Изобретение относится к новым производным арилзамещенных пиперидинов, обладающим антагонистической активностью к рецептору NK3 человека, к способу их получения и к их использованию в фармацевтических композициях.
В последние годы проводятся многочисленные исследования в отношении тахикининов и их рецепторов. Известно, что тахикинины находятся одновременно в центральной нервной системе и в периферической нервной системе. Рецепторы тахикининов также известны и классифицированы на три типа: NK1, NK2, NK3. Вещество P (SP) представляет собой эндогенный лиганд рецепторов NK2, нейрокинин A (NKA) представляет собой эндогенный лиганд рецепторов NK2 и нейрокинин B (NKB) представляет собой эндогенный лиганд рецепторов NK3.
Рецепторы NK1, NK2, NK3 были обнаружены у различных видов животных. Так, рецепторы NK3 найдены у морской свинки, крысы, обезьяны (Br. J. Pharmacol., 1990, 99, 767-773, Neurochem. Int., 1991, 18, 149-165), относительно недавние исследования показали наличие этих рецепторов у человека (FEBS Zetters, 1992, 299 (1), 90-95).
В последней публикации C.A.Maggi и сотр. рассматриваются рецепторы тахикининов, а также антагонисты этих рецепторов, представлены фармакологические исследования антагонистов и применение их в терапии (J. Autonomic Pharmacol. , 1993, 13, 23-93).
Из специфических антагонистов к рецептору NK1 можно назвать следующие непептидные соединения: CP-96345 (J. Med. Chem., 1992, 35, 2591-2600), RP-68651 (Proc. Natl. Acad. Sci., США, 1991, 88, 10208-10212), SR 140333 (Curr. J. Pharmacol., 1993, 250, 403-413).
В качестве селективного непептидного антагониста к рецептору NK2 можно назвать соединение SP 48968, подробно описанное в журнале Zife Sci., 1992, 50, PL 101-PL 106.
Что касается рецептора NK3, то некоторые непептидные соединения являющиеся антагонистами к ангиотензину II, обнаруживают сродство к рецептору NK3 головного мозга крысы и морской свинки, однако это сродство очень слабое и соответствует константе ингибирования K и порядка 10-5 М (FASEB J., 1993, 7(4), A 710, 4104). Известно, также и что пептидный антагонист (Trp7, Ala8) NKA, проявляет слабую специфичность к рецептору NK3 крысы (J. Autonomic. Pharmacol., 1993, 13, 23-93).
В европейской заявке на патент EP-512901 указывается, что хлоргидрат 5-[2-(4-гидрокси-4-фенилпиперид-1-ил)этил] -5-(3,4- дихлорфенил)-1-бензилпиперид-2-она, ниже называемый соединением A, антагонизирует связь эледоизина с Kи = 200 наномоль, причем эледоизин представляет собой пептид бактериального происхождения, эквивалентный нейрокинину B.
В европейской заявке на патент EP-474561 описаны антагонисты нейрокининов, более конкретно антагонисты рецепторов NK1 или NK2, в частности описан хлоргидрат N-метил-N-[2-(3,4- дихлорфенил)-5-(4-гидрокси-4-фенилпиперид-1-ил)-пентил]-бензамида.
Таким образом ни одно из пептидных или непептидных соединений, известных на сегодняшний день, не обладает высоким сродством к рецептору NK3 человека.
Фармакологические исследования антагонистов рецепторов NK1 и NK2 пептидного и непептидного характера показали, что их сродство к этим рецепторам, и их фармакологические активности, очень сильно зависят от вида живого существа по всей вероятности вследствие незначительных различий в аминокислотных последовательностях, индуктирующих таким образом очень тонкие структурные изменения этих рецепторов от одного вида к другому (J. Autonomic. Pharmacol. , 1993, 13, 23-93). Некоторые экспериментальные данные, подтвержденные фармакологическими показателями соединений согласно изобретению указывают на то, что аналогичное положение существует в отношении рецептора NK3. В частности рецептор NK3 человека отличается от рецептора NK3 крысы.
Задача изобретения - разработка новых соединений непептидного характера, которые обладали бы специфичным и высоким сродством к рецептору NK3 человека. Указанная активность может быть использована для получения лекарственных средств, пригодных для лечения психиатрических заболеваний или заболеваний психосоматического происхождения и любых заболеваний центральной или периферической нервной системы, при которых нейрокинин В и рецептор NK3 вмешиваются в интернейронные регуляции.
Под очень сильным сродством к рецептору NK3 человека понимают сродство, характеризующееся константой ингибирования Kи обычно ниже 5•10-9 М.
При излучении фиксации лиганды, константу ингибирования Kи определяют по отношению Ченга-Прусоф'а (в Receptor Binding in Drug Research, изд. R.A. O'Brien, Marcel Dekker, New York, 1986):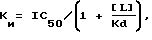
где [L] = концентрация лиганда,
Kd = константа диссоциации лиганда,
IC50 = концентрация, которая ингибирует 50% фиксации лиганда.
Под высокой специфичностью к рецептору NK3 человека понимают, что константа ингибирования для рецептора NK3 человека обычно по крайней мере в 100 раз меньше константы ингибирования для рецептора NK2 или константы ингибирования для рецептора NK1 различных видов, живых существ.
Под заболеванием психосоматического происхождения понимают заболевания, первопричина которых находится в центральной нервной системе (ЦНС) и патологические последствия которых находятся на периферическом уровне.
Так, согласно одному из своих аспектов, предметом настоящего изобретения являются производные арилзамещенных пиперидинов формулы (I):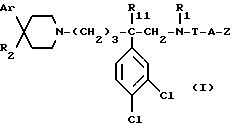
в которой: Ar обозначает пирид-2-ил или фенил, незамещенный или замещенный галогеном, метилом или (C1-C4)-алкоксилом,
R1 обозначает метильную группу,
R11 обозначает водород,
или R1 и R11 вместе обозначают группу -(CH2)3-,
R2 обозначает гидроксил, (C1-C7)алкоксил, (C1-C7)ацилоксигруппу, циано-группу, группу -NR6R7, группу -NR3COR4, группу -NR3COOR8, группу -NR3SO2R9, группу -NR3CONR10R12, (C1-C7)-ацильную группу, (C1-C7)алкоксикарбонильную группу, группу -CONR10R12, группу -CH2OH, (C1-C7)алкоксиметильную группу, (C1-C7)-ацилоксиметильную группу, (C1-C7)-алкиламинокарбонилоксиметильную группу, группу -CH2NR13R14, группу -CH2NR3COR4, группу -CH2NR3COOR8, группу -CH2NR3SO2R9, группу -CH2NR3CONR10R12, или
R2 представляет собой двойную связь между атомом углерода, с которым он связан, и соседним атомом углерода пиперидинового цикла,
или Ar и R2 вместе с атомом углерода, с которым они связаны, представляют собой группу формулы (а):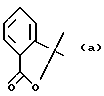
R3 обозначает водород или (C1-C4)-алкил,
R4 обозначает водород, (C1-C7)-алкил, фенил, бензил, пиридил или (C3-C7)циклоалкил, незамещенный или замещенный одной или несколькими метильными группами,
или R3 и R4 вместе обозначают группу -(CH2)n-:
"n" обозначает число 3 или 4,
T обозначает метилен, карбонил, группу -COO-, группу -CONR5-,
A обозначает простую связь, метилен, этилен, пропилен, винилен,
- или -T-A- обозначает группу -SO2-,
- Z обозначает фенил, незамещенный или замещенный одним или несколькими галогенами, (C1-C4)-алкилом, (C1-C4)-алкоксилом, нитро-группой,
- R5 обозначает водород или (C1-C4)-алкил,
- R6 и R7, каждый, обозначают, независимо друг от друга, водород, или (C1-C7)алкил; R7, кроме того, может обозначать (C3-C7)-циклоалкилметил, бензил или фенил; или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероцикл, выбираемый среди азетидина, пирролидина, пиперидина, морфолина, тиоморфолина или пергидроазепина,
- R8 обозначает (C1-C7)-алкил или фенил,
- R9 обозначает (C1-C7)-алкил, свободную или замещенную одним или двумя (C1-C7)-алкилами амино-группу; фенил, незамещенный или замещенный один или несколько раз заместителем, выбираемым в группе, включающей атом галогена, (C1-C7)алкил, трифторметил, гидроксил, (C1-C7)алкоксил, карбоксил, (C1-C7)-алкоксикарбонил, (C1-C7)-алкилкарбонилокси-группу, циано-группу, нитро-группу, свободную или замещенную одним или двумя (C1-C7)-алкилами амино-группу, причем вышеуказанные заместители идентичны или различны,
- R10 и R12, каждый, независимо друг от друга, обозначают водород или (C1-C7)-алкил; R12, кроме того, может обозначать (C3-C7)-циклоалкил, (C3-C7)циклоалкилметил, гидроксил, (C1-C4)алкоксил, бензил или фенил, или
R10 и R12 вместе с атомом азота, с которым они связаны, образуют гетероцикл, выбираемый среди азетидина, пирролидина, пиперидина, морфолина, тиоморфолина или пергидроазепина,
- R13 и R14, каждый, независимо друг от друга, обозначает водород или (C1-C7)алкил; R14, кроме того, может обозначать (C3-C7)-циклоалкилметил или бензил,
при условии, что:
1) когда Ar обозначает фенильную группу, R2 обозначает гидроксильную группу, T-A-Z обозначает бензоильную группу, R1 отличен от метильной группы,
2) когда Ar обозначает фенильную группу, R2 обозначает группу -NH-CO-CH3, T-A-Z обозначает бензоильную группу, R1 и R11 вместе не образуют группу -(CH2)3-,
3) когда Ar обозначает фенильную группу, R2 обозначает гидроксильную группу, T-A-Z обозначает 3-метоксибензильную группу, R1 и R11 вместе не образуют группу -(CH2)3-,
и их соли, а также их рацематы и энантиомеры.
В настоящем описании алкильные или алкоксильные группы являются линейными или разветвленными, под галогеном понимают атом фтора, хлора, брома или йода, предпочтительно фтор, хлор или йод.
В настоящем описании, под ацилом понимают формил или (C1-C6)-алкилкарбонил.
Соли соединений формулы (I) включают как соли с неорганическими или органическими кислотами, которые позволяют осуществлять соответствующие выделение или кристаллизацию соединений формулы (I), с такими, как пикриновая кислота или щавелевая кислота или оптически активная кислота, например, миндальная кислота или камфорсульфокислота, так и соли, которые представляют собой фармацевтически приемлемые соли.
Фармацевтически приемлемые соли представляют собой такие, как хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, малеат, фумарат, 2-нафталинсульфонат, бензолсульфонат, гликолят, глюконат, цитрат, изэтионат, пара-толуолсульфонат.
В зависимости от значения R1 и R11, соединения согласно изобретению относятся к одной из нижеописанных групп формул (II) или (III):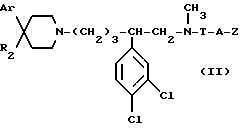
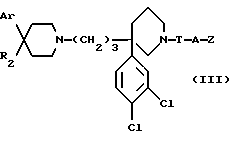
в которых: Ar, R2, T, A и Z имеют вышеуказанные для формулы (I) значения.
Предпочтительными соединениями являются соединения формулы (I), в которых:
- Ar обозначает пирид-2-ил или фенил, незамещенный или замещенный галогеном,
- R1 обозначает метильную группу,
- R11 обозначает водород,
- или R1 и R11 вместе обозначают группу -(CH2)3-,
- R2 обозначает гидроксил, (C1-C7)-алкоксил, аминогруппу, (C1-C7)-ацилокси-группу, группу -NR3COR4, или
R2 представляет собой двойную связь между атомом углерода, с которым он связан, и соседним атомом углерода пиперидинового цикла,
- R3 обозначает водород или (C1-C4)-алкил,
- R4 обозначает водород, (C1-C7)-алкил, фенил, пиридил или (C3-C7)-циклоалкил, незамещенный или замещенный одной или несколькими метильными группами,
- или R3 и R4 вместе обозначают группу -(CH2)n-,
- "n" обозначает число 3 или 4,
- T обозначает метилен, карбонил, группу -COO-, группу -CONR5-,
- A обозначает простую связь, метилен, этилен, пропилен, винилен,
- или -T-A- обозначает группа -SO2-,
- Z обозначает фенил, незамещенный или замещенный один или несколько раз галогеном, (C1-C4)-алкилом, (C1-C4)-алкоксилом, нитро-группой,
- R5 обозначает водород или (C1-C4)-алкил,
при условии, что:
1) когда Ar обозначает фенильную группу, R2 обозначает гидроксильную группу, T-A-Z обозначает бензоильную группу, R1 отличен от метильной группы,
2) когда Ar обозначает фенильную группу, R2 обозначает группу -NH-CO-CH3, T-A-Z обозначает бензоильную группу, R1 и R11 не образуют вместе группы -(CH2)3-,
3) когда Ar обозначает фенильную группу, R2 обозначают гидроксильную группу, T-A-Z обозначает 3-метоксибензильную группу, R1 и R11 не образуют вместе группы -(CH2)3-,
также, как их соли.
Соединения формулы (II), в которых R2 обозначает ацетамидо-группу, пропиониламино-, бутириламино-, изобутириламино-, ацетил-N-метиламино-, пропионил-N-метиламино-, бутирил-N-метиламино-, изобутирил-N-метиламино- группу и T-A-Z представляет собой бензилоксикарбонил, незамещенный или замещенный на фениле хлором или нитро-группой, также являются предпочтительными соединениями.
Соединения формулы (III), в которых Ar обозначает фенил, незамещенный или замещенный галогеном; R2 обозначает ациламино-группу с 1-8 C-атомами, ацил-N-метиламино-группу, в которой ацил содержит 1-8 C-атомов; T-A-Z обозначает бензоил, также представляют собой предпочтительные соединения.
Также особенно предпочтительны, в форме рацематов или в одной из их энантиомерных (+) - (-) - форм, следующие соединения:
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(ацетил-N- метиламино)-4-фенилпиперидин-1-ил)-пропил/-пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-пропиониламино-4- фенилпиперидин-1-ил)-пропил/-пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(пропионил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/-пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-бутириламино-4- фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3/3-(4-(бутирил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-изобутириламино-4- фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(изобутирил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-валериламино-4- фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(валерил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-изовалериламино-4- фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(изовалерил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-пивалоиламино-4- фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-пивалоил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
и их соли.
Преимущественно предпочтителен хлоргидрат 1-бензоил-3-(3,4- дихлорфенил)-3-/3-(4-(ацетил-N-метиламино)-4-фенилпиперидин-1- ил)-пропил/пиперидина в оптически чистой форме, предпочтительно в форме (+)-изомера.
Среди соединений формулы (II) те соединения, в которых:
- либо R2 обозначает (C5-C7)-алкоксил, (C5-C7)-ацилокси-группу, группу -NR3COR4, где R4 отличается от (C1-C6)-алкила, если R3 обозначает водород; группу -NR6R7, где R6 и R7 отличаются от H или (C1-C4)-алкила; группу -NR3COOR8; группу -NR3SO2R9; группу -NR3CONR10R12 (C5-C7)-ацил; (C5-C7)-алкоксикарбонил; группу -CONR10R12; (C1-C7)-алкоксиметил; (C1-C7)-ацилоксиметил; (C1-C7)-алкиламино-карбонилоксиметил; группу -CH2NR13R14, где R13 и R14 отличаются от водорода; группу -CH2NR3COR4, где R4 отличается от (C1-C3)-алкила, если R3 обозначает галоген; группу -CH2NR3COOR8, группу CH2NR3SO2R9; группу -CH2NR3CONR10R12;
- либо T обозначает метилен или группу -CONR5- с R5 другим, чем водород,
- либо T-A- обозначает группу -SO2-,
образуют предпочтительную группу соединений изобретения.
Среди соединений формулы (III), те соединения, в которых:
- либо R2 обозначает (C5-C7)-алкоксил; (C5-C7)-ацилокси-группу; группу -NR3COR4, где R4 отличается от водорода или (C1-C3)-алкила; группу -NR6R7, где R6 и R7 отличаются от водорода или (C1-C4)-алкила, или, если R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероцикл, отличающийся от пирролидинового, пиперидинового или морфолинового цикла; группу -NR3COOR8; группу -NR3SO2R9, группу -NR3CONR10R12; (C1-C7)-ацил; (C5-C7)-алкоксикарбонил; группу -CONR10R12; группу -CH2OH; (C1-C7)-алкоксиметил; (C1-C7)-ацилоксиметил; (C1-C7)-алкиламинокарбонилоксиметил; группу -CH2NR13R14; группу -CH2NR3COR4;
группу NR3COOR8; группу -CH2NR3COOR8; группу -CH2NR3SO2R9; группу -CH2NR3CONR10R12;
- либо T обозначает группу -CONR5-, группу -COO-,
- либо A обозначает винилен,
- либо T-A-Z обозначает группу -SO2-,
образуют другую предпочтительную группу соединений изобретения.
Особенно предпочтительны соединения формулы (IV):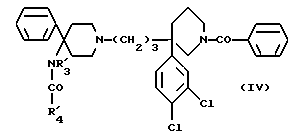
в которой R'3 обозначает водород или метил,
- R'4 обозначает (C4-C7)-алкил; фенил, бензил, пиридил или (C3-C7)-циклоалкил, незамещенный или замещенный одним или несколькими метильными группами,
- или R'3 и R'4 вместе обозначают группу -(CH2)n-,
- и "n" обозначает число 3 или 4,
также, как их соли.
Соли соединений формул (II), (III) и (IV) согласно настоящему изобретению включают как соли с неорганическими или органическими кислотами, которые позволяют осуществлять соответствующее выделение или кристаллизацию соединений формул (II), (III) и (IV), с такими, как пикриновая кислота или щавелевая кислота или оптически активная кислота, например, миндальная или камфорсульфокислота, так и соли, которые представляют собой фармацевтически приемлемые соли, такие, как описанные выше для соединений формулы (I).
1-Бeнзoил-3-(3,4-диxлopфeнил)-3-[3-(4-пивaлoилaминo-4- фенил-пиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-бензоиламино-4- фенил-пиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(ацетил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(пирид-2-ил) карбоксамидо-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(изобутил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-валериламино-4- фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(пропионил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(бутирил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(валерил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(изовалерил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(пивалоил-N- метиламино)-4-фенилпиперидин-1-ил)пропил/пиперидин, в форме рацематов или одного из них (+) - или (-) - энантиомеров, и их соли представляют собой особенно предпочтительные соединения согласно настоящему изобретению.
Соединения согласно изобретению получают известными способами, в особенности такими, которые описаны в европейских заявках на патент EP-474 561 и EP-512 901.
Один из способов, который пригоден для получения соединений формулы (I), описывается ниже. Согласно этому способу
а) соединение формулы: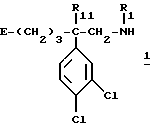
в которой R1, R11 имеют значения, указанные выше для соединений формулы (I), и E обозначает гидроксил или в случае необходимости O-защищенную группу, такую, как, например, тетрагидропиран-2-ил-окси-группа, бензилокси-группа или (C1-C4)алкилкарбонилокси-группа, или группу формулы: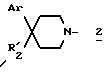
в которой Ar имеет вышеуказанное значение и R'2 обозначает R2, такой, как указан выше для формулы (I), или обозначает предшественник R2, имея в виду, что, когда R'2 обозначает гидроксил или амино-группу, то эти группы могут быть защищены,
обрабатывают:
- либо функциональным производным кислоты формулы:
HOCO - A - Z, 3
в которой A и Z имеют вышеуказанное для формулы (I) значение, если нужно получить соединение формулы (I), где Т обозначает -CO-,
- либо галогенированным производным формулы:
Гал - CH2 - A - Z 4
в которой Гал обозначает галоген, предпочтительно бром или хлор, если нужно получить соединение формулы (I), где T обозначает -CH2-,
- либо хлорформиатом формулы:
ClCOO - A - Z, 5
если нужно получить соединение формулы (I), где Т обозначает -COO-,
- либо изоцианатным производным формулы
O = C = N - A - Z, 6
если нужно получить соединение формулы (I), где T обозначает -CONH-
- либо карбамоилхлоридом формулы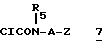
если надо получить соединение формулы (I), где Т обозначает -CONR5-, где R5 отличным от водорода,
- либо бензолсульфонилхлоридом формулы:
ClSO2 - Z 7а
если надо получить соединение формулы (I), в которой -T-A- обозначает -SO2-,
для получения соединения формулы: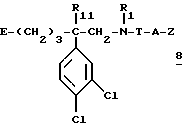
б) в случае необходимости удаляют O-защитную группу путем воздействия кислоты или основания,
в) полученный спирт формулы: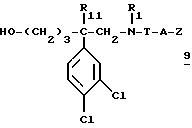
обрабатывают соединением формулы:
W - SO2 - Cl 9а
в которой W обозначает метильную, фенильную, толильную, трифторметильную группу;
г) полученный сульфаонат формулы: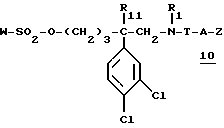
вводят во взаимодействие со вторичным амином формулы: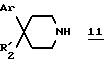
в которой Ar и R'2 имеют вышеуказанные значения,
д) после возможного удаления защиты с гидроксильной или амино-группы, обозначаемой R'2, или возможного превращения R'2 в R2, полученный продукт при необходимости превращают в одну из его солей.
Соединение 9а, получаемое на стадии В) предпочтительно представляет собой метансульфонилхлорид, и, следовательно, в соединении 10 стадии г) W предпочтительно обозначает метильную группу.
Если в соединении формулы 1 E обозначает группу: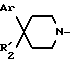
то способ включает только стадии а) и д).
Согласно одному варианту способа:
- а1) аминофункцию соединения формулы 1 защищают защитной группой с получением соединения формулы: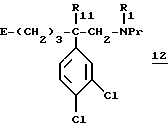
в которой E, R1, R11 имеют вышеуказанное значение и Pr обозначает азотзащитную группу, например трет-бутоксикарбонил (Boc), тритил, бензил,
- б1) в случае необходимости удаляют O-защитную группу путем воздействия кислоты или основания,
- в1) таким образом полученный спирт формулы: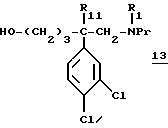
обрабатывают соединением вышеописанной формулы: (9а)
- г1) таким образом полученный сульфонат формулы: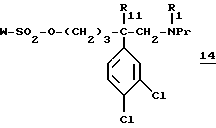
вводят во взаимодействие со вторичным амином: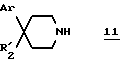
для получения соединения формулы: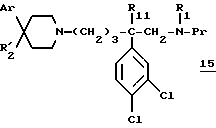
- д1) осуществляют удаление защиты с азота в кислой среде,
- е1) таким образом полученное соединение формулы: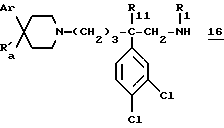
обрабатывают одним из вышеописанных соединений 3, 4, 5, 6, 7 или 7a,
- ж1) после возможного удаления защиты с гидроксильной или амино-группы, обозначаемой R'2, или возможного превращения R'2 в R2, полученный продукт формулы (I) при необходимости превращают в одну из его солей.
Соединение 9а, получаемое на стадии в1), предпочтительно представляет собой метансульфонилхлорид и, следовательно, в соединении 14 стадии г1) W предпочтительно обозначает метильную группу.
O-защитные группы, в случае необходимости используемые для получения соединения формулы (I), в которой R2 обозначает гидроксил, представляют собой хорошо известные специалисту классические O-защитные группы, такие, как указанные выше для E.
N-защитные группы, в случае необходимости используемые для получения соединения формулы (I), в которой R2 обозначает амино-группу, представляют собой классические, хорошо известные специалисту, N-защитные группы, такие, как например, тритильная группа, метокситритильная группа, трет-бутоксикарбонильная группа или бензилоксикарбонильная группа.
Более конкретно, если в качестве O-защитной группы используют ацетильную группу, то получают соединение формулы (I), в которой R2 обозначает ацетокси-группу, или, если в качестве N-защитной группы используют трет-бутоксикарбонильную группу, то получают соединение формулы (I), в которой R2 обозначает трет-бутоксикарбониламино-группу.
Конкретные предпочтительные условия осуществления вышеуказанных стадий указываются ниже и иллюстрируются в примерах.
На стадии а), функциональным производным кислоты 3 может быть сама кислота или одно из ее функциональных производных, которые реагируют с аминами, например, ангидрид, смешанный ангидрид, хлорангидрид, или активированный сложный эфир, например, сложный пара-нитрофенильный эфир.
Когда используют саму кислоту формулы 3, то работают в присутствии агента сочетания, используемого в химии пептидов, такого, как 1,3-дициклогексилкарбодиимид или гексафторфосфат бензотриазол-1-ил-окситрис(диметиламино)фосфония, в присутствии основания, такого, как триэтиламин или N,N-диизопропилэтиламин, в инертном растворителе, таком, как дихлорметан или N,N-диметилформамид, при температуре от 0oC до комнатной температуры.
Когда используют хлорангидрид кислоты, то реакцию проводят в инертном растворителе, таком, как дихлорметан или бензол, в присутствии основания, такого, как триэтиламин или N-метилморфолин, и при температуре от -60oC до комнатной температуры.
Когда используют галогенированное производное формулы 4, то реакцию проводят в инертном растворителе, таком, как тетрагидрофуран, N,N-диметилформамид или диметилсульфоксид, в присутствии основания, такого, как трет-бутилат калия, гидрид натрия или диизопропиламид лития, и при температуре 0-80oC.
Когда используют хлорформиат формулы 5, то реакцию осуществляют в инертном растворителе, таком, как дихлорметан, в присутствии основания, такого, как триэтиламин, и при температуре от 0oC до комнатной температуры.
Когда используют изоцианат формулы 6, то реакцию проводят в инертном растворителе, таком, как дихлорметан или бензол, и при температуре от -70oC до комнатной температуры.
Когда используют карбамоилхлорид формулы 7, то реакцию проводят в инертном растворителе, таком, как толуол или 1,2-дихлорэтан, при температуре 0-110oC и в присутствии основания, такого, как триэтиламин.
Когда используют бензолсульфонилхлорид формулы 7а, то реакцию проводят в инертном растворителе, таком, как дихлорметан, в присутствии основания, такого, как триэтиламин, и при температуре от -20oC до комнатной температуры.
Соединение формулы 8, полученное таким образом, на стадии б) подвергают при необходимости операции удаления защитных групп согласно известным специалисту способам. Например, когда E обозначает тетрагидропиран-2-ил-окси-группу, то удаление защиты осуществляют путем кислотного гидролиза, используя соляную кислоту в растворителе, таком, как простой эфир, метанол или смесь этих растворителей, или используя пиридиний-п-толуолсульфонат в растворителе, таком, как метанол, или используя смолу Amberlyst® в растворителе, таком, как метанол. Реакцию осуществляют при температуре от комнатной до температуры рефлюкса. Когда E обозначает бензоилокси-группу или (C1-C4)-алкилкарбонилокси-группу, то удаление защитных групп осуществляют путем гидролиза в щелочной среде, используя, например, гидроксид, щелочного металла, такой, как гидроксид натрия, гидроксид калия или гидроксид лития, в инертном растворителе, таком, как вода, метанол, диоксан или смесь этих растворителей, при температуре от 0oC до температуры рефлюкса.
На стадии в) реакцию спирта формулы 9 с сульфонилхлоридом формулы 9а предпочтительно осуществляют в присутствии основания, такого, как триэтиламин, пиридин, N,N-диизопропилэтиламин или N-метилморфолин, в инертном растворителе, таком, как дихлорметан, бензол или толуол, при температуре от -20oC до температуры рефлюкса.
Когда соединение формулы 10 вводят во взаимодействие с соединением формулы 11 (стадия г), то реакцию осуществляют предпочтительно в инертном растворителе, как N, N-диметилформамид, ацетонитрил, метиленхлорид, толуол или изопропанол, в присутствии или в отсутствие основания. Когда используют основание, то его выбирают среди органических оснований, таких, как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, или среди карбонатов или бикарбонатов щелочных металлов, таких, как карбонат калия, карбонат натрия или бикарбонат натрия. В отсутствие основания, реакцию проводят при использовании избытка соединения формулы 11 и в случае необходимости в присутствии йодида щелочного металла, такого, как йодид калия или йодид натрия. Реакцию осуществляют при температуре от комнатной до 100oC.
Таким образом, полученные продукты формулы (I) выделяют в форме свободного основания, или в виде соли, согласно классическим способам.
Когда соединение формулы (1) получают в форме свободного основания, то перевод его в соль осуществляют путем обработки выбранной кислотой в органическом растворителе. При обработке свободного основания, растворенного, например, в простом эфире, таком, как диэтиловый эфир, или в спирте, таком, как пропан-2-ол, или в ацетоне, с помощью раствора выбранной кислоты в том же растворителе, получают соответствующую соль, которую выделяют классическими способами.
Так, например, получают хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, оксалат, малеат, фумарат, нафталин-2-сульфонат, бензолсульфонат.
По окончании реакции, соединения формулы (1) могут быть выделены в виде одной из их солей, например, в виде хлоргидрата или оксалата в этом случае, если необходимо, свободное основание может быть получено путем нейтрализации вышеуказанной соли с помощью неорганического или органического основания, такого, как гидроксид натрия или триэтиламин, или с помощью карбоната или бикарбоната щелочного металла, такого, как карбонат или бикарбонат натрия или калия.
Замещенные пиперидины формулы 11 известны или их получают известными способами.
Соединения формулы 11 обычно получают в защищенной на азоте пиперидина форме, их можно получать путем удаления защиты с самих соединений формулы 11. Например, когда Ar обозначает пирид-2-ил, то 2-бром-пиридин вводят во взаимодействие с N-бензил-4-пиперидином в растворителе в присутствии бутиллития с целью получения N-бензил-4-гидрокси-4-пирид-2-ил-пиперидина, затем путем удаления защитной группы в основной среде получают 4-гидрокси-4-пирид-2-ил-пиперидин.
Соединения формулы 11, в которой R'2 обозначает гидроксил, и которые содержат защитную группу на азоте пиперидина, можно подвергать реакции Риттера путем воздействия ацетонитрила с получением соединений формулы 11, в которой R'2 представляет собой ацетамидо-группу, согласно методике, описанной в европейской заявке на патент EP-0474561. Путем гидролиза в кислой среде затем получают соединения формулы 11 в которой R'2 обозначает амино-группу. В случае необходимости можно осуществлять замещение амино-группы группой R3, представляющей собой (C1-C4)-алкил. Соединения формулы 11, в которой R'2 обозначает группу -NR3COR4, в которой R3 представляет собой водород или (C1-C4)-алкил и R4 обозначает водород или, соответственно, (C1-C7)-алкил, фенил, бензил, пиридил или (C3-C7)-цикло-алкил, в случае необходимости, замещенный, получают путем воздействия муравьиной кислоты в уксусном ангидриде или, соответственно, соответствующего ангидрида (R4CO)2O или соответствующего хлорангидрида R4COCl, в присутствии основания, такого, как триэтиламин, на соединение формулы 11, в которой R'2 обозначает группу -NHR3.
В частности, соединение формулы 11, в которой R'2 обозначает группу -NR3COR4, в которой R4 представляет собой этильный радикал, можно получать путем гидрирования, в присутствии катализатора, такого как палладий-на-угле, соединения формулы 11, в которой R'2 обозначает акрилоиламино-группу или акрилоил-N-(C1-C4)-алкиламино-группу.
Путем взаимодействия хлорформиата ClCOOR8 получают соединения формулы 11, в которой R'2 обозначает группу -NR3COOR8. Путем воздействия сульфонилхлорида ClSO2R9 получают соединения формулы 11, в которой R'2 обозначает группу -NR3SO2R9. Путем воздействия изоцианата R12N = C = O получают соединения формулы 11, в которой R'2 обозначает группу -NR3CONR10R12, где R10 = H. Путем воздействия карбамоилхлорида R12R10NCOCl получают соединения формулы 11, в которой R'2 обозначает группу -NR3CONR10R12.
Соединение формулы 11, в которой R'2 обозначает группу -NR3CONR10R12, также можно получать путем воздействия соединения HNR10R12 с соединением формулы 11, в которой R'2 обозначает группу -NR3COOR8, где R8 = фенил.
Само собой разумеется, что реакции получения соединений формулы 11, где R'2 обозначает -NHR3, -NR3COOR8, -NR3SO2R9 или -NR3CONR10R12, можно прямо переносить на получение соединений формулы 11, где R'2 обозначает -CH2NHR3, -CH2NR3COOR8, -CH2NR3SO2R9 или -CH2NR3CONR10R12.
Соединение формулы 11, в которой R'2 обозначает группу -NR6R7, в которой R6 и R7 вместе с азотом, с которым они связаны, образуют гетероцикл, получают по способу, описанному в Tetrahedron Letters, 1988, 29 (52), 6827.
Соединение формулы 11, в которой R'2 представляет собой гидроксиметильную группу, получают путем восстановления соединения формулы 11, в которой R'2 обозначает метоксикарбонил, с помощью такого восстановителя, как литийалюминийгидрид. Путем взаимодействия хлорангидрида кислоты с 2-7 C-атомами с соединением формулы 11, в которой R'2 обозначает гидроксилметил, получают соединения формулы 11, в которой R'2 обозначает (C2-C7)-ацилоксиметил. Путем воздействия муравьиной кислоты получают соединение формулы 11, в которой R'2 обозначает формилоксиметил. Путем воздействия карбамоилхлорида (C1-C7)-алкил-NHCOCl на соединение формулы 11, в которой R'2 обозначает гидроксиметил, получают соединения формулы 11, в которой R'2 обозначает (C1-C7)алкиламинокарбонилоксиметил.
Соединение формулы 11, в которой Ar и R'2 вместе с атомом углерода, с которым они связаны, образуют группу формулы: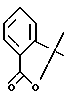
получают по способу, описанному в J. Heteroc. Chem., 1969, 6, 475.
Путем воздействия хлорангидрида кислоты с 2-7 C-атомами на соединения формулы 11, в которой R'2 обозначает гидроксил, получают соединения формулы 11, в которой R'2 обозначает (C2-C7)-ацилокси-группу, путем воздействия муравьиной кислоты получают соединения формулы 11, в которой R'2 обозначает формилокси-группу.
Для получения соединения формулы 11, в которой R'2 обозначает R4CONR3-группу, где R3 и R4, образующие вместе группу -(CH2)3- или -(CH2)4-, поступают согласно J. Med. Chem., 1985, 28, 46-50.
Превращение заместителя R2 = циано-группа в заместитель R2 = аминометильная группа можно осуществлять путем каталитического гидрирования либо соединения формулы 11, либо соединения формулы (I). Путем соответствующих реакций тогда получают соединения согласно изобретению, имеющие различные заместители на азоте аминометила.
Вышеуказанные методы хорошо известны и иллюстрируются нижеприводимыми приготовлениями, которые предшествуют примерам. Эти приготовления представляют собой адаптацию методов, описанных в европейских патентах EP-A-0428434, EP-A-0474561, EP-A-0512901, или в следующих публикациях: J. Heterocyclic. Chem., 1986, 23, 73-75, J. Chem. Soc., 1950, 1469, J. Chem. Soc., 1945, 917, J. Pharmaceutical Sci., 1972, 61, 1316-1317, J. Org. Chem., 1957, 22, 1484-1489.
Так, например, при получении соединения формулы 11, в которой R'2 обозначает группу -NR6R7, где R6 обозначает водород и R7 обозначает (C1-C7)алкил, или, соответственно, (C3-C7)-циклоалкилметил или бензил, можно осуществлять восстановление соединения формулы 11, в которой R'2 обозначает группу -NR3COR4, где R3 обозначает водород и R4 обозначает водород или (C1-C6)-алкил, или, соответственно, (C3-C7)-циклоалкил или фенил. Реакцию проводят с помощью восстановителя, такого, как литийалюминийгидрид, в растворителе, таком, как тетрагидрофуран, при температуре рефлюкса.
Путем идентичной реакции можно получать соединения формулы 11, в которой R'2 обозначает группу -NR6R7, где R6
обозначает (C1-C4)-алкил и R7 обозначает (C1-C7)-алкил, или, соответственно, (C3-C7)-циклоалкилметил или бензил, из соединения формулы 11, в которой R'2 обозначает группу -NR3COR4, где R3 обозначает (C1-C4)-алкил и R4 обозначает водород или (C1-C6)-алкил, или, соответственно, (C3-C7)-циклоалкил или) фенил. Точно также можно получать соединения формулы 11, в которой R'2 обозначает группу -NR6R7, где R6 обозначает (C5-C7)-алкил.
Точно также можно получать соединения формулы 11, в которой R'2 обозначает группу -CH2NR13R14, где R13 обозначает водород или (C1-C4)-алкил и R14 обозначает (C1-C7)-алкил, (C3-C7)-циклоалкил-метил или бензил, из соединения формулы 11, в которой R'2 обозначает группу -CH2NR3COR4, где R3 обозначает водород или (C1-C4)-алкил и R4 обозначает водород, (C1-C6)-алкил, (C3-C7)-циклоалкил или фенил. Точно также можно получать соединения формулы 11, в которой R'2 обозначает группу -CH2NR13R14, где R13 обозначает (C5-C7)-алкил.
Превращение заместителя R2 в значении гидроксил или гидроксиметил в заместитель R2 = (C1-C7)ацилокси-группа или (C1-C7)-ацилоксиметил, можно осуществлять при использовании либо соединения 11, либо соединения формулы (I).
В случае необходимости, превращение группы R2 из R'2 = гидроксил или амино-группа могут быть осуществлены при использовании соединения 15.
Производные пиперидина формулы (V):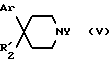
в которой Ar имеет вышеуказанное для формулы (I) значение:
- R'2 обозначает группу -NR3COR4, где R3 и R4 указаны выше для формулы (I), при условии, что, когда R3 обозначает водород, R4 другой, чем метил,
- Y обозначает водород или защитную группу, такую, как трет-бутоксикарбонил, тритил или бензил,
и их возможные соли являются новыми и составляют следующий аспект настоящего изобретения.
Из этих соединений, те соединения формулы (V), в которой Ar обозначает фенил, R'2 обозначает группу -NR3COR4, причем R3 обозначает метил, а R4 обозначает (C1-C7)-алкил, и Y обозначает водород или N-защитную группу, и их возможные соли, являются предпочтительными продуктами.
Соединения формулы (V), в которой Ar обозначает фенил, R'2 обозначает группу -NR3COR4, причем R3 и R4 оба обозначают метильные группы, и Y обозначает водород или защитную группу, в частности, трет-бутоксикарбонил, тритил или бензил, и их возможные соли, являются особенно предпочтительными согласно изобретению.
Соединения формулы 1, в которой E обозначает гидроксил, получают известными методами. В схеме I, данной ниже, кратко представлен один из этих методов получения соединений формулы 1'.
Соединения формул 3, 4, 5, 6, 7, 7а известны или получаются известными способами.
Эти соединения могут быть использованы в меченой форме, например, с помощью трития или радиоактивного йода, для получения меченых соединений формулы (I) согласно изобретению.
В этом случае работают исходя из соединения 3, 4, 5, 6, 7, 7а, где радикал Z замещен атомом йода, затем обменивают этот атом йода на тритий или на атом радиоактивного йода с получением меченного соединения 3*, 4*, 5*, 6*, 7*, 7а*, позволяющего получать меченое соединение формулы (I*), в частности меченое соединение формулы (IV*).
Спирт 1', получаемый согласно схеме I, является рацемическим. В случае необходимости можно осуществлять разделение этих оптических изомеров известными методами, например, путем хроматографии или перекристаллизации, затем получать соответствующий оптически чистый мезилат и таким образом приготовлять соединения согласно изобретению в оптически чистой форме.
Спирт формулы 1', который представляет собой ключевое промежуточное соединение в синтезе соединений формулы (IV), особенно предпочтительных в качестве сильных и селективных антагонистов рецептора NK3 человека, представляет собой новое соединение. Рацемическая форма этого спирта, два (+)- и (-) - энантиомера и соли этих соединений, следовательно, составляют другой аспект настоящего изобретения, причем особенно предпочтительны (+)-изомер и его соли присоединения кислот.
Согласно следующему аспекту, предметом настоящего изобретения является использование новых соединений, обладающих очень сильным сродством к вышеуказанному рецептору NK3 для получения лекарственных средств, пригодных для лечения любой патологии, в которой принимает участие нейрокинин B, в особенности для получения лекарственных средств, предназначенных для борьбы с психиатрическими заболеваниями, заболеваниями психосоматического происхождения, гипертонии, патологий, связанных с нарушениями нейромодуляции или нейротрансмиссии, зависимой от NK3.
Изобретение в особенности относится к использованию соединения формулы (I'):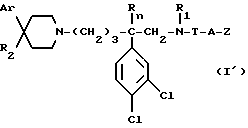
в которой: Ar обозначает пирид-2-ил или фенил, незамещенный или замещенный галогеном, метилом или (C1-C4)-алкоксилом,
- R1 обозначает метильную группу,
- R11 обозначает водород,
- или R1 и R11 вместе обозначают группу -(CH2)3-,
- R2 обозначает гидроксил, (C1-C7)-алкоксил, (C1-C7)-ацилоксигруппу, циано-группу, группу -NR6R7, группу -NR3COR4, группу -NR3COOR8, группу -NR3SO2R9, группу -NR3CONR10R12, (C1-C7)-ацильную группу, (C1-C7)-алкоксикарбонил, группу -CONR10R12, группу -CH2OH, (C1-C7)-алкоксиметил, (C1-C7)-ацилоксиметил, (C1-C7)-алкиламинокарбонилоксиметил, группу -CH2NR13R14, группу -CH2NR3COR4,
группу -CH2NR3COOR8, группу -CH2NR3SO2R9, группу -CH2NR3CONR10R12, или
R2 представляет собой двойную связь между атомом углерода, с которым он связан, и соседним атомом углерода пиперидинового цикла,
- или Ar и R2 вместе с атомом углерода, с которым они связаны, образуют группу формулы (а)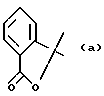
R3 обозначает водород или (C1-C4)-алкил,
- R4 обозначает водород, (C1-C7)-алкил, фенил, бензил, пиридил или (C3-C7)-циклоалкил, незамещенный или замещенный одной или несколькими метильными группами,
- или R3 и R4 вместе обозначают группу -(CH2)n-,
- "n" обозначает число 3 или 4,
- T обозначает метилен, карбонил, группу -COO-, группу - CONR5-,
- A обозначает прямую связь, метилен, этилен, пропилен, винилен,
- или -T-A- обозначает группу -SO2-,
- Z обозначает фенил, незамещенный или замещенный одно- или многократно галогеном, (C1-C4)-алкилом, (C1-C4)-алкоксилом, нитро-группой,
- R5 обозначает водород или (C1-C4)-алкил,
- R6 и R7, каждый, независимо друг от друга, обозначают водород или (C1-C7)-алкил; R7, кроме того, может обозначать (C3-C7)-циклоалкилметил, бензил или фенил; или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероцикл, выбираемый среди азетидина, пирролидина, пиперидина, морфолина, тиоморфолина или пергидроазепина,
- R8 обозначает (C1-C7)-алкил или фенил,
- R9 обозначает (C1-C7)-алкил; свободную или замещенную одним или двумя (C1-C7)-алкилами амино-группу; фенил, незамещенный или замещенный один или несколько раз заместителем, выбираемым в группе, включающей атом галогена, (C1-C7)-алкил, трифторметил, гидроксил, (C1-C7)-алкоксил, карбоксил, (C1-C7)- алкоксикарбонил, (C1-C7)-алкилкарбонилокси-группу, циано-группу, нитро-группу; свободную или замещенную одной или двумя (C1-C7)-алкильными группами амино-группу, причем вышеуказанные заместители идентичны или различны,
- R10 и R12, каждый, независимо друг от друга, обозначают водород, или (C1-C7)-алкил; R12, кроме того, может обозначать (C3-C7)-циклоалкил, (C3-C7)-циклоалкилметил, гидроксил, (C1-C4)-алкоксил, бензил или фенил,
или R10 и R12 вместе с атомом азота, с которым они связаны, образуют гетероцикл, выбираемый среди азетидина, пирролидина, пиперидина, морфолина, тиоморфолина или пергидроазепина,
- R13 и R14, каждый, независимо друг от друга, обозначают водород, или (C1-C7)-алкил; R14, кроме того, может обозначать (C3-C7)-циклоалкилметил или бензил, или его фармацевтически приемлемых солей для получения лекарственных указанных выше средств и предпочтительно для получения лекарственных средств, пригодных для лечения гипертонии.
Сродство соединений формулы (I') к рецепторам тахикининов оценивают ин витро путем нескольких биохимических испытаний, при использовании радиолигандов:
1*) Связывание /125I/ BH-SP (вещество P, меченное йодом-125 с помощью реактива Bolton-Hunter) с рецепторами NK1 коры головного мозга крысы, подвздошной кишки морской свинки и человеческих лимфобластических клеток,
2*) Связывание /125I/ His-NKA рецептора NK2 двенадцатиперстной кишки крысы или подвздошной кишки морской свинки,
3*) Связывание /125I/ His (MePhe7)NKB с рецепторами NK3 коры головного мозга крысы, коры головного мозга морской свинки и коры головного мозга песчанки, также, как с клонированными человеческими рецепторами NK3, выраженными клетками CHO (Buell и др., FEBS Letters, 1992, 299, 90-95).
Испытания проводили согласно X. Emonds-Alt и др. (Eur. J. Pharmacol., 1993, 250, 403-413).
Соединения согласно изобретению сильно ингибируют связывание /125I/ His (MePhe7)NKB с рецепторами NK3 коры головного мозга морской свинки и песчанки, также, как с клонированными человеческими рецепторами NK3, константа ингибирования обычно менее 5•10-9 М. Для тех же соединений констатируют, что константа ингибирования (Kи) для рецепторов NK3 коры головного мозга крысы выше 10-7 М и константа ингибирования (Kи) для рецептора NK2 двенадцатиперстной кишки крысы и рецепторов NK1 коры головного мозга крысы обычно выше или равна 10-7 М.
В качестве сравнения согласно условиям работы, описанным выше, измеряли константы ингибирования по отношению к различным рецепторам для соединения A. Антагонизм связывания с эледоизином, который описывается в европейской заявке на патент EP-512901, соответствует константе ингибирования в отношении рецептора NK3 крысы: Kи = 10-7 М.
Для человеческого рецептора NK3 константа ингибирования соединения A составляет Kи = 1•10-9 М.
Для рецептора NK2 двенадцатиперстной кишки крысы константа ингибирования соединения A составляет Kи = 1•10-10 М.
Таким образом, соединение A не является селективным в отношении человеческого рецептора NK3, в противоположность тому, что наблюдают для соединений формулы (I) согласно настоящему изобретению.
N-Метил-N-/2-(3,4-дихлорфенил)-5-(4-гидрокси-4-фенил- пиперидин-1-ил)пентил/бензамид-хлоргидрат, описанный в примере 22 европейского патента EP-474561, относится к семейству соединений формулы (I') согласно настоящему изобретению: его константы ингибирования показывают большую специфичность и сильное сродство этого соединения к человеческому рецептору NK3: человеческий рецептор NK3, Kи = 5•10-9 М, рецептор NK2 двенадцатиперстной кишки крысы, Kи = 5•10-7 М, рецептор NK1 коры головного мозга крысы, Kи = 5•10-7 М.
Соединения согласно настоящему изобретению также оценивают ин виво на двух животных моделях.
В случае песчанки, состояние вращения индуктируют путем интрастриатального введения специфического агониста рецептора NK3: Сенктида; констатируют, что одностороннее введение Сенктида в полосатое тело песчанки приводит к сильным противоположным вращениям, которые ингибируются соединениями согласно изобретению, вводимыми либо интраперитонеально, либо орально.
Этот результат показывает, что соединения согласно изобретению проходят гематоменингеальный барьер и способны блокировать, на уровне центральной нервной системы, присущее рецепторам NK3 действие. Таким образом, их можно применять для лечения любой NKB-зависимой центральной патологии, такой, как психиатрические заболевания, или любой другой патологии, вызываемой на центральном уровне рецептором NK3, такой, как психосоматические заболевания.
В случае морской свинки, инъекция Сенктида, внутривенно или интрацеребровентрикулярно, индуцирует гипертонию, которую ликвидируют за счет введения орально или внутривенно соединений согласно изобретению.
Этот результат показывает, что соединения согласно изобретению действуют на сердечно-сосудистом уровне и что они способны блокировать присущее рецепторам NK3 действие на этом уровне, особенно в отношении гипертонии (Nakayama и др. Brain Res. , 1992, 595, 339-342, Takano и Kamiya, Asia Pacific J. Pharmacol., 1991, 6, 341-346, Saigo и др. Neuroscience, 1993, 159, 187-190).
В этих тестах соединения согласно изобретению эффективны в дозах, изменяющихся в пределах 0,1-3 мг на кг, перорально, внутривенно или интраперитонеально.
Соединения, пригодные для получения лекарственных средств согласно изобретению, обычно вводятся в виде дозировочной единицы. Вышеуказанные дозировочные единицы предпочтительно формулируют в виде фармацевтических композиций, в которых действующее начало смешано с фармацевтическим эксципиентом.
Согласно другому из своих аспектов, настоящее изобретение относится к фармацевтическим композициям, включающим, в качестве действующего начала, соединение формулы (I), предпочтительно формулы (II) или (III), предпочтительно соединение формулы (IV), обладающее очень сильным сродством к человеческому рецептору NK3, характеризующееся константой ингибирования Kи ниже 5•10-9 М, согласно изучению фиксации лиганда.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в суточных дозах 0,01-100 мг на кг веса тела подвергаемых лечению млекопитающих, предпочтительно в суточных дозах 0,1-50 мг/кг. В случае человека, доза может изменяться предпочтительно от 0,5 до 4000 мг в день, преимущественно от 2,5 до 1000 мг, в зависимости от возраста подвергаемого лечению субъекта или от типа лечения: профилактика или терапия.
Заболевания, для лечения которых могут быть использованы соединения и их фармацевтически приемлемые соли, представляют собой, например, заболевания, связанные с дисфункцией допаминергических систем, например шизофрения, болезнь Паркинсона, заболевания, связанные с дисфункцией норадренергических систем, например тревога, расстройства бодрствования, эпилептические заболевания любой формы и в особенности Jrand Mal, деменция, невродегенеративные заболевания, и периферические заболевания, при которых участие центральной нервной системы и/или периферической нервной системы вызывается нейрокинином B, действующим как центральный нейропередатчик или нейромодулятор, например, боли, мигрень, острое или хроническое воспаление, сердечно-сосудистые расстройства, в частности гипертония, сердечная недостаточность и аритмия, респираторные нарушения (астма, насморк, кашель, бронхиты, аллергии, сверхчувствительность), нарушения желудочно-кишечной системы, такие, как язва пищевода, колит, нарушения, связанные со стрессом, раздражительный синдром ободочной кишки (IBS), воспаление ободочной кишки (JBD), гиперсекреция кислоты, нарушения мочевой системы (недержание мочи, неврологическое состояние мочевого пузыря), заболевания иммунной системы (ревматоидный артрит), и вообще любую патологию, зависящую от нейрокинина B.
В фармацевтических композициях настоящего изобретения для введения орально, подъязычно, в виде ингаляции, подкожно, внутримышечно, внутривенно, чрескожно, локально или ректально, действующие начала могут вводиться животным и людям в препаративных формах единичной дозировки, в смеси с классическими фармацевтическими носителями. Соответствующие препаративные единичные формы введения включают пероральные формы, такие, как таблетки, желатиновые капсулы, порошки, глануляты и оральные растворы или суспензии, формы введения подъязычно и через рот, подкожные, внутримышечные, внутривенные, внутриносовые или внутриглазные формы введения и ректальные формы введения.
Когда готовят твердую композицию в форме таблеток, то основное действующее начало смешивают с фармацевтическим эксципиентом, таким, как диоксид кремния, желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные вещества. На таблетки можно наносить покрытие из сахарозы, различных полимеров или других соответствующих материалов или обрабатывать их таким образом, чтобы они имели пролонгированную или замедленную активность и чтобы они непрерывно высвобождали заданное количество действующего начала.
Препарат в виде желатиновых капсул с лекарственным средством получают путем смешения действующего начала с разбавителем, таким, как гликоль или сложный эфир гликоля, и путем введения полученной смеси в мягкие или жесткие желатиновые капсулы.
Препарат в виде сиропа или эликсира может содержать действующее начало в комбинации с подслащивающим средством, предпочтительно некалорийным, метилпарабен и пропилпарабен в качестве антисептика, а также агент, придающий вкус, и соответствующий краситель.
Порошки или грануляты, диспергируемые в воде, могут содержать действующее начало в смеси с диспергаторами или смачивателями, или с суспендирующими агентами, как поливинилпирролидон, точно также, как вместе с подслащивающими средствами или улучшающими вкус средствами.
Для ректального введения прибегают к свечам, которые готовят со связующими, плавящимися при ректальной температуре, например, как масло какао или полиэтиленгликоли.
Для парентерального введения, внутриглазного или внутриносового введения используют водные суспензии, солевые изотонические растворы или растворы для инъекций, которые содержат диспергаторы и/или смачиватели, которые фармакологически приемлемы, например, как пропиленгликоль или бутиленгликоль.
Для введения путем ингаляции используют аэрозоль, содержащий, кроме того, например, сорбитантриолеат или олеиновую кислоту, также, как трихлорфторметан, дихлорфторметан, дихлортетрафторэтан или любой другой, биологически приемлемый, выталкивающий газ, также можно использовать систему, содержащую действующее начало, одно или в комбинации с эксципиентом, в виде порошка.
Действующее начало также может находиться в форме комплекса с циклодекстрином, например, как α,β,γ- циклодекстрин, 2-гидрокси-пропил -β- циклодекстрин, метил -β- циклодекстрин.
Действующее начало также может быть сформулировано в форме микрокапсул, в случае необходимости с одним или несколькими носителями или добавками.
В каждой единичной препаративной форме действующее начало формулы (I) находится в количествах, соответствующих предусмотренным суточным дозам. Обычно каждая единичная препаративная форма надлежащим образом подбирается в зависимости от суточной дозы и предусматриваемого типа введения, например, таблетки, желатиновые капсулы, пакетики, ампулы, сиропы, капли, так, чтобы такая единичная форма содержала 0,5-1000 мг действующего начала, предпочтительно, 2,5-250 мг перед введением ее от одного до четырех раз в день.
Вышеуказанные композиции также могут включать другие активные продукты, пригодные для терапевтических целей, например, бронхорасширяющие средства, антигистаминные средства или средства от кашля.
Благодаря своему очень сильному сродству к рецептору NK3 человека и благодаря своей большой селективности, соединения согласно изобретению могут быть использованы в меченой радиоактивным изотопом форме в качестве лабораторного реагента.
Например, они позволяют охарактеризовать, идентифицировать и локализовать рецептор NK3 человека в срезах тканей, или рецептор NK3 животного путем одной ауторадиографии.
Соединения согласно изобретению также позволяют осуществлять сортировку или скрининг молекул в зависимости от их сродства по отношению к рецептору NK3 человека. В этом случае осуществляют реакцию смещения меченного радиоактивным изотопом лиганда согласно настоящему изобретению, из его человеческого рецептора NK3.
В следующих примерах используют следующие аббревиатуры:
TA = комнатная температура,
Т.пл. = температура плавления
ТЭА = триэтиламин
Pd/C = 10%-ный палладий-на-угле
ДХМ = дихлорметан
ТГФ = тетрагидрофуран
ДБУ = 1,8-диазабицикло/5.4.0/ундец-7-ен
ДМАП = 4-диметиламинопиридин
AcOEt = этилацетат
MeOH = метанол
ВЭЖХ = высокоэффективная жидкостная хроматография
Me = метил
Et = этил
iPr = изопропил
Bu = н-бутил
C6H5 = фенил
HCl = соляная кислота
(Boc)2O = ди-трет-бутилдикарбонат
Boc = трет-бутоксикарбонил
BOP = бензотриазол-1-ил-окси-трис-диметиламинофосфоний-гексафторфосфат
NaCl = хлорид натрия
MgSO4 = сульфат магния
Na2SO4 = сульфат натрия
LiAlH4 = литийалюминийгидрид
NaOH = гидроксид натрия
NH4Cl = хлорид аммония
эфир = диэтиловый эфир
изо-эфир = диизопропиловый эфир
K2CO3 = карбонат калия
Солянокислый эфир = насыщенный раствор HCl в эфире
Диоксид кремния H = силикагель 60 H, выпускаемый в продажу фирмой Merck (DARMSTAD)
ЯМР = ядерный магнитный резонанс
с. = синглет, с.ш. = широкий синглет, д. = дублет,
т. = триплет, м. = массив, мт. = мультиплет, тд = расщепленный надвое дублет, сд = расщепленный надвое синглет.
Приготовления
Приготовление 1.1. 4-Фенил-4-пивалоил-аминопиперидин
А) 1-Бензил-4-гидрокси-4-фенил-пиперидин
Это соединение получают путем воздействия фениллития на 1-бензил-пиперидин-4-он.
Б) 4-Ацетамидо-1-бензил-4-фенил-пиперидин
Это соединение получают по реакции Риттера за счет присоединения ацетонитрила к полученному в стадии А) соединению.
В) 4-Амино-1-бензил-4-фенилпиперидин-дихлоргидрат
Полученное в стадии Б) соединение гидролизуют при кипячении с обратным холодильником в течение 3-х часов в 6 н HCl. После выпаривания досуха остаток растворяют в метаноле, кристаллизуют путем добавления ацетона, отфильтровывают и высушивают с получением целевого соединения.
Г) 1-Бензил-4-фенил-4-пивалоиламинопиперидин
70 г полученного в предыдущей стадии соединения растворяют в 150 мл диоксана, добавляют 85 мл ТЭА, затем 45 г пивалоилхлорида. После перемешивания в течение 2-х часов при 60oC, выпаривают, обрабатывают с помощью ДХМ, промывают разбавленным раствором гидроксида натрия, раствором NaCl, затем сушат над MgSO4 и выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя с помощью ДМХ. Получают 43 г целевого продукта в виде масла.
Д) 4-Фенил-4-пивалоиламинопиперидин
13 г полученного в предыдущей стадии соединения растворяют в 20 мл 95%-ного этанола, добавляют 1,5 Pd/C, затем гидрируют в течение 24-х часов при комнатной температуре и при атмосферном давлении, отфильтровывают, выпаривают, с получением масла, которое кристаллизуется, давая 8 г целевого продукта.
Приготовление 1.2. 4-Фенил-4-(пивалоил-п-метиламино) пиперидин
А) 1-Бензил-4-(N-трет-бутоксикарбониламино)-4-фенил-пиперидин
К раствору 14,5 г 4-амино-1-бензил-4-фенилпиперидин- дихлоргидрата, полученного в стадии В) приготовления 1.1, 12 мл ТЭА в 100 мл диоксана прикапывают раствор 12 г (Boc)2O в 50 мл диоксана и нагревают при 50oC в течение 18 часов. Реакционную смесь концентрируют, в вакууме остаток экстрагируют этилацетатом, промывают буферным раствором с pH 2, 1 н раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 13 г целевого продукта, после кристаллизации из смеси эфира с гептаном.
Б) 4-(N-трет-Бутоксикарбониламино)-4-фенилпиперидин
В течение 72-х часов при комнатной температуре и при атмосферном давлении гидрируют смесь 13 г полученного в предыдущей стадии соединения, 1,5 г 10%-ного палладия-на-угле в 300 мл 95o этанола. Катализатор отфильтровывают на Целите и фильтрат выпаривают в вакууме. Получают 9,7 г целевого продукта.
В) 4-(N-трет-Бутоксикарбониламино)-4-фенил-1-тритилпиперидин
13,25 г полученного в предыдущей стадии соединения и 5 г ТЭА растворяют в 150 мл ДХМ при 0oC и в атмосфере азота. Прикапывают 13,4 тритилхлорида и перемешивают в течение 1 часа. Выпаривают, остаток обрабатывают эфиром, промывают водой, буферным раствором с pH 2, раствором NaCl, затем сушат над сульфатом магния и выпаривают. Получают 23 г целевого продукта в виде масла.
Г) 4-(N-Метиламино)-4-фенил-1-тритилпиперидин
Суспензию 5 г литийалюминийгидрида в 100 мл ТГФ нагревают до 60oC в атмосфере азота и прикапывают раствор 23 г полученного в предыдущей стадии соединения в 100 мл ТГФ. После кипячения с обратным холодильником в течение 2-х часов, гидролизуют с помощью 25 мл воды, отфильтровывают, выпаривают. Получают 17 г целевого продукта, который кристаллизуется из метанола при нагревании. Т.пл. = 125oC.
Д) 4-(Пивалоил-N-метиламино)-4-фенил-1-тритилпиперидин
2,6 г полученного в предыдущей стадии соединения растворяют в 15 мл пиридина, добавляют 500 мг ДМАП и 4 мл пивалоилхлорида. Оставляют реагировать в течение 72-х часов при 70oC и в атмосфере азота, затем выпаривают, растворяют остаток в этилацетате, промывают водой, буферным раствором с pH 2, 5%-ным раствором гидроксида натрия, раствором NaCl, затем сушат над сульфатом магния. Остаток хроматографируют на диоксиде кремния, элюируя смесью пентана с этилацетатом. Получают целевой продукт (1,5 г) в форме масла.
Е) 4-Фенил-4-(пивалоил-N-метиламино)пиперидин
1,5 г полученного в предыдущей стадии продукта растворяют в смеси 20 мл муравьиной кислоты и 20 мл воды. После перемешивания в течение часа отфильтровывают, нейтрализуют фильтрат добавлением, при охлаждении, 40%-ного раствора NaOH, после чего экстрагируют 3 раза по 50 мл ДХМ, сушат над сульфатом магния и выпаривают. Получают 700 мг целевого продукта в виде пасты. Т.пл. 50-55oC.
Приготовление 1.3. 4-(Ацетил-N-метиламино)-4-фенилпиперидин
А) 4-(Ацетил-N-метиламино)-4-фенил-1-тритил-пиперидин
Охлаждают до 0oC, в атмосфере азота, раствор 2,8 г полученного в стадии Г) приготовления 1.2 соединения в 20 мл ДХМ и добавляют 1,5 мл ТЭА, затем 0,55 г ацетилхлорида. Перемешивают в течение 2-х часов и концентрируют реакционную смесь в вакууме. Остаток обрабатывают этилацетатом, промывают буферным раствором с pH 2, 5%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают растворитель в вакууме. Получают 3 г целевого продукта.
Б) 4-(Ацетил-N-метиламино)-4-фенилпиперидин
В течение 1 часа нагревают при 60oC раствор 3 г полученного в предыдущей стадии соединения, 30 мл муравьиной кислоты в 15 мл воды. Добавляют 50 мл воды к реакционной смеси, отфильтровывают, фильтрат промывают эфиром, водную фазу подщелачивают до pH > 10 путем добавки концентрированного раствора NaOH, экстрагируют с помощью ДХМ, сушат экстракт над сульфатом магния и растворитель выпаривают в вакууме. Получают 1 г целевого продукта в виде масла.
Приготовление 1.4. 4-/(Ацетил-N-метиламино)метил/-4- фенилпиперидин-п-толуолсульфонат
А) 4-(Аминометил)-1-бензил-4-фенилпиперидин
Охлаждают до 0oC суспензию 2,8 г литийалюминийгидрида в 50 мл ТГФ и прикапывают раствор 20 г 1-бензил-4-циано-4-фенилпиперидина в 50 мл ТГФ. Оставляют при перемешивании в течение 1 часа при комнатной температуре, затем нагревают при 40oC в течение 1 часа. Реакционную смесь охлаждают на ледяной бане, последовательно добавляют 3 мл воды, 3 мл 4 н раствора NaOH и 12 мл воды. Отфильтровывают неорганические соли и фильтрат выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол (100: 3 по объему)-(100 : 10 по объему). Получают 11 г целевого продукта.
Б) 1-Бензил-4-/(N-формиламино)-метил/-4-фенил-пиперидин
К смеси 11 г полученного в предыдущей стадии соединения в 76 мл муравьиной кислоты при комнатной температуре прикапывают 25 мл уксусного ангидрида, затем оставляют при перемешивании в течение 5 часов. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой, подщелачивают до pH 14 путем добавки концентрированного раствора NaOH, экстрагируют эфиром, промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 12 г целевого продукта.
В) 1-Бензил-4-/(N-метиламино)метил/-4-фенил-пиперидин
Нагревают до 40oC суспензию 3,9 г литийалюминийгидрида в 50 мл ТГФ, прикапывают раствор 12 г полученного в предыдущей стадии соединения в 50 мл ТГФ, затем кипятят в течение 3-х часов с обратным холодильником. После охлаждения на ледяной бане последовательно добавляют 4 мл воды, 4 мл 4 н раствора NaOH и 12 мл воды. Отфильтровывают неорганические соли и фильтрат концентрируют в вакууме. Остаток экстрагируют эфиром, экстракт сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 10 г целевого продукта.
Г) 4-/(Ацетил-N-метиламино)/метил/-1-бензил-4-фенил-пиперидин
К раствору из 3,3 г полученного в предыдущей стадии соединения, 1,4 г триэтиламина в 50 мл ДХМ добавляют 0,863 г ацетилхлорида и перемешивают 2 часа при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой, экстрагируют эфиром, экстракт промывают водой, сушат над сульфатом натрия и растворитель выпаривают. Остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (100/3 по объему). Получают 2,4 г целевого продукта.
Д) 4-/(Ацетил-N-метиламино)метил/-4-фенил-пиперидин-п- толуолсульфонат
Гидрируют, при комнатной температуре и при атмосферном давлении, смесь 2,3 г полученного в предыдущей стадии соединения, 1,2 г моногидрата п-толуолсульфокислоты, 0,23 г 10%-ного палладия-на-угле и 100 мл метанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Получают 2,7 г целевого продукта.
Приготовление 1.5. 4-/(N'-этил-N-метилуреидо)/метил/- 4-фенил-пиперидин
А) 1-Бензил-4-/N'-этил-N-метилуреидо)метил/-4-фенил- пиперидин
К раствору 2,7 г полученного в стадии В) приготовления 1.4 соединения в 50 мл ДХМ, при комнатной температуре, добавляют раствор 0,71 г этилизоцианата в 10 мл ДХМ и перемешивают в течение 1 часа. Реакционную смесь концентрируют в вакууме и остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (100/2,5 по объему). Получают 1,7 г целевого продукта.
Б) 4-/(N'-этил-N-метилуреидо)метил/-4-фенилпиперидин
При 40oC и атмосферном давлении гидрируют смесь 1,7 г полученного в предыдущей стадии соединения, 0,2 г 10%-ного палладия-на-угле и 50 мл метанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Получают 1,23 г целевого продукта.
Приготовление 1.6. 4-/(N',N'-Диэтил-N-метилуреидо)- метил/-4-фенил-пиперидин
А) 1-Бензил-4-/(N',N'-Диэтил-N-метилуреидо)метил/-4- фенил-пиперидин.
К раствору 4 г полученного в стадии В) приготовления 1.4 соединения, 1,55 триэтиламина в 30 мл 1,2-дихлорэтана, при комнатной температуре, добавляют 1,92 г N,N-диэтилкарбамоилхлорида и кипятят с обратным холодильником в течение 2-х часов. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (100/2 по объему). Получают 2,5 г целевого продукта.
Б) 4-/(N', N'-диэтил-N-метилуреидо)метил/-4-фенил- пиперидин-п-толуолсульфонат
При 30oC и при атмосферном давлении, гидрируют смесь 2,4 г полученного в предыдущей стадии соединения, 1 г моногидрата п-толуолсульфокислоты, 0,24 г 10%-ного палладия-на-угле и 50 мл метанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Получают 2,8 г целевого продукта.
Приготовление 1.7. 4-Фенил-4-(пиперид-1-ил)-пиперидин- дихлоргидрат-дигидрат
А) 1-Бензил-4-циано-4-(пиперид-1-ил)пиперидин
12,16 г Хлоргидрата пиперидина и 18,9 г 1-бензил-пиперид-4-она растворяют в 50 мл смеси метанола с водой (50/50 по объему). Прикапывают 5,3 г NaCN в виде раствора в 20 мл воды. После перемешивания в течение 48 часов образовавшийся осадок отфильтровывают, промывают водой и сушат с получением 27 г целевого продукта.
Б) 1-Бензил-4-фенил-4-(пиперид-1-ил)-пиперидин
Готовят раствор фенилмагнийбромида из 1,5 г магния, 12 г фенилбромида в 50 мл эфира. После перемешивания в течение 1 часа, при комнатной температуре, прикапывают раствор 10 г полученного в предыдущей стадии соединения в 100 мл эфира и перемешивают в течение 30 минут. Реакционную смесь выливают в 300 мл насыщенного раствора хлорида аммония, после декантации промывают водой, экстрагируют 2 н раствором HCl, водную кислую фазу промывают с помощью ДХМ, подщелачивают водную фазу путем добавки концентрированного раствора NaOH, экстрагируют с помощью ДХМ, сушат над сульфатом магния и растворитель выпаривают в вакууме. Полученное масло хроматографируют на диоксиде кремния, элюируя смесью ДХМ с метанолом и гидроксидом аммония (50/50/1 по объему). Получают 4,2 г целевого продукта, после кристаллизации из изо-эфира.
В) 4-Фенил-4-(пиперид-1-ил)пиперидин-дихлоргидрат- дигидрат
К раствору 4 г полученного в предыдущей стадии соединения в 25 мл ДХМ прикапывают раствор 1,6 г CNBr 20 мл хлороформа и кипятят с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют в вакууме, остаток обрабатывают 50 мл 6 н раствора HCl и в течение 4-х часов кипятят с обратным холодильником. Затем оставляют на ночь при перемешивании и при комнатной температуре, добавляют животный уголь, отфильтровывают, фильтрат подщелачивают путем добавления 40%-ного раствора NaOH, экстрагируют дважды эфиром, сушат над сульфатом магния и выпаривают в вакууме. Полученный продукт обрабатывают с помощью ДХМ, подкисляют путем добавления солянокислого эфира и выпаривают в вакууме. Получают 3 г целевого продукта.
Приготовление 1.8. 4-(формиламино)-4-фенил-пиперидин- хлоргидрат
А) 1-Бензил-4-(формиламино)-4-фенил-пиперидин-хлоргидрат
К раствору 2 г полученного в стадии В) приготовления 1.1) соединения, 0,9 г формиата натрия в 14 мл муравьиной кислоты прикапывают 4,5 мл уксусного ангидрида и перемешивают в течение 48 часов при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают водой, подщелачивают добавлением концентрированного раствора NaOH, экстрагируют с помощью ДХМ, экстракт сушат над сульфатом магния и растворитель выпаривают в вакууме. Полученный продукт обрабатывают с помощью ДХМ, подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 1,7 г целевого продукта, после кристаллизации из ацетона. Т.пл. = 225oC (разложение).
Б) 4-(Формиламино)-4-фенил-пиперидин-хлоргидрат
При комнатной температуре и при атмосферном давлении гидрируют смесь 1,7 г полученного в предыдущей стадии соединения, 0,2 г 10%-ного палладия-на-угле и 50 мл 95o этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Получают 1,1 г целевого продукта, после кристаллизации из ацетона. Т.пл. = 217oC.
Приготовление 1.9. 4-(Формил-N-этиламино)-4-фенилпиперидин- п-толуол-сульфонат
А) 1-Бензил-4-(N-этиламино)-4-фенилпиперидин
К суспензии из 1,5 г литийалюминийгидрида в 20 мл ТГФ добавляют раствор 5 г 4-ацетамидо-1-бензил-4-фенилпиперидина в 50 мл ТГФ и кипятят с обратным холодильником в течение 3-х часов. После охлаждения добавляют раствор 1 мл концентрированного раствора NaOH в 8 мл воды, отфильтровывают неорганические соли и фильтрат выпаривают в вакууме. Полученное масло растворяют в 50 мл ТГФ, этот раствор добавляют к суспензии 1,5 г литийалюминийгидрида в 20 мл ТГФ и кипятят с обратным холодильником в течение 1 часа. Гидролизуют путем добавления раствора 0,5 мл концентрированного раствора NaOH в 6 мл воды, отфильтровывают неорганические соли и фильтрат выпаривают в вакууме. Получают 4,8 г целевого продукта, который используют таким, какой есть, в следующей стадии.
Б) 1-Бензил-4-(формил-N-этиламино)-4-фенилпиперидин- п-толуолсульфонат
К раствору 4,8 г полученного в предыдущей стадии соединения в 40 мл муравьиной кислоты прикапывают 13 мл уксусного ангидрида и нагревают при 40oC в течение 24-х часов. Добавляют 30 мл муравьиной кислоты, затем 25 мл уксусного ангидрида и продолжают нагревание при 40oC в течение 24-х часов. Снова добавляют 30 мл муравьиной кислоты, затем 25 мл уксусного ангидрида и продолжают нагревание при 40oC в течение 24-х часов. Концентрируют в вакууме, остаток обрабатывают водой, подщелачивают водный раствор путем добавления концентрированного раствора NaOH, экстрагируют с помощью ДХМ, экстракт сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя с помощью ДХМ, затем смесью ДХМ с метанолом (97/3 по объему). Полученный продукт растворяют в 50 мл ацетона, добавляют 2,8 г моногидрата п-толуолсульфокислоты и оставляют кристаллизоваться. Получают 6,3 г целевого продукта. Т.пл. = 199oC.
В) 4-(Формил-N-этиламино)-4-фенилпиперидин-п-толуолсульфонат
При комнатной температуре и атмосферном давлении гидрируют смесь 6,3 г полученного в предыдущей стадии соединения, 0,7 10%-ного палладия-на-угле и 100 мл 95o этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Получают 4,78 г целевого продукта, после кристаллизации из смеси ацетона с эфиром. Т.пл. = 151oC.
Приготовление 1.10. 4-(Циклопропилкарбонил-N-метиламино)-4- фенил-пиперидин-п-толуолсульфонат
А) 1-Бензил-4-(N-метиламино)-4-фенилпиперидин
К суспензии 12,5 г литийалюминийгидрида в 100 мл ТГФ прикапывают раствор 38,8 г полученного в стадии А), приготовления 1.8 соединения (в виде свободного основания, т.пл. = 140oC) в 400 мл ТГФ и кипятят в течение 3-х часов с обратным холодильником. Гидролизуют путем добавки раствора из 5 мл концентрированного раствора NaOH в 45 мл воды, отфильтровывают неорганические соли и фильтрат концентрируют в вакууме. Получают 38 г целевого соединения, которое используют таким, какое есть.
Б) 1-Бензил-4-(циклопропилкарбонил-N-метиламино)-4- фенилпиперидин
Охлаждают до 0oC раствор 1,5 г полученного в предыдущей стадии соединения, 1,5 мл триэтиламина в 20 мл ДХМ, прикапывают 0,58 мл циклопропанкарбонилхлорида и оставляют при перемешивании на 2 часа, причем температура повышается до комнатной. Реакционную смесь промывают дважды водой, 1 н раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 1,8 г целевого продукта.
В) 4-(Циклопропилкарбонил-N-метиламино)-4-фенилпиперидин- п-толуолсульфонат
При комнатной температуре и атмосферном давлении гидрируют смесь 1,8 г полученного в предыдущей стадии соединения, 0,85 г моногидрата п-толуолсульфокислоты, 0,35 г 10%-ного палладия-на-угле и 100 мл этанола. Катализатор отфильтровывают на Целите и фильтрат выпаривают в вакууме. Получают 1,5 г целевого продукта, после кристаллизации из смеси ацетона с этилацетатом.
Приготовление 1.11. 4-(Циклопропилкарбониламино)-4-фенил- пиперидин-хлоргидрат
А) 1-Бензил-4-(циклопропилкарбониламино)-4-фенилпиперидин
Охлаждают до -20oC раствор 1 г полученного в стадии В), приготовления 1.1 соединения, 1,7 мл триэтиламина в 30 мл ДХМ, прикапывают 0,22 мл циклопропанкарбонилхлорида и перемешивают при повышении температуры до комнатной. Реакционную смесь экстрагируют с помощью ДХМ, промывают два раза водой, 0,5 н раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток обрабатывают с помощью этилацетата, образовавшиеся кристаллы отсасывают, промывают их этилацетатом, затем эфиром. Получают 0,77 г целевого продукта.
Б) 4-(Циклопропилкарбониламино)-4-фенилпиперидин-хлоргидрат
При 35oC и атмосферном давлении гидрируют смесь 0,77 г полученного в предыдущей стадии соединения, 0,14 г 10%-ного палладия-на-угле и 40 мл этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Остаток обрабатывают с помощью ДХМ, подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,6 г целевого продукта.
Приготовление 1.12. 4-(Циклобутилкарбониламино)-4-фенил- пиперидин-хлоргидрат
А) 1-Бензил-4-(циклобутилкарбониламино)-4-фенилпиперидин
Охлаждают до 0oC раствор 1,5 г полученного в стадии В), приготовления 1.1 соединения, 2,1 г триэтиламина в 30 мл ДХМ, прикапывают 0,45 мл циклобутанкарбонилхлорида и оставляют при перемешивании при повышении температуры до комнатной. Экстрагируют с помощью ДХМ, промывают 2 раза водой, 0,5 н раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 1,1 г целевого продукта, после кристаллизации из этилацетата, затем перекристаллизации из эфира.
Б) 4-(Циклобутилкарбониламино)-4-фенилпиперидин- хлоргидрат
При 35oC и при атмосферном давлении гидрируют смесь 1,1 г полученного в предыдущей стадии соединения, 0,18 г 10%-ного палладия-на-угле и 60 мл этанола. Катализатор отфильтровывают на Целите и фильтрат выпаривают в вакууме. Остаток обрабатывают с помощью ДХМ, подкисляют путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,92 г целевого продукта.
Приготовление 1.13. 4-(Циклогексилкарбониламино)-4- фенилпиперидин-хлоргидрат
А) 1-Бензил-4-(циклогексилкарбониламино)-4-фенилпиперидин
Это соединение получают согласно методике, описанной в стадии А) приготовления 1.12, исходя из 1,5 г полученного в стадии В) приготовления 1.1 соединения и 0,75 мл циклогексанкарбонилхлорида. Получают 1,3 г целевого продукта.
Б) 4-(Циклогексилкарбониламино)-4-фенилпиперидин- хлоргидрат
Это соединение получают по методике, описанной в стадии Б) приготовления 1.12. Получают 0,9 целевого продукта.
Приготовление 1.14. 4-Метоксикарбонил-4-фенилпиперидин- п-толуолсульфонат
К раствору 10 г 4-карбокси-4-фенилпиперидин-п-толуол- сульфоната в 300 мл метанола добавляют 1 г моногидрата п-толуолсульфокислоты и кипятят с обратным холодильником в течение 3-х дней. Реакционную смесь концентрируют в вакууме, остаток обрабатывают ацетоном и добавляют эфир вплоть до появления осадка. После отсасывания образовавшегося осадка получают 9,34 г целевого продукта.
Приготовление 1.15. 4-(N-Метилкарбамоил)-4-фенилпиперидин
А) 1-трет-Бутоксикарбонил-4-карбокси-4-фенилпиперидин
К смеси 30 г 4-карбокси-4-фенилпиперидин-п-толуолсульфоната в 300 мл диоксана добавляют 30 мл воды, 32,9 г K2CO3, после чего нагревают до 60oC и медленно добавляют 18,21 г ди-трет-бутилдикарбоната. Затем нагревают в течение 2-х часов при 60oC, после чего 30 минут кипятят с обратным холодильником. Реакционную смесь концентрируют в вакууме, остаток обрабатывают с помощью ДХМ, промывают буфером с pH 2, подкисляют до pH 4 путем добавления 2 н HCl, экстрагируют с помощью ДХМ, промывают буфером с pH 2, водой, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Получают 23,7 г целевого продукта.
Б) 1-трет-Бутоксикарбонил-4-(N-метилкарбамоил)-4-фенил- пиперидин
К раствору 1,5 г полученного в предыдущей стадии соединения в 5 мл ДХМ и 5 мл ДМФ добавляют 1,98 г триэтиламина, затем 0,49 г метиламин-хлоргидрата. Охлаждают на ледяной бане, добавляют 2,39 г BOP и оставляют в течение 24-х часов при перемешивании, причем температура повышается до комнатной. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, промывают экстракт водой, затем буферным раствором с pH 2, 10%-ным раствором NaOH, водой, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Получают 1,4 г целевого продукта.
В) 4-(N-Метилкарбамоил)-4-фенилпиперидин
К раствору 1,4 г полученного в предыдущей стадии соединения в 30 мл метанола добавляют 4 мл концентрированной HCl и перемешивают в течение 1 часа при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют с помощью ДХМ, промывают водой, затем два раза 10%-ным раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 0,6 г целевого продукта.
Приготовление 1.16. 4-(N-н-Бутилкарбамоил)-4-фенилпиперидин
А) 1-трет-Бутоксикарбонил-4-(N-н-бутилкарбамоил)-4-фенил- пиперидин
Это соединение получают по методике, описанной в стадии Б) приготовления 1.15, исходя из 1,0 г полученного в стадии А) приготовления 1.15 соединения и 0,24 г н-бутиламина. Получают 1,3 г целевого соединения, которое используют таким, какое есть, в следующей стадии.
Б) 4-(N-н-Бутилкарбамоил)-4-фенилпиперидин
Это соединение получают по методике, описанной в стадии В) приготовления 1.15. Получают 0,4 г целевого соединения.
Приготовление 1.17. 4-(N,N-Диэтилкарбамоил)-4-фенил- пиперидин-трифторацетат
А) 1-трет-Бутоксикарбонил-4-(N,N-диэтилкарбамоил)-4- фенилпиперидин
Это соединение получают по методике, описанной в стадии Б) приготовления 1.15, исходя из 1,5 г соединения, полученного в стадии А), приготовления 1.15 и 0,8 г диэтиламин-хлоргидрата. Получают 1,7 г целевого продукта.
Б) 4-(N,N-диэтилкарбамоил)-4-фенилпиперидин-трифторацетат
1,7 г полученного в предыдущей стадии соединения растворяют в 20 мл трифторуксусной кислоты и перемешивают 30 минут при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают эфиром и выпаривают в вакууме. Получают 2,8 г целевого продукта в форме масла.
Приготовление 1.18. 4-(Пирролидин-1-ил-карбамоил)-4- фенилпиперидин
А) 1-трет-Бутоксикарбонил-4-(пирролидин-1-ил-карбонил)- 4-фенилпиперидин
Это соединение получают по методике, описанной в стадии Б) приготовления 1.15, исходя из 1 г соединения, полученного в стадии А) приготовления 1.15, и 0,23 г пирролидина. Получают 1,0 г целевого продукта.
Б) 4-(Пирролидин-1-ил-карбонил)-4-фенил-пиперидин
К раствору 1,0 г полученного в предыдущей стадии соединения в 25 мл метанола добавляют 3 мл концентрированной HCl и перемешивают 1 час при 35-40oC. Концентрируют в вакууме, остаток обрабатывают метанолом и растворитель выпаривают в вакууме. Остаток экстрагируют эфиром, промывают два раза 10%-ным раствором NaOH, водой, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 0,43 г целевого продукта.
Приготовление 1.19. 4-(N-Метокси-N-метилкарбамоил)-4- фенилпиперидин
А) 1-трет-Бутоксикарбонил-4-(N-метокси-N-метилкарбамоил)- 4-фенил-пиперидин
Это соединение получают по методике, описанной в стадии Б) приготовления 1.15, исходя из 1,5 г соединения, полученного в стадии А) приготовления 1.15, и 0,71 г O-метил-N- метилгидроксиламин-хлоргидрата. Получают 1,71 г целевого продукта.
Б) 4-(N-Метокси-N-метилкарбамоил)-4-фенилпиперидин
Это соединение получают по методике, описанной в стадии В) приготовления 1.15 исходя из 1,7 г полученного в предыдущей стадии соединения. Получают 1,1 г целевого продукта.
Приготовление 1.20. 4-(Метилсульфонамидо)-4-фенил-пиперидин- хлоргидрат
А) 1-Бензил-4-(метилсульфонамидо)-4-фенилпиперидин-п- толуолсульфонат
Охлаждают до 0oC в атмосфере азота, смесь 5 г полученного в стадии В) приготовления 1.1 соединения, 10 мл триэтиламина в 100 мл ДХМ, прикапывают 2,68 г мезилхлорида и оставляют на 30 минут при перемешивании. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, промывают три раза водой, затем 10%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Полученное масло растворяют в 50 мл ацетона, добавляют 2,8 г моногидрата п-толуолсульфокислоты и, после перемешивания, выпаривают в вакууме. Получают 6,8 г целевого продукта.
Б) 4-(Метилсульфонамидо)-4-фенилпиперидин-хлоргидрат
К раствору 6,8 г полученного в предыдущей стадии соединения в воде добавляют 40%-ный раствор NaOH, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. К полученному остатку (3,69 г) добавляют 2,2 г 1-хлорэтил-хлорформиата, 1 мл 1,2,2,6,6-пентаметилпиперидина и оставляют стоять в течение ночи при перемешивании. Реакционную смесь выпаривают в вакууме, остаток растворяют в метаноле и нагревают при 60oC в течение 1 часа. Растворитель выпаривают в вакууме, остаток обрабатывают ацетоном и отсасывают выделившиеся кристаллы. Получают 3 г целевого продукта.
Приготовление 1.21. 4-(Метансульфонил-N-метиламино)-4- фенил-пиперидин-п-толуолсульфонат
А) 1-Бензил-4-(метансульфонил-N-метиламино)-4-фенил- пиперидин-п-толуолсульфонат
К раствору 2 г полученного в стадии А) приготовления 1.10 соединения в 30 мл ДХМ добавляют 1,6 мл триэтиламина, затем 0,9 мл мезилхлорида, затем перемешивают 30 минут при комнатной температуре. Реакционную смесь промывают два раза водой, 5%-ным раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток обрабатывают эфиром, отфильтровывают нерастворимую часть и фильтрат выпаривают в вакууме. Остаток растворяют в ацетоне, добавляют 1,4 г моногидрата п-толуолсульфокислоты, затем эфир вплоть до кристаллизации. После отсасывания, получают 2,3 г целевого продукта. Т.пл. = 175oC.
Б) 4-(Метансульфонил-N-метиламино)-4-фенилпиперидин-п- толуолсульфонат
При комнатной температуре и атмосферном давлении гидрируют смесь 2,3 г полученного в предыдущей стадии соединения, 0,25 г 10%-ного палладия-на-угле в 40 мл этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Получают 1,7 г целевого продукта в виде пены.
Приготовление 1.22. Ди-п-толуолсульфонат-4-(циклопропил- метиламино)-4-фенил-пиперидина
А) 4-(Циклопропилкарбониламино)-4-фенил-1-тритилпиперидин
Охлаждают до +5oC смесь 0,565 г 4-(циклопропилкарбониламино)-4- фенилпиперидин-хлоргидрата в 50 мл ДХМ, добавляют 0,565 г тритилхлорида, затем 0,7 мл триэтиламина и оставляют при перемешивании, причем температура повышается до комнатной. Реакционную смесь промывают водой, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 1 г целевого продукта, после кристаллизации из эфира.
Б) 4-(Циклопропилметиламино)-4-фенил-1-тритил-пиперидин
К суспензии 0,5 г литийалюминийгидрида в 50 мл ТГФ добавляют 0,9 г полученного в предыдущей стадии соединения и в течение 30 минут кипятят с обратным холодильником. Гидролизуют путем добавления раствора из 0,4 мл концентрированного раствора NaOH в 3 мл воды, отфильтровывают неорганические соли и фильтрат выпаривают в вакууме. Полученный продукт добавляют к суспензии 0,7 г литийалюминийгидрида в 50 мл ТГФ и кипятят с обратным холодильником в течение 1 часа. Гидролизуют путем добавления раствора из 0,5 мл концентрированного раствора NaOH в 4 мл воды, отфильтровывают неорганические соли и фильтрат выпаривают в вакууме. Получают 0,5 г целевого продукта, после кристаллизации из смеси ДХМ/изо-эфир.
В) 4-(Циклопропилметиламино)-4-фенил-пиперидин-ди-п- толуолсульфонат
В течение 1 часа нагревают при 50oC смесь 0,5 г полученного в предыдущей стадии соединения, 7,5 мл муравьиной кислоты и 7,5 мл воды. Реакционную смесь отфильтровывают, фильтрат подщелачивают путем добавления 40%-ного раствора NaOH, экстрагируют с помощью ДХМ, сушат над сульфатом магния и растворитель выпаривают в вакууме. Полученный продукт растворяют в ДХМ, добавляют 0,9 г моногидрата п-толуолсульфокислоты и кипятят с обратным холодильником. После охлаждения, отсасывают образовавшиеся кристаллы. Получают 0,35 г целевого продукта, после перекристаллизации из смеси ацетона с эфиром.
Приготовление 1.23. 4-Гидроксиметил-4-фенилпиперидин
Охлаждают до -20oC суспензию 1,16 г литийалюминийгидрида в 50 мл ТГФ, добавляют 4 г полученного в приготовлении 1.14, соединения и оставляют при перемешивании в течение ночи при повышении температуры до комнатной. Гидролизуют путем добавки 1,2 мл воды, затем 2,5 мл 10%-ного раствора NaOH и 2,5 мл воды. Разбавляют эфиром, отфильтровывают неорганические соли и фильтрат выпаривают в вакууме. Получают 1,8 г целевого продукта.
Приготовление 1.24 4-Спиро(фталид-3)-пиперидин-хлоргидрат
А) 1-Бензил-4-спиро(фталид-3)пиперидин
Охлаждают до -70oC раствор 10 г N-метилбензамида в 100 мл ТГФ и добавляют в атмосфере азота 100 мл 1,6 М раствора н-бутиллития в гексане. Перемешивают при повышении температуры до 0oC, затем снова охлаждают до -70oC и добавляют по каплям 7 г 1-бензил-пиперид-4-она. Выдерживают 30 минут при перемешивании, реакционную смесь выливают в 1 литр воды со льдом, экстрагируют два раза по 500 мл эфиром, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью ДХМ с метанолом (99/1 по объему). Получают 2,7 г целевого продукта.
Б) 4-Спиро(фталид-3)пиперидин-хлоргидрат
Охлаждают до 0oC раствор 1 г полученного в предыдущей стадии соединения в 20 мл ДХМ, добавляют в атмосфере азота 0,51 г 1-хлор-этил-хлорформиата и выдерживают 2 часа при перемешивании и при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают 10 мл метанола и нагревают при 50oC в течение 35 минут. Концентрируют в вакууме. Получают 0,66 г целевого продукта, после кристаллизации из ацетона. Т.пл. 280oC (разложение).
Приготовление 1.25. 4-Гидрокси-4-(пирид-2-ил)пиперидин- дихлоргидрат
А) 1-Бензил-4-гидрокси-4-(пирид-2-ил)пиперидин-хлоргидрат
25 г 2-Бромпиридина растворяют в 100 мл ТГФ при -70oC, в атмосфере азота, и прикапывают 1,6 М раствор н-бутиллития в гексане, затем раствор 30 г 1-бензил-пиперид-4-она в 25 мл ТГФ. Оставляют температуру повышаться до комнатной, и спустя час, частично испаряют растворители, остаток выливают в насыщенный раствор NH4Cl, экстрагируют эфиром, промывают водой, сушат над сульфатом магния и выпаривают. Получают масло, которое растворяют в 200 мл ДХМ и пропускают путем барботирования газообразный HCl с целью получения хлоргидрата. Целевой продукт (21 г) кристаллизуют из смеси метанола с эфиром. Т.пл. = 185oC.
Б) 4-Гидрокси-4-(пирид-2-ил)-пиперидин-дихлоргидрат
В минимальном количестве воды растворяют 17,7 г полученного в предыдущей стадии соединения, добавляют 40%-ный раствор гидроксида натрия, экстрагируют с помощью ДХМ, сушат над сульфатом магния и выпаривают. Тогда получают 15 г продукта стадии А), в форме свободного амина. Растворяют его в 150 мл метанола, добавляют 2,5 г 10%-ного Pd/C и 17 г формиата аммония. После перемешивания в течение 2-х часов при комнатной температуре, затем кипячения с обратным холодильником в течение 2-х часов, выпаривают, остаток обрабатывают хлороформом, промывают полученный раствор 5%-ным раствором гидроксида натрия, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают. Остаток растворяют в метаноле и) получают дихлоргидрат (10 г) путем добавления 4 н раствора HCl в эфире. Т.пл. = 230oC.
Приготовление 1.26. 4-Гидрокси-4-(2-метоксифенил)-пиперидин
А) 1 Бензил-4-гидрокси-4-(2-метоксифенил)пиперидин
Охлаждают до -70oC, в атмосфере азота, раствор 15 г 2-броманизола в 50 мл ТГФ, прикапывают 1,6 М раствор н-бутиллития в ТГФ и оставляют при перемешивании в течение 1 часа. Охлаждают до -70oC и прикапывают раствор 15,2 г 1-бензилпиперид-4- она в 50 мл ТГФ. Оставляют при перемешивании, причем температура повышается до комнатной, и спустя 1 час реакционную смесь концентрируют в вакууме. Остаток обрабатывают этилацетатом, органическую фазу промывают водой, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 14 г целевого продукта, после кристаллизации из смеси этилацетата с эфиром и гептаном.
Б) 4-Гидрокси-4-(2-метоксифенил)пиперидин
Выдерживают 2 часа при перемешивании смесь 5 г полученного в предыдущей стадии соединения, 1 г 5%-ного палладия-на-угле, 5 г формиата аммония, в 100 мл метанола. Реакционную смесь отфильтровывают и фильтрат выпаривают в вакууме. Остаток обрабатывают с помощью ДХМ, органическую фазу промывают 40%-ным раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 2,2 г целевого продукта, т.пл. = 200oC.
Приготовление 1.27. 4-(Этиламинокарбонилоксиметил)-4- фенил-пиперидин-хлоргидрат
А) 1-трет-Бутоксикарбонил-4-(гидроксиметил)-4-фенил- пиперидин
К раствору 22,8 г полученного в приготовлении 1,23 соединения в 250 мл 1,2-диметоксиэтана добавляют 26,05 г ди-трет-бутилдикарбоната и кипятят с обратным холодильником в течение 2-х часов. Реакционную смесь концентрируют в вакууме, остаток обрабатывают с помощью ДХМ, органическую фазу промывают буферным раствором с pH 2, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 17,86 г целевого продукта, после кристаллизации из эфира. Т.пл. = 134oC.
Б) 1-трет-Бутоксикарбонил-4-(этиламинокарбонилоксиметил)- 4-фенил-пиперидин
Выдерживают в течение ночи при перемешивании и при комнатной температуре смесь 2,91 г полученного в предыдущей стадии соединения, 2,4 г этилизоцианата, 2-х капель триэтиламина в 30 мл толуола. Затем в течение 24-х часов нагревают при 100oC и реакционную смесь концентрируют в вакууме. Остаток обрабатывают эфиром, органическую фазу промывают буферным раствором с pH 2, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 3,85 г целевого продукта в виде масла.
В) 4-(этиламинокарбонилоксиметил)-4-фенил-пиперидин- хлоргидрат
К раствору 3,85 г полученного в предыдущей стадии соединения в 50 мл метанола добавляют 10 мл концентрированной HCl и в течение 2-х часов нагревают при 60oC. Концентрируют в вакууме, остаток обрабатывают ацетоном и растворитель выпаривают в вакууме. Получают 2,6 г целевого продукта, после кристаллизации из смеси этилацетата с эфиром. Т.пл. = 240-242oC.
Приготовление 1.28. 4-Фенил-4-(пропионил-N-метиламино)- пиперидин-п-толуолсульфонат
А) 4-(Акрилоил-N-метиламино)-1-бензил-4-фенилпиперидин
Охлаждают до 0oC раствор 1,5 г полученного в стадии А) приготовления 1.10 соединения, 1,5 мл триэтиламина в 40 мл ДХМ, прикапывают 0,5 мл акрилоилхлорида и перемешивают при повышении температуры до комнатной. Реакционную смесь выливают в воду, экстрагируют с помощью ДХМ, органическую фазу промывают водой, 2 н раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 1,3 г целевого продукта, после кристаллизации из смеси эфира с пентаном.
Б) 4-(Акрилоил-N-метиламино)-1-бензил-4-фенилпиперидин- п-толуол-сульфонат
К раствору 1,15 г полученного в предыдущей стадии соединения в 10 мл ДХМ добавляют 0,59 г моногидрата п-толуолсульфокислоты и оставляют кристаллизоваться. Получают 1,65 г целевого продукта.
В) 4-Фенил-4-(пропионил-N-метиламино)пиперидин-п-толуолсульфонат
При комнатной температуре и атмосферном давлении гидрируют смесь 1,64 г полученного в предыдущей стадии соединения 0,2 г 10%-ного палладия-на-угле в 100 мл этанола. Катализатор отфильтровывают на Целите ® и растворитель выпаривают в вакууме. Получают 1,3 г целевого продукта.
Приготовление 1.29. 4-(Циклогексилкарбонил-N-метиламино)-4- фенил-пиперидин-п-толуолсульфонат
А) 1-Бензил-4-(циклогексилкарбонил-N-метиламино)-4- фенилпиперидин-п-толуолсульфонат
К раствору 1,5 г соединения, полученного в стадии А) приготовления 1.10 и 1,5 мл триэтиламина в 15 мл ДХМ, при комнатной температуре, прикапывают 0,78 мл циклогексанкарбонилхлорида и перемешивают в течение 2-х часов. Реакционную смесь промывают два раза водой, 2 н раствором NaOH, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток растворяют в ДХМ, добавляют 0,97 г моногидрата п-толуолсульфокислоты и концентрируют в вакууме. После кристаллизации из смеси этилацетата с эфиром получают 3,3 г целевого продукта.
Б) 4-(Циклогексилкарбонил-N-метиламино)-4-фенилпиперидин- п-толуол-сульфонат
При комнатной температуре и атмосферном давлении гидрируют смесь 3,3 г полученного в предыдущей стадии соединения 0,35 г 10%-ного палладия-на-угле в 100 мл этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Остаток обрабатывают ацетоном и растворитель выпаривают в вакууме. Получают 2,2 г целевого продукта, после кристаллизации из смеси этилацетата с эфиром. Т.пл. = 160oC.
Приготовление 1.30. 4-Карбамоил-4-фенил-пиперидин
А) 1-трет-Бутоксикарбонил-4-карбамоил-4-фенилпиперидин
Охлаждают до -20oC раствор 1,5 г полученного в стадии А) приготовления 1.15 соединения, 0,99 г триэтиламина, 2,39 г BOP в 10 мл ДХМ, затем в раствор пропускают путем барботирования газообразный аммиак. Оставляют температуру повышаться до комнатной и перемешивают в течение 2-х часов. Реакционную смесь концентрируют в вакууме, остаток экстрагируют эфиром, органическую фазу промывают водой, буферным раствором с pH 2, водой, 10%-ным раствором NaOH, водой, насыщенным раствором NaCl, затем сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 1,32 г целевого продукта.
Б) 4-Карбамоил-4-фенилпиперидин
Это соединение получают по методике, описанной в стадии В), приготовления 1.15, исходя из 1,32 г полученного в предыдущей стадии соединения. Получают 0,41 г целевого продукта.
Приготовление 1.31. 4-(N,N-Диметилкарбамоил-)-4- фенилпиперидин
А) 1-трет-Бутоксикарбонил-4-(N,N-диметилкарбамоил)-4- фенилпиперидин
Это соединение получают по методике, описанной в стадии Б) приготовления 1.15, исходя из 1,5 г полученного в стадии А) приготовления 1.15 соединения и 0,6 г диметиламин-хлоргидрата. Получают 1,6 г целевого продукта.
Б) 4-(N,N-Диметилкарбамоил)-4-фенилпиперидин
Это соединение получают по методике, описанной в стадии В) приготовления 1.15, исходя из 1,6 г полученного в предыдущей стадии соединения. Получают 1,1 г целевого продукта.
Приготовление 1.32. 4-(N-Изопропилкарбамоил)-4-фенил- пиперидин
А) 1-трет-Бутоксикарбонил-4-(N-изопропилкарбамоил)-4- фенилпиперидин
Это соединение получают по методике, описанной в стадии Б) приготовления 1.15, исходя из 1,5 г соединения, полученного в стадии А) приготовления 1.15 и 0,29 г изопропиламина. Получают 1,61 г целевого продукта.
Б) 4-(N-Изопропилкарбамоил)-4-фенилпиперидин
Это соединение получают по методике, описанной в стадии В) приготовления 1.15, исходя из 1,61 г полученного в предыдущей стадии соединения. Получают 1,1 г целевого продукта.
Приготовление 2.1. 2-(3,4-Дихлорфенил)-5-(тетрагидропиран- 2-ил-окси)-пентиламин
А) 2-(3,4-Дихлорфенил)-5-(тетрагидропиран-2-ил-окси)- пентан-нитрил
15 г 60%-ного гидрида натрия в масле суспендируют в 200 мл безводного ТГФ. В течение 30 минут прикапывают раствор 69,5 г 3,4-дихлорфенилацетонитрила в 500 мл ТГФ, затем реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Смесь охлаждают до -20oC, добавляют раствор 85 г 1-бром-3- (тетрагидропиран-2-ил-окси)пропана в 100 мл ТГФ. Смесь оставляют стоять до возвращения ее температуры к комнатной и еще 2 часа, выливают ее в раствор 50 г хлорида аммония в 3 л воды. Экстрагируют эфиром, экстракт промывают насыщенным раствором хлорида натрия, декантируют, сушат над сульфатом магния и концентрируют. Остаток хроматографируют на силикагеле, элюируя смесью толуола с градиентом этилацетата (3 ---> 5%). Фракции чистого продукта концентрируют, получая 77 г целевого продукта в виде масла.
Б) 2-(3,4-Дихлорфенил)-5-(тетрагидропиран-2-ил-окси)- пентиламин
77 г полученного в предыдущей стадии нитрила растворяют в 500 мл абсолютного этанола. Добавляют 200 мл концентрированного аммиака, затем никель Ренея (10% в расчете на количество исходного нитрила). Затем, в атмосфере водорода, при комнатной температуре и при атмосферном давлении, гидрируют, причем поглощаются 10,5 л водорода, катализатор отделяют путем отфильтровывания при использовании Целита. Фильтрат концентрируют в вакууме и остаток обрабатывают раствором NaCl. После экстракции эфиром и высушивания органической фазы над сульфатом магния, получают 75 г целевого продукта в виде масла.
Приготовление 2.2. N-Метил-2-(3,4-дихлорфенил)-5- гидроксипентиламин-хлоргидрат
А) Этил-N-/2-(3,4-дихлорфенил)-5-(тетрагидропиран- 2-ил-окси)пентил/-карбамат
20 г полученного в приготовлении 2.1 соединения растворяют в 200 мл ДХМ и добавляют 9,3 мл ТЭА. Охлаждают до -50oC и прикапывают раствор 6,3 мл этилхлорформиата. Спустя 15 минут, промывают водой, затем разбавленной HCl. Сушат над сульфатом магния и концентрируют досуха, получая 24 г масла.
Б) N-Метил-2-(3,4-дихлорфенил)-5-(тетрагидропиран- 2-ил-окси)пентил-амин
К 5 г LiAlH4 в виде суспензии в 150 мл ТГФ добавляют раствор 24 г полученного в предыдущей стадии карбамата в 100 мл безводного ТГФ. В течение 2-х часов реакционную смесь кипятят с обратным холодильником. Гидролизуют с помощью 20 мл воды и 5 мл концентрированного раствора гидроксида натрия, отфильтровывают неорганическую часть и концентрируют фильтрат досуха. Получают 20,1 г целевого продукта в виде масла.
В) N-Метил-2-(3,4-дихлорфенил)-5-гидроксипентиламин- хлоргидрат
20 г полученного в предыдущей стадии соединения растворяют в 200 мл абсолютного этанола. Добавляют 8 мл концентрированной HCl и перемешивают при комнатной температуре в течение 2-х часов 30 минут. Концентрируют досуха, добавляют этанол и толуол и снова концентрируют досуха. Остаток постепенно кристаллизуется из ацетона, к которому добавлен эфир. Отфильтровывают и высушивают полученные кристаллы. Получают 15,8 г целевого продукта. Т.пл. = 124oC.
Приготовление 2.3. 3-(3,4-Дихлорфенил)-3-(3-гидрокси- пропил)пиперидин-хлоргидрат
А) Метил-4-циано-4-(3,4-дихлорфенил)гептандиоат
В трехгорлой колбе, 37,2 г 3,4-дихлорфенилацетонитрила и 34,43 г метилакрилата растворяют в 20 мл диоксана, добавляют 1 мл ДБУ, нагревают 2 часа при 60oC, выпаривают, разбавляют с помощью 400 мл этилацетата, затем промывают разбавленной HCl, раствором NaCl, сушат над сульфатом магния и выпаривают. Целевой продукт кристаллизуется из 100 мл этилацетата, и 100 мл эфира со 100 мл гептана. Получают 47 г продукта.
Б) Метил-3-/5-(3,4-дихлорфенил)-2-оксопиперид-5-ил/ пропионат
40 г полученного в стадии А) продукта растворяют в 500 мл 2-метоксиэтанола, добавляют 2 г никеля Ренея и гидрируют при 40oC и при атмосферном давлении в течение 3-х дней. Отфильтровывают, фильтрат выпаривают, и получают целевой продукт в виде масла (39 г).
В) 3-/5-(3,4-Дихлорфенил)-2-оксопиперид-5-ил/пропановая кислота
17 г полученного в предыдущей стадии соединения растворяют в 250 мл метанола, добавляют 2,8 г гидроксида калия и 10 мл воды, затем кипятят с обратным холодильником в течение 2-х часов. Выпаривают досуха, полученное масло обрабатывают с помощью 200 мл воды и экстрагируют с помощью 100 мл этилацетата. Водную фазу подкисляют 30%-ным раствором HCl, затем отфильтровывают и высушивают образовавшийся осадок. Перекристаллизуют из горячего метанола и получают 18,3 г целевого соединения.
Г) 3-(3,4-Дихлорфенил)-3-(3-гидроксипропил)пиперидин- хлоргидрат
5 г полученного в предыдущей стадии соединения растворяют в 20 мл ТГФ, добавляют 75 мл борана (концентрация 1 М в ТГФ) и кипятят с обратным холодильником в течение 24-х часов в атмосфере азота. Добавляют 25 мл метанола, 50 мл 4 н HCl и перемешивают в течение 30 минут, затем добавляют 40%-ный раствор гидроксила натрия вплоть до pH выше 10. Экстрагируют 3 раза по 150 мл ДХМ, органическую фазу сушат над сульфатом магния и выпаривают. Остаток растворяют в ДХМ с 4 н раствором HCl в эфире. После выпаривания, получают пену, и целевой продукт (4,5 г) кристаллизуется из смеси этилацетата с эфиром.
Пример 1. N-Метил-N-/2-(3,4-дихлорфенил)-5-(4-гидрокси- 4-фенил-пиперид-1-ил)пентил/бензамид-хлоргидрат
А) N-/2-(3,4-Дихлорфенил)-5-(тетрагидропиран-2-ил- окси)пентил/-бензамид
15 г амина, полученного в приготовлении 2.1, растворяют в 200 мл ДХМ. Раствор охлаждают до 0oC, добавляют 7 мл ТЭА, затем 5,3 мл бензоилхлорида. Реакционную смесь затем перемешивают при комнатной температуре в течение 30 минут, затем концентрируют досуха. Остаток обрабатывают эфиром, органическую фазу промывают водой, затем буферным раствором с pH 2, также, как раствором Na2CO3. После высушивания и концентрирования, получают 19,5 г целевого продукта в виде масла.
Б) N-Метил-N-/2-(3,4-дихлорфенил)-5-(тетрагидропиран- 2-ил-окси)пентил/-бензамид
При комнатной температуре перемешивают смесь 19,5 г полученного в предыдущей стадии соединения и 3,1 г 60%-ного гидрида натрия в масле, в 200 мл безводного ТГФ. В течение 1 часа нагревают при 40oC и добавляют 7,7 мл метилиодида. После перемешивания в течение 1 часа при 40oC, концентрируют досуха, остаток обрабатывают водой, экстрагируют эфиром, экстракт промывают буферным раствором с pH 2, затем раствором Na2CO3. Сушат над сульфатом магния и выпаривают. Получают 21 г целевого продукта в виде масла.
В) N-Метил-N-/2-(3,4-дихлорфенил)-5-гидроксипентил/-бензамид
21 г полученного в предыдущей стадии соединения растворяют в 170 мл метанола в присутствии 5 мл смолы Amberlyst 15 и смесь кипятят с обратным холодильником в течение 2-х часов. Смесь фильтруют через Целит, фильтрат концентрируют в вакууме и остаток хроматографируют на силикагеле, элюируя ДХМ, затем смесью ДХМ/этилацетат, вплоть до чистого этилацетата. Получают 13,5 г целевого продукта в виде масла.
Г) N-Метил-N-/2-(3,4-дихлорфенил)-5-(мезилокси)- пентил/бензамид
При 0oC перемешивают 13,5 г полученного в предыдущей стадии спирта и 5,7 мл ТЭА в 150 мл ДХМ. Затем прикапывают 3,2 мл мезилхлорида. Спустя 15 минут, концентрируют досуха, остаток обрабатывают эфиром и органическую фазу промывают два раза водой. Сушат над сульфатом магния и растворитель выпаривают. Получают 16,2 г целевого продукта в виде масла.
Д) N-Метил-N-/2-(3,4-дихлорфенил)-5-(4-гидрокси- 4-фенилпиперид-1-ил)-пентил/бензамид
Смесь 1 г полученного в предыдущей стадии мезилата, 800 мг 4-гидрокси-4-фенилпиперидина и 3 мл ДМФ нагревают при 70oC в течение 3-х часов. После охлаждения, смесь выливают в воду и экстрагируют этилацетатом, промывая затем этилацетатный раствор с помощью раствора NaCl. Сушат над сульфатом магния и растворитель выпаривают. Продукт хроматографируют на диоксиде кремния, элюируя ДХМ с градиентом метанола (2 ---> 5%). Получают 400 мг целевого продукта.
Хлоргидрат этого соединения описан в европейской заявке на патент EP-474 561, в примере 22. Т.пл. = 148oC.
Пример 2. N-Метил-N-/2-(3,4-дихлорфенил)-5-(4- пропионилокси-4-фенил-пиперид-1-ил)пентил/бензамид-хлоргидрат
При комнатной температуре перемешивают 400 мг полученного в примере 1 соединения и 0,19 мл ТЭА в 10 мл ДХМ. Прикапывают 0,13 мл пропионилхлорида и смесь промывают водой, затем раствором бикарбоната. Сушат над сульфатом магния и растворитель выпаривают. Продукт хроматографируют на силикагеле, элюируя ДХМ с градиентом метанола (1% ---> 2%). После растворения в ДХМ и добавления солянокислого эфира образуется хлоргидрат. Растворитель выпаривают и хлоргидрат конкретизируют в эфире. Получают 320 мг целевого продукта. Т.пл. = 112oC.
Пример 3. N-Метил-N-/2-(3,4-дихлорфенил)-5-(4-ацетамидо-4- фенил-пиперид-1-ил)пентил/фенилацетамид-хлоргидрат
А) N-Boc-N-метил-2-(3,4-дихлорфенил)-5-гидроксипентиламин
15,8 г аминоспирта, полученного в приготовлении 2.2, растворяют в 150 мл диоксана. Добавляют 15 мл воды, затем 10 мл ТЭА и 12,7 г (Boc)2O. Смесь нагревают при 60oC в течение 1 часа. Затем концентрируют досуха, обрабатывают остаток эфиром, промывают органическую фазу водой, затем разбавленным раствором HCl. Сушат над сульфатом магния и растворитель выпаривают. Получают 19,2 г целевого продукта в виде масла.
Б) N-Boc-N-метил-2-(3,4-дихлорфенил)-5-мезилоксипентиламин
При 0oC перемешивают 19,2 г полученного в предыдущей стадии спирта и 9,8 мл ТЭА в 200 мл ДХМ. Затем прикапывают 5,4 мл мезилхлорида. Спустя 15 минут концентрируют досуха, остаток обрабатывают эфиром, органическую фазу промывают водой, затем раствором Na2CO3. Сушат над сульфатом магния и растворитель выпаривают. Получают 23,5 г целевого продукта в виде масла.
В) N-Boc-N-метил-5-(4-ацетамидо-4-фенилпиперид-1-ил)- -2-(3,4-дихлор-фенил)-пентиламин
Смесь 10 г полученного в предыдущей стадии мезилата, 14 г 4-ацетамидо-4-фенилпиперидина и 20 мл ДМФ нагревают при 70oC в течение 2-х часов. После охлаждения, реакционную смесь выливают на лед и экстрагируют этилацетатом, промывая органическую фазу разбавленным раствором гидроксида натрия и раствором NaCl. Сушат над сульфатом магния и растворитель выпаривают. Продукт хроматографируют на диоксиде кремния, элюируя с помощью ДХМ, содержащего метанол (в виде градиента вплоть до 10%). Получают 11,6 г целевого соединения.
Г) N-Метил-5-(4-ацетамидо-4-фенилпиперид-1-ил)-2- (3,4-дихлорфенил)-пентиламин-дихлоргидрат
11 г полученного в предыдущей стадии соединения растворяют в 50 мл метанола. Добавляют 20 мл концентрированной HCl и перемешивают в течение 1 часа. Смесь концентрируют досуха, обрабатывают минимальным количеством метанола и органическую фазу выливают в эфир. Отфильтровывают и высушивают, получая 11,5 г целевого продукта. Т.пл. = 170oC.
Д) N-Метил-N-/2-(3,4-дихлорфенил)-5-(4-ацетамидо-4- фенилпиперид-1-ил)пентил/фенилацетамид-хлоргидрат
К 1 г полученного в предыдущей стадии дихлоргидрата, растворенному в 20 мл ДХМ, добавляют 280 мг фенилуксусной кислоты, затем 0,92 мл ТЭА и 1 г BOP. После перемешивания в течение 15 минут при комнатной температуре, смесь концентрируют в вакууме, остаток обрабатывают этилацетатом и органическую фазу промывают последовательно водой, разбавленным раствором гидроксида натрия, раствором NaCl. Органическую фазу сушат над сульфатом магния, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на силикагеле, элюируя ДХМ с градиентом метанола (3% ---> 6%). После растворения в ДХМ и добавления эфира образуется хлоргидрат. Растворитель выпаривают и хлоргидрат конкретизируют (отверждают) в эфире. Получают 480 мг целевого продукта. Т. пл. = 137oC.
Также получены и описаны в нижеприведенной таблице 1 другие соединения согласно изобретению, принадлежащие к семейству соединений формулы (II).
Пример 15. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4- пивалоиламино-4-фенилпиперид-1-ил)пропил/пиперидин-хлоргидрат; (+)-изомер
А) 3-(3,4-Дихлорфенил)-3-(3-гидроксипропил)пиперидин- хлоргидрат, (+)-изомер
10 г соединения, полученного в приготовлении 2.3, растворяют в 20 мл воды, добавляют 5 мл 40%-ного раствора гидроксида натрия, экстрагируют 3 раза с помощью 50 мл ДХМ, органическую фазу сушат над сульфатом магния и выпаривают, получая 9 г масла. 2,7 г полученного масла растворяют в 50 мл изопропанола, добавляют 2,36 г (+)-изомера 10-камфорсульфокислоты, при нагревании, и оставляют охлаждаться. Образовавшиеся кристаллы (3,86 г) растворяют в 10%-ном растворе NaOH, экстрагируют хлороформом, хлороформный экстракт сушат над сульфатом магния и выпаривают. Получают 2,3 г продукта в виде масла, из которого получают хлоргидрат. Измеряют вращательную способность хлоргидрата. [α]
Вторая кристаллизация, осуществляемая из 2,12 г полученного масла и 1,84 г камфорсульфокислоты ((+)-изомер), дает 3,27 г кристаллов, которые, после подщелачивания с помощью раствора гидроксида натрия и экстракции, дают 2,10 г целевого продукта в виде масла, из которого получают хлоргидрат. [α]
После третьей кристаллизации получают ту же самую вращательную способность. Хиральная чистота, определенная с помощью ВЭЖХ с хиральной фазой, выше 98%.
Б) N-Boc-3-(3,4-дихлорфенил)-3-(3-гидроксипропил)- пиперидин, (+)-изомер
900 мг полученного в предыдущей стадии соединения, 600 мг ТЭА и 610 мг (Boc)2O растворяют в 100 мл ДХМ и оставляют реагировать, в атмосфере азота и при комнатной температуре, в течение 30 минут. Выпаривают, остаток растворяют в этилацетате, полученный раствор промывают буфером с pH 2, разбавленным раствором гидроксида натрия, раствором NaCl, затем сушат над сульфатом магния и выпаривают, получая целевой продукт в виде масла (1,1 г).
В) N-Boc-3-(3,4-дихлорфенил)-3-(3-мезилоксипропил) пиперидин, (+)-изомер
1,1 г полученного в предыдущей стадии соединения растворяют в 10 мл ДХМ, в атмосфере азота и при температуре 0oC, затем добавляют 700 мг ТЭА и 325 мг мезилхлорида, растворенных в 3 мл ДХМ. После перемешивания в течение 2-х часов, реакционную смесь выпаривают, остаточное масло обрабатывают этилацетатом, органическую фазу промывают буферным раствором с pH 2, водой, раствором NaCl, затем сушат и выпаривают. Получают 1,4 г целевого продукта в виде масла.
Г) N-Boc-3-(3,4-дихлорфенил)-3-/3-(4-пивалоиламино-4- фенилпиперид-1-ил)пропил/пиперидин, (+)-изомер
1,4 г полученного в предыдущей стадии соединения и 900 мг 4-фенил-4-пивалоиламинопиперидина, полученного в приготовлении 1,1, растворяют в 25 мл ацетонитрила. Добавляют 450 мг K2CO3 и перемешивают в течение 2-х часов при 60oC. Реакционную смесь выпаривают, остаток растворяют в этилацетате, этилацетатную фазу промывают буферным раствором с pH 2, разбавленным раствором NaOH, раствором NaCl, сушат над сульфатом магния и выпаривают, получая 1,65 г целевого продукта в виде масла.
Д) 3-(3,4-Дихлорфенил)-3-/3-(4-пивалоиламино-4-фенилпиперид-1- ил)-пропил/пиперидин-хлоргидрат, (+)-изомер
К 25 мл ДХМ добавляют 1,65 г полученного в предыдущей стадии соединения и 5 мл 4 н раствора HCl в эфире. После перемешивания в течение 1 часа, получают 1,12 г целевого продукта.
Е) 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4-пивалоиламино-4- фенил-пиперид-1-ил)пропил/пиперидин-хлоргидрат, (+)-изомер
1,10 г полученного в предыдущей стадии соединения, 800 мг ТЭА и 252 мг бензоилхлорида, в атмосфере азота и при 0oC, растворяют в 15 мл ДХМ. Спустя 15 минут, выпаривают, остаток растворяют в этилацетате, этилацетатную фазу промывают разбавленным раствором HCl, раствором NaCl, сушат над сульфатом магния и выпаривают. Полученную пену хроматографируют на диоксиде кремния, элюируя смесью ДХМ с метанолом при градиенте вплоть до 5%. Полученный продукт превращают в соль с помощью раствора хлороводорода в эфире. [α]
Пример 16. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4- пивалоиламино-4-фенил-пиперид-1-ил)-пропил/пиперидин-хлоргидрат, (-)-изомер
Это соединение получают, как в примере 15, используя камфорсульфокислоту, (-)-изомер, из стадии А).
Пример 17. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4- пивалоиламино-4-фенилпиперид-1-ил)пропил/пиперидин-хлоргидрат, рацемат
Это соединение получают, как в примере 15, без осуществления разделения на оптические антиподы согласно стадии А).
Пример 18. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4-(ацетил-N- метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат-моно- гидрат
А) N-Boc-3-(3,4-дихлорфенил)-3-(3-гидроксипропил)- пиперидин
Выдерживают в течение 1 часа при перемешивании, при комнатной температуре и в атмосфере азота, смесь 23 г соединения, полученного в приготовлении 2.3, 15 г триэтиламина, 16 г (Boc)2O в 100 мл ДХМ. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, этилацетатную фазу промывают буферным раствором с pH 2, 5%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 30 г целевого продукта в виде масла.
Г) N-Boc-3-(3,4-дихлорфенил)-3-(3-мезилоксипропил)- пиперидин
Охлаждают до 0oC раствор 30 г полученного в предыдущей стадии соединения, 15 мл триэтиламина в 200 мл ДХМ и, в атмосфере азота, прикапывают раствор 9 г мезилхлорида в 50 мл ДХМ. Перемешивают в течение 2-х часов и растворитель выпаривают в вакууме. Остаток экстрагируют этилацетатом, этилацетатную фазу промывают буферным раствором с pH 2, 5%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 34 г целевого продукта.
В) N-Boc-3-(3,4-дихлорфенил)-3-/3-/4-(ацетил-N- метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин
Нагревают при 60oC в течение 3-х часов смесь 1 г полученного в предыдущей стадии соединения, 2 г полученного в приготовлении 1.3 соединения, 0,6 г K2CO3 в 15 мл ДМФ, затем оставляют на ночь при перемешивании при комнатной температуре. К реакционной смеси добавляют этилацетат, органическую фазу промывают водой, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (95/5 по объему). Получают 0,78 г целевого продукта.
Г) 3-(3,4-Дихлорфенил)-3-/3-/4-(ацетил-N-метиламино)- 4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат
К раствору 0,78 г полученного в предыдущей стадии соединения в 5 мл ДХМ добавляют 2 мл насыщенного раствора HCl в эфире и перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь выпаривают в вакууме и получают 0,8 г целевого продукта, который используют таким, какой есть, в следующей стадии.
Д) 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-4-(ацетил- N-метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- моногидрат
Смесь 0,75 г полученного в предыдущей стадии соединения и 0,6 мл триэтиламина в 10 мл ДХМ охлаждают до 0oC и, в атмосфере азота, добавляют 0,18 г бензоилхлорида. Реакционную смесь перемешивают в течение 2-х часов при ОoC и концентрируют в вакууме. Остаток экстрагируют этилацетатом, этилацетатную фазу промывают буферным раствором с pH 2, 5%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (95/5 по объему). Полученный продукт обрабатывают с помощью ДХМ, полученную органическую фазу подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,5 г целевого хлоргидрата. Т.пл. = 171oC.
Пример 19. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-(ацетил-N- метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- моногидрат, (+)-изомер
А) 1-Бензоил-3-(3,4-дихлорфенил)-3-(3-гидроксипропил) пиперидин, (+)-изомер
Раствор 7,45 г полученного в стадии А) примера 15 (в
форме основания) соединения в 30 мл ДХМ охлаждают до -20oC и добавляют 4 мл триэтиламина, затем прикапывают 2,85 мл бензоилхлорида. Оставляют при перемешивании при повышении температуры до комнатной, затем реакционную смесь промывают 0,5 н раствором HCl, насыщенным раствором карбоната натрия, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя градиентом смеси ДХМ/этилацетата (90/10 по объему) вплоть до (80/20 по объему), затем смесью ДХМ/метанол (97/3 по объему). Получают 9,40 г целевого продукта.
Б) 1-Бензоил-3-(3,4-дихлорфенил)-3-(3-мезилоксипропил)- пиперидин, (+)-изомер
Раствор 9,4 г полученного в предыдущей стадии соединения и 5 мл триэтиламина в 50 мл ДХМ охлаждают до -10oC и прикапывают 2,24 мл мезилхлорида. Оставляют реакционную смесь при перемешивании при повышении температуры до комнатной и растворитель удаляют в вакууме. Остаток экстрагируют этилацетатом, этилацетатную фазу промывают два раза водой, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 10 г целевого продукта.
В) 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-(ацетил-N- метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- моногидрат, (+)-изомер
В течение 2-х часов, в атмосфере азота, нагревают при 100oC смесь 2 г полученного в предыдущей стадии соединения, 4 г 4-(ацетил-N-метиламино)-4-фенилпиперидин-п- толуолсульфоната, 1 г карбоната калия и 50 мл ацетонитрила и 50 мл ДМФ. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, этилацетатную фазу промывают водой, затем 0,5 н раствором HCl, 10%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол от (99/1 по объему) вплоть до (95/5 по объему). Полученный продукт обрабатывают ДХМ, полученный раствор подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 1,05 г целевого продукта. [α]
Пример 20. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-(ацетил-N- метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- моногидрат, (-)-изомер
А) 3-(3,4-Дихлорфенил)-3-(3-гидроксипропил)пиперидин- хлоргидрат, (-)-изомер
К раствору 4,7 г полученного в приготовлении 2.3 соединения, в форме свободного основания, в 100 мл изопропанола добавляют 3,8 г 10-камфорсульфокислоты, (-)-изомер, и все кипятят с обратным холодильником. После охлаждения, кристаллизации и отсасывания образовавшихся кристаллов (4,90 г), эти последние растворяют в 10%-ном растворе NaOH, экстрагируют хлороформом, хлороформный экстракт сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 2,7 г продукта в виде масла, из которого получают хлоргидрат. Измеряют вращательную способность хлоргидрата. [α]
Осуществляют вторую кристаллизацию, исходя из 2,6 г полученного масла, 2,77 г 10-камфорсульфокислот, (-)-изомер, и 40 мл изопропанола. После подщелачивания с помощью раствора гидроксида натрия, экстракции хлороформом, высушивания хлороформного экстракта над сульфатом магния и выпаривания, получают целевой продукт в виде масла, из которого получают хлоргидрат. Получают 2,4 г целевого продукта. [α]
Б) 1-Бензоил-3-(3,4-дихлорфенил)-3-(3-гидроксипропил)- пиперидин, (-)-изомер
Раствор 11 г полученного в предыдущей стадии соединения (в форме свободного основания) и 6 мл триэтиламина в 75 мл ДХМ охлаждают до -30oC и прикапывают к нему 4,2 мл бензоилхлорида. Оставляют при перемешивании при повышении температуры до комнатной, затем реакционную смесь выливают в воду. Экстрагируют с помощью ДХМ, органическую фазу промывают водой, 0,5 н раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя градиентом смеси ДХМ/этилацетат от (85/15 по объему) вплоть до (75/25 по объему), затем смесью ДХМ/метанол (97/3 по объему). Получают 10 г целевого продукта.
В) 1-Бензоил-3-(3,4-дихлорфенил)-3-(3-метоксипропил)- пиперидин, (-)-изомер
Раствор 10 г полученного в предыдущей стадии соединения и 5,5 мл триэтиламина в 100 мл ДХМ охлаждают до -30oC и прикапывают к нему 2,4 мл мезилхлорида. Оставляют стоять при перемешивании при повышении температуры до комнатной и концентрируют в вакууме. Остаток экстрагируют этилацетатом, этилацетатную фазу промывают 2 раза водой, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Получают 11 г целевого продукта.
Г) 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4- (ацетил-N-метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-моногидрат, (-)-изомер
Это соединение получают по методике, описанной в стадии В) примера 19, исходя из 0,5 г полученного в предыдущей стадии соединения и 0,8 г 4-(ацетил-N-метиламино)-4-фенилпиперидин-п- толуолсульфоната. Получают 0,375 г целевого продукта. [α]
Согласно методике, подобной таковой, которая описана в примере 15, получают соединения согласно изобретению, описанные в нижеприведенной таблице 2.
(1) Получение этих соединений описано в европейской заявке на патент EP-512901, в примерах 27 и 29.
(2) Дихлоргидрат
Пример 42. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4-фенил- (1,2,5,6-тетрагидропирид-1-ил))пропил/пиперидин-хлоргидрат
Это соединение получают по методике, описанной выше, исходя из 4-фенил-(1,2,5,6-тетрагидропиридина), который имеется в продаже.
Пример 43. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4-циано-4- фенилпиперид-1-ил)пропил/пиперидин-хлоргидрат
А) 1-Бензоил-3-(3,4-дихлорфенил)-3-(3-гидроксипропил)- пиперидин
Раствор 16,22 г полученного в приготовлении 2,3 соединения и 18,2 г триэтиламина в 250 мл ДХМ охлаждают на ледяной бане и к нему прикапывают 14,06 г бензоилхлорида в 10 мл ДХМ. Перемешивают в течение 1 часа при повышении температуры до комнатной. Избыток бензоилхлорида удаляют путем добавления метанола, затем реакционную смесь концентрируют в вакууме. Остаток обрабатывают метанолом и растворитель выпаривают в вакууме. Остаток экстрагируют эфиром, эфирную фазу промывают водой, 2 н раствором HCl, 5%-ным раствором NaHCO3, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Таким образом, промежуточно полученный 1-бензоил-3-(3,4-дихлорфенил)-3- (3-бензоилоксипропил)пиперидин растворяют в 150 мл метанола, добавляют 10%-ный раствор NaOH, нагревают в течение 1 часа при 50-60oC и концентрируют в вакууме. Остаток обрабатывают эфиром, эфирную фазу промывают водой, 2 н раствором HCl, 5%-ным раствором NaHCO3, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 18 г целевого продукта в виде масла.
Б) 1-Бензоил-3-(3,4-дихлорфенил)-3-(3-мезилоксипропил)- пиперидин
Раствор 16,8 г полученного в предыдущей стадии соединения и 5,18 г триэтиламина в 100 мл ДХМ охлаждают на ледяной бане и прикапывают раствор 5,40 г мезилхлорида в 10 мл ДХМ, затем оставляют при перемешивании в течение 30 минут, причем температура повышается до комнатной. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, этилацетатную фазу промывают водой, 2 н раствором HCl, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 19,6 г целевого продукта в виде масла. ЯМР-спектр (200 МГц, в ДМСО): м.д.: 1-2,35 (м, 8H), 3,15 (с, 3H), 3,2-4,6 (м, 6H), 6,8-7,8 (м, 8H).
В) 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4-циано-4-фенилпиперид-1-ил)-пропил/пиперидин-хлоргидрат
Кипятят с обратным холодильником в течение 2-х часов смесь 5,9 г соединения, полученного в предыдущей стадии, 3 г 4-циано-4-фенил-пиперидина, 6,9 г карбоната калия, в 20 мл ацетонитрила и 5 мл ДМФ. После охлаждения, реакционную смесь выливают в воду, экстрагируют эфиром, эфирную фазу промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол от (110/1 по объему) вплоть до (100/2 по объему). Полученный продукт обрабатывают с помощью ДХМ, полученный раствор подкисляют вплоть до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 4,5 г целевого хлоргидрата, после кристаллизации из смеси ДХМ с эфиром. Т. пл. = 239-241oC.
Пример 44. 3-/3-/4-(Аминометил)-4-фенилпиперид-1-ил/-пропил/- 1-бензоил-3-(3,4-дихлорфенил)-пиперидин-дихлоргидрат-дигидрат
При комнатной температуре и атмосферном давлении, гидрируют смесь 3,5 г соединения, полученного в примере 43, 10 мл концентрированного раствора NH4OH, 0,5 г никеля Ренея и 50 мл этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Остаток экстрагируют с помощью ДХМ, органическую фазу промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток обрабатывают с помощью ДХМ, полученный раствор подкисляют до pH 1 с помощью добавки солянокислого эфира и выпаривают в вакууме. Получают 2,8 г целевого продукта, после кристаллизации из смеси ДХМ/эфир. Т.пл. = 174oC.
Пример 45. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-/(N- этоксикарбонил-амино)метил/-4-фенилпиперидин-1- ил/пропил/пиперидин-хлоргидрат-полугидрат
Раствор 1 г соединения, полученного в примере 44, 0,535 г триэтиламина в 20 мл ДХМ охлаждают до 0oC и к нему добавляют 0,188 г этилхлорформиата. Перемешивают в течение 30 минут и концентрируют в вакууме. Остаток экстрагируют эфиром, эфирную фазу промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ с метанолом от (100/3 по объему) вплоть до (100:5 по объему). Полученный продукт растворяют в ДХМ, полученный раствор подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,4 г целевого продукта, после кристаллизации из смеси ДХМ/эфир. Т.пл. = 135-141oC (разложение).
Пример 46. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-/(N- пропиониламино)-метил/-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-моногидрат
К раствору 0,87 г соединения, полученного в примере 44, 0,3 г триэтиламина в 20 мл ДХМ добавляют 0,166 г пропионилхлорида и перемешивают в течение 30 минут при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, этилацетатную фазу промывают водой, сушат над сульфатом магния и выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол от (100/3 по объему) вплоть до (100/5 по объему). Остаток растворяют в ДХМ, подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,49 г целевого продукта, после кристаллизации из смеси ДХМ/эфир. Т. пл. = 143-149oC (разложение)
Пример 47. 3-/3-/4-/(Ацетил-N-метиламино)метил/-4- фенилпиперид-1-ил/-пропил/-1-бензоил-3-(3,4-дихлорфенил)пиперидин- хлоргидрат-моногидрат
Смесь 1,6 г полученного в стадии Б) примера 43, 2 г 4-/(ацетил-N-метиламино)метил/-4-фенилпиперидин-п-толуол- сульфоната, 2 г карбоната калия и 4 мл ДМФ нагревают при 100oC в течение 2-х часов. После охлаждения, реакционную смесь выливают в воду, экстрагируют эфиром, эфирный экстракт промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (100/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,75 г целевого хлоргидрата, после кристаллизации из смеси ДХМ/эфир. Т.пл. = 126oC.
Пример 48. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-/(N'-этил- N-метил-уреидо)метил/-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-полугидрат
Смесь 1,7 г соединения, полученного в стадии Б) примера 43, 1,2 г 4-/(N'-этил-N-метилуреидо)метил/-4- фенилпиперидина, 1 г карбоната калия и 5 мл ДМФ нагревают при 100oC в течение 2-х часов. После охлаждения, реакционную смесь выливают в воду, экстрагируют смесью эфира с этилацетатом, органическую фазу промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол от (100/3 по объему) до (100/7 по объему). Полученный продукт растворяют в ДХМ, подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 1,15 г целевого хлоргидрата, после кристаллизации из смеси ДХМ/эфир. Т.пл. = 160oC.
Пример 49. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-/(N',N'- диэтил-N-метилуреидо)метил/-4-фенил-пиперид-1- ил/пропил/пиперидин-хлоргидрат-полугидрат
Это соединение получают по методике, описанной в примере 48, исходя из 2 г полученного в стадии Б) примера 43 соединения, 2,85 г 4-/(N',N'-диэтил-N-метилуреидо)метил/-4-фенилпиперидин-п- толуолсульфоната, 2,5 г карбоната калия и 4 мл ДМФ. Получают 0,9 г целевого хлоргидрата, после кристаллизации из смеси ДХМ/эфир. Т.пл. = 117-132oC (разложение).
Пример 50. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-(пиперид-1- ил)-4-фенилпиперид-1-ил/пропил/пиперидин-дихлоргидрат-дигидрат
1,7 г 4-Фенил-4-(пиперид-1-ил)пиперидин-дихлоргидрата в виде дигидрата добавляют к 40%-ному раствору NaOH, экстрагируют с помощью ДХМ, раствор в ДХМ сушат над сульфатом магния и растворитель выпаривают досуха. Остаток растворяют в 20 мл ДМФ, добавляют 1,25 г соединения, полученного в стадии Б) примера 43 и нагревают при 100oC в течение 2-х часов. После охлаждения, реакционную смесь разбавляют с помощью ДХМ, органическую фазу промывают водой, 1 н раствором HCl, 5%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с метанолом (95/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют до pH 1 путем добавления солянокислого эфира и выпаривают в вакууме. Получают 1,2 г целевого продукта. Т.пл.= 192oC.
Пример 51. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4- (формиламино)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- моногидрат
0,55 г 4-(Формиламино)-4-фенилпиперидин-хлоргидрата растворяют в воде, подщелачивают путем добавления концентрированного раствора NaOH, экстрагируют с помощью ДХМ, экстракт сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток обрабатывают с помощью 20 мл ацетонитрила, добавляют 0,9 г полученного в стадии Б) примера 43 соединения, 1 г карбоната калия и кипятят с обратным холодильником в течение 2,5 часов. Концентрируют в вакууме, остаток экстрагируют этилацетатом, этилацетатную фазу промывают водой, 5%-ным раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя с помощью ДХМ, затем с помощью смеси ДХМ/метанол (90/10 по объему). Полученный продукт растворяют в ДХМ, подкисляют путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,53 г целевого продукта, после кристаллизации из изо-эфира. ЯМР-спектр (200 МГц, в ДМСО) δ, м.д.: 1,1-2,65 (м., 12H), 2,7-4,5 (м, 10H), 7,0-7,7 (м, 13H), 8,0 (с, 1H), 8,4 (с, 1H), 10,5 (с. ш, 1H).
Пример 52. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4- (циклопропилкарбонил-амино)-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-моногидрат
0,370 г 4-(Циклопропилкарбониламино)-4- фенилпиперидин-хлоргидрата растворяют в воде, подщелачивают путем добавления концентрированного раствора NaOH, экстрагируют с помощью ДХМ, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток растворяют в 10 мл ДМФ, добавляют 0,655 г соединения, полученного в стадии Б) примера 43, 0,192 г карбоната калия и смесь нагревают при 80oC в течение 30 минут. После перемешивания в течение ночи при комнатной температуре, реакционную смесь выливают в воду, экстрагируют этилацетатом, этилацетатную фазу промывают два раза водой, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя дихлорметаном, затем смесью ДХМ/метанол (95/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют раствор до pH 1 путем добавления солянокислото эфира и выпаривают в вакууме. Получают 0,27 г целевого продукта. ЯМР-спектр (200 МГц, в ДМСО) δ, м. д.: 0,6 (мт, 4H), 1,0-2,7 (м, 13H), 2,75-4,5 (м, 10H), 7,0-7,9 (м, 13H), 8,4 (с, 1H), 10 (сш, 1H).
Пример 53. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-(4- метоксикарбонил-4-фенилпиперид-1-ил)пропил/-пиперидин-хлоргидрат- полугидрат
В течение 3-х часов кипятят с обратным холодильником смесь 0,9 г 4-метоксикарбонил-4-фенилпиперидин-п-толуолсульфоната, 0,91 г соединения, полученного в стадии Б) примера 43, 1,06 г карбоната калия, в 5 мл ДМФ и 5 мл ацетонитрила. Реакционную смесь выливают в воду, экстрагируют этилацетатом, этилацетатную фазу промывают два раза водой, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя с помощью ДХМ, затем смеси ДХМ с метанолом (97/3 по объему). Полученный продукт растворяют в этилацетате, подкисляют раствор путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,54 г целевого продукта, после кристаллизации из смеси ацетона с эфиром. Т.пл. 123-125oC.
Пример 54. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-(N, N- диэтилкарбамоил)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- моногидрат
Это соединение получают по методике, описанной в примере 53, исходя из 1,7 г 4-(N,N-диэтилкарбамоил)-4- фенилпиперидин-трифторацетата, 1,77 г полученного в стадии Б) примера 43 соединения, 2,08 г карбоната калия, 5 мл ДМФ и 5 мл ацетонитрила. Получают 1,1 г целевого продукта. Т.пл. = 124-126oC.
Пример 55. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-(N-метокси- N-метилкарбамоил)-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-полугидрат
Это соединение получают по методике, описанной в примере 53, исходя из 1,1 г 4-(N-метокси-N- метилкарбамоил)-4-фенилпиперидина, 1,73 г соединения, полученного в стадии Б) примера 43, 1,52 г карбоната калия, 5 мл ДМФ и 5 мл ацетонитрила. Получают 0,99 г целевого продукта. Т.пл. = 155-157oC.
Пример 56. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4- (метилсульфонамидо)-4-фенилпиперид-1-ил)-/пропил/пиперидин- хлоргидрат-полугидрат
1,6 г 4-(метилсульфонамидо)-4-фенилпиперидин-хлоргидрата растворяют в минимальном количестве 40%-ного раствора гидроксида натрия, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток растворяют в 25 мл ДМФ, добавляют 1,29 г соединения, полученного в стадии Б) примера 43, и нагревают при 80oC в течение 2-х часов в атмосфере азота. Реакционную смесь выливают в воду, отсасывают образовавшийся осадок и промывают его водой. Осадок растворяют в ДХМ, органическую фазу промывают два раза водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя с помощью ДХМ, затем смеси ДХМ/метанол (95/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,7 г целевого продукта. Т. пл. = 210oC.
Пример 57. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4- (метансульфонил-N-метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-полугидрат
В течение 3-х часов кипятят с обратным холодильником смесь 0,79 г 4-(метансульфонил-N-метиламино)-4- фенилпиперидин-п-толуолсульфоната, 0,70 г полученного в стадии Б) примера 43 соединения, 0,80 г карбоната калия и 10 мл ацетонитрила. Концентрируют в вакууме, остаток обрабатывают водой, экстрагируют этилацетатом, этилацетатную фазу промывают 5%-ным раствором NaOH, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя с помощью ДХМ, затем смеси ДХМ с метанолом (95/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют раствор путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,71 г целевого продукта, после кристаллизации из эфира. ЯМР-спектр (200 МГц, в ДМСО) δ, м.д.: 1,0-2,45 (м, 15H), 2,45-4,50 (м, 13H), 7,0-7,9 (м, 13H), 10,75 (с, 1H).
Пример 58. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4- (пропионилокси)-4-фенилпиперид-1-ил/пропил/пиперидин-хлоргидрат- полугидрат
Раствор 0,95 г полученного в примере 39 соединения, 0,4 мл триэтиламина в 50 мл ДХМ охлаждают до 0oC, прикапывают к нему 0,15 мл пропионилхлорида и перемешивают при повышении температуры до комнатной. Реакционную смесь концентрируют в вакууме и остаток хроматографируют на диоксиде кремния, элюируя градиентом смеси ДХМ/метанол от (99/1 по объему) вплоть до (97/3 по объему). Полученный продукт растворяют в ДХМ, подкисляют путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,5 г целевого продукта. ЯМР-спектр (200 МГц, в ДМСО) δ, м.д.: 1,0 (т, 3H), 1,15-2,0 (м, 6H), 2,0-2,8 (м, 8H), 2,9-4,6 (м, 10H), 7,1-7,9 (м, 13H), 10,2 (сш, 1H).
Пример 59. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-спиро (фталид-3)пиперид-1-ил)пропил/пиперидин-хлоргидрат-моногидрат
Смесь 1,4 г 4-спиро(фталид-3)пиперидин-хлоргидрата, 0,715 г трет-бутилата калия и 15 мл ДМФ перемешивают в течение 5 минут. Затем добавляют 2,5 г полученного в стадии Б) примера 43 соединения, 2 г карбоната калия, нагревают при 100oC в течение 45 минут и оставляют на ночь при перемешивании и при комнатной температуре. Реакционную смесь разбавляют путем добавления этилацетата, органическую фазу промывают буферным раствором с pH 2, 5%-ным раствором NaOH, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол от (99/1 по объему) до (95/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют путем добавления солянокислого эфира и выпаривают в вакууме. Получают 0,77 г целевого продукта. Т.пл. = 165oC.
Согласно методикам, описанным в вышеприведенных примерах, получают соединения согласно изобретению, описанные в нижеприведенной таблице 3.
Примечания к таблице 3:
(а) это соединение получают по методике, описанной в примере 47,
(б) это соединение получают по методике, описанной в примере 52,
(с) это соединение получают по методике, описанной в примере 43, стадия В),
(d) это соединение получают по методике, описанной в примере 53.
ЯМР-спектр (200 МГц, в ДМСО) δ, м.д., соединения примера 60: 0,85 (тд, 3H), 1,0-2,5 (м, 12H), 2,5-4,5 (м, 12H), 7,0-7,9 (м, 13H), 8,3-8,6 (сд, 1H), 9,2-11,0 (сд, 1H).
ЯМР-спектр (200 МГц, в ДМСО) δ, м.д., соединения примера 61: 0,7-2,3 (м, 13H), 2,6-4,6 (м, 15H), 7,0-7,9 (с, 13H), 10,3 (сш, 1H),
ЯМР-спектр (200 МГц, в ДМСО) δ, м.д., соединения примера 62: 1,4-3,1 (м, 19H), 3,15-4,9 (м, 10H), 7,1-7,8 (м, 13H), 7,95 (с, 1H), 10,0 (сш, 1H).
ЯМР-спектр (200 МГц, в ДМСО) δ, м.д., соединения примера 63: 0,8-2,7 (м, 23H), 2,75-4,6 (м, 10H), 7,0-8,0 (м, 13H), 8,0 (с, 1H), 10,2 (сш, 1H).
Пример 76. 1-Бензоил-3-(3,4-дихлорфенил)-3-/3-/4-гидрокси-4- (2-метоксифенил)пиперид-1-ил/пропил/пиперидин-хлоргидрат • 1,5 H2O
Смесь 1,80 г 4-гидрокси-4-(2-метоксифенил)-пиперидина, 1,92 г полученного в стадии Б) примера 43 соединения и 10 мл ДМФ нагревают при 80oC в течение 2-х часов. Реакционную смесь выливают в воду, отсасывают образовавшийся осадок и промывают его водой. Осадок растворяют в хлороформе, органическую фазу промывают водой, буферным раствором с pH 2, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя градиентом смеси ДХМ/метанол от (99/1 по объему), до (97/3 по объему). Полученный продукт обрабатывают солянокислым эфиром и отсасывают образовавшийся осадок. Получают 0,7 г целевого продукта. Т. пл. = 180oC.
Пример 77. 1-(4-Йодбензоил)-3-(3,4-дихлорфенил)-3-/3-/4- (ацетил-N-метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-моногидрат, (+)-изомер
А) 1-(4-Йодбензоил)-3-(3,4-дихлорфенил)-3-(3-гидрокси- пропил)пиперидин, (+)-изомер
К смеси 5 г полученного в стадии А) примера 15 соединения, 3,9 г 4-йодбензойной кислоты и 100 мл ДХМ, при комнатной температуре, добавляют 6,5 мл триэтиламина, затем 8,2 г BOP и перемешивают в течение 15 минут при перемешивании и при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают эфиром, органическую фазу промывают водой, 1 н раствором NaOH, водой, 1 н раствором HCl, насыщенным раствором NaCl, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя с помощью ДХМ, затем смеси ДХМ с этилацетатом (50/50 по объему), затем с помощью этилацетата. Получают 8,5 г целевого продукта.
Б) 1-(4-Йодбензоил)-3-(3,4-дихлорфенил)-3-(3-мезил- оксипропил)-пиперидин, (+)-изомер
Раствор 8,5 г полученного в предыдущей стадии соединения, 2,7 мл триэтиламина в 150 мл ДХМ охлаждают до 0oC и прикапывают 1,5 мл мезилхлорида. Перемешивают при повышении температуры до комнатной и концентрируют в вакууме. Остаток экстрагируют эфиром, органическую фазу промывают два раза водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 10 г целевого продукта.
В) 1-(4-Йодбензоил)-3-(3,4-дихлорфенил)-3-/3-/4- (ацетил-N-метиламино)-4-фенилпиперид-1-ил/пропил/пиперидин- хлоргидрат-моногидрат, (+)-изомер
В течение 1 часа нагревают при 80oC смесь 10 г полученного в предыдущей стадии соединения, 8,2 4-(ацетил-N-метиламино)-4-фенилпиперидин-п-толуолсульфоната, 9,3 г карбоната калия и 80 мл ДМФ. Затем добавляют 4 г 4-(ацетил-N- метиламино)-4-фенилпиперидин-п-толуолсульфоната к реакционной смеси и продолжают нагревание при 80oC в течение 2-х часов. После охлаждения реакционную смесь выливают в воду со льдом, отсасывают образовавшийся осадок и промывают его водой. Осадок растворяют в ДХМ, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния H, элюируя с помощью ДХМ, затем с помощью смеси ДХМ/метанол (95/5 по объему). Полученный продукт растворяют в ДХМ, подкисляют солянокислым эфиром до pH 1 и выпаривают в вакууме. Получают 9,2 г целевого продукта, после кристаллизации из эфира. Т.пл. = 172oC (разложение), [α]
Фармацевтические композиции согласно изобретению могут быть представлены в виде желатиновых капсул следующего состава:
Желатиновая капсула с 5 мг активного начала:
активное начало - 5,00 мг
лактоза - 75,00 мг
крахмал кукурузы - 14,00 мг
повидон - 5,00 мг
стеарат магния - 1,00 мг
Для желатиновой капсулы, заполняемой до 100 мг
Желатиновая капсула с 25 мг активного начала:
активное вещество - 25,00 мг
лактоза - 170,00 мг
крахмал кукурузы - 50,00 мг
повидон - 6,00 мг
стеарат магния - 1,00 мг
Для желатиновой капсулы, заполняемой до 252 мг
Аналогично можно выпускать желатиновые капсулы с содержанием 50, 100 и 250 мг активного вещества, используя соответствующие количества упомянутых эксципиентов.
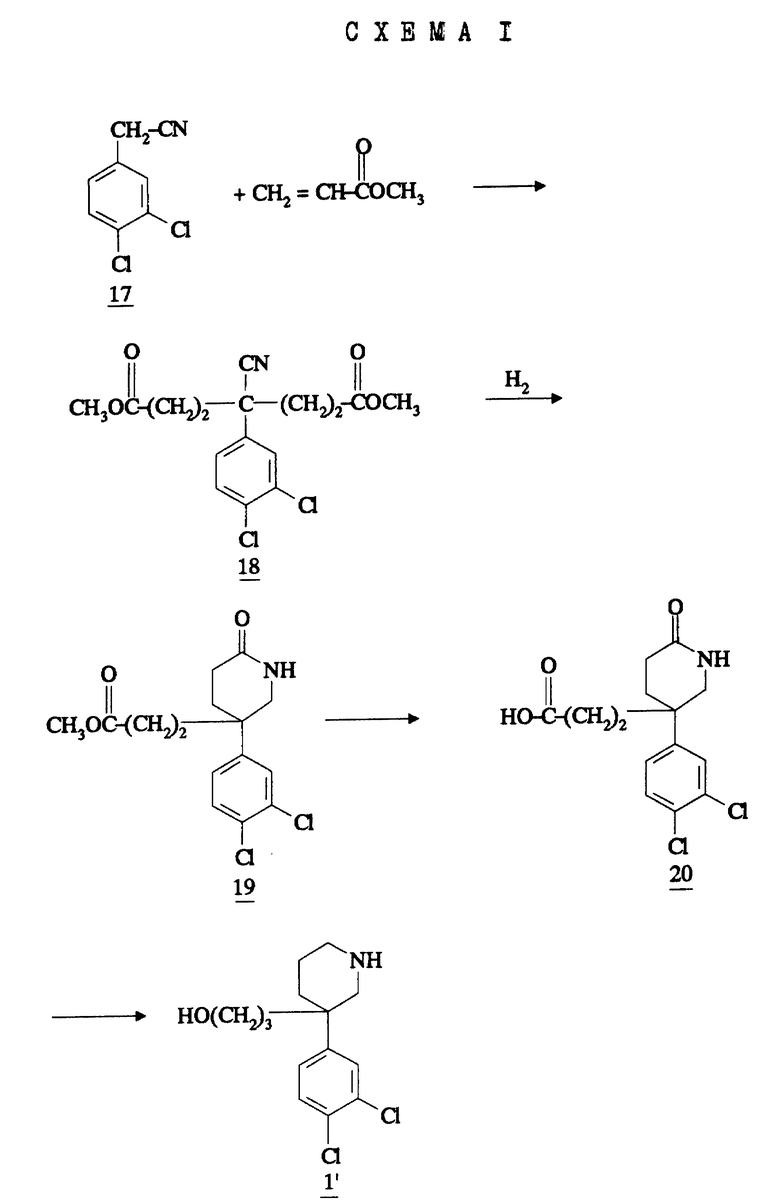
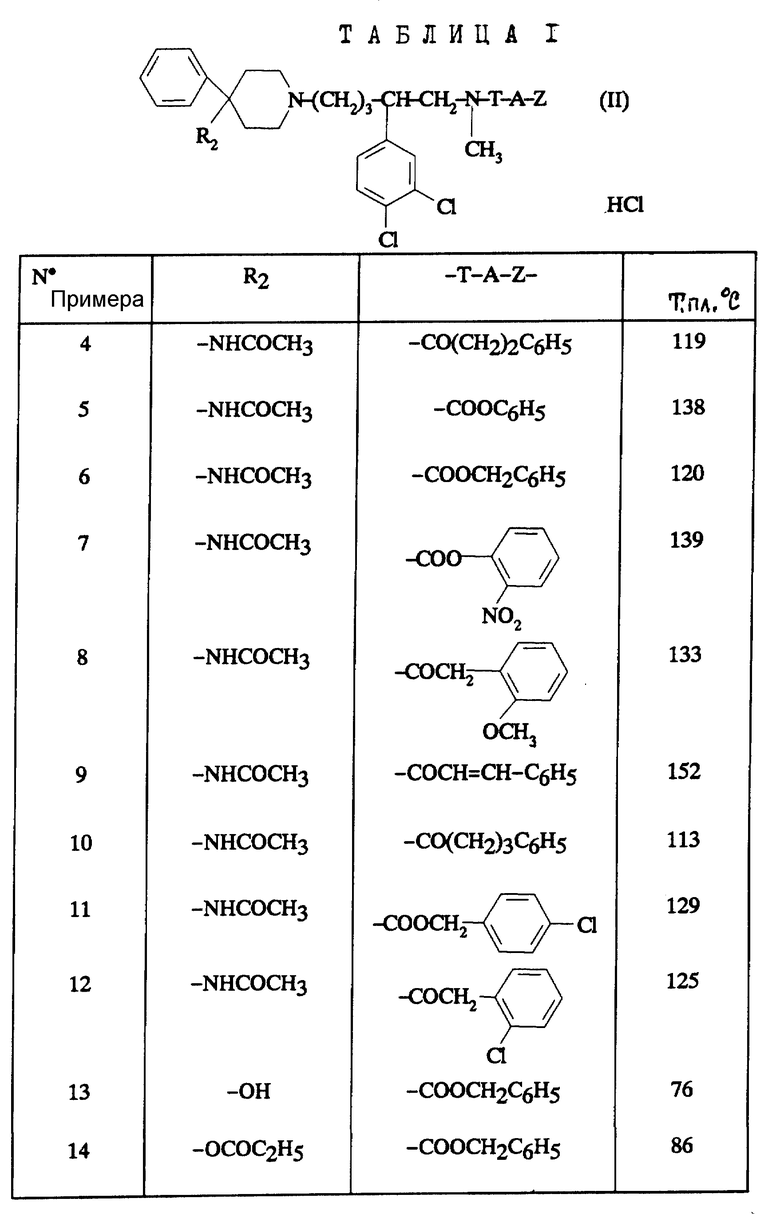
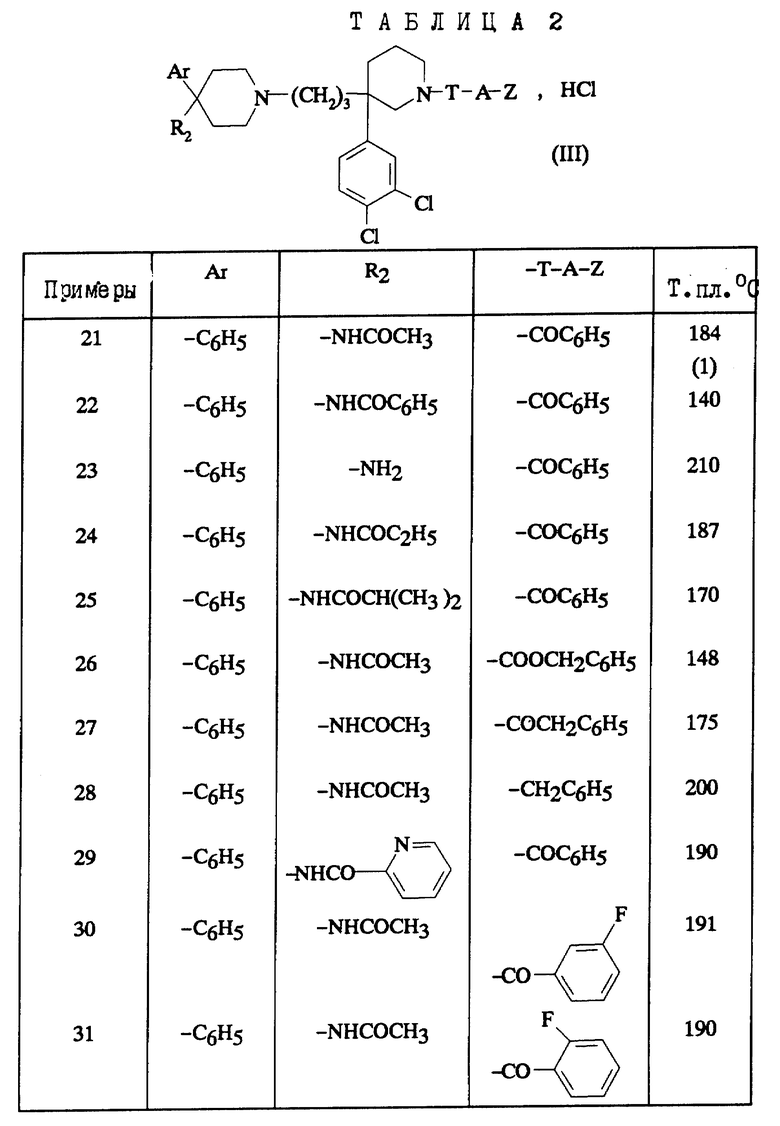
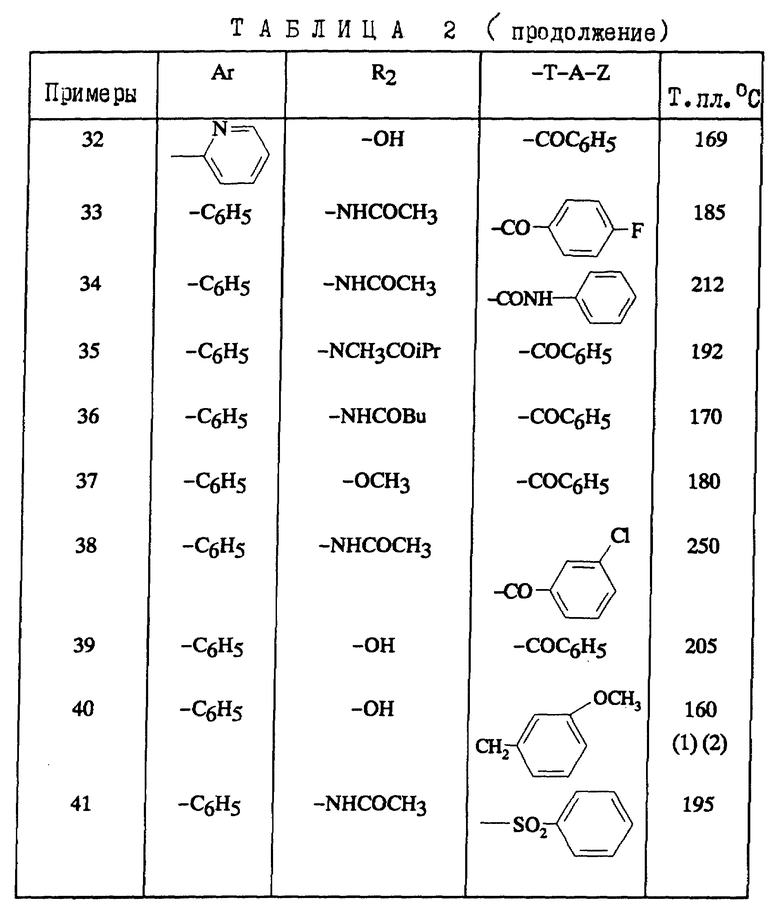
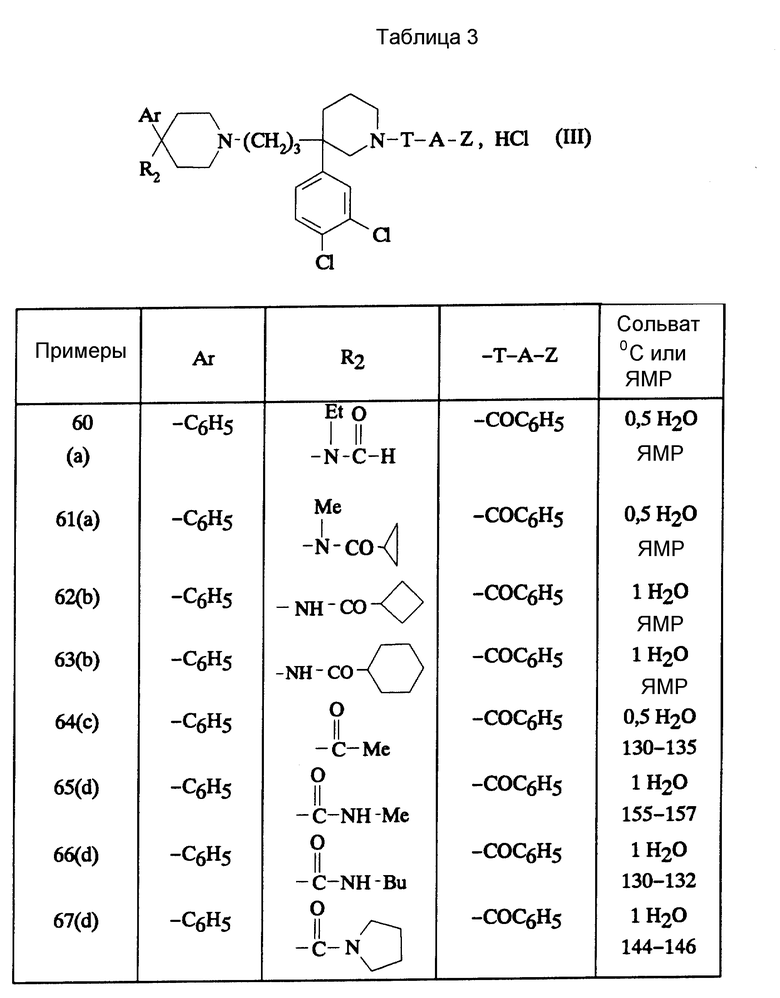
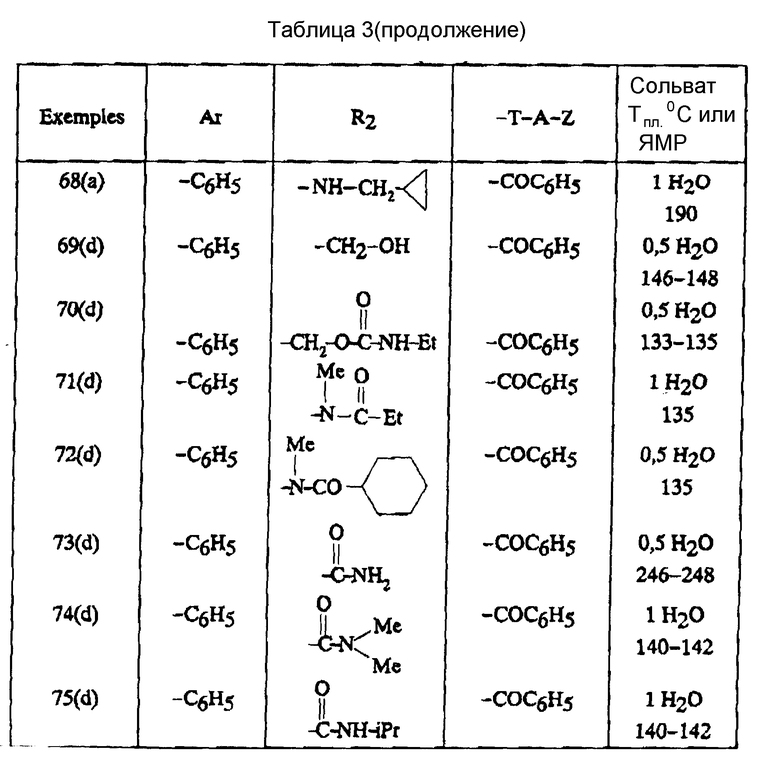
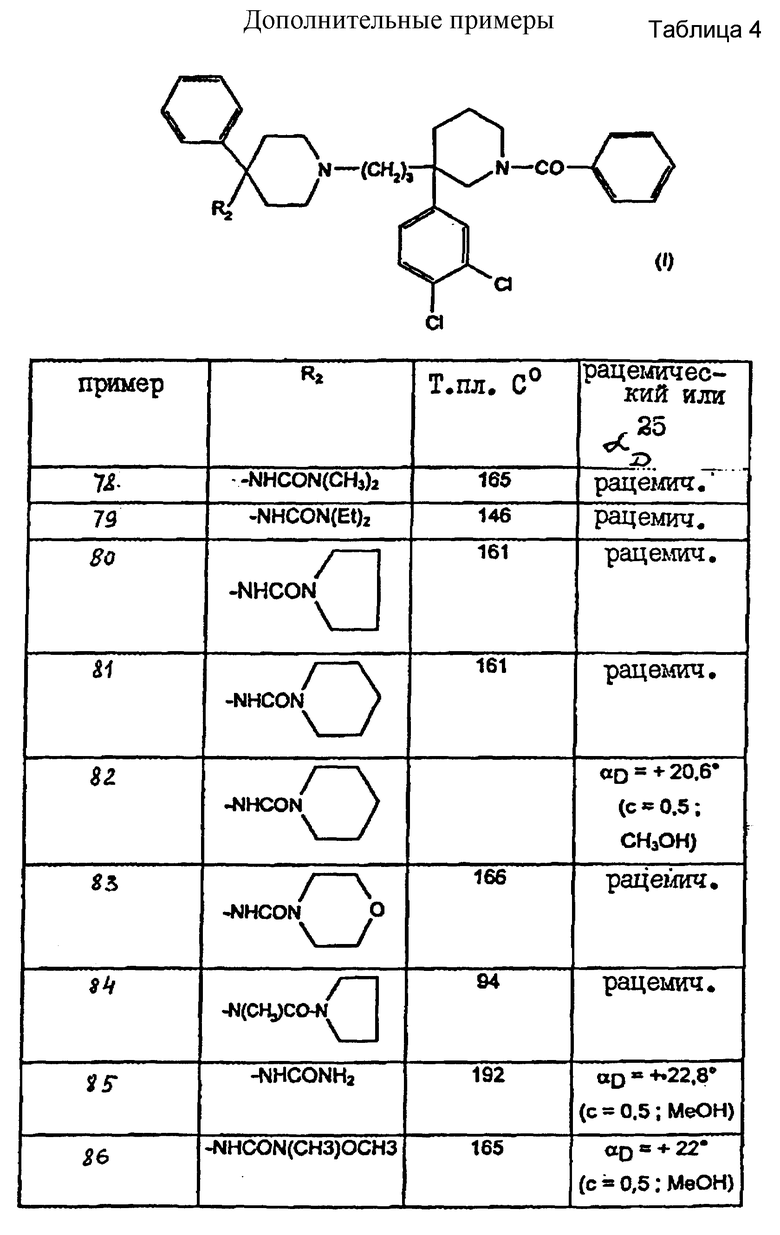
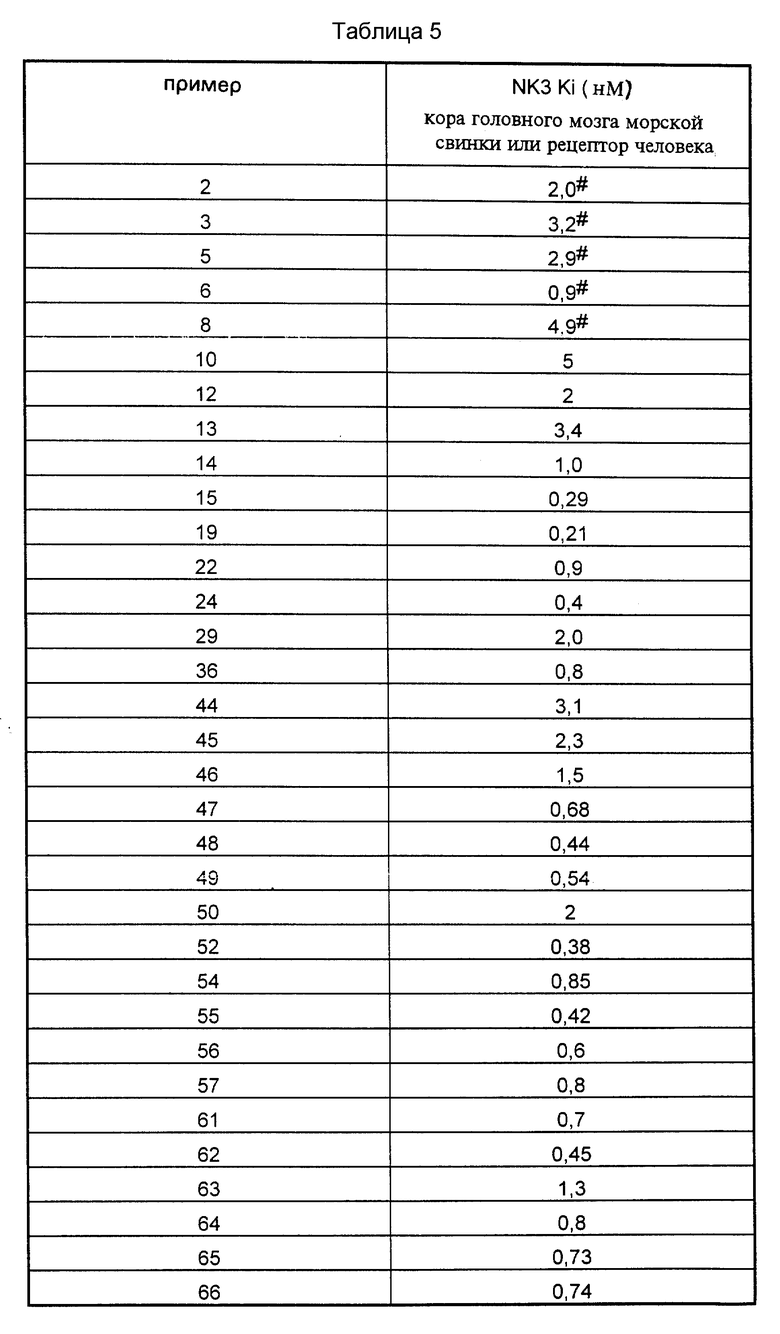

Производные пиперидина формулы, где Аr - пирид-2-ил или фенил, возможно замещенный галогеном, метилом или алкоксилом; R1 -CH3, R11 - Н или R1 и R11 вместе обозначают группу -(СН2)3; R2 - гидроксил, алкоксил, ацилокси, циано, -NR6R7, -NR3COR4, -NR3COOR8, -NR3SO2R9, -NR3CONR10R12 (другие обозначения см. в п. 1 ф-лы), а также их соли обладают антагонистической активностью к рецептору NK3 человека. 6 с. и 14 з.п. ф-лы, 5 табл.
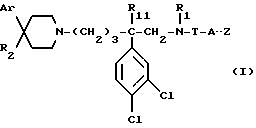
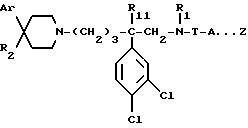
в которой Ar обозначает пирид-2-ил или фенил, незамещенный или замещенный галогеном, метилом или (С1-С4)-алкоксилом;
R1 обозначает метильную группу;
R11 обозначает водород или
R1 и R11 вместе обозначают группу -(СН2)3-;
R2 обозначает гидроксил, (С1-С7)-алкоксил, (С1-С7)-ацилоксигруппу, цианогруппу, группу -NR6R7, группу -NR3СОR4, группу -NR3СООR8, группу -NR3SO2R9, группу -NR3СОNR10R12, (С1-С7)-ацильную группу, (С1-С7)-алкоксикарбонил, группу -СОNR10R12, группу -СН2ОН, (С1-С7)-алкоксиметил, (С1-С7)ацилоксиметил, (С1-С7)-алкиламинокарбонилоксиметил, группу -СН2NR13R14, группу -СН2NR3СОR4, группу -СН2NR3СООR8, группу -СН2NR3SO2R9, группу -СН 2NR3СОNR10R12 или
R2 представляет собой двойную связь между атомом углерода, с которым он связан, и соседним атомом углерода пиперидинового цикла;
или Ar и R2 вместе с атомом углерода, с которым они связаны, представляют собой группу формулы а
R3 обозначает водород или (С1-С4)-алкил;
R4 обозначает водород, (С1-С7)-алкил, фенил, бензил, пиридил или (С3-С7)-циклоалкил, незамещенный или замещенный одной или несколькими метильными группами;
или R3 и R4 вместе обозначают группу -(СН2)n-;
n обозначает число 3 или 4;
Т обозначает метилен, карбонил, группу -СОО-, группу -СОNR5-,
А обозначает простую связь, метилен, этилен, пропилен, винилен;
или -Т-А обозначает группу -SO2-;
Z обозначает фенил, незамещенный или замещенный один или несколько раз галогеном, (С1-С4)-алкилом, (С1-С4)-алкоксилом, нитрогруппой,
R5 обозначает водород или (С1-С4)-алкил;
R6 и R7, каждый, независимо друг от друга, обозначают водород, или (С1-С7)-алкил, R7, кроме того, может обозначать (С3-С7)-циклоалкилметил, бензил или фенил; или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероцикл, выбираемый среди азетидина, пирролидина, пиперидина, морфолина, тиоморфолина или пергидроазепина;
R8 обозначают (С1-С7)-алкил или фенил;
R9 обозначает (С1-С7)-алкил; свободную или замещенную одним или двумя (С1-С7)-алкилами аминогруппу; фенил, незамещенный или одно- или многократно замещенный заместителем, выбираемым в группе, включающей атом галогена, (С1-С7)-алкил, трифторметил, гидроксил, (С1-С7)-алкоксил, карбоксил, (С1-С7)-алкоксикарбонил, (С1-С7)-алкилкарбонилоксигруппу, цианогруппу, нитро-группу, свободную или замещенную одним или двумя (С1-С7)-алкилами аминогруппу, причем вышеуказанные заместители одинаковые или разные,
R10 и R12, каждый, независимо друг от друга, обозначают водород, или (С1-С7)-алкил; R12, кроме того, может обозначать (С3-С7)-циклоалкил, (С3-С7)-циклоалкилметил, гидроксил, (С1-С4)-алкоксил, бензил или фенил, или
R10 и R12 вместе с атомом азота, с которым они связаны, образуют гетероцикл, выбираемый среди азетидина, пирролидина, пиперидина, морфолина, тиоморфолина или пергидроазепина,
R13 и R14, каждый, независимо друг от друга, обозначают водород или (С1-С7)-алкил; кроме того, R14 может обозначать (С3-С7)-циклоалкилметил или бензил, при условии, что
1) когда Ar обозначает фенильную группу, R2 обозначает гидроксильную группу, Т-А-Z обозначает бензоильную группу, R1 отличен от метильной группы,
2) когда Ar обозначает фенильную группу, R2 обозначает группу -NН-СО-СН3, Т-А-Z обозначает бензоильную группу, R1 и R11 не образуют вместе группу -(СН2)3-,
3) когда Ar обозначают фенильную группу, R2 обозначает гидроксильную группу, Т-А-Z обозначает 3-метоксибензильную группу, R1 и R11 не образуют вместе группу -(СН2)3-,
4) когда Ar означает фенил, R2означает группу -NН-СО-СН3, Т-А-Z означает бензилоксикарбонильную группу, R1 означает метильную группу; а также их соли.
R3 обозначает водород или (С1-С4)-алкил;
R4 обозначает водород, (С1-С7)-алкил, фенил, пиридил или (С3-С7)-циклоалкил;
Т обозначает метилен, карбонил, группу -СОО-, группу -СОNR5-;
А обозначает прямую связь, метилен, этилен, пропилен, винилен;
или -Т-А- обозначает группу -SO2-,
Z обозначает фенил, незамещенный или одно- или многократно замещенный галогеном, (С1-С4)-алкилом, (С1-С4)-алкоксилом, нитрогруппой;
R5 обозначает водород;
R6 и R7, каждый, независимо друг от друга, обозначает водород, или (С1-С7)-алкил; R7, кроме того, может обозначать (С3-С7)-циклоалкилметил или R6 и R7 вместе с атомом азота, с которым они связаны, образуют пиперидин;
R8 обозначает (С1-С7)-алкил или фенил,
R9 обозначает (С1-С7)-алкил;
R10 и R12, каждый, независимо друг от друга, обозначают водород или (С1-С7)-алкил; R12, кроме того, может обозначать (С1-С4)-алкоксил или
R10 и R12 вместе с атомом азота, с которым они связаны, образуют пирролидин, пиперидин или морфолин,
R13 и R14, каждый, означает водород,
а также их соли.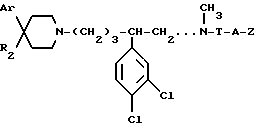
в которой Ar имеет указанное в п.1 значение;
R2 обозначает ацетамидо-, пропиониламино, бутириламино, изобутириламино, ацетил-N-метиламино-, пропионил-N-метиламино-, бутирил-N-метиламино-, изобутирил-N-метиламиногруппу;
Т-А-Z обозначает бензилоксикарбонил, незамещенный или замещенный на фениле хлором или нитрогруппой,
а также их соли.
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-(ацетил-N-метиламино)-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-пропиониламино-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-(пропионил-N-метиламино)-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-бутириламино-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-(бутирил-N-метиламино)-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-изобутириламино-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-(изобутирил-N-метиламино-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-валериламино-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-валерил-N-метиламино)-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-изовалериламино-4-фенилпиперин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-(изовалерил-N-метиламино)-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-пивалоиламино-4-фенилпиперидин-1-ил)пропил]пиперидин,
1-бензоил-3-(3,4-дихлорфенил)-3-[3-(4-(пивалоил-N-метиламино)-4-фенилпиперидин-1-ил)пропил]пиперидин,
в форме рацемата или (+) - или (-)-энантиомера и его фармацевтически приемлемые соли.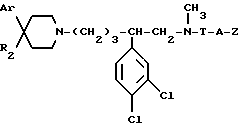
в которой Ar, Z имеют указанное в п.1 значение и либо R2 обозначает (С5-С7)-алкоксил; (С5-С7)-ацилоксигруппу, группу -NR3СОR4, где R4 отличается от (С1-С6)-алкила, если R3 обозначает водород; группу -NR6R7, где R6 и R7 отличаются от водорода или (С1-С4)-алкила; группу -NR3СООR8; группу -NR3SO2R9; группу -NR3СОNR10R12; (С5-С7)-ацил; (С5-С7)-алкоксикарбонил; группу -СОNR10R12; (С1-С7)-алкоксиметил; (С1-С7)-ацилоксиметил; (С1-С7)-алкиламинокарбонилоксиметил; группу -СН2NR13R14, где R13 и R14 другие, чем водород; группу -СН2NR3СОR4, где R4 отличается от (С1-С3)-алкила, если R3 обозначает водород; группу -СН2NR3СООR8; группу -СН2NR3SO2R9; группу -СН2NR3СОNR10R12, при этом Т и А имеют указанное в п.1 значение: либо Т обозначает метилен или группу -СОNR5-, где R5 отличается от водорода, при этом R2 и А имеют указанное в п.1 значение; либо -Т-А- обозначает группу -SO2-, при этом R2 имеет указанное в п.1 значение, в форме рацемата или (+) - или (-) -энантиомера, а также их соли.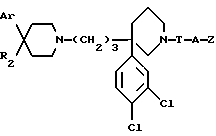
в которой Ar, Z имеют указанное в п.1 значение, и либо R2 обозначает (С5-С7)-алкоксил; (С5-С7)-ацилоксигруппу; группу -NR3СОR4, где R4 отличается от водорода или (С1-С3)-алкила; группу -NR6R7, где R6 и R7 отличаются от водорода или (С1-С4)-алкила, или, если R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероцикл, то он отличается от пирролидинового, пиперидинового или морфолинового цикла; группу -NR3СООR8; группу -NR3SO2R9; группу -NR3СОNR10R12; (С1-С7)-ацил; (С5-С7)-алкоксикарбонил; группу -СОNR10R12; группу -СН2ОН; (С1-С7)-алкоксиметил; (С1-С7)-ацилоксиметил; (С1-С7)алкиламинокарбонилоксиметил; группу -СН2NR13R14; группу -СН2NR3СОR4; группу -NR3СООR8; группу СН2NR3СООR8;
группу -СН2NR3SO2R9; группу -СН2NR3СОNR10R12, при этом Т и А имеют указанное в п. 1 значение; либо Т обозначает группу -СОNR5-; либо -СОО-, при этом R2 и А имеют указанное в п.1 значение; либо А обозначает винилен, при этом R2 и Т имеют указанное в п.1 значение; либо -Т-А- обозначает группу -SO2-, при этом R2 имеет указанное в п.1 значение,
в форме рацемата, или (+)-, или (-)-энантиомера, а также их соли.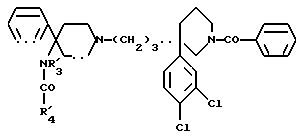
в которой R'3 обозначает водород или метил;
R'4 обозначает (С4-С7)-алкил, фенил, бензил, пиридил; (С3-С7)-циклоалкил, незамещенный или одно- или многократно замещенный метильными группами,
или R'3 и R'4 вместе обозначают группу -(СН2)n-,
n обозначает число 3 или 4,
а также их соли.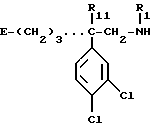
в которой R1, R11 имеют указанное в п.1 значение и Е обозначает гидроксил или в случае необходимости О-защищенную группу, такую, как тетрагидропиран-2-ил-окси-, бензоилокси- или (С1-С4)алкилкарбонилоксигруппу, или группу формулы 2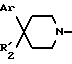
в которой Ar имеет указанное в п.1 значение и R'2 обозначает R2, имеющий значения, указанные в п.1, или предшественник R2, причем если R2 обозначает гидроксил или аминогруппу, то эти группы могут быть защищены,
обрабатывают либо функциональным производным кислоты формулы 3
НОСО - А - Z,
в которой А и Z имеют указанные для формулы I значения в случае получения соединений формулы I, где Т обозначает -СО-,
либо галогенированным производным формулы 4
Гал-СН2-А-Z,
в которой Гал обозначает галоген, предпочтительно бром или хлор, в случае получения соединений формулы I, где Т обозначает -СН2-,
либо хлорформиатом формулы 5
ClCOO-A-Z,
в случае получения соединений формулы I, где Т обозначает -СОО-, либо изоцианатным производным формулы 6
О = С = N - А - Z,
в случае получения соединений формулы I, где Т обозначает -СОNН-, либо карбамоилхлоридом формулы 7
в случае получения соединений формулы I, где Т обозначает -СОNR5-, где R5 отличается от водорода, либо бензолсульфонилхлоридом формулы 7а
ClSO2-Z,
в случае получения соединений формулы I, в которой -Т-А- обозначает -SO2-, для получения соединений формулы 8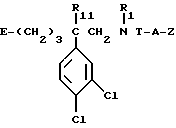
б) в случае необходимости удаляют О-защитную группу путем воздействия кислоты или основания; в) таким образом полученный спирт формулы 9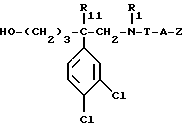
обрабатывают соединением формулы 9а
W-SO2-Cl,
в которой W обозначает метильную, фенильную, толильную, трифторметильную группу,
г) таким образом полученный сульфонат формулы 10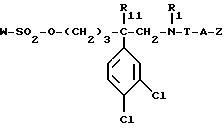
вводят во взаимодействие со вторичным амином формулы 11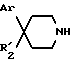
в которой Ar и R'2 имеют вышеуказанные значения,
д) после возможного удаления защиты с гидроксильной группы или аминогруппы, обозначаемой R'2, или после возможного превращения R'2 в R2, в случае необходимости полученный продукт превращают в одну из его солей.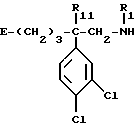
в которой R1, R11 имеют указанные в п.1 значения и Е обозначает гидроксил или в случае необходимости О-защищенную группу, такую, как тетрагидропиран-2-ил-оксигруппу, бензилоксигруппу или (С1-С4)-алкилкарбонилоксигруппу, или группу формулы 2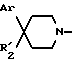
защищают защитной группой, чтобы получить соединение формулы 12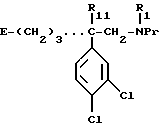
в которой Pr обозначает азотзащитную группу, например трет.-бутоксикарбонил (Вос), тритил, бензил,
б1) в случае необходимости удаляют О-защитную группу путем воздействия кислоты или основания, в1) таким образом полученный спирт формулы 13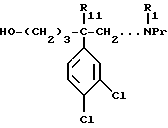
обрабатывают соединением формулы 9а
W-SO2-Cl,
в которой W обозначает метильную, фенильную, толильную, трифторметильную группу,
г1) таким образом полученный сульфонат формулы 14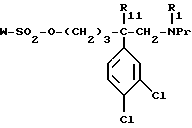
вводят во взаимодействие со вторичным амином формулы 11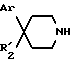
для получения соединения формулы 15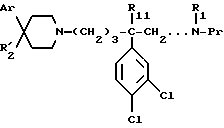
д1) осуществляют удаление защитной группы с азота в кислой среде,
е1) таким образом полученное соединение формулы 16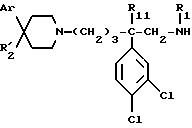
обрабатывают одним из соединений формулы 3, 4, 5, 6, 7 или 7а по п.9, ж1) после возможного удаления защитной группы с гидроксильной группы или аминогруппы, обозначаемой R'2, или после возможного превращения R'2 в R2, в случае необходимости, полученный продукт формулы I превращают в одну из его солей.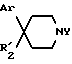
в которой Ar имеет указанное в п.1 значение;
R'2 обозначает группу -NR3СОR4 с R3 и R4 такими, как указаны в п.1, при условии, что когда R3 обозначает водород, R4 не является метилом,
Y обозначает водород или защитную группу,
а также их возможные соли в качестве промежуточных соединений при получении производных пиперидина по п.1.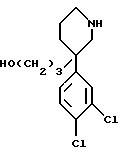
и его соли с неорганическими или органическими кислотами в качестве промежуточного соединения при получении производных пиперидина по п.1.
Приоритет по пунктам:
18.03.94 - по пп.3-5, 8-12 и 17;
29.07.94 - по пп.1-2, 6-7 и 13-16;
19.01.95 - по п.18.
| Устройство для охлаждения листов | 1970 |
|
SU474561A1 |
| Устройство для обработки поверхностей деталей | 1974 |
|
SU512901A2 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1987, ч.1, с.50. | |||

