Изобретение относится к медицине.
Сосудистый эндотелий составляет главный орган, действующий как регулятор свертывания крови, воспаления и обмена жидкостей и посредников между внутрисосудистым отделением и тканями паренхимы. По существу, правильная функция эндотелия является решающей для общего гомеостаза. Дисфункция эндотелия, возникающая в результате изменения экспрессии важных молекул поверхности, может привести к дефектам свертывания, местному и системному сосудистым воспалениям и усилению развития и разрыва атеросклеротических бляшек. Эти эффекты могут в дальнейшем привести к состояниям, включающим инфаркт миокарда, обширный венозный тромбоз, рассеянный внутрисосудистый тромбоз и удар.
Определенные белки поверхности клетки меняются в ответ на повреждение сосуда или инсульт и могут быть использованы в качестве маркеров дисфункции эндотелия. Критическим классом таких белков являются рецепторы/лиганды, являющиеся переносчиками в межклеточной адгезии, включающие интегрин, селектин (например, ELAM) и члены иммуноглобулинового семейства, такие, как ICAM и VCAM. Количество этих молекул увеличиваются в ответ на множество стимулов, включающих цитокины, и в дополнение к тому, что они являются важными маркерами дисфункции эндотелия, они играют ключевую роль в тромботических, воспалительных и атеросклеротических процессах в стенке сосуда. Другие функции, такие, как реакция на поверхностный антикоагулянт, также ослабевают в состоянии эндотелиальной дисфункции. Соединение, которое блокировало бы эндотелиальную дисфункцию, что определяется путем измерения его способности ингибировать межклеточную адгезию или экспрессии прокоагулянтных активностей, могло бы быть полезным при лечении таких состояний, как сепсис, нарушений, включающих повреждение и травму основной ткани, синдром системной воспалительной реакции, синдром сепсиса, септический шок и синдром сложной дисфункции органов (включающий DIC), а также разрыв атеросклеротической бляшки и их объединенное осложнение. Поскольку межклеточная адгезия является фундаментальным процессом большой биологической важности, способность специфично модулировать связывающие белки может быть использована во многих клинических применениях вне сосудистой ткани, включая ее использование в качестве противовоспалительного агента.
Настоящее изобретение обеспечивает способы ингибирования межклеточной адгезии, включающие введение для применения человеку эффективного количества соединения формулы 1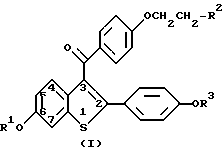
где R1 и R3 - независимо водород, -CH3, -C(O)(C1-C6 алкил), или -C(O)Ar, где Ar - необязательно замещенный фенил;
R2 выбирается из группы, состоящей из пирролидино, гексаметиленимино и пиперидиногрупп, их фармацевтически приемлемых солей и сольватов.
Настоящее изобретение касается открытия того, что некоторые группы 2-фенил-3-ароилбензотиофенов (бензотиофенов), представленных формулой 1, пригодны для ингибирования межклеточной адгезии, в частности сосудистой межклеточной адгезии.
Способы применения, обеспеченные настоящим изобретением, осуществляются на практике путем введения нуждающемуся в этом человеку дозы соединения формулы 1 или его фармацевтически пригодных соли или сольвата, которая эффективна для ингибирования межклеточной адгезии или ее последствий. Термин "ингибирование" включает его общепринятый смысл, который включает препятствование, предотвращение, ограничение и замедление, остановку и обращение. По существу, настоящий способ включает как медицинское терапевтическое, так и/или профилактическое назначение соответственно.
Ралоксифен, соединение настоящего изобретения, где оно представляет собой гидрохлорид соединения формулы I, R1 и R3- водород и R2 - 1 -пиперидинил, является ядерной регуляторной молекулой. Было показано, что ралоксифен связывается с рецептором эстрогена и, как полагали сначала, представляет собой молекулу, действие и фармакология которой являются антиэстрогенными в том, что она блокирует способность эстрогена активировать ткань матки и зависящий от эстрогена рак груди. На самом деле ралоксифен действительно блокирует действие эстрогена в некоторых клетках; однако, в клетках другого типа ралоксифен активирует те же гены, что и эстроген и проявляет ту же фармакологию, например, на остеопороз, гиперлипидемию. В результате этого ралоксифен называют антиэстрогеном со смешанными свойствами агонист-антагонист. Уникальное свойство, которое проявляет ралоксифен и в чем он отличается от эстрогена, как теперь полагают, осуществляется за счет уникальной активации и/или подавления различных функций гена комплексом ралоксифен-рецептор эстрогена, в противоположность активации и/или подавлению генов комплексом эстроген-рецептор эстрогена. Поэтому, хотя ралоксифен и эстроген используют и конкурируют за один и тот же рецептор, фармакологический результат регуляции гена ими обоими предсказать трудно и он является уникальным для каждого.
Как правило, соединение смешивается с обычными эксципиентами, разбавителями или носителями и прессуется в таблетки или составляется в виде эликсиров или растворов для обычного приема через рот, или вводится внутримышечным или внутривенным путями. Эти соединения могут вводиться через кожу и могут быть составлены в виде лекарственных форм с отсроченным высвобождением.
Соединения, используемые в способах настоящего изобретения, могут быть приготовлены известными способами, такими, как описанные в патентах США N 4133814, 4418068, и 4380635, которые все включены в описание в качестве ссылки. Обычно способ начинается с бензо[b]тиофена с 6-гидроксигруппой и 2-(4-гидроксифенильной) группой. Исходное соединение защищают, ацилируют, снимают защиту, при этом образуется соединение формулы I. Примеры получения таких соединений приведены в упомянутых выше патентах США. Необязательно замещенный фенил включает фенил и моно- и дизамещенный фенил, где заместителями являются C1-C6алкильные, C1-C6-алкокси, гидрокси, нитрогруппы, фтор или три(хлор- или фтор) метильная группы.
Соединения, используемые в способах настоящего изобретения, образуют фармацевтически приемлемые соли кислот и оснований с широким кругом органических и неорганических кислот и оснований и включают физиологически приемлемые соли, которые часто используются в фармацевтической химии. Такие соли также являются частью настоящего изобретения. Неорганические кислоты, используемые для образования таких солей, включают хлористоводородную, бромистоводородную, йодистоводородную, азотную, серную, фосфорную, гипофосфорную кислоты и т. п. Также могут быть использованы соли, полученные из таких органических кислот, как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые и гидроксидиалкановые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты. Такие фармацевтически приемлемые соли включают, таким образом, ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, нафталин-2-бензоат, бромид, изобутират, фенилбутират, β -гидроксибутират, бутин-1,4-диоат, гексин-1,4-диоат, капрат, каприлат, хлорид, циннамат, цитрат, формиат, фумарат, гликоллат, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, нитрат, оксалат, фталат, терефталат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, пропиолат, пропионат, фенилпропионат, салициллат, себацат, сукцинат, суберат, сульфат, бисульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензолсульфонат, п-бромфенилсульфонат, хлорбензолсульфонат, этансульфонат, 2-гидроксиэтансульфонат, метансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, п-толуолсульфонат, ксилолсульфонат, тартрат и т.п. Предпочтительной солью является гидрохлорид.
Фармацевтически приемлемые соли кислот обычно образуются при взаимодействии соединения формулы I с эквимолярным количеством кислоты или ее избытком. Реагенты, как правило, взаимодействуют в общем растворителе, таком, как диэтиловый эфир или бензол. Соль обычно выпадает из раствора в течение от одного часа до 10 дней и выделяется фильтрованием или растворитель может быть удален обычными способами.
Основания, обычно используемые для образования солей, включают гидроксид аммония и гидроксиды щелочных и щелочноземельных металлов, а также алифатические первичные, вторичные и третичные амины, алифатические диамины. Основания, особенно полезные при приготовлении солей, включают гидроксид аммония, карбонат калия, метиламин, диэтиламин, этилендиамин и циклогексиламин.
Фармацевтически приемлемые соли, как правило, имеют повышенные характеристики растворимости по сравнению с соединениями, из которых они были получены, и поэтому чаще более подходят для составов в виде жидкостей или эмульсий.
Фармацевтический состав может быть приготовлен известными в данной области способами. Например, соединения могут быть смешаны с обычными эксципиентами, разбавителями или носителями и образованы в форме таблеток, капсул, суспензий, порошков и т.п. Эксципиенты, разбавители и носители, подходящие для подобных составов, включают следующее: наполнители и разбавители, такие, как крахмал, сахара, маннит и кремнийорганические производные; связывающие агенты, такие, как карбоксиметилцеллюлоза и другие производные целлюлозы, альгинат, желатин и поливинилпирролидон; увлажняющие агенты, такие, как глицерин; разрыхляющие агенты, такие, как карбонат кальция и гидрокарбонат натрия; замедляющие растворение агенты, такие, как парафин; ускорители ресорбции, такие, как четвертичные аммониевые соединения; поверхностно-активные агенты, такие, как цетиловый спирт, моностеарат глицерина; адсорбционные носители, такие, как каолин и бентонит, и смазывающие вещества, такие, как тальк, стеараты кальция и магния и твердые полиэтилгликоли.
Эти соединения также могут быть составлены в виде эликсиров или растворов для обычного перорального приема или растворов, подходящих для парентерального приема, например, внутримышечным, подкожным или внутривенным путями. Кроме этого, эти соединения хорошо подходят для составов в виде лекарственных форм с отсроченным высвобождением. Составы могут быть приготовлены так, чтобы они высвобождали активный ингредиент только или предпочтительно на определенном участке кишечного тракта, возможно, через некоторый период времени. Покрытия, оболочки и защитные матрицы могут быть сделаны из полимерных веществ или парафина.
Определенная доза соединения формулы I, необходимая для ингибирования межклеточной адгезии или ее последствий, или любое другое раскрытое применение, согласно настоящему изобретению будет зависеть от тяжести состояния, способа приема и сопутствующих факторов, которые будут определяться лечащим врачом. Обычно принятые и эффективные дневные дозы составляют от около 0,1 до около 1000 мг/день и более обычно - от около 50 до около 200 мг/день. Такие дозировки будут вводиться нуждающемуся в этом субъекту от одного до трех раз каждый день или более часто, в случае необходимости для эффективного ингибирования межклеточной адгезии или ее последствий, или любого другого раскрытого применения.
Обычно соединение формулы I предпочитают вводить в виде его соли кислоты, как это принято при введении лекарств, имеющих основную группы, такую, как пиперидиновое кольцо. Такое соединение также вводят преимущественно перорально. Для этих целей пригодны дозировки вводимых перорально форм.
Составы
В следующих составах "активный ингредиент" означает соединение формулы I.
Состав 1: Желатиновые капсулы
Твердые желатиновые капсулы готовят, используя следующее:
Ингредиент - Количество (мг/капсулу)
Активный ингредиент - 0,1 - 1000
Крахмал, NF - 0 - 650
Крахмал, сыпучий порошок - 0 - 650
Силиконовая жидкость, 350 сантистокс - 0 - 15
Ингредиенты смешивают, пропускают через сито США N 45 меш и заполняют ими твердые желатиновые капсулы.
Примеры составов капсул ралоксифена включают приведенные ниже:
Состав 2: Капсула ралоксифена
Ингредиент - Количество (мг/капсулу)
Ралоксифен - 1
Крахмал, NF - 112
Крахмал, сыпучий порошок - 225,3
Силиконовая жидкость, 350 сантистокс - 1,7
Состав 3: Капсула ралоксифена
Ингредиент - Количество (мг/капсулу)
Ралоксифен - 5
Крахмал, NF - 108
Крахмал, сыпучий порошок - 225,3
Силиконовая жидкость, 350 сантистокс - 1,7
Состав 4: Капсула ралоксифена
Ингредиент - Количество (мг/капсулу)
Ралоксифен - 10
Крахмал, NF - 103
Крахмал, сыпучий порошок - 225,3
Силиконовая жидкость, 350 сантистокс - 1,7
Состав 5: Капсула ралоксифена
Ингредиент - Количество (мг/капсулу)
Ралоксифен - 50
Крахмал, NF - 150
Крахмал, сыпучий порошок - 397
Силиконовая жидкость, 350 сантистокс - 3,0
Приведенные выше составы могут меняться в согласии с необходимыми разумными изменениями.
Составы таблеток готовят с использованием следующих ингредиентов
Состав 6: Таблетки
Ингредиент - Количество (мг/таблетку)
Активный ингредиент - 0,1 - 1000
Целлюлоза, микрокристаллическая - 0 - 650
Двуокись кремния, коллоидальная - 0 - 650
Стеариновая кислота - 0 - 15
Компоненты смешивают и прессуют с образованием таблеток.
По другому способу таблетки, содержащие каждая 0,1-1000 мг активного компонента, готовят следующим образом
Состав 7: Таблетки
Ингредиент - Количество (мг/таблетку)
Активный ингредиент - 0,1-1000
Крахмал - 45
Целлюлоза микрокристаллическая - 35
Поливинилпирролидон (в виде 10% водного раствора) - 4
Натрий карбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Активный ингредиент, крахмал и целлюлозу пропускают через сито США N 45 меш и тщательно перемешивают. Раствор поливинилпирролидона смешивают с полученными порошками, которые затем пропускают через сито США N 14 меш. Полученные таким образом гранулы сушат при 50-60oC и пропускают через сито США N 18 меш. Затем натрий карбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито США N 60 меш, добавляют к гранулам, которые после смешения прессуют на таблеточной машине с образованием таблеток.
Суспензии, содержащие каждая 0,1-1000 мг лекарства на 5 мл дозы, готовят следующим образом
Состав 8: Суспензии
Ингредиент - Количество (мг/5 мл)
Активный ингредиент - 0,1-1000 мг
Натрий карбоксиметилцеллюлоза - 50 мг
Сироп - 1,25 мг
Бензойной кислоты раствор - 0,10 мл
Ароматизатор - g.v.
Краситель - g.v.
Очищенная вода до - 5 мл
Препарат пропускают через сито США N 45 меш и смешивают с натрий карбоксиметилцеллюлозой и сиропом до образования однородной пасты. Раствор бензойной кислоты, ароматизатор и краситель разбавляют некоторым количеством воды и прибавляют при перемешивании. Затем добавляют достаточное количество воды для обеспечения нужного объема.
Пробы на адгезию клетки in vitro
Клетки эндотелия человеческой пупочной вены (HUVEC) или человеческий аортальный эндотелий (HAE) были получены от Clonetics (Сан-Диего) и выращены в EBM среде, предоставленной Cloneties. Клетки были помещены на пластинку с 96 лунками с такой плотностью, чтобы получить сливающиеся монослои, и после этого выдерживались в термостате при 37oC в течение всей ночи. Добавили исследуемое соединение и выдерживали его в термостате в безсывороточной среде в течение 8-20 ч. Затем монослои выдерживали в термостате с или без 2 нг/мл IL-1 или с 20 нанограммами фактора некроза опухоли (TNF) в течение от 4 до 24 ч до связующей пробы в общем объеме от 75 до 100 микролитров в присутствии исследуемого соединения. После выдерживания в термостате, в объемах 50 микролитров были добавлены меченные тритием клетки U937 в количестве от 1 до 3 х 10 (6) клеток на углубление. Клетки U937 были мечены тритием при добавлении 3H-тимидина до конечной концентрации 1 микрокюри на 1 миллилитр, с последующим выдерживанием в термостате в течение от 18 до 20 ч. До использования клетки были промыты PBS, чтобы удалить избыток метки. После 20-минутного выдерживания в термостате меченых клеток U937 с клетками эндотелия лунки были очищены от жидкости и четыре раза промыты кальцийсодержащим PBS. Монослой и слившиеся клетки U937 были растворены при добавлении 0,25% раствора SDS/0,1 N NaOH при перемешивании в течение 5 мин. Уровень связывания был определен по счету сцинтилляций растворенных клеток.
Проба на антикоагулянтную активность
Слитые культуры обработанных IL-1 (2 нг/мл) или необработанных клеток человеческого эндотелия на пластинках с 96 лунками были один раз промыты HBSS для удаления белков сыворотки и выдержаны в термостате в безсывороточной среде (среда DMEM/F-12, 20 мМ-HEPES, pH 7,5, 50 мг/мл гентамицина, 1 мг/мл трансферрина человека и 1 мг/мл бычьего инсулина), содержащей 400 нМ рекомбинанта человеческого белка C и 10 нМ человеческого тромбина. Клетки выдерживали в термостате при 37oC и в разное время среду удаляли и добавляли к равному объему раствора 20 мМ Tris-HCl, pH 7,5, 150 мМ NaCl, 1 мг/мл BSA и 10 U/мл гирудина. Пробы выдерживали в термостате в гирудинсодержащем буфере в течение 5 мин для ингибирования активности тромбина. Количество генерированного активированного белка C было определено при добавлении хромогенного субстрата (S-2366) до конечной концентрации 0,75 мМ и при измерении изменения абсорбции единиц/минуту при 405 нм в считывающем устройстве кинетического тарелочного микротитратора ThermoMax (Molecular Devices). Во всех экспериментах пробы раствора белок C/тромбин выдерживались в термостате в углублениях в отсутствие клеток для определения базовых уровней тромбин-катализируемой активации белка C. Количество генерированного активированного белка C выражается адсорбцией (мОД) на минуту на микрограмм клеточного белка.
Результаты
Клетки эндотелия человеческой пупочной вены(HUVEC) были обработаны соединением A, где R1 и R3 - водород, а R2 - пирролидин, одновременно с индукцией TNF экспрессии адгезии молекул. Как показано в табл. 1, присутствие 100 нМ соединения A приводит к приблизительно 40% снижению уровня межклеточной адгезии в данной пробе. Когда клетки были предварительно обработаны только 10 нМ соединения A перед индукцией TNF, наблюдалось приблизительно 65% снижение адгезии (табл. 2). Мы также обработали как HUVEC, так и клетки человеческого аортального эндотелия (HAEC) IL1, другим медиатором воспаления, чтобы снизить экспрессию адгезии молекул в присутствии 10 нМ соединения A. Как показано в табл. 3, соединение A эффективно ингибируют IL1-снижение адгезии в обоих клеточных линиях. Таким образом, соединение A способно блокировать снижение экспрессии адгезии молекул, переносимой двумя независимыми способами как в венозных, так и в артериальных клетках.
В качестве дальнейшего свидетельства способности этого соединения модулировать функциональные свойства эндотелия, мы измерили способность клеток эндотелия активировать человеческий белок C, природную регуляторную функцию, которая регулируется в состояниях эндотелиальной дисфункции. Как показано в табл. 4, обработка этих клеток IL1 значительно снижает способность эндотелия обеспечивать генерацию белка C. Однако при последующей обработке этих клеток соединением A подавление этой функции IL1 было существенно уменьшено. Приведенные выше данные указывают на то, что соединение A защищает клетки от действия воспалительной и прокоагулирующей активности.



Изобретение относится к медицине. Предложены способ ингибирования межклеточной адгезии и способ ингибирования воспалительного процесса с использованием ралоксиорена. Изобретение позволяет снизить экспрессию адгезивных молекул в клетках эндотелия. 2 с. и 2 з.п. ф-лы, 4 табл.
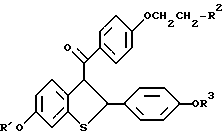
где R1 и R3 независимо представляют водород;
R2 представляет пиперидиногруппу,
или его фармацевтически приемлемых солей и сольватов.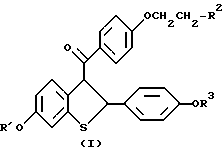
где R1 и R3 независимо водород;
R2 представляет пиперидиногруппу,
или его фармацевтически приемлемых солей и сольватов.
| US А 4133814, 09.01.1999 | |||
| БЕЛИКОВ В.Г | |||
| Фармацевтическая химия | |||
| - М.: Высшая школа, 1993, т.1, с.43-47 | |||
| Краткий курс молекулярной фармакологии/Под ред | |||
| Сергеева П.В | |||
| - М., 1975, с.10. |
