Настоящее изобретение направлено на нейрозащитные (противоишемические и блокирующие стимуляторные аминокислотные рецепторы) 2-пиперидино-1-алканольные производные, определенные формулами (I), (II) и (III), представленными ниже; фармацевтически приемлемые соли их; способ использования этих соединений при лечении приступов или дегенеративных заболеваний центральной нервной системы (ЦНС), таких как болезнь Альцгеймера, болезнь Хантингтона и болезнь Паркинсона; и на некоторые промежуточные производные для них.
Ифенпродил является рацемическим, так называемым dl-эритросоединением, имеющим относительную стереохимическую формулу ---(A) который подается в качестве гипотензивного агента, разделяя полезность с рядом близких аналогов; Carron et al, патент США 3509164; Carron и др., Drug Res., т.21, с. 1992-1999 (1971). Совсем недавно было показано, что ифенпродил обладает противоишемической активностью блокирования, стимулирующей аминокислотные рецепторы; Gotti и др. , J. Pharm. EXр Therap; том 247, с. 1211-21 (1988); то же, с. 1222-32 (1988). Целью в значительной степени достигнутой настоящим изобретением, был поиск соединений, обладающих таким нейрозащитным эффектом в удовлетворительной мере, и в то же время имеющих пониженный или никакого значительного гипотензивного эффекта.
---(A) который подается в качестве гипотензивного агента, разделяя полезность с рядом близких аналогов; Carron et al, патент США 3509164; Carron и др., Drug Res., т.21, с. 1992-1999 (1971). Совсем недавно было показано, что ифенпродил обладает противоишемической активностью блокирования, стимулирующей аминокислотные рецепторы; Gotti и др. , J. Pharm. EXр Therap; том 247, с. 1211-21 (1988); то же, с. 1222-32 (1988). Целью в значительной степени достигнутой настоящим изобретением, был поиск соединений, обладающих таким нейрозащитным эффектом в удовлетворительной мере, и в то же время имеющих пониженный или никакого значительного гипотензивного эффекта.
Также известно, что некоторые структурно-родственные 1-фенил-3-(4-арил-4-ацилоксипиперидино)-1-пропанолы являются полезными в качестве анальгетиков (патент США 3294804), и что 1-4-(амино- и гидроксиалкил)фенил-2-(4-гидрокси-4-толилпиперазино)-1-алканолы и алканоны обладают анальгетической противоги- пертензивной, психотропной или противовоспалительной активностью.
Настоящее изобретение направлено на соединения формулы:
 где R - H, C1-C6-алкил, С2-С6-алкенил или С2-С6-алкинил; Х - водород. С1-С3-алкил, галоид, OR1, OCOR1, CO2R1, SR1, NHR1, NHCOR1, CONH2 или CN;
где R - H, C1-C6-алкил, С2-С6-алкенил или С2-С6-алкинил; Х - водород. С1-С3-алкил, галоид, OR1, OCOR1, CO2R1, SR1, NHR1, NHCOR1, CONH2 или CN;
R1 - водород или C1-C3-алкил;
Q - сера или СН=СН;
Y и Y1 взяты вместе и представляют
=CH(CH2)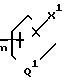
или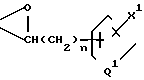
или Y и Y1 взяты отдельно, и
Y - водород или ОН, и Y1 - соединение ф-лы
-(CH2)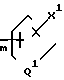
или Y представляет водород, и Y1 представляет собой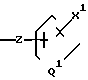
n - 0, 1, 2 или 3;
m - 0, 1, 2, 3 или 4;
Q1 представляет независимо значения Q, определенные выше;
Х1 представляет независимо значения Х, определенные выше;
представляет 0, S, SO или SO2;
соединения формулы: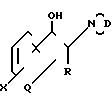

где D представляет собой радикал формул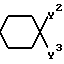 ,
,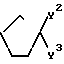 или
или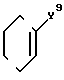
Y2 и Y3 взяты вместе, и представляют
=CH(CH2)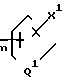
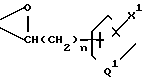
Y2 и Y3 взяты отдельно, и Y2 представляет собой ОН, а Y3представляет радикал формулы
-(CH2)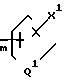
Y9 представляет собой
-(CH2)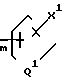
и R, R1, X, X1, Q, Q1, n и m имеют значения, определенные выше, и соединения формулы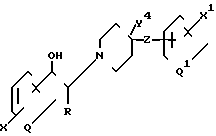
 где Y4 представляет собой Н; и
где Y4 представляет собой Н; и
R, R1, Q, Q1, X, X1 и Z имеют значения, определенные выше;
и фармацевтически приемлемые аддитивные соли кислот и этих соединений.
Выражение "фармацевтически приемлемые кислотно-аддитивные соли" или "аддитивные соли кислот" включает, но не ограничивается ими, такие соли как хлоргидрат, бромгидрат, йодгидрат, нитрат, кислый сульфат, первичный кислый фосфат, мезилат, малеат и сукцинат. Также соли обычно получаются при реакции формы свободного основания соединения (I), (II) или (III) с соответствующей кислотой, обычно с одним молярным эквивалентом, и в растворителе. Те соли, которые непосредственно не выпадают в осадок, обычно выделяются с помощью концентрирования растворителя и/или добавления нерастворителя.
Предпочтительные соединения настоящего изобретения обычно имеют заместитель R в виде метила и обладают 1S*, 2S* или трео относительной стереохимией в 1- и 2-положениях пропанольной цепи, т.е.

Кроме того, независимо от значения R, предпочтительными соединениями настоящего изобретения являются соединения формул (I) или (II), имеющие Y и Y1 или Y2 и Y3, взятые отдельно, дополнительно имеющие Y или Y2 в виде ОН, или Y1 в виде Z/C4H3Q1/X1; или соединения формулы (III). Предпочтительным значением Z во всех случаях является S.
Настоящее изобретение также направлено на фармацевтические композиции и способы лечения млекопитающих, особенно людей, страдающих расстройствами центральной нервной системы, которые предусматривают назначение указанным млекопитающим нейрозащитного эффективного количества соединения формулы (I), (II) или (III). Указанные композиции и способы являются особенно ценными при лечении ударов, болезни Альцгеймера, болезни Паркинсона, болезни Хантингтона и расстройств, связанных с центральной нервной системой.
Настоящее изобретение дополнительно направлено на промежуточные соединения формулы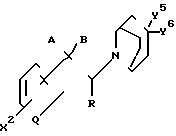 где А и В взяты вместе, и представляют собой кислород, образуя вместе с углеродом, к которому они прикреплены, карбонильную группу, или А и В взяты отдельно, и А является водородом, а R - гидрокси;
где А и В взяты вместе, и представляют собой кислород, образуя вместе с углеродом, к которому они прикреплены, карбонильную группу, или А и В взяты отдельно, и А является водородом, а R - гидрокси;
Х2 - водород, (С1-С3)алкил, галоид, OR1, OR2, COOR1, OCOR1, SR1, SR2, NHR1, NR1R3, NHСOR1, CONH2 или CN;
R1 - водород или (С1-С3)алкил;
R2 - обычная защитная группа гидроксила или меркаптана;
R3 представляет собой общепринятую аминозащищающую группу;
Y5 и Y6 взяты вместе, и представляют собой группу
=CH(CH2)
или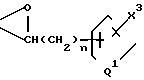
Y5 и Y6 взяты отдельно, и
Y5 представляет собой водород или ОН, и Y6 представляет
-(CH2)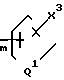
Y5 представляет водород, и Y6 представляет группу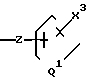
X3 представляет собой независимо значения радикала Х2, определенные выше; при условии, что когда А и В взяты отдельно (по крайней мере один из Х2 и Х3 представляет собой OR2, SR2 или NR1R3;
Z представляет собой 0, S, SO или SO2; или
R, Q, Q1 n и m имеют значения, определенные выше;
промежуточные соединения формулы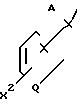 B
B
где Е представляет собой группу ,
, или
или ;
;
Y7 и Y8 взяты вместе и представляют собой
=CH(CH2) или
или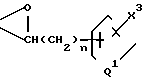 или
или
Y7 и Y8 взяты отдельно, и Y7 представляет собой ОН, а Y8 - группу формулы
-(CH2)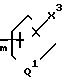
Y10 представляет собой группу формулы
-(CH2)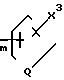
и А, В, R, R1, R2, R3, Q, Q1, X2, X3, n и m имеют значения, определенные выше, с тем же условием, касающимся А и В;
и промежуточное соединение формулы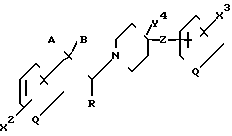
 где все группы имеют значения, определенные выше, при том же самом условии, касающемся А и В.
где все группы имеют значения, определенные выше, при том же самом условии, касающемся А и В.
Те соединения формулы (I) или (IV), указанные как эндо, имеют гидроксигруппу или оксирановый кислород на одной и той же стороне пиперидинового кольца, что и этиленовый мостик.
Очевидно можно заметить, что те соединения формулы (I) - (VI), которые являются 1-алканолами, обладают асимметричным С-1 атомом углерода, в то время как те соединения, в которых R является отличным от водорода, обладают еще одним асимметричным центром при С-2 углерода алканола. Аналогичным образом, в тех соединениях формул (VI) - (VI), которые являются 1-алканонами, в которых R является иным, чем водород, имеется С-2 асимметричный углерод. Специалистам в области органической химии должно быть очевидно, что такие соединения могут разделяться на оптические изомеры, показывающие равное, но противоположное вращение в плоскости поляризованного света. Например, все из этих соединений потенциально расщепляются с помощью фракционной кристаллизации их диастереомерных аддитивных солей с оптически активной кислотой, как проиллюстрировано примерами ниже; при этом спирты также потенциально расщепляются с помощью хроматографии или фракционной кристаллизации сложных эфиров, получаемых по реакции с активированными формами оптически активных кислот или с оптически активными изоцианатами. Альтернативно, оптически активные формы некоторых из настоящих соединений получаются с помощью взаимодействия соответствующего амина с оптически активным эпоксидом, как показано также в примерах, представленных ниже. Таким образом, настоящее изобретение не должно рассматриваться, как ограниченное рацемическими формами настоящих соединений.
Соединения настоящего изобретения, имеющие формулу (I), (II) и (III), определенные ниже, легко и обычно получаются с помощью нуклеофильного замещения с последующим восстановлением получающегося в результате кетона в спирт, как подробно изложено ниже.
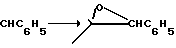
Предшествующие кетоны обычно сначала получаются с -ОН, SH и -NHR1-группами в защищенной форме, т.е. в виде -OR2, - SR2 или NR1R3-групп в соединениях формул (IV), (V) и (VI), где А и В взяты вместе в виде кислорода, образуя карбонильную группу. Такие защищенные кетоны обычно образуются с помощью нуклеофильного замещения соответствующим образом замещенного 2-галоид, 2-алкансульфонолокси- или 2-арилсульфонилокси-1-алканона, замещенным соответствующим образом пиперидиновым производным, например,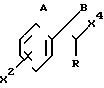 + H
+ H
где Х4 представляет собой типично хлор, бром, мезилокси или тозилокси. Данная реакция осуществляется в условиях, типичных для нуклеофильных замещений вообще. Когда два реагента являются примерно эквивалентными по доступности, могут использоваться количества в значительной степени близкие к молярным эквивалентам; хотя, когда один из них является более легкодоступным, обычно предпочитается использовать количество одного в избытке для того, чтобы способствовать данной биомолекулярной реакции для завершения ее в более короткий период времени. Реакция обычно осуществляется в присутствии по крайней мере 1 молярного эквивалента основания, самого производного пиперидина, если он является легкодоступным, но более обычно, третичного амина, который по крайней мере сравним по силе основания с нуклеофильным пиперидином; и в реакционно инертном растворителе, таком как этанол. При необходимости реакция катализируется добавлением до одного молярного эквивалента или более йодидной соли (например, NaI, KI). Температура не является критической, но обычно она является несколько повышенной для того, чтобы способствовать завершению реакции в более короткий период времени, но не такой высокой, чтобы приводить к чрезмерному разложению. Обычно удовлетворительной является температура 50-120оС, в удобном случае температура дефлегмации реакционной смеси.
В том смысле, как оно используется в предыдущем абзаце и далее в тексте, выражение "реакционно инертный растворитель" относится к любому растворителю, который не взаимодействует с исходными материалами, реагентами, промежуточными веществами или продуктами таким способом, который отрицательно воздействует на выход желаемого продукта.
Если необходимо, кетоновые промежуточные продукты, имеющие ОН-, SH- или NHR1-группы в защищенной форме (OR2, SR2 или NHR1R3), могут деблокироваться на данной стадии с помощью общепринятых методов. Например, когда R2 представляет собой триизопропилсилил или трет-бутилдиметилсилил, защитная группа удобным образом удаляется с помощью реакции с фтористым тетрабутиламмонием (обычно с 2 молярными эквивалентами) в реакционно инертном растворителе таком, как тетрагидрофуран. Когда R2 представляет бензил или R3 представляет бензилоксикарбонил, защитная группа обычно удаляется с помощью общепринятого гидрогенолиза над благородным металлически катализатором в реакционно инертном растворителе, например с использованием 10% Pd/C в качестве катализатора, предпочтительно при низких давлениях (например, 1-10 атм) и температурах (например, 20-75оС) и обычно в реакционно инертном растворителе, таком как метанол.
Обычно, исключая кетоновые промежуточные соединения, содержащие сложноэфирные группы или защитные группы, такие как бензилоксикарбонил (которые обычно удаляются перед восстановлением кетонов), и в иных отношениях с использованием или без предварительного удаления защитных групп, кетоновые промежуточные вещества удобным образом превращаются в соответствующие спирты с помощью обычного восстановления литийалюминийгидридом, обычно взятым в избытке (например, 1 моль на 1 моль), в реакционно инертном растворителе, таком как тетрагидрофуран, при пониженной температуре (например от -15 до 15оС). Альтернативно, кетоновые промежуточные соединения, особенно соединения, содержащие сложноэфирные группы, восстанавливаются более мягким гидридным восстанавливающим агентом, таким как NaBH4, снова - взятым в избытке, на сей раз в протонном растворителе, таком как метанол или этанол, обычно при несколько более высокой температуре, например 15-45оС.
Любые защитные группы, которые все еще имеются после восстановления кетона, затем удаляются согласно способам, описанным выше. Некоторые другие преобразования, такие как гидрирование олефинов, эпоксидирование и восстановление смесью натрий/жидкий аммиак эпоксидов, например
 ,
,
также необязательно осуществляются позднее в последовательности синтеза, например после сочетания с образованием кетона, после удаления защитных групп (если незащищенные группы не мешают преобразованиям) и/или после восстановления кетона в спирт.
Указанные реакции эпоксидирования легко выполняются, например, с помощью реакции метиленового соединения с одним молярным эквивалентом м-хлорнадбензойной кислоты в реакционном инертном растворителе, таком как СН2Сl2. Восстановление эпоксида в спирт легко достигается с помощью использования обычной смеси натрий/жидкий аммиак, обычно оно осуществляется при температурах ниже точки кипения жидкого аммиака (например, при -78оС, температуре бани из смеси ацетона и сухого льда) в присутствии реакционно инертного растворителя, такого как тетрагидрофуран.
Исходные вещества и реагенты, требуемые для синтеза соединений настоящего изобретения, являются легкодоступными, или получаемыми в промышленности, согласно методам, описанным в литературе, или по способам, показанным в примерах получения, приведенных ниже.
Настоящие соединения формул (I), (II) и (III) обладают селективно нейрозащитной активностью, основанной на их антиишемической активности и способности блокировать стимулирующие аминокислотные рецепторы, а при этом в то же самое время они обычно имеют пониженную, или вообще не имеют какой-либо значительной гипотензивной активности. Противоишемическая активность настоящих соединений определяется в соответствии с одним или более их методов, которые были подробно описаны в работах, приведенных выше, или с помощью аналогичных методов. Способность соединений настоящего изобретения блокировать стимулирующие аминокислотные рецепторы демонстрируется их способностью блокировать индуцированные N-метил-D-аспарагиновой кислотой (NMDA) повышения (элевации) cGMP в мозжечке неонатальных (новорожденных) крыс в соответствии со следующей процедурой. Мозжечки от 10 крыс Вистар в возрасте 8-14 дней быстро иссекали и помещали их в Кребс/бикарбонатный буфер с температурой 4оС, рН 7,4, а затем нарезали на кусочки размером 0,5 х 0,5 мм с использованием тканерезки Mc Нvain (фирмы "Никл Лаборатори Инжиниринг Ко"., Comshall, Surrey, Англия). Получающиеся кусочки мозжечка переносили в 100 мл Кребс/бикарбонатного буфера при 37оС, который непрерывно уравновешивали смесью 95: 5 02/СО2. Кусочки мозжечка инкубировали таким образом в течение 90 мин с тремя сменами буфера. Буфер затем декантировали, ткань центрифугировали (1 мин, 3200 об. /мин), и ткань повторно суспендировали в 20 мл Кребс/бикарбонатного буфера. Затем 250 мкл аликвоты (приблизительно 2 мг) удаляли и помещали в 1,5 мл микрофуги-трубки. В эти трубки добавляли 10 мкл исследуемого соединения из исходного раствора, с последующим добавлением после 10-минутного инкубационного периода 10 мкл 2,5 мМ раствора NMDA для начала реакции. Конечная концентрация NMDA составляла 100 мкМ. Контроль не содержит NMDA, т. е. NMDAне добавляли. Трубки (пробирки) инкубировали в течение 1 мин при 37оС на встряхиваемой водяной бане, а затем для прекращения реакции добавляли 750 мкл 50 мМ трис-Cl, 5 мМ ЕДТА раствора. Трубки помещали немедленно на кипящую водяную баню на 5 мин. Содержимое каждой пробирки затем подвергали обработке ультразвуком, в течение 15 с с использованием зондового гомогенатора, установленного на уровне энергии три. 10 мкл удаляли и белок определяли по методу Lowry, Anal. Biochem 100:201-220 (1979). Пробирки затем центрифугировали (5 мин, 10000х), 100 мкл поверхностного слоя удаляли и анализировали уровень циклического GMP (cGMP) с использованием анализа сGMP PIA New England nuclear (Бостон, Массачусетс), согласно методике поставщика. Данные сообщаются в виде п-мол, cGMP, генерируемого на 1 мг белка. С помощью известных методов также определяется нежелательная гипотензивная активность, например согласно методам Tarron и др., также приведенным выше.
Такая селективная нейрозащитная противоишемическая активность и активность блокирования стимуляторных аминокислотных рецепторов свидетельствуют о неоценимой полезности настоящих соединений при лечении дегенеративных ЦНС расстройств, таких как удар и болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона, не обладая при этом значительным потенциалом сопутствующего чрезмерного падения кровяного давления. При системном лечении этих заболеваний нейрозащитным количеством соединений формул (I), (II) или (III) доза в типичном случае составляет примерно 0,02 - 10 мг/кг/день (1-500 мг/день для человека, имеющего массу в типичном случае 50кг) в виде единственной или раздельных доз независимо от способа назначения. Конечно, в зависимости от точного вида соединения и конкретной природы индивидуальной болезни лечащими врачами могут предписываться дозы, находящиеся вне данного интервала. Обычно предпочитается оральный метод назначения препарата. Однако, если пациент не может проглатывать, или оральная абсорбция по каким-либо причинам нарушается, предпочтительным методом назначения будет парэнтеральный (внут- римышечный, внутривенный) и топический или местный способ назначения.
Соединения настоящего изобретения обычно назначаются в форме фармацевтических композиций, включающих по крайней мере одно из соединений формул (I), (II) или (III) вместе с фармацевтически приемлемым носителем или разбавителем. Такие композиции обычно формируются обычным образом с использованием твердых или жидких носителей или разбавителей, в зависимости от того, что соответствует способу необходимого назначения: для орального назначения - в форме таблеток, твердых или мягких желатиновых капсул, суспензий, грану, порошков и т.д., для парэнтерального назначения - в форме инъецируемых растворов или суспензий и т.д.; и для топического назначения - в форме растворов, лосьонов, мазей и т.д.
Настоящее изобретение иллюстрируется с помощью следующих примеров, но оно не ограничивается подробностями этих примеров.
Все неводные реакции проводили в атмосфере азота для удобства и, обычно, для увеличения выходов до максимума. Все системы растворители/разбавители подвергались сушке в соответствии со стандартными описанными в литературе процедурами, или приобретались на рынке в заранее высушенном виде. Все реакционные смеси перемешивали или магнитным, или механическим способом. ЯМР-спектры регистрируются при 300 МГц и приводятся в млн. долях. Растворителем ЯМР был CDCl3, если не указан иной. ИК-спектры даются в см-1, обычно указывающих только сильные сигналы.
П р и м е р 1. 2-(3-Фенилметилен-8-азабицикло/3-2-1/окт-8-ил)-1- [4-(триизопропилсилилокси)фенил]-1-пропанон
Смесь 3-фенилметилен-8-азабицикло/3.2.1/октана (1,09 г, 4,87 ммоль), 4-(триизопропилсилокси)- α-бромпропиофенона (1,88 г, 4,88 ммоль) и триэтиламина (1,5 мл, 10,76 ммоль) в этаноле (75 мл) нагревали с обратным холодильником в течение 22 ч. После охлаждения добавляли эфир (50 мл) и смесь фильтровали через диатомовую землю. Фильтрат концентрировали и хроматографировали на силикагеле (2 х 6 дюймов,/ 2,53 х 6 см), гексан, затем этилацетат (гексановый градиент), получая 0,56 г (23%) оранжевого продукта в виде масла;
ЯМР 8,18 (д, 2Н), 7,25 (т, 2Н), 7,12 (д, 3Н), 6,86 (д, 2Н), 6,29 (с, 1Н), 4,08 (м, 1Н), 3,47-3,26 (рм, 2Н), 2,75 (м, 1Н), (м, 1Н), 2,57-2,37 (м, 2Н), 2,03-1,72 (м, 4Н), 1,6 (м, частично ниже водного пика из ЯМР растворителя), 1,40 (д, 3Н), 1,25 (м, 3Н), 1,09 (д, 18Н).
П р и м е р 2. Смесь /1R*, 2S*/- и /1S*, 2S*/-2-(3-фенилметилен-8-азабицикло-(3-2-1)окт-8-ил)-1-4- (триизопропилсилилокси)фенил-1-пропанол
К суспензии литийалюминийгидрида (0,61 г, 16,07 ммоль) в тетрагидрофуране (50 мл) при 0оС добавляют целевой продукт предшествующего примера (8,04 г, 15,96 ммоль) в тетрагидрофуране (150 мл) на протяжении 15 мин. Смесь перемешивали 15,5 ч при комнатной температуре, затем осторожно гасили водой (1,2 мл), фильтровали через диатомовую землю и концентрировали, давая желтое масло. Данная смесь рацемических целевых продуктов использовалась непосредственно в следующей реакции без очистки.
П р и м е р 3. (1S*, 2S*)- и (1R*, 2S*)-1-(4-гидроксифенил)-2-(3-фенилметилен-8-азабицикло/3.2.1/окт-8- ил)-1-пропанол
Целевой продукт предыдущего примера (7,15 г, 14,14 ммоль) растворяли в тетрагидрофуране (250 мл) и добавляли фтористый тетрабутиламмоний (28,5 мл, 28,5 ммоль, 1 М в тетрагидрофуране) весь сразу. Раствор перемешивался при комнатной температуре в течение 18 ч, затем концентрировался и хроматографировали на силикагеле (4 х 6 дюймов, этилацетат/гексановый градиент с последующим метанол/этилацетатным градиентом), давая сначала рацемический (1S*, 2S)-целевой продукт (1,58 г), а затем более полярный рацемический (1R*, 2S*)-целевой продукт (2,88 г) (заметьте, что, вследствие асимметрии в 3-фенилметилен-8-азабицикло/3.2.1/окт-8-ильной боковой цепи, каждый из этих продуктов представляет фактическая смесь двух рацематов).
(1S*, 2S*)-продукт перекристаллизовывался из смеси этилацетат/гексан, давая 0,923 г белого твердого вещества; т.пл. 175-177оС;
ЯМР включает: 4,10 (т., J = 7,7 Гц, 1Н).
Анализ, %: С 78,77; Н 7,90; N 3,92.
Вычислено,%: С 79,05; Н 7,90; N 3,92.
(1R*, 2S*)-продукт далее очищали с помощью радиальной хроматографии с использованием для элюирования смеси 60% этилацетат/гексан, давая 0,24 г бесцветного масла. Данное масло кристаллизовалось из смеси эфир/гексан, давая 0,17 г мелкокристаллического твердого вещества; т.пл. 78,5-85оС. Последнее превращалось в его HCl-соль с помощью барботирования HCl-газа в эфирный раствор соединения в течение 3 мин. Белый осадок собирали и перекристаллизовывали из этанола, давая (1R*, 2S*)-хлоргидратную соль в виде ее полугидрата; т. пл. 215-218оС; ЯМР (DBCO-d6) включает: 5,17 (с., 1Н), 1Н и 4,60-3,93 (м., 2Н).
Найдено,%: С 70,00; Н 7,45; N 3,35.
Вычислено,%: С 69,95; Н 7,40; N 3,54.
П р и м е р 4. 2-[4-(фенилтио)пиперидино]-1-[4-(трет-бутилдиметилсилилокси)- фенил]-1-пропанон
Данный продукт приготавливали в соответствии с процедурой примера 1 с использованием 4-фенилтиопиперидина (1,13 г, 5,85 ммоль), триэтиламина (0,82 мл, 5,88 ммоль) и 4-(трет-бутилдиметилсилилокси)- α-бромпропиофенона (2,0 г, 5,83 ммоль) и времени нагревания с обратным холодильником 21,5 ч. Продукт выделяли с помощью мгновенной хроматографии на силикагеле с использованием этилацетат/гексанового градиентного элюирования. Выход был 1,29 г целевого продукта в виде желтого масла:
ЯМР 8,00 (д, J = 9 Гц, 2Н), 7,35 (д, J = 7,8 Гц, 2Н), 7,35 (м, 3Н), 6,81 (д, J = 8,4 Гц, 2Н), 4,00 (кв, J = 6,8 Гц, 1Н), 3,03 (м, 1Н), 2,91-2,87 (м, 1Н), 2,80-2,76 (м, 1Н), 2,44 (д.т, J = 9,5, 2,6 Гц), 2,29-2,19 (м, 1Н), 1093-1,85 (м, 2Н), 1,66-1,51 (м, 2Н - частично ниже водного пика из растворителя), 1,22 (д, J = 7,1 Гц, 3Н), 0,97 (с, 9Н), 0,22 (с, 6Н).
Более поздние фракции от хроматографии давали дополнительно 0,51 г продукта, который десилилировался во время реакции. Данный материал мог также превращаться в конечные целевые соединения с помощью процедур, представленных ниже.
С помощью тех же методов 4-(триизопропилсилилокси)- α-бромпропиофенон (9,97 г, 25,9 ммоль) превращался в хроматографированный 2-[4-(фенилтио)пиперидино] -1-4-(триизопропилсилилокси)фенил-1-пропан-он в виде светло-оранжевого масла:
8,32 д; ЯМР 8,00 (д, J = 8,8 Гц, 2Н), 7,37 (дд, J = 1,5, 8,4 Гц, 2Н), 7,29-7,18 (м, 3Н), 6,86 (д, J = 8,8 Гц, 2Н), 4,02 (кв, J = 6,8 Гц, 1Н), 3,08-3,00 (м, 1Н), 2,85 (д, J = 26,2 Гц, 1Н), 2,75 (д, J = 16,6 Гц, 1Н), 2,45 (д.т, J = 11, 2,6 Гц, 1Н), 2,23 (д.т, J = 9,8, 2,5 Гц, 1Н), 1,96-1,88 (м, 2Н), 1,71-1,50 (м, 5H), 1,09 (д, J = 7 Гц, 18Н).
П р и м е р 5. (1S*, 2S*)- и (1R*, 2S*)-2-(4-фенилтио)пиперидино-1,4-(триизопропи- лсилилокси)фенил-1-пропанол
С помощью метода примера 2 триизопропилсилилокси-продукт предыдущего примера (8,32 г, 16,7 ммоль) с использованием хроматографии на силикагеле с элюированием этилацетат/гексановым градиентом для отделения изомеров превращался в 6,06 г менее полярного (1S*, 2S*)-целевого продукта в виде масла:
ЯМР: 7,41 (д, J = 6,7 Гц, 2Н), 7,32-7,24 (м, 3Н), 7,15 (д, J = 8,5 Гц, 2Н), 6,82 (д, J = 8,4 Гц, 2Н), 4,14 (д, J = 9,7 Гц, 1Н), 3,11 (м, 1Н), 2,88 (м, 1Н), 2,69 (м, 1Н), 2,69-2,55 (м, 2Н), 2,21 (т, 1Н), 2,03 (м, 2Н), 1,85-1,58 (м, 3Н), 1,35-1,20 (м, 2Н), 1,23 (д, J = 15 Гц, 18Н), 1,07 (д, J = 7 Гц, 3Н), и до 0,2 г более полярного (1S*, 2S*)-целевого продукта ЯМР: 7,35 (д, J = 7 Гц, 2Н), 7,28-7,16 (м, 3Н), 7,08 (д, J = 9 Гц, 2Н), 6,78 (д, J = 9 Гц, 2Н), 4,68 (д, J = 5 Гц, 1Н), 3,10-2,98 (м, 1Н), 2,98-2,87 (м, 1Н), 2,75-2,60 (м.м, 2Н), 2,33 (т, J = 9 Гц, 2,16 (т, J = 11 Гц, 1Н), 1,91 (т, J = 15 Гц, 2Н), 1,70-1,48 (м, 2Н), 1,32-1,12 (м, 4Н), 1,06 (д, J = 9 Гц, 18Н), 0,88-0,78 (м, 3Н).
П р и м е р 6. (1S*, 2S*)-1-(4-Гидроксифенил)-2-[4-(фенилтио)пиперидино]-1-про-панол
Метод А
К суспензии литийалюминийгидрида (0,11 г, 2,9 ммоль) в тетрагидрофуране (25 мл), охлажденной до 0оС, добавляли целевой продукт примера 4 (1,29 г, 2,88 ммоль) в тетрагидрофуране (50 мл). Реакционную смесь подогревали до температуры окружающей среды в течение 2 ч, нагревали с обратным холодильником в течение 3 ч и оставляли перемешиваться в течение 72 ч. Смесь осторожно гасили водой и фильтровали через диатомовую землю. Фильтрат концентрировался до влажного твердого вещества, которое брали в этилацетат и промывали солевым раствором, сушили (сульфатом кальция), концентрировали, давая 0,41 г белого твердого вещества, и он перекристаллизовывался из смеси эфир/гексан, давая 0,16 г целевого продукта; т.пл. 155-157оС; ЯМР включает: 4,12 (д, 1Н).
Найдено,%: С 69,57; Н 7,28; N 3,95.
Вычислено,%: С 69,94; Н 7,34; N 4,08.
Окисно-алюминиевые соли из реакции экстрагировались в аппарате Сокслета этилацетатом в течение 24 ч. Концентрирование давало дополнительно 0,3 г продукта.
Метод В.
Тот же продукт получался из (1S*, 2S*)-целевого продукта предыдущего примера по способу примера 3.
П р и м е р 7. (1R*, 2S*)-1-(4-Гидроксифенил)-2-[4-(фенилтио)пиперидино] -1-про-панол
С использованием процедуры примера 3 настоящий целевой продукт получался из (1R*, 2S*)-целевого продукта примера 5 (0,2 г, 0,4 ммоль) и фтористого тетрабутиламмония (0,8 мл, 0,8 ммоль, 1 М в тетрагидрофуране) в тетрагидрофуране (5 мл) при температуре окружающей среды в течение 72 ч. Продукт получали с помощью мгновенной хроматографии на силикагеле (6 х 1 дюймов, элюирование этилацетат/гексановым градиентом), давая 0,14 г полутвердого вещества. Перекристаллизация из смеси метиленхлорид/гексан давала 0,056 г белого твердого вещества: т.пл. 124,5-126оС.
ЯМР включает: 4,72 (д, J = 4,1 Гц, 1Н).
Найдено,%: С 69,68; Н 7,24; N 4,14.
Вычислено,%: С 69,94; Н 7,34; N 4,08.
П р и м е р 8. (1S*, 2S*(-1-(4-Гидроксифенил)-2-4-(фенилсульфонил)пиперидино-1- пропанол
Целевой продукт примера 6 (0,13 г, 0,378 ммоль) растворяли в метиленхлориде (20 мл) и добавляли м-хлорнадоксибензойную кислоту (0,23 г, 1,133 ммоль) вся сразу. Раствор перемешивали в течение 23 ч, затем выпавший в осадок материал фильтровался, давая 0,154 г белого твердого вещества, которое представляло сырой промежуточный N-оксид. Последнюю гидрировали в аппарате Парра в метаноле (20 мл) с использованием 10% палладиевого катализатора на угле (0,03 г) при давлении водорода 50 фунт/кв.дюйм (3,515 кг/кв. см). Реакцию завершали через 6 ч, и смесь фильтровали через диатомовую землю и концентрировали, получая 0,166 г желтого масла. Данное масло брали в метиленхлорид и промывали насыщенным бикарбонатом натрия. Органическую фазу сушили (сульфатом магния) и концентрировали, получая 0,106 г желтого масла, которое кристаллизовалось из смеси этилацетата и гексана, давая 0,076 г белого твердого вещества; т.пл. 169-175оС. Порция продукта (0,045-0,050 г) далее очищалась с помощью энергичного смешения с насыщенным бикарбонатом натрия и этилацетатом в течение 15 мин. Фазы разделялись, и водная фаза далее экстрагировалась этилацетатом (2 раза). Объединенную органическую фазу сушили (сульфатом кальция), концентрировали, получая бесцветное масло; и после кристаллизации из этилацетата получали 0,02 г белого порошка; т.пл. 195-196оС.
ЯМР 7,88 включает: 4,15 (д, J = 9,7 Гц, 1Н).
Найдено,%: С 63,77; Н 6,61; N 3,61.
Вычислено,%: С 63,98; Н 6,71; N 3,73.
П р и м е р 9. (1S*, 2S*)-1-(4-гидроксифенил)-2-[4-(фенилсульфонил)пиперидино]-1- пропанол
Целевой продукт примера 6 (0,5 г, 1,46 ммоль) растворяли в метиленхлориде (40 мл) и добавляли м-хлорнадоксибензойную кислоту (0,3 г, 1,48 ммоль) всю сразу. После перемешивания на протяжении ночи при температуре окружающей среды смесь концентрировалась непосредственно на силикагеле и подвергалась мгновенной хроматографии (6 х 1 дюймов, этилацетат/гексановый градиент), давая 0,34 мг сырого продукта в виде белого твердого вещества, которое дополнительно очищалось с помощью распределения между насыщенным бикарбонатом натрия и этилацетатом при энергичном перемешивании в течение 20 мин. Фазы разделялись, и органический слой концентрировался, давая жирное твердое вещество, которое кристаллизовалось из смеси этилацетата и гексана, давая 0,122 г белого твердого вещества; т.пл. 110оС.
ЯМР включает: 4,16 (спаренные д. в длинной зоне, J = 9,7 Гц, 1Н); HPMS: 360.635.
Вычислено: 360.1626.
П р и м е р 10. 1-[4-(бензилокси)фенил]-2-(4-бензил-4-гидроксипиперидино)-1-про- панон
Данный продукт приготавливали, следуя процедуре примера 1, из 4-гидрокси-4-бензилпиперидина (2,0 г, 10,46 ммоль), триэтиламина (1,46 мл, 10,47 ммоль) и 4-бензилокси-α -бромпропиофенона (3,33 г, 10,43 ммоль) в этаноле (50 мл) при нагревании с обратным холодильником в течение 24 ч. Настоящий рацемический продукт получали после мгновенной хроматографии на силикагеле с элюированием этилацетат/гексановым градиентом. Выход был 2,88 г (64%) желтого твердого вещества.
ЯМР 8,06 (д, 2Н), 7,52-7,08 (м, 10Н), 6,97 (д, 2Н), 5,11 (с, 2Н), 4,00 (кв, 1Н), 2,72 (с, 2Н), 2,72-2,53 (м, 2Н), 2,43 (т, 1Н), 1,85-1,39 (м, 6Н), 1,23 (д, 3Н).
НРМ 412.2348.
Вычислено (-ОН) 412.2273.
П р и м е р 11. (1S*, 2S*(-1-4-(Бензилокси)фенил-2-(4-бензил-4-гидроксипипериди- но)-1-пропанол
Боргидрид натрия (0,25 г, 6,61 ммоль) добавляли весь сразу к раствору целевого продукта предыдущего примера (2,88 г, 6,70 ммоль) в этаноле (50 мл). Смесь перемешивали в течение 20 ч при комнатной температуре, образовывался осадок. Твердое вещество отфильтровывали и сушили, получая 60 г целевого продукта; т.пл. 147-148оС.
ЯМР включает: 4,17 (д, J = 10 Гц, 1Н).
ИК (кВ): 3387, 3024, 2936, 1611, 1513, 1453, 1239, 1026, 1011, 695.
Фильтрат от указанной выше реакции концентрировался, остаток распределялся между этилацетатом и водой, и фазы разделялись. Водная фаза экстрагировалась этилацетатом. Объединенные органические фазы промывались водой, сушились (сульфатом кальция) и концентрировались, давая 2,82 г дополнительного целевого продукта.
П р и м е р 12. (1S*, 2S*)-1-(4-Гидроксифенил)-2-(4-бензил-4- гидроксипиперидино)-1-пропанол
Целевой продукт предшествующего примера (0,49 г, 1,14 ммоль) и тетрагидрофуран (30 мл) охлаждались до -78оС, и аммиачный газ (30 мл) конденсировался в смесь. Добавлялся натрий (0,082 г, 3,57 ммоль) четырьмя частями. Реакционная смесь, которая становилась синей, перемешивалась в течение 15 мин, а затем гасилась хлористым аммонием (0,29 г). Реакционной смеси давали возможность подогреться до комнатной температуры, при этом аммиак выкипал. Реакционная смесь концентрировалась, и остаток брался в этилацетат и промывался водой и солевым раствором. Органическая фаза сушилась (сульфатом кальция) и концентрировалась, давая 0,39 г белого твердого вещества. Перекристаллизация из гексана давала 0,19 г целевого продукта.
ЯМР (DМCO-d6) 7,25-7,11 (м, 5Н), 7,08 (д, J = 8,2 Гц, 2Н), 6,68 (д, J = 8,6 Гц, 2Н), 4,14 (с, 1Н), 4,09 (д, J = 9,2 Гц, 1Н), 3,33 (с, 2Н), 3,30 (с, 1Н), 2,74 (м, 1Н - частично ниже водного пика из ЯМР растворителя), 2,60-2,35 (м, 4Н - частично ниже пика растворителя), 1,70-1,44 (м, 4Н), 7,63 (д, J = 6,7 Гц, 3Н).
Оксид дейтерия вымывал синглеты при 4,14 и 3,30 млн.дол.
Данный продукт перекристаллизовывался из этилацетата, давая очищенный целевой продукт; т.пл. 213-214оС.
ИК (кВ): 3263, 3023, 2940, 2917, 1615, 1517, 1453, 1273, 1221, 1186, 1020, 1011, 831, 687.
Анализ: С 73,73, Н 8,03, N 4,01.
Вычислено: С 73,87, Н 7,97, N 4,10.
П р и м е р 13. 1-(4-(трет-Бутилдиметилсилилокси)фенил)-2-(3-фенилтио-8-азабиц-икло /3.2.1/окт-8-ил)-1-пропанон
При использовании процедуры примера 1 данный продукт приготавливался на 4-трет-бутилдиметилсилилокси-альфа-бро- мпропиофенона (1,25 г, 3,65 ммоль), 3-фенилтио-8-азабицикло/3.2.1/октана (0,8 г, 3,65 ммоль) и триэтиламина (0,51 мл, 3,65 ммоль) в этаноле (30 мл) при нагревании с обратным холодильником на протяжении ночи. Продукт подвергался мгновенной хроматографии на силикагеле (10% этилацетат-гексан элюирование смесью); 0,889 г (51%).
ЯМР 8,13 (д, J = 9 Гц, 2Н), 7,38 (м, 2Н), 7,30-7,15 (м, 3Н), 6,83 (д, J = 9 Гц, 2Н), 3,93 (кв, J = 7 Гц, 1Н), 3,42-3,28 (м, 3Н), 2,05-1,56 (м, 9Н), 1,32 (д, J = 7 Гц, 3Н), 0,99 (с, 9Н), 0,25 (с, 6Н).
ИК: 2940, 2840, 1600, 1390-1290 (шир.), 910.
П р и м е р 14. Смесь (1R*, 2S*)- и (1S*, 2S*)-1-4-(трет-бутилдиметилсилилокси)-фен- ил-2-(3-фенилтио-8-азабицикло/3.2.1/окт-8- ил)-1-пропанола
Данный продукт приготавливался, как в примере 2, при перемешивании на протяжении ночи при температуре окружающей среды из продукта предыдущего примера (0,85 г, 1,77 ммоль) и алюминийгидрида лития (0,153 г, 4,0 ммоль) в тетрагидрофуране (24 мл). Продукт отделялся в виде желтого масла (0,78 г, 91%) в виде смеси рацемических целевых продуктов.
П р и м е р 15. (1S*, 2S*) - и (1R*, 2S)-1-(4-Гидроксифенил)-2- (3-фенилтио-8-азабицикло/3.2.1/окт-8-ил)-1-пропанол.
Целевой продукт предыдущего примера (0,78 г, 1,6 ммоль) десилилировался согласно процедуре примера 3 с помощью фтористого тетрабутиламмония (1,6 мл, 1,6 ммоль, 1М в тетрагидрофуране) в течение 5-минутной реакции. Получающаяся смесь рацематов разделялась с помощью мгновенной хроматографии на силикагеле (элюирование смесью 50% этилацетат/гексан). Первым элюировался (1S*, 2S*)-целевой продукт, 0,133 г.
ЯМР (DMCO-d6) 7,40-7,21 (м, 5Н), 7,06 (д, J = 8,5 Гц, 2Н), 6,63 (д, J = 8,0 Гц, 2Н), 4,35 (д, J = 5,0 Гц, 1Н), 3,54-3,40 (м, 2Н), 3,35-3,29 (м, 2Н), 2,70-2,62 (шир.т, 1Н), 2,50 (м, 1Н), 1,80-1,54 (м, 6Н), 0,63 (д, J = 6,5 Гц, 3Н).
Продолжительное элюирование дает (1R*, 2S*)-целевой продукт.
ЯМР 7,45-7,40 (м, 2Н), 7,32-7,22 (м, 3Н), 7,15 (д, 2Н), 6,76 (д, 2Н), 4,80 (д, 1Н), 3,60-3,52 (м, 2Н), 3,51-3,38 (м, 1Н), 2,80-2,72 (м, 1Н), 2,00-1,65 (м, 8Н), 0,68 (д, 3Н).
Данный продукт (80 мг) превращался в его HCl-соль с помощью растворения в 15 мл простого эфира, барботирования в него сухого НCl в течение 2 мин и растирания получающегося в результате маслянистого твердого вещества с эфиром, давая 30 мг в виде белого твердого вещества.
П р и м е р 16. 2-(4-Бензил-4-гидроксипиперидино)-1-4-(триизопропилсилилокси)-фенил-1-пропан он
Данный продукт получался в соответствии с процедурами примера 1 из 4-гидрокси-4-бензилпиперидина (2,72 г, 14,22 ммоль) 4-триизопропилсилилокси- α -бромпропиофенона (5,48 г, 14,22 ммоль) и триэтиламина (2,0 мл, 14,35 ммоль) в этаноле (50 мл) при времени нагревания с обратным холодильником 17 ч, давая 4,92 г (70%) хроматографического целевого продукта в виде оранжевого масла.
ЯМР 8,01 (д, J = 8,8 Гц, 2Н), 7,31-7,22 (м, 3Н), 7,18-7,15 (м, 2Н), 6,87 (д, J = 8,8 Гц,2Н), 4,03 (кв, J = 6,7 Гц, 1Н), 2,72 (с, 2Н), 2,68-2,57 (м, 3Н), 2,45 (д,т, 1Н), 1,78-1,42 (м, 4Н), 1,40-1,25 (м, 7Н), 1,10 (д, J = 7 Гц, 18Н).
П р и м е р 17. (1S*, 2S*)- и (1R*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1-4-трии- зопропилсилилокси)фенил-1-пропанол
Целевой продукт предыдущего примера (4,92 г, 9,33 ммоль) растворяли в этаноле (100 мл) и добавляли боргидрид натрия (0,38 г, 10 ммоль) весь сразу. После перемешивания на протяжении ночи при температуре окружающей среды выделяли (1S*, 2S*)-целевой продукт с помощью фильтрования; 2,11 г.
ЯМР 7,46-7,17 (м, 7Н), 6,84 (д, J = 7 Гц, 2Н), 4,18 (д, J = 11 Гц, 1Н), 2,86 (шир. т, 1Н), 2,77 (с, 2Н), 2,70-2,42 (м, 4Н), 1,89-1,55 (м, 6Н), 1,30-1,13 (м, 3Н), 1,10 (д. J = 8,6 Гц, 18Н), 0,75 (д, J = 6 Гц, 3Н).
Фильтрат концентрировался, и остаток растворялся в этилацетате, экстрагировался водой (2х) и солевым раствором, сушился (сульфатом кальция), концентрировался до 2,33 г светло-желтого твердого вещества, и подвергался мгновенной хроматографии на силикагеле (2 х 6 дюймов, элюированием с использованием этилацетат/гексанового градиента), давая сначала 1,4 г (1S*, 2S*)-продукта, а затем 0,46 г (1R*, 2S*)-продукта.
ЯМР 7,33-7,11 (м, 7Н), 6,82 (д, J = 8,6 Гц, 2Н), 4,77 (д, J = 4 Гц, 1Н), 2,80-2,39 (м, 5Н), 1,88-1,43 (м, 6Н), 1,31-1,13 (м, 8Н), 1,08 (д, J = 6,7 Гц, 18Н), 0,84 (д, J = 6 Гц, 3Н).
П р и м е р 18. (1R*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1- (4-гидроксифенил)-1-пропанол
По способу примера 3 при использовании при хроматографии элюирования градиентом этилацетат/гексан, (1R*, 2S*)-целевой продукт предыдущего примера (0,46 г, 0,92 ммоль) превращался в 0,24 г (74%) настоящего целевого продукта в виде моногидрата, т.пл. 173-174оС.
ЯМР (DМCO с добавлением D2О) 7,21-7,12 (м, 5Н), 7,04 (д, J = 7,6 Гц, 2Н), 6,64 (д. J = 8,6 Гц, 2Н), 4,64 (д, J = 8,6 Гц, 1Н), 2,59 (с, 2Н), 2,59-2,49 (м, 5Н-частично ниже ЯМР растворителя) 1,50-1,31 (м, 4Н), 0,82 (д, J = 7 Гц, 3Н).
Найдено,%: С 70,26; Н 7,96; N 3,85.
Вычислено для моногидрата: С 70,17; Н 8,13; N 3,90.
П р и м е р 19. 2-(4-бензил-4-гидроксипиперидино)-1-(4-фторфенил)-1-пропанол
Целевой продукт, полученный как в примере 10, с выходом 80%, перекристаллизовывался из простого эфира; т.пл. 119,5-120оС.
Найдено,%: С 73,41, Н 7,08; N 4,03.
Вычислено,%: С 73,87; Н 7,09; N 4,10.
П р и м е р 20. (1S*, 2S*)- и (1R*, 2S)-2-(4-бензил-4-гидрокси- пиперидино)-1-(4-фторфенил)-1-пропанол.
Получение, как в примере 17, целевые продукты разделялись с помощью мгновенной хроматографии на силикагеле (с использованием этилацетат/гексанового, а затем метанол/этилацетатного градиента). (1S*, 2S*)-продукт элюировался первым с выходом 84% в виде твердого вещества, которое перекристаллизовывалось из смеси этанола и простого эфира, т.пл. 153,5-154,5оС.
Найдено,%: С 73,53; Н 7,67; N 4,08.
Вычислено,%: С 73,44; Н 7,63; N 4,08.
П р и м е р 21. 2-(3-Фенилметилен-8-азабицикло/3.2.1/окт-8- ил)-1-(4-бензилоксифенил)-1-пропанон
Целевой продукт, полученный следуя процедуре примера 1, из 4-бензилокси- α-бромпропиофенона, получили с выходом 40-60% после мгновенной хроматографии на силикагеле.
ЯМР 8,34 (д, 2Н), 7,56-7,45 (м, 7Н), 7,20 (д, 3Н), 7,03 (д, 2Н), 6,36 (с, 1Н), 5,14 (с, 2Н), 4,13 (кв, 1Н), 3,56-3,30 (м, 2Н), 2,81 (т, 1Н), 2,70-2,40 (м, 2Н), 2,10-1,76 (м, 3Н), 1066 (м, 1Н), 1,45 (д, 3Н).
П р и м е р 22 (1S*, 2S*)-2-(3-Фенилметилен-8-азабицикло/3.2.1/ акт-8-ил)-1-(4-бензилоксифенил)-1-пропанол
Целевой продукт, приготовленный с помощью процедуры примера 3 при времени реакции 1,25 ч, получали в чистом виде с помощью мгновенной хроматографии на силикагеле, т.пл. 145-148оС.
Найдено,%: С 81,69; Н 7,46. N 3,02.
Вычислено,%: С 81,97; Н 7,57; N 3,19.
П р и м е р 23. (1S*, 2S*)-2-(3-бензил-8-азабицикло/3.2.1/окт- 8-ил)-1-(4-гидроксифенил)-1-пропанол
Целевой продукт предыдущего примера (0,23 г, 0,523 ммоль) растворяли в тетрагидрофуране (20 мл) и охлаждали до -78оС. В раствор конденсировался аммиак (30 мл). Добавляли натрий (0,06 г, 2,6 ммоль) тремя порциями, и постепенно образовывался голубой раствор. Спустя 10 мин реакция гасилась избытком хлористого аммония, и смесь оставляли подогреваться до комнатной температуры с выпариванием аммиака. Остаточную смесь концентрировали, и остаток экстрагировали этилацетатом. Фильтрование и концентрирование давали 0,24 г маслянистого твердого вещества, которое подвергалось мгновенной хроматографии на силикагеле (6 х 1 дюйм, элюированием с использованием этилацетат/гексанового градиента с последующим испарением метанола). Это давало сначала регенерированный исходный материал, а затем продукт (0,071 г). Продукт дополнительно очищался с помощью перекристаллизации (этилацетат/гексан). Во время данного процесса происходила утечка некоторой порции, но 0,005 г белого твердого продукта получалось в виде 1:3 смеси эпимеров бензильной группы.
ЯМР 7,30-7,10 (м, 7Н), 6,72 (J = 8,5 Гц) и 6,71 (J = 8,6 Гц), (пара перекрывающихся д. всего 2Н), 4,11 (д, J = 8,6 Гц, 1Н), 3,45 (с, 1Н), 3,31 и 3,24 (пара д, 2Н), 2,61 (квинтет, J = 7,6 Гц, 1Н), 2,11 (м, 1Н), 1,86-1,17 (м, 10Н), 0,81 (д, J = 6,7 Гц, 3Н).
НРМС: 352.2276.
Вычислено: для МН+: 352.2278.
П р и м е р 24. 2-(4-Бензил-4-гидроксипиперидино)-1-(4-хлорфенил)-1-пропанон
Целевой продукт, полученный из 4-хлор-α -бромпропиофенона, с использованием процедуры примера 10 с выходом 72%, очищался с помощью мгновенной хроматографии на силикагеле и перекристаллизовывался из простого эфира, т. пл. 135,5-136оС.
Найдено,%: С 70,11; Н 6,70; N 3,85.
Вычислено,%: С 70,48; Г 6,76; N 3,91.
П р и м е р 25. (1S*, 2S*)- и (1R*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1-(4-хлор- фенил)-1-пропанол
Целевые продукты, полученные с использованием процедуры примера 17, разделялись с помощью той же процедуры хроматографии. (1S*, 2S*)-продукт получали с выходом 70%, т.пл. 159,5-160,5оС; (этанол/простой эфир).
Найдено,%: С 70,13; Н 7,50; N 3,91.
Вычислено,%: С 70,08; Н 7,28; N 3,89.
1R*, 2S*-продукт получался с выходом 7%; т.пл. 150,5-151,5оС (этанол/простой эфир).
Найдено,%: С 69,92; Н 7,38; N 3,92.
Вычислено,%: С 70,08; Н 7,28; N 3,89.
П р и м е р 26. 2-(4-бензил-4-гидроксипиперидино)-1-(4-хлорфенил)-1-этанон
Целевой продукт приготавливался с использованием процедуры примера 10 из 4-хлор-α -бромацетофенона с выходом 76%.
ЯМР 7,93 (д, J = 8,5 Гц, 2Н), 7,39 (д,J = 8,6 Гц, 2Н), 7,31-7,16 (м, 5Н), 3,74 (с, 2Н), 2,74 (с, 2Н), 2,73-2,71 (м, 2Н), 2,43 (д.т, J = 11,5, 2.2.4 Гц, 2Н), 1,80 (д.т, J = 12,7, 4,3 Гц, 2Н), 1,50 (шир.д, J = 13,8 Гц, 2Н).
П р и м е р 27. 2-(4-Бензил-4-гидроксипиперидино)-1-(4-хлорфенил)этанол
Целевой продукт, полученный из 4-хлор-α -бромацетофенона с помощью процедуры примера 17 с выходом 83%, перекристаллизовывался из смеси этанол-эфир, т.пл. 151-152оС.
ЯМР 7,34-7,18 (м, 9Н), 4,67 (д.д, J = 10,5, 3,5 Гц, 1Н), 4,18 (шир.с, 1Н), 2,89-2,86 (м, 1Н), 2,76 (с, 1Н), 2,68-2,47 (м, 3Н), 2,41-2,31 (м, 2Н), 1,73 (д.кв, J = 13,3, 4,4 Гц, 2Н), 1,58-1,50 (м, 2Н), 1,24 (с, 1Н).
П р и м е р 28. 2-(4-Бензил-4-гидроксипиперидино)-1-(4-фторфенил)-1-этанон
Целевой продукт приготавливали с использованием процедуры примера 10 с выходом 59% из 4-фтор- α-бромацетофенона.
ЯМР 8,05-7,99 (м, 2Н), 7,33-7,04 (м, 7Н), 3,76 (с, 2Н), 2,83-2,71 (м, 4Н), 2,43 (д. т, J = 11,5, 2,1 Гц, 2Н), 1,82 (д.т, J = 12,7, 4,3 Гц, 3Н), 1,51 (шир.д, J = 11,5 Гц, 2Н).
П р и м е р 29. 2-(4-Бензил-4-гидроксипиперидино)-1-(4-фторфенил)-1-этанол
Целевой продукт, полученный следуя процедуре примера 17 с выходом 85%, перекристаллизовывали из смеси этанол/эфир; т.пл. 144,5-146оС.
ЯМР 7,35-7,25 (м, 5Н), 7,19 (д, J = 6,4 Гц, 2Н), 7,01 (т, J = 8,7 Гц, 2Н), 4,67 (д.д, J = 10,5, 3,5 Гц, 1Н), 4,18 (шир.с, 1Н), 2,88 (шир.д, J = 11,2 Гц, 1Н), 2,76 (с, 2Н), 2,68-2,31 (м, 5Н), 1,81-1,66 (м, 2Н), 1,58-1,50 (м, 2Н), 1,28 (с, 1Н).
П р и м е р 30. Эндо-1-4-(триизопропилсилилокси)фенил-2-(3' - фенилспиро 8-азабицикло/3.2.1/октан-3,2'-оксиран- 8-ил)- -пропанон
Целевой продукт примера получения 13, приведенного ниже (0,72 г, 3,34 ммоль), целевой продукт примера получения 10, приведенного ниже (1,29 г, 3,35 ммоль) и карбонат натрия (0,93 г, 6,7 ммоль) объединяли в тетрагидрофуране (80 мл), нагревали с обратным холодильником в течение 30 ч, охлаждали и фильтровали через диатомовую землю. Фильтрат концентрировался и хроматографировался на силикагеле (с элюированием с использованием этилацетат/гексанового градиента), давая 0,77 г (44%) целевого продукта в виде смеси диастереомеров. Эпоксидные протоны наблюдались при δ 3,65 и 3,60 млн. дол. в спектре ЯМР.
Соответствующий экзо-изомер приготавливали аналогичным образом из другого изомера примера получения 13 с выходом 37%. Эпоксидные протоны в смеси диастереомерных продуктов наблюдали при δ 3,80 и 3,86 млн.дол. в спектре ЯМР.
П р и м е р 31. Эндо- и экзо-(1S*, 2S*) и (1R*, 2S*)-1-4-(триизопропилсилилокси (-фенил-2-(3-фенилспиро 8-азабицикло/3.2.1/октан-3,2-оксиран-8-ил)-1-пропа-нол
Смесь настоящих эндо-целевых продуктов получалась из эндо- целевого продукта предыдущего примера с выходом 82% с помощью процедуры примера 17. Мгновенная хроматография давала чистый более быстрый идущий (1S*, 2S)-изомер.
ЯМР 7,34-7,23 (м, 5Н), 7,17 (д, J = 8,5 Гц, 2Н), 6,82 (д, J = 8,5 Гц, 2Н), 4,02 (д, J = 8 Гц, 1Н), 3,65 (с, 1Н), 3,45 (шир.с, 1Н), 3,28 (шир.с, 1Н), 2,65 (квинтет, J = 7,2 Гц, 1Н), 2,54 (д.д, J = 13,8, 3,2 Гц, 1Н), 2,15 (кв, J = 8,6 Гц, 2Н), 1,93-1,72 (м, 4Н), 1,40 (д, J = 13,8 Гц, 2Н).
Более медленно идущий (1R*, 2S*)-изомер, который показывает характерный ЯМР-сигнал при 4,81 млн.дол. (широкий с., 1Н), получался в виде смеси (1S*, 2S*)-изомера.
Соответствующие экзо-изомеры получались аналогичным образом из экзо-изомера предыдущего примера с выходом 82% в виде 3:1 смеси (1S*, 2S*)- и (1R*, 2S*)-изомеров. Эпоксидные протоны видны были при 3,86 и 3,82 млн.дол. в ЯМР-спектре.
П р и м е р 32. Эндо-(1S*, 2S*)-1-(4-гидроксифенил)-2-(3' -фенилспиро 8-азабицикло/3.2.1/октан-3,2' -оксиран- 8-ил)-1- -пропанол
С помощью способа примера 3 эндо-(1S*, 2S*)-целевой продукт предыдущего примера превращался в настоящий целевой продукт с выходом 62%; т.пл. 204,5-205оС (хлороформ/гексан).
ЯМР 7,32-7,25 (м, 5Н), 1,17 (д, J = 8,4 Гц, 2Н), 6,73 (д, J = 6,6 Гц, 2Н), 5,25 (шир.с, 1Н)-4,01 (д, J = 8,2 Гц, 1Н), 3,66 (с, 1Н), 3,47 (шир.с, 1Н), 3,31 (шир.с, 1Н), 2,65-2,54 (м, 2Н), 2,16 (д, J = 8 Гц, 2Н), 1,89-1,73 (м, 3Н), 1,44 (шир,д, J = 13,9 Гц, 1Н), 1,24 (шир.д. J = 14 Гц, 1Н), 0,83 (д, J = 6,7 Гц, 3Н).
П р и м е р 33. Эндо-(1S*, 2S*)-2-(3-бензил-3-гидрокси-8-азабицикло/3.2.1/окт-8-ил)-1-(4- триизопропилсилилокси)фенил-1-пропанол
Эндо-(1S*, 2S*) (1R*, 2S*) смесь примера 31 (0,19 г, 0,36 ммоль) растворялась в тетрагидрофуране (20 мл) и охлаждалась до -78оС. В раствор конденсировали аммиак (30 мл) и добавляли металлический натрий (0,08 г) в виде небольших кусочков на протяжении 1 ч. За данное время смесь становилась темно-голубой. Реакционную смесь перемешивали еще 10 мин, затем гасили твердым хлористым аммонием. Аммиаку давали возможность выпариваться, остаточная смесь распределялась между этилацетатом и водой, и водный слой экстрагировался свежим этилацетатом. Объединенные органические слои промывались солевым раствором, сушились над сульфатом кальция и концентрировались, давая 0,18 г (95%) светло-желтого масла, которое очищалось с помощью мгновенной хроматографии на силикагеле (с использованием элюирования этилацетат/гексановым градиентом), давая 0,1 г бесцветного масла, которое представляло (1S*, 2S*)-целевой продукт.
ЯМР 7,33-7,25 (м, 3Н), 7,18 (д, J = 8,3 Гц, 4Н), 6,84 (д, J = 8,4 Гц, 2Н), 4,08 (д, J = 7,5 Гц, 1Н), 3,42 (шир. с, 1Н), 3,13 (шир. с, 1Н), 2,70-2,58 (м, 3Н), 2,11-1,91 (м, 4Н), 1,73-1,51 (м, 4Н), 1,30-1,16 (м, 5Н), 1,09 (д, J = 6,9 Гц, 18Н), 0,86 (д, J = 6,7 Гц, 3Н).
П р и м е р 34. Эндо-(1S*, 2S*)-2-(3-бензил-3-гидрокси-8-азабицикло/3.2.1/окт-8-ил-1-(4-гидроксифенил)- 1- пропанол
С помощью процедуры примера 3 целевой продукт предыдущего примера превращался в настоящий целевой продукт с выходом 38% после мгновенной хроматографии и перекристаллизации из смеси этилацетат/гексан; т.пл. 162-163оС.
13С-ЯМР: 156,97; 138,80. 135,86, 131,73, 129,11, 128,70, 127,07, 115,58, 76,09, 71,74, 64,64, 62,36, 54,62, 52,97, 45,82, 45,68, 29,28, 28,85, 14,50.
С помощью процедуры предыдущего примера целевой продукт примера 32 превращается в тот же самый продукт.
П р и м е р 35. Экзо-(1S*, 2S*)- и (1R*, 2S*)-1-(4-гидроксифенил)-2-(3-фенилспиро--8-азабицикло/3.2.1/октан-3,2-оксир ан)-8-ил)-1-пропанол
С использованием процедуры примера 3 экзо-(1S*, 2S*)/(1R*,2S*) смесь примера 31 превращалась в смесь настоящих целевых продуктов с выходом 93%. (1S*, 2S*)-изомер отделялся с помощью мгно- венной хроматографии и перекристаллизовывался из смеси эфира и гексана; т.пл. 115-117оС.
(1R*, 2S*)-изомер, полученный в виде второстепенного компонента (примерно 25%) в более поздних фракциях процесса хроматографии, также перекристаллизовывался из смеси эфира и гексана; т.пл. 107-110оС.
П р и м е р 36. 1-[4-(Триизопропилсилилокси(фенил)-2-(4-гидрокси-4-фенилпипери- дино)-1-пропанон
С помощью процедуры примера 1 4-гидрокси-4-фенилпиперидин превращался в настоящий целевой продукт с выходом 37% в виде светлого масла.
ЯМР 8,03 (д, J = 8,5 Гц, 2Н), 7,47 (д, J = 8 Гц, 2Н), 7,33 (т, J = 7,5 Гц, 2Н), 7,26-7,24 (м, 1Н), 6,89 (д, J = 8,5 Гц, 2Н), 4,08 (кв, J = 7,5 Гц, 1Н), 2,90-2,60 (м, 2Н), 2,25-2,10 (м, 2Н), 1,85-1,75 (м, 2Н), 1,65-1,55 (м, 2Н), 1,32-1,22 (м, 6Н), 1,10 (д, J = 7 Гц, 18Н).
П р и м е р 37. (1S*, 2S*)-1-[4-(Tриизопропилсилилокси)фенил]-2- (4-гидрокси-4-фенилпиперидино)-1-пропанол
С использованием процедуры примера 17 целевой продукт предыдущего примера превращался в настоящий целевой продукт с выходом 87%; т.пл. 148-151оС.
ЯМР 7,52 (д, J = 7 Гц, 2Н), 7,38 (т, J = 7 Гц, 2Н), 7,30-7,25 (м, 1Н), 7,19 (д, J = 8,5 Гц, 2Н), 6,84 (д, J = 8,5 Гц, 2Н), 4,23 (д, J = 9,5 Гц, 1Н), 3,13-3,02 (м, 1Н), 2,80-2,58 (м, 3Н), 2,30-2,08 (м, 2Н), 1,90-1,78 (м, 2Н), 1,29-1,17 (м, 3Н), 1,09 (д, J = 7 Гц, 18Н), 0,79 (д, J = 6,5 Гц, 3Н).
П р и м е р 38. (1S*, 2S)-1-(4-Гидроксифенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанол
Целевой продукт, полученный с использованием процедуры примера 3 из целевого продукта предыдущего примера с выходом 65%, перекристаллизовывался из этанола; т.пл. 202-204оС.
Найдено,%: С 71,95, Н 8,09; N 4,26.
С2Н5ОН
Вычислено,%: С 71,97; Н 8,05; N 4,00.
П р и м е р ы 39-76. С использованием методик предыдущих примеров получались следующие дополнительные соединения, при этом выход указывается по очищенному материалу на конечной стадии:
39. 2-(4-Бензил-4-гидроксипиперидино)-1-(4-гидроксифенил)-этанол; 42%; т.пл. 98-99oС (из этанола);
40. (1S*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1-(4-метоксифенил)-1-пропанол; 36%; т.пл. 145,5-146оС (из смеси этанол/эфир);
41. (1S*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1-(4-гидроксифенил)-1-пентанол; 55%; т.пл. 158-159оС (из смеси этанола и эфира);
42. (1R*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1-(4-гидроксифенил)-1-пентанол; 37%; т.пл. 156-157оС (из эфира);
43. (1S*, 2S*)-2-(4-бензил-4-гидроксипиперидино)-1-(4-гидроксифенил)-1-бутанол; 53%; т.пл. 190-191оС (из этанола);
44. (1S*, 2S*(-2-(4-бензил-4-гидроксипиперидино)-1-(4-метоксифенил)-1-бутанол; 61%; т.пл. 143-144оС (очищенный с помощью мгновенной хроматографии на силикагеле с использованием этила- цетат/гексанового градиента для элюирования).
45. 2-(4-Бензил-4-гидроксипиперидино)-1-(4-цианофенил)-этанол; 52%; т. пл. 142-143оС (из смеси этанол/эфир/гексан);
46. 2-(4-Бензил-4-гидроксипиперидино)-1-(2-гидроксифенил)этанол; 43%; т.пл. 172-173,6оС (из этанола);
47. 2-(4-Бензил-3-гидроксипиперидино)-1-(3-гидроксифенил)этанол; 76%; т.пл. 198-199оС (из этанола);
48. 1-(4-Хлорфенил)-2-(4-гидрокси-4-фенилпиперидино)-этанол; 63%; т.пл. 155,5-157оС (из смеси этанол/эфир);
49. (1S*, 2S*)-2-4-(4-хлорфенил)-4-гидроксипиперидино)-1-(4- гидроксифенил)-1-пропанол; 59%; т.пл. 204-206оС (из этилацетата);
50. 2-(4-Гидрокси-4-фенилпиперидино)-1-(2-тиенил/этанол; 54% ; т.пл. 167-168оС (из этанола);
51. (1S*, 2S*)-экзо-1-(4-гидроксифенил)-2-[3-(2-тиенилтио)-8-азабицикло/3.2.1/окт- 8-ил)-1-пропанол; 30%; т.пл. 127,5-129оС (из смеси этилацетат/гексан);
52. (1R*, 2S*)-экзо-1-(4-гидроксифенил)-2-3-(2-тиенилтио)-8-азабицикло/3.2.1/окт-8-ил) -1-п19%; т.пл. 141-142оС (из этилацетата);
53. (1S*, 2S*)-экзо-2-3-(4-хлорфенилтио)-8-азабицикло/3.2.1/окт- 8-ил)-1-(4-гидроксифенил)-1-пропанол; 84%; т.пл. 181-182оС (из этилацетата);
54. (1R*, 2S*)-экзо-2-3-(4-хлорфенилтио)-8-азабицикло/3.2.1/окт-8- ил)-1-(4-гидроксифенил)-1-пропанол; 94%; т.пл. 154-156оС (из смеси этилацетата и гексана);
55. (1S*, 2S*)-Экзо-1-(4-гидроксифенил)-2-3-(4-метоксифенилтио)- 8-азабицикло/3.2.1/окт-8-ил)-1-пропанол; 55%; т.пл. 118-119оС (из смеси этилацетата и гексана);
56. (1R*, S*)-экзо-1-(4-гидроксифенил)-2-3-(4-метоксифенилтио)- 8-азабицикло/3.2.1/окт-8-ил)-1-пропанол; 37%; т.пл. 72-75оС (мгновенная хроматография на силикагеле с элюированием с использованием этилацетат/гексанового градиента и растирания с гексаном);
57. 1-(4-Трифторметилфенил)-2-(4-гидрокси-4-фенилпиперидино) этанол; 34%; т.пл. 152-153оС (из смеси этанола и эфира);
58. 1-(4-Ацетамидофенил)-2-(4-гидрокси-4-фенилпиперидино)этанол; 34%; т.пл. 217-218оС (из этанола);
59. (1S*, 2S*)-1-(4-гидроксифенил)-2-[4-гидрокси-4-(2-фенилэтил)-пиперидино]-1- пропанол; 51%; т.пл. 200-201оС (из этилацетата);
60. (1S*, 2S*)-1-(4-гидроксифенил)-2-[4-гидрокси-4-(3-фенилпропил) пиперидино]-1-пропанол; 46%; т.пл. 200,5-201оС (из этилацетата);
61. (1S*, 2S*)-2-[4-(4-фторфенил)-4-гидроксипиперидино]-1-(4-гидроксифенил)-1-пропанол ; 37%; т.пл. 197-198оС (из этилацетата);
62. 1-(4-Цианофенил)-2-(4-гидрокси-4-фенилпиперидино)этанол; 51%; т.пл. 140-140,5оС (из смеси этанола и эфира);
63. (1S*, 2S*)-1-(4-гидроксифенил)-2-(4-гидрокси-4-(4-метилфенил) пиперидино)-1-пропанол; 41%; т.пл. 188-189оС;
64. 1-(4-Карбамоилфенил)-2-(4-гидрокси-4-фенилпиперидино)этанол; 3%; т. пл. 213,5-215оС (из этанола);
65. (1S*, 2S*)-1-(4-гидроксифенил)-2-(3-эндо-гидрокси-3-фенил-8- азабицикло/3.2.1/октан-8-ил)-1-пропанол; 60% ; т.пл. 216-217оС (из смеси этанола и эфира);
66. (1S*, 2S*)-1-(4-фторфенил)-2-(4-гидрокси-4-фенилпиперидино-1- пропанол; 44%; т.пл. 177-179оС (из этанола);
67. (1R*, 2S*)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанол; 25%; т.пл. 162-155оС (из этанола);
68. (1S*, 2S*)-2-(4-гидрокси-4-фенилпиперидино)-1-фенил-1-пропанол; 50% ; т.пл. 149-152оС (из смеси этилацетата и гексана);
69. (1S*, 2S*)-1-(4-хлорфенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанол; 38%; т.пл. 192-194оС (из этанола);
70. 1-(4-Аминофенил)-2-(4-гидрокси-4-фенилпиперидино)-этанол; 47%; т. пл. 156,5-158оС (из смеси этилацетата/эфир);
71. (1S*, 2S*)-2-(3-бензил-3-гидроксипирролидино)-1-(4-гидроксифенил)-1-про- панол; 71%; т.пл. 134-136оС;
72. (1S*, 2S*)-1-(4-Гидроксифенил)-2-(3-гидрокси-3-фенилпирролидино)-1-пропа- нол; 56%; т.пл. 74-78оС;
73. (1S*, 2S*)-1-(4-Хлорфенил)-2-(4-гидрокси-4-(2-фенилэтил) пиперидино)-1-пропанол; 33%; т.пл. 152-154оС (из этанола);
74. (1S*, 2S*)-1-(4-гидроксифенил)-2-[4-гидрокси-4-(4-фенилбутил)пиперидино]-1-пропано л; 44%; т.пл. 191-192оС (из этанола);
75. 1-(4-Карбоксифенил)-2-(4-гидрокси-4-фенилпиперидино)этанол; 98%; т. пл. 254,5-255оС (из воды);
76. 2-(4-Гидрокси-4-фенилпиперидино)-1-(4-метоксикарбонил)фенил)этанол; 57%; т.пл. 138,5-139,5оС (из смеси этанола) и эфира.
П р и м е р 77. (R-1-(4-Хлорфенил)-2-(4-гидрокси-4-фенилпиперидино)этанол
4-Гидрокси-4-фенилпиперидин (177 мг, 1 ммоль) растворяли в сухом тетрагидрофуране (13 мл) и охлаждали до -15оС при перемешивании в атмосфере азота. По каплям на протяжении 3 мин добавляли бутиллитий (0,8 мл, 2 ммоль, 2,5 н.).Oксид (-)-R-1-(4-хлорфенил)этилена (155 мг, 1 ммоль), J. Am. Chem. cos.,  , 7925 (1987); J.Org. Chem.
, 7925 (1987); J.Org. Chem.  , 2861 (1988) растворяли в 1 мл тетрагидрофуране и добавляли к холодной реакционной смеси с прополаскиванием (1 мл). Смесь подогревали до комнатной температуры и наконец нагревали с обратным холодиль- ником на протяжении ночи. Смесь охлаждали до комнатной температуры и гасили твердым бикарбонатом натрия. Неочищенная реакционная смесь непосредственно хроматографировалась на силикагеле с использованием элюирования этилаце- тат/гексановым градиентом. Продукт-содержащие фракции осторожно повторно хроматографировались на силикагеле с элюированием смесью 50% этилацетат/гексан, давая 112 мг (33%) продукта в виде маслянистой пены. Растирание со смесью эфир-гексан давало 15,2 кремово-окрашенного продукта, который имел т.пл. 110-113оС; [α ]D = -18o.
, 2861 (1988) растворяли в 1 мл тетрагидрофуране и добавляли к холодной реакционной смеси с прополаскиванием (1 мл). Смесь подогревали до комнатной температуры и наконец нагревали с обратным холодиль- ником на протяжении ночи. Смесь охлаждали до комнатной температуры и гасили твердым бикарбонатом натрия. Неочищенная реакционная смесь непосредственно хроматографировалась на силикагеле с использованием элюирования этилаце- тат/гексановым градиентом. Продукт-содержащие фракции осторожно повторно хроматографировались на силикагеле с элюированием смесью 50% этилацетат/гексан, давая 112 мг (33%) продукта в виде маслянистой пены. Растирание со смесью эфир-гексан давало 15,2 кремово-окрашенного продукта, который имел т.пл. 110-113оС; [α ]D = -18o.
(S)-1-(4-хлорфенил)-2-(4-гидрокси-4-фе- нилпиперидино)этанол, имеющий те же физические свойства, за исключением знака вращения, приготавливали таким же образом из (+)-S-1-(4-хлорфенил)этиленоксида.
П р и м е р 78. Энантиомерные (1S, 2S)- и (1R, 2R)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанолы
(+)-Винную кислоту (300 мг, 2 ммоль) растворяли в 30 мл теплого метанола. Добавляли сразу весь рацемический 1S*, 2S*-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенил- пиперидин)пропанол (655 мг, 2 ммоль). При перемешивании и мягком подогреве получался бесцветный гомогенный раствор. После стояния при температуре окружающей среды в течение 24 ч получалось 319 мл (66% ) мелкокристаллического белого осадка. Данный продукт перекристаллизовывался из метанола, давая 263 мг (+)-тартратной соли правовращающего целевого продукта в виде белого твердого вещества; т.пл. 206,5-207,5оС; [ α]D = -36,2оС. Данную соль (115 мг) добавляли к 50 мл насыщенного бикарбоната натрия. Этилацетат (5 мл) и указанную смесь энергично перемешивали в течение 30 мин. Водная фаза повторно экстрагировалась этилацетатом. Органические соли объединялись и промывались солевым раствором, сушились над сульфатом кальция, и концентрировались. Рыжевато-коричневый остаток перекристаллизовывался из смеси этилацетат/гексан, давая 32 мг (39%) белого правовращающего целевого продукта; т.пл. 203-204оС; [ α]D = +56,9о.
Найдено,%: С 72,61; Н 7,45; N 4,21.
С20Н25NO3
Вычислено,%: С 73,37; Н 7,70; N 4,28.
Фильтрат после получения (+)-тартратной соли выше обрабатывали 100 мл насыщенного водного бикарбоната натрия и экстрагировали этилацетатом. Объединенные органические экстракты промывались солевым раствором, сушились над сульфатом кальция и концентрировались, давая 380 мг регенерированного исходного материала (частично расщепленного). Данный материал обрабатывался (-)-винной кислотой (174 мг) в 30 мл метанола, как указано выше. После стояния в течение 24 ч, фильтрование давало 320 мг (66%) продукта, который далее перекристаллизовывался из метанола, давая 239 мг (-)-тетратной соли левовращающего целевого продукта; т.пл. 206,5-207,5оС; [α ]D = +33,9о. Последняя превращалась в левовращающий целевой продукт описанным выше способом с выходом 49%; т.пл. 204-205оС; [ α]D = -58,4о.
Найдено,%: С 72,94; Н 7,64; N 4,24.
П р и м е р 79. (1S*, 2S*)-1-(4-Гидроксифенил)-2-(1,2,3,6-тетрагидро-4-фенилпири- до)-1- пропанол
С помощью способов примеров 1, 2 и 3 4-(триизопропилсилилокси)- -бромпропиофенон и 1,2,3,6-тетрагидро-4-фенилпиридин превращались в настоящий целевой продукт с выходом 14% на последней стадии; т.пл. 208-211оС (разл.).
Приготовление 1 8-(2,2,2-Трихлорэтоксикарбонил)-3-фе-нилметилен-8-азабицикло/3.2.1/октан
Раствор бензилтрифенилфосфонийхлорида (13,26 г, 43,1 ммоль) в тетрагидрофуране (400 мл) охлаждали до -78оС и добавляли бутиллитий (13,6 мл, 2,5М в гексане, 34 ммоль). Это давало в результате гетерогенную органическую смесь, которую перемешивали 5 мин при -78оС и затем подогревали до 0оС. Раствор становился почти гомогенным красным и добавляли 11-2,2,2-трихлорэтоксикарбонилпропинон (7,1 г, 23,3 ммоль; Montzka и др., Tetrahedron Letters, т.14, с. 1325, 1974) в тетрагидрофуране (20 мл с 20 мл жидкости после ополаскивания). Pеакционную смесь нагревали при температуре дефлегмации в течение 4 дней, охлаждали и фильтровали. Концентрирование фильтра давало вязкое коричневое масло. Мгновенная хроматография на силикагеле (3 х 6 дюймов) с использованием сначала гексана, а затем этила- цетат/гексанового градиента давало 7,87 г белого твердого вещества (74%); т.пл. 88-89оС; ИК (KBr): 3437, 2958, 1700, 1445, 1425, 1321, 1125, 711.
Найдено,%: С 54,89; Н 4,82; N 3,77.
Вычислено,%: С 54,9; Н 4,84; N 3,74.
Приготовление 2
3-Фенилметилен-8-азабицикло/3.2.1/октан
Смесь целевого продукта предыдущего приготовления (1,0 г, 2,67 ммоль), порошка цинка (0,88 г, 46 ммоль) и уксусной кислоты (50 мл) перемешивалась в течение ночи. Реакционную смесь затем нагревали при 70оС в течение 22 ч, затем охлаждали и концентрировали. Остаток распределяли между смесью простой эфир/этилацетат и насыщенным бикарбонатом натрия, и смесь перемешивалась в течение 30 мин и затем фильтровали через диатомовую землю. Водная фаза отделялась и затем экстрагировалась этилацетатом (2х). Объединенные органические слои промывались водой и солевым раствором, сушились (CaSO4) и концентрировались, давая 0,5 г светло-желтого масла. Дальнейшая очистка осуществлялась путем взятия масла в 10%-ную соляную кислоту. Этот кислотный раствор экстрагировали этилацетатом (2х). Кислотный слой нейтрализовали надо льдом гидроксидом натрия и вновь экстрагировали этилацетатом. Этот органический слой сушили (CaSO4) и концентрировали, получая 0,24 г (40%) светло-желтого масла;
ЯМР 7,24 (м, 2Н), 7,13 (д, J= 6,6 Гц, 3Н), 6,28 (с, 1Н), 3,52 (шир.д, J = 28,2 Гц, 2Н), 2,56 (т, J = 15,4 Гц, 3Н), 2,26 (д, J = 17,7 Гц, 1Н), 2,12 (д, J = 13,9 Гц, 1Н), 1,64 (м, 3Н), 1,40 (м, 1Н).
Альтернативно продукт превращается в его соль соляной кислоты путем обработки ацетонового раствора газообразным хлористым водородом.
Приготовление 3
4-(трет-Бутилдиметилсилилокси)пропи- офенон
4-Гидроксипропиофенон (15 г, 100 ммоль) и имидазол растворяли в диметилформамиде (50 мл). трет-Бутилдиметилсилилхлорид (19,6 г, 130 ммоль) в диметилформамиде (40 мл) добавляли по каплям при комнатной температуре. Смесь перемешивали в течение 18 ч, затем разбавляли водой (300 мл) и экстрагировали эфиром (4 х 200 мл). Объединенные эфирные слои промывались 1М LiCl и солевым раствором, сушились (CaSO4), концентрировались до получения твердого маслянистого вещества, мгновенная хроматография на силикагеле с использованием смеси 1: 10 этилацетат/гексан в качестве элюента давала 26 г целевого продукта, который затем очищался с помощью прогонки, давая 23,2 г (88%) очищенного целевого продукта в виде гигроскопического белого твердого вещества; т.пл. 30-31оС.
ЯМР 7,76 (д, J = Гц, 2Н), 6,74 (д, J = Гц, 2Н), 2,82 (кв, J = Гц, 2Н), 1,09 (т, J = Гц, 3Н), 0,87 (с, 9Н), 0,11 (с, 6Н).
Приготовление 4
4-(трет-Бутилдиметилсилилокси)-α -бром- пропиофенон
4-(трет-Бутилдиметилсилилокси(пропи- офенон (20 г, 75,8 ммоль) растворялся в уксусной кислоте (300 мл), и добавлялся бром (3,9 мл, 75,8 ммоль) в (30 мл) уксусной кислоты к раствору. Оранжевая окраска брома продолжала оставаться в течение приблизительно 1 мин и затем реакционная смесь быстро обесцвечивалась, как только добавляли бром. Реакционная смесь перемешивалась еще в течение 1 ч, затем концентрировалась, и мгновенная хроматография остатка на силикагеле (элюирование гексаном) давала 7,12 г маслянистого продукта.
ЯМР 7,92 (д, J = 9 Гц, 2Н), 6,86 (д, J = 9 Гц, 2Н), 5,22 (кв, J = 6,5 Гц, 1Н), 1,85 (д, J = =6,5 Гц, 3Н), 0,96 (с, 9Н), 0,22 (с, 6Н).
13С ЯМР: 76,73; 59,55; 40,47; 38,59; 37,68; 25,42.
Приготовление 5
О-метансульфонилтропин
Тропин (14,2 г, 100 ммоль) растворяли в метиленхлориде (210 мл) и триэтиламин (23 мл, 160 ммоль) добавляли к раствору. Метансульфонилхлорид (9,3 мл, 120 ммоль) быстро добавляли по каплям. Это вызывало мягкое дефлеглирование метиленхлоридного раствора. Далее смесь перемешивалась в течение 1 ч, затем экстрагировалась холодным 0,5 М гидроксидом натрия, водой, солевым раствором, сушилась путем фильтрации через бумагу для разделения фаз и концентрировалась, давая 13,8 г (65%) целевого продукта в виде желтого твердого вещества.
ЯМР: 4,88 (т, J = 5 Гц, 1Н), 3,10-3,05 (м, 2Н), 2,94 (с, 3Н), 2,22 (с, 3Н), 2,20-2,10 (м, 2Н), 2,02-1,88 (м, 6H).
Приготовление 6
3-Фенилтио-8-метил-8-азабицикло/3.2. 1/октан
NaH (60% в масле; 2,77 г, 69 ммоль) промывали трижды гексаном и затем суспендировали в тетрагидрофуране (300 мл). Тиофенол (6,5 мл), 63 ммоль) в тетрагидрофуране (25 мл) добавлялся по каплям в течение 5 мин. Молочно-белую суспензию, которая образовывалась с выделением водорода, перемешивали 10 мин и затем сразу весь добавляли О-метансульфонилтропин (13,8 г, 63 ммоль в 25 мл тетрагидрофурана). Смесь нагревали при температуре дефлегмации в течение ночи, охлаждали и фильтровали через диатомовую землю с промывкой простым эфиром. Фильтрат разбавляли этилацетатом и промывали холодным 1М NaOH, водой и солевым раствором, сушили (CaSO4) и концентрировали, получая 11,48 г (78%) целевого продукта в виде желтого твердого вещества.
ЯМР: 7,50-7,18 (м, 5Н), 3,32-3,21 (м, 1Н), 3,15-3,09 (м, 2Н), 2,25 (с, 3Н), 2,02-1,94 (м, 2Н), 1,79-1,72 (м, 4Н), 1,60-1,51 (м, 2Н).
13С ЯМР: 134,8; 132,3; 128,8; 126,9; 61,16; 39,21; 38,38; 37,72; 26,42.
Приготовление 7
3-Фенилтио-8-(2,2,2-трихлорэтоксикар- бонил)-8-азабицикло/3.2.1/октан
Целевой продукт предыдущего приготовления (11,48 г, 49,3 ммоль) и карбонат калия (0,75 г, 5,4 ммоль) смешивали с бензолом (200 мл), и быстро по каплям добавляли 2,2,2-трихлорэтилхлорформат (7,5 мл, 54,4 ммоль). Реакционную смесь нагревали при температуре дефлегмации 2 ч, охлаждали, фильтровали и концентрировали. Оранжевый маслянистый остаток растворяли в метиленхлориде, промывали насыщенным бикарбонатом натрия и затем солевым раствором, сушили (CaSO4) и концентрировали. Остаток очищался мгновенной хроматографией на силикагеле (элюирование гексаном и затем 5%-ной смесью этилацетат/гексан), давая сначала непрореагировавший тиофенол от предыдущей реакции и затем целевой продукт в виде желтого масла. (13 г, 67%).
ЯМР 7,42-7,23 (м, 5Н), 4,72 (АВ кв, J =12 Гц, 2Н), 4,35-4,30 J (м, 4Н), 2,73 (гептет, J = =6 Гц, 1Н), 2,05-1,68 (м, 6Н).
Масло отверждалось путем растирания с гексаном; т.пл. 83-84,5оС.
Найдено,%: С 48,47; Н 4,58; N 3,49.
Вычислено,%: С 48,58; Н 4,60; N 3,55.
Приготовление 8
3-Фенилтио-8-азабицикло/3.2.1/октан
Целевой продукт предыдущего приготовления (13,0 г, 33 ммоль) растворяли в уксусной кислоте (400 мл) и добавляли порошкообразный цинк (11 г, 168 ммоль). Смесь нагревали до 10оС в течение ночи, затем концентрировали и остаток разделяли между метиленхлоридом и насыщенным бикарбонатом натрия. Полученную в результате эмульсию очищали фильтрацией через диатомовую землю. Фазы разделялись и органический слой сушился с использованием фильтровальной бумаги для разделения фаз и концентрировался, давая 6,1 г (84%) целевого продукта в виде желтого масла, которое затвердевало при стоянии.
ЯМР: 7,38-7,36 (м, 2Н), 7,29-7,20 (м, 3Н), 3,52 (с, 2Н), 3,36 (гептет, J = 6 Гц, 1Н), 1,94-1,54 (м, 8Н).
13С ЯМР: 134,0; 132,43; 128,83; 127,06; 54,93; 40,81; 39,01; 28,98.
Приготовление 9.
4-(Триизопропилсилилокси)пропиофе-нон
С помощью метода приготовления 3 4-гидроксипропиофенон и триизопропилсилилхлорид превращался в настоящий целевой продукт в виде светлого масла со 100%-ным выходом.
ЯМР: 7,87 (д, J = 8,5 Гц, 2Н), 6,89 (д, J = 8,5 Гц, 2Н), 2,94 (кв, J = 7 Гц, 2Н), 1,32-1,15 (м, 3Н), 1,20 (т, J =7 Гц, 3Н), 1,09 (д, J = 7 Гц, 18Н).
Приготовление 10
4-(Триизопропилсилилокси)- α-бромпропиофенон
Целевой продукт предыдущего приготовления (60,63 г, 198 ммоль) растворяли в четыреххлористом углероде (1100 мл) и добавляли по каплям раствор брома (11 мл, 210 ммоль в 60 мл CCl4). После того, как была добавлена порция раствора брома, без какого-либо заметного обесцвечивания спустя 15 мин, двумя порциями добавляли уксусную кислоту (1,0 мл). Раствор обесцвечивался в течение 20 мин и добавление завершалось довольно быстро Смесь перемешивали еще 15 мин; затем летучий HBr частично удаляли с помощью потока азота. Реакционную смесь выливали в воду (600 мл), и фазы разделялись. Органический слой промывался водой, насыщался бикарбонатом натрия, водой и солевым раствором, сушился (CaSO4) и концентрировался, давая 76,2 г (100%) целевого продукта в виде прозрачно-желтого масла.
ЯМР 7,94 (д, J = 9 Гц, 2Н), 6,91 (д, J = 9 Гц, 2Н), 5,24 (кв, J = 6,5 Гц, 1Н), 1,87 (д, J = 6,5 Гц, 2Н), 1,33-1,17 (м, 3Н), 1,10 (д, J = 7 Гц, 18Н).
Приготовление 11
4-Фтор- α-бромпропиофенон
Целевой продукт, приготовленный, как в приготовлении 4, с 86%-ным выходом после дистилляции, перекристаллизовывался из этанола, давая более кристаллическое твердое вещество; т.пл. 33-34оМ.
Найдено,%: С 46,47; Н 3,38.
Вычислено,%: С 4,78; Н 3,49.
Приготовление 12
4-Хлор- α - бромпропиофенон
Продукт, приготовленный с помощью процедуры 10 с 98%-ным выходом, перекристаллизовывался из этанола; т.пл. 78-79оС.
Найдено,%: С 43,74; Н 3,17.
Вычислено,%: С 43,67; Н 3,26.
Приготовление 13
Эндо- и экзо-8-(2,2,2-трихлорэтоксикарбонил)-3-фенилспиро-8-азабицик- ло/3.2.1/октан-3,2-оксиран)
Целевой продукт приготовления 2 (5,0 г, 13,34 ммоль) растворялся в дихлорметане (80 мл) и добавлялась м-перхлорбензойная кислота (2,71 г, 13,35 ммоль), 85%-ная чистота). После перемешивания при подходящей температуре в течение ночи смесь экстрагировалась насыщенным бикарбонатом натрия, затем водой и солевым раствором, сушилась с помощью фазоразделительной бумаги и концентрировалась, давая 5,3 г стекловидного желтого масла, которое хроматографировалось на силикагеле (элюирование градиентом этилацетат/гексан). Выделенный исходный материал элюировался первым, а затем быстро продвигающийся эндо-эпоксидный продукт (2,23 г, 42,8%); т.пл. 78-79оС.
Найдено,%: С 52,75; Н 4,44; N 3,20.
Вычислено, % : С 52,26; Н 4,64; N 3,59; и наконец медленно двигающийся экзо-эпоксидный продукт (2,32 г, 44,5%); т.пл. 107-108оС.
Найдено,%: С 52,34; Н 4,40; N 3,54.
Вычислено как для эндо-изомера.
Приготовление 14
Эндо- и экзо-3-фенилспиро-8-азабицикло/3.2.1/октан-3,2-оксиран
Быстро продвигающийся эндо-продукт предыдущего примера (1,55 г, 3,97 ммоль) растворяли в тетрагидрофуране (30 мл, и добавляли цинковый порошок (9,3 г, 142 ммоль) и 1М монокалий фосфат (10 мл). После перемешивания на протяжении ночи смесь разбавляли водой (10 мл), величина рН доводилась до 10-11 с помощью NaCO3 и фильтровалась через диатомовую землю с промывкой этилацетатом и водой. Водный слой в объединенном фильтрате и промывные отделяли и промывали свежим этилацетатом. Объединенные органическое слои промывали солевым раствором, сушили (сульфатом кальция) и отпаривались, давая 0,85 г (100%) эндо-целевого продукта в виде желтого масла. Перегонка (температура бани 110-115оС, 0,5 мм) давала эндо-целевой продукт в виде прозрачного бесцветного масла.
ЯМР: 7,34-7,23 (м, 5Н), 3,63-3,60 (м, 2Н), 3,55 (м, 1Н), 2,36 (д.д, J = 14,1 и 3,5 Гц, 1Н), 2,17-2,12 (м, 2Н), 1,83-1,81 (м, 2Н), 1,70-1,61 (м, 2Н), 1,42 (дт, J =11,9, 2,2 Гц, 1Н), 1,18 (дт, J = 14,5, 2,3 Гц, 1Н).
НРМS 215.1301.
Вычислено: 215.1308.
С помощью такого же самого способа медленно продвигающийся изомер предшествующего приготовления превращался в соответствующий экзо-изомер с выходом 96%, очищался с помощью перегонки (110-125оС температура бани, 0,8 мм); т.пл. 114,5-116оС.
Найдено,%: С 77,97; Н 8,05; N 6,44.
Вычислено,%: С 78,10; Н 7,96; N 6,51.
Использование: в качестве лекарственных средств при лечении приступов или дегенеративных заболеваний центральной нервной системы. Сущность изобретения: продукт - 2-(8-азабицикло[3,2,1]окт-8-ил)алкалоны формулы 1, где R - H или C1-C3 Q - группа -S- или -CH-CH-; X - Н, галоген, OR1 COOR1 NH2 CONH2 или R1CONH где R1 - Н или C1-C3 D-группа формулы 2, где при Y-Н, Y1 -бензил, незамещенный или 4-галоген или 4-метоксизамещенный фенилтио или 2-тиенилтио, при Y-ОН, Y1 - фенил или бензил, или Y4 Y1 вместе образуют группу CH-C6H5 или группа формулы 3, или группа формулы 4, где при Y2 - водород, Y3 - бензил или (O)nSC6H5, где n = 0 или 2, при Y2 -ОН, Y3 - группа формулы 5, где m = 0,1,2,3 или 4 и R2-H - Н метил или галоген, или D - группа формулы 6, где l = 0 или 1, или D - группа формулы 7. Реагент 1: 3-фенилметилен-8-азабицикло [3,2,1] октан. Реагент 2: 4-(триизопропилсилокси)- α -бромпропиофенон. Реагент 3: триэтиламин. Условия реакции: нагревание с обратным холодильником. Реагент 4: литийалюминийгидрид. Условия реакции: в тетрагидрофуране при комнатной температуре. 16 з.п. ф-лы. Структура формулы 1,2,3,4,5,6 и 7: 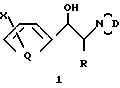
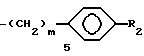

где R - водород или C1 - C3-алкил;
Q - группа - CH=CH - или - S -;
X - водород, галоген, OR1, COOR1, NH2, CONH2 или R1CONH, где R1 - водород или C1 - C3-алкил;
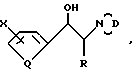
D - группа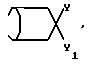
где при Y - водород
Y1 - бензил, незамещенный или 4-галоген, или 4-метоксизамещенный фенилтио или 2-тиенилтио, при Y - OH Y1 - фенил или бензил, или Y и Y1 вместе образуют группу =CH - C6H5 или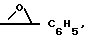 или группа
или группа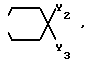
где при Y2 - водород Y3 - бензил или (O)nSC6H5, где n = 0 или 2, при Y2 - OH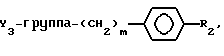 где m = 0,1, 2, 3, или 4,
где m = 0,1, 2, 3, или 4,
R2 - водород, метил или галоген,
или D - группа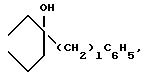
где l = 0 или 1,
или D - группа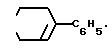
2. Соединение по п.1, отличающееся тем, что Q представляет - CH=CH - , X является заместителем в 4-положении фенильного кольца и представляет собой гидрокси, фтор или хлор, D представляет собой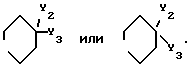
3. Соединение по п.2, отличающееся тем, что R представляет метил, имеющий 1S*, 2S* относительную стереохимию: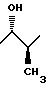
4. Соединение по п.3, отличающееся тем, что X представляет 4-гидрокси и Y3 - бензил.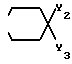
8. Соединение по п.7, отличающееся тем, что Y2 представляет собой гидрокси и Y3 - фенил.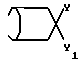
12. Соединение по п.11, отличающееся тем, что R представляет метил, имеющий 1S*,2S* относительную стереохимию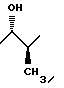
13. Соединение по п.12, отличающееся тем, что Q представляет собой - CH= CH - и X - гидрокси.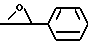
16. Соединение по п.13, отличающееся тем, что Y и Y1 взяты отдельно.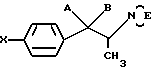
где X - галоген или защитная группа, такая, как бензилокси или тринизшийалкилсилилоксигруппа;
А и B вместе образуют = 0 или один из А или B - гидроксил, а другой - водород;
E - группа
где при Y - водород Y1 - C6H5S, при Y - OH Y1 - фенил или бензил, или Е - группа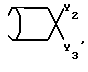
где при Y2 - водород Y3 - C6H5S, при Y2 - OH Y3 - бензил или Y2 и Y3 вместе образуют группу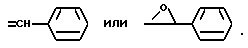
| Патент США N 3294804, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

