Настоящее изобретение относится к 2-замещенным 4,5-диарилимидазолам и к их применению при лечении заболеваний, опосредованных ФНО-α (фактор-α некроза опухоли) и ИЛ-1 (интерлейкин 1), таких как ревматоидный артрит, и болезней костного обмена веществ, например остеопороза.
Таким образом, по настоящему изобретению предлагаются новые 2-замещенные 4,5-диарилимидазолы, в которых:
I) атом азота в 1-м положении замещен триалкилсилилсодержащим заместителем или
II) заместителем во 2-м положении является арилалкил, арилсульфонил, арилтиогруппа, арилселен, арилтеллур, циклоалкил, циклоалкенил, алкилциклоалкил, алкилциклоалкенил, амино- или гидразиновая группа, моно- или бициклический N-гетероциклил, у которого N-содержащее кольцо является шестичленным, при условии, что заместителем во 2-м положении не является пиперидин-4-ил, остаток 4-бензилпиперидин-4-илтрет-бутилового эфира 1-карбоновой кислоты, 1,4-диметилпиперидин-4-ил, 4-бензилпиперидин-4-ил или пиперидинил, который дополнительно замещен только при атоме N, а также дополнительно при условии, что ни 4-, ни 5-арильные заместители не являются фенилами, замещенными радикалом, выбранным из алкилсульфонила и аминосульфонила,
их фармацевтически приемлемые кислотно-аддитивные соли и их физиологически расщепляемые эфиры.
4- или 5-арильным заместителем может служить любой из известных в данной области техники, например, такой как описанный в WO 95/03297 и WO 97/12876. Так, например, 4- и 5-арильными заместителями могут являться те, которые в дальнейшем указаны в качестве радикалов R1 и R2 в формуле I и включают гетероарильные заместители.
Когда атом азота в 1-м положении замещен триалкилсилилсодержащим заместителем, пригодным заместителем является триалкилсилилалкоксиалкильный заместитель.
Когда заместителем во 2-м положении является арилалкил, приемлем фенилалкил.
Когда заместитель во 2-м положении представляет собой арилалкил, арилсульфонил, арилтиогруппу, арилселен, арилтеллур, циклоалкил, циклоалкенил, алкилциклоалкил, алкилциклоалкенил, амино- или гидразиновую группу, моно- или бициклический N-гетероциклил, он может дополнительно иметь до 6 заместителей включительно, выбранных из галогена, ОН, С1-С4алкила, С2-С4алкенила, С2-С4алкинила, С1-С4алкоксигруппы, С1-С4тиоалкоксигруппы, нитрогруппы, аминогруппы, С1-С4алкилсульфинила, С1-С4алкилсульфонила, карбоксилата и сложных эфиров.
В вышеприведенной части настоящего описания и в дальнейшем термины "галоид" и "галоген" обозначают I, Br, Cl или F, предпочтительно F.
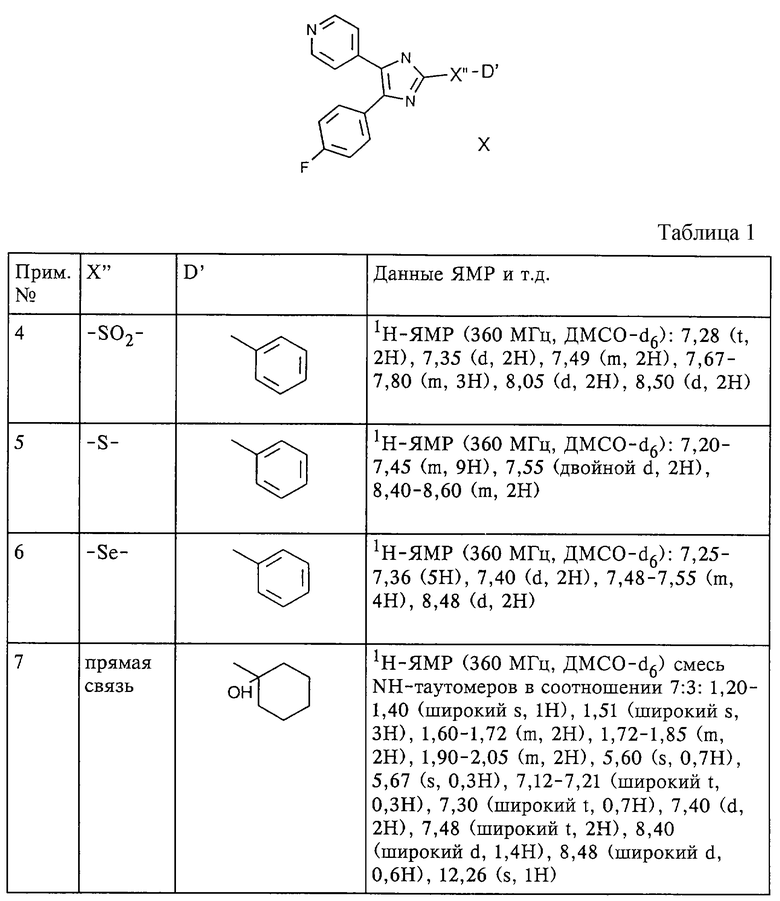
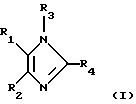
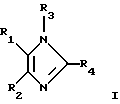
В конкретных вариантах выполнения изобретения предлагается соединение формулы I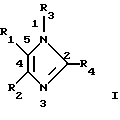
в которой R1 обозначает 4-пиридил, пиримидинил, хиназолин-4-ил, хинолил, изохинолил, 1-имидазолил или 1-бензамидазолил, который необязательно замещен одним или двумя заместителями, каждый из которых независимо друг от друга выбран из С1-С4алкила, галогена, С1-С4алкокси-, C1-С4алкилтиогруппы, NR5R6 или N-гетероциклила, содержащего 5-7 кольцевых атомов и необязательно содержащего дополнительный гетероатом, выбранный из О, S и N, где каждый из R5 и R6 независимо друг от друга обозначает С1-4алкил,
R2 обозначает фенил, нафт-1-ил или нафт-2-ил, который необязательно может иметь до 5 заместителей,
R3 обозначает водород, гетероциклил, гетероциклоС1-С10алкил, триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, необязательно галоидзамещенные С1-С10алкил, С2-С10алкенил, С2-С10алкинил, С3-С7циклоалкил, С3-С7циклоалкилС1-С10алкил, С5-С7циклоалкенил, арил, арилС1-С10алкил, гетероарил или гетероарилС1-С10алкил, необязательно моно- или диС1-С4алкилзамещенный С0-С10алкилоксикарбонил или -окситиокарбонил, необязательно замещенный C1-С10алкилом, С3-С7циклоалкилом, гетероциклилом, гетероциклилС1-С10алкилом, арилом, арилС1-С10алкилом, гетероарилом, гетероарилС1-С10алкилом, или моно- или диС1-С4алкилзамещенный С1-С10лалкил, необязательно замещенный цианогруппой, нитрогруппой, гидрокси-, С1-С10алкокси-, С3-С7циклоалкокси-, гетероциклокси-, гетероциклилС1-С10алкокси-, арилокси-, арилС1-С10алкокси-, гетероарилокси-, гетероарилС1-С10алкоксигруппой, необязательно замещенной аминогруппой, карбоксилатом, тиокарбоксилатом, карбонилом, тиокарбонилом, сульфинилом или сульфонилом,
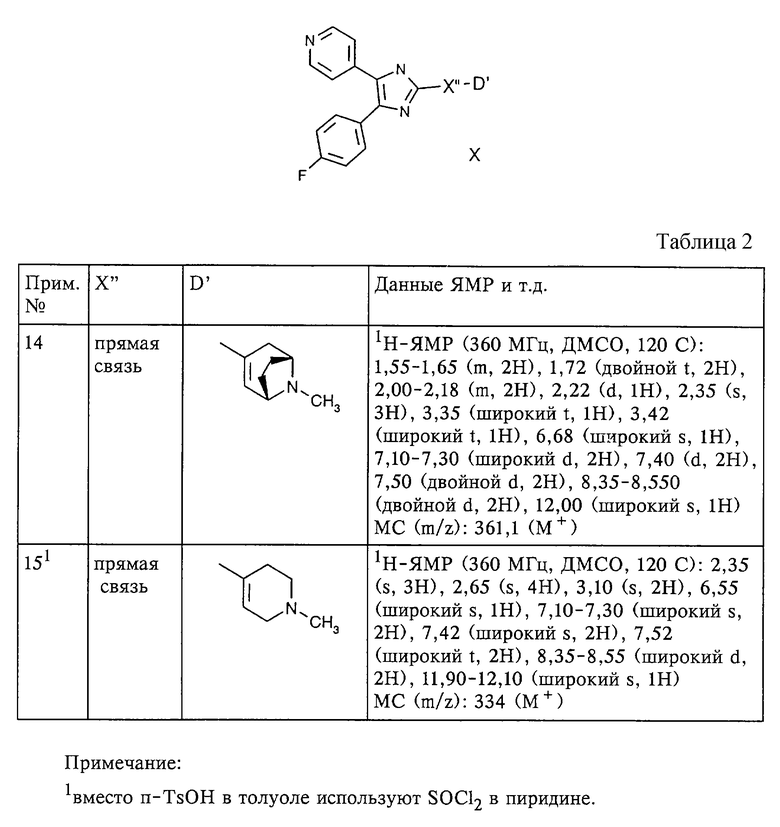
R4 обозначает моно или диС3-С7циклоалкилС0-С4алкил, необязательно замещенный галогеном, -ОН, С1-С4алкилом, С1-С4алкокси, С1-С4тиоалкокси, нитро-, аминогруппой, С1-С4алкилсульфинилом, С1-С4алкилсульфонилом, карбоксилатом или остатком сложного эфира, группу -NR7R8, NHNHR9, где каждый из R7, R8 и R9 независимо друг от друга обозначает С1-С4алкил, С2-С4алкенил, С2-С6алкинил, группу -Х-С5-С10арил (включая гетероарил), где Х обозначает S, SO2, Se, Те или С1-С4алкил, моно- или бициклический N-гетероциклил, у которого N-содержащее кольцо является шестичленным, или арил или гетероарил, необязательно содержащий до 4 заместителей,
при условии, что когда R3 не обозначает триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, R4 не обозначает арил или гетероарил, необязательно содержащий до 3 заместителей, за исключением случая, когда R4 обозначает моно- или бициклический N-гетероциклил, у которого N-содержащее кольцо является шестичленным, а также при дополнительном условии, что R4 не обозначает пиперидин-4-ил, остаток 4-бензилпиперидин-4-ил-трет-бутилового эфира 1-карбоновой кислоты, 1,4-диметилпиперидин-4-ил, 4-бензилпиперидин-4-ил или пиперидинил, который дополнительно замещен только при атоме N, и, кроме того, при дополнительном условии, что R2 не обозначает фенил, замещенный радикалом, выбранным из алкилсульфонила и аминосульфонила, его фармацевтически приемлемые кислотно-аддитивные соли и его физиологически расщепляемые сложные эфиры.
R2 содержит до 5 заместителей, которыми могут служить любые заместители, известные в данной области техники, например, такие как указанные для радикала R4 в WO 95/03297 и радикала R в WO 97/12876.
Когда R4 обозначает -Х-С5-С10арил, то С5-С10арил или X, представляющий собой С1-С4алкил, могут иметь до 6 заместителей включительно, выбранных из галогена, ОН, С1-С4алкила, С1-С4алкоксигруппы, С1-С4тиоалкоксигруппы, нитрогруппы, аминогруппы, С1-С4алкилсульфинила, С1-С4алкилсульфонила, карбоксилата и сложного эфира.
Когда R4 обозначает моноциклический или бициклический N-гетероциклил, в котором N-содержащее кольцо является шестичленым, он может быть насыщенным или ненасыщенным, например ароматическим, гетероциклилом.
Когда R4 обозначает арил или гетероарил, необязательно содержащий до 4 заместителей, R4 может включать один из коммерчески доступных арильных или гетероарильных заместителей, используемых в данной области техники, например, таких, которые указаны как заместители для радикала R3 в WO 93/03297.
Имидазолы с 2-заместителем, например R4, который описан выше, а также с арильными заместителями в положениях как 4, так и 5, например с такими, которые выше указаны как значения радикалов R1 и R2 и у которых атом азота в 1-м положении замещен триалкилсилилсодержащим заместителем, являются новыми.
Таким образом, в качестве еще одного объекта изобретения предлагаются соединение формулы I'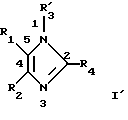
в которой R3' обозначает триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, a R1, R2 и R4 имеют указанные выше значения, его фармацевтически приемлемые кислотно-аддитивные соли и его физиологически расщепляемые сложные эфиры.
Соединения формулы I', в которой R4 обозначает Н, а значения R2, R3' и R4 указаны выше, являются ключевыми промежуточными продуктами для синтеза других соединений формулы I, в которых R3 не обозначает триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, как это изложено выше.
Ниже перечислены предпочтительные независимые друг от друга значения заместителей R1, R2, R3, R'3 и R4.
Предпочтительно R1 обозначает 4-пиридил или пиримидинил, прежде всего 4-пиридил.
Предпочтительно R2 обозначает фенил, включая замещенный фенил.
Наиболее предпочтительно R'3 обозначает триметилсилилэтоксиметил.
Когда R3 обозначает триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, R1 предпочтительно обозначает 4-пиридил.
Когда R3 обозначает триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, R2 предпочтительно обозначает 4-фторфенил.
Когда R3 обозначает триС1-С4алкилсилилС1-С10алкоксиС1-С4алкил, R4 предпочтительно обозначает Н.
В еще одном предпочтительном варианте выполнения изобретения R4 обозначает -Х-С5-С10арил, предпочтительно -Х-фенил, где Х имеет значения, указанные выше, как, например, в соединении формулы III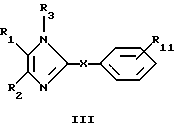
где R1, R2, R3 и Х имеют значения, указанные выше, a R11 обозначает заместитель (заместители) в положениях 1-4, независимо друг от друга выбранные из Н, галогена, ОН, С1-С4алкила, С1-С4алкоксигруппы, С1-С4тиоалкоксигруппы, нитрогруппы, аминогруппы, С1-С4алкилсульфинила, С1-С4алкилсульфонила, карбоксилатного или сложноэфирного остатка, в его фармацевтически приемлемых кислотно-аддитивных солях и их физиологически расщепляемых сложных эфирах.
В другом предпочтительном варианте выполнения изобретения R4 обозначает циклоалкил, циклоалкенил, алкилциклоалкил, алкилциклоалкенил, моно или бициклический N-гетероциклил, у которого N-содержащее кольцо является шестичленным, как, например, в соединении формулы IV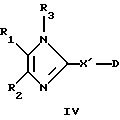
где R1, R2 и R3 имеют значения, указанные выше, D обозначает С3-С7циклоалкил, С3-С7циклоалкенил, моно- или бициклический N-гетероциклил, у которого N-содержащее кольцо является 6-членным, а X' обозначает прямую связь или группу -CR12R13-, где R12 обозначает Н или С1-С4алкил, a R13 обозначает Н или С1-С4алкил, необязательно замещенный С3-С7циклоалкилом или С3-С7циклоалкенилом, при условии, что X' обозначает прямую связь, когда D обозначает моно- или бициклический N-гетероциклил, у которого N-содержащее кольцо является 6-членным, в его фармацевтически приемлемых кислотно-аддитивных солях и их физиологически расщепляемых сложных эфирах.
В формуле IV радикалы X' и D могут дополнительно содержать до 6 заместителей, выбранных из галогена, ОН, C1-С4алкила, C1-С4алкоксигруппы, С1-С4тиоалкоксигруппы, нитрогруппы, аминогруппы, С1-С4алкилсульфинила, С1-С4алкилсульфонила, карбоксилатного или сложноэфирного остатка.
Предпочтительно D обозначает циклогексил, циклогексенил, циклопропил, пиридинил (например 4-пиридинил), пиперидинил (например пиперидин-4-ил), пипериденил (например пипериден-4-ил), азабицикло[3,2,1]октанил, азабицикло [3,3,1] нонил или тропанилбициклический N-гетероцикл (или ениловый аналог такого бициклического N-гетероцикла, например 8-азабицикло[3,2,1]окт-2-ен-3-ил). В особенно предпочтительных вариантах -X'-D обозначает 1-гидроксициклогексил, 1-аминоциклогексил, 1-циклогексенил, циклопропилметил, 1,2-дициклопропилэтил, 2,3,5,6-тетрафторпиридинил, 2-амино-3,5,6-трифторпиридинил, 2,6-диамино-3,5дифторпиридинил, тропан-3-олил, 4-гидрокси-1-метилпиперидинил, 4-С1-С6алкокси-1-метилпиперидинил, например 4-н-бутилокси-1-метилпиперидинил, 8-метил-8-азабицикло [3,2,1]окт-2-ен-3-ил, 1-метил-4-пиперидинил.
Когда R4 обозначает -Х-арил или -X'-D, R1 предпочтительно обозначает 4-пиридил.
Когда R4 обозначает -Х-арил или -X'-D, R2 предпочтительно обозначает замещенный галогеном фенил, прежде всего 4-фторфенил.
Когда R4 обозначает -Х-арил или -X'-D, R3 предпочтительно обозначает Н.
Особенно предпочтительны соединения формулы IV, в которых X' обозначает прямую связь, a D обозначает необязательно замещенный пиридинил, например 4-пиридинил или пиперидинил, например пиперидин-4-ил.
Новые 2-замещенные 4,5-диарилимидазолы по изобретению, в частности соединения формул I-IV и конкретные соединения из примеров 1-19, в дальнейшем в настоящем описании обозначены как "соединения по изобретению".
Соединения по изобретению, которые включают свободные гидроксильные группы, могут также существовать в форме фармацевтически приемлемых, физиологически расщепляемых сложных эфиров и как таковые включены в объем изобретения. Такие фармацевтически приемлемые сложные эфиры в предпочтительном варианте представляют собой пролекарственные сложноэфирные производные, такие, которые в физиологических условиях способны подвергаться превращению путем сольволиза или расщепления с образованием соответствующих соединений по изобретению, которые включают свободные гидроксильные группы. Приемлемыми фармацевтически приемлемыми пролекарственными сложными эфирами являются те, которые являются производными карбоновой кислоты, моноэфира карбоновой кислоты или карбаминовой кислоты, предпочтительно сложные эфиры, которые являются производными необязательно замещенной низшей алифатической кислоты или арилкарбоновой кислоты.
Соединения по изобретению могут также существовать в форме фармацевтически приемлемых солей и как таковые включены в объем изобретения. Эти фармацевтически приемлемые соли включают кислотно-аддитивные соли обычных кислот, например минеральных кислот, в частности соляной кислоты, серной или фосфорной кислоты, и органических кислот, например алифатических или ароматических карбоновых и сульфоновых кислот, в частности уксусной, пропионовой, янтарной, гликолевой, молочной, яблочной, винной, лимонной, аскорбиновой, малеиновой, фумаровой, гидроксималеиновой, пировиноградной, памовой, метансульфоновой, толуолсульфоновой, нафталинсульфоновой, сульфаниловой и циклогексилсульфаминовой кислот, а также аминокислот, таких как аргинин и лизин. Что касается соединений по изобретению, содержащих кислотные группы, например карбоксильные группы, то примерами фармацевтически приемлемых солей также являются соли металлов и аммония, такие как соли щелочных и щелочно-земельных металлов, например натриевые, калиевые, магниевые и кальциевые соли, равно как и аммониевые соли, которые образуются из аммиака и приемлемых органических аминов.
Соединения по изобретению формул III и IV, которые представлены выше, могут быть получены взаимодействием соединения формулы VIII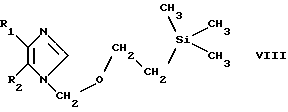
в которой R1 и R2 имеют значения, указанные выше, с соответствующим альдегидом, кетоном, дисульфониламином, дисульфидом, диселенидом, дителлуридом или галогенидом и при необходимости введением целевого заместителя R3 или дальнейшим превращением получаемого продукта с необязательным его переводом в форму свободного соединения или соли. Так, например, соединение формулы VIII обрабатывают соответствующим альдегидом, кетоном, дисульфониламином, дисульфидом, диселенидом, дителлуридом или галогенидом в присутствии н-бутиллития (н-BuLi), например в охлажденном (в частности до -40oС) ТГФ-раствором.
Когда соединение формулы VIII обрабатывают соответствующим альдегидом или кетоном, получаемый исходный продукт является 1-гидроксизамещенным по месту заместителя R4, например 1-гидроксициклогексил, когда кетоном служит циклогексанон. Может быть получено соответствующее дегидратированное соединение, например такое, у которого R4 обозначает 1-циклогексенил, в частности обработкой с помощью пара-TsOH кипячением с обратным холодильником в толуольном растворе.
В объем изобретения включен также способ получения соединения по изобретению или соли формул III и IV, как это изложено выше, который включает взаимодействие соединения формулы VIII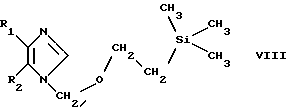
в которой R1 и R2 имеют значения, указанные выше, с соответствующим альдегидом, кетоном, дисульфониламином, дисульфидом, диселенидом, дителлуридом или галогенидом и при необходимости введением целевого заместителя R3 или дальнейшим превращением получаемого продукта с необязательным переводом соединения по изобретению в форму свободного соединения или соли.
Соединения формулы VIII могут быть получены путем обработки соответствующего 1H-имидазола формулы I, т.е. соответствующего соединения формулы I, в которой R4 обозначает Н, 2-(триметилсилил)этоксиметилгалогенидом (например, хлоридом), в частности в присутствии калийбис(триметилсилил)амида в охлажденном (например до -78oС) растворе ДМФ/ТГФ. В результате такого процесса образуется смесь соответствующих 1-2(триметилси-лил)этоксиметилимидазолов формул VIII и IX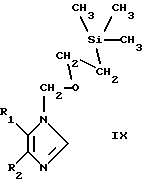
в которой R1 и R2 имеют значения, указанные выше.
Соединения формулы VIII представляют собой новые промежуточные продукты для получения других соединений по изобретению и как таковые включены в объем настоящего изобретения. Соединения формулы IX представляют собой соединения по изобретению.
В другом предпочтительном варианте вместо триметилсилилэтоксиметильной защитной группы в качестве группы, защищающей азотный атом, используют алкоксиалкильную группу, например диалкоксиалкильную защищающую азотный атом группу, в частности диэтоксиметильную защищающую азотный атом группу. Такую алкоксиалкильную защитную группу можно вводить обработкой соответствующего 1H-имидазола формулы I, т.е. соответствующего соединения формулы I, в которой R4 обозначает Н, триалкилортоформиатом, например триэтилортоформиатом, как это, в частности, описано ниже в примерах.
Синтез соединений по изобретению описан ниже в примерах.
ПРИМЕРЫ
Примеры 1 и 2. 4-(4-фторфенил)-5-(4-пиридил)-1-[2-(триметилсилил)этоксиметил] имидазол и 4-(4-пиридил)-5-(4-фторфенил)-1-[2-(триметилсилил) этоксиметил] имидазол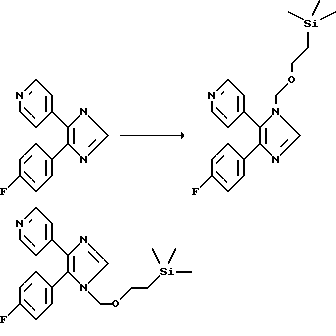
1 г (4,18 ммоль) 4-(4-фторфенил)-5-(4-пиридил)-1Н-имидазола (3) растворяют в ДМФ/ТГФ (50 мл/20 мл) и охлаждают до -78oС. При -78oС вводят 6,7 мл (5 ммоль) 15%-ного калийбис(триметилсилил)амида, перемешивают в течение 30 мин, затем добавляют 2-(триметилсилил)этоксиметилхлорид и реакционную смесь нагревают до комнатной температуры (КТ), по истечении 2 ч сливают в воду и 3 раза экстрагируют этилацетатом. Объединенные органические фазы сушат над Na2SO4, упаривают досуха и хроматографируют (SiO2, смесь ацетон/гексан в соотношении от 4/6 до 6/4) с получением 1-[2-(триметилсилил)этоксиметил]-4-(4-фторфенил)-5-(4-пиридил)имидазола, который элюируют первым в виде белых кристаллов (218 мг, 14%), а затем 4-(4-пиридил)-5-(4-фторфенил)-1-[2(триметилсилил)этоксиметил] имидазола в виде белых кристаллов (590 мг, 38%). Точное определение структур осуществляли с помощью ROESY-, HSQC- и НМВС-спектрометрии.
1H-ЯМР (360 МГц, СDСl3) 1-[2-(триметилсилил)этоксиметил]-4-(4-фторфенил)-5-(4-пиридил)имидазола (пример 1): 0,00 (s, 9H), 0,92 (t, 2H), 3,55 (t, 2H), 5,15 (s, 2H), 6,95 (t, 2H), 7,36 (d, 2H), 7,42 (двойной d, 2H), 7,72 (s, 1H), 8,68 (d, 2H); 4-(4-пиридил)-5-(4-фторфенил)-1-[2-(триметилсилил)этоксиметил]имидазола (пример 2): 0,00 (s, 9H), 0,90 (t, 2H), 3,48 (t, 2H), 5,10 (s, 2H), 7,20 (t, 2H), 7,35-7,45 (m, 4H), 7,75 (s, 1H), 8,45 (d, 2H).
В другом варианте 1H-имидазоловый исходный продукт можно превращать в соответствующие 4-(4-фторфенил)-5-(4-пиридил)-1-(1,1-диэтоксиметил)имидазол и 4-(4-пиридил)-5-(4-фторфенил)1-(1,1-диэтоксиметил)имидазол.
1-(1,1-диэтоксиметил)-4-(4-фторфенил)-5-(4-пиридинил) имидазол и 1 (1,1-диэтоксиметил)-5-(4-фторфенил)-4-(4-пиридинил)имидазол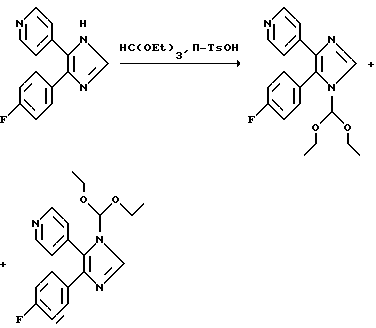
72,7 г (0,304 моль) 4-(4-фторфенил)-5-(4-пиридинил)имидазола и 1,1 г (5 ммоль) n-TsOH•H2O растворяют в 770 мл горячего триэтилортоформиата и кипятят с обратным холодильником, медленно отгоняя примерно 300 мл триэтилортоформиата и этанола. По истечении 2 ч реакционную смесь упаривают досуха и остаток растворяют в 500 мл трет-бутилметилового эфира. Осторожно добавляют 5 л гексана, осадок отфильтровывают и промывают смесью трет-бутилметиловый эфир/гексан в соотношении 1:9. Фильтрат промывают 1н. Na2CO3, сушат над Na2SO4 и упаривают. Дважды добавляют ксилол и вновь упаривают, получая 79,3 г желто-коричневатого вязкого масла (выход: 76% смеси в соотношении ~1:1), которое используют без дополнительной очистки.
Пример 3. 4-(4-фторфенил)-2-[(RS)-1-гидрокси-4'-(4-фторбензил)-5-(4-пиридил)имидазол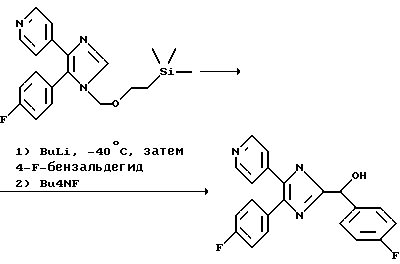
0,085 мл (0,13 ммоль) 1,6 М н-BuLi в гексане при -40oС вводят в раствор 50 мг (0,13 ммоль) 1-[2-(триметилсилил)этоксиметил]-4-(4-фторфенил)-5-(4-пиридил)имидазола (пример 2) в 1,4 мл ТГФ. После 15-минутной выдержки при -40oС в реакционную смесь добавляют 0,018 мл (0,18 ммоль) 4-фторбензальдегида в 0,4 мл ТГФ, нагревают до КТ и по истечении 10 мин выливают в воду и 3 раза экстрагируют этилацетатом. Объединенные органические фазы сушат над Na2SO4 и упаривают досуха, получая 64 мг целевого N-защищенного соединения. Для удаления СЭМ-защитной группы этот последний продукт растворяют в 2 мл ТГФ, в течение 1 ч при 60oС обрабатывают 4,3 мл 1 М Bu4NF в ТГФ, выливают в насыщенный раствор NаНСО3 и 3 раза экстрагируют этилацетатом. Объединенные органические фазы сушат над Na2SO4, упаривают досуха и хроматографируют (SiO2), толуол/ЕtOН/концентрированный раствор NH3 в соотношении 90/10/0,6) с получением в виде белых кристаллов 32 мг (выход на 2 стадиях: 67%) указанного в заголовке соединения.
1H-ЯМР (360 МГц, ДМСО-d6): 5,80 (s, 1H), 6,30 (широкий s, ОН), 7,15 (t, 2H), 7,20-7,30 (широкий s, 1H), 7,37 (d, 2H), 7,42-7,48 (m, 2H), 7,52-7,58 (m, 2H), 8,38-8,51 (широкий s, 2H), 12,50-12,60 (широкий s, NH).
В другом варианте для получения 4-(4-фторфенил)-5-(4-пиридил)-2-(2,3,5,6-тетрафторпиридинил)имидазола вместо 1-[2-(триметилсилил)этоксиметил] -4-(4-фторфенил)-5-(4-пиридинил)имидазола может быть использована смесь 1-(1,1-диэтоксиметил)-4-(4-фторфенил)-5-(4-пиридил)имидазола и 3-(1,1-диэтоксиметил)-4-(4-фторфенил)-5-(4-пиридинил)имидазола, полученная по описанной выше методике.
4-(4-фторфенил)-5-(4-пиридил)-2-(2,3,5,6-тетрафторпиридинил)имидазол
66 мл (45 ммоль) 1,6 М н-BuLi при -45oС добавляют к 15 г (43 ммоль) смеси 1-(1,1-диэтоксиметил)-4-(4-фторфенил)-5-(4-пиридинил)имидазола и 1-(1,1 -диэтоксиметил)-5-(4-фторфенил)-4-(4-пиридинил)имидазола в соотношении ~ 1: 1 в 210 мл ТГФ. После 15-минутной выдержки при -45oС реакционную смесь охлаждают до -55oС и быстро вводят в нее 5,1 мл (47 ммоль) пентафторпиридина. Охлаждающую баню удаляют, реакционной смеси дают нагреться до -15oС и выливают в 1 л воды, после чего подкисляют 100 мл 2н. НСl. После перемешивания в течение 5 мин смесь объединяют с насыщенным раствором Nа2СО3 и три раза экстрагируют этилацетатом. Объединенные органические фазы сушат над Na2SO4, фильтруют и упаривают досуха с получением в виде коричневатых кристаллов 16,3 г указанного в заголовке соединения. В результате хроматографии (SiO2, ацетон/гексаны в соотношении 1:1) получают указанное в заголовке соединение совместно с некоторым количеством непрореагировавшего и незащищенного имидазолового исходного материала, который можно было бы удалить промывкой ацетоном с получением 8,7 г (выход: 52,4%) указанного в заголовке соединения.
1H-ЯМР (360 МГц, CDCl3): 7,30-7,40 (широкий t, 2H), 7,45 (d, 2H), 7,58 (широкий q, 2H), 8,52 (широкий s, 2H). MC (m/z): 388,9 (МН+).
По методам, по существу аналогичным описанным в примере 3, и с использованием соответствующих исходных продуктов получают следующие соединения формулы X, которые представлены в таблице 1 (примеры 4-12).
Пример 13. 2-(1-циклогексенил)-4-(4-фторфенил)-5-(4-пиридил)имидазол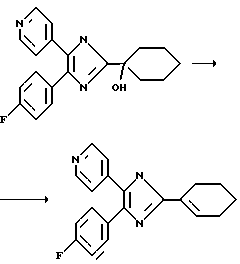
50 мг (0,15 ммоль) продукта из примера 7, т.е. 2-[(1-гидрокси)циклогексил]-4-(4-фторфенил)-5-(4-пиридил)-1Н-имидазола, растворяют в 100 мл толуола и в течение 15 мин кипятят совместно со 100 мг п-TsOH. Реакционную смесь выливают в насыщенный раствор NaHCO3 и 3 раза экстрагируют этилацетатом. Объединенные органические фазы сушат над Na2SO4, упаривают досуха и хроматографируют (SiO2, ацетон/гексан в соотношении 4:6) с получением в виде белых кристаллов 38 мг (выход: 81%) указанного в заголовке соединения.
lH-ЯMP (360 МГц, ДМСО-d6) смесь NH-таутомеров в соотношении 8:2: 1,60 (m, 2Н), 1,68 (m, 2H), 2,18 (широкий s, 2H), 2,50 (широкий s, 2H), 6,55 (широкий s, 0,8H), 6,62 (широкий s, 0,2H), 7,15-7,60 (m, 6H), 8,40 (d, 1,6H), 8,52 (d, 0,4H), 12,25 (широкий s, 0,2H), 12,37 (широкий s, 0,8H)
По методам, по существу аналогичным описанным в примере 13, и с использованием соответствующих исходных продуктов получают следующие соединения формулы X, которые представлены в таблице 2 (примеры 14 и 15).
Примеры 16 и 17: 4-(4-фторфенил)-5-(4-пиридил)-2-(2-амино-3,5,6-трифторпиридинил)имидазол и 4-(4-фторфенил)-5-(4-пиридил)-2-(2,6-диамино-3,5-дифторпиридинил)имидазол
2 г (5,15 ммоль) продукта из примера 10, т.е. 4-(4-фторфенил)-5-(4-пиридил)-2-(2,3,5,6-тетрафторпиридинил)имидазола, суспендируют в 200 мл концентрированного раствора NH3 (25%-ного) и в течение 5 ч выдерживают при 150oС в герметично закрытом стальном баллоне. Воду выпаривают и остаток хроматографируют (SiO2, трет-бутилметиловый эфир/МеОН/концентрированный раствор NH3 в соотношении 98/2/0,2) с получением в виде слегка окрашенных кристаллов указанных в заголовке соединений: 880 мг (выход: 44,4%) 4-(4-фторфенил)-5-(4-пиридил)-2-(2-амино-3,5,6-трифторпиридинил)имидазола и 650 мг (выход: 33%) 4-(4-фторфенил)-5-(4-пиридил)-2-(2,6-диамино-3,5-дифторпиридинил)имидазола.
4-(4-фторфенил)-5-(4-пиридил)-2-(2-амино-3,5,6-трифторпиридинил)имидазол:
1Н-ЯМР (400 МГц, ДМСО-d6) смесь таутомеров: 6,80 (s, 2H), 7,25 (широкий t, 0,6H), 7,38 (t, 1,4H), 7,45 (d, 2H), 7,58 (t, 2H), 8,49 (d, 1,4H), 8,62 (широкий d, 0,6H)
MC (m/z): 385 (М+)
4-(4-фторфенил)-5-(4-пиридил)-2-(2,6-диамино-3,5-дифторпиридинил) имидазол:
1Н-ЯМР (400 МГц, ДМСО-d6) смесь таутомеров: 5,75 (s, 2H), 7,22 (t, 0,6Н), 7,33 (t, 1,4H), 7,41-7,47 (m, 2H), 7,52-7,58 (m, 2H), 8,47 (d, 1,4H), 8,58 (d, 0,6H)
MC (m/z): 382 (M+)
Пример 18. 4-(4-фторфенил)-2-[(1-амино)циклогексил]-5-(4-пиридил)имидазол
а) Гидробромид 1-(4-фторфенил)-2-бром-2-(4-пиридил)этанона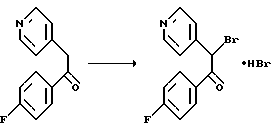
74,4 г (0,46 моль) брома в 160 мл уксусной кислоты в течение 10 мин при 21oС вводят в раствор 100 г (0,46 моль) 4-фторфенил-4-пиридилметилкетона (см. I. Lantos и др. , J. Med. Chem. 1984, 27, 72-75) в 800 мл уксусной кислоты. Желтые кристаллы отфильтровывают, промывают уксусной кислотой, диэтиловым эфиром и гексаном и затем сушат под пониженным давлением с получением 250 г (выход: 72%) гидробромида целевого соединения.
б) 4-(4-фторфенил)-2-(1-N-карбобензилоксициклогексил)-5-(4-пиридил)имидазол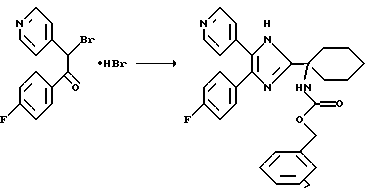
13,9 г (50 ммоль) 1-N-карбобензилокси-1-циклогексанкарбоновой кислоты [см. Е. Didier и др., Tetrahedron 1992, 48 (39), 8471] и 4,8 г (50 ммоль) карбоната аммония (фирма Fluka) растворяют в 50 мл ДМФ и выдерживают при 110oС в течение 10 мин до прекращения выделения газа. Реакционную колбу охлаждают до 60oС, добавляют в виде твердого вещества 3,75 г (10 ммоль) гидробромида 1-(4-фторфенил)-2-бром-2-(4-пиридил)этанона и в течение 2,5 ч выдерживают при 125oС. Реакционную смесь выливают в 1 М раствор Na2CO3 и трижды экстрагируют этилацетатом. Объединенные органические фазы промывают водой, сушат над Na2SO4 и упаривают досуха, получая 4,8 г сырого указанного в заголовке соединения, в результате хроматографии которого (SiO2, этилацетат) в виде светло-желтых кристаллов выделяют 1,4 г (выход: 30%) чистого указанного в заголовке соединения.
1H-ЯМР (400 МГц, СDСl3): 1,25-2,40 (m, 10Н), 5,12 (s, 2H), 7,08-7,16 (m, 2H), 7,30-7,50 (m, 9Н), 8,50 (d, 2H)
MC (m/z): 471,2 (МН+)
в) 4-(4-фторфенил)-2-(1-аминоциклогексил)-5-(4-пиридил)имидазол (243-653)
1,6 г (4 ммоль) 4-(4-фторфенил)-2-[(1-N-карбобензилокси)циклогексил]-5-(4-пиридил)имидазола под давлением 1 атм в присутствии 0,7 г 10%-ного Pd/C в течение 2 ч при комнатной температуре растворяют в 140 мл EtOH и гидрогенизируют. В результате фильтрования и выпаривания растворителя с последующей перекристаллизацией из этилацетата/диэтилового эфира в виде не совсем белых кристаллов получают 0,63 г (выход: 47%) целевого амина.
1H-ЯМР (400 МГц, ДМСО-d6): 1,25-1,78 (m, 8H), 1,95-2,10 (широкий t, 2H), 7,25-7,34 (широкий t, 2H), 7,40 (d, 2H), 7,47-7,52 (m, 2H), 8,43 (d, 2H)
MC (m/z): 336 (M+)
Пример 19. 4-(4-фторфенил)-5-(4-пиридил)-2-(4-н-бутилокси-1-метилпиперидин 4 ил) имидазол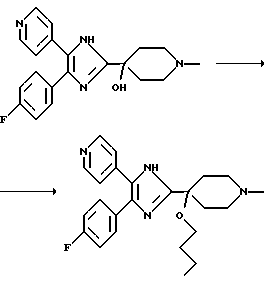
22,2 г (63 ммоль) продукта из примера 12, т.е. 4-(4-фторфенил)-5-(4-пиридил)-2-(4-гидрокси-1-метилпиперидин-4-ил)имидазола при нагревании до 40o С растворяют в 1 л 1-бутанола. По каплям добавляют 27,8 г (283 ммоль) концентрированной H2SO4 и первоначально образующуюся суспензию кипятят в течение 3,5 ч, одновременно отгоняя ~200 мл 1-бутанола. Реакционную смесь охлаждают до комнатной температуры и выливают в 500 мл насыщенного раствора Nа2СО3. Водную фазу экстрагируют этилацетатом и объединенные органические фазы сушат над Na2SO4, фильтруют и упаривают досуха. В результате хроматографии (SiO2, трет-бутилметиловый эфир/МеОН/концентрированный раствор NH3 в соотношении от 96/4/0,4 до 70/30/1) получают в виде желтых кристаллов 15,5 г (выход: 60,3%) указанного в заголовке соединения. Образец перекристаллизовывают из смеси CH2Cl2/трет-бутилметиловый эфир с получением бесцветных кристаллов; tпл: 177oС.
1H-ЯМР (400 МГц, ДМСО-d6): смесь таутомеров, которые дублируют сигналы ароматических групп: 0,80 (широкий t, 3Н), 1,25-1,35 (m, 2H), 1,38-1,48 (m, 2H), 2,12 (широкий s, 4Н), 2,18 (s, 3Н), 2,35 (m, 2H), 2,42 (m, 2H), 3,12 (t, 2H), 7,18 (t, 0,5H), 7,32 (t, 1,5H), 7,40 (m, 2H), 7,48 (m, 2H), 8,40 (d, 1,5H), 8,53 (d, 0,5H)
MC (m/z): 408 (М+, 20%); 351 (100%); 335 (95%)
Соединения по изобретению в форме свободных соединений, фармацевтически приемлемых кислотно-аддитивных солей или физиологически расщепляемых сложных эфиров, которые проявляют фармакологическое действие и могут быть использованы в виде фармацевтических препаратов, например, в терапии, при лечении заболеваний и состояний, как это изложено ниже, в дальнейшем называются как средства по изобретению.
Средства по изобретению обладают, в частности, способностью ингибировать МАР-киназу р38 (активированная митогеном протеинкиназа). Таким образом, действие средств по изобретению заключается в подавлении продуцирования воспалительных цитокинов, таких как ФНО_α и ИЛ-1, a также в способности блокировать воздействие этих цитокинов на их клетки-мишени. Это и другое фармакологическое действие средств по изобретению может быть проиллюстрировано на примере стандартных методов испытаний, как это, например, описано ниже.
Исследование с помощью МАР-киназы р38
Субстратом (GST-ATF-2; слитый протеин, включающий с 1 по 109 аминокислоту протеина ATF-2 и протеин GST, полученный экспрессией в Е.coli), покрывают лунки титрационных микропланшет (50 мкл/лунку; 1 мкг/мл в ЗФР/0,02% азида Na) и инкубируют в течение ночи при 4oС. На следующий день титрационные микропланшеты четырежды промывают смесью ЗФР/0,5% Твина 20/0,02% азида Na и блокируют смесью ЗФР/2% БСА/0,02% азида Na при 37oС в течение 1 ч. Планшеты вновь 4 раза промывают смесью ЗФР/0,5% Твина 20/0,02%. Затем начинают реакцию активации киназы добавлением к 10-микролитровым аликвотам следующих реагентов с доведением конечного объема реакционной смеси до 50 мкл.
1. Средства по изобретению, титрованные либо последовательными 10-кратными разведениями, либо растворителем (ДМСО), либо водой до концентрации от 10 до 0,001 мкМ.
2. Буфер для киназы (5-кратный); рН: 7,4; 125 мМ Hepes (исходный 1 М раствор; Gibco 15630-056), 125 мМ β_глицерофосфат (Sigma G-6251): 125 мМ MgCl2 (Merck 5833); 0,5 мМ ортованадат натрия (Sigma 5-6508), 10 мМ дитиотреитол (Boehringer Mannheim 708992). Буфер для киназы (5-кратный) должен быть свежеприготовленным в день испытания из исходных растворов 5-кратной концентрации, выдерживаемых при КТ. Дитиотреитол хранят при -20oС и добавляют в качестве последнего реагента.
3. МАР-киназа His-p38 (10 нг/лунку; Novartis - слитый протеин, включающий полноразмерную МАР-киназу р38 мыши и метаболитный гистидин, полученный экспрессией в Е.coli).
4. Охлажденный раствор АТФ (конечная концентрация 120 мкМ; Sigma А-9187).
5. Вода.
После инкубации в течение 1 ч при 37oС реакцию активации киназы останавливают четырехкратным промыванием планшетов, как это указано выше. Далее фосфорилированный GST-ATF-2 определяют добавлением:
1) антитела к ATF-2 PhosphoPlus (Thr 71) (50 мкл/лунку; конечное разведение 1/1000 в смеси ЗФР/2% БСА/0,02% азида Na; New England Biolabs 9221L) с инкубацией в течение 90 мин при КТ;
2) меченного биотином козьего антитела к кроличьему IgG (50 мкл/лунку; конечное разведение 1/3000 в смеси ЗФР/2% БСА/0,02% азида Na; Sigma В-9642) с инкубацией в течение 90 мин при КТ;
3) стрептавидин-щелочной фосфатазы (50 мкл/лунку; конечное разведение 1/5000 в смеси ЗФР/2% БСА/0,02% азида Na; Jackson Immunoresearch 016-050-084) с инкубацией в течение 30 мин при КТ;
4) субстрата [100 мкл/лунку; фосфатазный субстрат в таблетках Sigma 104, 5 мг/таблетку; 104-105; 1 мг/мл в буфере для субстрата, диэтаноламин (97 мл/л; Merck 803116) + MgCl2•6H2O (100 мг/л; Merck 5833) + азид Na (0,2 г/л) + 1M HC1 до рН 9,8] с инкубацией в течение 30 мин при КТ.
После стадий 1, 2 и 3 титрационные микропланшеты четыре раза промывают смесью ЗФР/0,5% Твина 20/0,02% азида Na. После стадии 4 планшеты считывают с помощью планшет-ридера фирмы Bio-Rad в двухволновом режиме (измерительный фильтр на 405 нм и эталонный фильтр на 490 нм). Вычитают фоновое значение (без АТФ) и с помощью компьютерной программы Origin (4-параметровая логистическая функция) рассчитывают значения ИК50.
Значения ИК50 средств по изобретению при ингибировании МАР-киназы р38, когда их определяют в ходе проведения вышеописанного испытания, как правило, находятся в интервале от примерно 1 мкМ до примерно 10 нМ или менее. Так, например, в таком испытании значение ИК50 для соединения из примера 17 составляет примерно 10 нМ.
Испытание на ингибирование выделения ФНО_α из hPBMC
Мононуклеарные клетки периферической крови человека (hPBMC) получают из периферической крови здоровых добровольцев путем разделения в градиенте плотности фиколл-гипак в соответствии с методом Hansell и др., J. Imm. Methods (1991) 145: 105, и используют в концентрации 105 клеток/лунку в среде RPMI 1640 плюс 10% фетальной телячьей сыворотки (ФТС). Перед добавлением γ_интерферона (IFN_γ) (100 ед/мл) и липополисахарида (ЛПС) (5 мг/мл) клетки инкубируют с серийными разведениями тестируемых соединений в течение 30 мин при 37o С и в дальнейшем дополнительно инкубируют в течение трех часов. Инкубацию завершают центрифугированием при 1400 об/мин в течение 10 мин. ФНО_α в супернатанте определяют с помощью коммерчески доступного набора для твердофазного иммуноферментного анализа [Innotest hTNFα, фирма Innogenetics N.V., Цвийнаарде, Бельгия). Средства по изобретению тестируют в концентрации 0-10 мкМ. В ходе проведения этого исследования приведенные в качестве примеров средства по изобретению, как правило, подавляют выделение ФНО при значениях ИК50 от примерно 1 мкМ до примерно 10 нМ или менее. Так, например, значение ИК50 для соединения из примера 17, когда его определяют в таком тесте, составляет примерно 90 нМ.
Испытание на ингибирование продуцирования ФНО_α, стимулированного ЛПС, у мышей
Инъекция липополисахарида (ЛПС) индуцирует быстрое высвобождение растворимого фактора некроза опухоли (ФНО_α) в периферию. Эту модель применяют для оценки высвобождения предполагаемых блокаторов ФНО in vivo.
Мышам линии OF1 (самки 8-недельного возраста) внутривенной инъекцией вводят 20 мг/кг ЛПС. Через 1 ч у животных берут кровь и в соответствии с методом твердофазного иммуноферментного анализа с использованием антитела к ФНО-α определяют уровень содержания ФНО в плазме. ЛПС при применении в концентрации 20 мг/кг обычно индуцирует высвобождение до 15 нг ФНО-α/мл плазмы. Тестируемые соединения вводят либо орально, либо подкожно за 1-4 ч до инъекции ЛПС. За точку начала отсчета принимают ингибирование высвобождения ФНО, вызванного ЛПС.
В ходе проведения вышеописанного испытания степень ингибирования продуцирования ФНО, проявляемая средствами по изобретению, когда их вводят орально в дозе 10 мг/кг, как правило, составляет от примерно 50 до примерно 90% или выше. Так, например, степень ингибирования продуцирования ФНО для соединения из примера 17 в таком тесте составляет примерно 80%.
Как указано в описании вышеприведенных испытаний, средства по изобретению являются потенциальными средствами подавления высвобождения ФНО-α. Таким образом, фармацевтическое применение новых соединений состоит в следующем.
Средства по изобретению могут быть использованы для профилактики и лечения заболеваний и патологических состояний, вызванных цитокинами, такими как ФНО-α и ИЛ-1, например воспалительных состояний, например аутоиммунных болезней, тяжелых инфекционных заболеваний, отторжения трансплантата органа или ткани, в частности для лечения реципиентов при пересадке сердца, легкого, сочетания сердца/легкого, печени, почки, поджелудочной железы, кожи или роговицы, и для профилактики гомологической болезни, такой как следствие пересадки костного мозга.
Средства по изобретению особенно эффективны при лечении, профилактике и облегчении аутоиммунных болезней и воспалительных состояний, в частности воспалительных состояний с этиологией, включающих аутоиммунный компонент, таких как артрит (например, ревматоидный артрит, хронические прогредиентные артриты и деформирующий артрит) и ревматизма. Конкретными примерами аутоиммунных болезней, для лечения которых можно применять средства по изобретению, являются аутоиммунные гематологические нарушения (к которым относятся, например, гемолитическая анемия, апластическая анемия, анемия красных клеток и идиопатическая тромбоцитопения), системная красная волчанка, полихондрит, склеродома, гранулематоз Вегенара, дерматомиозит, хронический активный гепатит, тяжелая миастения, псориаз, синдром Стивенса-Джонсона, идеопатическая белая диарея, аутоиммунные воспалительные кишечные заболевания (к которым относятся, например, неспецифический язвенный колит и болезнь Крона), эндокринная офтальмопатия, болезнь Гревса, саркоидоз, рассеянный склероз, первичный билиарный цирроз печени, ювенильный диабет (сахарный диабет типа I), увеит (передний и задний), сухой и весенний кератоконъюнктивит, интерстициальный легочный фиброз, псориатический артрит и гломерулонефрит (с нефротическим синдромом или без него, включая, например, идиопатический нефротический синдром и нефропатию с минимальными изменениями).
Средства по изобретению могут быть использованы также для лечения, профилактики и улучшения состояния при астме, бронхите, пневмокониозе, эмфиземе легких и других заболеваний, связанных с недостаточной проходимостью и воспалением дыхательных путей.
Средства по изобретению эффективны при лечении нежелательных острых и сверхострых воспалительных реакций, которые опосредованы ФНО, прежде всего ФНО-α, например при острых инфекциях, в частности при септическом шоке (например, при эндотоксиновом бактериально-токсическом шоке и респираторном дистресс-синдроме взрослых), менингите, пневмонии, а также при сильных ожогах и при лечении кахексии или синдрома истощения, связанного с патологическим высвобождением ФНО, инфекционных заболеваний, рака или дисфункции органа, прежде всего кахексии, связанной со СПИДом, в частности связанной с ВИЧ-инфекцией или являющейся ее следствием.
Средства по изобретению особенно эффективны при лечении болезни костного обмена веществ, включая остеоартрит, остеопороз и другие воспалительные артриты.
В случае вышеперечисленных показаний соответствующая дозу обычно варьируется, как очевидно, в зависимости, например, от конкретно используемого средства по изобретению, подвергаемого лечению пациента, пути введения, а также природы и серьезности состояния, при котором проводят лечение. Однако обычно удовлетворительных результатов на животных достигают оральным введением при ежедневной дозе от примерно 1 до примерно 10 мг/кг. В случае более крупных млекопитающих, например людей, рекомендуемая ежедневная доза составляет от примерно 50 до примерно 750 мг средства по изобретению, вводимого орально в виде однократной дозы либо предпочтительно в виде дробных доз для двух-четырехразового ежедневного приема.
Средства по изобретению можно вводить любым обычным путем, например перорально, в частности в форме растворов для питья, таблеток или капсул, либо парентерально, в частности в форме растворов или суспензий для инъекций. Обычно для системного введения предпочтительны пероральные дозированные препаративные формы, хотя при некоторых показаниях средства по изобретению можно также вводить локально или чрескожно, например в форме крема или геля для кожи или аналогичного препарата, или же эти средства можно вводить в форме крема для глаз, геля или препарата в виде капель для глаз с целью нанесения на глаза или их можно вводить путем ингаляции, например для лечения астмы. Пригодные препаративные формы в дозах на один прием для перорального введения включают, например, 25-250 мг нового соединения на одну дозу.
В соответствии с вышеизложенным объектами настоящего изобретения являются также следующие.
А. Способ ингибирования продуцирования растворимого ФНО, прежде всего ФНО-α, или ослабления воспалительного процесса у субъекта (т.е. млекопитающего, прежде всего человека), нуждающегося в таком лечении, включающий введение в организм этого субъекта эффективного количества средства по изобретению, или способ лечения при любом из вышеупомянутых состояний, в частности способ лечения воспалительного или аутоиммунного заболевания или состояния, например ревматоидного артрита, или ослабления одного или нескольких симптомов любого из вышеупомянутых состояний.
Б. Средство по изобретению для применения в виде фармацевтического препарата, например при использовании в качестве иммунодепрессивного или противовоспалительного средства или при использовании с целью профилактики, улучшения состояния или лечения любого заболевания или состояния, как это описано выше, например аутоаллергического или воспалительного заболевания или состояния.
В. Фармацевтическая композиция, включающая средство по изобретению в сочетании с фармацевтически приемлемым разбавителем или наполнителем, например для применения в качестве иммунодепрессивного или противовоспалительного средства или для использования с целью профилактики, улучшения состояния или лечения любого заболевания или состояния, как это описано выше, например аутоаллергического или воспалительного заболевания или состояния.
Г. Применение средства по изобретению при приготовлении лекарственного препарата, предназначенного для использования в качестве иммунодепрессивного или противовоспалительного средства или для использования с целью профилактики, улучшения состояния или лечения любого заболевания или состояния, как это описано выше, например аутоаллергического или воспалительного заболевания или состояния.
Пример 20. Композиция таблетки, которую получают непосредственно прессованием, приведена в табл. 3.
Таблетки, в которых доза активного вещества составляет от 5 до 125 мг, могут быть получены путем варьирования общего веса таблетки или соотношения первых трех ингредиентов. Обычно предпочтительно поддерживать соотношение микрокристаллическая целлюлоза: безводная лактоза, равное 1:1.

Изобретение относится к 2-замещенным 4,5-диарилимидазолам общей формулы (I), где R1 - 4-пиридил; R2 - фенил, нафт-1-ил или нафт-2-ил, который необязательно может содержать до 5 заместителей, выбранных из галогена; R3 - водород; R4 - пиридил, необязательно замещенный галогеном или аминогруппой. Также описан способ получения этих соединений и фармацевтические композиции на их основе. Технический результат: изобретение может быть использовано в медицине для лечения воспалительных или аутоиммунных заболеваний. 5 с. и 1 з.п.ф-лы, 3 табл.
в которой R1 - 4-пиридил;
R2 - фенил, нафт-1-ил или нафт-2-ил, который необязательно может содержать до 5 заместителей, выбранных из галогена;
R3 - водород;
R4 - пиридил, необязательно замещенный галогеном или аминогруппой,
в форме свободного соединения или фармацевтически приемлемой кислотно-аддитивной соли.
4-(4-фторфенил)-5-(4-пиридил)-2-(2,3,5,6-тетрафторпиридинил)имидазола,
4-(4-фторфенил)-5-(4-пиридил)-2-(2-амино-3,5,6-трифторпиридин-ил)имидазола,
4-(4-фторфенил)-5-(4-пиридил)-2-(2,6-диамино-3,5-дифторпиридин-ил)имидазола.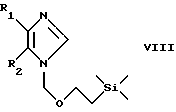
в которой R1 и R2 имеют значения, указанные выше, с соответствующим R4-галогенидом и при необходимости дальнейшее превращение полученного продукта с необязательным его переводом в форму свободного соединения или соли.
| WO 9503297 A1, 02.02.1995 | |||
| WO 9314081 A1, 22.07.1993 | |||
| WO 9712876 A1, 10.04.1997 | |||
| RU 94036003 A1, 10.08.1996 | |||
| WO 9314082 A1, 22.07.1993. |

