Изобретение относится к новым фармацевтическим препаратам замедленного высвобождения, содержащим в качестве действующего активного начала 2-[[1-[1-[(4-фторфенил)метил] -1Н- бензимидазол-2-ил]пиперид-4-ил]метиламино]-пиримидин-4-ол или 2-[[1-[1-[(4-фторфенил)метил] -1Н-бензимидазол-2-ил]пиперид-4- ил]метиламино]-пиримидин-4(1Н)-он, или мизоластин.
Мизоластин описан в европейском патенте ЕР 0217700.
Мизоластин связывается с H1 гистаминовым рецептором и ингибирует дегрануляцию мастоцитов in vitro и in vivo. Таким образом, он может применяться для лечения респираторных, кожных или глазных аллергий, а также разнообразных других проявлений аллергии.
При пероральном введении содержащих мизоластин фармацевтических препаратов немедленного высвобождения было отмечено нежелательное седативное действие, которое ассоциировано с наличием высокой пиковой концентрации в плазме.
Таким образом, было необходимо найти препараты для перорального введения, обладающие таким профилем высвобождения активного действующего начала, который позволял бы получать более низкую пиковую концентрацию в плазме без снижения биодоступности.
Авторы изобретения основывали свои исследования подобных препаратов на изучении кинетики растворения мизоластина. Обоснованием для этого послужил тот факт, что мизоластин является слабым основанием (рК 5,6), которое умеренно растворимо в воде (13 мг/л при нейтральном pH), но намного лучше растворимо при кислом pH (11 г/л при pH 3); при pH 2 первые желатиновые капсулы высвобождали в среду-растворитель 100% мизоластина в течение 30 мин, в то время как при pH 6,8 растворялось только 40%.
Более того, требовалось чтобы на высвобождение мизоластина из фармацевтической формы замедленного высвобождения по данному изобретению не влияли различия в pH в желудочно-кишечном тракте.
Задача данного изобретения заключается в том, чтобы предложить содержащие мизоластин препараты, обладающие следующим профилем растворения:
- около 30-70% мизоластина растворяется в течение 1 ч,
- 100% мизоластина растворяется в течение 3-5 ч,
- pH - независимый профиль.
Авторы изобретения показали, что полностью пригодны таблетки, содержащие ядро, образованное таблеткой замедленного высвобождения, содержащей мизоластин в сочетании с жировой основой и органической кислотой, причем указанная таблетка полностью покрыта оболочкой для предотвращения разрушения продукта под действием света.
Таблетки по данному изобретению содержат от 1 до 25 мг мизоластина. Эти дозы соответствуют концентрациям мизоластина от 0,5 до 12% по массе.
Жировая основа изготовлена из гидрогенизированного касторового масла или из гидрогенизированных лецитинов, или из длинноцепочечных жирных кислот, например жирных кислот с длиной цепи C12-C28, таких как бегеновая кислота, или из триглицеридов, этерифицированных жирными кислотами со средней длиной цепи, например C8-C18 жирными кислотами.
Органическая кислота, предпочтительно имеющая рК, равное 2 или более, выбрана из малеиновой, винной, яблочной, фумаровой, молочной, лимонной, адипиновой и янтарной кислот в форме рацематов или изомеров. Наиболее предпочтительной кислотой по данному изобретению является L-винная кислота.
Массовое соотношение между мизоластином и органической кислотой должно составлять от 0,3 до 1. Для L-винной кислоты это соотношение предпочтительно равняется 0,5.
Таблетки изготавливают посредством гранулирования с использованием активного действующего начала, агента, составляющего жировую основу, органической кислоты и других эксципиентов, таких как, например, лактоза, маннит и сахара или аналогичные спирты на основе сахаров, микрокристаллическая целлюлоза, крахмал, фосфаты и сульфаты кальция, поливидон и замещенные целлюлозы, такие как гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза или метилцеллюлоза.
Гранулирование может проводиться во влажной фазе, например в присутствии воды или спирта, или же может быть выполнено посредством плавления или прессования. Стадия гранулирования может при необходимости быть исключена, а таблетки изготавливают путем непосредственного таблетирования смеси мизоластина и эксципиентов.
К полученным гранулам добавляют безводный коллоидный диоксид кремния и стеарат магния, и смесь таблетируют. Затем таблетки покрывают пленкой-оболочкой путем их опрыскивания образующим оболочку раствором в аппарате со слоем псевдоожиженного воздуха или в турбине для наложения оболочки.
Нижеследующий пример иллюстрирует изобретение, но не ограничивает его:
Таблетка, % (по массе):
Мизоластин - 4,8
Гидрогенизированное касторовое масло - 12,0
Лактоза - 60,0
Микрокристаллическая целлюлоза - 9,6
L-винная кислота - 9,6
Поливидон - 2,9
Безводный коллоидный диоксид кремния - 0,2
Стеарат магния - 0,9
Очищенная вода - Q.S.
Итого - 100,0
Оболочка
Метилгидроксипропилцеллюлоза - 74,0
Диоксид титана (Е171) - 18,5
Пропиленгликоль - 7,5
Очищенная вода - Q.S.
Итого - 100,0
Q.S. - сколько требуется
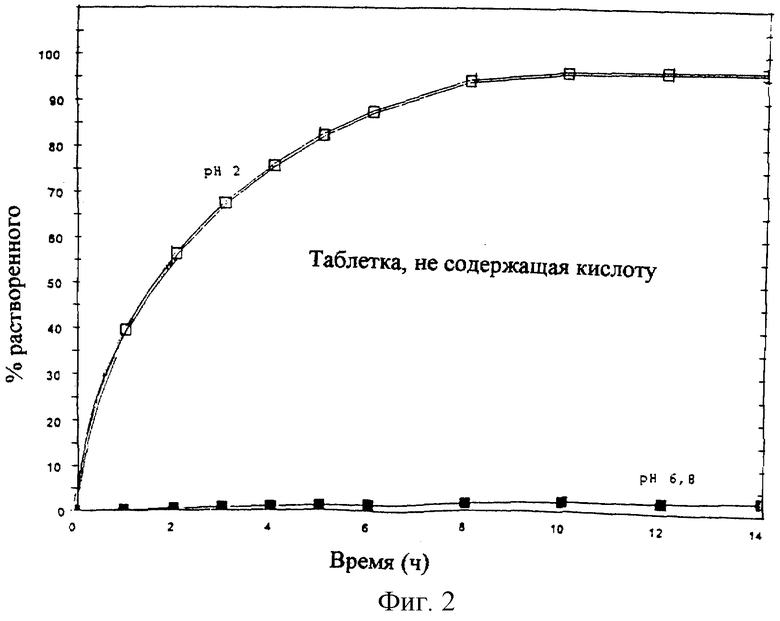
Профиль растворения, полученный для препарата по данному изобретению, приведен на фиг. 1.
При подобном профиле около 50% продукта растворяется в течение 1 ч, 100% продукта растворяется в течение 3-5 ч независимо от pH.
Профиль растворения, полученный для препарата, идентичного таковому по данному изобретению, но не содержащего L-винной кислоты, приведен на фиг. 2.
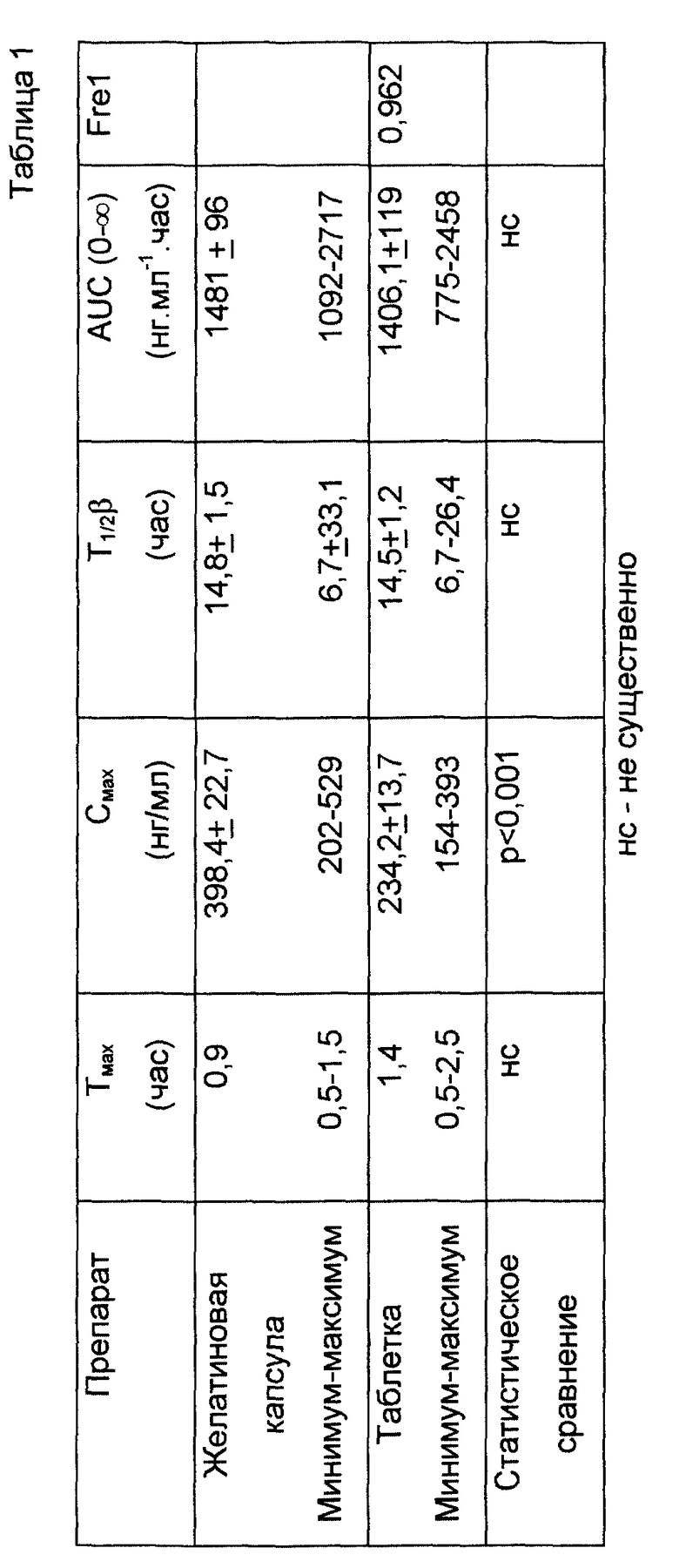
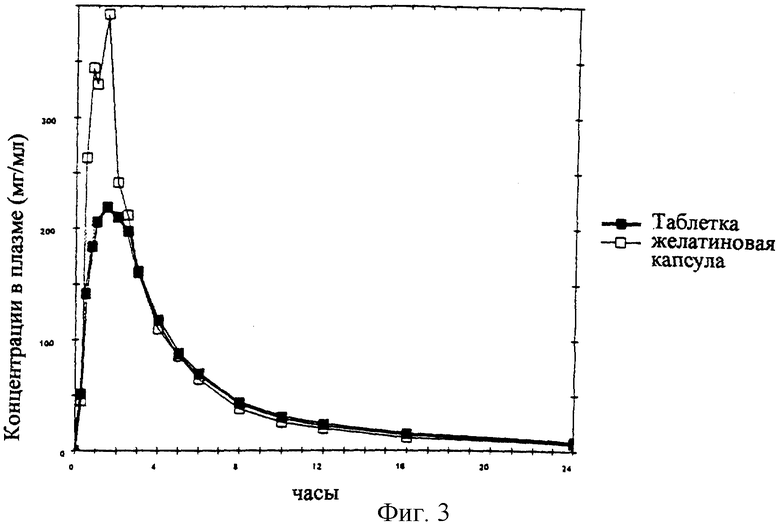
Плазменную кинетику содержащей 10 мг мизоластина фармацевтической формы по данному изобретению изучали на здоровом добровольце после однократного перорального введения в сравнении со стандартной желатиновой капсулой немедленного высвобождения, содержащей 10 мг мизоластина.
В табл. 1 представлены кинетические параметры, а на фиг. 3 показаны кривые плазменной кинетики, полученные соответственно для каждого из препаратов; плазменная кинетика, выявленная для фармацевтической формы по данному изобретению, позволяет предотвратить возникновение любого пика концентрации в плазме без потери биодоступности.
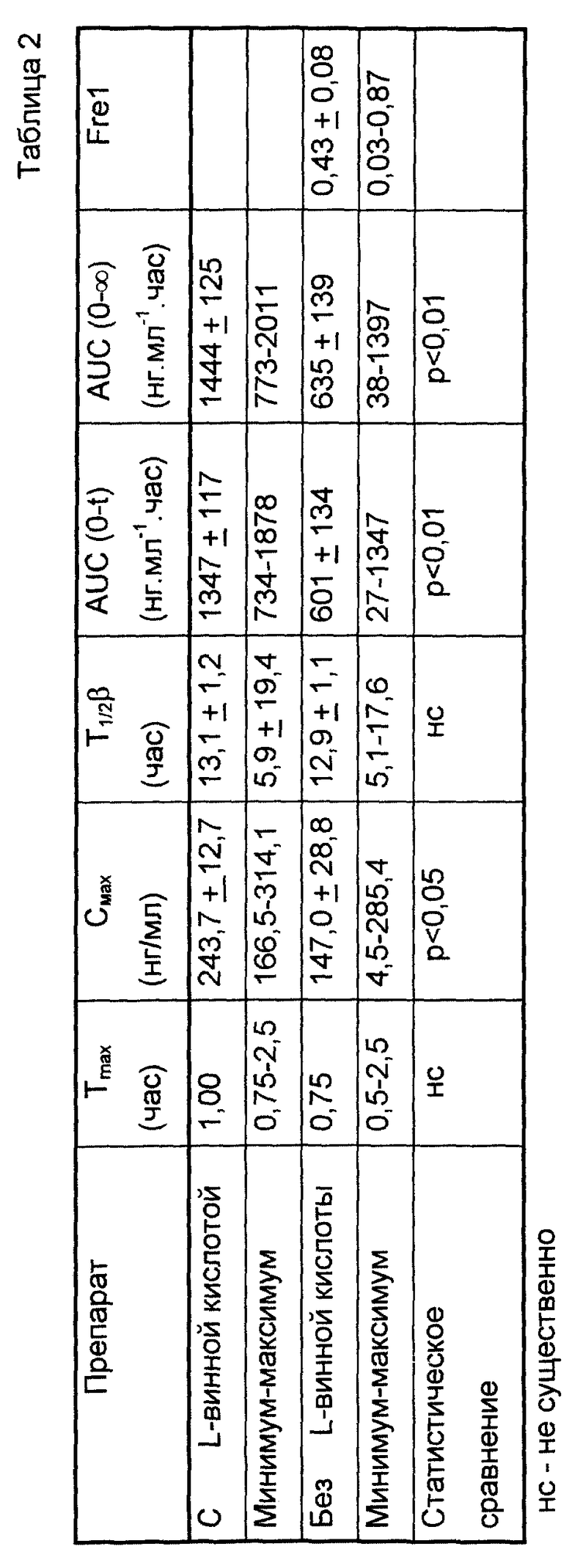
Плазменную кинетику фармацевтической формы по данному изобретению также исследовали в сравнении с таким же препаратом, не содержащим L-винной кислоты.
Исследования выполняли на 12 здоровых добровольцах после однократного перорального введения содержащей 10 мг мизоластина таблетки по данному изобретению или такой же таблетки, не содержащей L-винной кислоты.
Как показано в табл. 2, биодоступность препарата, не содержащего L-винную кислоту, составляет только 43% от биодоступности, наблюдаемой для препарата по данному изобретению, содержащего L-винную кислоту. Значения Cmax и AUC (площадь под кривой) (0-∞) соответственно в 1,5 и 2 раза выше для препарата, содержащего L-винную кислоту, по сравнению с препаратом, не содержащим ее.
К тому же для препарата с L-винной кислотой разница между минимальными и максимальными показателями гораздо ниже, что свидетельствует о значительной равномерности высвобождения.
В целом приведенные результаты свидетельствуют о том, что препараты по данному изобретению обладают:
- независимым от pH профилем растворения;
- in vivo высвобождением, предотвращающим возникновение любого пика концентрации в плазме;
- биодоступностью, не сниженной по отношению к препарату немедленного высвобождения;
- более низкой вариабельностью результатов плазменной кинетики.
Изобретение относится к химико-фармацевтической промышленности и касается фармацевтического препарата замедленного высвобождения. Изобретение заключается в том, что фармацевтический препарат содержит мизоластин и отличается тем, что он содержит ядро, состоящее из содержащей мизоластин таблетки замедленного высвобождения в сочетании с жировой основой и органической кислотой, причем указанная таблетка покрыта оболочкой. Изобретение обеспечивает высвобождение активного действующего вещества, которое позволяло бы получать более низкую пиковую концентрацию в плазме без снижения биодоступности. 7 з.п. ф-лы, 3 ил., 2 табл.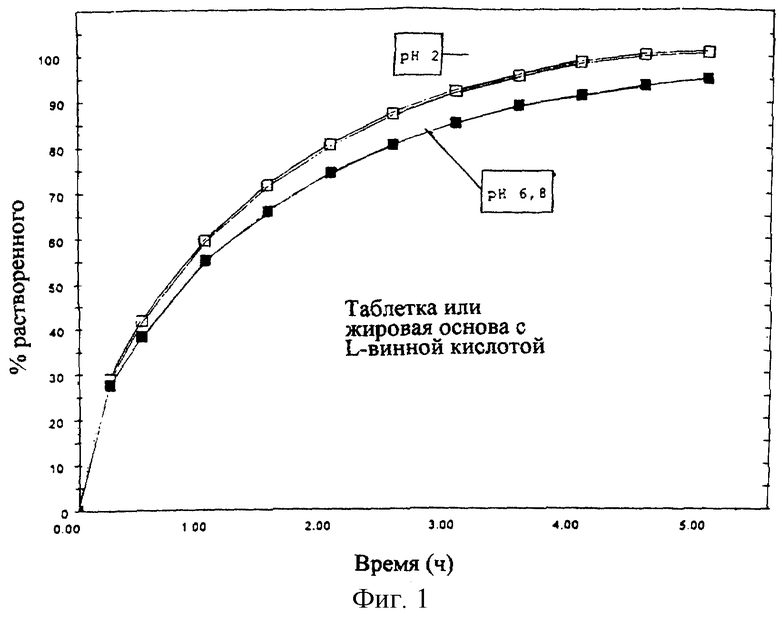
| ПРОИЗВОДНЫЕ 4-ОКСО-1,4-ДИГИДРОПИРИМИДИНА, ОБЛАДАЮЩИЕ АНТИАЛЛЕРГИЧЕСКОЙ И ИММУНОТРОПНОЙ АКТИВНОСТЬЮ | 1992 |
|
RU2015975C1 |
| DE 3719818 A, 23.12.1987 | |||
| УСТАНОВКА ДЛЯ АВТОМАТИЧЕСКОЙ ДУГОВОЙ СВАРКИ ПОВЕРХНОСТЕЙ С УЧАСТКАЛ1И РАЗНОЙ КРИВИЗНЫ | 0 |
|
SU269383A1 |

