Область, к которой относится изобретение
Изобретение относится к фармацевтическим композициям, применимым для лечения пациентов, страдающих от боли.
Предпосылки создания изобретения
В последнее десятилетие все больше осознается важность адекватного лечения пациентов, страдающих от боли.
Для лечения хронической боли от умеренной до сильной и даже пронзительной (острой), такой как у больных раком, за последние десятилетия все большей популярностью пользуются опиоидные анальгетики. Среди факторов, которые обусловливают это обстоятельство, введение препаратов с контролируемым высвобождением опиоидов, таких как морфин, гидроморфон и оксикодон, которые пациент может принимать реже, чем препараты этих агентов с мгновенным высвобождением, имевшиеся ранее.
Препараты опиоида оксикодона с контролируемым высвобождением, такие как таблетки Oxygesic® (оксигесик), который в качестве активного агента содержит оксикодона гидрохлорид, и таблетки Targin® (таргин), который содержит комбинацию оксикодона гидрохлорида и антагониста опиоидных рецепторов налоксона гидрохлорида, оказались успешными как с точки зрения промышленного производства, так и с точки зрения приемлемости их для пациентов.
Однако существуют ситуации, в которых препараты с контролируемым высвобождением опиоидов необязательно могут быть препаратами первого ряда при лечении пациентов, страдающих от боли.
Цель и сущность изобретения
Одной из целей настоящего изобретения является получение фармацевтических композиций, применимых для лечения боли и, в частности, для лечения хронической боли от умеренной до сильной и даже мучительной (жестокой, острой) боли, которые можно применять для титрования дозы для пациентов, страдающих от боли, и/или лечения прорывной боли у пациентов, страдающих от боли, в то же время избегая нежелательных побочных эффектов, которые могут проявляться при стандартном лечении боли.
Эти и другие цели, которые станут очевидными из нижеприведенного описания, позволяет достичь предмет изобретения по независимым пунктам Формулы изобретения. Представленные ниже зависимые пункты Формулы изобретения относятся к некоторым предпочтительным вариантам настоящего изобретения.
В одном варианте изобретения настоящее изобретение относится к пероральной фармацевтической композиции с мгновенным высвобождением, содержащей по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль).
Как правило, пероральные фармацевтические композиции с мгновенным высвобождением в соответствии с настоящим изобретением, помимо оксикодона или его фармацевтически приемлемой соли и налоксона или его фармацевтически приемлемой соли, могут содержать другие фармацевтически активные агенты. Однако именно предпочтительным вариантом настоящего изобретения может быть вариант, в котором оксикодон или его фармацевтически приемлемая соль и налоксон или его фармацевтически приемлемая соль являются единственными фармацевтически активными агентами в фармацевтических композициях по настоящему изобретению.
Обычно пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению могут содержать оксикодон в виде свободного основания или его соли и налоксон также в виде свободного основания или его соли. Однако может быть предпочтительно, чтобы пероральные фармацевтические композиции по настоящему изобретению с мгновенным высвобождением содержали оксикодон в виде оксикодона гидрохлорида и налоксон в виде налоксона гидрохлорида. Еще более предпочтительным может вариант изобретения, в котором оксикодона гидрохлорид и налоксона гидрохлорид являются единственными фармацевтически активными агентами фармацевтической композиции.
В первом аспекте изобретения пероральные фармацевтические композиции с мгновенным высвобождением по изобретению содержат некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-160 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида. В одном из предпочтительных вариантов изобретения пероральные фармацевтические композиции с мгновенным высвобождением по изобретению могут содержать некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-10 мг налоксона гидрохлорида. Эти фармацевтические композиции могут содержать фармацевтически активные агенты в примерном весовом соотношении 2:1.
Предпочтительный вариант данного первого аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов в примерном соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), причем оксикодона гидрохлорид присутствует в примерном количестве 2.5-40 мг, а налоксона гидрохлорид присутствует в примерном количестве 1.25-20 мг.
Еще более предпочтительный вариант данного первого аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов в примерном соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), причем оксикодона гидрохлорид присутствует в примерном количестве 2.5-20 мг, а налоксона гидрохлорид присутствует в примерном количестве 1.25-10 мг.
Второй аспект настоящего изобретения относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль) и отличающимся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥75% вес. оксикодона или его фармацевтически приемлемой соли и ≥75% вес. налоксона или его фармацевтически приемлемой соли.
Предпочтительный вариант данного второго аспекта относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль) и отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥80% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. налоксона или его фармацевтически приемлемой соли.
Более предпочтительный вариант этого второго аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль) и отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥90% вес. оксикодона или его фармацевтически приемлемой соли и ≥90% вес. налоксона или его фармацевтически приемлемой соли.
Еще более предпочтительный вариант этого второго аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль) и отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥95% вес. налоксона или его фармацевтически приемлемой соли.
Во всех вариантах второго аспекта изобретения пероральные фармацевтические композиции с мгновенным высвобождением могут содержать некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-160 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида.
Еще более предпочтительным может быть количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-80 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-40 мг налоксона гидрохлорида, количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-40 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-20 мг налоксона гидрохлорида, количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-20 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-10 мг налоксона гидрохлорида.
В третьем аспекте настоящего изобретения пероральная фармацевтическая композиция с мгновенным высвобождением может содержать оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую, причем композиция находится в твердом виде и отличается тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥75% вес. оксикодона или его фармацевтически приемлемой соли и ≥75% вес. налоксона или его фармацевтически приемлемой соли.
Предпочтительный вариант этого третьего аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥80% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. налоксона или его фармацевтически приемлемой соли.
Более предпочтительный вариант этого третьего аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥90% вес. оксикодона или его фармацевтически приемлемой соли и ≥90% вес. налоксона или его фармацевтически приемлемой соли.
Еще более предпочтительный вариант этого третьего аспекта может относиться к пероральным фармацевтическим композициям с мгновенным высвобождением, отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин высвобождается ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥95% вес. налоксона или его фармацевтически приемлемой соли.
Во всех вариантах третьего аспекта изобретения пероральные фармацевтические композиции с мгновенным высвобождением могут содержать некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-160 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида.
Еще более предпочтительным может быть количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-80 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-40 мг налоксона гидрохлорида, количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-40 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-20 мг налоксона гидрохлорида, количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-20 мг оксикодона гидрохлорида, или количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-10 мг налоксона гидрохлорида.
Во всех вариантах третьего аспекта изобретения фармацевтическая композиция может содержать разрыхлитель (дезинтегрант, вещество, способствующее механическому разрушению) и, необязательно, наполнитель и, необязательно, другие фармацевтически приемлемые эксципиенты. Предпочтительно, в качестве разрыхлителя можно применять, например, комбинацию крахмала и лактозы. Сама по себе лактоза может в то же самое время служить наполнителем. Особенно предпочтительный вариант изобретения содержит продукт Старлак (Starlac)®, комбинацию, включающую 85% лактозы и 15% крахмала, каждый из которых может выступать в роли как разрыхлителя, так и наполнителя. Ниже представлены различные типы разрыхлителей, наполнителей и других типов фармацевтически приемлемых эксципиентов. Смешанный наполнитель/разрыхлитель может содержаться в композиции в примерном количестве 40%-90%, предпочтительно, в примерном количестве 50%-85%, и еще более предпочтительно, в примерном количестве 60%-80% вес. от веса композиции. Эти значения особенно актуальны, если используется эксципиент, выполняющий двойную функцию - разрыхлителя и наполнителя, такой как Старлак®.
Во всех вариантах третьего аспекта изобретения фармацевтическая композиция может содержать оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль).
Во всех вариантах третьего аспекта изобретения фармацевтическая композиция может предусматриваться в виде таблеток, капсул, гранул, мультичастиц и т.п. Особенно предпочтительными являются таблетки.
Предпочтительными пероральными фармацевтическими композициями с мгновенным высвобождением по изобретению, содержащими некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-10 мг налоксона гидрохлорида, в примерном весовом соотношении 2:1, могут являться композиции, которые предусматривают AUCt оксикодона в примерном диапазоне 15 нг·час/мл - 500 нг·час/мл, предпочтительно, в примерном диапазоне 20 нг·час/мл - 400 нг·час/мл, более предпочтительно, в примерном диапазоне 25 нг·час/мл - 300 нг·час/мл, и, еще более предпочтительно, в примерном диапазоне 30 нг·час/мл - 250 нг·час/мл при исследовании на людях с введением разовой дозы здоровым добровольцам. Такие пероральные фармацевтические композиции с мгновенным высвобождением, предпочтительно, могут содержать хлористоводородные соли оксикодона и налоксона.
Предпочтительными пероральными фармацевтическими композициями с мгновенным высвобождением по изобретению, содержащими некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-10 мг налоксона гидрохлорида, в примерном весовом соотношении 2:1, могут являться композиции, которые предусматривают Cmax оксикодона в примерном диапазоне 1 нг/мл - 300 нг/мл, предпочтительно, в примерном интервале 2 нг/мл - 200 нг/мл, более предпочтительно, в примерном интервале 3 нг/мл - 100 нг/мл, еще более предпочтительно, в примерном интервале 4 нг/мл - 75 нг/мл, и еще более предпочтительно, в примерном интервале 6 нг/мл - 50 нг/мл при исследовании на людях с введением разовой дозы здоровым добровольцам. Такие пероральные фармацевтические композиции с мгновенным высвобождением, предпочтительно, могут содержать хлористоводородные соли оксикодона и налоксона.
Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению могут, без ограничения, быть в виде таблетки, капсулы, мультичастиц, например гранул, сфероидов или бусин, и жидкостей, например, в виде раствора, суспензии или эмульсии.
Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению в качестве фармацевтически приемлемых эксципиентов могут содержать по меньшей мере разбавитель и, необязательно, разрыхлитель. Кроме того, они могут содержать другие фармацевтически приемлемые эксципиенты, такие как смазки, наполнители, красители, корригенты, вещества, корректирующие рН, пластификаторы, вещества, предотвращающие слипание, связующие и т.п. Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению в качестве фармацевтически приемлемых эксципиентов могут также содержать по меньшей мере разрыхлитель и, необязательно, разбавитель. Помимо этого они могут содержать другие фармацевтически приемлемые эксципиенты, такие как смазки, наполнители, красители, корригенты, вещества, корректирующие рН, пластификаторы, вещества, предотвращающие слипание, связующие и т.п. Следует понимать, что, предпочтительно, можно применять эксципиенты, выполняющие две функции, как разрыхлителя, так и разбавителя, такие как продукт Старлак®.
В особенно предпочтительном варианте настоящего изобретения пероральная фармацевтическая композиция содержит, примерно, 2.5-20 мг оксикодона гидрохлорида и, примерно, 1.25-10 мг налоксона гидрохлорида в примерном весовом соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), причем эта композиция содержит оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов, имеет форму таблетки, в качестве фармацевтически приемлемых эксципиентов содержит по меньшей мере разбавитель и, необязательно, разрыхлитель и отличается тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, через 45 мин in vitro высвобождается ≥80% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. налоксона или его фармацевтически приемлемой соли. В другом предпочтительном аспекте этих особенно предпочтительных вариантов настоящего изобретения фармацевтическая композиция отличается тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, через 15 мин in vitro высвобождается ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥95% вес. налоксона или его фармацевтически приемлемой соли.
Другой аспект этих особенно предпочтительных вариантов настоящего изобретения относится к фармацевтическим композициям, которые предусматривают AUCt оксикодона в примерном диапазоне 25 нг·час/мл - 300 нг·час/мл, и, еще более предпочтительно, в примерном диапазоне 30 нг·час/мл - 250 нг·час/мл при исследовании на людях с введением разовой дозы здоровым добровольцам. Такие пероральные фармацевтические композиции с мгновенным высвобождением, предпочтительно, могут содержать хлористоводородные соли оксикодона и налоксона.
Другой аспект этих особенно предпочтительных вариантов настоящего изобретения относится к фармацевтическим композициям, которые предусматривают Cmax оксикодона в примерном диапазоне 4 нг/мл - 75 нг/мл, и еще более предпочтительно, в примерном диапазоне 6 нг/мл - 50 нг/мл при исследовании на людях с введением разовой дозы здоровым добровольцам. Такие пероральные фармацевтические композиции с мгновенным высвобождением, предпочтительно, могут содержать хлористоводородные соли оксикодона и налоксона. Особенно предпочтительными являются фармацевтические композиции, которые дают значения AUC, указанные в предыдущем абзаце.
Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению можно применять для титрования дозы для пациентов, страдающих от боли, в частности страдающих от хронической боли от умеренной до сильной и даже от острой боли.
Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению можно также применять для лечения прорывной боли у пациентов, страдающих от боли, в частности страдающих от хронической боли от умеренной до сильной и даже от острой боли.
Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению можно также применять для лечения боли у пациентов, страдающих от боли, в частности страдающих от хронической боли от умеренной до сильной и даже от острой боли.
Настоящее изобретение относится также к описанным выше пероральным фармацевтическим композициям с мгновенным высвобождением и к получению лекарственного препарата для титрования дозы для пациентов, страдающих от боли, в частности, страдающих от хронической боли от умеренной до сильной, и даже от острой боли.
Помимо этого настоящее изобретение относится также к применению описанных выше пероральных фармацевтических композиций с мгновенным высвобождением и к получению лекарственного препарата для лечения прорывной боли у пациентов, страдающих от боли, в частности страдающих от хронической боли от умеренной до сильной и даже от острой боли.
Далее настоящее изобретение относится также к применению описанных выше пероральных фармацевтических композиций с мгновенным высвобождением и к получению лекарственного препарата для лечения боли у пациентов, страдающих от боли, в частности страдающих от хронической боли от умеренной до сильной и даже от острой боли.
В другом аспекте настоящее изобретение к способу титрования дозы для пациента, страдающего от боли, в частности страдающего от хронической боли от умеренной до сильной и даже от острой боли, с помощью перорального введения описанной выше фармацевтической композиции с мгновенным высвобождением.
В другом аспекте настоящее изобретение относится к способу лечения прорывной боли у пациента, страдающего от боли, в частности страдающего от хронической боли от умеренной до сильной и даже от острой боли, с помощью перорального введения описанной выше фармацевтической композиции с мгновенным высвобождением.
Еще в одном аспекте настоящее изобретение относится к способу лечения боли у пациента, страдающего от боли, в частности страдающего от хронической боли от умеренной до сильной и даже от острой боли, с помощью перорального введения описанной выше фармацевтической композиции с мгновенным высвобождением.
Далее настоящее изобретение относится к способу получения описанной выше пероральной фармацевтической композиции с мгновенным высвобождением.
Описание чертежей
На Фиг.1 представлена блок-схема процесса получения фармацевтической композиции по изобретению
На Фиг.2 изображена схема лечения в клиническом испытании, описанном в Эксперименте 2.
На Фиг.3 показан график средней концентрации оксикодона в плазме крови по определению для IR OXN 2.5/1.25, IR OXN 5/2.5, IR OXN 10/5 и IR OXN 20/10 в Эксперименте 2 с участием 21 человека.
На Фиг.4 показан график средней концентрации налоксона в плазме крови по определению для IR OXN 2.5/1.25, IR OXN 5/2.5, IR OXN 10/5 и IR OXN 20/10 в Эксперименте 2 с участием 21 человека.
На Фиг.5 показан график средней концентрации налоксона-3-глюкуронида в плазме крови по определению для IR OXN 2.5/1.25, IR OXN 5/2.5, IR OXN 10/5 и IR OXN 20/10 в Эксперименте 2 с участием 21 человека.
На Фиг.6 изображена схема лечения в клиническом испытании, описанном в Эксперименте 3.
На Фиг.7 показан график средней концентрации оксикодона в плазме крови по определению для IR OXN 20/10, IR Oxycodone 20 мг и PR Oxycodone 20/10 (Targin®, таргин) в Эксперименте 3 с участием 21 человека.
На Фиг.8 показан график средней концентрации налоксона в плазме крови по определению для IR OXN 20/10, IR Oxycodone 20 mg and PR Oxycodone 20/10 (Targin®, таргин) в Эксперименте 3 с участием 21 человека.
На Фиг.9 показан график средней концентрации налоксона- 3- глюкуронида в плазме крови по определению для IR OXN 20/10, IR Oxycodone 20 mg and PR Oxycodone 20/10 (Targin®, таргин) в Эксперименте 3 с участием 21 человека.
Подробное описание изобретения
Настоящее изобретение, описываемое ниже, можно применять на практике в отсутствие любого элемента или любых элементов, ограничения или ограничений, конкретно не раскрываемых в данном описании.
Настоящее изобретение описывается на конкретных вариантах изобретения и со ссылкой на некоторые чертежи, но изобретение не ограничивается ими, оно ограничивается только Формулой изобретения. Представленные чертежи являются лишь схематическими и не ограничивающими. Как правило, представленные в данном описании термины употребляются в своем обычном значении, если не указано иначе.
Если термин "содержащий" ("включающий") употребляется в данном описании и в Формуле изобретения, он не исключает других элементов. Для целей настоящего изобретения термин "состоящий из" ("consisting of) рассматривается как предпочтительный вариант термина "содержащий" ("comprising"). Если в данном описании группа по определению содержит по меньшей мере определенное число вариантов изобретения, это следует также понимать, что раскрывается группа, которая, предпочтительно, состоит только из этих вариантов изобретения.
Если не указано иначе, применение существительного в единственном числе включает также множество таких существительных. Термины "около" или "примерно" в контексте настоящего изобретения обозначает интервал точности, который, как понимает специалист в данной области техники, все еще позволяет гарантировать технический эффект рассматриваемого признака. Термин, как правило, указывает на отклонение (девиацию) от указанного численного значения ±10% и, предпочтительно, ±5%.
Если ниже делается ссылка на оксикодон или налоксон, она всегда также включает ссылку на фармацевтически приемлемую соль свободного основания оксикодона или на фармацевтически приемлемую соль свободного основания налоксона или их производные, если специально не указывается, что термин "оксикодон" или "налоксон" относится только к свободному основанию
Выражение "с мгновенным высвобождением" (или "мгновенного высвобождения") относится к скорости высвобождения, с которой активные ингредиенты, т.е. оксикодон или его фармацевтически приемлемая соль и налоксон или его фармацевтически приемлемая соль, высвобождаются из фармацевтической композиции. В соответствии со своим основным значением выражение "с мгновенным высвобождением" относится к фармацевтическим композициям, высвобождение активного вещества (активных веществ) из которых намеренно не модифицируется с помощью специальных методов разработки и/или производства рецептуры.
Выражение "с мгновенным высвобождением" относится, в частности, к свойству фармацевтических композиций по изобретению высвобождать in vitro через 45 мин ≥75% вес. оксикодона или его фармацевтически приемлемой соли и ≥75% вес. налоксона или его фармацевтически приемлемой соли, при измерении в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм.
Скорости высвобождения in vitro определяют в соответствии с Method Paddle (мешалка с лопастью) Европейской Фармакопеи (European Pharmacopeia, Ph. Eur.), описанным в Ph. Eur. 2.9.3 6е издание. Скорость вращения лопастей устанавливают 100 об/мин в воспроизводимом желудочном соке (USP (United States Pharmacopeia, (Фармакопеи США) без пепсина)) в качестве растворителя (среды для растворения). Аликвоты растворителя отбирают через 15 минут и через 45 минут и анализируют методом обращенно-фазовой ВЭЖХ на колонке Merck LiChrospher 60 RP Select В, поддерживаемой при температуре 60°С. Подвижная фаза состоит из 85:15 об/об; рН 2.0 хлорида калия: метанола. УФ-детекцию проводят при 230 нм. Количественное определение оксикодона и налоксона проводят с помощью внешнего стандарта.
Если фармацевтическая лекарственная форма с мгновенным высвобождением содержит некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное количеству вплоть до 5 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное количеству вплоть до 2.5 мг налоксона гидрохлорида, Ph. Eur. paddle test (тест с лопастной мешалкой по Ph. Eur.) осуществляют в 500 мл 0.1 N соляной кислоты.
Если фармацевтическая лекарственная форма с мгновенным высвобождением содержит некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное количеству более 5 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное количеству более 2.5 мг налоксона гидрохлорида, Ph. Eur. paddle test (тест с лопастной мешалкой по Ph. Eur.) осуществляют в 900 мл 0.1 N соляной кислоты.
Термин "титрование" и его грамматические варианты, указываемые в данном описании, относится к процессу, с помощью которого терапевт (практикующий врач) определяет подходящую дозу конкретного опиоида, такого как оксикодона гидрохлорида, для пациента, страдающего от боли, и ,в частности, от хронической боли от умеренной до сильной и даже острой боли. Так как пациентам, страдающим от боли, требуются различные дозы опиоидных анальгетиков, определяемые от различным обменом веществ, врач, как правило, сначала пытается повышать дозы препарата с мгновенным высвобождением опиоида, чтобы определить количество опиоида, достаточное для того, чтобы снять боль у пациента или по меньшей мере уменьшить интенсивность боли, чтобы обеспечить контроль приемлемой (терпимой) боли.
Когда определяют дозу, подходящую для того, чтобы постоянно контролировать (хроническую) боль, испытываемую этими пациентами, пациента обычно переводят на препарат с контролируемым высвобождением в соответствующей дозе таким образом, чтобы препарат можно было принимать с меньшей частотой через определенное время, например через каждые 12 часов или только один раз в день. Тех пациентов, которые при отсутствии лечения боли страдали бы от ощущения хронической боли, обычно называют пациентами с контролируемой (регулируемой) фоновой болью.
В контексте настоящего изобретения термин "прорывная(-ые) боль(-и)" и его грамматические варианты относится к транзиторному (временному) усилению боли по сравнению с основной или фоновой болью, испытываемой пациентом, получающему опиоиды по часам (by-the clock).
Термин "фоновая боль" по данному описанию обычно обозначает боль, оцениваемую больным как боль средней интенсивности, испытываемая находящимся на лечении опиоидами больным, например, в течение последних 12 или более часов.
Интенсивность фоновой боли, как правило, определяют, применяя обычные методы, такие как цифровой тест по аналоговой шкале (NAS). Определение приступов прорывной боли и фоновой боли описано, например, в Portenoy et al. (1999) (The Journal of Pain 7 (8): 583-591 и Portenoy et al. (1999) (Pain 81: 129-134). Определение прорывной боли и контролируемой фоновой боли, данное в этих публикациях, вводится в данное описание в качестве ссылки. Таким образом, для указания (обозначения) "контролируемая фоновая боль" обычно требуется, чтобы два критерия совпали.
Во-первых, пациент должен утвердительно ответить на вопрос "Включает ли Ваша боль компонент, который Вы описали бы как "постоянный" или "почти постоянный" или "была бы постоянной или почти постоянной", если бы не проводимое лечение?". Во-вторых, пациенту может потребоваться лечение опиоидом по схеме, которая согласуется со сравнительно надежным контролем боли. Специалист в данной области техники поймет как определить контролируемую фоновую (базовую, основную) боль с учетом сведений, представленных в этих двух ссылочных материалах.
Затем определяют прорывную боль как внезапное обострение (вспышка) боли, испытываемое пациентом, превышающее уровень контролируемой фоновой боли. Интенсивность боли можно оценить по 5-балльной категорийной шкале, включающей пункты "нет (отсутствие)" боли, "легкая (слабая)", "умеренная", "жестокая, острая" и "мучительная" боль. Как правило, пациент испытывает приступ прорывной боли, если этот приступ оценивается им либо как острый, жестокий, либо как мучительный.
Термин "биоэквивалентность" и его грамматические варианты по данному описанию применяется в его обычном значении. В частности, говорят, что фармацевтическая композиция является биоэквивалентной эталонной фармацевтической композиции, если 90% доверительного интервала среднего значения площади под кривой (AUC) (AUCt или AUCinf) уровней в плазме крови тестируемой фармацевтической композиции составляет от 80% до 125% от соответствующего среднего значения эталонной фармацевтической композиции и если 90% доверительного интервала среднего значения максимальной концентрации (Cmax) в плазме крови тестируемой фармацевтической композиции составляет от 80% до 125% от соответствующего среднего значения эталонной фармацевтической композиции.
Величина Cmax обозначает максимальную концентрацию активного вещества, а именно оксикодона и/или налоксона (или их солей, таких как гидрохлорид) в плазме крови.
Величины tmax обозначают временную точку, в которой достигается величина Cmax. Другими словами, tmax представляет собой временную (время) точку, в которой достигается максимальная наблюдаемая концентрация в плазме.
Величина AUC (площадь под кривой) соответствует площади под кривой концентрации. Величина AUC пропорциональна общему количеству активных агентов, а именно оксикодона и налоксона (или их солей, таких как гидрохлорид), попадающих в кровоток, и, следовательно, является мерой биодоступности.
Величины AUCt представляет собой площадь под кривой зависимости концентрации от времени от времени введения до последней измеренной концентрации. Величины AUCt обычно рассчитывают, используя метод трапеций. Если это возможно, LambdaZ, константу скорости конечной фазы, определяют, используя эти точки на конечной линейной фазе. t1/2Z, кажущийся период полужизни для конечной фазы, обычно определяют из соотношения ln2 к LambdaZ. Площади под кривой концентрация в плазме - время между последней измеренной точкой и бесконечностью можно вычислить по отношению конечной наблюдаемой концентрации в плазме (Clast) к LambdaZ. Эту величину добавляют к AUCt, получают AUCinf, которая представляет собой площадь под кривой концентрация в плазме - время от времени введения до бесконечности.
Термин "биодоступность" для целей настоящего изобретения определяется как степень (величина) всасывания активных агентов, таких как оксикодон и налоксон (или их солей, таких как гидрохлорид) после перорального введения фармацевтической композиции.
Термин "стационарное (равновесное) состояние" можно описать следующим образом: в момент t=0, время введения первой дозы, концентрация С=0. Затем концентрация проходит первый максимум и затем падает до первого минимума. Перед тем, как концентрация упадет до 0, вводят вторую дозу, так что второе повышение концентрации начинается не с 0.
После прохождения этого первого минимума концентрации введение второй дозы кривая проходит второй максимум, который превышает первый максимум, и падает до второго минимума, превышающего первый минимум. Таким образом, кривая плазматической концентрации повышается за счет повторных (многократных) доз и ассоциированной с ними постадийной (постепенной) аккумуляции активного агента до тех пор, пока не дойдет до уровня, в которой не уравновесятся абсорбция и удаление (из организма). Состояние, при котором абсорбция (всасывание) и удаление уравновешены, а концентрация постоянно колеблется между определенным минимумом и определенным максимумом, называют стационарным (равновесным) состоянием.
Термины "поддерживающая терапия" и "продолжительная (длительная) терапия" для целей настоящего изобретения определяются как лекарственная терапия после титрования вводимой пациенту дозы опиоидного анальгетика до стационарного состояния по определению выше. Указанная выше фоновая боль относится к ощущению боли в течение поддерживающей/продолжительной терапии.
Показатели, описывающие кривую плазмы крови, можно получить в процессе клинических испытаний, прежде всего с помощью однократного (once-off) введения активного агента, такого как оксикодон и налоксон (или их солей, таких как гидрохлорид), большому числу испытуемых. Затем усредняют значения отдельных испытуемых, получают среднее значение AUC, Cmax и tmax.
Если не указано иначе, фармакокинетические параметры (показатели), такие как AUC, Cmax и tmax, в контексте настоящего изобретения относятся к средним значениям. Далее, если не указано иначе, показатели in vivo, такие как AUC, Cmax и tmax или анал(ь)гезирующая эффективность, в контексте настоящего изобретения относятся показателям или к значениям (величинам), полученным после введения в стационарном состоянии разовой дозы больным и/или здоровым людям.
Если фармакокинетические параметры, такие как средние значения tmax, Cmax и AUC, определяют для здоровых людей, как правило, их получают, измеряя показатели в плазме крови в во времени в тестируемой группе, состоящей из 16-24 здоровых людей.
Регулирующие органы, такие как Европейское агентство по оценке медицинских продуктов (ЕАЛС, ЕМЕА) или Управлению по контролю за пищевыми продуктами и лекарственными препаратами (FDA, США) обычно признают данные, полученные, например, от 20 или 24 человек. Однако в начальных испытаниях участвует меньше участников, например, может быть также приемлемым участие 5 тестируемых.
Термин "здоровый" человек (субъект) в данном контексте относится к типичному мужчине или к типичной женщине, обычно представителям белой расы, со средними параметрами, такими как рост, вес, и средними физиологическими показателями, такими как артериальное давление. Здоровых субъектов для настоящего изобретения отбирают согласно критериям включения и критериям исключения, которые основаны на рекомендациях и соответствуют рекомендациям международной организации "Международная конференция по гармонизации" (ICH). Для целей настоящего изобретения здоровых субъектов можно идентифицировать в соответствии с критериями включения и исключения, изложенными, например, в Примере 2 и в Примере 3.
Так, критерии включения охватывают, например, возраст от ≥18 до ≤60 лет; например, BMI (индекс массы тела) в интервале 19-29 кг/м2 и, например, вес в интервале 60-100 кг для мужчин и 55-90 кг для женщин; эти женщины не должны быть ни кормящими грудью, ни беременными и должны иметь отрицательную реакцию мочи на B-hCG тест на беременность в течение 24 часов до приема исследуемого лекарства; обычно хорошее здоровье, которое подтверждается отсутствием заметных аномальных данных в анамнезе, физикальным исследованием, клиническими лабораторными тестами, основными показателями жизненно важных функций и ЭКГ (ECG) и т.д.
Критерии исключения охватывают, например, прием любого исследуемого лекарства или плацебо в течение 3 месяцев до первой дозы исследуемого лекарства, любое серьезное заболевание в течение 30 дней до введения первой дозы исследуемого лекарства, любые клинически значимые аномалии, идентифицированные при скрининге перед исследованием в анамнезе, при физикальном исследовании или в лабораторных анализах, применение любого прописанного лекарственного средства (за исключением HRT (гормонозаместительной терапии) для женщин в период менопаузы и контрацептивов) в течение 21 дня или средства противотерапии, такого как регулятор кислоты, витаминов, лекарственных трав и/или минеральных добавок в течение 7 дней до приема первой дозы исследуемого лекарственного средства, наличие конкурентного медицинского состояния, заведомо препятствующего всасыванию лекарства в желудочно-кишечном тракте (например, замедляющего опорожнение (эвакуацию содержимого из) желудка, синдромы мальадсорбции), распределению (например, ожирение), метаболизму или экскреции (например, гепатит, гломерулонефрит), в анамнезе или конкурентное (одновременное) медицинское показание, которое, по мнению исследователя, сможет помешать субъекту без опасности для своего здоровья завершить исследование, эпилепсию в анамнезе, требующую фармакологического лечения, в анамнезе курение более 5 сигарет в день, субъекты с явной текущей или в анамнезе наркотической или алкогольной зависимостью по критериям DSM-IV, субъекты, уведомляющие о регулярном употреблении спиртного 2 или более раз в день, или в крови которых при обследовании (скрининге) содержание алкоголя составляет ≥0.5%, сдача более 500 мл донорской крови или продуктов крови или другая значительная потеря крови в течение 3 месяцев до первой дозы исследуемого лекарства, любые положительные результаты перед исследованием на этанол, опиаты, барбитураты, амфетамины, метаболиты кокаина, метадон, пропоксифен, фенциклидин, бензодиазепины и каннабиоиды в образце мочи, собранной при скрининге, заведомо чувствительном к оксикодону, налоксону или родственным соединениям и т.д.
Если фармакокинетические параметры, такие как средние значения tmax, cmax и AUC, определяют для больных людей (пациентов), то группы пациентов обычно включают от 10 до 200 пациентов. Корректное число пациентов составляет, например, 10, 20, 30, 40, 50, 75, 100, 125 или 150 человек. Пациентов отбирают в соответствии с симптомами состояния, подлежащего лечению. Пациенты могут быть в возрасте ≥18 лет, страдать хронической болью от умеренной до сильной или даже жестокой боли, вызванной или не вызванной опухолью, для которых недостаточно эффективны анальгетики, рекомендуемые ВОЗ на II или III ступени, и/или которые толерантны к этим анальгетикам, и т.д. Если у пациента в данное время имеются симптомы алкогольной или наркотической зависимости, симптомы тяжелого сердечнососудистого или респираторного заболевания, тяжелой печеночной или почечной недостаточности и т.д., то его фармакокинетические параметры можно не определять.
Следует понимать, что значения фармакокинетических показателей, указанные выше и ниже, определяют исходя из данных, которые получают в Примерах 2 и 3, все они относятся к клиническим исследованиям с введением однократной дозы ("однодозовым" исследованиям) на здоровых людях. Однако предполагается, что сравнимые результаты будут получены при введении в стационарном состоянии здоровым людям или при введении разовой дозы и в стационарном состоянии больным пациентам. Естественно, специалист в данной области техники знает, что Cmax стационарного состояния будет выше, чем Cmax после введения разовой дозы.
Расчет фармакокинетического показателя можно проводить некомпартментным (модельно независимым) методом, используя программу WinNonlin Enterprise Edition, Version 4.1.
Как указывается выше, настоящее изобретение относится к пероральной фармацевтической композиции с мгновенным высвобождением, содержащей по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль).
Как представлено ниже, такие пероральные фармацевтические композиции с мгновенным высвобождением, содержащие по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль), при испытании являются биоэквивалентными эталонной фармацевтической композиции с контролируемым высвобождением, содержащей по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль). Эталонная фармацевтическая композиция с контролируемым высвобождением, используемая в Примере 3, содержащая оксикодона гидрохлорид и налоксона гидрохлорид в примерном весовом соотношении 2:1, доступна на немецком рынке под торговой маркой Targin® в виде таблеток. Было показано, что этот препарат с контролируемым высвобождением, таблетки Targin®, не только обеспечивает прекрасный контроль (прекрасное регулирование) боли у пациентов, страдающих, например, от боли, от сильной до мучительной, жестокой боли, но также дает имеет хороший профиль побочных эффектов. Смягчение, ослабление побочных эффектов при использовании таблеток Targin® можно оценивать как улучшение функции кишечника, как снижение вызванной опиоидом констипации, как снижение вызванной опиоидом задержки мочи и как повышенную переносимость и предпочтение препарата пациентами, страдающими от боли (см., например, Vondrackova et al., J. Pain (2008), 9 (12): 1144-1154, Nastawek et al., Int. J. Clin. Pract. (2008), 62 (8): 1159-1167 и Meissner et al., Eur. J. Pain (2008), статья в печати).
Далее из Эксперимента 2 становится ясным, что пероральные фармацевтические композиции с мгновенным высвобождением по изобретению являются пропорциональны дозе, в частности, когда они содержат некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-10 мг налоксона гидрохлорида в примерном весовом соотношении 2:1.
Ввиду биоэквивалентности, которая в данном случае означает эквивалентность степени доставки фармацевтических композиций с мгновенным высвобождением, содержащих оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1, препарату Targin® таблетки с контролируемым высвобождением (и в особенности ввиду неожиданной степени биодоступности и того факта, что эффект налоксона после первого прохода сохраняется также для препарата с мгновенным высвобождением), кажется оправданным принять, что фармацевтические композиции с мгновенным высвобождением по изобретению не только обеспечивают эффективное облегчение боли у пациентов, страдающих от боли, в частности хронической боли от умеренной до сильной и даже до жестокой (мучительной) боли, но также у пациентов, пролеченных этими композициями, наблюдается сравнимое, если не такое же, улучшение в сравнении с профилем побочных эффектов, наблюдаемым для Targin® таблеток, в частности, по сравнению с пероральными препаратами с мгновенным высвобождением, известным из предыдущего уровня техники, которые содержат только агонист опиоидных рецепторов оксикодона гидрохлорид.
Поэтом полагают, что пероральные фармацевтические композиции с мгновенным высвобождением по изобретению будут эффективными при лечении боли и будут вызывать улучшение в том, что касается побочных эффектов, таких как констипация (обстипация), вызванная опиоидами, уменьшение вызванной опиоидами задержки мочи и повышение переносимости и предпочтения этого медицинского средства пациентами по сравнению с пероральными композициями с мгновенным высвобождением, известным из предыдущего уровня техники, которые содержат только агонист опиоидных рецепторов оксикодона гидрохлорид.
Эти свойства в сочетании с пропорциональностью дозы пероральных фармацевтических композиций с мгновенным высвобождением по изобретению позволяют врачу применять фармацевтические композиции по изобретению в тех случаях, в которых ранее фармацевтические композиции с контролируемым высвобождением, содержащие оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль, были предпочтительны по сравнению с препаратами с мгновенным высвобождением, содержащим только агонист опиоидных рецепторов оксикодон, вследствие лучшего профиля побочных эффектов препарата с контролируемым высвобождением.
Пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению применимы, в частности, для титрования дозы для пациентов, страдающих от боли, и/или для лечения прорывной боли у пациентов, страдающих от боли.
В частности, пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению можно применять для титрования дозы для пациента, страдающего от боли, и, в частности, от сильной до жестокой, (мучительной) (хронической) боли, тогда как ранее такое титрование с применением препаратов с контролируемым высвобождением оксикодона и налоксона означало бы неприятную, если не непереносимую боль вследствие задержанного действия препаратов с контролируемым высвобождением. Помимо этого пероральные препараты с мгновенным высвобождением в соответствии с настоящим изобретением можно использовать для лечения, например, приступов прорывной боли у пациентов, которые уже принимают препараты с контролируемым высвобождением оксикодона или его фармацевтически приемлемой соли и налоксона или его фармацевтически приемлемой соли, но тем не менее испытывают неожиданные приступы боли и поэтому принимают требуемый препарат с мгновенным высвобождением, содержащий только опиоид оксикодона гидрохлорид.
Как указывается, фармацевтические лекарственные формы по настоящему изобретению относятся также к пероральной фармацевтической композиции с мгновенным высвобождением, содержащей по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль, причем композиция предусматривает профиль с быстрым in vitro растворением, такой, что, например, более 90% или даже более 95% активных веществ высвобождается через 15 минут. Все эти свойства сообщают композиции то преимущество, что ее можно вводить в твердом виде, например в виде проглатываемой таблетки. В то же время группы пациентов, такие как группы детей или пожилых людей, у которых имеются проблемы с проглатыванием, могут принимать таблетку непосредственно перед приемом быстро растворенную в жидкости, или эту таблетку можно помещать непосредственно в рот с целью быстрого растворения ее в слюне. Это может быть очень важно и удобно в случае внезапного приступа боли, когда необходимо быстрое обезболивание.
Как уже упоминалось, настоящее изобретение в основном относится к пероральной фармацевтической композиции, содержащей по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль, его фармацевтически приемлемую соль в примерном весовом соотношении 2:1 (оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль).
В принципе пероральные фармацевтические композиции с мгновенным высвобождением помимо оксикодона и налоксона могут содержать другие фармацевтически активные агенты. Однако в предпочтительном варианте всех аспектов настоящего изобретения пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению содержат оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в качестве единственных фармацевтически активных агентов.
Обычно пероральные фармацевтические лекарственные формы с мгновенным высвобождением по настоящему изобретению могут содержать оксикодон и налоксон в виде свободного основания.
Однако оксикодон или налоксон могут также присутствовать в виде фармацевтически приемлемых солей. Такие соли включают, например, соли: гидрохлорид, сульфат, бисульфат, тартрат, нитрат, цитрат, битартрат, фосфат, малат, малеат, гидробромид, гидроиодид, фумарат, сукцинат и т.п.
Оксикодон и налоксон, как указано далее, могут находиться также в виде солей присоединения основания, таких как соли щелочных металлов, включающих литий, натрий и калий. Они могут присутствовать также в виде производных свободного основания. Такие производные могут включать, например, сложные эфиры.
В предпочтительном варианте в настоящем изобретении применяются оксикодона гидрохлорид и налоксона гидрохлорид. Еще более предпочтительным может вариант настоящего изобретения, в котором единственными фармацевтически активными агентами в пероральных фармацевтических композиций с мгновенным высвобождением являются оксикодона гидрохлорид и налоксона гидрохлорид, предпочтительно, в примерном весовом соотношении 2:1.
Пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению содержат оксикодон в количестве, достаточном для лечения боли, и, в частности, (хронической) боли у пациента от сильной до мучительной (жестокой). Как правило, фармацевтическая композиция по изобретению содержит некоторое количество оксикодона или его фармацевтически приемлемую соль, эквивалентную, примерно, 1-160 мг оксикодона гидрохлорида.
Количество налоксона в фармацевтических композициях по настоящему изобретению подбирается таким образом, чтобы это количество вводимого налоксона не оказывало заметного отрицательного воздействия на обезболивание, опосредуемое оксикодоном. Как правило, фармацевтическая композиция по настоящему изобретению содержит некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида.
Следует понимать, что количество оксикодона или его фармацевтически приемлемой соли и налоксона или его фармацевтически приемлемой соли можно подбирать таким образом, чтобы оксикодон или его фармацевтически приемлемая соль и налоксон или его фармацевтически приемлемая соль находились в примерном весовом соотношении 2:1 в расчете на оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль.
Таким образом, как правило, фармацевтические композиции по настоящему изобретению содержат некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-160 мг оксикодона гидрохлорида, эквивалентное, примерно, 1-80 мг оксикодона гидрохлорида, эквивалентное, примерно, 1-40 мг оксикодона гидрохлорида, эквивалентное, примерно, 1-20 мг оксикодона гидрохлорида, эквивалентное, примерно, 1-20 мг оксикодона гидрохлорида, эквивалентное, примерно, 1-5 мг оксикодона гидрохлорида, и эквивалентное, примерно, 1-2.5 мг оксикодона гидрохлорида.
Помимо этого, как правило, фармацевтические композиции по настоящему изобретению содержат некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида, эквивалентное, примерно, 0.5-40 мг налоксона гидрохлорида, эквивалентное, примерно, 0.5-20 мг налоксона гидрохлорида, эквивалентное, примерно, 0.5-10 мг налоксона гидрохлорида, эквивалентное, примерно, 0.5-5 мг налоксона гидрохлорида, эквивалентное, примерно, 0.5-2.5 мг налоксона гидрохлорида, и эквивалентное, примерно, 0.5-1.25 мг налоксона гидрохлорида.
Предпочтительные варианты настоящего изобретения относятся к фармацевтическим композициям, которые содержат некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-40 мг, предпочтительно, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-20 мг налоксона гидрохлорида, предпочтительно, примерно, 1.25-10 мг налоксона гидрохлорида. В этих композициях могут, в частности, применяться гидрохлориды оксикодона и налоксона.
Особенно предпочтительными пероральными фармацевтическими композициями с мгновенным высвобождением по настоящему изобретению являются препараты с предпочтительной эффективной дозой, которые содержат некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25 мг налоксона гидрохлорида, некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 5 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5 мг налоксона гидрохлорида, некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 10 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 5 мг налоксона гидрохлорида, некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 10 мг налоксона гидрохлорида. В этих композициях можно, в частности, применять хлористоводородные соли оксикодона и налоксона.
Особенно предпочтительные варианты настоящего изобретения относятся к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим 2.5 мг оксикодона гидрохлорида и 1.25 мг налоксона гидрохлорида, 5 мг оксикодона гидрохлорида и 2.5 мг налоксона гидрохлорида, 10 мг оксикодона гидрохлорида и 5 мг налоксона гидрохлорида, и 20 мг оксикодона гидрохлорида и10 мг налоксона гидрохлорида
Пероральные фармацевтические композиции с мгновенным высвобождением по изобретению, содержащие оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1, отличаются тем, что как правило, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 45 мин из них высвобождается ≥75% вес. оксикодона или его фармацевтически приемлемой соли и ≥75% вес. налоксона или его фармацевтически приемлемой соли. Более предпочтительные фармацевтические композиции по изобретению отличаются тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 45 мин высвобождается ≥80% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. налоксона или его фармацевтически приемлемой соли.
Еще более предпочтительные фармацевтические композиции, содержащие оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении 2:1, отличаются тем, что как правило, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин из них высвобождается ≥80% вес. и, предпочтительно, ≥90% вес, и еще более предпочтительно, ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. и, предпочтительно, ≥90% вес, и еще более предпочтительно, ≥95% вес. налоксона или его фармацевтически приемлемой соли.
Пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению могут выпускаться в виде лекарственной формы, обычной для препаратов с мгновенным высвобождением. Так, пероральные фармацевтические композиции с мгновенным высвобождением по настоящему изобретению могут быть в твердом виде, таком как таблетка, капсула, мультичастицы, сфероиды или бусины, или в жидком виде, например в виде раствора, суспензии или эмульсии. Предпочтительная лекарственная форма для пероральных фармацевтических композиций с мгновенным высвобождением по настоящему изобретению представляет собой таблетку.
При получении пероральных фармацевтических композиций с мгновенным высвобождением по настоящему изобретению помимо оксикодона или его фармацевтически приемлемой соли и налоксона или его фармацевтически приемлемой соли можно использовать фармацевтически приемлемые эксципиенты, обычные в области фармацевтики. Типичными фармацевтически приемлемыми эксципиентами являются разрыхлители, разбавители, смазки, вещества, способствующие проглатыванию - глиданты, вещества, предотвращающие слипание, связующие, вещества, корректирующие рН и т.п. Эти эксципиенты (за исключением разрыхлителей) следует выбирать таким образом, чтобы они не вызывали значительного изменения описанной выше скорости мгновенного высвобождения in vitro.
Предпочтительными могут быть фармацевтические композиции по настоящему изобретению, содержащие в качестве фармацевтически приемлемых эксципиентов по меньшей мере разбавитель и, необязательно, разрыхлитель, в частности, в случае, когда фармацевтические композиции по настоящему изобретению выпускаются в виде таблеток. Также предпочтительно, чтобы фармацевтические композиции по настоящему изобретению в качестве фармацевтически приемлемых эксципиентов содержали по меньшей мере один разрыхлитель и, необязательно, разбавитель, в частности, в случае, когда фармацевтические композиции по настоящему изобретению выпускаются в виде таблеток. Кроме того, предпочтительно, применять эксципиенты, которые действуют как разрыхлитель и как разбавитель.
Например, разрыхлитель нужен для обеспечения быстрого измельчения таблетки после введения, чтобы активные ингредиенты оксикодон или его фармацевтически приемлемая соль и налоксон или его фармацевтически приемлемая соль стали легкодоступными для всасывания.
Разбавители можно выбирать, но без ограничения, из лактозы, такой как моногидрат лактозы, безводная лактоза, крахмала, такого как кукурузный (маисовый) крахмал, прежелатинизированный крахмал, микрокристаллической целлюлозы, глюкозы, маннита, мальтита, StarLac® (85% высушенной распылением лактозы, 15% кукурузного крахмала), сахарозы, солей кальция, таких как гидрофосфат кальция, или любых комбинаций вышеприведенных веществ.
Разрыхлители можно выбирать, но без ограничения, в том числе из StarLac® (85% высушенной распылением лактозы, 15% кукурузного крахмала), кроскармеллозы, такой как кроскармеллоза натрия, крахмала гликолята натрия, кросповидона, альгиновой кислоты или гидроксипропилцеллюлозы с низкой степенью замещения.
Особенно предпочтительной может быть комбинация лактозы и крахмала, такая, как продукт StarLac®, поскольку он обладает свойствами как наполнителя, так и разрыхлителя (дезинтегранта).
Глиданты и смазки можно выбирать, но без ограничения, в том числе из высокодисперсного диоксида кремния, талька, оксида магния, стеарата магния, стеарилфумарата натрия и т.д.
Агенты, повышающие текучесть, и смазки включают, в том числе высокодисперсный диоксид кремния, тальк, оксид магния, стеарат магния, стеарилфумарат натрия и т.д.
Если фармацевтические композиции по настоящему изобретению предусматриваются в виде таблеток, с целью их идентификации они могут покрываться косметическим покрытием. Такие покрытия не оказывают заметного влияния на свойства, обуславливающие мгновенное высвобождение фармацевтических композиций по изобретению.
В предпочтительном варианте настоящее изобретение относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим, примерно, 2.5-160 мг оксикодона гидрохлорида и, примерно, 1.25-80 мг налоксона гидрохлорида в примерном весовом соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин из них высвобождается ≥90% вес. и, предпочтительно, ≥95% вес. оксикодона гидрохлорида и ≥90% вес. и, предпочтительно, ≥95% вес. налоксона гидрохлорида. Предпочтительно, такие композиции содержат разрыхлитель (дезинтегрант) и находятся в твердом виде, например в виде таблетки.
В другом предпочтительном варианте настоящее изобретение относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим, примерно, 2.5-80 мг оксикодона гидрохлорида и, примерно, 1.25-40 мг налоксона гидрохлорида в примерном весовом соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин из них высвобождается ≥90% вес. и, предпочтительно, ≥95% вес. оксикодона гидрохлорида и ≥90% вес. и, предпочтительно, ≥95% вес. налоксона гидрохлорида. Предпочтительно, такие композиции содержат разрыхлитель (дезинтегрант) и находятся в твердом виде, например в виде таблетки.
В другом предпочтительном варианте настоящее изобретение относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим, примерно, 2.5-40 мг оксикодона гидрохлорида и, примерно, 1.25-20 мг налоксона гидрохлорида в примерном весовом соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин из них высвобождается ≥90% вес. и, предпочтительно, ≥95% вес. оксикодона гидрохлорида и ≥90% вес. и, предпочтительно, ≥95% вес. налоксона гидрохлорида.
Предпочтительно, такие композиции содержат разрыхлитель (дезинтегрант) и находятся в твердом виде, например в виде таблетки.
В другом предпочтительном варианте настоящее изобретение относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим, примерно, 2.5-20 мг оксикодона гидрохлорида и, примерно, 1.25-10 мг налоксона гидрохлорида в примерном весовом соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин из них высвобождается ≥90% вес. и, предпочтительно, ≥95% вес. оксикодона гидрохлорида и ≥90% вес. и, предпочтительно, ≥95% вес. налоксона гидрохлорида. Предпочтительно, такие композиции содержат разрыхлитель (дезинтегрант) и находятся в твердом виде, например в виде таблетки.
В другом предпочтительном варианте настоящее изобретение относится к пероральным фармацевтическим композициям с мгновенным высвобождением, содержащим, примерно, 2.5-10 мг оксикодона гидрохлорида и, примерно, 1.25-5 мг налоксона гидрохлорида в примерном весовом соотношении 2:1 (оксикодона гидрохлорид: налоксона гидрохлорид), отличающимся тем, что, по измерению в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°С и с УФ-детекцией при 230 нм, in vitro через 15 мин из них высвобождается ≥90% вес. и, предпочтительно, ≥95% вес. оксикодона гидрохлорида и ≥90% вес. и, предпочтительно, ≥95% вес. налоксона гидрохлорида. Предпочтительно, такие композиции содержат разрыхлитель (дезинтегрант) и находятся в твердом виде, например в виде таблетки.
Предпочтительными пероральными фармацевтическими композициями с мгновенным высвобождением по изобретению, содержащими некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25 -10 мг налоксона гидрохлорида, в примерном весовом соотношении 2:1, могут являться композиции, которые предусматривают AUCt оксикодона в примерном диапазоне 15 нг·час/мл - 500 нг·час/мл, предпочтительно, в примерном диапазоне 20 нг·час/мл - 400 нг·час/мл, более предпочтительно, в примерном диапазоне 25 нг·час/мл - 300 нг·час/мл, и, еще более предпочтительно, в примерном диапазоне 30 нг·час/мл - 250 нг·час/мл при исследовании на людях с введением разовой дозы здоровым добровольцам. Такие пероральные фармацевтические композиции с мгновенным высвобождением, предпочтительно, могут содержать хлористоводородные соли оксикодона и налоксона.
Предпочтительными пероральными фармацевтическими композициями с мгновенным высвобождением по изобретению, содержащими некоторое количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-10 мг налоксона гидрохлорида, в примерном весовом соотношении 2:1, могут являться композиции, которые предусматривают Cmax оксикодона в примерном диапазоне 1 нг/мл - 300 нг/мл, предпочтительно, в примерном интервале 2 нг/мл - 200 нг/мл, более предпочтительно, в примерном интервале 3 нг/мл -100 нг/мл, еще более предпочтительно, в примерном интервале 4 нг/мл - 75 нг/мл, и еще более предпочтительно, в примерном интервале 6 нг/мл - 50 нг/мл при исследовании на людях с введением разовой дозы здоровым добровольцам. Такие пероральные фармацевтические композиции с мгновенным высвобождением, предпочтительно, могут содержать хлористоводородные соли оксикодона и налоксона.
Варианты настоящего изобретения относятся также к пероральным фармацевтическим композициям с мгновенным высвобождением, которые содержат оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль, причем фармацевтическая композиция после хранения в стрессовых условиях высвобождает фармацевтически активные агенты практически с такой же скоростью, как и перед хранением этой фармацевтической композиции в этих стрессовых условиях.
Другие варианты настоящего изобретения относятся к пероральным фармацевтическим композициям с мгновенным высвобождением, которые содержат оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль, причем фармацевтическая композиция после хранения в стрессовых условиях теряет менее 3.0% от общего количества веществ, относящихся к оксикодону или его фармацевтически приемлемой соли и/или к налоксону или его фармацевтически приемлемой соли (обусловленных оксикодоном или его фармацевтически приемлемой солью, и/или обусловленных налоксоном или его фармацевтически приемлемой солью).
В контексте настоящего изобретения хранение в стрессовых условиях означает, что фармацевтическая композиция подвергается действию повышенной температуры и/или относительной влажности (RH) в течение длительного периода времени. Например, понятие "типичные стрессовые условия" относится к хранению по меньшей мере в течение одного, двух, трех, четырех, пяти, шести, девяти или двенадцати месяцев при 25°С и при 60% RH. В другом случает понятие "стрессовые условия" относятся к хранению по меньшей мере в течение одного, двух, трех, четырех или пяти месяцев при 40°С и при 75% RH.
Такие стрессовые условия хранения применяют для того, чтобы определить, имеет ли фармацевтическая композиция срок годности, достаточный для длительного хранения ее пациентами в обычных домашних условиях при отсутствии отрицательного воздействия на ее (фармацевтической композиции) безопасность и эффективность. Это отрицательное воздействие (отрицательные эффекты) может заключаться в том, что in vitro скорость высвобождения меняется во времени, а на эффективность композиции влияет то, что после введения высвобождаются различные количества активных веществ. Аналогично, отрицательные эффекты (отрицательное воздействие) могут также возникать в результате расщепления фармацевтически активных агентов, что может либо уменьшать общее количество функционального фармацевтически активного агента, либо привести к образованию токсических побочных продуктов.
Если после хранения в стрессовых условиях наблюдаются изменения профиля высвобождения in vitro или количества активного агента (активных агентов) фармацевтической композиции, это может свидетельствовать о проблемах со стабильностью (устойчивостью). Если такие изменения не наблюдаются, это, напротив, означает, что фармацевтическая композиция устойчива при хранении.
Выражение "практически такая же скорость высвобождения" относится к ситуации, при которой in vitro скорость высвобождения для фармацевтической композиции после хранения в стрессовых условиях сравнима со скоростью высвобождения для эталонной композиции. Эталонная композиция представляет собой идентичную фармацевтическую композицию, но не подвергнувшуюся хранению в стрессовых условиях. Если профиль in vitro высвобождения композиции, хранившейся в стрессовых условиях, не отклоняется более чем на 20%, предпочтительно, не более чем на 15%, более предпочтительно, не более чем на 10%, и еще более предпочтительно, не более чем на 5% от профиля in vitro высвобождения эталонной композиции, in vitro скорость высвобождения считают практически такой же самой.
Выражение "все (тотальные) вещества, относящиеся к оксикодону и налоксону" относится к веществам, которые возникают в результате химических реакций этих фармацевтически активных агентов. Эти вещества включают, например, налоксон N-оксид и т.п.
Для оценки стабильности можно выдерживать фармацевтическую композицию в стрессовых условиях, указанных выше, и определять количество соединений, относящихся к оксикодону и/или налоксону. Затем определяют количество соединений, относящихся к оксикодону и/или налоксону, для идентичной фармацевтической композиции, которая не выдерживалась в стрессовых условиях. Эту композицию принимают за эталонную композицию. Обнаружение "веществ, относящихся к оксикодону" и "веществ, относящихся к налоксону" обычно проводят методом ВЭЖХ. Идентичность веществ можно определять, проведя такой же анализ чистых известных эталонных веществ.
Фармацевтическая композиция считается стабильной, если после выдерживания ее в стрессовых условиях она содержит не более, примерно, 3%, предпочтительно, не более, примерно, 2%, более предпочтительно, не более, примерно, 1%, и еще более предпочтительно, не более, примерно, 0.5% веществ, относящихся к оксикодону и/или налоксону, от количества гидроморфона или налоксона, присутствующего в композиции.
Следует понимать, что свойства композиций, имеющих практически такую же скорость высвобождения после хранения в стрессовых условиях, относятся также к вариантам изобретения, указанным выше, таким как композиции с весовым соотношением 2:1, композиции, содержащие указанные выше количества, и/или композиции, имеющие вышеуказанные профили высвобождения in vitro, в особенности, если композиции находятся в твердом виде, например, в виде таблеток.
Фармацевтические композиции по изобретению, предусматриваемые в твердом виде, например, в виде таблетки, можно получать методом, включающим, например, следующие стадии:
а) Приготовление смеси оксикодона или его фармацевтически приемлемой соли, налоксона или его фармацевтически приемлемой соли, причем оба реагента имеют подходящую дисперсность, разбавителя и, необязательно, дезинтегранта (разрыхлителя);
б) Необязательное добавление смазки в эту смесь;
в) Прямое прессование указанной смеси с целью получения таблеток.
Фармацевтические композиции по изобретению в твердом виде, например, в виде таблетки, можно получать методом, включающим следующие стадии:
а) Приготовление смеси оксикодона или его фармацевтически приемлемой соли, налоксона или его фармацевтически приемлемой соли, причем оба реагента имеют подходящую дисперсность, дезинтегранта (разрыхлителя) и, необязательно, разбавителя;
б) Необязательное добавление смазки в эту смесь;
в) Прямое прессование указанной смеси с целью получения таблеток.
Если полагают необходимым, в вышеописанный процесс приготовления однородной смеси может включать стадии скрининга с отбором проб, чтобы гарантировать отсутствие комкообразных компонентов.
Предпочтительно, размер частиц всех эксципиентов и, необязательно, активных ингредиентов, находится в одном и том же диапазоне (одинаковая дисперсность). Типичный размер частиц составляет около 100 мкм - 300 мкм.
Найдено, что полученные таким методом таблетки легко измельчаются и по измерению в приборе с лопастью (Ph. Eur. Paddle Method) через 15 мин высвобождают in vitro, например ≥90% вес. и, предпочтительно, ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥90% вес. и, предпочтительно, ≥95% вес. налоксона или его фармацевтически приемлемой соли. Например, в Примере 4 таблетки, полученные таким методом, устойчивы при хранении, это означает, что они практически не изменяют своего поведения при высвобождении in vitro после продолжительного хранения в стрессовых условиях.
Вышеописанные и полученные как указано выше фармацевтические композиции с мгновенным высвобождением по данному описанию можно применять для титрования дозы для пациентов, страдающих от боли, и, в частности, хронической боли от умеренной до мучительной (жестокой).
Применение пероральных фармацевтических композиций с мгновенным высвобождением для титрования дозы для пациентов и лечения прорывной боли у пациентов гарантирует быстрое облегчение боли (обезболивание) у пациентов вследствие мгновенного высвобождения, при этом отсутствуют типичные побочные эффекты, вызываемые опиоидами, такие как констипация и задержка мочи.
Далее изобретение описано на некоторых конкретных примерах. Однако эти примеры не следует считать ограничивающими.
Примеры
Пример 1: Приготовление фармацевтических композиций с мгновенным высвобождением, содержащим оксикодона гидрохлорид и налоксона гидрохлорид
Фармацевтические композиции с мгновенным высвобождением, содержащие 20 мг оксикодона гидрохлорида/10 мг налоксона гидрохлорида (IR-OXN 20/10), 10 мг оксикодона гидрохлорида/5 мг налоксона гидрохлорида (IR-OXN 10/5), 5 мг оксикодона гидрохлорида/2.5 мг налоксона гидрохлорида (IR-OXN 5/2.5) и 2.5 мг оксикодона гидрохлорида/1.25 мг налоксона гидрохлорида (IR-OXN 2.5/1.25), получают, как описано ниже. Их состав подробно показан в Таблице 1.
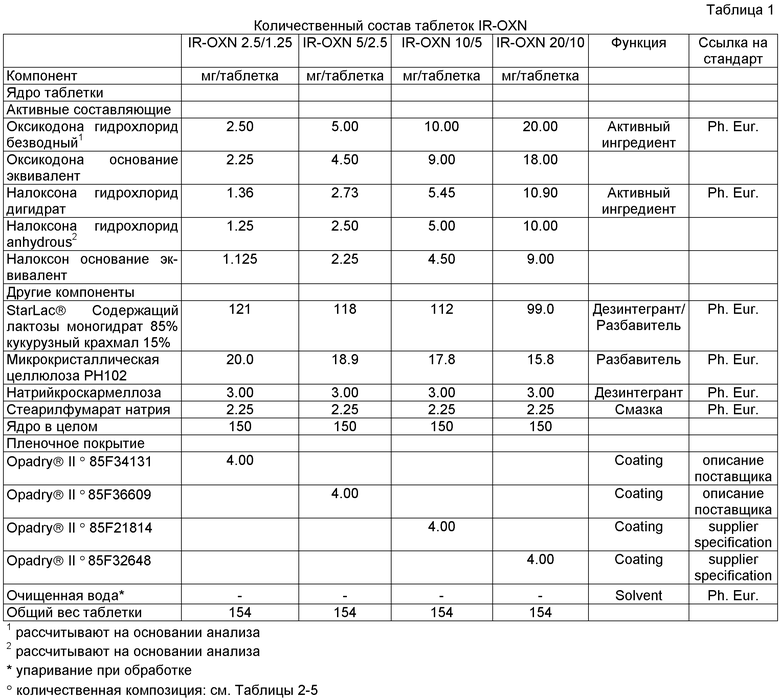
Компоненты (составляющие) в Таблицах 1-5 обрабатывают, как изображено на блок-схеме, представленной на Фигуре 1.
Говоря конкретнее, микрокристаллическую целлюлозу, кроскармеллозу натрия и активные ингредиенты просеивают через сита с размером отверстий 500 мкм для удаления любых агломератов, помещают в смеситель (блендер) со StarLack® и перемешивают до получения однородной смеси. StarLack® просеивать не требуется, так как он представляет собой легкосыпучий (свободно сыпучий) порошок. Смазку, стеарилфумарат натрия, просеивают через сита с размером отверстий 500 мкм, добавляют в смеситель и дополнительно перемешивают. Затем смесь прессуют в таблетки прямым прессованием. Для обеспечения приемлемой однородности смеси используют измельченный налоксона гидрохлорид, чтобы получить сравнимый интервал дисперсности оксикодона гидрохлорида и других эксципиентов.
Окрашенное поверхностное пленочное покрытие применяют для того, чтобы различать продукт с различной силой действия (эффективностью). Условия для получения пленочного покрытия оптимизируют, чтобы стабильно получать покрытые оболочкой таблетки соответствующего эстетически приемлемого качества.
Затем таблетки IR-OXN 20/10, IR-OXN 10/5, IR-OXN 5/2.5 и IR-OXN 2.5/1.25 проверяют, используя тест с лопастью Ph/Eur. Paddle test.
Во всех случаях через 15 мин высвобождается более 95% оксикодона гидрохлорида и налоксона гидрохлорида.
Пример 2: Однодозовое клиническое исследование с целью сравнения пропорциональности доз таблеток IR-OXN 20/10, IR-OXN 10/5, IR-OXN 5/2.5 и IR-OXN 2.5/1.25 у здоровых субъектов
1. Цель
Целью является оценка пропорциональности доз оксикодона и налоксона (или суррогатного налоксон-3-глюкуронида) из таблеток IR-OXN с силой действия (эффективность) 2.5/1.25 мг, 5/2.5 мг, 10/5 мг и 20/10 мг.
2. Тестируемая популяция
Планировалось всего рандомизировать 21 здоровых взрослых людей, мужчин и женщин, получающих исследуемый препарат, рассчитывая на то, что 18 человек завершат исследование и предоставят ценные фармакокинетические данные. Действительно, в исследование было включено и рандомизировано 21 человек и в случае 20 из них исследование доведено до конца.
Критерии включения
Субъектами, включенными в исследование, являются субъекты, отвечающие всем нижеприведенным критериям:
1. Мужчины и женщины в возрасте от 18 до 55 лет включительно.
2. Женщины, живущие половой жизнью или начавшие жить половой жизнью в процессе исследования, должны выразить желание применять высокоэффективные методы контрацепции в ходе исследования. Высокоэффективный метод контроля за рождаемостью определяют как метод, который дает очень низкий процент неудач (т.е. менее 1% в год) при постоянном и корректном применении, такой как стерилизация, имплантаты, инъецируемые, комбинированные пероральные контрацептивы, некоторые внутриматочные устройства или вазэктомизированный партнер.
3. Женщины в период менопаузы, длящийся менее года, должны иметь отрицательный сывороточный тест на беременность и не должны кормить грудью.
4. Женщины, период менопаузы у которых длится >1 года и имеющие повышенный уровень фолликулостимулирующего гормона (ФСГ, FSH) в сыворотке, пролеченные с помощью заместительной гормональной терапии (HRT).
5. Мужчины должны выразить желание применять контрацепцию при контакте со своими партнерами в ходе исследования и в течение 30 дней по завершении исследования и должны быть согласны информировать Исследователя, если партнерша забеременела в течение этого времени.
6. Вес (масса) теле в интервале 55-100 кг и BMI (индекс массы тела) в интервале ≥18 и ≤29.
7. Здоровые и показывающие отсутствие заметных аномальных показателей в анамнезе, при физикальном исследовании, подтвержденных основными показателями жизненно важных функций, клиническими лабораторными тестами и электрокардиограммой (ЭКГ).
8. Желание есть любую пищу в ходе исследования.
9. Личный врач (терапевт) субъекта подтверждает, что за последние 12 месяцев в анамнезе нет ничего, что препятствовало бы его включению в клиническое исследование.
Критерии исключения
Субъекты, которых следует исключить из исследования, отвечают любому из следующих критериев:
1. Любая наркотическая или алкогольная зависимость в анамнезе.
2. Любые состояния в анамнезе, которые могли бы препятствовать всасыванию, распределению, метаболизму или экскреции лекарства.
3. Применение лекарственного средства, содержащего опиод или антагонист опиоидного рецептора в предшествующие 30 дней.
4. Любые частые приступы тошноты и рвоты в анамнезе, вне зависимости от этиологии.
5. Любые эпилептические припадки или любая симптоматическая эпилепсия вследствие травмы головы в анамнезе.
6. Участие в клиническом исследовании лекарства в течение 90 дней, предшествующих введению начальной дозы в данном исследовании.
7. Любое серьезное заболевание в течение 4 недель перед включением в данное исследование.
8. Применение любого лекарственного средства, включая витамины, растительные и/или минеральные добавки, в течение 7 дней, предшествующих приему первой дозы или в ходе данного исследования (за исключением постоянного применения HRT (гормонозаместительной терапии) и контрацептивов).
9. Отказ, при каждом указании (ограничении) не принимать пищу в течение 8 часов до и 4 часов после введения исследуемого лекарства и отказ, при каждом указании (ограничении), совершенно воздержаться от напитков, содержащих кофеин или ксантин.
10. Еженедельное потребление алкоголя, превышающее эквивалент 14 минимальных порций/неделя для женщин и 21 минимальной порции/неделя для мужчин.
11. Потребление алкогольных напитков в течение 48 часов перед введением исследуемого лекарства и отказ воздержаться от алкоголя по меньшей мере в течение 48 часов после введения лекарства.
12. Курение в течение 45 дней перед введением исследуемого лекарства и отказ воздержаться от курения в ходе исследования.
13. Сдача крови или продуктов крови в течение 30 дней перед введением исследуемого лекарства или в любой момент в ходе исследования, за исключением того, что требуется данным протоколом.
14. Положительные результаты исследования мочи на лекарства, теста на алкоголь, на поверхностный (австралийский) антиген гепатита В (HBsAg), на антитело гепатита С или на вирус иммунодефицита человека (ВИЧ).
15. Заведомая чувствительность к оксикодону, налоксону, налтрексону или родственным соединениям.
16. Отказ информировать своего лечащего врача (терапевта, врача первой помощи).
Демографические данные о популяции субъектов показаны в Таблице 6. Исследуемая популяция включает 14 мужчин и семь женщин, средний возраст которых составляет 31 год (интервал: 21-53 года). Двадцать человек принадлежат к белой расе и один к черной расе.
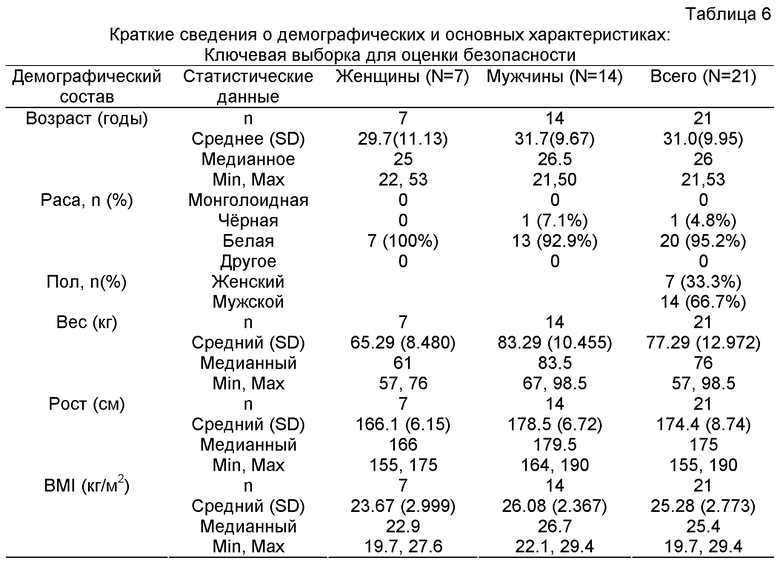
3. План (дизайн) исследования
Исследование является открытым, однодозовым, четырехпериодным рандомизированным перекрестным четырех лекарственных препаратов на здоровых мужчинах и женщинах, проводимым в одном исследовательском центре. Каждый человек получал разовые дозы таблеток IR OXN с силой действия (содержащие) 2.5/1.25 мг, 5/2.5 мг, 10/5 мг и 20/10 мг. Участники исследования получают один из четырех лекарственных препаратов по схеме случайного распределения (RAS), причем время выведения (вымывания) препарата из организма перед введением каждой дозы составляет 7 дней.
Испытуемые наносят "скрининговый визит" (визит отбора) в течение 21 дня перед введением первой дозы в Период Исследования 1. Затем субъектов, пригодных для исследования, регистрируют в исследовательском отделении за день до введения лекарства в каждый период исследования. На следующее утро участникам исследования натощак вводят соответствующее лекарство.
Пробы крови для фармакокинетических анализов отбирают в течение 36 часов после введения исследуемого лекарства в каждый период исследования и испытуемых выписывают через 36 часов после взятия пробы крови.
В течение каждого периода исследования перед введением дозы и через 1, 2, 4, 6, 8, 12, 24 и 36 часов после введения дозы лекарства проводят контроль жизненно важных функций (пульс, артериальное давление и частоту дыхания). Оральную температуру проверяют перед введением дозы и через 24 и 36 часов после введения дозы. Побочные явления регистрируют в ходе всего исследования. Испытуемые наносят визит после исследования через 7-10 после последней дозы исследуемого лекарства в случае завершения/прерывания (досрочного завершения) исследования.
Дизайн исследования представлен на Фигуре 2. Ниже даны сокращения на Фигуре 2:
R=рандомизация (случайное распределение)
P1, Р2, Р3, Р4 = Периоды 1-4, каждый из которых включает разовую дозу исследуемого лекарства в соответствии с RAS с последующим отбором проб крови и оценки безопасности вплоть до 36 часов после введения дозы. В каждый период исследования время выведения лекарства (отмывочный период между дозами) составляет минимум 7 дней.
PS med=продолжающееся наблюдение врача в течение "постисследовательских" 7-10 дней после введения последней дозы исследуемого лекарства в случае завершения/прерывания исследования.
VI-V6=визиты
4. Отбор проб для фармакокинетического исследования
Начало в 1, 8, 15 и 22 дни, серийный отбор образцов крови (6 мл каждый) проводят в следующие моменты времени:
перед введением дозы, через 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, 24, 28, 32 и через 36 часов после введения дозы (22 образца крови за период после введения лекарства).
Приемлемое временное окно (интервал времени) от номинального времени отбора пробы крови составляет ±5 минут.
Для фармакокинетических измерений у каждого участника испытаний отбирают около 528 мл крови (22 образцов по 6 мл для четырех периодов).
5. Вводимые лекарственные средства
Лекарство вводят перорально, натощак как указано ниже:
А: Одну таблетку IR OXN 2.5/1.25 мг
В: Одну таблетку 5/2.5 мг
С: Одну таблетку 10/5 мг
D: Одну таблетку 20/10 мг
Налтрексон HCl вводят перорально со всеми лекарственными средствами (А-D), чтобы снизить побочные явления, обусловленные опиоидами. Таблетка налтрексон HCl 50 мг (таблетки Налорекс®, Bristol-Myers Squibb Pharmaceuticals Ltd). Одну таблетку налтрексона вводят во время за 14 час (-13 час), за 1 час (-1 час) до и через 11 час (и +11 час) после введения исследуемого лекарства в каждом периоде исследования.
6. Фармакокинетические параметры
Следующие фармакокинетические параметры рассчитывают на основании плазматических концентраций оксикодона, нороксикодона, оксиморфона, нороксиморфона, налоксона, 6β-налоксола, налоксон-3-глюкуронида и 6β-налоксол-3-глюкуронида:
- Площадь под кривой зависимости концентрации в плазме (плазматической концентрации) от времени, измеренную с момента введения дозы лекарства до момента последнего определения концентрации (AUCt)
- Площадь под кривой зависимости концентрации в плазме (плазматической концентрации) от времени, измеренную с момента введения дозы лекарства и экстраполированную до бесконечности (AUCinf)
- Максимальную наблюдаемую (экспериментальную плазматическую концентрацию (Cmax)
- Время максимальной наблюдаемой плазматической концентрации (tmax)
- Константа скорости в конечной фазе (Лямбда (Lambda)Z)
- Период полужизни в конечной фазе (t1/2.Z)
Значения AUC для оксикодона, нороксикодона, оксиморфона, нороксиморфона и налоксон-3-глюкуронида даются в нг·час/мл, а значения Cmax даются в нг/мл. Для налоксона, 6β-налоксола и 6β-налоксол-3-глюкуронида значения AUC даются в пг.час/мл, а значения Cmax в пг/мл.
Величины AUCt рассчитывают, решая линейные уравнения методом трапеций. При возможности LambdaZ определяют, используя точки, определенные в конечной log-линейной фазе. t1/2Z определяют из отношения ln 2 r LambdaZ. Площади под кривой зависимости плазматической концентрации от времени между последней измеренной точкой и бесконечностью рассчитывают из отношения конечной экспериментальной концентрацией в плазме (Clast) к LambdaZ. Затем эту величину прибавляют к AUCt, получают AUCINF.
Расчет фармакокинетического показателя проводят, используя программу WinNonlin Enterprise Edition, Version 4.1.
В частности, при пониженных дозах плазматические концентрации налоксона чрезвычайно низки и полная фармакокинетическая характеристика невозможна. Ниже, в разделах Результаты и Обсуждения, наибольшее внимание уделяется его первичному метаболиту, налоксон-3-глюкурониду.
Основной целью исследования является определение пропорциональности доз оксикодона и налоксона (или суррогатного налоксон-3-глюкуронида) из таблеток IR OXN с силой действия (эффективностью, содержанием) 2.5/1.25 мг, 5/2.5 мг, 10/5 мг и 20/10 мг.
Корректированные по дозе величины относительной системной биодоступности рассчитывают на основании величины AUCt и, где это возможно, на основании величины AUCINF. Целевыми сравнениями являются:
- таблетка OXN IR 5/2.5 мг vs таблетка OXN IR 2.5/1.25 мг
- таблетка OXN IR 10/5 мг vs таблетка OXN IR 2.5/1.25 мг
- таблетка OXN IR 20/10 мг vs таблетка OXN IR 2.5/1.25 мг.
Максимальная экспериментальная (наблюдаемая) плазматическая концентрация (Cmax) и время Cmax (tmax) получают непосредственно из экспериментальных данных на плазматическая концентрация - время. Корректированные по дозе отношения Cmax рассчитывают из вышеуказанных сравнений.
7. Результаты
Результаты для фармакокинетических показателей для оксикодона, налоксона и налоксон-3-глюкуронида показаны на Фигурах 3-5, соответственно, для проверяемого 21 человека.
Оксикодон
Суммарные статистические данные для оксикодона показаны в Таблице 7.
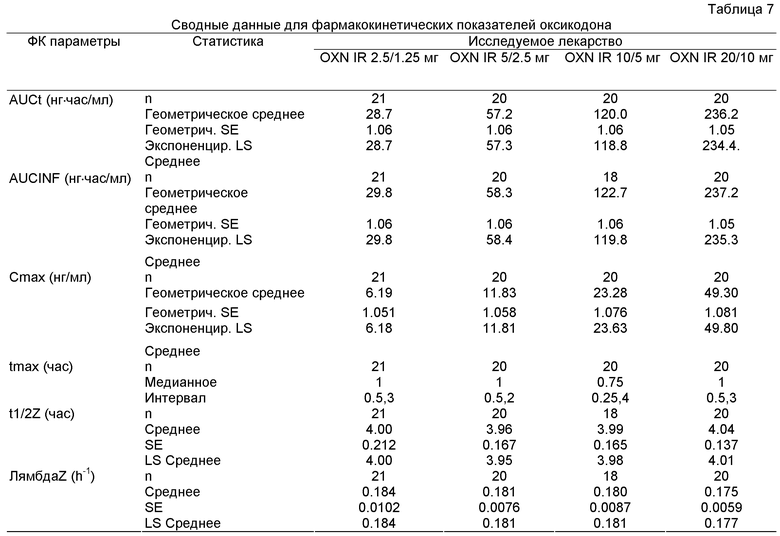
Аналогичные значения tmax и t1/2Z регистрируют для всех таблеток IR OXN с различной силой действия (с различным содержанием активных веществ)
Все таблетки IR OXN с различной силой действия проявляют биодоступность оксикодона, эквивалентную скорректированной по дозе биодоступности оксикодона таблетки IR OXN 2.5/1.25 мг, выраженную как AUCt, AUCINF и Cmax, с 90% доверительным интервалом для каждой из относительных величин сравнения, попадающих в интервал 80-125% приемлемости для биоэквивалентности. Результаты статистического анализа фармакокинетических показателей оксикодона представлены в Таблице 8.
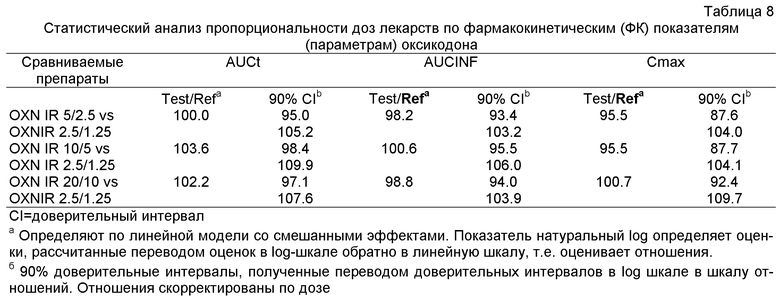
Налоксон-3-глюкуронид
Фармакокинетические параметры налоксон-3-глюкуронида анализируют как идентификаторы (заменители) налоксона.
Суммарные статистические данные для налоксон-3-глюкуронида показаны в Таблице 9.
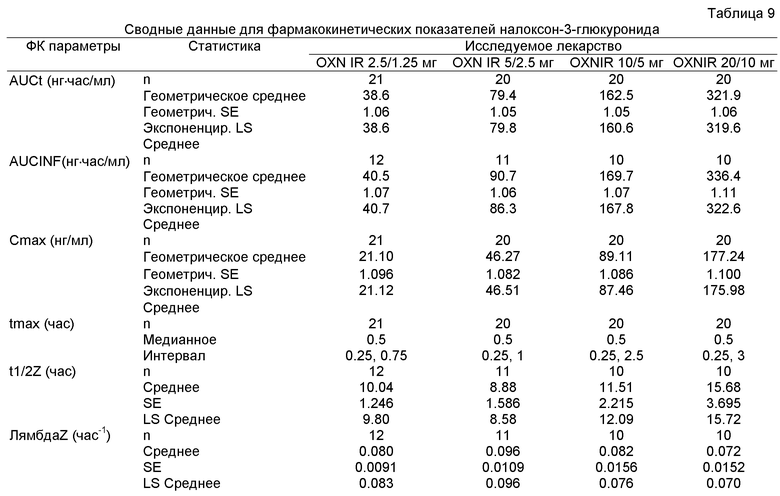
Аналогичные значения tmax регистрируют для всех таблеток IR OXN с различной силой действия (с разным содержанием активных веществ). Средние значения t1/2Z в интервале от 8.88 часов для таблеток IR OXN 5/2.5 мг (сила действия) до 15.68 час для таблеток IR OXN 20/10 мг.
Все таблетки IR OXN с различной силой действия проявляют биодоступность налоксон-3-глюкуронида, эквивалентную скорректированной по дозе биодоступности оксикодона таблетки IR OXN 2.5/1.25 мг, выраженную как AUCt, AUCINF и Cmax, с 90% доверительным интервалом для каждой из относительных величин сравнения, попадающих в интервал 80-125% приемлемости для биоэквивалентности, за исключением верхнего 90% доверительного интервала для отношения Cmax для IR OXN 5/2.5 мг vs. IR OXN 2.5/1.25, который составляет 131.9%. Результаты статистического анализа фармакокинетических показателей налоксон-3-глюкуронида представлены в Таблице 10.
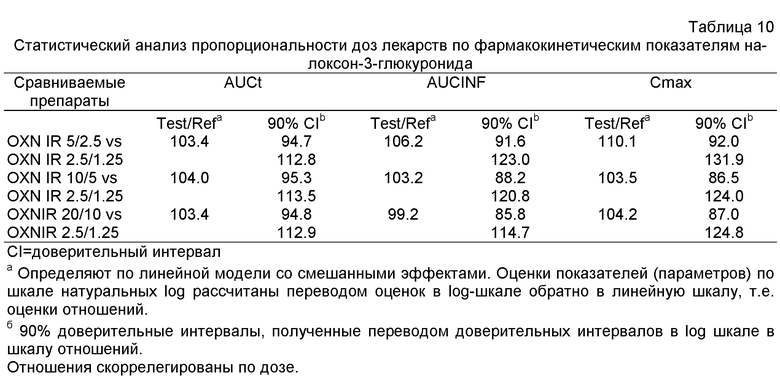
8. Выводы
Биодоступность скорректированной дозы получают для всех таблеток IR OXN с различной силой действия относительно таблеток IR OXN 2.5/1.25 мг для основных аналитов оксикодона и налоксон-3-глюкуронида. Пропорциональность доз подтверждают для таблеток IR OXN с силой действия в интервале от 2.5/1.25 мг до 20/10 мг. Концентрации в плазме (плазматические концентрации) налоксона чрезвычайно низки, в особенности при пониженных дозах, это подтверждает результаты предыдущих исследований и анализ суррогатного (идентификатора, заменителя) налоксон-3-глюкуронида.
Пример 3. Однодозовое клиническое исследование с целью сравнения биодоступности таблеток IR-OXN20/10 и таблеток оксикодона/налоксона 20/10 мг с пролонгированным высвобождением (Targin®) на здоровых субъектах
1. Цель
Целью данного исследования является оценка фармакокинетики, биодоступности и безопасности таблеток OXN IR 20/10 мг, таблеток OXN PR 20/10 мг и IR капсул оксикодона 20 мг, вводимых натощак здоровым субъектам.
2. Тестируемая популяция
Планировалось всего рандомизировать 21 здоровых взрослых людей, мужчин и женщин, получающих исследуемую лекарственную терапию, рассчитывая на то, что 18 человек завершат полное исследование и предоставят ценные фармакокинетические данные. Действительно, в исследование было включено и рандомизировано 22 человека и в случае 21 из них исследование доведено до конца. Один человек прервал исследование до введения исследуемого лекарства (т.е. OXN или оксикодона).
Критерии включения
Субъектами, включенными в исследование, являются субъекты, отвечающие всем нижеприведенным критериям:
1. Мужчины и женщины в возрасте от 18 до 55 лет включительно.
2. Женщины, живущие половой жизнью или начавшие жить половой жизнью в процессе исследования, должны выразить желание применять высокоэффективные методы контрацепции в ходе исследования. Высокоэффективный метод контроля за рождаемостью определяют как метод, который дает очень низкий процент неудач (т.е. менее 1% в год) при постоянном и корректном применении, такой как стерилизация, имплантаты, инъецируемые, комбинированные пероральные контрацептивы, некоторые внутриматочные устройства или вазэктомизированный партнер.
3. Женщины в период менопаузы, длящийся менее года, должны иметь отрицательный сывороточный тест на беременность и не должны кормить грудью.
4. Женщины, период менопаузы у которых длится >1 года и имеющие повышенный уровень фолликулостимулирующего гормона (ФСГ, FSH) в сыворотке, пролеченные с помощью заместительной гормональной терапии (HRT).
5. Вес (масса) тела в интервале 55-100 кг и BMI (индекс массы тела) в интервале ≥18 и ≤29.
6. Здоровые и не имеющие заметных аномальных показателей в анамнезе, при физикальном исследовании, что подтверждено основными показателями жизненно важных функций, клиническими лабораторными тестами и электрокардиограммой (ЭКГ).
7. Желание есть любую пищу в ходе исследования.
9. Личный врач (терапевт) субъекта подтверждает, что за последние 12 месяцев в анамнезе нет ничего, что препятствовало бы его включению в клиническое исследование.
Критерии исключения
Субъекты, которых следует исключить из исследования, отвечают любому из следующих критериев:
1. Любая наркотическая или алкогольная зависимость в анамнезе.
2. Любые состояния в анамнезе, которые могли бы препятствовать всасыванию, распределению, метаболизму или экскреции лекарства.
3. Применение лекарственного средства, содержащего опиод или антагонист опиоидного рецептора в предшествующие 30 дней.
4. Любые частые приступы тошноты и рвоты в анамнезе, вне зависимости от этиологии.
5. Любые эпилептические припадки или любая симптоматическая эпилепсия вследствие травмы головы в анамнезе.
6. Участие в клиническом исследовании лекарства в течение 90 дней, предшествующих введению начальной дозы в данном исследовании.
7. Любое серьезное заболевание в течение 4 недель перед включением в данное исследование.
8. Применение любого лекарственного средства, включая витамины, растительные и/или минеральные добавки, в течение 7 дней, предшествующих приему начальной дозы или в ходе данного исследования (за исключением постоянного применения HRT (гормонозаместительной терапии) и контрацептивов).
9. Отказ, при каждом указании (ограничении), не принимать пищу в течение 8 часов до и 4 часов после введения исследуемого лекарства и отказ, при каждом указании (ограничении), совершенно воздержаться от напитков, содержащих кофеин или ксантин.
10. Еженедельное потребление алкоголя, превышающее эквивалент 14 минимальных порций/неделя для женщин и 21 минимальной порции/неделя для мужчин.
11. Потребление алкогольных напитков в течение 48 часов перед введением исследуемого лекарства и отказ воздержаться от алкоголя по меньшей мере в течение 48 часов после введения лекарства.
12. Курение в течение 45 дней перед введением исследуемого лекарства и отказ воздержаться от курения в ходе исследования.
13. Сдача крови или продуктов крови в течение 30 дней перед введением исследуемого лекарства или в любой момент в ходе исследования, за исключением того, что требуется данным протоколом.
14. Положительные результаты исследования мочи на лекарства, теста на алкоголь, на поверхностный (австралийский) антиген гепатита В (HBsAg), на антитело гепатита С или на вирус иммунодефицита человека (ВИЧ).
15. Заведомая чувствительность к оксикодону, налоксону, налтрексону или родственным соединениям.
16. Отказ информировать своего лечащего врача (терапевта, врача первой помощи).
Демографические данные о популяции субъектов показаны в Таблице 11. Исследуемая популяция включает 11 мужчин и 10 женщин, средний возраст которых составляет 33 года (интервал: 20-52 года). Все субъекты относятся к белой расе.
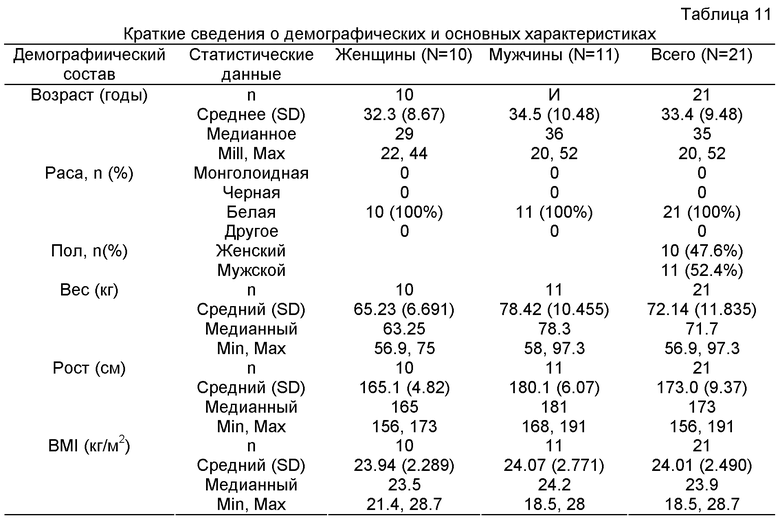
3. План (дизайн) исследования
Исследование является открытым, однодозовым, трехпериодным рандомизированным перекрестным исследованием трех лекарственных препаратов. Каждый человек получает разовые дозы: таблетку IR OXN 20/10 мг, IR капсулу оксикодона 20 мг и таблетку OXN PR 20/10 мг (Targin® (таргин)). Участники исследования получают один из трех лекарственных препаратов по схеме случайного распределения (RAS), причем время выведения (вымывания) препарата из организма перед введением каждой дозы составляет по меньшей мере 7 дней.
Испытуемые наносят "скрининговый визит" (визит отбора) в течение 21 дня перед введением первой дозы в Период Исследования 1. Затем субъектов, пригодных для исследования, регистрируют в исследовательском отделении за день до введения лекарства в каждый период исследования. На следующее утро участникам исследования натощак вводят соответствующее лекарство.
Пробы крови для фармакокинетических анализов отбирают в течение 36 часов после введения исследуемого лекарства в каждый период исследования, и испытуемых выписывают через 36 часов после взятия пробы крови.
В течение каждого периода исследования перед введением дозы и через 1, 2, 4, 6, 8, 12, 24 и 36 часов после введения дозы лекарства проводят контроль жизненно важных функций (пульс, артериальное давление и частоту дыхания). Оральную температуру проверяют перед введением дозы и через 24 и 36 часов после введения дозы. Побочные явления регистрируют в ходе всего исследования.
Испытуемые наносят визит после исследования через 4-7 после последней дозы исследуемого лекарства в случае завершения/прерывания (досрочного завершения) исследования.
Дизайн исследования представлен на Фигуре 6. Ниже даны сокращения на Фигуре 6:
R=рандомизация (случайное распределение)
P1, Р2, Р3 = Периоды 1-3, каждый из которых включает разовую дозу исследуемого лекарства с последующим отбором проб крови и оценки безопасности вплоть до 36 часов после введения дозы. В каждый период исследования время выведения лекарства (отмывочный период между дозами) составляет 7 дней. Налтрексон вводят также за 13 час (-13 час), за 1 час (-1 час) до и через 11 час (+11 час) после введения лекарства в каждый период исследования.
PS med=продолжающееся наблюдение врача в течение "постисследовательских" 4-7 дней после введения последней дозы исследуемого лекарства в случае завершения/прерывания исследования. Участники исследования, которые получают налтрексон только прерыванием (досрочным завершением) исследования, проходят медосмотр перед выпиской из стационара.
VI-V5=визиты
4. Отбор проб для фармакокинетического исследования
Начало в 1, 8 и 15 дни, серийный отбор образцов крови (6 мл каждый) проводят в следующие моменты времени:
Лечебная схема А и В:
Перед введением дозы, через 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, 24, 28, 32 и через 36 часов после введения дозы (22 образца крови за период введения лекарства).
Лечебная схема С:
Перед введением дозы, через 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, 24, 28, 32 и через 36 часов после введения дозы (19 образцов крови за период введения лекарства).
Если у пациента наблюдается тошнота в течение 12 часов после введения дозы таблетки OXN PR 20/10 или в течение 6 часов после введения таблетки OXN IR 20/10 или капсулы оксикодона IR 20 мг, то в оставшийся период исследования у этого субъекта образцы крови для фармакокинетического исследования не отбирают.
Приемлемое временное окно (интервал времени) от номинального времени отбора пробы крови составляет ±5 минут.
Для фармакокинетических измерений у каждого участника испытаний отбирают около 3788 мл крови (22 образца по 6 мл для в двух случаях и 19 образцов по 6 мл в одном случае).
5.Вводимые лекарственные средства
Лекарства, применяемые в данном исследовании, представлены ниже:
Испытуемые лекарственные средства: Таблетка IR OXN 20/10 мг. Лекарство вводят перорально, натощак, следующим образом:
Лечебная схема А: Одна OXN IR таблетка 20/10 мг
Эталонные (контрольные) лекарственные средства (препараты):
Капсула оксикодона IR 20 мг (OxyNorm®, Napp Pharmaceuticals Ltd, UK). Лекарство вводят перорально, натощак, как указано ниже:
Лечебная схема В: Одна капсула оксикодона IR 20 мг
Таблетка OXN PR 20/10 мг (Targin®). Производство Bard Pharmaceuticals Ltd, UK. Лекарство вводят перорально, натощак, как указано ниже:
Лечебная схема С: Одна таблетка OXN PR 20/10 мг
Введение исследуемого лекарства проводят "под защитой" налтрексона, чтобы снизить риск побочных явлений, обусловленных опиоидами, на каждой стадии исследования. Таблетки налтрексона гидрохлорида 50 мг (таблетки Налорекс®, Bristol-Myers Squibb Pharmaceuticals Ltd) принимают следующим образом:
Одну таблетку налтрексона, 50 мг, проглатывают целиком, запивая 100 мл воды, за 13 час (-13 час, Дни -1, 7 и 14), за 1 час (-1 час) до и через 11 час (и +11 час, Дни 1, 8 и 15) после введения исследуемого лекарства (всего 3 дозы).
6. Фармакокинетические параметры
Полученные плазматические концентрации и фармакокинетические параметры перечислены для зарегистрированной популяции.
Концентрацию в плазме (плазматическую концентрацию) для каждого аналита поясняют, описывая временной точкой и лекарством для субъектов в зарегистрированной популяции. Для каждой схемы лечения (лекарственной схемы) для каждого аналита строят также кривую зависимости индивидуальных и средних плазматических концентраций от времени.
Фармакокинетические параметры (AUCt, AUCINF, Cmax, tmax, LambdaZ (ЛямбдаZ и t1/2Z) для каждого аналита поясняют, описывая лекарственную схему для всех субъектов в популяции с полным анализом фармакокинетических показателей (параметров). Чтобы фармакокинетический показатель был достоверным, субъект не должен испытывать тошноту в течение 12 часов после введения таблетки OXN PR 20/10 или в течение 6 часов после введения таблетки OXN IR 20/10 или капсулы оксикодона IR 20 мг.
Трансформированные в log-шкале данные для аналитов оксикодона, нороксикодона, оксиморфона, нороксиморфона, налоксона, 6β- налоксола, налоксон-3-глюкуронида и 6β-налоксол-3-глюкуронида и фармакокинетические параметры AUCt, AUCINF и Cmax анализируют, используя линейную модель 18 со смешанными эффектами с фиксированными ограничениями для лечения и периода и случайный эффект для субъекта.
Отношения/различия схем лечения (лекарственные схемы) и связанные с ними 90% доверительные интервалы рассчитывают методом наименьших квадратов.
Ниже представлены сравниваемые лечебные схемы (терапия, лекарства):
- IR OXN 20/10 мг vs оксикодон 20 мг
- IR OXN 20/10 мг vs OXN PR 20/10 мг
Соотношения метаболит: исходное лекарство рассчитывают для каждой лечебной схемы, используя значения AUCt и, где это возможно, AUCINF.
Все расчеты фармакокинетических величин проводят, используя программу WinNonlin Enterprise Edition, Version 4.1.
7. Результаты
Результаты для фармакокинетических параметров (показателей) для оксикодона, налоксона и налоксон-3-глюкуронида показаны на Фигурах 7-9, соответственно, для тестируемых субъектов.
Оксикодон
Суммарные статистические данные для оксикодона показаны в Таблице 12.
По сравнению с таблеткой OXN PR 20/10 средняя пероральная биодоступность оксикодона в таблетке IR OXN IR 20/10 составляет 96.3% с доверительными интервалами 90%, которые отвечают критерию биодоступности. При сравнении препарата с мгновенным высвобождением и препарата с пролонгированным высвобождением Cmax для таблетки OXN IR 20/10 оказывается значительно выше, чем для таблетки OXN PR 20/10.
По сравнению с IR капсулой оксикодона средняя пероральная биодоступность оксикодона в таблетке IR OXN 20/10 составляет 99.2%, а среднее отношение Cmax составляет 100%. Все оценки биоэквивалентности для этих отношений (сравнений) имеют доверительный интервал 90%, который отвечает критериям биоэквивалентности.
Средние значения периода полужизни для оксикодона аналогичны во всех лечебных схемах и находятся в диапазоне 4.2-4.3 часа.
Медианные значения tmax для таблетки IR OXN 20/10 и IR капсулы оксикодона 20 мг аналогичны, tmax для OXN PR 20/10 наступает позже, чем при других лекарственных схемах (схемах терапии).
Результаты статистического анализа фармакокинетических показателей оксикодона представлены в Таблице 13.
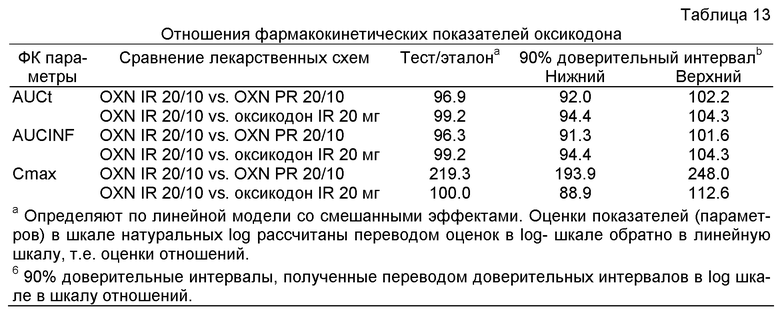
Налоксон-3-глюкуронид
Фармакокинетические параметры налоксон-3-глюкуронида анализируют как идентификаторы (заменители) налоксона.
Суммарные данные для налоксон-3-глюкуронида показаны в Таблице 14.
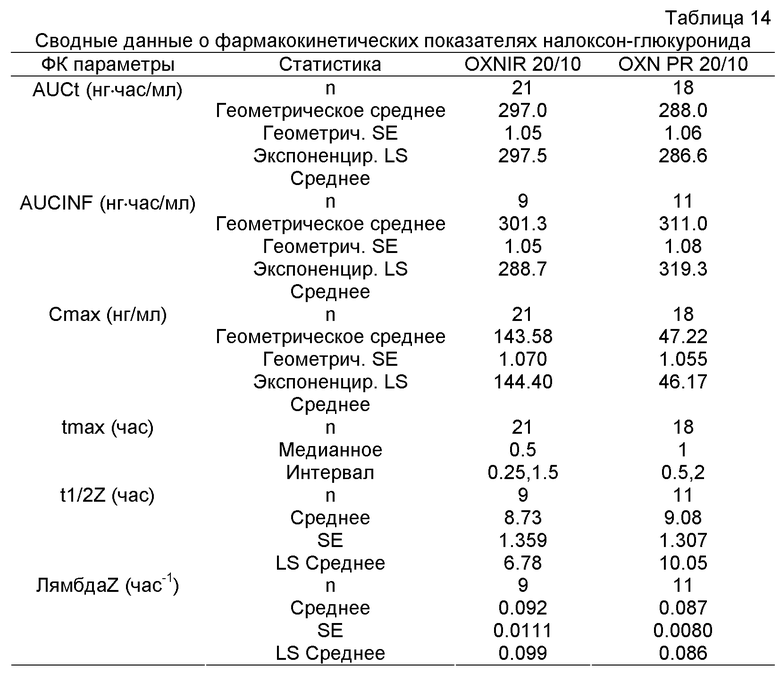
По сравнению с таблеткой OXN PR 20/10 средняя пероральная биодоступность налоксон-3-глюкуронида в таблетке IR OXN IR 20/10 составляет 90.4% с доверительными интервалами 90%, которые отвечают критерию биодоступности. При сравнении препарата с мгновенным высвобождением и препарата с пролонгированным высвобождением Cmax для таблетки OXN IR 20/10 оказывается значительно выше, чем для таблетки OXN PR 20/10.
Средние значения периода полужизни для таблеток IR OXN 20/10 (8.7 часов) и таблеток OXN PR (9.1 час) аналогичны.
Значения tmax для OXN PR 20/10 наступает позже, чем для таблетки IR OXN 20/10.
Результаты статистического анализа фармакокинетических показателей налоксон-3-глюкуронида представлены в Таблице 15.
8. Выводы
Результаты, полученные для фармакокинетических параметров, показывают хорошую эквивалентную биодоступность оксикодона и налоксона (или суррогатного налоксон-3-глюкуронида) из IR OXN 20/10 мг и OXN PR 20/10 мг и биоэквивалентность IR OXN 20/10 мг и IR капсулы оксикодона 20 мг в отношении оксикодона. Как и ожидалось, биодоступность налоксона очень низка, что подтверждает результаты предыдущих исследований и анализ суррогатного (идентификатора, заменителя) налоксон-3-глюкуронида.
Пример (Эксперимент) 4: Устойчивость (стабильность) таблеток IR-OXN 20/10, IR-OXN 10/5, IR-OXN 5/2.5 и IR-OXN 2.5/1.25 при хранении
Образцы таблеток IR-OXN 20/10, IR-OXN 10/5, IR-OXN 5/2.5 и IR-OXN 2.5/1.25 хранят в блистерных упаковках из PVC/PVdC с одной стороны и алюминиевой фольги - с другой при 25°C/60%RH и 40°C/75%RH.
Данные для устойчивости представлены для партий таблеток IR-OXN 20/10, IR-OXN 10/5, IR-OXN 5/2.5 и IR-OXN 2.5/1.25 после 3-месячного хранения при 25°C/60%RH и при 40°C/75%RH.
Представленные ниже данные подтверждают стабильность физических характеристик и растворимости таблеток оксикодона гидрохлорида/налоксона гидрохлорида с мгновенным высвобождением.
Средняя (кп)
Средний (мг)
Средняя (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Средний (мг)
Средняя (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Потеря от общего количества веществ после начального хранения составляет лишь около 0.3%. Эта величина становится максимальной - 0.5% - после хранения в течение 12 месяцев.
Оксикодона гидрохлорид (мг)
Оксикодона гидрохлорид (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Потеря от общего количества веществ после начального хранения составляет лишь около 0.3%. Эта величина становится максимальной - 2.3% - после хранения в течение 6 месяцев.
Средний (мг)
Средняя (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Потеря от общего количества веществ после начального хранения составляет лишь около 0.3%. Эта величина остается постоянной при хранении в течение 12 месяцев.
Средний (мг)
Средняя (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Потеря от общего количества веществ после начального хранения составляет лишь 0.3%. Эта величина становится максимальной - 1.7% - после хранения в течение 6 месяцев.
Средний (мг)
Средняя (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Потеря от общего количества веществ после начального хранения составляет лишь около 0.3%. Эта величина становится максимальной - 0.4% - после хранения в течение 12 месяцев.
Средний (мг)
Средняя (кп)
Налоксона гидрохлорид (мг/табл)
Оксикодона гидрохлорид (мг/табл)
Потеря от общего количества веществ после начального хранения составляет лишь 0.3%. Эта величина становится максимальной - 0.7% - после хранения в течение 6 месяцев.

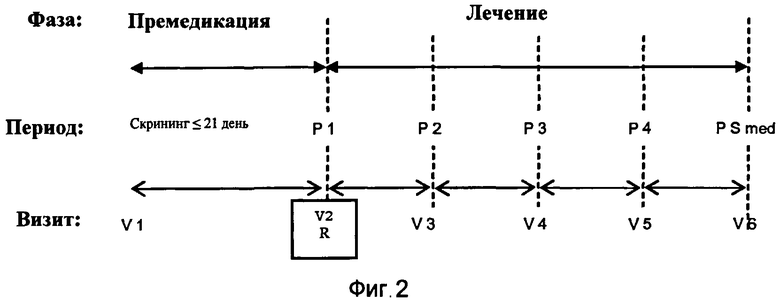
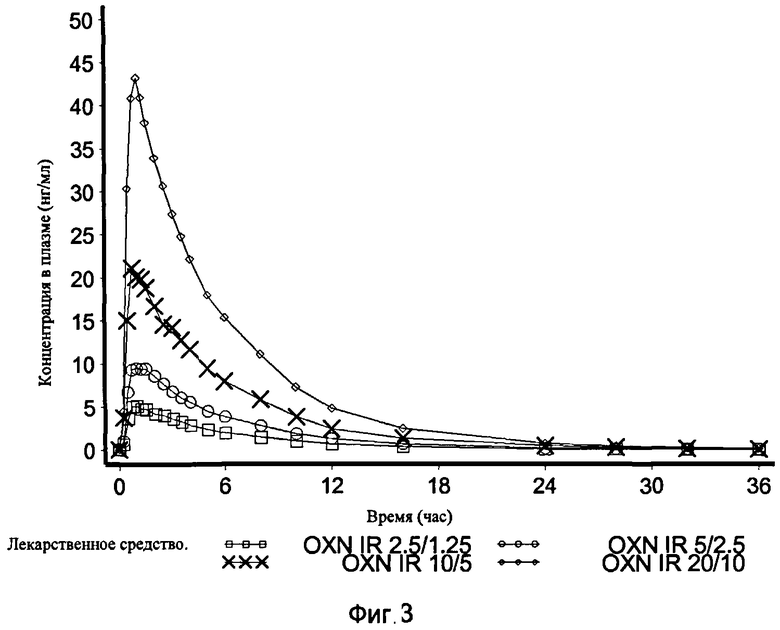
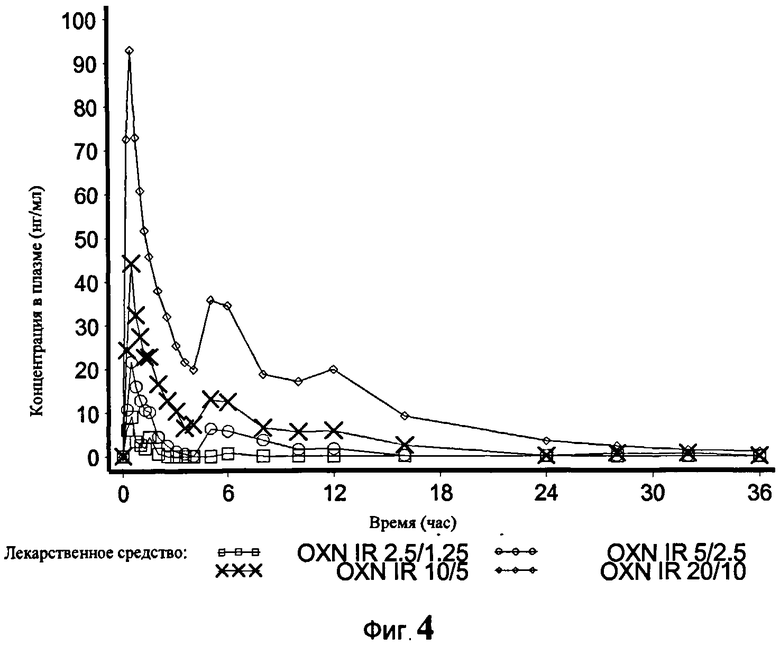
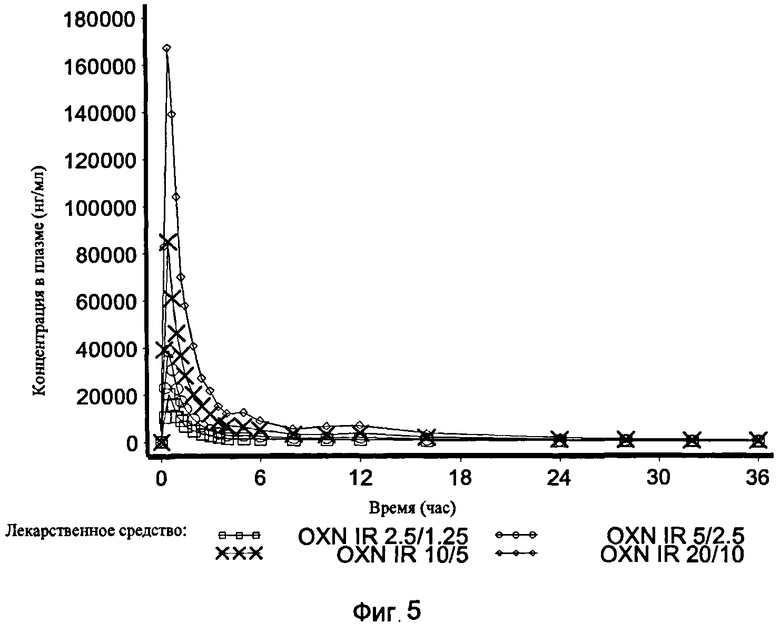
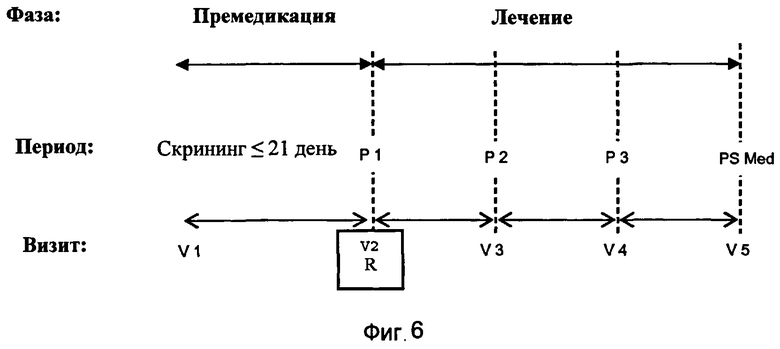
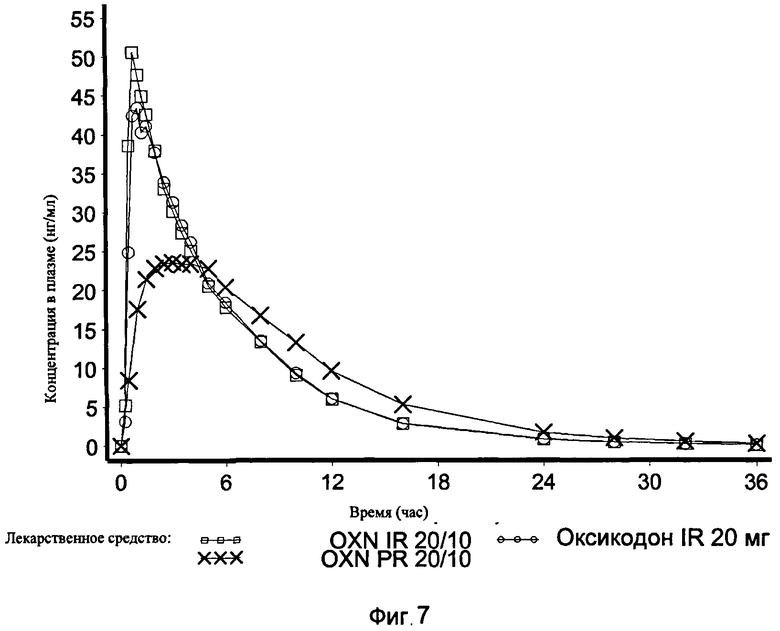
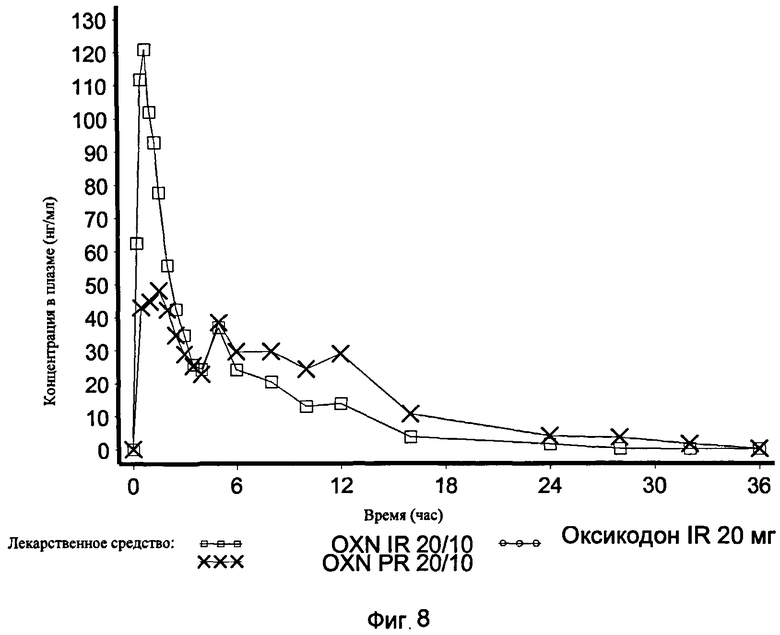
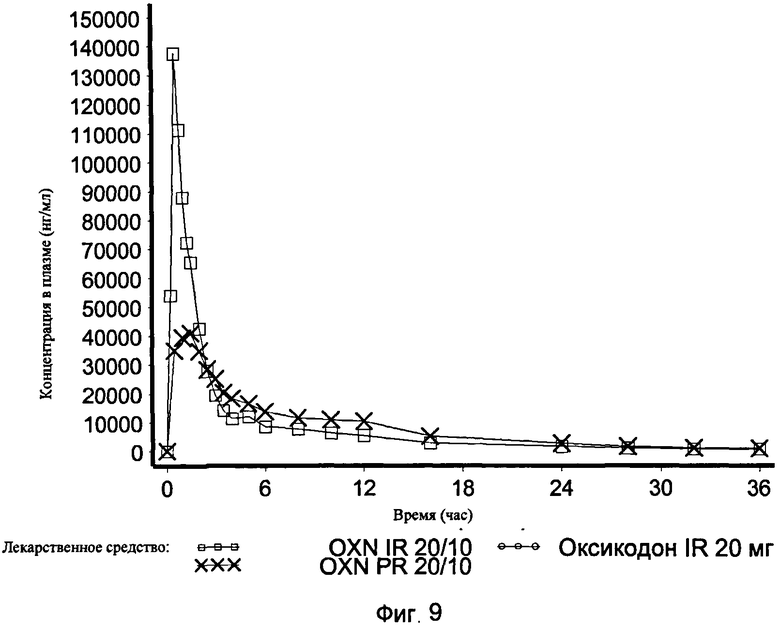
Предложены: пероральная фармацевтическая композиция с мгновенным высвобождением для лечения боли у пациентов или для титрования дозы для пациентов, страдающих от боли, содержащая по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении оксикодон или его фармацевтически приемлемая соль: налоксон или его фармацевтически приемлемая соль 2:1 (варианты), ее применение для получения лекарственного препарата для титрования дозы для пациентов, страдающих от боли, ее применение для получения лекарственного препарата для лечения прорывной боли, способ титрования дозы у пациента, страдающего от боли, и способ лечения прорывной боли. Показано достижение заявленных результатов при отсутствии нежелательных побочных эффектов опиоида, при этом уровни налоксона в плазме крови чрезвычайно низки, а биодоступность оксикодона составляет 96,3 или 99,2%, таблетки стабильны при хранении. 6 н. и 24 з.п. ф-лы, 9 ил., 15 табл.
1. Пероральная фармацевтическая композиция с мгновенным высвобождением для лечения боли у пациентов или для титрования дозы для пациентов, страдающих от боли, содержащая по меньшей мере оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении оксикодон или его фармацевтически приемлемая соль:налоксон или его фармацевтически приемлемая соль 2:1.
2. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.1, отличающаяся тем, что фармацевтически приемлемую соль оксикодона и фармацевтически приемлемую соль налоксона выбирают из гидрохлоридов оксикодона и налоксона.
3. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.2, отличающаяся тем, что эта композиция содержит оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов.
4. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.1, отличающаяся тем, что содержит количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-160 мг оксикодона гидрохлорида, и некоторое количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида.
5. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.4, отличающаяся тем, что содержит количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-40 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-20 мг налоксона гидрохлорида.
6. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.5, отличающаяся тем, что содержит количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 2.5-20 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1.25-10 мг налоксона гидрохлорида.
7. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.6, отличающаяся тем, что содержит, примерно, 2.5-20 мг оксикодона гидрохлорида и, примерно, 1.25-10 мг налоксона гидрохлорида.
8. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.7, отличающаяся тем, что содержит оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов.
9. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм эта композиция in vitro через 45 мин высвобождает ≥75% вес. оксикодона или его фармацевтически приемлемой соли и ≥75% вес. налоксона или его фармацевтически приемлемой соли.
10. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.9, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм эта композиция in vitro через 45 мин высвобождает ≥80% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. налоксона или его фармацевтически приемлемой соли.
11. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.9, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм эта композиция in vitro через 15 мин высвобождает ≥80% вес. оксикодона или его фармацевтически приемлемой соли и ≥80% вес. налоксона или его фармацевтически приемлемой соли.
12. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.11, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм эта композиция in vitro через 15 мин высвобождает ≥90% вес. оксикодона или его фармацевтически приемлемой соли и ≥90% вес. налоксона или его фармацевтически приемлемой соли.
13. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.12, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм эта композиция in vitro через 15 мин высвобождает ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥95% вес. налоксона или его фармацевтически приемлемой соли.
14. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8, отличающаяся тем, что имеет форму таблетки, капсулы, мультичастиц, гранул или в находится жидком виде.
15. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8, отличающаяся тем, что в качестве фармацевтически приемлемых эксципиентов содержит по меньшей мере разбавитель и, необязательно, дезинтегрант (разрыхлитель).
16. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8, отличающаяся тем, что в качестве фармацевтически приемлемых эксципиентов содержит по меньшей мере дезинтегрант (разрыхлитель) и, необязательно, разбавитель.
17. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8, отличающаяся тем, что содержит, примерно, 2.5-20 мг оксикодона гидрохлорида и, примерно, 1.25-10 мг налоксона гидрохлорида в примерном весовом соотношении 2:1, причем композиция содержит оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов, при этом композиция находится в твердом виде, необязательно, в виде таблетки, в качестве фармацевтически приемлемых эксципиентов содержит по меньшей мере дезинтегрант (разрыхлитель) и, необязательно, разбавитель, и по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм эта композиция через 45 мин in vitro высвобождает ≥80% вес. оксикодона гидрохлорида и ≥80% вес. налоксона гидрохлорида.
18. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.17, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм in vitro через 15 мин высвобождает ≥95% вес. оксикодона гидрохлорида и ≥95% вес. налоксона гидрохлорида.
19. Пероральная фармацевтическая композиция с мгновенным высвобождением для лечения боли у пациентов или для титрования дозы для пациентов, страдающих от боли, содержащая оксикодон или его фармацевтически приемлемую соль и налоксон или его фармацевтически приемлемую соль в примерном весовом соотношении оксикодон или его фармацевтически приемлемая соль:налоксон или его фармацевтически приемлемая соль 2:1, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм через 15 мин in vitro высвобождает ≥90% вес. оксикодона или его фармацевтически приемлемой соли и ≥90% вес. налоксона или его фармацевтически приемлемой соли.
20. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.19, отличающаяся тем, что, по измерению методом в приборе с лопастью (Ph. Eur. Paddle Method) при скорости вращения 100 об/мин в 0.1 N растворе соляной кислоты при 37°C и с УФ-детекцией при 230 нм через 15 мин in vitro высвобождает ≥95% вес. оксикодона или его фармацевтически приемлемой соли и ≥95% вес. налоксона или его фармацевтически приемлемой соли.
21. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.19, отличающаяся тем, что содержит количество оксикодона или его фармацевтически приемлемой соли, эквивалентное, примерно, 1-160 мг оксикодона гидрохлорида, и количество налоксона или его фармацевтически приемлемой соли, эквивалентное, примерно, 0.5-80 мг налоксона гидрохлорида.
22. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.19, отличающаяся тем, что находится в твердом виде.
23. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.19, отличающаяся тем, что дополнительно содержит по меньшей мере дезинтегрант (разрыхлитель).
24. Пероральная фармацевтическая композиция с мгновенным высвобождением по п.19, отличающаяся тем, что содержит оксикодона гидрохлорид и налоксона гидрохлорид в качестве единственных фармацевтически активных агентов.
25. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8 или 19-24, применяемая с целью титрования дозы (лекарства) для пациентов, страдающих от боли.
26. Пероральная фармацевтическая композиция с мгновенным высвобождением по любому из пп.1-8 или 19-24, применяемая для лечения прорывной боли у пациентов, страдающих от боли.
27. Применение пероральной фармацевтической композиции с мгновенным высвобождением по любому из пп.1-24 для получения лекарственного препарата для титрования дозы для пациентов, страдающих от боли.
28. Применение пероральной фармацевтической композиции с мгновенным высвобождением по любому из пп.1-24 для получения лекарственного препарата для лечения прорывной боли у пациентов.
29. Способ титрования дозы у пациента, страдающего от боли, путем введения пероральной фармацевтической композиции с мгновенным высвобождением по по любому из пп.1-24.
30. Способ лечения прорывной боли путем введения пациенту пероральной фармацевтической композиции с мгновенным высвобождением по любому из пп.1-24.
| Устройство для укладки рыбы в тару | 1989 |
|
SU1604666A1 |
| KOGAN A | |||
| et al | |||
| Early oral analgesia after fast-track cardiac anesthesia | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| BENZIGER DP et al | |||
| Differential effects of food on the bioavaila-bility of controlled-release oxycodone tablets and | |||

