Изобретение относится к области лечения злокачественных опухолей. В частности, настоящее изобретение относится к лечению раковых опухолей комбинациями химиотерапевтических агентов и оксидов 1,2,4-бензотриазина.
Большинство обычно применяемых противораковых лекарств являются более цитотоксичными по отношению к нормально снабжаемым кислородом опухолевым клеткам по сравнению с гипоксичными опухолевыми клетками. Широко известна также резистентность гипоксичных клеток к облучению.
Следовательно, гипоксия опухолей и, как результат, их резистентность к лечению имеют большое значение в проблеме лечения рака.
Солидные раковые опухоли содержат как адекватно оксигенированные клетки, так и различные количества неадекватно оксигенированных или гипоксичных клеток. Гипоксию обычно наблюдают в тех случаях, когда опухолевые клетки расположены вдали от кровеносных сосудов.
Такие клетки имеют также тенденцию к замедленной пролиферации. Считается, что резистентность гипоксичных клеток к противоопухолевым препаратам происходит вследствие неадекватного поглощения ими лекарственного агента, а также вследствие их медленного роста и удаленности от кровеносных сосудов, приносящих лекарственный агент, однако это явление еще не получило исчерпывающего объяснения.
Таким образом, относительная доля гипоксичных клеток в опухоли может иметь огромное значение для результативности лечения. Резистентные гипоксичные клетки, которые выживают после облучения или лекарственного лечения, могут быть реоксигенированы, восстанавливая, таким образом, чувствительность опухоли к дальнейшему лечению.
Тем не менее, вместо того чтобы рассчитывать на такие неопределенные явления, желательно разрабатывать такие способы лечения рака, при которых раковые клетки, включая гипоксичные, инактивируются или погибают непосредственно в ходе применяемой терапии.
Патент США 5175287 раскрывает применение оксидов 1,2,4-бензотриазина в сочетании с облучением для лечения опухолей.
Оксиды 1,2,4-бензотриазина сенсибилизируют опухолевые клетки к действию радиации, вследствие чего они становятся более восприимчивыми к этому способу лечения.
Holden и др. (1992) в статье "Enhancement of Alrylating Agent Activity by SR-4233 in the FSaLLC Murine Fibrosarcoma" JNCI 84: 187-193 раскрывают применение вещества SR-4233, известного также как тирапазамин, в сочетании с противоопухолевым алкилирующим агентом.
Каждый из четырех противоопухолевых алкилирующих агентов, цисплатин, циклофосфамид, кармустин и мелфалан, применялся для проверки способности тирапазамина преодолевать резистентность гипоксичных опухолевых клеток к противоопухолевым алкилирующим агентам.
Тирапазамин испытывали отдельно и в сочетании с различными количествами каждого из этих противоопухолевых алкилирующих агентов.
В случаях, когда тирапазамин применяли непосредственно перед воздействием разовой дозы циклофосфамида, кармустина или мелфалана, наблюдали заметное усиление действия этой дозы, ведущее к синергистическим цитотоксическим воздействиям на опухолевые клетки.
В случаях, когда тирапазамин применяли непосредственного перед введением разовой дозы цисплатина, усиление дозы приводило только к дополнительному эффекту, за исключением наивысших доз цисплатина.
Нитроимидазольные цититоксические агенты комбинировали с различными противораковыми лекарствами и обнаружили, что терапевтическое усиление эффекта достигалось, когда эти агенты применялись в сочетании с различными противораковыми лекарствами, в частности с алкилирующими агентами, циклофосфамидом и мелфаланом, и нитрозомочевинами, BCNU и CCNU.
Однако позже обнаружили, что дополнительный терапевтический эффект не являлся следствием избирательного действия нитроимидазолов на гипоксичные клетки, приводящего к гибели последних, а возникал благодаря механизму, усиливающему образование поперечных сшивок ДНК метаболитами нитроимидазолов, индуцированное алкилирующим агентом (Murray и др., (1983) Br. J.Cancer 47: 195-203).
В настоящем документе описан способ лечения раковых опухолей, в частности твердых, который включает введение млекопитающим, нуждающимся в таком лечении, эффективного количества соединения формулы I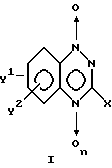
где X представляет водород; (1-4C) углеводородный радикал; (1-4C) углеводородный радикал, замещенный OH, NH2, NHR, NRR, (1- 4C) алкокси или галогеном; галоген; OH; (1-4C) алкокси, NH2; NHR или NRR; где каждый R независимо выбирают из следующих: низший алкил (1-4C); низший ацил (1-4C); низший алкил (1-4C) и низший ацил (1-4C), замещенный ОН, NH2, вторичными (1-4C) алкил и третичными (1-4C) диалкил аминогруппами, (1-4C) алкокси или галогеном; и когда X представляет NRR, оба R могут быть связаны между собой непосредственно или через кислородный мостик, образуя морфолиновое, пирролидиновое или пиперидиновое кольцо;
n = 0 или 1; и
Y1 и Y2 независимо друг от друга представляют водород; нитрогруппу, галоген; (1-4C) углеводородный радикал, включая циклические, ациклические и ненасыщенные углеводородные радикалы, необязательно замещенные одним или двумя заместителями, выбранными из следующей группы: галоген, гидрокси, эпокси (1-4C) алкокси, (1-4C) алкилтио, первичный амино (NH2), вторичный (1-4C) алкил амино, третичный (1- 4C) диалкил амино, третичный (1-4C) диалкил амино, в котором два алкила связаны вместе, образуя морфолиновое, пирролидиновое или пиперидиновое кольцо, (1-4C) ацилокси, (1- 4C) ациламидо и его тиоаналоги, ацетиламиноалкил (1-4C), карбокси, алкоксикарбонил (1- 4C), карбамил, (1-4C) алкилкарбамил, (1-4C) алкилсульфонил или (1- 4C) алкилфосфонил, где углеводородная цепь необязательно может прерываться одной эфирной (-O-) связью; и где Y1 и Y2 независимо друг от друга представляют морфолино, пирролидино, пиперидино, NH2, NHR', NR'R', O(CO)R', NH(CO)R', O(SO)R', или O(POR')R', в котором R' является (1-4C) углеводородным радикалом, который может быть замещен OH, NH2, вторичным (1-4C) алкил амино, третичным (1-4C) диалкил амино, морфолино, пирролидино, пиперидино, (1-4C) алкокси или галогеном, или фармакологически приемлемых солей названного соединения; и введение млекопитающим спустя примерно от получаса до 24 часов после введения соединения формулы I, определенного выше, эффективного количества химиотерапевтического агента, к которому чувствительна (восприимчива) данная опухоль.
Согласно настоящему изобретению предоставляется способ повышения цитотоксичности химиотерапевтического агента по отношению к твердой опухоли, восприимчивой к лечению названным химиотерапевтическим агентом, который включает введение млекопитающим с такими опухолями примерно от получаса до 24 часов до введения или примерно от 1 до 2 часов после введения химиотерапевтического агента увеличивающего цитотоксичность количества названного соединения формулы I, определенного выше.
Описан также способ лечения раковых опухолей млекопитающих, включающий введение млекопитающим соединения формулы I, определенного выше, спустя один - два часа после введения химиотерапевтического агента.
Было обнаружено, что введение вышеназванного соединения формулы I как до, так и после введения химиотерапевтического агента убивало раковые клетки в значительно большем количестве, чем введение каждого агента по отдельности или обоих агентов одновременно.
Например, когда тирапазамин вводили до 24 часов перед введением цисплатина, обнаружили, что раковых клеток погибало в 10-1000 раз больше, чем в том случае, когда тирапазамин и цисплатин вводили одновременно.
Наибольший синергистический эффект этого сочетания агентов обнаруживали, когда тирапазамин вводили за 2,5 часа до введения цисплатина.
Заявленный способ представляет огромное увеличение противоопухолевой эффективности химиотерапевтического агента (т.е. его цитотоксического действия на опухолевые клетки).
Помимо этого, при испытаниях на системную токсичность цисплатина (азот мочевины в сыворотке крови и острая токсичность) в комбинации при максимальной эффективности по отношению к опухоли обнаружили почти отсутствие усиления или полное отсутствие усиления системной токсичности по сравнению с цисплатином, который вводился один.
Таким образом, большая часть, если не вся, дополнительной гибели опухолевых клеток выражается в терапевтическом улучшении при использовании этого сочетания. Синергистическое взаимодействие тирапазамина и цисплатина также является важным, поскольку большое возрастание гибели опухолевых клеток достигалось при относительно низких дозах цисплатина.
Еще одним аспектом настоящего изобретения является применение соединения формулы I, способного усиливать цитотоксический эффект в отношении раковой опухоли, или его фармакологически приемлемой соли для производства лекарственного препарата, который применяется для лечения млекопитающих и вводится за 0,5 - 24 часа до начала лечения данной опухоли химиотерапевтическим агентом.
Настоящее изобретение более конкретно описывается в приводимых предпочтительных вариантах практического осуществления в последующем описании, которые, однако, не должны истолковываться как ограничительные.
Фиг. 1 графически изображает зависимость относительного количества клоногенных клеток на опухоль в экспериментальных опухолях RIF-I от времени (от -3 до +2 часов) введения тирапазамина по отношению к времени введения цисплатина.
Фиг. 2 графически изображает зависимость относительного количества клоногенных клеток на опухоль в экспериментальных опухолях RIF-I от времени (от -24 до 0 часов) введения тирапазамина по отношению к времени введения цисплатина.
Для способа лечения раковых опухолей млекопитающих, включая раковые опухоли человека, в частности твердые опухоли, термин "чувствительность опухоли к химиотерапевтическому агенту" означает, что названный химиотерапевтический агент способен демонстрировать терапевтический эффект в отношении этой опухоли с помощью любого механизма, такого как гибель опухолевых клеток, уменьшение клеточной пролиферации или уменьшение размера опухоли.
"Эффективное количество" соединения формулы I, упоминаемое здесь, относится к количеству, способному убивать опухолевые клетки, или к количеству, способному убивать опухолевые клетки в сочетании с химиотерапевтическим агентом.
"Эффективное количество" химиотерапевтического агента относится к такому его количеству, которое способно убивать раковые клетки или иным образом оказывать терапевтическое воздействие, например, уменьшая размер опухоли или замедляя рост и пролиферацию опухолевых клеток.
Для способа увеличения цитотоксичности химиотерапевтического агента по отношению к твердой опухоли, чувствительной к этому химиотерапевтическому агенту, термин "количество, усиливающее цитотоксичность" относится к такому количеству соединения формулы I, которое способно увеличивать цитотоксическое воздействие химиотерапевтического агента на клетки.
Предпочтительно количество, усиливающее цитотоксичность, является достаточным для проявления синергистического эффекта, т.е. эффект больше, чем сумма эффектов химиотерапевтического агента и соединения формулы I, когда они применяются по одному.
Количества соединения формулы I, усиливающие цитотоксичность, можно установить испытаниями подобных соединений с химиотерапевтическими агентами (агентом) ин виво и/или ин витро на экспериментальных моделях опухолей, описанных здесь, или на любой другой модели опухоли, известной специалистам.
Количество, усиливающее цитотоксичность, которое определялось на экспериментальных моделях опухолей ин виво и ин витро, затем используется как руководство для определения количеств двух агентов, которые будут применяться для лечения опухолей млекопитающих.
В настоящее время считается, что комбинация бензотриазинового химиотерапевтического агента формулы I, описанного выше, который является цитотоксичным по отношению к гипоксичным раковым клеткам, и химиотерапевтического агента, обладающего наибольшей активностью в отношении нормально оксигенируемых раковых клеток, обеспечивает усиленную или синергистическую гибель опухолевых клеток. Оксиды бензотриазинов формулы I, в частности, требуют более низкую, чем нормальная, концентрацию кислорода для проявления своего действия.
Эта потребность в гипоксии является большим преимуществом, поскольку она обеспечивает основу для опухолеспецифичного взаимодействия между двумя лекарственными агентами.
Обычно нормальные ткани существуют при концентрации кислорода свыше 10-15 мм рт. ст. При этих и более высоких парциальных давлениях кислорода цитотоксический эффект тирапазамина очень мал.
С другой стороны, многие опухоли имеют значительное количество клеток, существующих при концентрациях кислорода ниже 10 мм рт.ст.; при таких парциальных давлениях метаболизм тирапазамина и других бензотриазинов формулы I для цитотоксических видов сильно возрастает.
Применяемый здесь термин "гипоксические опухолевые клетки" относится к опухолевым клеткам, существующим при парциальном давлении кислорода менее 10 мм рт.ст.
Способы, описанные в настоящем документе, являются полезными для лечения раковых опухолей млекопитающих, включая человека, в частности для лечения твердых опухолей, имеющих гипоксичные участки.
Примеры таких опухолей включают, но не исчерпываются этим перечнем, адренокарциномы, глиобластомы (и другие опухоли мозга), опухоли груди, шейки матки, толстой и прямой кишки, эндометрия, желудка, печени, легких (мелкоклеточные и немелкоклеточные), лимфомы (включая не Ходжкина, Беркитта, диффузные гигантоклеточные, фолликулярные и диффузные Ходжкина), меланому (метастатическую), нейробластому, остеосаркому, опухоли яичника, ретинобластому, мягкие тканевые саркомы, опухоли семенника и другие опухоли, которые поддаются химиотерапии.
Таким образом, способы настоящего изобретения могут применяться для лечения раковых опухолей, включая экспериментально индуцированные раковые опухоли, у любого вида млекопитающих, включая человека, наиболее часто используемых лабораторных животных, таких как крысы, мыши, кролики и собаки, приматов, таких как обезьяны, лошадей, кошек и других животных.
Способы, описанные в настоящем описании, можно практиковать с любым типом химиотерапевтических агентов. В любом практическом варианте осуществления настоящего изобретения химиотерапевтический агент нужно подбирать с учетом многих факторов, таких как тип раковой опухоли или эффективность химиотерапевтического агента для лечения именно этого типа опухоли. Химиотерапевтический агент можно выбирать из алкилирующих агентов, антиметаболитов, природных продуктов, гормонов и антагонистов и других типов соединений.
Примеры алкилирующих агентов включают 2-хлорэтиламины, такие как, например, хлорметин, хлорамбуцил, мелфалан, урамустин, манномустин, экстрамустин фосфат, мехлор-таминоксид, циклофосфамид, ифозамид и трифосфамид; алкилирующие агенты с замещенной азиридиновой группой, такие как, например, третамин, тиотепа, триазиквон и митомицин; алкилирующие агенты алкилсульфонатного типа, такие как, например, бусульфан и пипосульфан; алкилирующие производные N-алкил-N-нитрозомочевины, такие как, например, кармустин, ломустин, семустин или стрептозотоцин; алкилирующие агенты типа митобронитола, дакарбазина и прокарбазина и платиновые комплексы, такие как, например, цисплатин и карбоплатин.
Примеры антиметаболитов включают производные фолиевой кислоты, такие как, например, метотрексат, аминоптерин и 3'-дихлорметотрексат; пиримидиновые производные, такие как, например, 5-фторурацил, флоксуридин, тегафур, цитарабин, идоксуридин и флуцитозин; пуриновые производные, такие как, например, меркаптопурин, тиогуанин, азатиоприн, тиамиприн, видарабин, пентостатин и пуромицин.
Примеры природных продуктов включают алкалоиды, такие как, например, винбластин и винкристин; эпиподофилотоксины, такие как, например, этопозид и тенипозид; антибиотики, такие как, например, адримицин, дауномицин, даунорубицин, дактиномицин, доксорубицин, митрамицин, блеомицин и митомицин; ферменты, такие как, например, L-аспарагиназа; модификаторы биологических ответных реакций, такие как, например, альфа-интерферон; камптотецин; таксол и ретиноиды, такие как ретиновая кислота.
Примеры гормонов и антагонистов включают адренокортикоиды, такие как, например, преднизон; прогестины, такие как, например, гидроксипрогестерона ацетат, медроксипрогестерона ацетат и мегестрола ацетат; эстрогены, такие как, например, диэтилстилбестрол и этинилэстрадиол; антиоэстрогены, такие как, например, тамоксифен; андрогены, такие как, например, тестостерона пропионат и флуоксиместрон; антиандрогены, такие как, например, флутамид; аналоги гонадотропин-высвобождающего фактора, такие как, например, лейпролид.
Примеры смешанных агентов включают антрацендионы, такие как, например, митоксантрон; замещенные мочевины, такие как, например, гидроксимочевины; адренокортикальные супрессанты, такие как, например, митотан и аминоглютетимид.
Помимо этого, химиотерапевтический агент может быть иммуносупрессором, таким как, например, циклоспорин, азатиоприн, сульфазалазин, метозален и талидомид.
Химиотерапевтические агенты, полезные для практики настоящего изобретения, являются коммерчески доступными или могут быть приготовлены с помощью стандартных методик. Химиотерапевтический агент можно применять отдельно или в комбинации с другими химиотерапевтическими агентами (одним или более).
Например, для лечения раковых опухолей можно применять сочетание трех различных химиотерапевтических агентов и одного или более соединений формулы I в соответствии со способом настоящего изобретения.
В соединениях формулы I
X представляет водород; незамещенный (1-4C) углеводородный радикал с прямой или разветвленной цепью, такой как метил, этил, втор-бутил или изопропил; галоген; гидрокси; (1-4C) алкокси, такой как метокси, этокси, пропокси и трет-бутокси; первичный амино (NH2); вторичный амино (NHR), в котором R представляет алкил или ацил с 1-4 атомами углерода, такой как метиламино или этиламино; третичный амино (NRR), в котором каждый R представляет алкил или ацил с 1-4 атомами углерода, например диэтиламино и т.п., или два R соединены, образуя морфолиновое, пирролидиновое или пиперидиновое кольцо.
В случае различных алкильных или ацильных R групп последние далее могут быть замещены OH, NH2, вторичным (1- 4C) алкил низшим амино и третичным (1-4C) диалкил амино, морфолино, пирролидино, пиперидино, (1-4C) алкокси или галогеном (фтором, хлором, бромом или йодом).
Углеводородные X группы могут быть в дальнейшем замещены OH, NH2, вторичным алкил амино, третичным диалкил амино, (1-4C) алкокси или галогеном (фтором, хлором, бромом или йодом).
Более предпочтительно, чтобы X представлял водород, первичный амин (NH2); незамещенный (1-4C) углеводородный радикал с прямой или разветвленной цепью; или замещенный (1-4C) углеводородный радикал с прямой или разветвленной цепью,
n = 0 или 1, предпочтительно 1,
Y1 и Y2 независимо друг от друга представляют водород; нитро; галоген (фтор, хлор, бром или йод); или (1-14C) углеводородный радикал.
Когда они являются углеводородными радикалами, Y1 и Y2 могут быть насыщенными и ненасыщенными, циклическими и ациклическими, и необязательно могут прерываться одной эфирной связью.
Так, незамещенными углеводородными формами Y1 или Y2 могут быть, например, метил, этил, н-пропил, втор-бутил, н-гексил, 2-метил-н-пентил, 2-этоксиэтил, 3-(н-пропокси)-н-пропил, 4-метоксибутил, циклогексил, тетрагидрофурфурил, фурфурил, циклогексенил, 3-(н-децилокси)-н-пропил и 4-метилоктил, 4,7-диметилоктил.
Углеводородные Y1 и Y2 группы необязательно могут быть замещены одним или двумя заместителями, выбранными из следующей группы: галоген (любой); гидрокси; эпоски; алкокси (1-4C), такой как, например, метокси, н-пропокси и трет-бутокcи; алкилтио; (1-4C) первичный амино (NH2); морфолино; пирролидино; пиперидино; вторичный амино (NHR'), где R' представляет (1-4C) алкил, такой как метиламин, пропиламин и т.п.; третичный амино (NR'R'); ацилокси и ациламидо, представленные группами R'COO- И R'CONH- соответственно, и их тиольные аналоги, представленные группами R'CSO- и R'CSNH- соответственно; карбокси (-C(O)OH); алкоксикарбонил (-C-(O)OR'); карбамил (-C(O)NH2); алкилкарбамил (1-4C) (-C(O)NHR'); алкилсульфонил (1-4C) (R'SO2) и алкилфосфонил (R'P(1-4C)(OR')O-).
Помимо этого, Y1 и Y2 каждый независимо может представлять -NH2, -NHR', -NR'R', -OCOR', -NH(CO)R', -O(SO)R' или -O(POR')R', в которых различные R' группы представляют низшие алкилы, которые, в свою очередь, могут быть замещены OH, NH2, вторичным и третичным алкил амино, пирролидино, пиперидино, (1-4C) алкокси или галогеном.
Более предпочтительно, Y1 и Y2 независимо представляют водород, нитро, карбокси, алкоксикарбонил или алкилсульфонил.
Особенно предпочтительные соединения формулы I для использования в настоящем изобретении включают:
1,2,4-бензотриазин 1,4-диоксид (где X представляет водород, Y1 и Y2 оба представляют водород, а n = 1);
3-амино-1,2,4-бензотриазин 1,4-диоксид (т.е. тирапазамин, где X представляет NH2, Y1 и Y2 оба представляют водород, a n = 1);
3-этил-1,2,4-бензотриазин 1,4-диоксид (где X представляет этил, Y1 и Y2 оба представляют водород, а n = 1);
3-пропил-1,2,4-бензотриазин 1,4-диоксид (где X представляет пропил, Y1 и Y2 оба представляют водород, а n = 1) и
3-(1-гидроксиэтил)-1,2,4-бензотриазин 1,4-диоксид (где X представляет 1-гидроксиэтил, Y1 и Y2 оба представляют водорода, а n = 1);
особенно предпочтителен 3-амино-1,2,4-бензотриазин 1,4-диоксид.
Фармацевтически приемлемые соли соединений формулы I включают соли, образованные неорганическими кислотами, такими как хлористо-водородная, бромисто-водородная или фосфорная кислоты; органическими кислотами, такими как уксусная, пировиноградная, янтарная, миндальная и н-толуолсульфоновая кислоты; соли, образованные неорганическими основаниями, такими как гидроксиды натрия, калия или кальция или органическими основаниями, такими как кофеин, этиламин или лизин.
Соединения формулы I могут применяться перорально или парентерально (внутривенно, подкожно, внутримышечно, интраспинально, интраперитонеально и т.п.).
Для парентерального применения эти соединения входят в состав форм для инъекций (раствор, суспензия, эмульсия), содержащих фармацевтически приемлемый носитель. Такие носители обычно нетоксичны и не являются лекарственными агентами.
Примеры таких носителей включают воду, физиологический раствор, раствор Рингера, раствор декстрозы, раствор Хенка, и неводные растворы, такие как жирные масла (например, кукурузное, хлопковое, арахисовое и кунжутное масла), этилолеат и изопропилмиристат. Стерильный физиологический раствор является предпочтительным.
Носитель может содержать небольшие количества добавочных веществ, таких как вещества, усиливающие растворимость, изотоничность и химическую стабильность, например антиоксиданты, буферы и консерванты.
Для перорального (или ректального) применения эти соединения входят в состав соответствующих фармацевтических форм, таких как таблетки, капсулы, суппозитории или пастилки. Такие рецептуры обычно включают твердый, полутвердый или жидкий носитель или растворитель.
Примерами таких растворителей и носителей являются лактоза, декстроза, сахароза, сорбитол, маннитол, крахмалы, аравийская камедь, фосфат кальция, нефтепродукты, масло какао, кокосовое масло, альгинаты, трагакант, желатин, метилцеллюлоза, полиоксиэтилен, сорбитана монолуарат, метилгидроксибензоат, пропилгидроксибензоат, тальк и стеарат магния.
Химиотерапевтический агент вводят млекопитающим стандартными способами, соответствующими конкретному химиотерапевтическому агенту. Химиотерапевтический агент и соединение формулы I можно вводить одним и тем же способом или разными способами, в зависимости от конкретной комбинации препарата и соединения формулы I. Млекопитающим можно вводить только одно соединение формулы I или же в сочетании с одним или более соединений формулы I.
Соединения формулы I вводят млекопитающим в количествах, достаточных для того, чтобы произвести цитотоксическое действие или же убить гипоксичные опухолевые клетки. Количество это будет зависеть от таких факторов, как тип раковой опухоли, возраст и состояние здоровья млекопитающего, максимально переносимая и/или летальная доза химиотерапевтического агента, вид соединения формулы I и характер взаимодействия между ним и химиотерапевтическим агентом.
В предпочтительном варианте осуществления настоящего изобретения тирапазамин вводят в количествах приблизительно от 10 мг/м2 до 450 мг/м2; более предпочтительно приблизительно от 20 мг/м2 до 350 мг/м2; наиболее предпочтительно приблизительно от 30 мг/м2 до 250 мг/м2.
Когда соединение формулы I вводят млекопитающим разделенными дозами, предпочтительной может быть более низкая доза, в зависимости от максимально переносимой дозы этого соединения и его взаимодействия с химиотерапевтическим агентом.
Химиотерапевтический агент вводят млекопитающим в количествах, эффективных для лечения чувствительных опухолей. Эти количества хорошо известны специалистам и могут быть уточнены по литературным научным источникам или из приложений-инструкций к препаратам.
В предпочтительных вариантах осуществления изобретения химиотерапевтический агент и соединение формулы I воздействуют на опухоль как синергисты, и поэтому становится возможным назначать химиотерапевтический агент в дозах, которые ниже, чем если бы применяли один этот препарат.
Такое снижение дозы может быть желательным, если данный химиотерапевтический агент имеет тяжелые побочные эффекты.
Если химиотерапевтический агент должен вводиться млекопитающему разделенными дозами, достаточные количества соединения формулы I вводят таким образом, чтобы синергистический эффект комбинации двух агентов поддерживался, например, до первоначальной дозы химиотерапевтического агента или до каждой отдельной дозы химиотерапевтического агента.
Способы настоящего изобретения также могут применяться совместно с другими видами лечения рака, такими как лучевая терапия и хирургическое удаление опухоли.
Соединение формулы I предпочтительно вводят млекопитающим приблизительно за полчаса и до 24 часов до введения химиотерапевтического агента.
В другом варианте соединение формулы I можно вводить приблизительно от одного до двух часов после введения химиотерапевтического агента.
Для некоторых комбинаций химиотерапевтического агента и соединения формулы I возможно введение последнего более чем за 24 часа до введения химиотерапевтического агента при сохранении преимуществ настоящего изобретения.
Разница во времени введения, обеспечивающая наиболее высокое увеличение цитотоксичности, может быть определена посредством испытаний комбинации химиотерапевтического агента и соединения формулы 1 на экспериментальных моделях опухолей ин витро и/или ин виво, подобных той, что описана ниже, или на любой другой модели опухоли.
Разница во времени введения препаратов, определенная на таких моделях, затем используется как ориентировочная при лечении опухолей млекопитающих и в процессе лечения корректируется, если это необходимо.
Для комбинации тирапазамина и цисплатина обнаружено, что наибольшее взаимодействие между этими двумя агентами наблюдалось, когда тирапазамин вводили между 1 и 18 часами, предпочтительно между 1 и 3 часами, наиболее предпочтительно между 2 и 3 часами перед введением цисплатина, причем наибольшая гибель клеток наблюдалась, когда тирапазамин вводили приблизительно за 2,5 часа до цисплатина.
Когда тирапазамин вводили 2,5 часа спустя после введения цисплатина, усиление цитотоксического эффекта также наблюдалось, однако не настолько большое. Для некоторых соединений формулы I может быть желательным их введение одновременно с химиотерапевтическими агентами.
Настоящее изобретение обеспечивает также наборы для лечения опухолей млекопитающих, включающие по меньшей мере один химиотерапевтический агент и по меньшей мере одно соединение формулы I.
Соединение формулы I предпочтительно представлено в наборе в количествах или дозах, усиливающих цитотоксичность.
Удобные фармацевтические формы соединения формулы I раскрываются в настоящем документе. Конкретная фармацевтическая форма химиотерапевтического агента и соединения формулы I зависит от типа раковой опухоли, предпочтительного пути введения и типа химиотерапевтического агента.
Химиотерапевтический агент и соединение формулы I предпочтительно помещают в отдельные контейнеры, чтобы облегчить введение того и другого в различное время согласно способу настоящего изобретения.
Соединения формулы I, полезные для практики настоящего изобретения, можно приготовить согласно способам, раскрытым в патенте США 5175287.
Основные способы приготовления 3-амино производных можно найти, например, в патенте США 3980779, Ley и др.
Эти соединения могут быть приготовлены из бензофуроксана формулы
путем реакции с солью цианамида с последующим подкислением реакционной смеси. Бензофуроксановый исходный материал не является симметричным в отношении его 5- и 6-положений (которые являются положениями 6 и 7 в полученном оксиде 3-амино бензотриазина).
Следовательно, в результате может получиться смесь 6- и 7-замещенных материалов. По желанию эту смесь можно разделить на отдельные компоненты с помощью стандартных методик, имеющие заместители в 6- или в 7-положении.
Диоксид можно также получить из исходного монооксида или 1,2,4-бензотриазина с помощью окисления над кислотой (см. Robbins и др., J.Chem.Soc. 3186 (1957) и Mason и др., J.Chem.Soc. В 911 (1970)).
Помимо этого, монооксид можно приготовить посредством:
(1) циклизации 1-нитро-2-аминобензольного соединения с помощью H2NCN•2HCl;
(2) окисления соединения следующей структуры
или контролируемого восстановления соответствующего диоксида (см. Mason, выше, и Wolf и др., J.Am. Chem.Soc. 76: 355 (1954)).
1,2,4-бензотриазины можно приготовить путем циклизации предшественников формазана, применяя BF3AcOH (см. схему I и Atallah and Nazer, Tetrahedron 38: 1793 (1982)).
3-амино-1,2,4- бензотриазины можно приготовить или циклизацией исходного соединения (см. схему II и Arndt, Chem. Ber. 3522 (1913)) или восстановлением монооксида или диоксида, как описано выше.
3-гидрокси-1,2,4-бензотриазина оксиды можно приготовить с использованием пероксида и вольфрамата натрия (схема III), процедуры синтеза для получения 3-гидрокси-1,4-диоксида или концентрированной серной кислоты и нитрата натрия (схема IV) для получения монооксида.
Схемы I-IV представлены в конце описания.
1,2,4-бензотразина оксиды, незамещенные в 3-положении (иногда здесь упоминаются как "3-дезамино" соединения), можно приготовить следующим способом, который включает воздействие на 1,2,4-бензотриазина оксид формулы I, в котором X представляет NH2, низшим алкилнитритом в условиях восстановительного дезаминирования.
Под "условиями восстановительного дезаминирования" подразумеваются такие условия реакции, которые обеспечивают по меньшей мере на 10%, предпочтительно на 50%, увеличение выхода незамещенного в 3-положении продукта реакции.
В этом методе в качестве низшего алкилнитрита предпочтительно использовать н-бутилнитрит. Примером условий восстановительного дезаминирования может служить реакция в подходящем растворителе, например диметилформамиде, при температуре по меньшей мере 60oC, обычно при температуре 60-65oC.
Эта реакция в общих чертах приведена на схеме V, представленной в конце описания.
Способы настоящего изобретения далее иллюстрируются примерами. Примеры 1-18 относятся к синтезу соединений формулы I, которые определены выше. Пример 19 относится к испытаниями ин виво и ин витро тирапазамина и цисплатина.
Пример 1. Получение 1,4 диоксида 3-гидрокси-1,2,4-бензотриазина.
Перемешиваемая смесь 1,50 г (9,25 ммоль) 1-оксида 3-амино- 1,2,4-бензотриазина, 100,0 мл кислоты и 30,0 мл 30% перекиси водорода обрабатывалась 3,05 г (9,25 ммоль) Na2WO4•2H2O. Смесь перемешивали в масляной бане при 60oC в течение 4 дней.
Желтовато-оранжевую смесь охлаждали приблизительно до 30oC и фильтровали для удаления светло-желтого твердого вещества, не поглощающего УФ. Оранжевый раствор пероксида водорода в уксусной кислоте выпаривали до полувысыхания, несколько раз добавляя воду и уксусную кислоту для удаления большей части пероксида. Концентрированный раствор оставляли при комнатной температуре, четырежды собирая образовавшееся оранжевое твердое вещество, 0,86 г, (42% выход натриевой соли конечного продукта).
УФ: λмакс (20% CH3OH/H2O): 262,2 (ε 39,460); 477 (ε 7,030).
ИК (ближняя область): 3530 м, 3150 м, 2650 м, 2180 м и 1635 м.
Анал. расч. для C7H4N3O3Na 1,25 H2O, 223,64: C 37,6; H 2,93; N 18,79.
Обнаружено: C 37,8; H 2,75; N 18,65.

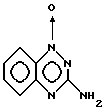
Пример 2. Получение 1-оксида 3-амино-7- трифторметил-1,2,4-бензотриазина.
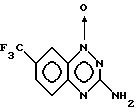
Смесь 4-хлор-3-нитробензотрифторида (Aldrich, 2,70 г, 12,9 ммоль) и дигидрохлорида цианамида (2,75 г, 24 ммоль) (предварительно полученного обработкой эфирного раствора цианамида газообразным HCl, после чего собирали выпадавшее в осадок твердое вещество) нагревали до 140oC и выдерживали при этой температуре в течение 1 часа. Остаток обрабатывали 2H NaOH (45 мл), нагревали еще в течение 5 минут, и позволяли смеси остыть. Осадок собирали, промывали водой, высушивали и порошковали с ацетон-толуолом, получая 1,32 г (45%) искомого продукта в виде светло-желтого твердого вещества с т. пл. 301-302oC.
ТСХ Rf = 0,60 (9:1 метиленхлорид:метанол на пластинках из силикагеля).
МС: m/z (относительная интенсивность) 230 (100, М+).
Пример 3. Получение 3-амино-7-децил -1,2,4- бензотриазина.
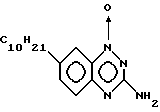
Получение 4-(1-децил)-2-нитроанилина. К перемешиваемому раствору 4-дециланилина (Aldrich, 80 г, 0,34 моль) в гексанах (2,41) в течение 30-минутного периода добавляли уксусный ангидрид (400 мл).
После перемешивания в течение 1 часа смесь охлаждали и обрабатывали ее в течение 30 минут 70% азотной кислотой (34 мл) при температуре 5-10oC. Перемешивание продолжали при 5-10oC в течение 1 часа и при 25oC в течение 16 часов. Смесь разбавляли водой (1:1), перемешивали 5 часов, выливали в открытую емкость и оставляли на 16 часов.
После дополнительного разбавления водой (1,51) твердое вещество собирали и перекристаллизовывали из 85% раствора этанола в воде, получая 92 г (84%) промежуточного агента в виде оранжевого твердого вещества, т. пл. 64oC.
Раствор (100 мл) 85% КОН (19 г, 0,288 моль) в воде объединяли с суспензией 4-(1-децил)-2-нитроанилина (89 г, 0,28 моль), полученного, как описано выше, в метаноле (900 мл). Смесь перемешивали 6 часов, нейтрализовали до pH 7-8 концентрированной HCl и выпаривали в вакууме почти досуха.
После разбавления водой (400 мл) твердое вещество собирали и сушили на воздухе, получая 77 г (100%) промежуточного реагента в виде оранжевого твердого вещества, т.пл. 59oC.
1,0 г (8,7 ммоль) дигидрохлорида цианамида (предварительно полученного обработкой эфирного раствора цианамида газообразным HCl) в течение 10 минут по частям добавляли к предварительно нагретому расплаву (190oC) 4-(1-децил)-2- нитроанилина, полученного на предыдущей стадии (500 мг, 1,8 ммоль). Реакционную смесь нагревали при 190oC в течение 5 минут, охлаждали до 25oC, обрабатывали 6 н. КОН (10 мл) и нагревали до 90- 95oC, выдерживая при этой температуре 1 час.
После охлаждения до 25oC твердое вещество собирали, промывали водой и этанолом и сушили на воздухе, получая 0,25 г (46%) конечного продукта в виде светло-желтого вещества, т.пл. 177oC (разлагается).
МС: m/z (относительная интенсивность) 285 (100, М+), 302 (13).
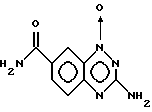
Пример 4. Получение 1-оксида 3-амино-7-карбамил -1,2,4-бензотриазина.
Получение 4-хлор-3-нитробензамида. 20,2 г (0,1 моль) 4-хлор-3-нитробензойной кислоты и 20 мл тионилхлорида объединяли, оставляли стоять в течение 16 часов и нагревали с обратным холодильником в течение 4 часов с получением прозрачного красного раствора. Раствор выпаривали в вакууме и азеотропно подгоняли с бензолом. Остаток растворяли в ацетонитриле (20 мл) и добавляли в течение 30 минут к холодному (-10oC) концентрированному гидроксиду аммония (100 мл).
После 3 часов при -10oC и 16 часов при 25oC смесь выливали в открытую емкость и составляли выпариваться досуха. Остаток суспендировали в воде, твердое вещество собирали и сушили на воздухе, получая 19,8 г (98%) промежуточного реагента в виде светло-желтого вещества, т.пл. 153oC.
Раствор натрия в этаноле (3,45 г, 0,15 моль) (75 мл) добавляли к раствору гидрохлорида гуанидина (15,8 г, 0,165 моль) в этаноле (75 мл). Спустя 1 час смесь фильтровали и фильтрат объединяли с суспензией 4-хлор-3-нитробензамида (10 мг, 0,05 моль), полученного, как описано выше, в этаноле (50 мл). Смесь перемешивали и нагревали с обратным холодильником 16 часов, охлаждали до 0-5oC и подкисляли концентрированной HCl (8 мл).
Собранное твердое вещество объединяли с K2CO3 (28 г, 0,2 моль) и водой (40 мл), и смесь перемешивали и нагревали при 100oC в течение 8 часов.
После охлаждения до 25oC собирали твердое вещество, промывали водой и сушили на воздухе. Твердое вещество суспендировали в кипящем этилацетате, собирали и промывали горячим этилацетатом.
Затем твердое вещество повторно суспендировали в кипящем диоксане и вновь собирали (6 х 100 мл). Объединенные фильтраты выпаривали в вакууме до твердого вещества. Твердое вещество суспендировали в 95% этаноле, собирали и сушили на воздухе, получая 0,44 г (4,3%) конечного продукта в виде светло-желтого твердого вещества, т.пл. 300oC.
ТСХ: Rf = 0,23 (метиленхлорид: ацетон 2:1, пластинки из силикагеля).
МС: m/z (относительная интенсивность) 205 (100, М+).
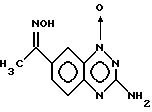
Пример 5. Получение оксима 1-оксида 7-ацетил-3-амино-1,2,4-бензотриазина.
Объединенную смесь 1-оксида 7-ацетил-3-амино-1,2,4- бензотриазина (50 мг, 0,25 ммоль), гидрохлорида гидроксиламина (200 мг, 2,88 ммоль), пиридина (1 мл) и этанола (1 мл) нагревали в течение часа при 90-95oC, а затем охлаждали до 25oC. Смесь разбавляли 95% этанолом (5 мл), а твердое вещество собирали и сушили на воздухе, получая 30 мг (56%) конечного продукта в виде светло-желтого твердого вещества, точка плавления 278oC (разлагается).
ТСХ: Rf = 0,60 (9:1 метиленхлорид:метанол).
МС: m/z (относительная интенсивность) 219 (100, М+).
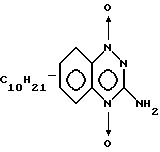
Пример 6. Получение 1,4-диоксида 3-амино-6(-7)-децил-1,2,4- бензотриазина.
Получение 5-(1-децил)бензофуроксана. Объединенную смесь 4- (1-децил)-2-нитроанилина (77 г, 0,28 моль), 5,25% NaOCl в воде (476 г, 0,34 моль), 85% КОН (20,3 г, 0,31 моль), nBu4NHSO4 (4,7 г, 0,014 моль) и CH2Cl2 (2,281) быстро перемешивали в течение 6 часов и разбавляли водой (500 мл) и CH2Cl2 (1: 1).
Отделенную органическую фазу последовательно промывали 1 н. HCl (1:1) и насыщенным солевым раствором (2 x 1:1) сушили (Na2SO4) и концентрировали в вакууме, получая красное масло, 70 г (92%).
Раствор 5-(1-децил)бензофуроксана (10 г, 0,036 моль), полученного, как описано выше, и бензилтриэтилхлорида аммония (0,36 г, 0,0016 моль) в ДМСО (180 мл) обрабатывали в течение нескольких часов постепенно цианамидом (13,0 г, 0,31 моль) и K2CO3 (36,8 г, 0,27 моль). Смесь перемешивали 48 часов и фильтровали. Фильтрат разбавляли водой (6:1) и ледяной уксусной кислотой (40 мл) и экстрагировали CH2Cl2 (4 х 500 мл).
Объединенный органический раствор последовательно промывали 5% NaHCO3 (1 х 500 мл) и солевым раствором (2 х 500 мл), сушили (Na2SO4) и выпаривали в вакууме досуха. Сырой продукт очищали с помощью хроматографии на силикагеле, применяя CH2Cl2: метанол (98,2), получая конечный продукт в виде красного твердого вещества, точка плавления 155oC (разлагается).
МС: m/z (относительная интенсивность) 318 (4, М+), 285 (100).
Пример 7. Получение 1,2,4-бензотриазина диоксида.
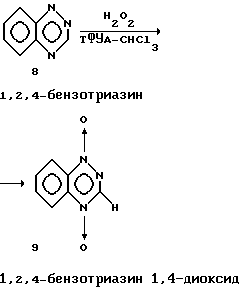
Смесь 1,80 г (13,73 ммоль 90% H2O2 (9 мл)), трифторуксусного ангидрида (13,5 мл) и Na2WO4•2H2O (12,50 г, 38 ммоль в CHCl3 (170 мл)) перемешивали при комнатной температуре 5 дней. Реакционную смесь разбавляли водой (100 мл) и экстрагировали CHCl3 (100 мл). Органический слой промывали водой (50 мл), сушили (Na2SO4), и растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле, используя EtOAc:CH2Cl2 (1:1), получая 0,30 г (13,4%) соединения 9 в виде желтого твердого вещества с точкой плавления 204-205oC.
Анал. расч. для C7H5N3O2 (163,13):
C 51,5; H 3,09; N 25,76.
Обнаружено:
C 51,6; H 3,36; N 26,01.
МС: m/z (относительная интенсивность) 163 (100, М+), 147 (50).
ТСХ: Rf = 0,27 (EtOAc-CH2Cl2 1:1, пластинки из силикагеля).
ИК (нуйол): 1600μ, 1460μ, 1300μ.
УФ: λмакс (H2O): 227 (ε 22,900); 252 (ε 12,950); 392 (ε 4,080).
Пример 8. Получение 1,4-диоксида 7-хлор-3- гидрокси-1,2,4-бензотриазина.
10
11
7-хлор-3-гидрокси-1,2,4-бензотриазин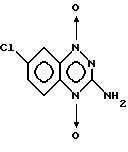
7-хлор-3-амино-1,2,4-бензотриазин 1,4-диоксид
Смесь 1,50 г (7,63 ммоль) соединения 10 в 100 мл уксусной кислоты обрабатывали 2,52 г (7,63 ммоль) Na2WO4•2H2O и 30 мл 30% H2O2. Смесь перемешивали и нагревали при 50oC в течение 6 дней, затем медленно выпаривали досуха для удаления H2O2. Остаток кипятили в 250 мл H2O и фильтровали, удаляя около 25 мг исходного материала 10. Водные растворы затем экстрагировали 2 х 250 мл этилацетата.
Образовавшийся темно-красный кристаллический материал, который по данным ТСХ и МС представлял соединение 12, собирали фильтрованием, получая 60,0 мг желтовато-оранжевого твердого вещества (3,7% выход), охарактеризованного как 12, которое показало хорошую растворимость в смеси горячего изопропилового спирта и воды.
МС: М+ = 212 (q = 100) (соединение 10).
ТСХ: Rf = 0,34 (ацетон, пластинки из силикагеля).
Описанные выше растворы в этилацетате, отделенные от водного слоя после фильтрации для удаления 12, выпаривали досуха. Остаток затем обрабатывали изопропиловым спиртом при комнатной температуре и получали оранжевое твердое вещество, 0,41 г (25% выход соединений 11). Масс. спектр.: М+ = 213 (q = 70); ТСХ: Rf = 22 (ацетон, пластинки из силикагеля). Этилацетатные растворы, отделенные от слоя воды после фильтрования для удаления 12, выпаривались досуха. Остаток затем обрабатывали изопропанолом при комнатной температуре, получая оранжеватое твердое вещество, 0,41 г (выход 25%) соединения 11. Масс. спектр: М+ = 213 (q = 70); ТСХ: Rf = 0,22 (ацетон, силикагельные пластинки). Соединение 11 охарактеризовалось как соль аммония, C7H4ClN3O3NH3, м.в. 230,61.
Свободную кислоту 11 растворяли в концентрированном NH4OH, охлаждали во льду и фильтровали для удаления следов нерастворимого 12. Красный фильтрат и смывы выпаривались досуха, давая красновато-оранжевое твердое вещество. Твердое вещество обрабатывали 50 мл кипящего 1,2-диметоксиэтана, собирали на фильтре и промывали дополнительно 25 мл горячего 1,2-диметилового эфира. Твердое вещество сушили над P2O5 при 56oC 1/1,0 мм, получая 0,244 г (87% выход) соединения 13.
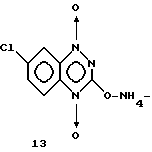
Анал. рассчитанный для C7H4ClN3O3NH3 (230,61): C 36,5; H 3,06; N 24,30.
Обнаружено: C 36,5; H 3,07; N 23,94.
УФ: λмакс (H2O): 219 (ε 12,580); 265,4 (ε 40,000); 4830486 (ε 6,640).
Пример 9. Получение 1,4-диоксида 7-нитро-3-амино-1,2,4-бензотриазина.

Получение 1-оксида 7-нитро-3-трифторацетамид-1,2,4- бензотриазина (15). Раствор 1-оксида 7-нитро-3-1,2,4-бензотриазина (14) (4,00 г, 19,3 ммоль; Parish Chemical Co.), CHCl3 (125 мл) и трифторуксусного ангидрида (12,0 мл, 85,0 ммоль) перемешивали при комнатной температуре 44 часа.
Полученное светло-желтое твердое вещество отфильтровывали, промывали CHCl3 (50 мл) и сушили, получая 5,35 г (91% выход) продукта в виде желтого твердого вещества.
Анал. рассчитанный для C9H4F3N5O4: C 35,7; H 1,33; N 23,10.
Обнаружено: C 35,7; H 1,23; N 23,06.
1,4-диоксид 7-нитро-3-амино-1,2,4-бензотриазина (1,6). К перемешиваемому раствору 1-оксида 7-нитро-3-трифторацетамид-1,2,4-бензотриазина, полученного выше (15) (2,50 г, 8,25 ммоль), в CHCl3 (200 мл) добавляли Na2WO4•2H2O (90 мг, 0,273 ммоль), а затем - 70% H2O2 (10 мл).
Через 15 минут раствор обрабатывали трифторуксусным ангидридом (8,0 мл, 56,7 ммоль), и перемешивание продолжали при комнатной температуре 64 часа. Реакционную смесь подвергали хроматографии (EtOAc, 20% метанол/ацетон и наконец 20% ДМФ/ацетон), затем перекристаллизовывали из ацетона, получая 1,20 г (65% выход) продукта (16) в виде оранжевого твердого вещества с точкой плавления 286-288oC (разлагается).
УФ: λмакс 259, 300, 345, 387, 472.
Анал. рассчитанный для C7H5N5O4: C 37,40; H 2,26; N 31,39.
Обнаружено: C 37,70; H 2,13; N 30,94.
Пример 10. Получение 1,4-диоксида 3-(3-N, N-диэтиламинопропиламино)- 1,2,4- бензотриазина.

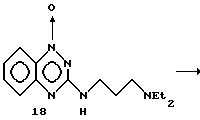
Получение 1-оксида 3-(3-N,N-диэтиламинопропиламино)- 1,2,4-бензотриазина (18). Раствор 1-оксида- 3-хлор-1,2,4- бензотриазина (17) (3,0 г, 16,5 ммоль) (полученного по способу Sasse и др., патент США 4289771) в CH2Cl2 (100 мл) обрабатывали N, N-диэтилпропилендиамином (9,5 мл, 88,3 ммоль). Спустя 20 часов при комнатной температуре смесь разбавляли 1,2-дихлорэтаном (50 мл) и промывали последовательно Na2CO3 и водой. Желтый раствор сушили (Na2SO4), фильтровали и выпаривали в вакууме, получая 3,93 г (87% выход) продукта в виде желтого твердого вещества.
После перекристаллизации (эфир/петролейный эфир) получали чистый материал с точкой плавления 47-48oC.
Анал. расч. для C14H21N5O (18): C 61,10; H 7,69; N 25,44.
Обнаружено: C 61,30; H 7,80; N 25,61.
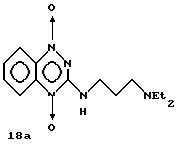
К перемешиваемому раствору 1-оксида 3-(3-N,N-диэтиламинопропиламино) -1,2,4-бензотриазина (18), полученного как описано выше (1,60 г, 6,10 ммоль), в CHCl3 (50 мл) добавляли трифторуксусный ангидрид (22,0 мл).
Спустя 15 минут смесь охлаждали до -10oC, добавляли 70% H2O2 (10 мл) и затем перемешивали при комнатной температуре 20 дней. Реакционную смесь сушили (Na2SO4), фильтровали и выпаривали досуха. Остаток растворяли в насыщенном растворе NaHCO3 (50 мл) и экстрагировали CH2Cl2 (3 х 150 мл). Органический слой сушили (Na2SO4), фильтровали и выпаривали, получая продукт 18a, 0,51 г (29% выход), в виде красного твердого вещества.
Точка плавления 92-94oC.
ЯМР: δ (400 МГц, CDCl3) 1,11 (6H, т. J = 7,1 Гц, CH3), 1,84-1,90 (2H, м. Н-2'), 2,48-2,64 (4H, м. NCH2CH3 и H-3'), 3,68 (2H, шир. т. J = 5,5 Гц, H-1'), 7,46 (1H, ддд, J = 7,1, 7,0 и 1,2 Гц, H-6), 7,85 (1H, ддд, J = 7,0, 6,9 и 1,2 Гц, H-7), 8,31 (2H, м, H-5 и 8), 8,80 (1H, шир. т., NH).
УФ: λмакс 220, 270, 476.
Анал. расч. для C14H21N5O5 (1/3 H2O): C 56,50; H 7,34; N 23,55.
Обнаружено: C 56,90; H 7,15; N 23,40.
Пример 11. Получение 1,4-диоксида 7-нитро-3-(2-N,N-диэтиламино- этиламино)-1,2,4-бензотриазина.

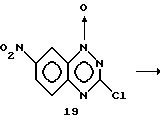


Получение гидрохлорида 1-оксида 7-нитро-3-2-N, N- диэтиламиноэтиламино)-1,2,4- бензотриазина (20). Раствор 1-оксида 7-нитро-3-хлор-1,2,4-бензотриазина (19) (1,60 г, 7,06 ммоль) (полученного, как описано в патенте США 4289771, Sasse и др., с использованием: (a) NaNO2 и H2SO4 при 40oC с последующим (в) хлорированием POCl3 при 106oC) в CH2Cl2 (50 мл) обрабатывали N, N-диэтилэтилендиамином (6,0 мл, 42,7 ммоль).
После 6 часов при комнатной температуре смесь выпаривали досуха в вакууме при 60oC. Желтое твердое вещество перемешивали в смеси 20% изопропанол эфир (150 мл) в течение 5 часов, фильтровали, промывали сначала изопропиловым спиртом, затем петролейным эфиром и сушили (80oC/1,0 мм рт.ст.), получая 1,80 г (74% выход) продукта 20 в виде желтых игольчатых кристаллов.
ЯМР δ (90 МГц, d6 - ДМСО/d4 метанол): 1,25 (6H м, J = 6,0 Гц, CH3), 3,25 (6H, м. , NCH2), 3,82 (2H, м., H-1'), 7,74 (1H, д, J = 7,0 Гц, H-5), 8,52 (1H, дд., J = 7,0 и 2,0 Гц, H-6), 8,91 (1H, д, J = 2,0 Гц, H-8).
Гидрохлорид 1,4-диоксида 7-нитро-3-(2-N,N-диэтиламиноэтиламино)-1,2,4-бензотриазина (21). К перемешиваемому раствору 1-оксида 7-нитро-3-(2-N,N- диэтиламиноэтиламино)-1,2,4-бензотриазина (20, полученного, как описано выше) (0,50 г, 1,46 ммоль), в CHCl3 (50 мл) при 0oC добавляли трифторуксусный ангидрид (9,0 мл).
Спустя 30 мин добавляли 70% H2O2 (4,0 мл), и смесь перемешивали при комнатной температуре 3 дня, затем сушили (Na2SO4), фильтровали и выпаривали в вакууме досуха, получая трифторацетат, 0,67 г, 45% выход. Продукт растворяли в насыщенном растворе NaHCO3 (30 мл) и экстрагировали CH2Cl2 (3 х 30 мл). Дихлорметан промывали водой, сушили (Na2SO4), фильтровали, насыщали газообразным HCl и выпаривали досуха, получая 0,35 г (63% выход, общий 28%) продукта в виде красного твердого вещества, точка плавления 194-195oC.
УФ: λмакс 260, 306, 388, 479.
Анал. расч. для C13H18N6O4HCl: C 43,50; H 5,34; N 23,43.
Обнаружено: C 43,20; H 5,37; N 23,11.
Следующие примеры 12-15 описывают реакции восстановительного дезаминирования для получения соединений формулы I, незамещенных в 3-положении, т.е. в которых заместитель X является водородом.
Пример 12. Получение 1,4-диоксида 1,2,4-бензотриазина путем восстановительного дезаминирования 1,4-диоксида 3-амино-1,2,4- бензотриазина.

К быстроперемешиваемому раствору трет-бутилнитрита (867 мг, 1,0 мл, 8,41 ммоль) в ДМФ (20 мл) при 60-65oC добавляли 1,4- диоксид 3-амино-1,2,4-бензотриазина (500 мг, 2,81 ммоль) (полученный по способу Seng и др., Anqew. Chem. Internal. Edit. II (1972)) малыми порциями в течение 5 минут.
После добавления и прекращения бурного выделения газа (приблизительно 5 минут) раствор собирали и выпаривали в вакууме до темного воскообразного вещества. Флэш хроматография (30% EtOAc/CH2Cl2) дала желтое твердое вещество т. пл. 188-189,5oC (разлагается), которое перекристаллизовывали из этанола, получая 195 мг (43% выход) продукта 9 в виде ярко-желтых пластинок, точка плавления 192-194oC (разлагается).
ЯМР: (400 МГц, d6-ацетон) 8,04 (1H, ддд, J = 8,5, 7, 1,5 Гц), 8,15 (1H, ддд. J = 8,5, 7, 1,5 Гц), 8,42 (1H, дд, J = 8,5, 1,5 Гц), 8,43 (1H, дд., J = 8,5, 1,5 Гц), 9,05 (1H, c., H-3).
УФ: λмакс 405, 300, 225.
MC: m/z (относительная интенсивность) 147 (13), 136 (19); интенсивность) 164 (9), 163 (100, M+), 147 (13), 136 (19), 90 (7), 78 (27), 76 (26), 75 (8), 64 (9), 63 (10), 52 (12), 51 (48), 50 (28), 38 (8), 37 (5), 30 (18), 28 (6), 27 (7).
Анал. расч. для C7H5N3O5: C 51,54; H 3,09; N 25,76.
Обнаружено: C 51,42; H 3,03; N 25,66.
Пример 13.
Получение 1,4-диоксида 7-аллилокси-1,2,4-бензотриазина путем восстановительного дезаминирования.

К перемешиваемому раствору трет-бутилнитрита (271 мг, 0,312 мл, 2,63 ммоль) в ДМФ (15 мл) при 60-65oC добавляли 1,4-диоксид 7-аллилокси-3-амино-1,2,4-бензотриазина (23) (205 мг, 0,875 ммоль) малыми порциями в течение 5 минут.
Спустя 30 минут добавляли дополнительное количество трет-бутилнитрита (271 мг, 0,31 мл, 2,63 ммоль), после чего очень скоро происходило бурное выделение газа из темно-красного раствора, который в течение нескольких минут заметно посветлел.
Спустя 30 минут полученный оранжевый раствор упаривали в вакууме до коричневого твердого вещества, которое последовательно подвергали флэш хроматографии (10% EtOAc/CH2Cl2) и перекристаллизации (CH2Cl2/петролейный эфир), получая 72 мг (38% выход) продукта (24) в виде светло-оранжевых кристаллов, точка плавления 147-148oC.
ЯМР: δ (400 МГц, d6-ацетон) 4,89 (2H, ддд, H-1', J1′.2′ = 5,5, J1′.3′цис= J1′.3′транс = 1,5 Гц), 5,36 (1H, ddd, H-3', J3′.2′цис = 10,5, J3′3′ = 1,5 Гц), 5,52 (1H, ддд. H-3', J3′.2′транс = 17,5, J3′.3′ = 3, J3′.1′ = 1,5 Гц), 6,14 (1H, ддд, H-2', J2′.3′цис = 10,5, J2′.1′ = 5,5 Гц), 7,70 (1H, д, H-8, J8.6 = 2,5 Гц), 7,74 (1H, дд. H-6, J6.5 = 9,5, J6.8 = 2,5 Гц), 8,33 (1H, д, H-5, J5.6 = 9,5 Гц), 8,93 (1H, с, H-3).
УФ: λмакс 425, 410, 365, 355, 320, 245, 200.
MC: m/z (относительная интенсивность) 220 (4), 219 (34 M+), 103 (4), 77 (4), 75 (4), 63 (13), 62 (4), 42 (3), 41 (100), 39 (16).
Анал. расч. для C10H9N3O3: C 54,79; H 4,14; N 19,17.
Обнаружено: С 54,73; H 4,16; N 19,15.
Пример 14. Получение 1,4-диоксида 7-(3-N-этилацетамидо-2-ацетоксипропокси) -1,2,4-бензотриазина путем восстановительного дезаминирования.
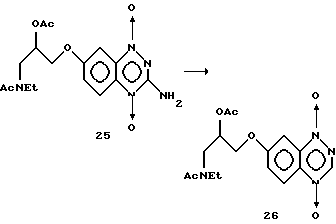
К перемешиваемому раствору трет-бутилнитрита (9185 мг, 1,79 ммоль) в ДМФ (5 мл) при 60oC добавляли через шприц раствор 1,4- диоксида 7-(3-N-этилацетамидо-2-ацетоксипропокси)-3-амино- 1,2,4-бензотриазина (25) (125 мг, 0,329 ммоль) в ДМФ (5 мл) в течение 1 минуты.
Спустя 5 минут добавляли еще трет-бутилнитрит (217 мг, 2,10 ммоль) и происходила немедленная реакция, о чем свидетельствовало выделение газа и изменение цвета раствора с красного на светло-оранжевый.
По истечении 10 минут отгоняли легкие фракции, после чего желто-коричневое твердое вещество элюировали через силикагель смесью 5% метанол/CH2Cl2, получали смесь CH2Cl2/лигроин, которая затем давала 90 мг желтого твердого вещества (75% выход), точка плавления 179-180,5oC.
ЯМР/: δ (400 МГц, d4-метанол, смесь ротамеров, приблизительное соотношение 2: 1) 1,12, 1,22 (триплеты 1: 2, 3H общий, J = 7 Гц), 2,0-6, 2,07 (синглеты, 2: 1, 3H общий), 2,11, 4,34- 4,48 (м. 2H), 5,48-5,58 (м. 1H), 7,76-7,85 (м. 2H), 8,36-8,42 (м. 1H), 9,04, 9,06, (с.н. 2:1, 1H общий).
УФ: λмакс 420, 405, 365, 350, 315, 240, 200.
MC: m/z (относительная интенсивность) 365 (0,5), 364 (1,4, M+), 349 (0,5), 348 (1,1), 347 (0,5), 332 (1,2), 331 (3,6), 187 (7), 186 (66), 102 (6), 100 (21), 84 (30), 63 (6), 58 (100), 56 (8), 43 (65), 42 (9), 41 (9), 41 (5), 30 (14), 29 (5), 28 (8).
Пример 15. Получение 1,4-диоксида 7-нитро-1,2,4-бензотриазина путем восстановительного дезаминирования.

К перемешиваемому раствору т.-бутилнитрита (988 мг, 0,85 ммоль) в ДМФ (5 мл) при 60oC добавляли 1,4-диоксид 7-нитро-3- амино-1,2,4-бензотриазина (14) (38 мг, 0,17 ммоль). Спустя 30 минут добавление дополнительного количества т. -бутилнитрита (175 мг, 1,70 ммоль) немедленно повлекло за собой изменение окраски реакционной смеси и бурное газообразование.
Спустя еще 10 минут оранжевый раствор упаривали в вакууме до красного твердого вещества и подвергали хроматографии с использованием смеси 1% уксусная кислота/CH2Cl2, получая 3 мг продукта (27) в виде желтого твердого вещества (10% выход).
ЯМР δ (90 МГц, d6-диметилсульфоксид) 7,68 (д. 1H, J = 9,2 Гц), 7,92 (дд, 1H, J = 9,2, 2,2 Гц), 8,10 (д. 1H, J = 2,2 Гц), 8,65 (c. 1H).
УФ: λмакс 420, 310, 240, 205.
MC: m/z (относительная интенсивность) 209 (9), 208 (100, M+), 192 (54), 181 (14), 162 (16), 105 (9), 77 (28), 75 (52), 74 (27), 63 (21), 62 (16), 30 (77), 18 (26).
Пример 16. 3-этил-1,2,4-бензотриазин-1,4-диоксид.

Гидразон (28), образованный путем конденсации пропионового альдегида и фенилгидразина, подвергали реакции с хлористым фенилдиазонием в смеси уксусной кислоты, нитрита натрия и HCl, получая формазан (29). Циклизация с использованием BF3 - AcOH (трифторид борауксусная кислота) при 90-95oC давала 3-этил-1,2,4-бензотриазин (30) в виде масла, которое затем очищали перегонкой. Окисление 70% перекисью водорода и трифторуксусным ангидридом (ТФУА) в CH2Cl2 давало 3-этил-1,2,4-бензотриазин-1,4-диоксид (31).
Названное соединение (31) очищали колоночной хроматографией с нормальной фазой и перекристаллизацией из водного этанола, получая материал чистотой 99,8%, точка плавления 141-142oC.
Пример 17. 3-пропил-1,2,4-бензотриазин-1,4-диоксид.
Данный продукт (32) получали и очищали по способу примера 16 (получение 13-этил-1,2,4- бензотриазин-1,4-диоксида), за исключением того, что в реакции использовали гидразон, образованный в результате конденсации масляного альдегида и фенилгидразина, вместо гидразона, образованного реакцией конденсации пропионового альдегида и фенилгидразина. Точка плавления 32 114-116oC.
Пример 18. 1-оксид 3-(1-гидроксиэтил)-1,2,4- бензотриазина.
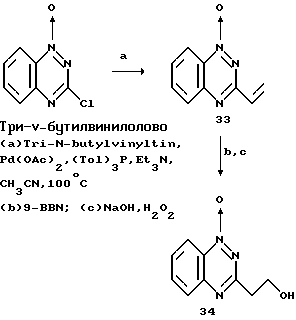
1-оксид 3-хлор-1,2,4-бензотриазина обрабатывали небольшим избытком три-N-бутилвинилолова в смеси ацетонитрила, тритолуолфосгена и триэтиламина, с использованием ацетата палладия II в качестве катализатора, в закрытой пробирке при 100oC в течение 48 часов. Удаление растворителя и очистка с помощью колоночной хроматографии давали 1-оксид 3-винил-1,2,4- бензотриазина (33). Восстановление 9-борабицикло [3.3.1] нонаном (9-BBN) с последующим окислением гидроксидом натрия и перекисью водорода давали названное соединение (34).
Пример 19. Тирапазамин и цисплатин испытывали ин виво на модели опухоли RIF-I. Эти препараты испытывали также ин витро с использованием клеток RIF-I в условиях гипоксии и аэробных условиях.
Животные и опухоли.
Фибросаркому RIF-I (культура лаборатории д-ра Martin Brown, Department of Radiation Oncology, Stanford University, Stanford, CA; Twentyman и др., J. Nat'l. Cancer Inst. 64: 595-604) мышей линии C3H/Km (выведенной и поддерживаемой в лаборатории радиационной биологии медицинской школы Стэнфордского университета), которых содержали в условиях определенной микрофлоры, поддерживали попеременно ин виво и ин витро согласно опубликованному ранее протоколу (Twentyman, см. выше). Монослои опухолевых клеток, которые росли в среде Waymouth, содержащей 15% плодной бычьей сыворотки, собирали с помощью 0,05% трипсина.
Из этой суспензии 2 • 105 клеток в 0,05 мл среды инокулировали каждой мыши внутрикожно в спину, приблизительно на 2 см выше хвоста. Эксперименты начинали спустя две недели, когда опухоли в среднем достигали объема 200 мм3.
Препараты.
Тирапазамин (SR 4233) поставлялся фирмой Стерлинг Драг Инк (Нью-Йорк). Для исследований на животных препарат растворяли в физиологическом растворе в концентрации 1 мг/мл и вводили интраперитонеально на основе ммоль/кг. Цисплатин (c-DDP), полученный из Bristol Laboratories (Принстон Нью Джерси), растворяли в стерильной воде и вводили интраперитонеально в количестве 0,01 мл/г веса тела.
Выживание клеток.
В исследованиях на животных выживание клеток RIF-I оценивали согласно анализу эксцизии ин виво/ин витро. Мышей умерщвляли спустя 24 часа после введения цисплатина; опухоли вырезали, измельчали и диссоциировали с помощью ферментного коктейля (Twentyman, см. выше), и клетки помещали в чашки для исследования клоногенности.
Полученные колонии опухолевых клеток окрашивали кристаллическим фиолетовым и подсчитывали после двух недель инкубации при 37oC в увлажняемой атмосфере, содержащей 5% CO2.
Относительное количество клоногенных клеток на опухоль рассчитывали по эффективности роста на чашках и по выходу опухолевых клеток для леченых опухолей по отношению к тем же показателям для контрольных нелеченых опухолей.
Для исследований на клетках ин витро, клетки RIF-I засевали на 60 мм стеклянные чашки Петри в среду Waymouth, содержащую 15% плодной бычьей сыворотки, в концентрации 2•104 клеток на чашку. Эксперименты производили спустя 4-5 дней, когда количество клеток достигало приблизительно 106 клеток на чашку ко времени воздействия препаратами. Среду заменяли на 2 мл среды без сыворотки, содержащей тирапазамин в концентрации 2 или 4 мкг/мл.
В каждый эксперимент включали группы, в которых воздействие тирапазамином и цисплатином осуществляли как одновременно, так и с интервалом.
В тех группах, в которых был интервал, немедленно после подвержения воздействию тирапазамина клетки дважды промывали и заменяли среду полной культуральной средой до тех пор, пока не наступало время второй обработки (цисплатином), которую также осуществляли в среде без сыворотки. Подвержение действию и тирапазамина, и цисплатина осуществляли в течение 1 часа в условиях гипоксии.
Для достижения гипоксии чашки помещали в специально сделанные предварительно подогретые алюминиевые камеры, которые помещались на специальный встряхивающий стол, и соединяли с системой трубопроводов, включающей выходной газопровод для создания вакуума и входные газопроводы для воздуха или азота (+5% CO2). Гипоксия в алюминиевых камерах достигалась серией из 5 откачиваний по 2-3 минуты до 0,1 атм попеременно с нагнетанием азота (+5% CO2).
После этого камеры герметизировали и инкубировали при 37oC в течение 1 часа. Измерение уровня кислорода в среде с помощью электрода Кларка показало, что гипоксия достигалась достаточно быстро (примерно за 10 минут, средний уровень кислорода pO2 в течение часа экспозиции составлял менее 200 частей на миллион).
Немедленно после воздействия цисплатином клетки трипсинизировали, подсчитывали и помещали в пластиковые чашки Петри в среду Waymouth с 15% плодной бычьей сыворотки, после чего инкубировали 14 дней при 37oC в увлажняемой атмосфере, содержащей 5% CO2, затем колонии окрашивали кристаллическим фиолетовым и подсчитывали.
Нормальная ткань.
Ответную реакцию нормальной ткани на тирапазамин и цисплатин оценивали по почкам и костному мозгу с помощью анализа крови на азот мочевины и количество лейкоцитов в периферической крови. Кровь брали из хвостовой вены или посредством пункции сердца, антикоагулянты не применяли. Количество лейкоцитов в периферической крови у каждой мыши определяли исходя из 20 мкл цельной крови, разбавленной в 0,280 мл 3% уксусной кислоты.
Для исследования азота мочевины образцы крови от двух мышей объединяли, коагулировали и центрифугировали при 830 g в течение 15 минут. Уровни азота мочевины в полученной сыворотке определяли в коммерческой ветеринарной клинической лаборатории. В другом эксперименте также фиксировали выживание до 30 дня.
Результаты.
(а) Результаты на опухолях ин виво. Фиг. 1 показывает объединенные результаты двух экспериментов, в которых 0,35 ммоль/кг тирапазамина (63 мг/кг) вводили мышам с опухолями в различные сроки, от 3 часов до введения и до 2 часов после введения 8 мг/кг цисплатина, а клоногенное выживание оценивали через 24 часа. Ось Y показывает относительное количество клоногенных клеток на опухоль, ось X показывает время введения тирапазамина по отношению к введению цисплатина (-2 представляет данные, полученные у мышей, которым вводили тирапазамин за 2 часа до введения цисплатина). Кружки обозначают только тирапазамин; квадраты - только цисплатин; треугольники обозначают тирапазамин и цисплатин вместе.
Как видно из фиг. 1, когда тирапазамин вводили между тремя часами до и 1-2 часами после цисплатина, относительные количества клоногенных клеток на опухоль снижались приблизительно с 10-4 до 10-7.
Эти данные представляют от 10 до 1000 раз снижение количества клоногенных клеток на опухоль по сравнению с количеством клоногенных клеток на опухоль, когда тирапазамин и цисплатин вводили одновременно. Синергизм тирапазамина и цисплатина был выражен в наибольшей степени, когда тирапазамин вводили от 3 до 1 часа до введения цисплатина, причем наибольший эффект наблюдался, если тирапазамин вводили за 2,5 часа до введения цисплатина.
Поскольку происходила гибель слишком большого количества клеток, что было на пределе возможностей метода, дозу тирапазамина в эксперименте уменьшили с 0,35 ммоль до 0,27 ммоль/кг (48,6 мг/кг) и эксперименты повторили.
В этом эксперименте промежуток времени между введениями цисплатина и тирапазамина увеличили до 24 часов. Результаты представлены на фиг. 2. Обозначения те же, что на фиг. 1.
Как видно из фиг. 2, усиленное взаимодействие между тирапазамином и цисплатином имело место, когда тирапазамин вводили вплоть до 24 часов до введения цисплатина.
Несмотря на сокращение дозы тирапазамина во второй серии экспериментов, данные от трех экспериментов показали те же результаты: существенную дополнительную токсичность, когда препараты вводились одновременно, и наибольшее цитотоксическое взаимодействие, когда препараты вводились раздельно, с максимальным сокращением числа клоногенных клеток на опухоль в случае, когда введение тирапазамина осуществилось примерно за 2,5 часа до введения цисплатина.
Дополнительно произвели эксперименты с различными дозами тирапазамина, даваемыми за 2,5 часа до введения 4 или 8 мг/кг цисплатина.
Наблюдали почти экспоненциальное уменьшение выживаемости опухолевых клеток при обеих дозах цисплатина, в зависимости от возрастания дозы тирапазамина.
(в) Результаты на нормальной ткани.
Предварительные исследования на мышах C3H показали, что количество лейкоцитов достигает наименьших уровней на третий день после лечения тирапазамином и цисплатином, а затем вновь увеличивается почти до контрольных уровней на пятый день.
Поэтому изучение ответов на дозы было предпринято на 3 день в случае цисплатина и в случае тирапазамина и цисплатина в сочетании при дозе тирапазамина (0,35 ммоль), которую вводили за 2,5 часа до цисплатина. Цисплатин вводили в трех различных дозах - 10, 14 и 18 мг/кг. И тирапазамин и цисплатин вызывали небольшую лейкопению, а их комбинация вызывала эффект, соответствующий предсказанному на основании сложения ответов на оба лекарства по отдельности.
Определение азота мочевины в сыворотке крови выполняли на 6 день после инъекции тирапазамина и цисплатина, основываясь на предварительном исследовании, которое выявило время максимального возрастания азота мочевины в крови после высоких доз цисплатина. Азот мочевины в крови мышей C3H определяли спустя 6 дней после инъекции только тирапазамина (0,27 ммоль/кг), только цисплатина (10, 14 или 18 мг/кг) или обоих лекарств вместе (причем тирапазамин вводили за 2,5 часа до инъекции цисплатина). Уровни азота мочевины для доз 10 и 14 мг/кг цисплатина были такими же, как у контрольных мышей (приблизительно 30 мг/дл).
Однако 18 мг/кг цисплатина повысили его уровень почти до 80 мг/дл. В противоположность этому, при каждой дозе комбинации препаратов уровень азота мочевины в эксперименте был ниже, чем у контрольных (нелеченых) мышей.
Эти результаты свидетельствуют о том, что тирапазамин в сочетании с цисплатином не увеличивает токсичность цисплатина по отношению к почкам и может даже защищать их при самой высокой из испытанных доз.
В качестве исследования, показывающего, усиливает ли тирапазамин системную токсичность цисплатина, выполнили эксперименты по определению LD50 только с цисплатином и с цисплатином, которому предшествовало (за 2,5 часа) введение тирапазамина. LD50 для мышей, которым вводили 0,35 ммоль/кг тирапазамина плюс цисплатин, составила 17,7 мг/кг (16,8-18,7 мг/кг - достоверность 95%), по сравнению с таковой для цисплатина в отдельности, которая составила 17,8 (17,1-18,5 мг/кг).
(c) Результаты экспериментов.
На клетки воздействовали в течение одного часа тирапазамином (2 или 4 мкг/мл) в условиях гипоксии, а также цисплатином (2 мкг/мл) в течение одного часа в разное время по отношению к введению тирапазамина. Концентрации каждого агента подбирали таким образом, чтобы они давали тот же уровень гибели гипоксичных клеток, что и для RIF-I опухолей ин виво: для тирапазамина 0,3 и 0,009 и 2 и 4 мкг/мл, соответственно, и для цисплатина (3,5 • 10-3).
В каждом эксперименте были группы, в которых не было интервала между воздействиями двух агентов (т.е. тирапазамин и цисплатин применяли одновременно в течение одного часа в условиях гипоксии), а также группы, в которых оба воздействия разделялись во времени от 1 до 4 часов. Результаты, полученные для лекарств, даваемых одновременно, не имели значимой разницы с теми, что получали для раздельно даваемых препаратов; в то время как при раздельном введении лекарств гибель клеток возрастала до 102 раз.
Здесь наблюдается та же кинетика увеличения степени гибели клеток, которая наблюдалась в результатах экспериментов ин виво, хотя абсолютная величина эффекта от разъединения двух доз была меньше, чем в экспериментах ин виво.
Чтобы проверить, действительно ли взаимодействие между двумя агентами зависит от наличия гипоксии, эксперименты повторили с тремя часами между экспозицией клеток с тирапазамином в аэробных условиях и экспозицией клеток с цисплатином в условиях гипоксии.
В этих экспериментах тирапазамин не проявлял цитотоксичности, и не наблюдалось потенцирования гибели клеток по сравнению с той, которая наблюдалась в таких же экспериментах только с цисплатином.



Изобретение относится к медицине, точнее к способу увеличения цитотоксичности химиотерапевтического агента в отношении твердых опухолей, чувствительных к лечению этим химиотерапевтическим агентом. Сущность изобретения: млекопитающему с такой опухолью вводят в срок от 0,5 до 24 ч до введения химиотерапевтического агента усиливающее цитотоксичность количество соединения, представляющего собой производное бензотриазина. Изобретение относится также к наборам для лечения таких опухолей, которые включают химиотерапевтический агент и усиливающее цитотоксичность количество оксида 1,2,4-бензотриазина. Изобретение также обеспечивает расширение арсенала средств для лечения твердых опухолей. 2 с. и 9 з.п. ф-лы, 2 ил.

где Х представляет водород, (1-4С) углеводородный радикал, (1-4С) углеводородный радикал, замещенный ОН, NH2, NHR, NRR (1-4С) алкокси или галогеном, галоген, ОН, (1-4С) алкокси, NH2, NHR или NRR, где каждый R независимо выбран из группы (1-4С) низший алкил и (1-4С) низший ацил, и (1-4С) низший алкил и (1-4С) низший алкил, замещенный ОН, NH2, вторичный (1-4С) алкил и третичный (1-4С) диалкил аминогруппами, (1-4С) алкокси или галогеном, и, когда Х представляет NRR, оба R, взятые вместе непосредственно или через мостиковый кислород, образуют морфолино кольцо, пирролидино кольцо или пиперидино кольцо;
n = 0 или 1;
Y1 и Y2 представляют независимо водород, нитро, галоген, (1-14С) углеводородные радикалы, включая циклические, ациклические и ненасыщенные углеводородные радикалы, необязательно замещенные 1 или 2 заместителями, выбранными из группы, состоящей из галогена, гидрокси, эпокси, (1-4С) алкокси, (1-4С) алкилтио, первичного амино (NH2), вторичного (1-4С) алкиламино, третичного (1-4С) диалкиламино, в котором две алкильные группы связаны вместе, образуя морфолино, пирролидино или пиперидино кольцо, (1-4С) ацилокси, (1-4С) ациламидо и их тиоаналогов, ацетиламино (1-4С) алкила, карбокси, (1-4С) алкоксикарбонила, карбамила, (1-4С) алкилкарбамила, (1-4С) алкилсульфонила или (1-4С) алкилфосфонила, где углеводородные радикалы могут необязательно прерываться одной эфирной (-О-) связью, или где Y1 и Y2 представляют независимо морфолино, пирролидино, пиперидино, NH2, NHR1, NR1R1, O(CO)R1, NH(CO)R1, O(SO)R1 или О(РОR1)R1, в которых R1 представляет (1-4С) углеводородный радикал, который может быть замещен OH, NH2, вторичным (1-4С) алкиламино, третичным (1-4С) диалкиламино, морфолино, пирролидино, пиперидино, (1-4С) алкокси, или галогеновым заместителями, или фармакологически приемлемой соли указанного соединения.
| СРАВНИВАЮЩЕЕ УСТРОЙСТВО | 0 |
|
SU371559A1 |
| С А РЕТЕНИЯ | 0 |
|
SU369696A1 |
| Murray et al | |||
| "British J.Cancer", 47, p.195-203, 1983. | |||

