Изобретение относится к полипептидам, которые ковалентно связаны с длинноцепными полимерами, такими как метоксиполиэтиленгликоль. Это изобретение также описывает способы и реагенты для взаимодействия активированных полимерных молекул с различными биологически важными полипептидами.
Обнаружено, что многие протеины, которые были идентифицированы и выделены из организма человека и животных, обладают многообещающими лекарственными и терапевтическими возможностями. Были сделаны большие успехи в методах идентификации и характеристики таких протеинов, в дополнение к способам получения таких протеинов в относительно чистом виде и в относительно больших количествах. По мере совершенствования методов использования таких потенциально ценных веществ, возникло много препятствий к получению этих соединений в форме, пригодной для использования в клинических моделях.
Например, было обнаружено, что многие такие протеины имеют исключительно короткий период полуведения из сыворотки крови. Большей частью протеины выводятся из сыворотки через почки. Систематическое введение относительно больших количеств протеинов, особенно чужеродных человеку, может вызвать иммуногенные реакции, которые, могут привести к таким проблемам, как быстрое выведение протеинов из организма через образование иммунных комплексов. В случае других протеинов проблемы растворимости и агрегации также мешают оптимальному дозированию протеина.
Одним из наиболее многообещающих способов решения этих проблем является ковалентное присоединение одной или более цепей инертного полимера к нужному полипептиду. Наиболее часто используемым полимером является полиэтиленгликоль (ПЭГ) или монометоксилполиэтиленгликоль (мПЭГ) /1/. ПЭГ является идеальным для этих целей благодаря своей доказанной нетоксичности. Другие исследователи используют для подобных целей полиоксиэтилированный глицерол (ПОГ). /2/.
Описаны многочисленные результаты того, как ковалентное модифицирование протеинов полиэтиленгликолями ("пегилирование") приводило к появлению у протеина желаемых характеристик. Например, было показано, что пэгилирование IL-2 уменьшает выведение IL-2, не влияя существенно на активность цитокина. Сниженное выведение приводит к повышенной эффективности по сравнению с не пегилированным веществом /3/.
Увеличение периода полувыведения перекиси дисмутазы (ПД) из сыворотки крови стало решающим препятствием к использованию ПД для лечения различных симптомов. Большое число исследований показало, что пегилирование ПД приведет к снижению скорости выведения /4/.
Агрегация иммуноглобулина G (IgG) считается фактором, который приводит к серьезным побочным эффектам у пациентов, которым вводили внутривенно IgG. Было показано, что пегилирование IgG снижает агрегацию протеинов, снимая эту проблему /5/.
Была также показана возможность через пегилирование влиять на иммуногенность протеина. Абуховский с сотрудниками изучал иммуногенность и период выведения из крови пегилированной каталазы бычьей печени /6/.
Присоединение ПЭГ-групп к этим разнообразным протеинам замедляет выведение благодаря увеличению размера молекулы пегилированного протеина. Вплоть до определенного размера скорость гломерулярной фильтрации протеинов обратно пропорциональна размеру протеина. Следовательно, способность пегилирования замедлять выведение вызвана, как правило, не тем, как много ПЭГ-групп присоединилось к протеину, а общей молекулярной массой измененного протеина. Это подтверждено исследованиями процесса выведения, в которых варьировали как размер боковых ПЭГ-цепей, так и число ПЭГ цепей, связанных с IL-2. /3/.
Многочисленные исследования процессов выведения, иммуногенности, агрегации и физических свойств пегилированных протеинов указывают на то, что ПЭГ образует гибкую гидрофильную оболочку вокруг протеина. Цепи ПЭГ становятся сильно гидратированными и придают пегилированным протеинам более высокую кажущуюся молекулярную массу, чем можно было ожидать, и экранируют заряды на протеине.
Благодаря многочисленным обнадеживающим результатам, которые были получены в этой области, создан каталог способов присоединения ПЭГ-единиц к полипептидам. Ключевой элемент этих способов заключается в "активации" концевой OH-группы полиэтиленгликоля. Такая активация необходима для того, чтобы образовать связь между ПЭГ-группой и полипептидом. В подавляющем большинстве таких методик активируют ПЭГ-составляющую для того, чтобы сделать возможной реакцию со свободными первичными аминогруппами полипептидов. Большинство этих свободных аминов обнаружено в аминокислотных остатках лизина.
Обычно, к протеинам присоединяют много ПЭГ-цепей. Например, было обнаружено, что для подавления иммуногенности, желательно использовать от 15 до 50 молей полимера на моль полипептида /7/.
Вследствие того, что к каждому полипептиду обычно присоединяют множество ПЭГ-цепей, и поскольку в каждом протеине обычно присутствует большое число остатков лизина, протеины легко пегилировать с получением гомогенных продуктов реакции /8/. Это отсутствие специфичности реакции приводит к многочисленным осложнениям. Среди них - то, что пегилирование часто приводит к значительной потере активности протеина. Предположительно, присоединение к имеющему значение остатку лизина могло бы изменить активный центр протеина, сделав его неактивным.
По крайней мере на одной системе было показано, что пегилирование может привести к возникновению стерических затруднений в активных центрах. Другими словами, относительно малые субстраты могут подойти к протеину, в то время как на активность протеинов, которые реагируют с большими субстратами, может резко повлиять беспорядочное пегилирование /7/. Селективное пегилирование активных центров таких протеинов могло бы привести к модифицированным веществам, приобретающим при пегилировании желаемые качества без потери своей активности. Вдобавок, если пегилированный протеин предназначен для терапевтического использования, то смесь разнообразных по составу протеинов, которая возникает при проведении неспецифического пегилирования, приводит к трудностям в получении продукта с воспроизводимыми и поддающимися определению свойствами. Это чрезвычайно затрудняет оценку лечения и получение информации о дозировке и эффективности.
Обнаружено, что в некоторых случаях введение многомерных комплексов, которые содержат больше чем один биологически активный полипептид или медикамент, может привести к синергическим положительным эффектам. Например, комплекс, содержащий два идентично связанных полипептида, может иметь существенно большее сродство к лиганду или активному центру, которые он связывает, по сравнению с одномерным полипептидом. По этой причине, желательно иметь многомерные комплексы протеинов, чтобы увеличить сродство протеина к этому лиганду в дополнение к увеличению молекулярной массы комплекса.
Протеины часто проявляют свой биологический эффект через взаимодействие с другими протеинами. В тех случаях, когда достаточно простого комплекса двух протеинов, чтобы достигнуть биологического эффекта, оказалось возможным сымитировать физиологическое действие эндогенных протеинов, вводя экзогенные протеины. Однако там, где биологическое воздействие требует составления комплекса, содержащего более чем два протеина, воспроизвести функцию эндогенных протеинов с помощью рекомбинантно полученных экзогенных эквивалентов оказывается более трудным из-за того, что комплексы более высокого порядка часто нестабильны. В таких случаях может быть выгодным использовать сшитые разновидности, содержащие два из компонентов комплекса, чтобы симулировать биологически активный комплекс.
Вслед за описываемым здесь изобретением, по меньшей мере три исследовательские группы сообщили о получении сшитых протеинов, в которых внеклеточные части одного из рецепторов ФНО присоединяют к тяжелой цепи иммуноглобулина IgG человека или мыши, которые затем сшивают дисульфидными связями /9/. В каждом случае, эти протеины экспрессировали в экспрессивных системах животной клетки и, обнаружили, что они являются значительно более эффективными при ингибировании ФНО, чем просто одномерный растворимый рецептор. Аналогичные способы использовали для получения похожих сшитых протеинов из CD4 протеина /10/, CR1 протеина /11/ и CR2 протеина /12/.
Показано, что эти сшитые протеины, построенные из двух полипептидных цепей и части антитела иммуноглобулина IgG, являются перспективными терапевтическими агентами. Сшитые протеины имеют увеличенную молекулярную массу, что способствует снижению скорости выведения комплекса из организма, в дополнение к явному усилению сродства протеинов к их лигандам. Однако протеины, сшитые таким способом, до сих пор были получены только экспрессией в экспрессивных системах животной клетки экспрессией слившихся генов. Это было необходимо для того, чтобы та часть протеина, которую представляет IgG, была правильно упакована после экспрессии. Кроме того, закрепленная тяжелой цепью часть антитела IgG, которая служит связующим звеном - спейсером или линкером - между полипептидными единицами, не позволяет изменить длину, размер и геометрию спейсера. При данном явном синергическом эффекте, достигнутом посредством двумерных протеинов, путем изменения пространственной ориентации полипептидов преимущества синергического эффекта можно, вероятно, оптимизировать. И, наконец, сшитые протеины могут быть антигенными и/или обладать пониженной растворимостью. Тяжелая цепь антител не является биологически инертной.
Описаны другие двумерные или "бивалентные" комплексы. Одна такая группа двумерных соединений названа "гирулоги". Эти соединения включают очень короткие полипептидные единицы, которые соединены коротким полиглициновым спейсером или линкером. Одна из полипептидных единиц представляет собой ингибитор тромбина - последовательность из 5 аминокислот, взятая из протеина гирудина, состоящего из 65 аминокислот - а другая является ингибитором узнавания анион-связывающего экзоцита (АСЭ) /13/.
C-реактивный протеин (CРП) представляет собой протеин острофазной сыворотки, построенный из пяти 23 к a суб-единиц. CРП может вызывать реакции осаждения и агглютинации, а также может взаимодействовать с Clg для активации классического пути комплемента.
С использованием бис(сульфосукцинимидил)субстрата или 3,3-дитио(сульфосукцинимидилпропионат) а в качестве сшивающих агентов, получены сшитые олигомеры CРП /14/.
Исследовано также образование двумерных бивалентных лигандов для конъюгирования опиодных рецепторов. Непептидные фармакофоры β- нальтексамин или оксиморфамин были соединены короткими этиленоксидными или глициновыми спейсерами /15/. Тетрапептидные энкефалины, соединенные короткими метиленовыми мостиками, также были предназначены для конъюгирования опиоидных рецепторов и, как было показано, обладали большей селективностью и сродством к дельта рецептору, чем исходный дельта лиганд /16/.
Посредством сшивки через остатки сахаров также был получен в многомерных формах гликопротеин поверхности клетки CD4. В качестве сшивающего агента использовали бисмальимидогексан (БМГ) /17/.
Антиген-3, ассоциированный с лимфоцитной функцией (ЛФА-3) представляет собой широко распространенный гликопротеин поверхности клетки, который является лигандом для T лимфоцита CD2. ЛФА-3 со своими ассоциированными лигандами образует протеиновые мицеллы из восьми мономеров, которые увеличили их способность взаимодействовать с клетками, на поверхности которых находится CD2 /18/.
Используя относительно близкую методику, одна группа изучала ингибирующее влияние синтетического полипептида, который включал повторяющуюся пентапептидную единицу. Полимер синтезировали полимеризацией в присутствии дифенилфосфорилазида до достижения около 10 000 дальтонов. Этот полимеризованный пентапептид является одной из важнейших структур в нескольких биологических реакциях /19/.
Дальнейшее препятствие в разработке эффективных экзогенных протеинов, которые бы увеличивали или конкурировали с эндогенными веществами, заключается в том, что экзогенные протеины должны предпочтительно вводиться систематически, чем быть локализованы в соответствующем месте. Это может приводить к более низкой эффективности и к увеличению побочных эффектов. Несколько исследовательских групп сообщают о попадании биоактивных протеинов в соответствующие сайты посредством связывания их с другими протеинами, которые естественным образом направляются на эти сайты. Часто такое связывание осуществляют через слияние генов активного и попадающего протеинов.
После появления настоящего изобретения поступило сообщение о том, что для создания связанных суперантигенов антител, были использованы спейсеры или линкеры из полиэтиленгликоля. Моноклональный антиген, реактивный по отношению к клеткам карциномы прямой кишки, был присоединен к стафилококковому энтеротоксину бактериального суперантигена. Эти комплексы предназначены скорее не для использования преимуществ, связанных с другими бивалентными комплексами (например, более высокая молекулярная масса; синергические эффекты бивалентности), а для конъюгирования определенных зон суперантигенов. Описанный для образования этих связанных суперантигенов способ пегилирования дает комплекс, который содержит широкую смесь веществ. Сочетание данного антитела и этого суперантигена осуществляли при использовании - сукцинимидил 3-(2-пиридилдитио) пропионата и гидрофильного спейсера на основе ПЭГ с цепочкой из 24 атомов. В соответствии с этим способом, к каждой единице антитела были присоединены от 7 до 18 связующих элементов, и на каждом из суперантигенов прореагировали один или два лизина /20/. Используя этот способ, было бы невозможно выделить один тип продукта реакции для того, чтобы оптимизировать продукт или способ.
Две группы протеиновых веществ, имеющих важные применения при лечении широкого разнообразия медицинских показаний, представляют собой ингибиторы фактора некрозоопухолей (ФНО) и антагонисты рецепторов интерлейкина-1 (IL-1ra). Показано, что эти вещества оказывают положительный эффект при лечении болезней, вызванных ФНО и IL-1 соответственно. Среди показаний, идентифицированных как принадлежащие к вызываемым либо ФНО, либо IL-1, оказываются синдром респираторного нарушения взрослых, легочный фиброз, ревматоидный артрит, воспаление кишечника и септический шок.
Находящаяся на рассмотрении заявка, ссылка на которую здесь приведена /21/, описывает класс естественно встречающихся протеиновых ингибиторов ФНО и способ получения их в существенных количествах с высокой степенью чистоты. В частности, в вышеупомянутой заявке детально описаны две субсерии ингибиторов ФНО, обозначенные как 30 кДа ингибитор ФНО и 40 кДа ингибитор ФНО. Кроме имеющего полную длину цепочки протеина 40 кДа ингибитора ФНО, получены также две урезанные, однако биологически активные формы 40 кДа ингибитора ФНО. Эти протеины, в которых из имеющего полную длину цепи протеина были убраны 51 и 53 аминокислоты с концевыми карбоксильными группами, обозначают соответственно как 40 кДа-ингибитор ФНО Δ 51 и 40 кДа ингибитор ФНО Δ 53.
Находящаяся на рассмотрении заявка /22/, ссылка на которую здесь приведена, описывает предпочтительный класс природных протеиновых ингибиторов IL-1 и способ получения их в существенных количествах с высокой степенью чистоты. В частности, в заявке подробно описаны три таких ингибитора интерлейкина-1, которыми являются антагонисты рецепторов интерлейкина-1 (IL-1ra's), а именно, IL-1ra α, IL-1ra β и IL-1rax.
Два дополнительных класса веществ, которые потенциально полезны для лечения при разнообразных медицинских показаниях, представляют собой ингибиторы интерлейкина-2 и ингибиторы комплемента. Потенциальные ингибиторы интерлейкина-2 включают рецепторы интерлейкина-2, внеклеточный домен рецепторов интерлейкина-2, антагонисты рецепторов интерлейкина-2, антитела, которые узнают интерлейкин-2, и фрагменты любых таких веществ, которые содержат связующую функцию IL-2. Потенциальные ингибиторы системы комплемента включают рецептор CR1, внеклеточный домен R1 и фрагмент CR1, который содержит функцию связывания комплемента.
В /23/ описаны рецептор интерлейкина-2 и способы его выделения. Описан также ген-кодирующий рецептор интерлейкина-2 и методы для его рекомбинантного получения /24/.
Можно допустить, что до определенной степени растворимый внеклеточный домен любого рецептора интерлейкина-2 будет действовать как ингибитор действия интерлейкина-2 цитокина. Интерлейкин-2 является одним из наиболее охарактеризованных цитокинов, о которых известно, что они играют центральную роль в антигенспецифическом клоновом росте T лимфоцитов. Было также показано, что интерлейкин-2 действует на разновидности других клеток в иммунной системе.
Существуют три дискретных формы рецептора интерлейкина-2, включающие две различные молекулы рецептора, обозначенные или как IL-2r α, или как IL-2r β.
Рецептор IL-2 с самым сильным сродством состоит из двух отличающихся рецепторов IL-2. Оба эти рецептора были клонированы и охарактеризованы. Рецептор IL-2 с низким сродством (IL-2r α ) был клонирован в 1984 г. и был подробно охарактеризован /25/. Внеклеточный домен этой молекулы имел молекулярную массу 24825 и имел два N - связанные сайта гликозилирования. Эта молекула содержит 11 цистеинов, 10 из которых участвуют во внутримолекулярных дисульфидных связях. Предполагаемые ломены связывания IL-2 с этой молекулой были нанесены на карту как с помощью матугенеза, так и эпитопным картированием. Рецептор IL-2 с промежуточным сродством (IL-2r β\ ) был клонирован в 1989 г. и не был так полно охарактеризован, как IL-2r α /26/. Внеклеточный домен IL-2r β имеет молекулярную массу 24693. Молекула содержит 8 цистеинов и 4 N-связанных сайта гликозилирования. Образование дисульфидных связей в данной молекуле неизвестно. IL-2r β имеет цитоплазменный домен из 286 аминокислот.
Для IL-2 рецепторов были определены константы диссоциации (Kд). Они составляют 10-8 М для IL-2r α, 10-9 М для IL-2r β и 10-11 М для рецептора с высоким сродством, который состоит из комплекса IL-2r α , IL-2r β и IL-2. Существующие модели указывают, что формирование комплекса с высоким сродством протекает сначала через присоединение IL-2 к IL-2r α, а затем к IL-2r β /27/.
Ингибитор IL-2 может быть ценным в предупреждении отторжения трансплантата, так же как и при аутоиммунных заболеваниях. В настоящее время моноклональное антитело, которое препятствует связыванию IL-2, испытывают при почечной трансплантации у человека /28/. В одном исследовании на 15 пациентах было показано, что данное антитело, в сочетании с иммунодепрессантами, является столь же эффективным в предупреждении отторжения аллографта, как в контрольной группе, получавшей более высокие дозы иммунодепрессантов. При многих заболеваниях, некоторых инфекциях, так же как и при трансплантации и отторжении обнаружено высокое содержание в крови растворимого IL-2r α. Это наводит на мысль об участии IL-2 в этих заболеваниях.
CRI представляет собой протеин, также обозначаемый как C3b/C4b рецептор. CRI присутствует на эритроцитах и ряде других типов клеток и специфически связывает C3b, C4b и iC3b. CRI может также ингибировать классический и альтернативный путь C3/C5 конвертаз и действовать как ко-фактор для расщепления C3b и C4b фактором 1 /29/. CRI - это гликопротеин, состоящий из одной полипептидной цепи, и он существенно в четырех аллотипичных формах. Известно, что CRI содержит повторяющиеся кодирующие последовательности, и этот факт используют, чтобы объяснить существование многочисленных аллотипов /30/.
Пониженную экспрессию CRI на эритроцитах связывали с красной волчанкой, и было также обнаружено, что число CRI обратно пропорционально содержанию сыворотки иммунных комплексов. Сам CRI протеин, CRI ген и способы получения CRI описаны в /31/. Как описано выше, были также описаны двумерные образования, содержащие CRI и домены антитела /32/.
Данное изобретение относится к способу модификации полипептидов и полученным в результате модифицированным полипептидам.
Данное изобретение включает по существу чистые соединения, охватываемые формулой R1-X-R2, в которой R1 и R2 представляют собой биологически активные группы, а X является непептидным полимерным спейсером. R1 и R2 могут быть одинаковыми или различными группами, и по крайней мере один из них, R1 или R2 является пептидным. В предпочтительных практических реализациях R1 и R2 представляют собой или антагонист рецептора интерлейкина-1; или 30 кДа ингибитор ФНО; или рецептор интерлейкина-2 или CRI, а X представляет собой или полиэтиленгликоль, или полиоксиэтилированный глицерол, или декстран, или кислоты прямой кишки, или поли -β- аминокислоты, или углеводные полимеры. Включены также фармацевтические составы, содержащие также по существу чистые соединения в смеси с фармацевтически приемлемым носителем. Далее включены способы лечения пациентов, нуждающихся в этом, такими терапевтическими составами. Соединения формулы R1-X-R2, изображенные на фиг. 19, в дальнейшем называют "гантелями".
Это изобретение также включает способ получения по существу чистых терапевтически ценных соединений, охватываемых формулой R1-X-R2, который включает взаимодействие непептидной полимерной группы, имеющей по крайней мере две реакционноспособные группы, способные образовывать ковалентные связи, с биологически активной группой R; и выделение указанного продукта.
В другой практической реализации, это изобретение включает способ получения по существу чистых терапевтически ценных соединений, охватываемых формулой R1-X-R2, где R1 и R2 различны, который заключается в: проведении взаимодействия непиптидной полимерной группы, способный образовывать ковалентные связи при реагировании с биологически активной группой R1, с образованием комплекса R1-X; проведении взаимодействия комплекса R1-X с биологически активной группой R2 с получением указанного соединения; и выделении и очистке указанного соединения.
В одной практической реализации, это изобретение относится к сайт-специфическому пегилированию ингибитора ФНО и ингибитора IL-1,
Для того чтобы обеспечить сайт-специфичность пегилирования, выбирают такие пегилирующие реагенты, которые будут реагировать почти исключительно со свободными SH-группами остатков цистеина в полипептидах. Примером реагента пегилирования, который почти исключительно связывается SH-группами цистеина, является O-(2-мальимидоэтил)-O'-метилполиэтиленгликоль.
Сайт-специфическое пегилирование можно осуществить либо на природных "свободных" цистеиновых остатках данного полипептида, либо на свободных цистеинах, содержащихся на мутеинах природных полипептидов. Цистеин можно как добавлять или вводить в аминокислотную последовательность природного полипептида, так и замещать ими другие аминокислотные остатки в выбранном месте.
В одной практической реализации этого изобретения полипептиды, которые подлежат пигелированию, получают посредством рекомбинантной ДНК-технологии из бактериальной клетки-хозяина. В большинстве случаев бактериально экспрессированный полипептид до стадии пегилирования должен быть повторно уложен, чтобы получить биологическую активность. В некоторых примерах данного изобретения нативный полипептид не содержит никаких свободных цистеиновых остатков, но тогда получают измененный полипептид, содержащий по крайней мере один свободный цистеин в биологически активном полипептиде. Согласно этому способу, повторную укладку бактериально экспрессированного полипептида облегчают присоединением, поочередно, сульфгидрила, содержащего такое соединение как цистеин, и дисульфида, содержащего такое соединение как цистеин. После повторной укладки и очистки этот полипептид обрабатывают ограниченным количеством мягкого восстановителя, такого как дитиотрейтол ("ДТТ"), чтобы регенерировать сульфгидрильную группу этого нового цистеинового остатка измененного полипептида. После диализа в условиях, рассчитанных на то, чтобы не допустить окисления, полипептид можно привести во взаимодействие с агентом специфического пегилирования цистеина, чтобы сайт-специфически получить ковалентно модифицированный полипептид.
Предпочтительные пегилированные полипептиды настоящего изобретения представляют собой сайт-специфически пегилированные ингибиторы ФНО и ингибиторы IL-1. Более конкретно данное изобретение описывает пегилированный 30 кДа ингибитор ФНО и пегилированный антагонист рецептора IL-1. Наиболее предпочтительные пегилированные ингибиторы ФНО включают 30 кДа ингибитор ФНО, в котором остаток аспарагиновой аминокислоты в позиции 105, нативного протеина человека заменяют на цистеин, используя in vitro мутагенез, и пегилирование происходит на свободном цистеина в позиции 105. Другие пегилированные производные мутированных 30 кДа ангибиторов ФНО включают мутации, при которых цистеин был присоединен в позициях 1, 14, 111 и 161. Кроме однократно пегилированных мутеинов, любая и всех комбинации различных мутаций могут быть включены в один мутеин для получения измененного 30 к а ингибитора ФНО с более чем одним свободным цистеиновыим остатком, способным к пегилированию.
Наиболее предпочтительный пегилированный IL-1ra включает природный или нативный IL-1ra, который содержит четыре свободных цистеина. Монопегилирование нативного IL-1ra приводят к сайт-специфическому пегилированию при цистеине в позиции 116. Другие пегилированные производные мутированного IL-1r α включают мутаины, имеющие цистеин, присоединенный к аминным концам полипептида, цистеин, присоединенный в позициях 6, 8, 9, 84 или 141, и замещение цистеина в позиции 116 серином. Кроме однократно пегилированных мутеинов, любая и все комбинации различных мутаций могут быть использованы для того, что получить измененный и IL-1ra с более чем одним свободным цистеином, способным к пегилированию
Другие аспекты и преимущества настоящего изобретения будут очевидны при рассмотрении последующего подробного описания изобретения, включающего иллюстративные примеры практического применения изобретения.
Фиг. 1 представляет последовательность аминокислот нативного IL-1ra.
Фиг. 2 представляет последовательность нативного 30 кДа ингибитора ФНО.
Фиг. 3 представляет результат электрофореза непегилированной и пегилированной форм IL-1ra и мутеинового c84s116 IL-1ra в системе ДСН-ПААГ )окрашивание Кумасси). Дорожки 2, 3, 5 и 6 содержат реакционные смеси пегилирования. Дорожки 1 и 4 представляют немодифицированные протеины:
Дорожка 1 - IL-1ra
Дорожка 2 - мПЭГ5000 * IL-1ra
Дорожка 3 - мПЭГ8500 * IL-1ra
Дорожка 4 - c84s116 IL-1ra
Дорожка 5 - мПЭГ5000c84s116 IL-1ra
Дорожка 6 - мПЭГ8500c84s116 IL-1ra
Фиг. 4 представляет результаты ионообменной хроматографии на сорбенте моно S: хроматограмма A - реакционная смесь пегилирования при получении мПЭГ5000 *IL-1ra, пик 1 относится к модифицированному, а пик 2 - к немодифицированному IL-1ra; хроматограмма B представляет очищенный мПЭГ5000 *IL-1ra.
Фиг. 5 изображает хроматограмму, полученную способом ВЭЖХ, показывающую профиль элюции нескольких веществ - стандартов размеров, и мПЭГ8500 *IL-1ra (фракция 7) и IL-1ra (фракция 13).
Фиг. 6 представляет результаты фракционирования методом обращенно-фазной ВЭЖХ продуктов триптического расщепления алкилированного мПЭГ5000 *IL-1ra, обработанного меченной тритием иодуксусной кислотой для того, чтобы поместить свободные цистеины. Разделение проводили на колонке Браунли C8 (2,1 х 220 мм) при комнатной температуре и скорости потока • 1000 мкл/мин с линейным градиентом. Растворитель A представлял собой 0,1%-ный раствор ТФУК (трифторуксусуной кислоты) в воде, а растворитель B - 0,085%-ный раствор ТФУК в смеси из 80% ацетонитрила и 20% воды.
Фиг. 7 представляет результаты фракционирования методом обращенно-фазной ВЭЖХ химотриптического расщепления пептида 18 на фиг. 6. Условия идентичны условиям для фиг 6. Пептиды 5 и 8 содержат тритиевые метки, и пептид 5 имеет аминокислотную последовательность LCTAMEADQPVSL. Цистеин был идентифицирован в виде карбоксиметилцистеинового производного. Этот цикл был единственным, обнаружившим радиоактивность выше уровня фона. Аминокислотная последовательность пептида 8 начиналась с серина 103 IL-1ra. Повторное расщепление этого пептида с химотрипсином позволило элиминировать все тритиевые метки из полипептида.
Фиг. 8 представляет профили изменения концентрации плазменного IL-1ra во времени для зрелого IL-1ra, пегилированного IL-1ra и нескольких пегилированных мутеинов IL-1ra.
Фиг. 9 представляет результат электрофореза в системе ДСН-ПААГ, показывающий с105 30 кДа ингибитор ФНО мПЭГ и разделение непрореагировавшего 30 кДа ингибитора ФНО от мПЭГ с105 30 кДа ингибитора ФНО методом ГПХ.
Фиг. 10 представляет кривые изменения внутривенной концентрации плазменной IL-1ra во времени для ряда образцов однократно пегилированного IL-1ra, двукратно пегилированного IL-1ra и гантелеобразных разновидностей IL-1ra ПЭГ.
Фиг. 11 представляет кривые изменения подкожной концентрации плазменного IL-1ra во времени для того же ряда соединений IL-1ra, как на фиг. 10.
Фиг. 12 показывает содержание плазменного IL-G во времени после введения мыши ФНО.
Фиг. 13 представляет сравнение содержания IL-6, индуцированное в мыши при пяти соотношениях с105s30 кДа ингибитора ФНО к ФНО (A) и пяти соотношениях соединения с105s30 кДа ингибитор ФНО ПЭГ2000 (гантель) к ФНО (B).
Фиг. 14 представляет содержание плазменной IL-6, индуцированное в мышах одним ФНО и смесями 1: 1 ФНО с гантелеобразными соединениями с105s30 кДа ингибитор ФНО ПЭГ3500 и ПЭГ10000.
Фиг. 15 представляет процент нейтрофилов, индуцированных при различных соотношениях ФНО к с105s30 кДа ингибитору ФНО (A); с105s30 30 кДа ингибитору ФНО ПЭГ3500 (гантель) (B); с105s30 кДа ингибитору ФНО ПЭГ10000 (гантель) (C); и с105s30 кДа ингибитору ФНО ПЭГ20000 (гантель) (D).
Фиг. 16 представляет зависимости изменения внутривенной концентрации плазменного 30 кДа ингибитора ФНО во времени для нативного 30 кДа ингибитора ФНО, с105s30 кДа ингибитора ФНО ПЭГ8500 и ПЭГ10000, и для гантелеобразных структур 30 кДа ингибитора ФНО ПЭГ3500, ПЭГ10000 и ПЭГ20000.
Фиг. 17 представляет зависимости изменения подкожной концентрации плазменного 30 кДа ингибитора ФНО от времени для того же ряда соединений 30 кДа ингибитора ФНО как на фиг. 16.
Фиг. 18 представляет растворимость для трех растворов нативного IL-1ra и с84 IL-1ra ПЭГ8500 по изменению оптической плотности при 450 нм во времени.
Фиг. 19 представляет основную структуру соединений в соответствии с настоящим изобретением, имеющих общую формулу R1-X-R2, которые называют соединения-гантели.
Это изобретение включает селективную модификацию фармацевтически полезных полипептидов, в частности, ингибиторов фактора некрозоопухолей ("ФНО") и ингибиторов интерлейкина-1 ("IL-1"). Конкретнее, это изобретение описывает селективную модификацию 30 кДа ингибитора ФНО и антагониста рецептора IL-1 ("IL-1ra") Селективные модификации служат как для усиления фармакокинетических свойств полипептидов, так и для получения гомогенных составов для терапевтического использования в применении к человеку.
Дополнительные полипептиды, которые могут быть селективно модифицированы согласно данному изобретению, включают рецепторы инетерлейкина-2 ("IL-2r") и CRI. Все ссылки на рецептор интерлейкина-2 следует понимать так, что они включают как α-, так и β- цепи IL-2r, если не оговариваются специально.
В предпочтительных практических реализациях данного изобретения модифицированные полипептиды и ДНК последовательности являются человеческими. Однако в той степени, в какой существует достаточная гомология между ДНК и пептидными последовательностями животных и такими же формами человека, их можно включать в сферу данного изобретения.
В одной конкретной реализации способ модификации согласно настоящему изобретению включает сайт-специфическое ковалентное связывание длинноцепных полимеров с представляющими интерес полипептидами. Выбранные полипептиды могут быть нативными или представляющими интерес природными полипептидами, или они могут представлять собой биологически активные мутеины полипептидов, которые были получены с целью усилить процесс модификации, описываемый здесь. Этот способ согласно настоящему изобретению включает селекцию, получение и отбор желаемых мутеинов, которые удовлетворяют целям настоящего изобретения. В других практических реализациях этого изобретения данный способ для модификации полипептидов требует только проведения модификации таким образом, чтобы полученный в результате продукт представлял собой по существу чистую форму, как этот термин определен в изобретении.
В некоторых практических реализациях изобретения, модифицированные полипептиды в соответствии с настоящим изобретением связывают с длинноцепными полимерами в специфических точках аминокислотной последовательности. Такие модифицированные полипептиды в соответствии с настоящим изобретением сохраняют значительную часть своей биологической активности. В предпочтительных практических реализациях модифицированные полипептиды сохраняют по крайней мере одну десятую биологической активности нативного полипептида при испытании связывания рецептора. В более предпочтительной конкретной реализации, модифицированный полипептид будет сохранять по крайней мере одну пятую биологической активности нативного полипептида, а в самой предпочитаемой конкретной реализации сохранит по крайней мере одну четвертую активности. Кроме того, модифицированный полипептид будет служить улучшению фармакокинетической характеристики нативного полипептида по крайней мере в одной из следующих областей:
1) увеличение кажущейся молекулярной массы нативного полипептида и, следовательно, уменьшение скорости выведения после подкожного или системного введения;
2) увеличение растворимости нативного полипептида в водных растворах; или
3) снижение антигенности нативного полипептида.
В многочисленных практических реализациях данного изобретения достигается каждая из этих целей. В предпочтительных практических реализациях данного изобретения длинноцепной полимер представляет собой полиэтиленгликоль или монометоксиполиэтиленгликоль. Структурная единица полиэтиленгликоля обозначена здесь как ПЭГ, а структурная единица монометоксиполиэтиленгликоля - как мПЭГ. Приблизительная молекулярная масса этих структурных единиц указана около их обозначений внизу. Например, единица монометоксиполиэтиленгликоля с приблизительной молекулярной массой 5000 будет обозначена как мПЭГ5000 или ПЭГ5000. Другими длинноцепными полимерами, включенными в сферу данного изобретения, являются полипропиленгликоль ("ППГ"), полиоксиэтилированный глицерол ("ПОГ"), декстран, кишечные кислоты или другие полимеры на основе углеводов, и полимеры β- аминокислот и производных биотина.
В другой практической реализации настоящего изобретения, единица длинноцепнонго полимера представляет собой дигидроксиполиэтиленгликоль, или HO-(CH2CH2O)n-H. Будучи активировано для последующего ковалентного присоединения к полипептидам или другим биологически активным соединениям, как описано ниже, такое дигидрокси-вещество будет содержать два реагирующих сайта.
В предпочтительных практических реализациях настоящего изобретения длинноцепные полимерные единицы связаны с полипептидом через ковалентное присоединение к сульфгидрильной группе (-SH) цистеинового остатка. Для достижения селективности реакции и гомогенности реакционной смеси, полезно использовать функциональные полимерные единицы, которые будут реагировать специфически с сульфгидрильными группами. Функциональную или реагирующую группу, соединенную с длинноцепным полимером, обозначают здесь как активирующую группу. Активирующую группы включают мальимидную группу, сульфгидрильную группу, тиол, трифлат, трезилат, азиридин, оксиран и 5-пиридил. Предпочтительными активирующими группами являются мальимидные.
Активированные дигидроксиполиэтиленгликоли из-за физической удаленности концов полимерной цепи являются почти одинаково реакционноспособными на каждом конце молекулы. При соответствующем выборе реакционных условий и полипептидов, активированные дигидроксиполиэтиленгликоли или любая другая многократноактивированная единица длинноцепного полимера будут реагировать с полипептидами с образованием гантелеобразных комплексов, в которых два полипептида соединяются длинноцепной полимерной структурной единицей.
Используя различия, которые были обнаружены в скоростях реакций между активированной полимерной связанной группой и различными цистеинсодержащими полипептидами, и особенности кинетики этих реакций, опытным исследованиям легко получить гантелеобразные комплексы, в которых по существу чистые соединения могут быть образованы включением двух различных полипептидных групп, или включением одной полипептидной группы и отличной биологически активной группы. Примеры таких гетеро-гантельных соединений даны ниже.
Пределы и доступность для реакции цистеинов резко меняется от полипептида к полипептиду. Поэтому в биологически активной форме многие полипептиды не имеют "свободных" цистеинов, или цистеинов, не связанных с другим цистеином. Кроме того, существование "свободных" цистеинов не означает, что цистеины доступны для связывания с реакционноспособными реагентами. Поскольку модификацию обычно осуществляют на активном или трехмерно упакованном полипептиде, реакция происходит незначительно или совсем не происходит в тех случаях, когда свободный цистеин находится внутри складчатой структуры. Дальнейшее ограничение при модификации полипептидов заключается в потенциальном воздействии, которое модификация может оказывать на активный сайт полипептида. Модификация цистеина, относительно близко расположенного от активного сайта, может фактически дезактивировать полипептид. Даже когда о выбранных полипептидах известно много, трудно, если не невозможно, точно предсказать, какие цистеиновые остатки можно эффективно модифицировать.
Те же самые факторы существуют также, когда получают мутированные полипептиды, содержащие дополнительные цистеиновые остатки. Когда такой полипептид рекомбинантно получают через бактериальную экспрессию, ненативные протеины могут вмешиваться в правильную упаковку полипептида. Кроме того, цистеин должен быть доступен для агента пегилирования, и пегилированный цистеин не должен в значительной степени мешать активному сайту полипептида.
На выбор в данном полипептиде потенциальных сайтов для введения ненативного цистеина можно влиять, основываясь на информации из различных источников. Например, хорошим место для мутации через включение свободного цистеина могут быть сайты гликозилирования. В этой степени, в которой имеется информация о связывании или активном сайте этого полипептида, ее также можно использовать для отбора потенциальных мутаинов. Присоединение или замещение цистеинового остатка на аминных или карбоксильных концах полипептида также может оказаться перспективным благодаря своему местоположению. И, наконец, можно рассматриваться мутацию лизиновых остатков в цистеин, основываясь на предположении, что лизины обычно находятся на поверхности биологически активного полипептида.
Хотя для любого данного полипептида можно выбрать много потенциальных мутаинов, которые могут удовлетворять желаемым характеристикам, только после синтеза, пегилирования и испытания таких измененных мутаинов станет известно, какой из них будет отвечать целям настоящего изобретения. В свете данного изобретения и при опыте и знаниях профессионалов, такой синтез, пегилирование и испытание можно провести без чрезмерного экспериментирования. Следует отметить, что даже если пегилирование полипептидов вызывает снижение, в некоторой степени, биологической активности полипептида, то улучшение фармакокинетического действия этого полипептида может значительно увеличить ценность данного нативного полипептида в различных терапевтических приложениях.
При выборе мутеинов мишени предпочтительный способ для получения этих мутаинов заключается в рекомбинантном экспрессировании кодирующего гена для данного мутаина. Полагая, что кодирование гена для нативного полипептида известно, измененный ген можно создать или стандартными способами сайт-специфического мутагенеза на нативном гене, или построением измененного гена стандартными способами генного синтеза. Такая техника хорошо известна специалистам в этой области.
Кодирующий ген для мутаина мишени может быть экспрессирован во многих экспрессивных системах, включая системы животных, насекомых и бактерий. Хотя такие системы были отработаны для экспрессии нативных полипептидов, в определенной степени те же системы можно использовать для мутеинов мишени. В предпочтительной конверсии реализации настоящего изобретения, кодирующие гены для мутеинов мишени получены посредством сайт-специфического мутагенеза нативного гена, а ген, кодирующий данный мутеин, экспрессирован из бактериальной экспрессивной системы. Ген, кодирующий нативный IL-1ra, и способ для экспрессирования указанного гена в E.Coli подробно описан в /33/.
Ген, кодирующий нативный 30 кДа ингибитор ФНО, и способ экспрессирования указанного гена в E.Coli детально описаны в заявке /34/. Каждая из заявок здесь включена с данной ссылкой,
Мутеины и пегилированные вещества согласно настоящему изобретению включают аллельные вариации в протеиновых последовательностях (вариации последовательности, обусловленные естественной изменчивостью от индивидуума к индивидууму) и по существу эквивалентные протеины. "По существу эквивалентные", как этот термин употребляют в описании и формуле изобретения, обозначает, что имеют в виду наличие очень высокой степени гомологии аминокислотных остатков ((/35/, ссылка включена здесь специально), так же как наличие сопоставительной биологической активности. В сферу данного изобретения включены также мутеины и пегилированные полипептиды, которые представляют собой частично урезанные версии нативных полипептидов.
В одной предпочтительной конкретной реализации способа настоящего изобретения в котором мутеин мишени получают путем рекомбинантной ДНК технологии в бактериальной экспрессивной системе, осуществляют следующие стадии:
1) Кодирующий ген для мутеина мишени создают посредством сайт-направленного мутагенеза кодирующего гена для нативного полипептида;
2) Кодирующий ген для мутеина мишени экспрессируют в бактериальной экспрессивной системе;
3) Отделяют от бактерий и очищают мутеин мишени;
4) Мутеин мишени подвергают повторной укладке в присутствии цистеина или другого содержащего сульфгидрильную группу соединения;
5) Повторно уложенный мутеин мишени выделяют и очищают;
6) Очищенный и повторно уложенный мутеин мишени обрабатывают мягким восстановителем;
7) Реакционную смесь подвергают диализу в отсутствие кислорода; и
8) Диализированную реакционную смесь обрабатывают длинноцепным полимером, содержащим активирующую группу.
В предпочтительной конкретной реализации для получения пегилированных мутеинов 30 кДа ингибитора ФНО, мягкий восстановитель представляет собой дитиотрейтол ("ДТТ"). В другой конкретной реализации модификации может происходить раньше повторной укладки экспрессированного протеина или мутеина.
В предпочтительной конкретной реализации настоящего изобретения пегилированные мутеины и пегилированные нативные полипептиды могут быть очищены и введены в фармацевтические составы обычными способами. В другой практической реализации, очищенные мутеины также можно ввести в фармацевтические составы.
Пегилированные полипептиды в соответствии с настоящим изобретением, образованные реакцией дезактивированного длинноцепного полимера, обладают дополнительными полезными свойствами. Эти гантелеобразные молекулы могут содержать два представляющих интерес полипептида, связанных одним полимерным отрезком. Такая структура придает определенную степень линейности полимерной молекуле и несколько ослабляет стерические затруднения, присущие использованию таких больших гидрофильных полимеров, как полиэтиленгликоль. Достигается цель получения молекул с увеличенной кажущейся молекулярной массой при сохранении высокой биологической активности. В сферу настоящего изобретения особенно включены бидентатные молекулы, где две молекулы IL-1ra или две молекулы ингибитора ФНО ковалентно связаны с одной полимерной цепью, или где два разных полипептида связаны с одной полимерной цепью, т.е. одна бидентатная молекула состоит из двух частей: ингибитора ФНО и IL-1ra-части.
Нативный IL-1ra (фиг. 1) и многочисленные мутеины IL-1ra пегилировали согласно настоящему изобретению. Пегилирование дикого типа IL-1ra по свободным сульфгидрильным группам способами, описанными ниже в примерах, приводит к присоединению мПЭГ к остатку цистеина в позиции 116 IL-1ra (с116). Другие три цистеина в полностью нативной молекуле недоступны для пегилирования. Для того чтобы присоединить молекулы мПЭГ к различным сайтам IL-1ra и получить продукты присоединения мПЭГ, содержащие более одной молекулы мПЭГ, по концевым аминогруппам этого протеина присоединяют IL-1ra, в котором нативные аминокислоты в IL-1ra были замещены цистеином, или дополнительные цистеины. Для того, чтобы получить продукты присоединения, в которых остаток 116 не пегилирован, с116 заменили не серин в большом числе мутеинов. Ниже приведен список мутеинов, которые были получены для взаимодействия с мПЭГ (нумерация остатков основана на последовательности, данной на фиг. 1; "с" относится к цистеину, а "s" - к серину):
c0s116 - c0c116
c84s116 - c84c116
c6s116 - c6c116
c8s116 - c8c116
c9s116 - c9c116
c141s116 - c141c116
Нативный 30 кДа ингибитор ФНО (фиг. 2) не содержит свободных остатков цистеина. Были получены следующие мутеины 30 кДа ингибитора ФНО (нумерация остатков основана на последовательности, приведенной на фиг. 2; "с" относится к цистеину):
c105 30 кДа ингибитор ФНО
c1 30 кДа ингибитор ФНО
c14 30 кДа ингибитор ФНО
c111 30 кДа ингибитор ФНО
c161 30 кДа ингибитор ФНО
В сферу данного изобретения включен целый класс соединений, показанных на фиг. 19, которые могут быть представлены формулой R1-X-R2, в которой R1 и R2 являются биологически активными группами, и по крайне мере одна из групп R1 или R2 является полипептидной, а X представляет собой непептидный полимерный сейсер или линкер. R1 и R2 могут быть одной и той же группой, или разными группами. В этих случаях, когда R1 и R2 представляют собой разные группы, как R1, так и R2 могут быть полипептидными группами, или R1 может быть полипептидным, а R2 может представлять собой любую биологическую активную группу. Соединения, имеющие такую структуру, которые были названы "гантельными" соединениями, отличаются тем, что являются по существу чистыми. "По существу чистыми" в данном контексте означает - быть гомогенным по составу.
Гомогенный по составу предполагает наличие одной молекулы линкера X, одной молекулы R1 и одной молекулы R2. Гомогенный по составу предполагает, но не обязательно, от факт, что и биологически активные группы R1 и R2 присоединены к линкеру точно в одинаковом месте этих групп в каждой молекуле такого соединения. В некоторых практических реализациях данного изобретения биологически активные группы связаны с линкером сайт-специфически. Например, в соединении c105 ингибитор ФНО ПЭГ3000 (гантель) две группы c105 30 кДа ингибитора ФНО связаны с линкером ПЭГ3000 при 105 цистеиновом остатке.
Когда говорят о "гомогенном по составу", это следует понимать так, что на уровне молекула-к-молекуле гантельное соединение совсем не обязательно гомогенно также относительно точной длины группы-спейсера. Профессионалам понятно, что любой процесс получения, в котором используют ПЭГ в данных пределах молекулярной массы или другой высокомолекулярный полимер, начинается с раствора, который содержит полимер "средней" молекулярной массы. Поэтому, когда звено ПЭГ с двумя реакционноспособными группами реагирует с полипептидной группой, это звено ПЭГ по определению является полидисперсным, и образующееся в результате гантельное соединение является гетерогенным в той степени, в какой подвержена изменениям длина линкера, о чем профессионалам известно. В итоге, "по существу чистый" в этом контексте относится к веществам, которые по существу свободны от соединений: 1) которые отклоняются по составу R1 или R2, или 2) которые связаны между собой более чем одним линкером X.
R1 и R2 определяют как биологически активные группы. Биологически активные группы включают любое соединение, которое может оказывать биологическое действие при взаимодействии с природной биологической молекулой. Биологически активные группы включают протеины, полипептиды, стероиды, углеводы, такие органические вещества как гепарин, металлосодержащие агенты, витамины или любые другие биологические активные вещества. По крайней мере одна из групп R1 или R2 является полипептидной. В предпочтительной практической реализации как R1, так и R2 являются полипептидными.
Полипептидный - определяют как любое соединение, которое является по существу протеиновым по своей природе. Однако полипептидная группа может содержать некоторые непептидные составные части. Например, в это определение включают гликозилированные полипептиды или синтетические модифицированные протеины.
Биологически активные группы R1 и R2 включают связывающие группы и целевые группы. Связывающие группы определяют по их сродству к данному биологическому лиганду. Целевые группы определяют по их сродству к данному биологическому лиганду. Целевые группы определяют по их способности направлять положение комплекса внутри биологической системы. R1 и R2 могут обладать сродством к одному и тому же лиганду, в этом случае "гантель" может усилить сродство к этому лиганду. R1 и R2 могут обладать сродством к разным лигандам, при этом R1 служит для того, чтобы направить данный комплекс в положение, где лиганд для R2 будет доминирующим.
Предпочтительными полипептидными группами являются рецепторы, внеклеточные части рецепторов, молекулы поверхности клеток, внеклеточные матричные молекулы, связывающие протеины и антагонисты рецептора. В полипептидные группы, которые могут быть использованы как R1 или R2, включают также следующие полипептиды и любые их фрагменты: антагонист рецептора IL-1, 30 кДа ингибитор ФНО, 40 кДа ингибитор ФНО, рецептор IL-2, (все ссылки на СК1 включают одиночный или комбинацию консенсусов повторяющихся последовательностей CR1), PDCF-рецептор, IL-2, MCSF-рецептор, EGF-рецептор, рецептор IL-5, рецептор IL-3, GMCSF рецептор, Т-клеточный рецептор, HLA-I, HLA-II, NGF-рецептор, IgG (VH, VI), CD40, CD27, рецептор IL-6, интегрины CR3, VLA4, ICAM и VCAM, CR2, GMP 140 Lec домен, ламинин-связывающий протеин, фрагменты ламинина, связывающий маннозу протеин, экзон 6 пептид PDGF, и протеазы (с 2 каталитическими доменами или целевым доменом и каталитическим доменом). Все ссылки на рецепторы включают все формы рецептора, если они существуют. В предпочтительных практических реализациях группы R1 и R2 представляют собой или антагонист рецептора IL-1, или 30 кДа ингибитор ФНО, или GRj, или рецептор IL-2 (как α-, так и β- цепи).
В предпочтительной практической реализации непептидный полимерный спейсер можно в дальнейшем определять следующим образом: X = -Y1 - (Z)n - Y2-, где Y1 и Y2 представляет собой остатки активирующих групп, которые реагируют с R1 и R2 так, чтобы связать спейсер с группами R1 и R2, а (Z)n представляет собой основную полимерную группу. В соответствии с настоящим изобретением, n больше 6 и предпочтительно больше чем 10.
Непептидной называют полимерную группу, которая по существу не является пептидной по своей природе. При включении менее 50 мас.% α- аминокислотных остатков в качестве составной части Y1, Y2 и Z считают непептидными по своей природе, и их рассматривают как непептидные. В предпочтительной практической реализации непептидный линкер X является неиммуногенным, биологически инертным и гидрофильным. Кроме того, предпочтительные линкеры способны придавать биологически активным полипептидным группам такие желаемые свойства, как пониженная иммуногенность, повышенная растворимость или пониженная скорость выведения из организма, - без значительного снижения сродства данных R1 и R2 - групп к их лиганду. В наиболее предпочтительных практических реализациях соединение R1-X-R2 (где R1-R2 и R1 и R2 являются связывающими группами) имеет сродство к своему лиганду, превышающее сродство немодифицированной связующей группы к этому лиганду. Например, по существу чистое соединение с105 30 кДа ингибитор ФНО ПЭГ3400-гантель обладает ингибиторной активностью по отношению к ФНО в 20 раз большей, чем ингибиторная активность, которую проявляет по отношению к ФНО с105 30 кДа ингибитор ФНО.
Активирующие группы Y1 и Y2, которые являются частью полимерного линкера X, могут включать любые из активирующих групп, включая мальимидную группу, сульфгидрильную группу, тиол, трифлат, трезилаи, азиридин, оксиран и 5-пиридил. Предпочтительными активирующими группами являются мальимидные.
Полимерная группа (Z)n преимущественно представляет собой группу, включающую полиэтиленгликоль, полипропиленгликоль, полиоксиэтилированный глицерол, декстран, поли -β- аминокислоты, кислоты толстой кишки или другие углеводные полимеры и полимеры производных биотина. В предпочтительных реализациях полимерная группа представляет собой полиэтиленгликоль. В сферу данного изобретения попадает также любая непептидная полимерная группа, которая выполняла бы описанные здесь функции.
Одно из преимуществ настоящего изобретения состоит в возможности изменять расстояние между группами R1 и R2, изменяя длину полимерной группы, соединяющей две связывающие группы. Предполагают, хотя и не обосновывая этого теоретически, что увеличение биологической активности, наблюдаемое для многомерных соединений данного изобретения, можно приписать многомерной природе клеточных рецепторов и лигандов in vivo.
По этой причине, профессионал легко определит, меняя размер спейсера X, оптимальное расстояние между звеньями R1 и R2 (которое обычно прямо пропорционально длине полимерного звена (Z)n).
В одной практической реализации настоящего изобретения, группы R1 R2 являются одинаковыми. Однако в другой практической реализации R1 и R2 представляют собой разные группы. Такие соединения можно предназначать для создания гетеродимера, в котором как R1, так и R2 действуют в пределах одних и тех же биологических систем. Например, считается, что как антагонист рецептора IL-1, так и ингибиторы ФНО обрывают каскад воспаления. Можно планировать также бифункциональные комплексы, в которых R1 или R2 является "целевой" группой, которая направляет комплекс в специфическое положение посредством своего сродства к связыванию с определенным субстратом, а противостоящая связывающая группа имеет нужную активность по отношению к данному локализованному сайту.
Примером гетеродимера, который обладает большими возможностями для того, чтобы быть эффективным ингибитором IL-2, является димер, в котором R1 представляет собой IL-2r α, а R2 - IL-2r β. Такой гетеродимер имитирует комплекс этого рецептора, который обладает самым высоким сродством к IL-2 (см. пример XYII). Дополнительным гетеродимером, который может действовать как ингибитор комплемента, является такой гетеродимер, в котором R1 представляет собой связующий домен C3b из CRI, и R2 представляет собой связующий домен C4b из CR1. См. пример XYIII. В дополнительном гетеродимере R1 представляет собой exon 6 PDGF и R2 представляет собой IL-1rd. См. пример XIX.
В предпочтительной практической реализации данного изобретения способы для получения бифункциональных комплексов R1-X-R2 являются по существу теми же самыми, как и способы, используемые для сайт-селективной реакции полипептидов как описано выше. Синтез соединения: с105 30 кДа ингибитор ФНО ПЭГ3400-гантель - описан ниже в примере XIII. Бис-реактивная полимерная группа реагирует с цистеинсодержащим полипептидом, в котором активирующая группа в бис-реактивной полимерной группе образует тиоэфирную связь с выбранным свободным цистеиновым остатком. Как это описано выше, данный цистеин может представлять собой свободный цистеин естественного происхождения на этой полипептидной группе, или ненативный цистеин, который был присоединен или замещен в естественной последовательности.
Предпочтительное бис-реактивное полимерное соединение в соответствии с данным изобретением представляет собой α- (2-мальимидо) ω- мальимидополи(оксиэтилен) или бис-мальимидо ПЭГ. Синтез бис-мальимидо ПЭГ описан в примере XII. В соответствии с предпочтительным способом, бис-мальимидо соединение получают из бис-гидроксил ПЭГ через бис-амино промежуточное соединение.
Обзор нескольких способов превращения концевых гидроксилов ПЭГ в соответствующие аминогруппы был сделан Гаррисом /36/. Это осуществляют получением реактивного промежуточного соединения путем либо галогенирования, либо окисления гидроксила, за которым следует замещение активированной концевой группы нуклеофилом.
Существуют другие практические альтернативы синтезу бис-мальимида ПЭГ, приведенному в примере XII. Реактивным промежуточным соединением в превращении данного гидроксила в соответствующий амин может быть галогенированное производное например ( α- (бромэтил) -ω- бромполи(оксиэтилен) промежуточного соединения /37/, за которым следует прямое замещение аммиаком /38/ или промежуточным веществом альдегидного характера (см. /36/). Бис-мальимид ПЭГ не является единственным сульфгидрил-специфическим реагентом, который можно использовать. Гласс с сотрудниками разработали другой способ для присоединения ПЭГ к сульфгидрилам /39/. Однако с тиолами эта реакция обратима. Другой способ для присоединения ПЭГ к цистеинилсульфгидрилам представляет собой производное бис-4-винил-пиридин ПЭГ.
Гаррис /36/ также приводит обзор способов синтеза различных электрофильных производных ПЭГ, которые можно использовать для модификации протеинов. Эти реагенты включают хлоркарбонаты, изоцианат, эпоксид, сукцинимидил сукцинат, хлорангидрид циануровой кислоты, смешанный ангидрид, карбодиимиды и сульфонаты. Последняя группа включает трезилат, тозилат и мезилаты. Некоторые из реагентов реагируют селективно с аминами (например, хлорангидрид циануровой кислоты и карбодиимиды), тогда как другие реагируют и с сульфгидрилами, и с аминами (например, эпоксид и трезилаты). Некоторые из этих реагентов были использованы для модифицирования протеинов, и это могло привести к потере активности в разной степени.
Предпочтительное получение комплексов R1-X-R2, в которых R1 и R2 разные, требует двухстадийного процесса, в котором бис-реактивную полимерную группу приводят во взаимодействие сначала с R1, а затем с R2. Получение таких гетеродимеров может быть выполнено профессионалом без лишнего экспериментирования. В некоторых случаях промежуточное соединение R1-X следует сначала выделить и очистить до того, как проводить реакцию с R2, а в других случаях очистка промежуточного соединения может быть ненужной.
Внеклеточные домены как IL-2r α, так и IL-2r β можно клонировать, используя PCR, и клонировать в векторе, способном к направляющей экспрессии в E. coli. Эти протеины можно повторно упаковать и очистить от E.coli и их способности ингибировать IL-2 активность, измеренную в биопробах. Можно использовать мутагенез in vitro для замещения цистеином нативных остатков в молекулах, чтобы обеспечить сайт-направленное присоединение ПЭГ. Мутеины как IL-2r α, так и IL-2r β можно затем идентифицировать, что предусматривает эффективное присоединение ПЭГ без потери активности пегилированного вещества. Связанный через ПЭГ гетеродимер может быть получен пегилированием сначала IL-2r α в присутствии избытка бис-мальимида ПЭГ. Однократно пегилированный IL-2r α можно очистить и к нему добавить IL-2r β для взаимодействия с активной мальимидной группой и образования гетеродимера. Эта молекула должна имитировать рецептор IL-2 с высоким сродством, найденный на поверхности клеток.
Гантельный комплекс, в котором R1 представляет собой IL-2, а R2 является IL-2r β, может также быть полезен как антагонист рецептора IL-2.
Пример I. Синтез реагентов для присоединения полиэтиленггликоля.
Приведены три реагента, чтобы показать различные средства, которые можно использовать для модификации полипептидов. Структуры промежуточных соединений и реагентов, описанных ниже, см. в приложении к примеру I.
А. Синтез реагента 1: мПЭГx-сложный эфир-малеимид
Сложноэфирное производное янтарной кислоты и мПЭГx (промежуточное соединение 1) было получено по методу, описанному в /40/. Полученный в результате продукт взвесили и растворили в минимальном объеме сухого диоксана при 60oC. После охлаждения раствора до комнатной температуры добавили эквимолярные количества три-н-бутиламина и изобутилового эфира хлормуравьиной кислоты. Реакцию проводили при перемешивании в течение 30 мин. В это время готовили боратный буфер, pH 8,8, титрованием 0,5 М раствора борной кислоты 1,6-гександиамином. Раствор, содержащий смешанный ангидрид, по каплям прибавляли к аликвоте боратного буфера, содержащего 10-кратный избыток (по молярному соотношению) 1,6-гександиамина к смешанному ангидриду. Реакционную смесь подвергали исчерпывающему диализу в деионизованной воде при 4oC и лиофилизировали. На это промежуточное полимерное соединение (промежуточное соединение 2) действовали избытком 2,5:1 (по молярному соотношению) эфира сульфосукцинимидил 4-(-малеимидоэтил)циклогексан-1 карбоновой кислоты (сульфо-СМЦК, производство Pierce Chemical Co, Rockford III) в 50 мМ буфера из фосфата натрия или HEPES Buffer, pH 7,0, в течение двух часов при комнатной температуре. Полученный в результате полимер очищали методом ГПХ, пропуская реакционную смесь через Sephadex G-25, используя 50 мМ фосфат натрия (или HEPES), pH 7,0, для элюции при 4oC. Малеимидополимер выходил при незаполненном объеме колонки, и его определяли, регистрируя поглощение при 260 нм. Реагент был использован для алкилирования полипептидов в пределах одного часа после его очистки. Поскольку мПЭГ, присоединенный в этой реакции, легко можно удалить основным гидролизом, этот реагент полезен для идентификации места присоединения мПЭГ к протеину.
B. Синтез реагента 2: мПЭГx-амид малеимид
мПЭГx-тозилат (промежуточное соединение 3) был получен как описано в /4/. Выход сульфированного промежуточного продукта оценивали спектрофотометрически как описано в /42/. Этот продукт превратили в производстве фтальимида (промежуточное соединение 4) и затем восстановили гидразингидратом в мПЭГx-NH2 (промежуточный продукт 5) способом, описанным в /41/. Содержание аминогрупп в эквивалентах на грамм продукта определяли микротитрованием соляной кислотой. Провели взаимодействие мПЭГx-NH2 с сульфо-СМЦК в HEPES или фосфатном буфере с pH 7,2 при комнатной температуре в течение двух часов. Были сделаны пробы при молярных соотношениях мПЭГx-амина к сульфо-СМЦК от 5: 1 до 1:5.
Для определения оптимальных условий в реакциях пегилирования использовали конечный реагент (реагент 2), и количество и качество полученного в этой реакции мПЭГx•IL-Ira оценивали методом электрофореза в системе ДСН-полиакриламидный гель (ЭПААГ) (обозначение мПЭГ IL-1ra будет использовано для продукта пегилирования IL-1ra, полученного в реакции с реагентом 2, а для продукта пегилирования IL-1ra, полученного в реакции с реагентом 3, описанным ниже, - обозначение мПЭГx • IL-1ra). Оптимальный результат наблюдали при отношении СМЦК к мПЭГx - NH2 1:1. Более высокие соотношения СМЦК вызывали появление многочисленных более высокомолекулярных производных IL-1ra, регистрируемых ЭПААГ, и множества пиков при аналитической ионообменной хроматографии, а более низкие соотношения приводили к снижению выхода пегилированного протеина. Реагент 2 очищали методом гель-проникающей хроматографии, используя смолу
C. Синтез реагента 3: мПЭГx-малеимид
мПЭГx-H2 (промежуточное соединение 5) можно модифицировать далее, для получения другого малеимидо-производного (реагент 3). Последнее осуществляют взаимодействием мПЭГ-NH2 с малеиновым ангидридом по способу Батлера и Хартли /43/, приспособленному к данному случаю, и циклизацией этого промежуточного соединения (промежуточное соединение 6) в соответствующий O-(2-малеимидоэтил)-O-метилполиэтиленгликоль, используя способ, описанный в /44/.
Приложение к примеру I
Синтез реагента 1
Структуры исходного вещества, промежуточных соединений и реагента из синтеза 1.
Исходное вещество:
Общая формула монометоксиполиэтиленгликоля (мПЭГx):
CH3O-(CH2CH2O)n-H,
где x означает среднюю молекулярную массу в килодальтонах, и n означает среднее число повторяющихся оксиэтиленовых групп.
Промежуточное соединение 1: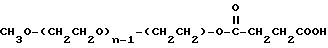
Промежуточное соединение 2: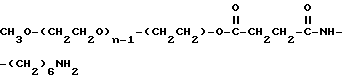
Реагент 1: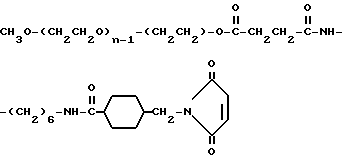
Синтез реагента 1
Структуры исходного вещества, промежуточных соединений и реагента из синтеза 2.
Исходное вещество:
Общая формула монометоксиполиэтиленгликоля (мПЭГx):
CH3O-(CH2CH2O)n-H
где x означает среднюю молекулярную массу в килодальтонах, и n означает среднее число повторяющихся оксиэтиленовых групп.
Промежуточное соединение 3: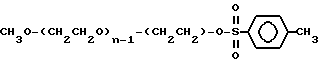
Промежуточное соединение 4: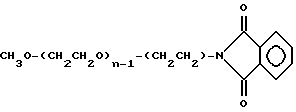
Промежуточное соединение 5 (мПЭГx - NH2:
CH3O-(CH2CH2O)n-1-(CH2CH2) -NH2
Реагент 2: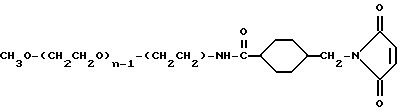
Синтез реагента 3
Структуры исходного вещества, промежуточных соединений и реагента из синтеза 3.
Исходное вещество:
Промежуточное соединение 5 (мПЭГx-NH2):
CH3O-(CH2CH2O)n-(CH2CH2) -NH2
Промежуточное соединение 6: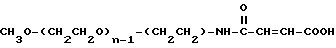
Реагент 3:
O-(2-малеимидоэтил)-O'-метилполиэтиленгликоль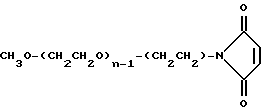
Пример II: Получение пегилированного нативного IL-1ra
Для оптимизации реакции пегилирования нативного были проверены различные параметры, причем удачное пегилирование определяли визуальным наблюдением по одной узкой полосе при 22 килодальтонах при электрофорезе в системе ДСН-ПААГ с окрашиванием Кумасси, и по одному острому пику в случае аналитической ионообменной хроматографии. Если не оговаривали иначе, реакции пегилирования проводили при концентрации 1 мг/мл нативного IL-1ra при комнатной температуре в буфере HEPES с pH 7,2 при соотношении мПЭГ-реагента к IL-1ra 2:1. Реагент, использованный в этих исследованиях, представлял собой мПЭГ-амидо-малеимид (реагент 2), и продукт обозначили как мПЭГх•IL-1ra, но данные результаты применимы ко всем трем реагентам.
A. Время
Анализировали реакции пегилирования, длившиеся от 0,5 до 24 ч, при комнатной температуре. Конверсия IL-1ra в пегилированную форму завершается (80%-90%) в течение 2 - 4 часов, и общее количество мПЭГ•IL-1ra не увеличивается и не уменьшается после более длительных периодов инкубации. Качество мПЭГ•IL-1ra, определяемое по электрофорезу в системе ДСН-ПААГ, снижается при увеличении времени, о чем свидетельствует появление дополнительных полос и пятен на окрашенном геле в области более высоких молекулярных масс.
B. Температура
Реакционные смеси для пегилирования инкубировали при 4, 25, 37 и 50oC, и затем анализировали по истечении 0,5; 1; 2; 4 и 17 ч. Реакции при 25 и 37oC привели к образованию большого количества (около 50 - 80%) пегилированного протеина в течение от одного до двух часов, а реакции при 4 и 50oC привели к значительно меньшему выходу (10 - 20%) даже при большей продолжительности реакции. Качество мПЭГ•IL-1ra, по-видимому, значительно не меняется с температурой.
C. Концентрация протеина
Реакция пегилирования проводили при концентрациях протеина (нативный IL-1ra) в пределах от 50 мкг/мл до 10 мг/мл. Все испытанные концентрации действовали хорошо, и разницы в качестве мПЭГ•IL-1ra не было.
D. pH
Нативный IL-1ra пегилировали в условиях реакции, которые были установлены выше, при pH ы пределах между 5,5 и 7,5. Качество мПЭГ•IL-1ra слегка лучше при более низком pH (5,5), как это установлено методами электрофореза в системе ДСН-ПААГ и ионного обмена, но процент конверсии при этом одинаков.
E. Отношение мПЭГ-амидо-малеимид к нативному
Были испытаны отношения мПЭГ-амидо-малеимид к нативному IL-1ra от 0,5:1 до 20:1. Отношения выше чем примерно 2:1 приводят к эффективному превращению в пегилированную форму IL-1ra (50 - 90%). Однако отношения выше чем 5:1 вызывают снижение качества мПЭГ•IL-1ra, что сказывается в увеличении количества полос в области очень высоких молекулярных масс при электрофорезе в системе ДСН-ПААГ и множества пиков при ионообменной хроматографии.
Оптимальными реакционными условиями как для количества полученного мПЭГ•IL-1ra, так и для качества полученного вещества, в пределах используемых параметров, являются следующие: 2:1 мПЭГ-амидо-малеимид /IL-1ra при 25oC в течение 2 - 4 часов, используя мПЭГ-амидо-малеимид, полученный при соотношении сульфо-СМЦК к мПЭГ-амин 1:1. В этих условиях в пегилированную форму превращают 80 - 90% IL-1ra, используя реагент, синтезированный либо с мПЭГ5000, либо с мПЭГ8500 в качестве исходного вещества (фиг. 3).
F. Получение соединений IL-1ra ПЭГ-гантель
Гантельные комплексы ПЭГ, содержащие IL-1ra, получают тем же способом, как другие пегилированные разновидности IL-1ra. Использование 2 - 4 молярный избыток бис-малеимидо ПЭГ к IL-1ra в буфере HEPES при pH 7,0. Использованная разновидность IL-1ra может представлять собой молекулу дикого типа, которая имеет свободный и доступный цистеиновый остаток, или мутеин, полученный как описано выше. IL-1ra присутствует в концентрации 2 - 5 мг/мл. Смесь для пегилирования инкубируют при комнатной температуре в течение от 4 до 6 ч. Соединения IL-1ra ПЭГ-гантель очищают от непегилированных и однократно пегилированных соединений катионно-обменной хроматографией на колонке MonoS при pH 5,5 в буфере MES концентрацией 20 - 50 мМ, используя градиент концентрации NaCl от 0 до 1000 мМ. Дальнейшей очистки можно добиться методом ГПХ, используя колонку BioRad или Superdex 75, как описано ниже.
Пример III: Очистка пегилированного нативного IL-1ra
Очистку мПЭГх•IL-1ra можно с успехом выполнять методами катионообменной или гель-проникающей хроматографии. Эти способы применены к пегилированному IL-1ra, полученному из всех трех реагентов, описанных выше.
A. Катионообменная хроматография
мПЭГx•IL-1ra можно очистить, используя колонку MonoS (Pharmacia) с буфером MES концентрацией 20 мМ при pH 5,5. Протеины вымывали из колонки, используя солевой градиент от 0 до 500 мМ NaCl в том же буфере. Например, немодифицированный IL-1ra выходит из колонки при 220 мМ NaCl, при этом чистоту проверяют, используя разные методы, включая аналитическую ионообменную хроматографию и электрофорез в системе ДСН-ПААГ. мПЭГ5000 IL-1ra выходит из колонки при 160 мМ (фиг. 4).
B. Гель-проникающая хроматография
мПЭГ5000•IL-1ra, который имеет протяженность около 52 кДа, и мПЭГ8500•IL-1ra, который имеет протяженность около 68 кДа (основано на калибровании колонки по стандартам с известным размером), можно легко отделить от немодифицированного IL-1ra (17 кДа) методом ГПХ на колонке с сорбентом Superdex 75 (Pharmacia) стандартным хроматографическим способом (фиг. 5).
Пример IV: характеристика
Очищенный мПЭГx•IL-1ra дает один симметричный пик при повторной хроматографии на сорбенте MonoS показывает себя чистым при электрофорезе в системе ДСН-ПААГ, так и при гель-проникающей хроматографии (фиг. 3 и 4). Сравнение триптических карт IL-1ra и МПЭГx•IL-1ra показывает один пик, соответствующий пептиду, содержащему C116 и C122, который отсутствует на карте конъюгата, с появлением нового широкого пика на этой карте. Дальнейшее расщепление этого нового пика, проведенное с химитрипсином, и последующий анализ аминокислотной последовательности показывает, что в использованных условиях был пегилирован С116 (фиг. 6).
Пример V: получение мутеинов IL-1ra
На однонитевом ДНК из гена IL-1ra, клонированном в бактериофаге M13, осуществили мутагенез. Использовали мутагенный набор BioRad, который применяли в способе, описанном Кункелем с сотр. в /45/. Вкратце, генерировали матрицу однонитевой ДНК, используя штамм E.coli, который содержит dut и ung мутации, приводящие к матрице, включающей урацил вместо тимидина. Мутагенные олигонуклеотиды длиной от 20 до 30 основных пар ренатурировали в матрицу и ресинтезировали вторую нить, используя ДНК полимеразы и ДНК лигазы. Данные реакционные смеси использовали для превращения штамма дикого типа E.coli, в котором урацил-содержащую нить разрушают под действием восстановительных механизмов ДНК, а мутантной нити позволяют воспроизводиться. Мутантный фаг сортировали и устанавливали последовательность аминокислотных остатков стандартными способами. Затем фрагмент, содержащий мутированный ген, субклонировали в векторе экспрессии pT5T /44/ и превращали в штамм с T7 экспрессирующей системой (E.coli B121DE3). Можно также использовать другие системы экспрессии E.Coli.
Экспрессирующие клоны выращены при 37oC на Luria Broth с добавкой 15 мкг/мл тетрациклина. Когда культуры достигали оптической плотности 0,8 при 600 нм, их температуру доводили до 30oC и добавляли IPTG до конечной концентрации 1 мМ, с целью вызвать экспрессию гена IL-1ra. Общее накопление протеина IL-1ra было максимальным через 4 - 6 часов и не изменялось существенно вплоть до 12 часов после индукции.
Пример VI: очистка мутеинов
Клеточную культуру, индуцированную как описано выше, собирали центрифугированием при 10000 г в течение 10 мин. Клетки вновь суспендировали в 20 - 50 мл 30 мМ буфера из ацетата натрия при pH 5,2. Лизиса достигали двукратным пропусканием через французскую установку для фильтровании под давлением при 18000 фунт/кв.дюйм. Клеточный лизат центрифугировали при 10000 г в течение 10 мин. Растворимую часть поместили в колонку с сорбентом S-sepharosa и промыли тем же буфером, содержащим 75 мМ NaCl•IL-1ra мутеин выходит из колонки с буфером, содержащим 200 мМ NaCl. Однократное прохождение над ионообменной смолой приводит к продукту достаточной чистоты (>95%) для изучения пегилирования. Дальнейшую очистку с успехом провести, используя другие ионообменные смолы, также как Q-Sepharose или MonoQ. Этот способ с равным успехом был использован для нескольких мутеинов IL-1ra. В некоторых случаях было необходимо слегка изменять pH и/или концентрации NaCl, чтобы очистить мутеины, которые имели небольшие отличия в заряде протеина из-за изменения в аминокислотной последовательности. С этими небольшими изменениями, которые легко может провести обычный специалист, этот способ широко применим ко всем из изученных мутеинов.
Пример VII: пегилирование мутеина IL-1ra
В дополнение к нативному IL-1ra, были пегилированы мутеины c84s116, c84c116, c0s116 и c9s116. Используя те же условия, которые были применены для нативного IL-1ra, получили и выделили в чистом виде пегилированные формы c84s116 и c84c116. Так как c84c116 содержит два реакционноспособных цистеина, пегилирование приводит к протеину с более высокой молекулярной массой, приблизительно 40 кДа при электрофорезе в системе ДСН-ПААГ. Этот протеин можно очистить методом катионообменной или гель-проникающей хроматографии, причем на последней, для случая, когда используют ПЭГ5000, он выходит при ожидаемой молекулярной массе около 68 кДа.
Пример VIII: эффективность мПЭГ•IL-1ra
Эффективность молекул пегилированного нативного IL-1ra была проверена стандартной пробой связывания конкурентного рецептора, используя S35-IL-1ra в качестве лиганда. Клетки мыши (EL4), содержащие рецептор IL-1 мышиного типа 1, или клетки хомяка (CHO), экспрессирующие рецептор человеческого типа 1 из клонированного гена, использовали при 1•106 клеток на лунку и 1•105 клеток, соответственно, в микролитровых плошках на 96 лунок. S35-IL-1ra с удельной активностью 4000 Ci /ммоль был добавлен так, чтобы конечная концентрация составляла 150 пМ. В серии разбавлений от 28 мМ до 13 пМ добавили холодный лиганд, и оставили инкубировать в течение 4 часов при 4oC. Затем клетки отфильтровывали на фильтре с миллилитровой фильтровальной пластиной (Миллипор, фильтр Дюрапор с размером пор 0,5 мкм), промыли до удаления радиоактивных примесей, фильтр с клетками сняли и замеряли активность, используя Ambis Radioanalytical Ivnaging Systein.
Были рассчитаны константы равновесной диссоциации (кД), и их использовали для сравнения пегилированной и немодифицированной форм IL-1ra. Немодифицированный дикий тип IL-1ra и c84s116 имеют равные кД мышиного рецептора типа 1, равные 150 - 300 пМ в нашем опыте, кД для пегилированной формы IL-1ra составляет около 400 - 800 пМ, а для пегилированного c84s116 - 500 - 10000, что в 2,5 и 3,5 раза выше, чем значения для соответствующих немодифицированных протеинов. кД для всех, кроме одного (c6s116) из непегилированных мутеинов находятся в пределах 65 - 150% от нативного протеина, в пределах ошибки опыта (см. таблицу 1).
Пример IX: фармакокинетика пегилированного нативного мутеина IL-1ra
Был проверен фармакокинетический характер молекул нескольких пегилированных нативных и мутеиновых IL-1ra после внутривенной инъекции этих молекул крысам. Нативный или пегилированный IL-1ra вводили в виде внутривенной болусдозы (3 мг/кг). Из хвостовой вены отобрали серию проб крови и проанализировали на нативный или пегилированный IL-1ra с помощью пробы на энзим-связанном иммуносорбенте (ПЭСИС). Из графика (фиг. 8) изменения концентрации плазматического IL-1ra во времени видно, что пегилирование оказывает заметное влияние на исчезновение IL-1ra из плазмы после внутривенной инъекции. Падение содержания в плазме IL-1ra и пегилированных производных IL-1ra лучше всего описывается тремя экспоненциальными компонентами. Эти данные указывают, что пегилирование продлевают у крыс полупериод удерживания этих экспоненциальных компонентов до 6 раз (таблица 2). Полупериод удерживания этих экспоненциальных компонентов увеличивается с увеличением размера молекул ПЭГ (таблица 2). Кроме того, существует доказательство того, что увеличение продолжительности полупериода удержания может определяться сайт-специфичностью пегилирования. Для интерпретации данных фиг. 8 использовали стандартный компартментальный анализ. Продление полупериода удерживания можно объяснить, основываясь на принятой фармакокинетической теории, которая утверждает, что плазматический полупериод удерживания для лекарства обратно пропорционально связан с плазменным очищением от лекарства и прямо пропорционально связан с кажущимся объемом распределения лекарства. Фармакокинетический анализ исчезновения пегилированных IL-1ra из плазмы указывает, что для пегилированных молекул по сравнению с нативным IL-1ra (таблица 2) продление полупериода удерживания обратно пропорционально снижению плазменного очищения. Снижение в плазменном очищении совпадает с ожиданием, связанным объемом, снижением гломерулярной фильтрации пегилированных молекул через почки. Продление полупериода пегилированием также пропорционально увеличению распределения (Vd стабильного состояния, таблица 2) пегилированных молекул. Увеличение в объеме распределения указывает на большее проникновение пегилированных молекул во внесосудистую систему. Через этот механизм пегилирование улучшает терапию с IL-1ra, увеличивая степень, до которой активные молекулы перемещаются из системной циркуляции во внесосудистую систему, систему, в которой, как ожидают, будут обнаружены рецепторы IL-1ra. Из-за сходства между крысами и человеком в механизмах как очищения, так и распределения для IL-1ra, очевидно, что пегилирование улучшит подобным образом и фармакокинетические свойства в человеческом организме.
I. Дополнительная внутривенная фармакокинетика для пегилированного IL-1ra
Для восьми дополнительных пегилированных мутеинов IL-1ra, используя ранее описанные способы, была описана внутривенная фармакокинетика. На фиг. 10 изображены зависимости изменения внутривенной плазматической концентрации IL-1ra во времени для каждого типа молекул. Из обзора всех данных внутривенной фармакокинетики (таблица 3) ясно, что с увеличением размера ПЭГ-единиц (однократное или двукратное пегилирование) происходит снижение плазматического очищения и, следовательно, возрастает среднее время внутривенного удержания и увеличивается полупериод исчезновения IL-1ra из плазмы. Сайт пеглирования является важным в определении степени, в которой уменьшается плазменное очищение и растет среднее время удерживания. Добавление двух единиц ПЭГ к IL-1ra продлевает среднее время внутривенного удерживания в 14 раз по сравнению с диким типом IL-1ra.
2. Подкожная фармакокинетика для пегилированного IL-1ra
Абсорбционную фармакокинетику пегилированных мутеинов IL-1ra охарактеризовали после подкожной инъекции данных молекул крысам. Из хвостовой вены отобрали серию проб крови и проанализировали на нативный или пегилированный IL-1ra с помощью пробы на энзим-связанном иммуносорбенте (ПЭСИС). Кривые изменения получающейся в результате подкожной плазматической концентрации IL-1ra во времени приведены на фиг. 11. Данные подкожной фармакокинетики (таблица 3) обнаруживают для пегилированных мутеинов изменчивую системную доступность, связанную с определенным сайтом и размером данного ПЭГ-звена, и связанную с подкожной инъекцией неоптимизируемыми зависимостями. Таблица 3 также показывает значительное положительное влияние пегилирования на среднее время удерживания для подкожного введенного IL-1ra. По мере того как увеличивается размер ПЭГ, среднее время удерживания обычно растет. Вероятно, что это увеличение является результатом более медленной абсорбции через лимфатическую циркуляцию (более длительные средние времена удерживания), что связано с размером молекул, так же как замедленного очищения после того, как пегилированная молекула достигла системной циркуляции (плазма). Это является существенным и улучшит фармакокинетический характер подкожно введенного IL-1ra для человека.
Пример X: получение мутеинов 30 кДа-ингибитора ФНО
Нативный остаток был замещен цистеином как при концевой аминогруппе, так и при концевой карбоксильной группе протеина, так же как и при всех трех сайтах гликозилирования (остатки 1; 14; 105; 111 и 161, как показывает фиг. 2). Мутагенез проводили на однонитевой ДНК из гена 30 кДа ингибитора ФНО, клонированного в бактериофаге М13. Этот ген подробно описан в /21/. Мутагенез проводили как описано в /43/ (см. пример V). Подвергнутый мутагенезу ген выделили и субклонировали в векторе экспрессии pT5T /44/, и превращали в штамм E. coli BL21DE3 с Т7 экспрессирующей системой. Мутеины 30 кДа-ингибитора ФНО очищали и подвергали повторной упаковке, как описано для нативного 30 кДа ингибитора ФНО /21/. Повторная упаковка включает добавление цистеина к раствору, содержащему очищенный протеин. Этот цистеин помогает в процессе повторной упаковки и "связывает" свободный цистеин в мутеине.
Пример XI: пегилирование мутеинов 30 кДа-ингибитора ФНО
Мутеин с105 30 кДа-ингибитора ФНО обрабатывали 6-кратным молярным избытком ДТТ в HEPES концентрацией 50 мМ с pH 7,0 в течение 30 мин при комнатной температуре, для удаления избыточного цистеина, связанного в ходе процесса повторной упаковки. Затем протеин подвергали диализу в дегазированном HEPES концентрацией 50 мМ с pH 7,0 в течение 2 часов для удаления ДТТ. После этого провели взаимодействие с105 30 кДа-ингибитора ФНО с 5-кратным молярным избытком пегилирующего реагента 1 (см. пример 1A) в течение 2 часов при комнатной температуре в HEPES концентрацией 50 мМ с pH 7,0. В пегилированную форму превращается приблизительно 60% мутеина.
Реакционную смесь с105 пегилирования загружали в колонку для скоростной жидкостной хроматографии протеинов (СЖХП) сорбентом Pharmacia, подавая 0,25 мл/мин 50 мМ Трис с pH 7,0, 100 мМ NaCl. Фракции, содержащие с105-ПЭГ 30 кДа-ингибитор ФНО собирали и загружали в TSK-200 SW ВЭЖХ колонку (Bio-Rad), подавая 0,2 мл/мин в том же буфере. Фракции, содержащие практически чистый с105-ПЭГ 30 кДа-ингибитор ФНО, что было определено по электрофорезу в системе ДСН-ПААГ с окрашиванием серебром, собрали и определили концентрацию протеина по Bio-Rad протеиновой пробе (см. фиг. 9).
Используя пробу на цитоксичность L929 - клетки ФНО, как описано в /21/, определили активность
Пример XII: получение бис-малеимидо ПЭГ
Синтез α- (2-аминоэтил) -ω- аминополи(оксиэтилен) производного ПЭГ (обозначено здесь и в последующем как бисамино-ПЭГ) состоит из трех стадий: 1) сульфирование гидроксильной группы, используя трезилхлорид, как описано Нильсоном и Мосбэком /40/, 2) замещение трезилированного промежуточного соединения фтальимидом /39/, и 3) восстановление промежуточного фтальимидного соединения в амин гидразингидратом /39/. Структуры исходного соединения, промежуточных соединений и продуктов приведены в приложении 1 к данному примеру. В оптимальных условиях получена приблизительно 80%-ная конверсия гидроксила в амин, как определено пробой с 2,4,6-тринитробензолсульфокислотой (ТНБСК). Бисамино ПЭГ можно выделить в чистом виде из реакционной смеси методом ионообменной хроматографии. Это - ключевая стадия для удаления реакционноспособных побочных продуктов, которые могут помешать при образовании димера.
Бисамино ПЭГ ацилировали малеиновым ангидридом /41/, и полученное промежуточное соединение циклизовали с образованием α- (2-мелеимидоэтил -ω- малеимидополи(оксиэтилен) а /42/. Это соединение реагирует с сульфгидрилом по реакции присоединения Микаэля с образованием стабильного тиоэфира.
Приложение к примеру XII
Исходное соединение
Общая формула для полиэтиленгликоля ПЭГx
HO-(CH2 CH2O)п-H
где x обозначает среднюю молекулярную массу в килодальтонах, а n среднее число повторяющихся оксиэтиленовых групп.
Промежуточное соединение 1
F3-CH2-SO2-O-(CH2CH2O)n-1- (CH2CH2)-O-SO2-CH2F3.
Промежуточное соединение 2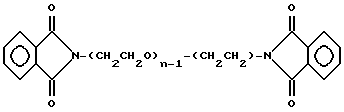
Промежуточное соединение 3
H2N-(CH2CH2O)n-1-(CH2CH2) -NH2
Промежуточное соединение 4 O-(2-малеимидоэтил)-O-метил-полиэтиленгликоль
O-(2-малеимидоэтил)-O-метил-полиэтиленгликоль
Пример XIII: результаты in vivo для комплексов с105 30 кДа-ингибитор ФНО ПЭГ
На двух различных стимулируемых ФНО физиологических действия испытали in vivo ингибирующий эффект четырех образцов, пегилированных с105 30 кДа-ингибитора ФНО. Одним из результатов было появление IL-6 в плазме мышей, которым внутривенно ввели рекомбинантный ФНО человека. Другим результатом явилось увеличение миграции нейтрофилов в брюшинную полость после введения внутрибрюшинно рекомбинантного ФНО человека.
Опыт один. Внутривенное введение с105 30 кДа ингибитора ФНО (ПЭГ2000, ПЭГ3500, ПЭГ10000) одновременно с рекомбинантным ФНО человека ингибирует индукцию IL-6 в плазме мышей.
Для измерения уровней индукции плазматического IL-6 с рекомбинантным ФНО человека использовали BALB/с самок мышей весом от 20 до 23 г. В предварительном опыте была получена временная зависимость появления IL-6 в плазме после инъекции внутривенно через хвостовую вену двух доз рекомбинантного ФНО человека (фиг. 12). Пик содержания IL-6 отмечали через два часа после стимуляции с помощью 10 или 20 мкг рекомбинантного ФНО человека на мышь. В последующих опытах использовали меньшую дозу.
Сравнили степень воздействия с105 30 кДа-ингибитора ФНО ПЭГ2000-гантель и непегилированного с105 30 кДа-ингибитора ФНО. Рекомбинантный ФНО человека вводили внутривенно дозой 10 мкг на мышь, один или одновременно с ингибиторами ФНО. Были проверены (фиг. 13) четыре разных отношения ингибиторов к ФНО. Отношения вычисляли, основываясь на содержании протеина. Каждая доза испытана на трех мышах. Кровь отбирали через два часа после внутривенных инъекций. Содержание IL-6 определили с помощью ПЭСИС.
Как с105 30 кДа-ингибитор ФНО, так и с103 30 кДа-ингибитор ФНО ПЭГ-гантель почти полностью ингибируют появление при введении при соотношениях ингибитора к ФНО 10: 1 и 5:1. При соотношении 1:1 с105 30 кДа-ингибитор ФНО ПЭГ2000-гантель вызывает 95%-ное снижение содержания IL-6, стимулированного одним ФНО, в то время как непегилированный с105 30 кДа-ингибитор ФНО снижает содержание IL-6 только примерно на 70%. Результаты этого опыта показывают, что для испытанных соотношениях, как с105 30 кДа-ингибитор ФНО, так и с105 30 кДа-ингибитор ФНО ФЭГ2000-гантель являются хорошими ингибиторами этого, стимулированного ФНО, физиологического параметра. При соотношении 1:1, с105 30 кДа-ингибитор ПЭГ2000-гантель оказывает более сильное ингибирующее действие, чем непегилированный ингибитор.
Были испытаны другие образцы пегилированных с105 30 кДа-ингибитор ФНО. Ингибирующие эффекты с105 30 кДа-ингибитор ФНО ПЭГ3500-гантель и с105 30 кДа-ингибитор ФНО ПЭГ10000-гантель испытывали на индукцию IL-6 в плазме. Ингибиторы вводили внутривенно одновременно с рекомбинантным ФНО человека при соотношении 1: 1 (с105 30 кДа-ингибитор ФНО-гантель: ФНО) (фиг. 14). В каждый из двух групп мышей, обработанных ингибитором, было по 3 особи. 10 мышей получили инъекции только ФНО. При соотношении 1:1, введение либо с105 30 кДа-ингибитора ФНО ПЭГ3500-гантель, либо с105 30 кДа-ингибитора ФНО ПЭГ10000-гантель не вызывает появления IL-6 в плазме мышей, тогда как после введения мышам одного рекомбинантного ФНО человека вызывает появление в плазме значительного количества IL-6.
Результаты этих двух опытов показывали, что с105 30 кДа-ингибитор ФНО ПЭГ2000, ПЭГ3500 и ПЭН10000-гантели являются хорошими ингибиторами индукции плазматического IL-6 рекомбинантным ФНО человека, при введении в низком соотношении (1:1) относительно стимулуса.
Опыт два. Введение подкожно с105 30 кДа-ингибитор ФНО (ПЭГ3500, ПЭГ10000 и ПЭГ20000) одновременно с внутрибрюшинной инъекцией рекомбинантного ФНО человека ингибирует миграцию нейтрофилов в брюшинную полость.
Для измерения миграции нейтрофилов в брюшинную полость после стимулирования рекомбинантным ФНО человека, использовали BALB/c самок мышей, весящих от 20 до 23 г. Использовали такую же методику как Ким МакИнтайр с сотр. /45/, которая кратко описана здесь. Прямо в брюшинную полость мышам вводили 0,1 мл ФНО. Через 4 ч мышей убивали и немедленно после этого промывали брюшинную полость и вспрыскивали в нее 4 мл сбалансированного солевого раствора Хэнка (СРХ) (не содержащего кальция и магния). Низ живота осторожно массировали. Брюшинную жидкость вытягивали с помощью иглы и шприца. На счетчике Коултера подсчитывали общее число брюшинных клеток. Аликвоту клеточной суспензии сушили на стекле и окрашивали красителем Diff-Kwik. Эти клетки подсчитывали по дифференциальному способу при прямом микроскопическом наблюдении. Сто клеток исследовали и классифицировали либо как нейтрофилы, либо как лимфоциты, либо как макрофаги.
В предварительном эксперименте сравнили относительный клеточный состав промывной жидкости после введения внутрибрюшинно или свободного от пирогена физиологического раствора, или 7,5 нг рекомбинантного ФНО человека. ФНО вызвал увеличение как в процентном содержании нейтрофилов, так и в абсолютном числе нейтрофилов, присутствующих в брюшинной промывной жидкости. В мышах, подвергнутых действию физиологического раствора, из промывной жидкости собрали 9,4 • 104 нейтрофилов, что составило только 2,3% от общего числа внутрибрюшинных клеток. В мышах, подвергнутых действию ФНО (7,5 нг), общее число нейтрофилов возросло до 12,9•105, а процентное содержание нейтрофилов возросло до 19,7%.
Сравнили также активности непегилированного с105 30 кДа-ингибитора ФНО и трех пегилированных образцов с105 30 кДа-ингибитора ФНО (ПЭГ3500, ПЭГ10000 и ПЭГ2000-гантели). Сохраняя стимул ФНО постоянным при 7,5 нг на мышь, провели испытания ингибиторов при соотношения 100:1; 10:1 и 1:1 (образец с105 30 кДа-ингибитор ФНО : ФНО). Отношения расчитывали, основываясь на содержании протеина. Мышам вводили подкожно с105 30 кДа-ингибитор ФНО одновременно с внутрибрюшинным введением ФНО. Каждую дозу испытывали на шести мышах. Через четыре часа брюшинную промывную жидкость собирали и анализировали. Величины, приведенные на фиг. 15, представляют собой процентное содержание нейтрофилов в брюшинной промывной жидкости. Самым низким соотношением, при котором непегилированный с105 30 кДа-ингибитора ФНО и с105 30 кДа-ингибитор ФНО ПЭГ3500-гантель в значительной степени ингибируют миграцию нейтрофилов, является 100: 1. с105 30 кДа-ингибитор ФНО ПЭГ10000- и ПЭГ20000-гантель значительное ингибируют миграцию нейтрофилов при отношении 10:1.
Результаты этого эксперимента показывают, что с105 30 кДа-ингибитор ФНО ПЭГ3500- ПЭГ10000- и ПЭГ20000-гантель являются хорошими ингибиторами стимулированной ФНО миграции нейтрофилов в брюшинную полость. с105 30 кДа-ингибиторы ФНО ПЭГ10000- и ПЭГ20000-гантель являются более активными, чем непегилированный с105 30 кДа-ингибитор ФНО и с105 30 кДа-ингибитор ФНО ПЭГ3500.
Пример XIV: получение и биоактивность с105 30 кДа-ингибитор ФНО ПЭГ-гантель
Синтез
2-3 мл/г рекомбинантного с105 30 кДа-ингибитора ФНО обрабатывают 4-кратным молярным избытком ДТТ в течение 2 ч при комнатной температуре. Затем ингибитор ФНО подвергают диализу в дегазированном HEPES концентрацией 50 мМ с pH 7,0 в течение 3 ч при 4oC. Для получения гантели, связанной через ПЭГ, ингибитор ФНО приводят во взаимодействие с бис-малеимидо ПЭГ, при разных молярных соотношениях, в HEPES концентрацией 50 мМ с pH 7,0. Ингибитор ФНО вступает в реакцию с бис-малеимидо ПЭГ в эквимолярном соотношении. Все реакции инкубировали в течение 3-12 ч при комнатной температуре. После инкубации связанную через ПЭГ гантель ингибитора ФНО отделяли от непегилированного и однократно пегилированного ФНО, используя СЖХП на сорбенте MONO-S в растворе уксусной кислоты концентрацией 50 мМ с pH 4,0, применяя ступенчатый градиент 260 мМ, 310 мМ и 350 мМ NaCl. Связанная молекулой ПЭГ гантель ингибитора ФНО выходит из колонки при градиенте 310 мМ NaCl. Оставшийся непегилированный ингибитор ФНО удаляют хроматографически на сорбенте Superdex 75.
Ступенчатое присоединение реагента
После обработки ДТТ и диализа в HEPES концентрацией 50 мМ с pH 7,0 добавляют эквимолярное количество бис-малеимидо ПЭГ, через 1,5 ч инкубации снова добавляют эквимолярное количество бис-малеимидо ПЭГ. Смесь инкубируют в течение 1,5 ч. Это приводит к оптимальному содержанию ПЭГ-связанного гантельного соединения. Затем добавляют 2-кратный избыток ПЭГ-реагента, получая конечное соотношение ПЭГ /ингибитор ФНО 4:1. Все это инкубируют в течение 2 ч, и затем смесь подвергают диализу в ацетате концентрацией 50 мМ с pH 4,0, используемом для хроматографирования на сорбенте Mono-S. Образуется смесь, которая содержит главным образом димер, связанный через ПЭГ, и однократно пегилированный ингибитор ФНО. Это позволяет более эффективно провести очистку ПЭГ-связанной гантели, поскольку имеет место большее разделение между однократно пегилированным ингибитором ФНО и гантелью, чем между гантелью и непегелированным ингибитором ФНО.
Этот способ оптимизирует образование гантельного соединения, и способствует более эффективной очистке.
Ступенчатая реакция:
После обработки ДТТ и диализа в HEPES концентрацией 50 мМ с pH 7,0 добавляют 8-кратный молярный избыток бис-малеимидо ПЭГ. Смесь инкубируют в течение 2 ч при комнатной температуре. Это превращает по существу весь ингибитор ФНО в однократно пегилированную форму. Однократно пегилированный ингибитор ФНО отделяют от ПЭГ-реагента и непрореагировавшего ингибитора ФНО, используя ВЭЖХ на сорбенте Mono-S в ацетате концентрацией 50 мМ с pH 4,0 при градиенте NaCl. Однократно пегелированное соединение подвергают диализу HEPES концентрацией 50 мМ с pH 7,0, и концентрируют до 2-4 мг/мл. Затем добавляют обработанный ДТТ ингибитор ФНО, для того чтобы получить ПЭГ-связанную гантель. Через 2 ч ПЭГ-связанное гантельное соединение очищают, используя ВЭЖХ на сорбенте Mono-S. Данный способ можно использовать для получения ПЭГ-связанной гетерогантели путем добавления второго, иного протеинового соединения.
Этот способ оптимизирует образование гантельного соединения и может быть использован для получения гетерогантельных соединений. Однако этот способ несколько трудоемок и длителен.
Биоактивность ПЭГ-связанных гантельных соединений ингибитора ФНО
Способность гантельных соединений с105 30 кДа-ингибитора ФНО ингибировать цитотоксичность ФНО была измерена в пробе цитотоксичности клетки murine L 929. Это позволило определить для этих молекул ED50, которые имеют следующие значения, нг/мл:
Дикий тип ингибитора ФНО r - 220
ВМН-связанные гантели - 220
ПЭГ1900-гантели - 4,1
ПЭГ3500-гантели - 4,8
ПЭГ10000-гантели - 4,6
ПЭГ20000-гантели - 4,2
Гантельные соединения ингибитора ФНО обладают также сильно увеличенной активностью в ингибировании цитотоксичности ФНО β в биопробе L 929. Значение ED50 по отношению к ФНО β являются следующими:
Дикий тип ингибитора ФНО r, мкг/мл - 70
ПЭГ3400-гантели, нг/мл - 80
ПЭГ2000-гантели, нг/мл - 22
Пример XV: фармакокинетика пегилированного 30 кДа-ингибитора ФНО
1. Внутривенная фармакокинетика для пегилированного 30 кДа-ингибитора ФНО
Определяли фармакокинетическое поведение нескольких типов пегилированных молекул 30 кДа ингибитора ФНО после внутривенного введения крысам. Нативный или пегилированный ингибитор ФНО вводили внутривенно в виде болус-дозы. Затем их хвостовой вены отбирали серию проб крови и анализировали на присутствие непегилированного или пегелированного ингибитора ФНО пробой на энзим-связанном иммуносорбенте (ПЭСИС). Полученные в результате временные зависимости внутривенной плазматической концентрации ингибитора ФНО (фиг. 16) показывают, что пегилирование оказывает значительное воздействие на исчезновение ингибитора ФНО из плазмы после внутривенной инъекции. Для интерпретации данных фиг. 16 использовали статистическую теорию момента (площадь под кривой и площадь под кривой первого момента). Эти данные показывают, что пегилирование продлевает в крысе среднее время внутривенного удерживания ингибитора ФНО почти в 50 раз (таблица 4). Среднее время внутривенного удерживания увеличивается с увеличением размеров связанной молекулы ПЭГ (таблица 4). Хотя теоретически это не подтверждено, продление среднего времени удерживания можно объяснить на основе обычной фармакокинетической теории, которая утверждает, что время внутривенного удерживания для лекарства обратно пропорционально связано с плазменным очищением для этого лекарства, и прямо пропорционально связано с кажущимся объемом распределения для данного лекарства. Фармакокинетический анализ исчезновения пегилированных молекул ингибитора ФНО из плазмы показывает, что продление полупериода удерживания обратно пропорционально связано с пониженным плазменным очищением для пегилированных молекул, по сравнению с непегилированным ингибитором ФНО (таблица 4). Снижение плазменного очищения согласуется с ожидаемым, определяемым размерами, снижением гломерулярной фильтрации пегилированных молекул почками. Из-за вероятного качественного сходства между механизмами плазменного очищения для ингибитора ФНО у крыс и у человека, очевидно, что пегилирование сходным образом улучшит фармакокинетические свойства ингибитора ФНО у человека.
2. Подкожная фармакикинетика для пегилированного 30 кДа-ингибитора ФНО
Изучали фармакокинетику абсорбции пегилированного ингибитора ФНО после инъекции его подкожно крысам. Из хвостовой вены отбирали серию проб крови и анализировали, определяя изменение концентрации непегилированного и пегелированного ингибитора ФНО во времени; результаты приведены на фиг. 17. Данные подкожной фармакокинетики (таблица 4) обнаруживают для пегелированных молекул изменчивую системную доступность, связанную с размером ПЭГ, и связанную с подкожной инъекцией неоптимизируемыми зависимостями. Таблица 4 показывает также положительное влияние пегилирования на среднее время удерживания для подкожно введенного ингибитора ФНО. С ростом размера ПЭГ обычно возрастает среднее время удерживания. Хотя теорией не подтверждено, это увеличение, вероятно, является результатом связанного с размером молекул замедления абсорбции через лимфатическую циркуляционную систему (более длительные средние времена удерживания), так же как и замедленного очищения после того, как пегилированная молекула достигнет плазмы. Эта пролонгация является значительной и улучшит фармакокинетический характер подкожного ингибитора ФНО для человека.
Пример XVI: растворимость пегилированных протеинов IL-1 ra
Результаты изучения растворимости приведены на фиг. 18. Приведены кривые растворимости для трех различных препаратов
IL-1ra и с84 IL-1ra ПЭГ8500. Опыты проводили при 37oC в микролитровых чашках с концентрацией протеинов 160 мг/мл. Чашки закрывали крышкой и затем просматривали в специальном устройстве при 405 нм через определенные промежутки времени. Увеличение абсорбции указывало на высаживание полимера. Имеется явное уменьшение количества выпадающего из раствора протеина для пегилированного образца по сравнению с нативным IL-1ra.
30 кДа-ингибитор
Нативный 30 кДа-ингибитор ФНО нельзя сконцентрировать больше, чем до 5 мг/мл. После пегилирования растворимость увеличивается по крайней мере в 5 раз.
Пример XVII: получение гетерогантели ингибитора IL-2
ПЭГ-связанная гетерогантель может быть получена путем первого пегелирования IL-2ra α в присутствии избытка бисмалеимидо ПЭГ. Одонократно пегилированный IL-2ra α модно очистить и добавить к нему IL-2ra β для проведения реакции с оставшейся реакционноспособной малеимидной группой с образованием гетеродимера.
Потенциальные сайты для пегилирования IL-2ra α включают остатки как с амино-, так и с карбоксильными концевыми группами, два N-связанных сайта гликозирования, так же как и нативный свободный цистеиновый остаток в этой молекуле. Было установлено, что цистеиновой остаток 192 в растворимом внеклеточном домене α не участвует в дисульфидном связывании /46/. Этот цистеиновый остаток расположен в эпитоне анти-IL-2r α моноклонального антитела, которое не влияет на связывание IL-2 с IL-2r α /47/. Это указывает, что данный остаток является вероятным кандидатом для пегилирования без того, чтобы влиять на активность IL-2r α.
Для IL-2r β потенциальные сайты включают амино- и карбоксильные концевые группы, N-связанные сайты гликозирования и область 108-118, которая подобна биологически значимой области в рецепторе murine erythropoietin /48/. Анализ точек мутации других остатков в рецепторах может также дать возможность обнаружить другие сайты пегилирования, что приведет к оптимальным свойствам в гетерогантельной молекуле.
Пример XVIII: получение гетерогантелей, которые ингибируют классический путь системы комплемента
Были идентифицированы и клонированы многие протеины, которые регулируют систему комплемента. Некоторые из них являются мембранными протеинами. Один из мембранных протеинов назван CR1 (рецептор 1 комплемента). Растворимую форму CR1 исследовали in vivo на моделях болезней. Ингибитор комплемента ингибирует пост-ишемическое воспаление миокарда и некроз /49/, обратную пассивную артус-реакцию /50/ и отторжение аллографта /51/.
Растворимый CR1 связан с C3b и C4b. Он состоит из 30 коротких консенсусов повторяющихся последовательностей (КПП). Большинство КПП содержит один сайт возможного гликозилирования и четыре цистеина. Вероятно, все эти цистеины участвуют в дисульфидном связывании. Установлено, что КПП 1-4 участвуют в связывании C4b. Две отдельные порции CR1, КПП 8-11 и КПП 15-18 участвуют в связывании C3b /52/, /11/. согласно данному изобретению, возможно получить гетерогантель, которая содержит C4b-связывающий домен и C3b-связывающий домен из CR1. Используя PCR, можно клонировать КПП, которые содержат C4b-связывающие и C3b-связывающие домены из CR1. Эти КПП будут представлять собой КПП 1 через 5 (C4b-связывание) и КПП 8 через 12 (C3b-связывание). Ген, кодирующий эти КПП, можно клонировать в векторе экспрессии E.coli. E.coli-экспрессированные протеины могут быть повторно упакованы и очищены. Успех повторной упаковки можно ценить по способности связывать полиC3b или полиC4b. Можно провести in vitro мутагенез этих генов, чтобы заместить в цистеине нативные аминокислотные остатки. Эти цистеины затем можно использовать для связывания ПЭГ-молекулы. Возможными сайтами для пегелирования будут сайт гликозилирования или концевая карбоксильная группа остатка КПП 5 или КПП 12. Для связывания ПЭГ-молекулы можно получить и использовать C4b-связывающий и C3b-связывающий домены, которые содержат дополнительный цистеин при концевой карбоксильной группе остатка. ПЭГ-связанное гетерогантельное соединение можно получить в двухступенчатом способе из примеры XIV. Очистку можно провести методом ионообменной хроматографией.
Пример XIX: синтез IL-1ra бис(матеимид)-(тромбоцитарного фактора роста)-пептид ПЭГ-гетерогантель
Пептид тромбоцитарного фактора роста (PDGF) YGRPRESGKKRKRKRLKPT описан в /53/. Чтобы сделать возможным связывание с малеимидом, добавили концевой C.
Гетерогантель синтезировали в две стадии. На первой стадии 1,6 наномолей IL-1ra, суспендированного в 3 мкл буфера HEPES концентрацией 0,05 м с pH 7,5, смешивали с 6,4 наномолями бис-малеимидо ПЭГ1900, растворенного в 11 мкл того же буфера. Эту реакцию проводили в течение 30 мин при 20oC. На второй стадии, к этому продукту, полученному на первой стадии, добавили 32 наномоля пептида PDGF, растворенного в 4 мкл 0,2 М буфера фосфата натрия, pH 7,0. Реакцию проводили в течение 1 ч при 20oC. Затем реакцию обрывали введением равного объема буфера для электрофореза в системе ДСН-ПААГ, содержащего 30 мкмолей 2-меркаптоэтанола.
Образцы продуктов первой стадии данной реакции и продукты полной двухстадийной реакции, так же как и соответствующие маркеры молекулярной массы были разделены электрофорезом в системе ДСН-ПААГ на 15%-ном полиакриламидном геле с последующим окрашиванием синим Кумасси. Продукт двухстадийной реакции дает дополнительную полосу, соответствующую гантельному соединению ожидаемого размера. В гетерогантельное соединение путем двухстадийной реакции превращают приблизительно 33% исходного IL-1ra.
Продукты первой стадии реакции можно выделить катионообменной хроматографией на смоле S-Sepharose. Гетеродимер возможно выделить в с помощью катионообменной хроматографии, благодаря обилию основных аминогрупп в этом пептиде.
Следует понимать, что применение рекомендаций настоящего изобретения к конкретной экспрессирующей системе или пегилирующему реагенту, в свете приведенных здесь рекомендаций, будет находиться в пределах возможностей специалиста. Таким образом, очевидно, что возможны различные модификации и варианты способа и продуктов настоящего изобретения. Подразумевается, что настоящее изобретение включает эти модификации и варианты, если они находятся в объеме приведенной формулы с учетом возможности эквивалентной замены указанных в ней признаков.
Литература
1. Davis et al. Biomedical Polymers: Plymeric Materials and Pharmaceuticals for Biomedical Use, p.p. 441-451 (1980).
2. Knauf et al. J. of Biolog. Chem., V. 263, p. 15064 (1988).
3. Katre et al. Proc. Natl. Acad. Sci. USA, V. 84, p. 1487 (1987).
4. Conforti et al. Pharm. Research Commun., V. 19, p. 287 (1987).
5. Suzuki et al. Biochem, Biophys. Acta. V. 788, p. 248 (1984).
6. Abuchowski et al. J. Biol. Chem., V.252, p. 3582 (1977). 7. Patent ISA N 4179337, Davis et al.
8. Goodson et al. Biotechnology, V. 8, p.343 (1990).
9. a) Peppel et al. J. Exp. Med., V. 174, p. 1483 (1991);
б) Ashkenazi et al. Proc. Natl. Acad. Sci. USA, V. 88, p. 10535 (1991).
в) Zoetscher et al. J. Biol. Ghem. V. 266, p. 18324 (1991).
10. Byrn et al. Nature (London). V. 344, p. 667 (1990).
11. a) Kalli et al. J. Exp. Med., V. 174, p. 1451 (1991);
б) Hebell et al. WO 91/16437 (1991).
12. Hebell et al. Science, V. 254, p. 102 (1991).
13. a) Maragonore et al. Biochemistry, V. 29, p. 7085 (1990);
б) Bourdon et al. FEBS, V. 294, p. 163 (1991).
14. Jiang et al. Immunology, V. 74, p. 735 (1991).
15. a) Erez et al. J. Med. Ghem., V.25, p. 847 (1982);
б) Portoghese et al. J. Med. Chem., V. 29. p. 1855 (1986).
16. Shimohigashi et al. Nature. V. 197, p. 333 (1982).
17. Ghen et al. J. Biol. Chem. V. 266, p. 18237 (1991).
18. Dustin et al. J. Exp. Med. V.169, p. 503 (1989).
19. Morata et al. Inst. J. Biol. Macromol., V. 11, p. 97 (1989).
20. Dohlsten et al. Proc. Natl. Acad. Sci. USA. V.88, p. 9287 (1991)
21. U.S. Patent Application N 555274 (19.07.91).
22. U.S. Patent Application N 07/506522 (6.04.1990).
23. a) US Patent N 4578335;
б) US Patent N 4816565.
24. a) European Patent Application N 89104023;
б) European Patent Application N 90104246.6;
в) Honjo et al. Nature. V. 311. p. 631 (1984);
г) Taniguchi et al. Science, V. 244, p. 551 (1989). (01) (19) (11) (21) (51) 6 (54) (57) (08) (09) (00)
25. Nakaido et al. Nature, V. 311, p. 631 (1984).
26. Hatakayama et al. Science, V. 244, p. 551 (1989).
27. Ogura et al. Mol. Biol. Med., V. 5, p. 123 (1988).
28. Heisse et al. La Presse Mediocle. V. 20, p. 2036 (1991).
29. Fierson et al. Proc. Natl. Acad. Scie. USA. V. 75, p. 5867 (1979).
30. Krickstein et al. Coriipleinent. V. 2, p. 44 (1985).
31. WO 91/05047, WO 89/09220, Pearson et al.
32. M. Dayhoff, Atlas of Protein. Sequences and Structure. V. 5, p. 124 (1972), National Biochemical Research Foundation, Washington, D.G.
33. Harris et al. J. Polym. Sci. V, 22, p.341 (1984).
34. Harris et al. Rev. Macromol. Chem. V. C25(3). p. 325 (1985).
35. Johannson, Biochim. et Biophy. V. 222, p. 381 (1970).
36. Buckmann et al. Macromol. Chem. V. 182. p. 1379 (1981).
37. Glass et al. J. Biopolymers. V. 18, p. 383 (1979).
38. Wie et al. Int. Archs. Allergy App. Immun., V. 64, pp. 84-99 (1981).
39. Phillai et al. J. Ong. Gheni. V.45, p. 5364 (1980).
40. Nilson and Mosbach in Methods of Enzymology. V. 104, pp. 56-69, Academic Press. Inc., N.Y., N.Y. (1984).
41. Butler Hartley, Methods of Enzymology, V. XXV, p. 191- 199, Academic Press. Inc., N.Y., N.Y. (1972).
42. Wunsch et al. Biol. Chem. Hoppe-Seyler, V. 366, p. 53 (1985).
43. Kunkel et al. Methods in Enzymology. V. 154, pp. 367-382 (1987).
44. Eisenberg et al. Nature. V. 343, p.341 (1989).
45. Kim Mclntyre et al. J. Exp. Med. V. 173, p. 931 (1991).
46. Niedel et al. BBRC, V. 154, p. 372 (1988).
47. Lorenzo et al. J. Immunology. V. 147, p. 2970 (1991).
48. Yoshimura, Longmore, Jodish, Nature. V. 348, p. 647 (1990).
49. Weisman et al. Science. V. 149, p. 145 (1990).
50. Yet et al. J. Immunology. V. 146, p. 250 (1991).
51. Pruitt et al. J. Surgical Research. V. 50, p. 350 (1991).
52. Klickstein et al. J. Exp. Med. V. 168, p. 1699 (1988).
53. Khachigian L. et al. Biol. Chem. V. 267, p. 1660 (1991). Я.

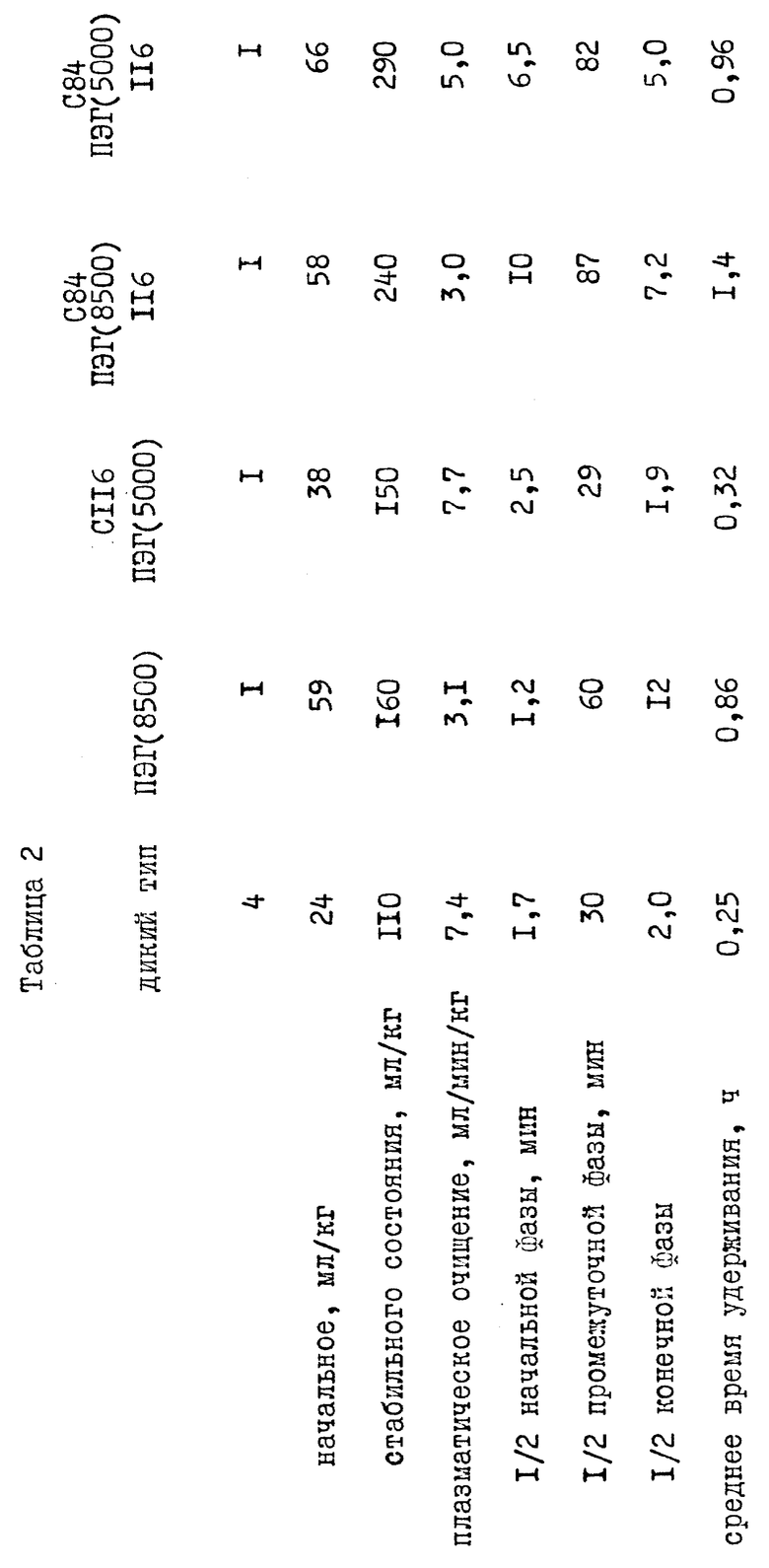
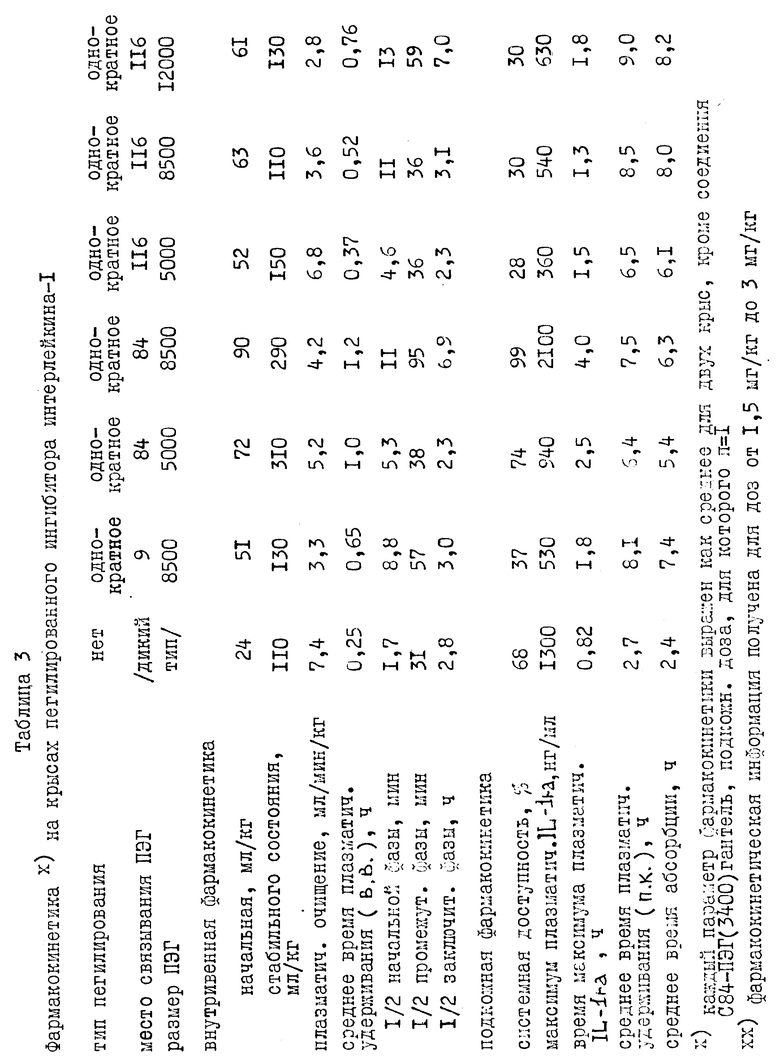
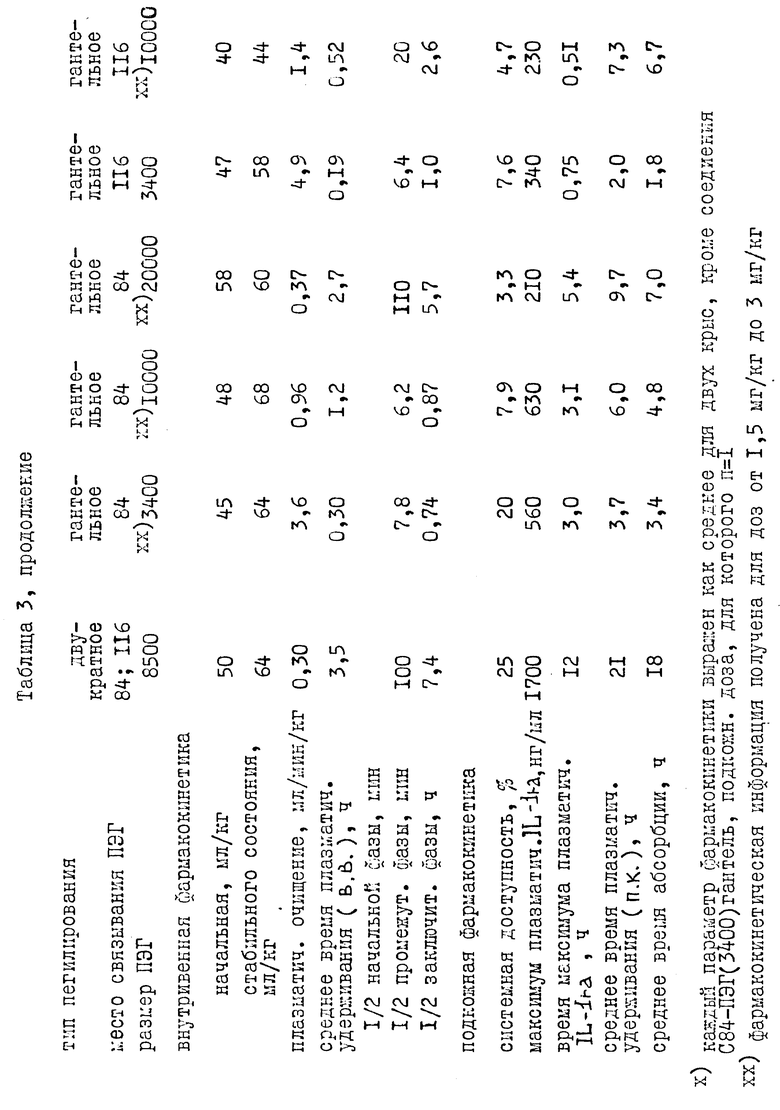
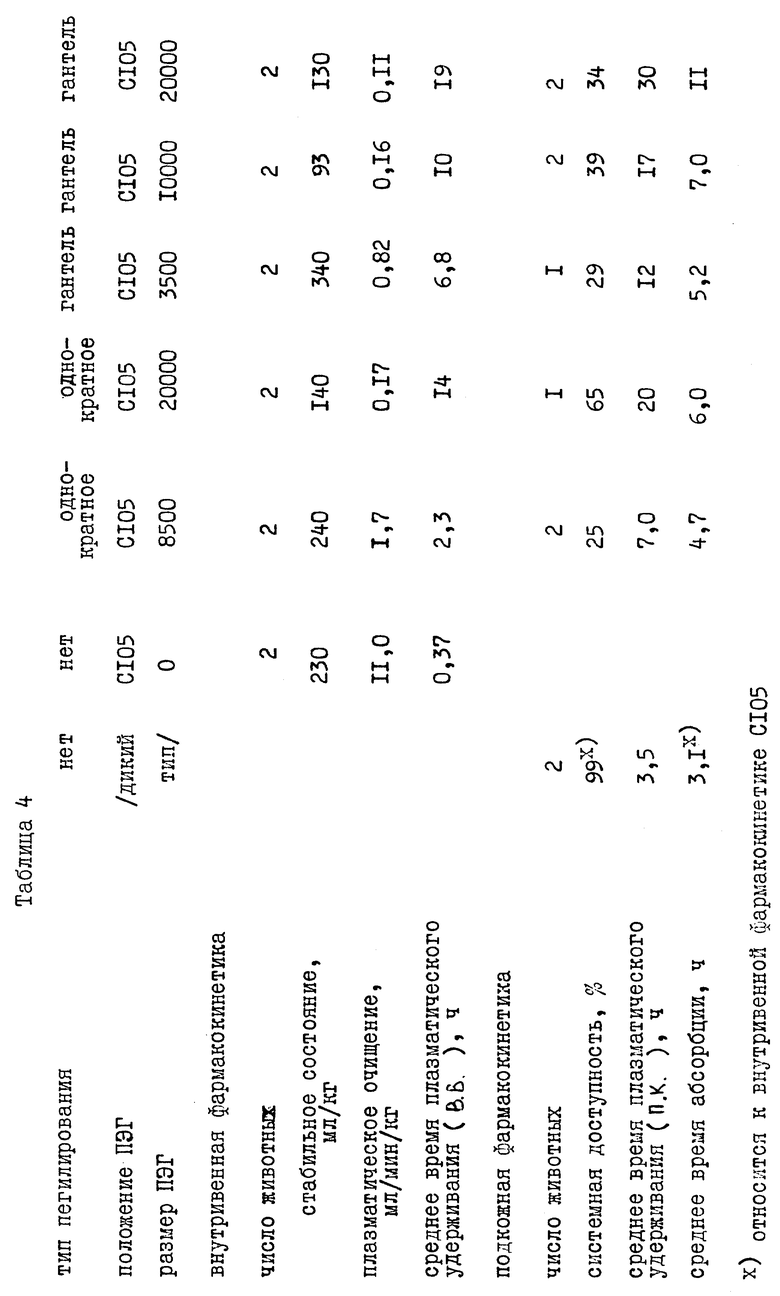


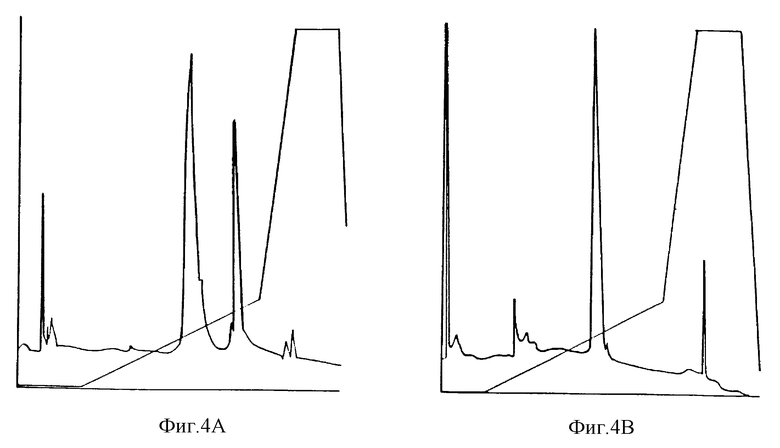
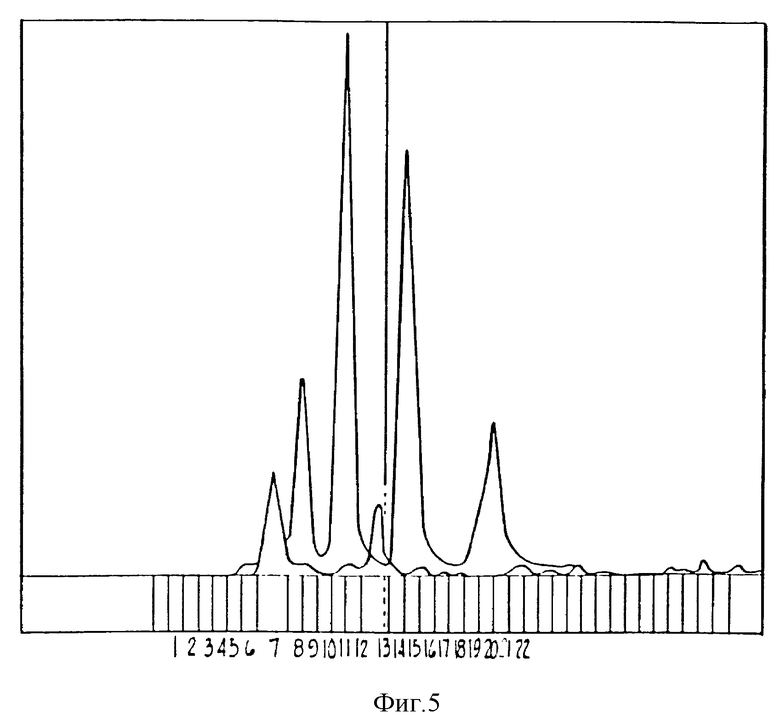
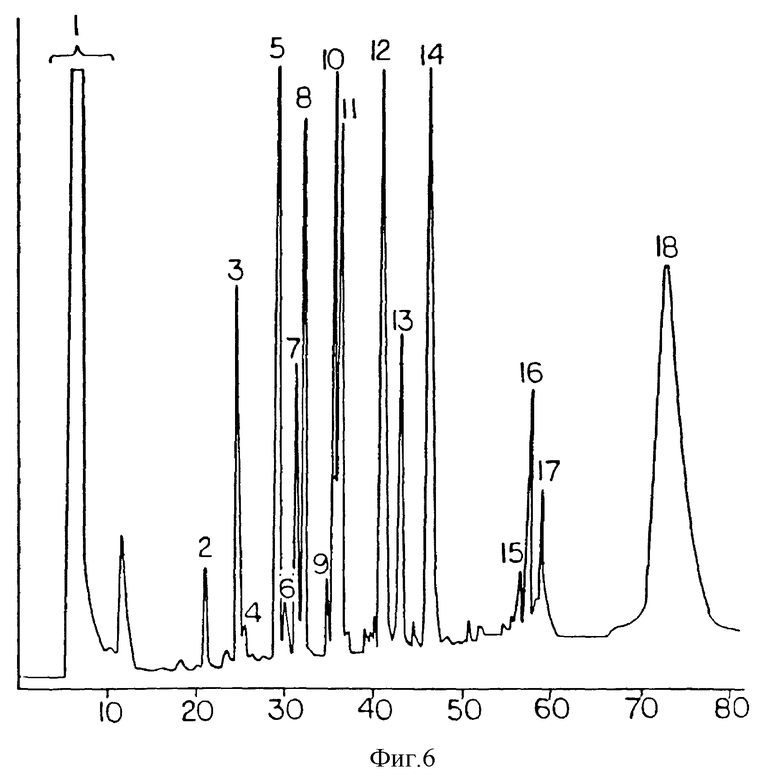
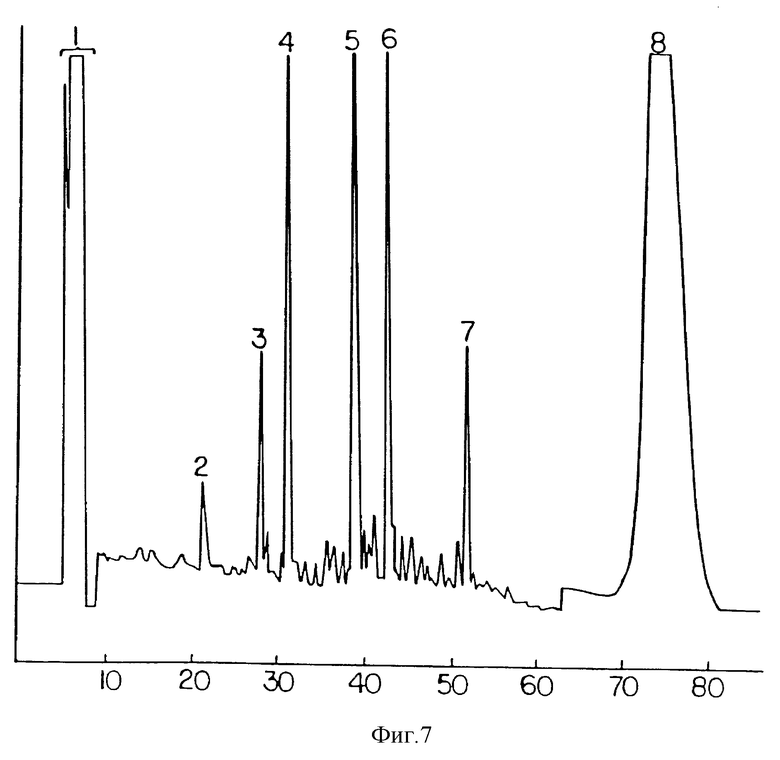
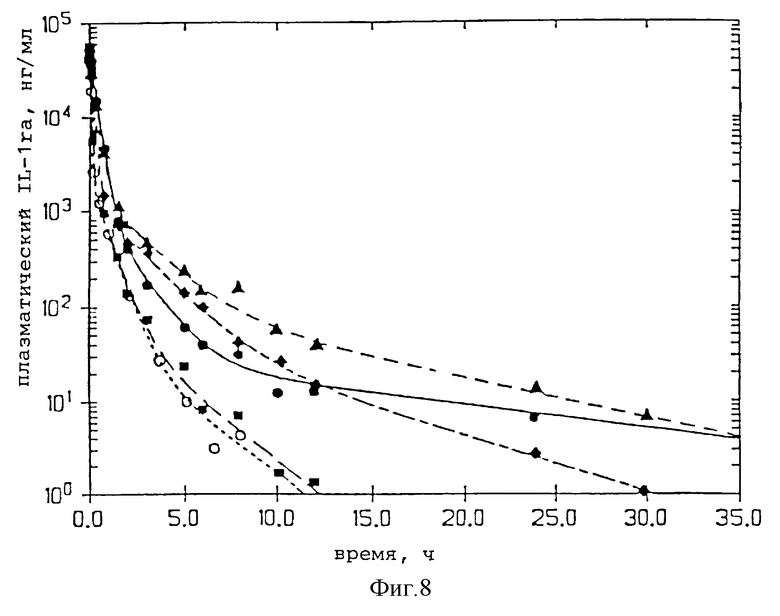

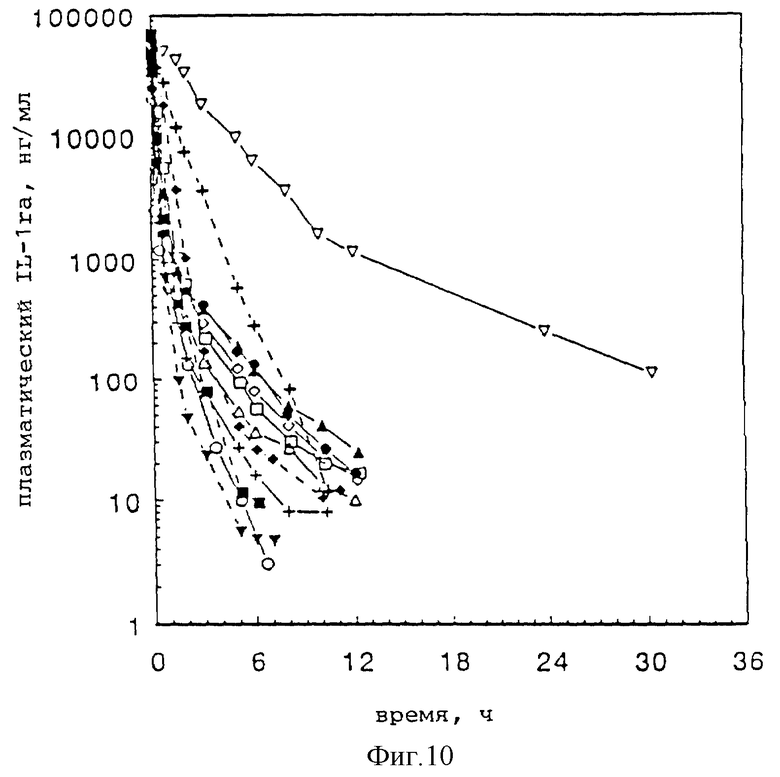
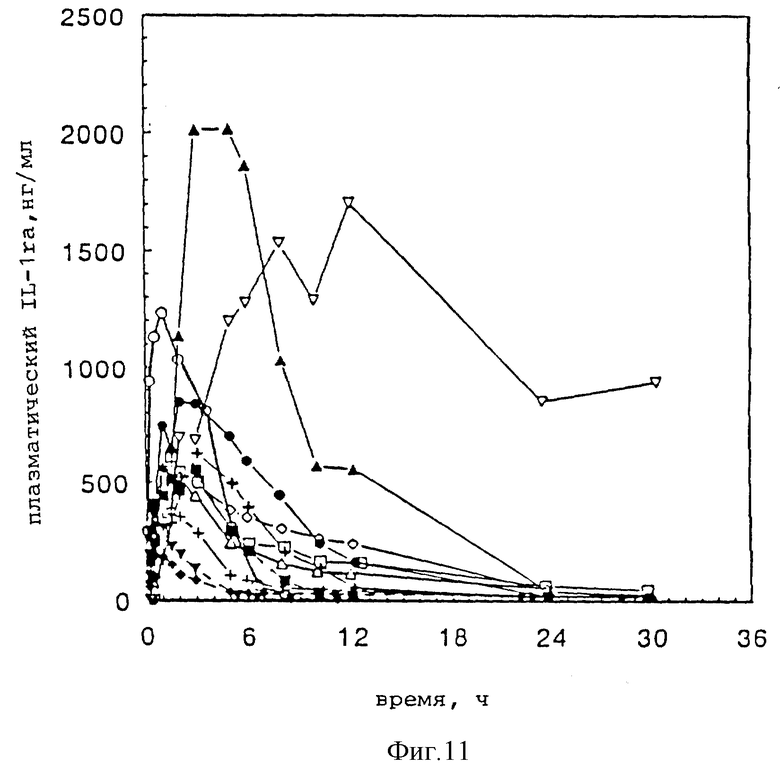
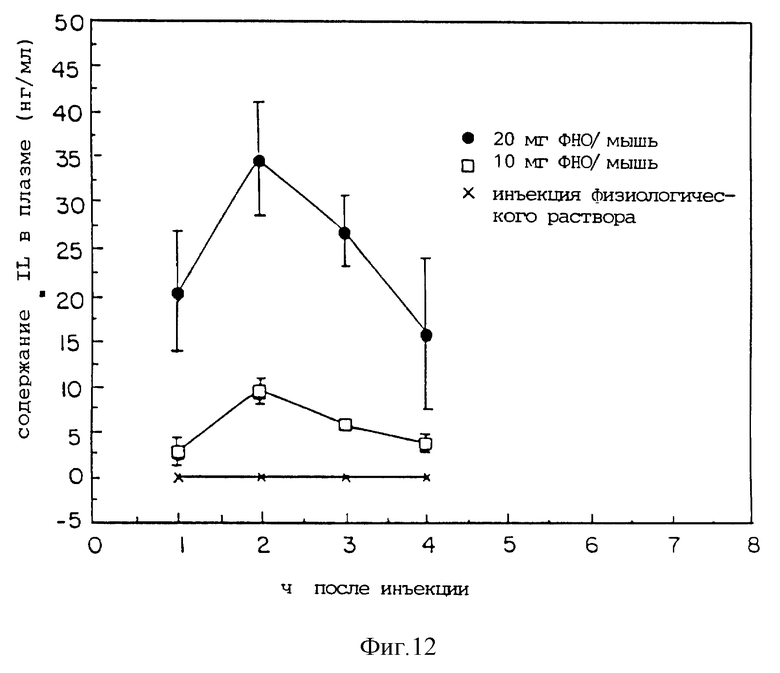
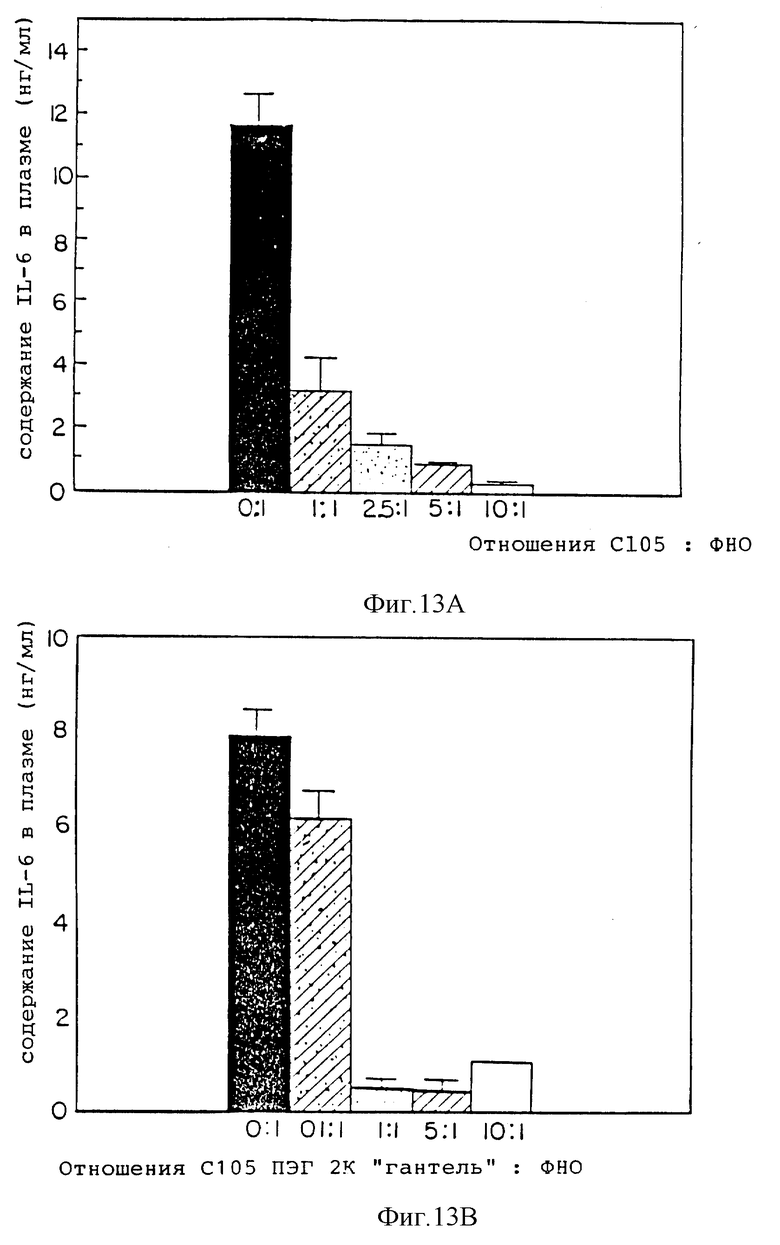
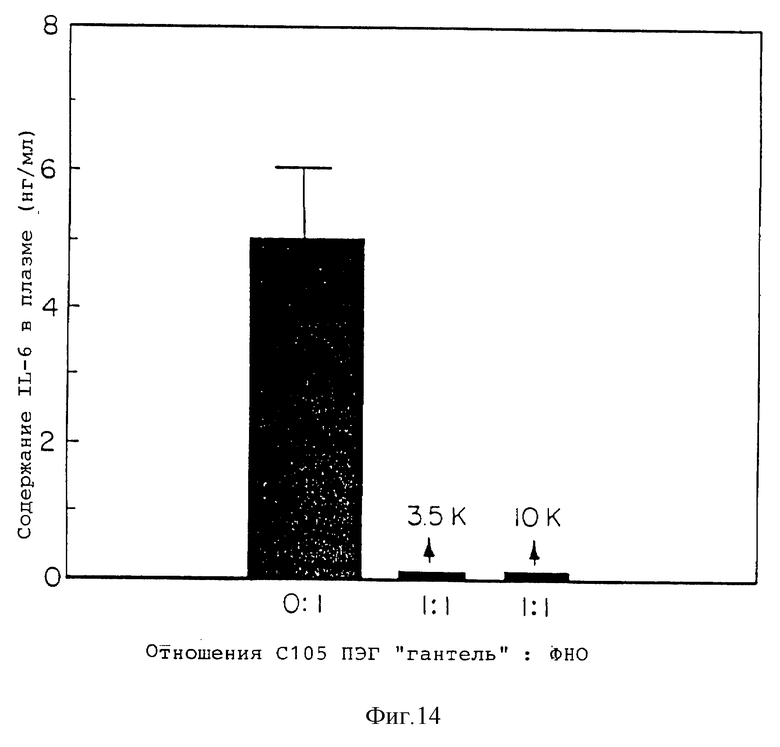
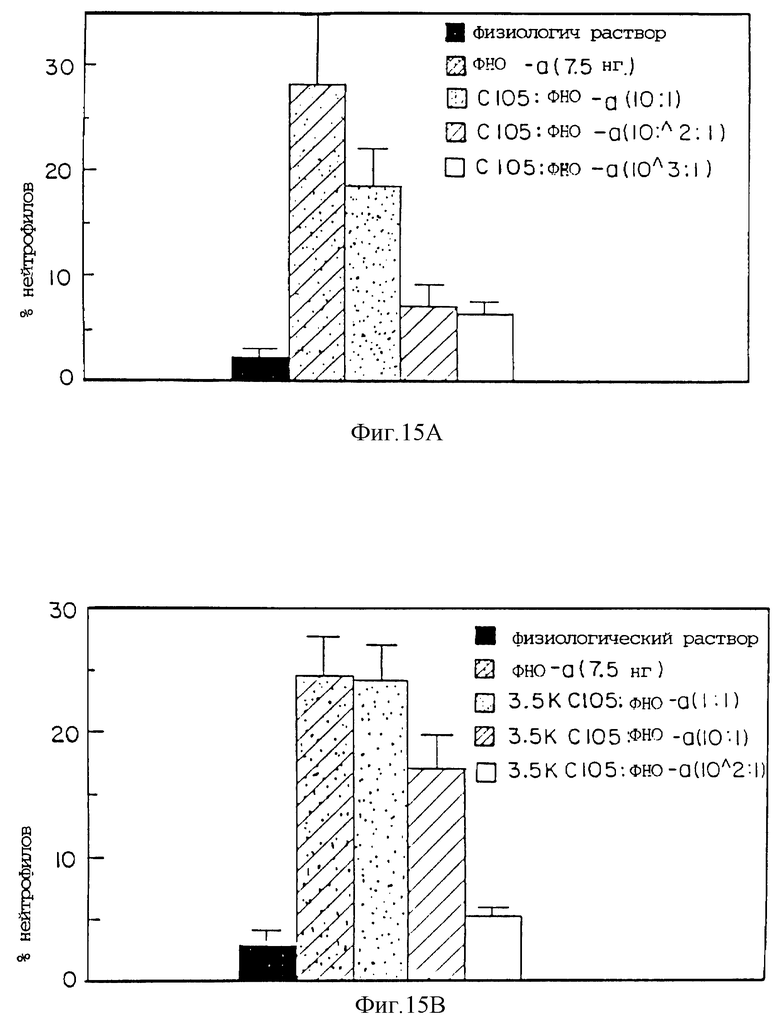
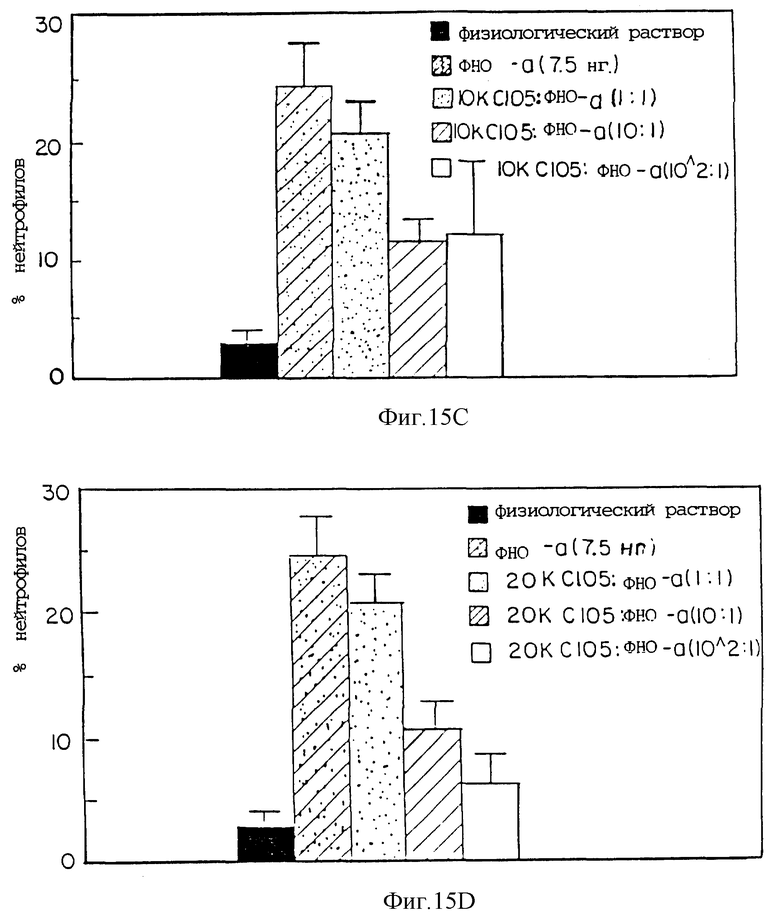
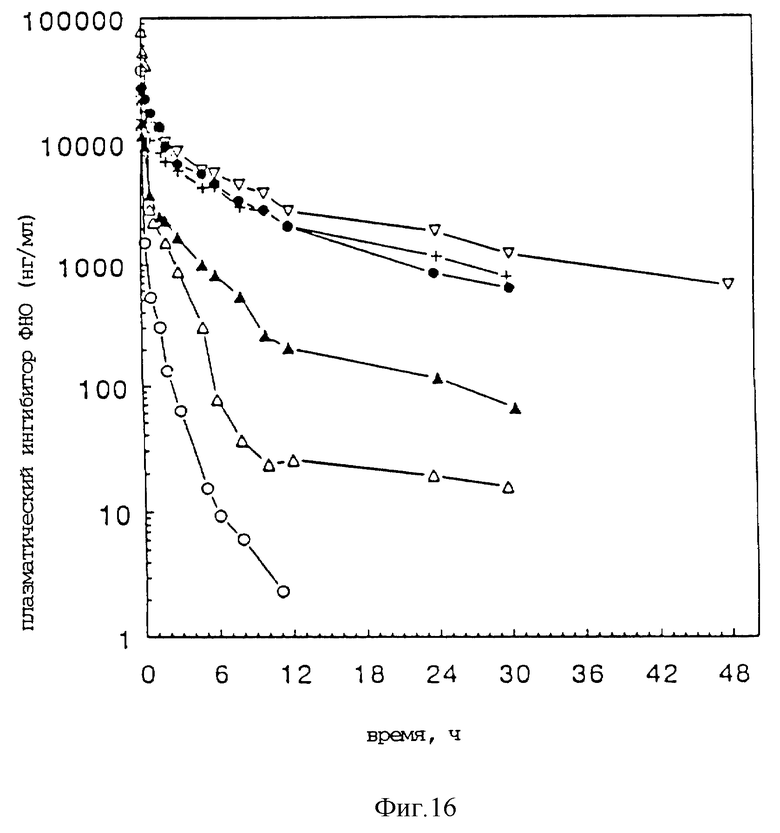
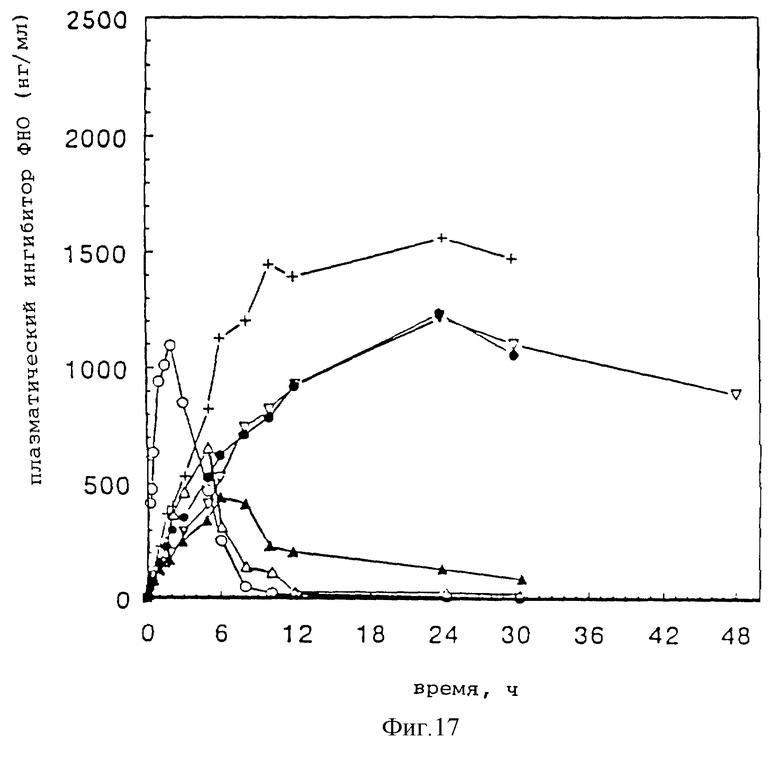
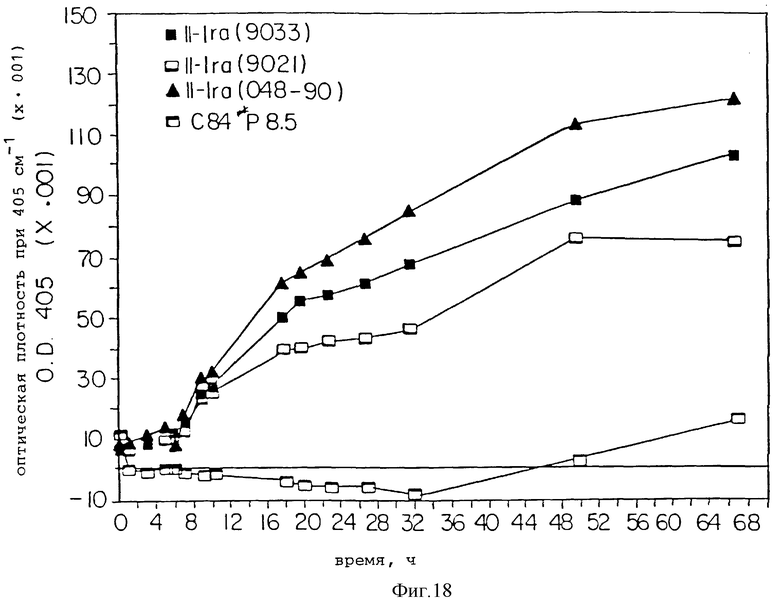
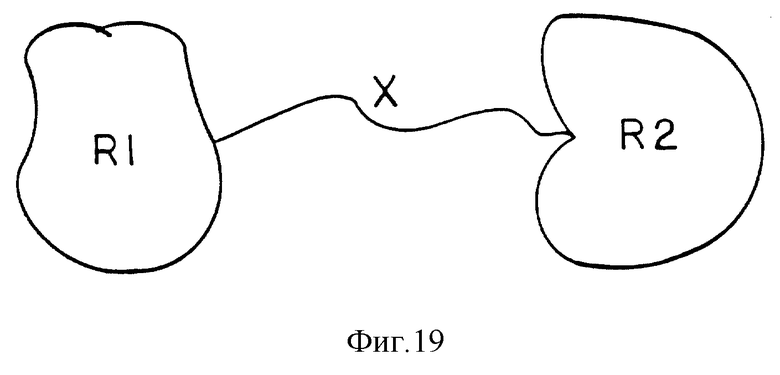
Изобретение относится к биотехнологии и медицине. Соединения, имеющие общую формулу R1 - X - R2, где Х - непептидный полимерный спейсер, а R1 и R2 - антагонист рецептора интерлейкина-1 или ингибитор ФНО, получают путем взаимодействия R1 и R2, содержащих или модифицированных так, что они содержат реакционноспособный цистеин, с непептидным спейсером, способным образовывать тиоэфирные связи. Мутеины получают рекомбинантным путем. Использование соединений позволяет снизить скорость выведения из организма терапевтического агента. 12 с. и 24 з.п. ф-лы, 24 ил., 4 табл.
R1 - X - R2,
в котором R1 и R2 каждый представляет собой пептидную связывающую группу, представляющую собой ингибитор фактора некроза опухоли (ингибитор ФНО) или его биологически активную часть, которые естественным образом содержат или модифицированы так, что они содержат реакционноспособную серу;
Х - непептидный терапевтически приемлемый водорастворимый полимерный спейсер.
R1 - X - R2,
где R1 и R2 каждый представляет собой полипептидную группу, выбранную из ингибитора ФНО, антагониста рецептора ИЛ-1 и их биологически активных частей, которые естественным образом содержат или модифицированы так, что они содержат реакционноспособный цистеин;
Х - непептидный терапевтически приемлемый водорастворимый полимерный спейсер, являющийся биологически инертным и имеющий общую формулу Y1 - (Z)n - Y2, где Y1 и Y2 представляют собой те остатки активирующих групп, которые реагируют с R1 и R2 для связывания спейсера с группами R1 и R2, и (Z)n представляет собой основную полимерную группу, причем n > 6,
отличающийся тем, что осуществляют взаимодействие R1 и R2 с непептидной терапевтически приемлемой полимерной группой, имеющей по меньшей мере две реакционноспособные группы, способные к образованию тиоэфирных связей при реакциях с цистеиновыми аминокислотными остатками с образованием указанного R1 - X - R2 соединения, и проводят выделение и очистку указанного соединения R1 - X - R2.
R1 - X - R2,
где R1 и R2 каждый представляет собой полипептидную группу, выбранную из ингибитора ФНО, антагониста рецептора ИЛ-1 и их биологически активных частей, которые естественным образом содержат или модифицированы так, что они содержат реакционноспособный цистеин;
Х - непептидный терапевтически приемлемый водорастворимый полимерный спейсер, являющийся биологически инертным и имеющий общую формулу Y1 - (Z)n - Y2, где Y1 и Y2 представляют собой те остатки активирующих групп, которые реагируют с R1 и R2 для связывания спейсера с группами R1 и R2, и (Z)n представляет собой основную полимерную группу, причем n > 6,
отличающийся тем, что осуществляют взаимодействие R1 с непептидной терапевтически приемлемой полимерной группой, содержащей по меньшей мере две реакционноспособные группы, способные образовывать тиоэфирные связи при взаимодействии с цистеиновыми аминокислотными остатками с образованием комплекса R1 - X, затем осуществляют взаимодействие комплекса R1 - X с R2 с образованием указанного соединения R1 - X - R2, и проводят выделение и очистку указанного соединения R1 - X - R2.
Y1 - (Z)n - Y2,
где Y1 и Y2 представляют остатки активирующих групп, способные реагировать с полипептидами, содержащими реакционноспособную серу;
(Z)n представляет собой основную полимерную группу, причем n > 6.
Y1 - (Z)n - Y2,
где Y1 и Y2 представляют остатки активирующих групп, способные реагировать с полипептидами, содержащими реакционноспособную серу; (Z)n представляет собой основную полимерную группу, причем n > 6.
Y1 - (Z)n - Y2,
где Y1 и Y2 представляют остатки активирующих групп, способные реагировать с полипептидами, содержащими реакционноспособную серу;
(Z)n представляет собой основную полимерную группу, причем n > 6.
| Davis et al | |||
| Biomedical Polymers: Polymeri Materials and Pharmaceuticals for Biomedical Use, p | |||
| Кинематографический аппарат | 1918 |
|
SU441A1 |
| US 4766106, 23.09.88 | |||
| US 4935233, 19.06.90 | |||
| US 4847325, 11.07.89 | |||
| 0 |
|
SU154316A1 | |
