Изучение суточных доз болеутоляющих опиумных средств показало, для устранения боли приблизительно у 90% пациентов необходимо использовать суточные дозы в диапазоне с восьмикратными отклонениями. Такой чрезмерно широкий диапазон необходимых доз требует применения способа титрования, который является достаточно трудоемким как в отношении временных, так и в отношении материальных затрат, в результате чего пациент остается без требуемого болеутоляющего средства в течение недопустимо продолжительного периода времени.
При исследовании традиционных способов снятия боли с использованием опиумных болеутоляющих средств было установлено, что у отдельных индивидуумов имеются значительные отклонения в восприимчивости к данной дозе данного лекарственного средства, а поэтому среди пациентов наблюдается значительная вариабельность в отношении дозировки опиумных аналгетиков, необходимых для снятия боли без нежелательных побочных эффектов. Указанная выше необходимость прибегать к способу титрования вынуждает некоторых врачей-клиницистов устанавливать соответствующую дозу для каждого отдельного пациента, на что уходит значительное количество времени, поскольку такой способ требует тщательной оценки как терапевтического, так и побочного действия дозы, а также ее корректировки в течение нескольких дней или более, прежде чем будет установлена окончательная приемлемая доза.
В The American Pain Society's 3rd Edition of Principles of Analgetic Use in the Treatment of Acute Pain and Cancer Pain объясняется, "что следует сознавать, что оптимальная доза анальгетика широко варьируется среди пациентов. Исследования показывают, что для всех возрастных групп имеются огромные различия в дозах опиоидных анальгетиков, необходимых для устранения боли, даже среди пациентов с идентичными хирургическими поражениями... Это огромное различие указывает на необходимость установления режима приема анальгетиков, который включал бы меру предосторожности для дополнительных доз, и использования внутривенных болюсов и внутривенного вливания, для быстрого снятия острой боли. . . Дайте каждому аналгетику соответствующее испытание путем титрования дозы. . . перед тем, как переключиться на другой лекарственный препарат".
Лечение опиоидными аналгетиками с приемлемым ослаблением боли с существенно более узким диапазоном суточных доз будет способствовать улучшению эффективности и качества снятия боли.
Как известно из литературы, композиции опиоидных аналгетиков с контролируемым выделением, такие как морфин, гидроморфин или их соли, могут быть приготовлены в соответствующей матрице. Например, в патенте США N 4990341 (Goldie), который также был передан правопреемнику настоящей заявки, описываются композиции гидроморфина, где степень растворения in vitro лекарственной формы, измеренная the USP Paddle методом при 100 об/мин в 900 мл водного буфера (pH между 1,6 и 7,2), при 37oС составляет от 12,5 до 42,5 мас. %) гидроморфина, выделенного через 1 ч после ее введения от 25 до 55 мас.%, выделенного через 2 ч, от 45 до 75 мас.%, выделенного после 4 ч, и от 55 до 85 мас.% выделенного через 6 ч после ее введения.
Целью настоящего изобретения является способ, существенно улучшающий эффективность и качество снятия боли.
Другой целью настоящего изобретения является опиоидный аналгезирующий препарат, который обладает значительно более высокой эффективностью и качеством при устранении боли.
Следующей целью настоящей заявки является способ и препарат (препараты), которые позволяют существенно снизить уровень суточных доз приблизительно с восьмикратным превышением требуемых норм снятия боли приблизительно у 90% пациентов.
Другой целью настоящей заявки является способ и препараты, способствующие значительному снижению отклонений в суточных дозах и требований к препаратам, необходимым для снятия боли у практически всех пациентов.
Другой целью настоящей заявки является способ, который позволяет существенно снижать время и средства, необходимые для проведения титрования доз для пациентов, нуждающихся в опиоидных аналгетиках.
Еще одной целью настоящей заявки являются препараты опиоидных композиций с контролирующим выделением, которые имеют значительно меньшие индивидуальные отклонения с точки зрения дозы опиоидного аналгетика, требуемой для снятия боли без нежелательных побочных явлений.
Приведенные выше и другие цели достигаются с помощью настоящего изобретения, которое относится к твердым оральным лекарственным формам с контролируемым выделением, и с дозой, включающей в себя от 10 до 40 мг оксикодона или его соль в матрице, где степень растворения in vitro лекарственной формы, измеренная the USP Paddle методом при 100 об/мин в 900 мл водного буффера (pH от 1,6 до 7,2) при 37oС, составляет от 12,5 до 42,5 мас.% оксикодона, выделенного через 1 ч, от 25 до 56 мас.% оксикодона, выделенного через 2 ч, от 45 до 75 мас.% оксикодона, выделенного после 4 ч и от 55 до 85 мас. % оксикодона, выделенного через 6 ч после введения, причем скорость выделения не зависит существенно от pH, так что уровень максимальной концентрации оксидона в плазме, полученный in vivo, имеет место через 2-4,5 ч после введения лекарственной формы.
USP Paddle метод является the Paddle методом, описанным, например, в U. S.Pharmacopoeia XXII (1990).
В настоящем описании "не зависит существенно от pH" означает, что при любом данном времени разница между количеством оксикодона, выделенным при, например, pH 1,6, и количеством, выделенным при любом другом pH, например 7,2 (когда для измерений используют USP Paddle метод при 100 об/мин в 900 мл водного буффера), составляет 10 мас.% или меньше. Полученные выделенные количества во всех случаях являются средними по крайней мере по трем экспериментам.
Далее настоящая заявка относится к способу, который существенно снижает диапазон суточных доз, необходимых для снятия боли приблизительно у 90% пациентов, и который заключается во введении перорально твердого препарата с контролируемым выделением содержащего от 10 до 40 мг оксикодона или его соль, причем указанный препарат обеспечивает среднюю максимальную концентрацию оксикодона в плазме от 6 до 60 нг/мл в среднем через 2-4,5 ч после введения и среднюю минимальную концентрацию плазмы от 3 до 30 нг/мл в среднем через 10 до 14 ч после повторного введения g12h (т.е. каждые 12 ч) в стационарных условиях.
Далее настоящая заявка относится к способу, который значительно снижает объем суточных доз, необходимых для снятия боли у практически всех пациентов и который заключается во введении перорального твердого препарата с контролируемым выделением дозы, содержащего около 160 мг оксикодона или его соли; причем указанный препарат обеспечивает среднемаксимальную концентрацию оксикодона в плазме около 240 нг/мл в среднем через 2-4,5 ч после введения, среднеминимальную концентрацию плазмы около 120 нг/мл в среднем через 10-14 ч после повторного введения g12h (т.е. каждые 12 ч) приема в стационарных условиях.
Далее настоящая заявка относится к препаратам оксикодона с контролируемым выделением, содержащим от 10 до 40 мг оксикодона или его соли, причем указанные препараты обеспечивают среднемаксимальную концентрацию оксикодона в плазме от 6 до 60 мг/мл в среднем через 2-4,5 ч после приема и среднеминимальную концентрацию в плазме от 3 до 30 нг/мл в среднем через 10-14 ч после повторного введения g12h приема в стационарных условиях.
Далее настоящая заявка относится к препаратам оксикодоновым рецептурам с контролируемым выделением, содержащим около 160 мг оксикодона или его соли, причем указанные препараты обеспечивают среднемаксимальную концентрацию оксикодона в плазме около 240 нг/мл, в среднем через от 2 до 4,5 ч после приема и среднеминимальную концентрацию около 120 нг/мл в среднем через 10-14 ч после повторного введения g12h приема в стационарных условиях.
Представленные чертежи иллюстрируют сущность заявки, но не ограничивают существо заявки, изложенное в формуле изобретения.
На фиг. 1-4 приведены кривые временной зависимости для разницы болевой интенсивности и снятия боли для примера 17.
На фиг. 5 приведена зависимость средней концентрации оксикодона в плазме для 10 мг оксикодоновой композиции с контролируемым выделением, приготовленной согласно настоящей заявке, и сравнительные исследования по сравнению с известными препаратами.
Неожиданно было установлено, что оксикодоновые композиции настоящего изобретения с контролируемым выделением дают хороший эффект ослабления боли при существенно более меньшем уровне доз (примерно в 4 раза) (от 10 до 40 мг каждые 12 ч - около 1 ч дозирования) у приблизительно 90% пациентов. Это резко отличается от обычного приблизительно восьмикратного отклонения в диапазоне доз, необходимых для приблизительно 90% пациентов.
Применение от 10 до 40 мг доз оксикодона с 12-часовым контролируемым выделением для снятия боли у приблизительно 90% пациентов по сравнению с более широким диапазоном доз других mμ- агонистов-аналгетиков, применяемых при умеренных и сильных болях, является примером отличительных особенностей настоящего изобретения. Необходимо также подчеркнуть, что остальные 10% пациентов возможно будут успешно применять оксикодон с 12-часовым контролируемым выделением с относительно более низким объемом дозы, чем при использовании других подобных аналгетиков. В основном все из оставшихся 10% пациентов, оказавшиеся не восприимчивыми к оксикодону с контролируемым выделением от 10 до 40 мг каждые 12 ч, могут успешно использовать дозы, превышающие 40 мг (каждые 12 ч), вплоть по 160 мг (каждые 12 ч), используя любую дозу с количественным содержанием 10, 20, 40, 80 и 160 мг единичных доз или их комбинации. В противоположность этому использование других подобных аналгетиков, таких как морфин, будут требовать более широкого диапазона доз для снятия боли у оставшихся 10% пациентов. Например, известно, что дневная доза перорального морфина находится в пределах от 1 до более 20 г. Подобно этому будет требоваться широкий спектр доз орального гидроморфина.
Морфин, который можно рассматривать как прототип опиоидного аналгетика, изготавливается в виде готового препарата с 12-часовым контролируемым выделением (например, MS Contin⊗ таблетки, производства Purdue Pharma). Несмотря на то что как оксикодон с контролируемым выделением, так и морфин с контролируемым выделением, которые принимаются каждые 12 ч круглосуточно, обладают качественно сравнимыми клиническими фармакокинетическими характеристиками, однако в случае оксикодоновых препаратов, защищаемых настоящей заявкой, могут быть использованы в дозе, составляющей 1/2 по сравнению с дозами морфиновых препаратов с контролируемым выделением, выпускаемых промышленностью (таких как MS Contin⊗ ) для контроля приблизительно у 90% пациентов сильной боли.
Исследования повторных доз оксикодоновых препаратов с контролируемым выделением, применяемых каждые 12 ч, по сравнению с пероральным оксикодоном с немедленным выделением, принимаемым каждые 6 ч при той же самой суточной дозе, дали сравнимые результаты по абсорбции, а также по максимальным и минимальным концентрациям. Период для максимальной концентрации составляет приблизительно 2 - 4,5 ч после орального приема продукта с контролируемым выделением и 1 ч для продукта с немедленным выделением. Подобные исследования повторяемых доз с MS Contion⊗ таблетками по сравнению с морфином с немедленным выделением приводят к относительно сравнимым результатам, полученными для препаратов с контролируемым выделением настоящей заявки.
Существует незначительное отличие в параллелизме кривых дозовой зависимости для оксикодона либо в виде препарата с контролируемым выделением настоящей заявки, или перорального препарата с немедленным выделением, либо парентерального препарата оксикодона по сравнению с оральными и парентеральными опиоидами, с которыми оксикодон сравнивали при исследованиях дозового эффекта и относительной аналгезирующей активности доз.
- Beaver, et al., "Analgesic Studies of Codeine and
- Oxycodone in Patients with Cancer. II. Comparisons
- of Intra muscular Oxycodone With Intramuscular
- Morphine and Cadeine", J. Pharmacol. and Exp. Jher., vol. 207, N 1, p. 101 - 108, сообщает о сравнении кривых дозовой зависимости для парентерального оксикодона с кривыми для парентерального морфина и сравнении кривых дозовой зависимости для перорального и парентерального оксикодонов.
Обзор исследований дозового эффекта и оценок родственных аналгезирующих препаратов - mu-агонистов опиоидных аналгетиков, которые включают всего оксикодон, морфин, гидроморфин, леворфапол, метадон, меперидин, героин, указывает на то, что нет существенного отклонения от параллелизма в отношениях их дозовых зависимостей. Это установлено настолько хорошо, что становится основным критерием для установления относительных показателей аналгетической активности и соотношения доз, которые просто применяют при переходе пациента от одного mμ-агонистирующего аналгетика к другому, не обращая внимания на дозу первого. Если кривые дозовых реакций не параллельны, то показатели конверсии не будут иметь силу для широкого диапазона включенных доз, когда одно лекарство заменяется другим.
Клиническое значение, которое обеспечивается оксикодоновыми лекарственными препаратами с контролируемым выделением настоящей заявки при дозе от 10 до 40 мг каждые 12 ч для приемлемого снятия боли у приблизительно 90% пациентов от умеренной до сильной боли наряду с другими опиоидными анальгетиками, требующими двойной дозы, заключается в обеспечении эффективного для человека способа повторного позирования, необходимого для удаления боли. Экспертиза и время врачей и сестер наряду с продолжительностью недопустимой боли у пациентов, которую они должны выдерживать в течение титрования описанных аналгетиков, значительно снижаются благодаря эффективности оксикодонового препарата с контролируемым выделением настоящего изобретения.
Далее клиническое значение состоит в том, что доза приблизительно в 80 мг, контролируемая выделение оксикодона и принимаемая каждые 12 ч, будет обеспечивать приемлемое устранение боли от умеренной до тяжелой приблизительно у 90% пациентов, а при 160 мг оксикодона с контролируемым выделением при приеме каждые 12 ч будет обеспечиваться приемлемое удаление боли от умеренной до тяжелой приблизительно у всех пациентов.
Для того чтобы получить лекарственную дозу с контролируемым выделением, обладающую по крайней мере 12-часовым терапевтическим действием, обычно в фармацевтике получают препарат, который дает пиковый уровень лекарственного средства в плазме между 4 - 8 ч после введения (для исследования одной дозы). Заявители настоящего изобретения неожиданно нашли, что в случае оксикодона его пиковый уровень в плазме через 2 - 4,5 ч после введения дает по крайней мере 12-часовое ослабление боли, и что самое неожиданное, что это облегчение боли, полученное благодаря указанному препарату, большее, чем то, которое достигается при помощи препаратов, дающих пиковые уровни плазмы (оксикодона) в обычный период (до 2 ч после приема).
Кроме того, преимущество настоящей композиции заключается в том, что она выделяет оксикодон со скоростью, которая существенно не зависит от pH, что позволяет избежать дозовый демпинг при оральном приеме. Другими словами, оксикодон равномерно высвобождается в желудочно-кишечном тракте.
Оральная лекарственная форма может быть изготовлена, например, в виде гранул, сфероидов или шариков в капсуле или в другой приемлемой твердой форме. Однако предпочтительно, чтобы пероральная лекарственная форма представляла собой таблетки.
Настоящая пероральная лекарственная форма предпочтительно содержит от 1 до 500 мг, наиболее предпочтительно от 10 до 160 мг оксикодона гидрохлорида. Кроме того, лекарственная доза может содержать эквивалентные количества других оксикодоновых солей или оснований.
Используемая матрица может быть любой матрицей, при которой скорости растворения in vitro оксикодона находятся в необходимых пределах, и освобождение оксикодона происходит независимо от pH. Предпочтительно, чтобы матрица представляла собой матрицу с контролируемым выделением, хотя могут быть использованы и обычные матрицы, имеющие покрытия, которые контролируют выделение лекарства. Приемлемыми веществами для включения в матрицу с контролируемым выделением являются:
(а) усвояемые полимеры, такие как смолы, эфиры, целлюлоза, акриловые смолы и материалы белкового происхождения. Из указанных полимеров особенно предпочтительными являются гидроксиалкилцеллюлоза и карбоксиалкилцеллюлоза. Оральная лекарственная форма может содержать от 1 до 80 мас.% по крайней мере одного гидрофильного или гидрофобного полимера.
(б) гидролизуемые длинноцепочечные (C8-C50, особенно C12-C40), замещенные или незамещенные углеводороды, такие как жирные кислоты, жирные спирты, глицериновые эфиры жирных кислот, минеральные и растительные масла и парафины. Предпочтительны углеводороды, имеющие точку кипения от 25 до 90oC. Из этих длинноцепочечных углеводородов предпочтительны жирные (алифатические) спирты. Оральная лекарственная форма может содержать до 60 мас.% по крайней мере одного гидролизуемого длинноцепочечного углеводорода;
(с) полиалкиленгликоли. Oральная лекарственная форма может содержать до 60 мас.% по крайней мере одного полиалкиленгликоля.
Одна отдельная приемлемая матрицы содержит по крайней мере одну водорастворимую гидроксиалкильную целлюлозу по крайней мере один C12-C36, предпочтительно C14-C22, алифатический спирт и необязательно по крайней мере один полиалкиленгликоль.
Предпочтительно, чтобы по крайней мере одна гидроксиалкилцеллюлоза представляла собой гидрокси-(C1 до C6)-алкилцеллюлозу, такую как гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и особенно гидроксиэтилцеллюлозу. Количество по крайней мере одной алкилцеллюлозы в настоящей оральной лекарственной форме будет определяться inter alia точной скоростью требуемого выделения. Однако предпочтительно, чтобы оральная лекарственная доза содержала от 5 до 25%, особенно от 6,25 до 15 вес.%, по крайней мере одну гидроксиалкил целлюлозу.
По крайней мере один алифатический спирт может быть, например, лауриловым спиртом, миристиловым или стеариловым спиртами. Особенно предпочтительным решением настоящей лекарственной формы, однако, является по крайней мере один алифатический спирт, такой как цетиловый или цетостеариловый спирты. Количество по крайней мере одного алифатического спирта в настоящей оральной лекарственной форме будет определяться, как указано выше, точной скоростью требуемого выделения оксикодона. Это будет также зависеть от того, будет ли по крайней мере один полиалкиленовый спирт присутствовать или отсутствовать в оральной лекарственной форме. В отсутствие по крайней мере одного полиалкиленового спирта оральная лекарственная форма содержит от 20 до 50 вес.% по крайней мере одного алифатического спирта. Если по крайней мере один полиалкиленовый спирт присутствует в оральной лекарственной форме, то полнaя масса по крайней мере одного алифатического спирта и по крайней мере одного полиалкиленового спирта предпочтительно составляет от 20 до 50 мас.% от общей дозы.
В предпочтительном варианте осуществления настоящего изобретения композиция с контролируемым выделением содержит от 5 до 25 мас.% акриловой смолы и от 8 до 40 мас.% алифатического спирта от полной массы лекарственной формы. Наиболее предпочтительной акриловой смолой является Eudragit⊗ RSPM, выпускаемая в промышленности Rohm Pharma.
Для настоящей предпочтительной лекарственной формы отношение, например, по крайней мере одной гидроксиалкилцеллюлозы или акриловой смолы и по крайней мере одному алифатическому спирту/полиалкиленгликолю определяет в значительной степени скорость выделения оксикодона из лекарственного препарата. Предпочтительное отношение по крайней мере одной гидроксиалкил целлюлозы к по крайней мере одному алифатическому спирту/полиалкиленгликолю составляет от 1:2 до 1:4, особенно предпочтительно от 1:3 до 1:4.
По крайней мере одним из полиалкиленгликолей может быть, например, полипропиленгликоль или предпочтительно полиэтиленгликоль. Предпочтительно, чтобы средний молекулярный вес по крайней мере одного полиалкиленгликоля составлял от 1000 до 15000, лучше от 1500 до 12000.
Другая приемлемая матрица с контролируемым выделением может содержать алкилцеллюлозу (предпочтительно этилцеллюлозу), C12-C36 алифатический спирт и оптимально полиалкиленгликоль.
Дополнительно к вышеуказанным ингредиентам матрица с контролируемым выделением может также содержат приемлемые количества других веществ, например разбавители, смазки, связующие, гранулирующие добавки, красители, ароматизирующие вещества и средства для повышения скольжения, которые обычно применяются в фармацевтике.
Как альтернатива матрицы с контролируемым выделением настоящая матрица может быть обычно выделяющей матрицей, имеющей покрытие, которое контролирует выделение лекарства. В особенно предпочтительном варианте осуществления настоящего изобретения лекарственная форма представляет собой сфероиды с пленочным покрытием, содержащие активный ингредиент и водонерастворимый сферообразующий агент. Термин "сфероид" известен в фармацевтике и означает сферическую гранулу с диаметром от 0,5 до 2,5 мм, предпочтительно от 0,5 до 2 мм.
Сферообразующий агент может быть любым фармацевтически приемлемым веществом, который вместе с активным ингредиентом может быть преобразован в форму сфероида. Предпочтительна микрокристаллическая целлюлоза.
Приемлемая микрокристаллическая целлюлоза представляет собой, например, твердое вещество, такое как Avicel PH 101 (торговая марка FHC Corporation). Согласно предпочтительному варианту настоящей заявки сфероиды с пленочным покрытием содержат от 70 до 99 вес.%, предпочтительно от 80 до 95 вес.% сферообразующего агента, предпочтительно микрокристаллическую целлюлозу.
Кроме активного ингредиента и сферообразующего агента сфероиды могут также содержать связующее вещество. Приемлемые связующие, такие как водорастворимые полимеры с низкой вязкостью хорошо известны в фармацевтической литературе, однако предпочтительной является водорастворимая гидроксиалкилцеллюлоза, такая как гидроксипропилцеллюлоза. Кроме (или вместо) этого, сфероиды могут содержать водонерастворимый полимер, предпочтительно акриловый полимер, акриловый сополимер, такой как сополимер метакриловой кислоты и этилакрилата или этилцеллюлозы.
Предпочтительно, чтобы сфероиды были покрыты пленкой из материала, который позволяет выделяться оксикодону (или соли) с контролируемой скоростью в водной среде. Пленочное покрытие выбирают таким образом, чтобы достичь в сочетании с другими ингредиентами указанной выше скорости выделения in vitro (от 12,5 до 42,5 вес.% выделения через 1 ч после введения и т.п.).
Пленочное покрытие обычно включает водонерастворимый материал, такой как
а) парафин, либо один, либо в смеси с жирным спиртом;
б) шеллак или зеин;
в) водонерастворимую целлюлозу, предпочтительно этилцеллюлозу;
г) полиметакрилат, предпочтительно Eudragit⊗.
Предпочтительно, чтобы пленочное покрытие включало смесь водонерастворимого и водорастворимого материалов. Наряду с другими факторами отношение водонерастворимого к водорастворимому материалу определяется скоростью выделения, которая необходима, и характеристиками растворимости выбранного материала.
Водорастворимым материалом может быть, например, поливинилпирролидон или предпочтительно водорастворимая целлюлоза, особенно гидроксипропилметилцеллюлоза.
Приемлемая комбинация водонерастворимого и водорастворимого материалов для пленочного покрытия включает шеллак и поливинилпирралидон или, что является предпочтительным, этилцеллюлозу и гидроксипропилметилцеллюлозу.
Согласно настоящему изобретению для облегчения приготовления твердой пероральной лекарственной формы с контролируемым выделением еще один вариант настоящей заявки относится к способу приготовления твердой с контролируемым выделением оральной лекарственной формы, который включает в себя введение гидроморфина или его соли в матрицу с контролируемым выделением. Введение в матрицу может быть осуществлено, например, путем:
a) формирования гранул, включающих по крайней мере одну водорастворимую гидроалкилцеллюллозу и оксикодон или соль оксикодона;
b) смешивания гидроксиалкилцеллюлозы, содержащей гранулы по крайней мере с одним C12-C36 алифатическим спиртом и
c) если необходимо, прессования и формирования гранул.
Предпочтительно, чтобы гранулы формировались путем мокрого гранулирования гидроксиалкилцеллюлозы/оксикодона с водой. В особенно предпочтительном варианте этого способа количество воды, добавляемой во время стадии мокрого гранулирования, составляет предпочтительно от 1,5 до 5 частей, особенно от 1,75 до 3,5 частей от сухого веса оксикодона.
Настоящая твердая с контролируемым выделением лекарственная форма может быть также приготовлена в виде сфероидов с пленочным покрытием путем:
а) смешивания смеси, включающей оксикодон или соль оксикодона или водонерастворимый сферонизующий агент,
б) экструзии перемешанной смеси для получения экструдата;
в) сферонизации экструдата до получения сфероидов и
г) покрытия сфероидов пленочным покрытием.
Настоящая твердая пероральная лекарственная форма с контролируемым высвобождением и способ ее приготовления будут лишь проиллюстрированы в примерах.
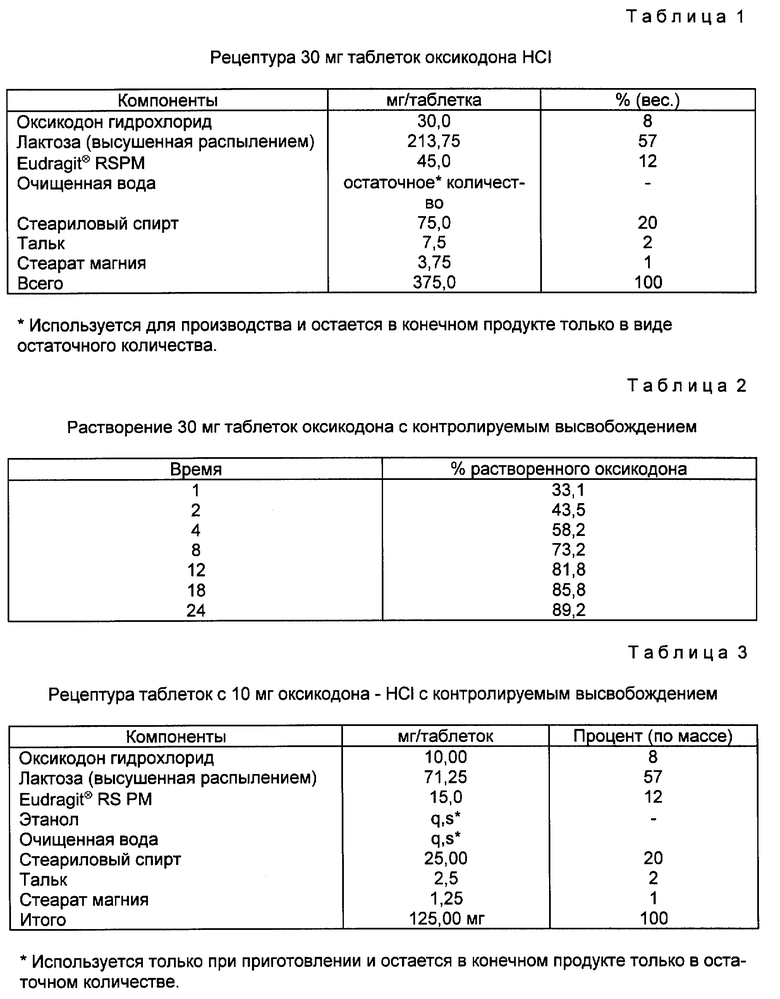
Пример 1. Таблетки с 30 мг оксикодона HCl с контролируемым выделением - водное получение.
Требуемые количества оксикодона гидрохлорида, осушенного распылением лактозы и Eudragit⊗ RSPM, помещают в смеситель соответствующего размера, и смешивают приблизительно 5 мин. При смешивании порошка смесь гранулируют с достаточным количеством воды и получают мокрую гранулированную массу. Затем гранулы высушивают в сушилке с псевдоожиженным слоем при 60oC и затем пропускают через сито 8 меш. После этого гранулы подсушивают и пропускают через сито 12 меш. Требуемое количество стеарилового спирта расплавляют приблизительно при 60-70oC и добавляют, перемешивая при этом, расплавленный стеариловый спирт. Теплые гранулы возвращают в смеситель.
Гранулы с покрытием удаляют из смесителя и охлаждают. Затем гранулы пропускают через сито 12 меш. После этого гранулят смазывают путем смешивания с требуемым количеством талька и стеарата магния в соответствующем смесителе. Прессуют таблетки по 375 мг по весу с помощью приемлемой таблитирующей машины. Рецептура таблеток примера 1 дана в табл. 1.
Затем таблетки по примеру 1 исследуют на растворение методом с использованием Basket Method при 37oC, 100 об/мин; в первый час в 700 мл желудочной жидкости при pH 1,2 с последующей заменой на 900 мл при 7,5. Результаты представлены в табл. 2.
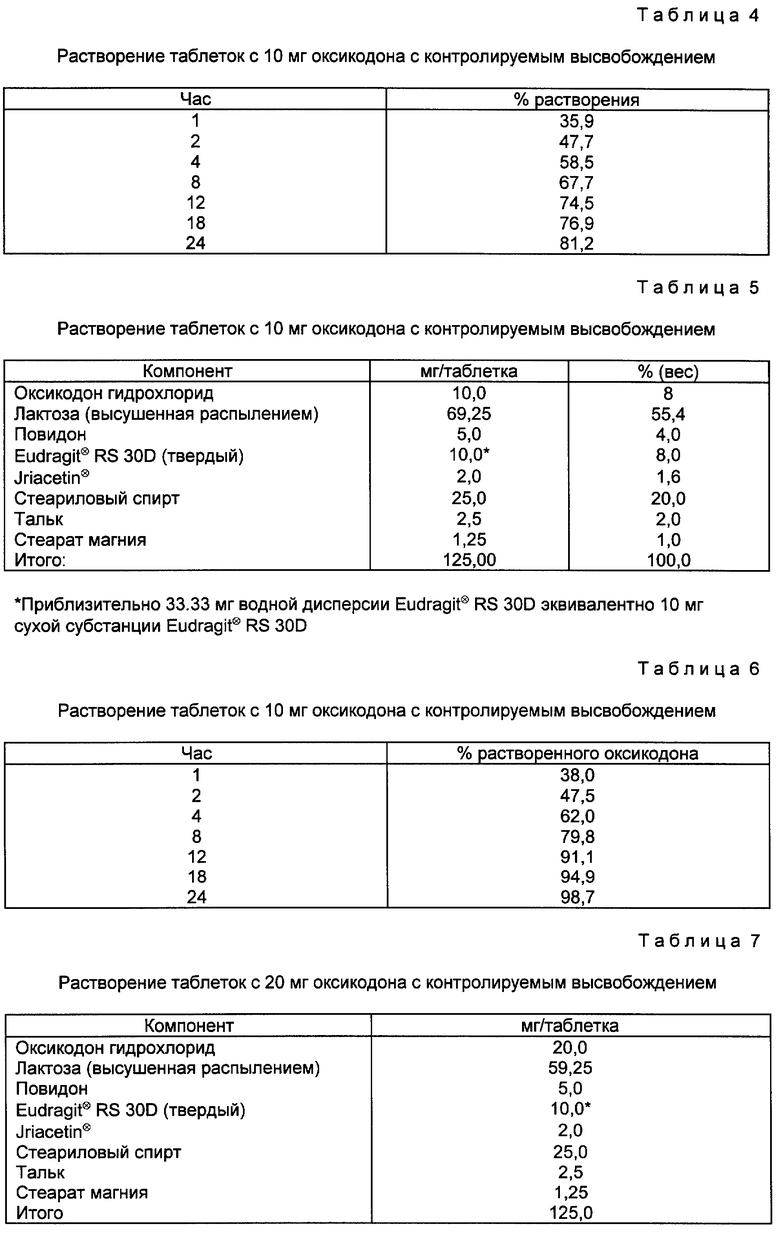
Пример 2. 10-мг таблетки с контролируемым высвобождением оксикодона HCl - органическое получение.
Требуемые количества оксикодона гидрохлорида и высушенной распылением лактозы помещают в смеситель соответствующего размера и перемешивают приблизительно 6 мин. Приблизительно 40% требуемого Eudragit⊗ RSPM порошка диспергируют в этаноле. Размешивая, порошки гранулируют с дисперсией, и смешивание продолжают до тех пор, пока не образуется мокрая гранулированная масса. Если необходимо, то добавляют этанол для того, чтобы достичь конечной точки гранулирования. Гранулированную массу помещают в осушитель с псевдоожиженным слоем и высушивают при 30oC, затем пропускают через сито 12 меш. Оставшийся Eudragit⊗ RSPM диспергируют в растворителе из 90 частей этанола и 10 частей очищенной воды и распыляют в гранулы в грануляторе/осушителе с жидким слоем при 30oC. Далее гранулы пропускают через 12-меш сито. Требуемое количество стеарилового спирта расплавляют приблизительно при 60-70oC. Теплые гранулы возвращают в смеситель. При перемешивании добавляют расплавленный стеариловый спирт. Покрытые гранулы удаляют из смесителя и охлаждают. После этого их пропускают через сито 12 меш.
Затем гранулы смазывают смесью из требуемого количества талька и стеарата магния в подходящем смесителе. После этого гранулы прессуют в таблетки по 126 мл на соответствующей таблетирующей машине.
Рецептура таблеток по примеру 2 (10 мг оксикодона с контролируемым высвобождением) представлена в табл. 3.
Таблетки примера 2 затем тестируют на растворение via USP Basket методом при 37oC, 100 об/мин; первый час в 700 мл желудочной жидкости (pH 1,2), а затем в 900 мл при pH 7,5.
Результаты представлены в табл. 4.
Примеры 3-4. Таблетки с 10 и 20 мг оксикодона с контролируемым высвобождением (водное приготовление).
Eudragit⊗ RS 3ОD и Triacetin вместе пропускают через сито 60 меш и смешивают с небольшим сдвигом приблизительно 5 мин или до тех пор не образуется однородная дисперсия.
Затем соответствующие количества оксикодона HCl, лактозы и повидона помещают в емкость гранулятора/осушителя с псевдоожиженным слоем (ГВД) и суспензию распыляют в порошок в жидком слое. После распыления гранулирующую массу пропускают через сито 12 меш, если необходимо удалить крупные комки. Затем сухую гранулирующую массу помещают в смеситель.
Тем временем требуемое количество стеарилового спирта расплавляют при температуре приблизительно 70oC. Расплавленный стеариновый спирт вливают в гранулируемую массу при перемешивании. Мокрую гранулируемую массу помещают в гранулятор/осушитель с псевдоожиженным слоем или противни и охлаждают при комнатной температуре или ниже. Охлажденную гранулируемую массу пропускают через сито 12 меш. После этого парафинированную гранулирующую массу помещают в миксер/смеситель и смазывают требуемым количеством талька и стеарата магния приблизительно 3 мин, затем гранулят прессуют в таблетки до 125 мл в приемлемой таблетирующей машине.
Рецептура для таблеток примера 3 представлена в табл. 5.
Затем таблетки примера 3 тестируют на растворение the USP Basket методом при 37oC, 100 об/мин, в первый час в 700 мл моделированной желудочной жидкости при PH 1,2, а затем в 900 мл при pH 7,5. Результаты представлены в табл. 6.
Рецептура для таблеток по примеру 4 представлена в табл. 7.
Затем таблетки примера 4 исследуют на растворение - the USP Basket методом при 37oC, 100 об/мин, в первый час в 700 мл желудочной жидкости, при pH 1,2, затем в 900 мл при pH 7,5.
Результаты представлены в табл. 8.
Примеры 5-6.
В примере 5 таблетки с 3 мг оксикодона гидрохлорида с контролируемым высвобождением готовят согласно методике, приведенной в примере 1.
В примере 6 таблетки с 10 мг оксикодонa гидрохлорида готовят с контролируемым высвобождением согласно методике, приведенной в примере 2.
После этого исследования растворимости таблеток примеров 5 и 6 проводят при различных значениях pH, а именно pH 1,3; 4,56; 6,88 и 7,5.
Результаты представлены в табл. 9 и 10.
Примеры 7-12.
В примерах 7-12 таблетки с 4 мг и 10 мг оксикодона HCl готовят по рецептурам и методикам, приведенным в описании патента США N 4990341.
В примере 7 оксикодон гидрохлорид (10,00 мг) подвергается мокрому гранулированию с моногидратом лактозы (417,5 мг) и гидроэтилцеллюлозы (100,0 мг), и гранулы просеивают через сито 12 меш. Затем гранулы высушивают в сушилке с псевдоожиженным слоем при 50oC и просеивают через 16-меш сито.
Добавляют расплавленный цетостеариловый спирт (300,0 мг) к теплым оксикодонсодержащим гранулам и тщательно перемешивают. Смесь охлаждают на воздухе, повторно гранулируют и пропускают через сито 16 меш.
Затем добавляют очищенный тальк (15,0 мг) и стеарат магния (7,5 мг) и смешивают с гранулами. Затем гранулы прессуют в таблетки.
Методика приготовления препарата примера 8 аналогична примеру 7, за исключением того, что данный препарат включает 10 мг оксикодона HCl на таблетку. Рецептуры примера 7 и 8 представлены в табл. 11 и 12.
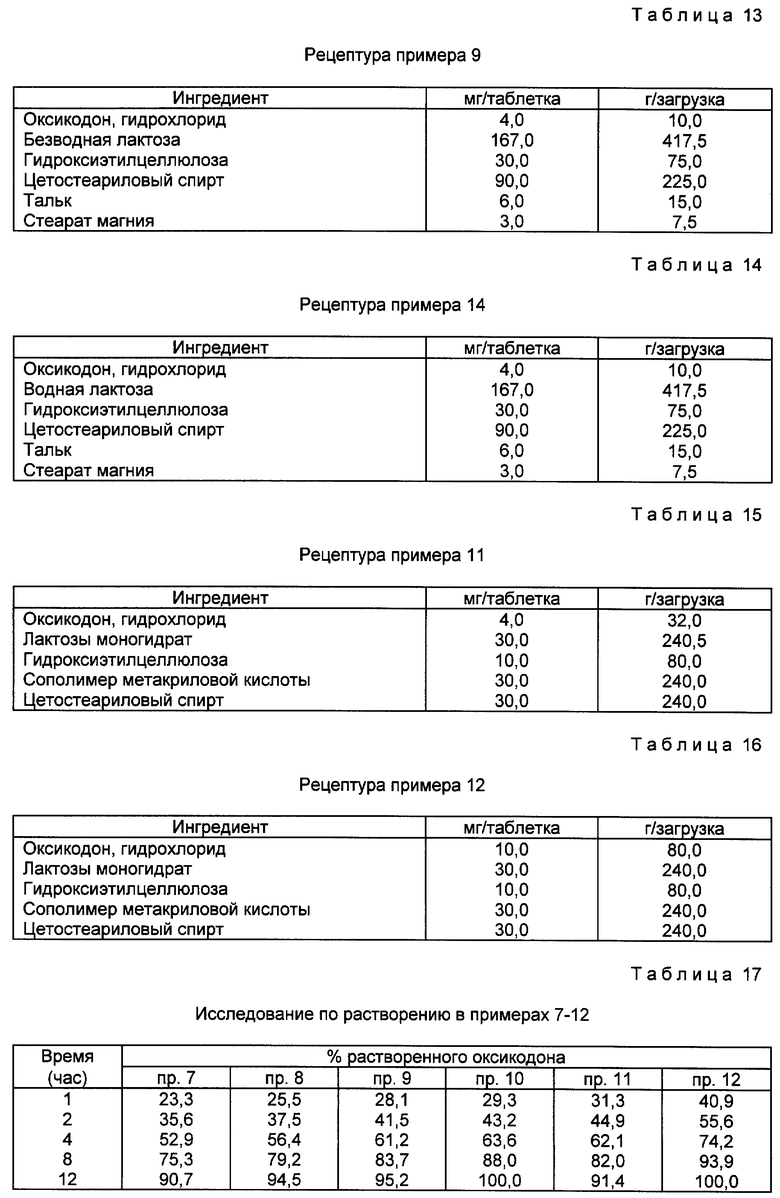
В примере 9 таблетки с 4 мг оксикодона HCl с контролируемым высвобождением готовят согласно рецептуре, приведенной в примере 2 патента США N 4990341. Методика приготовления аналогична приведенной в примерах 7 и 8. Методика приготовления примера 10 аналогична примеру 9, за исключением того, что на таблетку приходится 10 мг оксикодона HCl. Рецептуры примеров 9 и 10 приведены в табл. 13 и 14.
В примере 11 таблетки с 4 мг оксикодона с контролируемым высвобождением готовят в соответствии с рецептурой, описанной в примере 3 патента США N 4990341.
Оксикодон гидрохлорид (32,0 мг) подвергают мокрому гранулированию с моногидратом лактозы (240,0 мг), гидроэтилцеллюлозой (80,0 мг) и сополимером метакриловой кислоты (240,0 мг, Eudragit⊗ L-100-55) и гранулы просеивают через 12-меш сито. Затем гранулы сушат в сушилке с псевдоожиженным слоем при 50oC и пропускают через 16-меш сито.
Теплые оксикодонсодержащие гранулы добавляют к расплавленному цетостеариловому спирту (240,0 мг) и все тщательно перемешивают. Смесь охлаждают на воздухе, еще раз гранулируют и просеивают через 16-меш сито. Затем гранулы прессуют в таблетки.
В примере 12 препараты получают способом, аналогичным способу примера 11, за исключением того, что таблетки включают 10 мг оксикодона HCl, Рецептуры примеров 11 и 12 приведены в табл. 15 и 16.
Затем были проведены исследования на растворимость таблеток примеров 7-12 USP Basket методом, как описано в U.S.Pharmacoloeia XXII (1990). Скорость составляла 100 об/мин, для первого часа использовали среду, имитирующую желудочную жидкость, а затем ее заменяли на жидкостьб имитирующую кишечную жидкость при 37oC. Результаты приведены в табл. 17.
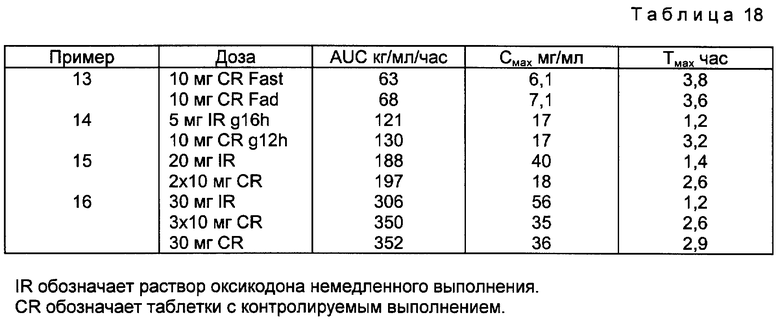
В примерах 13-16 методом перекрестной рандомизации были проведены исследования биологической доступности препаратов, полученных в соответствии с описанными в примере 2 органическим способом получения и в примере водным способом получения.
В примере 13 исследование быстродействующей разовой дозы проводили на 24 пациентах, используя при этом таблетки оксикодона, полученные в соответствии с описанием примера 3.
В примере 14 проводили стационарное исследование 23 пациентов через 12 ч после введения им таблеток оксикодона, полученных в соответствии с описанием примера 2, и данные сравнивали с данными, полученными для раствора 5 мг оксикодона с быстрым высвобождением.
В примере 15 исследование однократной дозы проводили на 22 субъектах с использованием таблеток оксикодона, приготовленных согласно примеру 3, и сравнивали с раствором 20 мг оксикодона немедленного выделения.
В примере 16 исследование однократной дозы проводили на 12 субъектах с использованием 3/10 мг таблеток оксикодона, приготовленных согласно примеру 3, и сравнивали с раствором 30 мг оксикодона немедленного выделения.
Результаты примеров 12-16 представлены в табл. 18.
Пример 17. Клинические исследования.
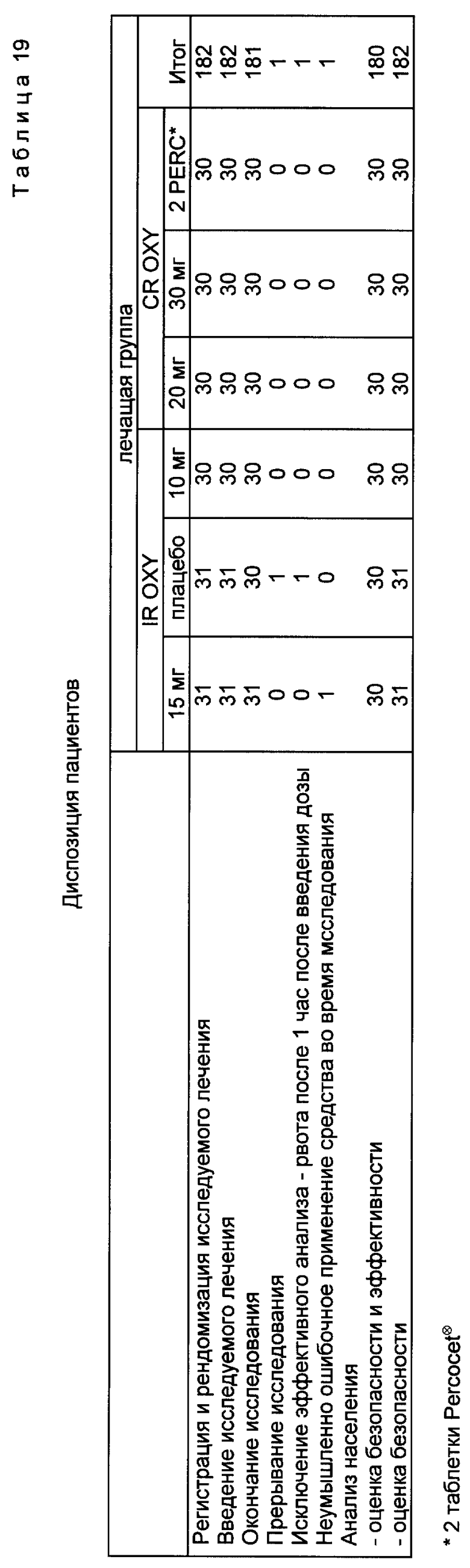
В примере 17 путем рандомизированного исследования разовой дозы с использованием двойного контроля определяли относительную аналгезирующую эффективность, доступность и относительное время действия при пероральном введении 10, 20 и 30 мг оксикодона с контролируемым выделением, приготовленного согласно настоящей заявке (CR OXY) по сравнению с 15-мг оксикодоном с немедленным выделением (IR OXY), 10-мг оксикодоном с немедленным выделением в сочетании с 650 мг ацетаминофена (IR OXY/APAP) плацебо на 180 пациентах с умеренной и тяжелой болью после брюшной или гинекологической операций. Пациенты оценивали интенсивность и облегчение боли ежечасно в течение 12 ч после введения дозы. Лечение сравнивалось с использованием стандартных шкал для интенсивности и стихания боли и данных о начале и продолжительности ослабления боли.
Все активные препараты по своему действию сильно превосходили плацебо для большинства ежечасных измерений как по разнице суммарной болевой интенсивности (SPID), так и полному стиханию боли (TOTPAR). Эффект дозы был получен на 3 дозовых уровнях CR OXY для стихания боли и для разницы интенсивности пиковой боли (PID), причем для CR OXY 20 мг и 30 мг этот эффект был значительно лучше, чем для 10-мг дозы. IR OXY значительно превосходил CR OXY 10 мг на 1 и 2 ч. IR OXY/APAP значительно превосходил 3 дозы CR OXY на 1 ч и CR OXY 10 мг через 2-5 ч. Начальное время было значительно короче для IR OXY и IR OXY/APAP для исследуемых групп по сравнению с 3 CR OXY. Функции распределения для периода стихания боли были значительно более продолжительными для трех CR OXY доз, чем для IR OXY и IR OXY/APAP. Эксперименты, не оказывающие неблагоприятного воздействия на пациента, были повторены. Результаты представлены в табл. 19.
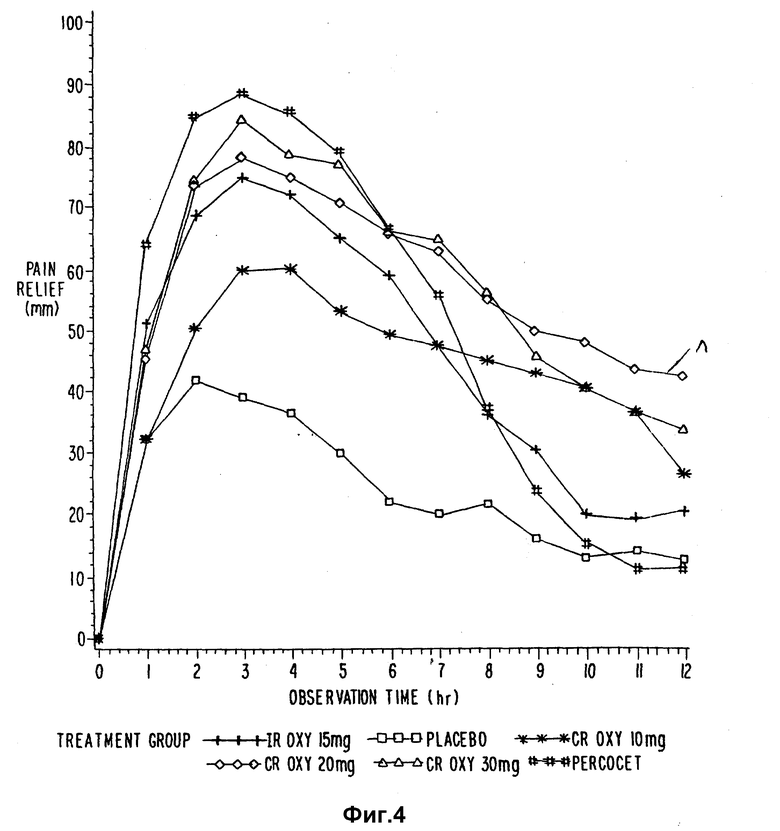
Кривые временных зависимостей интенсивности боли, разности болевой интенсивности и ослабления бoли представлены на фиг. 1-4. CR OXY 10 мг дали значительно (p<15) меньшие показатели интенсивности боли по шкале оценок, чем обработанные плацебо пациенты через 3-11 ч, и меньшие показатели боли, чем IR OXY 15 мг и Percocet⊗ через 10 ч. CR OXY 20 мг дали значительно (p<0,05) меньшие показатели интенсивности боли по сравнению с плацебо через 2-11 ч, и значительно меньшие показатели (p<0,05) боли, чем CR OXY 10 мг, IR OXY 15 мг и Percocet⊗ при 9-11 ч. CR OXY 30 мг обладает значительно меньшими показателями (p<0,05) боли, чем плацебо через 2-11 ч и меньшим уровнем боли, чем CR OXY 10 мг при 2, 3 и 5 ч и ниже, чем Percocet⊗ через 10 ч.
Для ежечасных значений ослабления боли безусловно и с визуальным подтверждением было установлено, что (CAT и VAS) CR OXY 10 мг имеют значительно (p<. 05) большую величину стихания боли, чем плацебо через 3-11 ч и большую величину стихания боли, чем IR OXY и Percocet⊗ через 10 ч и Percocet⊗ через 11 ч. CR OXY 20 мг имеет значительно (p .05) больший уровень стихания боли, чем плацебо через 2-12 и большую, чем Percocet⊗ при 9-12 ч. Кроме того, CR OXY имеет значительно (p < .05) больший уровень стихания боли, чем IR OXY при 10-12 ч. CR OXY 30 мг имеет значительно (p < .05) большее облегчение боли, чем плацебо при 2-12 ч, и большее, чем Percocet⊗, при 9-12 ч и IR OXY 15 мг при 10 ч.
Каждая исследуемая группа показала (p < 0.5) лучшие результаты по сравнению с плацебо-группой как в отношении суммы разницы болевой интенсивности (SPID), так и в отношении полного устранения боли (TOTРAR).
Продолжительность стихания боли, которая измерялась the patient stopwatch методом, показывает что CR OXY 10 мг, 20 мг и 30 мг обладают значительно (p < .05) большим временем действия по сравнению с IR OXY 15 мг и 2 таблетками. Кроме того, три препарата с контролируемым выделением обладают значительно (p < .05) большей продолжительностью действия, чем Percocet⊗.
Перед вылечиванием 104 (57%) пациентов сообщили о неблагоприятных явлениях. Наиболее обычными являются сонливость, жар, головокружение и головная боль.
На основании результатов исследования было сделано заключение, что оксикодоновые препараты с контролируемым выделением, полученным в соответствии с настоящим изобретением, позволяют снимать умеренную и сильную послеоперационную боль, например, при брюшных и гинекологических операциях у женщин. Существует эффект дозы, где placebo < 10 мг < 20 мг < 30 мг OR OXY при введении разовой дозы. Отмечено, что начало действия имеет место через 1 ч после введения, а максимальный эффект достигается через 2-5 ч, а продолжительность действия составляет от 10 до 12 ч. Для ситуации с хронической болью при стационарном дозировании срок этого действия может быть удлинен. Наблюдаются побочные эффекты. Головная боль может быть следствием введенной дозы. Имеются данные о возникновении головокружения и сонливости.
IR OXY 15 мг обладает промежуточным пиковым действием по сравнению с контролируемо выделяемым оксикодоном. Время его действия короче (6-8 ч). Percocet⊗ вполне эффективен с точки зрения начала действия, максимальной эффективности и безопасности. Время действия составляет 6-8 ч.
Итак, CR OXY очевидно является эффективным пероральным аналгетиком с меньшим началом действия, но с большей продолжительностью действия, чем IR OXY или IR/APAP.
Пример 18. Клинические исследования.
В примере 18 стационарное перекрестное испытание было проведено на 21 пациентах-мужчинах со следующей схемой приема:
а) прием CR ОХУ 10 мг каждые 12 ч (q12h);
б) пероральный прием раствора Roxicodone⊗ 5 мг (ROX) каждые 6 ч (q6h).
Лечение (б) было проведено по стандарту. Средний возраст составлял 34 года, рост 176 см и вес 76 кг. Отдельных отклонений внутри группы не отмечалось.
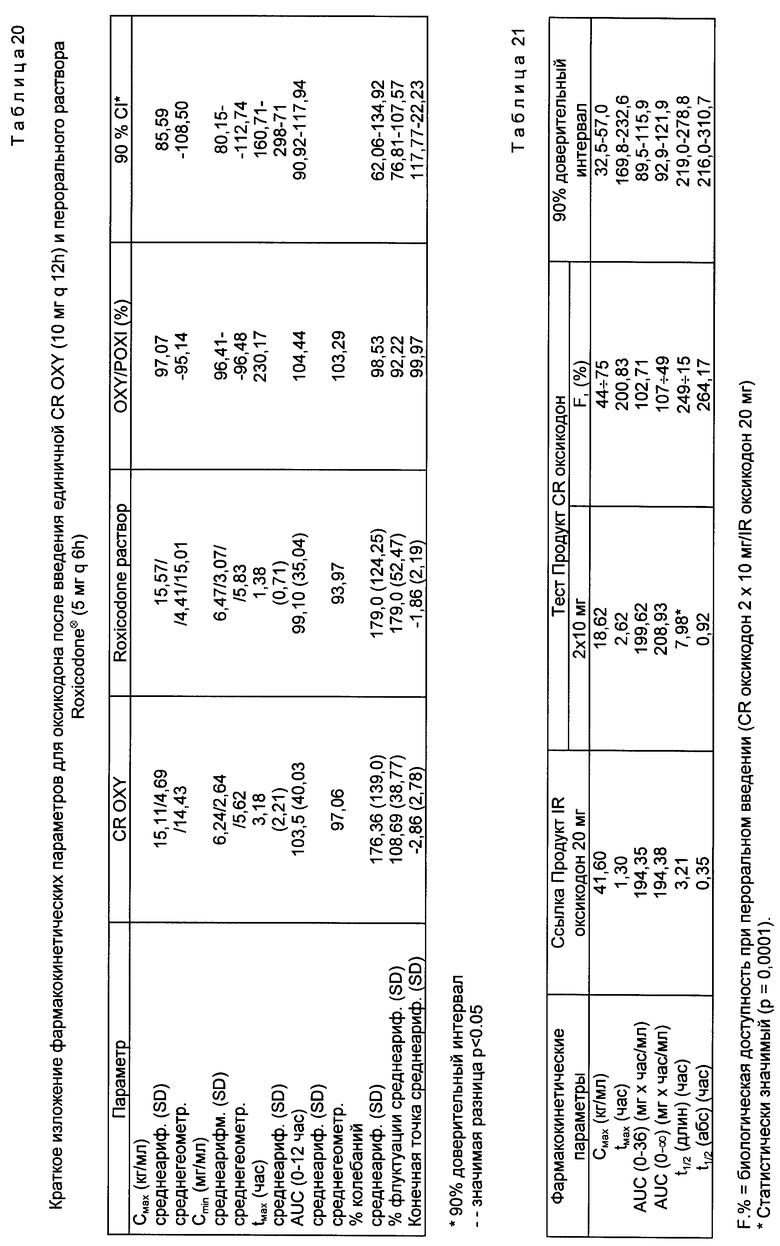
На фиг. 5 представлены средние концентрации оксикодона в плазме для двух препаратов в течение 12-часового дозирования. Результаты приведены в табл. 18 исходя из средних значений, отношений средних значений и 90%-ных доверительных интервалов.
Как видно из табл. 20 (за одним лишь исключением), значительных различий между двумя препаратами не наблюдается. Единственным отличием является среднее tmax для CR ОХУ за 3,18 ч, которое, как ожидалось для препарата с контролируемым выделением, значительно превышает ROX среднее значение за 1,38 ч. Средняя биодоступность исходя из AUC (ROX = 100%) составляет 104,4% с 90%-ными доверительными пределами от 90,9 до 117,9%. Таким образом, FDA спецификация ±20% является достаточно удовлетворительной, чтобы по полученным результатам было можно утверждать о равной пригодности оксикодона.
Пример 19. Клинические исследования.
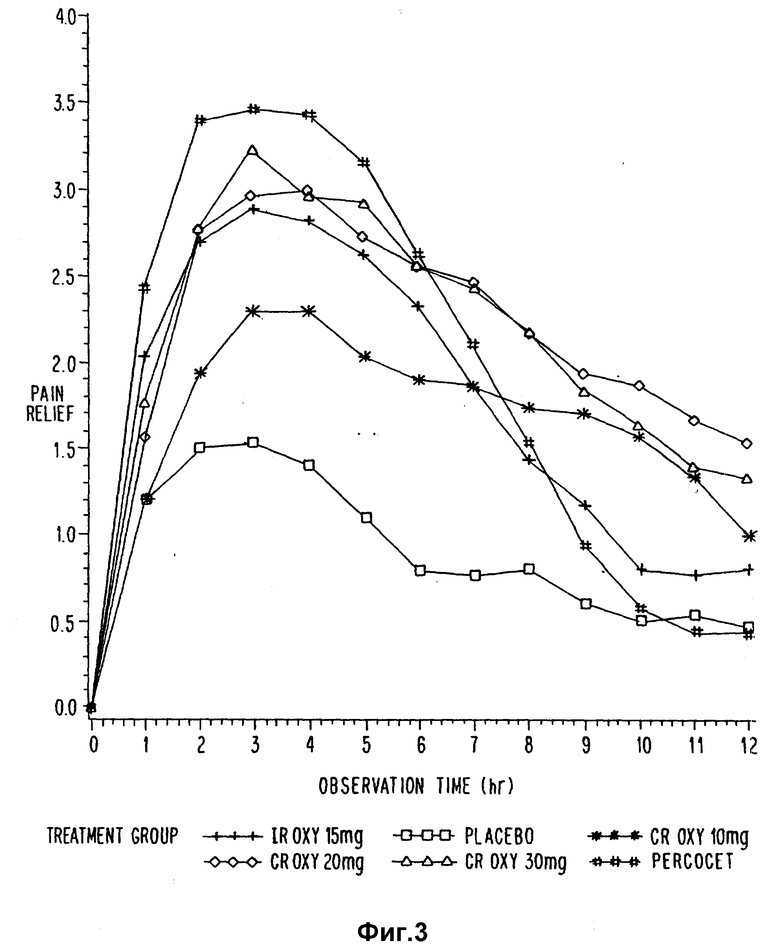
В примере 19 двадцать четыре здоровых мужчин были подвергнуты двухстадийному перекрестному рандомизированному исследованию разовой дозы в целях сравнения концентраций в плазме оксикодона после введения двух таблеток с 10 мг контролиуемо выделяемым оксикодоном и раствора 20 мг (20 мл, 5 мг/5 мл) оксикодона гидрохлорида с быстрым выделением. Двадцать три объекта закончили исследования и были пригодными для анализа.
Концентрация оксикодона в плазме была определена с помощью жидкостной хроматографии высокого разрешения. Среднеарифетические Cmax, tmax, AUC и полупериоды, вычисленные для каждой концентрации оксикодона в плазме в зависимости от времени, представлены в табл. 21.
Для Cmax, tmax, t1/2(элин) и t1/2(обс) существуют статистически значимые различия между CR ОХУ и IR ОХУ. Нет статистически значимых различий в степенях абсорбции [AUC/0,36). AUC (0,∞)] для двух испытаний. 90%-ный доверительный интервал от CR ОХУ до IR ОХУ составляет 89,5 - 115,9% для AUC (0,36) и 92,9 - 121,9% для AUC (0,∞), Исходя из анализа 90%-ного доверительного интервала можно видеть, что контролируемо выделяемые оксикодоновые таблетки эквивалентны до степени абсорбции (AUC 0,36) раствору немедленно выделяемого оксикодона. Абсорбция контролируемо выделяемого оксикодона медленнее приблизительно на 1,3 ч. Не было замечено статистически значимых различий между двумя испытаниями в отношении нежелательных явлений, клинических отклонений для препарата в описанном типе исследования не наблюдалось.
Вышеприведенные исследования показывают значительную дозовую зависимость при использовании оксикодоновых препаратов настоящего изобретения с контролируемым выделением при дозах 10, 20 и 30 мг, которые не отклоняются от параллелизма кривыми дозовых зависимостей для MS Contin в подобном исследовании хорошо контролируемой аналгетической эффективности MS Contin, описанной Kaiko R.S., Van Wagoner D., Brown J., et al., "Controlled-Release Oral Morphine (M. S. Contin ⊗ Tablets, MSC) in Postoperative Pain". Pain Suppl., 5: S 149, 1990, которые сравнивают 30, 60, 90 и 120 мг MS Contin с 10 мг внутримышечного морфина и placebo и Bloomfield, et al., "Analgesic Efficacy and Potency of Two Oral Controlled-Release Morphine Preparations", Clinical Pharmacology & Therapeutics (в печ.), которые сравнивали 30 и 90 мг MS Contin с от 30 до 90 мг другим контролируемо выделяемым оральным морфином препаратом, Oramorph SR 30-мг таблетками.
Примеры, которые приведены выше, не являются исключительными.
Настоящее изобретение может быть осуществлено и в других вариантах, не выходящих за рамки нижеследующей формулы изобретения.



Способ включает введение пероральной фармацевтической композиции с контролируемым высвобождением. Композиция содержит от 10 до 160 мг оксикодона или его соли и обеспечивает среднемаксимальную концентрацию оксикодона в плазме in vivo до 240 нг/мл через 2-4,5 ч после введения и среднеминимальную концентрацию оксикодона в плазме in vivo до 120 нг/мл через 10-14 ч после повторного введения каждые 12 ч в стационарных условиях. Новая пероральная фармацевтическая композиция выполнена в виде твердой лекарственной формы и включает аналгетически эффективное количество сфероидов, сферообразующий агент и пленочное покрытие, контролирующее высвобождение оксикодона или соли оксикодона с регулируемой скоростью. Предпочтительно выполнение твердой пероральной лекарственной формы в виде таблетки. Новые композиции обеспечивают ослабление боли у 90% пациентов при существенно меньшем уровне доз. 4 с. и 7 з.п. ф-лы, 21 табл., 5 ил.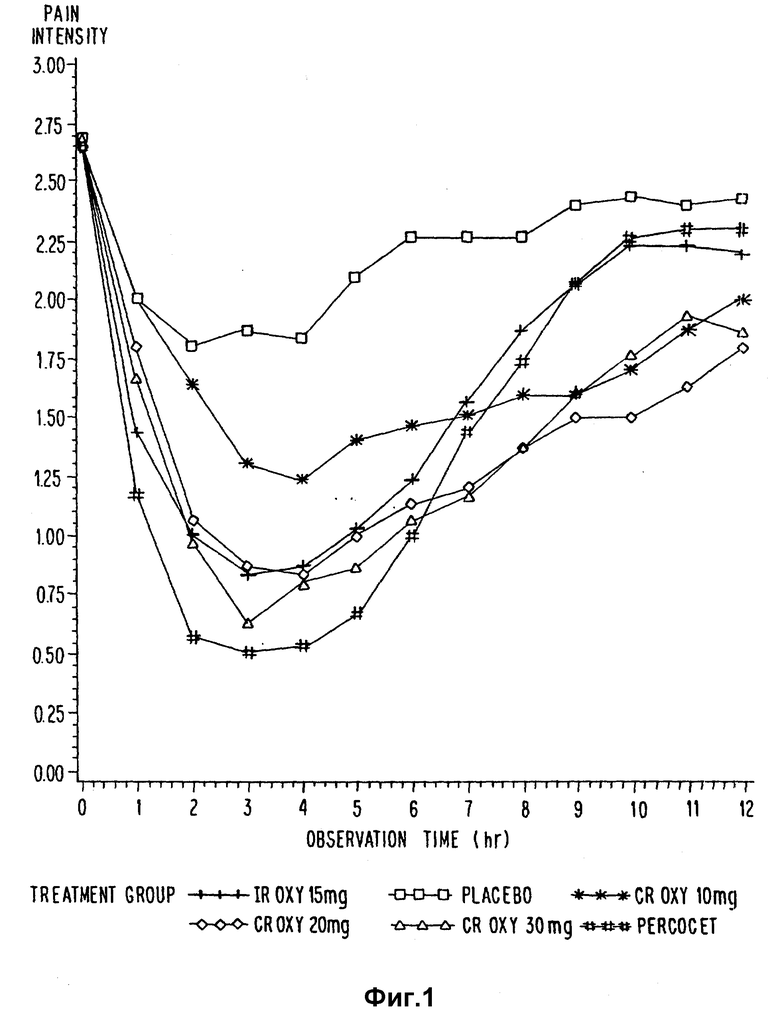
| US 4990341 A, 05.02.91. |
