Изобретение относится к способу разделения 1- азабицикло[2.2.2]октан-3-амин, 2- (дифенилметил) -N- [[2-метокси- 5-(1-метилэтил)фенил]метила].
Вышеуказанное соединение (далее обозначаемое как "рацемат") и (2S,3S) энантиомер такого соединения (далее обозначен как (2S,3S)энантиомер) являются антагонистами рецептора P вещества, что полезно для лечения и предотвращения широкого ряда заболеваний центральной нервной системы, желудочно-кишечных, воспалительных и других заболеваний. Рацемат и (2S, 3S)энантиомер, а также способ их получения, рассмотрены в заявке на патент США 08/211,120, поданной 23.05.94 на национальную фазу и соответствующей заявке PCT/US 92/03317, поданной 28.04.92 г. Заявка на патент США 08/211,120 включена в настоящее описание в качестве ссылки. Как вышеуказанные соединения, так и способ их получения раскрыты, в общем виде, в патенте США 5,162,339, выданном 10.11.92. Этот патент также включен сюда в качестве ссылки.
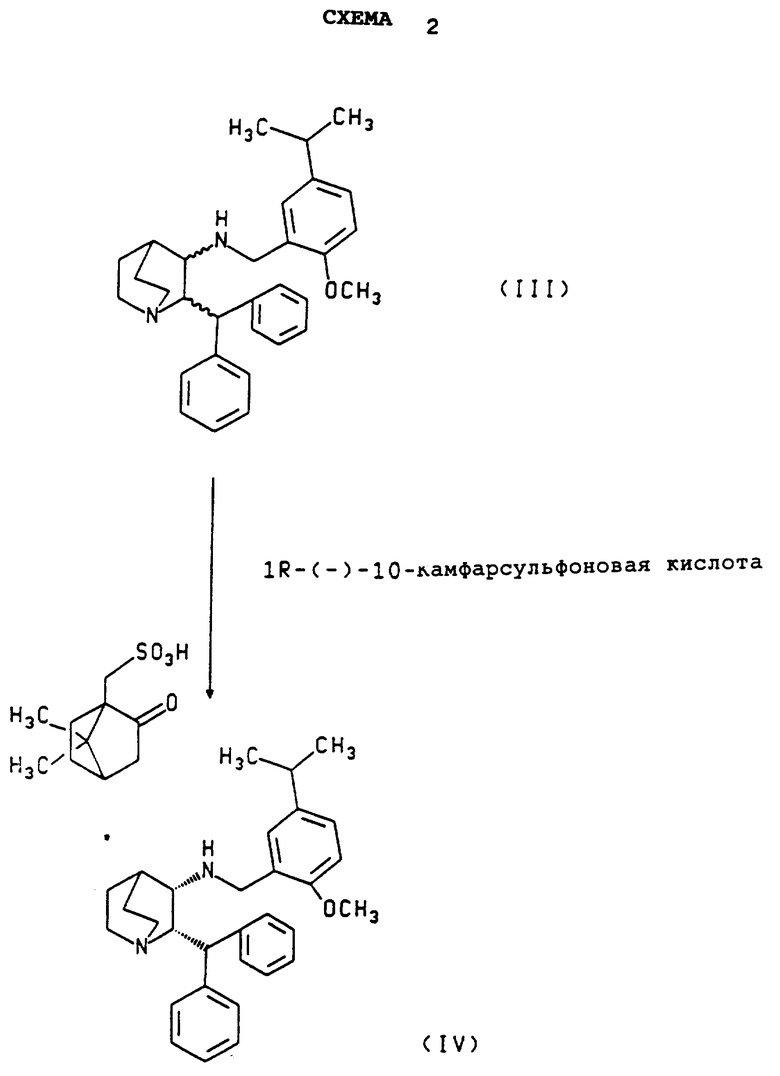
Настоящее изобретение относится к способу разделения 1-азабицикло [2.2.2] октан-3-амин, 2-(дифенилметил)-N- [[2-метокси-5-(1-метилэтил)фенил] метила] , который включает взаимодействие 1-азабицикло [2.2.2] октан-3-амин 2-(дифенилметил)-N- [[2 -метокси-5-(1-метилэтил)фенил]метила] с 1R-(-)-10-камфарсульфоновой кислотой в соответствующем растворителе с образованием соли камфарсульфоновой кислоты с (2S,3S)-1-азабицикло [2.2.2] октан-3- амин, 2-(дифенилметил)-N-[ [2-метокси-5-(1-метилэтил) фенил] метилом] и затем необязательный гидролиз такой соли с получением свободного основания (2S,3S) энантиомера.
Растворителем для проведения вышеуказанного разделения может быть выбран любой растворитель, способный растворять как рацемат, так и разделяющий агент - камфарсульфоновую кислоту, и избирательно растворяющий соль камфарсульфоновой кислоты с соответствующим (2R,3R) энантиомером по сравнению с (2S, 3S)энантиомером. Примерами таких растворителей являются ацетонитрил, ацетон и этанол. Ацетонитрил предпочтительнее. Соль камфарсульфоновой кислоты и (2S, 3S)энантиомера, полученная в вышеописанном способе разделения, может быть необязательно репульпирована, как показано в части B, абзац 2 Примера, для улучшения оптической чистоты продукта.
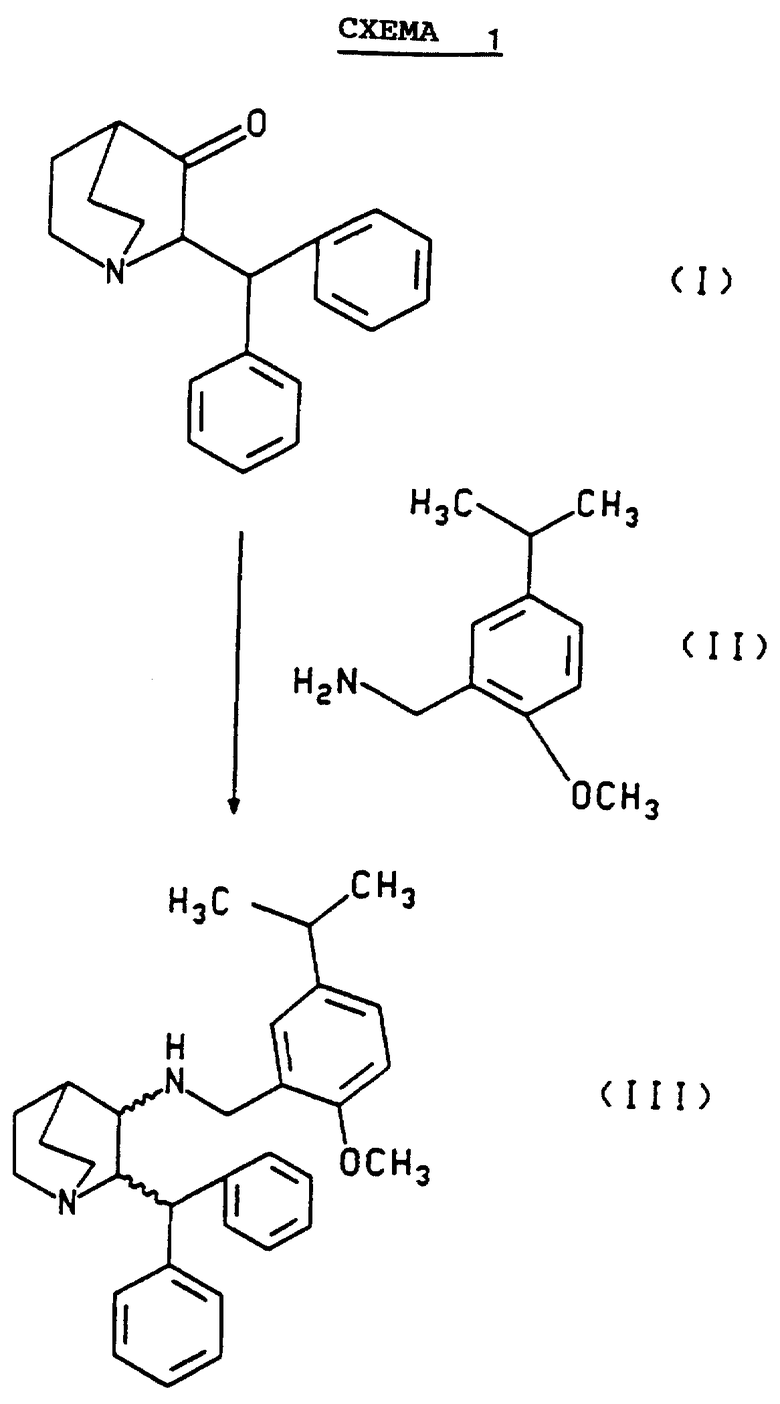
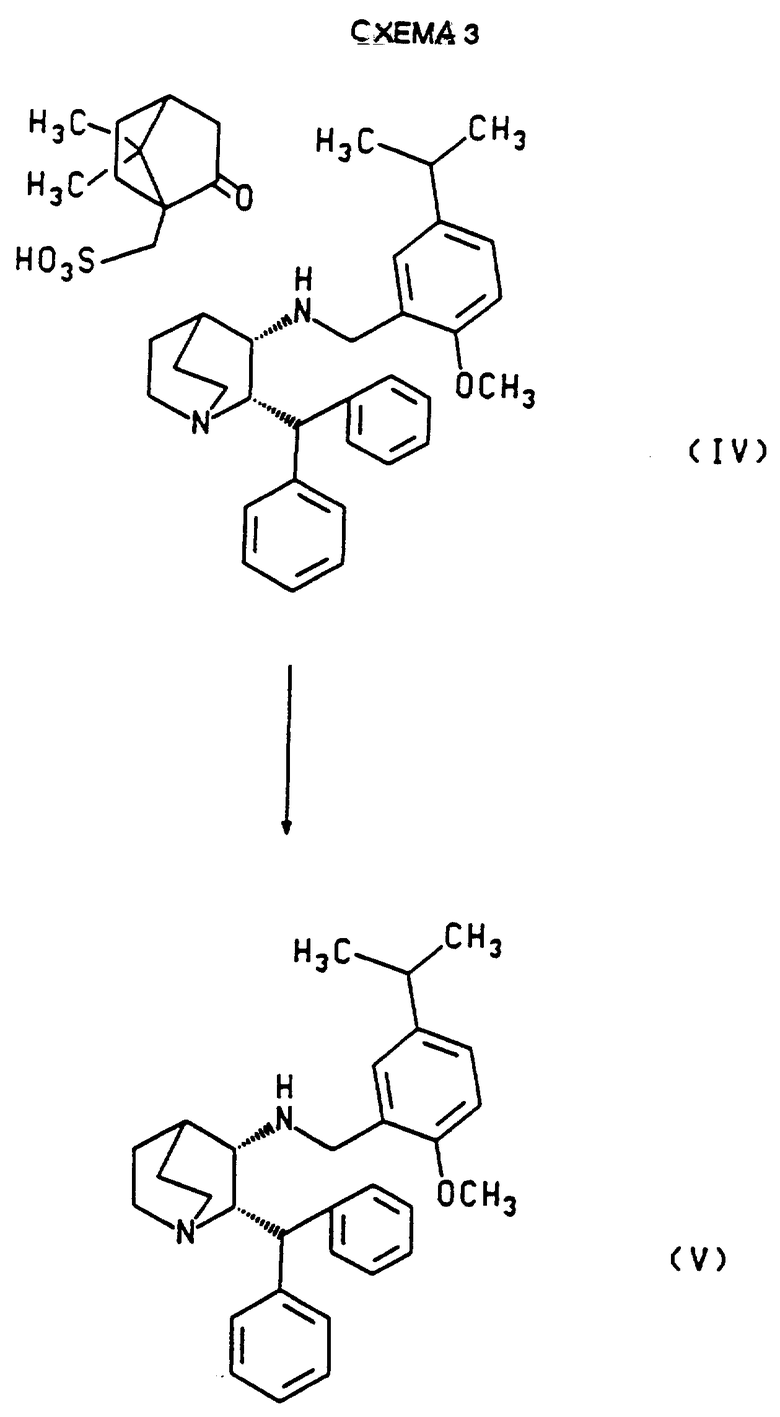
Схема 1, приведенная ниже, иллюстрирует метод, которым может быть получен рацемат. Схема 2, приведенная ниже, иллюстрирует разделение рацемата с получением соли камфарсульфокислоты (2S,3S)- энантиомера. Схема 3 иллюстрирует разложение соли камфарсульфокислоты (2S,3S)-энантиомера с образованием оптически активного свободного основания этого энантиомера.
Показанный на Схеме 1 рацемат может быть получен в соответствии со следующей двухстадийной методикой. Первая стадия включает дегидратацию соединения формулы 1 взаимодействием соединения формулы II в присутствии каталитического количества камфарсульфокислоты и осушающего агента или в аппарате, предназначенном для азеотропной отгонки образующейся воды (например, в присутствии молекулярных сит или в аппарате с ловушкой Дина-Старка) с получением иминового промежуточного соединения формулы VIII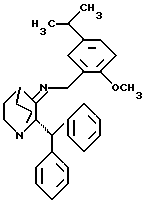
Подходящие растворители для этой реакции включают толуол, дихлорметан, бензол и ксилолы. Подходящие системы осушающий агент/растворитель включают сульфат магния, четыреххлористый титан/дихлорметан, изопропоксид титана/дихлорметан и молекулярные сита/ТГФ. Предпочтительным является сульфат магния. Если используется ловушка Дина-Старка, то предпочтительным растворителем является толуол. Эта реакция может проводиться при температуре от примерно 25oC до примерно 110oC. Предпочтительной температурой является температура кипения растворителя с обратным холодильником.
Примерами других катализаторов, которые могут использоваться вместо камфарсульфокислоты, являются метансульфокислота и паратолуолсульфокислота.
Иминовое промежуточное соединение может вступать в реакцию in situ (как описано в Примере) или после выделения с восстанавливающим агентом, таким как триацетоксиборгидрид натрия, цианоборгидрид натрия, боргидрид натрия, водород, и металлическим катализатором, цинком и соляной кислотой, борандиметилсульфидом или муравьиной кислотой с получением рацемата. Подходящие инертные растворители для этой реакции включают растворители, не содержащие кетоны, такие как низшие спирты (например, метанол, этанол и изопропанол), уксусная кислота, хлороформ, изопропиловый эфир, метиленхлорид, тетрагидрофуран (ТГФ), и смеси вышеперечисленных растворителей, например уксусная кислота в ТГФ или уксусная кислота в метиленхлориде. Реакцию обычно проводят при температуре от примерно 0oC до примерно 30oC, предпочтительно от примерно 0oC до примерно 10oC. Если в качестве восстанавливающего агента используется триацетоксиборгидрид, предпочтительно, чтобы растворителем не был низший спирт. Предпочтительно, восстанавливающим агентом является триацетоксиборгидрид и растворителем является уксусная кислота в ТГФ.
Стадия разделения оптических изомеров, которая показана на Схеме 2, включает реакцию 1-азабицикло [2.2.2] октан-3- амин, 2-(дифенилметил)-N-[[2-метокси-5-(1-метилэтил)-фенил]- метила] с 1R-(-)-10-камфарсульфокислотой в растворителе, способном растворять оба вышеназванных соединения и селективно (то есть, преимущественно) растворять соль камфарсульфокислоты соответствующего (2R, 3R)-энантиомера по сравнению с солью (2S,3S)-энантиомера, и перемешивание смеси с образованием оптически активной соли камфарсульфокислоты и (2S, 3S)-1-азабицикло [2.2.2] октан-3-амин, 2-(дифенилметил)-N-[[2-метокси-5-(1-метилэтил) фенил]метила]. Соль может быть затем выделена с использованием обычных методов (например, как описано в разделе B, параграф 1 Примера, перемешиванием в течение нескольких часов, выделением осадка фильтрованием, промывкой лепешки на фильтре и вакуумной сушкой).
Вышеописанное разделение предпочтительно проводят в атмосфере азота. Температура реакции может меняться от примерно 10oC до примерно 50oC, где более высокие температуры в этом интервале благоприятствуют улучшению оптической чистоты по сравнению с выходом, а температуры нижней границы интервала благоприятствуют лучшему выходу по сравнению с оптической чистотой.
Соль камфарсульфокислоты (2S,3S)-энантиомера, полученная вышеописанным способом разделения, может быть необязательно повторно пульпирована, как показано в разделе B, параграф 2 Примера, чтобы увеличить оптическую чистоту продукта.
Соль камфарсульфокислоты (2S,3S)-1-азабицикло [2.2.2] октан-3-амин, 2-(дифенилметил)-N-[ [2-метокси-5-(1-метилэтил)-фенил] метила] может быть также необязательно гидролизована, как показано на Схеме 3, чтобы получить свободное основание (2S,3S)-энантиомера. Такой гидролиз может быть осуществлен реакцией соли с подходящим щелочным агентом с использованием методов, хорошо известных специалистам. Например, оптически активный осадок может быть распределен между дихлорметаном и водным основанием, таким как гидроокись натрия или калия или карбонат калия, или спиртовый раствор осадка может быть перемешан с основной ионообменной смолой. Свободное основание, полученное в растворе, может быть затем выделено или переведено в раствор для получения соответствующей соли с соляной кислотой или другой желаемой соли присоединения кислоты.
Другой способ получения рацемата описан ниже. (Этот способ может быть также использован для получения (2S,3S) или (2R,3R) энантиомера).
Соединение формулы XI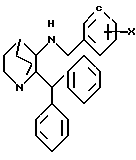
где X представляет водород или метокси, имеющее такую же абсолютную стереохимию, как желаемый продукт, подвергается гидролитическому удалению бензильной или метоксибензильной группы с получением соответствующего соединения формулы VI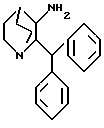
имеющего такую же желаемую стереохимию с последующей реакцией полученного таким образом вышеуказанного соединения с альдегидом формулы VII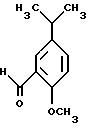
в присутствии восстанавливающего агента.
Гидролитическое удаление бензильной или метоксибензильной группы обычно проводят в присутствии сильной минеральной кислоты, такой как соляная, бромистоводородная или йодисто-водородная кислота, при температуре от комнатной до температуры кипения кислоты с обратным холодильником. Предпочтительно реакцию проводят в бромистоводородной кислоте при температуре кипения с обратным холодильником. Эту реакцию обычно проводят в течение около 2 часов.
Альтернативно, гидролитическое удаление бензильной или метоксибензильной группы по вышеописанной методике может быть заменено на гидрогенолитическое удаление этой группы. Гидрогенолитическое удаление обычно осуществляется с использованием водорода в присутствии металл-содержащего катализатора, такого как платина или палладий. Реакцию обычно проводят в реакционно инертном растворителе, таком как уксусная кислота или низший спирт, при температуре от примерно 0oC до примерно 50oC. Бензильная или метоксибензильная группа может быть удалена, альтернативно, обработкой соединения формулы II растворимым металлом, таким как литий или натрий или аммоний, при температуре от примерно -30oC до примерно 78oC, или солью муравьиной кислоты в присутствии палладия, или циклогексаном в присутствии палладия.
Предпочтительно бензильную или метоксибензильную группу удаляют обработкой соединения формулы XI водородом в присутствии гидроокиси палладия на угле в содержащем соляную кислоту метаноле при температуре около 25oC.
Полученное соединение формулы VI может быть превращено в желаемый рацемат (или энантиомер) реакцией с альдегидом формулы VII в присутствии восстанавливающего агента. Реакцию обычно проводят с использованием такого восстанавливающего агента как цианборгидрид натрия, триацетоксиборгидрид натрия, боргидрид натрия, водород и металлический катализатор, цинк и соляная кислота, диметилсульфидборан или муравьиная кислота при температуре от примерно -60oC до примерно 50oC. Подходящие инертные растворители для этой реакции включают растворители, не содержащие кетоны, такие как низшие спирты (например, метанол, этанол и изопропанол), уксусная кислота, хлороформ, изопропиловый эфир, метиленхлорид, тетрагидрофуран (ТГФ), и комбинации вышеперечисленных растворителей. Предпочтительно растворителем является метиленхлорид, температура около 25oC, и восстанавливающим агентом является триацетоксиборгидрид.
Альтернативно, реакция соединения формулы VI с соединением формулы VII может быть проведена в присутствии осушающего агента или с использованием аппарата, предназначенного для азеотропного удаления образующейся воды, чтобы получить имин формулы VIII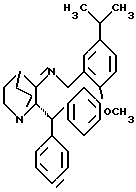
которое затем реагирует с восстанавливающим агентом, как описано выше, предпочтительно с триацетоксиборгидридом натрия при комнатной температуре. Получение имина обычно проводят в реакционно инертном растворителе, таком как бензол, ксилолы или толуол, предпочтительно в толуоле, при температуре от примерно 25oC до примерно 110oC, предпочтительно при температуре кипения растворителя с обратным холодильником. Подходящие системы осушающий агент/растворитель включают четыреххлористый титан/дихлорметан, изопропоксид титана/дихлорметан и молекулярные сита/ТГФ. Предпочтительным является четыреххлористый титан/дихлорметан.
Рацемат (и оба энантиомера) могут быть также получены из соединения формулы VI, имеющей такую же стереохимию, реакцией соединения формулы VI с соединением формулы IX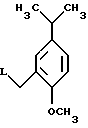
где L представляет подходящую отщепляемую группу (например, хлоро, бромо, йодо или мезилатную группу).
Эту реакцию обычно проводят в реакционно инертном растворителе, таком как дихлорметан или ТГФ, предпочтительно в дихлорметане, при температуре от примерно 0oC до примерно 60oC, предпочтительно при примерно 25oC.
Рацемат (и оба энантиомера) могут быть также получены из соединения формулы VI, имеющей такую же стереохимию, реакцией соединения формулы VI с соединением формулы X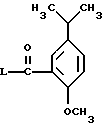
где L является такой, как описано выше, или представляет имидазол,
с последующим восстановлением полученного амида. Эту реакцию обычно проводят в реакционно инертном растворителе, таком как ТГФ или дихлорметан, при температуре от примерно -20oC до примерно 60oC, предпочтительно в дихлорметане при примерно 0oC. Восстановление полученного в результате амида осуществляют обработкой восстанавливающим агентом, таким как комплекс борандиметилсульфид, литийалюминийгидрид или диизобутилалюминийгидрид в инертном растворителе, таком как этиловый эфир или ТГФ. Температура реакции может меняться от примерно 0oC до температуры кипения растворителя с обратным холодильником. Предпочтительно восстановление осуществляют с использованием комплекса боран-диметилсульфид в ТГФ при примерно 60oC.
Рацемат и (2S, 3S)-энантиомер являются основными по природе и поэтому способны образовывать целый ряд различных солей с разнообразными неорганическими и органическими кислотами. Хотя такие соли могут быть фармацевтически приемлемыми для введения животным, на практике часто желательно вначале выделить активное соединение из реакционной смеси в виде фармацевтически неприемлемой соли, затем просто превратить последнюю опять в соединение в виде свободного основания обработкой щелочным реагентом, после чего превратить это свободное основание в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислот рацемата и (2S,3S)-энантиомера могут быть легко получены обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в водной среде растворителей или в подходящем органическом растворителе, таком как метанол или этанол. Желаемая твердая соль легко получается при осторожном выпаривании растворителя.
Рацемат и (2S,3S)-энантиомер и их фармацевтически приемлемые соли (ниже называемые также "активными соединениями") проявляют связывающую активность по отношению к рецептору вещества P и поэтому имеют ценность для лечения и профилактики клинических состояний или заболеваний млекопитающих, включая человека, на лечение или профилактику которых можно воздействовать или облегчать снижением нейротрансмиссии, медиируемой веществом P. Такие состояния включают воспалительные заболевания (например, артриты, псориаз, астма и воспалительное заболевание кишечника) беспокойство, депрессию или дистимические заболевания, колиты, рвоты, психозы, боли, аллергии, такие как экзема и риниты, хронические обструктивные заболевания дыхательных путей, заболевания, связанные с повышенной чувствительностью, такие как вызванные ядовитым плющом, гипертония, заболевания, связанные со спазмом сосудов, такие как грудная жаба, мигрень и болезнь Рейно (Reynaud), фиброзы и колагенные заболевания, такие как склеродерма и эозинофильный фасциолазис (eosinophilic fascioliasis), рефлексосимпатическую дистрофию, такую как синдром плечо/рука, болезни, связанные с привыканием, такие как алкоголизм, связанные со стрессом соматические заболевания, периферийную невропатию, невралгию, невропатологические заболевания, такие как болезнь Альцгеймера, связанное со СПИД'ом слабоумие, диабетическая невропатия и множественный склероз, солнечные ожоги, удар, глазные болезни, болезни, связанные с усилением или подавлением иммунной системы, такие как системная красная волчанка, заболевания, вызванные или медиируемые ангиогенезисом, или для которых ангиогенезис является симптомом, и ревматические заболевания, такие как фиброзиты.
Активные соединения могут быть введены перорально, парентерально или местно. В общем, эти соединения наиболее желательно вводить в дозах в интервале от 0,5 мг до 500 мг в сутки, хотя могут меняться в зависимости от веса и состояния субъекта и конкретно выбранного способа введения. Изменения могут иметь место в зависимости от вида животного и от его индивидуальной реакции на указанное лекарство, а также от выбранного типа фармацевтической препаративной формы, периода времени и интервалов, при которых проводят введение лекарства. В некоторых случаях уровень дозы ниже нижнего предела вышеуказанного интервала может быть более приемлем, тогда как в других случаях только большие дозы могут применяться без каких-либо вредных побочных эффектов, при условии, что такие большие дозы первоначально делят на несколько маленьких доз для введения в течение суток.
Активные соединения могут вводиться в чистом виде или в комбинации с фармацевтически приемлемыми носителями или разбавителями любым из трех ранее указанных способов, и такое введение может быть проведено в виде одной или нескольких доз. Более предпочтительно такие соединения могут быть введены в виде многочисленных различных дозированных форм, например они могут быть соединены с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, лепешек, драже, конфет, порошков, спреев, кремов, мазей, свечей, желе, гелей, паст, лосьонов, притираний, водных суспензий, растворов для инъекций, эликсиров, сиропов и тому подобное. Такие носители включают твердые разбавители или наполнители, стерильную водную среду и различные нетоксичные органические растворители, и т.п. Кроме того фармацевтические композиции для перорального применения могут быть подходящим образом подслащены и/или ароматизированы. В общем случае активное соединение или его фармацевтически приемлемая соль присутствуют в таких дозированных формах в диапазоне концентраций от 5,0% до 70% по массе.
Для перорального введения таблетки, содержащие различные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальцийфосфат и глицин, могут применяться с различными разрыхлителями, такими как крахмал (предпочтительно кукурузный, картофельный крахмал или крахмал из тапиоки), альгиновая кислота и некоторые сложные силикаты, вместе со связывающими агентами для грануляции, такими как поливинилпирролидон, сукроза, желатина и камедь. Кроме того, смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк, часто оказываются очень полезными для целей таблетирования. Твердые композиции подобного типа могут также применяться как наполнители желатиновых капсул; предпочтительные материалы включают также лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Если для перорального введения желательны водные суспензии и/или эликсиры, активный ингредиент может быть объединен с различными подслащивающими или ароматизирующими агентами, окрашивающими веществами или красителями и, если требуется, с эмульгирующими и/или суспендирующими агентами, а также с такими разбавителями как вода, этанол, пропиленгликоль, глицерин и различными их комбинациями.
Для парентерального введения могут применяться растворы активного соединения в конопляном или арахисовом масле или в водном пропиленгликоле. Водные растворы должны быть соответствующим образом буферированы (предпочтительно pH больше 8), если это необходимо, и жидкий разбавитель прежде всего должен быть изотоничным. Такие водные растворы являются подходящими для внутривенных инъекций. Масляные растворы являются подходящими для внутрисуставных, внутримышечных и подкожных инъекций. Приготовление всех этих растворов в стерильных условиях легко осуществляют с помощью стандартных фармацевтических методов, хорошо известных специалистам.
Кроме того, возможно также вводить активные соединения местно при лечении воспалительных заболеваний кожи и это может, предпочтительно, осуществляться с помощью кремов, желе, гелей, паст, мазей и т.п., в соответствии со стандартной фармацевтической практикой.
Активность активных соединений в качестве антагонистов рецептора вещества P может быть определена по их способности ингибировать связывание вещества P по его рецепторным сайтам в тканях бычьего хвоста с использованием радиоактивных лигандов, чтобы сделать видимыми рецепторы тахикинина с помощью авторадиографии. Антагонистическая активность таких соединений по отношению к веществу P может быть оценена с использованием стандартного метода анализа, описанного М.А. Cascieri et al. в Journal of Biological Chemistry, Vol. 258, p. 5158 (1983). Этот метод в основном включает определение концентрации активного соединения изобретения или его фармацевтически приемлемой соли, необходимой для того, чтобы снизить на 50% количество радиоактивномеченых лигандов вещества P по их рецепторным сайтам в указанной выделенной ткани коровы, тем самым позволяя получить характеристические значения IC50 для испытуемого соединения.
По этой методике ткань бычьего хвоста извлекают из -70oC морозильника и гомогенизируют в 50 объемах (масс./об.) охлажденного льдом 50 мМ Трис (т.е. триметиламина, которым является 2-амино-2-гидроксиметил-1,3- пропандиол) солянокислого буфера, имеющего pH 7,7. Гомогенат центрифугируют при 30.000хG в течение 20 минут. Осадок вновь суспендируют в 50 объемах Трис-буфера, гомогенизируют и затем вновь центрифугируют при 30.000хG еще двадцать минут. Осадок затем повторно суспендируют в 40 объемах охлажденного льдом 50 мМ Трис-буфера (pH 7,7), содержащего 2 мМ хлорида кальция, 2 мМ хлорида магния, 40 г/мл бакитракина, 4 мкг/мл лейпептина, 2 мкг химостатина и 200 г/мл бычьего сывороточного альбумина. Эта стадия завершает приготовление препарата ткани.
Радиолигандное связывание проводят затем следующим образом, а именно, инициированием реакции путем добавления 100 мкл испытуемого соединения, приготовленного в концентрации 1 мкМ, последующего добавления 100 мкл радиоактивного лиганда, приготовленного с конечной концентрацией 0,5 мМ, и в конце добавления 800 мкл препарата ткани, получение которого описано выше. Конечный объем составляет 1,0 мл. Реакционную смесь затем взбалтывают и инкубируют при комнатной температуре (20oC) в течение 20 минут. Содержимое пробирок затем фильтруют, используя сборщик клеток (cell harvester) и фильтры из стекловолокна (Whatman GF/B), промывают четыре раза 50 мМ Трис-буфера (pH 7,7), причем фильтры предварительно пропитывают в течение двух часов перед процедурой фильтрации. Радиоактивность затем определяют с помощью счетчика бета-частиц при 53%-ной эффективности счета, и вычисляют величину IC50, используя стандартные статистические методы.
Антипсихотическая активность активных соединений, как нейролептических агентов для лечения различных психических расстройств, может быть определена, в первую очередь, изучением его способности подавлять вызванную веществом P или вызванную агонистом вещества P гиперподвижность у морских свинок. Этот опыт проводят следующим образом: вначале морским свинкам вводят дозу контрольного соединения или соответствующее тестируемое соединение по настоящему изобретению, затем морским свинкам вводят вещество P или агонист вещества P путем внутримозгового введения через канюлю, после чего измеряют их индивидуальную локомоторную реакцию на указанный стимул.
Настоящее изобретение иллюстрируется далее Примером. Понятно, однако, что изобретение не ограничивается конкретными деталями этого примера.
Пример.
A. 1-азабицикло [2 .2.2] октан-3-амин, 2-(дифенилметил) -N-[[2-метокси-5-(1-метилэтил)-фенил]метил].
В трехгорлую 125 см3 колбу, снабженную механической мешалкой, подводом для азота, ловушкой Дина-Старка и обратным холодильником, загружали 10 г 2-дифенилметил-1-азабицикло- [2.2.2]октан-3-оксида (34,3 ммоль, 1 эквивалент), 6,89 г 1-метокси-2- аминометил-4-изопропилбензола (38,43 ммоль, 1,12 эквив.), 16 мг 1R-(-) -10-камфарсульфоновой кислоты (0,069 ммоль, 0,002 эквив. ) и 45 см3 толуола. Полученную суспензию нагревали на масляной бане до кипения с обратным холодильником (110oC). Реакционную смесь кипятили с обратным холодильником в течение 3 часов, и приблизительно 0,6 см3 воды собиралось в ловушке Дина-Старка. Смеси давали остыть до комнатной температуры и перемешивали в течение 14 часов. Смесь переносили в одногорлую колбу и упаривали на роторном испарителе до объема примерно 24 см3. Концентрат добавляли по каплям в 200 см3 трехгорлую колбу, снабженную механической мешалкой, термометром и подводом для азота и содержащую 18,18 г (85,77 ммоль, 2,5 эквив.) триацетоксиборгидрида натрия и 10,3 г (171,55 ммоль, 5 эквив.) уксусной кислоты в 60 см3 тетрагидрофурана, предварительно охлажденного на бане лед/вода до 0oC. Добавление толуольного концентрата завершали за 7 минут, и температура внутри колбы достигала +10oC.
Ледяную баню удаляли, полученной гетерогенной реакционной смеси давали нагреться до комнатной температуры (24oC) и перемешивали в течение 14 часов. За ходом реакции следили с помощью тонкослойной хроматографии (ТСХ), используя 100%-ный этилацетат и смесь этилацетат/метанол (2/1).
Реакционную смесь затем упаривали на роторном испарителе до объема примерно 40 см3 и затем разбавляли 150 см3 дихлорметана. К смеси добавляли 200 см3 воды при перемешивании с помощью магнитной мешалки и полученную смесь перемешивали 15 минут. Показатель pH смеси составлял 4,0, и его доводили до pH 11,0, добавляя по порциям 25%-ный водный раствор гидроокиси натрия. Затем органический и водный слои разделяли и основной органический слой экстрагировали (1х70 см3) дихлорметаном, после чего объединенные органические слои сушили над безводным сульфатом магния в течение одного часа. Осушающий агент отфильтровывали и фильтрат выпаривали на роторном испарителе до объема примерно 100 см3. К этому концентрату добавляли 160 см3 2-пропанола и смесь вновь выпаривали на роторном испарителе до объема примерно 100 см3. Конечный концентрат перемешивали с помощью магнитной мешалки при комнатной температуре, и спустя 15 минут образовывался белый осадок. Суспензия гранулировалась в течение 2 часов. Белое твердое вещество отфильтровывали и лепешку на фильтре промывали 2-пропанолом и сушили в вакууме, получая 7,68 г (выход 49%) соединения, указанного в заголовке. Температура плавления = 111-115oC.
Анализ ЖХВР твердого вещества проводили на жидкостном хроматографе Hewlett Packard series 2, используя колонку Zorbax CN, УФ-детектор 203 нм и подвижную фазу из 55% ацетонитрила и 45% воды (с 0,1% H3PO4 + 0,2% триэтиламина (ТЭА)) при скорости потока 1 мл/мин. Анализ показал, что присутствует только транс-диастереомер с чистотой 90%.
B. (2S, 3S)-1-азабицикло[2.2.2] октан-3-амин, 2- (дифенилметил)-N-[[2-метокси-5-(1-метилэтил)фенил]метил], соль (1R)-(-)-10-камфарсульфокислоты
В трехгорлую 125 см3 колбу, снабженную магнитной мешалкой и подводом для азота, загружали 5,11 г 1-азабицикло-[2.2.2] октан-3-амин, 2-(дифенилметил)-N-[[2-метокси-5-(1-метилэтил)фенил] метила] (11,24 ммоль, 1 эквивалент) и 51 см3 ацетонитрила, чтобы получить частичную суспензию. Затем добавляют 2,61 г (1R)-(-)-10- камфарсульфоновой кислоты (11,24 ммолей, 1 эквивалент) одной порцией и реакционная масса становится гомогенной. После перемешивания при комнатной температуре в течение 5 минут образовывался осадок. После этого добавляли еще 5 см3 ацетонитрила и реакционную смесь перемешивали в течение 4 часов. Твердый осадок отфильтровывали, лепешку промывали (2х6 см3) ацетонитрилом и сушили в вакууме, получая 2,97 г белого твердого вещества (общий выход 38,5%, выход желаемой энантиомерной соли 77%). Температура плавления = 177-182oC.
Анализ ЖХВР сырой соли (2,97 г) проводили на колонке Chrom Tech Chiral-AGP. Подвижная фаза - 0,01М KH2PO4 (pH 5,5):ацетонитрил (85:15 об/об). Определение проводилось в УФ-свете, 229 нм, скорость потока 1 мл/мин, объем впрыскивания 20 мкл. Анализ показал содержание 95,7% желаемого энантиомера и 4,3% нежелательного энантиомера.
2,87 г вышеописанной неочищенной соли и 20 см3 ацетонитрила загружали в 35 см3 колбу, снабженную магнитной мешалкой, и образовавшуюся суспензию перемешивали при комнатной температуре в течение 5 часов. Твердую фазу затем отфильтровывали, промывали ацетонитрилом (2х3 см3) и затем сушили в вакууме, получая твердое белое вещество. Вес = 2,8 г (извлечение 97% масс.). Температура плавления = 180-185oC.
Анализ ЖХВР репульпированной соли (2,8 г) проводили на колонке Chrom Tech Chiral-AGP. Подвижная фаза - 0,01М KH2PO4 (pH 5,5):ацетонитрил (85:15 об/об). Определение проводили в УФ-свете, 229 нм, скорость потока 1 мл/мин, объем впрыскивания 20 мкл. Анализ показал содержание 96,6% желаемого энантиомера и 3,4% нежелательного энантиомера.
Оптическое вращение репульпированной соли измеряли на поляриметре Perkin Elmer 241, с использованием источника света Натрий 589. Репульпированную соль (44,9 мг) растворяли в 10 см3 метанола и использовали 5 см3 для заполнения ячейки 10 дм.
[α]
C. (2S, 2S)-1-азабицикло[2.2.2] октан-3-амин, 2- (дифенилметил)-N-[[2-мeтoкcи-5-(1-метилэтил)фенил]метил]
В 100 см3 колбу Эрленмейера, снабженную магнитной мешалкой, загружали 2,63 г (3,83 ммоль) репульпированной соли с вышеописанной стадии B, 32 см3 дихлорметана и 16 см3 воды, получая гомогенный двухфазный раствор. Показатель pH водного слоя составлял 4,0, и его доводили до pH 11,0, добавляя по каплям 25%-ный водный раствор гидроокиси натрия. После подщелачивания два слоя перемешивали в течение 15 минут. Слои разделяли, органический слой промывали (1х16 см3) водой, слои разделяли, органический слой сушили над безводным сульфатом натрия один час и осушающий агент отфильтровывали. Органический слой перемешивали до получения смеси пена/масло, которая кристаллизовалась при стоянии при комнатной температуре в течение двух дней. Вес = 1,659 г (выход 95,3%). Температура плавления = 100-103oC.
Анализ хиральной ЖХВР проводили на колонке Chrom Tech Chiral-AGP (100 мм x 4,0 мм, 5 мкм). Подвижная фаза - 0,01М KH2PO4 (pH 5,5):ацетонитрил (85:15 об/об). Определение проводили в УФ-свете, 229 нм, скорость потока 1 мл/мин, объем впрыскивания 20 мкл.
Анализ показал содержание 99,5% желаемого энантиомера и 0,5% нежелательного энантиомера.
Анализ чистоты по ЖХВР проводили на колонке Zorbax Rx C-8 (15 см x 4,6 мм внутр. диаметр). Подвижной фазой являлась смесь ацетонитрил: вода: триэтиламин: фосфорная кислота (650: 350: 3:1 об/об). Определение проводили в УФ-свете, 229 нм, скорость потока 2 мл/мин, объем впрыскивания 20 мкл. Анализ показал чистоту продукта 99,5%.
Оптическое вращение оптически активного свободного основания, конечного продукта, измеряли на поляриметре Perkin Elmer 241, с использованием источника света Натрий 589. Соединение (52,4 мг) растворяли в 10 см3 метанола и использовали 5 см3 раствора для заполнения ячейки длиной 10 дм.
[α]
Разделение рацемического (±)-(2R, 3R, 2S, 3S)- 1-азабицикло-[2,2,2] октан-3-амин, 2-(дифенилметил)-N-[[2-метокси-5-(1-метилэтил)фенил] метила] включает взаимодействие указанного рацемата с 1R-(-)-10-камфарсульфоновой кислотой в растворителе и получение 2S, 3S-энантиомера селективным осаждением и выделение его в виде камфарсульфокислотной соли общей формулы I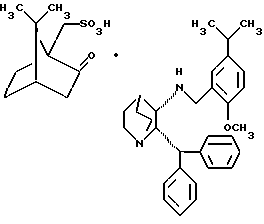
или в виде свободного основания. 2 с. и 8 з.п. ф-лы.
отличающийся тем, что включает 1) реакцию указанного рацемата с 1R-(-)-10-камфарсульфоновой кислотой общей структурной формулы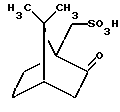
в растворителе и 2) получение 2S,3S-энантиомера, по существу, селективным осаждением и выделением его камфарсульфокислотной соли общей структурной формулы (IV)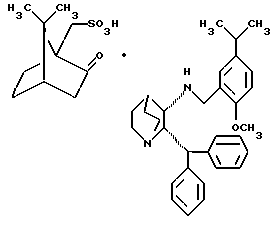
или в виде свободного основания.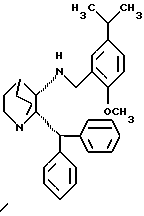
3. Способ по п.1, в котором указанный растворитель представляет собой ацетонитрил.
включающий:
А. Реакцию рацемического (±) - (2S,2R)-1-азабицикло-[2,2,2]октан-3-оксид, 2-дифенилметила общей структурной формулы I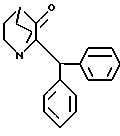
с 1-метокси-2-аминометил-4-изопропилбензолом общей структурной формулы II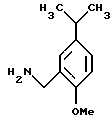
с получением рацемического (±) - (2R,3R,2S,3S)-1-азабицикло-[2,2,2]октан-3-амин, 2-(дифенилметил)-N-[[2-метокси-5-(1-метилэтил)фенил]метила] общей структурной формулы III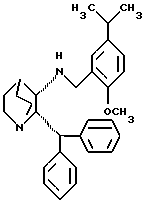
В. разделение указанного рацемата, полученного на предыдущей стадии, реакцией его с 1R-(-)-10-камфарсульфокислотой общей структурной формулы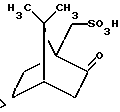
в системе растворителя, способного растворять реагенты указанного способа, включающего рацемат и камфарсульфокислоту, в то же время селективно растворяя только полученную оптически активную соль камфарсульфокислоты указанного (2R, 3R) энантиомера рацемата, таким образом проводя выделение (2S, 3S) энантиомера из рацемата путем его осаждения и извлечения в виде оптически активной соли камфарсульфокислоты общей структурной формулы IV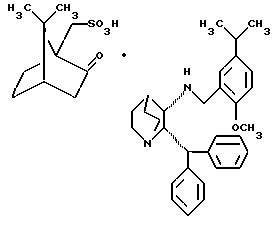
и
С. гидролиз указанной оптически активной соли, которая осадилась из раствора с получением указанного (2S,3S) энантиомера в виде свободного основания общей структурной формулы V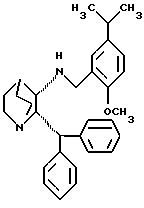
9. Способ по п. 8, в котором после указанного выделения осаждением и извлечения и перед вышеуказанным гидролизом до свободного основания (2S,3S) энантиомер дополнительно очищают до чистоты по меньшей мере 96,6%.
| 5162339, 1992 | |||
| Способ получения производных пиридина | 1985 |
|
SU1507211A3 |

