Изобретение относится к новому способу получения 2-замещенных 5-хлоримидазол-4-карбальдегидов общей формулы I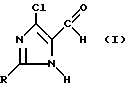
где
R обозначает водород, алкильную группу, алкенильную группу, циклоалкильную группу, бензиловую группу, фенильную группу либо арильную группу.
2-Замещенные 5-хлоримидазол-4-карбальдегиды общей формулы I представляют собой исходные продукты для изготовления снижающих кровяное давление фармацевтических препаратов (см. патент США 4355040) или для получения обладающих гербицидным действием соединений (см. выложенную заявку ФРГ 2804435).
Существует целый ряд способов получения названных соединений общей формулы I.
Так, например, в патенте США 4355040 описывается способ, согласно которому 2-амино-3,3-дихлоракрилнитрил с помощью альдегида преобразуют сначала в соответствующий азометиновый промежуточный продукт, а затем с помощью галогеноводорода и воды преобразуют в 2-замещенный 5-галогенимидазол-4-карбальдегид. Однако в названном патенте отсутствуют экспериментальные данные. Существенный недостаток описанного в патенте синтеза состоит в том, что применяемый 2-амино-3,3-дихлоракрилнитрил должен быть получен исходя из дихлорацетонитрила путем преобразования этого последнего с помощью синильной кислоты/цианида натрия. По причине крайней высокой токсичности участвующих в реакции веществ и связанных с этим мер по технике безопасности, необходимых уже на стадии получения исходного продукта, способ в целом не пригоден для применения в промышленном масштабе.
В патенте США 4355040 в качестве одного из вариантов дается описание 3-ступенчатого способа, в котором на первой ступени осуществляют циклизацию амидингидрохлорида с помощью диоксиацетона при высоком давлении NH3, затем галогенируют имидазоловый спирт и наконец окисляют до альдегида.
Было найдено, что для осуществления реакции замыкания цикла требуется давление свыше 20 бар.
Окисление спирта может проводиться согласно патенту США 4355040 в присутствии окиси хрома.
Совершенно ясно, что окисление с помощью окислов тяжелых металлов, которые, как правило, не рециркулируемы, с существующей ныне экологической точки зрения более неприемлемо.
Исходя из вышесказанного, была поставлена задача создать такой способ, который позволил бы устранить указанные недостатки.
Поставленная задача была решена благодаря новому способу по п.1.
На первой ступени способа гидрогалогенид сложного глицинового эфира общей формулы II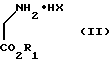
где
R1 обозначает алкильную группу, а X представляет собой атом галогена, преобразуют с помощью сложного имидоэфира общей формулы III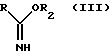
где
R имеет указанное выше значение, а R2 обозначает алкильную группу, в присутствии какого-либо основания в 2-замещенный 3,5-дигидроимидазолин-4-он общей формулы IV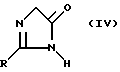
где
R имеет указанное выше значение.
Общие обозначения групп в заместителях R, R1 и R2 имеют следующие значения.
Под алкильной группой имеются в виду неразветвленные либо разветвленные C1-C6-алкильные группы, как, например, метиловая группа, этиловая группа, пропиловая группа, изопропиловая группа, н-бутиловая группа, втор-бутиловая группа, трет-бутиловая группа, пентиловая группа либо гексильная группа. Предпочтительной алкильной группой для R является н-пропиловая группа либо н-бутиловая группа. Под алкенильной группой имеется в виду неразветвленная либо разветвленная C1-C6-алкенильная группа, как, например, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, пентенил и его изомеры или же гексенил и его изомеры. Предпочтительной алкенильной группой для R является 2 - либо 3-бутениловая группа.
В качестве представителей циклоалкильных групп следует назвать циклопропиловую группу, циклобутиловую группу, циклопентиловую группу или же циклогексильную группу.
Как бензиловая группа, так и фенильная группа может содержать в качестве заместителей названные выше алкильные группы, атомы галогена, нитрогруппы либо аминогруппы.
Под галогеном подразумевается предпочтительно хлор, бром либо йод, в первую очередь хлор.
Целесообразно работать таким образом, чтобы гидрогалогенид сложного глицинового эфира общей формулы II реагировал в присутствии основания предпочтительно при pH 7-12, прежде всего при pH 9-11, со сложным имидоэфиром общей формулы III.
Гидрогалогениды сложного глицинового эфира общей формулы II представляют собой устойчивые соединения, имеющиеся в продаже.
Пригодными для осуществления описанных выше реакций основаниями являются гидроокиси щелочных металлов, как, например, гидроокись натрия либо гидроокись калия, или алкоголяты щелочных металлов, как, например, метилат натрия либо метилат калия, этилат натрия либо этилат калия, или трет-бутилат натрия либо трет-бутилат калия.
Предпочтительно основание применяют в растворенном виде, для чего используют соответствующий растворитель. В качестве такового особенно пригодны алифатические спирты, как метанол либо этанол. Сложный имидоэфир добавляют предпочтительно также в виде раствора, для чего используют инертный растворитель. Как правило, для этой цели особенно пригодны ароматические растворители, как, например, толуол либо хлорбензол, или упомянутые выше алифатические спирты.
Преобразование участвующих в реакции гидрогалогенида сложного глицинового эфира, сложного имидоэфира и основания осуществляют предпочтительно в стехиометрическом соотношении 1:1:1.
Температуру реакции выбирают предпочтительно в диапазоне от -20 до 50oC, прежде всего от 0 до 25oC.
После окончания реакции, продолжающейся в течение нескольких часов, известным образом, как правило, путем обычной фильтрации может быть выделен соответствующий 2-замещенный 3,5-дигидроимидазолин-4-он общей формулы IV, при этом выход продукта составляет более 95%.
Предпочтительно получаемую реакционную смесь используют без выделения 2-замещенного 3,5-дигидроимидазолин-4-она для последующего преобразования в 5-хлоримидазол-4-карбальдегид (так называемый способ получения в одном аппарате).
Получаемые при осуществлении этой первой стадии способа соединения общей формулы IV, где R обозначает н-пропил, н-бутил, 2- либо 3-бутенил, являются особенно предпочтительными.
Эти соединения не известны из опубликованной литературы и поэтому также являются составной частью изобретения.
Эта первая ступень предлагаемого согласно изобретению способа позволяет существенным образом улучшить известный способ, описанный в R.Jacguier et al. Bull. Soc. Chim. France, 1971, 1040, согласно которому осуществляют преобразование свободного сложного глицинового эфира с помощью имидоэтилового эфира общей формулы III (значения R ограничиваются метилом, фенилом, бензилом) при отсутствии растворителя в соответствующий 3,5-дигидроимидазол-4-он. Недостаток этого известного способа заключается в том, что свободный глициновый эфир очень нестабилен и по этой причине его необходимо для каждой реакции всякий раз снова синтезировать и выделять. Другим существенным недостатком этого способа является то, что реакция продолжается в течение длительного времени (24 часа и более) и выход продукта составляет лишь 30-48%.
Реакцию преобразования в требуемый 2-замещенный 5-хлоримидазол-4-карбальдегид общей формулы I осуществляют согласно изобретению с помощью фосфороксихлорида в присутствии N,N-диметилформамида.
Молярное соотношение реагентов, как то: 2-замещенного 3,5-дигидроимидазолин-4-она, фосфороксихлорида и N,N-диметилформамида, выбирают в пределах от 1:1:1 до 1:5:5, предпочтительно приблизительно 1:3:3.
Температура реакции лежит предпочтительно в диапазоне между 50 и 130oC.
При определенных условиях реакцию можно проводить в присутствии дополнительного инертного растворителя.
Выделение получаемого 2-замещенного 5-хлоримидазол-4-карбальдегида из реакционной смеси осуществляют известным образом, предпочтительно путем его экстракции с помощью соответствующего растворителя.
Пример 1. Получение 2-н-бутил-3,5-дигидроимидазол-4-она.
В раствор, содержащий 10,1 г (0,25 моль) гидроокиси натрия в метаноле, при 0oC добавляли 31,71 г (0,25 моль) гидрохлорида сложного глицинметилового эфира. Через 15 мин в суспензию белого цвета в течение 5 мин по каплям добавляли 126,5 г 22,8%-ного раствора сложного метилового эфира пентанимидокислоты в хлорбензоле. Светло-желтую суспензию перемешивали в течение 4 ч при комнатной температуре и разбавляли хлорбензолом (100 мл). Метанол отгоняли при температуре 26oC и давлении порядка 30-50 мбар, после чего суспензию оранжевого цвета разбавляли хлористым метиленом (100 мл) и затем фильтровали. После удаления растворителя из фильтрата получили 34,09 г (97%) соединения, указанного в заголовке (содержание > 95% согласно газовой хроматографии и 1H-ЯМР).
1Н-ЯМР (CDCl3, 300 МГц) δ в част./млн: 0,95 (t, 3H); 1,45 (m, 2H); 1,68 (m, 2H); 2,48 (t, 2H); 4,1 (m, 2H); 9,3 (шир. s, 1H).
Пример 2. Получение 2-н-бутил-5-хлоримидазол-4-карбальдегида.
В раствор, содержащий фосфороксихлорид (26,83 г, 175 ммоль) в хлорбензоле (50 мл), при комнатной температуре добавляли 2-н-бутил-2-имидазолин-5-он (9,81 г, 70 ммоль). Суспензию оранжевого цвета нагревали в течение 5 мин до 100oC, после чего в течение 3 мин производили добавку N,N-диметилформамида (12,79 г, 175 ммоль). Черную суспензию выдерживали в течение 2 ч при 100oC, затем охлаждали до 40oC и сливали на воду (84 мл). После добавки уксусного эфира (42 мл) смесь перемешивали в течение 15 мин при 26-28oC и затем с помощью добавки 30%-ного натрового щелока (67 мл) устанавливали на pH 7. Фазы разделяли и водную фазу дважды экстрагировали уксусным эфиром порциями по 70 мл. Соединенные органические фазы сушили с помощью MgSO4, фильтровали и концентрировали с помощью ротационно-вакуумного испарителя. Указанное в заголовке соединение получали с выходом 8,31 г (64%) по отношению к 2-н-бутил-2-имидазолин-5-ону.
Пример 3. Получение 2-н-бутил-5-хлоримидазол-4-карбальдегида (способ без выделения 2-н-бутил-2-имидазолни-5-она).
В охлажденный до 0oC раствор, содержащий 10,17 г (0,25 моль) гидроокиси натрия в метаноле (80 мл), добавляли одной порцией 31,71 г (0,25 моль) гидрохлорида сложного глицинметилового эфира в виде твердого вещества, причем белая суспензия (NaCl выпадает в осадок) охлаждалась до -8oC. В течение последующих 15 мин температура повышалась до 0oC, после чего в течение 15 мин добавляли 127,0 г 22,68%-ного раствора метилового эфира пентанимидокислоты в хлорбензоле (0,25 моль метилового эфира пентанимидокислоты). Температура в течение одного часа повышалась до 21oC, затем желто-коричневую суспензию в течение 3,5 ч перемешивали при 21oC. После добавки хлорбензола (400 мл) отгоняли приблизительно 240 г смеси из метанола, воды и хлорбензола при 30oC. В оставшуюся часть суспензии (приблизительно 420 г) добавляли фосфороксихлорид (107,33 г, 0,7 моль) (температура повышалась до 35oC). Мутную смесь оранжевого цвета нагревали до 100oC, после чего в течение 5 мин добавляли по каплям N,N-диметилформамид (51,71 г, 0,70 моль) (температура повышалась до 108oC). Через 2 ч при 100oC черную смесь охлаждали до 75oC и сливали в 300 г охлажденной до 10oC воды. Смесь разбавляли уксусным эфиром (150 мл), перемешивали в течение 15 мин при 50oC и путем добавки 160 мл 30%-ного натрового щелока устанавливали на pH 1. Затем фазы разделяли и водную фазу дважды экстрагировали уксусным эфиром порциями по 250 мл. Соединенные органические фазы сушили с помощью MgSO4, фильтровали и концентрировали с помощью ротационно-вакуумного испарителя. Указанное в заголовке соединение получали с выходом 34,3 г (73%) по отношению к гидрохлориду сложного глицинметилового эфира.
Пример 4. Получение 2-н-бутил-хлоримидазол-4-карбальдегида (способ без выделения 2-н-бутил-2-имидазолин-5-она).
В охлажденный до 0oC раствор, содержащий 10,15 г (0,25 моль) гидроокиси натрия в метаноле (80 мл), добавляли одной порцией 31,72 г (0,25 моль) гидрохлорида сложного глицинметилового эфира в виде твердого вещества, причем белая суспензия (NaCl выпадает в осадок) охлаждалась до -9oC. В течение последующих 15 мин температура повышалась до 0oC, после чего в течение 15 мин добавляли 127,0 г 22,68%-ного раствора метилового эфира пентанимидокислоты в хлорбензоле (0,25 моль метилового эфира пентанимидокислоты). Температура в течение одного часа повышалась до 21oC, затем желто-коричневую суспензию перемешивали в течение 3,5 ч при 21oC. После добавки хлорбензола (200 мл) отгоняли приблизительно 210 г смеси из метанола, воды и хлорбензола при 30oC. В оставшуюся часть суспензии (приблизительно 226 г) добавляли фосфороксихлорид (107,33 г, 0,70 моль) (температура повышалась до 35oC). Мутную смесь оранжевого цвета нагревали до 100oC, после чего в течение 5 мин добавляли по каплям диметилформамид (51,71 г, 0,70 моль) (температура повышалась до 108oC). Через 2 ч при 100oC черную смесь охлаждали до 40oC и сливали в 300 г охлажденной до 10oC воды. Смесь разбавляли уксусным эфиром (150 мл), перемешивали в течение 15 мин при 50oC и путем добавки 160 мл 30%-ного натрового щелока устанавливали на pH 1. Затем фазы разделяли и водную фазу дважды экстрагировали уксусным эфиром порциями по 250 мл. Соединенные органические фазы сушили с помощью MgSO4, фильтровали, концентрировали с помощью ротационно-вакуумного испарителя до 125 г и охлаждали до -10oC. Выпавший в осадок продукт отфильтровывали, вымывали холодным уксусным эфиром и сушили при 50oC. Выход составлял 19,01 г (40% по отношению к гидрохлориду сложного глицинметилового эфира). Маточный раствор содержал согласно газовой хроматографии с внутренним стандартом еще 11,66 г указанного в заголовке соединения, тем самым общий выход продукта составлял 65% по отношению к гидрохлориду сложного глицинметилового эфира.
Пример 5. Получение 2-н-пропил-3,5-дигидроимидазол-4-она.
В раствор, содержащий гидроокись натрия (5,56 г, 139 ммоль) в метаноле (55 мл), при 0oC добавляли 17,39 г (138 ммоль) гидрохлорида сложного глицинметилового эфира. Через 15 мин в белую суспензию в течение 8 мин добавляли по каплям 14,50 г (содержание 96,3%, 138 ммоль) метилового эфира бутанимидокислоты. Смесь перемешивали в течение 3 ч при комнатной температуре, после чего концентрировали с помощью ротационно-вакуумного испарителя. Остаток обрабатывали CH2Cl2 (250 мл) и полученную суспензию фильтровали. Затем фильтрат концентрировали с помощью ротационно-вакуумного испарителя, вторично обрабатывали CH2Cl2 (250 мл) и повторно фильтровали. После удаления растворителя получали соединение, указанное в заголовке, (15,13 г, содержание > 95% согласно 1Н-ЯМР, выход 83%).
1H-ЯМР (CDCl3, 400 МГц) δ в част./млн: 1,03 (t,3H); 1,75 (m, 2H); 2,46 (t, 2H); 4,12 (s, 2H); 9,98 (шир. s, 1H).
Пример 6. Получение 2-н-пропил-5-хлоримидазол-4-карбальдегида.
В смесь, содержащую 2-н-пропил-3,5-дигидроимидазол-4-он (4,49 г, 35,6 ммоль) и POCl3 (14,76 г, 96,3 ммоль) в хлорбензоле (40 мл), при 100oC добавляли N,N-диметилформамид (7,04 г, 96,3 ммоль). Смесь нагревали в течение 2 ч при 100oC, затем охлаждали и сливали на 40 г льда. Путем добавки 30%-ного натрового щелока (22,5 мл) смесь устанавливали на pH 1 и фазы разделяли. Водную фазу дважды экстрагировали с помощью уксусного эфира порциями по 40 мл и соединенные органические фазы вымывали водой (20 мл) и фильтровали с помощью силикагеля. После удаления растворителя из фильтрата получали 2,79 г твердого вещества светло-коричневого цвета. После перекристаллизации из уксусного эфира/петролейного эфира получали соединение, указанное в заголовке (2,04 г, 33%). Тпл.: 133,3 - 137,5oC.
1Н-ЯМР (CDCl3, 400 МГц) δ в част./млн: 1,01 (t, 3H); 1,84 (m, 2H); 2,83 (t, 2H); 9,64 (s, 1H); 11,56 (шир.s, 1H).
Пример 7. Получение 2-бут-2-енил-3,5-дигидроимидазол-4-она.
Аналогично тому, как это описано в примере 1, путем взаимодействия гидрохлорида сложного глицинметилового эфира и метилового эфира 3-пентанимидокислоты получали вещество, указанное в заголовке.
1Н-ЯМР (CDCl3, 400 МГц) δ в част./млн: 1,75 (d, 3H); 3,19 (d, 2H); 4,12 (s, 2H); 5,54 (m, 1H); 5,75 (m, 1H); 9,20 (шир. s, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННЫХ 5-ХЛОРИМИДАЗОЛ-4-КАРБАЛЬДЕГИДОВ | 1993 |
|
RU2103262C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННЫХ 4,6-ДИАЛКОКСИПИРИМИДИНОВ,2-N-БУТИЛАМИНО-4,6-ДИМЕТОКСИПИРИМИДИН И СПОСОБ ПОЛУЧЕНИЯ ГАЛОГЕНПРОИЗВОДНЫХ ПИРИМИДИНА | 1992 |
|
RU2117007C1 |
| 3-ОКСИ-2-ЦИКЛОБУТЕН-1-ОН-ОСНОВНЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2078757C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННЫХ 4-(3-ТРЕТ-БУТИЛ-4-ГИДРОКСИФЕНИЛ)-ТИАЗОЛОВ | 1990 |
|
RU2021264C1 |
| ПРОИЗВОДНОЕ 1-ЗАМЕЩЕННОГО 4-НИТРОИМИДАЗОЛА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2324682C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННЫХ 4-(3-ТРЕТ-БУТИЛ-4-ГИДРОКСИФЕНИЛ)-ТИАЗОЛОВ | 1990 |
|
RU2017739C1 |
| ЗАМЕЩЕННЫЕ 2-ТИО-3,5-ДИЦИАНО-4-ФЕНИЛ-6-АМИНОПИРИДИНЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2002 |
|
RU2315757C9 |
| 1-ГЕТЕРОЦИКЛИЛСУЛЬФОНИЛ, 2-АМИНОМЕТИЛ, 5-(ГЕТЕРО-)АРИЛ ЗАМЕЩЕННЫЕ 1-Н-ПИРРОЛ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СЕКРЕЦИИ КИСЛОТЫ | 2006 |
|
RU2415838C2 |
| БИЦИКЛИЧЕСКИЕ АМИДИНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2132850C1 |
| 2-ЗАМЕЩЕННЫЕ 4,5-ДИАРИЛИМИДАЗОЛЫ, СПОСОБ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2214408C2 |
В заявке описывается новый способ получения 2-замещенных 5-хлоримидазол-4-карбальдегидов общей формулы I. Эти соединения представляют собой ценные промежуточные продукты для изготовления снижающих кровяное давление фармацевтических препаратов или для получения соединений, обладающих гербицидным действием 2 с. и 7 з.п. ф-лы.
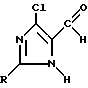
где R - водород, алкильная группа, алкенильная, циклоалкильная, бензиловая группы либо арильная группа, отличающийся тем, что гидрогалогенид сложного глицинового эфира общей формулы II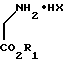
где R1 - алкильная группа;
Х - галоген,
подвергают взаимодействию с имидоэфиром общей формулы III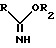
где R имеет указанное значение;
R2 - алкильная группа,
в присутствии основания с получением 2-замещенного 3,5-дигидроимидазолин-4-она общей формулы IV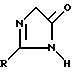
где R имеет указанное значение,
который затем вводят во взаимодействие с фосфороксихлоридом в присутствии N,N-диметилформамида.
| US, патент, 4355040, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
