Предлагаемое изобретение используется при проведении работ в области радиоэкологического мониторинга для измерения содержания радионуклидов в различных компонентах окружающей среды, при обработке результатов измерений в комплексе аппаратно-программных средств, позволяющих оперировать с большими массивами радиоэкологической информации. Кроме того, предлагаемый способ применяется при накоплении, хранении, обновлении и передачи радиоэкологических данных с последующей математической обработкой с применением компьютерного отображения результатов на электронной картографической основе. В частности, способ применяется для определения активности альфа-излучающих радионуклидов, таких как U-232, Pu-236, Pu-239, Am-241, Po-210 и др., в подготовленных жидких пробах с помощью обработки аппаратных спектров в диапазоне активностей от 1 Бк/проба с использованием жидкостного сцинтилляционного счетчика.
Известны способы измерения активности проб, содержащих несколько радионуклидов, в которых используют жидкостной сцинтилляционный счетчик. Для предполагаемых N радионуклидов выбирают как минимум N+1 окон анализатора высот импульсов и измеряют счет в каждом окне. Далее для предполагаемого уровня тушения выбирают полученные ранее эффективности счета отдельных радионуклидов для каждого окна и составляют N+1 уравнений, из которых с помощью метода наименьших квадратов получают искомые значения активностей отдельных радионуклидов. Далее выбирают другой уровень тушения, и для него заново рассчитывают значения активностей. Этот цикл повторяют до тех пор, пока не будет достигнуто минимальное отклонение суммы этих активностей от измеренной активности [1].
Кроме того, известен способ, в котором измеряют спектр исследуемого образца с помощью многоканального амплитудного анализатора, подключенного к аналого-цифровому преобразователю, определяют его уровень тушения, рассчитывают нормированные спектры отдельных радионуклидов для данного уровня тушения и с использованием метода наименьших квадратов определяют, на какие множители необходимо умножить спектры единичных образцов, чтобы получить суммарный спектр, наиболее близкий к исследуемому. Эти множители пропорциональны искомому содержанию радионуклидов в образце [2].
Наиболее близким к предлагаемому способу по технической сущности и достигнутому эффекту является способ идентификации радионуклидов в жидком сцинтилляционном образце, в котором измеряют спектр исследуемого образца, после чего для соответствующего уровня гашения из библиотеки базовых спектров отдельных радионуклидов для различных уровней гашения методом интерполяции и экстраполяции определяют нормированные модельные спектры отдельных радионуклидов. Далее методом наименьших квадратов минимизируют разницу между спектром образца Pi и суммой модельных спектров отдельных радионуклидов Mij, умноженных на коэффициенты cj, определяющие активность отдельных радионуклидов. Минимизируемое выражение при этом выглядит следующим образом: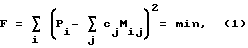
где i - номер канала анализаторa, j - индекс радионуклида [3].
Однако известные способы не позволяют идентифицировать альфа-активные радионуклиды на жидкосцинтилляционном счетчике. Это обусловлено следующим: альфа-спектры различных радиоизотопов при измерении на жидкосцинтилляционном счетчике имеют близкие по форме и практически полностью перекрывающиеся спектры. Спектр пробы, состоящей из нескольких альфа-активных нуклидов по форме практически ничем не отличается от спектра отдельного радионуклида, в связи с чем существующие методы не способны различить вклады различных альфа-излучателей. Это приводит к тому, что существующие методы проводят только контроль наличия альфа-активных радионуклидов, не определяя радиоизотопный состав.
Техническим результатом предложенного способа является повышение достоверности измерения и возможность осуществления жидкостного сцинтилляционного анализа альфа-излучающих радионуклидов при наличии в пробах мешающих радионуклидов.
Для получения технического результата предложен способ идентификации радионуклидов в пробах с использованием жидкостного сцинтилляционного счетчика, включающий отбор проб окружающей среды и технологических проб, подготовку проб к измерению на жидкостном сцинтилляционном счетчике, измерение и запись спектра пробы и параметров измерения, сворачивание аппаратного спектра пробы в группы, создание модельного спектра пробы на основе библиотеки базовых спектров, минимизацию отклонения модельного спектра от спектра пробы и определение содержания радионуклидов в пробе. При этом идентифицируют альфа-излучающие радионуклиды при наличии в отобранных пробах мешающих радионуклидов. Для этого при подготовке твердых проб перед вскрытием пробы проводят ее отжиг, осуществляют кислотное вскрытие с помощью концентрированной азотной кислоты и перекиси водорода, концентрируют упариванием до состояния влажных солей, переводят влажный остаток в солянокислый раствор путем добавления горячего раствора 1M соляной кислоты и концентрируют упариванием при периодическом добавлении не менее двух раз растворов концентрированной перекиси водорода и дистиллированной воды до объема не менее 2 мл. Затем в полученный раствор добавляют концентрированную ортофосфорную кислоту, концентрируют упариванием при периодическом добавлении не менее двух раз концентрированной перекиси водорода и горячей дистиллированной воды до образования бесцветного вязкого раствора объемом не менее 2 мл, представляющего собой смесь пирофосфатных комплексов основных компонентов твердой пробы. Раствор охлаждают до комнатной температуры, добавляют в него насыщенный раствор хлорида двухвалентного олова и переносят в сцинтилляционный коктейль, добавляя 1 мл эмульгатора "Triton X-100", и смесь тщательно перемешивают, получая счетный образец, подготовленный для спектрометрического анализа, измеряют его на жидкосцинтилляционном счетчике, и записывают полученный спектр образца. Сворачивают спектр образца в группы, граничные значения Ni в которых являются квазиарифметической прогрессией вида Ni+1 = Ni + [(i+1)/2] , где i=1,2,..n,. Осуществляют минимизацию отклонения модельного спектра от спектра пробы путем составления модельного спектра  где Mij - модельные спектры радионуклидов, cj - относительные вклады спектров радионуклидов в модельный спектр пробы, i - номер группы, j - индекс радионуклида. Для определения относительных вкладов спектров радионуклидов в модельный спектр пробы минимизируют разницу между модельным спектром и спектром пробы по выражению
где Mij - модельные спектры радионуклидов, cj - относительные вклады спектров радионуклидов в модельный спектр пробы, i - номер группы, j - индекс радионуклида. Для определения относительных вкладов спектров радионуклидов в модельный спектр пробы минимизируют разницу между модельным спектром и спектром пробы по выражению  где Pi - спектр пробы, Mi - модельный спектр, δ - коэффициент устойчивости процесса минимизации. Минимизацию проводят в несколько этапов, выбирая сначала большую величину δ и нулевые значения cj, определяют минимум, после чего δ уменьшают, повторяя процесс минимизации, при этом исходными значениями вкладов cj для каждого следующего шага минимизации являются результаты предыдущего шага. Расчет останавливают, когда процесс минимизации становится неустойчивым. Завершают идентификацию альфа-радионуклидов в исследуемой пробе пересчетом полученных значений cj в значения абсолютных активностей радионуклидов в пробе.
где Pi - спектр пробы, Mi - модельный спектр, δ - коэффициент устойчивости процесса минимизации. Минимизацию проводят в несколько этапов, выбирая сначала большую величину δ и нулевые значения cj, определяют минимум, после чего δ уменьшают, повторяя процесс минимизации, при этом исходными значениями вкладов cj для каждого следующего шага минимизации являются результаты предыдущего шага. Расчет останавливают, когда процесс минимизации становится неустойчивым. Завершают идентификацию альфа-радионуклидов в исследуемой пробе пересчетом полученных значений cj в значения абсолютных активностей радионуклидов в пробе.
На фиг. 1 представлена блок-схема реализации способа идентификации альфа-радионуклидов в пробах с использованием жидкостного сцинтилляционного счетчика.
На фиг. 2 представлен фрагмент библиотеки базовых спектров.
На фиг. 3 показан спектр пробы, состоящей из смеси альфа- и бета-излучающих радионуклидов.
На фиг. 4 показан спектр пробы, состоящей из смеси альфа-излучающих радионуклидов.
На фиг. 5 показан пример определения радионуклидного состава пробы, состоящей из смеси бета- и альфа-излучающих изотопов.
Способ осуществляют в соответствии с блок-схемой на фиг. 1 следующим образом.
I. Отбор и подготовка проб к измерению.
Спектрометрический анализ на основе жидкосцинтилляционного счета можно проводить для любых проб (окружающей среды и технологических) после их соответствующей подготовки. Как правило, это водные пробы, твердые пробы (почва, грунт, донные осадки и др. ), пробы растительности (древесная и травяная) и пробы воздуха.
I.1. Отбор проб.
Водные пробы отбирают в чистые емкости объемом 1 л в количестве не менее 3-х штук на контролируемый объект. Подкисляют до pH 3-4. Емкости герметично закрывают. Указывают название пробы, время и место отбора и доставляют в лабораторию.
Твердые пробы, в частности пробы почвы, отбирают с участка размером 10х10 м, площадь отбора размечают конвертом, с узлов конверта (5 точек) отбирают пробы монолитным куском размером 10х10 см, толщиной 5 см, с которого предварительно удаляют травяной покров.
Пробу очищают от корней и упаковывают в полиэтиленовый пакет с указанием названия пробы, места отбора, даты отбора, массы пробы, мощности дозы в месте отбора пробы и доставляют в лабораторию.
Пробы растительности для травянистых проб отбирают с элементарных площадок площадью от 0.25 до 1 м2 в зависимости от густоты травянистого покрова. Смешанную пробу составляет из 4-5 индивидуальных проб, причем отбирают надземные части растений. Пробы древесной растительности составляют путем объединения нескольких индивидуальных проб от различных видов деревьев пропорционально их доле в составе древостояния. В качестве пробы отбирают побег последнего годового прироста (хвойные) или листву с одной ветки из нижней части кроны. Отобранные пробы растительности упаковывают в полиэтиленовый пакет с указанием названия пробы, места отбора, даты отбора, массы пробы и доставляют в лабораторию.
Пробы воздуха отбирают в помощью аспирационных установок с производительностью не менее 1000 м3/ч на тонковолокнистый фильтр Петрянова. После прокачивания необходимого объема воздуха фильтр снимают и упаковывают в чистую плотную бумагу, указывают название пробы, место и дату отбора, объем прокаченного воздуха и доставляют в лабораторию.
I. 2. Подготовка проб к измерению с помощью жидкосцинтилляционного счета (ЖСС).
I.2.1. Подготовка водных проб.
I.2.1.1. Технологические пробы.
Если предполагаемая объемная активность радионуклида в пробе составляет более 102 Бк/л, то пробу не концентрируют, а анализируют сразу. Для этого отбирают по 5 мл каждой из трех параллельных проб, проверяют pH раствора и при необходимости доводят его до нейтрального (концентрированными соляной кислотой или аммиаком), затем вносят нейтральные растворы во флаконы для жидкосцинтилляционного счета, предварительно заполненные 10 мл сцинтилляционного коктейля. Энергично перемешивают содержимое каждого флакона для получения гомогенного раствора, выдерживают флаконы в темноте не менее 12 ч (для затухания возможных процессов хемо- и фотолюминесценции) и затем устанавливают в жидкосцинтилляционый анализатор для дальнейших измерений. Одновременно с анализируемой пробой по аналогии готовят фоновую, в которую вносят такой же объем деионизованной воды, как и подготовленной пробы, но не более 10 мл.
I.2.1.2. Низкоактивные пробы.
Пробы с низкой объемной активностью радионуклидов (<102 Бк/л) перед анализом обязательно концентрируют. Для этого отбирают три параллельных пробы объемом 1 л каждая, при необходимости подкисляют концентрированной соляной кислотой до pH 3-4 и упаривают на электрической плитке до объема 50-70 мл, переносят в термостойкий стакан объемом 0.1 л и упаривают до объема 5-8 мл. Далее раствор охлаждают, нейтрализуют концентрированным раствором аммиака до pH 5-7 (объем подготовленной пробы не должен превышать 10 мл) и вносят во флаконы для жидкосцинтилляционного счета, предварительно заполненные 10 мл сцинтилляционного коктейля. Содержимое флаконов тщательно перемешивают и выдерживают с темноте не менее 12 ч перед измерением. Одновременно с анализируемой пробой по аналогии готовят фоновую, в которую вносят такой же объем деионизованной воды, как и подготовленной пробы.
При концентрировании проб упариванием могут быть утеряны некоторые радионуклиды, например углерод-14, сера-35, находящиеся в воде в виде растворенных газов или летучих органических соединений, поэтому их наличие и количество необходимо оценить измерением 10 мл неконцентрированной пробы на жидкосцинтилляционном анализаторе.
I.2.2. Подготовка твердых проб (почвы, грунта и др.).
Отобранную твердую пробу, например, почвы весом 1 кг высушивают в течение 6-8 ч при температуре 60oC в сушильном шкафу. В зависимости от вида пробы (песок, глина) и ее твердости ее либо предварительно измельчают в фарфоровой ступке, либо сразу рассеивают на вибростоле с набором сит различного диаметра. Самую мелкую фракцию (0.5 мм и менее) делят методом квартования и отбирают для анализа навеску 20 г. Дальнейшую подготовку твердых проб проводят с помощью кислотного разложения или микроволновой технологии.
I.2.2.1. Подготовка твердых проб путем кислотного разложения.
Тушение (I) наряду с люминесценцией (II) является нежелательным процессом.
При подготовке твердой пробы и ее смешении с жидким сцинтиллятором для измерения с помощью ЖСС возникают мешающие процессы, такие как тушение (I) и люминесценция (II), проявляющиеся в снижении числа регистрируемых фотоэлектронными умножителями (ФЭУ) световых импульсов, изменении формы и амплитуды спектра радионуклида с одновременным сдвигом спектра в низкоэнергетическую область. Вследствие этого значительно уменьшается вероятность правильной идентификации радионуклида и эффективность регистрации его излучения ФЭУ.
I. Тушение подразделяют на два основных вида: (1) химическое и (2) оптическое.
I. 1. Химическое тушение обусловлено присутствием в сцинтилляционной смеси химических соединений, которые способны возбуждаться подобно сцинтилляторам, присутствующим в жидкосцинтилляционном коктейле, но не высвечиваться, а рассеивать энергию возбуждения целиком в виде тепла. Они также являются "ловушками" энергии, мигрирующей в растворителе, конкурируя с молекулами сцинтиллятора, поэтому эффект тушения проявляется даже при низких концентрациях тушителя. Примером химических тушителей служат вода, кислород воздуха, перекиси, некоторые кислоты и щелочи, ряд органических соединений и растворителей (ацетон, этанол, четыреххлористый углерод, тетранитрометан и т. п. ), высокомолекулярные биологически активные соединения (белки, нуклеиновые кислоты и т.п.) и др.
I. 2. Оптическое (или цветовое) тушение обусловлено поглощением сцинтилляционных вспышек окрашенными веществами ("оптические ловушки"), присутствующими в подготовленном для измерения на ЖСС растворе. Примером оптических тушителей являются различные пигменты, органические красители, хромофоры, окрашенные неорганические соединения, содержащие катионы Fe, Co, Ni, Cu и др. Из всех окрашенных соединений лишь имеющие голубой цвет не вызывают оптического тушения, так как имеют минимум адсорбции света сцинтилляционных вспышек в области максимальной спектральной чувствительности ФЭУ (390-460 нм). Растворы, имеющие желтое или красное окрашивание, вызывают наибольшее оптическое тушение.
II. Люминесценцию можно подразделить на хемолюминесценцию (1) и фотолюминесценцию (2).
II. 1. Хемолюминесценция представляет собой свечение, обусловленное не взаимодействием радиоактивного излучения с молекулами сцинтилляторов, а вызванное возбуждением при протекании химических реакций в подготовленном образце или сцинтилляционном коктейле. Возникновению процесса хемолюминесценции способствует присутствие в счетном растворе молекул окислителей, щелочей, перекисей и др. Энергетический спектр хемолюминесценции близок к тритиевому и может служить источником серьезных ошибок при идентификации и расчета активности радионуклидов.
II.2. Фотолюминесценция представляет собой свечение, возникающее в счетном образце в результате поглощения энергии ультрафиолетового излучения и также может служить источником ошибок.
Все рассмотренные выше процессы, возникающие при подготовке образца и смешении его с сцинтилляционным коктейлем, являются нежелательными для проведения ЖСС, и их необходимо свести к минимуму. При подготовке к ЖСС проб воды и воздуха влияние перечисленных явлений не столь существенно, как для твердой пробы (почва, грунт, донные осадки), химический состав которых и традиционные методы вскрытия обуславливают протекание полного набора нежелательных процессов (химическое /HNO3/ и цветовое /Fe3+/ тушение, хемолюминесценция /H2O2/).
Современные жидкосцинтилляционные счетчики оборудованы устройствами автоматического определения уровня люминесценции и затушенности анализируемого жидкого образца, смешанного с сцинтилляционным коктейлем. Так, жидкосцинтилляционный счетчик "Tri-Carb 2550 TR/AB" фирмы "Canberra Packard" (США), на котором осуществлен данный способ, достаточно точно и воспроизводимо определяет степень люминесценции (Lum) и два параметра (или индекса) тушения, значения которых предоставляются пользователю. Первый параметр тушения - "спектральный индекс образца" (SIS) представляет собой значение максимальной энергии радионуклида. Так, для практически не затушенного образца трития параметр SIS = 16-18, углерода-14 SIS=140-15. Полученное значение параметра SIS ниже значений максимальной энергии анализируемого радионуклида свидетельствует о наличии тушителя в подготовленном образце.
Второй параметр тушения - "трансформированный спектральный индекс внешнего стандарта" (tSIE) также характеризует степень затушенности измеряемого образца и вычисляется на основе анализа спектра, обусловленного взаимодействием с препаратом "комптоновских электронов" встроенного внешнего стандарта (Ba-133). Принято, что совершенно не затушенный образец (например, раствор образцового источника в запаянной ампуле, приготовленный в заводских условиях) имеет параметр тушения tSIE=950-1050. В реальных же условиях совершенно не затушенных образцов приготовить не удается, так как даже кислород воздуха, попадающий в образец в процессе его подготовки, вызывает довольно сильное тушение сцинтилляций. Так, например, даже чистый сцинтилляционный коктейль (марки Ultima Gold AB, Canberra) из только что вскрытой заводской фасовки без внесенного в него образца уже изначально имеет параметр tSIE=700. При внесении в коктейль 1 мл дистиллированной воды tSIE понижается до значения приблизительно 660, 5 мл дистиллированной воды - до значения приблизительно 400, 10 мл дистиллированной воды - до значения 300. Известно, что тушение с tSIE ≤ 100 приводит к недостоверной регистрации излучения, и результаты измерений имеют недопустимо большую ошибку.
Наиболее близким техническим решением является способ подготовки твердых проб, заключающийся в том, что из навески отбирают 3 г почвы и проводят ее кислотное вскрытие. Пробу нагревают на электрической плитке в течение 1 ч с 40 мл концентрированной азотной кислоты и 2 мл 30%-ной перекиси водорода при периодическом перемешивании в термостойком стакане, накрытом часовым стеклом. Затем смесь отфильтровывают на воронке с фильтром "синяя лента", остаток с фильтром переносят в тот же стакан, вносят такие же объемы азотной кислоты и перекиси водорода и процесс повторяют. Осадок на фильтре промывают 20 мл 6 н азотной кислоты, все три кислотные вытяжки объединяют. Объединенную кислотную вытяжку упаривают до состояния влажных солей [4].
Для подготовки к ЖСС остаток растворяют в 5 мл горячей 2%-ной азотной кислоты. Раствор подготовленной пробы имеет интенсивное желтое окрашивание, поэтому для ЖСС взяли не всю пробу (5 мл), а аликвоту 0.5 мл, что соответствовало навеске 0.3 г. При внесении аликвоты в 10 мл сцинтилляционного коктейля марки "Ultima Gold AB" счетный образец имеет желтое окрашивание, измеренный параметр тушения tSIE равен 60. Однако такую пробу подвергать спектрометрическому анализу не имеет смысла ввиду огромного тушения счетного образца. В другую аликвоту пробы 0.5 мл добавили 1 мл свежеприготовленного раствора хлорида двухвалентного олова и смешали с 10 мл сцинтилляционного коктейля марки "Ultima Gold AB". Измеренный параметр тушения tSIE в этом случае равен 180, т.е. на пределе возможностей программы обсчета и расшифровки спектров, и, таким образом, лишь небольшая (1/10) часть подготовленного образца, соответствующая 0.3 г почвы, была подвергнута спектрометрическому анализу.
Таким образом, подготовка подходящего для спектрометрии на основе ЖСС образца твердой пробы (грунта, почвы, донных осадков) достаточно трудная задача, поскольку традиционные методы вскрытия пробы предусматривают применение сильных окислителей (концентрированной азотной кислоты, перекиси водорода и др.), вызывающих сильное химическое тушение. Кроме того, в подготовленном образце присутствуют ионы железа (3+), придающие раствору в зависимости от концентрации цвет от желтого до бурого, что вызывает критическое для спектрометрии оптическое тушение наряду с химическим. При этом кислотная вытяжка из 0,2 - 0,3 г средней по составу пробы почвы вызывает настолько сильное тушение (tSIE ≤ 70-80), что не позволяет достоверно расшифровать спектр сложного радионуклидного состава.
Техническим результатом предлагаемого способа подготовки твердых проб путем кислотного разложения является повышение достоверности измерения радиоактивности этих проб за счет увеличения веса счетного образца, направляемого на спектрометрический анализ.
Для достижения технического результата при подготовке твердых проб для спектрометрического анализа с использованием жидкосцинтилляционного счетчика осуществляют следующее.
Перед вскрытием пробы проводят ее отжиг от возможно присутствующих органических примесей, которые могут вызывать дополнительное окрашивание подготовленного образца и служить причиной оптического тушения. Отжиг ведут в течение 5-7 ч при температуре 4500-5500oC. Возможную потерю изотопов цезия и иода предварительно оценивают гамма-спектрометрией неотожженной пробы. Для отжига берут 10-20 г тонкоизмельченной пробы. После отжига берут навеску 3-5 г пробы и проводят ее кислотное вскрытие с помощью концентрированной азотной кислоты HNO3 и концентрированной перекиси водорода H2O2. Навеску пробы переносят в термостойкий стеклянный стакан объемом 150-200 мл, приливают 40 мл концентрированной азотной кислоты, 2 мл 30%-ной перекиси водорода, смесь перемешивают, накрывают часовым стеклом и нагревают на электрической плитке в течение 1 ч. Затем смесь фильтруют на воронке с фильтром "синяя лента", остаток с фильтром переносят в тот же стакан, вносят такие же объемы азотной кислоты и перекиси водорода и процесс повторяют. Осадок на фильтре промывают 20 мл 6 н азотной кислоты, все три кислотные вытяжки объединяют. Объединенную кислотную вытяжку объемом 40-60 мл концентрируют упариванием. Упаривают вытяжку до состояния влажных солей.
При подготовке пробы к жидкосцинтилляционному счету с минимизацией нежелательных процессов химического и оптического тушения и хемолюминесценции проводят следующие операции.
Для снижения степени химического тушения переводят азотнокислый раствор в солянокислый, поскольку соляная кислота, во-первых, является по сравнению с азотной гораздо более слабым тушителем и, во-вторых, хлориды более растворимы, чем нитраты.
К влажному остатку приливают 20 мл горячей дистиллированной воды, растворяют остаток и упаривают до состояния влажных солей. При этом обеспечивается частичная отгонка окислов азота. К остатку приливают 20 мл раствора горячей 1 н соляной кислоты до полного растворения осадка и упаривают до состояния влажных солей. Этим обеспечивается перевод нитратов в хлориды и окончательная отгонка окислов азота. Далее к остатку приливают 10 мл горячей дистиллированной воды и вновь упаривают до состояния влажных солей для удаления избытка хлора, являющегося сильным тушителем. Остаток растворяют в 3-5 мл горячей соляной кислоты. Как правило, в зависимости от состава пробы и содержания в ней железа раствор имеет интенсивное желтое или красно-бурое окрашивание.
Для снижения степени оптического тушения раствора переводят составляющие его хлориды в пирофосфатные комплексы, которые для большинства катионов - компонентов твердых проб, в частности почвы (Al3+, Ca2+, Fe3+, Mg2+) бесцветны и обладают высокой константой устойчивости.
Для этого в солянокислый раствор (охлажденный) при перемешивании вносят 2 мл концентрированной ортофосфорной кислоты (H3PO4). Известно, что в 1 кг средней по составу пробы почвы (грунта) может содержаться до 50 г железа - основного элемента, придающего вытяжкам окрашивание. Таким образом, для навески почвы в 5 г содержание в ней железа может составлять до 250-300 мг.
В пирофосфатном комплексе катион Fe3+ имеет координационное число, равное 6, и состав комплекса можно представить в виде [Fe(H2P2O7)3]3-.
Тогда для количественного образования комплекса из 300 мг Fe3+ необходимо ≈ 2,8 г дигидропирофосфат-аниона, т.е. ≈ 3,1 г концентрированной H3PO4 или 1,7 мл (ρ = 1,8 г/мл). По реакции необходимо взять избыток H3PO4 для образования пирофосфатных комплексов с другими катионами (Ca2+ и др.).
Для образования пирофосфорной кислоты (H4P2O7) по реакции
2H3PO4 ---> H4P2O7 + H2O
солянокислый раствор c добавленной H3PO4 нагревают до температуры ≥ 200oC.
При нагревании и упаривании соляная кислота удаляется, раствор H3PO4 постепенно концентрируется и образуется пирофосфорная кислота, с которой в свою очередь реагируют катионы металлов с образованием устойчивых пирофосфатных комплексов. Раствор упаривают до образования вязкой массы. В процессе упаривания в кипящий раствор этой массы 2 - 3 раза (T ≥ 200oC) добавляют небольшими порциями (5 - 10 мл) горячую дистиллированную воду для удаления остатка хлора (химический и оптический тушитель) и по каплям 300 - 500 мкл 30%-ной H2O2 для дополнительного разложения органических соединений, возможно присутствующих в растворе после отжига пробы.
Раствор при этом обесцвечивается вследствие образования бесцветных пирофосфатных комплексов. Упаривание ведут до объема не менее 1,5 - 2 мл, так как более низкие объемы при охлаждении приводят к застекловыванию полученной вязкой жидкости. Далее раствор охлаждают до температуры приблизительно 30oC и при необходимости (если начался процесс стеклования) добавляют 1 мл горячей дистиллированной воды и раствор тщательно перемешивают до однородной по консистенции массы.
Для подавления процессов хемолюминесценции в раствор при перемешивании добавляют 1 мл насыщенного свежеприготовленного раствора хлорида двухвалентного олова.
При варке пирофосфорной кислоты, полученной при упаривании H3PO4, образуются перекисные и надперекисные соединения фосфора переменного состава, вызывающие сильную хемолюминесценцию, достигающую 70% и продолжительную во времени (≥2 суток), что значительно затрудняет измерение подготовленного образца. Добавление SnCl2 полностью подавляет люминесценцию (Lum=0).
Для подготовки счетного образца полученный раствор переносят в измерительный флакон для ЖСС, предварительно заполненный 10 мл сцинтилляционного коктейля марки "Ultima Gold AB" фирмы "Canberra Packard" (США) и добавляют 1 мл концентрированного раствора эмульгатора-октилфенолдекаэтиленгликолевого эфира (C34H62O11) - "TRIOR-X-100", повышающего эмульгирующую способность сцинтилляционного коктейля и растворимость в нем солей. В результате проведенных исследований установлено, что добавление 1 мл тритона X-100 в состав коктейля "Ultima Gold AB" лишь незначительно увеличивает его тушение (≈ 5%0), не влияет на форму спектра радионуклида и не приводит к ошибке в его идентификации и количественном определении.
Для подавления фотолюминесценции подготовленную таким образом пробу выдерживают не менее 12 ч в затемненном месте и затем измеряют на жидкосцинтилляционном счетчике.
В предложенном способе подготовки пробы почвы среднего состава весом до 5 г и смешанные со сцинтилляционным коктейлем практически не вызывают оптического тушения, а степень тушения определяется в основном объемом вносимого в счетный флакон приготовленного бесцветного раствора (tSIE=250-400).
Таким образом, предлагаемый способ подготовки твердых проб путем кислотного разложения позволяет повысить достоверность идентификации радионуклидов при измерении радиоактивности твердых проб с помощью ЖСС путем снижения степени тушения и люминесценции раствора.
I.2.2.2. Подготовка твердых проб с помощью микроволновой технологии.
Мелкодисперсную фракцию отквартованной пробы измельчают в планетарной микромельнцие до размера 0.1 мм и менее, отбирают навеску 5-10 г и вносят во фторопластовые стаканы ротора микроволновой печи. В каждый из стаканов вносят также по 10 мл смеси концентрированной азотной кислоты и 30%-ной перекиси водорода в объемном соотношении 9:1. Стаканы закрывают специальными крышками с перепускными клапанами, помещают в ротор и устанавливают в микроволновую печь. Процесс разложения пробы проводится в следующих режимах: 5 мин при 250 Вт импульсного микроволнового излучения, 10 мин - 400 Вт и 5 мин - 500 Вт. Благодаря равномерному микроволновому нагреву пробы в закрытом объеме в реакционной среде создаются высокие температура и давление, позволяющие на порядок сократить время вскрытия пробы и способствующие в большинстве случаев ее полному разложению.
После окончания процесса и охлаждения пробы ее фильтруют (в случае неполного разложения), остаток на фильтре промывают 2 раза по 5 мл 5 M азотной кислоты, кислотные фракции объединяют и подготавливают для жидкосцинтилляционного счета, как описано в п. 1.2.2.1.
I.2.3. Подготовка проб растительности.
Пробы растительности высушивают в течение 6-8 ч при температуре 60oC в сушильном шкафу. Высушенные пробы измельчают с помощью мельницы, взвешивают, помещают в фарфоровые тигли с крышками и озоляют в муфельной печи в течение 6-7 ч при 400-450oC. После завершения озоления и охлаждения тиглей в них заливают 10-20 мл концентрированной азотной кислоты (в зависимости от количества полученной золы) и проводят кислотное выщелачивание и подготовку для жидкосцинтилляционного счета, как описано для проб почвы.
I.2.4. Подготовка проб воздуха.
Фильтры, состоящие из ткани Петрянова, измельчают ножницами, помещают в фарфоровые тигли с крышками и озоляют в муфельной печи в течение нескольких часов при 400-450oC. После завершения озоления и охлаждения тиглей в них заливают 10-20 мл концентрированной азотной кислоты (в зависимости от количества полученной золы) и проводят кислотное выщелачивание и подготовку для жидкосцинтилляционного счета, как описано для проб почвы.
I.2.5. Подготовка фоновых образцов.
При анализе проб воды, почвы, растительности и воздуха в качестве фоновых используют образцы, приготовленные смешением во флаконах для жидкосцинтилляционного счета (объемом 20 мл) 10 мл сцинтилляционного коктейля с объемом 1 н соляной кислоты (или ее смеси с раствором хлорида олова), равным объему подготовленной и внесенной в измерительный флакон пробы.
II. Измерение и запись спектра пробы.
При измерении спектра пробы выполняют следующие операции.
В специальную кассету помещают фоновую пробу и подготовленные анализируемые. Кассету устанавливают в анализатор. Слева в кассету вставляют клипсу c кодом, соответствующим программе измерения образца в режиме подсчета зарегистрированных импульсов в 1 мин. В программе измерения устанавливают время измерения и энергетический диапазон от 0 до 2000 КэВ. После этого анализатор переводят в режим счета, в котором все помещенные в кассету флаконы с образцами автоматически просчитываются. Записываются все заданные в программе параметры, а именно индекс пробы, время измерения, счет, погрешность, параметр тушения, и сохраняется полученный спектр образца в виде файла с распределением количества зарегистрированных импульсов по энергетическим каналам многоканального анализатора. После этого по специально разработанной программе анализируют полученный энергетический спектр образца и определяют качественный состав пробы и объемные (удельные) активности ее компонентов.
III. Сворачивание аппаратного спектра пробы в группы.
В данном способе используют сворачивание аппаратного спектра пробы в группы, граничные значения в которых представлены в табл. 1 (см. в конце описания) и являются квазиарифметической прогрессией вида
Ni+1=Ni+[(i+1)/2], (2)
где i = 1,2...n (квадратные скобки здесь обозначают целую часть).
В табл. 1 приведено сворачивание аппаратного спектра в 90 групп для 2115 каналов анализатора. Количество групп выбирается в зависимости от детальности аппаратного спектра.
IV. Cоздание библиотеки базовых спектров.
Для создания библиотеки базовых спектров выполняют следующее. Для каждого из контрольных источников готовят серии из флаконов c известным одинаковым количеством радиоактивной метки. В каждый из флаконов, начиная со второго, вносят возрастающее количество химического тушителя, после чего просчитывают всю серию на жидкосцинтилляционном анализаторе с записью всех спектров в нуклидную библиотеку, при этом каждому значению параметра тушения соответствует определенная эффективность регистрации излучения радионуклида. На фиг. 2 представлены энергетические спектры некоторых альфа-излучающих радионуклидов с различным химическим тушением. При увеличении тушения в образце регистрируемая анализатором спектральная кривая располагается в области более низких энергий, причем эффективность регистрации, как правило, сильно уменьшается.
В библиотеку вводят пересчитанные в групповые спектры отдельных радионуклидов для 10 различных тушений.
В библиотеку также включают значения эффективности регистрации для каждого тушения.
Все библиотечные спектры нормированы на единичную активность.
Полученная библиотека спектров наиболее распространенных нуклидов и кривых тушения позволяет анализировать реальные образцы в широком диапазоне тушений для данной марки сцинтилляционного коктейля.
V. Пересчет базовых спектров к тушению пробы.
B библиотеке приведены базовые спектры для конечного набора тушений. Тушение исследуемой пробы может не совпадать с библиотечными. Поэтому при обработке пробы с определенным тушением базовые спектры приводят к этому тушению путем интерполяции между библиотечными спектрами. К данному тушению интерполируют также эффективность регистрации для каждого радионуклида. Полученные спектры нормируют и используют далее при создании модельного спектра.
VI. Минимизация отклонения модельного спектра от спектра пробы.
На следующем этапе составляют модельный спектр
где Mij - модельные спектры отдельных радионуклидов,
cj - относительные вклады в модельный спектр этих радионуклидов,
i - номер группы,
j - индекс радионуклида.
Для нахождения вкладов cj минимизируют разницу между модельным спектром и спектром пробы Pi.
При решении задач минимизации наиболее важным моментом является правильный выбор критерия минимизации.
Использование минимизации абсолютного отклонения спектра пробы от спектра модели (см. формулу (1)), как это сделано в прототипе, не позволяет "чувствовать" малые активности на фоне больших.
Для того чтобы выделить в пробах альфа-радионуклиды c малой активностью на фоне радионуклидов с большой активностью, следует минимизировать следующее выражение
где Pi - спектр пробы,
Mi - модельный спектр,
δ - коэффициент устойчивости процесса минимизации.
Здесь деление на min (Pi, Mi) (переход к относительному отклонения), а не просто на Pi, используют для повышения чувствительности к "низкоактивным" областям спектра, так как это позволяет еще более увеличить роль слагаемых с малыми значениями Mi.
Коэффициент устойчивости δ является малой добавкой, необходимой для исключения деления на ноль при min (Pi, Mi)=0 и исключения неустойчивости решения (4) из-за слагаемых, в которых член min (Pi, Mi) меньше статистического разброса значений Pi.
Выбор коэффициента δ одновременно влияет как на устойчивость решения (чем больше δ, тем лучше устойчивость), так и на чувствительность способа к малоактивным элементам (чем больше δ, тем хуже чувствительность). В крайнем варианте при δ → ∞, минимизируемое выражение сводится к виду (1), используемому в прототипе. При расчете сначала выбирают большую величину коэффициента δ, находят минимум выражения (4), после чего δ уменьшают и повторяют процесс минимизации. При этом исходными значениями вкладов cj для каждого следующего шага являются результаты предыдущего шага. Расчет останавливают, когда процесс минимизации выражения (4) становится неустойчивым.
VII. Расчет активности радионуклидов.
Полученные значения cj определяют относительный вклад радионуклидов в активность пробы. Переход к абсолютной активности проводят по формуле
Aj = cj • A/Ej, (5)
где A - интегральный счет пробы (сумма аппаратного спектра),
Ej - эффективность регистрации.
Таким образом, в результате измерения и обработки спектра пробы идентифицирован радионуклидный состав и получены активности каждого входящего в состав пробы радионуклида.
Преимущества данного способа показаны на фиг. 3 и 4.
На фиг. 3 показаны аппаратный спектр пробы (синяя линия), состоящей из сложной смеси бета-активных изотопов - H-3, C-14, Sr-90, Cs-137 по 5 Бк каждого из изотопов и 1 Бк альфа-активного Pu-239, а также рассчитанные по предлагаемому способу вклады этих изотопов. Как видно из фиг. 3 и табл. 2 (см. в конце описания), на фоне спектра сложной смеси бета-активных изотопов достаточно точно определяется альфа-активный радионуклид.
На фиг. 4 показаны аппаратный спектр пробы (синяя линия), состоящей из смеси альфа-активных изотопов - U-232, Pu-239, Am-241, по 5 Бк каждого из изотопов, а также рассчитанные по предлагаемому способу вклады этих изотопов. Как видно из фиг. 4 и табл. 3 (см. в конце описания), способ позволяет достаточно четко разделять альфа-активныe радионуклиды в их смеси.
Пример осуществления способа.
Проводят отбор технологической пробы почвы. Отобранную пробу весом 1 кг высушивают в течение 6-8 ч при температуре 60oC в сушильном шкафу. В зависимости от вида пробы (песок, глина) и ее твердости пробу либо предварительно измельчают в фарфоровой ступке, либо рассеивают на вибростоле с набором сит различного диаметра. Самую мелкую фракцию (0.5 мм и менее) делят методом квартования и отбирают для анализа навеску 10 г.
Проводят ее отжиг в течение 6 ч при температуре 450oC. После отжига отбирают 3 г пробы и проводят ее кислотное вскрытие традиционным способом с помощью концентрированной азотной кислоты и концентрированной перекиси водорода. Для этого пробу нагревают на электрической плитке в течение 1 ч с 40 мл концентрированной азотной кислоты и 2 мл 30%-ной перекиси водорода при периодическом перемешивании в термостойком стакане, накрытом часовым стеклом. Затем смесь отфильтровывают на воронке с фильтром "синяя лента", остаток с фильтром переносят в тот же стакан, вносят такие же объемы азотной кислоты и перекиси водорода и процесс повторяют. Осадок на фильтре промывают 20 мл 6 н азотной кислоты, все три кислотные вытяжки объединяют. Объединенную кислотную вытяжку упаривают до состояния влажных солей.
После этого к влажному азотнокислому остатку приливают 20 мл горячей дистиллированной воды, растворяют остаток и упаривают до состояния влажных солей. К остатку добавляют 20 мл раствора горячей 1 н соляной кислоты до полного растворения осадка и упаривают до состояния влажных солей. Далее к остатку приливают 10 мл горячей дистиллированной воды и вновь упаривают до состояния влажных солей. Остаток растворяют в 4 мл горячей 1 н соляной кислоты. Полученный раствор имеет интенсивное желтое окрашивание. Для снижения степени окрашивания раствора в охлажденный солянокислый раствор при перемешивании вносят 2 мл концентрированной ортофосфорной кислоты и упаривают на электрической плитке с периодическим добавлением по каплям 500 мкл 30%-ной H2O2 и 20 мл кипящей дистиллированной воды до образования бесцветной вязкой массы объемом 2 мл. Далее раствор охлаждают до температуры приблизительно 30oC и при перемешивании добавляют 1 мл свежеприготовленного насыщенного раствора хлорида двухвалентного олова.
Полученный раствор переносят в измерительный флакон для ЖСС, предварительно заполненный 10 мл сцинтилляционного коктейля марки "Ultima Gold AB", добавляют 1 мл эмульгатора-"TRITON X-100" и тщательно перемешивают.
Затем измеряют тушение подготовленного счетного образца. Параметр тушения tSIE в этом случае равен 300, люминесценция отсутствует (Lum=0).
После этого анализируют полученный энергетический спектр образца и определяют качественный состав пробы и объемные активности ее компонентов.
Из библиотеки базовых спектров для измеренного уровня тушения формируют модульный спектр  для чего выбирают набор радиоизотопов, характерных для данного пункта захоронения. Здесь cj - относительные вклады в модельный спектр радионуклидов, i - номер группы, j - индекс радионуклидов. Далее, установив δ = 1 и cj=0 для всех j, минимизируют следующее выражение
для чего выбирают набор радиоизотопов, характерных для данного пункта захоронения. Здесь cj - относительные вклады в модельный спектр радионуклидов, i - номер группы, j - индекс радионуклидов. Далее, установив δ = 1 и cj=0 для всех j, минимизируют следующее выражение 
Далее уменьшают δ в 10 раз и заново проводят минимизацию, используя в качестве начальных значений вкладов cj, полученные на предыдущем шаге. Повторяют процедуру до тех пор, пока процесс минимизации не становится неустойчивым (изменение вкладов cj не приводит к уменьшению минимизируемого выражения).
Полученные таким образом коэффициенты cj определяют относительный вклад радионуклидов в активность пробы. Переход к абсолютной активности проводят по формуле
Aj = cj • A/Ej,
где A - интегральный счет пробы (сумма аппаратного спектра),
Ej - эффективность регистрации.
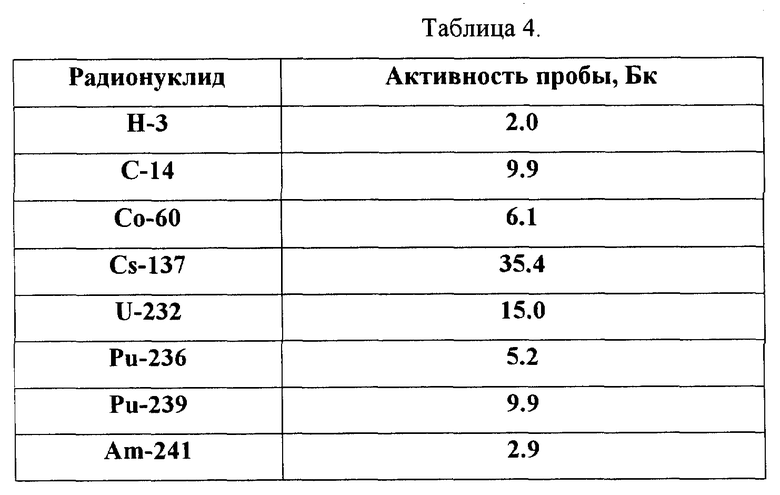
На фиг. 5 показан спектр пробы, состоящей из смеси альфа-активных изотопов - U-232, Pu-236, Pu-239, Am-241 и мешающих бета-активных радионуклидов - H-3, C-14, Co-60, Cs-137. На фиг. 5 видно полное совпадение спектра пробы с рассчитанным спектром модели, а в табл. 4 представлены результаты идентификации радиоизотопного состава, полностью согласующиеся с исходным составом пробы (см. в конце описания).
В результате проведенного отбора, пробоподготовки, измерения и обработки спектров пробы идентифицирован радионуклидный состав и получены активности каждого входящего в состав пробы радионуклида.
Техническая эффективность предложенного способа по сравнению с прототипом заключается в увеличении чувствительности спектрометрического анализа, в возможности получения образца с приемлемой степенью тушения, что значительно повышает достоверность идентификации радионуклидов в твердых пробах с использованием ЖСС, а также в том, что данный способ позволяет выделять и идентифицировать с достаточной точностью альфа-активные радионуклиды на фоне мешающих радионуклидов.
Источники информации
1. Патент США N 4,918,310 МПК G 01 T 1/204.
2. Патент США N 5,134,294 МПК G 01 T 1/204.
3. PCT N 91/10922 МПК G 01 T 1/204.
4. Бок. Р. Методы разложения в аналитической химии. М.: Химия, 1984 г.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИДЕНТИФИКАЦИИ АЛЬФА-ИЗЛУЧАЮЩИХ РАДИОНУКЛИДОВ В ПРОБАХ С ИСПОЛЬЗОВАНИЕМ ЖИДКОСТНОГО СЦИНТИЛЛЯЦИОННОГО СЧЕТЧИКА | 2000 |
|
RU2191409C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ РАДИОНУКЛИДОВ В ПРОБАХ С ИСПОЛЬЗОВАНИЕМ ЖИДКОСТНОГО СЦИНТИЛЛЯЦИОННОГО СЧЕТЧИКА | 1997 |
|
RU2120646C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ АЛЬФА-ИЗЛУЧАЮЩИХ РАДИОНУКЛИДОВ | 2004 |
|
RU2267800C1 |
| СПОСОБ РАДИОЭКОЛОГИЧЕСКОГО МОНИТОРИНГА ПРОМЫШЛЕННОГО РЕГИОНА | 1997 |
|
RU2112999C1 |
| Способ определения активности бета-, альфа- излучающих нуклидов в пробах аэрозолей воздуха | 2023 |
|
RU2811788C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РАДИАЦИОННОГО ФОНА ПОЧВ ПРИ ПРОВЕДЕНИИ РАДИОЭКОЛОГИЧЕСКОГО МОНИТОРИНГА ПРОМЫШЛЕННОГО РЕГИОНА | 2001 |
|
RU2209445C2 |
| СПОСОБ РАДИАЦИОННОГО МОНИТОРИНГА ЭКОСИСТЕМ ПО ИЗМЕРЕНИЮ РАДИОАКТИВНОСТИ СНЕЖНОГО ПОКРОВА ПРИ ПРОВЕДЕНИИ СНЕГОМЕРНОЙ СЪЕМКИ | 1999 |
|
RU2188442C2 |
| СПОСОБ РАДИАЦИОННОГО МОНИТОРИНГА ЭКОСИСТЕМ ПО РАДИОАКТИВНОСТИ АТМОСФЕРНЫХ ОСАДКОВ, ОТБОРА И ПРИГОТОВЛЕНИЯ ПРОБ | 1999 |
|
RU2178159C2 |
| СПОСОБ РАДИОЭКОЛОГИЧЕСКОГО МОНИТОРИНГА СОДЕРЖАНИЯ ТРИТИЯ В ОКРУЖАЮЩЕЙ СРЕДЕ ПРОМЫШЛЕННОГО ПРЕДПРИЯТИЯ | 2002 |
|
RU2223517C2 |
| СПОСОБ ИЗОЛЯЦИИ ТВЕРДЫХ РАДИОАКТИВНЫХ ОТХОДОВ ОТ ОКРУЖАЮЩЕЙ СРЕДЫ | 1999 |
|
RU2153720C1 |






Способ применяется при проведении работ в области радиоэкологического мониторинга для измерения содержания радионуклидов в различных компонентах окружающей среды, в частности для определения активности альфа излучающих радионуклидов. Способ включает отбор проб, подготовку проб к измерению на жидкостном сцинтилляционном счетчике, измерение и запись спектра пробы и параметров измерения, сворачивание аппаратного спектра пробы в группы, создание модельного спектра пробы на основе библиотеки базовых спектров, минимизацию отклонения модельного спектра от спектра пробы и определение содержания радионуклидов в пробе. При этом идентифицируют альфа-излучающие радионуклиды при наличии в отобранных пробах мешающих радионуклидов. Для этого при подготовке твердых проб перед вскрытием пробы проводят ее отжиг, осуществляют кислотное вскрытие с помощью концентрированной азотной кислоты и перекиси водорода, концентрируют упариванием до состояния влажных солей, переводят влажный остаток в солянокислый раствор и концентрируют упариванием. Затем в полученный раствор добавляют концентрированную ортофосфорную кислоту, концентрируют упариванием. Раствор охлаждают и переносят в сцинтилляционный коктейль. Измеряют образец на жидкосцинтилляционном счетчике, записывают полученный спектр образца и сворачивают его в группы. Осуществляют минимизацию отклонения модельного спектра от спектра пробы. Технический результат заключается в увеличении чувствительноcти спектрометрического анализа, в возможности получения образца с приемлемой степенью тушения, что значительно повышает достоверность идентификации радионуклидов в твердых пробах с использованием жидкостного сцинтилляционного счетчика, а также в том, что способ позволяет выделять и идентифицировать с достаточной точностью альфа-активные радионуклиды на фоне сопутствующих радионуклидов. 4 табл., 5 ил.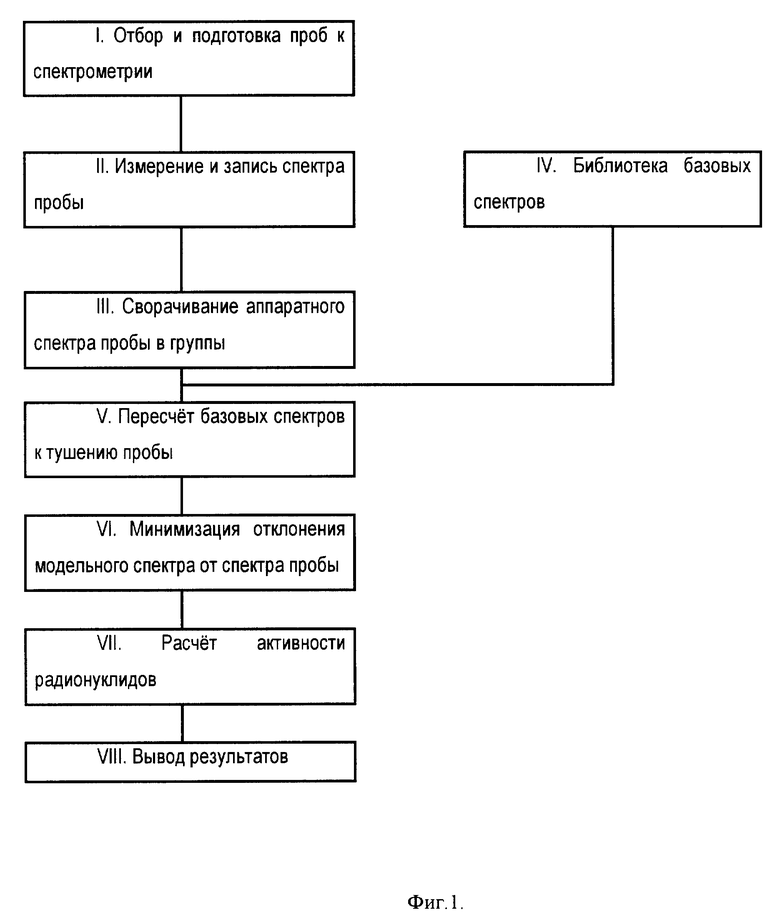
Способ идентификации радионуклидов в пробах с использованием жидкостного сцинтилляционного счетчика, включающий отбор проб окружающей среды и технологических проб, подготовку проб к измерению на жидкостном сцинтилляционном счетчике, измерение и запись спектра пробы и параметров измерения, сворачивание аппаратного спектра пробы в группы, создание модельного спектра пробы на основе библиотеки базовых спектров, минимизацию отклонения модельного спектра от спектра пробы и определение содержания радионуклидов в пробе, отличающийся тем, что идентифицируют альфа-излучающие радионуклиды при наличии в отобранных пробах мешающих радионуклидов, для этого при подготовке твердых проб перед вскрытием пробы проводят ее отжиг, осуществляют кислотное вскрытие с помощью концентрированной азотной кислоты и перекиси водорода, концентрируют упариванием до состояния влажных солей, переводят влажный остаток в солянокислый раствор путем добавления горячего раствора 1М соляной кислоты и концентрируют упариванием при периодическом добавлении не менее двух раз растворов концентрированной перекиси водорода и дистиллированной воды до объема не менее 2 мл, затем в полученный раствор добавляют концентрированную ортофосфорную кислоту, концентрируют упариванием при периодическом добавлении не менее двух раз концентрированной перекиси водорода и горячей дистиллированной воды до образования бесцветного вязкого раствора объемом не менее 2 мл, представляющего собой смесь пирофосфатных комплексов катионов основных компонентов твердой пробы, раствор охлаждают до комнатной температуры, добавляют в него насыщенный раствор хлорида двухвалентного олова и переносят в сцинтилляционный коктейль, добавляя 1 мл эмульгатора "Triton X-100" и смесь тщательно перемешивают, получая счетный образец, подготовленный для спектрометрического анализа, измеряют его на жидкосцинтилляционном счетчике и записывают полученный спектр образца, сворачивают спектр образца в группы, граничные значения Ni в которых являются квазиарифметической прогрессией вида Ni+1 = Ni + [(i + 1) / 2], где i = 1, 2 ... n, осуществляют минимизацию отклонения модельного спектра от спектра пробы путем составления модельного спектра
где Mij - модельные спектры радионуклидов;
cj - относительные вклады спектров, радионуклидов в модельный спектр пробы;
i - номер группы;
j - индекс радионуклида,
а для определения относительных вкладов спектров радионуклидов в модельный спектр пробы минимизируют разницу между модельным спектром и спектром пробы по выражению
где Pi - спектр пробы;
Mi - модельный спектр;
δ - коэффициент устойчивости процесса минимизации,
минимизацию проводят в несколько этапов, выбирая сначала большую величину δ и нулевые значения cj, определяют минимум, после чего δ уменьшают, повторяя процесс минимизации, при этом исходными значениями вкладов cj для каждого следующего шага минимизации являются результаты предыдущего шага, расчет останавливают, когда процесс минимизации становится неустойчивым, и завершают идентификацию альфа-радионуклидов в исследуемой пробе пересчетом полученных значений cj в значения абсолютных активностей радионуклидов в пробе.
| Огнетушитель | 0 |
|
SU91A1 |
| Способ проведения абсолютных измерений альфа-активности нуклидов с помощью счетчика с жидким сцинтиллятором | 1980 |
|
SU845615A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РАДИОАКТИВНОСТИ БИОЛОГИЧЕСКИХ ПРЕПАРАТОВ | 0 |
|
SU286092A1 |
| Радиометр для измерения активности радионуклидов в жидком сцинтилляторе | 1979 |
|
SU807807A1 |
| GB 1145713 A, 19.03.69 | |||
| US 5412216 A, 02.05.95 | |||
| Устройство для измерения оборотов | 1974 |
|
SU487407A2 |
| СУЛЬФАТОУБОРОЧНАЯ МАШИНА | 0 |
|
SU221626A1 |
| Способ определения пирамидальности угловой меры | 1985 |
|
SU1384943A1 |
| US 4555629 A, 26.11.85. | |||

