является анионом;
m равен 2 или 3;
Ar1 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители могут быть одинаковыми или разными; тиенил; бензотиенил; нафтил или индолил, который не замещен или N-замещен (C1-C4)-алкилом или бензилом;
T обозначает группу, выбираемую из -CH2Z, -CH(C6H5)2 и -C(C6H5)3; T может также быть группой -CO-B-Z, если A является двухвалентным радикалом, выбираемым из -O-CH2-CH2 и -N(R1)-CH2-CH2-;
B обозначает прямую связь или метилен; и
Z обозначает:
- фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена; трифторметил; циано; гидроксил; нитро; фенил, который не замещен либо моно- или полизамещен галогеном, трифторметилом, (C1-C4)-алкилом, гидроксилом или (C1-C4)-алкокси, причем указанные заместители могут быть одинаковыми или разными; аминогруппу, которая не замещена либо моно- или дизамещена (C1-C4)-алкилом; бензиламино; карбоксил; (C1-C10)-алкил; (C1-C7)-циклоалкил, который не замещен либо моно- или полизамещен метилом; (C1-C10)-алкокси; (C3-C7)-циклоалкокси, который не замещен либо моно- или полизамещен метилом; меркапто; (C1-C10)-алкилтио; (C1-C6)-алкилкарбонилокси; формиламино; (C1-C6)-алкилкарбониламино; бензоиламино; (C1-C4)-алкоксикарбонил; (C3-C7)-циклоалкилкарбонил, карбамоил, который не замещен либо моно- или дизамещен (C1-C4)-алкилом; уреидо, который не замещен либо моно- или дизамещен в 3-положении (C1-C4)-алкилом или (C3-C7)-циклоалкилом; и (пирролидин-1-ил)карбониламино, причем указанные заместители являются одинаковыми или разными;
- нафтил, который не замещен либо моно- или полизамещен галогеном, трифторметилом, (C1-C4)-алкилом, гидроксилом или (C1-C4)-алкокси; или
- пиридил, тиенил, индолил, хинолил, бензотиенил или имидазолил,
и при желании их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Кроме того, предпочтение отдается соединениям формулы I, в которой:
A является двухвалентным радикалом, выбираемым из:
A1) -O-CO-
A2) -CH2-O-CO
A3) -O-CH2-CO
A4) -O-CH2-CH2-
A5) -N(R1)-CO-
A6) -N(R1)-CO-CO-
A7) -N(R1)-CH2-CH2-
где R1 обозначает водород или (C1-C4)-алкил;
m равен 2 или 3;
Ar1 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил, трифторметил и метилендиокси, причем указанные заместители являются одинаковыми или разными; тиенил, который не замещен или замещен атомом галогена; бензотиенил, который не замещен или замещен атомом галогена; нафтил, который не замещен или замещен атомом галогена; индолил, который не замещен или N-замещен (C1-C4)-алкилом или бензилом; имидазолил, который не замещен или замещен атомом галогена; пиридил, который не замещен или замещен атомом галогена; или бифенил;
T обозначает группу, выбираемую из CH2-Z, -CH(C6H5)2 и -C(C6H5)3; T может также быть группой -CO-B-Z, если A является двухвалентным радикалом, выбираемым из -O-CH2-CH2 и -N(R1)-CH2-CH2-;
B обозначает прямую связь или метилен;
Z обозначает необязательно замещенную моно-, ди- или трициклическую ароматическую или гетероароматическую группу; и
Am обозначает:
i - либо группу Am1 формулы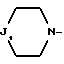
где J1 является:
i1 - или группой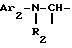
где Ar2 обозначает пиридил; фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил, трифторметил, нитро и метилендиокси, причем указанные заместители являются одинаковыми или разными; тиенил; пиримидил; или имидазолил, который не замещен или замещен (C1-C4)-алкилом; и
R2 обозначает водород; (C1-C7)-алкил; бензил; формил; или (C1-C7)-алкилкарбонил;
i2 - или группой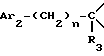
где Ar2 имеет указанные выше значения;
n равен 0 или 1; и
R3 является группой, выбираемой из:
(1) водорода;
(2) (C1-C7)-алкила;
(3) формила;
(4) (C1-C7)-алкилкарбонила;
(5) циано;
(6) -(CH2)q-OH;
(7) -(CH2)q-O-(C1-C7)-алкила;
(8) -(CH2)q-OCHO;
(9) -(CH2)q-OCOR17;
(10) -(CH2)q-OCONH-(C1-C7)-алкила;
(11) -NR4R5;
(12) -(CH2)q-NR6C(=W1)R7;
(13) -(CH2)q-NR6COOR8;
(14) -(CH2)q-NR6SO2R9;
(15) -(CH2)q-NR6C(=W1)NR10R11;
(16) -CH2-NR12R13;
(17) -CH2-CH2-NR12-R13;
(18) -COOH;
(19) (C1-C7)-алкоксикарбонила;
(20) -C(=W1)NR10R11;
(21) -CH2-COOH;
(22) (C1-C7)-алкоксикарбонилметила;
(23) -CH2-C(=W1)NR10R11;
(24) -O-CH2CH2-OR18;
(25) -NR6COCOR19;
(26) -CO-NR20-NR21R22;
(27)
(28)
или R3 образует двойную связь между атомом углерода, к которому он присоединен, и смежным атомом углерода пиперидинового кольца;
q равен 0, 1 или 2;
W1 обозначает атом кислорода или атом серы;
R4 и R5 независимо друг от друга обозначают водород или (C1-C7)-алкил; R5 может также быть (C3-C7)-циклоалкилметилом, бензилом или фенилом; либо R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина, пергидроазепина и пиперазина, который не замещен или замещен в 4-положении (C1-C4)-алкилом;
R6 обозначает водород или (C1-C7)-алкил;
R7 обозначает водород; (C1-C7)-алкил; винил; фенил; бензил; пиридил; (C3-C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами; фурил; тиенил; пирролил или имидазолил;
либо R6 и R7 вместе обозначают группу -(CH2)p-;
p равен 3 или 4;
R8 обозначает (C1-C7)-алкил или фенил;
R9 обозначает (C1-C7)-алкил; аминогруппу, которая является свободной или замещена одним или двумя (C1-C7)-алкилами; или фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, (C1-C7)-алкил, трифторметил, гидроксил, (C1-C7)-алкокси, карбоксил, (C1-C7)-алкоксикарбонил, (C1-C7)-алкилкарбонилокси, циано, нитро и аминогруппу, которая является свободной или замещена одним или двумя (C1-C7)-алкилами, причем указанные заместители являются одинаковыми или разными;
R10 и R11 независимо друг от друга обозначают водород или (C1-C7)-алкил; R11 может также быть (C3-C7)-циклоалкилом, (C3-C7)-циклоалкилметилом, гидроксилом, (C1-C4)-алкокси, бензилом или фенилом; либо R10 и R11 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина и пергидроазепина;
R12 и R13 независимо друг от друга обозначают водород или (C1-C7)-алкил; R13 может также быть (C3-C7)-циклоалкилметилом или бензилом;
R17 обозначает (C1-C7)-алкил; (C3-C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами; фенил или пиридил;
R18 обозначает водород; (C1-C7)-алкил; формил или (C1-C7)-алкилкарбонил;
R19 обозначает (C1-C4)-алкокси;
R20 обозначает водород или (C1-C7)-алкил;
R21 и R22 независимо друг от друга обозначают водород или (C1-C7)-алкил;
либо альтернативно R21 и R22 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из пирролидина, пиперидина и морфолина;
R23 обозначает водород или (C1-C7)-алкил; и
R24 и R25 независимо друг от друга обозначают водород или (C1-C7)-алкил; R25 может также быть формилом или (C1-C7)-алкилкарбонилом;
i3 - либо группой
где R3 имеет указанные выше значения;
R14 обозначает (C1-C7)-алкил или (C3-C7)-циклоалкил; R14 может также быть группой -CONR15R16, если R3 является водородом, или группой -NR15R16, если R3 является циано, карбоксилом, (C1-C7)-алкоксикарбонилом или группой -C(=W1)NR10R11; и
R15 и R16 независимо друг от друга обозначают (C1-C7)-алкил; либо R15 и R16 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из азетидина, пирролидина, пиперидина, морфолина, тиоморфолина и пергидроазепина;
ii - либо группу Am2 формулы
где J2 является:
ii2 - или группой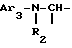
где Ar3 обозначает фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными; и
R2 имеет значения, указанные выше для J1;
ii2 - или группой
где Ar3 имеет указанные выше значения;
n равен 0 или 1;
Q обозначает (C1-C6)-алкил или бензил, причем указанный заместитель занимает аксиальное или экваториальное положение; и
X⊖ является анионом;
iii - либо группу Am3 формулы
где Ar2 имеет указанные выше значения;
n равен 0 или 1; и
X⊖ является анионом;
iv - либо группу Am4 формулы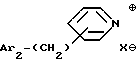
где Ar2 имеет указанные выше значения;
n равен 0 или 1;
X⊖ является анионом;
и при желании их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Другую группу предпочтительных соединений по настоящему изобретению составляют соединения формулы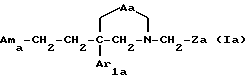
где Aa является двухвалентным радикалом, выбираемым из -O-CO-; -CH2-O-CO-; -O-CH2-CO-; -N(R1)-CO- и -N(R1)-CO-CO-, где R1 обозначает водород или (C1-C4)-алкил;
Ama обозначает:
* либо группу Am2a формулы
* либо группу Am3 формулы
n равен 0 или 1;
Q имеет значения, указанные выше для соединения формулы I, и занимает аксиальное положение;
X⊖ является фармацевтически приемлемым анионом;
Ar2 и Ar3 имеют значения, указанные выше для соединения формулы I;
Ar1a обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными; и
Za обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1-C10)-алкил, (C1-C10)-алкокси и гидроксил, причем указанные заместители являются одинаковыми или разными.
Среди указанных соединений наиболее предпочтительными являются соединения формулы
где Aa и Ama имеют значения, указанные выше для соединения формулы Ia;
Ar'1a обозначает 3,4-дихлорфенил или 3,4-дифторфенил; и
Z'a обозначает 3,5-бис(трифторметил)фенил, 3,5-диметилфенил или 2,4-бис(трифторметил)фенил.
Еще одну группу предпочтительных соединений по настоящему изобретению составляют соединения формулы
где Ab является двухвалентным радикалом -O-CH2-CH2-; -N(R1)-CH2-CH2- или -O-CH2-, где R1 обозначает водород или (C1-C4)-алкил;
Amb обозначает:
* либо группу Am2m формулы
* либо группу Am3 формулы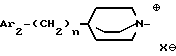
* либо группу Am1a формулы
n равен 0 или 1;
Q имеет значения, указанные выше для соединения формулы I, и занимает аксиальное положение;
X⊖ является фармацевтически приемлемым анионом;
Ar2, Ar3 и R3 имеют значения, указанные выше для соединения формулы I; и
Ar1a и Za имеют указанные выше значения;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Среди указанных соединений наиболее предпочтительными являются соединения формулы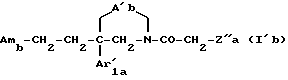
где Amb имеет значения, указанные выше для соединения формулы Ib;
A'b является двухвалентным радикалом -O-CH2-CH2- или -N(R1)-CH2-CH2;
Ar'1a имеет значения, указанные выше для соединения формулы I'a; и
Z''a обозначает фенил, замещенный в 3-положении галогеном или (C1-C10)-алкоксильной группой,
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Следующую группу предпочтительных соединений по настоящему изобретению составляют соединения формулы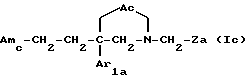
где Ac является двухвалентным радикалом, выбираемым из -O-CH2-CO-, -CH2-O-CO- и -O-CO-;
Amc обозначает группу Am1a формулы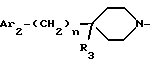
где n равен 0 или 1;
Ar2 и R3 имеют значения, указанные выше для соединения формулы I; и
Ar1a и Za имеют указанные выше значения;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Среди указанных соединений наиболее предпочтительными являются соединения формулы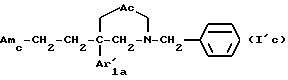
где Ac, Amc и Ar'1a имеют указанные выше значения,
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Следующую группу предпочтительных соединений по данному изобретению составляют соединения формулы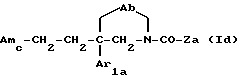
где Ab, Amc, Ar1a и Za имеют указанные выше значения;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Среди указанных соединений наиболее предпочтительными являются соединения формулы
где A'b, Amc и Ar'1a имеют указанные выше значения,
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Среди соединений формул Ia и I'a наиболее предпочтительными являются соединения, в которых:
Ar2 обозначает пиридил или фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными; и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Среди соединений формул I'b, Ic, I'c и I'd наиболее предпочтительными являются соединения, в которых:
Ar2 обозначает пиридил или фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными, и
R3 обозначает водород; (C1-C7)-алкил; формил; (C1-C7)-алкилкарбонил; циано; группу -(CH2)q-OH; группу (C1-C7)-алкил-O-(CH2)q-; группу HCOO-(CH2)q-; группу (C1-C7)-алкил-COO-(CH2)q-; группу (C1-C7)-алкил-NHCOO-(CH2)q-; группу -NR4R5; группу -(CH2)q-NR6COR7; группу -(CH2)q-NR6COOR8; группу -(CH2)q-NR6SO2R9; группу -(CH2)q-NR6CONR10R11; группу -CH2-NR12R13; группу -CH2-CH2-NR12R13; карбоксил; (C1-C7)-алкоксикарбонил; группу -CONR10R11; карбоксиметил; (C1-C7)-алкоксикарбонилметил; группу -CH2-CONR10R11 или 2-аминотиазол-4-ил, где аминогруппа является свободной или замещена одним или двумя (C1-C7)-алкилами;
или R3 обозначает двойную связь между атомом углерода, к которому он присоединен, и смежным атомом углерода пиперидинового кольца;
q равен 0, 1 или 2;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Среди соединений формул Ib и Id наиболее предпочтительными являются соединения, в которых:
Ab является двухвалентным радикалом -O-CH2-CH2- или -N(R1)-CH2-CH2-,
Ar2 обозначает пиридил или фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1-C4)-алкокси, (C1-C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными, и
R3 обозначает водород; (C1-C7)-алкил; формил; (C1-C7)-алкилкарбонил; циано; группу -(CH2)q-OH; группу (C1-C7)-алкил-O-(CH2)q-; группу HCOO-(CH2)q-; группу (C1-C7)-алкил-COO-(CH2)q-; группу (C1-C7)-алкил-NHCOO-(CH2)q-; группу -NR4R5; группу -(CH2)q-NR6COR7; группу -(CH2)q-NR6COOR8; группу -(CH2)q-NR6SO2R9; группу -(CH2)q-NR6CONR10R11; группу -CH2-NR12R13; группу -CH2-CH2-NR12R13; карбоксил; (C1-C7)-алкоксикарбонил; группу -CONR10R11; карбоксиметил; (C1-C7)-алкоксикарбонилметил; группу -CH2-CONR10R11 или 2-аминотиазол-4-ил, где аминогруппа является свободной или замещена одним или двумя (C1-C7)-алкилами;
или R3 обозначает двойную связь между атомом углерода, к которому он присоединен, и смежным атомом углерода пиперидинового кольца;
q равен 0, 1 или 2;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Еще одну группу предпочтительных соединений по данному изобретению составляют соединения формулы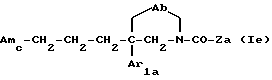
где Ab, Amc, Ar1a и Za имеют указанные выше значения;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
Еще одну группу предпочтительных соединений по данному изобретению составляют соединения формулы
где Ab, Amc, Ar1a и Za имеют указанные выше значения;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
4-Бензоил-2- (3,4-дифторфенил)-2-[2-[4- (N',N'-диметилуреидо)-4- фенилпиперид-1-ил] этил] морфолин в оптически чистой форме, предпочтительно в виде (+) изомера, и его соли, получаемые при взаимодействии с минеральными или органическими кислотами, являются наиболее предпочтительными.
5-(3,4-Дифторфенил)-5-[2- [4-фенил-1-азониабицикло[2.2.2] окт-1- ил]-этил]-3-[3,5-бис(трифторметил) бензил]оксазолидин-2-он с фармацевтически приемлемым анионом в оптически чистой форме, предпочтительно в виде (+) изомера, является наиболее предпочтительным.
Другим объектом настоящего изобретения является способ получения соединений формулы I и их солей, который заключается в том, что:
1) осуществляют обработку соединения формулы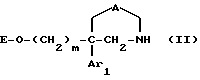
где m, Ar1 и A имеют значения, указанные выше для соединения формулы I и E обозначает водород или O-защитную группу,
или функциональным производным кислоты формулы
HOCO-B-Z (III)
где B и Z имеют значения, указанные выше для формулы I, если необходимо получить соединение формулы I, в которой T обозначает -CO-B-Z,
или галогензамещенным производным формулы
Hal-CH2-Z (IV)
где Z имеет указанные выше значения и Hal обозначает галоген, предпочтительно бром или хлор, если необходимо получить соединение формулы I, в которой T обозначает -CH2-Z,
или галогензамещенным производным формулы
Hal-CH(C6H5)2 (V)
если необходимо получить соединение формулы I, в которой T обозначает группу -CH(C6H5)2,
или галогензамещенным производным формулы
Hal-C-(C6H5)3 (VI)
если необходимо получить соединение формулы I, в которой T обозначает группу -C(C6H5)3,
в результате чего получают соединение формулы
2) при необходимости удаляют O-защитную группу в результате взаимодействия с кислотой или основанием с получением спирта формулы
где m, Ar1, A и T имеют указанные выше значения;
3) полученный спирт (VIII) обрабатывают соединением формулы
Y-SO2-Cl (IX)
где Y обозначает метил, фенил, толил или трифторметил, с получением соединения формулы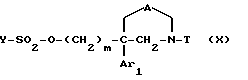
4) полученное соединение (X) подвергают взаимодействию:
либо с циклическим вторичным амином формулы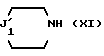
где J'1 является:
* или группой
где Ar2 имеет значения, указанные для формулы I, и R'2 аналогичен R2 в формуле I или является предшественником R2;
* или группой
где Ar2 и n имеют значения, указанные для формулы I, и R'3 аналогичен R3 в формуле I или является предшественником R3, причем если R'3 является гидроксилом или амино, то эти группы могут быть защищены;
* или группой
где R14 имеет значения, указанные для формулы I, и R'3 имеет указанные выше значения;
либо с третичным амином формулы
где J2 и Q имеют значения, указанные для формулы I;
либо с циклическим третичным амином формулы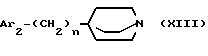
где Ar2 и n имеют значения, указанные для формулы I;
либо с соединением формулы
где Ar2 и n имеют значения, указанные для формулы I;
5) и, либо в случае использования циклического вторичного амина формулы XI и после возможного снятия защиты с гидроксильной группы или с аминогруппы, обозначенной R'3, или после возможного превращения R'2 в R2 или R'3 в R3 при желании превращают полученный продукт в одну из его солей;
либо в случае использования третичного амина формулы XII, циклического третичного амина формулы XIII или соединения формулы XIV выделяют полученный продукт в виде сульфоната или соли сульфоновой кислоты, или же при желании заменяют полученный анион или полученную соль кислоты на другой фармацевтически приемлемый анион или на другую фармацевтически приемлемую соль с минеральной или органической кислотой.
В соответствии с одним вариантом осуществления этого способа, если Am обозначает группу Am1,
1') соединение приведенной выше формулы XIII окисляют с получением соединения формулы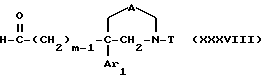
где m, Ar1, A и T имеют значения, указанные для соединения формулы I;
2') соединение формулы XXXVIII подвергают взаимодействию с соединением приведенной выше формулы XI в присутствии кислоты, после чего соль иминия, полученную в виде промежуточного продукта, восстанавливают с помощью восстановителя; и
3') после возможного снятия защиты с гидроксильной группы или с аминогруппы, или после возможного превращения R'2 в R2 или R'3 в R3 полученный продукт при желании превращают в его соль.
Соединения формулы
в виде чистых энантиомеров или рацемической смеси являются новыми соединениями и входят в объем данного изобретения.
Соединения формулы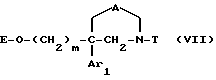
в виде чистых энантиомеров или рацемической смеси являются новыми соединениями и входят в объем данного изобретения.
Соединения формул II и VII, в которых E обозначает водород, являются наиболее предпочтительными.
Соединения формулы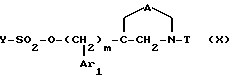
в виде чистых энантиомеров или рацемической смеси являются новыми соединениями и входят в объем данного изобретения.
Соединения формулы
в виде чистых энантиомеров или рацемической смеси являются новыми соединениями и входят в объем данного изобретения.
В формулах II, VII, X и XXXVIII, m и группы E, A, Ar1, T и Y имеют указанные выше значения.
Таким образом, другим объектом настоящего изобретения являются соединения формулы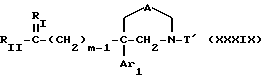
где m, Ar1 и A имеют значения, указанные для соединения формулы I;
RI обозначает два атома водорода и RII обозначает:
или группу -O-E, где E является атомом водорода или O-защитной группой,
или группу -O-SO2-Y, где Y является метилом, фенилом, толилом или трифторметилом;
или же RI обозначает атом кислорода и RII обозначает атом водорода; и
T' аналогичен T, значения которого приведены для соединения формулы I; T' может также быть водородом, если RI обозначает 2 атома водорода и RII одновременно является группой -O-E-,
в виде чистых энантиомеров или рацемической смеси.
Таким образом, если E является O-защитной группой, то ее выбирают из обычных O-защитных групп, хорошо известных специалистам в этой области, таких как, например, тетрагидропиран-2-ил, бензоил или (C1-C4)-алкил-карбонил.
O-защитные группы, которые можно использовать для получения соединения формулы I, в которой R3 обозначает гидроксил, являются классическими O-защитными группами, хорошо известными специалистам в этой области, которые представлены выше для E.
N-защитные группы, которые можно использовать для получения соединения формулы I, в которой R3 обозначает амино, являются классическими N-защитными группами, хорошо известными специалистам в этой области, такими как, например, тритил, метокситритил, трет-бутоксикарбонил или бензилоксикарбонил.
В частности, если используемой O-защитной группой является ацетильная группа, то полученное соединение формулы I представляет собой конечный продукт, в котором R3 является ацетокси, или если используемой N-защитной группой является трет-бутоксикарбонильная группа, полученное соединение формулы I представляет собой конечный продукт, в котором R3 является трет-бутоксикарбониламино.
На стадии 1) используемым функциональным производным кислоты (III) является сама кислота или же одно из функциональных производных, которое взаимодействует с аминами, например ангидрид, смешанный ангидрид, хлорангидрид или активированный сложный эфир, такой как паранитрофениловый эфир.
Если использована кислота формулы III, реакцию осуществляют в присутствии агента сочетания, применяемого в химии пептидов, такого как 1,3-дициклогексилкарбодиимид или гексафторфосфат бензотриазол-1-ил-окситрис(диметил-амино)фосфония, в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в инертном растворителе, таком как дихлорметан или N,N-диметилформамид, при температуре от 0oC до комнатной температуры.
Если использован хлорангидрид, реакцию осуществляют в инертном растворителе, таком как дихлорметан или бензол, в присутствии основания, такого как триэтиламин или N-метилморфолин, и при температуре от -60oC до комнатной температуры.
Если использовано галогензамещенное производное формулы IV, V или VI, реакцию осуществляют в инертном растворителе, таком как тетрагидрофуран, N, N-диметилформамид или диметилсульфоксид, в присутствии основания, такого как трет-бутилат калия, гидрид натрия или диизопропиламид лития, и при температуре от 0oC до 80oC.
У полученного соединения формулы VII при желании удаляют защитную группу на стадии 2) с помощью способов, хорошо известных специалистам в этой области. Например, если E является тетрагидропиран-2-ильной группой, защитную группу удаляют путем кислотного гидролиза, используя хлористо-водородную кислоту в растворителе, таком как простой эфир или метанол или смесь этих растворителей, или используя паратолуолсульфонат пиридиния в растворителе, таком как метанол, или же используя смолу Amberlyst® в таком растворителе, как метанол. Эту реакцию осуществляют при температуре от комнатной до температуры кипения растворителя с обратным холодильником. Если E является бензоильной или (C1-C4)-алкилкарбонильной группой, защитную группу удаляют путем гидролиза в щелочной среде, используя, например, гидроксид щелочного металла, такой как гидроксид натрия, гидроксид калия или гидроксид лития, в инертном растворителе, таком как вода, метанол, этанол или диоксан, или в смеси этих растворителей при температуре от 0oC до температуры кипения растворителя с обратным холодильником.
На стадии 3) спирт формулы VIII подвергают взаимодействию с сульфонилхлоридом формулы IX) в присутствии основания, такого как триэтиламин, пиридин, N, N-диизопропилэтиламин или N-метилморфолин, в инертном растворителе, таком как дихлорметан, бензол или толуол, при температуре от -20oC до температуры кипения растворителя с обратным холодильником.
Полученное соединение формулы X подвергают взаимодействию на стадии 4) с соединением формулы XI, XII, XIII или XIV, используя различные способы.
Если соединение формулы X подвергают взаимодействию с соединением формулы XI, то эту реакцию осуществляют в инертном растворителе, таком как N,N-диметилформамид, ацетонитрил, метиленхлорид, толуол или изопропанол, в присутствии основания или без него. Если использовано основание, его выбирают из органических оснований, таких как триэтиламин, N,N-диизопропилэтиламин и N-метилморфолин, или из карбонатов или бикарбонатов щелочных металлов, таких как карбонат калия, карбонат натрия и бикарбонат натрия. При отсутствии основания эту реакцию осуществляют при использовании избыточного количества соединения формулы XI и в присутствии йодида щелочного металла, такого как йодид калия или йодид натрия. Если -A- в соединении формулы X является двухвалентным радикалом -O-CO- или -CH2-O-CO-, реакцию осуществляют при температуре от комнатной до 80oC. Если -A- в соединении формулы X является двухвалентным радикалом -O-CH2-CO-, -O-CH2-CH2-, -N(R1)-CO-CO-, -N(R1)-CH2-CH2-, -N(R1)-CO- или -O-CH2-, реакцию осуществляют при температуре от комнатной до 100oC.
Если соединение формулы X подвергают взаимодействию с соединением формулы XII или XIII, эту реакцию осуществляют в полярном апротонном растворителе, таком как ацетонитрил, N,N-диметилформамид или N,N-диметилфенилацетамид, в простом эфире, таком как тетрагидрофуран, диоксан или метил-трет-бутиловый эфир, или в кетоне, таком как метилэтилкетон. Если -A- в соединении формулы X является двухвалентным радикалом -O-CO- или -CH2-O-CO-, реакцию осуществляют при температуре от комнатной до 60oC. Если -A- в соединении формулы X является двухвалентным радикалом -O-CH2-CO- , -O-CH2-CH2-, -N(R1)-CO-CO-, -N(R1)-CH2-CH2-, -N(R1)-CO- или -O-CH2-, реакцию осуществляют при температуре от комнатной до 100oC.
Если соединение формулы X подвергают взаимодействию с соединением формулы XIV, эту реакцию осуществляют в полярном апротонном растворителе, таком как ацетонитрил, N,N-диметилформамид или N,N-диметилфенилацетамид, в простом эфире, таком как тетрагидрофуран, диоксан или метил-трет-бутиловый эфир, или в кетоне, таком как метилэтилкетон. Реакцию осуществляют при температуре от комнатной до 100oC.
На стадии 1') варианта осуществления заявленного способа спирт формулы VIII окисляют до альдегида формулы XXXVIII. Реакцию окисления осуществляют, используя, например, оксалилхлорид, диметилсульфоксид и триэтиламин, в растворителе, таком как дихлорметан, при температуре от -78oC до комнатной температуры.
Затем на стадии 2') соединение формулы XI подвергают взаимодействию с альдегидом формулы XXXVIII в присутствии кислоты, такой как уксусная кислота, в спиртовом растворителе, таком как метанол, с получением in situ промежуточного имина, который восстанавливают химическим путем, используя, например, цианоборогидрид натрия, или каталитическим путем, используя водород и такой катализатор, как палладий на угле или никель Ренея.
Соединения формулы I согласно изобретению окончательно получают после возможного снятия защиты с гидроксильной группы или с аминогруппы, или после возможного превращения R'2 в R2 и R'3 в R3.
Полученные продукты формулы I выделяют:
- либо в виде свободного основания или соли согласно традиционным способам, если Am является Am1,
- либо, если Am является Am2, Am3 или Am4, в виде сульфатного аниона (YSO
Если соединение формулы I, в котором Am является Am1, получают в виде свободного основания, производят солеобразование путем обработки выбранной кислотой в органическом растворителе. В результате обработки свободного основания, растворенного, например, в эфире, таком как диэтиловый эфир, в спирте, таком как пропан-2-ол, в ацетоне или дихлорметане, раствором выбранной кислоты в таком же растворителе получают соответствующую соль, которую отделяют известными способами.
Таким способом получают, например, гидрохлорид, гидробромид, сульфат, бисульфат, первичный кислый фосфат, метансульфонат, оксалат, малеат, фумарат, нафталин-2-сульфонат и бензолсульфонат.
После завершения реакции соединения формулы I, в которой Am является Am1, можно выделить в виде одной из их солей, например, гидрохлорида или оксалата; в этом случае свободное основание при необходимости можно получить путем нейтрализации указанной соли минеральным или органическим основанием, таким как гидроксид натрия или триэтиламин, либо карбонатом или бикарбонатом щелочного металла, таким как карбонат или бикарбонат натрия или калия.
Анион сульфоната YSO
После завершения реакции соединения формулы I, в которой Am является Am2, получают в виде смеси аксиальных и экваториальных изомеров. Изомеры разделяют обычными способами, например посредством хроматографии или перекристаллизации.
Соединения формулы II получают разными способами.
Соединения формулы II, в которой -A- является двухвалентным радикалом -CH2-O-CO- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 1 (см. в конце описания), в которой m и Ar1 имеют значения, указанные для соединения формулы I, и Pr является O-защитной группой, указанной выше для E.
На стадии  схемы 1 соединение формулы XV подвергают взаимодействию с соединением формулы XVI по способу, описанному в заявках на европейский патент N 0428434 и N 0474561.
схемы 1 соединение формулы XV подвергают взаимодействию с соединением формулы XVI по способу, описанному в заявках на европейский патент N 0428434 и N 0474561.
Полученное соединение XVII подвергают взаимодействию на стадии  с водным раствором формальдегида в присутствии основания, такого как 1,8-диазабицикло[5.4.0] -ундец-7-ен, в растворителе, таком как 1,2-диметоксиэтан, и при температуре от комнатной до температуры кипения растворителя с обратным холодильником.
с водным раствором формальдегида в присутствии основания, такого как 1,8-диазабицикло[5.4.0] -ундец-7-ен, в растворителе, таком как 1,2-диметоксиэтан, и при температуре от комнатной до температуры кипения растворителя с обратным холодильником.
Производное нитрила формулы XVIII восстанавливают на стадии  с получением первичного амина формулы XIX. Реакцию восстановления осуществляют с помощью водорода в присутствии катализатора, такого как никель Ренея, оксид платины или палладий на угле, в инертном растворителе, таком как спирт, например этанол, который можно использовать отдельно или в смеси с водным раствором аммиака, либо с помощью восстановителя, такого как алюмогидрид лития, гидрид диизобутилалюминия или боран в тетрагидрофуране, в растворителе, таком как толуол, гексан, петролейный эфир, ксилол или тетрагидрофуран. Эту реакцию осуществляют при температуре от 0oC до 70oC.
с получением первичного амина формулы XIX. Реакцию восстановления осуществляют с помощью водорода в присутствии катализатора, такого как никель Ренея, оксид платины или палладий на угле, в инертном растворителе, таком как спирт, например этанол, который можно использовать отдельно или в смеси с водным раствором аммиака, либо с помощью восстановителя, такого как алюмогидрид лития, гидрид диизобутилалюминия или боран в тетрагидрофуране, в растворителе, таком как толуол, гексан, петролейный эфир, ксилол или тетрагидрофуран. Эту реакцию осуществляют при температуре от 0oC до 70oC.
На стадии  соединение (XIX) подвергают взаимодействию с реакционноспособным производным угольной кислоты, таким как фосген, растворенный в толуоле, или 1,1'-карбонилдиимидазол, в присутствии основания, такого как триэтиламин, N, N-диизопропилэтиламин или N-метилморфолин, в хлорированном растворителе, таком как дихлорметан или 1,2-дихлорэтан, или в эфире, таком как тетрагидрофуран, при температуре от -70oC до комнатной температуры, с получением соединения формулы II, в которой E является O-защитной группой.
соединение (XIX) подвергают взаимодействию с реакционноспособным производным угольной кислоты, таким как фосген, растворенный в толуоле, или 1,1'-карбонилдиимидазол, в присутствии основания, такого как триэтиламин, N, N-диизопропилэтиламин или N-метилморфолин, в хлорированном растворителе, таком как дихлорметан или 1,2-дихлорэтан, или в эфире, таком как тетрагидрофуран, при температуре от -70oC до комнатной температуры, с получением соединения формулы II, в которой E является O-защитной группой.
Используя описанные выше способы, O-защитную группу удаляют гидролизом (стадия  ) с получением соединения формулы II, в которой E является водородом.
) с получением соединения формулы II, в которой E является водородом.
Соединения формулы II, в которой -A- является двухвалентным радикалом -O-CH2-CO- и E является водородом или O-защитной группой, получают в соответствии со схемой 2 (см. в конце описания), в которой m и Ar1 имеют значения, указанные для соединения формулы I. Pr1 и Pr2 являются O-защитной группой Pr, представленной выше для E; в частности, Pr1 является O-защитной группой, гидролизуемой в кислотной среде, а Pr2 является O-защитной группой, гидролизуемой в щелочной среде.
На стадии  схемы 2 осуществляют синтез цианогидрина формулы XXI из альдегида формулы XX с помощью способов, хорошо известных специалистам в этой области, таких как, например, способ, описанный в издании Organic Syntheses; Wiley, New York, 1932; Collect. vol. 1, p. 336, или варианта этого способа с использованием метабисульфита натрия и цианида калия в водном растворе.
схемы 2 осуществляют синтез цианогидрина формулы XXI из альдегида формулы XX с помощью способов, хорошо известных специалистам в этой области, таких как, например, способ, описанный в издании Organic Syntheses; Wiley, New York, 1932; Collect. vol. 1, p. 336, или варианта этого способа с использованием метабисульфита натрия и цианида калия в водном растворе.
На стадии  гидроксильную группу соединения формулы (XXI) защищают с помощью способов, известных специалистам в этой области.
гидроксильную группу соединения формулы (XXI) защищают с помощью способов, известных специалистам в этой области.
Полученное соединение формулы XXII обрабатывают на стадии  сильным основанием, таким как диизопропиламид лития, трет-бутилат калия или гидрид натрия, с получением карбаниона, который подвергают взаимодействию с соединением формулы Hal-(CH2)m-O-Pr2, в которой Hal обозначает галоген, предпочтительно бром или хлор, с получением соединения формулы XXIII. Эту реакцию осуществляют в инертном растворителе, таком как эфир (например, тетрагидрофуран, диэтиловый эфир или 1,2-диметоксиэтан), амид (например, N,N-диметилформамид) или ароматический углеводород (например, толуол или ксилол), при температуре от -70oC до +60oC.
сильным основанием, таким как диизопропиламид лития, трет-бутилат калия или гидрид натрия, с получением карбаниона, который подвергают взаимодействию с соединением формулы Hal-(CH2)m-O-Pr2, в которой Hal обозначает галоген, предпочтительно бром или хлор, с получением соединения формулы XXIII. Эту реакцию осуществляют в инертном растворителе, таком как эфир (например, тетрагидрофуран, диэтиловый эфир или 1,2-диметоксиэтан), амид (например, N,N-диметилформамид) или ароматический углеводород (например, толуол или ксилол), при температуре от -70oC до +60oC.
Производное нитрила формулы XXIII восстанавливают на стадии  с помощью описанных выше способов с получением первичного амина формулы XXIV.
с помощью описанных выше способов с получением первичного амина формулы XXIV.
На стадии  соединение формулы XXIV подвергают взаимодействию с соединением формулы Hal-CO-CH2-Hal, в которой Hal обозначает галоген, предпочтительно хлор или бром, в присутствии основания, такого как третичный амин (например, триэтиламин, N-метилморфолин или пиридин), с получением соединения формулы XXV. Эту реакцию осуществляют в инертном растворителе, таком как хлорированный растворитель (например, дихлорметан, дихлорэтан или хлороформ), эфир (например, тетрагидрофуран или диоксан) или амид (например, N, N-диметилформамид), при температуре от -70oC до комнатной температуры.
соединение формулы XXIV подвергают взаимодействию с соединением формулы Hal-CO-CH2-Hal, в которой Hal обозначает галоген, предпочтительно хлор или бром, в присутствии основания, такого как третичный амин (например, триэтиламин, N-метилморфолин или пиридин), с получением соединения формулы XXV. Эту реакцию осуществляют в инертном растворителе, таком как хлорированный растворитель (например, дихлорметан, дихлорэтан или хлороформ), эфир (например, тетрагидрофуран или диоксан) или амид (например, N, N-диметилформамид), при температуре от -70oC до комнатной температуры.
На стадии  O-защитную группу Pr1 удаляют из соединения формулы XXV кислотным гидролизом с помощью описанных выше способов.
O-защитную группу Pr1 удаляют из соединения формулы XXV кислотным гидролизом с помощью описанных выше способов.
Альтернативно, O-защитную группу Pr1 удаляют из соединения формулы XXIV кислотным гидролизом на стадии  , после чего полученное соединение XXVII подвергают взаимодействию на стадии
, после чего полученное соединение XXVII подвергают взаимодействию на стадии  с соединением формулы Hal-CO-CH2-Hal с помощью способов, описанных выше на стадии
с соединением формулы Hal-CO-CH2-Hal с помощью способов, описанных выше на стадии 
Полученное соединение формулы XXVI подвергают циклизации в присутствии основания с получением соединения целевой формулы II. Если нужно получить соединение формулы II, в которой E является защитной группой Pr2, то используют основание, такое как карбонат щелочного металла (например, карбонат калия), гидрид щелочного металла (например, гидрид натрия) или трет-бутилат калия в инертном растворителе, таком как ароматический углеводород (например, ксилол или толуол), амид (например, N,N-диметилформамид) или эфир (например, тетрагидрофуран), при температуре от -30oC до температуры кипения растворителя с обратным холодильником (стадия 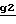 ). Если необходимо получить соединение формулы II, в которой E является водородом, то используют основание, такое как гидроксид щелочного металла (например, гидроксид натрия или гидроксид калия), в виде концентрированного водного раствора в таком растворителе, как алканол (например, пропан-2-ол) или амид (например, N,N-диметилформамид), или в смеси этих растворителей при температуре от комнатной до температуры кипения растворителя с обратным холодильником (стадия
). Если необходимо получить соединение формулы II, в которой E является водородом, то используют основание, такое как гидроксид щелочного металла (например, гидроксид натрия или гидроксид калия), в виде концентрированного водного раствора в таком растворителе, как алканол (например, пропан-2-ол) или амид (например, N,N-диметилформамид), или в смеси этих растворителей при температуре от комнатной до температуры кипения растворителя с обратным холодильником (стадия  ).
).
При желании соединение формулы II, в которой E является O-защитной группой Pr1, получают на стадии  с помощью способов, известных специалистам в этой области.
с помощью способов, известных специалистам в этой области.
Соединения формулы II, в которой -A- является двухвалентным радикалом -O-CH2-CH2- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 3 (см. в конце описания), где m и Ar1 имеют значения, указанные для соединения формулы I, и Pr1 и Pr2 имеют значения, представленные на схеме 2.
На стадии  схемы 3 производят восстановление соединения формулы II, в которой -A- является двухвалентным радикалом -O-CH2-CO- и E обозначает водород или O-защитную группу, которое получено в соответствии со схемой 2. Восстановление осуществляют с помощью восстановителя, такого как алюмогидрид лития, гидрид диизобутилалюминия, боргидрид натрия или боран в тетрагидрофуране, в инертном растворителе, таком как тетрагидрофуран, диэтиловый эфир, 1,2-диметоксиэтан или толуол, при температуре от комнатной до температуры кипения растворителя с обратным холодильником.
схемы 3 производят восстановление соединения формулы II, в которой -A- является двухвалентным радикалом -O-CH2-CO- и E обозначает водород или O-защитную группу, которое получено в соответствии со схемой 2. Восстановление осуществляют с помощью восстановителя, такого как алюмогидрид лития, гидрид диизобутилалюминия, боргидрид натрия или боран в тетрагидрофуране, в инертном растворителе, таком как тетрагидрофуран, диэтиловый эфир, 1,2-диметоксиэтан или толуол, при температуре от комнатной до температуры кипения растворителя с обратным холодильником.
Соединения формулы I, в которой -A- является двухвалентным радикалом -O-CO- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 4 (см. в конце описания), в которой m и Ar1 имеют значения, указанные для соединения формулы I, и Pr1 и Pr2 имеют значения, представленные в схеме 2.
На стадии 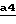 схемы 4 с помощью кислотного гидролиза, выполняемого в соответствии с описанными выше способами, удаляют O-защитную группу Pr1 соединения формулы XXIV, полученного на стадии
схемы 4 с помощью кислотного гидролиза, выполняемого в соответствии с описанными выше способами, удаляют O-защитную группу Pr1 соединения формулы XXIV, полученного на стадии  схемы 2.
схемы 2.
Полученное соединение формулы XXVII на стадии 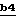 подвергают взаимодействию с реакционноспособным производным угольной кислоты, таким как 1,1'-карбонилдиимидазол, фосген в толуоле или паранитрофенилхлорформиат, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, с получением соединения целевой формулы II, в которой E обозначает O-защитную группу. Эту реакцию осуществляют в инертном растворителе, таком как хлорированный растворитель (например, 1,2-дихлорэтан или дихлорметан), эфир, такой как тетрагидрофуран, амид, такой как N,N-диметилформамид, или ароматический растворитель, такой как толуол, при температуре от -60oC до комнатной температуры.
подвергают взаимодействию с реакционноспособным производным угольной кислоты, таким как 1,1'-карбонилдиимидазол, фосген в толуоле или паранитрофенилхлорформиат, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, с получением соединения целевой формулы II, в которой E обозначает O-защитную группу. Эту реакцию осуществляют в инертном растворителе, таком как хлорированный растворитель (например, 1,2-дихлорэтан или дихлорметан), эфир, такой как тетрагидрофуран, амид, такой как N,N-диметилформамид, или ароматический растворитель, такой как толуол, при температуре от -60oC до комнатной температуры.
Используя описанные выше способы, O-защитную группу Pr2 удаляют щелочным гидролизом (стадия 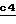 ) с получением соединения формулы II, в которой E обозначает водород.
) с получением соединения формулы II, в которой E обозначает водород.
Соединения формулы II, в которой -A- является двухвалентным радикалом -N(R1)-CO-CO- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 5 (см. в конце описания), в которой m, Ar1 и R1 имеют значения, указанные для соединения формулы I, и Pr1 имеет приведенные выше значения.
На стадии  схемы 5 соединение α_аминонитрила формулы XXVIII получают из альдегида формулы XX по способу, описанному в журнале Tetrahedron Letters, 1986, 25. (41), 4583-4586, используя амин формулы H2N-R1.
схемы 5 соединение α_аминонитрила формулы XXVIII получают из альдегида формулы XX по способу, описанному в журнале Tetrahedron Letters, 1986, 25. (41), 4583-4586, используя амин формулы H2N-R1.
Аминогруппу соединения формулы XXVIII защищают на стадии  N-защитной группой, такой как трет-бутоксикарбонил (Boc) или бензилоксикарбонил, например, с помощью способов, известных специалистам в этой области. Трет-бутоксикарбонильная группа представлена на схеме 5.
N-защитной группой, такой как трет-бутоксикарбонил (Boc) или бензилоксикарбонил, например, с помощью способов, известных специалистам в этой области. Трет-бутоксикарбонильная группа представлена на схеме 5.
Полученное соединение формулы XXIX обрабатывают на стадии 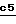 сильным основанием с образованием карбаниона, который подвергают взаимодействию с соединением формулы Hal-(CH2)m-O-Pr1 с получением соединения формулы XXX. Эту реакцию осуществляют по способу, описанному на стадии
сильным основанием с образованием карбаниона, который подвергают взаимодействию с соединением формулы Hal-(CH2)m-O-Pr1 с получением соединения формулы XXX. Эту реакцию осуществляют по способу, описанному на стадии  схемы 2.
схемы 2.
Производное нитрила формулы XXX восстанавливают на стадии  с помощью описанных выше способов с получением первичного амина формулы XXXI.
с помощью описанных выше способов с получением первичного амина формулы XXXI.
На стадии 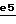 O-защитную группу и N-защитную группу удаляют из соединения формулы XXXI кислотным гидролизом, используя хлористо-водородную кислоту или трифторуксусную кислоту, например, в таком растворителе, как спирт (например, метанол), эфир (например, диэтиловый эфир, диоксан или тетрагидрофуран) или хлорированный растворитель (например, дихлорметан), при температуре от 0oC до температуры кипения реакционной смеси с обратным холодильником.
O-защитную группу и N-защитную группу удаляют из соединения формулы XXXI кислотным гидролизом, используя хлористо-водородную кислоту или трифторуксусную кислоту, например, в таком растворителе, как спирт (например, метанол), эфир (например, диэтиловый эфир, диоксан или тетрагидрофуран) или хлорированный растворитель (например, дихлорметан), при температуре от 0oC до температуры кипения реакционной смеси с обратным холодильником.
На стадии  соединение целевой формулы II получают с помощью способа, описанного R. Granger, H. Orzalesi и Y. Robbe в журнале Trav. Soc. Pharm. Montpellier, 1965, 25, Fasc. 4, 313-317, или его варианта, осуществляя взаимодействие соединения формулы XXXII с диэтилоксалатом в спиртовом растворителе, таком как этанол, в ароматическом растворителе, таком как толуол, или в смеси этих растворителей при температуре от комнатной до температуры кипения реакционной смеси с обратным холодильником.
соединение целевой формулы II получают с помощью способа, описанного R. Granger, H. Orzalesi и Y. Robbe в журнале Trav. Soc. Pharm. Montpellier, 1965, 25, Fasc. 4, 313-317, или его варианта, осуществляя взаимодействие соединения формулы XXXII с диэтилоксалатом в спиртовом растворителе, таком как этанол, в ароматическом растворителе, таком как толуол, или в смеси этих растворителей при температуре от комнатной до температуры кипения реакционной смеси с обратным холодильником.
Соединение формулы II, в которой E обозначает O-защитную группу Pr1, при желании получают на стадии  с помощью способов, известных специалистам в этой области.
с помощью способов, известных специалистам в этой области.
Соединения формулы II, в которой -A- является двухвалентным радикалом -N(R1)-CH2-CH2- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 6 (см. в конце описания), в которой m, R1 и Ar1 имеют значения, указанные для соединения формулы I, и Pr1 является O-защитной группой, представленной выше для E.
На стадии 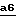 схемы 6 производят восстановление соединения формулы II, в которой -A- является двухвалентным радикалом -N(R1)-CO-CO- и E обозначает O-защитную группу, полученного на стадии
схемы 6 производят восстановление соединения формулы II, в которой -A- является двухвалентным радикалом -N(R1)-CO-CO- и E обозначает O-защитную группу, полученного на стадии 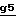 схемы 5. Восстановление осуществляют с помощью такого восстановителя, как алюмогидрид лития, в инертном растворителе, таком как эфир (например, тетрагадрофуран, 1,2-диметоксиэтан или диэтиловый эфир), или ароматический растворитель, такой как толуол, при температуре от комнатной до температуры кипения растворителя с обратным холодильником.
схемы 5. Восстановление осуществляют с помощью такого восстановителя, как алюмогидрид лития, в инертном растворителе, таком как эфир (например, тетрагадрофуран, 1,2-диметоксиэтан или диэтиловый эфир), или ароматический растворитель, такой как толуол, при температуре от комнатной до температуры кипения растворителя с обратным холодильником.
O-защитную группу при желании удаляют на стадии  кислотным гидролизом, используя описанные выше способы, с получением соединения формулы II, в которой E обозначает водород.
кислотным гидролизом, используя описанные выше способы, с получением соединения формулы II, в которой E обозначает водород.
Соединения формулы II, в которой -A- является двухвалентным радикалом -N(R1)-CO- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 7 (см. в конце описания), в которой m, R1 и Ar1 имеют значения, указанные для соединения формулы I, и Pr1 имеет значения, представленные в схеме 2.
На стадии  в соединении формулы XXXII, полученном на стадии
в соединении формулы XXXII, полученном на стадии 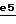 схемы 5, защищают гидроксил, используя способы, известные специалистам в этой области.
схемы 5, защищают гидроксил, используя способы, известные специалистам в этой области.
На стадии  полученное соединение формулы XXXIII подвергают взаимодействию с реакционноспособным производным угольной кислоты, таким как 1,1'-карбонилдиимидазол, фосген в толуоле или паранитрофенилхлорформиат, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, с получением целевого соединения формулы II, в которой E обозначает O-защитную группу. Эту реакцию осуществляют в инертном растворителе, таком как хлорированный растворитель (например, 1,2-дихлорэтан или дихлорметан), эфир (например, тетрагидрофуран), амид (например, N,N-диметилформамид) или ароматический растворитель (например, толуол), при температуре от -60oC до 60oC.
полученное соединение формулы XXXIII подвергают взаимодействию с реакционноспособным производным угольной кислоты, таким как 1,1'-карбонилдиимидазол, фосген в толуоле или паранитрофенилхлорформиат, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, с получением целевого соединения формулы II, в которой E обозначает O-защитную группу. Эту реакцию осуществляют в инертном растворителе, таком как хлорированный растворитель (например, 1,2-дихлорэтан или дихлорметан), эфир (например, тетрагидрофуран), амид (например, N,N-диметилформамид) или ароматический растворитель (например, толуол), при температуре от -60oC до 60oC.
Соединение формулы II, в которой E обозначает водород, при желании получают на стадии 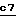 с помощью способов, известных специалистам в этой области.
с помощью способов, известных специалистам в этой области.
Соединения формулы II, в которой -A- является двухвалентным радикалом -O-CH2- и E обозначает водород или O-защитную группу, получают в соответствии со схемой 8 (см. в конце описания), в которой m и Ar1 имеют значения, указанные для соединения формулы I, и Pr2 имеет значения, представленные в схеме 2.
На стадии 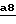 схемы 8 соединение формулы XXVII подвергают взаимодействию с водным раствором формальдегида в инертном растворителе, таком как тетрагидрофуран, при температуре от комнатной до температуры кипения растворителя с обратным холодильником с получением соединения целевой формулы II, в которой E обозначает O-защитную группу.
схемы 8 соединение формулы XXVII подвергают взаимодействию с водным раствором формальдегида в инертном растворителе, таком как тетрагидрофуран, при температуре от комнатной до температуры кипения растворителя с обратным холодильником с получением соединения целевой формулы II, в которой E обозначает O-защитную группу.
Используя описанные выше способы, O-защитную группу Pr2 удаляют щелочным гидролизом (стадия  ) с получением соединения формулы II, в которой E обозначает водород.
) с получением соединения формулы II, в которой E обозначает водород.
Рассматриваемые ниже соединения формул XVIII, XIX, XXIII, XXIV, XXVII, XXXI, XXX, XXXII и XXXIII и соединения формулы XXXIV являются новыми продуктами, которые представляют собой основные промежуточные соединения, используемые для получения соединений формулы I.
Таким образом, соединения формулы
где m и Ar1 имеют значения, указанные для соединения формулы I;
E имеет значения, указанные для соединения формулы II; и
L является цианогруппой или аминометильной группой,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой, являются новыми и входят в объем данного изобретения.
Соединения формулы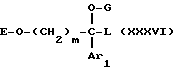
где m, E и L имеют указанные выше значения; и
G является водородом или O-защитной группой,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой, являются новыми и входят в объем данного изобретения.
Соединения формулы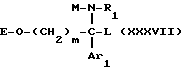
где m, Ar1, E и L имеют указанные выше значения;
R1 имеет значения, указанные для соединения формулы I; и
M является водородом или N-защитной группой,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой, являются новыми и входят в объем данного изобретения.
Соединения формул XXXV, XXXVI и XXXVII получают в соответствии с описанными выше способами. В частности, соединения с удаленными защитными группами (E = G = M = H) получают после удаления O-защитной или N-защитной групп с помощью способов, известных специалистам в этой области.
Таким образом, другим объектом данного изобретения являются соединения формулы
где m и Ar1 имеют значения, указанные для соединения формулы I;
E является водородом или O-защитной группой;
L является цианогруппой или аминометильной группой;
D является группой, выбираемой из
G является водородом или O-защитной группой;
M является водородом или N-защитной группой; и
R1 имеет значения, указанные для соединения формулы I,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой.
Пиперидины формулы XI являются известными соединениями или могут быть получены известными способами, которые описаны в заявках на европейский патент N 0428434, N 0474561, N 0512901 и N 0515240.
Пиперидины формулы XI можно также получить в соответствии со способами, хорошо известными специалистам в этой области, которые, например, описаны в следующих публикациях:
J. Heterocyclic Chem., 1986, 22, 73-75;
J. Chem. Soc., 1950, 1469;
J. Chem. Soc., 1945, 917;
J. Pharmaceutical Sci., 1972, 61, 1316-1317;
J. Org. Chem., 1957, 22, 1484-1489;
Chem. Ber., 1975, 108, 3475-3482.
Соединения формулы XI обычно получают в защищенном виде по азоту пиперидина; а после снятия защиты получают сами соединения формулы XI.
В частности, чтобы получить, например, соединение формулы XI, в которой J'1 является группой
где Ar2 обозначает пирид-2-ильный радикал, n равен 0 и R'3 обозначает гидроксил, 2-бромпиридин подвергают взаимодействию с 1-бензилпиперид-4-оном в присутствии основания, такого как бутиллитий. Целевой 4-гидрокси-4-(пирид-2-ил)-пиперидин получают после удаления N-защитной группы.
Чтобы получить соединение формулы XI, в которой R'3 является группой -NR4R5, где каждый из R4 и R5 обозначает водород, соединение формулы XI, в которой R'3 является ацетамидогруппой, гидролизуют в кислотной среде.
Соединение формулы XI, в которой R'3 является группой -NR4R5, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, получают с помощью реакции Бруйлантса или ее модификации (Bull. Soc. Chim. Belges, 1924, 22, 467 и Tetrahedron Letters, 1988, 22 (52), 6827-6830).
Чтобы получить соединение формулы XI, в которой R'3 является группой -CH2-NR12R13, где каждый из R12 и R13 обозначает водород, восстанавливают соединение формулы XI, в которой R'3 является циано. Восстановление
осуществляют в соответствии с способами, хорошо известными специалистам в этой области.
Соединение формулы XI, в которой R'3 является группой -CH2-CH2-NR12R13, где каждый из R12 и R13 обозначает водород, получают из соединения формулы XI, в которой R'3 является группой -CH2-CH2-OH, с помощью способа, описанного в журнале J. Med. Chem., 1989, 32, 391-396, или его модификации.
Соединение формулы XI, в которой R'3 является группой -NR4R5, где R4 обозначает водород и R5 обозначает (C1-C4)-алкил или соответственно (C3-C7)-циклоалкилметил или бензил, можно получить путем восстановления соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6COR7, где q равен нулю, R6 обозначает водород и R7 обозначает водород или (C1-C6)-алкил либо соответственно (C3-C7)-циклоалкил или фенил. Эту реакцию осуществляют с помощью восстановителя, такого как алюмогидрид лития, в растворителе, таком как тетрагидрофуран, при температуре кипения растворителя с обратным холодильником.
В результате осуществления аналогичной реакции получают соединения формулы XI, в которой R'3 является группой -NR4R5, где R4 обозначает (C1-C7)-алкил и R5 обозначает (C1-C7)-алкил или соответственно (C3-C7)-циклоалкилметил или бензил, исходя из соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6COR7, где q равен нулю, R6 обозначает (C1-C7)-алкил и R7 обозначает водород или (C1-C6)-алкил, или соответственно (C3-C7)-циклоалкил или фенил.
Аналогичным образом, соединения формулы XI в которой R'3 является группой -CH2-NR12R13 или соответственно -CH2CH2NR12R13, где R12 обозначает водород или (C1-C7)-алкил и R13 обозначает (C1-C7)-алкил, (C3-C7)-циклоалкилметил или бензил, можно получить из соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6COR7, где q равен соответственно 1 или 2, R6 обозначает водород или (C1-C7)-алкил и R7 обозначает водород, (C1-C6)-алкил, (C3-C7)-циклоалкил или фенил.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-NR6COR7, где R6 и R7 вместе обозначают группу -(CH2)3- или -(CH2)4-, получают с помощью способа, описанного в журнале J. Med. Chem., 1985, 28, 46-50, или его модификации.
Соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6C(= W1)R7, где W1 обозначает атом кислорода, q равен 0, 1 или 2, R6 обозначает водород или (C1-C7)-алкил и R7 обозначает водород или соответственно (C1-C7)-алкил, фенил, бензил, пиридил, (C3-C7)-циклоалкил, возможно замещенный фурил, тиенил, пирролил или имидазолил, получают в результате взаимодействия муравьиной кислоты в уксусном ангидриде или соответственно соответствующего ангидрида (R7CO)2O или хлорангидрида R7COCl, с соединением формулы XI, в которой R'3 является группой -NHR6, -CH2-NHR6 или -CH2-CH2-NHR6, в присутствии такого основания, как триэтиламин.
Специалистам в этой области также должно быть понятно, что соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6-C(=W1)R7, где q, R6 и W1 имеют указанные выше значения и R7 обозначает винильную группу, можно получить в результате взаимодействия с акрилоилхлоридом.
Аналогичным образом, соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6COOR8, получают в результате взаимодействия соединения формулы XI, в которой R'3 является группой -NHR6, -CH2-NHR6 или -CH2-CH2-NHR6, с хлорформиатом формулы ClCOOR8 в присутствии такого основания, как триэтиламин.
Соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6SO2R9, получают в результате взаимодействия соединения формулы XI, в которой R'3 является группой -NHR6, -CH2-NHR6 или -CH2-CH2-NHR6, с сульфонилхлоридом формулы ClSO2R9 в присутствии такого основания, как триэтиламин.
Соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6C(= W1)NR10R11, где R10 обозначает водород и W1 - атом кислорода, получают в результате взаимодействия соединения формулы XI, в которой R'3 является группой -NHR6, -CH2-NHR6 или -CH2-CH2-NHR6, с изоцианатом формулы R11N=C=O в присутствии такого основания, как триэтиламин.
Соединения формулы XI, в которой R'3 является группой -(CH2)q-NR6C(= W1)NR10R11, где R10 обозначает (C1-C7)-алкил и W1 - атом кислорода, получают в результате взаимодействия соединения формулы XI, в которой R'3 является группой -NHR6, -CH2-NHR6 или -CH2-CH2-NHR6, с карбамоилхлоридом формулы ClCONR10R11 в присутствии такого основания, как триэтиламин.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-NR6CONR10R11, можно также получить в результате взаимодействия соединения HNR10R11 с соединением формулы XI, в которой R'3 является группой -(CH2)q-NR6COOR8, где R8 обозначает фенил.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-NR6COOR8, где q = 0 и R6 обозначает водород, можно также получить в результате взаимодействия соединения R8OH с соединением формулы XI, в которой R'3 является изоцианатогруппой -N=C=O.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-NR6CONR10R11, где q = 0 и R6 обозначает водород, можно также получить в результате взаимодействия соединения NHR10R11 с соединением формулы XI, в которой R'3 является изоцианатогруппой.
Соединение формулы XI, в которой R'3 является изоцианатогруппой, получают из соединения формулы XI, в которой R'3 является карбоксилом, в соответствии со способом, описанным в Organic Synthesis, 51, 48-52.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-NR6C(= W1)R7, где W1 обозначает атом серы, получают из соответствующего соединения формулы XI, в которой атом азота пиперидина защищен защитной группой и W1 является атомом кислорода, в результате взаимодействия с пентасульфидом фосфора или реактивом Лавессона, [2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфидом], с последующим удалением группы, защищающей атом азота пиперидина.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-NR6C(= W1)NR10R11, где W1 обозначает атом серы, получают в результате взаимодействия соединения формулы XI, в которой атом азота пиперидина защищен защитной группой и R'3 является группой -(CH2)q-NR6CONR10R11, с пентасульфидом фосфора или реактивом Лавессона.
Соединение формулы XI, в которой R'3 является группой -(CH2)q-OH, где q соответственно равен 1 или 2, получают путем восстановления соединения формулы XI, в которой R'3 является соответственно метоксикарбонилом или метоксикарбонилметилом, в соответствии со способом, описанным в Chem. Ber., 1975, 108, 3475-3482.
Соединения формулы XI, в которой R'3 является группой -(CH2)q-OCOR17, получают в результате взаимодействия хлорангидрида R17COCl с соединениями формулы XI, в которой R'3 является группой -(CH2)q-OH, в присутствии основания, такого как триэтиламин; соединения формулы XI, в которой R'3 является группой HCOO-(CH2)q-, получают в результате взаимодействия с муравьиной кислотой.
Соединения формулы XI, в которой R'3 является группой (C1-C7)-алкил-NHCOO-(CH2)q-, получают в результате взаимодействия карбамоилхлорида (C1-C7)-алкил-NHCOCl с соединениями формулы XI, в которой R'3 является группой -(CH2)q-OH.
Те же соединения получают в результате взаимодействия изоцианата (C1-C7)-алкил-N= C=O с соединениями формулы XI, в которой R'3 является группой -(CH2)q-OH.
Соединение формулы XI, в которой R'3 является карбоксилом, можно получить путем гидролиза соединения формулы XI, в которой R'3 является циано, с помощью способов, известных специалистам в этой области.
Соединение формулы XI, в которой R'3 является карбоксиметилом, можно получить в соответствии со способом, описанным в Chem. Ber., 1975, 108, 3475-3482.
Соединение формулы XI, в которой R'3 является соответственно (C1-C7)-алкоксикарбонилом или (C1-C7)-алкоксикарбонилметилом, можно получить из соединения формулы XI, в которой R'3 является соответственно карбоксилом или карбоксиметилом, с помощью реакции этерификации, используя способы, хорошо известные специалистам в этой области.
Чтобы получить соединение формулы XI, в которой J'1 является группой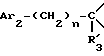
где Ar2 является возможно замещенным фенильным радикалом, n равен 1 и R'3 является (C1-C7)-алкоксикарбонилом, то защищенный 4-(C1-C7)-алкоксикарбонилпиперидин подвергают взаимодействию с возможно замещенным бензилгалогенидом в присутствии основания, такого как гидрид натрия, трет-бутилат калия или диизопропиламид натрия, в растворителе, таком как тетрагидрофуран, N,N-диметилформамид или диметилсульфоксид, при температуре от -78oC до комнатной температуры. Целевое соединение формулы XI получают после стадии удаления защитной группы.
Чтобы получить соединение формулы XI, в которой R'3 является соответственно группой -CONR10R11 или группой -CH2CONR10R11, то соединение формулы XI, в которой R'3 является соответственно карбоксилом или карбоксиметилом, подвергают взаимодействию с соединением формулы HNR10R11 в соответствии со способами, хорошо известными специалистам в этой области.
Используя описанные выше методы, получают соединения формулы XI, в которой R'3 является группой -C(=W1)NR10R11 или группой -CH2-С-(=W1)-NR10R11, в которой W1 обозначает атом серы, исходя из соединения соответствующей формулы XI, в которой W1 обозначает атом кислорода.
Чтобы получить соединение формулы XI, в которой R'3 является группой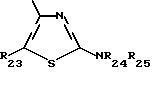
где R24 и R25 независимо друг от друга обозначают водород или (C1-C7)-алкил, соединение формулы XI, в которой R'3 является группой
где Hal обозначает атом галогена, предпочтительно бром, подвергают взаимодействию с тиомочевиной, в которой одна из аминогрупп является свободной или замещена одним или двумя (C1-C7)-алкилами.
Соединение формулы XI, в которой R'3 является группой
где R25 обозначает формил или соответственно (C1-C7)-алкилкарбонил, получают в результате взаимодействия муравьиной кислоты в уксусном ангидриде или соответственно хлорангидрида (C1-C7)-алкил-COCl с вышеуказанным соединением формулы XI, в которой атом азота пиперидина защищен и R25 является водородом, в присутствии такого основания, как триэтиламин. Целевое соединение получают после стадии удаления защитной группы.
Соединение формулы XI, в которой R'3 является группой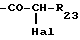
где Hal обозначает атом брома, получают путем бромирования соединения формулы XI, в которой R'3 является группой -CO-CH2-R23, с помощью известных методов.
Соединение формулы XI, в которой J'1 является группой
где R14 обозначает группу -CONR15R16 и R'3 обозначает водород, можно получить в результате взаимодействия защищенного 4-карбоксипиперидина с соединением формулы HNR15R16 в соответствии с методами, хорошо известными специалистам в этой области. Целевое соединение формулы XI получают после стадии удаления защитной группы.
Чтобы получить соединение формулы XI, в которой J'1 является группой
где R14 обозначает группу -CONR15R16 и R'3 обозначает циано, выполняют реакцию Стрекера, в ходе которой происходит взаимодействие 1-бензилпиперид-4-она с соединением формулы HNR15R16 в присутствии цианида натрия, цианида калия или метабисульфита натрия. Целевое соединение формулы XI получают после стадии удаления защитной группы.
Соединение формулы XI, в которой R'3 является группой -O-CH2-CH2-OR18, где R18 обозначает водород, можно получить в результате взаимодействия соединения формулы XI, в которой R'3 является бензоилокси, с этиленгликолем в присутствии такой кислоты, как серная кислота.
Соединения формулы XI, в которой R'3 является группой -O-CH2-CH2-OR18, где R18 обозначает (C1-C7)-алкил, получают в результате осуществления аналогичной реакции, используя 2-(C1-C7)-алкоксиэтанол.
Соединения формулы XI, в которой R'3 является группой -O-CH2-CH2-OR18, где R18 обозначает формил, получают в результате взаимодействия муравьиной кислоты с соединением формулы XI, в которой R'3 является группой -O-CH2-CH2-OH. Соединения формулы XI, в которой R'3 является группой -O-CH2-CH2-OR18, где R18 обозначает (C1-C7)-алкилкарбонил, получают в результате взаимодействия хлорангидрида кислоты C2-C8 в присутствии такого основания, как триэтиламин.
Соединение формулы XI, в которой R'3 является группой -NR6CO-COR19, где R19 обозначает (C1-C4)-алкокси, получают в результате взаимодействия соединения формулы Cl-COCOR19 с соединением формулы XI, в которой R'3 является группой -NHR6.
Соединение формулы XI, в которой R'3 является группой -CO-NR20-NR21R22, получают в результате взаимодействия соединения HNR20-NR21R22 с соединением формулы XI, в которой R'3 является хлорформилом.
Соединение формулы XI, в которой R'3 является группой
можно получить в результате взаимодействия защищенного соединения формулы XI, в которой R'3 является карбазоильной группой (-CONH-NH2), с бромцианом в соответствии со способом, описанным в журнале J. Org. Chem., 1961, 26, 88-95. Соединение формулы XI, в которой R'3 является карбазоильной группой, получают в результате взаимодействия гидразина с соединением формулы XI, в которой R'3 является хлорформилом, которое в свою очередь получают в результате взаимодействия тионилхлорида с соединением формулы XI, в которой R'3 является карбоксилом.
Пиперидины формулы XII являются известными соединениями или могут быть получены известными способами.
Хинуклидины формулы XIII являются известными соединениями или могут быть получены известными способами, описанными T. Perrine, J. Org. Chem., 1957, 22, 1484-1489.
Пиридины формулы XIV являются известными соединениями или могут быть получены известными способами.
Энантиомеры соединений по данному изобретению формулы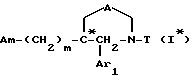
где "*" означает, что атом углерода, меченный таким образом, имеет (+) или (-) абсолютную конфигурацию; и
Am, m, Ar1, A и T имеют значения, указанные для соединений формулы I,
и их возможные соли с минеральными или органическими кислотами являются новыми соединениями, которые входят в объем данного изобретения.
Разделение рацемических смесей соединений формулы I позволяет выделить энантиомеры формулы I*.
Однако предпочтительно разделять рацемические смеси исходя из соединения формулы II или из промежуточного соединения, пригодного для получения соединения формулы II.
Таким образом, если желательно получить энантиомеры (I*) соединений формулы I, в которой -A- является двухвалентным радикалом -CH2-O-CO-, производят разделение рацемической смеси промежуточного соединения формулы
где m и Ar1 имеют значения, указанные для соединения формулы I, полученного после удаления O-защитной группы Pr из соединения формулы XIX с помощью вышеописанных методов.
Если желательно получить энантиомеры (I*) соединений формулы I, в которой -A- является двухвалентным радикалом -O-CO-, -O-CH2-CO-, -O-CH2-CH2- или -O-CH2-, производят разделение рацемической смеси промежуточного соединения формулы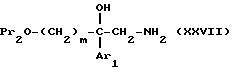
где m и Ar1 имеют значения, указанные для соединения формулы I, и Pr2 имеет значения, приведенные в схеме 2.
Если желательно получить энантиомеры (I*) соединений формулы I, в которой -A- является двухвалентным радикалом -O-CH2-CH2-, можно также произвести разделение рацемической смеси соединения формулы
где m и Ar1 имеют значения, указанные для соединения формулы I.
Если желательно получить энантиомеры (I*) соединений формулы I, в которой -A- является двухвалентным радикалом -N(R1)-CO-, -N(R1)-CO-CO- или -N(R1)-CH2-CH2-, производят разделение рацемической смеси промежуточного соединения формулы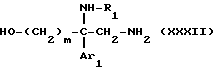
где m, Ar1 и R1 имеют значения, указанные для соединения формулы I.
Когда осуществляют разделение рацематов из промежуточных соединений формулы XXXIV, XXVII, XXXII или II [-A- = -O-CH2-CH2 и E = H], то это разделение проводят с помощью известных методов, включающих образование соли с оптически активными кислотами, например с (+) или (-) винной кислотой. Затем, применяя известные методы, такие как кристаллизация или хроматография, отделяют диастереоизомеры, после чего гидролизом получают оптически чистые энантиомеры.
Соединения вышеуказанной формулы I включают соединения, в которых один или несколько атомов водорода, углерода или йода заменены их радиоактивным изотопом, например, тритием, углеродом-14 или йодом-125. Такие меченые соединения находят применение в научно-исследовательских работах, метаболических или фармакокинетических исследованиях и биохимических испытаниях в качестве лигандов рецепторов.
Сродство соединений с рецепторами тахикинина определяли in vitro в результате выполнения нескольких биохимических испытаний с использованием радиоактивных лигандов:
1) Связывание [125I]BH-SP (вещество P, меченное йодом-125, с использованием реактива Болтона-Хантера) с рецепторами NK1 лимфобластов человека.
2) Связывание [125I]His-NKA с рецепторами NK2 двенадцатиперстной кишки или мочевого пузыря крыс.
3) Связывание [125I]His[MePhe7]NKB с рецепторами NK3 коры головного мозга крыс, морских свинок и песчанок, а также с клонированными рецепторами NK3 человека, экспрессированными в клетках яичника китайского хомячка (Buell et al., FEBS Letters, 1992, 299, 90-95).
Испытания выполняли по методу X. Emonds-Alt et al. (Eur. J. Pharmacol., 1993, 250, 403-413).
Соединения по данному изобретению обладают сродством с вышеуказанными рецепторами тахикинина при ингибиторной константе Ki ниже 10-8 М (табл. I).
В частности, соединения по настоящему изобретению являются активными веществами в фармацевтических композициях, токсичность которых находится в пределах, соответствующих требованиям, предъявляемым к применению в качестве лекарственных средств.
Соединения по настоящему изобретению обычно вводят в единичных дозах. Указанные единичные дозы предпочтительно вводят в состав фармацевтических композиций, содержащих активное вещество в смеси с фармацевтическими экципиентами.
Таким образом, другим объектом настоящего изобретения являются фармацевтические композиции, содержащие соединение формулы I или его фармацевтически приемлемую соль в качестве активного вещества в эффективном количестве.
Вышеуказанные соединения формулы I и их фармацевтически приемлемые соли можно использовать в виде суточных доз от 0,01 до 100 мг на килограмм веса тела нуждающегося млекопитающего, предпочтительно в виде суточных доз от 0,1 до 50 мг/кг. Для людей доза может предпочтительно изменяться в пределах от 0,5 до 4000 мг в день, более предпочтительно от 2,5 до 1000 мг в зависимости от возраста субъекта или типа воздействия: профилактического или лечебного.
Активные вещества, содержащиеся в фармацевтических композициях по настоящему изобретению, которые предназначены для перорального, подъязычного, ингаляционного, подкожного, внутримышечного, внутривенного, чрескожного, местного или ректального применения, можно вводить животным и людям в дозированной форме в смеси с обычными фармацевтическими наполнителями. Соответствующие дозированные формы включают таблетки, желатиновые капсулы, порошки, гранулы и растворы или суспензии для перорального введения, подъязычные и трансбуккальные препараты, аэрозоли, имплантаты, лекарственные препараты для подкожного, внутримышечного, внутривенного, назального введения и препараты для ректального введения.
При получении твердой композиции в виде таблеток основное активное вещество смешивают с фармацевтическим носителем, таким как диоксид кремния, желатин, крахмал, лактоза, стеарат магния, тальк, аравийская камедь или тому подобным. Таблетки могут быть покрыты оболочкой из сахарозы, различных полимеров или других приемлемых веществ, либо они могут быть подвергнуты обработке, сообщающей таблеткам пролонгированное или отсроченное действие, в результате чего достигается постоянное высвобождение заранее определенного количества активного вещества.
Препарат в виде желатиновых капсул получают путем смешивания активного вещества с разбавителем, таким как гликоль или эфир глицерина, и введения полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в виде сиропа или эликсира может содержать активное вещество в смеси с подсластителем, который предпочтительно является бескалорийным, метилпарабеном и пропилпарабеном в качестве антисептика, ароматизатором и соответствующим красителем.
Диспергируемые в воде гранулы или порошки могут содержать активное вещество в смеси с диспергаторами, смачивающими веществами или суспендирующими агентами, такими как поливинилпирролидон, а также подсласлителями или веществами, улучшающими вкус.
Для ректального применения предназначены свечи, получаемые при использовании связующих веществ, которые плавятся при температуре в прямой кишке, например масла какао или полиэтиленгликолей.
Для парентерального, назального или глазного применения предназначены водные суспензии, изотонические физиологические растворы или стерильные растворы для инъекций, которые содержат фармацевтически пригодные диспергаторы и/или смачивающие вещества, например пропиленгликоль или бутиленгликоль.
Для ингаляции используют аэрозоль, который содержит, например, сорбитан-триолеат или олеиновую кислоту, а также трихлорфторметан, дихлорфторметан, дихлортетрафторэтан или любой другой биологически пригодный пропеллент; кроме того, можно использовать систему, содержащую активное вещество в виде порошка, применяемого отдельно или в смеси с наполнителем.
Активное вещество при желании может также входить в состав микрокапсул вместе с одним или несколькими наполнителями или добавками.
В каждой дозированной форме активное вещество формулы I присутствует в количествах, достаточных для предполагаемых суточных доз. Как правило, каждую дозированную форму получают с учетом необходимой дозы и предполагаемого способа введения, например таблетки, желатиновые капсулы и тому подобное, саше, ампулы, сиропы и тому подобное, а также капли, так чтобы такая дозированная форма содержала от 0,5 до 1000 мг активного вещества, предпочтительно от 2,5 до 250 мг, при введении от одного до четырех раз в день.
Вышеуказанные композиции могут также содержать другие активные вещества, такие, например, как бронхолитические средства, противокашлевые средства, антигистаминные средства, противовоспалительные средства, противорвотные средства и химиотерапевтические средства.
Еще одним аспектом настоящего изобретения предусматривается применение соединений формулы I для получения лекарственных средств, предназначенных для лечения физиологических нарушений, связанных с избытком тахикининов, и всех вызываемых нейрокинином патологических состояний респираторной, желудочно-кишечной, мочевой, иммунной, сердечно-сосудистой и центральной нервной систем, а также болевых ощущений и мигрени.
Примеры, не ограничивающие объем данного изобретения, включают:
- острые и хронические боли, связанные, например, с мигренью, раком и стенокардией, а также с хроническими воспалительными процессами, такими как остеоартрит и ревматоидный артрит;
- воспаления, такие как нейрогенные воспаления, хронические воспалительные заболевания, например, обструктивные хронические респираторные заболевания, астма, аллергия, ринит, кашель, бронхит, аллергическая реакция, например, на пыльцу и клещей, ревматоидный артрит, фиброз, остеоартрит, псориаз, неспецифический язвенный колит, болезнь Крона, воспаление кишечника (слизистый колит), простатит, воспаленный мочевой пузырь, недержание мочи, цистит, уретрит и нефрит, болезни глаз, такие как конъюнктивит и витреоретинопатия, и кожные заболевания, такие как контактный дерматит, атонический дерматит, крапивница, экзема, зуд и ожоги, особенно солнечная эритема;
- заболевания иммунной системы, связанные с подавлением или стимулированием функций иммунокомпетентных клеток, например, ревматоидный артрит, псориаз, болезнь Крона, диабет, люпоидный туберкулез кожи и реакции отторжения после трансплантации органов;
- мелкоклеточные раковые заболевания легких и демиелинизация, такая как рассеянный склероз или боковой амиотрофический склероз;
- заболевания центральной нервной системы нейропсихиатрического или неврологического типа, такие как страх, бессонница, плохое настроение, депрессия, психоз, шизофрения, маниакальное состояние, старческое слабоумие, эпилепсия, болезнь Паркинсона, болезнь Альцгеймера, лекарственная зависимость, хронический алкоголизм, болезнь Дауна и хорея Хантингтона, а также нейродегенеративные заболевания и вызываемые стрессом соматические расстройства;
- заболевания желудочно-кишечного тракта, такие как тошнота, рвота любого происхождения, слизистый колит, язва желудка и двенадцатиперстной кишки, язва пищевода, понос и гиперсекреция;
- заболевания сердечно-сосудистой системы, такие как гипертензия, сосудистые мигрени, отек, тромбоз, стенокардия, спазмы сосудов, заболевания системы кровообращения вследствие расширения кровеносных сосудов, симметричная гангрена, фиброз и коллагенозы; и
- нарушения частоты и ритма сердечных сокращений, в частности, вызываемые болью или стрессом.
В объем настоящего изобретения входит также способ лечения указанных заболеваний лекарственными препаратами по данному изобретению в указанных выше дозах.
В способах получения и примерах использованы следующие аббревиатуры:
Me, OMe: метил, метокси
Et, OEt: этил, этокси
EtOH: этанол
MeOH: метанол
Эфир: диэтиловый эфир
ДМФ: диметилформамид
ДМСО: диметилсульфоксид
ДХМ: дихлорметан
ТГФ: тетрагидрофуран
AcOEt: этилацетат
Na2CO3: карбонат натрия
NaHCO3: бикарбонат натрия
NaCl: хлорид натрия
Na2SO4: сульфат натрия
MgSO4 сульфат магния
NaOH: гидроксид натрия
HCl: хлористо-водородная кислота
TFA: трифторуксусная кислота
KCN: цианид калия
Na2S2O5: метабисульфит натрия
ДБУ: 1,8-диазабицикло[5.4.0]ундец-7-ен
NH4Cl: хлорид аммония
Т.п.: температура плавления
Силикагель: силикагель 60Н, поставляемый фирмой Merck (Дармштадт)
ЯМР: ядерный магнитный резонанс
δ: химический сдвиг
с: синглет
ш.с.: широкий синглет
д: дублет
т: триплет
к: квадруплет
м: мультиплет
н.с.: неразделенные сигналы
Способ получения 1.1
5-(3,4-Дихлорфенил)-5-[2- (тетрагидропиран-2-илокси) этил] тетрагидро- 2H-1,3-оксазин-2-он
A) 2-(3,4-Дихлорфенил)-4- (тетрагидропиран-2- илокси)бутаннитрил
Суспензию 17,75 г гидрида натрия (80% дисперсия в масле) в 750 мл ТГФ охлаждают на ледяной бане, добавляют по каплям раствор 100 г 3,4-дихлорфенилацетонитрила в 250 мл ТГФ, после чего реакционную смесь перемешивают в течение двух часов при комнатной температуре. Полученную смесь охлаждают до -20oC, по каплям добавляют раствор 112,36 г 1-бром-2-(тетрагидропиран-2-илокси)этана в 120 мл ТГФ и реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу дважды промывают буферным раствором с pH 4, буферным раствором с pH 7 и дважды насыщенным раствором NaCl, сушат над Na2SO4 и выпаривают растворитель в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента толуол, а затем смесь толуола и AcOEt (100/3 в объемном отношении), с получением 113,5 г целевого продукта, который используют без дальнейшей очистки.
B) 2-(3,4-Дихлорфенил)-2- (гидроксиметил)-4-(тетрагидропиран-2- илокси)бутаннитрил
Смесь 12,56 г соединения, полученного на предыдущей стадии, 9,6 г 37% водного раствора формальдегида и 0,3 г ДБУ в 25 мл 1,2-диметоксиэтана нагревают с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу дважды промывают водой, дважды буферным раствором с pH 4, дважды водой и дважды насыщенным раствором NaCl, сушат над Na2SO4 и выпаривают растворитель в вакууме с получением 17 г целевого продукта, который используют без дальнейшей очистки.
C) 2-(3,4-Дихлорфенил)-2- (гидроксиметил)-4-(тетрагидропиран-2- илокси)бутиламин
Смесь 17 г соединения, полученного на предыдущей стадии, 6 г никеля Ренея в 300 мл EtOH и 40 мл 20% водного раствора аммиака гидрируют в течение 5 часов при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в ДХМ, органическую фазу промывают водой и насыщенным раствором NaCl, сушат над MgSO4 и выпаривают растворитель в вакууме с получением 26,5 г целевого продукта в виде масла, которое используют без дальнейшей очистки.
D) 5-(3,4-Дихлорфенил)-5-[2- (тетрагидропиран-2-илокси) этил]тетрагадро- 2H-1,3-оксазин-2-он
24,6 г 20% раствора фосгена в толуоле, разбавленном 150 мл ДХМ, охлаждают до -70oC, по каплям добавляют раствор 16,5 г соединения, полученного на предыдущей стадии, и 5,7 г триэтиламина в 100 мл ДХМ, после чего реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток растворяют в смеси воды и AcOEt и продукт, который кристаллизуется на поверхности раздела фаз, центрифугируют с получением первого сбора целевого продукта. После декантации фильтрата органическую фазу промывают водой, буферным раствором с pH 4 и насыщенным раствором NaCl, сушат над MgSO4 и выпаривают растворитель в вакууме. Остаток растворяют в AcOEt и образовавшийся кристаллический продукт центрифугируют с получением второго сбора этого продукта. Получено в общей сложности 4,5 г целевого продукта.
Способ получения 1.2
5-(3,4-Дихлорфенил)-5-[3- (тетрагидропиран-2-илокси) пропил]тетрагидро- 2H-1,3-оксазин-2-он
A) 2-(3,4-Дихлорфенил)-5- (тетрагадропиран-2-илокси)пентаннитрил
К суспензии 12 г гидрида натрия (55% дисперсия в масле) в 175 мл ТГФ при температуре ниже 20oС по каплям добавляют раствор 50,8 г 2,4-дихлорфенилацетонитрила в 250 мл ТГФ, после чего реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Полученную смесь охлаждают до -20oC, по каплям добавляют раствор 62,5 г 1-бром-3-(тетрагидропиран-2-илокси)пропана в 60 мл ТГФ и реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Эту смесь выливают в раствор 31 г хлорида аммония в 1,4 литра воды и экстрагируют эфиром, соединенные органические фазы промывают насыщенным раствором HCl и сушат над MgSO4, растворители выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента толуол, а затем смесь толуола и AcOEt (95/5 в объемном отношении), с получением 64 г целевого продукта, который используют без дальнейшей очистки.
B) 2-(3,4-Дихлорфенил)-2- (гидроксиметил)-5-(тетрагидропиран-2- илокси)пентаннитрил
Смесь 15 г соединения, полученного на предыдущей стадии, 11,2 г 37% водного раствора формальдегида и 0,35 г ДБУ в 30 мл 1,2-диметоксиэтана нагревают с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой, дважды промывают буферным раствором с pH 4, дважды водой и дважды насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента толуол, а затем смесь толуола и AcOEt (80/20 в объемном отношении), с получением 15,5 г целевого продукта, который используют без дальнейшей очистки.
C) 2-(3,4-Дихлорфенил)-2- (гидроксиметил)-5-(тетрагидропиран-2- илокси)пентиламин
Смесь 15,5 г соединения, полученного на предыдущей стадии, 5 г никеля Ренея в 200 мл EtOH и 40 мл 20% водного раствора аммиака гидрируют в течение 5 часов при температуре 30oC и атмосферном давлении. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в ДХМ, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 14,9 г целевого продукта в виде масла, которое используют без дальнейшей очистки.
D) 5-(3,4-Дихлорфенил)-5-[3- (тетрагидропиран-2- илокси) пропил]тетрагидро-2H- 1,3-оксазин-2-он
21,4 г 20% раствора фосгена в толуоле, разбавленном 120 мл ДХМ, охлаждают до -70oC, по каплям добавляют раствор 14,9 г соединения, полученного на предыдущей стадии, и 4,98 г триэтиламина в 80 мл ДХМ, после чего реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 12,5 г целевого продукта, который используют без дальнейшей очистки.
Способ получения 1.3
6-(3,4-Дихлорфенил)-6-[2- (тетрагидропиран-2-илокси) этил]морфолин-3-он
A) 2-(3,4-Дихлорфенил)- 2-гидроксиацетонитрил
Смесь 70 г 3,4-дихлорбензальдегида и 90 г Na2S2O5 в 300 мл воды перемешивают в течение ночи при комнатной температуре. Реакционную смесь охлаждают до 0oC, по каплям добавляют раствор 52 г цианида калия (KCN) в 100 мл воды и реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Смесь экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 76 г целевого продукта, который используют без дальнейшей очистки.
B) 2-(3,4-Дихлорфенил)-2- (тетрагидропиран-2-илокси)ацетонитрил
Раствор 76 г соединения, полученного на предыдущей стадии, и 0,25 г моногидрата паратолуолсульфокислоты в 300 мл ДХМ охлаждают до 0 С, по каплям добавляют раствор 39 г 3,4-дигидро-2H-пирана в 50 мл ДХМ, после чего реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Смесь промывают насыщенным раствором NaHCO3 и водой, органическую фазу сушат над Na2SO4, растворитель выпаривают в вакууме и после кристаллизации из пентана при 0oC получают 33 г целевого продукта. Т.п. = 61oC.
C) 4-(Бензоилокси)-2-(3,4 -дихлорфенил)-2-(тетрагидропиран-2- илокси)бутаннитрил
56 мл 2 М раствора диизопропиламида лития и ТГФ охлаждают до -60oC, по каплям добавляют раствор 32 г соединения, полученного на предыдущей стадии, в 50 мл ТГФ, после чего смесь перемешивают в течение 1 часа при температуре -60oC. Затем при температуре -60oC по каплям добавляют раствор 25,4 г 2-бромэтилбензоата в 50 мл ТГФ и реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и буферным раствором с pH 4 и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь толуола и AcOEt (100/5 в объемном отношении), с получением 34 г целевого продукта, который используют без дальнейшей очистки.
D) 4-(Бензоилокси)-2-(3,4- дихлорфенил)-2-(тетрагидропиран-2- илокси)бутиламин
Смесь 34 г соединения, полученного на предыдущей стадии, 10 г никеля Ренея в 400 мл EtOH и 40 мл концентрированного раствора аммиака гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси ДХМ и MeOH (от 100/1 до 100/3 в объемном отношении), с получением 16 г целевого продукта, который используют без дальнейшей очистки.
E) N-(2-Бромацетил)-4- (бензоилокси)-2-(3,4- дихлорфенил)-2- гидроксибутиламин
Раствор 16 г соединения, полученного на предыдущей стадии, и 4,8 г триэтиламина в 100 мл ДХМ охлаждают до -60oC, по каплям добавляют раствор 5,68 г бромацетилхлорида в 20 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут. Смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и буферным раствором с pH 4 и сушат над Na2SO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в минимальном количестве MeOH и подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, и растворители выпаривают в вакууме. Остаток растворяют в воде и экстрагируют AcOEt, органическую фазу промывают насыщенным раствором NaHCO3 и водой, сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 16 г целевого продукта, который используют без дальнейшей очистки.
F) 6-(3,4-Дихлорфенил) -6-(2-гидроксиэтил) морфолин-3-он
Смесь 16 г соединения, полученного на предыдущей стадии, 50 мл пропан-2-ола, 15 мл 10 н. раствора NaOH и 10 мл ДМФ перемешивают в течение 4 часов. Реакционную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси ДХМ и MeOH (от 100/3 до 100/5 в объемном отношении), с получением 6,1 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,05 части на миллион: м: 2H
3,0 - 4,4 части на миллион: н.с.: 6H
4,5 части на миллион: т: 1H
7,3 - 8,3 части на миллион: н.с.: 4H
G) 6-(3,4-Дихлорфенил)-6-[2- (тетрагидропиран-2-илокси) этил]морфолин-3-он
Раствор 1,7 г соединения, полученного на предыдущей стадии, и 0,003 г моногидрата паратолуолсульфокислоты в 50 мл ДХМ охлаждают до 0oC, по каплям добавляют 0,588 г 3,4-дигидро-2H-пирана в 10 мл ДХМ, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают насыщенным раствором NaHCO3 и водой, сушат над Na2SO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь ДХМ и MeOH (100/2 в объемном отношении), с получением 1,8 г целевого продукта, который используют без дальнейшей очистки.
Способ получения 1.4
6-[2-(Бензоилокси) этил]-6-(3,4-дихлорфенил) морфолин-3-он
Смесь 4,8 г соединения, полученного на стадии E способа получения 1,3, и 1,4 г K2CO3 в 100 мл ксилола нагревают в течение ночи при температуре 130oC. После охлаждения до комнатной температуры реакционную смесь фильтруют и фильтрат концентрируют в вакууме. Остаток экстрагируют эфиром, органическую фазу промывают буферным раствором с pH 2 и водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь ДХМ и MeOH (100/1 в объемном отношении), с получением 1,4 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,45 части на миллион: м: 2H
3,75 части на миллион: система AB: 2H
3,9 - 4,5 части на миллион: н.с.: 4H
7,4 - 7,9 части на миллион: н.с.: 8H
8,25 части на миллион: ш.с.: 1H
Это соединение можно также получить с помощью трехстадийного способа, описанного ниже.
A') Гидрохлорид 4-(бензоилокси)-2- (3,4-дихлорфенил)-2- гидроксибутиламина
Это соединение описано на стадии A способа получения 1.7.
B') N-(2-Хлорацетил)-4- (бензоилокси)-2-(3,4- дихлорфенил)-2- гидроксибутиламин
Раствор 20 г соединения, полученного на предыдущей стадии, и 10,3 г триэтиламина в 100 мл ДХМ охлаждают до 0oC, по каплям добавляют 5,8 г хлорацетилхлорида и реакционную смесь перемешивают в течение 30 минут. Смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой, буферным раствором с pH 2 и водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 22 г целевого продукта, который используют без дальнейшей очистки.
C') 6-[2-(Бензоилокси) этил]-6-(3,4-дихлорфенил) морфолин-3-он
Раствор 22 г соединения, полученного на предыдущей стадии, в 600 мл ТГФ охлаждают до -10oC, добавляют 11,42 г трет-бутилата калия и реакционную смесь перемешивают до полного растворения. Смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают буферным раствором с pH 2 и водой и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из эфира получают 12,8 г целевого продукта.
Способ получения 1.5
2-(3,4-Дихлорфенил)-2- (2-гидроксиэтил)морфолин
Суспензию 1,6 г алюмогидрида лития в 25 мл ТГФ нагревают до 60oC, по каплям добавляют раствор 4 г соединения, полученного на стадии F способа получения 1.3, в 20 мл ТГФ, после чего смесь перемешивают при температуре кипения с обратным холодильником в течение 30 минут. После охлаждения добавляют 1,6 мл воды, 1,5 мл 4 н. раствора NaOH, а затем еще 4,5 мл воды. Минеральные соли отфильтровывают на целите Celite®, фильтрат декантируют и органическую фазу упаривают в вакууме. Остаток растворяют в эфире и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 3,6 г целевого продукта.
Способ получения 1.6
2-(3,4-Дихлорфенил)-2-(3- гидроксипропил)морфолин
A) 5-(Бензоилокси)-2-(3,4 -дихлорфенил)-2-(тетрагидропиран-2- илокси)пентаннитрил
47 мл 1,5 М раствора диизопропиламида лития в ТГФ охлаждают до -60oC, по каплям добавляют раствор 19,3 г соединения, полученного на стадии B способа получения 1.3, в 100 мл ТГФ, после чего смесь перемешивают в течение 30 минут при -60oC. При температуре -60oC по каплям добавляют 17 г 3-бромпропилбензоата и реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме и после кристаллизации из гексана получают 21 г целевого продукта.
B) 5-(Бензоилокси)-2-(3,4 -дихлорфенил)-2-(тетрагидропиран-2- илокси)пентиламин
Смесь 20 г соединения, полученного на предыдущей стадии, и 7 г скелетного никелевого катализатора гидрирования Raney® в 300 мл MeOH гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой, сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 20 г целевого продукта, который используют без дальнейшей очистки.
C) N-(2-Хлорацетил)-5- (бензоилокси)-2-(3,4-дихлорфенил)- 2-(тетрагидропиран-2-илокси) пентиламин
Раствор 9 г соединения, полученного на предыдущей стадии, и 2,4 г триэтиламина в 100 мл ДХМ охлаждают до 0oC, по каплям добавляют раствор 2,23 г хлорацетилхлорида в 20 мл ДХМ, после чего реакционную, смесь перемешивают в течение 30 минут. Смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 9,5 г целевого продукта, который используют без дальнейшей очистки.
D) N-(2-Хлорацетил)-5- (бензоилокси)-2-(3,4- дихлорфенил)-2- гидроксипентиламин
К раствору 9 г соединения, полученного на предыдущей стадии, в 50 мл ДХМ и 50 мл MeOH добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, после чего смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaHCO3 и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь ДХМ и MeOH (100/3 в весовом отношении), с получением 4,7 г целевого продукта, который используют без дальнейшей очистки.
E) 6-(3,4-Дихлорфенил)-6- (3-гидроксипропил)морфолин-3-он
Смесь 2,95 г соединения, полученного на предыдущей стадии, 40 мл пропан-2-ола и 3 мл 10 н. раствора NaOH перемешивают в течение 2 часов при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси ДХМ и MeOH (от 100/2 до 100/5 в объемном отношении), с получением 0,5 г целевого соединения. Т.п. = 130-132oC.
F) 2-(3,4-Дихлорфенил)-2- (3-гидроксипропил)морфолин
Суспензию 0,82 г алюмогидрида лития в 10 мл ТГФ нагревают до 60oC, по каплям добавляют раствор 2 г соединения, полученного на предыдущей стадии, в 20 мл ТГФ, после чего смесь перемешивают в течение 30 минут при температуре кипения с обратным холодильником. После охлаждения добавляют 1 мл воды, 1 мл 4 н. раствора NaOH, а затем еще 3 мл воды. Минеральные соли отфильтровывают на целите Celite®, фильтрат декантируют и органическую фазу упаривают в вакууме. Остаток растворяют в эфире и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 2 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 0,8 - 2,2 части на миллион: н.с.: 4H
2, 5 - 3,7 части на миллион: н.с.: 8H
4,3 части на миллион: т: 1H
7,2 - 7,7 части на миллион: н.с.: 3H
Способ получения 1.7
5-[2-(Бензоилокси) этил]-5-(3,4-дихлорфенил) оксазолидин-2-он
A) Гидрохлорид 4-(бензоилокси)-2-(3,4 -дихлорфенил)-2-гидроксибутиламина
К раствору 12 г соединения, полученного на стадии D способа получения 1.3, в 50 мл MeOH при комнатной температуре добавляют насыщенный раствор газообразного HCl в эфире, пока показатель pH не становится равным 1, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Смесь концентрируют в вакууме, остаток растворяют в ДХМ, образовавшийся осадок центрифугируют и промывают эфиром и после перекристаллизации из пропан-2-ола получают 3,4 г целевого продукта. Т.п. = 200-204oC.
B) 5-[2-(Бензоилокси) этил]-5-(3,4-дихлорфенил) оксазолидин-2-он
К раствору 3 г соединения, полученного на предыдущей стадии, и 0,85 г триэтиламина в 30 мл 1,2-дихлорэтана при комнатной температуре добавляют 1,4 г 1,1'-карбонилдиимидазола, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре, а затем нагревают при температуре 50oC в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют дихлорметаном, органическую фазу промывают буферным раствором с pH 2 и водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 3 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,6 части на миллион: м: 2H
3,75 части на миллион: система AB: 2H
4,35 части на миллион: м: 2H
7,4-7,8 части на миллион: н.с.: 8H
7,9 части на миллион: с: 1H
Способ получения 1.8
6-(3,4-Дихлорфенил) -1-метил-6-[2-(тетрагидропиран -2-илокси)этил]- пиперазин-2,3-дион
A) Гидрохлорид 2-(3,4-дихлорфенил) -2-(метиламино)ацетонитрила
Смесь 10 г 3,4-дихлорбензальдегида и 9,5 мл цианотриметилсилана охлаждают на ледяной бане, добавляют 10 мг йодида цинка и перемешивают смесь в течение 30 минут при комнатной температуре. Затем добавляют 20 мл 33% раствора метиламина в EtOH и нагревают смесь при температуре 40oC в течение 2 часов. Растворитель концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу сушат над MgSO4 и фильтруют. Насыщенный раствор газообразного HCl в эфире добавляют к фильтрату до тех пор, пока pH не становится равным 1, после чего добавляют ацетон, пока не происходит осаждение продукта. Образовавшийся осадок центрифугируют, промывают эфиром и сушат с получением 12,8 г целевого продукта. Т.п. = 172oC.
B) 2-(трет-Бутоксикарбонил -N-метиламино)-2-(3,4- дихлорфенил)ацетонитрил
К водной суспензии 12,8 г соединения, полученного на предыдущей стадии, добавляют концентрированный раствор NaOH, пока pH не становится равным 13, после чего смесь экстрагируют эфиром, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток растворяют в 20 мл 1,4-диоксана, добавляют 12,5 г ди-трет-бутилдикарбоната и реакционную смесь нагревают при температуре 60oC в течение 2 часов. Смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают буферным раствором с pH 2, насыщенным раствором NaCl и 10% раствором Na2CO3 и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента гептан, а затем смесь гептана и AcOEt (96/4 в объемном отношении), с получением 12,7 г целевого продукта, который используют без дальнейшей очистки.
C) 2-(трет-Бутоксикарбонил -N-метиламино)-2-(3,4-дихлорфенил)- 4-(тетрагидропиран-2 -илокси)бутаннитрил
Раствор 11,1 г соединения, полученного на предыдущей стадии, в 60 мл ДМФ по каплям добавляют к суспензии 1,5 г гидрида натрия (60% дисперсия в масле) в 50 мл ДМФ, поддерживая температуру равной 25oC, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Затем добавляют раствор 8,1 г 1-бром-2-(тетрагидропиран-2-илокси)этана в 20 мл ДМФ и реакционную смесь нагревают при 60oC в течение 4 часов. После охлаждения до комнатной температуры полученную смесь выливают в смесь льда и буферного раствора с pH 2 и экстрагируют эфиром, органическую фазу дважды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента гептан, а затем смесь гептана и AcOEt (75/25 в объемном отношении), с получением 13,2 г целевого продукта, который используют без дальнейшей очистки.
D) 2-(трет-Бутоксикарбонил -N-метиламино)-2-(3,4- дихлорфенил)- 4-(тетрагидропиран-2-илокси) бутиламин
Смесь 13,2 г соединения, полученного на предыдущей стадии, 4 г никеля Ренея в 150 мл EtOH и 50 мл 20% водного раствора аммиака гидрируют при температуре 30oC и атмосферном давлении. Через 5 часов катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток экстрагируют эфиром, органическую фазу дважды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 12,6 г целевого продукта, который используют без дальнейшей очистки.
E) Гидрохлорид 2-(3,4-дихлорфенил) -4-гидрокси-2-(метиламино)бутиламина
Смесь 4,6 г соединения, полученного на предыдущей стадии, и 10 мл концентрированного раствора HCl в 40 мл MeOH нагревают при 70oC в течение 1 часа. Затем растворитель концентрируют в вакууме, остаток растворяют в ацетоне, образовавшийся осадок центрифугируют, промывают эфиром и сушат с получением 2,79 г целевого продукта. Т.п. = 240oC (разложение).
F) 6-(3,4-Дихлорфенил)-6- (2-гидроксиэтил)-1-метилпиперазин -2,3-дион
К водной суспензии 5,3 г соединения, полученного на предыдущей стадии, добавляют концентрированный раствор NaOH, пока pH не становится равным 13, после чего эту смесь экстрагируют эфиром, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Полученный продукт (4 г) растворяют в 50 мл EtOH, добавляют 2,57 г диэтилоксалата и реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в 60 мл толуола и нагревают с обратным холодильником в течение 70 часов. Эту смесь концентрируют в вакууме и после кристаллизации из дихлорметана получают 2,8 г целевого продукта. Т.п. = 260oC.
G) 6-(3,4-Дихлорфенил)-1- метил-6-[2- (тетрагидропиран-2-илокси) этил] пиперазин-2,3-дион
К суспензии 2,8 г соединения, полученного на предыдущей стадии, в 50 мл ДХМ добавляют 0,1 г моногидрата паратолуолсульфокислоты, а затем 1,26 мл 3,4-дигидро-2H-пирана, после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь промывают 10% раствором Na2CO3 и водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента AcOEt, а затем смесь AcOEt и MeOH (93/7 в объемном отношении), с получением 3,1 г целевого продукта, который используют без дальнейшей очистки.
Способ получения 1.9
2-(3,4-Дихлорфенил)-1- метил-2-[2-(тетрагидропиран-2- илокси)этил] пиперазин
Раствор 2 г соединения, полученного в соответствии со способом получения 1.8, в 20 мл ТГФ по каплям добавляют к суспензии 1,2 г алюмогидрида лития в 20 мл ТГФ, после чего реакционную смесь нагревают с обратным холодильником в течение 1 часа. После охлаждения добавляют 5 мл воды, минеральные соли отфильтровывают, фильтрат концентрируют в вакууме, остаток растворяют в эфире, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 1,9 г целевого продукта в виде масла.
Способ получения 1.10
6-[2-(Бензоилокси)этил]-6-(3,4 -дифторфенил)морфолин-3-он
A) 2-(3,4-Дифторфенил) -2-гидроксиацетонитрил
Раствор 80,2 г Na2S2O5 в 250 мл воды нагревают до 50oC и добавляют 50 г 3,4-дифторбензальдегида, после реакционную смесь перемешивают в течение 1 часа при температуре 50oC и оставляют для выстаивания в течение ночи при комнатной температуре. Полученную смесь охлаждают до 0oC, по каплям добавляют раствор 77,7 г цианида калия в 100 мл воды, смесь перемешивают, позволяя ей нагреваться до комнатной температуры, после чего продолжают ее перемешивать в течение 1 часа при комнатной температуре. Реакционную смесь экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 48 г целевого продукта, который используют без дальнейшей очистки.
B) 2-(3,4-Дифторфенил)-2- (тетрагидропиран-2-илокси)ацетонитрил
Раствор 48 г соединения, полученного на предыдущей стадии, и 0,2 г моногидрата паратолуолсульфокислоты в 500 мл ДХМ охлаждают до 0oC, по каплям добавляют раствор 28,6 г 3,4-дигидро-2H-пирана в 50 мл ДХМ, смесь перемешивают, позволяя ей нагреваться до комнатной температуры, после чего продолжают ее перемешивать в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают 10% раствором Na2CO3 и водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь толуола и AcOEt (100/15 в объемном отношении), с получением 43 г целевого продукта, который используют без дальнейшей очистки.
C) 4-(Бензоилокси)-2- (3,4-дифторфенил)-2-(тетрагидропиран-2- илокси)бутаннитрил
133 мл 1,5 М раствора диизопропиламида лития в ТГФ охлаждают до -60oC, по каплям добавляют раствор 43 г соединения, полученного на предыдущей стадии, в 250 мл ТГФ, после чего смесь перемешивают в течение 30 минут при температуре -60oC. Затем при температуре - 60oC по каплям добавляют раствор 45,8 г 2-бромэтилбензоата в 100 мл ТГФ и реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и буферным раствором с pH 4 и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь толуола и AcOEt (100/5 в объемном отношении), с получением 47 г целевого продукта, который используют без дальнейшей очистки.
D) 4-(Бензоилокси)-2-(3,4- дифторфенил)-2-(тетрагидропиран-2- илокси)бутиламин
Смесь 47 г соединения, полученного на предыдущей стадии, и 10 г никеля Ренея в 400 мл EtOH гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 45 г целевого продукта, который используют без дальнейшей очистки.
E) Гидрохлорид 4-(бензоилокси) -2-(3,4-дифторфенил)-2-гидроксибутиламина
К раствору 45 г соединения, полученного на предыдущей стадии, в 250 мл MeOH при комнатной температуре добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в эфире, образовавшийся осадок центрифугируют и промывают эфиром и после перекристаллизации из пропан-2-ола получают 15 г целевого продукта. Т.п. = 202-204oC.
F) N-(2-Хлорацетил)-4- (бензоилокси)-2-(3,4-дифторфенил)-2- гидроксибутиламин
Раствор 12,2 г соединения, полученного на предыдущей стадии, и 7,88 г триэтиламина в 100 мл ДХМ охлаждают до 0oC, по каплям добавляют раствор 3,85 г хлорацетилхлорида в 100 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют смесью эфира и AcOEt (50/50 в объемном отношении), органическую фазу промывают водой, буферным раствором с pH 4, еще раз водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 13,5 г целевого продукта, который используют без дальнейшей очистки.
G) 6-[2-(Бензоилокси) этил]-6-(3,4-дифторфенил) морфолин-3-он
Смесь 13,5 г соединения, полученного на предыдущей стадии, и 20,7 г K2CO3 в 100 мл толуола нагревают с обратным холодильником в течение ночи. Реакционную смесь концентрируют в вакууме, остаток растворяют в эфире и смесь фильтруют. Фильтрат промывают водой, буферным раствором с pH 2 и водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь ДХМ и MeOH (100/1 в объемном отношении), с получением 4,9 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,35 части на миллион: м : 2H
3,65 части на миллион: система AB : 2H
3,8 - 4,35 части на миллион: н.с. : 4H
7,1 -7,8 части на миллион: н.с. : 8H
8,2 части на миллион: ш.с. : 1H
Способ получения 1.11
2-(3,4-Дифторфенил)-2- (2-гидроксиэтил)морфолин
К раствору 2,8 г соединения, полученного в соответствии со способом получения 1.10, в 10 мл ТГФ при комнатной температуре по каплям добавляют суспензию 1,8 г алюмогидрида лития в 20 мл ТГФ, после чего эту смесь нагревают с обратным холодильником в течение 5 часов. После охлаждения добавляют 2 мл воды, 2 мл 4 н. раствора NaOH, а затем еще 6 мл воды. Минеральные соли отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в ДХМ, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, и экстрагируют водой, водную фазу промывают эфиром, подщелачивают до pH 8, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 0,77 г целевого продукта, который используют без дальнейшей очистки в примере 27, стадия A.
Способ получения 1.12
5-[2-(Бензоилокси) этил]-5-(3,4-дифторфенил) оксазолидин-2-он
К раствору 2,1 г соединения, полученного на стадии E способа получения 1.10, и 0,63 г триэтиламина в 50 мл 1,2-дихлорэтана, при комнатной температуре добавляют 1 г 1,1'-карбонилдиимидазола, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре и нагревают в течение 2 часов при 50oC. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой, буферным раствором с pH 2 и водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 1,5 г целевого продукта.
Способ получения 1.13
5-(3,4-Дихлорфенил)-1-метил -5-[2-(тетрагидропиран-2- илокси) этил]имидазолидин-2-он
A) 2-(3,4-Дихлорфенил)-2- (метиламино)-4-(тетрагадропиран-2- илокси)бутиламин
Смесь 3,4 г соединения, полученного на стадии E способа получения 1.8, 0,05 г моногидрата паратолуолсульфокислоты и 2,1 мл 3,4-дигидро-2H-пирана в 30 мл ДМФ нагревают при температуре 60oC в течение 45 минут. Реакционную смесь выливают на лед, подщелачивают, добавляя концентрированный раствор NaOH, и экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 4,3 г целевого продукта, который используют без дальнейшей очистки.
B) 5-(3,4-Дихлорфенил)-1-метил -5-[2-(тетрагидропиран-2- илокси) этил] имидазолидин-2-он
К раствору 3,2 г соединения, полученного на предыдущей стадии, в 100 мл 1,2-дихлорэтана добавляют 1,6 г 1,1'-карбонилдиимидазола, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Затем эту смесь нагревают при температуре 60oC в течение 2 часов и концентрируют в вакууме. Остаток экстрагируют AcOEt, органическую фазу промывают буферным раствором с pH 2, насыщенным раствором NaCl и 10% раствором Na2CO3 и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (98/2 в объемном отношении), с получением 2,4 г целевого продукта в виде масла.
Способ получения 1.14
6-[3-(Бензоилокси) пропил]-6-(3,4-дихлорфенил) морфолин-3-он
Смесь 7,5 г соединения, полученного на стадии D способа получения 1.6, 9,3 г K2CO3 и 3,75 г йодида натрия в 100 мл метилэтилкетона нагревают с обратным холодильником в течение ночи. После охлаждения реакционную смесь концентрируют в вакууме, остаток растворяют в эфире, минеральные соли отфильтровывают, фильтрат промывают буферным раствором с pH 2 и водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем градиент смеси дихлорметана и MeOH (от 99/1 до 98/2 в объемном отношении), с получением 1,3 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,1 - 2,3 части на миллион: н.с.: 4H
3,65 части на миллион: система AB: 2H
3,8 - 4,4 части на миллион: н.с.: 4H
7,2 - 8,05 части на миллион: н.с.: 8H
8,2 части на миллион: с: 1H
Способ получения 1.15
6-[2-(Бензоилокси) этил]-6-(3,4-дифторфенил) морфолин-3-он, (-) изомер
A) 4-(Бензоилокси)-2-(3,4 -дифторфенил)-2-гидроксибутиламин, (+) изомер
Раствор 41 г L-(+)-винной кислоты в 1200 мл MeOH нагревают при температуре кипения с обратным холодильником, затем в один прием добавляют раствор 81,4 г соединения, полученного на стадии E способа получения 1.10, в виде свободного основания в 200 мл MeOH, после чего эту смесь оставляют для кристаллизации на 48 часов, позволяя смеси охладиться до комнатной температуры. Образовавшиеся кристаллы центрифугируют и промывают эфиром с получением 42,5 г соли винной кислоты.
[α]
Полученную соль перекристаллизовывают из 1450 мл 70o EtOH и после центрифугирования и промывки образовавшихся кристаллов эфиром получают 35 г соли винной кислоты.
[α]
Полученную соль растворяют в 2000 мл AcOEt, добавляют 40 мл 10% раствора Na2CO3 и после перемешивания и декантации органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диизопропилового эфира и пентана получают 23,5 г целевого продукта. Т.п. = 100,5-100,6oC.
[α]
B) N-(2-Хлорацетил)-4- (бензоилокси)-2-(3,4- дифторфенил)-2- гидроксибутиламин, (+) изомер
Раствор 23,5 г соединения, полученного на предыдущей стадии, и 8,1 г триэтиламина в 300 мл дихлорметана охлаждают до 0oC, по каплям добавляют раствор 8,3 г хлорацетилхлорида в 50 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут при 0oC. Полученную смесь концентрируют в вакууме, остаток экстрагируют смесью AcOEt и эфира (50/50 в объемном отношении), органическую фазу промывают буферным раствором с pH 2 и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диизопропилового эфира и пентана получают 24,8 г целевого продукта.
[α]
C) 6-[2-(Бензоилокси) этил]-6-(3,4-дифторфенил) морфолин-3-он, (-) изомер
Раствор 23,6 г соединения, полученного на предыдущей стадии, в 750 мл ТГФ охлаждают до 0oC, добавляют 13,7 г трет-бутилата калия и реакционную смесь перемешивают в течение 15 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают буферным раствором с pH 2 и водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/1 до 100/1,5 в объемном отношении), и после кристаллизации из пентана получают 14,5 г целевого продукта.
[α]
Способ получения 1.16
6-[2-(Бензоилокси) этил]-6-(3,4- дифторфенил)морфолин-3-он, (+) изомер
A) 4-(Бензоилокси)-2-(3,4 -дифторфенил)-2-гидроксибутиламин, (-) изомер
Жидкости, полученные после центрифугирования и промывки в процессе кристаллизации и перекристаллизации соли винной кислоты, полученной на стадии A способа получения 1.15, концентрируют в вакууме. Остаток обрабатывают 10% раствором Na2CO3 и экстрагируют AcOEt, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 49 г аминоспирта в виде смеси изомеров. Аминоспирт растворяют в 120 мл MeOH и этот раствор в один прием добавляют к нагреваемому с обратным холодильником раствору 24,1 г D-(-)-винной кислоты в 730 мл MeOH. Эту смесь оставляют для кристаллизации на 48 часов, позволяя смеси охладиться до комнатной температуры. Образовавшиеся кристаллы центрифугируют и промывают эфиром с получением 40,7 г соли винной кислоты.
Полученную соль перекристаллизовывают из 1445 мл 70o EtOH и после центрифугирования и промывки образовавшихся кристаллов эфиром получают 35 г соли винной кислоты. Полученную соль растворяют в 2000 мл AcOEt, добавляют 10% раствор Na2CO3 и после перемешивания и декантации органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 27 г целевого продукта. Т.п. = 102oC.
[α]
B) N-(2-Хлорацетил)-4- (бензоилокси)-2-(3,4- дифторфенил)-2- гидроксибутиламин, (-) изомер
Раствор 27 г соединения, полученного на предыдущей стадии, и 13 мл триэтиламина в 500 мл ДХМ охлаждают до -30oC, по каплям добавляют 5,8 г хлорацетилхлорида, после чего реакционную смесь перемешивают в течение 15 минут. Затем полученную смесь промывают водой, 1 н. раствором HCl и 10% раствором Na2CO3, органическую фазу сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диизопропилового эфира и пентана получают 28,6 г целевого продукта. Т.п. = 63oC.
[α]
C) 6-[2-(Бензоилокси) этил] -6-(3,4-дифторфенил) морфолин-3-он, (+) изомер
Раствор 28,5 г соединения, полученного на предыдущей стадии, в 250 мл ТГФ охлаждают до -30oC, в один прием добавляют 18,5 г трет-бутилата калия и реакционную смесь перемешивают в течение 45 минут. Полученную смесь выливают в 1000 мл буферного раствора с pH 2 и экстрагируют эфиром, органическую фазу сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диэтилового эфира и диизопропилового эфира получают 20,5 г целевого продукта. Т.п. = 92oC.
[α]
Способ получения 1.17
2-[2-(Бензоилокси) этил]-2-(3,4- дифторфенил)морфолин, (-) изомер
Раствор 12 г соединения, полученного в соответствии со способом получения 1.15, ((-) изомер), в 75 мл ТГФ при комнатной температуре по каплям добавляют к 100 мл 1 М раствора борана в ТГФ, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Затем полученную смесь нагревают с обратным холодильником в течение 3 часов, добавляют 40 мл 1 М раствора борана в ТГФ и продолжают нагревать с обратным холодильником еще 30 минут. Добавляют 80 мл кипящего MeOH и продолжают нагревать с обратным холодильником в течение 30 минут. Реакционную смесь охлаждают на ледяной бане, добавляют 30 мл насыщенного раствора газообразного HCl в эфире и реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в 10% растворе Na2CO3 и экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 11,2 г целевого продукта в виде масла.
Способ получения 1.18
2-[2-(Бензоилокси) этил]-2-(3,4- дифторфенил)морфолин, (+) изомер
Смесь 19,9 г соединения, полученного в соответствии со способом получения 1.16, ((+) изомер), и 300 мл 1 М раствора борана в ТГФ нагревают при температуре 60oC в течение 2 часов. Добавляют 60 мл кипящего MeOH и продолжают нагревать с обратным холодильником еще 30 минут. Реакционную смесь охлаждают до 10oC, добавляют 50 мл раствора газообразного HCl в эфире, после чего реакционную смесь оставляют для выстаивания в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в 300 мл 10% раствора Na2CO3, добавляют 300 мл эфира и смесь перемешивают в течение 30 минут. После декантации органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 20 г целевого продукта в виде масла.
Способ получения 1.19
2-[2-(Бензоилокси) этил]-2-(3,4- дихлорфенил)морфолин
Раствор 6 г соединения, полученного в соответствии со способом получения 1.4, в 30 мл ТГФ по каплям добавляют при комнатной температуре к 76 мл 1 М раствора борана в ТГФ, после чего смесь нагревают с обратным холодильником в течение 4 часов. По каплям добавляют 30 мл MeOH и продолжают нагревать с обратным холодильником еще 30 минут. Реакционную смесь охлаждают до 0oC, добавляют насыщенный раствор газообразного HCl в эфире и реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в 10% растворе Na2CO3 и экстрагируют эфиром, органическую фазу промывают 10% раствором Na2CO3 и водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 5 г целевого продукта.
Способ получения 1.20
5-[2-(Бензоилокси) этил]-5-(3,4- дифторфенил)оксазолидин-2-он, (-) изомер
Смесь 4 г соединения, полученного на стадии A способа получения 1.16, ((-) изомер), и 2,2 г 1,1'-карбонилдиимидазола в 40 мл ДХМ перемешивают в течение 30 минут при комнатной температуре, а затем нагревают с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь дважды промывают 1 н. раствором HCl, органическую фазу сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из диизопропилового эфира получают 4,18 г целевого продукта. Т.п. = 147oC.
[α]
Способ получения 1.21
2-[2-(Бензоилокси) этил]-2-(3,4- дифторфенил)морфолин
48 г соединения, полученного в соответствии со способом получения 1.10, добавляют порциями при комнатной температуре к 700 мл 1 М раствора борана в ТГФ, после чего полученную смесь нагревают при 60oC в течение 2 часов. По каплям добавляют 150 мл MeOH и продолжают нагревать еще 30 минут. Реакционную смесь охлаждают до 10oC, добавляют 120 мл насыщенного раствора хлористо-водородной кислоты в эфире и оставляют реакционную смесь для выстаивания в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в 600 мл насыщенного раствора Na2CO3 и 500 мл эфира и перемешивают полученную смесь в течение 1 часа при комнатной температуре. После декантации органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток растворяют в 400 мл пропан-2-ола, добавляют 15 г фумаровой кислоты, смесь перемешивают в течение 30 минут и образовавшийся осадок центрифугируют. Осадок растворяют в 400 мл 10% раствора Na2CO3 и экстрагируют эфиром, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 22 г целевого продукта в виде масла.
Способ получения 1.22
5-{2-(Бензоилокси) этил]-5-(3,4- дихлорфенил)оксазолидин
К раствору 9 г соединения, полученного на стадии A способа получения 1.7 (в виде свободного основания), в 100 мл ТГФ добавляют 4 г 27% водного раствора формальдегида, после чего эту смесь нагревают с обратным холодильником в течение 30 минут и перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 9 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,4 части на миллион: м: 2H
3,2 части на миллион: м: 2H
3,8 - 5,0 частей на миллион: н.с.: 4H
7,0 - 8,0 частей на миллион: н.с.: 8H
Способ получения 1.23
5-[2-(Бензоилокси)этил]-5-(3,4-дифторфенил)оксазолидин-2-он, (+) изомер
Это соединение получают в соответствии с процедурой, описанной в способе получения 1.20, из соединения, полученного на стадии A способа получения 1.15, ((+) изомер).
[α]
Способ получения 2.1
Полугидрат 4-фенил-4- (пирролидин-1-илкарбонил)пиперидина
A) 1-трет-Бутоксикарбонил- 4-карбокси-4-фенилпиперидин
К смеси 30 г паратолуолсульфоната 4-карбокси-4-фенилпиперидина и 300 мл диоксана добавляют 30 мл воды и 32,9 г K2CO3, полученную смесь нагревают до 60oC и по каплям добавляют 18,2 г дитрет-бутилдикарбоната. Затем реакционную смесь нагревают при 60oC в течение 2 часов и продолжают нагревать с обратным холодильником еще 30 минут. После охлаждения до комнатной температуры полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают буферным раствором с pH 2, подкисляют до pH 4, добавляя 2 н. раствор HCl, промывают буферным раствором с pH 2, водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 23,7 г целевого продукта.
B) 1-трет-Бутоксикарбонил -4-(пирролидин-1-илкарбонил) -4-фенилпиперидин
К раствору 14 г соединения, полученного на предыдущей стадии, в 200 мл ДХМ добавляют 9,29 г триэтиламина, а затем 3,27 г пирролидина. Эту смесь охлаждают на ледяной бане, добавляют 22,4 г бензилоктилфталата (BOP), после чего реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой, трижды 10% раствором NaOH, водой и трижды насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 16,4 г целевого продукта.
C) Полугидрат 4-фенил-4-(пирролидин -1-илкарбонил)пиперидина
К раствору 16,4 г соединения, полученного на предыдущей стадии, в 200 мл MeOH добавляют концентрированный раствор HCl, пока pH не становится равным 1, после чего реакционную смесь перемешивают в течение 5 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в ацетоне и растворитель выпаривают в вакууме с получением белого твердого вещества, которое перекристаллизовывают из пропан-2-ола. Полученный продукт растворяют в 10% растворе NaOH и экстрагируют дихлорметаном, органическую фазу промывают 10% раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из эфира получают 7 г целевого продукта. Т.п. = 126oC.
Способ получения 2.2
Полугидрат паратолуол-сульфоната 4-(N',N'-диметилуреидо)-4- фенилпиперидина
A) 4-Ацетамидо-1- бензил-4-фенилпиперидин
Это соединение получают в результате взаимодействия ацетонитрила с 1-бензил-4-гидрокси -4-фенилпиперидином в соответствии со способом, описанным в заявке на европейский патент N 474561.
B) Дигидрохлорид 4-амино-1-бензил -4-фенилпиперидина
Смесь 30 г соединения, полученного на предыдущей стадии, и 58 мл концентрированного раствора HCl в 135 мл воды нагревают с обратным холодильником в течение 48 часов. Реакционную смесь концентрируют в вакууме, остаток растворяют в смеси EtOH и толуола и растворители выпаривают в вакууме. Остаток растворяют в 50 мл MeOH и кристаллизуют, добавляя 250 мл ацетона, и после центрифугирования и сушки получают 30,5 г целевого продукта.
C) 1-Бензил-4-(N',N'-диметилуреидо) -4-фенилпиперидин
К раствору 6 г соединения, полученного на предыдущей стадии, и 7,14 г триэтиламина в 50 мл 1,2-дихлорэтана при комнатной температуре по каплям добавляют раствор 1,9 г N,N-диметилкарбамоилхлорида в 10 мл 1,2-дихлорэтана, после чего реакционную смесь нагревают с обратным холодильником в течение 8 часов. Добавляют еще несколько капель N,N-диметилкарбамоилхлорида и продолжают нагревать с обратным холодильником еще 3 часа. Реакционную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой, 10% раствором NaOH, водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 99/1 до 96/4 в объемном отношении), с получением 1,8 г целевого продукта.
D) Полугидрат паратолуолсульфоната 4-(N',N'-диметилуреидо)-4- фенилпиперидина
Смесь 1,8 г соединения, полученного на предыдущей стадии, 1,11 г моногидрата паратолуолсульфокислоты и 0,2 г 10% палладированного угля в 150 мл 95o EtOH гидрируют при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат упаривают в вакууме. Остаток растворяют в ацетоне и растворитель выпаривают в вакууме. Полученный продукт растворяют в 25 мл ацетона, этот раствор медленно добавляют к 200 мл эфира и образовавшийся кристаллический продукт центрифугируют с получением 1,86 г целевого продукта. Т.п. = 120-122oC.
Способ получения 2.3
Паратолуолсульфонат 4-(ацетил-N-метиламино) -4-фенилпиперидина
A) 1-Бензил-4-(формиламино) -4-фенилпиперидин
К раствору 48,9 г соединения, полученного на стадии B способа получения 2.2, и 25 г формиата натрия в 340 мл муравьиной кислоты по каплям добавляют 110 мл уксусного ангидрида, после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде, подщелачивают, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, органическую фазу сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диизопропилового эфира и пентана получают 38,8 г целевого продукта. Т.п. = 140oC.
B) 1-Бензил-4-(метиламино)-4-фенилпиперидин
Раствор 38,8 г соединения, полученного на предыдущей стадии, в 400 мл ТГФ медленно добавляют к суспензии 12,5 г алюмогидрида лития в 100 мл ТГФ, после чего полученную смесь нагревают с обратным холодильником в течение 3 часов. После охлаждения к реакционной смеси добавляют 5 мл концентрированного раствора NaOH в 45 мл воды, минеральные соли отфильтровывают и фильтрат концентрируют в вакууме с получением 38 г целевого продукта.
C) 4-(Ацетил-N-метиламино)-1-бензил-4-фенилпиперидин
Раствор 30 г соединения, полученного на предыдущей стадии, и 16,5 мл триэтиламина в 300 мл ДХМ охлаждают до 0-5oC, по каплям добавляют 8 мл ацетилхлорида и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь дважды промывают водой и 2 н. раствором NaOH, органическую фазу сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диизопропилового эфира и пентана получают 31,6 г целевого продукта. Т.п. = 104oC.
D) Паратолуолсульфонат 4-(ацетил-N-метиламино)-4-фенилпиперидина
Смесь 5 г соединения, полученного на предыдущей стадии, 2,9 г моногидрата паратолуолсульфокислоты, 0,5 г 10% палладированного угля и 80 мл EtOH гидрируют в течение 3 часов при температуре 25oC и атмосферном давлении. Катализатор отфильтровывают, фильтрат концентрируют в вакууме и после кристаллизации из ацетона получают 5,7 г целевого продукта. Т.п. = 165oC.
Способ получения 2.4
Трифторацетат 4-(этоксикарбониламино)-4-фенилпиперидина
A) 1-трет-Бутоксикарбонил-4-изоцианато-4-фенилпиперидин
Раствор 25 г соединения, полученного на стадии A способа получения 2.1, и 10,35 г триэтиламина в 100 мл ацетона охлаждают до 0-5oC, при температуре ниже 5oC по каплям добавляют раствор 8,7 г метилхлорформиата в 30 мл ацетона, после чего смесь перемешивают в течение 30 минут при температуре 5oC. Затем при температуре ниже 5oC по каплям добавляют раствор 10,66 г азида натрия в 30 мл воды и реакционную смесь перемешивают в течение 30 минут при температуре 5oC. Полученную смесь выливают в 500 мл смеси воды со льдом и четырежды экстрагируют толуолом, органическую фазу дважды промывают буферным раствором с pH 2 и насыщенным раствором NaCl, сушат над MgSO4 и фильтруют. Фильтрат нагревают при температуре 90oC в течение 1 часа и растворитель выпаривают в вакууме с получением 18,9 г целевого продукта в виде масла.
B) 1-трет-Бутоксикарбонил-4-(этоксикарбониламино)-4-фенилпиперидин
Раствор 6,28 г соединения, полученного на предыдущей стадии, в 100 мл EtOH нагревают с обратным холодильником в течение 5 часов 30 минут. Добавляют две капли триэтиламина, затем смесь перемешивают в течение ночи при комнатной температуре и концентрируют в вакууме с получением 7,25 г целевого продукта.
C) Трифторацетат 4-(этоксикарбониламино)-4-фенилпиперидина
Раствор 7,25 г соединения, полученного на предыдущей стадии, в 20 мл трифторуксусной кислоты перемешивают в течение 30 минут при комнатной температуре, а затем концентрируют в вакууме. Остаток растворяют в ацетоне, растворитель выпаривают в вакууме и после кристаллизации из смеси ацетона и эфира получают 6,02 г целевого продукта. Т.п. = 173oC.
Способ получения 2.5
4-Бензилхинуклидин
A) 1,4-Дибензил-4-цианопиперидин
Раствор 15 г 4-цианопиперидина в 250 мл ТГФ охлаждают до -50oC, по каплям добавляют 190 мл 1,5 М раствора диизопропиламида лития в циклогексане, после чего смесь перемешивают в течение 30 минут при температуре -50oC. Затем добавляют 34 мл бензилбромида и полученную смесь перемешивают в течение 3 часов после того, как она нагрелась до комнатной температуры. Реакционную смесь выливают в смесь льда с концентрированной HCl, добавляют эфир и образовавшийся осадок центрифугируют и промывают водой. Осадок растворяют в воде, подщелачивают до pH 12, добавляя концентрированный раствор NaOH, и экстрагируют эфиром, органическую фазу сушат над MgSO4, растворитель выпаривают и после кристаллизации из пентана получают 31,7 г целевого продукта. Т.п. = 92oC.
B) Гидрохлорид 4-ацетил-1,4-дибензилпиперидина
К раствору 20 г соединения, полученного на предыдущей стадии, в 400 мл эфира добавляют 55 мл 1,6 М раствора метиллития в эфире и перемешивают реакционную смесь в течение 3 часов при комнатной температуре. Полученную смесь выливают в смесь воды со льдом, органическую фазу декантируют и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток растворяют в 400 мл воды и 40 мл концентрированной HCl и нагревают с обратным холодильником в течение 2 часов. После выстаивания в течение ночи при комнатной температуре образовавшиеся кристаллы центрифугируют, промывают ацетоном, а затем эфиром и сушат с получением 17,6 г целевого продукта. Т.п. = 246oC.
C) Гидробромид 1,4-дибензил-4-(2-бромацетил)пиперидина
К раствору 10 г соединения, полученного на предыдущей стадии, в 40 мл уксусной кислоты добавляют 1,6 мл брома и реакционную смесь перемешивают в течение ночи при комнатной температуре. После этого добавляют 50 мл эфира, образовавшиеся кристаллы центрифугируют и промывают смесью ацетона и эфира, а затем эфиром с получением 12,5 г целевого продукта. Т.п. = 205oC.
D) Бромид 1,4-дибензил-3-оксохинуклидин
К водной суспензии 12,5 г соединения, полученного на предыдущей стадии, добавляют концентрированный раствор NaOH, пока pH не становится равным 12, затем смесь экстрагируют эфиром, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток растворяют в ацетоне и перемешивают в течение 2 часов при комнатной температуре. Осадок центрифугируют, промывают эфиром и сушат с получением 10,08 г целевого продукта. Т.п. = 234oC.
E) 4-Бензил-3-оксохинуклидин
Смесь 10 г соединения, полученного на предыдущей стадии, и 1 г 10% палладированного угля в 200 мл MeOH гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают и фильтрат упаривают в вакууме. Остаток растворяют в эфире и образовавшийся осадок центрифугируют. Осадок растворяют в воде и подщелачивают до pH 12, добавляя концентрированный раствор NaOH, после чего образовавшийся осадок центрифугируют, промывают водой и сушат с получением 5 г целевого продукта. Т.п. = 111oC.
F) 4-Бензилхинуклидин
Смесь 5 г соединения, полученного на предыдущей стадии, 2,5 г гидразингидрата и 4,3 г KOH в 25 мл этиленгликоля нагревают при температуре 175oC в течение 2 часов. Реакционную смесь выливают в смесь воды со льдом и дважды экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток растворяют в ацетоне и подкисляют до pH 1, добавляя насыщенный раствор хлористо-водородной кислоты в эфире, образовавшийся осадок центрифугируют и промывают смесью ацетона и эфира (50/50 в объемном отношении), а затем эфиром. Осадок растворяют в воде, подщелачивают до pH 12, добавляя концентрированный раствор NaOH, и экстрагируют эфиром, экстракт сушат над MgSO4 и растворитель выпаривают в вакууме с получением 1,8 г целевого продукта, Т.п. = 48oC.
Способ получения 2.6
Бензолсульфонат 4-фенил-4-(пирролидин-1-илкарбониламино)пиперидина
A) 1-(Бензилоксикарбонил)-4-карбокси-4-фенилпиперидин
Смесь 37,7 г паратолуолсульфоната 4-карбокси-4-фенилпиперидина, 53,3 г 30% водного раствора NaOH и 250 мл воды охлаждают до 5oC. При температуре 5oC быстро добавляют раствор 18 г бензилхлорформиата в 60 мл ацетона, после чего реакционную смесь перемешивают в течение ночи, позволяя ей нагреваться до комнатной температуры. Полученную смесь дважды промывают эфиром и после декантации водную фазу подкисляют до pH 1, добавляя концентрированную HCl, а затем 2 н. раствор HCl. Образовавшийся осадок центрифугируют, сушат, растворяют в эфире и еще раз центрифугируют с получением 30,6 г целевого продукта, Т.п. = 142-144oC.
B) 1-(Бензилоксикарбонил)-4-фенил-4-(пирролидин-1- илкарбониламино)пиперидин
Смесь 33,9 г соединения, полученного на предыдущей стадии, и 47,6 г тионилхлорида в 200 мл 1,2-дихлорэтана нагревают с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток растворяют в ацетоне и растворитель выпаривают в вакууме. Остаток растворяют в 200 мл ацетона и охлаждают до 5oC, по каплям добавляют раствор 13 г азида натрия в 50 мл воды и перемешивают смесь в течение 1 часа. 100 мл ацетона выпаривают в вакууме при комнатной температуре, к оставшемуся раствору добавляют насыщенный раствор NaHCO3, смесь экстрагируют толуолом, органическую фазу промывают водой и насыщенным раствором NaCl, сушат над Na2SO4 и выпаривают 50 об.% растворителя. Оставшийся раствор в толуоле нагревают с обратным холодильником в течение 30 минут, а затем концентрируют в вакууме. Остаток растворяют в 200 мл эфира, по каплям добавляют раствор 7,1 г пирролидина в 20 мл эфира и перемешивают смесь в течение 5 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают 2 н. раствором HCl, 5% раствором NaHCO3 и насыщенным раствором NaCl, сушат над Na2SO4 и растворитель выпаривают в вакууме. Остаток растворяют в 40 мл AcOEt, добавляют 200 мл эфира и образовавшийся кристаллический продукт центрифугируют с получением 31,4 г целевого продукта.
C) Бензолсульфонат 4-фенил-4-(пирролидин-1-илкарбониламино)пиперидина
Смесь 29 г соединения, полученного на предыдущей стадии, 11,27 г бензолсульфокислоты, 2 г 10% палладированного угля и 250 мл EtOH гидрируют при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме. Остаток кристаллизуют из смеси EtOH и ацетона с получением 29,4 г целевого продукта. Т.п. = 185oC.
Способ получения 2.7
Паратолуолсульфонат 4-[2-(диметиламино)тиазол-4-ил]-4-фенилпиперидина
A) 1,1-Диметилтиомочевина
Раствор 10,67 г тиоцианата калия в 100 мл ацетона охлаждают на ледяной бане, по каплям добавляют раствор 12,06 г пивалоилхлорида в 30 мл ацетона, после чего реакционную смесь перемешивают в течение 30 минут, позволяя ей нагреваться до комнатной температуры. Полученную смесь охлаждают до -10oC, по каплям добавляют 17,9 мл 5,6 н. раствора диметиламина в MeOH и перемешивают смесь, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в 50 мл концентрированного раствора HCl и нагревают с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь дважды промывают эфиром, водную фазу подщелачивают до pH 9, добавляя 30% раствор NaOH, и экстрагируют дихлорметаном, органическую фазу промывают 5% раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток растворяют в эфире и образовавшийся осадок центрифугируют с получением 6,7 г целевого продукта.
B) Гидробромид 4-(2-бромацетил)-4-фенилпиперидина
К суспензии 14 г гидробромида 4-ацетил-4-фенилпиперидина в 200 мл ДХМ быстро добавляют 7,9 г брома при комнатной температуре, после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь разбавляют, добавляя 200 мл эфира, образовавшийся осадок центрифугируют и промывают эфиром и после сушки в вакууме получают 16,7 г целевого продукта.
C) Паратолуолсульфонат 4-[2-(диметиламино)тиазол-4-ил]-4- фенилпиперидина
Смесь 7,26 г соединения, полученного на стадии B), и 2,08 г соединения, полученного на стадии A), в 150 мл EtOH нагревают с обратным холодильником в течение 1 часа 30 минут. После охлаждения до комнатной температуры эту смесь концентрируют в вакууме, остаток растворяют в воде, подщелачивают до pH 10, добавляя 10% раствор NaOH, и экстрагируют дихлорметаном, органическую фазу промывают 10% раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в ацетоне, по каплям добавляют раствор 3,31 г моногидрата паратолуолсульфокислоты в 10 мл ацетона и образовавшийся кристаллический продукт центрифугируют с получением 6,1 г целевого продукта. Т.п. = 164oC.
Способ получения 2.8
Моногидрат паратолуолсульфоната 4-(морфолин-4-илкарбониламино)-4- фенилпиперидина
A) 1-трет-Бутоксикарбонил-4-(морфолин-4-илкарбониламино)-4- фенилпиперидин
К раствору 6 г соединения, полученного на стадии A способа получения 2.4, в 100 мл ацетона при комнатной температуре по каплям добавляют раствор 1,74 г морфолина в 10 мл ацетона. Реакционную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают буферным раствором с pH 2 и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 7,1 г целевого продукта.
B) Моногидрат паратолуолсульфоната 4-(морфолин-4-илкарбониламино)-4- фенилпиперидина
К раствору 7,1 г соединения, полученного на предыдущей стадии, в 100 мл MeOH добавляют 15 мл концентрированного раствора HCl, после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде, подщелачивают до pH 10, добавляя концентрированный раствор NaOH, и трижды экстрагируют дихлорметаном, соединенные органические фазы промывают насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в ацетоне, по каплям добавляют раствор 3,46 г моногидрата паратолуолсульфокислоты в 10 мл ацетона и реакционную смесь концентрируют в вакууме. Остаток растворяют в эфире и образовавшийся осадок центрифугируют с получением 7 г целевого продукта. Т.п. = 93oC.
Способ получения 2.9
Бензолсульфонат 4-фенил-4-(пирролидин-1-иламинокарбонил)пиперидина
A) Бензолсульфонат 1-(бензилоксикарбонил)-4-фенил-4-(пирролидин-1- иламинокарбонил)пиперидина
Смесь 11,54 г соединения, полученного на стадии A способа получения 2.6, и 100 мл 1,2-дихлорэтана нагревают до температуры кипения с обратным холодильником, затем добавляют 16,18 г тионилхлорида, после чего реакционную смесь нагревают с обратным холодильником в течение 1 часа и перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в 100 мл ДХМ и охлаждают до 5oC, затем последовательно добавляют 10,3 г триэтиламина и 5 г гидрохлорида 1-аминопирролидина. После перемешивания в течение 1 часа реакционную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в ацетоне, добавляют эфир до появления осадка и образовавшийся осадок центрифугируют. Осадок растворяют в EtOH, добавляют 2,61 г бензолсульфокислоты и образовавшийся осадок центрифугируют с получением 9,34 г целевого продукта.
B) Бензолсульфонат 4-фенил-4-(пирролидин-1-иламинокарбонил)пиперидина
Смесь 9,34 г соединения, полученного на предыдущей стадии, 1 г 5% палладированного угля и 200 мл EtOH гидрируют в течение 3 часов при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают, фильтрат концентрируют в вакууме и после кристаллизации из смеси EtOH и ацетона получают 6,13 г целевого продукта.
Способ получения 2.10
Паратолуолсульфонат 4-бензил-4-(пирролидин-1-илкарбониламино)пиперидина
A) 4-Цианопиперидин
25 г изонипекотамида (или пиперидин-4-карбоксамида) добавляют небольшими порциями к 70 мл POCl3, после чего реакционную смесь нагревают с обратным холодильником в течение 4 часов. Полученную смесь концентрируют в вакууме, остаток растворяют на льду, подщелачивают до pH 13, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, а затем 4 раза эфиром, после чего объединенные органические фазы сушат над MgSO4 и растворители выпаривают в вакууме. Полученное масло перегоняют при пониженном давлении с получением 6,4 г целевого продукта. Температура кипения = 108-110oC при давлении 2400 Па.
B) 4-Циано-1,4-дибензилпиперидин
Раствор 15 г соединения, полученного на предыдущей стадии, в 250 мл ТГФ охлаждают до -50oC, по каплям добавляют 190 мл 1,5 М раствора диизопропиламида лития в циклогексане и перемешивают смесь в течение 30 минут при температуре -50oC. Затем добавляют 34 мл бензилбромида и реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. После выстаивания в течение 3 часов при комнатной температуре реакционную смесь выливают в смесь льда с концентрированной HCl, добавляют эфир и образовавшийся осадок центрифугируют и промывают водой. Осадок растворяют в воде, подщелачивают до pH 13, добавляя концентрированный раствор NaOH, и экстрагируют эфиром, органическую фазу сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из пентана получают 31,7 г целевого продукта. Т.п. = 92oC.
C) 1,4-Дибензил-4-карбоксипиперидин
6 г соединения, полученного на предыдущей стадии, добавляют к раствору 25 мл воды, 25 мл концентрированной H2SO4 и 25 мл AcOH, после чего реакционную смесь нагревают при температуре 140oC в течение 5 часов. После охлаждения полученную смесь выливают на лед, показатель pH доводят до 6,5, добавляя концентрированный раствор NaOH, и перемешивают смесь до тех пор, пока не происходит кристаллизация. Кристаллический продукт центрифугируют и промывают водой. Полученный продукт растворяют в MeOH, центрифугируют и промывают эфиром с получением 3 г целевого продукта. Т.п. = 262oC.
D) 1,4-Дибензил-4-изоцианатопиперидин
Смесь 2 г соединения, полученного на предыдущей стадии, и 1,6 г пентахлорида фосфора в 40 мл хлороформа нагревают при температуре 60oC в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток растворяют в 40 мл ацетона, добавляют раствор 2 г азида натрия в 5 мл воды и перемешивают смесь в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме при комнатной температуре, остаток растворяют в эфире, органическую фазу промывают насыщенным раствором Na2CO3 и водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток растворяют в 40 мл толуола и нагревают с обратным холодильником в течение 1 часа. Полученную смесь концентрируют в вакууме с получением 2 г целевого продукта в виде масла.
E) 1,4-Дибензил-4-(пирролидин-1-илкарбониламино)пиперидин
К раствору 5 г соединения, полученного на предыдущей стадии, в 50 мл ДХМ добавляют 1,5 мл пирролидина при комнатной температуре, после чего реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в пентане, образовавшийся осадок центрифугируют и промывают пентаном и после сушки получают 4,5 г целевого продукта. Т.п. = 126oC.
F) Паратолуолсульфонат 4-бензил-4-(пирролидин-1- илкарбониламино)пиперидина
Смесь 4,2 г соединения, полученного на предыдущей стадии, 2,1 г моногидрата паратолуолсульфокислоты, 0,4 г 10% паллидированного угля и 50 мл EtOH гидрируют в течение 48 часов при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме. Остаток растворяют в эфире и образовавшийся осадок центрифугируют с получением 4,99 г целевого продукта. Т.п. > 180oC.
Способ получения 2.11
Полугидрат паратолуолсульфоната 4-(метоксикарбониламино)-4- фенилпиперидина
Раствор 6,05 г соединения, полученного на стадии A способа получения 2.4, в 100 мл MeOH нагревают с обратным холодильником в течение 5 часов. Добавляют 1 каплю триэтиламина и смесь перемешивают в течение ночи при комнатной температуре. Концентрированный раствор HCl добавляют до тех пор, пока pH не становится равным 1, и реакционную смесь концентрируют в вакууме. Остаток растворяют в 10% растворе NaOH и экстрагируют дихлорметаном, органическую фазу промывают 10% раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в ацетоне, по каплям добавляют раствор 3,57 г моногидрата паратолуолсульфокислоты в 10 мл ацетона и концентрируют эту смесь в вакууме. Полученный продукт растворяют в эфире и растворитель выпаривают в вакууме с получением 7,17 г целевого продукта. Т.п. = 159oC.
Способ получения 2.12
Полугидрат паратолуолсульфоната 4-(2-амино-1,3,4-оксадиазол-5- ил)-4-фенилпиперидина
A) 1-(Бензилоксикарбонил)-4-(хлорформил)-4-фенилпиперидин
Смесь 17,1 г соединения, полученного на стадии A способа получения 2.6, и 24 г тионилхлорида в 150 мл 1,2-дихлорэтана нагревают с обратным холодильником в течение 1 часа. Полученную смесь концентрируют в вакууме, остаток растворяют в хлороформе и растворитель выпаривают в вакууме. Остаток растворяют в смеси эфира и пентана и растворители выпаривают в вакууме с получением 20 г целевого продукта в виде смолы, которую используют без дальнейшей очистки.
B) 1-(Бензилоксикарбонил)-4-карбазоил-4-фенилпиперидин
Раствор 16 г гидразинмоногидрата в 40 мл EtOH охлаждают до -50oC, по каплям добавляют раствор 11,44 г соединения, полученного на предыдущей стадии, в 20 мл 1,2-диметоксиэтана, после чего смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют дихлорметаном, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток растворяют в смеси EtOH и бензола и растворители выпаривают в вакууме с получением 11,2 г целевого продукта в виде смолы, который используют без дальнейшей очистки.
C) 4-(2-Амино-1,3,4-оксадиазол-5-ил)-1-(бензилоксикарбонил)-4- фенилпиперидин
К раствору 11,2 г соединения, полученного на предыдущей стадии, в 60 мл EtOH при комнатной температуре добавляют раствор 3,39 г бромциана в 10 мл EtOH и нагревают реакционную смесь с обратным холодильником в течение 1 часа. Полученную смесь концентрируют до 50 мл EtOH, затем по каплям добавляют воду, пока объем реакционной смеси не достигнет 400 мл.
Образовавшийся кристаллический продукт центрифугируют и промывают водой, а затем дихлорметаном, AcOEt и эфиром с получением 8 г целевого продукта.
D) Полугидрат паратолуолсульфоната 4-(2-амино-1,3,4-оксадиазол-5- ил)-4-фенилпиперидина
Смесь 7,85 г соединения, полученного на предыдущей стадии, 3,95 г моногидрата паратолуолсульфокислоты, 0,8 г 10% палладированного угля, 350 мл 95o EtOH и 10 мл воды гидрируют при температуре 50oC и атмосферном давлении. Через 3 часа катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме. Остаток растворяют в ацетоне, образовавшийся кристаллический продукт центрифугируют и промывают ацетоном, а затем эфиром с получением 7,65 г целевого продукта. Т.п. = 183-185oC.
Способ получения 2.13
Паратолуолсульфонат 4-[(ацетил-H-метиламино)метил]-4-фенилпиперидина
A) 4-(Аминометил)-1-бензил-4-фенилпиперидин
Суспензию 2,8 г алюмогидрида лития в 50 мл ТГФ охлаждают до 0oC и по каплям добавляют 20 г 1-бензил-4-циано-4-фенилпиперидина в 50 мл ТГФ. Реакционную смесь перемешивают в течение 1 часа при комнатной температуре, а затем нагревают в течение 1 часа при температуре 40oC. Полученную смесь охлаждают на ледяной бане и последовательно добавляют 3 мл воды, 3 мл 4 н. раствора NaOH и 12 мл воды. Минеральные соли отфильтровывают и фильтрат упаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 100/3 до 100/10 в объемном отношении), с получением 11 г целевого продукта.
B) 1-Бензил-4-[(N-формиламино)метил]-4-фенилпиперидин
К смеси 11 г соединения, полученного на предыдущей стадии, в 76 мл муравьиной кислоты при комнатной температуре по каплям добавляют 25 мл уксусного ангидрида, после чего реакционную смесь перемешивают в течение 5 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в воде, подщелачивают до pH 14, добавляя концентрированный раствор NaOH, и экстрагируют эфиром, экстракт промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 12 г целевого продукта.
C) 1-Бензил-4-[(N-метиламино)метил]-4-фенилпиперидин
Суспензию 3,9 г алюмогидрида лития в 50 мл ТГФ нагревают до 40oC, по каплям добавляют раствор 12 г соединения, полученного на предыдущей стадии, в 50 мл ТГФ, после чего смесь нагревают с обратным холодильником в течение 3 часов. После охлаждения на ледяной бане последовательно добавляют 4 мл воды, 4 мл 4 н. раствора NaOH и 12 мл воды. Минеральные соли отфильтровывают и фильтрат концентрируют в вакууме. Остаток экстрагируют эфиром, экстракт сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 10 г целевого продукта.
D) 4-[(Ацетил-N-метиламино)метил]-1-бензил-4-фенилпиперидин
К раствору 3,3 г соединения, полученного на предыдущей стадии, и 1,4 г триэтиламина в 50 мл ДХМ добавляют 0,863 г ацетилхлорида, после чего реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, экстракт промывают водой и сушат над Na2SO4, растворитель выпаривают. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении), с получением 2,4 г целевого продукта.
E) Паратолуолсульфонат 4-[(ацетил-N-метиламино)метил]-4- фенилпиперидина
Смесь 2,3 г соединения, полученного на предыдущей стадии, 1,2 г моногидрата паратолуолсульфокислоты, 0,23 г 10% палладированного угля и 100 мл MeOH гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают, фильтрат упаривают в вакууме и после растирания в порошок в эфире и центрифугирования получают 2,7 г целевого продукта.
Способ получения 2.14
Паратолуолсульфонат 4-[(этоксикарбонил-N-метиламино)метил] -4- фенилпиперидина
A) 1-Бензил-4-[(этоксикарбонил-N-метиламино)метил]-4-фенилпиперидин
К раствору 2,3 г соединения, полученного на стадии C способа получения 2.13, и 1,03 г триэтиламина в 50 мл ДХМ при комнатной температуре по каплям добавляют раствор 0,85 г этилхлорформиата в 10 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют водой и экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении), с получением 2,3 г целевого продукта.
B) Паратолуолсульфонат 4-[(этоксикарбонил-N-метиламино)метил]-4- фенилпиперидина
Смесь 2,3 г соединения, полученного на предыдущей стадии, 1,19 г моногидрата паратолуолсульфокислоты, 0,7 г 5% палладированного угля и 50 мл ДХМ гидрируют при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме с получением 2,7 г целевого продукта.
Способ получения 2.15
Паратолуолсульфонат 4-[(N', N'-диметил-N-метилуреидо)метил]-4- фенилпиперидина
A) 1-Бензил-4-[N',N'-диметил-N-метилуреидо)метил]-4-фенилпиперидин
К раствору 2,5 г соединения, полученного на стадии C способа получения 2.13, и 1,11 г триэтиламина в 50 мл ДХМ при комнатной температуре по каплям добавляют раствор 0,92 г N,N-диметилкарбамоилхлорида в 20 мл ДХМ, после чего реакционную смесь нагревают с обратным холодильником в течение 3 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении), с получением 2,92 г целевого продукта.
B) Паратолуолсульфонат 4-[(N', N'-диметил-N-метилуреидо)метил] -4- фенилпиперидина
Смесь 2,92 г соединения, полученного на предыдущей стадии, 1,52 г моногидрата паратолуолсульфокислоты, 0,3 г 10% палладированного угля и 50 мл MeOH гидрируют при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite®, фильтрат концентрируют в вакууме и после растирания остатка в порошок в смеси пентана и диизопропилового эфира с последующим центрифугированием получают 2,6 г целевого продукта.
Способ получения 2.16
Дигидрохлорид 4-карбамоил-4-(пиперид-1-ил)пиперидина
A) 1-Бензил-4-циано-4-(пиперид-1-ил)пиперидин
К раствору 18,9 г 1-бензилпиперид-4-она и 12,16 г гидрохлорида пиперидина в 25 мл MeOH и 25 мл воды при комнатной температуре по каплям добавляют раствор 5,3 г цианида натрия в 20 мл воды, после чего смесь перемешивают в течение 49 часов при комнатной температуре. Образовавшийся осадок центрифугируют, промывают водой и сушат в вакууме с получением 27 г целевого продукта.
B) 1-Бензил-4-карбамоил-4-(пиперид-1-ил)пиперидин
10 г соединения, полученного на предыдущей стадии, добавляют к 50 мл 95% серной кислоты, после чего реакционную смесь нагревают при температуре 100oC в течение 45 минут. После охлаждения до комнатной температуры полученную смесь выливают на 100 г льда, добавляют при охлаждении 250 мл ДХМ, органическую фазу декантируют и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный твердый продукт перекристаллизовывают из 300 мл смеси ацетонитрила и толуола (65/35 в объемном отношении) с получением 9,7 г целевого продукта. Т.п. = 150-160oC.
C) Дигидрохлорид 4-карбамоил-4-(пиперид-1-ил)пиперидина
К раствору 9,7 г соединения, полученного на предыдущей стадии, в 200 мл MeOH добавляют 10 г формиата аммония и 2,5 г 5% палладированного угля, после чего смесь перемешивают в течение 2 часов при комнатной температуре. Полученную смесь фильтруют на целите Celite® и фильтрат упаривают в вакууме. Остаток растворяют в 2 н. растворе HCl, подщелачивают до pH 13, добавляя 40% раствора NaOH, и экстрагируют хлороформом, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Полученный продукт растворяют в смеси MeOH и дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор хлористо-водородной кислоты в эфире, и упаривают в вакууме с получением 5 г целевого продукта. Т.п. = 185oC.
Способ получения 2.17
Бензолсульфонат 4-фенил-4-уреидопиперидина
A) 1-(Бензилоксикарбонил)-4-изоцианато-4-фенилпиперидин
Смесь 50,89 г соединения, полученного на стадии A) способа получения 2.6, и 71,4 г тионилхлорида в 400 мл 1,2-дихлорэтана нагревают с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток растворяют в ацетоне и растворитель выпаривают в вакууме. Остаток растворяют в 200 мл ацетона и охлаждают до 5oC, по каплям добавляют раствор 19,5 г азида натрия в 50 мл воды и смесь перемешивают в течение 2 часов при комнатной температуре. Ацетон выпаривают в вакууме при комнатной температуре, к оставшемуся раствору добавляют насыщенный раствор NaHCO3, смесь экстрагируют толуолом, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4, выпаривают 50 об.% растворителя. Оставшийся раствор в толуоле нагревают с обратным холодильником в течение 1 часа, а затем концентрируют в вакууме с получением 54 г целевого продукта в виде оранжевого масла, которое кристаллизуется.
B) 1-(Бензилоксикарбонил)-4-фенил-4-уреидопиперидин
Избыточное количество газообразного аммиака барботируют при комнатной температуре в раствор 29 г соединения, полученного на предыдущей стадии, в 300 мл эфира и 300 мл ДХМ, после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в горячем ацетоне и позволяют раствору охладиться до комнатной температуры. Сразу же после кристаллизации продукта добавляют смесь эфира и AcOEt и образовавшиеся кристаллы центрифугируют с получением 26,4 г целевого продукта.
C) Бензолсульфонат 4-фенил-4-уреидопиперидина
Смесь 25 г соединения, полученного на предыдущей стадии, 11,2 г бензолсульфокислоты, 3 г 5% палладированного угля и 300 мл EtOH гидрируют при температуре 40oC и атмосферном давлении. Реакционную смесь разбавляют, добавляя воду и MeOH, катализатор отфильтровывают на целите  фильтрат концентрируют в вакууме и после кристаллизации из ацетона получают 26,45 г целевого продукта. Т.п. = 235oC.
фильтрат концентрируют в вакууме и после кристаллизации из ацетона получают 26,45 г целевого продукта. Т.п. = 235oC.
Способ получения 2.18
Бензолсульфонат 4-(N'-метилуреидо)-4-фенилпиперидина
A) 1-(Бензилоксикарбонил)-4-(N'-метилуреидо)-4-фенилпиперидин
Раствор 25 г соединения, полученного на стадии A способа получения 2.17, в 300 мл эфира охлаждают до 5oC и барботируют в этот раствор избыточное количество газообразного метиламина. Реакционную смесь разбавляют 150 мл ДХМ и перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в горячем AcOEt и позволяют раствору охладиться до комнатной температуры. Эфир добавляют до тех пор, пока не происходит осаждение, после чего образовавшийся осадок центрифугируют с получением 24 г целевого продукта.
B) Бензолсульфонат 4-(N'-метилуреидо)-4-фенилпиперидина
Смесь 23 г соединения, полученного на предыдущей стадии, 9,9 г бензолсульфокислоты, 3 г 5% палладированного угля и 300 мл 95o EtOH гидрируют при температуре 40oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме. Остаток растворяют в ацетоне и образовавшийся осадок центрифугируют с получением 22,36 г целевого продукта. Т.п. = 227oC.
Способ получения 2.19
Паратолуолсульфонат 4-(3,3-диметилкарбазоил)-4-фенилпиперидина
A) Паратолуолсульфонат 1-(бензилоксикарбонил)-4-(3,3- диметилкарбазоил)-4-фенилпиперидина
Раствор 7,15 г соединения, полученного на стадии A способа получения 2.12, в 60 мл ДХМ охлаждают до 5oC, по каплям добавляют раствор 1,44 г 1,1-диметилгидразина и 4,04 г триэтиламина в 20 мл ДХМ, после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют AcOEt, органическую фазу промывают насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в 100 мл ацетона, быстро добавляют раствор 3,59 г моногидрата паратолуолсульфокислоты в 10 мл ацетона и концентрируют смесь в вакууме. Остаток растворяют в смеси эфира и дихлорметана и образовавшийся осадок центрифугируют с получением 9,65 г целевого продукта.
B) Паратолуолсульфонат 4-(3,3-диметилкарбазоил)-4-фенилпиперидина
Смесь 9,5 г соединения, полученного на предыдущей стадии, и 0,9 г 10% палладированного угля в 100 мл 95o EtOH гидрируют при температуре 30oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме. Остаток растворяют в 100 мл ацетона, образовавшийся кристаллический продукт центрифугируют и промывают эфиром с получением 7 г целевого продукта. Т.п. = 210-212oC.
Способ получения 2.20
Дигидрохлорид 4-[(этиламинокарбонилокси)метил]-4-фенилпиперидина
A) Паратолуолсульфонат 4-метоксикарбонил-4-фенилпиперидина
К раствору 10 г паратолуолсульфоната 4-карбокси-4-фенилпиперидина в 300 мл MeOH добавляют 1 г моногидрата паратолуолсульфокислоты, после чего реакционную смесь нагревают с обратным холодильником в течение 3 дней. Полученную смесь концентрируют в вакууме, остаток растворяют в ацетоне и добавляют эфир до тех пор, пока не происходит осаждение. После центрифугирования образовавшегося осадка получают 9,34 г целевого продукта.
B) 4-Гидроксиметил-4-фенилпиперидин
Суспензию 1,16 г алюмогидрида лития в 50 мл ТГФ охлаждают до -20oC, добавляют 4 г соединения, полученного на предыдущей стадии, после чего смесь перемешивают в течение ночи, позволяя ей нагреваться до комнатной температуры. Полученную смесь гидролизуют, добавляя 1,2 мл воды, а затем 2,5 мл 10% раствора NaOH и 2,5 мл воды. Смесь разбавляют эфиром, отфильтровывают минеральные соли и фильтрат упаривают в вакууме с получением 1,8 г целевого продукта.
C) 1-трет-Бутоксикарбонил-4-(гидроксиметил)-4-фенилпиперидин
К раствору 22,8 г соединения, полученного на предыдущей стадии, в 250 мл 1,2-диметоксиэтана добавляют 26,05 г ди-трет-бутилдикарбоната, после чего реакционную смесь нагревают с обратным холодильником в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в дихлорметане, органическую фазу промывают буферным раствором с pH 2 и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из эфира получают 17,86 г целевого продукта. Т.п. = 134oC.
D) 1-трет-Бутоксикарбонил-4-[(этиламинокарбонилокси)метил] -4- фенилпиперидин
Смесь 2,91 г соединения, полученного на предыдущей стадии, 2,4 г этилизоцианата и 2 капли триэтиламина в 30 мл толуола перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь нагревают при температуре 100oC в течение 24 часов и концентрируют в вакууме. Остаток растворяют в эфире, органическую фазу промывают буферным раствором с pH 2 и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 3,85 г целевого продукта в виде масла.
E) Гидрохлорид 4-[(этиламинокарбонилокси)метил]-4-фенилпиперидина
К раствору 3,85 г соединения, полученного на предыдущей стадии, в 50 мл MeOH добавляют 10 мл концентрированной HCl, после чего смесь нагревают при температуре 60oC в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в ацетоне и после кристаллизации из смеси AcOEt и эфира получают 2,6 г целевого продукта. Т.п. = 240-242oC.
Способ получения 2.21
4-(Ацетоксиметил)-4-фенилпиперидин
К раствору 2,91 г соединения, полученного на стадии C способа получения 2.20, и 1,31 г триэтиламина в 50 мл ДХМ добавляют при комнатной температуре 0,785 г ацетилхлорида, после чего смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Маслянистый остаток растворяют в 20 мл трифторуксусной кислоты и перемешивают смесь в течение 10 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде, водную фазу подщелачивают до pH 11, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, органическую фазу пять раз промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 1,6 г целевого продукта.
Способ получения 2.22
Бензолсульфонат 2-бензил-4-[(этоксикарбонил-N- метиламино)метил]пиперидина
A) 1-Бензил-4-карбамоилпиперидин
К смеси 58,2 г изонипекотамида и 69 г K2CO3 в 275 мл ДМФ при комнатной температуре по каплям добавляют 85,5 г бензилбромида, после чего реакционную смесь перемешивают в течение 2 часов при температуре 50oC. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме и после кристаллизации из 300 мл воды и сушки в вакууме получают 62 г целевого продукта.
B) 1-Бензил-4-цианопиперидин
Смесь 62 г соединения, полученного на предыдущей стадии, и 200 мл POCl3 нагревают с обратным холодильником в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в воде, подщелачивают до pH 13, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Полученное масло перегоняют при пониженном давлении с получением 52 г целевого продукта. Температура кипения = 120oC при давлении 10 Па.
C) 1,4-Дибензил-4-цианопиперидин
200 мл 1,5 М раствора диизопропиламида лития в циклогексане, разбавленном 100 мл ТГФ, охлаждают до -50oC, по каплям добавляют раствор 52 г соединения, полученного на предыдущей стадии, в 100 мл ТГФ и перемешивают смесь в течение 30 минут при температуре -50oC. Затем при температуре от -30oC до -25oC по каплям добавляют 51,3 г бензилбромида в 100 мл ТНФ, после чего смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме и после кристаллизации из пентана получают 64 г целевого продукта.
D) 4-(Аминометил)-1,4-дибензилпиперидин
Раствор 14,5 г соединения, полученного на предыдущей стадии, в 50 мл ТГФ при комнатной температуре по каплям добавляют к суспензии 1,95 г алюмогидрида лития в 50 мл ТГФ, после чего смесь нагревают с обратным холодильником в течение 6 часов. После охлаждения до комнатной температуры добавляют 2 мл воды, 2 мл 4 н. раствора NaOH и 6 мл воды. Минеральные соли отфильтровывают на целите Celite®, фильтрат декантируют, органическую фазу сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 14,7 г целевого продукта.
E) 1,4-Дибензил-4-[(формиламино)метил]пиперидин
К раствору 14,7 г соединения, полученного на предыдущей стадии, в 126 мл муравьиной кислоты при комнатной температуре по каплям добавляют 42 мл уксусного ангидрида, после чего смесь перемешивают в течение 3 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде, подщелачивают до pH 12, добавляя концентрированный раствор NaOH, и экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 14,5 г целевого продукта.
F) 1,4-Дибензил-4-[(метиламино)метил]пиперидин
Раствор 14,5 г соединения, полученного на предыдущей стадии, в 100 мл ТГФ при температуре 40oC по каплям добавляют к суспензии 5 г алюмогидрида лития в 100 мл ТГФ, после чего смесь нагревают с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры добавляют 5 мл воды, 5 мл 4 н. раствора NaOH и 15 мл воды. Минеральные соли отфильтровывают на целите Celite®, фильтрат декантируют, органическую фазу сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 12,8 г целевого продукта.
G) 1,4-Дибензил-4-[(этоксикарбонил-N-метиламино)метил]пиперидин
К раствору 3 г соединения, полученного на предыдущей стадии, и 1,18 г триэтиламина в 50 мл 1,2-дихлорэтана при комнатной температуре по каплям добавляют 1,26 г этилхлорформиата, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу сушат над Na2SO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении), с получением 1,6 г целевого продукта.
H) Бензолсульфонат 4-бензил-4-[(этоксикарбонил-N- метиламино)метил]пиперидина
Смесь 1,6 г соединения, полученного на предыдущей стадии, 0,66 г бензолсульфокислоты, 0,2 г 10% палладированного угля и 30 мл MeOH гидрируют при температуре 27oC и атмосферном давлении. Катализатор отфильтровывают на целите Celite® и фильтрат концентрируют в вакууме с получением 1,44 г целевого продукта.
Способ получения 2.23
Бензолсульфонат 4-бензил-4-[(метансульфонил-N- метиламино)метил] пиперидина
A) 1,4-Дибензил-4-[(метансульфонил-N-метиламино)метил]пиперидин
К раствору 3,2 г соединения, полученного на стадии F способа получения 2.22, и 1,26 г триэтиламина в 50 мл 1,2-дихлорэтана при комнатной температуре по каплям добавляют 1,26 г метансульфонилхлорида, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют AcOEt, органическую фазу сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 3,8 г целевого продукта.
B) Бензолсульфонат 4-бензил-4-[(метансульфонил-N- метиламино)метил]пиперидина
Смесь 3,8 г соединения, полученного на предыдущей стадии, 1,58 г бензолсульфокислоты, 0,7 г 10% палладированного угля и 30 мл MeOH гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают на целите Celite®, фильтрат концентрируют в вакууме и после кристаллизации из смеси дихлорметана и эфира получают 4,6 г целевого продукта.
Способ получения 2.24
Гидрохлорид 4-бензил-4-[(N',N'-диметил-N-метилуреидо)метил]пиперидина
A) 1,4-Дибензил-4-[(N',N'-диметил-N-метилуреидо)метил]пиперидин
К раствору 3,2 г соединения, полученного на стадии F способа получения 2.22, и 1,2 г триэтиламина в 40 мл 1,2-дихлорэтана при комнатной температуре по каплям добавляют 1,12 г N,N-диметилкарбамоилхлорида, после чего смесь нагревают с обратным холодильником в течение 4 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу сушат над Na2SO4 и растворитель выпаривают в вакууме с получением 3,6 г целевого продукта.
B) Гидрохлорид 4-бензил-4-[(N',N'-диметил-N-метилуреидо)метил]пиперидина
Смесь 3,6 г соединения, полученного на предыдущей стадии, 3,1 г формиата аммония и 0,8 г 5% палладированного угля в 50 мл MeOH перемешивают в течение 1 часа 30 минут. Катализатор отфильтровывают и растворитель выпаривают в вакууме. Остаток растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и растворители выпаривают в вакууме с получением 3,2 г целевого продукта.
Способ получения 2.25
4-Карбамоил-4-(морфолин-4-ил)пиперидин
A) 1-Бензил-4-циано-4-(морфолин-4-ил)пиперидин
К смеси 5 г 1-бензилпиперид-4-она и 1,9 г цианида калия в 50 мл смеси EtOH и воды (50/50 в объемном отношении) добавляют 2,5 мл морфолина, а затем 5,1 г Na2S2O5, после чего эту смесь нагревают при температуре 60oC в течение 2 часов. Добавляют еще 2,5 мл морфолина и реакционную смесь перемешивают в течение ночи при комнатной температуре. Добавляют воду и образовавшийся кристаллический продукт центрифугируют с получением 5,5 г целевого продукта.
B) 1-Бензил-4-карбамоил-4-(морфолин-4-ил)пиперидин
Смесь 14 г соединения, полученного на предыдущей стадии, и 50 мл 95% серной кислоты нагревают при температуре 100oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают на 100 г льда, доводят смесь до pH 7, добавляя концентрированный раствор NH4OH, и экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/5 до 100/10 в объемном отношении), и после кристаллизации из диизопропилового эфира получают 3,4 г целевого продукта.
C) 4-Карбамоил-4-(морфолин-4-ил)пиперидин
К раствору 3,4 г соединения, полученного на предыдущей стадии, в 50 мл MeOH добавляют 3,1 г формиата аммония и 0,8 г 5% палладированного угля, после чего смесь перемешивают в течение 2 часов при комнатной температуре. Катализатор отфильтровывают на целите Celite®, фильтрат упаривают в вакууме и после кристаллизации из пропан-2-ола получают 2,2 г целевого продукта.
Пример 1
Моногидрат гидрохлорида 3-бензил-5-[2-(4-бензилпиперид-1- ил)этил]-5-(3,4-дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
A) 3-Бензил-5-(3,4-дихлорфенил)-5-[2-(тетрагидропиран-2- илокси)этил]-тетрагидро-2H-1,3-оксазин-2-он
Раствор 4 г соединения, полученного в соответствии со способом получения 1.1, в 60 мл ТГФ охлаждают до +5oC, добавляют 1,37 г трет-бутилата калия и по каплям добавляют раствор 1,92 г бензилбромида в 15 мл ТГФ. Реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры, и концентрируют в вакууме. Остаток экстрагируют AcOEt, органическую фазу трижды промывают водой и дважды насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 4,2 г целевого продукта, который используют без дальнейшей очистки.
B) 0,25 Гидрат 3-бензил-5-(3,4-дихлорфенил)-5-(2- гидроксиэтил)тетрагидро-2H-1,3-оксазин-2-она
К раствору 4,2 г соединения, полученного на предыдущей стадии, в 40 мл MeOH добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, после чего реакционную смесь перемешивают в течение 48 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в насыщенном растворе газообразного HCl в MeOH и растворитель выпаривают в вакууме. Остаток растворяют в эфире и образовавшийся осадок центрифугируют с получением 3,44 г целевого продукта. Т.п. = 143oC.
C) 3-Бензил-5-(3,4-дихлорфенил)-5-[2-(метансульфонилокси)этил] тетрагидро-2H-1,3-оксазин-2-он
Раствор 1,4 г соединения, полученного на предыдущей стадии, и 0,45 г триэтиламина в 50 мл ДХМ охлаждают до +5oC и по каплям добавляют раствор 0,46 г метансульфонилхлорида в 5 мл ДХМ. После окончания добавления реакционную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 1,7 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,03 части на миллион: т: 2H
2,98 части на миллион: с: 3H
3,78 части на миллион: система AB: 2H
3,83 части на миллион: т: 2H
4,1 - 4,8 части на миллион: н.с.: 4H
7,0 - 7,6 части на миллион: н.с.: 8H
D) Моногидрат гидрохлорида 3-бензил-5-[2-(4-бензилпиперид-1- ил)этил]-5-(3,4-дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
Смесь 1,6 г 4-бензилпиперидина, 1,7 г соединения, полученного, на предыдущей стадии, и 0,6 г йодида калия в 10 мл ДМФ нагревают при температуре 60oC в течение 2 часов 30 минут. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан. Полученный продукт растворяют в насыщенном растворе газообразного HCl в эфире и образовавшийся осадок центрифугируют с получением 0,9 г целевого продукта. Т.п. = 122oC.
Пример 2
Гидрохлорид 3-бензил-5-(3,4-дихлорфенил)-5-[2-(4-гидрокси-4- фенилпиперид-1-ил)этил]тетрагидро-2H-1,3-оксазин-2-она
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 1, из 1,32 г 4-гидрокси-4-фенилпиперидина, 1,55 г соединения, полученного на стадии C примера 1, и 0,56 г йодида калия в 10 мл ДМФ. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт растворяют в дихлорметане и подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, растворители выпаривают в вакууме и после кристаллизации из эфира получают 0,72 г целевого соединения. Т.п. = 172oC.
Пример 3
1,5 Гидрат гидрохлорида 5-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил]-3-бензил-5-(3,4-дихлорфенил) тетрагидро-2H-1,3-оксазин-2-она
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 1, из 2,77 г 4-ацетамидо-4-фенилпиперидина, 1,7 г соединения, полученного на стадии C примера 1, и 0,61 г йодида калия в 10 мл ДМФ. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (97/3 в объемном отношении). Остаток растворяют в насыщенном растворе газообразного HCl в эфире, растворитель выпаривают в вакууме и после кристаллизации из AcOEt получают 0,6 г целевого продукта. Т.п. = 168oC.
Пример 4
Полугидрат гидрохлорида 3-[3,5-бис(трифторметил)бензил]-5-(3,4- дихлорфенил)-5-[2-(4-фенилпиперид-1-ил) этил]тетрагидро-2H-1,3-оксазин-2-она
A) 3-[3,5-Бис(трифторметил) бензил] -5-(3,4-дихлорфенил)- 5-[2-(тетрагидропиран-2-илокси) этил]тетрагидро-2H-1,3-оксазин-2-он
Раствор 3,7 г соединения, полученного в соответствии со способом получения 1.1, в 60 мл ТГФ охлаждают до +5oC, добавляют 1,27 г трет-бутилата калия, после чего по каплям добавляют раствор 2,72 г 3,5-бис(трифторметил)бензилхлорида в 15 мл ТГФ. Реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры, и концентрируют в вакууме. Остаток растворяют в воде и экстрагируют AcOEt, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 5,9 г целевого продукта, который используют без дальнейшей очистки.
B) 3-[3,5-Бис(трифторметил) бензил]-5-(3,4-дихлорфенил)-5-(2- гидроксиэтил)тетрагидро-2H-1,3-оксазин-2-он
К раствору 5,9 г соединения, полученного на предыдущей стадии, в 50 мл MeOH добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, после чего реакционную смесь перемешивают в течение 15 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток дважды растворяют в насыщенном растворе газообразного HCl в MeOH и растворитель выпаривают в вакууме. Остаток растворяют в гексане и образовавшийся осадок центрифугируют с получением 3,9 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,9 части на миллион: т: 2H
2,9 - 3,6 части на миллион: н.с.: 4H
3,8 части на миллион: система AB: 2H
4,35 части на миллион: с: 2H
5,45 части на миллион: ш.с.: 1H
7,1 -7,8 части на миллион: н.с.: 3H
8,0 - 8,5 части на миллион: н.с.: 3H
8,9 части на миллион: ш.с.: 1H
C) 3-[3,5-Бис(трифторметил )бензил]-5-(3,4-дихлорфенил)-5-[2- (метансульфонилокси) этил]тетрагидро-2H-1,3-оксазин-2-он
Раствор 3 г соединения, полученного на предыдущей стадии, и 0,7 г триэтиламина в 50 мл ДХМ охлаждают до +5oC, по каплям добавляют раствор 0,66 г метансульфонилхлорида в 6 мл ДХМ, после чего реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют AcOEt, органическую фазу промывают водой, 2 н. раствором HCl, водой, 5% раствором NaHCO3, водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 3,4 г целевого продукта.
D) Полугидрат гидрохлорида 3-[3,5-бис(трифторметил) бензил]-5-(3,4- дихлорфенил)-5-[2-(4-фенилпиперид-1-ил) этил]тетрагидро-2H-1,3-оксазин-2-она
Смесь 0,32 г 4-фенилпиперидина, 1 г соединения, полученного на предыдущей стадии, и 0,28 г йодида калия в 5 мл ДМФ нагревают при температуре 60oC в течение 3 часов. После охлаждения реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (98/2 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире и образовавшийся осадок центрифугируют с получением 0,22 г целевого продукта. Т.п. = 174oC.
Пример 5
1,5 Гидрат гидрохлорида 5-[2-(4-бензилпиперид-1-ил) этил] -3-[3,5- бис(трифторметил) бензил]-5-(3,4-дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 4, из 0,3 г 4-бензилпиперидина, 0,8 г соединения, полученного на стадии C примера 4, 0,24 г йодида калия и 5 мл ДМФ. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире и образовавшийся осадок центрифугируют с получением 0,13 г целевого продукта. Т.п. = 124oC.
Пример 6
Моногидрат дигидрохлорида 5-[2-(4-анилинопиперид -1-ил)этил] -3-[3,5- бис(трифторметил)бензил]-5-(3,4- дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
Смесь 0,3 г 4-анилинопиперидина, 0,8 г соединения, полученного на стадии C примера 4, и 0,69 г карбоната калия в 5 мл ацетонитрила нагревают при температуре 50-60oC в течение 5 часов. Реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире и образовавшийся осадок центрифугируют с получением 0,12 г целевого продукта. Т.п. = 158oC.
Пример 7
2,5 Гидрат хлорида 5-(3,4-дихлорфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2] окт-1-ил] этил] -3-[3,5-бис(трифторметил)бензил] тетрагидро-2H-1,3-оксазин-2-она
Смесь 0,53 г 4-фенил-1-азабицикло[2.2.2]октана и 1,4 г соединения, полученного на стадии C примера 4, в 5 мл ДМФ нагревают при температуре 60oC в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу дважды промывают 2 н. раствором HCl и трижды насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (92/8 в объемном отношении). Полученный продукт поглощают эфиром и образовавшийся осадок центрифугируют с получением 0,21 г целевого продукта. Т.п. = 138oC.
Пример 8
Гидрохлорид 3-бензил-5-[3-(4-бензилпиперид-1-ил)пропил]-5-(3,4- дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
A) 3-Бензил-5-(3,4-дихлорфенил)-5-[3-(тетрагидропиран-2- илокси) пропил] тетрагидро-2H-1,3-оксазин-2-он
Раствор 12,5 г соединения, полученного в соответствии со способом получения 1.2, в 150 мл ТГФ охлаждают до +5oC, добавляют 4,13 г трет-бутилата калия, а затем по каплям добавляют раствор 5,78 г бензилбромида в 30 мл ТГФ. Смесь перемешивают в течение 1 часа при комнатной температуре и концентрируют в вакууме. Остаток растворяют в воде и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением целевого продукта, который используют без дальнейшей очистки.
B) 3-Бензил-5-(3,4-дихлорфенил)-5-(3-гидроксипропил)тетрагидро- 2H-1,3-оксазин-2-он
Соединение, полученное на предыдущей стадии, поглощают насыщенным раствором газообразного HCl в MeOH, после чего реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток дважды растворяют в насыщенном растворе газообразного HCl в MeOH и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении), с получением 10,6 г целевого продукта, который используют без дальнейшей очистки.
C) 3-Бензил-5-(3,4- дихлорфенил)-5-[3-(метансульфонилокси)пропил] тетрагидро-2H-1,3-оксазин-2-он.
Раствор 10 г соединения, полученного на предыдущей стадии, и 3,07 г триэтиламина в 350 мл ДХМ охлаждают до +5oC и по каплям добавляют раствор 3,19 г метансульфонилхлорида в 35 мл ДХМ. Окончив добавление, реакционную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме с получением 11 г целевого продукта, который используют без дальнейшей очистки.
D) Гидрохлорид 3-бензил-5-[3-(4-бензилпиперид-1-ил)пропил]-5-(3,4- дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
Смесь 1,86 г 4-бензилпиперидина, 2 г соединения, полученного на предыдущей стадии, и 0,7 г йодида калия в 10 мл ДМФ нагревают при температуре 50oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (98/2 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире, растворитель выпаривают в вакууме и после кристаллизации из AcOEt получают 1,4 г целевого продукта. Т.п. = 219oC.
Пример 9
Полугидрат гидрохлорида 3-бензил-5-(3,4-дихлорфенил)-5-[3-(4- гидрокси-4-фенил-пиперид -1-ил]тетрагидро-2H-1,3-оксазин-2-она
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 8, из 1,6 г 4-гидрокси-4-фенилпиперидина, 2 г соединения, полученного на стадии C примера 8, и 0,75 г йодида калия в 10 мл ДМФ. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире и образовавшийся осадок центрифугируют с получением 1,34 г целевого продукта. Т.п. = 125oC.
Пример 10
1,5 Гидрат гидрохлорида 5-[3-(4-ацетамидо-4-фенилпиперид-1- ил)пропил] -3-бензил-5-(3,4- дихлорфенил)тетрагидро-2H-1,3-оксазин-2-она
2,02 г гидрохлорида 4-ацетамидо-4-фенилпиперидина растворяют в 5 мл воды, подщелачивают до pH 13, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, органическую фазу сушат над MgSO4, и растворитель выпаривают в вакууме. Остаток поглощают 10 мл ДМФ, добавляют 1,5 г соединения, полученного на стадии C примера 8, и 0,53 г йодида калия, после чего реакционную смесь нагревают при температуре 50oC в течение 2 часов 30 минут. Полученную смесь выливают в воду и экстрагируют AcOEt, органическую фазу трижды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразногого HCl в эфире, растворитель выпаривают в вакууме и после кристаллизации из AcOEt получают 1,34 г целевого продукта. Т.п. = 145oC.
Пример 11
Гидрохлорид 3-бензил-5-(3,4-дихлорфенил)-5-[3-[4-(N- фенилацетамид)пиперид-1-ил] пропил]тетрагидро-2H-1,3-оксазин-2-она
Смесь 1,76 г 4-анилинопиперидина, 2 г соединения, полученного на стадии C примера 8, и 0,75 г йодида калия в 10 мл ДМФ нагревают при температуре 50oC в течение 1 часа 30 минут. Реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу трижды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт растворяют в 10 мл уксусного ангидрида и реакционную смесь перемешивают в течение 48 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу дважды промывают 20% водным раствором аммиака, дважды водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире, растворитель выпаривают в вакууме и после кристаллизации из AcOEt получают 1,4 г целевого соединения. Т.п. = 195oC.
Пример 12
Полугидрат хлорида 3-бензил-5-(3,4- дихлорфенил)-5-[3-[4-фенил-1- азониабицикло[2.2.2]окт-1-ил] пропил]тетрагидро-2H-1,3-оксазин-2-она
Смесь 1,2 г 4-фенил-1-азабицикло[2.2.2]октана и 2 г соединения, полученного на стадии C примера 8, в 10 мл ДМФ нагревают при температуре 50oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу промывают водой, насыщенным раствором NaCl, дважды 1 н. раствором HCl, дважды водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток поглощают AcOEt и образовавшийся осадок центрифугируют с получением 1,56 г целевого продукта. Т.п. = 235oC.
Пример 13
Дигидрат хлорида 3-бензил-5-(3,4-дихлорфенил)-5-[3-[4- бензилпиридинио-1)пропил]тетрагидро-2H-1,3-оксазин-2-она
Смесь 1,07 г 4-бензилпиридина и 2 г соединения, полученного на стадии C примера 8, в 10 мл ДМФ нагревают при температуре 90oC в течение 6 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу промывают водой и трижды насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении). Полученный продукт растворяют в минимальном количестве ацетона, добавляют эфир, пока не происходит осаждение, и образовавшийся осадок центрифугируют с получением 0,8 г целевого продукта. Т.п. = 84oC.
Пример 14
Полугидрат гидрохлорида 4-бензил-6-(3,4-дихлорфенил)-6-[2-[4- гидрокси-4-фенил-пиперид-1-ил)этил]морфолин-3-она
A) 4-Бензил-6-(3,4-дихлорфенил)-6-(2-гидроксиэтил)морфолин-3-он
К раствору 1,7 г соединения, полученного в соответствии со способом получения 1.3, в 30 мл ТГФ при комнатной температуре добавляют 0,51 г трет-бутилата калия, затем медленно добавляют 0,77 г бензилбромида и реакционную смесь нагревают в течение 2 часов при температуре 50oC. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/0,5 в объемном отношении). Полученный продукт (1 г) растворяют 10 мл MeOH и подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, растворители выпаривают в вакууме. Остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,65 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,9 части на миллион: м: 2H
2,7 - 3,3 части на миллион: м: 2H
3,65 части на миллион: система AB: 2H
4,0 части на миллион: система AB: 2H
4,3 части на миллион: т: 1H
4,45 части на миллион: система AB: 2H
7,0 - 7,6 части на миллион: н.с.: 11H
Это соединение можно также получить в соответствии с описанным ниже способом.
A') 4-Бензил-6-(3,4-дихлорфенил)-6-(2-гидроксиэтил)морфолин-3-он
Смесь 6 г соединения, полученного в соответствии со способом получения 1.4, и 1,87 г трет-бутилата калия в 100 мл ТГФ перемешивают в течение двух часов при комнатной температуре, затем медленно добавляют 2,85 г бензилбромида и реакционную смесь нагревают с обратным холодильником в течение 3 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в буферном растворе с pH 2 и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в MeOH, добавляют 1,3 г моногидрата гидроксида лития и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1 в объемном отношении), с получением 4,6 г целевого продукта.
B) 4-Бензил-6-(3,4-дихлорфенил)-6-[2-(метансульфонилокси)этил] морфолин-3-он
Раствор 0,65 г соединения, полученного на предыдущей стадии, и 0,34 г триэтиламина в 30 мл ДХМ охлаждают до 0oC и добавляют 0,25 г метансульфонилхлорида. Окончив добавление, реакционную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,71 г целевого продукта, который используют без дальнейшей очистки.
C) Полугидрат гидрохлорида 4-бензил-6-(3,4-дихлорфенил)-6-[2-(4- гидрокси-4-фенилпиперид-1-ил)этил]морфолин-2-она
Смесь 0,7 г соединения, полученного на предыдущей стадии, 0,378 г 4-гидрокси-4-фенилпиперидина и 0,8 г K2CO3 в 5 мл ацетонитрила нагревают с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, и образовавшийся осадок центрифугируют с получением 0,43 г целевого продукта. Т.п. = 160-162oC.
Пример 15
Моногидрат гидрохлорида 6-(3,4-дихлорфенил)-6-[2- (4-гидрокси-4- фенилпиперид-1-ил)этил]-4-(4-фенилбензил)морфолин-3-она
A) 6-[2-(Бензоилокси)этил] -6-(3,4-дихлорфенил)-4- (4- фенилбензил)морфолин-3-он
Смесь 1,3 г соединения, полученного в соответствии со способом получения 1.4, и 0,36 г трет-бутилита калия в 30 мл ТГФ перемешивают в течение 30 минут при комнатной температуре, добавляют 0,79 г 4-(бромметил)бифенила, после чего реакционную смесь нагревают с обратным холодильником в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (100/0,5 в объемном отношении), с получением 1,1 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,3 части на миллион: м: 2H
3,55 части на миллион: система AB: 2H
3,9 - 4,4 части на миллион: н.с.: 4H
4,5 части на миллион: с: 2H
7,0 - 7,8 части на миллион: н.с.: 17H
B) 6-(3,4-Дихлорфенил)-6-(2-гидроксиэтил)-4-(4-фенилбензил)морфолин-3-он
Смесь 1,1 г соединения, полученного на предыдущей стадии, и 2 мл концентрированного раствора NaOH в 20 мл MeOH перемешивают в течение 30 минут при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,84 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,85 части на миллион: м: 2H
2,7 - 3,3 части на миллион: м: 2H
3,5 части на миллион: система AB: 2H
3,95 части на миллион: система AB: 2H
4,35 части на миллион: т: 1H
4,4 части на миллион: система AB: 2H
7,0 - 7,6 части на миллион: н.с.: 12H
C) 6-(3,4-Дихлорфенил)-6-[2-(метансульфонилокси)этил] -4-(4- фенилбензил)морфолин-3-он
К раствору 0,84 г соединения, полученного на предыдущей стадии, и 0,223 г триэтиламина в 30 мл ДХМ при комнатной температуре добавляют 0,252 г метансульфонилхлорида, после чего смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,89 г целевого продукта, который используют без дальнейшей очистки.
D) Моногидрат гидрохлорида 6-(3,4-дихлорфенил)-6-[2-(4-гидрокси-4- фенилпиперид-1-ил)этил]-4-(4-фенилбензил)морфолин-3-она
Смесь 0,89 г соединения, полученного на предыдущей стадии, 0,875 г паратолуолсульфоната 4-гидрокси-4-фенилпиперидина и 0,483 г K2CO3 в 2 мл ДМФ нагревают при температуре 80-100oC в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 100/2 до 100/5 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, и образовавшийся осадок центрифугируют с получением 0,45 г целевого продукта. Т.п. = 143-150oC.
Пример 16
Моногидрат хлорида 6-(3,4-дихлорфенил)-6-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-4-[3,5-бис(трифторметил) бензил]морфолин- 3-она
A) 6-[2-(Бензоилокси) этил]-6-(3,4-дихлорфенил)-4-[3,5- бис(трифторметил) бензил]морфолин-3-он
Смесь 2,1 г соединения, полученного в соответствии со способом получения 1.4, и 0,616 г трет-бутилата калия в 50 мл ТГФ перемешивают в течение 30 минут при комнатной температуре, добавляют 1,44 г 3,5-бис(трифторметил)бензилхлорида, после чего реакционную смесь нагревают с обратным холодильником в течение 1 часа. Полученную смесь концентрируют в вакууме, остаток растворяют в буферном растворе с pH 4 и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/0,5 в объемном отношении), с получением 1,8 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,4 части на миллион: м: 2H
4,05 части на миллион: система AB: 2H
4,15 - 4,6 части на миллион: н.с.: 4H
4,75 части на миллион: система AB: 2H
7,3 - 8,2 части на миллион: н.с.: 11H
B) 6-(3,4-Дихлорфенил)-6-(2- гидроксиэтил)-4-[3,5- бис(трифторметил) бензил]морфолин-3-он
Смесь 1,8 г соединения, полученного на предыдущей стадии, и 4 мл концентрированного раствора NaOH в 30 мл MeOH перемешивают в течение 30 минут при 0oC, после чего продолжают перемешивать еще 1 час при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1,1 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,0 части на миллион: т: 2H
2,9 - 3,45 части на миллион: м: 2H
3,9 части на миллион: система AB: 2H
4,2 части на миллион: система AB: 2H
4,45 части на миллион: т: 1H
4,7 части на миллион: система AB: 2H
7,1 - 8,2 части на миллион: н.с.: 6H
C) 6-(3,4-Дихлорфенил)-6-[2- (метансульфонилокси)этил] -4-[3,5- бис-(трифторметил) бензил]морфолин-3-он
К раствору 1,1 г соединения, полученного на предыдущей стадии, и 0,22 г триэтиламина в 30 мл ДХМ при комнатной температуре добавляют 0,25 г метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 1 часа. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1,2 г целевого продукта, который используют без дальнейшей очистки.
D) Моногидрат хлорида 6-(3,4- дихлорфенил)-6-[2-[4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-4-[3,5-бис(трифторметил)бензил]морфолин- 3-она
Смесь 1,2 г соединения, полученного на предыдущей стадии, и 0,45 г 4-фенил-1-азабицикло[2.2.2]октана в 4 мл ДМФ нагревают при температуре 100oC в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу промывают 2 н. раствором HCl, насыщенным раствором NaCl и водой и сушат над Na2SO4, растворитель выпаривают в вакууме и после кристаллизации из смеси дихлорметана, диизопропилового эфира и пентана получают 0,9 г целевого продукта. Т. п. = 160oC.
Пример 17
Моногидрат гидрохлорида 2-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил]-4-бензоил-2-(3,4-дихлорфенил)морфолина
A) 4-Бензоил-2-(3,4-дихлорфенил)-2-(2-гидроксиэтил)морфолин
Раствор 3,6 г соединения, полученного в соответствии со способом получения 1.5, и 2,9 г триэтиламина в 50 мл ДХМ охлаждают до -60oC, по каплям добавляют раствор 1,83 г бензоилхлорида в 20 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и 10% раствором NaOH и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 100/1 до 100/5 в объемном отношении), с получением 1,2 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,95 части на миллион: м: 2H
2,8 - 4,8 части на миллион: н.с.: 9H
6,8 - 7,9 части на миллион: н.с.: 8H
B) 4-Бензоил-2-(3,4-дихлорфенил)-2-[2- (метансульфонилокси)этил]морфолин
Раствор 1,2 г соединения, полученного на предыдущей стадии, и 0,637 г триэтиламина в 50 мл ДХМ нагревают до 60oC, по каплям добавляют раствор 0,414 г метансульфонилхлорида в 10 мл ДХМ, после чего реакционную смесь перемешивают в течение 1 часа при 60oC. Полученную смесь концентрируют в вакууме, остаток экстрагируют смесью AcOEt и эфира (50/50 в объемном отношении), органическую фазу промывают водой и насыщенным раствором NaHCO3 и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1 г целевого продукта, который используют без дальнейшей очистки.
C) Моногидрат гидрохлорида 2-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил] -4-бензоил-2-(3,4-дихлорфенил)морфолина
Смесь 1 г соединения, полученного на предыдущей стадии, 1 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 1 г K2CO3 в 10 мл ацетонитрила и 5 мл ДМФ нагревают с обратным холодильником в течение 4 часов. Реакционную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, и образовавшийся осадок центрифугируют с получением 0,4 г целевого продукта. Т.п. = 184-187oC.
Пример 18
Моногидрат хлорида 2-(3,4-дихлорфенил)-4-[(3-изопропоксифенил)ацетил]- 2-[2-[4-фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
A) 2-(3,4-Дихлорфенил)-2-(2-гидроксиэтил)-4-[(3- изопропоксифенил) ацетил]морфолин
Раствор 1,2 г соединения, полученного в соответствии со способом получения 1.5, 0,83 г 3-изопропоксифенилуксусной кислоты и 0,86 г триэтиламина в 15 мл ДХМ охлаждают до 0oC, добавляют 2 г бензилоктилфталата и реакционную смесь перемешивают в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой, буферным раствором с pH 2 и водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 100/0,5 до 100/2 в объемном отношении), с получением 0,25 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,2 части на миллион: д: 6H
1,8 части на миллион: т: 2H
2,8 - 4,8 части на миллион: н.с.: 11H
6,4 - 7,8 части на миллион: н.с.: 7H
B) 2-(3,4-Дихлорфенил)-4-[(3-изопропоксифенил)ацетил]- 2-[2-(метансульфонилокси) этил]морфолин
К раствору 0,25 г соединения, полученного на предыдущей стадии, и 0,061 г триэтиламина в 10 мл ДХМ при комнатной температуре добавляют 0,069 г метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,285 г целевого продукта, который используют без дальнейшей очистки.
C) Моногидрат хлорида 2-(3,4-дихлорфенил)-4-[(3- изопропоксифенил) ацетил]-2-[2-[4-фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
Смесь 0,28 г соединения, полученного на предыдущей стадии, и 0,15 г 4-фенил-1-азабицикло[2.2.2]октана в 2 мл ДМФ нагревают при температуре 100oC в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой, 10% раствором HCl и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме и после кристаллизации из смеси ацетона и эфира получают 0,17 г целевого продукта. Т.п. = 125-130oC.
Пример 19
Моногидрат гидрохлорида 2-[3-(4-ацетамидо-4-фенилпиперид-1- ил)пропил] -4-бензоил-2-(3,4-дихлорфенил)морфолина
A) 4-Бензоил-2-(3,4-дихлорфенил)-2-(3-гидроксипропил)морфолин
Раствор 2 г соединения, полученного в соответствии со способом получения 1.6, и 0,83 г триэтиламина в 30 мл ДХМ охлаждают до -10oC, по каплям добавляют раствор 0,97 г бензоилхлорида в 10 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и 10% раствором NaOH и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1 в объемном отношении), с получением 1,7 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,9 - 2,2 части на миллион: н.с.: 4H
3,1 - 4,6 части на миллион: н.с.: 9H
7,0 - 7,9 части на миллион: н.с.: 8H
B) 4-Бензоил-2-(3,4-дихлорфенил) -2-[3-(метансульфонилокси)пропил] морфолин
Раствор 1,7 г соединения, полученного на предыдущей стадии, и 0,52 г триэтиламина в 30 мл ДХМ охлаждают до 0oC, добавляют 0,6 г метансульфонилхлорида и перемешивают смесь в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1,7 г целевого продукта, который используют без дальнейшей очистки.
C) Моногидрат гидрохлорида 2-[3(4-ацетамидо-4-фенилпиперид-1- ил)пропил] -4-бензоил-2-(3,4-дихлорфенил)морфолина
Смесь 1,7 г соединения, полученного на предыдущей стадии, 2,18 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 2,5 г K2CO3 в 10 мл ДМФ и 10 мл ацетонитрила нагревают с обратным холодильником в течение 3 часов. Реакционную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 1 г целевого продукта. Т.п. = 170-173oC.
Пример 20
Гидрохлорид 3-бензил-5-(3,4-дихлорфенил)-5-[2-(4-гидрокси-4- фенилпиперид-1-ил) этил]оксазолидин-2-она
A) 5-[2-(Бензоилокси) этил]-3-бензил-5-(3,4-дихлорфенил)оксазолидин-2-он
К раствору 1,5 г соединения, полученного в соответствии со способом получения 1.7, в 30 мл ТГФ при комнатной температуре добавляют 0,44 г трет-бутилата калия, после чего смесь перемешивают в течение 1 часа при комнатной температуре. По каплям добавляют 0,667 г бензилбромида и реакционную смесь нагревают с обратным холодильником в течение 3 часов. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают буферным раствором с pH 2 и водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (99,5/0,5 в объемном отношении), с получением 0,9 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,6 части на миллион: м: 2H
3,5 - 4,0 части на миллион: система AB: 2H
4,2 - 4,7 части на миллион: н.с.: 4H
7,2 - 8,0 частей на миллион: н.с.: 13H
B) 3-Бензил-5-(3,4-дихлорфенил)-5-(2-гидроксиэтил)оксазолидин-2-он
Смесь 0,9 г соединения, полученного на предыдущей стадии, и 0,5 мл концентрированного раствора NaOH в 30 мл MeOH и 15 мл ДХМ перемешивают в течение 30 минут при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,7 г целевого продукта, который используют без дальнейшей очистки.
C) 3-Бензил-5-(3,4-дихлорфенил)-5-[2-(метансульфонилокси)этил] оксазолидин-2-он
К раствору 0,7 г соединения, полученного на предыдущей стадии, и 0,21 г триэтиламина в 20 мл ДХМ при комнатной температуре добавляют 0,24 г метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,76 г целевого продукта, который используют без дальнейшей очистки.
D) Гидрохлорид 3-бензил-5-(3,4-дихлорфенил)-5-[2-(4-гидрокси-4- фенилпиперид-1-ил)этил]оксазолидин-2-она
Смесь 0,76 г 4-гидрокси-4-фенилпиперидина и 0,282 г йодида калия в 2 мл ДМФ нагревают при температуре 50-60oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 100/2 до 100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH, добавляя насыщенный раствор газообразного HCl в эфире, и образовавшийся осадок центрифугируют с получением 0,36 г целевого продукта. Т.п. = 223-225oC.
Пример 21
Дигидрат хлорида 5-(3,4-дихлорфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-3-[3,5-бис(трифторметил)бензил] оксазолидин-2-она
A) 5-(3,4-Дихлорфенил)-5-(2-гидроксиэтил)-3-[3,5- бис(трифторметил)бензил]оксазолидин-2-он
Смесь 1,4 г соединения, полученного в соответствии со способом получения 1.7, и 0,45 г трет-бутилата калия в 30 мл ТГФ перемешивают в течение 30 минут при комнатной температуре, добавляют 1,05 г 3,5-бис(трифторметил)бензилхлорида, после чего смесь нагревают с обратным холодильником в течение 5 часов. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт растворяют в 30 мл MeOH, добавляют 0,18 г моногидрата гидроксида лития и 1 мл воды и перемешивают смесь в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/0,5 в объемном отношении), с получением 0,86 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,2 части на миллион: м: 2H
3,0 - 3,55 части на миллион: м: 2H
3,57 - 3,9 части на миллион: система AB: 2H
4,35 - 4,7 части на миллион: н.с.: 3H
7,2 - 8,1 части на миллион: н.с.: 6H
B) 5-(3,4-Дихлорфенил)-5-[2-(метансульфонилокси)этил]-3-[3,5- бис(трифторметил)бензил]оксазолидин-2-он
К раствору 0,95 г соединения, полученного на предыдущей стадии, и 0,21 г триэтиламина в 20 мл ДХМ при комнатной температуре добавляют 0,24 г метансульфонилхлорида, после чего реакционную смесь перемешивают
в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1,1 г целевого продукта, который используют без дальнейшей очистки.
C) Дигидрат хлорида 5-(3,4-дихлорфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил]этил]-3-[3,5-бис (трифторметил)бензил] оксазолидин-2-она
Смесь 0,46 г 4-фенил-1-азабицикло[2.2.2]октана и 1,1 г соединения, полученного на предыдущей стадии, в 2 мл ДМФ нагревают при температуре 60oC в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу промывают водой, насыщенным раствором NaCl, 2 н. раствором HCl и водой и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси дихлорметана и пентана получают 0,6 г целевого продукта. Т.п. = 127-130oC (разложение).
Пример 22
Гидрохлорид 5-[2-(4-ацетамидо-4-фенилпиперид -1-ил)этил]-1- бензил-5-(3,4-дихлорфенил) -4-метилпиперазин-2,3-диона
A) 1-Бензил-5-(3,4-дихлорфенил)-4 -метил-5-[2-(тетрагидропиран-2- илокси)этил]пиперазин-2,3-дион
К раствору 0,6 г соединения, полученного в соответствии со способом получения 1.8, в 10 мл ТГФ добавляют 0,2 г трет-бутилата калия и перемешивают смесь в течение 1 часа при комнатной температуре. Затем добавляют 0,21 г бензилбромида и реакционную смесь нагревают при температуре 40oC в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из эфира получают 0,58 г целевого продукта. Т.п. = 178oC.
B) 1-Бензил-5-(3,4-дихлорфенил)-5- (2-гидроксиэтил)-4- метилпиперазин-2,3-дион
Смесь 0,56 г соединения, полученного на предыдущей стадии, 10 мл MeOH и 1 мл насыщенного раствора газообразного HCl в эфире перемешивают в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток растворяют в диизопропиловом эфире и образовавшийся осадок центрифугируют с получением 0,45 г целевого продукта. Т.п. = 198oC.
C) 1-Бензил-5-(3,4-дихлорфенил)-5-[2- (метансульфонилокси)этил]-4- метилпиперазин-2,3-дион
Раствор 0,44 г соединения, полученного на предыдущей стадии, и 0,24 мл триэтиламина в 10 мл ДХМ охлаждают до 0oC, медленно добавляют 0,12 мл метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 15 минут. Полученную смесь дважды промывают водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 0,5 г целевого продукта, который используют без дальнейшей очистки.
D) Гидрохлорид 5-[2-(4-ацетамидо-4- фенилпиперид-1-ил)этил]-1- бензил-5-(3,4-дихлорфенил)-4- метилпиперазин-2,3-диона
Смесь 0,5 г соединения, полученного на предыдущей стадии, 0,85 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 0,6 г K2CO3 в 5 мл ДМФ нагревают при температуре 80oC в течение 8 часов 30 минут. После охлаждения до комнатной температуры реакционную смесь выливают в воду, образовавшийся осадок центрифугируют и промывают водой. Осадок растворяют в дихлорметане, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (93/7 в объемном отношении). Полученный продукт растворяют в дихлорметане, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, концентрируют в вакууме и после кристаллизации из эфира получают 0,105 г целевого продукта. Т.п. = 220oC (разложение).
Пример 23
Полугидрат дигидрохлорида 3-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил] -1-бензоил-3-(3,4- дихлорфенил)-4-метилпиперазина
A) 1-Бензоил-3-(3,4-дихлорфенил)-4-метил -3-[2-(тетрагадропиран-2- илокси)этил]пиперазин
К раствору 1 г соединения, полученного в соответствии со способом получения 1.9, в 15 мл ДХМ добавляют 0,45 мл триэтиламина, после чего реакционную смесь охлаждают до 0oC и по каплям добавляют 0,32 мл бензоилхлорида. Затем реакционную смесь промывают водой и 1 н. раствором NaOH, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента гептан, а затем смесь гептана и AcOEt (50/50 в объемном отношении), с получением 0,95 г целевого продукта, который используют без дальнейшей очистки.
B) Гидрохлорид 1-бензоил-3-(3,4-дихлорфенил) -3-(2-гидроксиэтил)-4- метилпиперазина
К раствору 0,95 г соединения, полученного на предыдущей стадии, в 15 мл MeOH добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, затем смесь оставляют для выстаивания в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток поглощают эфиром, образовавшийся осадок центрифугируют и сушат с получением 0,8 г целевого соединения.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,1 - 1,9 части на миллион: н.с.: 2H
2,1 - 5,1 части на миллион: н.с.: 12H
7,1 - 8,5 части на миллион: н.с.: 8H
C) Полугидрат дигидрохлорида 3-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил]-1-бензоил-3-(3,4- дихлорфенил)-4-метилпиперазина
К раствору 0,8 г соединения, полученного на предыдущей стадии, в 15 мл ДХМ добавляют 0,78 мл триэтиламина, после чего смесь охлаждают до -20oC и по каплям добавляют 0,25 мл метансульфонилхлорида. Реакционную смесь перемешивают в течение 10 минут, а затем промывают водой и 10% раствором Na2CO3, органическую фазу сушат над MgSO4 и фильтруют. К фильтрату добавляют 1,5 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и смесь концентрируют в вакууме. Остаток поглощают 10 мл ДМФ, добавляют 1,5 г K2CO3 и реакционную смесь нагревают при температуре 80oC в течение 2 часов 30 минут. После охлаждения полученную смесь выливают в смесь воды со льдом и экстрагируют AcOEt, органическую фазу промывают 1 н. раствором NaOH и водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (93/7 в объемном отношении). Полученный продукт поглощают насыщенным раствором газообразного HCl в эфире и образовавшийся осадок центрифугируют с получением 0,86 г целевого продукта. Т.п. = 210oC (разложение).
Пример 24
Гидрохлорид хлористого 3-(3,4-дихлорфенил)-1-[(3- изопропоксифенил)ацетил]-4-метил-3-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]пиперазина
A) 3-(3,4-Дихлорфенил)-1-[(3-изопропоксифенил) ацетил]-4- метил-3-[2-(тетрагидропиран-2-илокси) этил]пиперазин
К раствору 1 г соединения, полученного в соответствии со способом получения 1.9, в 15 мл ДХМ добавляют 0,57 мл триэтиламина, а затем 0,52 г 3-изопропоксифенилуксусной кислоты и 1,3 г бензилоктилфталата. Смесь перемешивают в течение 15 минут при комнатной температуре и концентрируют в вакууме. Остаток экстрагируют AcOEt, органическую фазу промывают водой, 1 н. раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента гептан, а затем смесь гептана и AcOEt (50/50 в объемном отношении), с получением 1,1 г целевого продукта, который используют без дальнейшей очистки.
B) Гидрохлорид 3-(3,4-дихлорфенил)-3- (2-гидроксиэтил)-1-[(3- изопропоксифенил) ацетил]-4-метилпиперазина
К раствору 1,1 г соединения, полученного на предыдущей стадии, в 15 мл MeOH добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, после чего реакционную смесь оставляют для выстаивания в течение 2 часов 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток поглощают эфиром, образовавшийся осадок центрифугируют и сушат с получением 1,05 г целевого продукта.
C) Гидрохлорид хлористого 3-(3,4-дихлорфенил)-1-[(3- изопропоксифенил) ацетил]-4-метил-3-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]пиперазина
К раствору 1,05 г соединения, полученного на предыдущей стадии, в 15 мл ДХМ добавляют 0,8 мл триэтиламина, затем смесь охлаждают до -20oC и по каплям добавляют 0,22 мл метансульфонилхлорида. Через 5 минут реакционную смесь промывают водой и 10% раствором Na2CO3, органическую фазу сушат над MgSO4 и фильтруют. Затем к фильтрату добавляют 1 г 4-фенилхинуклидина и смесь концентрируют в вакууме. Остаток растворяют в 1 мл ДМФ и нагревают при температуре 80oC в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь разбавляют дихлорметаном и дважды промывают 2 н. раствором HCl и насыщенным раствором NaCl, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении). Полученный продукт поглощают диизопропиловым эфиром и образовавшийся осадок центрифугируют с получением 1,05 г целевого продукта. Т.п. = 168oC (разложение).
Пример 25
Полугидрат гидрохлорида 6-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил]-4-бензил-6-(3,4-дифторфенил) морфолин-3-она
A) 6-[2-(Бензоилокси)этил]-4-бензил-6-(3,4-дифторфенил)морфолин-3-он
Смесь 1,1 г соединения, полученного в соответствии со способом получения 1.10, и 0,38 г трет-бутилата калия в 50 мл ТГФ перемешивают в течение 30 минут при комнатной температуре, добавляют 0,51 г бензилбромида и реакционную смесь нагревают с обратным холодильником в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и буферным раствором с pH 2, сушат над Na2SO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (100/2 в объемном отношении), с получением 0,6 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,3 части на миллион: м: 2H
3,75 части на миллион: система AB: 2H
3,9 - 4,4 части на миллион: н.с.: 4H
4,5 части на миллион: с: 2H
7,0 - 8,0 частей на миллион: н.с.: 13H
B) 4-Бензил-6-(3,4-дифторфенил)-6-(2-гидроксиэтил)морфолин-3-он
Смесь 0,6 г соединения, полученного на предыдущей стадии, и 0,5 мл концентрированного раствора NaOH в 10 мл MeOH перемешивают в течение 30 минут при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,42 г целевого продукта, который используют без дальнейшей очистки.
C) 4-Бензил-6-(3,4-дифторфенил)-6-[2-(метансульфонилокси)этил] морфолин-3-он
К раствору 0,42 г соединения, полученного на предыдущей стадии, и 0,146 г триэтиламина в 20 мл ДХМ при комнатной температуре добавляют 0,137 г метансульфонилхлорида, после чего эту смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,49 г целевого продукта, который используют без дальнейшей очистки.
D) Полугидрат гидрохлорида 6-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил] -4-бензил-6-(3,4- дифторфенил)морфолин-3-она
Смесь 0,49 г соединения, полученного на предыдущей стадии, 0,94 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 0,48 г K2CO3 в 1 мл ДМФ нагревают при температуре 80oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, и образовавшийся осадок центрифугируют с получением 0,5 г целевого продукта. Т.п. = 273-275oC.
Пример 26
Дигидрат хлорида 6-(3,4-дифторфенил)-6-[2-[4-фенил-1- азониабицикло[2.2.2]окт-1-ил]этил]-4-[3,5-бис(трифторметил)бензил] морфолин-3-она
A) 6-[2-(Бензоилокси) этил]-6-(3,4-дифторфенил)-4-[3,5- бис(трифторметил) бензил]морфолин-3-он
Смесь 1 г соединения, полученного в соответствии со способом получения 1.10, и 0,32 г трет-бутилата калия в 20 мл ТГФ перемешивают в течение 1 часа при комнатной температуре, затем добавляют 0,735 г 3,5-бис(трифторметил)бензилхлорида и реакционную смесь нагревают с обратным холодильником в течение 5 часов. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением целевого продукта, который используют без дальнейшей очистки.
B) 6-(3,4-Дифторфенил) -6-(2-гидроксиэтил)-4-[3,5- бис(трифторметил) бензил]морфолин-3-он
Продукт, полученный на предыдущей стадии, растворяют в 15 мл MeOH, добавляют 0,235 г моногидрата гидроксида лития и перемешивают реакционную смесь в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1 в объемном отношении), с получением 0,84 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,95 части на миллион: т: 2H
2,95 - 3,5 части на миллион: м: 2H
3,95 части на миллион: система AB: 2H
4,25 части на миллион: система AB: 2H
4,5 части на миллион: т: 1H
4,7 части на миллион: система AB: 2H
7,0 - 8,2 части на миллион: н.с.: 6H
C) 6-(3,4-Дифторфенил)-6-[2-(метансульфонилокси) этил]-4-[3,5- бис(трифторметил) бензил]морфолин-3-он
К раствору 0,8 г соединения, полученного на предыдущей стадии, и 0,2 г триэтиламина в 15 мл ДХМ при комнатной температуре добавляют 0,22 г метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 0,9 г целевого продукта, который используют без дальнейшей очистки.
D) Дигидрат хлорида 6-(3,4-дифторфенил)-6-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-4-[3,5-бис(трифторметил)бензил] морфолин-3-она
Смесь 0,9 г соединения, полученного на предыдущей стадии, и 0,38 г 4-фенил-1-азабицикло[2.2.2] октана в 2 мл ДМФ нагревают при температуре 80oC в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают 2 н. раствором HCl, водой, насыщенным раствором NaCl и водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,41 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,0 - 2,6 части на миллион: н.с.: 8H
2,7 - 3,7 части на миллион: н.с.: 8H
3,9 части на миллион: система AB: 2H
4,1 - 5,1 части на миллион: н.с.: 4H
7,0 - 8,2 части на миллион: н.с.: 11H
Пример 27
Моногидрат гидрохлорида 2-[2-[(4-ацетамидо-4-фенилпиперид-1- ил)этил]-4-бензоил-2-(3,4-дифторфенил)морфолина
A) 4-Бензоил-2-(3,4-дифторфенил)-2-(2-гидроксиэтил)морфолин
К раствору 0,77 г соединения, полученного в соответствии со способом получения 1,11 и 0,7 г триэтиламина в 30 мл ДХМ при комнатной температуре по каплям добавляют раствор 0,9 г бензоилхлорида в 10 мл ДХМ, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и растворитель выпаривают в вакууме. Остаток растворяют в MeOH, добавляют 0,53 г моногидрата гидроксида лития и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/0,5 в объемном отношении), с получением 0,5 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,95 части на миллион: м: 2H
2,8 - 4,9 части на миллион: н.с.: 9H
6,8 - 7,8 части на миллион: н.с.: 8H
B) 4-Бензоил-2-(3,4-дифторфенил)-2-[2-(метансульфонилокси)этил]морфолин
К раствору 0,5 г соединения, полученного на предыдущей стадии, и 0,17 г триэтиламина в 20 мл ДХМ при комнатной температуре по каплям добавляют раствор 0,19 г метансульфонилхлорида в 5 мл ДХМ, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 0,56 г целевого продукта, который используют без дальнейшей очистки.
C) Моногидрат гидрохлорида 2-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил] -4-бензоил-2-(3,4-дифторфенил)морфолина
Смесь 0,56 г соединения, полученного на предыдущей стадии, 1 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 0,7 г K2CO3 в 2 мл ДМФ нагревают при температуре 80oC в течение 3 часов. Реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, концентрируют в вакууме и после кристаллизации из смеси EtOH и эфира получают 0,4 г целевого продукта. Т.п. = 179-182oC.
Пример 28
1,5 Гидрат хлорида 5-(3,4-дифторфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-3-[3,5-бис(трифторметил)бензил] оксазолидин-2-она
A) 5-(3,4-Дифторфенил)-5-(2-гидроксиэтил)-3-[3,5-бис(трифторметил) бензил]оксазолидин-2-он
Смесь 1,5 г соединения, полученного в соответствии со способом получения 1.12, и 0,48 г трет-бутилата калия в 15 мл ТГФ перемешивают в течение 1 часа при комнатной температуре, добавляют 1,1 г 3,5-бис(трифторметил)бензилхлорида, после чего эту смесь нагревают с обратным холодильником в течение 5 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в буферном растворе с pH 2 и экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Полученный продукт поглощают 20 мл MeOH, добавляют 0,2 г моногидрата гидроксида лития и смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют дихлорметаном, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1 в объемном отношении), с получением 1,3 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,2 части на миллион: м: 2H
3, 0 - 3,55 части на миллион: м: 2H
3,75 части на миллион: система AB: 2H
4,4 - 4,6 части на миллион: н.с.: 3H
7,1 - 8,2 части на миллион: н.с.: 6H
B) 5-(3,4-Дифторфенил) -5-[2-(метансульфонилокси) этил] -3-[3,5- бис(трифторметил) бензил]оксазолидин-2-он
К раствору 1,3 г соединения, полученного на предыдущей стадии, и 0,3 г триэтиламина в 20 мл ДХМ при комнатной температуре добавляют 0,34 г метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1,1 г целевого продукта, который используют без дальнейшей очистки.
C) 1,5 Гидрат хлорида 5-(3,4-дифторфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-3-[3,5-бис(трифторметил)бензил] оксазолидин-2-она
Смесь 0,46 г 4-фенил-1-азабицикло[2.2.2]октана и 1,1 г соединения, полученного на предыдущей стадии, в 2 мл ДМФ нагревают при температуре 60oC в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу промывают водой, насыщенным раствором NaCl, 2 н. раствором HCl и водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 0,6 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,1 части на миллион: м: 6H
2,7 части на миллион: т: 2H
2,85 - 4,0 части на миллион: н.с.: 10H
4,6 части на миллион: система AB: 2H
7,1 - 8,2 части на миллион: н.с.: 11H
Пример 29
Гидрохлорид 4-[2-(4-ацетамидо-4-фенилпиперид-1-ил) этил]-1- бензил-4-(3,4-дихлорфенил) -3-метилимидазолидин-2-она
A) 1-Бензил-4-(3,4-дихлорфенил) -3-метил-4-[2-(тетрагидропиран-2-илокси) этил]имидазолидин-2-он
К раствору 1,45 г соединения, полученного в соответствии со способом получения 1.13, в 20 мл ТГФ добавляют 0,5 г трет-бутилата калия и перемешивают смесь в течение 1 часа при комнатной температуре. Затем добавляют 0,48 г бензилбромида и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель концентрируют в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь гептана и AcOEt (40/60 в объемном отношении), с получением 1,5 г целевого продукта, который используют без дальнейшей очистки.
B) 1-Бензил-4-(3,4-дихлорфенил)-4-(2-гидроксиэтил)-3- метилимидазолидин-2-он
К раствору 1,5 г соединения, полученного на предыдущей стадии, в 20 мл MeOH добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в MeOH, растворитель выпаривают в вакууме и после кристаллизации из диизопропилового эфира получают 1,05 г целевого продукта. Т.п. = 118oC.
C) 1-Бензил-4-(3,4- дихлорфенил)-4-[2-(метансульфонилокси) этил] -3- метилимидазолидин-2-он
Раствор 0,5 г соединения, полученного на предыдущей стадии, и 0,34 мл триэтиламина в 10 мл ДХМ охлаждают до 0oC, добавляют 0,17 мл метансульфонилхлорида и реакционную смесь перемешивают в течение 15 минут. Полученную смесь промывают водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением целевого продукта, который используют без дальнейшей очистки.
D) Гидрохлорид 4-[2-(4-ацетамидо -4-фенилпиперид-1-ил)этил]-1- бензил-4-(3,4-дихлорфенил)- 3-метилимидазолидин-2-она
Смесь соединения, полученного на предыдущей стадии, 0,8 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 0,8 г K2CO3 в 5 мл ДМФ нагревают при температуре 80oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают 1 н. раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (95/5 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, концентрируют в вакууме и после кристаллизации из диизопропилового эфира получают 0,3 г целевого продукта. Т.п. = 206oC.
Пример 30
Хлорид 4-(3,4-дихлорфенил)-4-[2- [4-фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]-3-метил-1-[3,5-бис(трифторметил) бензил]имидазолидин-2-она
A) 4-(3,4-Дихлорфенил)-3-метил-4-[2-(тетрагидропиран-2- илокси) этил]-1-[3,5-бис(трифторметил) бензил]имидазолидин-2-он
Смесь 1,45 г соединения, полученного в соответствии со способом получения 1.13, и 0,5 г трет-бутилата калия в 20 мл ТГФ перемешивают в течение 1 часа при комнатной температуре, затем добавляют 1,04 г 3,5-бис(трифторметил)бензилхлорида и реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента гептан, а затем смесь гептана и AcOEt (50/50 в объемном отношении), с получением 2,1 г целевого продукта, который используют без дальнейшей очистки.
B) 4-(3,4-Дихлорфенил)-4-(2- гидроксиэтил)-3-метил-1-[3,5- бис(трифторметил) бензил]имидазолидин-2-он
К раствору 2,1 г соединения, полученного на предыдущей стадии, в 20 мл MeOH добавляют 1 мл насыщенного раствора газообразного HCl в эфире, после чего эту смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток растворяют в MeOH, растворитель выпаривают в вакууме и после кристаллизации из смеси эфира и пентана получают 1,7 г целевого продукта. Т.п. = 96oC.
C) 4-(3,4-Дихлорфенил)-4-[2-(метансульфонилокси) этил] -3- метил-1-[3,5-бис(трифторметил) бензил]имидазолидин-2-он
Раствор 0,56 г соединения, полученного на предыдущей стадии, и 0,3 мл триэтиламина в 10 мл ДХМ охлаждают до 0oC, добавляют 0,15 мл метансульфонилхлорида и реакционную смесь перемешивают в течение 15 минут. Полученную смесь промывают водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением целевого продукта, который используют без дальнейшей очистки.
D) Хлорид 4-(3,4-дихлорфенил)-4-[2- [4-фенил-1-азониабицикло[2.2.2]окт- 1-ил]этил]-3-метил-1-[3,5-бис(трифторметил) бензил]имидазолидин-2-она
Смесь соединения, полученного на предыдущей стадии, и 0,35 г 4-фенил-1-азабицикло[2.2.2] октана в 1 мл ДМФ нагревают при температуре 80oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в смесь 50 мл воды, 50 мл ДХМ и 3 мл концентрированной HCl и перемешивают в течение 5 минут. Образовавшийся осадок центрифугируют, промывают водой, дихлорметаном, а затем эфиром и сушат с получением 0,3 г целевого продукта. Т.п. = 290oC.
Пример 31
Гидрохлорид 6-[3-(ацетамидо-4- фенилпиперид-1-ил)пропил]-4- бензил-6-(3,4- дихлорфенил)морфолин-3-она
A) 4-Бензил-6-[3-(бензоилокси) пропил]-6-(3,4-дихлорфенил)морфолин-3-он
К раствору 1,34 г соединения, полученного в соответствии со способом получения 1.14, в 20 мл ТГФ добавляют 0,369 г трет-бутилата калия, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Затем добавляют 0,564 г бензилбромида и реакционную смесь нагревают с обратным холодильником в течение 2 часов. Полученную смесь концентрируют в вакууме, остаток растворяют в буферном растворе с pH 2 и экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением целевого продукта, который используют без дальнейшей очистки.
B) 4-Бензил-6-(3,4-дихлорфенил)-6-(3-гидроксипропил)морфолин-3-он
К раствору соединения, полученного на предыдущей стадии, в 20 мл MeOH добавляют 0,277 г моногидрата гидроксида лития, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 0,9 г целевого продукта, который используют без дальнейшей очистки.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 0,9 - 2,1 части на миллион: н.с.: 4H
3,3 части на миллион: к: 2H
3,7 части на миллион: система AB: 2H
4,15 части на миллион: система AB: 2H
4,4 части на миллион: т: 1H
4,5 части на миллион: система AB: 2H
7,1 - 7,8 части на миллион: н.с.: 8H
C) 4-Бензил-6-(3,4-дихлорфенил)-6-[3-(метансульфонилокси)пропил] морфолин-3-он
К раствору 0,9 г соединения, полученного на предыдущей стадии, и 0,288 г триэтиламина в 50 мл ДХМ при комнатной температуре добавляют 0,316 г метансульфонилхлорида, после чего смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 1,08 г целевого продукта, который используют без дальнейшей очистки.
D) Гидрохлорид 6-[3-(4-ацетамидо -4-фенилпиперид-1-ил)пропил]-4- бензил-6-(3,4-дихлорфенил)морфолин-3-она
Смесь 1,08 г соединения, полученного на предыдущей стадии, 1,4 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 1,3 г K2CO3 в 5 мл ДМФ нагревают при температуре 80oC в течение 3 часов. После охлаждения реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, растворитель выпаривают в вакууме и после кристаллизации из смеси EtOH и эфира получают 0,6 г целевого продукта. Т.п. = 151-153oC.
Пример 32
Гидрохлорид 4-(3,4-дихлорфенил)-3 -метил-4-[2-(4-фенилпиперид-1- ил)этил]-1-[3,5-бис(трифторметил) бензил]имидазолидин-2-она
Смесь 0,55 г соединения, полученного на стадии C примера 30, 0,47 г 4-фенилпиперидина и 0,5 г K2CO3 в 4 мл ДМФ нагревают при температуре 80oC в течение 5 часов. После охлаждения реакционную смесь выливают в смесь воды со льдом и экстрагируют AcOEt, органическую фазу промывают 1 н. раствором NaOH и водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (98/2 в объемном отношении). Полученный продукт растворяют в эфире, добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 0,25 г целевого продукта. Т.п. = 208oC.
Пример 33
Моногидрат гидрохлорида 4-бензил-6-(3,4- дихлорфенил)-6-[2-[4- фенил-4-(пирролидин-1-илкарбонил)пиперид-1-ил] этил]морфолин-3-она
Смесь 0,98 г соединения, полученного на стадии B примера 14, 0,645 г 4-фенил-4-(пирролидин-1-илкарбонил)пиперидина и 0,69 г K2CO3 в 3 мл ДМФ нагревают при температуре 80oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют смесью AcOEt и эфира, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/2 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана и подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, образовавшийся осадок центрифугируют с получением 0,42 г целевого продукта. Т.п. = 155-178oC.
Пример 34
Моногидрат гидрохлорида 6-[2-[4-(ацетил-N- метиламино)-4- фенилпиперид-1-ил] этил]-4-бензил-6-(3,4-дихлорфенил)морфолин-3-она
Смесь 1,1 г соединения, полученного на стадии B примера 14, 1,85 г паратолуолсульфоната 4-(ацетил-N-метиламино)-4-фенилпиперидина и 1,3 г K2CO3 в 3 мл ДМФ нагревают при температуре 80-100oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/1 до 100/4 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 0,62 г целевого продукта. Т.п. = 148-150oC.
Пример 35
Моногидрат гидрохлорида 4-бензил-6-[2-[4-(этоксикарбониламино)-4- фенилпиперид-1-ил]этил]-6-(3,4- дихлорфенил)морфолин-3-она
Смесь 1,1 г соединения, полученного на стадии B примера 14, 1 г трифторацетата 4-(этоксикарбониламино)-4-фенилпиперидина и 0,7 г K2CO3 в 3 мл ДМФ нагревают при температуре 80-100oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и перемешивают в течение 30 минут, образовавшийся осадок центрифугируют, промывают водой и сушат в вакууме при температуре 60oC. Осадок хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/1 до 100/4,5 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 0,52 г целевого продукта. Т.п. = 148-150oC.
Пример 36
Моногидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4-(N',N'-диметилуреидо)-4 -фенилпиперид-1-ил]этил]морфолина, (-) изомер
A) 4-Бензоил-2-[2-(бензоилокси) этил]-2-(3,4-дифторфенил)морфолин, (-) изомер
Раствор 8,9 г соединения, полученного в соответствии со способом получения 1.17 ((-) изомер), и 3 г триэтиламина в 100 мл ДХМ охлаждают до 0oC, по каплям добавляют раствор 3,59 г бензоилхлорида в 20 мл ДХМ и реакционную смесь перемешивают в течение 30 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/0,5 в объемном отношении), с получением 9,3 г целевого продукта.
[α]
B) 4-Бензоил-2-(3,4-дифторфенил)-2-(2-гидроксиэтил)морфолин, (-) изомер
К раствору 11,5 г соединения, полученного на предыдущей стадии, в 60 мл MeOH при комнатной температуре по каплям добавляют 6,6 г 30% водного раствора NaOH и перемешивают реакционную смесь в течение 1 часа. Полученную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/1 до 100/5 в объемном отношении), и после кристаллизации из диизопропилового эфира получают 8,4 г целевого продукта. Т.п. = 80oC.
[α]
C) 4-Бензоил-2-(3,4- дифторфенил)-2-[2-(метансульфонилокси)этил] морфолин, (-) изомер
К раствору 1 г соединения, полученного на предыдущей стадии, и 0,3 г триэтиламина в 25 мл ДХМ при комнатной температуре по каплям добавляют раствор 0,36 г метансульфонилхлорида в 3 мл ДХМ, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой, насыщенным раствором NaHCO3 и водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 1,05 г целевого продукта.
[α]
D) Моногидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4- (N', N'-диметилуреидо)-4-фенилпиперид-1-ил] этил]морфолина, (-) изомер
2 г соединения, полученного в соответствии со способом получения 2.2, растворяют в воде, подщелачивают, добавляя концентрированный раствор NaOH, и экстрагируют дихлорметаном, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 1,08 г свободного основания. Полученный продукт поглощают 3 мл ДМФ, добавляют 0,95 г соединения, полученного на предыдущей стадии, и реакционную смесь нагревают при температуре 80oC в течение 3 часов. После охлаждения полученную смесь выливают в воду, образовавшийся осадок центрифугируют и промывают водой. Осадок растворяют в дихлорметане, органическую фазу промывают 10% раствором NaOH и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/1 до 100/7,5 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, растворители выпаривают в вакууме и после кристаллизации из смеси дихлорметана и пентана получают 0,3 г целевого продукта. Т.п. = 168-172oC.
[α]
Пример 37
Полугидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4-(N',N'- диметилуреидо)-4-фенилпиперид-1-ил]этил]морфолина, (+) изомер
A) 4-Бензоил-2-[2-(бензоилокси)этил] -2-(3,4-дифторфенил)морфолин, (+) изомер
Раствор 20 г соединения, полученного в соответствии со способом получения 1.18 ((+) изомер), и 8 мл триэтиламина в 200 мл ДХМ охлаждают до 0oC, по каплям добавляют 6 мл бензоилхлорида и перемешивают реакционную смесь в течение 15 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой, 1 н. раствором HCl и 10% раствором Na2CO3 и сушат над MgSO4 растворитель выпаривают в вакууме с получением 22 г целевого продукта в виде масла.
[α]
B) 4-Бензоил-2-(3,4-дифторфенил)-2-(2-гидроксиэтил)морфолин, (+) изомер
Смесь 22 г соединения, полученного на предыдущей стадии, 9 мл 30% водного раствора NaOH и 200 мл 95o EtOH перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу трижды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси эфира и диизопропилового эфира получают 13,7 г целевого продукта. Т.п. = 80oC.
[α]
C) 4-Бензоил-2-(3,4-дифторфенил) -2-[2-(метансульфонилокси) этил] морфолин, (+) изомер
Раствор 1 г соединения, полученного на предыдущей стадии, и 0,45 мл триэтиламина в 10 мл ДХМ охлаждают до 0oC, добавляют 0,25 мл метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 15 минут. Полученную смесь промывают водой и 10% раствором Na2CO3, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 1,2 г целевого продукта в виде масла.
D) Полугидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4-(N', N'-диметилуреидо) -4-фенилпиперид-1-ил] этил]морфолина, (+) изомер
Смесь 1,2 г соединения, полученного на предыдущей стадии, 3 г соединения, полученного в соответствии со способом получения 2.2, и 2 г K2CO3 в 10 мл ДМФ нагревают при температуре 80oC в течение 3 часов. Реакционную смесь выливают в смесь воды со льдом и экстрагируют AcOEt, органическую фазу промывают 1 н. раствором NaOH и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, растворитель выпаривают в вакууме и после кристаллизации из диизопропилового эфира получают 0,66 г целевого продукта.
[α]
Пример 38
Гидрохлорид 5-(3,4-дихлорфенил)-5-[2- (4-фенилпиперид-1- ил) этил]-3-[3,5-бис(трифторметил) бензил]оксазолидин-2-она
Смесь 1,5 г соединения, полученного на стадии B примера 21, и 1,03 г 4-фенилпиперидина в 3 мл ДМФ нагревают при температуре 80oC в течение 3 часов. После охлаждения реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, растворитель выпаривают в вакууме и после кристаллизации из смеси дихлорметана и пентана получают 0,7 г целевого продукта. Т.п. = 112-114oC.
Пример 39
Моногидрат гидрохлорида 5-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил]-5-(3,4-дихлорфенил)-3- [3,5-бис(трифторметил) бензил] оксазолидин-2-она
Смесь 1,5 г соединения, полученного на стадии B примера 21, 1,5 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 1,6 г K2CO3 в 4 мл ДМФ нагревают при температуре 80oC в течение 3 часов. После охлаждения реакционную смесь выливают в воду и экстрагируют смесью AcOEt и эфира (50/50 в объемном отношении), органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор газообразного HCl в эфире, пока pH не становится равным 1, растворитель выпаривают в вакууме и после кристаллизации из смеси дихлорметана и пентана получают 0,66 г целевого продукта. Т.п. = 165-170oC.
Пример 40
Моногидрат хлорида 5-[2-[4-бензил-1-изониабицикло[2.2.2]окт-1- ил]этил] -5-(3,4-дихлорфенил)-3-[3,5-бис(трифторметил) бензил] оксазолидин-2-она
Смесь 1,4 г соединения, полученного на стадии B примера 21, и 0,6 г 4-бензилхинуклидина в 3 мл ДМФ нагревают при температуре 80oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают водой, 300 мл 10% раствора HCl и 300 мл насыщенного раствора NaCl и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из AcOEt получают 1,35 г целевого продукта. Т.п. = 172-175oC.
Пример 41
Моногидрат гидрохлорида 4-бензил-6-(3,4-дихлорфенил)-6-[2-[4- фенил-4-(пирролидин-1- илкарбониламино)пиперид-1-ил) этил]морфолин-3-она
Смесь 1,1 г соединения, полученного на стадии B примера 14, 1,2 г бензолсульфоната 4-фенил-4-(пирролидин-1-илкарбониламино)пиперидина и 0,95 г K2CO3 в 3 мл ДМФ нагревают при температуре 80-100oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду, образовавшийся осадок центрифугируют, промывают водой и сушат в вакууме. Осадок хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/2 до 100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана и подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, образовавшийся осадок центрифугируют с получением 0,54 г целевого продукта. Т.п. = 158-160oC.
Пример 42
Дигидрохлорид 4-бензил-6-(3,4-дихлорфенил)-6-[2- [4-(2- диметиламинотиазол-4-ил)-4-фенилпиперид-1-ил] этил]морфолин-3-она
Это соединение получают в соответствии с процедурой, описанной в примере 41, из 1,1 г соединения, полученного на стадии B примера 14, 1,3 г паратолуолсульфоната 4-[2-(диметиламино)тиазол-4-ил] -4- фенилпиперидина, 0,97 г K2CO3 и 4 мл ДМФ, что дает 0,76 г целевого продукта. Т.п. = 155-160oC.
Пример 43
Моногидрат гидрохлорида 4-бензоил-2-(3,4-дихлорфенил)- 2-[2- [4-(морфолин-4-илкарболниламино)-4-фенилпиперид-1-ил] этил]морфолина
A) 4-Бензоил-2-[2-(бензоилокси)этил]-2-(3,4-дихлорфенил)морфолин
К раствору 5 г соединения, полученного в соответствии со способом получения 1.19, и 1,4 г триэтиламина в 100 мл ДХМ при комнатной температуре по каплям добавляют 2,03 г бензоилхлорида, после чего реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Полученный раствор концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1 в объемном отношении), с получением 4 г целевого продукта.
B) 4-Бензоил-2-(3,4-дихлорфенил)-2-(2-гидроксиэтил)морфолин
Смесь 4 г соединения, полученного на предыдущей стадии, и 0,7 г моногидрата гидроксида лития в 50 мл MeOH перемешивают в течение 30 минут при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 3,1 г целевого продукта.
Это соединение получают также в соответствии с процедурой, описанной на стадии A примера 17.
C) 4-Бензоил-2-(3,4-дихлорфенил)-2-[2- (метансульфонилокси)этил]морфолин
К раствору 3,1 г соединения, полученного на предыдущей стадии, и 0,95 г триэтиламина в 50 мл ДХМ при комнатной температуре по каплям добавляют 1,07 г метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют смесью AcOEt и эфира (50/50 в объемном отношении), органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 3,7 г целевого продукта, который используют без дальнейшей очистки.
Это соединение получают также в соответствии с процедурой, описанной на стадии B примера 17.
D) Моногидрат гидрохлорида 4-бензоил-2-(3,4-дихлорфенил)- 2-[2-[4-(морфолин-4-илкарбониламино)-4-фенилпиперид-1-ил] этил]морфолина
Смесь 1,3 г соединения, полученного на предыдущей стадии, 1,5 г паратолуолсульфоната 4-(морфолин-4-илкарбониламино)-4-фенилпиперидина и 1,2 г K2CO3 в 3 мл ДМФ нагревают при температуре 100oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и образовавшийся осадок центрифугируют и сушат в вакууме. Осадок хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/2 до 100/3 в объемном отношении). Полученный продукт растворяют в минимальном количестве дихлорметана, подкисляют до pH 1, добавляя насыщенный раствор газообразного HCl в эфире, концентрируют в вакууме и после кристаллизации из эфира получают 0,13 г целевого продукта.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 1,8 - 4,0 части на миллион: н.с.: 26H
6,5 части на миллион: с: 1H
6,9 - 7,8 части на миллион: н.с.: 13H
10,65 части на миллион: с: 1H
Пример 44
Дигидрат дигидрохлорида 4-бензоил-2-(3,4-дихлорфенил)- 2-[2-[4-фенил-4-(пирролидин -1-иламинокарбонил)пиперид-1-ил]этил]морфолина
Смесь 1,3 г соединения, полученного на стадии C примера 43, 1,46 г бензолсульфоната 4-фенил-4-(пирролидин-1-иламинокарбонил)пиперидина и 1,17 г K2CO3 в 5 мл ацетонитрила и 5 мл ДМФ нагревают при температуре 100oC в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу дважды промывают водой и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (97/3 в объемном отношении). Полученный продукт растворяют в ацетоне, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 1,01 г целевого продукта. Т.п. = 170-174oC.
Пример 45
Моногидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4-(метоксикарбониламино)-4-фенилпиперид-1-ил] этил]морфолина, (+) изомер
Смесь 1,2 г соединения, полученного на стадии C примера 37, 1,3 г паратолуолсульфоната 4-(метоксикарбониламино)-4-фенилпиперидина и 1,2 г K2CO3 в 3 мл ДМФ нагревают при температуре 80-100oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду, образовавшийся осадок центрифугируют и сушат в вакууме, осадок хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (от 100/1,5 до 100/3 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 0,8 г целевого продукта. Т.п. = 170oC.
[α]
Пример 46
Моногидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4-фенил-4-(пирролидин-1 -илкарбониламино)пиперид-1-ил]этил]морфолина, (+) изомер
Это соединение получают в соответствии с процедурой, описанной в примере 45, из 1,2 г соединения, полученного на стадии C примера 37, 1,4 г бензолсульфоната 4-фенил-4-(пирролидин-1-илкарбониламино)пиперидина, 1,2 г K2CO3 и 2 мл ДМФ, что дает 0,3 г целевого продукта. Т.п. = 163-168oC.
[α]
Пример 47
1,5 Гидрат гидрохлорида 4-бензоил-2-[2-[4-бензил-4-(пирролидин-1- илкарбониламино)пиперид-1-ил] этил]-2-(3,4-дифторфенил)морфолина, (+) изомер
Это соединение получают в соответствии с процедурой, описанной в примере 45, из 1,2 г соединения, полученного на стадии C примера 37, 1,5 г паратолуолсульфоната 4-бензил-4-(пирролидин-1- илкарбониламино)пиперидина, 1,2 г K2CO3 и 3 мл ДМФ, что дает 0,83 г целевого продукта. Т.п. = 140oC.
[α]
Пример 48
Полугидрат дигидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2-[4-[2-(диметиламино) тиазол-4-ил]-4-фенилпиперид-1-ил]этил]морфолина, (+) изомер
Это соединение получают в соответствии с процедурой, описанной в примере 45, из 1,2 г соединения, полученного на стадии C примера 37, 1,55 г паратолуолсульфоната 4-[2-(диметиламино)тиазол-4-ил] -4- фенилпиперидина, 1,2 г K2CO3 и 3 мл ДМФ, что дает 0,7 г целевого продукта. Т.п. = 147oC.
[α]
Пример 49
Дигидрохлорид 2-[2-[4-(2-амино-1,3,4 -оксадиазол-5-ил)-4- фенилпиперид-1-ил] этил]-4-бензоил-2-(3,4-дифторфенил)морфолина, (+) изомер
Это соединение получают в соответствии с процедурой, описанной в примере 45, из 1,2 г соединения, полученного на стадии C примера 37, 1,4 г паратолуолсульфата 4-(2-амино-1,3,4-оксадиазол-5-ил)-4- фенилпиперидина, 1,2 г K2CO3 и 3 мл ДМФ, что дает 0,6 г целевого продукта. Т.п. = 153oC.
[α]
Пример 50
Моногидрат хлорида 5-(3,4-дифторфенил)-5-[2-[4-фенил-1- азониабицикло [2.2.2]окт-1-ил]этил]-3-[3,5-бис(трифторметил)бензил] оксазолидин-2-она, (-) изомер
A) 5-[2-(Бензоилокси)этил]-5-(3,4-дифторфенил)-3-[3,5- бис(трифторметил) бензил]оксазолидин-2-он, (-) изомер
К раствору 3,5 г соединения, полученного в соответствии со способом получения 1.20 ((-) изомер), в смеси 50 мл ТГФ и 10 мл ДМФ при комнатной температуре добавляют 1,2 г трет-бутилата калия, после чего смесь перемешивают в течение 30 минут при комнатной температуре. Затем добавляют 2,7 г 3,5-бис(трифторметил)бензилхлорида и реакционную смесь нагревают при температуре 60oC в течение 6 часов. Полученную смесь выливают в 200 мл буферного раствора с pH 2 и экстрагируют эфиром, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, что дает 3,6 г целевого продукта.
B) 5-(3,4-Дифторфенил)-5-(2-гидроксиэтил)-3-[3,5- бис(трифторметил) бензил]оксазолидин-2-он, (-) изомер
Смесь 3,6 г соединения, полученного на предыдущей стадии, и 0,32 г моногидрата гидроксида лития в 50 мл MeOH и 5 мл воды перемешивают в течение 2 часов при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток растворяют в воде и экстрагируют эфиром, органическую фазу дважды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (98/2 в объемном отношении), с получением 2,6 г целевого продукта.
C) 5-(3,4-Дифторфенил)-5-[2- (метансульфонилокси)этил]-3-[3,5- бис(трифторметил)бензил]оксазолидин-2-он, (-) изомер
Раствор 2,6 г соединения, полученного на предыдущей стадии, в 50 мл ДХМ охлаждают до 0oC, добавляют 1,14 мл триэтиламина, а затем 0,62 мл метансульфонилхлорида и реакционную смесь перемешивают в течение 10 минут. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу дважды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 3 г целевого продукта, который используют без дальнейшей очистки.
D) Моногидрат хлорида 5-(3,4-дифторфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2] окт-1-ил] этил]-3-[3,5-бис(трифторметил) бензил] оксазолидин-2-она, (-) изомер
Смесь 2 г соединения, полученного на предыдущей стадии, и 1 г 4-фенил-1-азабицикло[2.2.2] октана в 1 мл ДМФ нагревают при температуре 90oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в 1 н. раствор HCl и экстрагируют дихлорметаном, органическую фазу промывают 1 н. раствором HCl и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении), и после кристаллизации из диизопропилового эфира получают 1,45 г целевого продукта. Т. п. = 130oC (разложение).
[α]
Пример 51
Фумарат 4-бензоил-2-(3,4-дифторфенил)-2-[2- [4-(N',N'- диметилуреидо)-4-фенилпиперид-1-ил] этил]морфолина
A) 4-Бензоил-2-(3,4-дифторфенил)-2-(формилметил)морфолин
Раствор 0,32 мл оксалилхлорида в 7,3 мл ДХМ охлаждают до -78oC в атмосфере азота, по каплям добавляют раствор 0,51 мл ДМСО в 3,5 мл ДХМ, а затем раствор 1 г соединения, полученного на стадии A примера 27, в 7,5 мл ДХМ и 0,7 мл ДМСО. Реакционную смесь перемешивают в течение 30 минут при температуре -60oC, добавляют 2 мл триэтиламина, после чего реакционную смесь перемешивают, позволяя ей нагреваться до комнатной температуры. Полученную смесь промывают 1 н. раствором HCl, насыщенным раствором Na2CO3 и водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 1,03 г целевого продукта в виде масла.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,7 - 4,8 части на миллион: н.с.: 8H
7,0 - 8,0 частей на миллион: н.с.: 8H
9,6 части на миллион: с: 1H
B) Фумарат 4-бензоил-2-(3,4-дифторфенил)-2-[2- [4-(N',N'- диметилуреидо)-4-фенилпиперид-1-ил] этил]морфолина
Раствор 1 г соединения, полученного на предыдущей стадии в 10 мл MeOH, добавляют при комнатной температуре к раствору 0,72 г 4-(N',N'-диметилуреидо)-4-фенилпиперидина (свободное основание) и 0,16 мл уксусной кислоты в 10 мл MeOH, после чего смесь перемешивают в течение 5 минут при комнатной температуре. Затем в один прием добавляют раствор 0,18 г цианоборогидрида натрия в 10 мл MeOH и реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Полученную смесь нейтрализуют, добавляя 10% раствор Na2CO3, и экстрагируют дихлорметаном, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (96/4 в объемном отношении). Полученный продукт (1,1 г) растворяют в 20 мл ацетона, добавляют 0,24 г фумаровой кислоты, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Образовавшийся кристаллический продукт центрифугируют и промывают ацетоном, а затем эфиром с получением 1,35 г целевого продукта. Т.п. = 204oC.
Соединения по настоящему изобретению, которые представлены в таблице II получают в соответствии с процедурой, описанной в примере 34, из соединения, полученного на стадии B примера 14, и пиперидинов, описанных в способах получения.
Спектр протонного ЯМР для примера 57 при 200 МГц в ДМСО-d6
δ: 1,6 - 3,7 части на миллион: н.с.: 17H
3,75 - 4,8 части на миллион: н.с.: 6H
7,0 - 7,6 части на миллион: н.с.: 13H
10,2 - 11 частей на миллион: 2 с: 1H
Спектр протонного ЯМР для примера 58 при 200 МГц в ДМСО-d6
δ: 0,8 - 4,8 части на миллион: н.с.: 30H
7,0 - 7,8 части на миллион: н.с.: 13H
10,0 частей на миллион: с: 1H
Спектр протонного ЯМР для примера при 200 МГц в ДМСО-d6
δ: 1,1 - 3,8 части на миллион: н.с.: 24H
3,9 - 4,75 части на миллион: н.с.: 4H
6,9 - 7,7 части на миллион: н.с.: 13H
9,7 части на миллион: с: 1H
Пример 63
Дигидрат хлорида 6-[2-[4-бензил-1- азониабицикло[2.2.2]окт-1- ил]этил] -6-(3,4-дифторфенил)-4-[3,5-бис(трифторметил) бензил]морфолин-3-она
Смесь 0,86 г соединения, полученного на стадии C примера 26, и 0,47 г 4-бензил-1-азабицикло[2.2.2] октана (или 4-бензилхинуклидина) в 1 мл ДМФ нагревают при температуре 90oC в течение 9 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют дихлорметаном, органическую фазу дважды промывают 1 н. раствором HCl и дважды насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении), и после кристаллизации из эфира получают 0,4 г целевого продукта. Т.п. = 135oC (разложение).
Пример 64
Полугидрат дигидрохлорида 6-(3,4-дифторфенил)-4-[3,5- бис(трифторметил) бензил]-6-[2-[4-(пиперид-1-ил)пиперид-1- ил] этил]морфолин-3-она
Смесь 0,86 г соединения, полученного на стадии C примера 26, и 0,7 г 4-(пиперид-1-ил)пиперидина в 3 мл ДМФ нагревают при температуре 70oC в течение 4 часов 30 минут. После охлаждения до комнатной температуры реакционную смесь выливают в воду и экстрагируют AcOEt, органическую фазу промывают 1 н. раствором NaOH и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении). Полученный продукт поглощают насыщенным раствором хлористо-водородной кислоты в эфире и образовавшийся осадок центрифугируют с получением 0,49 г целевого продукта. Т.п. = 260oC (разложение).
Соединения по настоящему изобретению, которые представлены в таблице III, получают в соответствии с процедурами, описанными в предшествующих примерах:
(a) Это соединение получают в виде свободного основания в соответствии с процедурой, описанной в примере 44, из соединения, полученного на стадии C примера 43, и соединения, полученного в соответствии со способом получения 2.16.
(b) Это соединение получают в соответствии с процедурой, описанной на стадии B примера 51, из соединения, полученного на стадии A примера 51, и соединения, полученного в соответствии со способом получения 2.17 (свободное основание). После хроматографирования полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и центрифугируют целевой гидрохлорид.
(c) Это соединение получают в соответствии с процедурой, описанной на стадии B примера 51, из соединения, полученного на стадии A примера 51, и соединения, полученного в соответствии со способом получения 2.18 (свободное основание). После хроматографирования полученный продукт поглощают насыщенным раствором хлористо-водородной кислоты в эфире и центрифугируют целевой гидрохлорид.
Спектр протонного ЯМР для примера 65 при 200 МГц в ДМСО-d6
δ: 0,8 - 4,2 части на миллион: н.с.: 28H
7,0 - 7,8 части на миллион: н.с.: 8H
7,9 - 8,6 части на миллион: 2 с: 2H
10,4 - 11,6 части на миллион: н.с.: 2H
Спектр протонного ЯМР для примера 67 при 200 МГц в ДМСО-d6
δ: 2,0 - 4,2 части на миллион: н.с.: 21H
5,9 части на миллион: м: 1H
6,75 части на миллион: с: 1H
7,0 - 7,8 части на миллион: н.с.: 8H
10,95 части на миллион: с: 1H
Пример 68
Дигидрат гидрохлорида 4-бензоил-2-(3,4-дифторфенил)-2-[2-(4- фенил-4-уреидопиперид-1-ил) этил]морфолина, (+) изомер
A) 4-Бензоил-2-(3,4-дифторфенил)-2-(формилметил)морфолин, (+) изомер
Раствор 2,6 мл оксалилхлорида в 59 мл ДХМ охлаждают до -78oC в атмосфере азота и по каплям добавляют раствор 4,6 мл ДМСО в 29,5 мл ДХМ, затем раствор 8,2 г соединения, полученного на стадии B примера 37, в 59 мл ДХМ и 5,7 мл ДМСО. Реакционную смесь перемешивают в течение 30 минут при температуре -60oC, добавляют 16 мл триэтиламина и продолжают перемешивать реакционную смесь, позволяя ей нагреваться до комнатной температуры. Полученную смесь промывают 1 н. раствором HCl, водой, насыщенным раствором NaHCO3 и водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 7,5 г целевого продукта в виде масла.
B) Дигидрат гидрохлорида 4-бензоил-2-(3,4- дифторфенил)-2-[2-(4- фенил-4-уреидопиперид-1-ил) этил]морфолина, (+) изомер
Раствор 2,5 г соединения, полученного на предыдущей стадии, в 26 мл MeOH добавляют при комнатной температуре к раствору 1,6 г 4-фенил-4-уроеидопиперидина (свободное основание) и 0,4 мл уксусной кислоты в 26 мл MeOH, после чего смесь перемешивают в течение 5 минут при комнатной температуре. Затем в один прием добавляют раствор 0,5 г цианборогидрида натрия в 26 мл MeOH и реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Полученную смесь выливают в 200 мл 10% раствора Na2CO3 и экстрагируют AcOEt, органическую фазу промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента градиент смеси дихлорметана и MeOH (от 100/5 до 100/7,5 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 1,6 г целевого продукта. Т.п. = 187-190oC.
[α]
Пример 69
Моногидрат гидрохлорида 4-бензоил-2-(3,4- дифторфенил)- 2-[2- [4-(N'-метилуреидо)-4-фенилпиперид-1-ил] этил]морфолина, (+) изомер
Это соединение получают в соответствии с процедурой, описанной на стадии B примера 68, из 1,85 г 4-(N'-метилуреидо)-4-фенилпиперидина (свободное основание), 0,44 мл уксусной кислоты и 26 мл MeOH с последующим добавлением 2,75 г соединения, полученного на стадии A примера 68, в 26 мл MeOH и 0,55 г цианборогидрида натрия в 26 мл MeOH, что дает 2 г целевого продукта. Т.п. = 170-173oC.
[α]
Пример 70
1,5 Гидрат дигидрохлорида 4-бензоил-2-(3,4-дифторфенил)- 2-[2- [4-(3,3-диметилкарбазоил)-4-фенилпиперид-1-ил] этил]морфолина, (+) изомер
Это соединение получают в соответствии с процедурой, описанной на стадии B примера 68, из 1,55 г 4-(3,3-диметилкарбазоил)-4-фенилпиперидина (свободное основание) и 0,38 мл уксусной кислоты в 26 мл MeOH с последующим добавлением 2,4 г соединения, полученного на стадии A примера 68, в 26 мл MeOH и 0,47 г цианборогидрида натрия в 26 мл MeOH, что дает 1,82 г целевого продукта.
[α]
Пример 71
Моногидрат хлорида 2-(3,4-дифторфенил)-4-[2- (3- изопропоксифенил)ацетил]-2-[2- [4-фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
A) 2-[2-(Бензоилокси) этил]-2-(3,4-дифторфенил)-4-[2-(3- изопропоксифенил) ацетил]морфолин
К раствору 2,1 г соединения, полученного в соответствии со способом получения 1.21, 1,2 г 2-(3-изопропоксифенил)уксусной кислоты и 2,2 мл триэтиламина в 50 мл ДХМ при комнатной температуре добавляют 3,2 г бензилоктилфталата, после чего реакционную смесь перемешивают в течение 15 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают 1 н. раствором HCl, водой и 10% раствором Na2CO3 и сушат над MgSO4, растворитель выпаривают в вакууме с получением 3,2 г целевого продукта в виде масла.
B) 2-(3,4-Дифторфенил)-2-(2-гидроксиэтил)-4- [2-(3- изопропоксифенил) ацетил]морфолин
Смесь 3,2 г соединения, полученного на предыдущей стадии, 2 мл концентрированного раствора NaOH и 50 мл MeOH перемешивают в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу дважды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме с получением 2,3 г целевого продукта в виде масла.
C) 2-(3,4-Дифторфенил)-4-[2- (3-изопропоксифенил) ацетил] -2-[2-(метансульфонилокси) этил]морфолин
К раствору 2,3 г соединения, полученного на предыдущей стадии, в 50 мл ДХМ добавляют 0,93 мл триэтиламина, а затем 0,51 мл метансульфонилхлорида, после чего смесь перемешивают в течение 15 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и 10% раствором Na2CO3 и сушат над MgSO4, растворитель выпаривают в вакууме с получением 2,7 г целевого продукта в виде масла.
D) Моногидрат хлорида 2-(3,4-дифторфенил)-4-[2-(3- изопропоксифенил)ацетил]-2-[2- [4-фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
Смесь 1,3 г соединения, полученного на предыдущей стадии, и 0,72 г 4-фенил-1-азабицикло[2,2,2] октана в 1,5 мл ДМФ нагревают при температуре 90oC в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь выливают в 1 н. раствор HCl и экстрагируют дихлорметаном, органическую фазу промывают 1 н. раствором HCl и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении), и после кристаллизации из диизопропилового эфира получают 0,38 г целевого продукта. Т.п. = 136oC (разложение).
Пример 72
Полугидрат дигидрохлорида 2-(3,4-дифторфенил)-4-[2-(3- изопропоксифенил)ацетил]-2-[2- [4-(пиперид-1-ил)пиперид-1-ил] этил]морфолина
Смесь 1,3 г соединения, полученного на стадии C примера 71, и 1,1 г 4-(пиперид-1-ил)пиперидина в 4 мл ДМФ нагревают при температуре 70oC в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь выливают в смесь воды со льдом и экстрагируют AcOEt, органическую фазу промывают 1 н. раствором NaOH и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используют в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении). Полученный продукт поглощают насыщенным раствором хлористо-водородной кислоты в эфире и образовавшийся осадок центрифугируют с получением 0,49 г целевого продукта. Т.п. = 270oC (разложение).
Пример 73
1,5 Гидрат хлорида 2-(3,4-дифторфенил)-2-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-4-[3,5-бис(трифторметил) бензоил]морфолина
A) 2-[2-(Бензоилокси)этил]-2-(3,4- дифторфенил)-4-[3,5- бис(трифторметил) бензоил]морфолин
Раствор 2,1 г соединения, полученного в соответствии со способом получения 1.21, и 1 мл триэтиламина в 50 мл ДХМ охлаждают до 0oC, по каплям добавляют 1,24 г 3,5-бис(трифторметил)бензоилхлорида, после чего реакционную смесь перемешивают в течение 5 минут. Полученную смесь дважды промывают водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 3,5 г целевого продукта в виде масла.
B) 2-(3,4-Дифторфенил)-2-(2 -гидроксиэтил)-4-[3,5- бис(трифторметил) бензоил]морфолин
Смесь 3,5 г соединения, полученного на предыдущей стадии, 2 мл концентрированного раствора NaOH и 50 мл MeOH перемешивают в течение 2 часов при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу дважды промывают водой и сушат над MgSO4, растворитель выпаривают в вакууме и после кристаллизации из смеси диизопропилового эфира и пентана получают 0,84 г целевого продукта. Т.п. = 101oC.
C) 2-(3,4-Дифторфенил)-2-[2- (метансульфонилокси)этил]-4-[3,5- бис(трифторметил) бензоил]морфолин
К раствору 0,84 г соединения, полученного на предыдущей стадии, в 10 мл ДХМ добавляют 0,5 мл триэтиламина, смесь охлаждают до 0oC, добавляют 0,26 мл метансульфонилхлорида, после чего реакционную смесь перемешивают в течение 5 минут при комнатной температуре. Полученную смесь дважды промывают водой, органическую фазу сушат над MgSO4 и растворитель выпаривают в вакууме с получением 0,98 г целевого продукта.
D) 1,5 Гидрат хлорида 2-(3,4-дифторфенил)-2-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-4-[3,5-бис(трифторметил) бензоил]морфолина
Смесь 0,98 г соединения, полученного на предыдущей стадии, и 0,65 г 4-фенил-1-азабицикло[2.2.2]октана в 1 мл ДМФ нагревают при температуре 100oC в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь выливают в 1 н. раствор HCl и экстрагируют дихлорметаном, органическую фазу промывают 1 н. раствором HCl и насыщенным раствором NaCl и сушат над MgSO4, растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, используя в качестве элюента дихлорметан, а затем смесь дихлорметана и MeOH (90/10 в объемном отношении), и после кристаллизации из диизопропилового эфира получают 0,42 г целевого продукта. Т.п. = 170oC (разложение).
Пример 74
Моногидрат гидрохлорида 5-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил]-3-бензоил-5-(3,4- дихлорфенил)оксазолидина
A) 3-Бензоил-5-(3,4-дихлорфенил)-5-(2-гидроксиэтил)оксазолидин
Раствор 1,23 г бензоилхлорида в 10 мл ДХМ при комнатной температуре по каплям добавляют к раствору 3,2 г соединения, полученного в соответствии со способом получения 1.22, и 1 г триэтиламина в 50 мл ДХМ, после чего смесь перемешивают в течение 1 часа при комнатной температуре. Затем добавляют еще 1,76 г триэтиламина и 2,46 г бензоилхлорида и перемешивают смесь еще 1 час при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток поглощают водой и экстрагируют AcOEt, органическую фазу промывают буферным раствором с pH 2 и насыщенным раствором NaCl и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток поглощают 30 мл MeOH, добавляют 2 мл 30% раствора NaOH и перемешивают смесь в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме. Остаток хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/1,5 в объемном отношении), с получением 1,6 г целевого продукта.
B) 3-Бензоил-5-(3,4-дихлорфенил)-5- [2-(метансульфонилокси) этил] оксазолидин
К раствору 1,6 г соединения, полученного на предыдущей стадии, и 0,485 г триэтиламина в 50 мл ДХМ при комнатной температуре по каплям добавляют раствор 0,55 г метансульфонилхлорида в 5 мл ДХМ, после чего смесь перемешивают в течение 30 минут при комнатной температуре. Полученную смесь концентрируют в вакууме, остаток экстрагируют AcOEt, органическую фазу промывают водой и сушат над Na2SO4, растворитель выпаривают в вакууме с получением 1,9 г целевого продукта.
C) Моногидрат гидрохлорида 5-[2-(4-ацетамидо-4-фенилпиперид-1- ил)этил] -3-бензоил-5-(3,4- дихлорфенил)оксазолидина
Смесь 1,8 г соединения, полученного на предыдущей стадии, 1,7 г паратолуолсульфоната 4-ацетамидо-4-фенилпиперидина и 1,7 г K2CO3 в 4 мл ДМФ нагревают при температуре 80-100oC в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь выливают в воду, образовавшийся осадок центрифугируют и сушат в вакууме. Осадок хроматографируют на кислом силикагеле, используя в качестве элюента смесь дихлорметана и MeOH (100/3 в объемном отношении). Полученный продукт растворяют в дихлорметане, добавляют насыщенный раствор хлористо-водородной кислоты в эфире, пока pH не становится равным 1, и образовавшийся осадок центрифугируют с получением 1 г целевого продукта. Т.п. = 165-170oC.
Пример 75
Моногидрат хлорида 5-(3,4-дифторфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2]окт-1-ил] этил]-3-[3,5-бис(трифторметил) бензил] оксазолидин-2-она, (+) изомер
A) 5-[2-(Бензоилокси)этил]-5-(3,4-дифторфенил)-3-[3,5- бис(трифторметил)бензил]оксазолидин-2-он, (+) изомер
Это соединение получают в соответствии с процедурой, описанной на стадии A примера 50, из соединения, полученного в соответствии со способом получения 1.23 ((+) изомер).
B) 5-(3,4-Дифторфенил)-5-(2- гидроксиэтил)-3-[3,5- бис(трифторметил) бензил]оксазолидин-2-он, (+) изомер
Это соединение получают в соответствии с процедурой, описанной на стадии B примера 50, из соединения, полученного на предыдущей стадии.
C) 5-(3,4-Дифторфенил)-5-[2- (метансульфонилокси)этил]-3-[3,5- бис(трифторметил) бензил]оксазолидин-2-он, (+) изомер
Это соединение получают в соответствии с процедурой, описанной на стадии C примера 50, из соединения, полученного на предыдущей стадии.
D) Моногидрат хлорида 5-(3,4-дифторфенил)-5-[2- [4-фенил-1- азониабицикло[2.2.2] окт-1-ил] этил]-3-[3,5-бис(трифторметил)бензил] оксазолидин-2-она, (+) изомер
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 50, из соединения, полученного на предыдущей стадии, и 4-фенил-1-азабицикло[2.2.2]октана.
[α]
Пример 76
Дигидрохлорид 4-бензоил-2-[3-[4-карбамоил-4-(пиперид -1- ил)пиперид-1-ил]пропил]-2-(3,4- дихлорфенил)морфолина
Это соединение получают в соответствии с процедурой, описанной на стадии C примера 19, из соединения, полученного на стадии B примера 19, и соединения, полученного в соответствии со способом получения 2.16.
Пример 77
Хлорид 2-(3,4-дихлорфенил)-4-[2- (3-хлорфенил)ацетил]-2-[2-[4- фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
A) 2-[2-(Бензоилокси)этил] -2-(3,4- дихлорфенил)-4-[2-(3- хлорфенил)ацетил]морфолин
Это соединение получают в соответствии с процедурой, описанной на стадии A примера 71, из соединения, полученного в соответствии со способом получения 1.19, и 2-(3-хлорфенил)уксусной кислоты.
B) 2-(3,4-Дихлорфенил)-4-[2-(3-хлорфенил)ацетил] -2-(2- гидроксиэтил)морфолин
Это соединение получают в соответствии с процедурой, описанной на стадии B примера 71, из соединения, полученного на предыдущей стадии.
C) 2-(3,4-Дихлорфенил)-4-[2-(3-хлорфенил) ацетил]- 2-[2-(метансульфонилокси) этил]морфолин
Это соединение получают в соответствии с процедурой, описанной на стадии C примера 71, из соединения, полученного на предыдущей стадии, и метансульфонилхлорида.
D) Хлорид 2-(3,4-дихлорфенил)-4-[2-(3- хлорфенил)ацетил]-2-[2-[4- фенил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 71, из соединения, полученного на предыдущей стадии, и 4-фенил-1-азабицикло[2.2.2]октана.
Спектр протонного ЯМР при 200 МГц в ДМСО-d6
δ: 2,0 - 2,5 части на миллион: н.с.: 8H
2,55 - 4,6 части на миллион: н.с.: 16H
7,0 - 8,0 частей на миллион: н.с.: 12H
Пример 78
Хлорид 2-(2,3-дихлорфенил)-4-[2- (3-хлорфенил)ацетил]-2-[2-[4- бензил-1-азониабицикло[2.2.2]окт-1-ил] этил]морфолина
Это соединение получают в соответствии с процедурой, описанной на стадии D примера 71, из соединения, полученного на стадии C примера 77, и 4-бензил-1-азабицикло[2.2.2]октана.


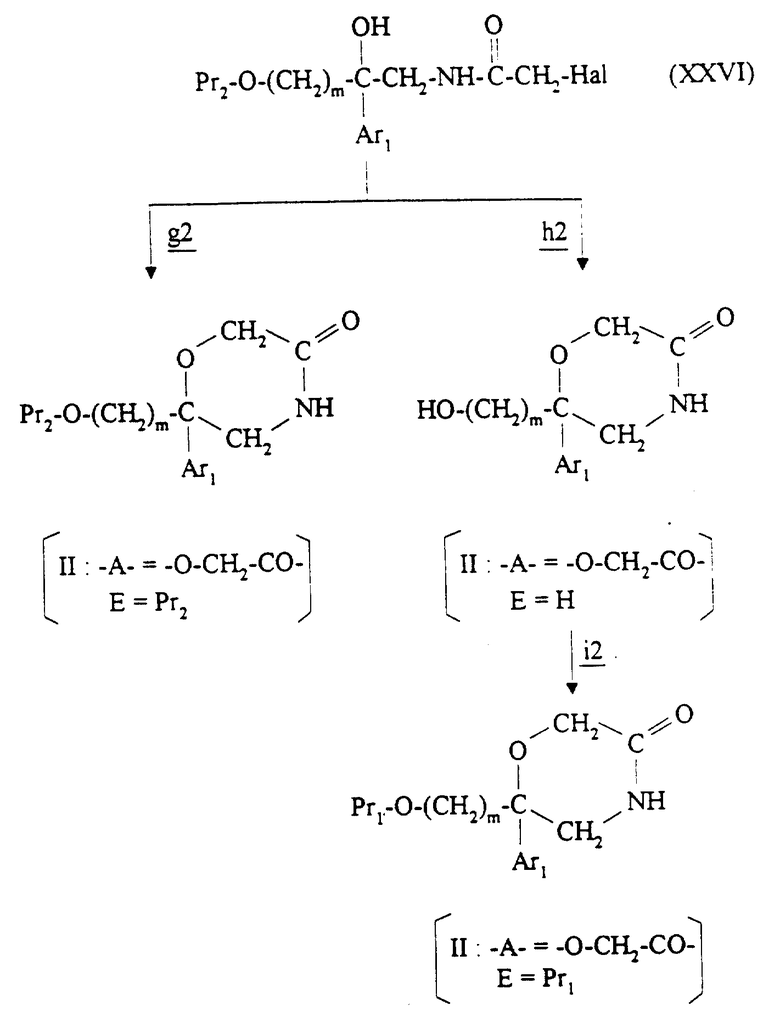



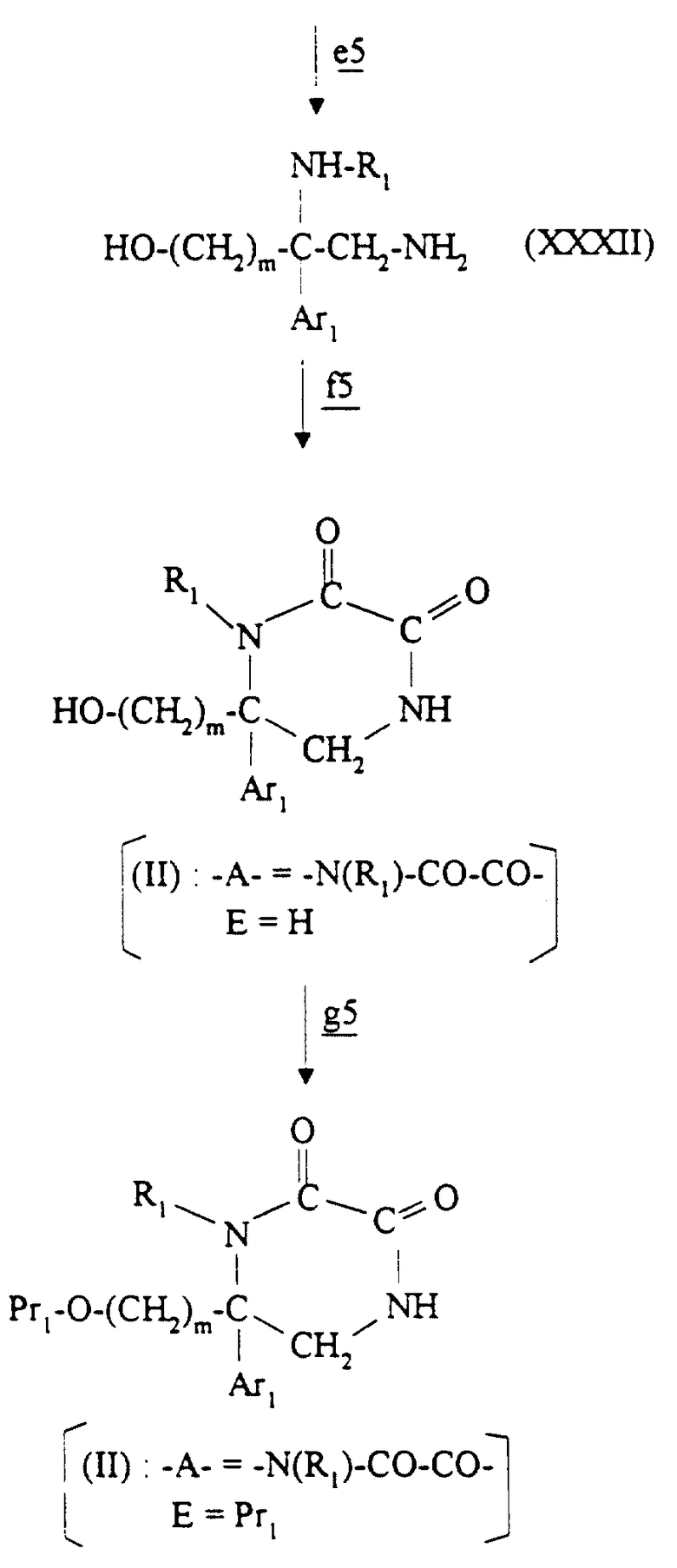





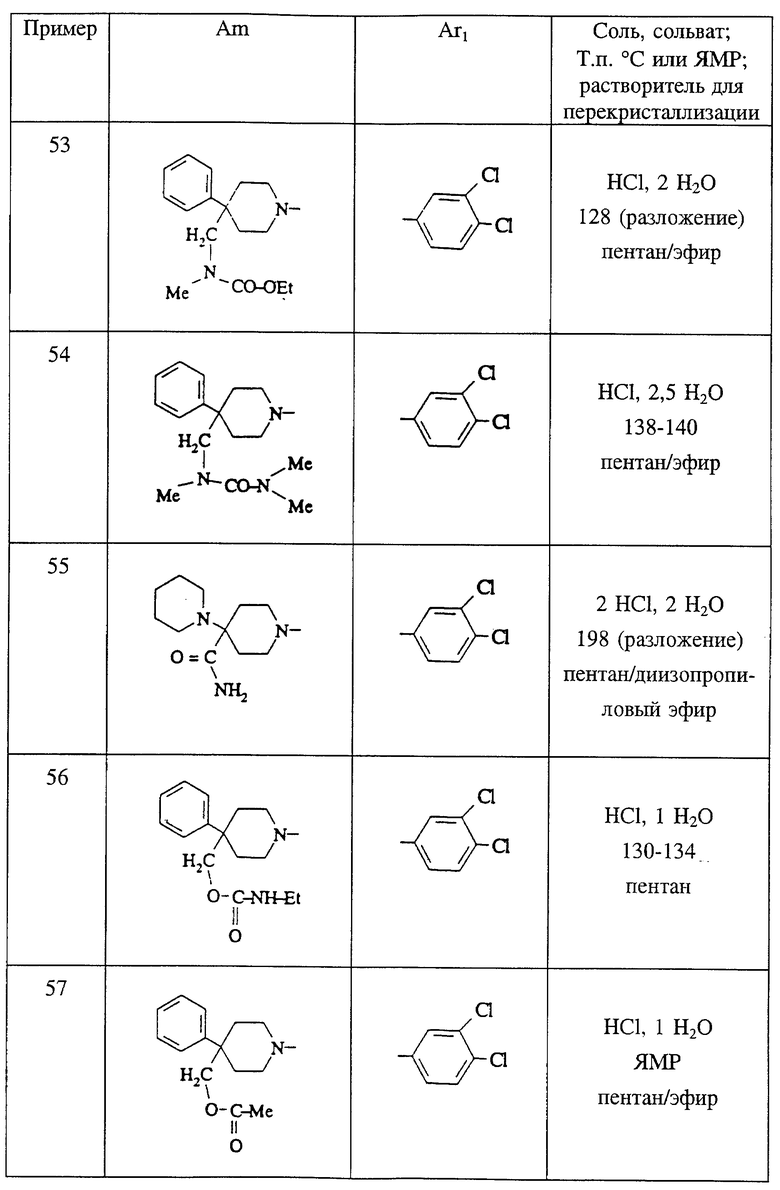


Описываются новые замещенные гетероциклические соединения формулы I, где А означает двухвалентный радикал, выбранный из -О-СО-, -CH2O-CO- и др., как указано в п.1 формулы, R1 означает водород или C1-C4-алкил; m - 2, 3; Ar1 означает фенил, возможно замещенный галогеном, гидроксилом, C1-C4-алкокси, C1-C4-алкилом, трифторметилом, метилендиокси; Т означает -CH2-Z, -CH(C6H5)2, -C(C6H5)3. Соединения формулы I обладают сродством к рецепторам тахикинина при ингибиторной константе Ki ниже 10-8 М. Описывается способ получения соединений формулы I и промежуточные соединения, используемые в нем. Описывается также фармацевтическая композиция, обладающая антагонистической активностью к рецепторам нейрокининов, содержащая в качестве активного вещества соединение формулы I. 12 с. и 13 з.п.ф-лы, 3 табл. 
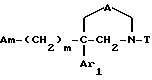
где A является двухвалентным радикалом, выбираемым из A1) -O-CO-; A2) -CH2-O-CO-; A3) -O-CH2-CO-; A4) -O-CH2-CH2; A5) -N(R1)-CO-; A6) -N(R1)-CO-CO-; A7) -N(R1)-CH2-CH2-; A8) -O-CH2-, где R1 обозначает водород или (C1 - C4)-алкил;
m = 2 или 3;
Ar1 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил, трифторметил и метилендиокси, причем указанные заместители являются одинаковыми или разными;
T обозначает группу, выбираемую из CH2 - Z, -CH(C6H5)2 и -С(C6H5)3; T может также быть группой -CO-B-Z, если A является двухвалентным радикалом, выбираемым из -O-CH2-CH2-, -N(R1)-CH2-CH2- и -O-CH2;
B обозначает прямую связь или метилен;
Z обозначает: фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена; трифторметил; гидроксил; фенил, который не замещен, либо моно- или полизамещен галогеном, трифторметилом, (C1 - C4)-алкилом, гидроксилом или (C1 - C4)-алкокси, причем указанные заместители являются одинаковыми или разными; (C1 - C4)-алкокси, уреидо, который не замещен либо моно- или дизамещен в 3-положении (C1 - C4)-алкилом или (C3 - C7)-циклоалкилом;
Am обозначает:
i - либо группу Am1 формулы
где J1 является:
i1 - или группой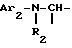
где Ar2 обозначает пиридил; фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил, трифторметил, нитро и метилендиокси, причем указанные заместители являются одинаковыми или разными;
R2 обозначает водород; (C1 - C7)-алкил; бензил; формил; или (C1 - C7)-алкилкарбонил;
i2 - или группой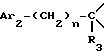
где Ar2 имеет указанные выше значения;
n = 0 или 1;
R3 является группой, выбираемой из: (1) водорода; (2) (C1 - C7)-алкила; (3) формила; (4) (C1 - C7)-алкилкарбонила; (5) циано; (6) -(CH2)q-OH; (7) -(CH2)q-O-(C1 - C7)-алкила; (8) -(CH2)q-OCHO; (9) -(CH2)q-OCOR17; (10) -(CH2)q-OCONH-(C1 - C7)-алкила; (11) -NR4R5; (12) -(CH2)q-NR6C(= W1)R7; (13) -(CH2)q-NR6COOR8; (14) -(CH2)q-NR6SO2R9; (15) -(CH2)q-NR6C(= W1)NR10R11; (16) -CH2-NR12R13; (17) -CH2-CH2-NR12-R13; (18) -COOH; (19) (C1 - C7)-алкоксикарбонила; (20) -C(= W1)NR10R11; (21) -CH2-COOH; (22) (C1 - C7)-алкоксикарбонилметила; (23) -CH2-C(= W1)NR10R11; (24) -O-CH2CH2-OR18; (25) -NR6COCOR19; (26) -CO-NR20-NR21R22;
(27)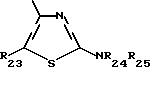
(28)
или R3 образует двойную связь между атомом углерода, к которому он присоединен, и смежным атомом углерода пиперидинового кольца;
q = 0, 1 или 2;
W1 обозначает атом кислорода;
R4 и R5 независимо друг от друга обозначают водород или (C1 - C7)-алкил; R5 может также быть (C3 - C7)-циклоалкилметилом, бензилом и фенилом;
R6 обозначает водород или (C1 - C7)-алкил;
R7 обозначает водород; (C1 - C7)-алкил; фенил; бензил; (C3 - C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами;
либо R6 и R7 вместе обозначают группу -(CH2)p-;
p = 3 или 4;
R8 обозначает (C1 - C7)-алкил или фенил;
R9 обозначает (C1 - C7)-алкил;
R10 и R11 независимо друг от друга обозначают водород или (C1 - C7)-алкил; R11 может также быть (C3 - C7)-циклоалкилом, (C3 - C7)-циклоалкилметилом, гидроксилом, (C1 - C4)-алкокси, бензилом или фенилом; либо R10 и R11 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из пирролидина и морфолина;
R12 и R13 независимо друг от друга обозначают водород или (C1 - C7)-алкил; R13 может также быть (C3 - C7)-циклоалкилметилом или бензилом;
R17 обозначает (C1 - C7)-алкил; (C3 - C7)-циклоалкил, который не замещен или замещен одним или несколькими метилами; фенил или пиридил;
R18 обозначает водород; (C1 - C7)-алкил; формил или (C1 - C7)-алкилкарбонил;
R19 обозначает (C1 - C4)-алкокси;
R20 обозначает водород или (C1 - C7)-алкил;
R21 и R22 независимо друг от друга обозначают водород или (C1 - C7)-алкил;
либо R21 и R22 вместе с атомом азота, к которому они присоединены, образуют пирролидин;
R23 обозначает водород;
R24 и R25 независимо друг от друга обозначают водород или (C1 - C7)-алкил;
i3 - либо группой
где R3 имеет указанные выше значения;
R14 обозначает (C1 - C7)-алкил или (C3 - C7)-циклоалкил; R14 может быть группой -CONR15R16, если R3 обозначает водород, или группой -NR15R16, если R3 обозначает водород, карбоксил, (C1 - C7)-алкоксикарбонил или группу -C(= W1)NR10R11;
R15 и R16 независимо друг от друга обозначают (C1 - C7)-алкил; либо R15 и R16 вместе с атомом азота, к которому они присоединены, образуют гетероцикл, выбираемый из пиперидина и морфолина;
ii - либо группу Am2 формулы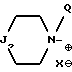
где J2 является
ii2 - или группой
где Ar3 обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
R2 имеет значения, указанные выше для J1;
ii2 - или группой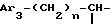
где Ar3 имеет указанные выше значения;
n = 0 или 1;
Q обозначает (C1 - C6)-алкил или бензил, причем указанный заместитель занимает аксиальное или экваториальное положение; и
X⊖ является фармацевтически приемлемым анионом;
iii - либо группу Am3 формулы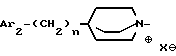
где Ar2 имеет указанные выше значения;
n = 0 или 1;
X⊖ является фармацевтически приемлемым анионом;
iv - либо группу Am4 формулы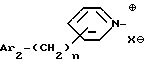
где Ar2 имеет указанные выше значения;
n = 0 или 1;
X⊖ является фармацевтически приемлемым анионом,
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.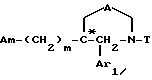
где "*" означает, что атом углерода, меченый таким образом, имеет (+) или (-) абсолютную конфигурацию;
Am, m, Ar1, A и T имеют значения, указанные для соединений формулы (I) в п.1;
и их соли, получаемые при взаимодействии с минеральными или органическими кислотами.
где Aa является двухвалентным радикалом, выбираемым из -O-CO-; -CH2-O-CO-; -O-CH2-CO-; -N(R1)-CO- и -N(R1)-CO-CO-, где R1 обозначает водород иди (C1 - C4)-алкил;
Ama обозначает:
* либо группу Am2a формулы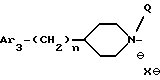
* либо группу Am3 формулы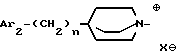
n = 0 или 1;
Q имеет значения, указанные выше для соединения формулы I в п.1 и занимает аксиальное положение;
X⊖ является фармацевтически приемлемым анионом;
Ar2 и Ar3 имеют значения, указанные выше для соединения формулы I в п.1;
Ar1a обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
Za обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1 - C4)-алкокси и гидрокси;
и их соли с минеральными или органическими кислотами.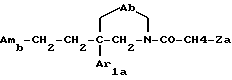
где Ab является двухвалентным радикалом -O-CH2-CH2-; -N(R1)-CH2-CH2- или -O-CH2-, где R1 обозначает водород или (C1 - C4)-алкил;
Amb обозначает:
* либо группу Am2m формулы
* либо группу Am3 формулы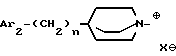
* либо группу Am1a формулы
n = 0 или 1;
Q имеет значения, указанные выше для соединения формулы I в п.1 и занимает аксиальное положение;
X⊖ является фармацевтически приемлемым анионом;
Ar2, Ar3 и R3 имеют значения, указанные выше для соединения формулы I в п.1;
Ar1a обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
Za обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1 - C4)-алкокси и гидроксил;
и их соли с минеральными или органическими кислотами.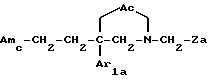
где Ac является двухвалентным радикалом, выбираемым из -O-CH2-CO-, -CH2-O-CO- и -O-CO-;
Amc обозначает группу Am1a формулы
где n = 0 или 1;
Ar2 и R3 имеют значения, указанные для соединения формулы I в п.1;
Ar1a обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
Za обозначает фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1 - C4)-алкокси и гидроксил,
и их соли с минеральными или органическими кислотами.
где Ab является двухвалентным радикалом -O-CH2-CH2-, -N(R1)-CH2-CH2- или -O-CH2-;
R1 обозначает водород или (C1 - C4)-алкил;
Amc обозначает группу Am1a формулы
n = 0 или 1;
Ar2 и R3 имеет значения, указанные для соединения формулы I в п.1;
Ar1a обозначает фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
Za обозначает фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1 - C4)-алкокси и гидрокси;
и их соли с минеральными или органическими кислотами.
где Ab является двухвалентным радикалом -O-CH2-CH2-, -N(R1)-CH2-CH2- или -O-CH2-;
R1 обозначает водород или (C1 - C4)-алкил;
Amc обозначает группу Am1a формулы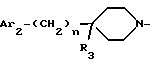
n = 0 или 1;
Ar2 и R3 имеют значения, указанные выше для соединения формулы I в п.1;
Ar1a обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
Za обозначает фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1 - C4)-алкокси и гидроксил,
и их соли с минеральными или органическими кислотами.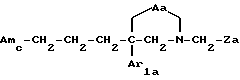
где Aa является двухвалентным радикалом -O-CO-, -CH2-O-CO-, -O-CH2-CO-, -N(R1)-CO- или -N(R1)-CO-CO-;
R1 обозначает водород иди (C1 - C4)-алкил;
Amc обозначает группу Am1a формулы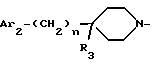
n = 0 или 1;
Ar2 и R3 имеет значения, указанные выше для соединения формулы I в п.1;
Ar1a обозначает фенил, который не замещен либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, гидроксил, (C1 - C4)-алкокси, (C1 - C4)-алкил и трифторметил, причем указанные заместители являются одинаковыми или разными;
Za обозначает фенил, который не замещен, либо моно- или полизамещен заместителем, выбираемым из группы, включающей атом галогена, трифторметил, (C1 - C4)-алкокси и гидроксил,
и их соли с минеральными или органическими кислотами.
1) осуществляют обработку соединения формулы II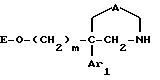
где m, Ar1 и A имеют значения, указанные для соединения формулы I в п.1 и E обозначает водород или O-защитную группу,
или функциональным производным кислоты формулы III
HOCO-B-Z
где B и Z имеют значения, указанные для формулы I в п.1, если необходимо получить соединение формулы I, в которой T является -CO-B-Z,
или галогензамещенным производным формулы IV
Hal-CH2-Z,
в которой Z имеет значения, указанные в п.1, и Hal обозначает галоген, если необходимо получить соединение формулы I, в которой T является -CH2-Z,
или галогензамещенным производным формулы V
Hal-CH(C6H5)2
если необходимо получить соединение формулы I, в которой T является группой -CH(C6H5)2
или галогензамещенным производным формулы VI
Hal-C-(C6H5)3
если необходимо получить соединение формулы I, в которой T является группой -C(C6H5)3,
с получением соединение формулы VII
где E, m, Ar1, A и T имеют указанные выше значения;
2) возможно, удаляют O-защитную группу путем обработки кислотой или основанием с получением спирта формулы VIII
где m, Ar1, A и T имеют указанные выше значения;
3) полученный спирт VIII обрабатывают соединением формулы IX
Y-SO2-Cl
где Y обозначает метил, фенил, толил или трифторметил, с получением соединения формулы X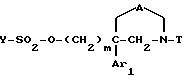
где Y, m, Ar1, A и T имеют указанные выше значения;
4) полученное соединение X подвергают взаимодействию:
либо с циклическим вторичным амином формулы XI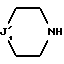
где J'1 обозначает
* или группу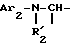
где Ar2 имеет значения, указанные для формулы I в п.1, и R'2 аналогичен R2 в формуле I по п.1 или является предшественником R2;
* или группу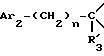
где Ar2 имеет значения, указанные для формулы I в п.1, и R'3 аналогичен R3 в формуле I по п.1 или является предшественником R3, причем если R'3 является гидроксилом или амино, то эти группы могут быть защищены;
* или группу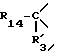
где R14 имеет значения, указанные для формулы I в п.1, и R'3 имеет указанные выше значения;
либо с третичным амином формулы XII
где J3 и Q имеют значения, указанные для формулы I в п.1;
либо с циклическим третичным амином формулы XIII
где Ar2 и n имеют значения, указанные для формулы I в п.1;
либо с соединением формулы XIV
где Ar2 и n имеют значения, указанные для формулы I в п.1;
5) и либо в случае использования циклического вторичного амина формулы XI и после возможного снятия защиты с гидроксильной группы или с аминогруппы, обозначенной R'3 или после возможного превращения R'2 в R2 или R'3 в R3, полученный продукт, при желании, превращают в одну из его солей; либо выделяют полученный продукт в виде сульфоната или в виде соли сульфоновой кислоты, или же, при необходимости, заменяют полученный анион или, соответственно, соль кислоты на другой фармацевтически приемлемый анион или, соответственно, на другую фармацевтически приемлемую соль с минеральной или органической кислотой, в случае использования третичного амина формулы XII, циклического третичного амина формулы XIII или соединения формулы XIV.
1') соединение формулы VIII, приведенной в п.12, окисляют с получением соединения формулы XXXVIII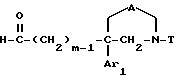
где m, Ar1, A и T имеют указанное в п.1 значение;
2') полученное соединение подвергают взаимодействию с соединением формулы XI, описанной в п.12, в присутствии кислоты, затем восстанавливают промежуточно полученную соль иминия с помощью агента восстановления, и
3') после возможного снятия защиты с гидроксильных или аминогрупп или после возможного превращения радикала R'2 в R2 или радикала R'3 в R3, переводят, при необходимости, полученное соединение в одну из его солей.
где m, A и Ar1 имеют значения, указанные для соединения формулы I в п.1, и E обозначает водород или O-защитную группу, в виде чистых энантиомеров или рацемической смеси.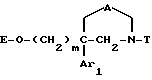
где m, A, Ar1 и T имеют значения, указанные для соединений формулы I в п. 1, и E обозначает водород или O-защитную группу, в виде чистых энантиомеров или рацемической смеси.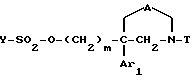
где m, A, Ar1 и T имеют значения, указанные для соединения формулы I в п. 1, и Y обозначает метильную, фенильную, толильную или трифторметильную группу, в виде чистых энантиомеров или рацемической смеси.
где m, Ar1, A и T имеют значения, указанные для соединения формулы I в п.1, в виде чистых энантиомеров или рацемической смеси.
где m, Ar1 и A имеют значения, указанные для соединения формулы I в п.1;
RI обозначает два атома водорода и RII является: или группой -O-E, в которой E обозначает атом водорода или O-защитную группу, или группой -O-SO2-Y, в которой Y обозначает метильную, фенильную, толильную или трифторметильную группу; или же RI обозначает атом кислорода и RII обозначает атом водорода;
T' аналогичен T, значения которого указаны для соединения формулы I; T' может быть также водородом, если R1 обозначает 2 атома водорода;
RII одновременно является группой -O-E-,
в виде чистых энантиомеров или рацемической смеси.
где m и Ar1 имеют значения, указанные для соединения формулы I в п.1;
E обозначает водород или O-защитную группу;
L обозначает цианогруппу или аминометильную группу;
D обозначает группу, выбираемую из: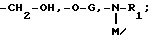
G обозначает водород или O-защитную группу;
M обозначает водород или N-защитную группу;
R1 имеет значения, указанные для соединения формулы I в п.1,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой.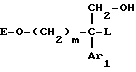
где m и Ar1 имеют значения, указанные для соединения формулы I в п.1;
E обозначает водород или O-защитную группу;
L обозначает цианогруппу или аминометильную группу,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой.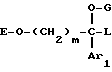
где m и Ar1 имеют значения, указанные для соединения формулы I в п.1;
E обозначает водород или O-защитную группу;
G обозначает водород или O-защитную группу;
L обозначает цианогруппу или аминометильную группу,
в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой.
где m, Ar1 и R1 имеют значения, указанные для соединения формулы I в п. 1;
E обозначает водород или O-защитную группу;
M обозначает водород или N-защитную группу;
L обозначает цианогруппу или аминометильную группу в виде чистых энантиомеров или рацемической смеси, если L является аминометильной группой.
Приоритет по пунктам и признакам:
30.01.95 по п. 1, где A означает A8, T означает -O-CH2-, R3 означает (CH2)q-OCOR17, для R17-C1-C7 алкил, R14 - водород, Ab означает -O-CH2-, п. 17, где T' - H, если R1 означает водород, R11 одновременно является -O-E-, п.25;
04.07.95 по пп.9 и 13, где соединения формулы II в виде чистых энантиомеров, п. 15, где соединения формулы X в виде чистых энантиомеров, п.19, где соединения XXXV в виде чистых энантиомеров, если L является аминометильной группой, п.20, где соединения XXXVI в виде чистых энантиомеров, если является аминометильной группой, п.21, где соединения XXXVII в виде чистых энантиомеров, если L является аминометильной группой;
03.11.95 по п.1, где Ar1 - метилендиокси, Ar2 - нитро и метилендиокси, R3-(CH2)q-OCOR17 для остальных значений R17, -O-CH2CH2-OR18, -NR6COCOR19, -CO-NR20-NR21R22, 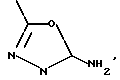 пп.12, 16 - 18.
пп.12, 16 - 18.
| Устройство для обработки поверхностей деталей | 1974 |
|
SU512901A2 |
| Устройство для управления реверсивным широтно-импульсным тиристорным преобразователем | 1973 |
|
SU515240A1 |
| EP 0591040 A1, 06.04.1994 | |||
| Способ получения производных 1,3-оксазолидин-2-она или их кислотно-аддитивных солей | 1986 |
|
SU1431679A3 |
| Устройство для изготовления волокнистого слоя под основу для длинноволокнистой бумаги и т.п. | 1948 |
|
SU85227A1 |
| 0 |
|
SU291244A1 | |

