Это изобретение относится к способу получения некоторых 3,4,4-трехзамещенных пиперидинил-N-алкилкарбоксилатов, новых промежуточных соединений и родственных им соединений. В конечном счете, это изобретение дает получение стабильных кристаллических соединений и лекарственных препаратов, используемых в качестве периферических опиодных антагонистов.
Многочисленные данные показывают, что периферические опиодные пептиды и их рецепторы играют важную физиологическую роль в сократительной способности кишечника. Следовательно, желудочно-кишечные расстройства, такие как идиопатический запор и синдром болезненной раздражимости кишечника, могут быть отнесены к дисфункции синдрома промежуточного регулирования опиодного рецептора, и агенты, действующие как антагонисты по отношению к таким рецепторам, могут быть полезными для лечения пациентов, страдающих от такой дисфункции.
N-замещенные пиперидины, полученные с использованием способа и промежуточных соединений данного изобретения, применяются в качестве периферически-селективных опиодных антагонистов. В частности, одним из наиболее подходящих для этого является 3,4,4-трехзамещенный пиперидинил-N-алкилкарбоксилат-(2S, 3R,4R)([[2-[[4-(3-гидроксифенил- 3,4-диметил-1-пиперидинил]метил]-1-оксо-3-фенилпропил]амино]уксусная кислота (I).
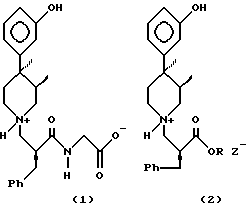 (I)
(I)
Характерный способ получения ( α S,3R,4R)-4-(3-гидроксифенил-3,4-диметил- α -(фенилметил)-1- пиперидин пропионовой кислоты в виде этилового эфира (2), являющегося промежуточным соединением для получения соединения (1), является известным для специалиста. Zimmerman описывает этот способ в патенте США 5250542 (включенный в ссылку). Однако этот способ дает смесь стереоизомерных продуктов, что мешает их использованию на практике в коммерческом способе. Получение нужного соединения формулы (I) требует длительного хроматографического разделения с выходом изомерного продукта всего лишь в 13%. Кроме того, каждое промежуточное соединение выделяется в виде "смолообразного" продукта из-за присутствия нежелательного изомера. "Смолообразный" продукт мешает очистке любого промежуточного соединения без хроматографии и поэтому является крайне нежелательным для коммерческих целей.
Способ данного изобретения предоставляет в настоящее время синтетический способ, обеспечивающий получение кристаллических промежуточных соединений без эпимеризации с целью облегчения коммерческого получения соединения (1) и его C1-C6 алкиловых эфиров.
Кроме того, способ данного изобретения дает возможность получать твердое кристаллическое вещество (1) и его C1-C6 алкиловые эфиры с приличными выходами. Наконец, способ синтеза данного изобретения включает получение промежуточных кристаллических соединений, обеспечивая как обогащение, так и очистку нужного продукта.
Данное изобретение обеспечивает создание крайне желательного стабильного кристаллического вещества - (2S,3R,4R) ([[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил]метил]-1- оксо-3-фенилпропил]амино]уксусной кислоты (1), которая является дигидратом.
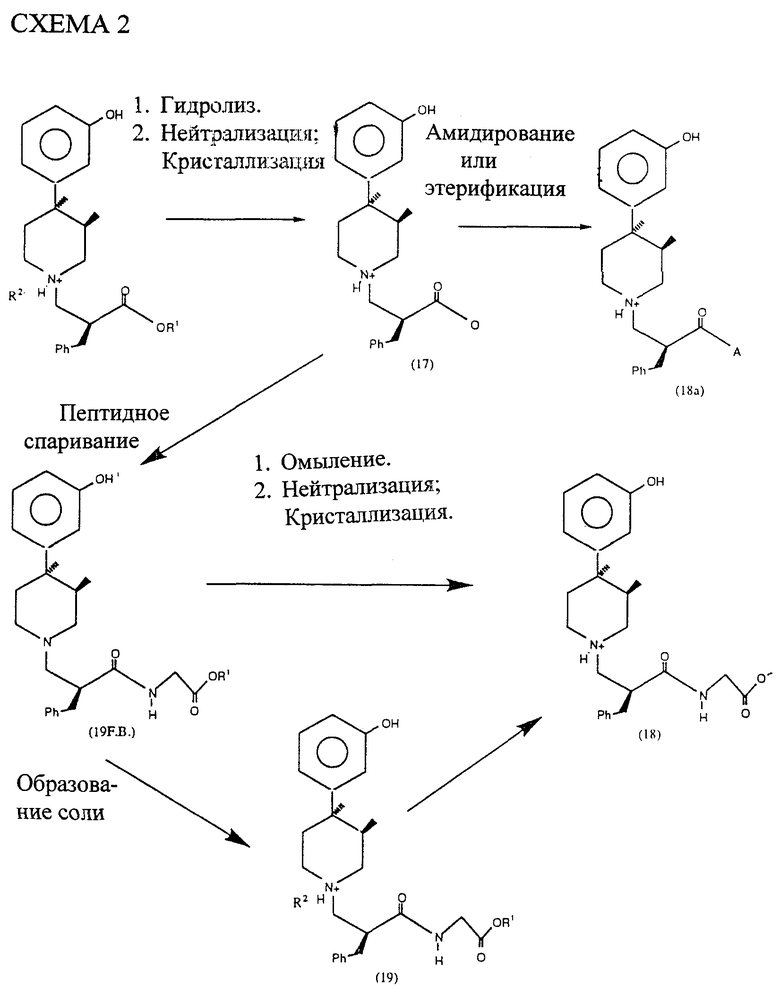
Новые кристаллические промежуточные соединения и метод кристаллизации являются особенно важными для промышленного усовершенствования получения фармацевтически активных 3-4,4-трехзамещенных пиперидинил-N-алкилкарбоксилатов (18 и 18a ниже).
Заявляемое изобретение дает новые кристаллические соли формулы 2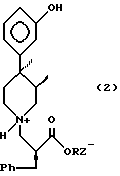
в которой R является C1-C6 алкилом; Z- представляет собой группу хлора, брома, сукцината, a (+) - дибензоилтартрат; с приемлемыми выходами.
Изобретение дает способ получения кристаллического моногидратного соединения 3
включающий кристаллизацию соединения 3 из растворителя, содержащего приблизительно от 50% до 75% низшего спирта и от 50% до 25% воды (по весу).
Кроме того, данное изобретение обеспечивает получение кристаллических соединений формулы 4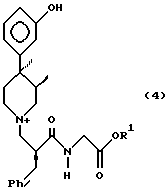
в которой R1 является C1-C6 алкилом; соединение представляет собой соль, такую как гидрохлорид и L-малат. Гидрохлоридная форма представляет собой единственную кристаллическую форму, существующую в виде ацетонового моносольвата. L-малатные соли также представляют собой единственные соли, так как их стехиометрия зависит от растворителя кристаллизации. Стехиометрическое соотношение может включать либо по 1 молю-эквиваленту L-яблочной кислоты и соединения 4 или может содержать 3 моль-эквивалента L-яблочной кислоты и 2 моль-эквивалента соединения 4. Как здесь принято, выражение "полуторомалат" относится к отношению 3:2 L-яблочной кислоты к соединению 4.
Наконец, данное изобретение относится к кристаллическому дигидратному соединению формулы 5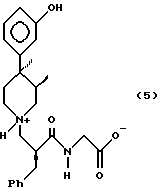
Выращивание "C1-C5 алкил", используемое здесь, относится к разветвленной или линейной алкильной группе, содержащей от одного до шести углеродных атомов. Обычно C1-C6 алкильные группы представляют собой метильную, этильную, н-пропильную, изо-пропильную, бутильную, изо-бутильную, вт-бутильную, трет-бутильную, пентильную, гексильную и т.п. Другие аналогичные термины относятся к прямым или разветвленным алкильным группам с определенным числом углеродных атомов. Например, "C1-C3 алкил" представляет собой метил, этил, н-пропил и изопропил.
Выражение "низший спирт" относится к метанолу, этанолу, 1-пропанолу и 2-пропанолу.
Термин "инертная атмосфера" и "инертные условия" относится к условиям реакции, в которых смесь покрывается слоем инертного газа, такого как азот или аргон.
Термин "практически чистый" используется здесь для обозначения по крайней мере 90-процентного молярного содержания необходимого абсолютного стереоизомера и/или полиморфного соединения. Более предпочтительным является содержание по крайней мере 95 молярных процентов и наиболее предпочтительным - 98 молярных процентов необходимого абсолютного стереозомера и/или полиморфного соединения.
Наиболее предпочтительным является, чтобы продукт и соединения данного изобретения представляли собой соединения, существующие в виде 3R,4R-изомера, как показано в формуле 3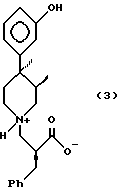
или 3S,4S-изомера формулы (6)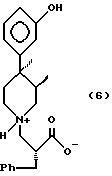
Кроме того, специалисту в данной области понятно, что бензиловый заместитель располагается в хиральном центре. Данное изобретение включает как ( α S,3R,4R), так и ( α R,3S,4S) диастереомеры. Особенно предпочтительными соединениями данного изобретения являются такие соединения формулы 2, 3, 4 и 5, в которых конфигурация заместителей в пиперидиновом кольце представляет собой 3R, 4R и углеродный атом, соединенный с бензиловой группой, представляет собой S-конфигурацию. Специалист может выбирать соответствующие реагенты для получения противоположного энантиомера.
Термины "R" и "S" используется здесь как обычно принято использовать в органической химии для обозначения специфической конфигурации хирального центра. См. Organic Chemistry, R.T. Morrison and R.N. Boyd, 4th ed., Allyn & Bacon, Inc. , Boston (1983) стр. 138-139 и The Vocabulary of Organic Chemistry, Orchin и др. John Wiley and Sons Inc., стр. 126.
Выражение "гидролиз", используемый здесь, включает все соответствующие известные методы эфирного гидролиза, включающие кислотный, основной и энзиматические процессы.
Предложенные методы описываются ниже.
Как здесь принято, фраза "кристаллизация 3" относится к нейтрализации продукта реакции гидролиза формулы 7, в которой M+ является натрием, литием или калием, с обозначенными реагентами и/или растворителями и кристаллизации с использованием известных методик. Смешивание может осуществляться с использованием обычных методов перемешивания, таких как размешивание, встряхивание и т.п. Кроме того, специалисту понятно, что процессы кристаллизации могут включать затравку, резкое охлаждение, "скребление" стекла реакционного сосуда и другие, обычно используемые для этого способы.
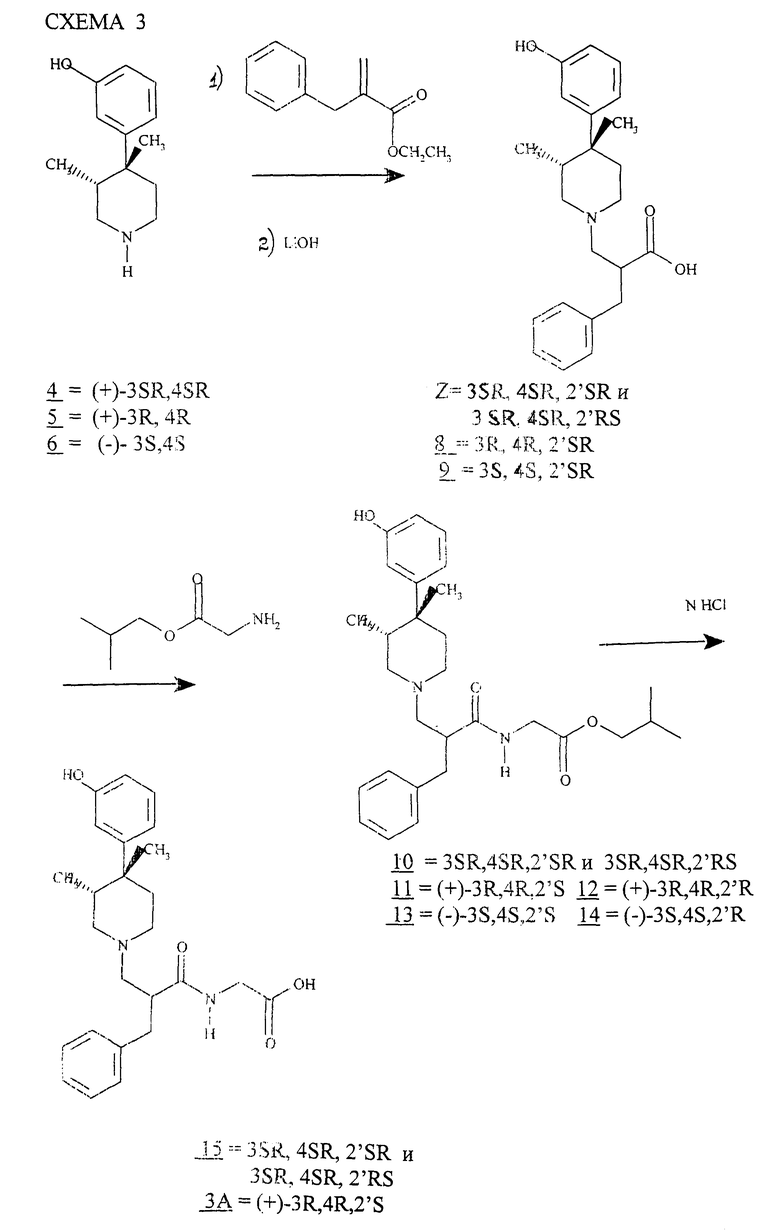
Исходные материалы для данного изобретения могут быть получены множеством разнообразных методов, хорошо известных специалистам данной отрасли. 3-замещенные-4-метил-4-(3-гидрокси- или алканоилоксифенил)пиперидиновые производные, используемые в качестве исходных веществ в способе данного изобретения, могут быть получены с применением основного метода, предложенного Zimmerman-ом в патенте США N 4115400 (1978) и Zimmerman-ом и др. в патенте США N 4891379 (1990). Патенты США N 4115400 и 4891379 объединены здесь ссылкой. Исходное вещество для синтеза соединений данного изобретения (3R,4R)-4-(3-гидроксифенил)-3,4-диметилпиперидин может быть получено по методу Zimmerman-а патент США 5250542, включенному здесь в ссылку. Специалист должен обратить особое внимание на примере 1 ссылки Zimmerman-а 5250542.
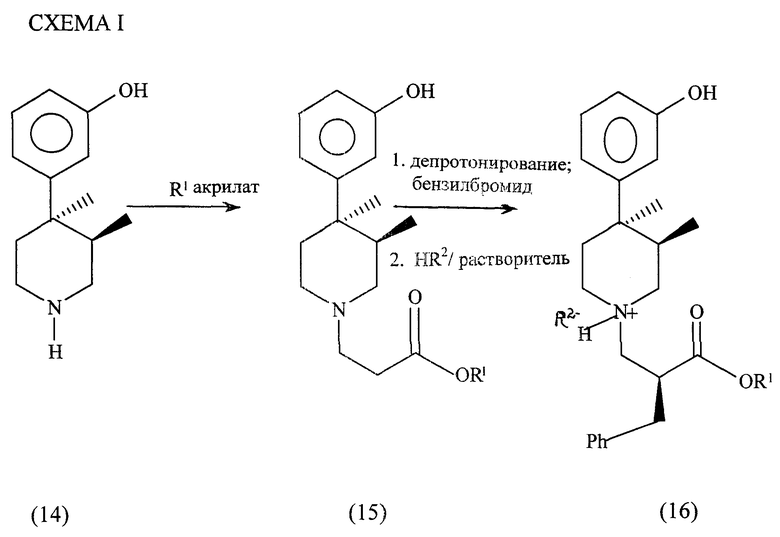
Исходное вещество 14, полученное как описано в методике, может быть использовано в способе по схеме 1 (см. в конце описания),
в которой R1 определен выше. R2 является хлоридом, бромидом, (+) - дибензоилтартратом или сукцинатом.
Как проиллюстрировано в схеме 1, соединение 14 контактирует с алкилакрилатом (R1 акрилат) с образованием соединения 15, R1 определен выше. Подходящими растворителями являются метанол, тетрагирофуран, этанол и другие. Наиболее подходящими растворителями являются метанол и тетрагидрофуран.
Соединение 15 подвергается депротонированию и контактированию с бензилбромидом. Депротонирование может быть осуществлено с использованием соответствующего основания. Примерами подходящих основных реагентов являются литий диизопропиламид или литий гексаметилдисилазид. Предпочтительными растворителями для реакции с основанием являются тетрагидрофуран и 1,2-диметоксиэтан. Специалисту понятно, что могут быть походящими и другие растворители. При использовании в качестве основания литий диизопропиламида (ЛДА) наиболее предпочтительным является присутствие 2 эквивалентов бензилбромида. Продукт алкилирования представляет собой смесь 1:1 ( α S,3R,4R)-изомера и ( α R,3R, 4R)-изомера.
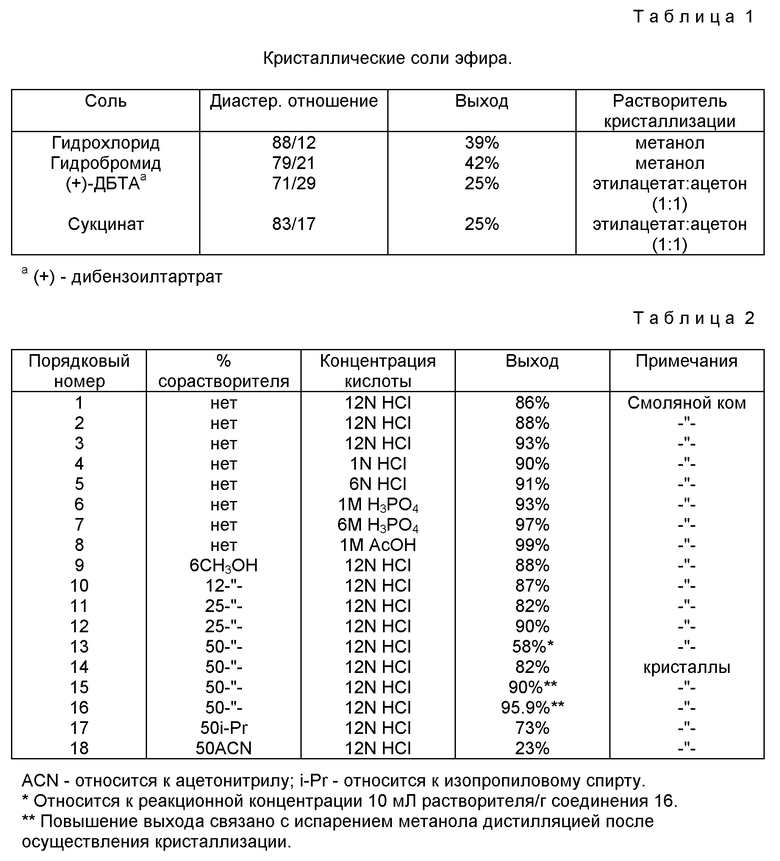
Кристаллические соединения формулы 16 являются новыми уникальными. Только четыре конкретные соли формулы 16 являлись как стабильными кристаллическими солями, так и характеризовались желательным диастереомерным обогащением. Были исследованы каждая из следующих кислот при использовании четырех различных систем растворителей: HCl, HBr, (+)-дибензоил винная, янтарная, (-)ди-п-толуилвинная, (+)-ди-п-толуилвинная (-)-дибензоилвинная, (1R,3S)-(+)-камфарная, гиппуровая, бензойная, L-яблочная, D-яблочная, малоновая, D-аспаркановая, (-)-винная, (+)-винная, (-)-миндальная, (+)-миндальная, L-аскорбиновая, малеиновая, серная, уксусная, фосфорная, лимонная, молочная, п-толуолсульфоновая, D-арабаскорбиновая и L-аспартиковая. Таким образом, среди исследованных 110 кристаллических соединений только четыре стабильные кристаллические соли характеризовались как обогащенные. Обогащение и выход четырех стабильных кристаллических солей иллюстрируется в таблице 1.
Как иллюстрируется схемой 2 (см. в конце описания), соединение 16 подвергается гидролизу с образованием соединения 17. Специалисту понятно, что соединение 17 может быть использовано для получения других полезных соединений, как это иллюстрируется соединением 18a. Соединения 18a описаны в основном в патенте США 5250542 как полезные в качестве опиодных антагонистов. Прежде всего, возможно получение абсолютно чистых стереохимических изомеров (18 и 18a) без утомительного хроматографического разделения, используя новые промежуточные соединения данного изобретения.
В схеме 2 R1 и R2 определены выше. A является OR4 или R5R6;
в которой:
R4 является водородом, C1-C10 алкилом, C2-C10 алкенилом, циклоалкилом, C5-C8-циклоалкенилом, циклоалкилзамещенным C1-C3 алкилом, C5-C8 циклоалкенилзамещенным C1-C3 алкилом или фенилзамещенным C1-C3 алкилом;
R5 является водородом или C1-C3 алкилом;
R6 является водородом, C1-C10 алкилом, C3-C10 алкенилом, циклоалкилом, фенилциклоалкилзамещенным C1-C3 алкилом, C5-C8 циклоалкенилом, C5-C8 циклоалкенилзамещенным C1-C3 алкилом, фенилзамещенным C1-C3 алкилом или (CH2)q-B; или
R5 и R6 вместе с N образуют насыщенное неароматическое 4-6-членное гетероциклическое кольцо;
B представляет собой 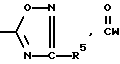 или NR7R8;
или NR7R8;
R7 является водородом или C1-C3 алкилом;
R8 является водородом, C1-C10 алкилом, C3-C10 алкенилом, циклоалкилзамещенным C1-C3 алкилом, циклоалкилом, C5-C8 циклоалкенилом, C5-C8 циклоалкенилзамещенным C1-C3 алкилом, фенилом или фенилзамещенным C1-C3 алкилом; или
R7 и R8 вместе с N образуют насыщенное неароматическое 4-6-членное гетероциклическое кольцо;
W представляет собой OR9M, MR10R11 или OE;
R9 является водородом, C1-C10 алкилом, C2-C10 алкенилом, циклоалкилом, C5-C8 циклоалкенилом, циклоалкилзамещенным C1-C3 алкилом, C5-C8 циклоалкенилзамещенным C1-C3 алкилом или фенилзамещенным C1-C3 алкилом;
R10 является водородом или C1-C3 алкилом;
R11 является водородом, C1-C10 алкилом, C3-C10 алкенилом, фенилом, циклоалкилом, C5-C8 циклоалкенилом, циклоалкилзамещенным C1-C3 алкилом, фенилзамещенным C1-C3 алкилом или  ; или
; или
R10 и R11 вместе с N образуют насыщенное неароматическое 4-6-членное гетероциклическое кольцо;
E является 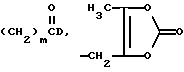 или
или 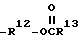
R12 является C1-C3 алкилзамещенным метиленом,
R13 является C1-C10 алкилом;
D представляет собой OR14 или NR15R16;
R14 является водородом, C1-C10 алкилом, C2-C10 алкенилом, циклоалкилом, C5-C8 циклоалкенилом, циклоалкилзамещенным C1-C3 алкилом или C5-C8-циклоалкенилзамещенным C1-C3 алкилом или фенилзамещенным C1-C3 алкилом;
R15 является водородом, C1-C10 алкилом, C3-C10 алкенилом, фенилом, фенилзамещенным C1-C3 алкилом, циклоалкилом, C5-C8-циклоалкенил, циклоалкилзамещенным C1-C3 алкилом или C5-C8 циклоалкенилзамещенным C1-C3 алкилом и
R16 является водородом или C1-C3 алкилом; или
R15 и R16 вместе с N образуют насыщенное неароматическое 4-6-членное тетроциклическое кольцо;
Y является OR17 или NR18R19;
R17 является водородом, C1-C10 алкилом, C2-C10 алкенилом, циклоалкилом, C5-C8 циклоалкенилом, циклоалкилзамещенным C1-C3 алкилом, C5-C8 циклоалкенилзамещенным C1-C3 алкилом или фенилзамещенным C1-C3 алкилом;
R18 является водородом или C1-C3 алкилом и
R19 является водородом, C1-C10 алкилом, C3-C10 алкенилом, фенилом, циклоалкилом, C5-C8 циклоалкенилом, циклоалкилзамещенным C1-C3 алкилом, C5-C8 циклоалкенилзамещенным C1-C3 алкилом или фенилзамещенным C1-C3 алкилом; или
R18 и R19 вместе с N образуют насыщенное неароматическое 4-6-членное гетероциклическое кольцо;
q равняется 1-4;
m равняется 1-4.
Заместитель "A" описывается в патенте США 5250542.
Реакция гидролиза может быть завершена при использовании известных методик кислотного гидролиза. Примером одного из таких методов кислотного гидролиза является действие водного раствора кислоты в диоксане в условиях дефлегмирования. Более предпочтительным является проведение реакции гидролиза с использованием условий омыления с тем, чтобы избежать эпимеризации. Примерами омыляющих реагентов являются гидроокись лития, гидроокись натрия, гидроокись калия и т.п.
Продукт реакции гидролиза (карбоксилатная соль) подвергают регулированию до изоэлектрической точки аминокислоты, используя водную кислоту для получения амфотерного соединения 17. Кристаллизация моногидрата 17 должна быть завершена с использованием 50-75% низшего спирта и 50-25% воды.
Таблица II иллюстрирует критическую зависимость кристаллизации от приблизительного состава растворителя 1:1 для низшего спирта и воды. Выражение "смоляной ком" относится к коагуляции липкого полутвердого продукта в аморфную массу.
Как это проиллюстрировано в схеме 2, продукт 17 может быть непосредственно использован в этапе амидирования/эстерификации. Когда требуется амидирование, должна быть выбрана аминокислота с целью образования необходимых соединений формул 18 и 18a. Аминокислота подвергается действию глицинового эфира в растворителе, таком как диметилформамид или тетрагидрофуран.
В качестве сопряженного реагента используется дициклогексилкарбодиимид. В качестве вспомогательного нуклеофильного вещества добавляется N-гидроксибензотриазол. Реакция сопряжения может осуществляться в инертных условиях. Более предпочтительным является использование в качестве растворителя тетрагидрофурана в реакции пептидного сопряжения. Специалисту понятно, что могут быть эффективно использованы также и другие способы пепидного сопряжения.
Или же кристаллическая соль соединения 19 СО (свободное основание) может быть получена, как это иллюстрируется в схеме 2. Были проведены исследования по кристаллизации с использованием 17 различных кислот с тремя растворителями: этилацетат, ацетон и этанол. Из этих 51 экспериментов только L-яблочная и хлористоводородная кислоты дали кристаллические соли, стабильные при 25oC.
Кристаллическая хлористоводородная соль получается контактированием 19 CO с безводной HCl в ацетоне. Капиллярный газохроматографический анализ показал, что соль образуется в виде моносольвата в ацетоне. Эта единственная моносольватная кристаллическая форма позволяет выделить соединение 19 в существенно чистом виде. Контактирование соединения 19 CO с безводным HCl в других растворителях дает аморфное твердое вещество без существенной очистки.
Заявители открыли, что соль L-яблочной кислоты может быть получена в виде стабильного кристаллического твердого вещества, имеющего два отношения 19 CO по отношению к L-яблочной кислоте в зависимости от растворителя кристаллизации. В том случае, когда кристаллизация осуществляется в растворителях, таких как метилэтилкетон, ацетон, ацетон/т-бутилметиловый эфир или ацетон/гептан была установлена предполагаемая стехиометрия 1:1 в 19 CO по отношению к L-яблочной кислоте. Однако, когда кристаллизация проводится в системе растворителей ацетон/этилацетат, ацетон/толуол или этанол/толуол, кристаллическая соль дает единственное стехиометрическое отношение 3:2 L-яблочной кислоты по отношению к 19 CO (полуторный малат). Такой результат является отчасти неожиданным, когда полуторный малат получается даже при отношении 1: 1 яблочной кислоты и 19 СО в определенных растворителях. Действительно, когда соединяются эквимолярные количества L-яблочной кислоты и 19 СО в растворителе или системе растворителей, образующих полуторный малат, этот полуторный малат является единственной солью, образованной с выходом около 67% в массовом равновесии 19 CO в маточном растворе. Конечно специалист ожидает, что отношение составит 1:1. Кроме того, кристаллизация полуторомалатной соли в "необразующем полуторомалата" растворителе или системах растворителей дает только кристаллическая мономалатная соль с выходом, близким к количественному, при этом избыток L-яблочной кислоты остается в маточной жидкости.
Кристаллизация полуторомалатной соли дает продукт фармацевтически приемлемой чистоты при высоком выходе, и этот продукт состоит из кристаллов с веской равномерностью кристаллической формы и величиной кристаллов. Гидрохлоридные и полуторомалатные соли могут быть использованы в качестве пролекарства, так как изобутиловый эфир легко расщепляется in vivo.
Кислотами, которые не образуют кристаллических солей, стабильных при 25oC, являются HBr, H2SO4, гиппуровая, d-винная, l-винная, малоновая, янтарная, уксусная, арабаскориновая, аскорбиновая, лимонная, бензойная, молочная, (S)-(+)-миндальная и (R)-(-)-миндальная кислоты.
Продукт реакции амидирования/эстерификации или солевые формы могут быть гидролизованы с использованием стандартных методик. Предпочтительно используются методы основного гидролиза. Предпочтительными реагентами омыления являются гидроокись натрия, гидроокись калия, гидроокись лития и т.п. Наиболее предпочтительным является осуществление этапа омыления с использованием гидроокиси натрия и растворителя. Особенно предпочтительными растворителями (1:1) являются метанол:вода и (2:1) этанол:вода. Реакция прекращается при использовании кислоты, такой как хлористоводородная кислота. После нейтрализации (pH 6) твердый кристаллический дигидратный продукт 18 выделяется непосредственно фильтрацией. Выделенный продукт 18 обладает фармацевтически приемлемой чистотой без последующих этапов очистки.
Особенно ценным является дигидрат 18, так как это соединение является стабильным кристаллическим твердым веществом с равномерной кристаллической формой и размером частиц, что обеспечивает воспроизводимые скорости растворения, оно характеризуется фармацевтически желаемыми качествами.
Соединения формул 4 и 5, приведенных выше, используются для блокирования периферических опиодных рецепторов и для предотвращения периферических индуцированных препаратами опия побочных эффектов. Эти побочные эффекты индуцируются приемом млекопитающим препарата опия, такого как морфин. Индуцированные препаратом опия побочные эффекты могут включать запор, тошноту и рвоту. Таким образом, соединения данного изобретения используются для лечения одного или нескольких индуцированных препаратами опия побочных эффектов. Эти соединения могут использоваться также для лечения синдрома болезненной раздражимости кишечника, неязвенной диспепсии и идиопатического запора. Эти соединения незначительно преодолевают гематоэнцефалический барьер и поэтому не снижают опиодного эффекта на центральные (мозговые и позвоночных тяжей) опиодные рецепторы. Следовательно, эти характеристики показывают, что соединения свободны в основном от других центральных промежуточных эффектов.
Для того, чтобы определить in vivo антагонизм опиодного рецептора, была использована мышь, находящаяся под воздействием анальгезиса. Испытуемые соединения оценивались по их способности к блокированию индуцированной морфином анальгезии.
Пять мышей мужской особи CF-1 (Charles River, Portage, MI), весом приблизительно в 20 г, после голодания в течение ночи подвергались одновременно наблюдениям с точки зрения реакции на сокращение. Ответ на сокращение определялся как сокращение брюшной мускулатуры, за которым следует вытяжение задних конечностей, и индуцировался внутрибрюшным приемом 0,6% уксусной кислоты в объеме 1 мл/100 г веса тела. Период наблюдения длился 10 минут, начиная с 5-и минут после инъекции уксусной кислоты. Процент торможения сокращения высчитывался, исходя из среднего числа сокращений в контрольной (не подвергнутой лекарственному воздействию) группе. ED50 определялась как доза вещества-агониста, имеющего ингибирующее значение на сокращение 50%. AD50 определялась как доза антагониста, которая снижает ингибирование сокращения, создаваемое дозой сульфата морфина 1,25 мг/кг, до 50%. Каждая мышь подвергалась испытанию только один раз. Все лекарственные препараты вводились подкожно (1 мл/100 г веса) за 20 минут до инъекции уксусной кислоты.
Проводились определения периферической опиодной активности. Мыши выдерживались как минимум в течение 10 дней на 0.01 М сахариновой воде, содержащей 1 г/л сульфата морфина при среднем потреблении 3,0 г воды на мышь в день в течение по крайней мере 3 дней. Морфиновая вода удалялась за 45 минут до инъекции предполагаемого опиодного антагониста.
После приема опиодного антагониста мыши помещались в пластмассовые цилиндры, дно которых было выстлано белыми бумажными полотенцами.
Мыши подвергались визуальному наблюдению в течение 30 минут после инъекции - на наличие подергивания и поноса.
Подергивание расценивалось как позитивное, если происходило по крайней мере одно подергивание в 30 минут. Понос расценивался положительно, когда экскременты были достаточно влажными и оставляли пятна на белой бумаге, выстилающей дно цилиндра. Через 30 минут испытаний мыши возвращались в свои первоначальные клетки, снова сажались на морфиновую воду и не подвергались повторным испытаниям в течение 48 часов. Были испытаны более низкие дозы соединений антагонистов, пока не были определены пороговые дозы поноса. Понос представляет собой периферически усредненное значение стремительного, вызванного препаратом опия воздержания.
Степень воздействия на периферическую активность представленных соединений по сравнению с воздействием на центральную активность может быть определена сравнением AD50 для мыши, подвергшейся испытанию на сокращение, с ED50 для мыши, подвергшейся испытанию на понос. При более высоком отношении наблюдается более высокий относительный антагонизм периферических опиодных рецепторов, обусловленный конкретным соединением.
Значения AD50 для соединений данного изобретения составляют свыше 8 мг/кг, в то время как значения ED50 составляют ниже 1.
Кроме того, было установлено, что соединения формул 5 и 4, приведенные выше, показывают прекрасную активность в анализе связывания опиодного рецептора, которая оценивается сродством соединений к связыванию Мю-рецепторов. Этот анализ проводился в соответствии со следующей методикой.
Мужские особи крыс Spraque Dawley были умерщвлены обезглавливанием и их мозг был извлечен. Мозговая ткань с выделенным мозжечком гомогенизировалась в тефлоне и в стеклянном гомогенизаторе для тканей. Всплывшая 1, шариковая IV фракция была заморожена в азотном замораживателе при концентрации 1,33 г/мл и хранилась до использования не более пяти недель.
Увелививающиеся концентрации экспериментального соединения (от 0,1 до 1000 наномол (нМ)), буфер Krebs-Hepes pH 7,4 и тритиевый налоксон (0,5 нМ) (3H лиганд) помещались в полистирольные пробирки при комнатной температуре.
Реакция инициировалась добавлением ресуспензированной ткани, которая была предварительно термостатирована при 37oC в течение 20 минут. Реакционная смесь термостатировалась при 37oC на водяной бане в течение 20 минут. Реакция заканчивалась быстрой фильтрацией (Brandel cell harvester) через стеклянные фильтры Wratman GF/B, которые были предварительно пропитаны буфером Krebs-Hepes с pH 7,4. Затем фильтры промывались 2 раза 5 мл ледяного буфера Krebs-Hepes с pH 7,4. Промытые фильтры помещались в сцинциляционные ампулы, было добавлено 10 мл (Brandel) и образцы подвергались обсчету на бета-счетчике Searle D-300. Время термостатирования реакционной смеси составляло 20 минут при 37oC. Значения Kj и KD рассчитывались с использованием стандартных методик.
Соединения данного изобретения выявляют высокие значения желаемой активности. Значение процента замещения испытуемыми соединениями при концентрации 10 нМ было выше 75% и свыше 80% при 100 нМ. Это особенно желательно в свете значений AD50 и ED50, приведенных выше. Результаты показывают, что соединения данного изобретения характеризуются благоприятной активностью для использования их в лечении синдрома раздражимости кишечника и условий, относящихся к связыванию мю рецепторов.
Хотя и возможен прием соединения данного изобретения непосредственно без каких-либо составов, предпочтительно использование таких соединений в виде фармацевтических композиций, включающих фармацевтически приемлемый наполнитель и по крайней мере одно из соединений изобретения. Эффективный интервал дозировок соединений данного изобретения достаточно широк. Так, эти композиции содержат приблизительно от 0,1 процента по весу до 90,0 процентов по весу заявленных здесь соединений. Само по себе, представленное изобретение предоставляет также фармацевтические композиции, включающие соединение данного изобретения с фармацевтически приемлемым наполнителем.
При приготовлении композиций данного изобретения активный ингредиент обычно смешивается с наполнителем, который может быть носителем или разбавителем, или может быть разбавлен носителем, или включен в носитель, который может представлять собой капсулу, пакетный, бумажный или другой контейнер. В том случае, когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким веществом, служащим связующим, наполнителем или средой для активного ингредиента. Таким образом, композиции могут иметь вид таблеток, пилюль, порошков, лепешек, пакетиков, облаток, элексиров, эмульсий, растворов, сиропов, суспензий, аэрозолей (как в твердой, так и в жидкой среде), суппозиториев, мягких и твердых желатиновых капсул.
Соединения данного изобретения могут вводиться трансдермально, если это желательно. Специалисту хорошо известны трансдермальные усилители проницаемости и доставляющие системы, включающие аппликации и т.п.
Примерами соответствующих носителей, наполнителей и разбавителей являются лактоза, декстроза, сахароза, сорбит, маннит, крахмалы, аравийская камедь, фосфат кальция, альгинаты, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, трагакант, желатин, сироп, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния, вода и минеральное масло. Композиции могут включать также увлажняющие агенты, эмульгирующие и суспендирующие агенты, консервирующие агенты, подслащивающие вещества или облагораживающие агенты. Композиции изобретения могут быть приготовлены как для быстрого, так и отложенного или замедленного выделения активного компонента после приема его пациентом - применением хорошо известных специалисту приемов.
Композиции данного изобретения могут вводиться трансдермально с использованием известных трансдермальных поставляющих систем и наполнителей. Более предпочтительным является смешение соединения данного изобретения с усилителями проницаемости, включающими, но не ограниченными пропиленгликоль, полиэтиленгликоль, монолаурат и азациклоалкан-2-оны и с включенными в аппликацию аналогичными поставляющими системами. Дополнительными наполнителями являются желатинирующие агенты, эмульгаторы и буферные вещества, которые могут быть добавлены в случае необходимости в трансдермальную композицию.
Для орального приема соединения данного изобретения могут быть в идеальном случае смешаны с носителями и разбавителями и сплавлены в таблетки или помещены в желатиновые капсулы. Соединения данного изобретения могут быть приготовлены в виде микрочастиц или микрошариков. Микрочастицы могут быть приготовлены с использованием полигликолида, полилактида или других полимеров для облегчения непрерывного выделения активного соединения или пролекарства.
Композиции готовятся предпочтительно в единичной дозированной форме, при этом каждая дозировка содержит приблизительно от 1 до 500 мг, более предпочтительно от 5 до 300 мг активного ингредиента. Другой предпочтительный интервал составляет приблизительно от 0,5 мг до 60 мг активного ингредиента на единичную дозированную форму. Термин "единичная дозированная форма" относится к физическим дискретным единицам, подходящим в качестве стандартных дозированных форм для человека и других млекопитающих, при этом каждая единица содержит предварительно определенное количество активного вещества, рассчитанная для обеспечения желаемого терапевтического эффекта в сочетании с соответствующим фармацевтическим носителем.
Специалисту понятно, что соединения данного изобретения могут быть составлены с применением других известных лекарственных препаратов. Такие составляющие могут сообщать синергический терапевтический эффект. Например, антацид может быть соединен с соединениями данного изобретения для создания желаемого желудочно-кишечного эффекта.
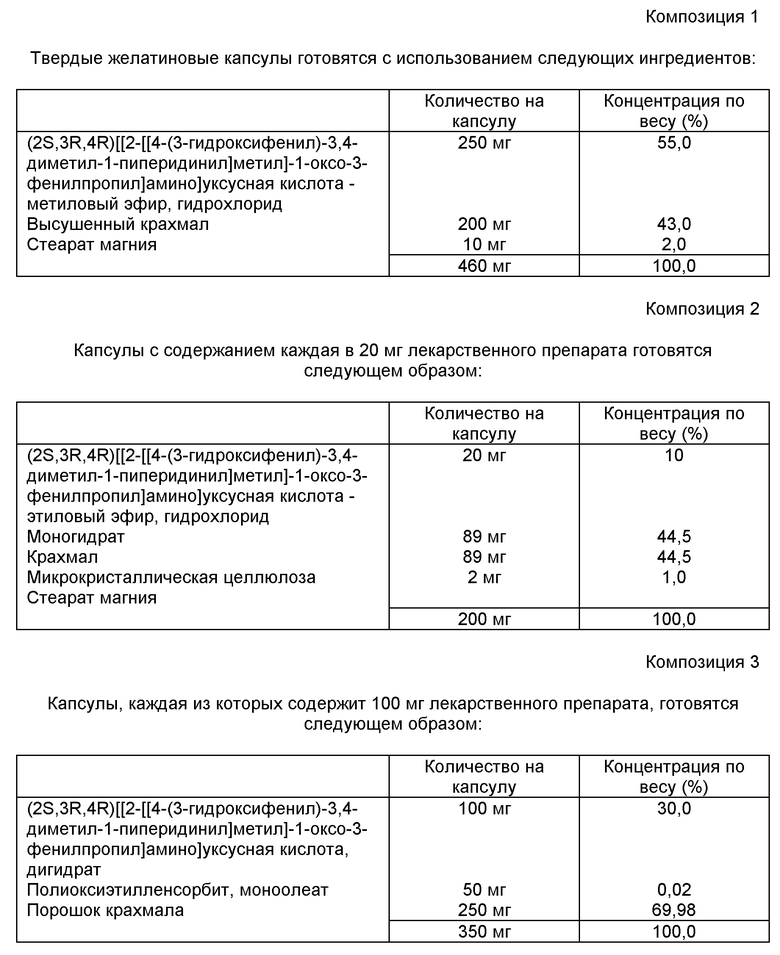
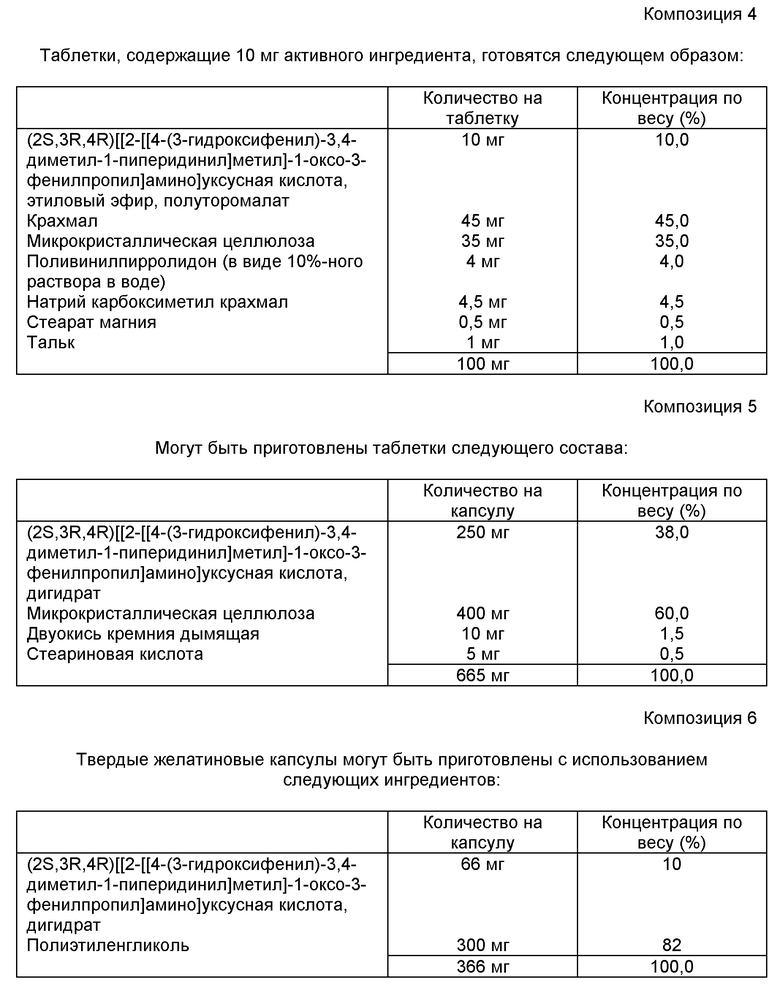
Для более полной иллюстрации действия данного изобретения ниже приводятся примеры составов. Примеры только иллюстрируют, но не ограничивают объем изобретения. Композиции могут использовать в качестве активных соединений любые соединения данного изобретения (композиции 1-6 см. в конце описания).
В композиции 1 ингредиенты смешиваются и помещаются в твердую желатиновую капсулу в количестве 460 мг.
В композиции 2 активный ингредиент, целлюлоза, крахмал и стеарат магния смешиваются, пропускаются через сита США с размером N 45 меш и помещаются в твердую желатиновую капсулу.
В композиции 3 ингредиенты тщательно перемешиваются и помещаются в пустую желатиновую капсулу.
В композиции 4 активный ингредиент, крахмал и целлюлоза пропускаются через сито США размером отверстий 45 меш и тщательно перемешиваются. С образующимся порошком смешивается раствор поливинилпирролидона, после чего смесь пропускается через сито США с размером отверстий 14 меш. Полученные таким образом гранулы сушатся при 50-60oC и пропускаются через сита США в 18 меш. Натрий карбоксиметил крахмал, стеарат магния и тальк, предварительно пропущенные через сито США с отверстиями в 60 меш, затем добавляются к гранулам, которые после смешения прессуются на таблетирующей машине с получением таблеток весом 100 мг.
В композиции 5 компоненты смешиваются и прессуются в виде таблеток весом 665 мг.
В композиции 6 все твердые ингредиенты пропускают через сито. Полиэтиленгликоль расплавляют и поддерживают в расплавленном состоянии. Лекарственный препарат помещают в расплавленное связующее. Сплавленная гомогенная суспензия помещается в твердую желатиновую капсулу соответствующего веса или объема с использованием подходящего для наполнения маслообразной пасты оборудования.
Капсулы, содержащие 6 мг активного вещества, могут быть приготовлены точно, как описано выше; однако количество дигидратного соединения должно быть снижено до 6,6 мг на капсулу. Капсулы, содержащие 0,6 мг активного вещества, могут быть приготовлены, как описано выше; однако количество дигидрата должно быть снижено до 0,66 мг с 200 мг полиэтиленгликоля на капсулу.
Промежуточные соединения и способы, соответствующие данному изобретению, могут использоваться для получения соединений, характеризующихся успешной периферической опиодной антагонистической активностью. В объеме данного изобретения предлагаются определенные соединения и условия. Следующие далее условия осуществления изобретения и характеристики соединений, приведенные в виде таблицы, независимо могут комбинироваться для создания множества предлагаемых соединений и условий способа. Следующий далее перечень осуществлений данного изобретения не имеет в виду каким-либо образом ограничить объем данного изобретения.
A) Кристаллическое соединение 2 представляет собой метиленовый сложный эфир.
B) Кристаллическое соединение 2 представляет собой ( α S,3R,4R)-3-[[4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовую кислоту, гидрохлорид.
C) Кристаллическое соединение 2 представляет собой этиловый сложный эфир.
D) Кристаллическое соединение 2 представляет собой HBr-соль.
E) Низший спирт является метанолом.
F) Соотношения низшего спирта и воды составляют: низшего спирта 50-60% и 50-40% воды.
G) R1 представляет собой C1-C4 алкил.
H) Кристаллические соединения формулы 4 представляют собой полуторомалатную соль.
I) Кристаллические соединения формулы 4 представляют собой гидрохлорид ацетоновый моносольват.
J) Практически чистый дигидрат формулы 5 представляет собой 97% или более 2S,3R,4R дигидрата.
K) Фармацевтическая композиция включает дигидратное соединение формулы 5 и один или несколько фармацевтически приемлемых наполнителя.
L) Фармацевтическая композиция включает полуторомалатную соль соединения формулы 4.
M) Способ использования соединения формулы 5 состоит в лечении синдрома раздражимости кишечника.
N) Способ использования одного или нескольких соединений формулы 4 состоит в лечении синдрома раздражимости кишечника.
O) Способ связывания мю-рецептора состоит в приеме эффективного количества соединения формулы 5.
P) Способ связывания мю-рецептора состоит в приеме эффективного количества одного или нескольких соединений формулы 4.
Предложенные осуществления данного изобретения представлены пунктами A-P.
Следующие далее примеры даются с целью иллюстрации и не могут быть рассмотрены в качестве ограничивающих объем притязаний изобретения.
Концентрации реагентов не являются критическими для осуществления изобретения. Специалист в данной области может изменить концентрации реагентов с целью достижения желаемой скорости реакции и выхода продукта.
Продолжительность времени осуществления описанных процессов не является критической. Как во всех случаях химических процессов, скорость реакции зависит от множества факторов, таких как температура и точные соединения, которые должны быть приготовлены. За ходом реакции можно следить, используя такие методы, как тонкослойная хроматография (ТСХ), высокоэффективная жидкостная хроматография (ВЭЖХ), газовая хроматография (ГХ) и ядерная магнитная резонансная спектроскопия (ЯМР) - с целью определения степени завершения реакции. Исполнитель может получить максимальные выходы, увеличивая время реакции. Или же, исполнитель может пожелать получить максимальную производительность, прекращая реакцию в точке, в которой достигается максимальное завершение с экономической точки зрения.
Как принято в дальнейших примерах, следующие выражения имеют такие значения. "ГОБТ" относится к 1-гидроксибензотриазол гидрату. "ТГФ" означает тетрагидрофуран. "ДМФ" относится к диметилформамиду. "ТЭА" означает триэтиламин. "ДЦК" означает дициклогексилкарбодиимид.
Препарат 1
(3R, 4R)-4-(3-гидроксифенил)-3,4-диметил-1- пиперидинпропоновая кислота, метиловый эфир
Круглодонная колба загружалась ТГФ (1000 мл) и (+)-3-(3,4-диметил-4-пиперидинил)фенолом (70,46 г, 0,343 мол). Суспензия нагревалась до 40-45oC и в течение 3 минут добавлялся метилакрилат (46,4 мл, 0,515 мол, 1,5 экв.). Изменения температуры не наблюдалось
Реакционная смесь перемешивалась при 45oC и развитие ее наблюдали, используя метод ВЭЖХ. Реакционная смесь осталась мутной. Через 4 часа реакционную смесь охлаждали до комнатной температуры и фильтровали через диатомовую землю. Растворитель и избыток метилакрилата удаляли концентрированием раствора посредством ротационного испарения при 40oC до общего веса 120 г. Неочищенный продукт подвергали повторному растворению в ДМФ (180 г) с получением раствора в 33,3 вес.% для использования его в способе примера 2. Количественный выход по ВЭЖХ. [ α ]/S (20, D) 75,3o (C 1,01, MeOH), [α]
ИКС (CHCl3): 3600, 3600-3100, 1732, 1440 см-1.
1H-ЯМР (CDCl3): δ 0,72 (д, 3H, J = 7,0 Гц), 1,30 (с, 3H), 1,59 (уш.д., 1H), 1,90 - 2,03 (м, 1H), 2,25 - 2,50 (м, 2H), 2,50 - 2,90 (м, 7H), 3,66 (с, 3H), 6,63 (дд, 1H, J = 7,8, 2,0 Гц), 6,73 (уш.с., 1H), 6,81 (д, 1H, J = 7,8 Гц), 7,15 (т, 1H, J = 8,0 Гц).
13C-ЯМР (CDCl3): δ 16,1, 27,4, 30,8, 32,0, 38,4, 38,9, 49,9, 51,7, 53,9, 55,7, 55,8, 112,5, 112,6, 113,0, 113,2, 117,6, 117,7, 129,2, 151,6, 156,1, 173,4.
УФ (EtOH): λmax 274 нм, ε 2028; 202 нм, ε 17350.
МС (FAB): m/z 292 (100%, M+1), 292 (18%, M+), 218 (65%).
Препарат 2
Изобутилглицин, соль п-толуолсульфоновой кислоты
Круглодонная колба наполнялась толуолом (600 мл), глицином (22,53 г, 0,30 мол, моногидратом п-толуолсульфоновой кислоты (62,76 г, 0,33 мол, 1,1 экв) и изобутиловым спиртом (60 мл, 0,65 мол, 2,17 экв). Гетерогенная реакционная смесь перемешивалась и нагревалась при дефлегмировании с наружным обогревом для азеотропного удаления воды по мере ее образования. Через два часа реакционная смесь была гомогенной. Спустя еще 1,5 часа реакционная смесь охлаждалась до 50oC и концентрировалась с применением ротационного испарения при 60oC до конечного веса 135 г.
Остаток (гомогенное масло) растворялся в этилацетате (450 мл) пока был еще теплым и раствор переносился в 3-х горлую круглодонную колбу, снабженную механической мешалкой и конденсатором дефлегматорного типа. К раствору при комнатной температуре при перемешивании добавлялся гексан (450 мл). Пастообразная смесь затем нагревалась с обратным холодильником для повторного растворения твердого вещества и раствору давали медленно охлаждаться при перемешивании. Затем раствор затравливали при 38oC для инициирования кристаллизации. После охлаждения до комнатной температуры смесь охлаждали до 5oC и перемешивали еще час. Продукт выделяли фильтрацией через воронку со стеклянным фильтром, сушили на воздухе в течение 1/2 часа и затем сушили в течение ночи в вакуумной печи (40oC, 5 мм рт. ст.). Общий выход белого кристаллического вещества составлял 89,1 г (97,9%).
Т.пл. 77,2 - 79,6oC.
pKa (67% водн. ДМФ) = 7,68.
ИК (CHCl3): 3300 - 2600, 3018, 2970, 1752, 1213, 1125, 1033, 1011 ом-1.
1H-ЯМР (300 МГц, CDCl3): δ 0,82 (д, 6H, J = 6,9 Гц), 1,79 (септ, 1H, J = 6,8), 2,33 (с, 3H), 3,66 (уш.р.с, 2H), 3,78 (д, 2H, J = 6,6 Гц), 7,10 (д, 2H, J = 8,1 Гц), 7,72 (д, 2H, J = 8,2 Гц), 8,03 (уш.с., 3H).
13C-ЯМР (CDCl3): δ 18,9, 21,3, 27,4, 40,3, 72,0, 126,1, 128,9, 140,3, 141,4, 167,5.
Анализ C13H21NO5S:
Рассчитано: C 51,47, H 6,98, N 4,62, S 10,57.
Установлено: C 51,74, H 6,77, N 4,76, S 10,73.
Пример 1
(2S, 3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил] метил]-1-оксо-3-фенилпропил]-амино]-уксусная кислота, 2-метилпропиловый эфир
Круглодонная колба наполнялась ( α S,3R,4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил- 1- пиперидинпропионовой кислотой (20,11 г, 0,0522 мол, 1 экв), соединением препарата 2 (17,60 г, 0,058 мол, 1,11 экв), гидроксибензотриазол моногидратом (7,83 г, 0,058 мол, 1,11 экв) и сухим тетрагидрофураном (144 мл). К смеси добавлялся триэтиламин (8,08 мл, 0,058 мол, 1,11 экв), затем следовало добавление дициклогексилкарбодиимида (11,97 г, 0,058 мол, 1,11 экв), растворенного в тетрагидрофуране (60 мл). Смесь перемешивалась в течение двух дней при 25oC в атмосфере азота. Завершение реакции определяли с использованием ВЭЖХ. Пастообразный осадок охлаждали при 0oC в течение двух часов и затем фильтровали. Затем фильтрат выпаривали до состояния, близкого к сухому, при пониженном давлении (10 Торр, 1333,22 Па) при 40oC. Масло растворяли в 250 мл этилацетата. Органический слой промывался 250 мл 0,5 М этилацетата. Органический слой промывался 250 мл 0,5 М буферным раствором CO3 -2/HCO3 -1 с pH 9,8. pH доводили до 9,5 - 9,8. Органический раствор промывался 250 мл насыщенного раствора соли. Органический слой сушили над Na2SO4, охлаждали при перемешивании до -20oC и оставляли стоять без перемешивания при -20oC на ночь (16 часов). Осажденный DCU удалялся фильтрованием. Этилацетат испаряли при пониженном давлении (10 Торр, 1333,22 Па), получая 25,0 г (95%) аморфного твердого вещества.
ИКС (CH3Cl3) 2897, 1740, 1659 см-1;
1H-ЯМР (300 МГц, CDCl3) δ 8,94 (дд, 1H, J = 2,0 Гц), 8,40 (уш.с., 1H), 7,20 - 6,93 (м, 4H), 6,60 - 6,50 (м, 3H), 4,04, 3,95 (м, 2H), 3,80 - 3,65 (м, 2H), 3,16 (дд, 1H, J = 13,8 Гц, J = 4,4 Гц), 2,69 (уш.д., 1H, J = 10,2 Гц, 2,63 - 2,41 (м, 4H), 2,40 - 2,15 (м, 4H), 1,84 - 1,71 (м, 2H), 1,42 (уш. д., 1H, J = 12,4 Гц), 1,10 (с, 3H), 0,77 (д, 6H, J = 6,9 Гц), 0,57 (д, 3H, J = 6,9 Гц);
1H-ЯМР (300 МГц, ДМСО-d6): δ 9,17 (уш.с., 1H), 8,40 (уш.т., 1H, J = 2,0 Гц), 7,26 - 7,14 (м, 4H), 7,04 (т, 1H, J = 7,8 Гц), 6,63 (м, 2H), 6,52 (д, 1H, J = 8,1 Гц), 3,81 - 3,79 (м, 4H), 2,90 - 2,43 (м, 6H), 2,37 (д, 1H, J = 12,4 Гц), 2,33 - 2,03 (м, 2H), 1,95 - 1,65 (м, 2H), 1,43 (д, 1H, J = 12,4 Гц), 1,17 (с, 3H), 0,85 (д, 6H, J = 6,7 Гц), 0,65 (д, 3H, J = 6,8 Гц);
13C ЯМР (75,4 МГц, ДМСО-d6) δ 174,03, 169,78, 157,05, 151,71, 140,08, 128,80, 128,71, 125,77, 115,93, 112,36, 112,06, 69,96, 59,73, 54,95, 49,87, 44,24, 40,59, 38,03, 37,83, 35,61, 29,93, 27,19, 27,08, 18,72, 15,72,15,79;
МС (FD) m/z 481 (M+);
[α]
УФ (MeOH) 274,4 нм ( ε = 2093), 202,8 нм.
Анализ C29H40N2O4:
Рассчитано: C 72,47, H 8,39, N 5,83
Найдено: C 72,49, H 8,59, N 5,63.
Пример 2
( α S, 3R,4R)-4-(3-Гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, метиловый эфир гидрохлорид
Круглодонная колба продувалась азотом и загружалась ТГФ (100 мл) и 2 М раствором LDA (17,6 мл, 35,18 ммол, 2,05 экв.). Раствор охлаждали до -30oC и к нему добавлялся раствор соединения препарата 1 (15,24 г, 17,6 ммол, 1,0 экв, 32,8 вес.% в ТГФ) - в течение 20 минут при поддержании температуры между -26o и -28oC.
После перемешивания в течение 15 минут при -25oC медленно добавлялся бензилбромид (5,81 г, 34,32 ммол, 2,0 экв.) при поддержании температуры между -17o и -20oC. Реакционная смесь перемешивалась в течение трех часов при температуре от -15o до -20oC. Отношение ( α S,3R,4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, метиловый эфир/ ( α R, 3R,4R)-4-(3-гидроксифенил)-3,4-диметил-1- пиперидинпропионовая кислота, метиловый эфир составляло 97/3.
Реакционная смесь резко охлаждалась 1 N HCl (22 мл, 22 ммол). pH регулировалось от 10,6 до 9,5 с помощью 12 N HCl (2,3 мл), после чего низкотемпературная баня удалялась. Добавлялся гептан (50 мл) и слои разделялись. К органическому слою добавлялся метанол (25 мл) и раствор охлаждался. К нему добавлялась безводная HCl (1,3 г) при поддержании температуры ниже 5oC до тех пор, пока смесь не становилась кислой. В процессе добавления осаждалась соль хлористоводородной кислоты. Смесь подвергалась концентрированию до конечного веса 32,58 г. Затем к масляному концентрату добавлялся метанол (36 мл) и через несколько минут образовался осадок. Смесь перемешивалась в течение ночи при комнатной температуре.
После охлаждения до 0oC в течение 1,25 часа осадок фильтровали, колбу промывали 10 мл фильтрата, а отжатый осадок промывали холодным метанолом (10 мл). Твердое вещество высушивали с получением 2,93 г (выход 40,9%) белого порошка.
Анализ, проведенный с применением ВЭЖХ, показал, что продукт представлял собой смесь 86:14 стереоизомеров.
Неочищенная хлористоводородная соль (2,75 г) помещалась в метанол (13,75 мл) и полученный пастообразный продукт нагревался при дефлегмировании в течение двух часов. Смесь охлаждалась до температуры приблизительно 0oC. Осадок фильтровался, колба промывалась фильтратом, а отжатый осадок промывали холодным метанолом (1,5 мл). Продукт сушили с получением 2,32 г белого твердого продукта (выход 84,4%).
Общий выход: 34,5% (алкилирование и выделение осадка)
Чистота: 96,2% ( α S, 3R, 4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, метиловый эфир гидрохлорид, 2,9% ( α R,3R,4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, метиловый эфир гидрохлорид и 0,7% ( α S,3R,4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, моногидрат (ВЭЖХ поверх.%).
Т.пл. 230 - 232oC (разл.).
ИКС (KBr): 3174, 1732, 1620, 1586, 1276, 785, 749, 706 см-1
1H-ЯМР (ДМСО-d6): [0,78 (д, 0,85 • 3H, J = 7,2 Гц) и 1,02 (д, 0,15 • 3H, J = 7,2 Гц), диастереомерные соли], [1,28 (с, 0,15 • 3H), 1,34 (с, 0,85 • 3H), диастереомерные соли] , 1,76 (уш.д., 1H), 2,10 - 2,48 (м, 2H), 2,75 - 3,65 (м, 12H), 6,60 - 6,90 (м, 3H), 7,11 (т, 1H, J = 7,8 Гц), 7,15 - 7,35 (м, 5H), 9,43 (уш.с., 1H), 9,75 (уш.с., 1H)
МС (FD): m/z 381 (100%, M - HCl).
Анализ C24H32ClNO3:
Рассчитано: C 68,97, H 7,72, N 3,35, Cl 8,48
Найдено: C 68,27, H 7,84, N 3,42, Cl 8,38
Пример 3
(+)-( α S, 3R,4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, моногидрат
В круглодонную колбу помещалась дистиллированная вода (230 мл) вместе с 50 вес. % раствора гидроокиси натрия (20,02 г, 250 ммол, 4,2 экв). В колбу добавлялась одна порция продукта примера 2 (25,0 г, 60 ммол, 1 экв). Смесь перемешивалась при комнатной температуре и фильтровалась. Фильтровальная бумага промывалась 33 мл 1 N раствора гидроокиси натрия. Раствор переносился в круглодонную колбу, пригодную для вакуумной перегонки. К раствору добавлялся метанол (240 мл). pH раствора доводили до 6,0, используя концентрированную хлористоводородную кислоту (32,14 г). Метанол удаляли при пониженном давлении (100 - 200 мм рт.ст.) и температуре (45 - 50oC). Метанол испаряли до тех пор, пока вес концентрата не достиг приблизительно 313 г. Пастообразный осадок перемешивали в течение четырех часов. pH раствора регулировали до 6,0 и затем осадок охлаждали в течение 1,5 часов при 0 - 5oC. Нужный продукт отфильтровывали и три раза промывали 50 мл дистиллированной воды. Затем продукт сушили. Нужный моногидратный продукт выделяли в виде белого гранулированного твердого вещества 21,3 г с выходом 92%.
Т.пл. 178 - 180oC (разлож.)
1H-ЯМР (300 МГц, ДМСО) δ 0,64 (д, 3H, J = 6,9 Гц), 1,19 (с, 3H), 1,51 (д, 1H, J = 13,1 Гц), 1,97 - 2,00 (м, 1H), 2,11 (тд, 1H, J = 3,6 Гц, 12,7 Гц), 2,34 - 2,95 (м, 9H), 6,54 (д, 1H, J = 8,1 Гц), 6,66 (м, 2H), 7,06 (т, 1H, J = 7,9 Гц), 7,14 - 7,28 (м, 5H), 9,22 (уш.с., 1H).
13C ЯМР (75,5 МГц, ДМСО) δ 15,5, 26,9, 29,5, 35,2, 37,5, 37,7, 42,7, 49,7, 53,7, 58,8, 112,2, 112,3, 115,9, 126,0, 128,2, 128,7, 128,9, 139,4, 151,2, 157,1, 175,1.
УФ (MeOH) λmax 203, ε 17,860; 275, ε 2356.
МС (FD) m/z 368.
ИКС (KBr) 3360, 3272, 2967, 1622, 1585, 1363, 844 см-1.
[α]
Анализ C23H31NO4:
Рассчитано: C 71,66, H 8,10, N 3,63
Найдено: C 72,29, H 8,10, N 3,71.
Пример 4
(2S, 3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил] метил]-1-оксо-3-фенилпропил] -амино] уксусная кислота, 2-метилпропиловый эфир полуторомалатная соль (1:1,5)
Соединение примера 1 (2,5 г, 5,2 ммол, 1 экв) растворялось в 50 мл этилацетата. К смеси добавлялась L-яблочная кислота (1,03 г, 7,8 ммол, 1,5 экв). После растворения L-яблочной кислоты при перемешивании раствор нагревали до 70oC и добавляли 4 мл ацетона. Раствор кристаллизовался. Продукт выделяли фильтрацией. Фильтрат в виде спекшегося осадка промывали этилацетатом. Соль сушили до остаточного уровня этилацетата 1%. Указанное в заглавии соединение выделяли в виде белого кристаллического вещества. Образец анализировали с применением метода диффракции рентгеновских лучей.
Т.пл. 94 - 95oC.
ИКС (KBr) 3346,92, 2972,68, 1741,94, 1601,12 см-1;
1H-ЯМР (300 МГц, ДМСО-d6) δ 9,70 (уш.с., 1H), 8,47 (т, 1H, J = 1,9 Гц), 7,27 - 7,13 (м, 4H), 7,06 (т, 1H, J = 7,9 Гц), 6,67 (д, 1H, J = 8,0 Гц), 6,63 (с, 1H), 6,53 (дд, 1H, J = 8 Гц, J = 1,7 Гц), 4,18 (т, 1,5H, J = 5,8 Гц), 3,82 - 3,78 (м, 3H), 3,33 - 1,8 (м, 16H), 1,48 (уш.д., 1H, J = 13,0 Гц), 1,18 (с, 3H), 0,85 (д, 6H, J = 6,7 Гц), 0,64 (д, 3H, J = 6,9 Гц);
13C ЯМР (75,4 МГц, ДМСО-d6) δ 175,63, 175,42, 171,44, 158,66, 138,63, 138,60, 130,50, 130,23, 129,66, 128,02, 114,07, 114,05, 114,01, 113,94;
МС (FD) m/z 481 (M+);
УФ (MeOH) 272,8 нм (e = 1797), 202,4 нм ( ε = 20576);
Анализ C70H98N4O23:
Рассчитано: C 61,65, H 7,38, N 4,10, O 26,98
Найдено: C 61,40, H 7,23, N 4,1, O 26,66
Пример 5
(2S, 3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил] метил]-1-оксо-2-фенилпропил]амино]уксусная кислота, дигидрат
Раствор соединения примера 1 (12,5 г, 0,026 мол, 1,0 экв) в 315 мл 3А этанола загружался в круглодонную колбу. К смеси добавлялась вода (74,0 мл). По каплям в течение 10-15 минут добавлялся водный раствор гидроокиси натрия (1,0 М), (0,77 мол,3,0 экв) при температуре 25-30oC. Раствор перемешивался и затем фильтровался. pH раствора доводилось с 12,5 до 6,0 добавлением концентрированной хлористоводородной кислоты. Раствор затравливали и в течение 10-15 минут начинала осаждаться (2S,3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил] метил]-1-оксо-3-фенилпропил]амино]уксусная кислота. Кристаллизация осуществлялась при перемешивании в течение двух часов при 25oC и затем (2S,3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил] метил] -1-оксо-3-фенилпропил] амино] уксусная кислота фильтровалась при применении легкого отсасывания к влажному осадку. Кристаллы разбавлялись 60 мл воды и отфильтровывались при отсасывании до образования твердого осадка. Кристаллы высушивались в течение ночи (16 часов) на воздухе при 25oC и относительной влажности 33% при прокачивании воздуха через продукт на фильтровальной воронке и легком отсасывании. Озаглавленный продукт выделялся с 85%-ным выходом (10,2 г) из ( α S,3R,4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидин пропионовой кислоты моногидрата в виде белого кристаллического вещества с четкой точкой плавления при 208oC. Образец анализировался с применением метода дифракции рентгеновских лучей.
ИКС (KBr) 3419, 3204, 3028, 1684, 1591 см-1;
1H ЯМР (300 МГц, ДМСО-d6) δ 9,18 (уш.с, 1H), 8,34 (т, 1H, J = 5,5 Гц), 7,26 - 7,12 (м, 6H), 7,05 (т, 1H, J = 7,9 Гц), 6,67 (д, 1H, J = 8,0 Гц), 6,63 (с, 1H), 6,52 (дд, 1H, J = 8,0 Гц, J = 1,8 Гц), 3,65 (д, 2H, J = 5,7 Гц), 2,89 - 2,10 (м, 14H), 1,91 (уш.д, 1H, J = 6,7 Гц), 1,18 (с, 3H), 0,64 (д, 3H, J = 6,9 Гц),
13C ЯМР (75,4 МГц, ДМСО-d6) δ 173,54, 71,30, 157,05, 151,28, 139,83, 148,83, 128,73, 128,05, 125,82, 115,97, 112,14, 59,62, 54,59, 49,92, 43,75, 41,12, 39,95, 39,67, 39,39, 39,12, 38,84, 37,80, 37,73 35,42, 29,68, 27,04, 15,54;
МС (FD) m/z (425 (M+ - 2H2O): УФ (MeOH) 275,0
( ε = 2246), 202,6 ( ε = 22709,4);
[α]
Анализ C25H36N2O6:
Рассчитано: C 65,20, H 7,88, N 6,08, O 20,84
Найдено: C 64,96, H 7,74, N 6,10, O 20,82,
Пример 6
(2S, 3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1- пиперидинил]метил]-1-оксо-3-фенилпропил]амино]уксусная кислота, дигидрат
Этанол (2400 мл, 3А) и соединение примера 4 (146 г с 5% EtOAc, 138,7 г чистого вещества (0,203 мол, 1,0 экв., 0,085 мольн.) помещались в круглодонную колбу. Добавлялся 1,0 М водный раствор гидроокиси натрия (1200 мл, 1,2 мол., 5,9 экв) - по каплям в течение 20 минут при 25-30oC. Раствор перемешивался, затем фильтровался. pH раствора доводилось с 12,96 до 6,00 добавлением концентрированной хлористоводородной кислоты. Раствор затравляли и через 10-15 минут начинала осаждаться (2S,3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1- пиперидинил] метил] -1-оксо-3-фенилпропил] -амино] уксусная кислота. Кристаллизация происходила при перемешивании в течение двух часов. Пастообразный осадок охлаждался до 0oC и перемешивался. Продукт в виде (2S, 3R, 4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1- пиперидинил]метил]-1-оксо-3-фенилпропил]амино]уксусной кислоты отфильтровывался при легком отсасывании влажного осадка. Кристаллы соединялись с 500 мл воды при 25oC при перемешивании, затем следовало легкое отсасывание, промывание 500 мл воды и фильтрование с отсасыванием до твердого осадка. Кристаллы сушились до дигидрата на воздухе при 35%-ной относительной влажности при 25oC и продувании воздуха через продукт на фильтровальной воронке при слабом отсасывании. Озаглавленное соединение выделялось с выходом > 93% (88 г) в виде белых кристаллов с четкой точкой плавления при 208oC.
Анализ C25H36N2O6;
Рассчитано: C 65,20, H 7,88, N 6,08, O 20,84
Найдено: C 65,38, H 7,87, N 6,25, O 20,90
Пример 7
(2S, 3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1- пиперидинил]метил]-1-оксо-3-фенилпропил] -амино] уксусная кислота, 2-метилпропиловый эфир гидрохлорид ацетон моносольват
6 г образца соединения примера 1 растворяли в 60,0 мл сухого ацетона. Порция в 0,45 г (0,98 экв) газообразного хлористого водорода, растворенная в 30 мл сухого ацетона, по каплям при 25oC добавлялась к первому раствору. Газообразный HCl в ацетоне добавлялся по каплям, пока pH не достигало 3. Когда pH раствора достигло 3,0, добавлялась вторая аликвотная часть 1,0 мл соединения примера 1 при такой же концентрации, как в исходном растворе. Образовался осадок. Реакционная смесь перемешивалась при 25oC в течение ночи, затем охлаждалась до 0oC. Затем перемешивалась при 0oC еще два часа. Желаемый продукт (2S,3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1- пиперидинил] метил] -1-оксо-3-фенилпропил]амино]уксусная кислота, 2-метилпропиловый эфир - гидрохлоридная соль - отфильтровывался фильтрованием под давлением с использованием азота. (2S,3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1- пиперидинил] метил] -1-оксо-3-фенилпропил] амино]уксусная кислота, 2-метилпропиловый эфир - гидрохлоридная соль - сушилась потоком азота над фильтратом с образованием моносольвата ацетона. Ацетоновый моносольват характеризовался с помощью капиллярного газохроматографического анализа, который выявил 9,3 - 9,97% (по весу) ацетона (теоретическое значение 10 процентов). Продукт характеризовался удалением молекулы ацетона из сольвата.
Гидрохлорид ацетонового моносольвата сушился далее в вакуумной печи в течение 2-3 дней при 50oC. Образование гидрохлорид моногридрата было затронуто распространением кристаллов на большую поверхность при 25oC при 40%-ной относительной влажности в течение 2 дней.
Выход составлял > 85% с чистотой около 99,3% по оценке ВЭЖХ с обращенными фазами.
Т.пл. 70 - 75oC;
ИКС (KBr) 3217,7, 3063,4, 2965,0, 1749,7, 1671,5
1H ЯМР (300 МГц, ДМСО-d6)  9,45 (уш.с, 1H), 9,37 (с, 1H), 8,94 (т, 0,85H, J = 1,5 Гц), 8,92 (т, 0,15H, J = 1,5 Гц, 7,28 - 7,20 (м, H), 7,09 (т, 1H, J = 7,8 Гц), 6,67 - 6,65 (м, 3H), 3,83 - 3,76 (м, 4H), 3,47 - 3,10 (м, 5H), 2,83 (д, кв., 2H, J = 18,0 Гц, J = 5,5 Гц, J = 2,0 Гц), 2,7 - 2,0 (м, 5H), 1,82 (септ, 1H, J = 6,7 Гц), 1,70 (д, 1H, J = 12,0 Гц), 1,29 (с, 0,85H), 1,24 (с, 0,15H), 0,99 (д, 0,45H, J = 7,4 Гц), 0,85 (д, 6H, J = 6,6 Гц), 0,71 (д, 2,55H, J = 7,3 Гц);
9,45 (уш.с, 1H), 9,37 (с, 1H), 8,94 (т, 0,85H, J = 1,5 Гц), 8,92 (т, 0,15H, J = 1,5 Гц, 7,28 - 7,20 (м, H), 7,09 (т, 1H, J = 7,8 Гц), 6,67 - 6,65 (м, 3H), 3,83 - 3,76 (м, 4H), 3,47 - 3,10 (м, 5H), 2,83 (д, кв., 2H, J = 18,0 Гц, J = 5,5 Гц, J = 2,0 Гц), 2,7 - 2,0 (м, 5H), 1,82 (септ, 1H, J = 6,7 Гц), 1,70 (д, 1H, J = 12,0 Гц), 1,29 (с, 0,85H), 1,24 (с, 0,15H), 0,99 (д, 0,45H, J = 7,4 Гц), 0,85 (д, 6H, J = 6,6 Гц), 0,71 (д, 2,55H, J = 7,3 Гц);
13C ЯМР (75,4 Гц, ДМСО-d6) δ 172,7, 169,8, 157,4, 149,4, 129,3, 128,3, 121,6, 118,6, 115,7, 112,9, 112,3, 53,9, 57,1, 70,2, 48,1, 46,4, 40,8, 37,3, 36,9, 27,3, 27,0, 26,5, 18,8, 15,1;
УФ (MeOH) 274, ( ε = 2738), 202,2 ( ε = 28413);
МС (FD) 481 (M+-HCl - H2O)
Анализ C29H41N2O4-H2O
Рассчитано: C 65,09, H 8,10, N 5,23, O 14,95, Cl 6,63
Найдено: C 66,06, H 7,92, N 5,27, O 15,19, Cl 6,92
Пример 8
(αS, 3R, 4R)-4-(3-гидроксифенил)-3,4-диметил- α -(фенилметил)-1-пиперидинпропионовая кислота, этиловый эфир гидрохлорид
Образец (+)-3-(3,4-диметил-4-пиперидинил)фенола (50,0 г, 243,5 ммол, 1 эквивалент) помещался в круглодонную колбу. Добавлялись тетрагидрофуран (1 Л) и этилацетат (33,0 мл, 304,4 ммол, 1,25 эквивалентов) и гетерогенная реакционная смесь перемешивалась в течение нескольких дней при комнатной температуре. Реакционная смесь фильтровалась через диатомовую землю и прозрачный раствор выделял вязкое янтарное масло весом 75,0 г. Порция аминоэфира (1,16 г, 3,80 ммол, 1,0 эквивалент) растворялась в 10 мл тетрагидрофурана (ТГФ) и добавлялась при -75oC к раствору лития диизопропиламида (3,90 мл, 7,80 ммол, 2,05 эквивалентов) в ТГФ (20 мл). Дополнительно выдерживали смесь приблизительно пять минут. Затем пастообразный осадок перемешивался при -70oC в течение 15 минут и добавлялся бензилбромид (0,47 мл, 3,99 ммол, 1,05 эквивалентов). Реакционной смеси дали нагреться до -25 - -30oC и перемешивали ее в течение 3 часов. Затем реакционная смесь быстро заливается 10 мл насыщенного раствора хлорида аммония, 10 мл H2O и 20 мл этилацетата. Водный слой отделялся.
Органический слой промывали насыщенным солевым раствором. Затем органический слой сушили над MgSO4. Смесь фильтровали и образующийся раствор подвергали выпариванию на ротационном испарителе с получением желтого масла весом 1,80 г. Смесь продукта и исходного вещества затем подвергали флеш-хроматографированию смесью этилацетата и гексана с выделением 1,07 г (71%) этилового эфира.
Приведенный выше этиловый эфир (14,8 г, 37,4 ммол) растворяли затем в 150 мл этанола. В раствор пропускали безводный хлористый водород, а этанол удаляли испарением на ротационном испарителе. Затем твердое вещество растирали в порошок с 50 мл этилацетата и фильтровали. Твердое вещество сушили в течение ночи при 30oC с выделением 12,25 г гидрохлорида (76%, точка плавления 179-181oC). Соотношение диастереомеров составляло 49% α S,3R,4R (нужный диастереомер) и 51% α R,3R,4R (ненужный диастереомер).
Гидрохлоридная соль (1,02 г) смешивалась с 5 мл этанола и нагревалась с обратным холодильником в течение 3 часов. Смеси давали охлаждаться до комнатной температуры и перемешивали. Затем смесь перемешивали в течение ночи при 0oC и отфильтровывали. Соль сушили в течение ночи при 40oC. Выделяли белое твердое вещество весом 0,42 г (47%). Соотношения диастереомеров было: 76% α S, 3R, 4R и 24% α R, 3R, 4R.
Гидрохлоридная соль (0,42 г) смешивалась с 6 мл этанола и нагревалась с обратным холодильником в течение 2 часов, после чего ее охлаждали до комнатной температуры и перемешивали. Пастообразный осадок охлаждали до 0oC в течение ночи и фильтровали. Твердое вещество сушили с получением палочки соли весом 0,31 г (74%). Соотношение диастереомеров составляло: 92% α S, 3R, 4R и 8% α R, 3R, 4R.
Часть соли (0,24 г) смешивали в третий раз с 2,5 мл этанола. Смесь нагревали в течение трех часов с обратным холодильником, затем давали ей охладиться до комнатной температуры и перемешивали. Пастообразный осадок охлаждали до 0oC в течение 1 часа и затем фильтровали. Твердое вещество сушили. Диастереомерно чистая (98% α S, 3R, 4R) гидрохлоридная соль этилового эфира имела вес 0,23 г (96%).
Далее приводятся более подробные результаты биологических испытаний согласно методикам, приведенным выше, для соединения формулы 5 - дигидрата LY 246736 - ключевого соединения настоящего изобретения, а далее - для одного из эфиров формулы 4, а именно для 2-метилпропилового эфира, который обозначен в этих испытаниях как соединение 11.
Дигидрат LY246736 Антагонист опиоидного рецептора μ
Дигидрат (+)-2-[2(S)-бензил-3-[4(R)-(3-гидроксифенил)- 3(R),4-диметилпиперидин-1-ил]пропионамидо]уксусной кислоты
Соединение формулы 5
C25H32N2O4 • 2H2O Мол. масса: 460,58
Белый порошок, т.пл. 210-3oC, [α]
Фармакологическое действие
Аффинности LY246736 в отношении опиоидных рецепторов определяли при помощи исследований связывания радиолиганда и они показаны в таблице III. Эти величины были получены в Lilli Research Laboratories и в Stanford Research Institute
с использованием различных радиоактивно меченных лигандов. LY246736 имеет очень высокую аффинность в отношении опиоидного рецептора μ (Ki < 1 нМ), более низкую аффинность в отношении рецептора δ и самую низкую аффинность в отношении рецептора κ. Он обладает высокой избирательностью в отношении связывания μ в противоположность связыванию κ. Связывание с рецепторами κ может быть дополнительно подразделено на связывание с сайтами κ1 и κ2. LY246736 имеет относительно низкую аффинность в отношении сайта κ1, но даже еще более низкую аффинность в отношении сайта κ2.
LY246736 оценивали также на аффинность при различных неопиоидных рецепторах с использованием гомогенатов мозга крыс. Не была обнаружена существенная аффинность (Ki < 100 мкМ) в отношении адренергических ( α1, и α2, и β ), допаминергических (D1 и D2) рецепторов, бензодиазепина, 5HT2, гистаминного-1, GABA или мускариновых рецепторов (см. табл. 3, 4, 5).
Антагонистические в отношении опиоидного рецептора активности LY246736 определяли измерением его способности ингибировать эффекты избирательных опиоидных агонистов на подвздошной кишке морской свинки (рецепторы μ и κ) и семявыводящих протоках (vas deferens) мыши (рецептор δ ). Эти результаты представлены в таблице IV. Как было предсказано на основании исследований связывания радиолиганда, LY246736 имеет наивысшую антагонистическую активность при рецепторе μ . Он приблизительно в 100 раз менее активен при рецепторе κ и в 10 раз менее активен в качестве антагониста рецептора δ . LY246736 не имел доступных для измерения опиоидных агонистических эффектов ни на подвздошной кишке морской свинки, ни на vas deferens мыши. Опиоидные антагонистические активности LY246736 в этих периферических тканях очень хорошо коррелируют с аффинностями рецепторов, определенными с применением гомогенатов мозга.
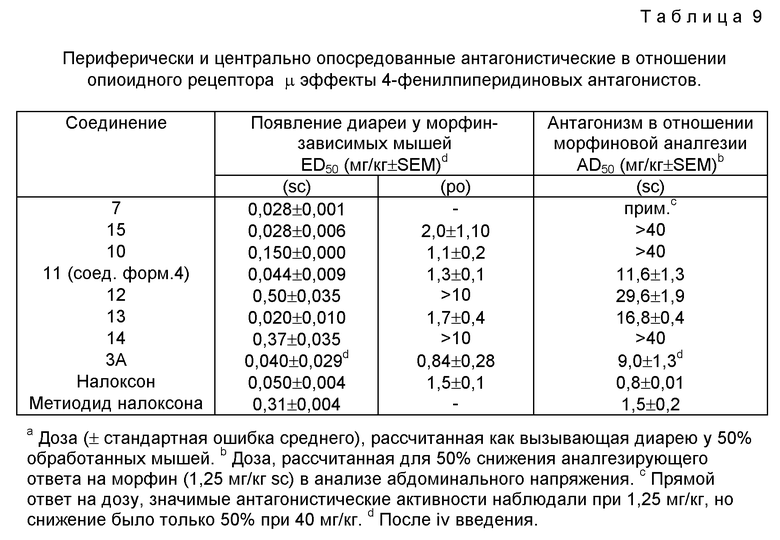
LY246736 оценивали в мышах и крысах на его периферически и центрально медиируемые антагонистические активности в отношении опиоидного рецептора μ . Для измерения периферической антагонистической активности определяли способность соединения вызывать диарею в морфинзависимых животных. В мышах антагонистическую активность в отношении рецептора μ центральной нервной системы измеряли определением способности соединения действовать антагонистически на индуцируемую морфином аналгезию. Таблица V суммирует результаты этих анализов. LY246736 вызывает диарею в морфинзависимых мышах с ED50 0,04 мг/кг после внутривенного введения, и только при относительно очень высоких дозах он пересекает гематоэнцефалический барьер и действует антагонистически на индуцированную морфином аналгезию. Его периферическая избирательность против его центральной избирательности в 200 раз выше согласно этим измерениям. Эта степень избирательности является замечательной при сравнении ее со степенью избирательности метиодида налоксон (таблица V); согласно этим измерениям метиодид налоксона имеет избирательность ~ 5. LY246736 также высокоактивен в вызывании диареи в морфинзависимых мышах и крысах после перорального введения. Рассчитанная ED50-доза LY246736 для вызывания диареи в обоих видах была менее 1 мг/кг.
Действие LY246736 на индуцированное морфином ингибирование прохождения по пищеварительному тракту угольной муки показало, что подкожное введение морфина вызывало зависимое от дозы ингибирование прохождения по пищеварительному тракту, причем доза 3 мг/кг уменьшала прохождение по пищеварительному тракту до ~25% от контроля. Пероральное введение LY246736 противодействовало ингибирующему действию морфина на прохождение по пищеварительному тракту зависимым от дозы образом. Эффекты 3 мг/кг морфина почти полностью обращались (снимались) 3 мг/кг LY246736. Было рассчитано, что ED50 LY246736 для обращения (снятия) эффектов 3 мг/кг морфина была 1,1 мг/кг р.о.
Изучение наступления и продолжительности антагонистической активности 3 мг/кг р. о. LY246736 против морфина (1 мг/кг, s.c.), показало, что LY246736 обнаруживает быстрое возникновение активности, давая почти максимальную активность через 30 минут после введения, и был полностью эффективен в предотвращении индуцируемого морфином ингибирования прохождения по пищеварительному тракту в течение по меньшей мере 8 часов после введения.
У собак после внутривенного введения дигидрата LY246736 наблюдали зависимое от дозы увеличение максимальных концентраций LY246736 в плазме и AUC плазмы (таблица VI). Было определено, что средний период полувыведения из плазмы был 10 минут. Пероральные дозы дигидрата LY246736 (до 100 мг/кг) давали низкие концентрации в плазме, приводя к пероральной системной биодоступности приблизительно 0,03%.
Внутривенные исследования на кроликах на протяжении периода дозирования 13 дней показали фармакокинетику, сходную с фармакокинетикой, полученной на собаках. Величины полувыведения находились в диапазоне 7-9 минут и не изменялись на протяжении периода дозирования, также не было накопления лекарственного средства в компартменте плазмы.
Самцы и самки крыс, получавшие пероральную дозу 200 мг/кг дигидрата LY246736, давали среднюю максимальную концентрацию в плазме 20,5 нг/мл при Tмакс 1 час. Крысы, получавшие перорально 10 мг/кг дигидрата 14C-LY246736, имели входные концентрации радиоактивности в плазме приблизительно 140 нг-эквивалентов 14C/мл после введения дозы. Системные концентрации радиоактивности в плазме были ниже определяемого количественно предела 10 нг-эквивалентов 14C/мл.
Исследования баланса массы у крыс, получавших одну пероральную или внутривенную дозу дигидрата 14C-LY246736 10 мг/кг, показали, что приблизительно 0,4% и 22% введенного радиоактивного углерода экскретировались в мочу соответственно. Исследования билиарной секреции после перорального или внутривенного введения 14C-LY246736 показали, что приблизительно 15% введенной дозы секретировалось в желчь. Исследование желчи показало присутствие LY246736, а также основных полярных метаболитов.
Авторадиографическое исследование всего тела проводили на крысах, получавших перорально 10 мг/кг дигидрата 14C-LY246736. Это исследование показало, что основная часть радиоактивности была локализована в желудочно-кишечном тракте. Радиоактивный углерод присутствовал в стенке желудочно-кишечного тракта. Не наблюдали доказательства системного распределения в кровь или другие ткани.
В целом, эти результаты показывают, что после перорального введения системное поглощения дигидрата LY246736 является слабым во всех видах исследованных до настоящего времени животных, приводящим к появлению низких и вариабельных концентраций исходного лекарственного средства. Однако результаты из исследований билиарной секреции на крысах показывают как поглощение, так и метаболизацию LY246736. Низкая пероральная биодоступность LY246736 является, возможно, результатом слабого всасывания, высокой первичной печеночной экстракцией и метаболизмом.
Токсикология
Исследования с одной дозой с применением перорального введения проводили на крысах и мышах (500 мг/кг) и исследования с внутривенным введением одной дозы (20 мг/кг) проводили на крысе. LY246736 не обнаружил летальности или острой токсичности в этих исследованиях.
Внутривенные и пероральные исследования подострой токсичности (30 дней) проводили на собаке и крысе. Собакам вводили внутривенно дозы до 2 мг/кг. Максимальные уровни LY246736 в плазме были пропорциональны дозе и превышали 4000 нг/мл при 2 мг/кг. Пероральные дозы LY246736 вводили крысам (до 200 мг/кг) и собакам (до 100 мг/кг) (см. табл. 6).
Ни в исследованиях с внутривенным введением, ни в исследованиях с пероральным введением не обнаружили важных токсикологических открытий.
LY246736 оценивали также в серии тестов генетической токсикологии in vitro и in vivo. Ни в одном из этих тестов не обнаружили положительных результатов. Таким образом, можно считать, что LY246736 не будет опасен для людей с точки зрения генетической токсикологии.
Обнаружение сильнодействующего периферически избирательного опиоидного антагониста транс-3,4-диметил-4-(3-гидроксифенил)пиперидина для лечения нарушений перистальтики желудочно-кишечного тракта (схему 3 см. в конце описания).
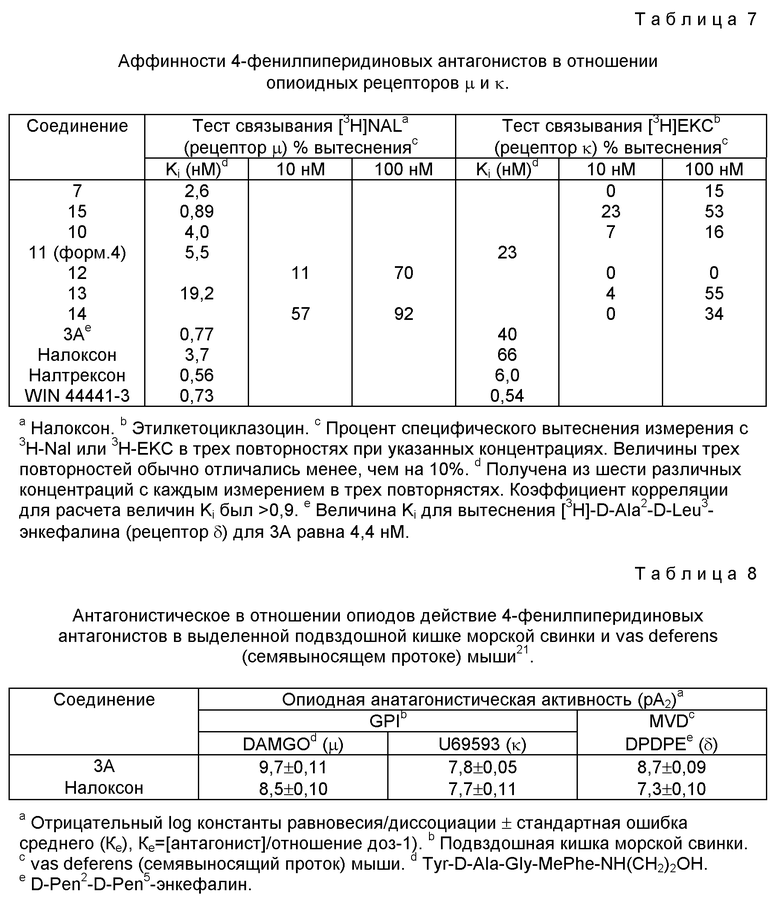
Соединение 3A первоначально было оценено в виде рацемической, диастереомерной смеси 15 (схема 3). В попытках увеличения пероральной активности были получены несколько эфиров 15. Из них 10 был наиболее обещающим. Были синтезированы четыре изомера 10, и наивысшая активность in vitro была обнаружена с (+)-3R, 4R, 2S-изомером 11 (соединение формулы 4). Основательное сравнение активностей 11 и 3A не выявило значимых различий в их антагонистических в отношении μ активностей после перорального введения (см. табл. 7, 8).
Фармакология
Аффинности в отношении опиоидных рецепторов определяли при помощи тестов связывания радиолигандов.
Для соединения 3A аффинности в отношении опиоидного рецептора определяли также в выделенных подзвдошной кишке морской свинки и vas deferens мыши путем измерения их способности ингибировать эффекты рецепторселективных агонистов.
Опосредованную рецептором μ опиоидную антагонистическую активность в центральной нервной системе определяли при помощи мышиного теста абдоминального напряжения. Измеряли способность тестируемого соединения обращать полностью эффективную дозу морфина (1,25 мг/кг sc).
Периферическую антагонистическую в отношении μ активность оценивали в морфинзависимых мышах путем измерения возникновения диареи. Этот тест был разработан специально для этого исследовательского проекта и детали его даны в табл. 9.
Измерение периферической опиодионй антагонистической активности: Появление диареи у морфинзависимых мышей. Колонию приблизительно 50 самцов мышей CF-A (Charles River, Portage, MI) содержали с доступом к морфину (1 мг/мл) в 0,01 М водном растворе Na-сахарина в качестве единственного источника жидкости. В этих экспериментах мышей выдерживали минимально в течение 10 дней на растворе морфина, и среднее потребление должно было быть 3 г раствора на мышь в день в течение последовательных 3 дней перед использованием животных для целей тестирования. При таких условиях мыши становились физически зависимыми от морфина. Для тестирования на периферическую опиодиную антагонистическую активность пять морфинзависимых мышей удаляли из свободного доступа к раствору морфина на 45 минут, инъецировали тест-соединением и помещали в цилиндрические камеры для наблюдения. При 30 минутах после инъекции мышей оценивали в баллах на присутствие или отсутствие диареи. Как правило, тестирование начинали с достаточно высокой дозы и затем тестировали более низкие дозы пока не переставала появляться диарея. После тестирования мышей возвращали в клетки их колонии с доступом к раствору морфина. Результаты рассчитывали в виде "процента мышей при каждой дозе с диареей" и по меньшей мере пять мышей тестировали для каждой дозы соединения. ED50 определяли как дозу антагониста, которая вызывала появление диареи в 50% тестированных мышей. ED50 и стандартную ошибку средних величин рассчитывали по способу, разработанному в Eli Lilly, который использует построение нелинейных кривых с программным обеспечением JMP.

Описывается новый кристаллический сложный эфир (αS, 3R , 4R )-4-(3-гидроксифенил)-3,4-диметил-α-(фенилметил)-1-пиперидин пропионовой кислоты формулы (2), где R - C1-C6-алкил, Z - группа, выбранная из гидрохлорида, гидробромида, сукцината и (+)-дибензоилтартрата, который может использоваться для связывания периферического опиоидного рецептора у пациента. Соединение используется для лечения таких состояний, как синдром раздражимости кишечника, идиопатический запор и диспепсия неязвенного происхождения. 5 с. и 7 з.п. ф-лы, 9 табл. 
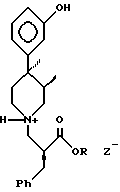
где R является C1 - C6-алкилом;
Z является группой, выбранной из гидрохлорида, гидробромида, сукцината и (+)-дибензоилтартрата.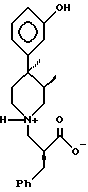
отличающийся тем, что соединение формулы 2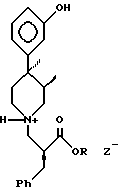
где R является C1 - C6-алкилом;
Z является группой, выбранной из гидрохлорида или гидробромида,
подвергают гидролизу и выделяют целевой продукт кристаллизацией из растворителя, состоящего из 50 - 75% низшего спирта и 50 - 25% воды по весу.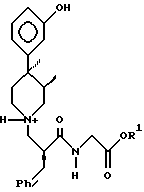
где R1 является C1 - C6-алкилом,
и соединение представляет собой соль, такую, как гидрохлорид ацетон моносольват или полуторомалат (3 : 2).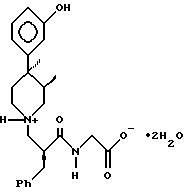
9. Кристаллическое дигидратное соединение по п.8, представляющее собой соединение, состоящее по крайней мере из 97% (2S, 3R, 4R)дигидрата.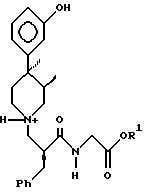
где R1 представляет собой C1 - C6-алкил.
| US 5250542, 05.10.1993 | |||
| US 4891379, 1990 | |||
| Способ изготовления керамико-металлического материала | 1973 |
|
SU506468A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1984, ч.2, с.10, 20, 21. | |||

