Область изобретения
Данное изобретение относится к способам и фармацевтическим соединениям для лечения сахарного диабета и связанных с ним симптомов.
Предпосылки изобретения
Инсулиннезависимый сахарный диабет (ИНСД, диабет II типа) характеризуется аномалиями секреции инсулина и действия инсулина. В Соединенных Штатах больные ИНСД составляют 90-95% из приблизительно 6 миллионов диагностированных больных сахарным диабетом. ИНСД характеризуется гипергликемией, являющейся результатом невосприимчивости к инсулину в периферических тканях (скелетной мускулатуре и жировой ткани), где снижаются стимулируемые инсулином потребление/утилизация глюкозы, и в печени, где наблюдается недостаточность подавления инсулином выхода глюкозы. Данные нарушения действия инсулина играют важную роль в развитии повышенного содержания глюкозы в крови натощак и непереносимости глюкозы.
Первоочередным лечением для больных ИНСД являются диета и физические упражнения. В целях снижения содержания глюкозы в крови больные ИНСД также принимают пероральные лекарственные средства, снижающие содержание глюкозы в крови. Наиболее широко применяемые средства, снижающие содержание глюкозы в крови, представляют различные составы инсулина и сульфонилмочевин. Основной недостаток данных способов лечения заключается в наличии потенциально угрожающей жизни гипогликемии, возникающей благодаря гиперинсулинемии.
Гиперинсулинемия, которая может возникнуть вследствие применения данных способов лечения, также связана с повышенным риском развития сердечно-сосудистых заболеваний, являющихся основной причиной смерти больных сахарным диабетом. Таким образом, существует потребность в антидиабетических лекарственных средствах, которые не повышают концентрацию инсулина в циркуляции.
Было подтверждено, что новый класс соединений, тиазолидиндионы, обладает антигипергликемической активностью по механизму усиления действия инсулина, а не стимуляции секреции инсулина. Тиазолидиндионы устраняют инсулинорезистентность и нормализуют содержание глюкозы и инсулина в плазме (в случае исходно повышенного содержания) без вызывания гипогликемического состояния, даже в очень больших дозах. Тиазолидиндионовые сенсибилизаторы инсулина, например, циглитазон (ciglitazone), энглитазон (englitazone), пиоглитазон (pioglitazone), BRL 49653 (5-[[4-[2-(метил-2-пиридиниламино)этокси]фенил] метил]-2,4-тиазолидиндион) и троглитазон (troglitazone) усиливают подавление выхода глюкозы из печени, опосредованное инсулином, и потребление и утилизацию глюкозы жировой ткани, стимулируемые инсулином. Тиазолидиндионы также изменяют экспрессию переносчика глюкозы (например, Glut 4), что способствует увеличению восприимчивости к инсулину.
Краткое изложение сущности
Заявитель обнаружил, что агонисты RXR имитируют или усиливают антидиабетические эффекты тиазолидиндионовых соединений. Агонисты RXR стимулируют активность транскрипции гетеродимеров RXR/PPARγ, повышают потребление глюкозы, стимулированное инсулином, снижают содержание триглицеридов, снижают содержание инсулина и повышают ЛВП холестерина. Было показано, что два агониста RXR снижают содержание глюкозы, триглицеридов и инсулина на двух устоявшихся животных моделях ИНСД, т.е. на мышах ob/ob и db/db. Таким образом, агонисты RXR могут быть применены в качестве сенсибилизаторов к инсулину или миметиков инсулина в лечении ИНСД и связанных с ним симптомов.
В дополнение к этому, применение сочетания агониста RXR и агониста PPARγ, такого как тиазолидиндиона, позволяет добиться синергичной активации гетеродимеров RXR/PPARγ, заключающейся в усилении адипогенного и антидиабетического эффектов PPARγ. На мышах db/db было показано, что введение сочетания агониста RXR и агониста PPARγ снижает содержание глюкозы на величину, большую, нежели каждое из данных соединений.
Таким образом, настоящее изобретение относится к способам и композициям для лечения субъекта, страдающего ИНСД, или сахарным диабетом, обусловленным невосприимчивостью к инсулину, путем введения субъекту композиции, содержащей фармацевтически эффективное количество активатора гетеродимера RXR/PPARγ, включая, но не ограничиваясь, агонист RXR. Субъект может представлять собой пациента-человека или животную модель ИНСД у человека. Композиции по данному изобретению приспособлены для лечения, облегчения проявления или предупреждения развития одного или более симптомов в организме субъекта. Предпочтительное лекарственное средство является сильнодействующим и избирательным с низким уровнем токсичности. В этом отношении, специалисты в данной области сочтут ИНСД как пример болезни обмена веществ, которая может подвергаться лечению соединениями и композициями по настоящему изобретению, содержащими агонист RXR. Другие примеры болезней обмена веществ, которые могут подвергаться лечению соединениями и композициями по настоящему изобретению, включают, но не ограничиваются, ожирение и аномалии выработки тиреоидного гормона.
Под термином "фармацевтически эффективное количество" подразумевают количество фармацевтического соединения или композиции, обладающей терапевтическим эффектом на течение ИНСД. Терапевтическим эффектом считается облегчение до некоторой степени проявления одного или более симптомов ИНСД у пациента или либо частичный, либо полный возврат к норме одного или более физиологических или биохимических показателей, связанных или вызываемых ИНСД, например, увеличение клеточной восприимчивости к циркулирующему инсулину, устранение, уменьшение проявления или предотвращение развития одного или более клинических симптомов ИНСД, включая, но не ограничиваясь, гипергликемию, гиперинсулинемию и гипертриглицеридемию. В предпочтительном осуществлении термин фармацевтически эффективное количество соединения или композиции обозначает количество, которое повышает потребление глюкозы жировой тканью или мышечной тканью. В другом предпочтительном осуществлении термин фармацевтически эффективное количество соединения или композиции обозначает количество, которое повышает потребление триглицеридов жировой тканью.
Под "активатором гетеродимера RXR/PPARγ" подразумевается соединение или композиция, которые, будучи объединены с гетеродимером RXR/PPARγ, повышают регуляторную активность транскрипции гетеродимера, измеряемую с помощью анализов, известных специалистам в данной области, включая, но не ограничиваясь, "котрансфекцию" или "цис-транс" анализ, описанные или раскрытые в патентах США 4981784, 5071773, 5298429, 5506102, WО 89/05355, WО 91/06677, WО 92/05447, WО 93/11235, WО 95/18380, PCT/US93/04399, PCT/US94/03795 и СА 2034220, которые включены в качестве ссылки. Данный термин включает, но не ограничивается, соединения, которые связываются с RXR, PPARγ или и с тем, и с другим.
Под "агонистом RXR" подразумевается соединение или композиция, которые, будучи объединены с гомодимерами или с гетеродимерами RXR, повышают регуляторную активность транскрипции RXR, измеряемую с помощью анализов, известных специалистам в данной области, включая, но не ограничиваясь, "котрансфекцию" или "цис-транс" анализ, описанные или раскрытые в патентах США 4981784, 5071773, 5298429, 5506102, WO89/05355, WO91/06677, WO92/05447, WO93/11235, WO95/18380, PCT/US93/04399, PCT/US94/03795 и СА 2034220, которые включены сюда в качестве ссылки. Данный термин включает, но не ограничивает ими соединения, которые преимущественно по сравнению с RAR активируют RXR (т.е. специфические агонисты RXR) и соединения, которые активируют как RXR, так и RAR (т.е. панагонисты). Он также включает соединения, которые активируют RXR при определенных, но не при каких других, условиях в клетке (т.е. частичные агонисты). Соединения, описанные в следующих статьях, патентах и заявках на патент, которые обладают агонистической RXR активностью, включены сюда в качестве ссылок: Патенты США 5399586 и 5466861, WO96/Q,5165, PCT/US95/16842, PCT/US95/16695, PCT/US93/10094, WO94/15901, PCT/US92/11214, WO93/11755, PCT/US93/10166, PCT/US93/10204, WO94/15902, PCT/US93/03944, WO93/21146, предварительные заявки 60004897 и 60009884, Boehm. et al., J. Med. Chem., 38(16): 3146-3155, 1994, Boehm, et al., J. Med. Chem., 37 (18):2930-2941, 1994, Antras et al., J. Biol. Chem., 266:1157-1161 (1991). Salazar-Olivo et al. , Biochem. Biophys. Res. Commun., 204:157-263 (1994) и Safanova, Mol. Cell. Endocrin., 104:201-211 (1994). Специфические агонисты RXR включают, но не ограничиваются, LG 100268 (т.е. 2-[1-(3, 5, 5, 8, 8-пентаметил-5,6,7,8-тетрагидро-2-нафтил)циклопропил] пиридин-5-карбоновую кислоту) и LGD 1069 (т. е. 4-[ (3,5,5,8,8-пентаметил-5,6,7,8-тетрагидро-2-нафтил)-2-карбонил]бензойную кислоту) и их аналоги, производные и фармацевтически приемлемые соли. Строение и методики синтеза LG 100268 и LGD 1069 описаны в Boehm, et al. , J. Med. Chem., 38 (16):3146-3155, 1994, включенной сюда в качестве ссылки. Панагонисты включают, но не ограничиваются, ALRT 1057 (т.е. 9-цис-ретиноевая кислота) и ее аналоги, производные и фармацевтически приемлемые соли.
В предпочтительном осуществлении фармацевтическая композиция также содержит фармацевтически эффективное количество агониста PPARγ. Альтернативно, отдельно субъекту вводится вторая композиция, содержащая фармацевтически эффективное количество агониста PPARγ. В другом предпочтительном осуществлении применяют соединение, обладающее агонистической активностью по отношению как к RXR, так и к PPARγ.
Под "агонистом PPARγ" подразумевается соединение или композиция, которые, будучи объединены с PPARγ, повышают активность характерного для рецептора взаимодействия, что измеряют с помощью анализов, известных специалистам в данной области, включая, но не ограничиваясь, "котрансфекцию" или "цис-транс" анализ, описанные или раскрытые в патентах США 4981784 и 5071773 и Lehmann, et al. , J. Biol. Chem., 270:12953-12956 (1995), которые включены сюда в качестве ссылки. Предпочтительный агонист PPARγ представляет собой тиазолидиндионовое соединение, включая, но не ограничиваясь, BRL 49653, троглитазон, пиоглитазон, циглитазон, WAY-120744, энглитазон, AD 5075, дарглитазон и их аналоги, производные и фармацевтически приемлемые соли. Соединения, описанные Tontonez et al., Genes & Develop., 8:1224-1234 (1994), Tontonez et al., Cell. 79:1147-1156 (1994), Lehmann, et al., J. Biol. Chem., 270(22): 1-4, 1995, Amri et al., J. Lipid Res., 32:1449-1456 (1991), Amri et al., J. Lipid Res.. 32:1457-1463 (1991) и Grimaldi et al., Proc. Natl. Acad. Sci. USA, 89:10930-10934 (1992), включены сюда в качестве ссылки.
В другом предпочтительном осуществлении фармацевтическая композиция также содержит фармацевтически эффективное количество инсулина, производного инсулина, вещества, усиливающего секрецию инсулина, вещества, повышающего восприимчивость к инсулину, или миметика инсулина. Альтернативно, отдельно хозяину вводится композиция, содержащая фармацевтически эффективное количество инсулина, производного инсулина, вещества, усиливающего секрецию инсулина, вещества, повышающего восприимчивость к инсулину, или миметика инсулина.
Композиция, содержащая фармацевтически эффективное количество активного ингредиента, может быть введена хозяину перорально или системно. В предпочтительном осуществлении она вводится перорально.
В другом аспекте, данное изобретение описывает фармацевтическую композицию для лечения ИНСД, содержащую фармацевтически эффективное количество агониста RXR; и фармацевтически приемлемый носитель, приемлемый для введения субъекту, страдающему ИНСД. В предпочтительном осуществлении фармацевтическая композиция также включает фармацевтически эффективное количество инсулина, производного инсулина, вещества, усиливающего секрецию инсулина, вещества, повышающего восприимчивость к инсулину, миметика инсулина или агониста PPARγ.
В предпочтительном осуществлении композицию содержат в контейнере, который включает этикетку, свидетельствующую о том, что композиция одобрена FDA в Соединенных Штатах (или аналогичным регламентирующим органом в другом государстве) для лечения гипсргликемии, гиперинсулинемии или гипертриглицеридемии. Такой контейнер содержит терапевтически эффективное количество активного ингредиента, подлежащего введению субъекту.
В другом аспекте, данное изобретение описывает методики скрининга соединений, применимых для лечения ИНСД. Данные методики позволяют отбирать соединения или композиции, которые, будучи объединены с гетеродимером RXR/PPARγ, повышают регуляторную активность транскрипции гетеродимера, что измеряют с помощью анализов, известных специалистам в данной области, включая, но не ограничиваясь, "котрансфекцию" или "цис-транс" анализ, описанные или раскрытые в патентах США 4981784, 5071773, 5298429, 5506102, WО 89/05355, WО 91/06677, WО 92/05447, WО 93/11235, WО 95/18380, PCT/US93/04399, PCT/US94/03795 и СА 2034220.
В одном примере претендующее соединение, такое как потенциальный агонист RXR вводят в адипоцит или преадипоцит. Измеряют содержание липида в клетке, и повышенное накопление липида после обработки претендующим соединением указывает на то, что претендующее соединение применимо для лечения ИНСД. В предпочтительных осуществлениях содержание липидов измеряют с помощью окрашивания масляным красным О (oil red О staining) или с помощью определения содержания триглицеридов в клетке.
В другом примере претендующее соединение, такое как потенциальный агонист RXR, вводят в адипоцит или в преадипоцит и измеряют интенсивность транскрипции специфического гена адипоцитов (например, гена липопротеинлипазы или ген PPARγ). Повышенная интенсивность транскрипции специфического гена адипоцитов после обработки претендующим соединением указывает на то, что претендующее соединение применимо для лечения ИНСД.
Еще в одном примере претендующее соединение, такое как потенциальный агонист RXR, вводят в адипоцит или в преадипоцит и измеряют уровень потребления глюкозы. Повышенный уровень потребления глюкозы после обработки претендующим соединением указывает на то, что претендующее соединение применимо для лечения ИНСД. Альтернативно, в клетку вводят как претендующее соединение, так и инсулин, и уровень потребления глюкозы сранивают с таковым в такой же клетке, обрабатываемой только инсулином. Более высокий уровень потребления глюкозы в клетке, обрабатываемой претендующим соединением и инсулином, указывает на то, что претендующее соединение представляет собой вещество, повышающее восприимчивость к инсулину, и является применимым для лечения ИНСД.
Другие свойства и преимущества данного изобретения станут очевидными из следующего подробного описания изобретения и из формулы изобретения.
Фиг. 1а представляет собой график, показывающий степень дифференцировки адипоцитов в клетках 3T3-L1, которая измерялась по содержанию триглицерида в преадипоцитах 3T3-L1, обрабатываемых различными комбинациями агониста RXR (LG 100268), агониста PPARγ (тиазолидиндионовое соединение BRL 49653) и инсулина. Ретиноид и BRL 49653 применяли в концентрации 1 мкМ.
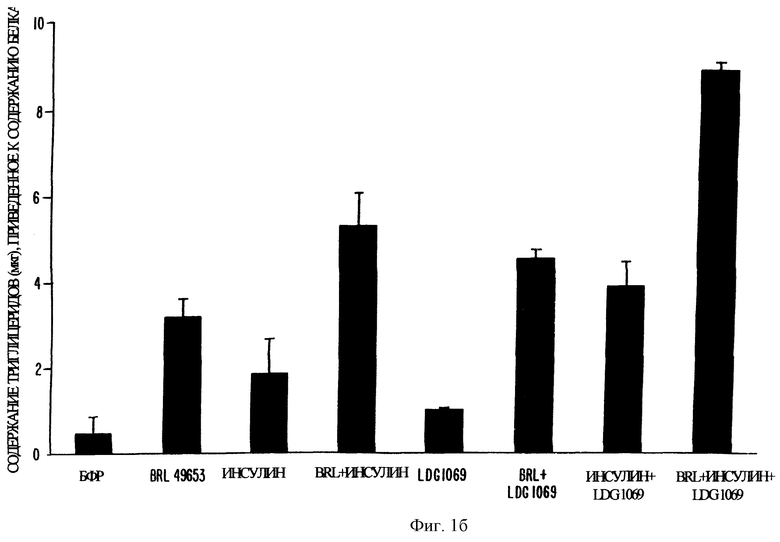
Фиг. 1б представляет собой график, показывающий степень дифференцировки адипоцитов в клетках 3T3-L1, измеряемую по содержанию триглицерида в преадипоцитах 3T3-L1, обрабатываемых различными комбинациями агониста RXR (LGD 1069), агониста PPARγ (тиазолидиндионовое соединение BRL 49653) и инсулина. Ретиноид и BRL 49653 применяли в концентрации 1 мкМ.
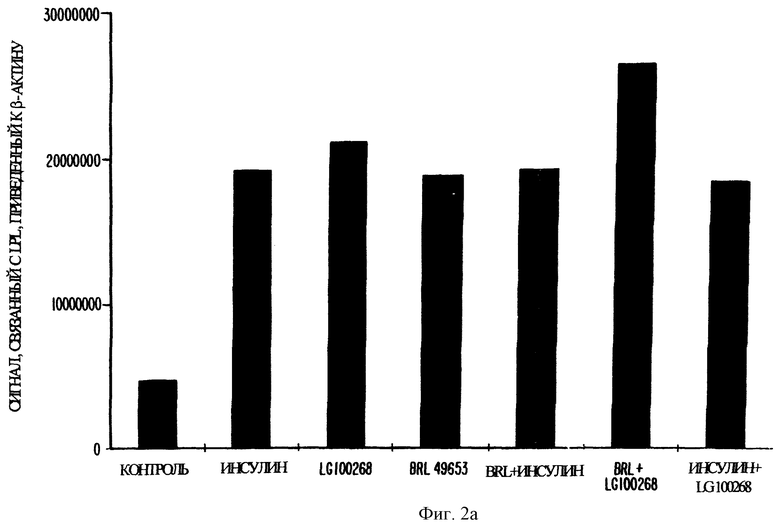
Фиг. 2а представляет собой график, показывающий содержание мРНК LPL в клетках 3T3-L1, обрабатываемых различными комбинациями агониста RXR (LG 100268), агониста PPARγ (тиазолидиндионовое соединение BRL 49653) и инсулина. Ретиноид и BRL 49653 применяли в концентрации 1 мкМ.
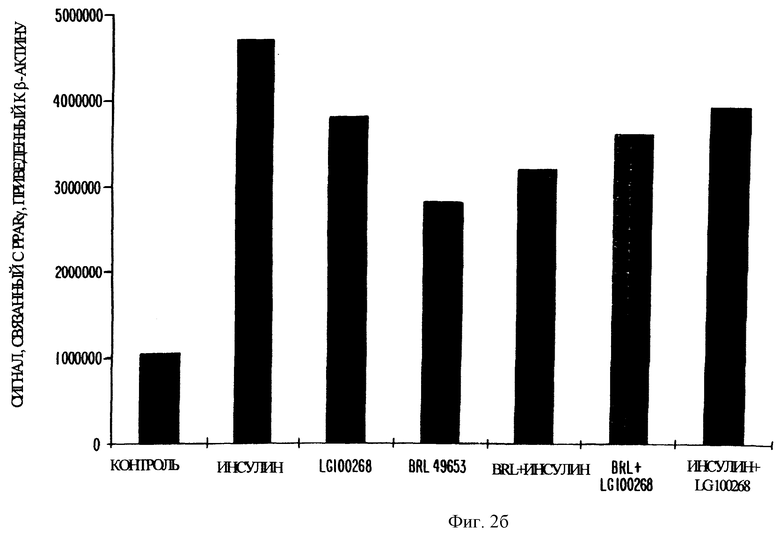
Фиг. 2б представляет собой график, показывающий содержание мРНК PPARγ в клетках 3T3-L1, обрабатываемых различными комбинациями агониста RXR (LG 100268), агониста PPARγ (тиазолидиндионовое соединение BRL 49653) и инсулина. Ретиноид и BRL 49653 применяли в концентрации 1 мкМ.
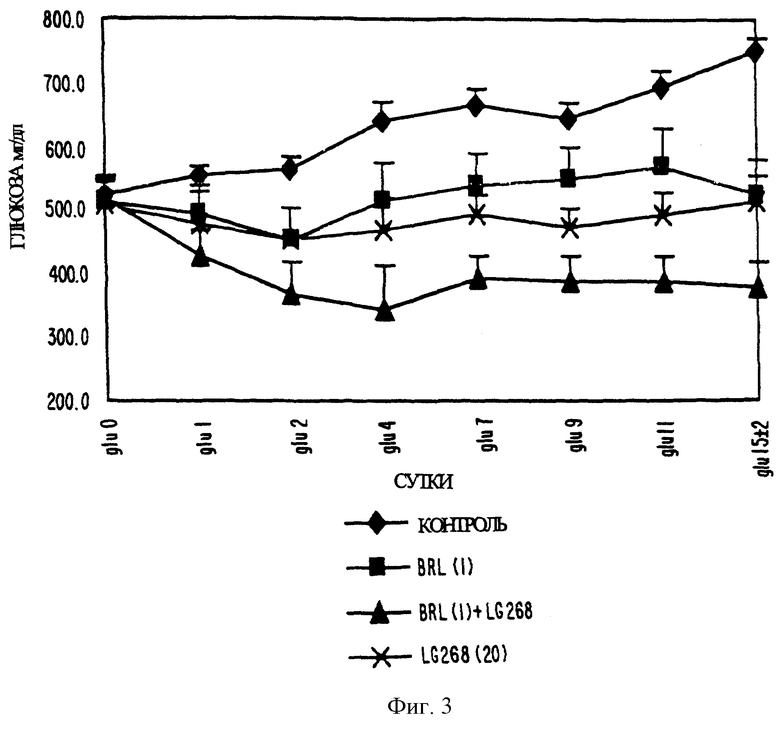
Фиг. 3 представляет собой график, показывающий содержание глюкозы у мышей db/db, обрабатываемых LG 100268, BRL 49653 и сочетанием LG 100268 и BRL 49653, соответственно.
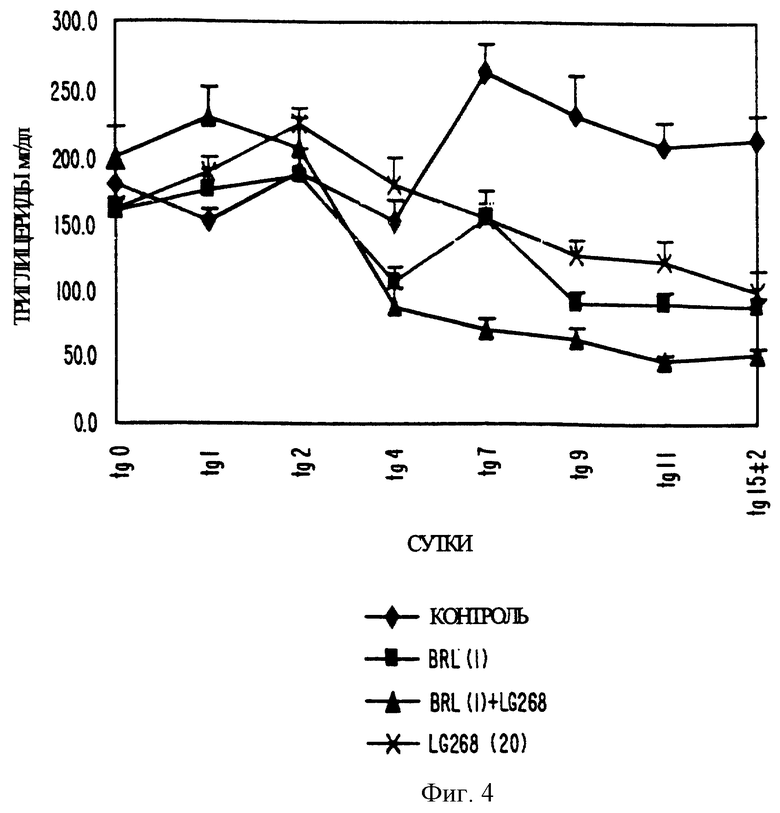
Фиг. 4 представляет собой график, показывающий содержание триглицеридов у мышей db/db, обрабатываемых LG 100268, BRL 49653 и сочетанием LG 100268 и BRL 49653, соответственно.
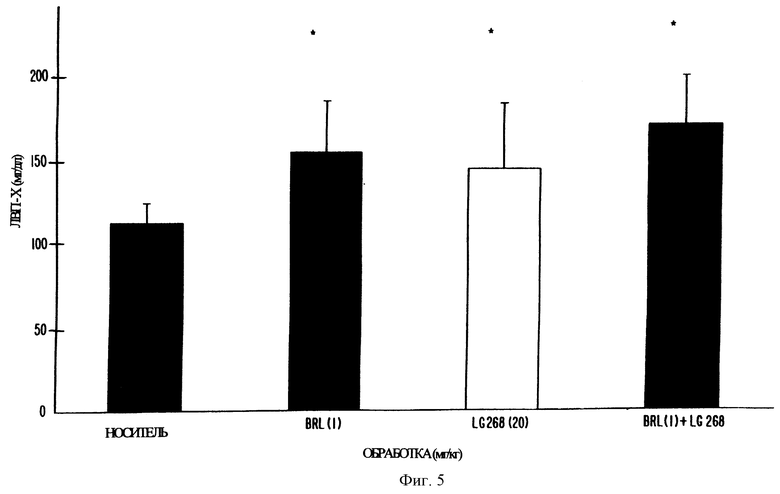
Фиг. 5 представляет собой график, показывающий отношение содержания ЛВП к содержанию холестерина у мышей db/db, обрабатываемых LG 100268, BRL 49653 и комбинацией LG 100268 и BRL 49653, соответственно.
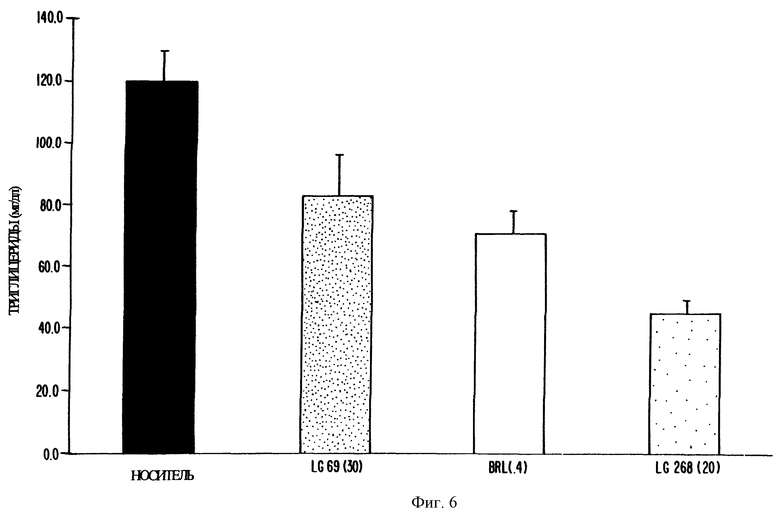
Фиг. 6 представляет собой график, показывающий содержание триглицеридов у мышей ob/ob, обрабатываемых LGD 1069, LG 100268 и BRL 49653, соответственно.
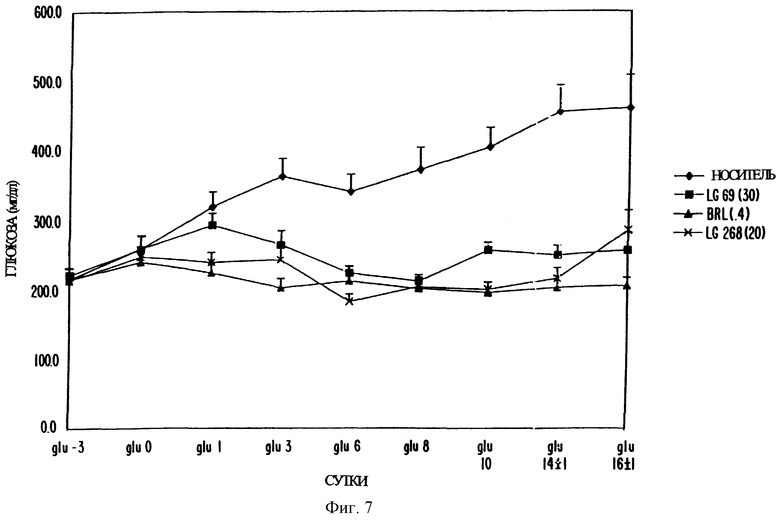
Фиг. 7 представляет собой график, показывающий содержание глюкозы у мышей ob/ob, обрабатываемых LGD 1069, LG 100268 и BRL 49653, соответственно.
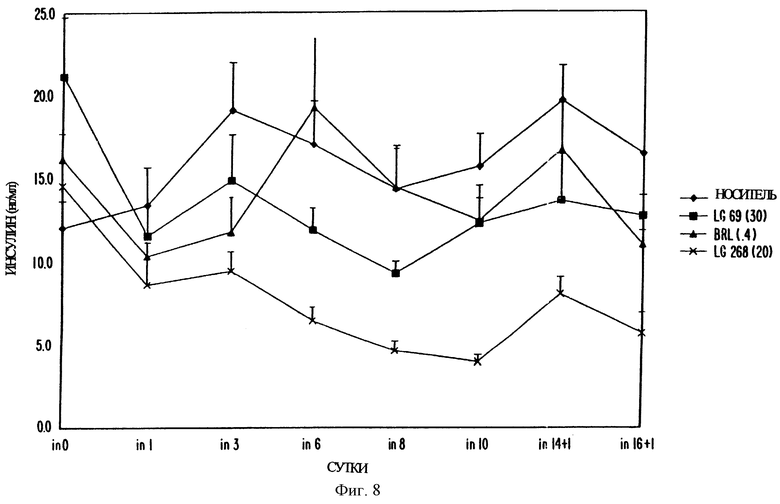
Фиг. 8 представляет собой график, показывающий содержание инсулина у мышей ob/ob, обрабатываемых LGD 1069, LG 100268 и BRL 49653, соответственно.
Подробное описание изобретения.
ТЗД оказывают антидиабетические и липогенные эффекты посредством PPARγ.
Тиазолидиндионы представляют собой вещества, усиливающие восприимчивость к инсулину, которые значительно снижают содержание глюкозы и липидов на животных моделях ИНСД и ожирения (Kees, et al., J. Medicinal Chem.. 38 (4): 617-628, 1995; Willson, et al., J. Medicinal Chem., 39 (3):665-668, 1996; Young, et al., Diabetes 44:1087-1092, 1995). Тиазолидиндионы повышают уровень утилизации глюкозы без стимулирования секреции инсулина.
Например, неоднократное введение BRL 49653 мышам, страдающим ожирением, приводит к нормализации содержания глюкозы в крови путем повышения восприимчивости тканей-мишеней к инсулину. BRL 49653 усиливает стимулированный инсулином транспорт глюкозы в адипоциты мышей с ожирением и невосприимчивостью к инсулину как путем повышения числа инсулиновых рецепторов, так и путем способствования перемещению GLUT4 из увеличенного внутриклеточного пула на клеточную поверхность.
Тиазолидиндионы также являются избирательными агонистами PPARγ (Lehmann, et al. , J. Biol. Chem., 270(22):1-4, 1995). Сравнение ЕС50 для активации PPARγ с минимальной эффективной дозой (МЭД) для антигипергликемической активности выявило значительную корреляцию. Корреляция между активностью PPARγ in vitro и антигипергликемической активностью тиазолидиндионов in vivo представляет PPARγ как молекулярную мишень для антидиабетических эффектов тиазолидиндионов.
PPARγ является членом надсемейства ядерных рецепторов, активируемых лигандами факторов транскрипции. Он специфично экспрессируется в жировой ткани, и его экспрессия индуцируется на ранних стадиях дифференцировки некоторых клеточных линий преадипоцитов. Усиленная экспрессия PPARγ в фибробластах приводила к дифференцировке адипоцитов.
Помимо активности, повышающей восприимчивость к инсулину, тиазолидиндионы обладают выраженным адипогенным эффектом на преадипоциты и мезенхимальные стволовые клетки (Tontonoz et al., Cell. 79:1147-1156 (1994). Обработка клеток С3Н/10Т1/2 BRL 49653 приводила к интенсивной дифференцировке адипоцитов, что показывает, что опосредованная лигандом активация PPARγ является достаточной для запуска каскада адипогенных сигналов в мезенхимальной стволовой клеточной линии. PPARγ представляет собой молекулярную мишень для адипогенного эффекта тиазолидиндионов.
Липогенез играет роль в развитии ИНСД, который характеризуется не только разбалансированным гомеостазом глюкозы, но также и повышенным содержанием циркулирующих липидов. Было показано, что повышение содержания липидов влияет на распределение глюкозы.
Адипоциты представляют собой высоко специализированные клетки, которые играют принципиальную роль в метаболизме липидов и энергетическом гомеостазе. Их главная задача состоит в накоплении триглицеридов в периоды избытка калорий и в мобилизации данных резервов в периоды лишения питания.
Дифференцировка адипоцитов характеризуется координированным увеличением уровня экспрессии адипоцит-специфичных генов. PPARγ специфично экспрессируется в адипоцитах. Его экспрессия индуцируется на ранних стадиях дифференцировки некоторых клеточных линий преадипоцитов. Усиленная экспрессия PPARγ в фибробластах приводила к дифференцировке адипоцитов.
Синергические эффекты RXR и PPARγ на липогенез.
Экспрессия PPARγ индуцируется на ранних стадиях дифференцировки культивируемых клеточных линий адипоцитов, и он специфично экспрессируется в жировой ткани с очень высоким содержанием. PPARγ регулирует липогенез путем модулирования транскрипции других адипоцит-специфичных генов, например, ген Р2 адипоцита (ген аР2). Ген аР2 кодирует внутриклеточный липид-связывающий белок и экспрессируется исключительно в клетках жировой ткани.
Фрагмент ДНК 518-bр с 5'-конца гена аР2 идентифицировали как энхансер, который направляет высокопродуктивную экспрессию адипоцит-специфичных генов как в культивируемых клетках, так и у трансгенных мышей. Пара элементов энхансера аР2, ARE6 и ARE7, связывают ядерный фактор, называемый ARF6, который определяется только в экстрактах ядер, полученных из адипоцитов. ARF6-связывающие участки являются как необходимыми, так и достаточными для экспрессии адипоцит-специфичных генов, что позволяет предположить, что взаимодействующий фактор ARF6 функционирует как зависимый от стадии дифференцировки и тканеспецифичный переключатель энхансера аР2 (Tontonoz, et al., Genes & Development, 8:1224-1234, 1994).
Узнаваемая последовательность ARF6 напоминает тип связывающего участка для ядерного рецептора гормонов, известный как DR-1 (прямой повтор с 1-нуклеотидным спейсером). Было показано, что данный мотив преимущественно связывать гетеродимеры RXR и COUP-TF и гетеродимеры RXR и PPAR. В экспериментах по изучению уменьшения подвижности ДНК с применением различных последовательностей HRE в качестве контроля было продемонстрировано, что ARF6 преимущественно распознает участки DR-1.
ARF6 идентифицировали как гетеродимерный комплекс RXRα и PPARγ. Было показано, что in vitro PPARγ и RXRα образуют гетеродимеры в АRF6-связывающих участках. Усиленная экспрессия данных факторов при нестойкой трансфекции является достаточной для активации адипоцит-специфичного энхансера аР2 в нежировых клетках, как, например, в фибробластах. Данная активация потенциируется пероксисомными пролифераторами, жирными кислотами и 9-цис-ретиноевой кислотой. Антисыворотка к RXRα специфично ингибирует активность ARF6 в экстрактах ядер из адипоцитов.
Котрансфекция вектором, экспрессирующим RXRα, и вектором, экспрессирующим PPARγ, оказывает синергический эффект на активацию энхансера аР2 в нежировых клетках. Максимальный уровень активации энхансера аР2 наблюдается, когда присутствуют и PPARγ и RXRα, и их агонисты.
Не ограничиваясь какой-либо теорией, заявитель предполагает, что агонист RXR влияет на утилизацию глюкозы в тканях посредством синергических эффектов гетеродимеров RXR и PPARγ. Гетеродимеры RXR/PPARγ, будучи активированными агонистом RXR или сочетанием агониста RXR и агониста PPARγ, индуцируют липогенез и модулируют уровни потребления глюкозы и триглицеридов. Альтернативно или в дополнение к этому, гетеродимеры RXR/PPARγ, будучи активированными агонистом RXR или комбинацией агониста RXR и агониста PPARγ, регулируют продукцию сигнальных молекул, секретируемых жировой тканью, таких как фактор некроза опухоли-α или лептин, которые, в свою очередь, модулируют метаболизм глюкозы в других тканях.
Применение агонистов RXR для имитации или усиления антидиабетического эффекта тиазолидиндиона.
Каждый из PPARα, β и γ образует гетеродимеры с формами RXR. Данные гетеродимеры RXR/PPARγ связываются с ДНК и регулируют активность транскрипции. Активаторы RXR содействуют активаторам PPARγ в стимулировании активности белка PPARγ (Kliewer, et а1., Nature, 358:771-774 (1992) и Mukherjee, et al., Steroid Biochem. Molec. Biol., 51:157-166 (1994)).
В данном изобретении сходная синергическая активация наблюдалась у активатора PPARγ и некоторых активаторов RXR.
В соответствии с данным изобретением агонисты RXR, например, LGD 1069, ALRT 1057 и LG 100268, могут быть применены для лечения сахарного диабета. Авторы заявки исследовали четыре независимых показателя эффектов агонистов RXR, т.е. морфологические изменения, накопление липидов, регуляцию экспрессии генов и повышенное потребление глюкозы.
Для проверки предположения об активации RXR в гетеродимере PPARγ/RXR применяли две клеточные линии преадипоцитов. Клетки 3T3-L1 и С3Н/10Т1/2 получали из АТСС; они выделены из мышиного эмбриона. Они контактно заингибированы и могут быть простимулированы на дифференцировку в адипоциты, содержащие в цитоплазме большие липидные капли. Дифференцировку адипоцитов можно пронаблюдать с помощью окрашивания oil red О, при котором липидные капли в цитоплазме окрашиваются в красный цвет. Степень дифференцировки адипоцитов может отслеживаться с помощью микроскопии.
В целях более количественного анализа и быстрого скрининга соединений для количественной оценки количества продуцируемых дифференцирующимися адипоцитами триглицеридов был разработан анализ на 96-луночном планшете. В данном анализе клетки выращивают в виде монослоя, полностью покрывающего лунки 96-луночного планшета, и обрабатывают BRL 49653, инсулином и ретиноидами по отдельности или в различных комбинациях. Данные способы обработки стимулируют дифференцировку как клеток 3T3-L1, так и клеток С3Н/10Т1/2 в различной степени. Уровень накопления триглицеридов может быть затем измерен с помощью ферментативной цветной реакции, изменения цвета в ходе которой могут быть зарегистрированы с помощью многоканального спектрофотометра для прочтения планшетов (планшет-ридера).
Третьим измерением для оценки дифференцировки адипоцитов является исследование регуляции экспрессии генов. Было показано, что уровни экспрессии мРНК, кодирующих как PPARγ, так и липопротеинлипазы (LPL), изменялись в ходе дифференцировки адипоцитов. Для анализа молекулярных аспектов влияния ретиноидов на гены-мишени претерпевающих дифференцировку адипоцитов применяли нозерн-блотирование. Содержание мРНК, кодирующих PPARγ, липопротеинлипазу (LPL) и β-актин (loading контроль), мониторировали после того, как клетки обрабатывали тиазолидиндионами и ретиноидами.
Четвертый показатель применимости соединения в лечении ИНСД или инсулинзависимого сахарного диабета представляет собой способность соединения усиливать стимулируемое инсулином потребление глюкозы. На клеточной линии преадипоцитов в присутствии инсулина и претендующего соединения проводили анализ с применением меченой 2-дезоксиглюкозы (2-ДОГ, аналога глюкозы) в целях измерения уровня включения 2-ДОГ.
А) Модулирование ретиноидами накопления липидов в клетках 3T3-L1, окрашенных oil red 0.
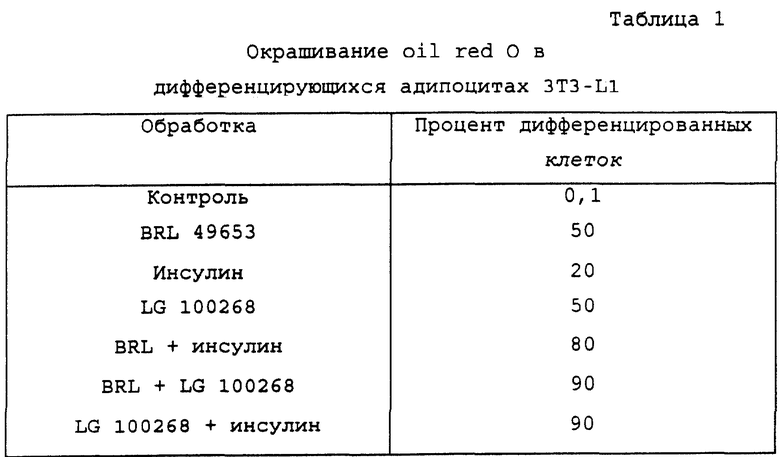
В таблице 1 показано процентное соотношение клеток 3T3-L1, которые претерпели дифференцировку в адипоциты, что выясняли с помощью анализа с применением окрашивания oil red О. BRL 49653 и LG 100268 применяли в концентрациях 1 мкМ, инсулин применяли в концентрации 0,01 мг/мл. Клетки, обработанные LG 100268, содержали в цитоплазме более крупные и красные липидные капли.
Обработка BRL 49653 и LG 100268 по отдельности стимулировала дифференцировку в адипоциты в 50% клеток. Данный процент резко возрастал при добавлении инсулина. Инсулин в сочетании с BRL стимулировал дифференцировку в адипоциты в 80% клеток 3T3-L1, тогда как комбинация инсулина и LG 100268 стимулировала дифференцировку в 90% клеток. Если BRL 49653 применяли в комбинации с LG 100268, агонистом RXR, количество дифференцированных адипоцитов также резко увеличивалось. Другие агонисты RXR имитируют активность LG 100268. Например, добавление ALRT 1057 (панагониста) или LGD 1069 (специфичного агониста RXR) в комбинации с BRL 49653 увеличивало интенсивность дифференцировки, хотя и в меньшей степени, нежели сильный агонист RXR LG 100268. Комбинация BRL 49653 и LGD 1069 обладала сильным стимулирующим дифференцировку действием (95%) на клетки 3T3-L1.
Б) Модулирование ретиноидами содержания триглицеридов в дифференцированных адипоцитах.
Модулирование ретиноидами образования липидов количественно учитывали путем мониторирования образования триглицеридов. На фиг. 1а и 1б показано накопление триглицеридов в клетках 3T3-L1, обработанных ретиноидом (LG 100268 или LGD 1069) отдельно или в комбинации с тиазолидиндионом и инсулином. Во всех экспериментальных комбинациях ретиноиды и BRL 49653 применяли в концентрациях 1 мкМ, инсулин применяли в концентрации 0,01 мг/мл.
Каждый из инсулина, BRL 49653 и ретиноидов стимулировал накопление некоторого количества триглицеридов, будучи применены по отдельности, причем наибольший эффект давал LG 100268. Добавление в анализируемые образцы ретиноидов (LGD 1069, LG 100268) с тиазолидиндионом (BRL 49653) повышало уровень накопления триглицеридов в дифференцирующихся адипоцитах 3T3-L1. Это также наблюдалось, когда BRL 49653 или LG 100268 применяли в комбинации с инсулином. Наиболее интенсивное накопление триглицеридов наблюдалось в клетках, обработанных совместно LG 100268, BRL 49653 и инсулином. Сходные результаты наблюдались, когда в анализе LG 100268 заменяли на LGD 1069. Данные результаты совпадают с таковыми, полученными в анализах с применением окрашивания oil red О.
В) Модулирование содержания мРНК, кодирующих PPARγ и LPL в процессе дифференцировки клеток 3Т3-L1.
(Продукты? ) адипоцит-специфичных генов мониторировали с помощью нозерн-блотирования. На фиг. 2а и 2б показан профиль экспрессии мРНК LPL (липопротеинлипазы) и мРНК PPARγ в клетках, которые в течение 7 суток обрабатывали BRL 49653 (1 мкМ), LG 100268 (1 мкМ) и инсулином (0,01 мг/мл) по отдельности или в комбинации.
Нозерн-блотирование показывает увеличение относительного сигнала по отношению к контролю в виде β-актина экспрессии мРНК как LPL, так и PPARγ, в клетках, обработанных каждым соединением по отдельности. Отмечалось от трех- до пятикратного увеличения содержания мРНК данных генов-мишеней адипоцитов, что указывает на то, что при обработке инсулином, BRL 49653 и LG 100268 происходит регуляция транскрипции. Комбинация инсулина, BRL 49653 и LG 100268 не увеличивала далее содержание мРНК.
Эти данные демонстрируют, что в клетках 3T3-L1 агонисты RXR стимулируют дифференцировку адипоцитов сами по себе или в комбинации с тиазолидиндионами или инсулином. Агонисты RXR усиливают активность тиазолидиндионов или инсулина. Три независимых измерения подтверждают представление о том, что агонисты RXR способствуют модулированию гетеродимера RXR/PPARγ в регуляции дифференцировки адипоцитов и являются применимыми для лечения ИНСД.
Г) LG 100268 усиливает стимулируемое инсулином потребление глюкозы клетками 3T3-L1.
Для исследования потребления глюкозы, липогенеза широко применяют мышиную клеточную линию преадипоцитов 3T3-L1, и ее применили для охарактеризования тиазолидиндионов и других активаторов PPARγ. Стимулируемое инсулином потребление меченой 2-дезоксиглюкозы (2-ДОГ, аналога глюкозы) наблюдали на клетках 3T3-L1, обрабатываемых BRL 49653 или LG 100268.
Клетки 3T3-L1 обрабатывали BRL 49653 (10 мкМ) или LG 100268 (1 мкМ) в течение 10 суток. К клеткам добавляли инсулин в концентрации 0,01 мг/мл в течение 5 суток, по прошествии которых инсулин больше не добавляли. Проводили анализ потребления меченой 2-дезоксиглюкозы (Szalkowski, et al., J. Endocrin., 136:1474-1481, 1995). Содержание включенной меченой 2-ДОГ определяли по отношению к контролю в виде общего количества клеточного белка. На клетках 3T3-L1, обработанных только BRL 49653, наблюдали приблизительно 5-кратное увеличение потребления 2-ДОГ. На клетках 3T3-L1, обработанных только LG 100268, наблюдали приблизительно 2-кратное увеличение потребления 2-ДОГ.
В данном эксперименте показано, что агонист RXR увеличивает опосредованное инсулином потребление глюкозы клетками 3T3-L1, как и известное вещество, усиливающее восприимчивость к инсулину, тиазолидиндион. Это прямо показывает, что агонисты RXR могут быть применены для лечения основного симптома ИНСД, т. е. невосприимчивости к инсулину. Другие агонисты RXR, применимые для лечения ИНСД, могут быть установлены с применением данного анализа.
Д) Модулирование ретиноидами накопления липидов в клетках С3Н/10Т1/2, окрашенных oil red О.
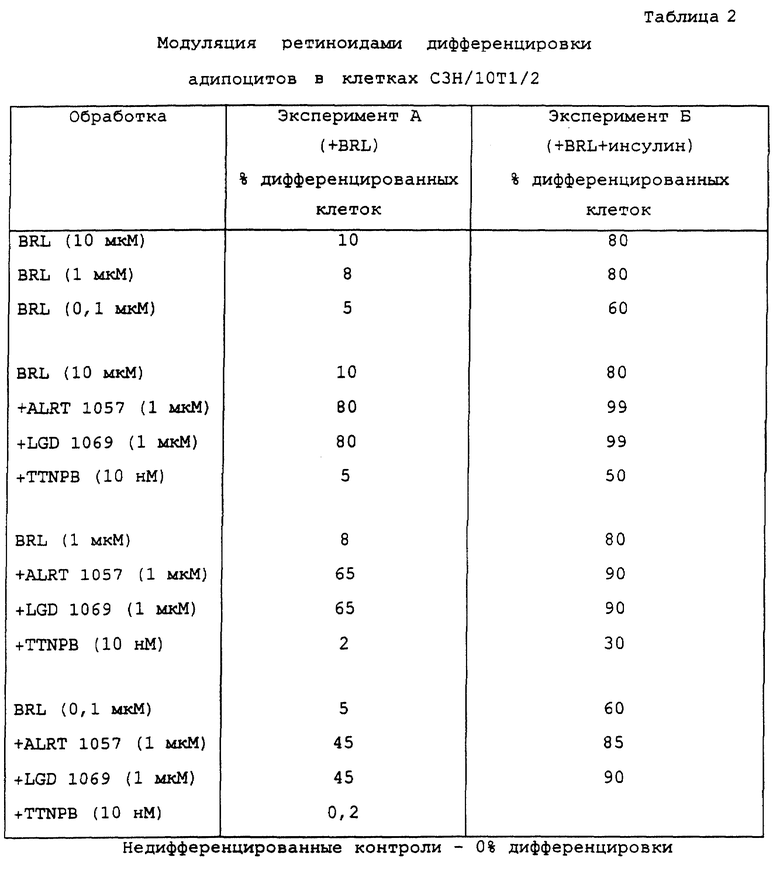
Для дальнейшей оценки регуляции ретиноидами функции адипоцитов мы исследовали клетки С3Н/10Т1/2, которые представляли собой фибробласты/мультипотентные стволовые клетки мышиного эмбриона, в которых может быть стимулирована дифференцировка в адипоциты или мышечные клетки. В таблице 2 показано процентное соотношение клеток С3Н/10Т1/2, которые дифференцировались в адипоциты, о чем судили с помощью анализа с применением окрашивания oil red О. Эксперимент А проводили в присутствии BRL 49653. Эксперимент Б проводили в присутствии как BRL 49653, так и инсулина. ALRT 1057 и LGD 1069 применяли в концентрации 1 мкМ, a TTNPB (избирательное RAR-соединение) применяли в концентрации 10 нМ. Инсулин применяли в концентрации 0,01 мг/мл; а BRL 49653 применяли в концентрации 0,1, 1 и 10 мкМ. Клетки С3Н/10Т1/2 обрабатывали в течение 7 сут, клетки окрашивали oil red О и изучали с помощью микроскопии.
Морфологические изменения наблюдались, когда клетки обрабатывали только ретиноидами, даже несмотря на то, что клетки не полностью дифференцировались в адипоциты. В отдельности BRL 49653 вызывал дифференцировку в адипоциты не более чем в 10% клеток. Однако, когда BRL 49653 применяли в комбинации с агонистом RXR, LGD 1069 или ALRT 1057, отмечалось большое увеличение интенсивности дифференцировки, тот же эффект наблюдали, когда BRL 49653 применяли в комбинации с инсулином.
Агонист RAR, TTNPB (который не активирует RXR), не оказывал того же эффекта, какой оказывал агонист RXR LGD 1069 и ALRT 1057. Действительно, TTNPB ингибировал дифференцировку, вызванную BRL 49653 или BRL/инсулином.
Указанные выше эксперименты показывают, что в отдельности тиазолидиндион (BRL 49653) вызывает минимальную интенсивность дифференцировки клеток С3Н/10Т1/2. Однако, когда BRL 49653 применяют в комбинации с ретиноидом, таким как LG 100268, LGD 1069 или ALRT 1057, которые являются агонистами RXR, интенсивность дифференцировки резко возрастает. Это не наблюдается при применении чистого агониста RAR, TTNPB. Эти данные подтверждают представление о том, что гетеродимеры PPARγ/RXR, которые направляют процесс дифференцировки адипоцитов, могут быть активированы и усилены путем присоединения агониста RXR. Агонисты RXR применимы для модулирования уровней потребления глюкозы и триглицеридов.
Применение агонистов RXR для снижения уровней содержания глюкозы и триглицеридов в животных моделях ИНСД.
(А) Эксперимент in vivo на мышах db/db.
Животные: Линия Diabetic C57BLKS/J - m +/+db, 82 мыши.
Источник: Jackson Lab.
Номер партии 000642.
Генотип m +/+db х m +/+db.
DOB 6/19/96 ± 3 сут, DOA 7/23/96 - возраст 34 сут.
Дата исследования: 8/5/96 - 8/21/96, возраст 44-63 сут.
Мышей различали по номеру на ушной метке ( 1 - 82) и разделяли на 8 групп (А-З). Каждая группа состояла из 10 мышей, размещенных в 4 клетках по 2-3 мыши/клетку. Несколько мышей в течение исследования погибли от попадания в легкие жидкости, после чего сформировались 2 группы по 9 мышей/клетку. Контрольная группа В состояла из 12 мышей. Мышей кормили гранулированным кормом Purina Lab 5015 с теплотворной способностью 3,83 ккал/г, содержащим 56% углеводов, 26% жира и 18% белка. Пищу и воду давали ad libitum. Потребление пищи/клетку измеряли за выбранные промежутки времени и выражали как г потребленной пищи/100 г массы мышей/сутки.
В дни исследования пищу удаляли из клеток в выбранный промежуток времени между 6: 15 и 7:00 утра. Массу тела животных фиксировали через 2 часа после лишения пищи и образцы крови отбирали через 3 часа после лишения пищи. Кровь отбирали из надреза на конце хвоста и собирали в гепаринизированный капилляр (приблиз. 75 мкл объема). После центрифугирования с помощью микрокапиллярного ридера определяли значение гематокрита, регистрировали и капилляр надламывали для взятия плазмы на анализ концентрации глюкозы, триглицеридов и инсулина.
Образцы крови отбирали на 0-1, 0, 3, 7, 10 сутки, и на последние сутки исследования, 13-15 сутки. После отбора образцов крови на 0 сутки животным восстанавливали питание кормом Purina и впоследствии вводили через зонд контрольный раствор в группе В и один из семи опытных растворов в группах, обозначаемых А, Б, Г-З. Вводимый раствор был эквивалентным в каждой группе, составляя в среднем 0,6 мл/42 г мышь (0,01429 мл/г). Различные растворы ежедневно вводили в соответстующих им группах, основанных на массе тела животных, измеренной данным утром, или на массе животного, измеренной в предыдущий день взвешивания. Для оценки изменений в содержании в плазме СЖК на 10 сутки отбирали дополнительный образец крови объемом 75 мкл в покрытый ЭДТА капилляр непосредственно после отбора основного образца в гепаринизированный капилляр.
В заключительный день исследования мышам не вводили через зонд опытные растворы. Последнее введение через зонд проводили на день, предшествующий заключительному дню, т.е. на 12 сутки животным, забитым на 13 сутки, на 13 сутки животным, забитым на 14 сутки, и на 14 сутки животным, забитым на 15 сутки. При забое кровь отворяли методом декапитации для получения сыворотки для оценки содержания ЛВП.
Результаты:
На фиг. 3 показано, что каждый из BRL 49653 и LG 100268 независимо снижал содержание глюкозы у мышей db/db. В дополнение к этому, комбинация BRL 49653 и LG 100268 снижала содержание глюкозы на большее значение, нежели каждое соединение в отдельности. BRL 49653 (1 мг/кг) и LG 100268 (20 мг/кг) к 15 суткам снижали содержание глюкозы на 40% по сравнению с контролем. Тиазолидиндион BRL 49653 в концентрации 1 мг/кг проявлял сходную эффективность. Комбинация активатора RXR и активатора PPAR проявляла большую эффективность, приводя почти к 50% снижению содержания глюкозы. Эффект комбинации развивался быстро, содержание глюкозы снижалось к 1 суткам исследования и принимало постоянное значение к 4 суткам. Это указывает на быстрое повторное установление постоянного уровня метаболизма глюкозы. Таким образом, активаторы RXR увеличивают эффективность активаторов ΠΠΑΡγ и наоборот.
На фиг. 4 показано, что активаторы RXR снижали содержание триглицеридов у мышей db/db. LG 100268 (20 мг/кг) к 15 суткам исследования снижал содержание триглицеридов на 40%. BRL 49653 (1 мг/кг) проявлял сходную эффективность. Комбинация данных двух соединений проявляла еще большую эффективность.
На фиг. 5 показано, что модуляторы активаторов RXR увеличивали отношение содержания ЛВП к содержанию холестерина у мышей db/db. LG 100268 (20 мг/кг) повышал отношение содержания ЛВП к содержанию холестерина (20%) по сравнению с контролем. BRL 49653 (1 мг/кг) вызывал эквивалентный уровень увеличения. Комбинация данных двух соединений проявляла большее увеличение. Содержание ЛВП-Х измеряли по методу преципитации, применяя наборы, полученные от Bohringer-Mannheim (каталог 543004 и 427578).
(Б) Эксперимент in vivo на мышах ob/ob.
Животные: Линия Obese C57 BL/6J-Lepоb (4), 121 мышь.
Источник: Jackson Lab.
Номер партии 000632.
Генотип Lepob(4)/+ • Lepob(4)/+.
DOB 5/22/96 ± 3 сут, DOA 7/2/96 - возраст 41 сут.
Дата исследования: 7/21/96 - 8/2/96, возраст 49-70 сут.
Мышей различали по номеру на ушной метке ( 1 - 121) и разделяли на 12 групп. Каждая группа состояла из 10 мышей. (Одну мышь забили до начала исследования по причине нездоровых зубов, что вызывало исходную потерю в весе). Как и в предыдущих исследованиях, мышей из каждой группы размещали в 4 клетках (2-3 мыши/клетку) и обеспечивали водой и кормом Purina Lab 505/15 ad libitum.
В дни исследования пищу удаляли из клеток в выбранный промежуток времени между 6: 15 и 7:00 утра. Массу тела животных фиксировали через 2 часа после лишения пищи и образцы крови отбирали через 3 часа после лишения пищи. Кровь отбирали из надреза на конце хвоста и собирали в гепаринизированный капилляр (приблиз. 75 мкл объема). После центрифугирования с помощью микрокапиллярного ридера определяли значение гематокрита, регистрировали и капилляр надламывали для взятия плазмы на анализ концентрации глюкозы, триглицеридов и инсулина.
Образцы крови отбирали на 0-3, 0, 3, 6, 8, 10, 14±1 сутки, и последний отбор проводили на 15, 16 или 17 сутки. Образцы для определения содержания СЖК отбирали на 10 сутки, отбирая второй образец крови объемом 75 мкл в негепаринизированный покрытый ЭДТА капилляр. Отбор в данный капилляр проводили непосредственно после отбора в гепаринизированный капилляр.
Животным из группы З ежедневно через зонд вводили контрольный раствор (0,6 мл/42 г), начиная с 0 суток и заканчивая на день, предшествующий заключительному дню. Животным из групп А-Ж, И-М вводили 11 опытных растворов. Все растворы вводили через зонд после восстановления кормления в дни, когда мышей не кормили по причине отбора образцов крови.
Результаты:
На фиг. 6 показано, что модуляторы RXR снижали содержание триглицеридов у мышей ob/ob. Активаторы RXR LGD 1069 (30 мг/кг) и LG 100268 (20 мг/кг) снижали содержание триглицеридов на 34 и 60% соответственно у мышей ob/ob к 14 суткам исследования. BRL 49653 также был способен снижать содержание триглицеридов, хотя и не так эффективно, как LG 100268.
На фиг. 7 показано, что модуляторы RXR снижали содержание глюкозы у мышей ob/ob. Активаторы RXR LGD 1069 в концентрации 30 мг/кг и LG 100268 в концентрации 20 мг/кг снижали содержание глюкозы почти на 50% по сравнению с контролем. Содержание глюкозы сокращалось до почти нормального уровня к 14 суткам исследования. BRL 49653 (0,4 мг/кг) проявлял сходную эффективность.
На фиг. 8 показано, что модуляторы RXR, LGD 1069 (30 мг/кг) и LG 100268 (20 мг/кг) снижали содержание инсулина у мышей ob/ob. LG 100268 снижал содержание инсулина к 14 суткам исследования на 66%. LGD 1069 проявлял меньшую эффективность. Эффект соединений развивался чрезвычайно быстро, так как содержание инсулина начинало уменьшаться на 1 сутки.
Фармацевтические составы и способы введения.
Конкретное соединение, которое оказывает влияние на течение интересующих заболеваний и состояний, может быть введено пациенту либо само по себе, либо в фармацевтических составах, где оно смешивается с приемлемыми носителями или наполнителем(ями). При лечении пациента с проявлениями интересующего заболевания вводят терапевтически эффективное количество средства или средств, таких как данные. Термин "терапевтически эффективная доза" относится к тому количеству соединения, введение которого приводит к уменьшению проявления симптомов или к продлению жизни пациента.
Соединения также могут быть приготовлены в виде фармацевтически приемлемых солей. Примеры фармацевтически приемлемых солей включают соли добавления кислот, такие как содержащие гидрохлорид, сульфат, фосфат, сульфамат, ацетат, цитрат, лактат, тартрат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, циклогексилсульфамат и хинат. (Смотри, например, PCT/US92/03736). Такие соли могут быть получены с применением таких кислот, как соляная кислота, серная кислота, фосфорная кислота, сульфамовая кислота, уксусная кислота, лимонная кислота, молочная кислота, винная кислота, малоновая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, циклогексилсульфамовая кислота и хинная кислота. Данные соли могут быть получены в соответствии со стандартными методиками. Например, соединение в форме свободного основания сначала растворяют в подходящем растворителе, таком как водный или водно-спиртовой раствор, содержащий подходящую кислоту. Соль затем выделяют путем выпаривания раствора. В другом примере соль получают путем проведения реакции свободного основания и кислоты в органическом растворителе.
В целях облегчения введения соединения могут быть применены носители и наполнители, например, для увеличения растворимости соединения. Примеры носителей и наполнителей включают карбонат кальция, фосфат кальция, различные сахара или типы крахмала, производные целлюлозы, желатин, растительные масла, полиэтиленгликоли и физиологически совместимые растворители.
В дополнение к этому, тестируемые молекулы могут быть применены для определения структурных характеристик, которые делают их способными воздействовать на гетеродимер RXR/PPARγ, и, таким образом, выбирать молекулы, применимые в данном изобретении. Специалисты в данной области знают, как можно сконструировать лекарственные средства из первичных (lead) молекул, применяя методики, такие как описанные в публикации РСТ WO 94/18959, включенной сюда в качестве ссылки.
Токсичность и терапевтическая эффективность таких соединений могут быть определены в соответствии со стандартными фармацевтическими методиками в клеточных культурах или на экспериментальных животных, например, для определения ЛД50 (летальной дозы для 50% популяции) и ЭД50 (терапевтически эффективной дозы для 50% популяции). Отношение доз между токсическим и терапевтическим эффектами представляет собой терапевтический индекс, и он может быть выражен как отношение ЛД50/ЭД50. Предпочтительными являются соединения, которые проявляют большие терапевтические индексы. Данные, полученные в данных анализах с применением клеточных культур и исследованиях на животных, могут быть применены в установлении интервала дозировок для применения на людях. Дозировка таких соединений, предпочтительно, лежит в пределах интервала циркулирующих концентраций, который включает ЭД50 с низкой или отсутствующей токсичностью. Дозировка может варьировать в пределах данного интервала в зависимости от применяемой дозированной формы и применяемого пути введения. Уровни содержания в плазме могут быть измерены, например, по методу ЖХВР.
Конкретный состав, путь введения и дозировка могут быть выбраны конкретным терапевтом в зависимости от состояния пациента. (Смотри, например, Fingl et al., в The Pharmacological Basis of Therapeutics, 1975, Гл. 1, стр. 1). Следует заметить, что принимать решения о том, как и когда следует прекратить, прервать или приспособить способ введения из-за токсичности или дисфункции органов, должен лечащий врач. И наоборот, лечащий врач должен также принимать решения о том, как приспосабливать лечение для повышения уровней содержания, если клинический ответ неадекватен (в предотвращении токсичности). Величина вводимой дозы при лечении интересующего заболевания будет варьировать в зависимости от тяжести подлежащего лечению состояния и пути введения. Тяжесть состояния может быть отчасти оценена, например, в соответствии со стандартными прогностическими оценочными методиками. Далее, доза и, возможно, частота введения дозы будут также варьировать в зависимости от возраста, массы тела и реакции отдельного пациента. Программа, сравнимая с таковой, обсуждавшейся выше, может быть применена в ветеринарной медицине.
В зависимости от конкретных подвергающихся лечению состояний такие средства могут быть составлены и введены системно или местно. Методики приготовления составов и введения могут быть найдены в Remington's Pharmacetical Sciences, 18th ed.. Mack Publishing Co., Easton, PA (1990). Подходящие пути могут включать пероральное, ректальное, чрескожное, вагинальное, чресслизистое или энтеральное введение; парентеральное введение, включая, например, внутримышечные, подкожные, интрамедуллярные инъекции, так же, как и интратекальные, прямые внутрижелудочковые, внутривенные, внутрибрюшинные, интраназальные или внутриглазные инъекции.
Для введения в виде инъекции средства по данному изобретению могут быть включены в состав в водных растворах, предпочтительно, в физиологически совместимых буферах, таких как раствор Хенкса, раствор Рингера или физиологический раствор. Для такого чресслизистого введения в составе должны быть применены смачивающие вещества, адекватные тому барьеру, через который необходимо проникнуть. Такие смачивающие вещества широко известны в данной области.
Применение фармацевтически приемлемых носителей для приготовления составов с описываемыми здесь соединениями для осуществления на практике данного изобретения в дозированных формах, подходящих для системного введения, находится в сфере данного изобретения. При условии правильного выбора носителя и подходящей методики получения композиции по настоящему изобретению, в частности, таковые, приготовленные в виде растворов, могут быть введены парентерально, как, например, путем внутривенной инъекции. Соединения могут легко быть приготовлены с применением фармацевтически приемлемых носителей, хорошо известных в данной области, в виде дозированных форм, подходящих для перорального введения. Такие носители делают возможным готовить соединения по данному изобретению в виде таблеток, пилюль, капсул, жидких составов, гелей, сиропов, взвесей, суспензий и тому подобного для перорального введения подлежащему лечению пациенту.
Средства, подлежащие введению в клетку, могут быть введены с применением методик, хорошо известных специалистам в данной области. Например, такие средства могут быть инкапсулированы в липосомы, а затем введены, как описано выше. Липосомы представляют собой сферические липидные двухслойные структуры с водным содержимым. Все молекулы, находящиеся в водном растворе во время образования липосом, включены в водное содержимое. Содержимое липосом защищено от факторов внешнего микроокружения и, поскольку липосомы сливаются с клеточными мембранами, эффективно доставляются в цитоплазму. В дополнение к этому, благодаря их гидрофобности, малые органические молекулы могут быть введены непосредственно в клетку.
Фармацевтические композиции, подходящие для применения по настоящему изобретению, включают композиции, где активные ингредиенты содержатся в количестве, эффективном для достижения предназначенной цели. Определение эффективных количеств полностью лежит в пределах возможностей специалистов в данной области, особенно в свете приводимого здесь подробного описания. Помимо активных ингредиентов, данные фармацевтические композиции могут содержать подходящие фармацевтически приемлемые носители, включая наполнители и вспомогательные средства, которые способствуют переработке активных соединений в препараты, которые могут быть применены в фармацевтических целях. Препараты, составленные для перорального применения, могут находиться в форме таблеток, драже, капсул или растворов. Фармацевтические композиции по настоящему изобретению могут производиться в соответствии с методикой, которая известна сама по себе, например, посредством традиционных методик смешивания, растворения, гранулирования, производства драже, levitating, эмульгирования, инкапсулирования, захвата или лиофилизации.
Фармацевтические составы для парентерального введения включают в себя водные растворы активных соединений в водорастворимой форме. В дополнение к этому, могут быть приготовлены суспензии активных соединений в виде подходящих масляных суспензий для инъекций. Подходящие лиофильные растворители и носители включают нелетучие масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые повышают вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия может также содержать стабилизаторы или средства, которые повышают растворимость соединений для обеспечения возможности получения высококонцентрированных растворов.
Фармацевтические составы для перорального применения могут быть получены путем объединения активных соединений с твердым наполнителем, необязательно, растирая полученную смесь, и обработки смеси гранул, при желании, после добавления подходящих вспомогательных средств с получением центральной части таблеток или драже. Подходящими наполнителями являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; производные целлюлозы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, смола трагаканта, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий-карбоксиметилцеллюлоза и/или поливинилпирролидон (ПВП). При желании могут быть добавлены дезинтеграторы, такие как перекрестносшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Ядро драже снабжают подходящими покрытиями. Для данной цели могут быть применены концентрированные растворы сахара, которые могут необязательно содержать гуммиарабик, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль и/или диоксид титана, лакирующие растворы и подходящие органические растворители или смеси растворителей. В целях различения или характеризации различных комбинаций доз активных соединений в покрытия таблеток или драже могут быть добавлены красители или пигменты.
Фармацевтические препараты, которые могут быть применены перорально, включают как push-fit капсулы, изготовленные из желатина, так и мягкие запечатанные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Push-fit капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связывающими веществами, такими как крахмалы, и/или скользящими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как нелетучие масла, жидкий парафин или жидкие полиэтиленгликоли. В дополнение к этому, могут быть добавлены стабилизаторы. Для доставки активного вещества в инкапсулированном виде могут быть применены липосомы.
Фармацевтические составы, раскрытые или описанные в Boehm, et al., WО 94/15902, включены сюда в качестве ссылки.
Все процитированные публикации включены сюда в качестве ссылки, включая последовательности нуклеиновых кислот и аминокислотные последовательности, приведенные в каждой публикации. Все соединения, которые были описаны или на которые ссылались в публикациях, указанных выше, включены сюда в качестве ссылки, включая соединения, которые были описаны или на которые ссылались в статьях, процитированных в публикациях, указанных выше.
Другие варианты осуществления данного изобретения описаны в следующей формуле изобретения.
Изобретение относится к медицине, в частности к эндокринологии, и касается лечения инсулиннезависимого сахарного диабета (ИНСД). Способ заключается во введении агониста ретиноид-Х-рецептора (RXR) отдельно или в сочетании с агонистом пероксисом-пролиферативно-активирующим рецептором (PPARγ). К последним относят тиазолидиндионовые производные. Для этого предлагается использовать также и фармацевтические композиции, содержащие указанные компоненты. Изобретение обеспечивает повышение потребления глюкозы тканями, уменьшая содержание глюкозы, триглицеридов, холестерина в крови. 3 с. и 29 з.п. ф-лы, 2 табл., 8 ил.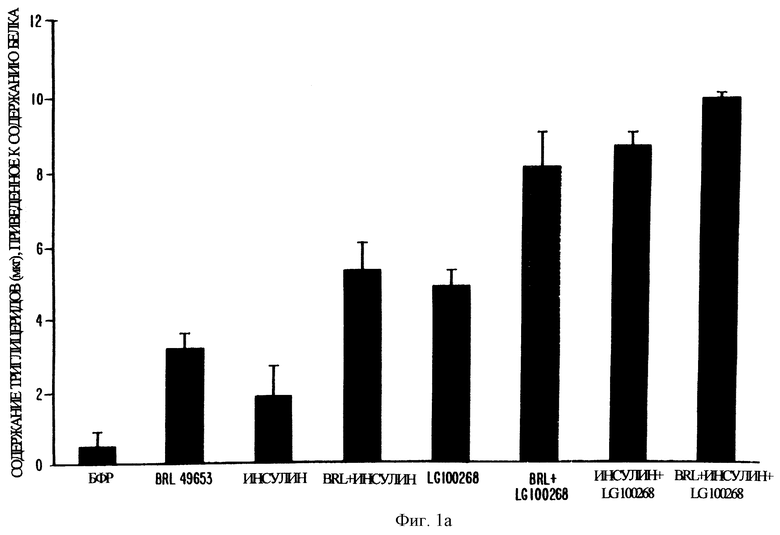
3. Способ по п.1, отличающийся тем, что дополнительно указанная композиция содержит фармацевтически эффективное количество агониста PPARγ.
4. Способ по п. 1, отличающийся тем, что указанный агонист RXR представляет собой специфический агонист RXR.
15. Композиция по п.13, отличающаяся тем, что указанный агонист RXR представляет собой LG 100268.
23. Способ по п.21, отличающийся тем, что указанная композиция дополнительно содержит фармацевтически эффективное количество агониста PPARγ.
24. Способ по п.21, отличающийся тем, что указанный агонист RXR представляет собой специфический агонист RXR.
| ЕР 0698392, А1, 28.02.1996 | |||
| Клиническая эндокринология | |||
| Под ред | |||
| Старковой Н.Т | |||
| - М., 1991, с.198. |

