Изобретение относится к области получения органических веществ и может быть использовано в производстве биологически активных соединений и нитропроизводных 1,2,4-триазола, имеющих разнообразное применение.
Известен способ получения солей 3,5-динитро-1,2,4-триазола диазотированием 3,4,5-триамино-1,2,4-триазола (гуаназина) в избытке нитрита натрия в условиях реакции Зандмейера (Пат. 3054800 США, МКИ С 06 В. 3,5-Dinitro-l, 2,4-triazoles /Н. Burchfield, D. K. Gullstrom. - Заявл. 17.09.1949, опубл. 18.09.1962. // С.А. - 1963 - V.58, 10220). Недостатками этого способа являются использование труднодоступного 3,4,5-триамино-1,2,4-триазола, применение дорогостоящего нитрата серебра и солей меди.
Наиболее близким по технической сущности и достигаемому результату является способ получения натриевой соли 3,5-динитро-1,2,4-триазола диазотированием 3,5-диамино-1,2,4- триазола в избытке нитрита натрия при температуре 0 . . . -5oC с последующим некаталитическим нуклеофильным замещением диазониевых групп в образовавшейся соли диазония на нитрогруппы при нагревании реакционной смеси до 60oC и последующим взаимодействием образовавшегося 3,5-динитро-1,2,4-триазола с гидрокарбонатом натрия в ацетоне (Багал Л.И., Певзнер М.С., Фролов А.Н., Шелудякова Н.И.// ХГС. - 1970. N 2.-С. 259-264). Аналогично по этому способу взаимодействием с карбонатами или гидрокарбонатами калия и лития в ацетоне могут быть получены калиевая и литиевая соли 3,5-динитро-1,2,4-триазола (Sitzmann М.Е.// J. Org. Chem. - 1978. -V.43. N 17. - P. 3389-3391). Недостатком данного способа является использование дорогостоящего 3,5-диамино-1,2,4-триазола и взрывоопасность солей диазония, образующихся на стадии диазотирования.
Задачей изобретения является разработка дешевого и безопасного способа получения солей 3,5-динитро-1,2,4-триазола, пригодных для применения в качестве полупродуктов при синтезе различных нитропроизводных 1,2,4-триазола и биологически активных веществ за счет использования вместо 3,5-диамино-1,2,4- триазола доступного и дешевого сырья - дициандиамида, гидразингидрата и соляной кислоты, а также совмещения стадий диазотирования и нуклеофильного замещения диазогруппы на нитрогруппу при диазотировании в интервале температур 40-50oC.
Поставленная задача достигается за счет того, что в предлагаемом способе получения солей 3,5-динитро-1,2,4-триазола, включающем взаимодействие с карбонатами или гидрокарбонатами щелочных металлов в ацетоне, к гидразингидрату при перемешивании приливают соляную кислоту и затем добавляют дициандиамид при мольном соотношении гидразин : HCl : дициандиамид 1:2:1, полученный раствор нагревают при 45-50oC, затем к раствору добавляют водный раствор серной кислоты и полученную смесь приливают при температуре 40-50oC и интенсивном перемешивании к водному раствору нитрита натрия при мольном соотношении дициандиамид : NaNO2 = 1:6-1:7.
К гидразингидрату при перемешивании и охлаждении приливают водный раствор соляной кислоты, затем прибавляют дициандиамид, образовавшийся раствор нагревают при 45-50oC в течение 60 минут. Затем к раствору добавляют водный раствор серной кислоты и полученную смесь приливают при температуре 40-50oC и интенсивном перемешивании к водному раствору нитрита натрия. Полученный раствор подкисляют серной кислотой, обрабатывают активированным углем, отфильтровывают и экстрагируют этилацетатом. Этилацетат отгоняют в вакууме, остаток растворяют в ацетоне и при перемешивании обрабатывают карбонатом или гидрокарбонатом щелочного металла. Избыток карбоната или гидрокарбоната отфильтровывают, ацетон отгоняют, полученный остаток кристаллизуют из воды или спирта с добавлением активированного угля и высушивают.
Первая стадия синтеза - взаимодействие дициандиамида с гидразингидратом и соляной кислотой - должна осуществляться при температуре 45-50oC. При более высокой температуре происходит снижение выхода целевого продукта, а при более низкой реакция протекает слишком медленно. Концентрация кислоты и гидразингидрата не оказывает существенного влияния на выход продукта, однако наиболее целесообразно применение производимых промышленностью гидразингидрата (концентрация ~ 80%) и соляной кислоты (концентрация ~ 30%). Мольное соотношение гидразин : соляная кислота : дициандиамид= 1:2:1 является стехиометрическим, его изменение приводит либо к снижению выхода целевого продукта, либо к неполному расходованию одного из реагентов и экономически нецелесообразно.
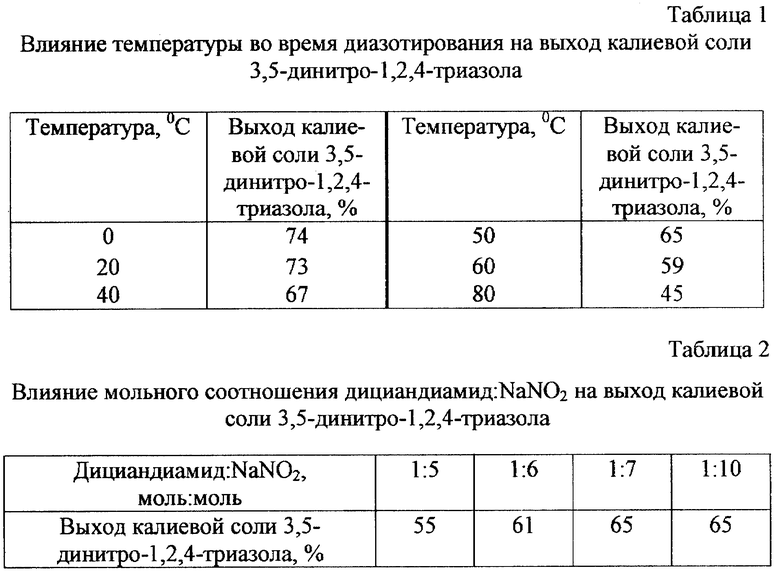
Повышение температуры на стадии диазотирования до 40-50oC хотя и вызывает снижение выхода 3,5-динитро-1,2,4- триазола на 7-10% по сравнению с проведением синтеза при температуре 0 ... -5oC, однако приводит к совмещению стадий диазотирования и нуклеофильного замещения диазогруппы на нитрогруппу. При этом взрывоопасные соли диазония не накапливаются в реакционной смеси, а по мере образования превращаются в стабильный анион 3,5-динитро-1,2,4-триазола, что повышает безопасность процесса. Повышение температуры до 60oC и более вызывает значительное снижение выхода целевого продукта и поэтому нецелесообразно.
Мольное соотношение дициандиамид : NaNO2 = 1:6 - 1:7 является наиболее оптимальным. Изменение мольного соотношения приводит либо к уменьшению выхода целевого продукта, либо (при значительном избытке нитрита натрия) к выделению большого количества оксидов азота, что требует дополнительных затрат на их утилизацию.
Пример 1
К 12,5 г (0,2 моль) 80%-ного гидразингидрата при перемешивании и охлаждении приливали 0,4 моль 30%-ной соляной кислоты, затем добавляли 16,8 г (0,2 моль) дициандиамида, нагревали при 45-50oC 60 мин, и затем к раствору добавляли 240 мл 2,5 н. водного раствора серной кислоты. Полученную смесь приливали к раствору 110 г NaNO2 в 135 мл H2O при необходимой температуре (таблица 1) и интенсивном перемешивании в течение 60 мин. При диазотировании в интервале температур 0-40oC реакционную смесь после диазотирования дополнительно выдерживали 30 мин при 60oC для обеспечения полного протекания реакции замещения диазогруппы на нитрогруппу. Полученный после диазотирования раствор подкисляли 62 мл 80%-ной H2SO4 (при этом происходит выделение оксидов азота), обрабатывали 4 г активированного угля и экстрагировали этилацетатом (5 раз по 60 мл). Этилацетат отгоняли в вакууме, остаток растворяли в 300 мл ацетона, обрабатывали 30 г карбоната калия и отфильтровывали, ацетон отгоняли, остаток высушивали при 60-80oC. Выход калиевой соли 3,5-динитро-1,2,4-триазола указан в таблице 1. После двукратной перекристаллизации из воды с добавлением 4 г активированного угля продукт имеет т.пл. 225-226oC. По литературным данным, т. пл. 223-225oC (Sitzmann М.Е. // J.Org. Chem. - 1978. - V. 43. N 17. - P. 3389-3391). Вещество не дает депрессии температуры плавления с калиевой солью 3,5-динитро- 1,2,4-триазола, полученной по известному способу, ИК- и УФ-спектры идентичны.
Пример 2
К 12,5 г (0,2 моль) 80%-ного гидразингидрата при перемешивании и охлаждении приливали 0,4 моль 30%-ной соляной кислоты, затем добавляли 16,8 г (0,2 моля) дициандиамида, нагревали при 45-50oC 60 мин, и затем к раствору добавляли 240 мл 2,5 н. водного раствора серной кислоты. Полученную смесь приливали к раствору необходимого количества NaNO2 (таблица 2) в 135 мл H2O при температуре 50oC и интенсивном перемешивании в течение 60 мин, дальнейшие операции проводили аналогично примеру 1. Выход калиевой соли 3,5-динитро-1,2,4-триазола представлен в таблице 2. Свойства продукта после очистки (т. пл. , ИК-, УФ-спектры) идентичны калиевой соли 3,5-динитро-1,2,4- триазола, полученной по известному методу.
Пример 3
К 12,5 г (0,2 моль) 80%-ного гидразингидрата при перемешивании и охлаждении приливали 0,4 моль 30%-ной соляной кислоты, затем добавляли 16,8 г (0,2 моль) дициандиамида, нагревали при 45-50oC 60 мин, и затем к раствору добавляли 240 мл 2,5 н. водного раствора серной кислоты. Полученную смесь приливали к раствору 110 г NaNO2 в 135 мл H2O при температуре 50oC и интенсивном перемешивании в течение 60 мин. Затем подкисляли 62 мл 80%-ной H2SO4, обрабатывали 4 г активированного угля и экстрагировали этилацетатом (5 раз по 60 мл). Этилацетат отгоняли в вакууме, остаток растворяли в 300 мл ацетона, обрабатывали 30 г карбоната или гидрокарбоната натрия, отфильтровывали, ацетон отгоняли, остаток трижды кристаллизовали из этанола. Выход дигидрата натриевой соли 3,5-динитро-1,2,4-триазола 16,5 г (38%), т. пл. 121-125o с разложением [по литературным данным (Багал Л.И. и др. // ХГС. - 1970. N 2. - С. 259-264) т. пл. 123oC с разл.]. Свойства продукта после очистки (ИК-, УФ-спектры) идентичны дигидрату натриевой соли 3,5-динитро-1,2,4-триазола, полученной по известному способу. После сушки при 60-80oC вещество превращается в безводную натриевую соль 3,5-динитро-1,2,4-триазола с т. пл. 294oC с разл.
Найдено (%): N 39,1.
C2N5O4Na. Вычислено (%): N 38,7.
Пример 4
Литиевую соль 3,5-динитро-1,2,4-триазола получали аналогично примеру 3 при использовании карбоната лития взамен карбоната натрия. Полученный продукт высушивали в вакууме при 100oC. Выход 11,6 г (35%), т. пл. 314o с разл. По литературным данным т. пл. 315oC (Sitzmann M.E. // J.Org.Chem. - 1978. - V. 43. N 17. - P. 3389-3391). Вещество не дает депрессии температуры плавления с литиевой солью 3,5-динитро-1,2,4-триазола, полученной по известному способу, ИК- и УФ-спектры идентичны.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ НИТРАТА 3,5-ДИАМИНО-1,2,4-ТРИАЗОЛА | 1998 |
|
RU2152389C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-АЦЕТИЛ-3,5-ДИАМИНО-1,2,4-ТРИАЗОЛА | 1998 |
|
RU2148579C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИГИДРОХЛОРИДА 5-АМИНО-3-АМИНОМЕТИЛ-1,2,4-ТРИАЗОЛА | 2011 |
|
RU2476428C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3(5)-ПИРИДИЛЗАМЕЩЕННЫХ 5(3)-АМИНО-1,2,4-ТРИАЗОЛОВ | 2009 |
|
RU2412180C1 |
| СПОСОБ ПОЛУЧЕНИЯ R-МЕТИЛПРОИЗВОДНЫХ 3,5-ДИАМИНО-1,2,4-ТРИАЗОЛА | 2005 |
|
RU2292340C1 |
| ЭЛЕКТРОЛИТ ДЛЯ ОСАЖДЕНИЯ КОМПОЗИЦИОННОГО ПОКРЫТИЯ НИКЕЛЬ-БОР-ФТОРОПЛАСТ | 2002 |
|
RU2213812C1 |
| СПОСОБ СТАБИЛИЗАЦИОННОЙ ОБРАБОТКИ ПЕРЕСЫЩЕННЫХ ВОД | 2000 |
|
RU2200712C2 |
| ГАЛЬВАНИЧЕСКИЙ КОМПОЗИЦИОННЫЙ МАТЕРИАЛ НА ОСНОВЕ НИКЕЛЯ | 2002 |
|
RU2213813C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОГИДРАТА 5-АМИНО-1,2,4-ТРИАЗОЛ-3-ИЛУКСУСНОЙ КИСЛОТЫ | 2006 |
|
RU2313522C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА-4-АМИНО-3-ФЕНИЛБУТАНОВОЙ КИСЛОТЫ | 1993 |
|
RU2072984C1 |
Изобретение может найти применение при производстве биологически активных соединений и нитропроизводных 1,2-4-триазола, имеющих разнообразное применение. Получение солей 3,5-динитро-1,2,4-триазола осуществляется взаимодействием карбонатов или гидрокарбонатов щелочных металлов с раствором 3,5-динитро-1,2,4-триазола в ацетоне. Для получения 3,5-динитро-1,2,4-триазола нагревают гидразингидрат, соляную кислоту и дициандиамид при мольном соотношении 1 : 2 : 1 и температуре 45 - 50°С, образовавшийся раствор приливают к водному раствору нитрита натрия при 40 - 50°С и интенсивном перемешивании. Технический результат - удешевление процесса, а также повышение безопасности процесса. 2 табл.
Способ получения солей 3,5-динитро-1,2,4-триазола путем взаимодействия последнего с карбонатами или гидрокарбонатами щелочных металлов в ацетоне, отличающийся тем, что 3,5-динитро-1,2,4-триазол получают путем приливания при перемешивании соляной кислоты к гидразингидрату с последующим добавлением дициандиамида при мольном соотношении гидразингидрат : соляная кислота : дициандиамид 1 : 2 : 1, полученный раствор нагревают при 45 - 50°С, затем к раствору добавляют водный раствор серной кислоты и полученную смесь приливают при 40 - 50°C и интенсивном перемешивании к водному раствору нитрита натрия при мольном соотношении дициандиамид : нитрит натрия 1 : (6-7).
| БАГАЛ Л.И | |||
| и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ХГС | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 3054800 A1, 18.09.1962 | |||
| US 4236014 А, 25.11.1980. | |||
