ПЕРЕКРЕСТНАЯ СВЯЗЬ С ПРИОРИТЕТНЫМИ ЗАЯВКАМИ
По настоящему изобретению испрашивается приоритет на основании предварительных заявок на патент США № 60/971654, поданной 12 сентября 2007 года; 60/953610, поданной 2 августа 2007 года; 60/953613, поданной 2 августа 2007 года, и 60/953614, поданной 2 августа 2007 года, каждая из которых включена в данное изобретение путем ссылки во всей своей полноте.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду, его новым солевым формам, способам их получения, новым промежуточным соединениям и способам лечения большого числа состояний и нарушений, включая те, которые связаны с дисфункцией центральной и вегетативной нервной системы.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Было показано, что нейронные никотиновые рецепторы (NNR), характерные для центральной нервной системы (ЦНС), существуют в виде нескольких подтипов, наиболее обычными среди которых являются подтипы α4β2 и α7. См., например, Schmitt, Current Med. Chem. 7:749 (2000), включенную в данное описание путем ссылки. Предполагалось, что лиганды, которые взаимодействуют с подтипом α7 NNR, могут использоваться при лечении множества болезненных состояний и нарушений. Основное представление в отношении подтипа α7 нейронного никотинового рецептора раскрыто в публикации Mazurov et al., Curr. Med. Chem. 13: 1567-1584 (2006) и процитированных там ссылках, включенных в данное описание путем ссылки. Известными аналогами таких состояний и нарушений являются нарушения сознания, шизофрения, воспаление, ангиогенез, невротическая боль и фибромиалгия.
При посмертном исследовании в ткани мозга пациентов с шизофренией обнаружено пониженное число гиппокампальных NNR. Также существует улучшенный психологический эффект у курящих пациентов с шизофренией по сравнению с некурящими. Никотин улучшает дефицит сенсорного отбора у животных и шизофреников. Блокада подтипа α7 NNR индуцирует дефицит отбора, аналогичный наблюдаемому при шизофрении. См., например, Leonard et al., Schizophrenia Bulletin 22(3): 431 (1996), включенную в данное описание путем ссылки. Биохимические, молекулярные и генетические исследования сенсорного процессинга у пациентов с Р50 вызванным слухом потенциальным дефицитом отбора дают основание предположить, что подтип α7 NNR может функционировать в качестве ингибирующего нейронного пути. См., например, Freedman et al., Biological Psychiatry 38(1): 22 (1995), включенную в данное описание путем ссылки.
Позднее было предположено, что α7 NNR являются медиаторами ангиогенеза, как описано в публикации Heeschen et al., J. Clin. Invest., 100:527 (2002), включенной путем ссылки. В данных исследованиях было показано, что ингибирование подтипа α7 уменьшает воспалительный ангиогенез. Рецепторы α7 NNR также были предложены в качестве мишеней контролируемого нейрогенеза и роста опухоли (Utsugisawa et al., Molecular Brain Research 106(1-2): 88 (2002) и патентная заявка США 2002/0016371, каждая из которых включена в данное изобретение путем ссылки). Наконец, недавно была выявлена роль подтипа α7 в когнитивной способности (Levin and Rezvani, Current Drug Targets: CNS and Neurological Disorders 1(4): 423 (2002)), нейропротекции (O'Neill et al., Current Drug Targets: CNS and Neurological Disorders 1(4): 399 (2002) и Jeyarasasingam et al., Neuroscience 109(2): 275 (2002)) и невропатической боли (Xiao et al., Proc. Nat. Acad. Sci. (US) 99(12): 8360 (2002)), каждое цитирование включено в данное описание путем ссылки.
Сообщалось, что различные соединения взаимодействуют с рецепторами α7 NNR и на данном основании они были предложены в качестве лечения. См., например, РСТ WO 99/62505, PCT WO 99/03859, PCT WO 97/30998, PCT WO 01/36417, PCT WO 02/15662, PCT WO 02/16355, PCT WO 02/16356, PCT WO 02/16357, PCT WO 02/16358, PCT WO 02/17358, Stevens et al., Psychopharm. 136: 320 (1998), Dolle et al., J. Labelled Comp. Radiopharm. 44: 785 (2001) и Macor et al., Bioorg. Med. Chem. Lett. 11:319 (2001) и процитированные там ссылки, данные ссылки включены в настоящее описание путем ссылки касательно предшествующих указаний по поводу α7 NNR и предлагаемого лечения. Среди данных соединений общим структурным мотивом является замещенный третичный бициклический амин (например, хинуклидин). Также сообщалось, что аналогичные замещенные хинуклидиновые соединения связываются с мускариновыми рецепторами. См., например, патенты США № 5712270, Sabb, и РСТ WO 02/00652 и WO 02/051841, каждый из которых включен в данное описание путем ссылки касательно таких соединений.
Ограничение некоторых никотиновых соединений заключается в том, что они связаны с различными нежелательными побочными действиями, например, со стимулированием мышечных и ганглионарных рецепторов. Это приводит к продолжающейся необходимости в соединениях, композициях и способах предупреждения или лечения различных состояний или нарушений, таких как нарушения ЦНС, включая облегчения протекания симптомов данных нарушений, где соединения проявляют характерное для никотиновых соединений фармакологическое действие вместе с благоприятным эффектом, а именно на функционирование ЦНС, но без значительных связанных с этим побочных действий. Существует необходимость в соединениях, композициях и способах, которые влияют на функцию ЦНС без существенного затрагивания тех подтипов никотиновых рецепторов, которые способны вызывать нежелательные побочные действия, такие как заметная активность в сердечно-сосудистых сайтах и сайтах скелетной мускулатуры. В настоящем изобретении разработаны такие соединения, композиции и способы.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном аспекте настоящее изобретение относится к (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду или его фармацевтически приемлемой соли. Другой аспект относится к (2S,3R)-N-(2-((3- пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду по существу в чистом виде или к его фармацевтически приемлемой соли. Следующий аспект относится к (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду, по существу не содержащему (2S,3S), (2R,3S) или (2R,3R) изомеров, или к его фармацевтически приемлемой соли.
Дополнительно, другой аспект относится к стереохимически обогащенному (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду или его фармацевтически приемлемой соли. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 90% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 95% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 98% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 99% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 99,5% или больше.
Другой аспект настоящего изобретения относится к соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида с кислотой, где кислоту выбирают из хлористоводородной кислоты, серной кислоты, фосфорной кислоты, малеиновой кислоты, п-толуолсульфоновой кислоты, галактаровой (слизевой) кислоты, D-миндальной кислоты, D-винной кислоты, метансульфоновой кислоты, R- и S-10-камфорсульфоновой кислоты, кетоглутаровой кислоты или гиппуровой кислоты. В одном варианте осуществления стехиометрическое соотношение (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида к кислоте составляет 2:1, 1:1 или 1:2. В одном варианте осуществления стехиометрическое соотношение составляет 1:1. Один вариант осуществления настоящего изобретения представляет собой гидрохлорид (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его гидрат или сольват, включая частичные гидраты или сольваты. Следующий вариант осуществления представляет собой моногидрохлорид (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его гидрат или сольват, включая частичные гидраты или сольваты.
Настоящее изобретение также относится к масштабируемым синтезам (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида и новых промежуточных соединений.
Объем настоящего изобретения включает все комбинации аспектов, вариантов осуществления и описанных в данном изобретении преимуществ.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг.1А1-1А4 проиллюстрированы ответные реакции рецепторов α7 крысы, экспрессированных в клетках GH4C1 млекопитающих, на (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид; рацемат, т.е. смесь (2S,3R), (2R,3S), (2R,3R) и (2S,3S); индивидуальные стереоизомеры и ацетилхолин (ACh).
На фиг.1В проиллюстрировано сравнение функциональных ответных реакций рецепторов α7 крысы, экспрессированных в клетках GH4C1 млекопитающих, на (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид; рацемат, т.е. смесь (2S,3R), (2R,3S), (2R,3R) и (2S,3S); и индивидуальные стереоизомеры в рамках эффективного диапазона концентраций в плазме крови.
На фиг.2А проиллюстрированы ответные реакции рецепторов α7 человека, экспрессированных в ооцитах Xenopus, на (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид.
На фиг.2В проиллюстрированы контрольные ответные реакции рецепторов α7 человека после применения соединения в указанных концентрациях. Данные были нормализованы к результирующему заряду контрольных ответных реакций на 300 мкМ Ach, полученных за 5 минут перед экспериментальными ответными реакциями, вызванными агонистом. Каждая точка представляет собой среднее значение ± статистическая ошибка (SEM) нормализованного ответа по крайней мере для 4 ооцитов.
На фиг.3 проиллюстрирована оценка познавательной способности в парадигме объекта распознавания (OR), демонстрирующая, что (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид оказывает положительное действие при дозе 0,3 и 1 мг/кг, введенной i.p. (внутрибрюшинно), p*<0,5.
На фиг.4 проиллюстрирована оценка познавательной способности в парадигме OR, демонстрирующая, что (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид, введенный перорально, оказывает положительное действие в широком диапазоне концентраций (0,3-10 мг/кг), p*<0,5.
На фиг.5 проиллюстрировано действие (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, введенного внутрибрюшинно, на предотвращение когнитивного дефицита, вызванного МК-801, также известного как дизоцилпин, коммерчески доступный неконкурентный антагонист рецептора NMDA, в задаче OR.
На фиг.6 проиллюстрировано, что среднее время, проведенное у объекта А относительно объекта В в задаче OR в испытании для группы, обработанной носителем, через 30 минут, 6 часов или 24 часа после конечного подострого введения (перорально), существенно не различалось (р=0,17, р=0,35 и р=0,12, соответственно). Альтернативно, через 30 минут, 2 часа, 6 часов или 18 часов после конечного подострого введения (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида в дозе 0,3 мг/кг, субъекты проводили существенно больше времени (P<0,05), исследуя объект В (новый), чем объект А (знакомый). Более того, в момент времени 2 часа (75%) и 6 часов (71%) индекс распознавания был существенно лучше у животных, обработанных (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамидом в дозе 0,3 мг/кг, по сравнению с индексом распознавания (54%) у группы, обработанной носителем, через 30 минут после конечного введения.
На фиг.7 проиллюстрирована оценка познавательной способности в парадигме лабиринта с угловым рычагом (RAM). (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид (0,1, 0,3 и 1,0 мг/кг) вводили перорально за 30 минут перед дневными занятиями. Улучшение решения задачи было очевидно в группе, получавшей 0,3 мг/кг (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида во время второй недели введения.
На фиг.8 проиллюстрировано исследование антипсихотического действия, измеренного как гиперактивное поведение, вызванное допаминовой сверхстимуляцией, показывающее, что (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид (0,3 и 1,0 мг/кг; подкожно) ослабляет локомоторную гиперактивность, вызванную апоморфином (1,0 мг/кг) после подкожного введения крысам.
На фиг.9 проиллюстрирована антипсихотическая оценка, предымпульсное ингибирование, указывающая, что вызванный апоморфином дефицит является обратимым при предварительной обработке (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамидом после подкожного введения.
На фиг.10А проиллюстрированы результаты рентгенодифракционного кристаллографического анализа моногидрохлорида (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, установившего абсолютную стереохимию вещества. Показанное соединение представляет собой частично гидратированную гидрохлоридную соль, что показано с помощью полностью разупорядоченного хлоридного аниона и частичного размещения молекулы воды в асимметрической ячейке.
На фиг.10В проиллюстрированы результаты рентгенодифракционного кристаллографического анализа моногидрохлорида (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, установившего абсолютную стереохимию вещества, изображенные со схемой нумерации атомов для ссылки. Представлен вид вниз относительно кристаллографической b-оси элементарной ячейки. Межмолекулярные водородные связи показаны пунктирными линиями.
На фиг.11А проиллюстрированы результаты рентгенодифракционного кристаллографического анализа п-хлорбензоата (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, установившего абсолютную стереохимию вещества.
На фиг.11В проиллюстрированы результаты рентгенодифракционного кристаллографического анализа п-хлорбензоата (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, установившего абсолютную стереохимию вещества, изображенные со схемой нумерации атомов для ссылки.
На фиг.12 проиллюстрирована полная хроматограмма, характеризующая четыре стереоизомера N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, где 2S,3R-изомер демонстрирует пик со временем удерживания 5,3 минуты, 2R,3S-изомер демонстрирует пик со временем удерживания 7,3 минуты, 2R,3R-изомер демонстрирует пик со временем удерживания 8,2 минуты и 2S,3S-изомер демонстрирует пик со временем удерживания 12,4 минуты. Как описано в данном изобретении, подвижная фаза, необходимая в анализе для обеспечения адекватного разделения, приводит к композиции гексаны:этанол:ди-н-бутиламин в соотношении 60:40:0,2, при скорости потока 1,0 мл/мин, при температуре колонки 20°С и УФ-детектировании при 270 нм.
На фиг.13 приведена порошковая рентгенограмма моногидрохлорида (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, иллюстрирующая как наблюдаемые (более светлый), так и рассчитанные пики (более темные). В обоих случаях характер распределения пиков согласуется по отношению к значениям 2θ, а минимальное различие в интенсивности и ширине пиков может быть связано с разрешением приборов и предпочтительными ориентационными эффектами. Как описано в данном изобретении, дополнительное минимальное различие может быть связано с температурным сдвигом, поскольку наблюдаемые данные получены при комнатной температуре, а данные расчета получены для структуры при 120 К.
На фиг.14 приведена порошковая рентгенограмма монотозилата (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Конкретное соединение, (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид, обладающее аффинностью (значение Ki≤1 нМ) и селективностью в отношении подтипа α7 NNR, проявляет эффективность на животных моделях сознания (усиление сознания) и психоза (антипсихотическое действие).
В одном аспекте настоящее изобретение относится к (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду или его фармацевтически приемлемой соли. Другой аспект относится к (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду по существу в чистом виде или к его фармацевтически приемлемой соли. Следующий аспект относится к (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду, по существу не содержащему (2S,3S), (2R,3S) или (2R,3R) изомеров, или к его фармацевтически приемлемой соли.
Дополнительно, другой аспект относится к стереохимически обогащенному (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамиду или его фармацевтически приемлемой соли. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 90% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 95% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 98% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 99% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 99,5% или больше.
Другой аспект настоящего изобретения относится к соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида с кислотой, где кислоту выбирают из хлористоводородной кислоты, серной кислоты, фосфорной кислоты, малеиновой кислоты, п-толуолсульфоновой кислоты, галактаровой (слизевой) кислоты, D-миндальной кислоты, D-винной кислоты, метансульфоновой кислоты, R- и S-10-камфорсульфоновой кислоты, кетоглутаровой кислоты или гиппуровой кислоты. В одном варианте осуществления стехиометрическое соотношение (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида к кислоте составляет 2:1, 1:1 или 1:2. В одном варианте осуществления стехиометрическое соотношение составляет 1:1. Один вариант осуществления настоящего изобретения представляет собой гидрохлорид (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его гидрат или сольват, включая частичные гидраты или сольваты. Следующий вариант осуществления представляет собой моногидрохлорид (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его гидрат или сольват, включая частичные гидраты или сольваты.
Другой аспект настоящего изобретения относится к (2S,3R)-N-(2-((3-пиридинил)метил)-3-амино-1-азабицикло[2.2.2]октану.
Другой аспект настоящего изобретения относится к фармацевтической композиции, включающей соединение по настоящему изобретению и один или несколько фармацевтически приемлемых носителей.
Другой аспект настоящего изобретения относится к способу лечения или предупреждения нарушения центральной нервной системы, воспаления, боли или неоваскуляризации, включающему введение соединения по настоящему изобретению. В одном варианте осуществления нарушение центральной нервной системы характеризуется изменением нормального высвобождения нейротрансмиттера. В одном варианте осуществления нарушение центральной нервной системы выбирают из слабого когнитивного ухудшения, связанного с возрастом ухудшения памяти, предстарческого слабоумия, раннего начала болезни Альцгеймера, старческого слабоумия, слабоумия по типу Альцгеймера, болезни Альцгеймера, деменции с тельцами Леви, слабоумия в результате микроинфаркта, связанного со СПИД слабоумия, ВИЧ-слабоумия, множественных церебральных инфарктов, паркинсонизма, болезни Паркинсона, болезни Пика, прогрессирующего супрануклеарного паралича, хореи Хантингтона, поздней дискинезии, гиперкинезии, мании, нарушения, связанного с дефицитом внимания, гиперактивности, связанной с дефицитом внимания, тревоги, депрессии, дислексии, шизофрении, когнитивной дисфункции при шизофрении, депрессии, обсессивно-компульсивных нарушений или синдрома Туретта. В одном варианте осуществления нарушение центральной нервной системы выбирают из болезни Альцгеймера, мании, нарушения, связанного с дефицитом внимания, гиперактивности, связанной с дефицитом внимания, тревоги, дислексии, шизофрении, когнитивной дисфункции при шизофрении, депрессии, обсессивно-компульсивных нарушений или синдрома Туретта.
Другой аспект настоящего изобретения включает применение соединения по настоящему изобретению для получения лекарственного средства для лечения или предупреждения нарушения центральной нервной системы, воспаления, боли или неоваскуляризации. В одном варианте осуществления нарушение центральной нервной системы характеризуется изменением нормального высвобождения нейротрансмиттера. В одном варианте осуществления нарушение центральной нервной системы выбирают из слабого когнитивного ухудшения, связанного с возрастом ухудшения памяти, предстарческого слабоумия, раннего начала болезни Альцгеймера, старческого слабоумия, слабоумия по типу Альцгеймера, болезни Альцгеймера, деменции с тельцами Леви, слабоумия в результате микроинфаркта, связанного со СПИД слабоумия, ВИЧ-слабоумия, множественных церебральных инфарктов, паркинсонизма, болезни Паркинсона, болезни Пика, прогрессирующего супрануклеарного паралича, хореи Хантингтона, поздней дискинезии, гиперкинезии, мании, нарушения, связанного с дефицитом внимания, гиперактивности, связанной с дефицитом внимания, тревоги, депрессии, дислексии, шизофрении, когнитивной дисфункции при шизофрении, депрессии, обсессивно-компульсивных нарушений или синдрома Туретта. В одном варианте осуществления нарушение центральной нервной системы выбирают из болезни Альцгеймера, мании, нарушения, связанного с дефицитом внимания, гиперактивности, связанной с дефицитом внимания, тревоги, дислексии, шизофрении, когнитивной дисфункции при шизофрении, депрессии, обсессивно-компульсивных нарушений или синдрома Туретта.
Другой аспект настоящего изобретения относится к соединению по настоящему изобретению для применения при лечении или предупреждения нарушения центральной нервной системы, воспаления, боли или неоваскуляризации. В одном варианте осуществления нарушение центральной нервной системы характеризуется изменением нормального высвобождения нейротрансмиттера. В одном варианте осуществления нарушение центральной нервной системы выбирают из слабого когнитивного ухудшения, связанного с возрастом ухудшения памяти, предстарческого слабоумия, раннего начала болезни Альцгеймера, старческого слабоумия, слабоумия по типу Альцгеймера, болезни Альцгеймера, деменции с тельцами Леви, слабоумия в результате микроинфаркта, связанного со СПИД слабоумия, ВИЧ-слабоумия, множественных церебральных инфарктов, паркинсонизма, болезни Паркинсона, болезни Пика, прогрессирующего супрануклеарного паралича, хореи Хантингтона, поздней дискинезии, гиперкинезии, мании, нарушения, связанного с дефицитом внимания, гиперактивности, связанной с дефицитом внимания, тревоги, депрессии, дислексии, шизофрении, когнитивной дисфункции при шизофрении, депрессии, обсессивно-компульсивных нарушений или синдрома Туретта. В одном варианте осуществления нарушение центральной нервной системы выбирают из болезни Альцгеймера, мании, нарушения, связанного с дефицитом внимания, гиперактивности, связанной с дефицитом внимания, тревоги, дислексии, шизофрении, когнитивной дисфункции при шизофрении, депрессии, обсессивно-компульсивных нарушений или синдрома Туретта.
В вышеуказанных способах и применениях в одном варианте изобретения эффективные дозы составляют между 1 мг и 10 мг в течение 24-часового периода.
Другой аспект настоящего изобретения относится к способу получения (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его фармацевтически приемлемой соли путем последовательного динамического оптического разделения и стереоселективного восстановительного аминирования (2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-она.
Объем настоящего изобретения включает все комбинации аспектов, вариантов осуществления и описанных в данном изобретении преимуществ.
Коммерческая разработка потенциальных лекарственных средств включает много стадий, включая масштабирование химического синтеза и очистки, выявление оптимальных солевых форм и тому подобное. В рецептуре лекарственных композиций лекарственное вещество предпочтительно находится в таком виде, с которым удобно работать и обрабатывать. Анализ включает коммерческую жизнеспособность, а также устойчивость при получении. Кроме того, при получении лекарственных композиций важно, чтобы в плазме крови обеспечивался надежный, воспроизводимый и постоянный профиль концентрации лекарственного средства после его введения пациенту.
Химическая стабильность, стабильность в твердом состоянии и продолжительность хранения активных ингредиентов также являются очень важными факторами. Лекарственное вещество и содержащая его композиция предпочтительно должны быть способными эффективно храниться на протяжении существенного периода времени без значительного изменения физико-химических характеристик активного компонента (например, его химического состава, плотности, гигроскопичности и растворимости). Кроме того, также важно иметь возможность получения лекарственного средства в форме, которая является чистой, насколько это возможно. Эти отличительные особенности изобретения обсуждаются ниже более подробно.
I. Соединения
Соединение по настоящему изобретению представляет собой (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид, представленный ниже как соединение А, или фармацевтически приемлемые солевые формы соединения А.
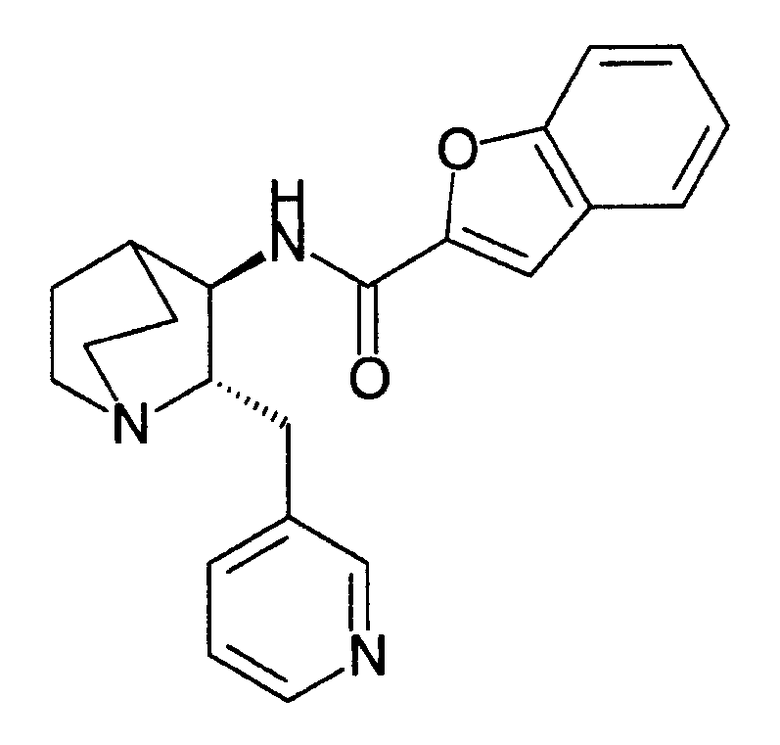
Соединение А
Рацемическое соединение - N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид, его синтез и применение в медицинских назначениях описаны в патенте США № 6953855, Мазурова и др., включенном в данное описание путем ссылки.
Рацемический N-(2-((3-пиридинил)метил)-1- азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид представляет собой высокоаффинный лиганд для подтипа α7 нейронного никотинового рецептора (NNR). Рацемический N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид содержит два асимметрически замещенных атома углерода. Таким образом, рацемический N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид существует в виде четырех стереоизомерных форм, а именно, (S,S), (S,R), (R,R) и (R,S). Изомер (S,R), а именно (2S,3R), представляет собой соединение А.
Ранее предполагалось, что доминирующие стереоизомерные формы, получаемые в сообщенных синтезах, включая патент США № 6953855, характеризуются относительной цис-конфигурацией в 2 и 3 положениях 1-азабицикло[2.2.2]октанового (хинуклидинового) кольца. Другими словами, существовало мнение, что цис-диастереомер ((2R,3R) и (2S,3S), пара энантиомеров) являются предоминирующими формами, которые образуются при получении сообщавшимися способами. Данное определение о предоминировании цис-синтеза было основано на (I) сравнении констант 1Н спин-спинового взаимодействия ядер водорода во 2 и 3 положениях хинуклидинового кольца и выделенных диастереомерных (цис и транс) промежуточных соединений с константами спин-спинового взаимодействия, сообщавшимися в литературе; и на (II) ожидаемом стереохимическом выходе синтетической химии, использованной для получения смеси соединений по аналогии с литературой, со ссылкой на Warawa et al., J. Med. Chem. 18(6): 587-593 (1975) и Viti et al., Tetrahedron Lett., 35(32): 5939-5942 (1994), обе из которых включены в данное описание путем ссылки. Таким образом, ожидалось, что будет образовываться цис-конфигурация. Как таковое, биологическое тестирование с использованием рацемата приводило к результатам, которые приписывались как присущие доминирующей цис-конфигурации.
В настоящий момент было установлено, с помощью рентгенодифракционного анализа кристаллических солевых форм и аналогов, что доминирующий диастереомер, образовывавшийся в первоначальном синтезе, представлял собой, в действительности, транс-диастереомер. Кроме того, было установлено, что два энантиомера с относительной транс-стереохимией, а именно (2S,3R) и (2R,3S), существенно отличаются друг от друга по своей способности взаимодействовать с подтипом α7 NNR. Имеющее конфигурацию (2S,3R) соединение А обладает большей активностью.
С использованием дополнительного анализа было установлено, что соединение А обладает фармакологическими свойствами, которые отличаются от (i) свойств каждого из других трех стереоизомеров, взятых по отдельности; (ii) свойств смеси всех четырех стереоизомеров, а именно, рацемата; и (iii) свойств других лигандов рецептора NNR α7, о которых сообщалось в литературе.
(2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид (соединение А) представляет собой высокоселективный полный агонист NNR рецептора α7 с замечательно низким значением ЕС50 (для активации) и хорошей разницей между ЕС50 и IC50 (для остаточного ингибирования), обеспечивая функциональный агонизм в широком диапазоне терапевтически полезных концентраций.
II. Масштабируемый синтез (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Конкретные синтетические стадии различаются по своей способности поддаваться масштабированию. Установлено, что реакции плохо поддаются масштабированию по множеству причин, включая соображения безопасности, дороговизну реагентов, трудную обработку или очистку, энергетические параметры реакции (термодинамика или кинетика) и выход реакции.
Описанный в данном изобретении синтез (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида был использован для получения килограммовых количеств вещества, и реакции компонентов были проведены в многокилограммовом масштабе с высоким выходом.
В масштабируемом синтезе использовано как динамическое оптическое разделение рацемизуемого кетона (2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она), так и стереоселективное восстановление иминного производного (R)-α-метилбензиламина (восстановительное аминирование) разделенного на изомеры кетона. Синтетическая последовательность, которая здесь описана, является легко масштабируемой и не включает хроматографические стадии очистки.
III. Получение новых солевых форм (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
(2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3- ил)бензофуран-2-карбоксамид в виде свободного основания представляет собой аморфный порошок с очень ограниченной растворимостью в воде. Свободное основание будет реагировать как с неорганическими, так и с органическими кислотами с получением определенных кислотно-аддитивных солей, которые обладают физическими и химическими свойствами, которые являются выигрышными для получения фармацевтических композиций, включая, но не ограничиваясь указанным, кристалличность, растворимость в воде и стабильность. Стехиометрия солей по настоящему изобретению может изменяться.
В зависимости от способа, с использованием которого образуются описанные в данном изобретении соли, они могут иметь кристаллическую структуру, в которой окклюдированы растворители, которые присутствуют во время получения соли. Таким образом соли могут существовать в виде гидратов и других сольватов с переменной стехиометрией растворителя относительно (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида.
Способ получения солевых форм может изменяться. Получение солевых форм (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида обычно включает
(i) смешивание свободного основания или раствора свободного основания, а именно (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, в подходящем растворителе с кислотой без растворителя или в виде раствора кислоты в подходящем растворителе;
(iia) охлаждение полученного раствора соли, при необходимости, для того, чтобы вызвать образование осадка; или
(iib) добавление подходящего «анти»-растворителя, для того, чтобы вызвать образование осадка; или
(iic) упаривание первого растворителя и добавление нового растворителя и повторение либо стадии (iia), либо стадии (iib); и
(iii) фильтрование и сбор полученной соли.
В одном варианте осуществления (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид является стереохимически обогащенным. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 90% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 95% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 98% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 99% или больше. В одном варианте осуществления энантиомерный и/или диастереомерный избыток составляет 99,5% или больше.
Стехиометрия, смесь растворителей, концентрация растворенного вещества и используемая температура могут изменяться. Иллюстративные растворители, которые можно использовать для получения или перекристаллизации солевых форм включают, без ограничения, этанол, метанол, изопропиловый спирт, ацетон, этилацетат и ацетонитрил.
Примеры подходящих фармацевтически приемлемых солей включают аддитивные соли неорганических кислот, такие как хлорид, бромид, сульфат, фосфат и нитрат; аддитивные соли органических кислот такие, как ацетат, галактарат, пропионат, сукцинат, лактат, гликолят, малат, тартрат, цитрат, малеат, фумарат, метансульфонат, п-толуолсульфонат и аскорбат; и соли с аминокислотами, такие как аспартат и глутамат. Соли в некоторых случаях могут представлять собой гидраты или сольваты с этанолом. Предусмотрены иллюстративные соли, как описано в патентах США № 5597919, Dull et al., 5616716, Dull et al., и 5663356, Ruecroft et al., каждый из которых включен в данное описание путем ссылки.
Скрининг солей свободного основания (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида показал, что хотя могут быть получены многие соли с фармацевтически приемлемыми кислотами, только некоторые из этих солей обладают приемлемыми свойствами для коммерческого производства. Способность предсказать характеристики, иллюстрирующие коммерчески жизнеспособную соль, соответственно, не существует. Кислоты, которые давали соли, которые были кристаллическими, а именно соли, которые проявляли некоторую степень кристалличности в зависимости от способа, с помощью которого их получают, включают хлористоводородную кислоту, серную кислоту, фосфорную кислоту, п-толуолсульфоновую кислоту, галактаровую (слизевую) кислоту, D-миндальную кислоту, D-винную кислоту, метансульфоновую кислоту, R- и S-10-камфорсульфоновую кислоту, малеиновую кислоту, кетоглутаровую кислоту и гиппуровую кислоту. Среди данных солей каждая из солей хлористоводородной кислоты, фосфорной кислоты, малеиновой кислоты и п-толуолсульфоновой кислоты проявляла дополнительные желательные свойства, включая высокие температуры плавления, хорошую растворимость в воде и низкую гигроскопичность. Такие характеристики для данных солей были неожиданными.
IV. Фармацевтические композиции
Фармацевтические композиции по настоящему изобретению включают описанные здесь соли, в чистом состоянии или в виде композиции, в которой соединения объединены с любым другим фармацевтически совместимым продуктом, который может быть инертным или физиологически активным. Полученные фармацевтические композиции можно использовать для предупреждения состояния или нарушения у субъекта, подверженного такому состоянию или нарушению, и/или для лечения субъекта, страдающего от данного состояния или нарушения. Описанные здесь фармацевтические композиции включают соединение по настоящему и/или его фармацевтически приемлемые соли.
Способ, с помощью которого вводят соединения, может изменяться. Композиции предпочтительно вводят перорально (например, в жидком виде в растворителе, таком как водная или неводная жидкость, или в твердом носителе). Предпочтительные композиции для перорального введения включают пилюли, таблетки, капсулы, каплеты, сиропы и растворы, включая твердые желатиновые капсулы и капсулы с высвобождением во времени. Стандартные эксципиенты включают связующие вещества, наполнители, красители, солюбилизаторы и тому подобные. Рецептура композиций может быть составлена в виде единичной препаративной лекарственной формы или в виде множества доз или подъединиц. Предпочтительные композиции находятся в жидком или полутвердом виде. Можно использовать композиции, включающие жидкий фармацевтически инертный носитель, такой как вода или другие фармацевтически совместимые жидкости, или полутвердые вещества. Применение таких жидкостей и полутвердых веществ хорошо известно специалистам в данной области.
Композиции также можно вводить с помощью инъекции, т.е. внутривенно, внутримышечно, подкожно, внутрибрюшинно, внутриартериально, внутритекально и внутрицеребровентикулярно. Внутривенное введение является предпочтительным способом инъекции. Подходящие носители для инъекций хорошо известны специалистам в данной области и включают 5% растворы декстрозы, физиологический раствор и забуференный фосфатом физиологический раствор. Лекарственный продукт также можно вводить в виде инфузии или инъекции (например, в виде суспензии или в виде эмульсии в фармацевтически приемлемой жидкости или смеси жидкостей).
Препараты также можно вводить с использованием других средств, например, с помощью ректального введения. Препараты, используемые для ректального введения, такие как суппозитории, хорошо известны специалисту в данной области. Лекарственный продукт также можно вводить путем ингаляции (например, в виде аэрозоля либо назально, либо с использованием устройств для доставки такого типа, который указан в патенте США № 4922901, Brooks et al., описание которого включено в данное изобретение во всей своей полноте); наружно (например, в виде лосьона); чрескожно (например, с использованием чрескожного пластыря) или ионтофоретически; или путем сублингвального или буккального введения. Хотя возможно введение соединения в виде основной массы активного химического вещества, предпочтительно, чтобы лекарственный продукт находился в виде фармацевтической композиции или препарата для действенного и эффективного введения.
Иллюстративные способы введения соединений будут очевидны квалифицированному специалисту. Применимость данных препаратов может зависеть от конкретной используемой композиции и конкретного субъекта, получающего лечение. Данные препараты могут содержать жидкий носитель, который может быть масляным, водным, эмульгированным или содержать определенные растворители, подходящие для данного способа введения.
Композиции можно вводить путем дробного введения или с постепенной, непрерывной, постоянной или контролируемой скоростью теплокровному животному (например, млекопитающему, такому как мышь, крыса, кошка, кролик, собака, свинья, корова или обезьяна), но преимущественно их вводят человеку. Кроме того, может изменяться время в течение дня и число раз в день, когда вводят фармацевтическую композицию.
Другие подходящие методы введения соединений по настоящему изобретению описаны в патенте США, 5604231 Smith et al., содержание которого включено в данное описание путем ссылки.
В варианте осуществления настоящего изобретения и как будет принято во внимание специалистами в данной области соединение по настоящему изобретению можно вводить в сочетании с другими лекарственными соединениями. Например, соединение по данному изобретению можно использовать в сочетании с другими лигандами NNR (такими как варениклин), антиоксидантами (такими как средства, улавливающие свободные радикалы), противомикробными средствами (такими как антибиотики пенициллинового ряда), противовирусными средствами (такими как аналоги нуклеозидов зидовудина и ацикловира), антикоагулянтами (такими как варфарин), противовоспалительными средствами (такими как НСПВС), жаропонижающими средствами, анальгетиками, анестетиками (такими как используемые в хирургии), ингибиторами ацетилхолинэстеразы (такими как донепезил и галантамин), антипсихотическими препаратами (такими как галоперидол, клозапин, оланзапин и кветипин), иммуносуппресантами (такими как циклоспорин и метотрексат), нейропротекторными средствами, стероидами (такими как стероидные гормоны), кортикостероидами (такими как дексаметазон, предизон и гидрокортизон), витаминами, минералами, пищевыми добавками, антидепрессантами (такими как имипрамин, флуоксетин, пароксетин, эсциталопрам, сертралин, венлафаксин и дулоксетин), анксиолитические средства (такие как алпразолам и баспирон), антиконвульсантами (такими как фенитоин и габапентин), сосудорасширяющими средствами (такими как празозин и силденафил), стабилизаторами настроения (такими как валпроат и арипипразол), противораковыми лекарственными средствами (такими как антипролиферативные средства), антигипертензивными средствами (такими как атенолол, клонидин, амлопидин, верапамил и олмезартан), слабительными средствами, препаратами для разжижения стула, диуретиками (такими как фуросемид), антиспазмолитиками (такими как дицикломин), антидискинетическими средствами и противоязвенными лекарственными средствами (такими как эзомепразол).
Соединения по настоящему изобретению можно использовать сами по себе или в сочетании с другими терапевтическими средствами. Такую комбинацию фармацевтически активных агентов можно вводить совместно или по отдельности и, при введении по отдельности, введение можно проводить одновременно или последовательно в любом порядке. Количества соединений или агентов и относительное распределение введения по времени будут выбираться для достижения желаемого терапевтического действия. Введение в комбинации можно проводить как сопутствующее введение в виде (1) единой фармацевтической композиции, включающей множество соединений; или (2) отдельных фармацевтических композиций, каждая из которых включает одно из соединений. Альтернативно, комбинацию можно вводить по отдельности последовательным образом, где один лекарственный агент вводят первым, а другой - вторым, или наоборот. Такое последовательно введение может быть близким по времени или разделено во времени. Соединения по настоящему изобретению можно использовать для лечения множества нарушений и состояний и, как таковые, соединения по настоящему изобретению можно использовать в сочетании с множеством других подходящих терапевтических агентов, используемых для лечения или профилактики данных нарушений или состояний.
Подходящая доза соединения представляет собой количество, эффективное для предотвращения появления симптомов нарушения или для лечения некоторых симптомов нарушения, от которого страдает пациент. Как отмечено, под терминами «эффективное количество», «терапевтическое количество» или «эффективная доза» подразумевается, что количество является достаточным для оказания желаемого фармакологического или терапевтического действия, таким образом, приводят к эффективному предупреждению или лечению нарушения.
При лечении нарушения ЦНС эффективное количество соединения представляет собой количество, достаточное для прохождения через гематоэнцефалический барьер субъекта для связывания с релевантными рецепторными сайтами в мозгу субъекта и для модулирования активности релевантных подтипов NNR (например, обеспечивая секрецию нейротрансмиттера, приводя тем самым к эффективному предупреждению или лечению нарушения). Пример предупреждения нарушения проявляется во временной отсрочке начала проявления симптомов заболевания. Пример лечения нарушения проявляется в уменьшении симптомов, связанных с нарушением, или облегчения рецидива симптомов нарушения. Предпочтительно, эффективное количество является достаточным для получения желаемого результата, но недостаточным для того, чтобы вызвать ощутимые побочные действия.
Эффективная доза может изменяться в зависимости от таких факторов, как состояние пациента, тяжесть симптомов заболевания и способа, с помощью которого вводят фармацевтическую композицию. Для пациентов-людей эффективная доза типичных соединений обычно требует введения соединения в количестве, достаточном для модулирования активности релевантных NNR, но количество должно быть недостаточным для того, чтобы вызвать действие в скелетных мышцах или ганглии в какой-либо значимой степени. Эффективная доза, конечно, будет изменяться от пациента к пациенту, но, в общем, включает количества, начинающиеся с момента появления действия на ЦНС или других желательных терапевтических действий, но ниже количества, при которых наблюдаются мышечные эффекты.
Описанные здесь соединения при использовании в эффективных количествах в соответствии с описанными здесь способами могут обеспечивать определенную степень предотвращения прогрессирования или облегчения симптомов или облечения, в некоторой степени рецидива нарушений ЦНС или других нарушений. Эффективные количества этих соединений обычно находятся ниже порога концентрации, требуемого для того, чтобы вызвать какие-либо ощутимые побочные действия, например, те действия, которые относятся к скелетным мышцам или ганглию. Соединения можно вводить в терапевтическом окне, в котором некоторые нарушения ЦНС и другие нарушения подвергаются лечению, а определенные подобные действия исключены. Идеально, эффективная доза описанных здесь соединений является достаточной для обеспечения желаемого действия на нарушения, но недостаточной (т.е. находится не на таком достаточно высоком уровне) для того, чтобы вызвать нежелательные побочные действия. Предпочтительно, соединения вводят в дозировке, которая является эффективной для лечения нарушений ЦНС или других нарушений, но меньшей чем, часто меньшей 1/5, и часто меньшей, чем 1/10, количества, необходимого для того, чтобы вызвать определенные подобные действия в какой-либо существенной степени.
Наиболее предпочтительно, эффективные дозировки находятся в очень низких концентрациях, при которых происходят максимальное действие с минимальными побочными действиями. Обычно эффективная доза таких соединений обычно требует введения соединения в количестве меньшем, чем 5 мг/кг веса пациента. Часто соединения по настоящему изобретению вводят в количестве меньшем, чем примерно 1 мг/кг веса пациента и обычно меньшем чем примерно 100 мкг/кг веса пациента, но чаще в диапазоне от примерно 10 мкг до меньше 100 мкг/кг веса пациента. Вышеуказанные эффективные дозы обычно представляют количество, вводимое в виде одной дозы или в виде одной или нескольких доз, вводимых в течение 24-часового промежутка времени.
Для пациентов-людей эффективная доза обычных соединений обычно требует введения соединения в количестве по меньшей мере примерно 1, часто по меньшей мере примерно 10 и чаще по меньшей мере примерно 100 мг/24 часа/пациент. Для пациентов-людей эффективная доза обычных соединений обычно требует введения соединения в количестве, которое обычно не превышает примерно 500, часто не превышает примерно 400 и чаще не превышает примерно 300 мг/24 часа/пациент. Дополнительно, композиции преимущественно вводят в такой эффективной дозе, что концентрация соединения в плазме крови пациента обычно не превышает 50 нг/мл, часто не превышает 30 нг/мл, и чаще не превышает 10 нг/мл. В одном варианте осуществления настоящего изобретения эффективная доза составляет между примерно 1 и 10 мг в течение 24-часового промежутка времени.
IV. Способ применения фармацевтических композиций
Как использовано в данном описании, термин «агонист» относится к веществу, которое стимулирует своего партнера связывания, обычно рецептор. Стимуляция определяется в контексте определенного анализа, или она может быть очевидна из обсуждавшейся здесь литературы, которая сравнивает фактор или вещество, которые приняты в качестве «агониста» или «антагониста» определенного партнера связывания при по существу аналогичных обстоятельствах, как это признано специалистами в данной области. Стимуляция может быть определена по увеличению определенного эффекта или функции, которые вызваны взаимодействием агониста или частичного агониста с партнером связывания и могут включать аллостерические эффекты.
Как использовано в данном описании, термин «антагонист» относится к веществу, которое ингибирует своего партнера связывания, обычно рецептор. Ингибирование определяется в контексте определенного анализа, или оно может быть очевидно из обсуждавшейся здесь литературы, которая сравнивает фактор или вещество, которые приняты в качестве «агониста» или «антагониста» определенного партнера связывания при по существу аналогичных обстоятельствах, как это признано специалистами в данной области. Ингибирование может быть определено по уменьшению определенного эффекта или функции, которые вызваны взаимодействием антагониста с партнером связывания и могут включать аллостерические эффекты.
Как использовано в данном описании, термин «частичный агонист» или «частичный антагонист» относятся к веществу, которое обеспечивает уровень стимуляции или ингибирования, соответственно, своего партнера связывания, который не является полностью или совершенно агонистическим или антагонистическим, соответственно. Будет понятно, что стимуляция и соответственно ингибирование определены по сути для любого вещества или категории веществ, которые будут определяться как агонисты, антагонисты или частичные агонисты.
Как использовано в данном описании, термин «внутренняя активность» или «эффективность» относится к определенной мере биологической эффективности комплекса связывания партнеров. В отношении фармакологии рецепторов, контекст, в котором внутреннюю активность или эффективность будут определять, будет зависеть от контекста комплекса связывания партнеров (например, рецептор/лиганд) и рассмотрения активности, относящейся к конкретному биологическому результату. Например, при определенных обстоятельствах, внутренняя активность может изменяться в зависимости от вовлеченной системы вторичного мессенджера. См. Hoyer D and Boddeke H., Trends Pharmacol. Sci. 14(7): 270-5 (1993), включенной в данное описание путем ссылки по поводу таких указаний. Где такие контекстуально специфические оценки являются значимыми, и насколько они могут быть значимыми в контексте настоящего изобретения будет очевидно для обычного специалиста в данной области.
Как использовано в данном описании, модулирование рецептора включает агонизм, частичный агонизм, антагонизм, частичный антагонизм или обратный агонизм рецептора.
Как использовано в данном описании, нейротрансмиттеры, чье высвобождение опосредуется описанными здесь соединениями включают, но не ограничиваются указанным, ацетилхолин, допамин, норепинефрин, серотонин и глутамат, и описанные здесь соединения функционируют в качестве модуляторов подтипа α7 NNR ЦНС.
Как использовано в данном описании, термин «предупреждение» или «профилактика» включает любую степень уменьшения прогрессирования или отсрочки начала заболевания, нарушения или состояния. Термин включает обеспечение защитного действия в отношении конкретного заболевания, нарушения или состояния, а также ослабления рецидива заболевания, нарушения или состояния. Таким образом, в другом аспекте изобретение относится к способу лечения субъекта, подверженного риску развития или появления рецидива нарушения, опосредованного NNR или nAChR. Соединения и фармацевтические композиции по изобретению можно использовать для достижения благоприятного терапевтического или профилактического действия, например, у субъекта с дисфункцией ЦНС.
Как отмечено выше, соединения по настоящему изобретению в виде свободного основания и соли модулируют подтип α7 NNR, характерный для ЦНС, и могут использоваться для предупреждения или лечения различных состояний или нарушений, включая состояния или нарушения ЦНС, у субъектов, которые имеют или которые подвержены риску появления таких состояний или нарушений, путем модулирования α7 NNR. Данные соединения обладают способностью селективно связываться с α7 NNR и проявлять никотиновую фармакологию, например, действовать в качестве агонистов, частичных агонистов, антагонистов, как это описано. Например, соединения по настоящему изобретению при введении в эффективных количествах нуждающимся в этом пациентам обеспечивают некоторую степень предупреждения развития нарушения ЦНС, а именно обеспечивают протекторное действие, облегчение симптомов нарушений ЦНС или ослабление рецидива нарушения ЦНС или сочетание этих действий.
Соединения по настоящему изобретению можно использовать для лечения или предупреждения тех типов состояний и нарушений, для которых другие типы никотиновых соединений были предложены или было показано, что их можно использовать в качестве лекарственных средств. См., например, перечисленные ранее в данном описании ссылки, а также Williams et al., Drug News Perspec. 7(4): 205(1994), Arneric et al., CNS Drug Rev. 1(1): 1-26 (1995), Arneric et al., Exp. Opin. Invest. Drugs 5(1): 79-100 (1996); Bencherif et al., J. Pharmacol. Exp. Ther. 279:1413 (1996), Lippiello et al., J. Pharmacol. Exp. Ther. 279: 1422 (1996), Damaj et al., J. Pharmacol. Exp. Ther. 291: 390 (1999); Chiari et al., Anesthesiology 91: 1447 (1999), Lavand'homme and Eisenbach, Anesthesiology 91: 1455 (1999), Holladay et al., J. Med. Chem. 40(28): 4169-94 (1997), Bannon et al., Science 279: 77 (1998), PCT WO 94/08992, PCT WO 96/31475, PCT WO 96/40682 и патенты США № 5583140, Bencherif et al., 5597919, Dull et al., 5604231, Smith et al., и 5852041, Cosford et al., описание которых, касающееся таких терапевтических указаний, включено в данное изобретение путем ссылки.
Соединение и их фармацевтические композиции можно использовать для лечения или предупреждения множества нарушений ЦНС, включая нейродегенеративные нарушения, нейропсихиатрические нарушения, неврологические нарушения и пагубные привычки. Соединения и их фармацевтические композиции можно использовать для лечения или предупреждения когнитивного дефицита и дисфункций, связанных с возрастом и другими факторами; нарушений внимания и слабоумия, включая те, которые возникают под действием инфекционных агентов или метаболических нарушений; для обеспечения нейропротекции; для лечения конвульсий и множественных церебральных инфарктов; для лечения нарушений настроения, маниакальных и связанных с вредными привычками поведенческих реакций; для обеспечения обезболивания; для контроля воспаления, такого как опосредованное цитокинами и ядерным фактором каппа В; для лечения воспалительных нарушений; для обеспечения облегчения боли; для лечения метаболических нарушений, таких как диабет или метаболический синдром; и для лечения инфекций в качестве противоинфекционных средств для лечения бактериальных, грибковых и вирусных инфекций.
Нарушения ЦНС
К числу нарушений, заболеваний и состояний, для лечения или профилактики которых можно использовать соединения и фармацевтические композиции по настоящему изобретению, относятся связанное с возрастом ухудшение памяти (AAMI), слабое когнитивное ухудшение (MCI), связанное с возрастом когнитивное угасание (ARCD), предстарческое слабоумие, раннее начало болезни Альцгеймера, старческое слабоумие, слабоумие по типу Альцгеймера, болезнь Альцгеймера, когнитивное ухудшение без слабоумия (CIND), деменция с тельцами Леви (ДТЛ), ВИЧ-слабоумие, комплекс связанного со СПИД слабоумия, мультиинфарктная деменция, синдром Дауна, травма головы, травматическое повреждение мозга (TBI), слабоумие в результате травмы, болезнь Крейцфельда-Якоба и прионовые болезни, удар, ишемия, нарушение дефицита внимания, связанная с дефицитом внимания гиперактивность, дислексия, шизофрения, нарушение шизофренической формы, шизофреноподобное нарушение аффективного типа, когнитивная дисфункция при шизофрении, когнитивный дефицит при шизофрении, такой как дефицит памяти, включая рабочую память, функции организации, внимания, вигильность, обработки информации и обучаемости, связанное с шизофренией слабоумие (любое из слабого, среднего или тяжелого), паркинсонизм, включая болезнь Паркинсона, постэнцефалический паркинсонизм, паркинсонизм-слабоумие Гаума, лобно-височное слабоумие паркинсонового типа (FTDP), болезнь Неймана-Пика (Niemann-Pick), болезнь Хантингтона, хорея Хантингтона, поздняя дискинезия, гиперкинезия, прогрессирующий супрануклеарный паралич, прогрессирующий супрануклеарный парез, синдром усталых ног, рассеянный склероз, амиотрофический латеральный склероз (ALS), заболевание двигательных нейронов (MND), множественная системная атрофия (MSA), кортикобазальная дегенерация, синдром Гийома-Барре (GBS), хроническая воспалительная демиелинизирующая полиневропатия (CIDP), эпилепсия, аутосомальная доминантная ночная лобно-височная эпилепсия, мания, тревожное состояние, депрессия, предменструальная дисфория, панические нарушения, булимия, анорексия, нарколепсия, избыточная сонливость в дневное время, биполярные нарушения, генерализованное тревожное состояние, обсессивно-компульсивное нарушение, вспышки ярости, нарушение оппозиционного неповиновения, синдром Туретта, аутизм, наркомания и хронический алкоголизм, склонность к курению, ожирение, кахексия, псориаз, волчанка, острый ангиохолит, афтозный стоматит, язвы, астма, язвенный колит, воспалительное заболевание кишечника, болезнь Крона, постоперационная кишечная непроходимость, спазматическая дистония, диарея, запор, резервуарный илеит, панкреатит, вирусная пневмония, артрит, включая ревматоидный артрит и остеоартрит, эндотоксемия, сепсис, атеросклероз, идиопатический фиброз легких, острая боль, хроническая боль, невропатии, недержание мочи, диабет и неоплазия.
Когнитивные ухудшения или дисфункции могут быть связаны с психиатрическими нарушениями или состояниями, такими как шизофрения и другие психотические нарушения, включая, но не ограничиваясь указанным, психотическое нарушение, шизофреноподобное нарушение, шизоаффективное нарушение, бредовое нарушение, кратковременное психотическое нарушение, острое психотическое нарушение и психотические нарушение вследствие одного или более общих медицинских состояний, слабоумие разного вида и другие когнитивные нарушения, включая, но не ограничиваясь указанным, слабое когнитивное ухудшение, предстарческое слабоумие, болезнь Альцгеймера, старческое слабоумие, слабоумие по типу Альцгеймера, связанное с возрастом ухудшение памяти, деменцию с тельцами Леви (ДТЛ), сосудистое слабоумие, связанный со СПИД комплекс слабоумия, дислексия, паркинсонизм, включая болезнь Паркинсона, когнитивное ухудшение и слабоумие при болезни Паркинсона, когнитивное ухудшение при рассеянном склерозе, когнитивное ухудшение, вызванное травматическим повреждением мозга, слабоумие вследствие других общих медицинских состояний, тревожные состояния, включая, но не ограничиваясь указанным, паническое состояние без агорафобии, паническое состояние с сопутствующей агорафобией, агорафобию без истории панического нарушения, специфические фобии, социальную фобию, обсессивно-компульсивный синдром, посттравматический стресс, острое стрессовое нарушение, генерализованное тревожное состояние и генерализованное тревожное состояние вследствие общего медицинского состояния, нарушения настроения, включая, но не ограничиваясь указанным, глубокую депрессию, психическую депрессию, биполярную депрессию, биполярную манию, биполярное нарушение I типа, связанную с манией депрессию, депрессивные или смешанные эпизоды, биполярное нарушение II типа, циклотимическое нарушение и нарушения настроения вследствие общего медицинского состояния, нарушения сна, включая, но не ограничиваясь указанным, бессонницу, первичную бессонницу, первичную повышенную сонливость, нарколепсию, парасомнию, нарушения, связанные с ночными кошмарами, нарушения ужаса перед сном и нарушения, связанные с хождением во сне, ментальное угасание, нарушения обучаемости, нарушения двигательных навыков, нарушения общения, нарушения, связанные с развитием роста, дефицит внимания и нарушения, связанные с разрушительным поведением, нарушение, связанное с дефицитом внимания, гиперактивность, связанная с нарушением внимания, нарушения кормления и питания младенцев, детей или взрослых, тики, элиминационные нарушения, нарушения, связанные с веществами, включая, но не ограничиваясь указанным, злоупотребление наркотиками, интоксикацию наркотиками, прекращение приема наркотиков, связанные с алкоголем нарушения, связанные с амфетамином или амфетаминоподобными соединениями нарушения, связанные с кофеином нарушения, связанные с марихуанной нарушения, связанные с кокаином нарушения, связанные с галлюциногенами нарушения, связанные с ингаляционными средствами нарушения, связанные с никотином нарушения, связанные с опиоидами нарушения, связанные с фенциклидином или фенциклидиноподобными средствами нарушения, и связанные с седативными, гипнотическими или анксиолитическими средствами нарушения, нарушения личности, включая, но не ограничиваясь указанным, обсессивно-компульсивные нарушения личности и импульсно-контрольные нарушения.
Симптомы шизофрении обычно подразделяют на три категории: положительные, отрицательные и когнитивные. Положительные симптомы также могут упоминаться как «психотические» симптомы и включают мании и галлюцинации. Положительные симптомы упоминаются как имеющиеся явные симптомы. Отрицательные симптомы включают эмоциональную подавленность или недостаток выражения, неспособность начать и продолжать активную деятельность, речь, которая является краткой и лишенной содержания, недостаток удовольствия и интереса к активной деятельности. Отрицательные симптомы относятся к недостатку определенных характеристик, которые в противном случае присутствовали бы у здорового человека. Когнитивные симптомы относятся к процессам мышления. Когнитивные симптомы включают недостаток познавательных способностей, таких как память, включая рабочую память, организующая функция, внимание, вигильность, обработка информации и обучаемость, со ссылкой на публикацию Sharma et al., Cognitive Function in Scizophrenia: Deficits, Functional Consequences and Future Treatment, Psychiatr. Clin. N. Am. 26 (2003) 25-40, включенную в данное описание путем ссылки. Шизофрения также влияет на настроение. Хотя многие индивидуумы, пораженные шизофренией, подвержены депрессии, у некоторых также имеются заметные колебания настроения и даже биполярно-подобные состояния.
Вышеуказанные состояния и нарушения обсуждаются более подробно, например, в справочнике «American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders», четвертое издание с редакцией, Вашингтон, DC, American Psychiatric Association, 2000; включенном в данное описание путем ссылки для определения таких состояний и нарушений. Данный справочник также можно упомянуть для более подробного описания симптомов и диагностических особенностей, связанных с применением, злоупотреблением и зависимостью от различных веществ.
Предпочтительно, лечение или предупреждение заболеваний, нарушений и состояний происходят без ощутимых побочных действий, включая, например, значительное повышение давления крови и сердечного ритма, существенное отрицательное влияние на желудочно-кишечный тракт и значительное воздействие на мышцы скелета.
Предполагается, что соединения по настоящему изобретению при использовании в эффективных количествах модулируют активность α7 NNR без заметного взаимодействия с другими подтипами никотиновых рецепторов, которые характерны для ганглия человека, как продемонстрировано недостатком способности вызывать никотиновую функцию в адреналиновой хромаффиновой ткани или мышце скелета, дополнительно продемонстрировано недостатком способности вызывать никотиновую функцию в препаратах клеток, экспрессирующих никотиновые рецепторы мышечного типа. Таким образом, можно полагать, что данные соединения способны лечить или предотвращать заболевания, нарушения и состояния, не вызывая заметных побочных действий, связанных с активностью ганглионовых и нейромышечных сайтов. Таким образом, предполагается, что введение соединений обеспечит терапевтическое окно, обеспечивающее лечение некоторых заболеваний, нарушений и состояний, при котором исключены некоторые побочные действия. То есть, полагается, что эффективная доза соединения является достаточной для обеспечения желаемого воздействия на заболевание, нарушение или состояние, но она полагается недостаточной, а именно не такого высокого уровня, чтобы вызвать нежелательные побочные воздействия.
Таким образом, настоящее изобретение относится к применению соединения по настоящему изобретению или его фармацевтически приемлемой соли для применения в терапии, такой как описанная выше терапия.
Еще в одном аспекте настоящее изобретение относится к применению соединения по настоящему изобретению или его фармацевтически приемлемой соли для получения лекарственного средства для использования при лечении нарушения ЦНС, такого как описанное выше нарушение, заболевание или состояние.
Воспаление
Известно, что нервная система, главным образом посредством блуждающего нерва, регулирует величину присущего иммунного ответа путем ингибирования высвобождения макрофагового фактора некроза опухоли (TNF). Данный физиологический механизм известен как «холинергический противовоспалительный путь» (см., например, Tracey, “The inflammatory reflex”, Nature 420:853-9 (2002), включенную в данное описание путем ссылки). Избыточное воспаление и синтез фактора некроза опухоли вызывают заболеваемость и даже смертность при множестве заболеваний. Данные заболевания включают, но не ограничиваются указанным, эндотоксемию, ревматоидный артрит, остеоартрит, псориаз, астму, атеросклероз, идиопатический фиброз легких и воспалительное заболевание кишечника.
Воспалительные состояния, которые можно лечить или для предупреждения которых можно использовать введение описанных в данном изобретении соединений, включают, но не ограничиваются указанным, хроническое и острое воспаление, псориаз, эндотоксемию, подагру, острую псевдоподагру, острый подагрический артрит, артрит, ревматоидный артрит, остеоартрит, полимиозит, дерматомиозит, анкилозирующий спондилит, болезнь Стилле, начало болезни Стилле у взрослых, отторжение аллотрансплантата, хроническое отторжение трансплантата, астму, атеросклероз, мононуклеарное-фагоцитзависимое повреждение легкого, идиопатической фиброз легких, атопический дерматит, хроническое обструктивное заболевание легких, респираторный дистресс-синдром у взрослых, острый легочный синдром при болезни серповидных клеток, воспалительное заболевание кишечника, болезнь Крона, язвенный колит, острый ангиохолит, афтозный стоматит (молочница), резервуарный илеит, гломерулонефрит, люпузный нефрит, тромбоз и реакцию трансплантат-против-хозяина.
Воспалительные ответные реакции, связанные с бактериальной
и/или вирусной инфекцией
Многие бактериальные и/или вирусные инфекции (например, менингит, гепатит и нефрит) связаны с побочными действиями, оказываемыми образованием токсинов, и природным ответом организма на бактерии или вирусы и/или токсины. Как обсуждалось выше, ответная реакция организма на инфекции часто включает образование значительного количества TNF и/или других цитокинов. Сверхэкспрессия таких цитокинов может нанести значительный вред, такой как септический шок (когда бактерия представляет собой сепсис), эндотоксический шок, уросепсис и синдром токсического шока.
Экспрессия цитокинов опосредуется NNR, и ее можно ингибировать путем введения агонистов или частичных агонистов данных рецепторов. Те соединения, которые описаны в данном изобретении, являются агонистами или частичными агонистами данных рецепторов, и, следовательно, могут использоваться для сведения к минимуму воспалительного ответа, связанного с бактериальной инфекцией, а также вирусными и грибковыми инфекциями. Примеры таких бактериальных инфекций включают карбункулы, ботулизм и сепсис. Некоторые из данных соединений также могут обладать антибактериальными свойствами.
Данные соединения также можно использовать в качестве вспомогательной терапии в сочетании с существующими методами лечения бактериальных, вирусных и грибковых инфекций, такими как антибиотики, антивирусные препараты и противогрибковые препараты. Также можно использовать антитоксины для связывания токсинов, продуцируемых инфицирующими агентами, и возможности проходить через организм в виде связанных токсинов, не вызывая воспалительного ответа. Примеры антитоксинов описаны, например, в патенте США № 6310043, Bundle et al., включенному в данное описание путем ссылки. Могут быть эффективными другие агенты, эффективные против бактерий и других токсинов, и их терапевтическое действие может быть дополнено совместным введением с описанными здесь соединениями.
Боль
Соединения можно применять для лечения и/или предупреждения боли, включая острую, неврологическую, воспалительную, невропатическую и хроническую боль. Анальгетическая активность описанных здесь соединений может быть продемонстрирована на модели постоянной воспалительной боли и невропатической боли, осуществленной, как описано в опубликованной заявке на патент США № 20010056084 А1 (Allgeier et al.), включенной в данное изобретение путем ссылки, в которой продемонстрирована гипералгезия на крысиной модели воспалительной боли с полным адъювантом Фрейнда и механическая гипералгезия невропатической боли на мышиной модели частичного лигирования седалищного нерва.
Анальгетическое действие является подходящим для лечения боли различного происхождения или этиологии, в частности для лечения воспалительной боли и связанной с этим гипералгезии, невропатической боли и связанной с этим гипералгезии, хронической боли (например, тяжелой хронической боли, послеоперационной боли и боли, связанной с различными состояниями, включая рак, стенокардию, почечную или желчную колику, менструацию, мигрень и подагру). Воспалительная боль может иметь различное происхождение, включая артрит и ревматоидное заболевание, синовит сухожильных влагалищ и васкулит. Невропатическая боль включает тригеминальную или герпетическую невралгию, диабетическую невропатическую боль, жгучую боль, боль в нижней части спины и синдромы деафферентации, такие как разрыв плечевого сплетения.
Неоваскуляризация
Рецептор α7 NNR связан с неоваскуляризацией. Ингибирование неоваскуляризации, например, путем введения антагонистов (или при некоторых дозировках частичных агонистов) α7 NNR может лечить или предотвращать состояния, характеризуемые нежелательной неоваскуляризацией или ангиогенезом. Такие состояния могут включать те, которые характеризуются воспалительным ангиогенезом и/или вызванным ишемией ангиогенезом. Неоваскуляризацию, связанную с ростом опухоли, также можно ингибировать путем введения таких соединений, как описанные в данном изобретении, которые функционируют в качестве антагонистов или частичных агонистов α7 NNR.
Специфический антагонизм специфической активности α7 NNR уменьшает ангиогенный ответ на воспаление, ишемию и неоплазию. Указания, касающиеся подходящих систем на модели животных для оценки описанных здесь соединений, могут быть найдены, например, в публикации Heeschen C. et al., «A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors”, J. Clin. Invest. 110(4): 527-36 (2002), включенной в данное описание путем ссылки для описания α7-специфического ингибирования ангиогенеза и клеточного (in vitro) и на модели животных моделирования ангиогенной активности, значимой для болезни человека, в особенности модели опухоли Леви легкого человека (in vivo, на мышах см., в частности, страницу 529 и 532-533).
Иллюстративные типы опухолей, которые можно лечить с использованием описанных здесь соединений, включают NSCLC (немелкоклеточная карцинома легких), рак яичника, рак поджелудочной железы, карциному молочной железы, карциному толстой кишки, карциному прямой кишки, карциному легких, карциному ротовой части глотки, карциному подглоточника, карциному пищевода, карциному желудка, карциному поджелудочной железы, карциному печени, карциному желчного пузыря, карциному желчных протоков, карциному тонкого кишечника, карциному мочевыводящих путей, карциному почки, карциному мочевого пузыря, уротелиальную карциному, карциному женских половых путей, карциному шейки матки, карциному матки, карциному яичника, трофобластическую опухоль, гестационное (относящиеся к беременности) трофобластическое заболевание, карциному мужских половых путей, карциному предстательной железы, карциному семенных пузырьков, карциному яичек, опухоли эмбриона, карциному эндокринной железы, карциному щитовидной железы, карциному надпочечника, карциному гипофиза, карциному кожи, геманглиомы, меланомы, саркомы, саркому кости и мягкой ткани, саркому Капоши, опухоли мозга, опухоли нервов, опухоли глаз, опухоли оболочек мозга, астроцитомы, глиомы, глиобластомы, ретинобластомы, нейромы, нейробластомы, шванномы, менингиомы, плотные опухоли, возникающие из гематопоэтических злокачественных новообразований (таких как лейкемии, хлорлейкемии, плазмацитомы и грибовидные бляшки и опухоли слизистой и лимфома/лейкемия кожных Т-клеток), и плотные опухоли, возникающие из лимфомы.
Соединения также можно вводить в сочетании с другими формами противораковой терапии, включая совместное введение с антинеопластическими противоопухолевыми агентами, такими как цисплатин, адриамицин, дауномицин и тому подобные и/или анти-VEGF (сосудистый эндотелиальный фактор роста) агентами, такими, как известно в данной области.
Соединения можно вводить таким образом, что они будут нацелены на место опухоли. Например, соединения можно вводить в микросферах, микрочастицах или липосомах, конъюгированных с различными антителами, которые направляют частицы в опухоль. Кроме того, соединения могут присутствовать в микросферах, микрочастицах или липосомах, которым придан подходящий размер для прохождения через артерии и вены, но которые застревают в капиллярном ложе, окружающем опухоль, и вводить соединения в опухоль локальным образом. Такие устройства для доставки лекарственных средств известны в данной области.
Другие нарушения
В дополнение к лечению нарушений ЦНС, воспаления, неоваскуляризации и боли, соединения по настоящему изобретению также можно использовать для предупреждения или лечения некоторых других состояний, заболеваний и нарушений, роль в которых играют NNR. Примеры включают аутоиммунные нарушения, такие как волчанка, нарушения, связанные с высвобождением цитокинов, вторичная кахексия после инфекции (например, как наблюдается при СПИД, связанном со СПИД комплексе и неоплазии), метаболические нарушения, включая диабет типа I, диабет типа II, метаболический синдром, ожирение или гиперкальцемию, пемфигус, недержание мочи, заболевания сетчатки, инфекционные заболевания, миастению, синдром Итона-Ламберта, повышенное давление крови, остеопороз, сужение кровеносных сосудов, дилатацию сосудов, сердечную аритмию, булимию, анорексию, а также показания, указанные в опубликованной заявке РСТ WO 98/25619, включенной в данной описание путем ссылки по вопросу, касающемуся таких нарушений. Соединения по данному изобретению также можно применять для лечения конвульсий, таких как являющиеся симптоматическими при эпилепсии, и для лечения состояний, таких как сифилис и болезнь Крейтцфельда-Якоба.
Диагностическое применение
Соединения можно использовать в диагностических композициях, таких как зонды, в особенности, если они модифицированы таким образом, что включают подходящие метки. Зонды можно использовать, например, для определения относительного числа и/или функции определенных рецепторов, в особенности подтипа α7 рецепторов. Для этой цели в соединения по настоящему изобретению предпочтительно вводят метку радиоактивного изотопа, такого как 11С, 18F, 76Br, 123I или 125I.
Введенные соединения можно обнаруживать с использованием известных методов обнаружения, подходящих для используемой метки. Примеры способов обнаружения включают позитронную эмиссионную томографию (РЕТ) и однофотонную эмиссионную компьютерную томографию (SPECT). Описанные выше радиоактивные метки можно использовать при РЕТ (например, 11С, 18F или 76Br) и SPECT (например, 123I) визуализации изображения с периодами полураспада, составляющими примерно 20,4 минуты для 11С, примерно 109 минут для 18F, примерно 13 часов для 123I, и примерно 16 часов для 76Br. Для визуализации выбранных подтипов рецепторов при концентрациях, не достигающих насыщения, желательна высокая специфическая активность. Вводимые дозы обычно находятся ниже токсического диапазона и обеспечивают изображения с высокой контрастностью. Ожидается, что соединения можно будет применять при нетоксичных уровнях. Определение дозы проводят способом, известным специалисту в области визуализации с применением радиоактивных меток. См., по поводу введения таких соединений, например, патент США № 5969144, London et al., включенный в данное изобретение путем ссылки.
Соединения можно вводить с использованием известных методов. См, например, патент США № 5969144, London et al., как отмечено, включенный в данное изобретение путем ссылки по вопросам, касающимся такого введения. Соединения можно вводить в рецептурных композициях, которые включают другие ингредиенты, такие как, например, те типы ингредиентов, которые используют при составлении рецептуры диагностических композиций. Соединения, используемые в соответствии с осуществлением настоящего изобретения, наиболее предпочтительно применяют в формах с высокой степенью чистоты. См, по поводу такого анализа патент США № 5853696, Elmalch et al., включенный в данное изобретение путем ссылки.
После введения соединений субъекту (например, человеку) присутствие такого соединения в организме субъекта можно отобразить визуально и оценить количественно с помощью подходящих методов для выявления присутствия, количества и функциональности выбранных подтипов NNR. Помимо людей, соединения также можно вводить животным, таким как мыши, крысы, собаки и обезьяны. SPECT и РЕТ визуализация может быть выполнена с использованием любого подходящего метода и прибора. Для описания иллюстративных методов визуализации см., Villemagne et al., в Arneric et al. (Eds.) Neuronal Nicotinic Receptors: Pharmacology and Therapeutic Opportunities, 235-250 (1998) и патент США № 5853696, Elmalch et al., каждый из которых включен в данное описание путем ссылки.
Радиоактивно меченные соединения связываются с высокой аффинностью с селективными подтипами NNR (например, α7) и предпочтительно проявляют пренебрежимо малое неспецифическое связывание в отношении других подтипов никотиновых холинергических рецепторов (например, те подтипы рецепторов, которые связаны с мускулатурой и нервными узлами (ганглии)). Как таковые, соединения можно использовать в качестве агентов для неинвазивной визуализации подтипов никотиновых холинергических рецепторов в организме субъекта, в частности в мозге, для диагностики, связанной с множеством заболеваний и нарушений ЦНС.
В одном аспекте диагностические композиции можно использовать в способе диагностирования заболевания у субъекта, такого как человек. Способ включает введение такому пациенту соединения с обнаруживаемой меткой, как описано в данном изобретении, и обнаружение связывания такого соединения с выбранными подтипами NNR (например, подтип α7 рецептора). Специалисты в области использования диагностических инструментов, таких как РЕТ и SPECT, могут использовать описанные здесь радиоактивно меченные соединения для диагностики большого числа состояний и нарушений, включая состояния и нарушения, связанные с дисфункцией центральной и вегетативной нервной системы. Такие нарушения включают множество заболеваний и нарушений ЦНС, включая болезнь Альцгеймера, болезнь Паркинсона и шизофрению. Можно провести оценки этих и других иллюстративных заболеваний и нарушений, включая те, которые указаны в патенте США № 5952339, Bencherif et al., включенному в данное описание путем ссылки.
В другом аспекте диагностические композиции можно использовать в способе контроля селективных подтипов никотиновых рецепторов у субъекта, такого как пациент-человек. Способ включает введение такому пациенту соединения с обнаруживаемой меткой, как описано в данном изобретении, и обнаружение связывания такого соединения с выбранными подтипами никотиновых рецепторов, а именно подтипа α7 рецептора.
Связывание с рецепторами
Соединения по данному изобретению можно использовать в качестве лигандов сравнения в анализах связывания для соединений, которые связываются с подтипами NNR, в частности с подтипом α7 рецептора. С этой целью в соединения по изобретению предпочтительно вводят радиоактивную метку с использованием радиоактивного изотопного фрагмента, такую как 3Н или 14С.
V. Синтетические примеры
Следующие синтетические примеры приведены для иллюстрации настоящего изобретения и их не следует рассматривать как ограничивающие объем изобретения. В данных экспериментах все части и проценты приведены по весу, если не указано другого. Все растворы представляют собой водные растворы, если не указано другого.
Пример 1
Синтез в небольшом масштабе (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамида (соединение А) и его энантиомера (2R,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамида
2-((3-Пиридинил)метилен)-1-азабицикло[2.2.2]октан-3-он
Гидроксид калия (56 г, 0,54 моль) растворяли в метаноле (420 мл). Добавляли гидрохлорид 3-хинуклидинона (75 г, 0,49 моль) и смесь перемешивали в течение 30 минут при температуре окружающей среды. Добавляли 3-пиридинкарбоксальдегид (58 г, 0,54 моль) и смесь перемешивали в течение 16 часов при температуре окружающей среды. За это время реакционная смесь приобретала желтый цвет, и твердый осадок образовывался на стенках колбы. Твердые вещества соскребали со стенок и крупные куски разрушали. При быстром перемешивании добавляли воду (390 мл). Когда твердые вещества растворялись, смесь охлаждали до 4°С и выдерживали в течение ночи. Кристаллы собирали фильтрованием, промывали водой и сушили на воздухе, получая 80 г желтого твердого вещества. Вторую порцию (8 г) получали путем концентрирования фильтрата до ~10% его первоначального объема и выдерживания при охлаждении до 4°С в течение ночи. Обе порции были достаточно чистыми для последующего превращения (88 г, 82% выход).
2-((3-Пиридинил)метил)-1-азабицикло[2.2.2]октан-3-он
2-((3-Пиридинил)метилен)-1-азабицикло[2.2.2]октан-3-он (20 г, 93 ммоль) суспендировали в метаноле (200 мл) и обрабатывали 46 мл 6М соляной кислоты. Добавляли 10% палладий-на-угле (1,6 г) и смесь встряхивали при давлении водорода 25 фт/кв.дюйм в течение 16 часов. Смесь фильтровали через диатомовую землю и растворитель удаляли из фильтрата путем упаривания на роторном испарителе. Это давало неочищенный гидрохлорид 2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она в виде белой смолы (20 г), который впоследствии обрабатывали 2М гидроксидом натрия (50 мл) и хлороформом (50 мл) и перемешивали в течение часа. Слой хлороформа отделяли и водную фазу обрабатывали 2М гидроксидом натрия (~5 мл, достаточно до повышения рН до 10) и насыщенным водным раствором хлорида натрия (25 мл). Водную смесь экстрагировали хлороформом (3×10 мл) и объединенный хлороформный экстракт сушили (безводный сульфат магния) и концентрировали путем упаривания на роторном испарителе. Остаток (18 г) растворяли в теплом простом эфире (320 мл) и охлаждали до 4°С. Белое твердое вещество отфильтровывали, промывали небольшой порцией холодного простого эфира и сушили на воздухе. Концентрирование фильтрата до ~10% его первоначального объема и охлаждение до 4°С давали вторую порцию. Получали объединенный выход в 16 г (79%).
3-Амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан
К перемешиваемому раствору 2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она (3,00 г, 13,9 ммоль) в сухом метаноле (20 мл) в атмосфере азота добавляли 1М раствор хлорида цинка в простом эфире (2,78 мл, 2,78 ммоль). После перемешивания при температуре окружающей среды в течение 30 минут данную смесь обрабатывали твердым формиатом аммония (10,4 г, 167 ммоль). После перемешивания в течение еще одного часа при температуре окружающей среды добавляли порциями твердый цианоборгидрид натрия (1,75 г, 27,8 ммоль). Реакционную смесь затем перемешивали при температуре окружающей среды в течение ночи и прекращали реакцию путем добавления воды (~5 мл). Реакционную смесь после гашения реакции распределяли между 5М гидроксидом натрия (10 мл) и хлороформом (20 мл). Водный слой экстрагировали хлороформом (20 мл) и объединенные органические слои сушили (сульфат натрия), фильтровали и концентрировали. Это приводило в остатке к 2,97 г желтой смолы. ГЖХ-масс-спектрометрический (GCMS) анализ показал, что продукт представлял собой смесь цис- и транс-аминов в соотношении 1:9 наряду со следами соответствующего спирта (98% выделение по общей массе).
(2R,3S) и (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан
Ди-п-толуоил-D-винную кислоту (5,33 г, 13,8 ммоль) добавляли к перемешиваемому раствору неочищенного 3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октана (6,00 г, 27,6 ммоль цис/транс в соотношении 1:9) в метаноле (20 мл). После завершения растворения прозрачный раствор затем концентрировали до получения твердой массы путем упаривания на роторном испарителе. Твердое вещество затем растворяли в минимальном количестве кипящего метанола (~5 мл). Раствор медленно охлаждали, первоначально до температуры окружающей среды (1 ч), затем выдерживали в течение ~4 часов при 5°С и, наконец, при -5°С в течение ночи. Выпавшую в осадок соль собирали фильтрованием с отсасыванием растворителя в вакууме и перекристаллизовывали из 5 мл метанола. Сушка на воздухе давала 1,4 г белого твердого вещества, которое распределяли между хлороформом (5 мл) и 2М гидроксидом натрия (5 мл). Хлороформный слой и 5 мл хлороформного экстракта водного слоя объединяли, сушили (безводный сульфат натрия) и концентрировали, получая бесцветное масло (0,434 г). Энантиомерную чистоту для данного свободного основания определяли путем преобразования его части в соответствующий N-(трет-бутоксикарбонил)-L-пролинамид, для которого затем анализировали диастереомерную чистоту (98%) с использованием ЖХМС.
Маточный раствор после первоначальной кристаллизации делали основным (~рН 11) с помощью 2М гидроксида натрия и дважды экстрагировали хлороформом (10 мл). Хлороформные экстракты сушили (безводный сульфат натрия) и концентрировали, получая масло. Данный амин (3,00 г, 13,8 ммоль) растворяли в метаноле (10 мл) и обрабатывали ди-п-толуоил-L-винной кислотой (2,76 г, 6,90 ммоль). Смесь нагревали для того, чтобы способствовать растворению и затем медленно охлаждали до -5°С, оставляя при этой температуре на ночь. Осадок собирали фильтрованием с отсасыванием растворителя в вакууме, перекристаллизовывали из метанола и сушили. Это давало в остатке 1,05 г белого твердого вещества. Данную соль преобразовывали в свободное основание (выход = 0,364 г) и энантиомерную чистоту (97%) оценивали с использованием пролинамидного способа, как описано выше для другого энантиомера.
Транс-энантиомер А N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамида
Дифенилхлорфосфат (0,35 мл, 0,46 г, 1,7 ммоль) добавляли по каплям к раствору бензофуран-2-карбоновой кислоты (0,28 г, 1,7 ммоль) и триэтиламина (0,24 мл, 0,17 г, 1,7 ммоль) в сухом дихлорметане (5 мл). После перемешивания при температуре окружающей среды в течение 30 минут добавляли раствор (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октана (0,337 г, 1,55 ммоль) (который был получен из соли ди-п-толуолил-D-винной кислоты) и триэтиламина (0,24 мл, 0,17 г, 1,7 ммоль) в сухом дихлорметане (5 мл). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды и затем обрабатывали 10% раствором гидроксида натрия (1 мл). Двухфазную смесь разделяли и органический слой концентрировали на центрифужном испарителе Genevac. Остаток растворяли в метаноле (6 мл) и очищали с помощью ВЭЖХ на колонке С18 с силикагелем с использованием градиента смеси ацетонитрил/вода, содержащей 0,05% трифторуксусной кислоты, в качестве элюента. Концентрирование выбранных фракций, распределение полученного остатка между хлороформом и насыщенным водным раствором бикарбоната натрия и упаривание хлороформа давали 0,310 г (42% выход) белого порошка (95% чистота по данных ГЖХ-масс-спектрометрии). 1Н ЯМР (300 МГц, CDCl3) δ 8,51 (д, 1H), 8,34 (дд, 1H), 7,66 (д, 1H), 7,58 (дт, 1H), 7,49 (д, 1H), 7,44 (с, 1H), 7,40 (дд, 1H), 7,29 (т, 1H), 7,13 (дд, 1H), 6,63 (д, 1H), 3,95 (т, 1H), 3,08 (м, 1H), 2,95 (м, 4H), 2,78 (м, 2H), 2,03 (м, 1H), 1,72 (м, 3H), 1,52 (м, 1H).
Позднее было определено с помощью хирального хроматографического анализа, что данное вещество (транс-энантиомер А) идентично веществу, абсолютная конфигурация которого 2S,3R (определено с помощью рентгенодифракционного кристаллографического анализа).
Транс-энантиомер В N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамида
Дифенилхлорфосфат (96 мкл, 124 мг, 0,46 ммоль) добавляли по каплям к раствору бензофуран-2-карбоновой кислоты (75 мг, 0,46 ммоль) и триэтиламина (64 мкл, 46 мг, 0,46 ммоль) в сухом дихлорметане (1 мл). После перемешивания при температуре окружающей среды в течение 45 минут добавляли раствор (2R,3S)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октана (0,10 г, 0,46 ммоль) (который был получен из соли ди-п-толуолил-L-винной кислоты) и триэтиламина (64 мкл, 46 мг, 0,46 ммоль) в сухом дихлорметане (1 мл). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды и затем обрабатывали 10% раствором гидроксида натрия (1 мл). Двухфазную смесь разделяли и органический слой и хлороформный экстракт (2 мл) водного слоя концентрировали с помощью роторного испарителя. Остаток растворяли в метаноле и очищали с помощью ВЭЖХ на колонке С18 с силикагелем с использованием градиента смеси ацетонитрил/вода, содержащей 0,05% трифторуксусной кислоты, в качестве элюента. Концентрирование выбранных фракций, распределение полученного остатка между хлороформом и насыщенным водным раствором бикарбоната натрия и упаривание хлороформа давали 82,5 мг (50% выход) белого порошка. Спектр ЯМР был идентичен спектру, полученному для 2S,3R-изомера. Поскольку непосредственный предшественник данного вещества (транс-энантиомер В) является энантиомерным непосредственному предшественнику 2S,3R-соединения (транс-энантиомер А), абсолютную конфигурацию транс-энантиомера В предполагали как представляющую собой 2R,3S.
Пример 2. Крупномасштабный синтез (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамида и п-толуолсульфонатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)-1-бензофуран-2-карбоксамида.
2-((3-Пиридинил)метилен)-1-азабицикло[2.2.2]октан-3-он
Гидрохлорид 3-хинуклидинона (8,25 кг, 51,0 моль) и метанол (49,5 л) добавляли, в атмосфере азота, в стеклянную реакционную колбу емкостью 100 л, оборудованную механической мешалкой, температурным датчиком и обратным холодильником. Через воронку для порошка добавляли гидроксид калия (5,55 кг, 99,0 моль) в течение примерно 30 минут, что приводило к повышению температуры реакционной смеси от 50°С до 56°С. К реакционной смеси на протяжении примерно 2 часов добавляли 3-пиридинкарбоксальдегид (4,80 кг, 44,9 моль). Полученную смесь перемешивали при 20°С±5°С в течение как минимум 12 часов по мере контроля за ходом реакции с помощью тонкослойной хроматографии (ТСХ). После завершения реакции реакционную смесь фильтровали через фильтр из пористого стекла и осадок на фильтре промывали метанолом (74,2 л). Фильтрат концентрировали, переносили в реакционную колбу и добавляли воду (66,0 л). Суспензию перемешивали в течение как минимум 30 минут, фильтровали и осадок на фильтре промывали водой (90 л) до тех пор пока рН раствора после промывки не достигал 7-9. Твердое вещество сушили в вакууме при 50°С±5°С в течение как минимум 12 часов, получая 8,58 кг (89,3%) 2-((3-пиридинил)метилен)-1-азабицикло[2.2.2]октан-3-она.
ди-п-Толуоил-D-тартратная соль (2S)-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она
2-((3-Пиридинил)метилен)-1-азабицикло[2.2.2]октан-3-он (5,40 кг, 25,2 моль) и метанол (40,5 л) в реакционную колбу емкостью 72 л с инертной атмосферой, оборудованную механической мешалкой, температурным датчиком, регулирующей системой низкого давления газа и прибором для измерения давления. Пространство над реакционной смесью заполняли азотом и смеси перемешивали до получения прозрачного желтого раствора. В колбу добавляли 10% палладий-на-угле (50% влажный) (270 г). Атмосферу из реактора удаляли с использованием вакуумного насоса и пространство над реакционной смесью заменяли на водород до давления 10-20 дюймов водяного столба. Удаление газовой атмосферы из колбы и подачу водорода под давлением повторяли еще 2 раза, оставляя реактор под давлением газообразного водорода в 20 дюймов водяного столба после третьей подачи газа под давлением. Реакционную смесь перемешивали при 20°С±5°С в течение как минимум 12 часов и контроль за ходом реакции проводили с помощью ТСХ. После завершения реакции суспензию фильтровали через слой Celite®545 (1,9 кг) на фильтре из пористого стекла и осадок на фильтре промывали метанолом (10,1 л). Фильтрат концентрировали до получения полутвердого вещества, которое переносили, в атмосфере азота, в реакционную колбу емкостью 100 л, оборудованную механической мешалкой, обратным холодильником и температурным датчиком. Полутвердое вещество растворяли в этаноле (57,2 л) и добавляли ди-п-толуоил-D-винную кислоту (DTTA) (9,74 кг, 25,2 моль). Перемешиваемую реакционную смесь нагревали при кипении с обратным холодильником в течение как минимум 1 часа и перемешивали дополнительно в течение как минимум 12 часов по мере охлаждения реакционной смеси до температуры между 15°С и 30°С. Суспензию фильтровали через настольный фильтр и осадок на фильтре промывали этанолом (11,4 л). Продукт сушили в вакууме при температуре окружающей среды, получая 11,6 кг (72% выход, 59,5% факторизируемый по чистоте) ди-п-толуоил-D-тартратной соли (2S)-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она.
ди-п-Толуоил-D-тартратная соль (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октана
Воду (46,25 л) и бикарбонат натрия (4,35 кг, 51,8 моль) добавляли в колбу емкостью 200 л. После завершения растворения добавляли дихлорметан (69,4 л). Добавляли ди-п-толуоил-D-тартратную соль (2S)-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она (11,56 кг, 19,19 моль) и реакционную смесь перемешивали в течение от 2 минут до 10 минут. Слои оставляли для отделения как минимум на 2 минуты (добавляли дополнительно воду (20 л), если это было необходимо для распределения слоев). Органическую фазу удаляли и сушили над безводным сульфатом натрия. К оставшейся водной фазе добавляли дихлорметан (34,7 л) и суспензию перемешивали в течение от 2 минут до 10 минут. Слои оставляли для отделения в течение от 2 минут до 10 минут. Опять органическую фазу удаляли и сушили над безводным сульфатом натрия. Экстракцию водной фазы дихлорметаном (34,7 л) повторяли еще один раз, как описано выше. Образцы, полученные в каждой экстракции, анализировали с помощью хирального ВЭЖХ анализа. Сульфат натрия удаляли путем фильтрования и фильтраты концентрировали, получая (2S)-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-он (4,0 кг) в виде твердого вещества.
(2S)-2-((3-Пиридинил)метил)-1-азабицикло[2.2.2]октан-3-он (3,8 кг) переносили в атмосфере азота в чистую стеклянную реакционную колбу емкостью 100 л, оборудованную механической мешалкой и температурным датчиком. Добавляли безводный тетрагидрофуран (7,24 л) и (+)-(R)-α-метилбензиламин (2,55 л, 20,1 моль). К перемешиваемой реакционной смеси в течение 1 часа добавляли изопропоксид титана(IV) (6,47 л, 21,8 моль). Реакционную смесь перемешивали в атмосфере азота в течение как минимум 12 часов. К реакционной смеси добавляли этанол (36,17 л). Реакционную смесь охлаждали до температуры ниже -5°С и добавляли порциями боргидрид натрия (1,53 кг, 40,5 моль), поддерживая температуру реакционной смеси ниже 15°С (такое добавление занимало несколько часов). Реакционную смесь затем перемешивали при 15°С±10°С в течение минимум 1 часа. Контроль за ходом реакции проводили с помощью ВЭЖХ и после завершения реакции (на что показывало присутствие менее 0,5% оставшегося (2S)-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-она) добавляли 2М гидроксид натрия (15,99 л) и смесь перемешивали в течение как минимум 10 минут. Реакционную смесь фильтровали через слой Celite®545 на настольной воронке. Осадок на фильтре промывали этанолом (15,23 л) и фильтрат концентрировали, получая масло.
Концентрат переносили в чистую стеклянную реакционную колбу емкостью 100 л, оборудованную механической мешалкой и температурным датчиком, в инертной атмосфере. Добавляли воду (1 л) и смесь охлаждали до 0°С±5°С. К смеси добавляли 2М соляную кислоту (24 л) для доведения рН смеси до рН 1. Затем смесь перемешивали в течение как минимум 10 минут и водную фазу экстрагировали дихлорметаном (3×15,23 л). Органические фазы сушили над безводным сульфатом натрия (2,0 кг), фильтровали и концентрировали, получая (2S,3R)-N-((1R)-фенилэтил)-3-амино-2- ((3-пиридинил)метил)-1-азабицикло[2.2.2]октан (4,80 кг, 84,7% выход).
(2S,3R)-N-((1R)-Фенилэтил)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан переносили в стеклянную колбу емкостью 22 л, оборудованную механической мешалкой и температурным датчиком, в инертной атмосфере. Добавляли воду (4,8 л) и перемешиваемую смесь охлаждали до 5°С±5°С. В реакционную колбу медленно добавляли концентрированную соляную кислоту (2,97 л), поддерживая температуру смеси ниже 25°С. Полученный раствор переносили в реакционную колбу емкостью 72 л, содержащую этанол (18 л), оборудованную механической мешалкой, температурным датчиком и обратным холодильником, в инертной атмосфере. В колбу добавляли 10% палладий-на-угле (50% влажный) (311,1 г) и циклогексен (14,36 л). Реакционную смесь нагревали при температуре, близкой к температуре кипения с обратным холодильником, в течение как минимум 12 часов и ход реакции контролировали с помощью ТСХ. После завершения реакции реакционную смесь охлаждали до температуры ниже 45°С и фильтровали через слой Celite® 545 (1,2 кг) на воронке со стеклянным пористым фильтром. Осадок на фильтре промывали этанолом (3 л) и фильтрат концентрировали, получая водную фазу. К концентрированному фильтрату добавляли воду (500 мл) и объединенный водный слой промывали метил-трет-бутиловым эфиром (МТВЕ) (2×4,79 л). К водной фазе добавляли 2М гидроксид натрия (19,5 л) для доведения рН смеси до рН 14. Смесь затем перемешивали в течение как минимум 10 минут. Водную фазу экстрагировали хлороформом (4×11,96 л) и объединенные органические фазы сушили над безводным сульфатом натрия (2,34 кг). Фильтрат фильтровали и концентрировали, получая (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан (3,49 кг > чем количественный выход) в виде масла.
(2S,3R)-3-Амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан переносили в чистую реакционную колбу емкостью 100 л, оборудованную механической мешалкой, обратным холодильником и температурным датчиком, в инертной атмосфере. Добавляли этанол (38,4 л) и ди-п-толуоил-D-винную кислоту (3,58 кг, 9,27 моль). Реакционную смесь нагревали при несильном кипении с обратным холодильником в течение как минимум 1 часа. Реакционную смесь затем перемешивали в течение как минимум 12 часов по мере ее охлаждения до температуры между 15°С и 30°С. Полученную суспензию фильтровали и осадок на фильтре промывали этанолом (5,76 л). Осадок на фильтре переносили в чистую стеклянную реакционную колбу емкостью 100 л, оборудованную механической мешалкой, обратным холодильником и температурным датчиком, в инертной атмосфере. Добавляли раствор этанол/вода в соотношении 9:1 (30,7 л) и полученную суспензию нагревали при несильном кипении с обратным холодильником в течение как минимум 1 часа. Реакционную смесь затем перемешивали в течение как минимум 12 часов по мере ее охлаждения до температуры между 15°С и 30°С. Смесь фильтровали, и осадок на фильтре промывали этанолом (5,76 л). Собирали продукт и сушили в вакууме при 50°С±5°С в течение как минимум 12 часов, получая 5,63 кг (58,1% выход) ди- п-толуоил-D-тартратной соли (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октана.
(2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамид
ди-п-Толуоил-D-тартратную соль (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октана (3,64 кг, 5,96 моль) и 10% водный раствор хлорида натрия (14,4 л, 46,4 моль) добавляли в стеклянную реакционную колбу емкостью 72 л, оборудованную механической мешалкой, в инертной атмосфере. К перемешиваемой смеси добавляли 5М гидроксид натрия (5,09 л) для доведения рН смеси до рН 14. Смесь затем перемешивали в течение как минимум 10 минут. Водный раствор экстрагировали хлороформом (4×12,0 л) и объединенные органические слои сушили над безводным сульфатом натрия (1,72 кг). Объединенные органические слои фильтровали и фильтрат концентрировали, получая (2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан (1,27 кг) в виде масла.
(2S,3R)-3-амино-2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан переносили в стеклянную реакционную колбу емкостью 50 л, оборудованную механической мешалкой, в инертной атмосфере. К реакционной смеси добавляли дихлорметан (16,5 л), триэтиламин (847 мл, 6,08 моль), бензофуран-2-карбоновую кислоту (948 г, 5,85 моль) и гексафторфосфат О-(бензотриазол-1-ил)-N,N,N,1-тетраметилурония (HBTU) (2,17 кг, 5,85 моль). Смесь перемешивали в течение как минимум 4 часов при температуре окружающей среды и ход реакции контролировали с помощью ВЭЖХ. После завершения реакции к реакционной смеси добавляли 10% водный раствор карбоната калия (12,7 л, 17,1 моль) и смесь перемешивали в течение как минимум 5 минут. Слои отделяли и органическую фазу промывали 10% раствором поваренной соли (12,7 л). Слои отделяли и органическую фазу охлаждали до 15°С±10°С. К реакционной смеси медленно добавляли 3М соляную кислоту (8,0 л) для доведения рН смеси до рН 1. Смесь затем перемешивали в течение как минимум 5 минут и слои оставляли для распределения как минимум на 5 минут. Твердые вещества отфильтровывали с использованием настольного фильтра. Слои фильтрата отделяли и водную фазу и твердые вещества из воронки переносили в реакционную колбу. В колбу медленно добавляли порциями 3М гидроксид натрия (9,0 л) для доведения рН смеси до рН 14. Водную фазу экстрагировали дихлорметаном (2×16,5 л). Объединенные органические фазы сушили над безводным сульфатом натрия (1,71 кг). Смесь фильтровали и фильтрат концентрировали, получая (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамид (1,63 кг, 77,0% выход) в виде желтого твердого вещества.
п-Толуолсульфонат (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид
(2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]октан-3-ил)бензофуран-2-карбоксамид (1,62 кг, 4,48 моль) и дихлорметан (8,60 кг) добавляли в оплетенную бутыль. Проценты вес/вес вещества в растворе определяли с помощью ВЭЖХ анализа. Раствор концентрировали до масла, добавляли ацетон (4 л) и смесь концентрировали, получая маслянистое твердое вещества. Дополнительно добавляли ацетон (12 л) к маслянистому веществу в колбе роторного испарителя и полученную суспензию переносили в стеклянную реакционную колбу емкостью 50 л с механической мешалкой, обратным холодильником, температурным датчиком и обратным холодильником в инертной атмосфере. Реакционную смесь нагревали до 50°С±5°С. К раствору добавляли воду (80,7 г) и перемешивали его в течение как минимум 10 минут. К реакционной смеси порциями добавляли п-толуолсульфоновую кислоту (853 г, 4,44 моль) в течение примерно 15 минут. Реакционную смесь нагревали до кипения с обратным холодильником и выдерживали при данной температуре в течение как минимум 30 минут для получения раствора. Реакционную смесь охлаждали до 40°С±5°С в течение приблизительно 2 часов. В течение примерно 1,5 часов добавляли изопропилацетат (14,1 л). Реакционную смесь медленно охлаждали до температуры окружающей среды в течение как минимум 10 часов. Смесь фильтровали и осадок на фильтре промывали изопропилацетатом (3,5 л). Выделенный продукт сушили в вакууме при 105°С±5°С в течение от 2 до 9 часов, получая 2,19 кг (88,5% выход) п-толуолсульфоната (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, т.пл. 226-228°С. 1Н ЯМР (500 МГц, D2O) δ 8,29 (с, 1H), 7,78 (м, J=5,1, 1H), 7,63 (д, J=7,9, 1H), 7,54 (д, J=7,8, 1H), 7,49 (д, J=8,1, 2H), 7,37 (м, J=8,3, 1H), 7,33 (м, J=8,3, 6,9, 1,0, 1H), 7,18 (м, J=7,8, 6,9, 1,0, 1H), 7,14 (д, J=8,1, 2H), 7,09 (с, 1H), 6,99 (дд, J=7,9, 5,1, 1H), 4,05 (м, J=7,7, 1H), 3,74 (м, 1H), 3,47 (м, 2H), 3,28 (м, 1H), 3,22 (м, 1H), 3,15 (дд, J=13,2, 4,7, 1H), 3,02 (дд, J=13,2, 11,5, 1H), 2,19 (с, 3H), 2,02 (м, 2H), 1,93 (м, 2H), 1,79 (м, 1H). 13C ЯМР (126 МГц, D2O) δ 157,2, 154,1, 150,1, 148,2, 146,4, 145,2, 138,0, 137,0, 130,9, 128,2 (2), 126,9, 126,8, 125,5 (2), 123,7, 123,3, 122,7, 111,7, 100,7, 61,3, 50,2, 48,0, 40,9, 33,1, 26,9, 21,5, 20,8, 17,0.
Образцы данного вещества преобразовывали в свободное основание соединения А (для применения в исследованиях по выбору соли) путем обработки водным раствором гидроксида натрия и экстракцией хлороформом. По мере упаривания хлороформа оставался не совсем белый порошок, т.пл. 167-170°С, имеющий следующие спектральные характеристики: электрораспыление в режиме положительных ионов, масс-спектр [M+H]+ ион m/z=362. 1Н ЯМР (500 МГц, ДМСО-d6) δ 8,53 (д, J=7,6 Гц, 1H), 8,43 (д, J=1,7 Гц, 1H), 8,28 (дд, J=1,6, 4,7 Гц, 1H), 7,77 (д, J=7,7 Гц, 1H), 7,66 (д, J=8,5 Гц, 1H), 7,63 (дт, J=1,7, 7,7 Гц, 1H), 7,52 (с, 1H), 7,46 (м, J=8,5, 7,5 Гц, 1H), 7,33 (м, J=7,7, 7,5 Гц, 1H), 7,21 (дд, J=4,7, 7,7 Гц, 1H), 3,71 (м, J=7,6 Гц, 1H), 3,11 (м, 1H), 3,02 (м, 1H), 2,80 (м, 2H), 2,69 (м, 2H), 2,55 (м, 1H), 1,80 (м, 1H), 1,77 (м, 1H), 1,62 (м, 1H), 1,56 (м, 1H), 1,26 (м, 1H). 13C ЯМР (126 МГц, ДМСО-d6) δ 158,1, 154,1, 150,1, 149,1, 146,8, 136,4, 135,4, 127,1, 126,7, 123,6, 122,9, 122,6, 111,8, 109,3, 61,9, 53,4, 49,9, 40,3, 35,0, 28,1, 26,1, 19,6.
Моногидрохлоридную соль соединения А (см. пример 5) подвергали рентгенодифракционному кристаллографическому анализу. Полученная кристаллическая структура (показана на фиг.10А и 10В, соответственно) позволяет установить 2S,3R абсолютную конфигурацию соединения А.
Пример 3. Синтез фосфатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
В круглодонную колбу добавляли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид (8,18 г, 22,6 ммоль) и 2-пропанол (180 мл). Смесь перемешивали и нагревали при 65-70°С до тех пор, пока не растворяли все твердые вещества. Раствор интенсивно перемешивали при 65-70°С и добавляли по каплям с помощью пипетки фосфорную кислоту (1,65 мл, 24,3 ммоль). Сразу же образовывалось белое гранулированное твердое вещество. Смесь перемешивали при 65-70°С в течение 30 минут, охлаждали до температуры окружающей среды (23°С) и дополнительно перемешивали в течение 24 часов. Белое твердое вещество собирали фильтрованием с отсасыванием осадка в вакууме и осадок на фильтре промывали 2-пропанолом (20 мл) и твердое вещество сушили на воздухе по крайней мере в течение 1 часа. Твердое вещество дополнительно сушили в вакуумной печи при 75°С в течение ночи (16 часов), получая 10,7 г продукта (> количественный выход), т.пл. 265-273°С с разложением, с изменением кристалличности, наблюдаемым при ~180°С. Спектр 1Н ЯМР (ДМСО-d6) показывал на присутствие 2-пропанола (прочный сольват), что может объяснить выход, превышающий количественный. Анализ методом хиральной ЖХ показывал чистоту 97,1% (270 нм).
Пример 4. Синтез малеатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Малеиновую кислоту (0,067 г, 0,630 ммоль) добавляли к горячей суспензии (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (0,203 г, 0,560 ммоль) в изопропилацетате (2 мл), с осаждением мелкого белого твердого вещества вместе со смолоподобным осадком. Дополнительно добавляли изопропилацетат (3 мл) и малеиновую кислоту (0,006 г) и смесь нагревали до кипения с обратным холодильником. При кипении с обратным холодильником добавляли изопропанол (5 мл). Полученную смесь белых твердых веществ охлаждали до температуры окружающей среды, фильтровали и твердые вещества промывали изопропилацетатом (2 мл). Продукт сушили в вакууме при 60°С в течение 18 часов получая 0,228 г не совсем белого хлопьевидного твердого вещества (выход 84,7%), т.пл.180-182°С. Спектр 1Н ЯМР (ДМСО-d6) указывал на стехиометрический состав моносоли. Вычислено для C22H23N3O2 .C4H4O4: C, 65,40; H, 5,70; N, 8,80. Найдено: С, 65,35, 65,29; H, 5,86, 5,68; N, 8,69, 8,78.
Пример 5. Синтез гидрохлоридных солей (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Моногидрохлорид: Получали раствор соляной кислоты в ТГФ путем добавления по каплям концентрированной соляной кислоты (1,93 мл 12М раствора, 23,2 ммоль) к 8,5 мл охлажденного ТГФ. Раствор нагревали до температуры окружающей среды. В круглодонную колбу добавляли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид (8,49 г, 23,5 ммоль) и ацетон (85 мл). Смесь перемешивали и нагревали при 45-50°С до получения абсолютного раствора. Полученный выход раствор соляной кислоты в ТГФ добавляли по каплям в течение 5 минут, используя дополнительное количество ТГФ (1,5 мл) для переноса. Во время добавления раствора кислоты начинало образовываться гранулированное белое твердое вещество. Смесь охлаждали до температуры окружающей среды и перемешивали в течение ночи (16 часов). Твердое вещество собирали фильтрованием с отсасыванием осадка в вакууме, осадок на фильтре промывали ацетоном (10 мл) и твердое вещество сушили на воздухе с отсасыванием в вакууме в течение 30 минут. Твердое вещество дополнительно сушили в вакуумной печи при 75°С в течение 2 часов, получая 8,79 г мелких белых кристаллов (выход 94%), т.пл. 255-262°С. Анализ методом хиральной ЖХ давал чистоту в 98,8% (270 нм). Спектр 1Н ЯМР (ДМСО-d6) не показал присутствия остаточных растворителей и подтверждал моностехиометрический состав. 1Н ЯМР (300 МГц, ДМСО-d6) δ 10,7 (широкий с, 1Н - четвертичный аммоний), 8,80 (широкий с, 1Н - амидный Н), 8,54 (с, 1H), 8,23 (д, 1H), 7,78 (д, 1H), 7,74 (д, 1H), 7,60 (д, 1H), 7,47 (м, 2H), 7,33 (м, 1H), 7,19 (м, 1H), 4,19 (м, 1H), 4,08 (м, 1H), 3,05-3,55 (м, 6H), 2,00-2,10 (м, 3H), 1,90 (м, 1H), 1,70 (м, 1H). Рентгеноструктурный кристаллографический анализ данной соли установил стереохимическое отнесение и стехиометрию (см. фиг.10А и 10В).
Дигидрохлорид: Газообразный хлористый водород медленно барботировали в охлаждаемый льдом раствор (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2- карбоксамида (1,9 г, 5,3 ммоль) в безводном простом эфире (25 мл). Летучие компоненты удаляли, первоначально в токе азота и затем в высоком вакууме (газопромыватель с гидроксидом натрия встраивали в высоковакуумную линию). Остаток растирали несколько раз с небольшими порциями безводного простого эфира (отбрасывали) и оставшееся твердое вещество сушили в высоком вакууме. Это давало 2,17 г (выход 94%) не совсем белого порошка, т.пл. 210-212°С (гигроскопично). Анализ методом хиральной ЖХ давал чистоту 93,7% (270 нм). Масс-спектр методом электрораспыления в режиме положительных ионов давал ион [M+H]+ m/z=362. 1Н ЯМР (300 МГц, CD3OD) δ 9,15 (с, 1H), 8,84 (д, 1H), 8,63 (д, 1H), 7,97 (т, 1H), 7,75 (д, 1H), 7,61 (д, 1H), 7,52 (м, 2H), 7,35 (т, 1H), 4,50 (м, 1H), 4,32 (м, 1H), 3,40-3,85 (м, 6H), 1,95-2,40 (м, 5H).
Пример 6. Синтез гемигалактаратной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Галактаровую (слизевую) кислоту (36,3 мг, 0,173 ммоль) добавляли к раствору (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (125 мг, 0,346 ммоль) в горячем этаноле (1 мл). Смесь нагревали при кипении с обратным холодильником после добавления воды (8 капель); затем горячую смесь фильтровали через хлопковую пробку, которую впоследствии промывали этанолом (1 мл). Охлаждение не приводило к образованию осадка. Летучие компоненты удаляли упариванием с использованием роторного испарителя и остаток (белое вспененное вещество) растирали с изопропанолом (отбрасывали) и оставшееся твердое вещество растворяли в ипящей смеси ацетон/вода (4 мл в соотношении 7:1). Медленное охлаждение до 5°С приводило к образованию белого твердого вещества, которое отфильтровывали, промывали изопропанолом (3×1 мл) и сушили в высоком вакууме. В остатке получали 118 мг (выход 73%) тонких белых пластин, т.пл. 134-139°С. 1Н ЯМР (300 МГц, D2O) δ 8,29 (с, 1H), 7,78 (д, 1H), 7,62 (д, 1H), 7,54 (д, 1H), 7,35 (м, 2H), 7,18 (т, 1H), 7,10 (с, 1H), 6,98 (м, 1H), 4,08 (с, 1H, галактаровая кислота), 3,98 (д, 1H), 3,77 (с, 1H, галактаровая кислота), 3,66 (м, 1H), 3,35 (м, 1H), 2,95-3,30 (м, 4H), 1,65-2,05 (м, 5H).
Пример 7. Синтез D-тартратной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Винную кислоту (25,6 мг, 0,173 ммоль) добавляли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (125 мг, 0,346 ммоль) в горячем этаноле (1 мл). Полученный раствор медленно охлаждали до температуры окружающей среды. Твердое вещество не выпадало в осадок, поэтому раствор концентрировали, получая белое вспененное вещество. Попытки кристаллизации в изопропаноле оказались неудачными. Вспененное вещество растворяли в метаноле и добавляли вторую порцию половины эквивалента винной кислоты (25,6 мг, 0,173 ммоль). Смесь концентрировали, получая белую пену, которая не закристаллизовывалась из смесей метанола и изопропанола. Концентрированное вещество (смесь твердого вещества и смолообразной жидкости) затем суспендировали в этилацетате (1 мл), получая белое твердое вещество. Его выделяли фильтрованием (промывали этилацетатом) и сушили в вакуумной печи (18 часов при 40°С), получая 141 мг (выход 79,7%) соли с моностехиометрическим составом (ЯМР), т.пл. 136-140°С. Анализ методом хиральной ЖХ давал чистоту 98,1% (270 нм). 1Н ЯМР (300 МГц, D2O) δ 8,50 (с, 1H), 8,01 (д, 1H), 7,86 (д, 1H), 7,75 (д, 1H), 7,56 (м, 2H), 7,38 (т, 1H), 7,32 (с, 1H), 7,21 (т, 1H), 4,34 (с, 2H, винная кислота), 4,26 (д, 1H), 3,95 (м, 1H), 3,64 (м, 2H), 3,15-3,55 (м, 4H), 1,90-2,30 (м, 5H).
Пример 8. Синтез метансульфонатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Метансульфоновую кислоту (33,2 мг, 0,346 ммоль) добавляли к раствору (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (125 мг, 0,346 ммоль) в горячем этаноле (1 мл). Охлаждение приводило к образованию осадка. Смесь нагревали до кипения с обратным холодильником и горячую смесь фильтровали через хлопковую пробку, которую впоследствии промывали метанолом (1 мл). Летучие компоненты удаляли с помощью роторного испарителя и остаток (светло-желтая пена) растворяли в горячем изопропаноле (1 мл). Опять, охлаждение приводило к образованию осадка. Изопропанол упаривали и остаток суспендировали в ацетоне (1 мл). Фильтрование и сушка в вакуумной печи (18 часов при 50°С) давали 146 мг (выход 92,5%) светло-бежевого твердого вещества, т.пл. 240-243°С. 1Н ЯМР (300 МГц, D2O) δ 8,32 (с, 1H), 7,82 (д, 1H), 7,66 (д, 1H), 7,57 (д, 1H), 7,38 (м, 2H), 7,20 (м, 1H), 7,12 (с, 1H), 7,01 (м, 1H), 4,09 (д, 1H), 3,75 (м, 1H), 3,47 (м, 2H), 3,00-3,40 (м, 4H), 2,67 (с, 3H, метансульфоновая кислота), 1,75-2,15 (м, 5H).
Пример 9. Синтез D-манделатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
D-миндальную кислоту (52,6 мг, 0,346 ммоль) добавляли к раствору (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (125 мг, 0,346 ммоль) в горячем этаноле (1 мл). Разбавление этилацетатом (4 мл) и охлаждение не приводили к образованию осадка. Летучие компоненты удаляли упариванием с использованием роторного испарителя и остаток (белая пена) растворяли в горячем изопропаноле (0,5 мл). Охлаждение до 5°С приводило к образованию белых кристаллов, которые собирали фильтрованием с отсасыванием в вакууме. Сушка в вакуумной печи (18 часов при 45°С) давала 111 мг (выход 62,4%) светло-бежевого твердого вещества, т.пл. 188,5-193°С. 1Н ЯМР (300 МГц, D2O) δ 8,33 (с, 1Н), 7,83 (с, 1Н), 7,67 (д, 1Н), 7,60 (д, 1Н), 7,27 (м, 8Н, включает миндальную кислоту), 7,12 (с, 1Н), 7,01 (м, 1Н), 4,85 (с, 1Н, миндальная кислота), 4,10 (д, 1Н), 3,75 (м, 1Н), 3,48 (м, 2Н), 3,00-3,40 (м, 4Н), 1,75-2,15 (м, 5Н).
Пример 10. Синтез R-камфорсульфонатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
R-10-Камфорсульфоновую кислоту (80,3 мг, 0,346 ммоль) добавляли к раствору (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (125 мг, 0,346 ммоль) в горячем этаноле (1 мл). Охлаждение не приводило к осаждению какого-либо осадка. Летучие компоненты удаляли упариванием с использованием роторного испарителя и остаток (белая пена) растворяли в горячем изопропаноле (0,5 мл). Охлаждение до 5°С приводило к образованию отдельных белых кристаллов и молочной суспензии. При царапании стенок колбы шпателем со временем приводило к преобразованию смеси в густую массу мелких белых кристаллов. Дополнительно добавляли 0,5 мл изопропанола и кристаллы собирали фильтрованием с отсасыванием осадка в вакууме. Сушка в вакуумной печи (5 часов при 70°С, затем 2 часа при 110°С) давали 193 мг (выход 93,8%) белого твердого вещества, т.пл. 149,5-156оС. 1Н ЯМР (300 МГц, D2O) δ 8,30 (с, 1Н), 7,79 (д, 1Н), 7,64 (д, 1Н), 7,55 (д, 1Н), 7,36 (м, 2Н), 7,18 (м, 1Н), 7,11 (с, 1Н), 6,99 (м, 1Н), 4,07 (д, 1Н), 3,73 (м, 1Н), 3,45 (м, 2Н), 3,95-3,35 (м, 5Н, включает камфорсульфоновую кислоту), 2,64 (д, 1Н, камфорсульфоновая кислота), 2,22 (м, 2Н), 1,70-2,10 (м, 8Н, включает камфорсульфоновую кислоту), 1,45 (м, 1Н, камфорсульфоновая кислота), 1,25 (м, 1Н, камфорсульфоновая кислота), 0,85 (с, 3Н, камфорсульфоновая кислота), 0,68 (с, 3Н, камфорсульфоновая кислота).
Пример 11. Синтез S-камфорсульфонатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
S-10-Камфорсульфоновую кислоту (80,3 мг, 0,346 ммоль) добавляли к раствору (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (125 мг, 0,346 ммоль) в горячем этаноле (1 мл). Разбавление этилацетатом (4 мл) и охлаждение не приводили к осаждению какого-либо осадка. Летучие компоненты удаляли упариванием с использованием роторного испарителя и остаток (белая пена) растворяли в горячем изопропаноле (1,5 мл). Охлаждение до 5°С приводило к образованию белых кристаллов. Смесь концентрировали до ~0,5 мл и опять охлаждали до 5°С. Затем твердое вещество собирали фильтрованием с отсасыванием осадка в вакууме и сушили в вакууме, первоначально 18 часов при 45°С, но затем постепенно при более высокой температуре (в конце при 110°С) для удаления остаточного изопропанола. Это давало 143 мг (выход 69,7%) белого твердого вещества, т.пл. 153,5-157°С. 1Н ЯМР (300 МГц, D2O) δ 8,29 (с, 1Н), 7,79 (д, 1Н), 7,63 (д, 1Н), 7,54 (д, 1Н), 7,34 (м, 2Н), 7,18 (м, 1Н), 7,10 (с, 1Н), 6,99 (м, 1Н), 4,05 (д, 1Н), 3,73 (м, 1Н), 3,44 (м, 2Н), 3,95-3,35 (м, 5Н, включает камфорсульфоновую кислоту), 2,67 (д, 1Н, камфорсульфоновая кислота), 2,23 (м, 2Н), 1,70-2,10 (м, 8Н, включает камфорсульфоновую кислоту), 1,46 (м, 1Н, камфорсульфоновая кислота), 1,25 (м, 1Н, камфорсульфоновая кислота), 0,84 (с, 3Н, камфорсульфоновая кислота), 0,64 (с, 3Н, камфорсульфоновая кислота).
С использованием способов, аналогичных описанным выше (примеры 3-11) было охарактеризовано несколько других солевых форм. Результаты получения таких соединений приведены в примерах 12-14.
Пример 12. Синтез сульфатной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Сульфатную соль осаждали из смеси изопропилацетата и воды. Т.пл. 278°С. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,28 (широкий с, 1Н, амид), 8,56 (дд, 1H), 8,24 (т, 1H), 7,77 (д, 1H), 7,74 (д, 1H), 7,60 (с, 1H), 7,40 (м, 2H), 7,35 (с, 1H), 7,21 (м, 1H), 4,21 (м, 1H), 3,93 (м, 2H), 3,10-3,60 (м, 5H), 2,05 (м, 3H), 1,92 (м, 1H), 1,73 (м, 1H).
Пример 13. Синтез кетоглутаратной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
α-Кетоглутаратную соль осаждали из изопропилацетата. Т.пл. 177°С. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,64 (с, 1Н, амид), 8,50 (д, 1H), 8,20 (д, 1H), 7,74 (д, 1H), 7,70 (д, 1H), 7,60 (м, 1H), 7,45 (м, 1H), 7,32 (м, 2H), 7,18 (м, 1H), 4,10 (м, 1H), 3,78 (м, 2H), 3,00-3,45 (м, 5H), 2,81 (м, 2H, кетоглутаровая кислота), 2,41 (м, 2H, кетоглутаровая кислота), 1,96 (м, 3H), 1,83 (м, 1H), 1,60 (м, 1H).
Пример 14. Синтез гиппуратной соли (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Гиппуратную соль осаждали из ацетона (слишком гигроскопична для получения температуры плавления). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,79 (с, 1Н, амид), 8,56 (д, 1Н), 8,44 (с, 1Н, гиппуровая кислота), 8,29 (м, 1Н), 7,87 (м, 2Н, гиппуровая кислота), 7,76 (д, 1Н), 7,65 (м, 1Н), 7,54 (м, 1Н), 7,49 (м, 4Н, включает гиппуровую кислоту), 7,34 (м, 2Н), 7,21 (м, 1Н), 3,91 (м, 1Н), 3,74 (м, 2Н), 3,00-3,50 (м, 5Н), 2,80 (м, 2Н, гиппуровая кислота), 1,79 (м, 2Н), 1,60 (м, 2Н), 1,30 (м, 1Н).
Пример 15. Выделение (2R,3R)- и (2S,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида и преобразование в соли галактаровой кислоты
Образец надосадочной жидкости из опыта по выделению п-толуолсульфоната (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида (пример 2) концентрировали путем упаривания на роторном испарителе, доводили до рН 10 с помощью 10% водного гидроксида натрия и экстрагировали дихлорметаном. Дихлорметановый экстракт упаривали и остаток (1,8 г) растворяли в абсолютном этаноле (55 мл), содержащем 0,5% ди-н-бутиламина. Этот раствор впрыскивали порциями по 0,25 мл на 25 см × 2,1 см колонку Chiralpak® AD хиральной ВЭЖХ, элюируя смесью 60:40:0,2 гексан/этанол/ди-н-бутиламин (скорость потока = 30 мл/мин), проводя мониторинг при 270 нм. Выделение эффлюента, элюированного при ~7,5 минуты, и затем элюирование при ~13,5 минуты давало после упаривания растворителя 0,48 г (98% хиральная чистота) и 0,47 г (99% хиральная чистота) соответственно бесцветного масла. Два спектра ЯМР были одинаковыми. 1Н ЯМР (300 МГц, CDCl3) δ 8,49 (с, 1H), 8,45 (д, 1H), 7,74 (д, 1H), 7,52 (м, 4H), 7,35 (т, 1H), 7,20 (дд, 1H), 7,05 (д, 1H), 4,55 (дт, 1H), 3,43 (м, 1H), 3,22 (м, 1H), 2,90 (м, 5H), 2,09 (м, 1H), 1,88 (м,4H).
Теплый раствор каждого из образцов свободного основания в абсолютном этаноле (10 мл) обрабатывали одним эквивалентом галактаровой кислоты. Полученные смеси нагревали при 75°С в течение 5 минут и охлаждали при перемешивании до температуры окружающей среды. Полученные твердые вещества собирали фильтрованием с отсасыванием осадка в вакууме и сушили в вакууме, получая 0,65 г (выход 87%) и 0,62 г (выход 85%) соответственно белого гранулированного твердого вещества (т.пл. 200-205оС в каждом случае). 1Н ЯМР (300 МГц, D2O) δ 8,38 (с, 1H), 8,28 (д, 1H), 7,94 (д, 1H), 7,70 (д, 1H), 7,59 (д, 1H), 7,48 (т, 1H), 7,40 (м, 1H), 7,32 (м, 2H), 4,42 (м, 1H), 4,21 (с, 2H), 3,87 (с, 2H), 3,68 (м, 1H), 3,35 (м, 6H), 2,25 (м, 2H), 2,02 (м, 3H).
Пример 16. Синтез п-хлорбензоатной соли (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида
Твердую п-хлорбензойную кислоту (46,8 мг, 0,299 ммоль) добавляли в виде одной порции к раствору ранее выделенного изомера N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида из примера 15 (108 мг, 0,299 ммоль) в ацетоне (10 мл). Эту смесь нагревали почти при кипении с обратным холодильником в течение 30 минут и охлаждали до температуры окружающей среды. Осадок не образовывался, поэтому раствор концентрировали примерно до 20% от его первоначального объема (горячая чашка), в этот момент начинали образовываться кристаллы. Смесь охлаждали и разбавляли изопропанолом (2 мл).
Данную смесь концентрировали путем медленного упаривания растворителя при температуре окружающей среды и полученное твердое вещество собирали и сушили. Данный продукт, 145 мг (выход 94%) представлял собой светло-желтые кристаллы, т.пл. 150-152°С. 1H ЯМР (300 МГц, CDCl3) δ 8,49 (с, 1H), 8,38 (д, 1H), 7,93 (д, 2H, п-хлорбензойная кислота), 7,67 (м, 2H), 7,57 (д, 1H), 7,45 (м, 1H), 7,36 (д, 2H, п-хлорбензойная кислота), 7,30 (м, 1H), 7,27 (с, 1H), 7,16 (м, 1H), 7,00 (д, 1H, амид), 6,90 (широкий с, четвертичный аммоний), 4,62 (м, 1H), 3,85 (дд, 1H), 3,36 (м, 1H), 2,95-3,25 (м, 5H), 2,16 (с, 1H), 1,70-2,10 (м, 4H).
По данным рентгенодифракционного исследования данного образца его абсолютная стереохимия 2R,3R (см. фиг.11A и 11B). Соответственно, путем исключения, последний элюированный изомер в примере 15 имеет абсолютную конфигурацию 2S,3S.
Пример 17. Хиральный хроматографический метод анализа стереоизомеров
Создание хирального хроматографического способа отделения четырех стереоизомеров, одного от другого, представлялось очень многообещающим. Первоначальные попытки (с использованием подвижной фазы гексан/изопропанол/триэтиламин) приводили к перекрыванию пиков и меньшей, чем оптимальная, форме пиков. Переход от изопропанола к этанолу и от триэтиламина к ди-н-бутиламину улучшило разделение и форму пика и сократило время прохода. Подробности способа следующие:
Аналитическая колонка: Chiralpak® AD (250х4,6 мм, 5 мкм)
Подвижная фаза: 60:40:0,2 гексаны/этанол/ди-н-бутиламин
Впрыскиваемый объем: 10 мкл
Скорость потока: 1,0 мл в минуту
Температура: 20°С
Детектирование: УФ при 270 нм
Общее время прохода: ~25 минут
Порядок элюирования (время удерживания): 2S,3R (5,3 мин); 2R,3S (7,3 мин); 2R3R (8,3 мин); 2S,3S (12,1 мин)
Иллюстративная хроматограмма стереоизомерных аналогов показана на фиг.12.
Пример 18. Рентгенодифракционный анализ порошка (XRPD)
Рентгенодифракционный анализ порошка (XRPD) был проведен для нескольких образцов описанных здесь солей. Приведен характер распределения пиков для гидрохлоридной (фиг.13) и тозилатной (фиг.14) солей.
Рентгенодифракционный анализ порошка (XRPD)
Пики на рентгенодифрактограмме порошка получены на каждом из или на обоих из двух приборов. В ряде случаев рентгенограммы были получены на дифрактометре Siemens D5000 с использованием CuKα излучения (40 кВ, 40 мА), θ-θ гониометр, V20 дивергенция и принимающие щели, графитовый вторичный монохроматор и сцинтилляционный счетчик. Работу прибора проверяли с использованием сертифицированного корундового стандарта (NIST 1976). Проход образцов в условиях окружающей среды проводили на плоской пластине для образцов с использованием порошков в том виде, как они были получены. Приблизительно 35 мг образца осторожно помещали в полость, вырезанную в полированной силиконовой подложке с нулевым фоном (510). Образец вращали в его собственной плоскости во время анализа, сканируя от 2° до 42° с шагом с 0,05°, из расчета 4 секунды на шаг, с использованием CuKα1 (λ=1,5406 Å).
В ряде случаев рентгенограммы были получены на дифрактометре Bruker AXS C2 GADDS с использованием CuKα излучения (40 кВ, 40 мА), автоматизированная стадия XYZ, лазерный видеомикроскоп для автоподачи образца и 2-мерного детектора площади HiStar. Оптика дифрактомера состоит из одного многослойного зеркала Гебеля, соединенного с коллиматором с точечной апертурой в 0,3 мм. Дивергенция луча (т.е. эффективный размер рентгеновского пучка на образце) составляла примерно 4 мм. Использовали непрерывный режим сканирования θ-θ с расстоянием образец-детектор, равным 20 см, что давало эффективный диапазон 2θ в 3,2°-30,0°. Обычно образец подвергали воздействию рентгеновского луча в течение 120 секунд. Проход образцов в условиях окружающей среды проводили на плоской пластине для образцов с использованием порошков в том виде, как они были получены без измельчения. Приблизительно 1-2 мг образца слегка спрессовывали на силиконовой подложке для получения плоской поверхности. Образцы для проведения эксперимента не в условиях окружающей среды устанавливали на силиконовой подложке с использованием теплопроводящего соединения. Образец затем нагревали до подходящей температуры со скоростью примерно 10°С/мин и затем выдерживали в изотермических условиях в течение примерно 5 минут перед началом сбора данных.
Дифференциальная сканирующая калориметрия (ДСК)
Данные ДСК получали на приборах TA Q1000, оборудованных автоматическим устройством для подачи образца на 50 положений. Прибор был откалиброван для калибровки энергии и температуры с использованием сертифицированного индия. Обычно 0,5-1,5 мг каждого образца в алюминиевой чашке с отверстиями под штифт нагревали со скоростью 10оС/мин от 25°С до 175-200°С. Над образцами поддерживали ток азота со скоростью 30 мл/мин.
Термогравиметрический анализ (ТГА)
Данные ТГА получали на приборе TA Q500 (TGA), оборудованных автоматическим устройством для подачи образца на 16 положений. Прибор был температурно откалиброван с использованием сертифицированного алюмеля. Обычно 5-10 мг каждого образца загружали в предварительно взвешенные платиновый тигель и алюминиевую чашку для ДСК и нагревали со скоростью 10°С/мин от температуры окружающей среды до 350°С. Над образцами поддерживали ток азота со скоростью 60 мл/мин.
Микроскопия в поляризованном свете (МПС)
Образцы исследовали в поляризованном свете на микроскопе Leica LM/DM с цифровой видеокамерой для фиксации изображения. Небольшое количество каждого образца помещали на покровное стекло в иммерсионном масле и покрывали покровным стеклом, отдельные частицы отделяли друг от друга насколько это возможно. Образцы смотрели при подходящем увеличении в частично поляризованном свете, соединенном с λ спектрозональным фильтром.
Высокотемпературная микроскопия
Высокотемпературную микроскопию проводили с использованием микроскопа Leica LM/DM в поляризованном свете, соединенном с горячей пластиной Mettler-Toledo MTFP82HT и цифровой видеокамерой для фиксации изображения. Небольшое количество каждого образца помещали на покровное стекло и отдельные частицы отделяли друг от друга насколько это возможно. Образцы смотрели при подходящем увеличении в частично поляризованном свете, соединенном с λ спектрозональным фильтром, по мере его нагревания от температуры окружающей среды со скоростью обычно в 10°С/мин.
Гравиметрическая сорбция пара (ГСП)
Сорбционные изотермы определяли на любом из двух или на обоих приборах. Ряд экспериментов проводили с использованием анализатора сорбции влаги VTI Corporation SGA-100, контролируемого программным обеспечением VTI FlowSystem 4. Температуру образца поддерживали при 25°С с помощью постоянной термостатируемой бани Polyscience. Влажность контролировали путем смешивания потоков сухого и влажного азота. Изменения веса как функцию % относительной влажности (ОВ) контролировали с использованием цифрового записывающего устройства Cahn Digital Recording Balance D200 с точностью +/- 0,0001 г.
Обычно образцы в 10-20 мг помещали на уравновешенную чашку весов в условиях окружающей среды. Образец сушили при 50°С в течение 1 часа. Стандартную изотерму адсорбции получали при 25°С с 5% интервалами ОВ в диапазоне 5-95% ОВ и изотерму десорбции аналогично получали при 25°С с 5% интервалами ОВ в диапазоне 95- 5% ОВ. Критерий уравновешивания образца включал 0,0100% вес. в течение 5 минут при максимальном времени наступления равновесия в 180 минут для каждой точки % ОВ.
Некоторые сорбционные изотермы получены с использованием сорбционного анализатора влаги Hiden IGASorp, контролируемого программным обеспечением CFRSorp. Температуру образца поддерживали при 25°С с использованием рециркулирующей водяной бани Хубера. Влажность контролировали путем смешивания потоков сухого и влажного азота, при общей скорости потока в 250 мл/мин. Относительную влажность измеряли с помощью калиброванного зонда Vaisala RH (динамический диапазон 0-95% ОВ), расположенного рядом с образцом. Изменения веса, (релаксацию массы) образца как функцию % ОВ постоянно контролировали с помощью микробаланса (точность ±0,001 мг). Обычно 10-20 мг образца помещали в уравновешенную корзину с ячейками из нержавеющей стали в условиях окружающей среды. Образец загружали и выгружали при 40% ОВ и 25°С (типичные условия окружающей среды). Изотерму сорбции влаги получали, как кратко описано ниже (2 скана давали 1 полный цикл). Стандартную изотерму получали при 25°С с интервалами в 10% ОВ в диапазоне 0-90% ОВ.
Обобщенные параметры способа ГСП
В программном обеспечении использован метод минимизации наименьших квадратов вместе с моделью релаксации массы. Измеренная величина релаксации массы должна быть в пределах 5% от той, которая предсказана с использованием программного обеспечения, перед выбором следующего значения в % ОВ. Минимальное время уравновешивания устанавливали в 1 час, а максимальное - в 4 часа. Обычно образцы после завершения получения изотермы повторно анализировали методом рентгенодифракционного анализа порошка (XRPD).
Определение воды по Карлу Фишеру
Содержание воды в каждом образце измеряли на кулонометре Mettler Toledo DL39 с использованием реагента Hydranal Coulomat AG и аргонового насоса. Взвешенные образцы твердого вещества вводили в сосуд на платиновой чашке для ТГА, которую соединяли с резиновой септой для исключения доступа воды. Использовали примерно 10 мг образцы на титрование и проводили определение в двукратной повторности.
Определение термодинамической водной растворимости методом ВЭЖХ
Водную растворимость определяли путем суспендирования достаточного количества соединения в 0,25 мл воды для получения максимальной конечной концентрации ≥10 мг/мл исходной свободной формы соединения. Суспензию уравновешивали при 25°С в течение 24 часов и затем измеряли рН. Суспензию фильтровали через стеклянный пористый фильтр С в 96-луночные планшеты. Фильтрат затем разбавляли с фактором 101. Количественную оценку проводили с помощью ВЭЖХ со ссылкой на стандартный раствор с концентрацией приблизительно 0,1 мг/мл в ДМСО. Проводили впрыскивание стандарта, разбавленного и неразбавленного раствора образца. Растворимость рассчитывали с использованием площадей пиков, определенных путем интегрирования пика, обнаруженного при том же времени удерживания, что и основной пик при впрыскивании стандарта. Если имелось достаточно твердого вещества на чашке фильтра, то получали данные порошковой дифрактометрии (XRPD)
Общие подробности способа для определения термодинамической водной растворимости
50×4,6 мм
Длина волны, ширина полосы (нм):
Определение химической чистоты методом ВЭЖХ
Анализ на чистоту проводили на системе серии Agilent HP1100, оборудованной детектором с диодной матрицей, и с помощью программного обеспечения ChemStation. Использовали один из двух способов, подробно описанных ниже.
Способ 1
Длина волны, ширина полосы (нм):
Способ 2
Длина волны, ширина полосы (нм):
Ионная хроматография
Данные получали на приборе Metrohm 861 Advanced Compact IC с использованием программного обеспечения IC Net, версия 2.3. Образцы получали в виде исходных растворов с концентрацией 1000 частей на миллион (ч/млн) в воде. Когда растворимость образцов была низкой, использовали подходящий сорастворитель, такой как ДМСО. Образцы разбавляли до концентрации 50 ч./млн или 100 ч./млн с использованием подходящего растворителя перед тестированием. Количественную оценку проводили путем сравнения со стандартными растворами с известными концентрациями анализируемого иона.
Метод ионной хроматографии для анионов
1,0 мМ гидрокарбонат натрия в воде
Метод ионной хроматографии для катионов
0,75 мМ дипиколиновая кислота в воде
Приблизительно 50 мг гидрохлорида (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида взвешивали в стеклянном пузырьке и нагревали до 50°С. К твердому веществу добавляли порции по 100 мкл смеси 1-бутанол/вода (5% воды по объему) до тех пор пока не образовывался прозрачный раствор (всего 500 мкл). Образец перемешивали при 50°С в течение 1 часа и проводили наблюдение. После нагревания при 50°С в течение часа образец продолжал оставаться прозрачным раствором и его охлаждали от 50°С до 25°С со скоростью 1,4°С в час. Образец оставался прозрачным раствором при охлаждении и его накрывали парапленкой со сделанным булавкой отверстием и оставляли упариваться при температуре окружающей среды. Через 2 недели в частично упаренном образце были видны большие кристаллы. На фиг.13 показана порошковая дифрактограмма (XRPD) моногидрохлорида (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, иллюстрирующая как наблюдаемые (более светлые), так и расчетные (более темные) пики.
Экспериментальный характер распределения пиков получен для образца моногидрохлорида (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, тогда как рассчитанные примеры пиков получены из рентгеноструктурного эксперимента для монокристалла, как здесь описано и показано на фиг.10А и 10В. Оба набора пиков согласуются в отношении значений 2θ, и незначительные различия в интенсивностях и ширине пиков могут быть связаны с разрешением приборов и предпочтительными ориентационными эффектами. Кроме того, минимальные различия могут быть связаны с температурным сдвигом, поскольку наблюдаемые данные получены при комнатной температуре, а данные расчета получены для структуры при 120 К.
Было подтверждено строение тозилатной соли, в частности кристаллической моносоли, и характер распределения пиков на дифрактограмме показа на фиг.14, данные получены с использованием CuKα излучения (40 кВ, 40 мА), θ-θ гониометр, V20 дивергенция и принимающие щели, графитовый вторичный монохроматор и сцинтилляционный счетчик. Дифрактограмма XRPD тозилатной соли через 1 неделю хранения при 40°С/75% относительной влажности выявила изменения, но образец все еще представляет собой форму 1. Вероятно, изменения связаны с более гидратированной формой.
VII. Биологические анализы
Способность соединения А и его стереоизомеров связываться с различными подтипами NNR и модулировать их функцию была оценена, как описано в патенте США 6953855 Mazurov et al., содержание которого включено в данное описание путем ссылки. Оценку профиля селективности в отношении рецепторов для соединения А (включая 5НТ3 и мускариновый) проводили с помощью корпорации NovaScreen® Bioscience.
Электрофизиологические измерения ответной реакции α7 NNR проводили в двух экспрессирующих системах: крысиные α7 NNR в клетках GH4C1 млекопитающих и α7 NNR человека в ооцитах Xenopus.
Клетки GH4C1, экспрессирующие α7 NNR крысы, получали, как описано в публикации Placzek et al., Mol. Pharm. 68(6): 1863-1876 (2005), включенной в данное описание путем ссылки. Электрофизиологические измерения агонистической активности проводили с использованием быстрой перфузионной системы Dynaflow и метода пэтч-клэмп с использованием экспрессирующей системы клеток GH4C1. Как ацетилхолин, так и никотин вызывали концентрационно-зависимую активацию α7-опосредованного тока. Агонистические значения ЕС50 из литературы были сопоставимы с полученными данным способом (см., Biochem Pharmacol, в печати (2007) и доступные он-лайн материалы Dynaflow (www.cellectricon.com), обе из которых включены в данное описание путем ссылки касательно данного способа).
Токи для целых клеток, регистрируемые с использованием усилителя Axopatch 700A, фильтровали при 1 кГц и испытывали при 5 кГц с использованием карты PCI (National Instrument). По сравнению с предшествующими исследованиями физиологические растворы модифицировали, как указано, для увеличения стабильности тока. Клетки регистрировали при комнатной температуре в следующей межклеточной среде: 130 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2, 2 мМ MgCl2, 10 мМ HEPES, доведенной до pH 7,4 водным NaOH. Боросиликатные электроды (3-5 МΩ) заполняли следующей средой: 130 мМ Трис-фосфата, 5 мМ NaCl, 2 мМ MgCl2, 10 мм HEPES, 10 мМ EGTA, доведенной до pH 7,4 водным КОН (см. Wu et al., J. Physiol. 576: 103-118 (2006), включенную в данное описание путем ссылки для описания таких указаний). В данных условиях активность макротока, полученная с использованием регистрации NNR в целых клетках, продолжается вплоть до 60 минут, когда она вызвана концентрацией ацетилхолина (ACh) в 1000 мкМ.
Способы работы с клетками были адаптированы по указаниям по применению Cellectricon для Dynaflow. Кратко, после удаления из инкубатора клетки тщательно промывали три раза средой для регистрации и помещали на подставку микроскопа Цейса. В среднем 5 минут требовалось перед установлением конфигурации регистрации для целых клеток. Для исключения модификации клеточных условий проводили регистрацию для одной клетки на нагрузку клеток в силиконовом чипе Dynaflow. Для экспериментальных условий различия во фракции клеток, дающих ответную реакцию, не могло быть обнаружено. Более 95% клеток давало ответную реакцию на Ach, и в расчет принималась каждая клетка, присутствующая в измеряемом токе. Клетки выдерживали при -60 мВ на протяжении эксперимента. Все тестируемые растворы для работы делали ежедневно из исходных растворов. Свежий исходный раствор ацетилхолина (ACh) готовили ежедневно в растворе Рингера и разводили. Кривые доза-ответная реакция были описаны одним уравнением Хилла с использованием программного обеспечения Prism 5,0.
Ооциты Xenopus, экспрессирующие α7 NNR человека, получали, как описано Papke и Papke, Brit. J. Pharmacol. 137: 49-61 (2002), включенным в данное описание путем ссылки. Зрелых самок (>9 см) африканских жаб Xenopus laevis (Nasco, Ft. Atkinson, WI) использовали в качестве источника ооцитов. Перед хирургическим вмешательством проводили анестезию жаб, помещая животное в раствор 1,5 г/л этилового эфира 3-аминобензойной кислоты на 30 минут. Ооциты удаляли через разрез, сделанный в брюшной полости.
Для удаления фолликулярного клеточного слоя собранные ооциты обрабатывали 1,25 мг/мл коллагеназой от Worthington Biochemical Corporation (Freehold, NJ) в течение 2 часов при комнатной температуре в не содержащем кальция растворе Барта (88 мМ NaCl, 1 мМ KCl, 15 мМ HEPES, pH 7,6, 0,81 мМ MgSO4, 2,38 мМ NaHCO3, 0,1 мг/мл сульфата гентамицина). Затем выделяли ооциты 5 стадии и вводили в каждый инъекцию 50 нл (5-20 нг) cRNA α7 человека. Регистрацию проводили на 2-7 день после инъекции. Свежие исходные растворы ацетилхолина (ACh) получали ежедневно в растворе Рингера.
Эксперименты проводили с использованием OpusXpress 6000A (Axon Instruments, Union City, CA). OpusXpress представляет собой интегрированную систему, которая обеспечивает автоматическое насаживание и фиксацию напряжения параллельно для восьми ооцитов. Электроды как напряжения, так и тока наполняли 3М KCl. Для клеток фиксировали напряжение при удерживающем потенциале -60 мВ. Данные получали при 50 Гц и фильтровали при 20 Гц. Клетки заливали в бане раствором Рингера и растворы агонистов доставляли с 96-луночного планшета посредством одноразовых наконечников, что исключало какую-либо возможность перекрестного загрязнения. Скорости потоков устанавливали при 2 мл/мин. Применение лекарственного средства чередовали между ACh контролями и экспериментальными агонистами. Применение средства составляло по продолжительности 12 секунд с последующим периодом промывания в течение 181 секунды.
Ответные реакции рассчитывали как результирующий заряд (см. Papke и Papke, Brit. J. Pharmacol. 137: 49-61 (2002), как процитировано выше) для рецепторов α7. Для каждого ооцита проводили первоначальное контрольное нанесение ACh, затем наносили экспериментальное лекарственное средство, а затем проводили последующее контрольное нанесение ACh (300 мкМ). Ответные реакции на нанесение экспериментального лекарственного средства рассчитывали относительно предшествующей контрольной реакции на ACh для нормализации данных, компенсируя изменения уровней экспресии каналов среди ооцитов. Отметим, что 300 мкМ ACh вызывал максимальные ответные реакции результирующего заряда от рецепторов α7, так что нормализация на контроли ACh эффективно нормализовала данные к максимальным ответным реакциям на ACh. Средние значения и стандартные ошибки (SEM) рассчитывали из нормализованных ответных реакций для по меньшей мере четырех ооцитов для каждой экспериментальной концентрации. Для соотношений концентрация-ответная реакция, для данных, полученных из анализа результирующих зарядов строили график с использованием программного обеспечения Kaleidagraph 3.0.2 (Abelbeck программное обеспечение, Reading, PA) и генерировали кривые из уравнения Хилла.
Поведенческую характеризацию соединения А проводили в соответствии со следующими протоколами. Задачу распознавания объекта проводили в соответствии с описанием Ennaceur и Delacour, Behav. Brain Res. 100: 85-92 (1988), включенным в данное описание путем ссылки. Парадигму лабиринта с угловым рычагом (RAM) проводили в соответствии с описанием Levin et al., Behav. Pharm. 10: 675-680 (1999), включенным в данное описание путем ссылки. Анализ предымпульсного ингибирования (PPI) проводили в соответствии с описанием Suemaru et al., Brit. J. Pharmocol. 142(5): 843-850 (2004). Анализ обратимости индуцированной апоморфином двигательной активности (APO LOCO) проводили в соответствии с описанием Roux et al., Curr. Protocols in Pharmacol. Unit 5.17 (1999).
Краткое изложение биологической активности in vitro
Соединение А конкурентно ингибирует связывание радиоактивно меченного MLA с α7 NNR гиппокампа мозга крысы со значениями константы равновесия (Ki) ~1 нм, показывающими, что оно обладает очень высокой аффинностью в отношении подтипа α7 NNR. Стереоизомеры соединения А имеют следующие значения Ki для α7 NNR крысы: 2R,3S (42 нМ) [ранее сообщалось как 28 нМ]; 2R,3R (1 нМ); 2S,3S (25 мкМ) (см. фиг.1А). Как проиллюстрировано на фиг.1А2, соединение А, энантиомер 2S,3R, проявляет активность в отношении подтипа альфа7 в противоположность его трем другим энантиомерным аналогам, которые представлены в виде перекрывающихся точек со слабой активностью. Соединение А не связывается с α4β2 NNR с какой-либо существенной аффинностью (значения Ki>2 мкМ).
Функциональная активность (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его фармацевтически приемлемой соли (соединение А) и его стереоизомеров была исследована с использованием пэтч-кламп (фиксация потенциала) электрофизиологического метода для α7 NNR крысы, стабильно экспрессируемого в клетках (млекопитающих) GH4C1. В данных экспериментах соединение А давало поразительно отличный функциональный профиль по сравнению с другими индивидуальными изомерами и рацемической смесью четырех изомеров. Как можно видеть на фиг.1А и 1В, соединение А является намного более сильным и эффективным по вызову функционального ответа (Емакс=93% относительно ацетилхолина (ACh); EC50=14 нМ), чем любой другой изомер или смесь четырех изомеров. Действительно, соединение А (изомер 2S,3R) является единственным изомером N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3- ил)бензофуран-2-карбоксамида, который способен обеспечить сильный агонизм в концентрационном диапазоне 1-50 нМ, где концентрация в 10 нМ относится к описанной здесь активности in vivo, как показано на фиг.1В.
Функциональная активность соединения А была также оценена электрофизиологически в ооцитах Xenopus, кратковременно экспрессирующих α7 NNR человека. В такой системе соединение А имеет значение ЕС50, составляющее 33 нМ, и Емакс, составляющее 100% для ответной реакции ACh. Наблюдалось уменьшение последующей контрольной ответной реакции в отношении ACh после применения (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида в концентрациях, превышающих 100 нМ (IC50=200 нМ). В противоположность ранее описанным полным агонистам α7 (см. публикацию Astles et al., Current Drug Targets CNS Neurological Disorders 1(4): 337-348 (2002), включенную в данное описание путем ссылки и касающееся такого сообщения), разделение между значениями ЕС50 и IC50 показывает, что концентрации, которые вызывают полумаксимальный функциональный ответ α7, приводят скорее к минимальному, а не полному остаточному ингибированию. Не имелось обнаруживаемой активации, когда (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид наносили на ооциты, экспрессирующие подтип α4β2 человека, и не имелось существенного понижения в последующих контрольных ответных реакциях на ACh, что указывает на то, что (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3- ил)бензофуран-2-карбоксамид не является ни агонистом, ни антагонистом в отношении α4β2.
Соединения проявляют незначительную или не проявляют агонистической активности на функциональных моделях, в которых использованы рецепторы мышечного типа (подтип α1β1γδ в клонированных клетках TE671/RD человека) или рецепторы ганглиевого типа (подтип α3β4 подклона Шутера клеток РС12 феохромоцитомы крысы и клонированные клетки SHSY-5Y человека), обеспечивая ≤10% (мышцы человека), ≤20% (ганглий крысы) и ≤10% (ганглий человека) никотиновой ответной реакции для данных подтипов. Эти данные показывают селективность в отношении подтипов ЦНС по сравнению с другими подтипами ПНС (периферической нервной системы).
Вследствие близкой гомологии последовательностей и структурной гомологии между рецепторами α7 и 5-гидрокситриптамином (5НТ3) и перекрестной реакционной способности в отношении данных 2 рецепторов, наблюдаемой для других никотиновых рецепторов, была исследована аффинность соединения А в отношении рецепторов 5НТ3. Соединение А (10 мкМ) проявило 59% ингибирование мышиного рецептора 5НТ3 и 25% ингибирование рецептора человека в анализе радиоактивного связывания. Исследование функциональной активации на рецепторах 5НТ3 человека позволяет предполагать минимальную активацию или отсутствие активации (т.е. максимальная ответная реакция в 15% активации наблюдалась при 100 мкМ).
Мускариновые рецепторы представляют собой другой предмет рассмотрения вследствие взаимодействия, которое наблюдалось для других никотиновых лигандов. Соединение А показало минимальное взаимодействие или отсутствие взаимодействия при исследовании в анализах ингибирования при конкурентном связывании для неселективных центральных и неселективных периферических мускариновых рецепторов М1, М2.
Данные показывают, что соединение А является селективным по отношению к лигандам α7 NNR. Соединение А плохо связывается с теми подтипами никотиновых рецепторов, которые являются характерными для периферической нервной системы или с мускариновыми, или 5НТ3 серотонергическими рецепторами. Таким образом соединение А обладает терапевтическим потенциалом для лечения нарушений центральной нервной системы, не вызывая побочных действий связанных с взаимодействием с периферической нервной системой.
Краткое изложение биологической активности in vivo
Соединение А, (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид или его фармацевтически приемлемая соль, проявляют значительную эффективность в двух поведенческих моделях сознания. Соединение А продемонстрировало высокую активность в парадигме распознавания объекта у крыс после как i.p. (внутрибрюшинно, фиг.3), так и p.o. (пероральное введение, фиг.4) введения и также продемонстрировало активность в широком диапазоне дозировки после перорального введения (фиг.4). При внутрибрюшинном введении при такой же низкой дозе (0,3 и 1 мг/кг) соединение А имеет тенденцию обращать вызванный МК-801 дефицит в задаче РО (распознавание объекта) (фиг.5) и при пероральном введении в дозе 0,3 мг/кг когнитивное действие соединения А продолжается по меньшей мере 18 часов (фиг.6). В парадигме лабиринта с угловым рычагом (RAM) (фиг.7), исследующей рабочую память, соединение А существенно увеличивало число правильных выборов перед ошибкой. Данные результаты показывают потенциал (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида для лечения когнитивного дефицита и дисфункций, связанных с шизофренией, включая дисфункции рабочей памяти.
Чтобы соединение можно было использовать для лечения когнитивной дисфункции при шизофрении, оно не должно снижать эффекты классических и атипичных антипсихотических средств на положительные симптомы шизофрении. Таким образом, неопровержимо, что, в дополнение к его свойствам усиления когнитивных функций, соединение А также проявляет эффективность в обращении индуцированной апоморфином двигательной активности (APO LOCO) (фиг.8) и предымпульсное ингибирование (PPI) (фиг.9) на моделях положительных симптомов шизофрении. Таким образом, можно ожидать, что (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамид обеспечит дополнительные преимущества в отношении положительных, а также когнитивных симптомов, связанных с шизофренией.
Специфические наблюдаемые фармакологические ответные реакции могут изменяться в соответствии и в зависимости от конкретного выбранного активного соединения, от того, присутствуют ли в нем фармацевтические носители, а также от типа препарата и используемого способа введения, и такие ожидаемые отклонения или различия в результатах рассматриваются в соответствии с практическим осуществлением настоящего изобретения.
Хотя специфические варианты осуществления настоящего изобретения проиллюстрированы и описаны здесь подробно, изобретение ими не ограничено. Вышеуказанные подробные описания приведены в качестве иллюстрации настоящего изобретения и их не следует рассматривать как составляющие какое-либо ограничение изобретения. Модификации будут очевидны специалистам в данной области, и предполагается, что все модификации, которые не отступают от сути изобретения, включены в объем прилагаемой формулы изобретения.

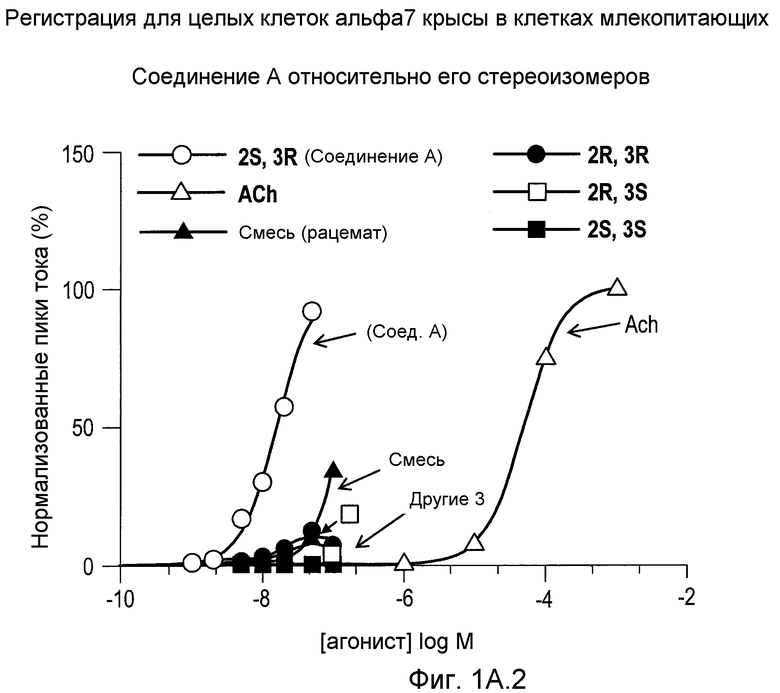
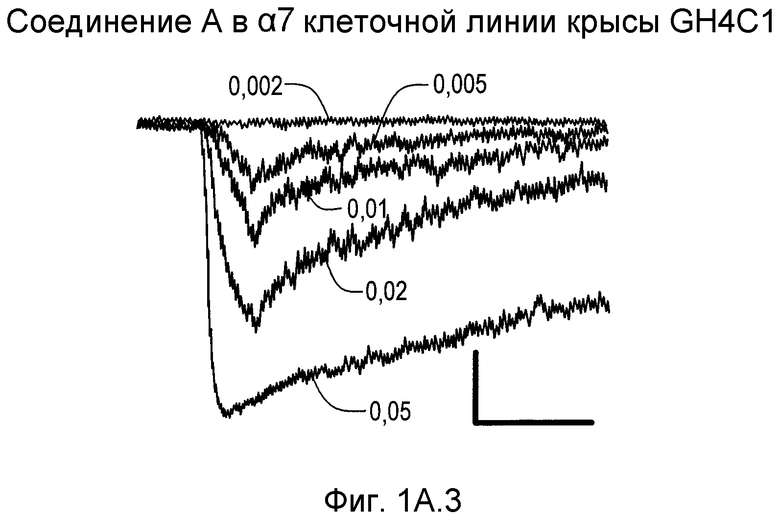
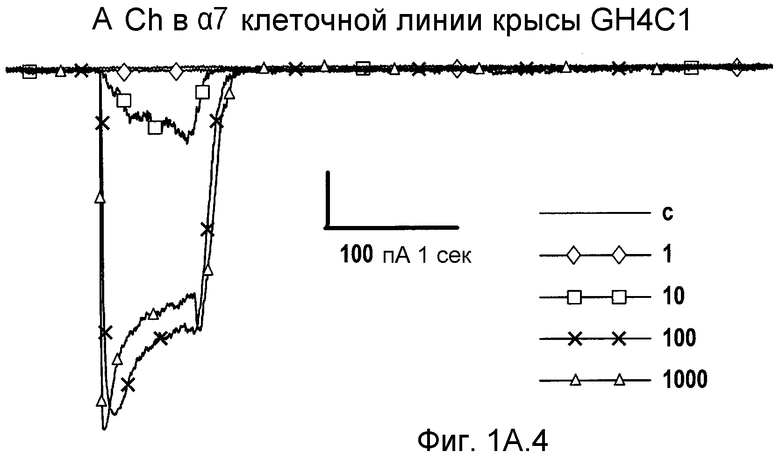
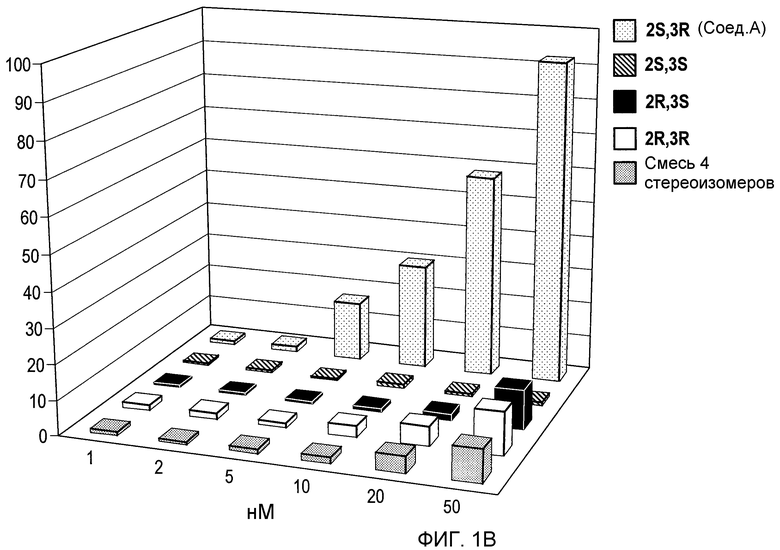
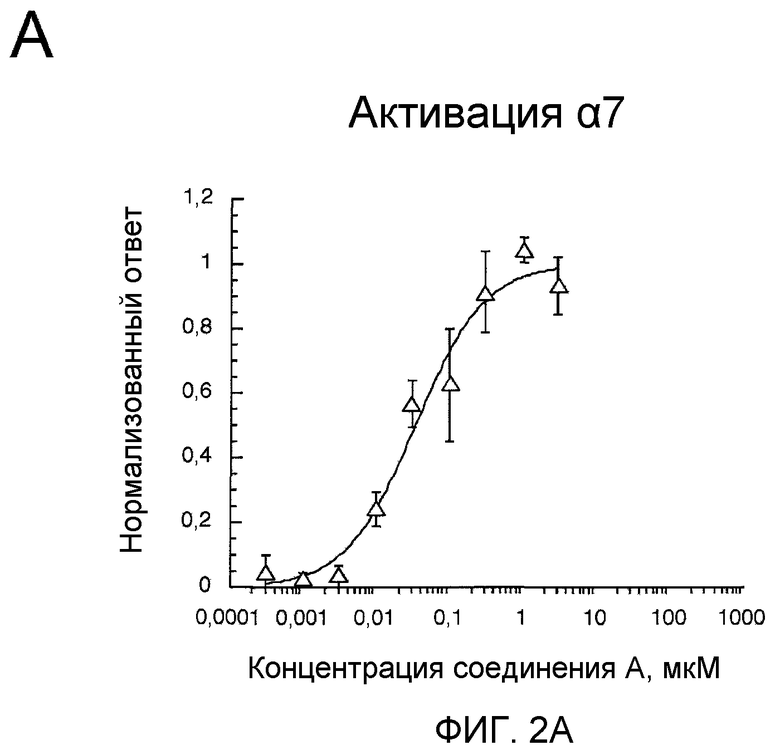
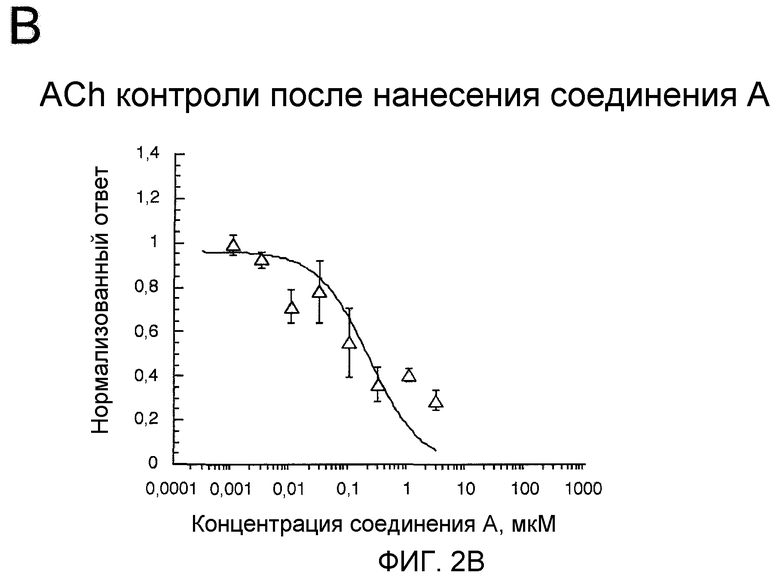
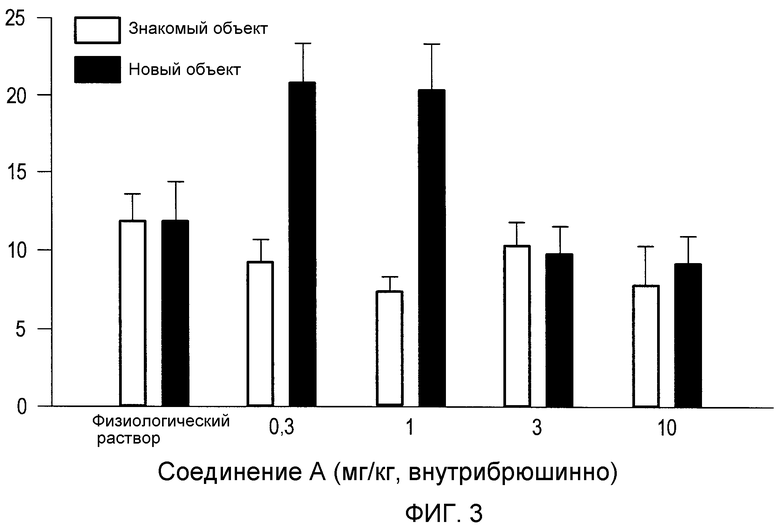
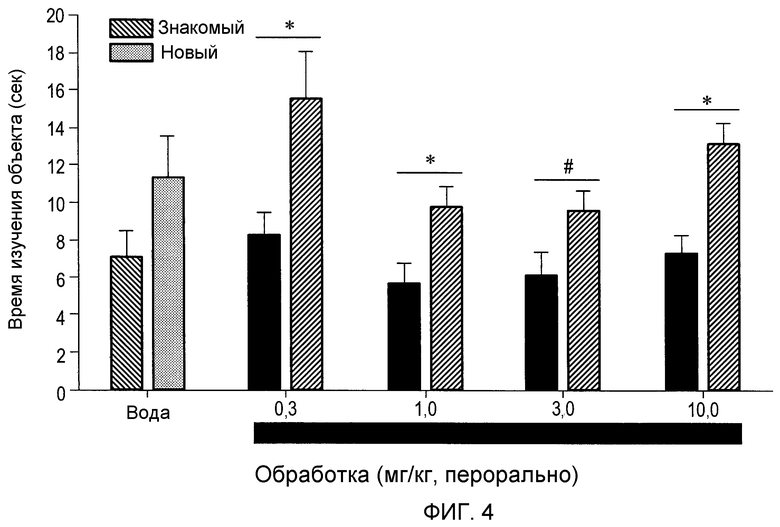
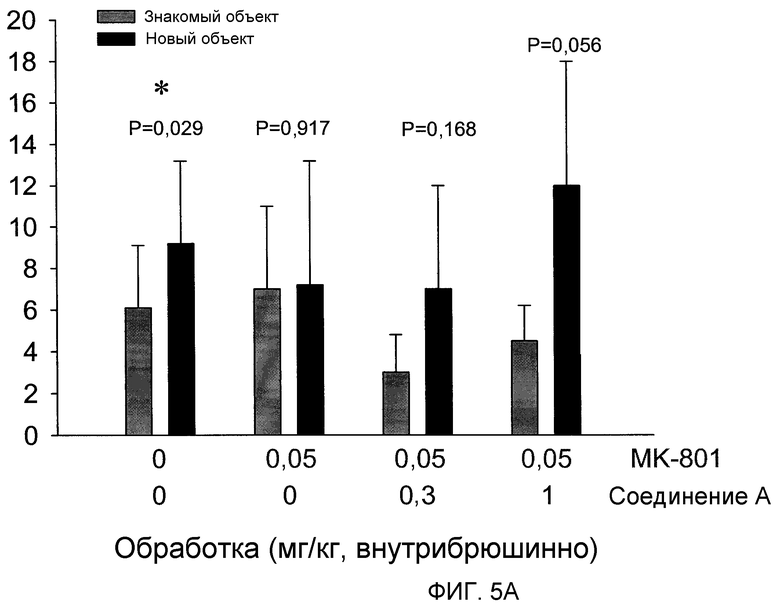
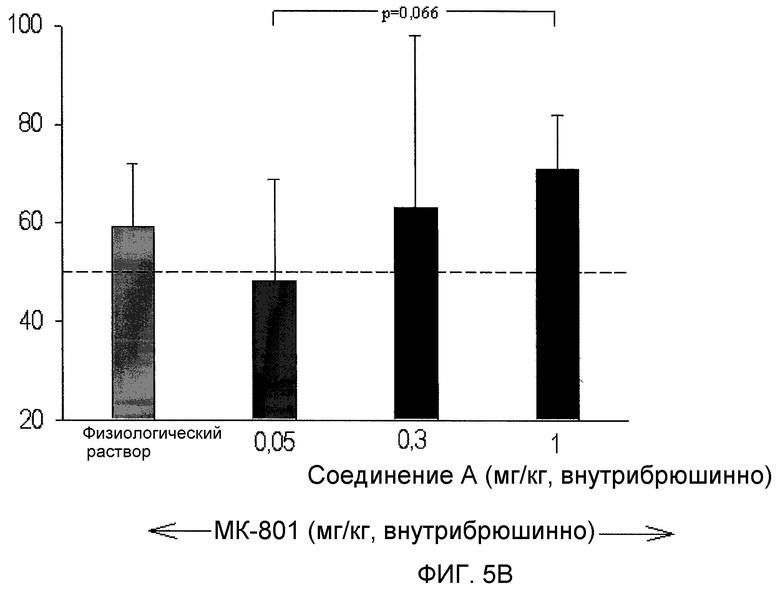
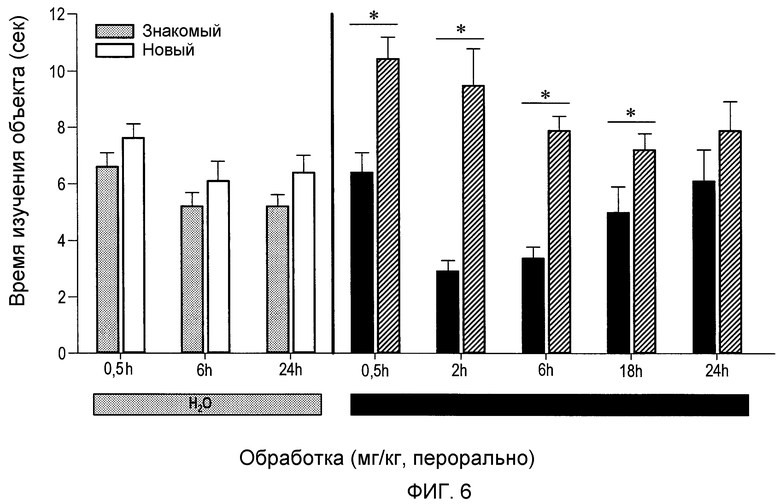
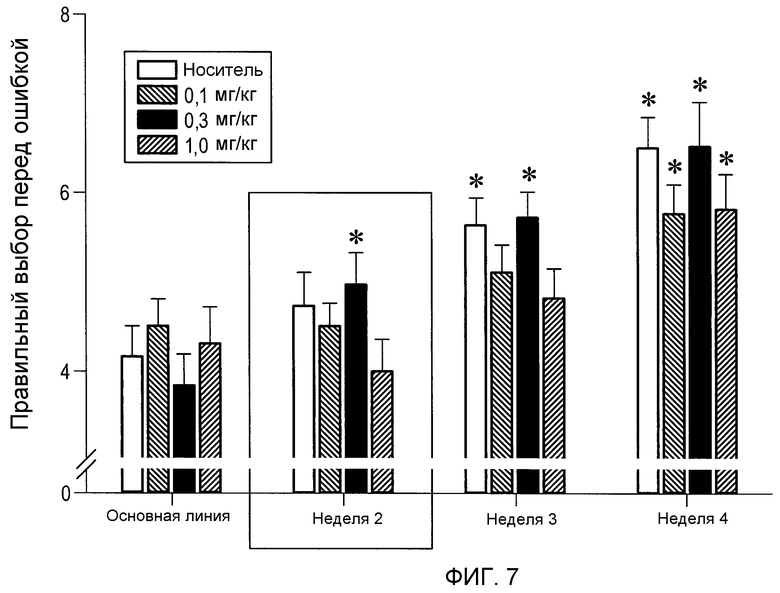
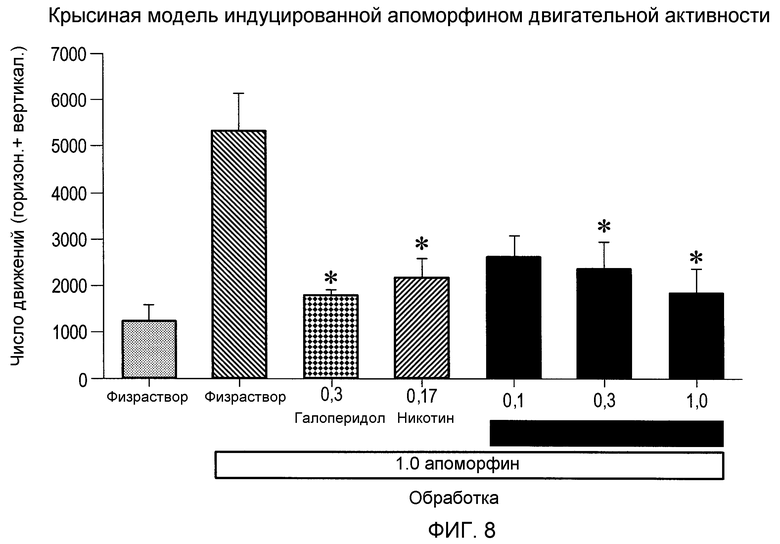
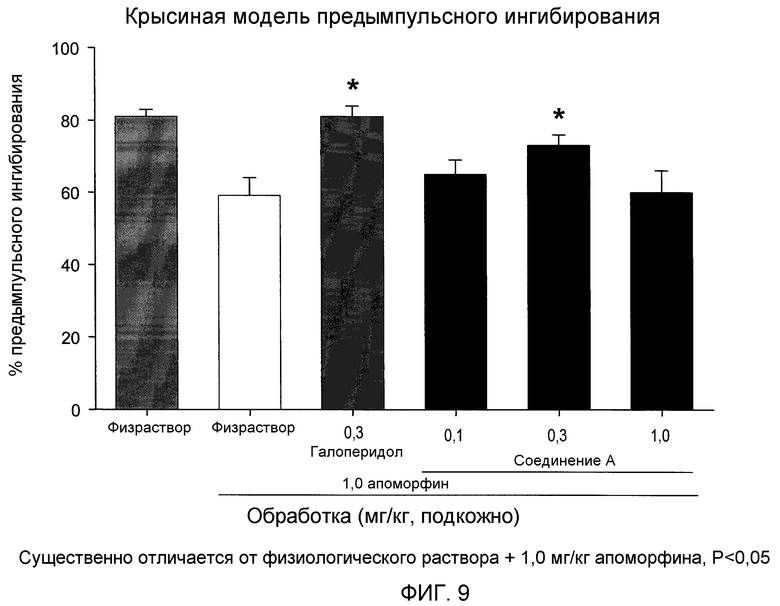
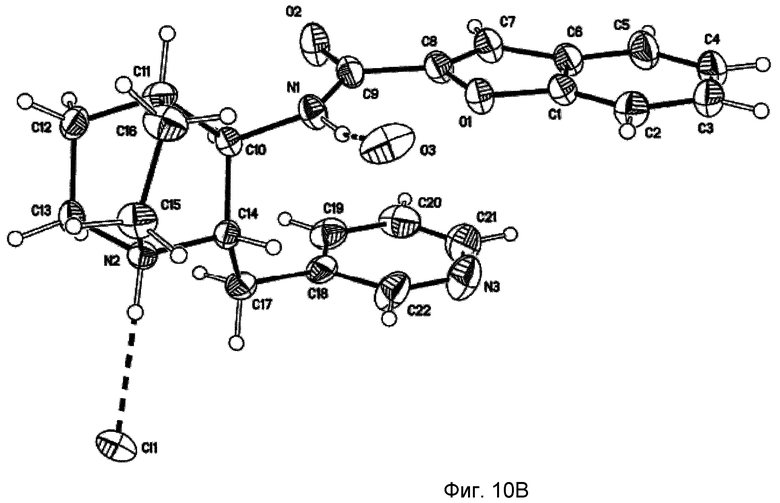
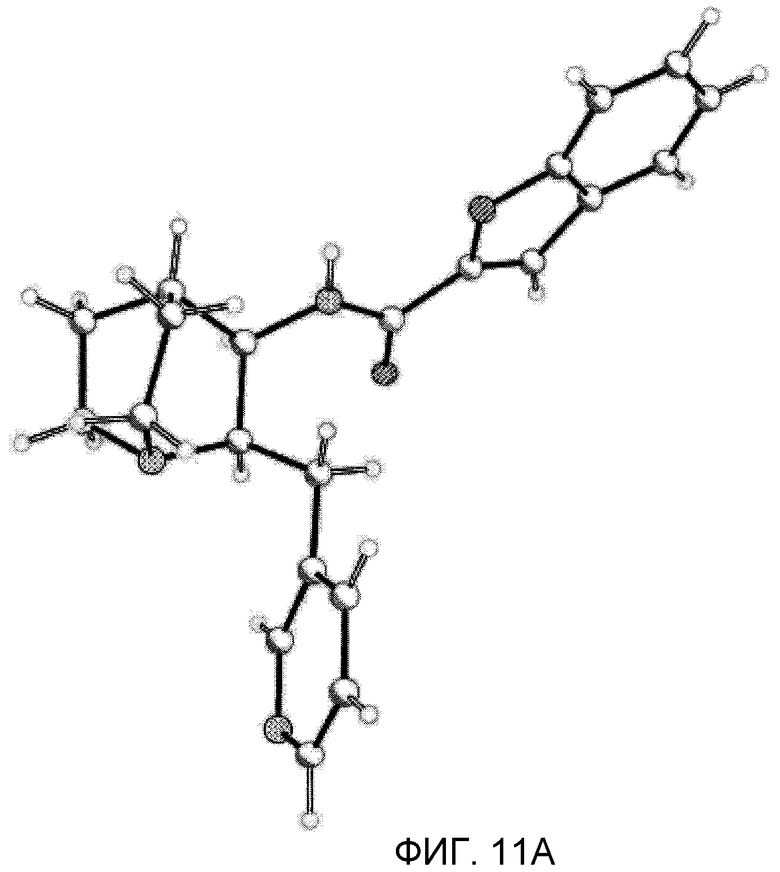
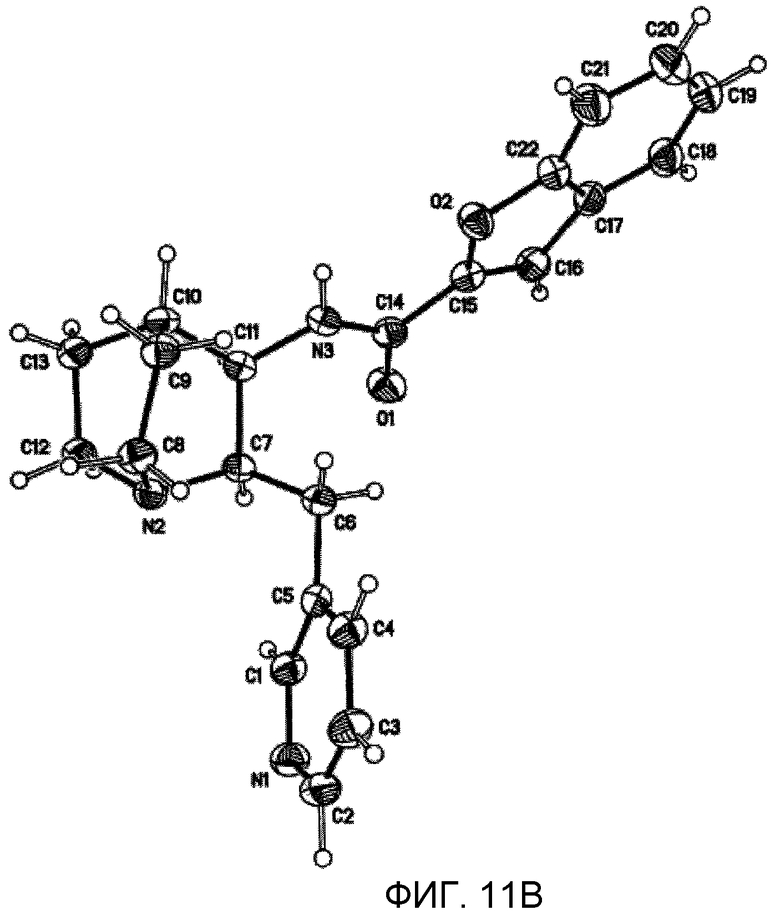
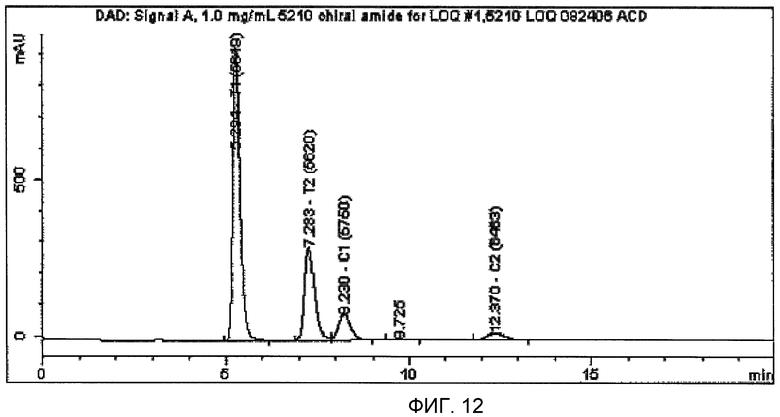
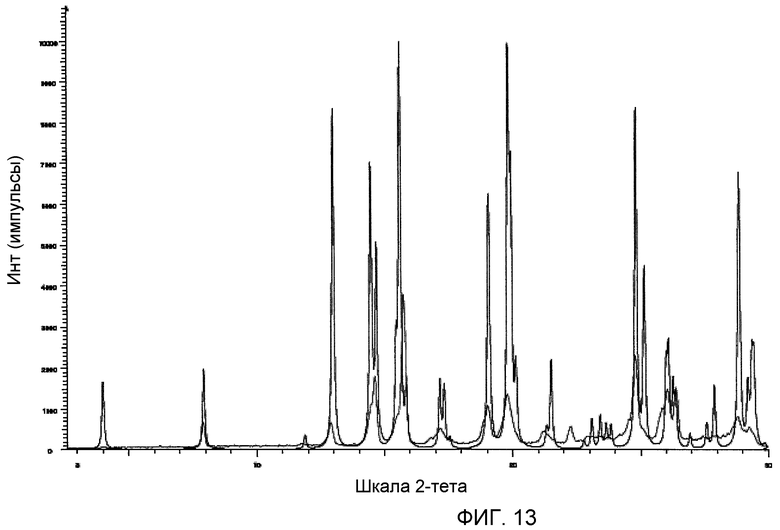
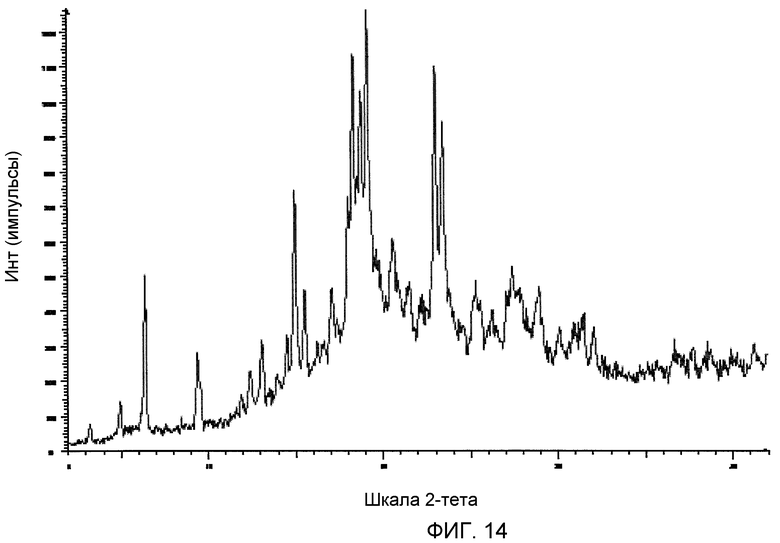
Настоящее изобретение относится к области органической химии и медицины и касается солей (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, по существу свободных от (2S,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, (2R,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида и (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, которые пригодны для лечения состояний и нарушений, связанных с дисфункцией центральной и вегетативной нервной системы. 3 н. и 2 з.п. ф-лы, 14 ил., 7 табл., 18 пр.
1. Соль кислоты и (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, по существу, свободная от (2S,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, (2R,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида и (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, где энантиомерный или диастереомерный избыток (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида выбирают из 90% или более; 95% или более; 98% или более; 99% или более; или 99,5% или более, и где кислоту выбирают из хлористоводородной кислоты, серной кислоты, фосфорной кислоты, малеиновой кислоты, толуолсульфоновой кислоты, галактаровой (слизевой) кислоты, D-миндальной кислоты, D-винной кислоты, метансульфоновой кислоты, R- и S-10-камфорсульфоновой кислоты, кетоглутаровой кислоты или гиппуровой кислоты.
2. Соединение по п.1, где стехиометрическое соотношение (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида к кислоте составляет 2:1, 1:1 или 1:2.
3. Соединение по п.2, где стехиометрическое соотношение составляет 1:1.
4. Гидрохлорид (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, по существу, свободный от гидрохлорида (2S,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, гидрохлорида (2R,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида и гидрохлорида (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, где энантиомерный или диастереомерный избыток (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида выбирают из 90% или более; 95% или более; 98% или более; 99% или более; или 99,5% или более.
5. Моногидрохлорид (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида или его гидрат или сольват, по существу, свободный от моногидрохлорида (2S,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил) бензофуран-2-карбоксамида, моногидрохлорида (2R,3S)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида и моногидрохлорида (2R,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида, где энантиомерный или диастереомерный избыток (2S,3R)-N-(2-((3-пиридинил)метил)-1-азабицикло[2.2.2]окт-3-ил)бензофуран-2-карбоксамида выбирают из 90% или более; 95% или более; 98% или более; 99% или более; или 99,5% или более.
| WO 2004076449 А, 10.09.2004 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2005109913 A, 10.01.2006. | |||

