Данная заявка притязает на льготы предварительной заявки на патент США 60/078180, поданной 3/16/98, озаглавленной как 1-оксо- и 1,3-диоксоизоиндолины и способ понижения уровней воспалительных цитокинов, включенной в эту заявку путем ссылки.
Предпосылки изобретения
Фактор некроза опухоли α или TNFα представляет собой цитокин, который высвобождается главным образом мононуклеарными фагоцитами в ответ на множество иммуностимуляторов. Он представляет собой ключевой провоспалительный цитокин в каскаде воспаления, вызывающий продуцирование и/или высвобождение других цитокинов и агентов. При введении животным или людям он вызывает воспаление, жар, сердечно-сосудистые эффекты, кровотечение, коагуляцию и ответы острой фазы, похожие на те, что наблюдаются во время острых инфекций и состояний шока. Таким образом, избыточное или неконтролируемое продуцирование TNFα вовлечено во множество болезненных состояний. Они включают в себя наличие эндотоксинов в крови и/или синдром токсического шока {Тrасеy et al., Nature 330, 662-664 (1987) и Hinshaw et al., Circ. Shock 30, 279-292 (1990)} ; кахексию {Dezube et al., Lancet, 335 (8690), 662 (1990)} и респираторный дистресс-синдром у взрослых (РДСВ), при котором в легочных материалах, полученных путем аспирации у пациентов, страдающих РДСВ, была обнаружена концентрация TNFα свыше 12000 пг/мл {Millar et al., Lancet 2(8665), 712-714 (1989)}. Системная инфузия рекомбинантного TNFα также дает в результате изменения, типично наблюдаемые при РДСВ {Ferrai-Baliviera et al., Arch. Surg. 124(12), 1400-1405 (1989)}.
По-видимому, TNFα вовлечен в заболевания, связанные с резорбцией костей, включающие в себя артрит. Если лейкоциты активированы, они будут вызывать резорбцию костей; данные наводят на предположение, что этой активности способствует TNFα. {Bertolini et al., Nature 319, 516-518 (1986) и Johnson et al. , Endocrinology 124(3), 1424-1427 (1989)}. Также было показано, что TNFα стимулирует резорбцию костей и подавляет образование костей in vitro и in vivo путем стимуляции образования и активации остеокластов, сочетаемых с подавлением функции остеобластов. Хотя TNFα может быть вовлечен во множество заболеваний, связанных с резорбцией костей, включающих в себя артрит, наиболее неопровержимую связь с заболеванием представляет собой связь между продуцированием TNFα опухолевыми тканями или тканями организма хозяина и гиперкальцемией, связанной со злокачественностью { Calci. Tissue Int. (US) 46(Suppl.), S3-10 (1990)}. В реакции "трансплантат против хозяина" повышенные уровни TNFα в сыворотке крови связываются со значительным осложнением, сопровождающим острые аллогенные трансплантаты костного мозга {Holler et al. , Blood, 75(4), 1011-1016 (1990)}.
Церебральная малярия представляет собой летальный гиперострый неврологический синдром, связанный с высокими уровнями в крови TNFα, и наиболее тяжелое осложнение, возникающее у пациентов, страдающих малярией. Уровни TNFα в сыворотке крови прямо коррелируют с тяжестью заболевания и прогнозом у пациента, страдающего острыми приступами малярии {Grau et al., N. Engl. J. Med. 320(24), 1586-1591 (1989)}.
Известно, что ангиогенез, вызванный макрофагами, опосредован TNFα. Лейбович и др. (Leibovich et al.) {Nature, 329, 630-632 (1987)} продемонстрировал, что TNFα in vivo в очень низких дозах вызывает образование капиллярных кровеносных сосудов в роговице крысы и развитие мембран хориоаллантоиса цыпленка, и предположил, что TNFα представляет собой кандидата на вызывание ангиогенеза при воспалении, заживлении ран и росте опухоли. Продуцирование TNFα также связывается с раковыми состояниями, в частности с индуцированными опухолями {Ching et al., Brit. J. Cancer, (1955) 72, 339-343, и Koch, Progress in Medicinal Chemistry, 22, 166-242 (1985)}.
TNFα также играет роль в области хронических легочных воспалительных заболеваний. Отложение частиц диоксида кремния приводит к силикозу, представляющему собой заболевание, связанное с прогрессирующей респираторной недостаточностью, вызванной фиброзной реакцией. Антитела к TNFα полностью блокировали вызванный диоксидом кремния фиброз легких у мышей {Pignet et al. , Nature, 344, 245-247 (1990)}. Высокие уровни продуцирования TNFα (в сыворотке крови и в изолированных макрофагах) были продемонстрированы на животных моделях фиброза, вызванного диоксидом кремния и асбестом {Bissonnette et al. , Inflammation 13(3), 329-339 (1989)}. Также было обнаружено, что альвеолярные макрофаги пациентов, страдающих саркоидозом легких, спонтанно высвобождают огромные количества TNFα по сравнению с макрофагами нормальных доноров {Baughman et al., J. Lab. Clin. Med. 115(1), 36-42 (1990)}.
TNFα также вовлечен в воспалительную реакцию, которая следует за реперфузией, называемую реперфузионным поражением, и представляет собой главную причину повреждения ткани после утраты кровотока {Vedder et al., PNAS 87, 2643-2646 (1990)}. TNFα также изменяет свойства эндотелиальных клеток и обладает различными прокоагулянтными активностями, такими как вызывание увеличения прокоагулянтной активности тканевого фактора и угнетение метаболизма антикоагулянтного протеина С, а также регуляция по типу обратной связи экспрессии тромбомодулина {Sherry et al., J. Cell Biol. 107, 1269-1277 (1988)}. TNFα обладает активностями, способствующими воспалению, которые вместе с его ранним продуцированием (во время начальной стадии воспалительного события) делают его вероятным медиатором повреждения ткани при некоторых важных расстройствах, включающих в себя инфаркт миокарда, удар и циркуляторный шок, но не ограничивающихся ими. Особую важность может представлять вызванная TNFα экспрессия факторов адгезии, таких как фактор межклеточной адгезии (IKAM) или фактор адгезии эндотелиальных лейкоцитов (ELAM) на эндотелиальных клетках {Munro et al., Am. J. Path. 135(1), 121-132 (1989)}.
Было продемонстрировано, что блокирование TNFα при помощи моноклональных антител к TNFα является полезным при ревматоидном артрите {Elliot et al., Int. J. Pharmac. 1995 17(2), 141-145}. Высокие уровни TNFα связываются с болезнью Крона {von Dullemen et al., Gastroenterology, 1995 109(1), 129-135}, и клиническая польза была достигнута при обработке антителами к TNFα.
Кроме того, в настоящее время известно, что TNFα представляет собой сильный активатор репликации ретровирусов, включая активацию ВИЧ-1. {Duh et al. , Proc. Nat. Acad. Sci. 86, 5974-5978 (1989); Poll et al., Proc. Nat. Acad. Sci. 87, 782-785 (1990); Monto et al., Blood 79, 2670 (1990); Clouse et al., J. Immunol. 142, 431-438 (1989); Poll et al., AIDS Res. Hum. Retrovirus, 191-197 (1992)}. СПИД является результатом инфекции Т-лимфоцитов вирусом иммунодефицита человека (ВИЧ). Были идентифицированы по меньшей мере три типа штаммов ВИЧ, т.е. ВИЧ-1, ВИЧ-2 и ВИЧ-3. Как следствие ВИЧ-инфекции иммунитет, опосредованный Т-клетками, ослабляется, и у инфицированных индивидуумов обнаруживаются инфекции, вызванные условно-патогенными микроорганизмами, и/или необычные новообразования. Проникновение ВИЧ в Т-лимфоцит требует активации Т-лимфоцита. Другие вирусы, такие как ВИЧ-1, ВИЧ-2 инфицируют Т-лимфоциты после активации Т-клеток, и экспрессия белков такого вируса и/или репликация опосредуется или поддерживается путем такой активации Т-клеток. После того как активированный Т-лимфоцит инфицируется ВИЧ, Т-лимфоцит должен продолжать поддерживаться в активированном состоянии для того, чтобы дать возможность экспрессии генов ВИЧ и/или репликации ВИЧ. Цитокины, в частности TNFα, вовлечены в опосредованную активированными Т-клетками экспрессию белков ВИЧ и/или репликацию вирусов посредством того, что они играют роль в поддержании активации Т-лимфоцитов. Поэтому нарушение активности цитокинов, такое как предотвращение или ингибирование продуцирования цитокинов, особенно TNFα, у индивидуумов, инфицированных ВИЧ, способствует ограничению поддержания Т-лимфоцитов, вызванного ВИЧ-инфекцией.
Моноциты, макрофаги и родственные клетки, такие как клетки Купфера и глиальные клетки, также вовлечены в поддержание ВИЧ-инфекции. Эти клетки подобно Т-клеткам представляют собой мишени для вирусной репликации, и уровень вирусной репликации зависит от состояния активации клеток. {Rosenberg et al., The Immunopathogenesis of HIV Infection, Advances in Immunology, 57 (1989)} . Было продемонстрировано, что цитокины, такие как TNFα, активируют репликацию ВИЧ в моноцитах и/или макрофагах {Poli et al., Proc. Natl. Acad. Sci. , 87, 782-784 (1990)}, следовательно, предотвращение или ингибирование продуцирования или активности цитокинов способствует ограничению развития ВИЧ в Т-клетках. Дополнительные исследования идентифицировали TNFα как общий фактор в активации ВИЧ in vitro и предложили ясный механизм действия через ядерный регуляторный белок, обнаруженный в цитоплазме клеток (Osborn, et al. , PNAS 86 2336-2340). Эти данные означают, что уменьшение синтеза TNFα может оказывать противовирусное действие при ВИЧ-инфекциях путем уменьшения транскрипции и, таким образом, продуцирования вируса.
Вирусная репликация латентного ВИЧ при СПИД в линиях Т-клеток и макрофагов может быть вызвана TNFα {Folks et al., PNAS 86, 2365-2368 (1989)}. Молекулярный механизм активности, индуцирующей вирус, предполагается исходя из способности TNFα активировать регуляторный белок генов (NFкB), обнаруженный в цитоплазме клеток, который активирует репликацию ВИЧ путем связывания с вирусной последовательностью регуляторных геннов (LTR). {Osborn et at., PNAS 86, 2336-2340 (1989)}. О TNFα при кахексии, связанной со СПИДом, предполагают по увеличению TNFα в сыворотке крови и высоким уровням спонтанного продуцирования TNFα в моноцитах в периферической крови пациентов {Wright et al. , J. Immunol. 141(1), 99-104 (1988)}. TNFα вовлечен в различных ролях в другие вирусные инфекции, такие как вирус цитомегалии (CMV), вирус гриппа, аденовирус и семейство вирусов герпеса по причинам, подобным тем, которые были указаны.
Ядерный фактор кВ (NFкB) представляет собой плейотропный активатор транскрипции (Lenardo, et al., Cell 1989, 58, 227-29). NFкB вовлечен в качестве активатора транскрипции во множество заболеваний и воспалительных состояний и, как полагают, регулирует уровни цитокинов, включающих в себя TNFα, не ограничивающихся им, и также является активатором транскрипции ВИЧ (Dbaibo, et al. , J. Biol. Chem. 1993, 17762-66; Dun et al., Proc. Natl. Acad. Sci. 1989, 86, 5974-78; Bachelerie et at., Nature 1991, 350, 709-12; Boswas et al., J. Acquired Immune Deficiency Syndrome 1993, 6, 778-786; Suzuki et al., Biochem. And Biophys. Res. Comm. 1993, 193, 277-83; Suzuki et al., Biochem. And Biophys. Res Comm. 1992, 189, 1709-15; Suzuki et al., Biochem. Mol. Bio. Int. 1993, 31(4), 693-700; Shakhov et al., Proc. Natl. Acad. Sci. USA 1990, 171, 35-47; и Staal et al., Proc. Natl. Acad. Sci. USA 1990, 87, 9943-47). Таким образом, ингибирование связывания NFкB может регулировать транскрипцию гена(ов)
цитокинов, и через это модулирование и другие механизмы быть полезным при подавлении множества болезненных состояний. Соединения, описанные здесь, могут ингибировать действие NFкB в ядре и, таким образом, являются полезными при лечении множества заболеваний, включающих в себя ревматоидный артрит, ревматоидный спондилит, остеоартрит, другие состояния, относящиеся к артриту, септический шок, сепсис, эндотоксиновый шок, заболевание "трансплантат против хозяина", атрофию, болезнь Крона, неспецифический язвенный колит, рассеянный склероз, системную красную волчанку, узловатую эритему при лепре (ENL), ВИЧ, СПИД и инфекции, вызванные условно-патогенными микроорганизмами, при СПИД, но не ограничивающихся ими. Уровни TNFα и NFкB находятся под влиянием петли взаимной обратной связи. Как указано выше, соединения по настоящему изобретению влияют на уровни как TNFα, так и NFкB.
Многие клеточные функции опосредованы уровнями аденозин-3',5'-циклофосфата (цАМФ). Такие клеточные функции могут способствовать воспалительным состояниям и заболеваниям, включающим в себя астму, воспаление и другие состояния (Lowe и Cheng, Drugs of the Future, 17(9), 799-807, 1992). Было показано, что увеличение цАМФ в лейкоцитах при воспалении подавляет их активацию и последующее высвобождение медиаторов воспаления, включающих в себя TNFα и NFкB. Повышенные уровни цАМФ также приводят к расслаблению гладкой мускулатуры дыхательных путей. Фосфодиэстеразы контролируют уровень цАМФ путем гидролиза, и было показано, что ингибиторы фосфодиэстераз повышают уровни цАМФ.
Понижение уровней TNFα и/или повышение уровней цАМФ, таким образом, составляет ценную терапевтическую стратегию лечения многих воспалительных, инфекционных, иммунологических или злокачественных заболеваний. Эти заболевания включают в себя септический шок, сепсис, эндотоксиновый шок, гемодинамический шок и септический синдром, постишемическое реперфузионное повреждение, малярию, микобактериальную инфекцию, менингит, псориаз, застойную сердечную недостаточность, фиброзное заболевание, кахексию, отторжение трансплантата, рак, аутоиммунное заболевание, инфекции, вызванные условно-патогенными микроорганизмами, при СПИД, ревматоидный артрит, ревматоидный спондилит, остеоартрит, другие состояния, относящиеся к артриту, болезнь Крона, неспецифический язвенный колит, рассеянный склероз, системную красную волчанку, узловатую эритему при лепре, лучевое поражение и гипероксическое альвеолярное повреждение, но не ограничиваются ими. Предшествующие попытки, направленные на подавление действий TNFα, простираются от использования стероидов, таких как дексаметазон и преднизолон, до использования как поликлональных, так и моноклональных антител {Beutler et al., Science 234, 470-474 (1985); WO 92/11383}.
Подробное описание изобретения
Настоящее изобретение основано на открытии того, что некоторые классы неполипептидных соединений, более полно описанных здесь, уменьшают уровни TNFα, повышают уровни цАМФ и ингибируют воспалительные цитокины. Настоящее изобретение, таким образом, относится к 1-оксо- и 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)изоиндолинам, замещенным в положении 4 изоиндолинового кольца и, возможно, дополнительно замещенным в положении 3 2,6-диоксопиперидинового кольца, к способу понижения уровней фактора некроза опухоли α и других воспалительных цитокинов у млекопитающего путем введения таких производных и к фармацевтическим композициям, содержащим такие производные.
В частности, данное изобретение относится к
(а) 2-(2,6-диоксопиперидин-3-ил)-изоиндолину формулы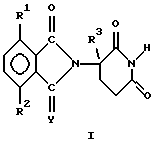
в которой
Y представляет собой кислород или H2,
один из R1 и R2 представляет собой галогено, алкил, алкокси, алкиламино, диалкиламино, циано или карбамоил,
второй из R1 и R2, независимо от первого, представляет собой водород, галогено, алкил, алкокси, алкиламино, диалкиламино, циано или карбамоил, и
R3 представляет собой водород, алкил или бензил, и
(б) солям присоединения кислот указанных 2-(2,6-диоксопиперидин-3-ил)-изоиндолинов, которые содержат атом азота, способный к присоединению протона.
Если не оговорено особо, термин алкил обозначает одновалентную насыщенную разветвленную или прямоцепочечную углеводородную цепь, содержащую от 1 до 4 атомов углерода. Представителями таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Алкокси относится к алкильной группе, связанной с оставшейся частью молекулы через эфирный атом кислорода. Представителями таких алкоксигрупп являются метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси и трет-бутокси.
Галогено включает в себя бромо, хлоро, фторо и йодо.
Соединения формулы I используют под руководством квалифицированных профессионалов для ингибирования нежелательных эффектов TNFα и других воспалительных цитокинов, включающих в себя интерлейкины IL-1, IL-6 и IL-12. Соединения можно вводить перорально, ректально или парентерально, сами по себе или в сочетании с другими терапевтическими агентами, включающими в себя антибиотики, стероиды, химиотерапевтические агенты и т.д., млекопитающему, нуждающемуся в таком лечении, например при лечении рака, ревматоидного артрита, воспалительного кишечного заболевания, мышечной дистрофии, болезни Крона и т.д.
Соединения по настоящему изобретению также можно использовать местно при лечении или профилактике болезненных состояний, опосредованных или обостряемых избыточным продуцированием TNFα, соответственно таких как вирусные инфекции, такие как инфекции, вызываемые вирусами герпеса, или вирусный конъюнктивит, псориаз, диффузный нейродермит и т.д.
Соединения также можно использовать в ветеринарном лечении млекопитающих, не являющихся людьми, при необходимости предотвращения или ингибирования продуцирования TNFα. Заболевания, опосредованные TNFα, в отношении которых предполагается осуществлять терапевтическое или профилактическое лечение животных, включают в себя болезненные состояния, такие как состояния, указанные выше, но в особенности вирусные инфекции. Примеры включают в себя вирус иммунодефицита кошек, вирус инфекционной анемии у лошадей, вирус артрита коз, вирус висны, вирус мэди, а также другие лентивирусы.
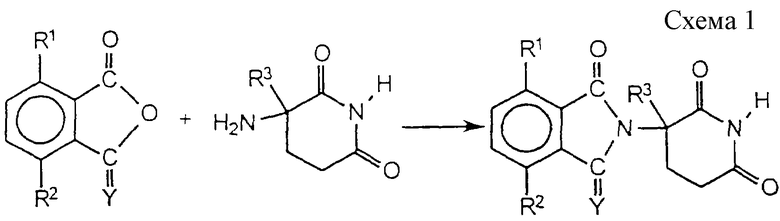
Соединения формулы 1 легко получить множеством путей. В первом воплощении ангидриду или лактону дают возможность реагировать с 3-амино-2,6-диоксо-пиперидином (см. схему 1 в конце описания).
В вышеуказанных реакциях каждый из R1, R2, R3 и Y является таким, как определено выше.
3-Амино-2,6-диоксопиперидин можно получить из соответствующего ангидрида глутаминовой кислоты путем обычного амидирования или путем циклизации подходящих глутаминовых производных.
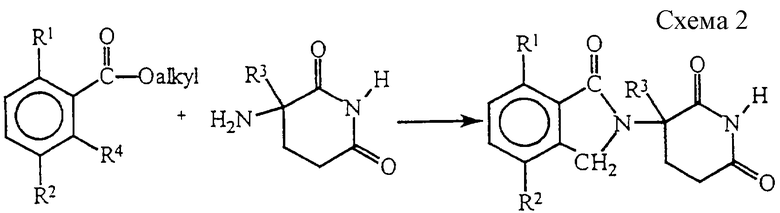
Соединения, в которых Y представляет собой Н2, в качестве альтернативы можно получить из промежуточного соединения двузамещенного бензоата в соответствиии с реакциями, приведенными в схеме 2, в которых R4 представляет собой СНО или СН2Вr, в присутствии акцептора кислот, такого как диметиламинопиридин или триэтиламин.
Промежуточные соединения двузамещенные бензоаты известны или могут быть получены обычными способами. Например, эфир низшего алкила и 3,6-двузамещенной орто-толуиловой кислоты бромируют N-бромсукцинимидом под воздействием света с получением 2-(бромметил)-3,6-двузамещенного бензоата низшего алкила.
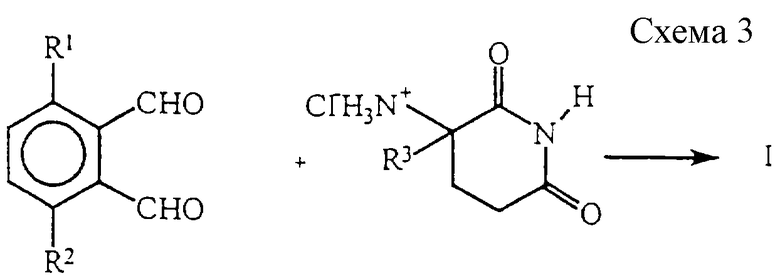
В качестве альтернативы диальдегиду дают возможность реагировать с хлоридом 2,6-диоксопиперидин-3-аммония с получением соединений формулы I, в которой Y представляет собой H2 (см. схему 3).
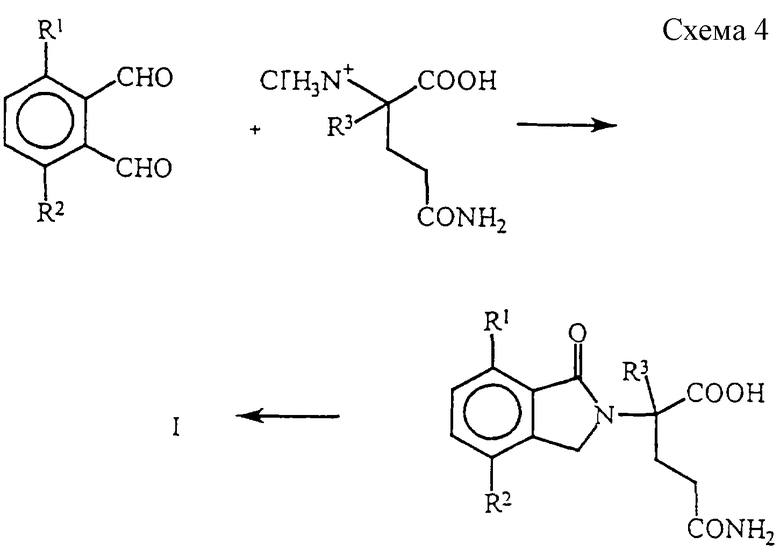
Наконец, диальдегиду дают возможность реагировать с глутамином и полученную в результате 2-(1-оксоизоиндолин-2-ил)глутаровую кислоту затем подвергают циклизации с получением 4,7-двузамещенного 1-оксо-2-(2,6-диоксопиперидин-3-ил)-изоиндолина формулы I, в которой Y представляет собой Н2 (см. схему 4).
Атом углерода, с которым связан R3 в соединениях формулы I, является центром хиральности, таким образом имея результатом оптические изомеры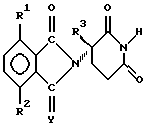
И рацематы этих изомеров и сами отдельные изомеры, а также диастереомеры, когда присутствует второй хиральный центр, входят в объем настоящего изобретения. Рацематы можно использовать как таковые или можно разделить на их отдельные изомеры механически как путем хроматографии, используя хиральный абсорбент. В качестве альтернативы отдельные изомеры можно получить в хиральной форме или выделить химически из смеси путем образования солей с хиральной кислотой или основанием, такими как отдельные энантиомеры 10-камфорсульфоновой кислоты, камфорной кислоты, α-бромкамфорной кислоты, метоксиуксусной кислоты, винной кислоты, диацетилвинной кислоты, яблочной кислоты, пирролидин-5-карбоновой кислоты и т.п., и затем удаления одного или обоих растворенных оснований, возможно повторяя способ так, чтобы получить один из двух или оба, по существу свободные один от другого, т.е. в форме, имеющей оптическую чистоту > 95%.
Настоящее изобретение также относится к физиологически приемлемым нетоксичным солям присоединения кислот соединения формулы I, которые содержат группу, способную к присоединению протона, например амино. Такие соли включают в себя соли, образованные с органическими и неорганическими кислотами, такими как, без ограничения, соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота, метансульфоновая кислота, уксусная кислота, винная кислота, молочная кислота, янтарная кислота, лимонная кислота, яблочная кислота, малеиновая кислота, сорбиновая кислота, аконитовая кислота, салициловая кислота, фталевая кислота, эмбоновая кислота, энантовая кислота и т.п.
Особенно предпочтительные соединения включают в себя 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-этилизоиндолин, 1,3-диоксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-метилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин, 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-этилизоиндолин, 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-метилизоиндолин, 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-7-этилизоиндолин, 1 -оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-7-метилизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-пропилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-хлоризоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-карбамоилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метоксиизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-метил-7-этилизоиндолин и 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диэтоксиизоиндолин. Из них 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин и 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин являются особенно предпочтительными.
Пероральные лекарственные формы включают в себя таблетки, капсулы, драже и похожей формы прессованные фармацевтические формы, содержащие от 1 до 100 мг лекарства на стандартную дозу. Можно использовать изотонические солевые растворы, содержащие от 20 до 100 мг/мл, для парентерального введения, которое включает в себя внутримышечный, внутриоболочечный, внутривенный и внутриартериальный пути введения. Ректальное введение можно осуществить путем использования суппозиториев, приготовленных из обычных носителей, таких как масло какао.
Фармацевтические композиции, таким образом, включают в себя одно или более чем одно соединение формул I, объединенное с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. При приготовлении таких композиций активные ингредиенты, как правило, смешивают с эксципиентом или разбавляют им или заключают внутри такого носителя, который может быть представлен в форме капсулы или саше. Когда эксципиент используется в качестве разбавителя, он может быть твердым, полутвердым или жидким веществом, которое действует в качестве разбавителя, носителя или среды для активного ингредиента. Таким образом, композиции могут быть представлены в форме таблеток, пилюль, порошков, эликсиров, суспензий, эмульсий, растворов, сиропов, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъекционных растворов и стерильных упакованных порошков. Примеры подходящих эксципиентов включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, аравийскую камедь, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу, препараты могут дополнительно включать в себя смазывающие вещества, такие как тальк, стеарат магния и минеральное масло, увлажнители, эмульгаторы и суспендирующие агенты, консерванты, такие как метил- и пропилгидроксибензоаты, подслащивающие вещества или корригенты.
Композиции предпочтительно готовят в виде стандартной лекарственной формы, подразумевающей под собой физически отдельные единицы, пригодные в качестве однократной дозировки, или заданную фракцию однократной дозы, которую нужно ввести людям и другим млекопитающим по схеме однократного или многократного приема лекарственного средства, причем каждая единица содержит заданное количество активного вещества, рассчитанного таким образом, чтобы оказывать желаемый терапевтический эффект, совместно с подходящим фармацевтическим эксципиентом. Композиции можно приготовить таким образом, чтобы обеспечить немедленное, длительное или замедленное высвобождение активного ингредиента после введения пациенту путем применения процедур, хорошо известных в данной области техники.
Иммуноферментные анализы для TNFα можно проводить обычным образом. Мононуклеарные клетки перифирической крови (МКПК) выделяют из нормальных доноров путем центрифугирования в градиенте плотности Ficoll-Hypaque. Клетки выращивают в среде RPMI, дополненной 10% сывороткой АВ+, 2 мМ L-глутамином, 100 единиц/мл пенициллином и 100 мг/мл стрептомицином. Лекарства растворяют в диметилсульфоксиде (Sigma Chemical), и дополнительные разведения осуществляются в дополненной RPMI. Конечная концентрация диметилсульфоксида в присутствии или отсутствие лекарства в суспензиях МКПК составляет 0,25 мас.%. Лекарства анализируют при полулогарифмических разведениях, начиная с 50 мг/мл. Лекарства добавляют к МКПК (106 клеток/мл) в 96-луночных планшетах за один час перед добавлением липополисахаридов (ЛПС). МКПК (106 клеток/мл) в присутствии или отсутствие лекарства стимулируют путем обработки 1 мг/мл ЛПС из Salmonella minnesota R595 (List Biological Labs, Campbell, CA). Клетки затем инкубируют при 37oС в течение 18-20 часов. Супернатанты собирают и немедленно анализируют в отношении уровней TNFα или держат замороженными при -70oС (в течение не более 4 дней) до анализа. Концентрацию TNFα в супернатанте определяют при помощи наборов для анализа TNFα человека путем ИФА (Endogen, Boston, MA) в соответствии с указаниями производителя.
Следующие примеры служат для дополнительного представления сущности данного изобретения, но их не следует рассматривать как ограничение объема изобретения, который определен исключительно прилагаемой формулой изобретения.
Пример 1
2-(2,6-Диоксопиперид-3-ил)-4-метилизоиндолин-1,3-дион
Перемешиваемый раствор 3-метилфталевого ангидрида (2,96 г, 18,2 ммоль), гидрохлорида 3-аминопиперидин-2,6-диона (3,00 г, 18,2 ммоль) и ацетата натрия (1,57 г, 19,1 ммоль) в уксусной кислоте (30 мл) нагревают с обратным холодильником в течение 23 часов. Растворитель удаляют в вакууме с получением твердого вещества, которое перемешивают с водой (40 мл) в течение 1 часа, фильтруют, промывают водой (30 мл) и затем нагревают с обесцвечивающим древесным углем (1 г) в ацетоне (2 л) при температуре дефлегмации в течение 30 мин. Суспензию фильтруют через слой Целита с получением чистого раствора. Растворитель фильтрата удаляют в вакууме с получением 2-(2,6-диоксопиперид-3-ил)-4-метилизоиндолин-1,3-диона в виде белого твердого вещества (4,08 г, выход 82%) т.пл. 290,0-292,0oС.
1Н ЯМР (ДМСО-d6); δ 2,03-2,09 (m, 1H, СНН), 2,50-2,60 (m, 2H, СН2), 2,63 (s, 3H, СН3), 2,83-2,95 (m, 1H, СНН), 5,13 (dd, J=5,4, 12,3 Гц, 1H, NCH), 7,65-7,79 (m, 3H, Ar), 11,13 (шир. s, 1H, NH).
13C ЯМР (ДМСО-d6) δ 17,04, 21,99, 30,93, 48,76, 121,05, 127,89, 131,63, 134,37, 136,91, 137,61, 167,04, 167,83, 169,87, 172,74.
Для C14H12N2O4:
Рассчитано: С, 61,76; Н, 4,44; N, 10,29.
Обнаружено: С, 61,68; Н, 4,37; N, 10,17.
Пример 2
Путем замещения эквивалентных количеств 3-этилфталевого ангидрида, 3-фторфталевого ангидрида, 3-хлорфталевого ангидрида, 3-карбамоилфталевого ангидрида и 3-метоксифталевого ангидрида в способе Примера 1 соответственно получают 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-этилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-фторизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-хлоризоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-карбамоилизоиндолин, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метоксиизоиндолин.
Пример 3
Путем замещения эквивалентных количеств гидрохлорида 3-амино-3-метилпиперидин-2,6-диона на гидрохлорид 3-аминопиперидин-2,6-диона в способе Примера 1 получают 1,3-диоксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-метилизоиндолин.
Пример 4
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин
Смесь 16,25 г хлорида 2,6-диоксопиперидин-3-аммония и 30,1 г метил-2-бромметил-3-метилбензоата и 12,5 г триэтиламина в 100 мл диметилформамида перемешивают при комнатной температуре в течение 15 часов. Смесь затем концентрируют в вакууме и остаток смешивают с метиленхлоридом и водой. Водный слой отделяют и экстагируют из него метиленхлоридом. Объединенные метиленхлоридные растворы сушат над сульфатом магния и концентрируют в вакууме с получением 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолина.
Похожим образом получают 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-этилизоиндолин и 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-метоксиизоиндолин путем замещения эквивалентных количеств метил-2-бромметил-3,6-диметилбензоата, метил-2-бромметил-3-этилбензоата и метил-2-бромметил-3-метоксибензоата соответственно на метил-2-бромметил-3-метилбензоат.
Пример 5
2-(2,6-Диоксопиперидин-3-ил)-4,7-диметилизоиндолин-1,3-дион
2-(2,6-Диоксопиперид-3-ил)-4,7-диметилизоиндолин-1,3-дион получают при помощи способа Примера 1 из 3,6-диметилфталевого ангидрида (220 мг, 1,25 ммоль), гидрохлорида 3-аминопиперидин-2,6-диона (204 мг, 1,24 ммоль) и ацетата натрия (110 мг, 1,34 ммоль) в уксусной кислоте (10 мл). Продукт представляет собой белое твердое вещество (200 мг, выход 56%): т.пл. 263,0-265,0oС.
1Н ЯМР (ДМСО-d6) δ 2,01-2,07 (m, 1H, СНН), 2,50-2,89 (m, 9H, СН3, СHН, СН2), 5,10 (dd, J=5,1, 12,4 Гц, 1H, NCH), 7,52 (s, 2H, Ar), 11,12 (шир. s, 1H, NH).
13C ЯМР (AMCO-d6) δ 16,82, 22,02, 30,97, 48,59, 128,01, 135,04, 136,58, 167,68, 169,98, 172,83.
Пример 6
2-(2,6-Диоксо(3-пиперидил))-4-этилизоиндолин-1,3-дион
2-(2,6-Диоксо(3-пиперидил))-4-этилизоиндолин-1,3-дион получают при помощи способа Примера 1 из 3-этилфталевого ангидрида (0,860 г, 4,89 ммоль), гидрохлорида 3-аминопиперидин-2,6-диона (0,803 г, 4,88 ммоль) и ацетата натрия (0,420 г, 5,12 ммоль) в уксусной кислоте (10 мл). Продукт представляет собой белое твердое вещество (1,06 г, выход 76%); т.пл. 235,0-236,5oС.
1H ЯМР (ДМСО-d6) δ 1,22 (t, J=7,4 Гц, 3Н, СН3), 2,04-2,10 (m, 1H, СНН), 2,47-2,63 (m, 2H, CH2), 2,83-2,98 (m, 1H, СНН), 3,07 (q, J=7,5 Гц, 2H, CH2), 5,13 (dd, J= 5,4, 12,5 Гц, 1H, NCH), 7,70-7,82 (m, 3H, Ar), 11,13 (шир. s, 1H, NH).
13С ЯМР (ДМСО-d6) δ 14,84, 21,95, 23,69, 30,90, 48,77, 121,09, 127,26, 131,76, 134,63, 135,39, 143,87, 166,99, 167,58, 169,85, 172,72.
Для C15H14N2O4:
Рассчитано: С, 62,93; Н, 4,93; N, 9,79.
Обнаружено: С, 62,74; Н, 4,84; N, 9,54.
Пример 7
4-Метокси-2-(2,6-диоксо(3-пиперидил)изоиндолин-1,3-дион
4-Метокси-2-(2,6-диоксо(3-пиперидил)изоиндолин-1,3-дион получают при помощи способа Примера 1 из 3-метоксифталевого ангидрида (1,0 г, 5,6 ммоль) { Rao. A. V. R. et al., Indian J. Chem. 1981, 20 (В), 248}, гидрохлорида 3-аминопиперидин-2,6-диона (0,92 г, 5,6 ммоль) и ацетата натрия (0,48 г, 6,0 ммоль) в уксусной кислоте (20 мл). Продукт представляет собой белое твердое вещество (0,44 г, выход 27%); т.пл. 281,5-282,5oС.
1H ЯМР (ДМСО-d6) δ 2,00-2,08 (m, 1Н, СНН), 2,56-2,62 (m, 2Н, СH2), 2,82-2,91 (m, 1Н, СНН), 3,97 (s, 3Н, СH3), 5,08 (dd, J=5,3, 12,8 Гц, 1Н, NCH), 7,46 (d, J=7,2 Гц, 1Н, Ar), 7,52 (d, J=8,5 Гц, 1Н, Ar), 7,84 (d, J=7,8 Гц, 1Н, Ar), 11,10 (шир. s, 1Н, NH).
13С ЯМР (ДМСО-d6) δ 21,97, 30,92, 48,73, 56,33, 115,24, 116,11, 119,01, 133,19, 137,15, 156,49, 165,37, 166,84, 169,94, 172,79.
Для C14H12N2O5:
Рассчитано: С, 58,33; Н, 4,20; N, 9,72.
Обнаружено: С, 58,23; Н, 3,90; N, 9,53.
Пример 8
4-Диметиламино-2-(2,6-диоксо(3-пиперидил))изоиндолин-1,3-дион
4-Диметиламино-2-(2,6-диоксо(3-пиперидил))изоиндолин-1,3-дион получают при помощи способа Примера 1 из 3-диметиламинофталевого ангидрида (1,34 г, 7,0 ммоль), гидрохлорида 3-аминопиперидин-2,6-диона (1,15 г, 7,0 ммоль) и ацетата натрия (0,60 г, 7,3 ммоль) в уксусной кислоте (20 мл). Продукт представляет собой желтое твердое вещество (1,59 г, выход 75%); т.пл. 214,5-216,5oС.
1H ЯМР (ДМСО-d6) δ 1,98-2,09 (m, 1H, СНН), 2,49-2,62 (m, 2Н, CH2), 2,81-2,95 (m, 1H, СНН), 3,04 (s, 6H, СН3), 5,08 (dd, J=5,5, 12,7 Гц, 1H, NCH), 7,23 (d, J=6,6 Гц, 1H, Ar), 7,26 (d, J=8,1 Гц, 1H, Ar), 7,63 (dd, J= 6,9, 8,6 Гц, 1H, Ar), 11,09 (шир. s, 1H, NH).
13C ЯМР (ДМСО-d6) δ 22,10, 30,96, 42,95, 48,77, 112,99, 113,41, 122,59, 133,90, 135,22, 149,88, 166,29, 167,13, 170,06, 172,83.
Для С15Н15N3О4:
Рассчитано: С, 59,80; Н, 5,02; N, 13,95.
Обнаружено: С, 59,60; Н, 4,94; N, 13,80.
Пример 9
2-(2,6-Диоксо(3-пиперидил))-4-хлоризоиндолин-1,3-дион
2-(2,6-Диоксо(3-пиперидил))-4-хлоризоиндолин-1,3-дион получают при помощи способа Примера 1 из 3-хлорфталевого ангидрида (0,40 г, 2,2 ммоль), гидрохлорида 3-аминопиперидин-2,6-диона (0,36 г, 2,2 ммоль) и ацетата натрия (0,19 г, 2,4 ммоль) в уксусной кислоте (10 мл). Продукт представляет собой белое твердое вещество (0,44 г, выход 69%); т.пл. 290,0-291,5oС.
1H ЯМР (ДМСО-d6) δ 2,05-2,11 (m, 1Н, СНН), 2,49-2,64 (m, 2Н, СH2), 2,64-2,92 (m, 1H, СНН), 5,17 (dd, J=5,2, 12,7 Гц, 1H, NCH), 7,86-7,94 (m, 3H, Ar), 11,17 (шир. s, 1H, NH).
13С ЯМР (ДМСО-d6) δ 21,83, 30,91, 49,12, 122,41, 126,94, 129,84, 133,52, 136,11, 136,39, 164,77, 165,76, 169,73, 172,77; .
Для C13H9N2O4Cl:
Рассчитано: С, 53,35; Н, 3,10; N, 9,57; Cl, 12,11.
Обнаружено: С, 53,37; Н, 2,94; N, 9,30; Cl, 11,97.
Пример 10
4-Метил-2-(2,6-диоксо-3-метил-(3-пиперидил))изоиндолин-1,3-дион
4-Метил-2-(2,6-диоксо-3-метил-(3-пиперидил))изоиндолин-1,3-дион получают при помощи способа Примера 1 из 3-метилфталевого ангидрида (0,27 г, 1,7 ммоль), гидрохлорида 3-амино-3-метилпиперидин-2,6-диона (0,30 г, 1,7 ммоль) и ацетата натрия (0,15 г, 1,8 ммоль) в уксусной кислоте (10 мл). Продукт представляет собой белое твердое вещество (0,13 г, выход 27%); т.пл. 248,0-250,0oС.
1H ЯМР (ДМСО-d6) δ 1,89 (s, 3H, СН3), 2,01-2,08 (m, 1H, СНН), 2,49-2,70 (m, 3H, СНН, CH2), 2,55 (s, 3H, СН3), 7,62-7,74 (m, 3H, Ar), 10,99 (шир. s, 1H, NH).
13C ЯМР (ДМСО-d6) δ 17,0, 21,0, 28,6, 29,1, 58,6, 120,7, 127,5, 131,5, 134,2, 136,8, 137,2, 167,7, 168,6, 172,1, 172,3.
Для C15H14N2O4 + 0,3 Н2О:
Рассчитано: С, 61,77; Н, 5,05; N, 9,60.
Обнаружено: С, 62,05; Н, 4,94; N, 9,20.
Пример 11
Таблетки, каждая из которых содержит 50 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолина, можно приготовить следующим образом:
Составные части (на 1000 таблеток), г
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин - 50,0
Лактоза - 50,7
Пшеничный крахмал - 7,5
Полиэтиленгликоль 6000 - 5,0
Тальк - 5,0
Стеарат магния - 1,8
Деминерализованная вода - Достаточное количество
Твердые ингредиенты сначала просеивают через сито с размером ячейки 0,6 мм. Затем смешивают активный ингредиент, лактозу, тальк, стеарат магния и половину крахмала. Другую половину крахмала суспендируют в 40 мл воды и эту суспензию добавляют к кипящему раствору полиэтиленгликоля в 100 мл воды. Полученный в результате клейстер добавляют к порошкообразным веществам и смесь гранулируют, если необходимо - с добавлением воды. Гранулят сушат в течение ночи при 35oС, продавливают через сито с размером ячейки 1,2 мм и прессуют с образованием таблеток диаметром приблизительно 6 мм, которые являются вогнутыми с обеих сторон.
Пример 12
Желатиновые капсулы с сухим наполнением, каждая из которых содержит 100 мг 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолина, можно получить следующим образом:
Композиция (на 1000 капсул),г:
1,3-Диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин - 100,0
Микрокристаллическая целлюлоза - 30,0
Лаурилсульфат натрия - 2,0
Стеарат магния - 8,0
Лаурилсульфат натрия просеивают в 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-метилизоиндолин через сито с размером ячейки 0,2 мм и два компонента хорошо смешивают в течение 10 минут. Затем добавляют микрокристаллическую целлюлозу через сито с размером ячейки 0,9 мм и всю смесь снова хорошо перемешивают в течение 10 минут. Наконец, добавляют стеарат магния через сито с размером ячейки 0,8 мм и после смешивания в течение следующих 3 минут смесь вносят порциями по 140 мг каждая в желатиновые капсулы с сухим наполнением размера 0 (удлиненные).
Пример 13
0,2% Раствор для инъекций или инфузий можно получить, например, следующим образом:
1,3-Диоксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин - 5,0 г
Хлорид натрия - 22,5 г
Фосфатный буфер рН 7,4 - 300,0 г
Деминерализованная вода - До 2500,0 мл
1-Диоксо-2-(2,6-диоксопиперидин-3-ил)-4,7-диметилизоиндолин растворяют в 1000 мл воды и фильтруют через микрофильтр. Добавляют буферный раствор и всю смесь доводят до 2500 мл водой. Для приготовления стандартных лекарственных форм порции 1,0 или 2,5 мл каждого вводят в стеклянные ампулы (каждая содержит соответственно 2,0 или 5,0 мг имида).
Данные по биологической активности заявленных соединений
Соединение, имеющее структуру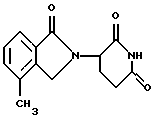
(3-(4-метил-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион), имеет ИК50 в отношении TNFα, равную 10 нМ.
Соединение, имеющее структуру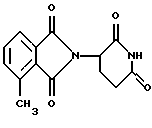
(2-(2,6-диоксопиперид-3-ил)-4-метилизоиндолин-1,3-дион), имеет ИК50 в отношении TNFα, равную 100 нМ.
Пример 14. Получение 3-(4-метил-1-оксоизоиндолин-2-ил)пиперидин-2,6-диона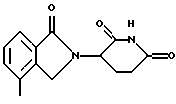
N,N-диэтил-2,3-диметилфенилкарбоксамид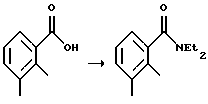
Суспензию 2,3-диметилбензойной кислоты (10,2 г, 67,9 ммоль) в тионилхлориде (50 мл) нагревали до температуры дефлегмации в течение 30 минут. Тионилхлорид удаляли посредством дистилляции. Остаток растворяли в метиленхлориде (20 мл) при 0oС. К раствору при 0oС добавляли диэтиламин (21 мл, 203 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Раствор выливали в HCl (1н., 50 мл) и метиленхлорид (20 мл). Водный слой отделяли и экстрагировали метиленхлоридом (50 мл). Объединенные органические слои промывали HCl (1н., 50 мл), рассолом (50 мл) и сушили над MgSО4. Удаление растворителя дало N,N-диэтил-2,3-диметилфенилкарбоксамид в виде коричневого масла (13,5 г, выход 97%, чистота 90%), который использовали на следующей стадии без дополнительной очистки.
N,N-диэтил-[2-(иодметил)-3-метилфенил]карбоксамид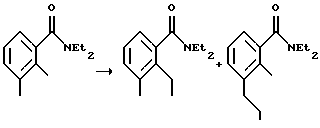
К раствору втор-BuLi (24 мл, 1,3 М, 31,2 ммоль) в тетрагидрофуране (ТГФ) (60 мл) при -78oС добавляли раствор N,N-диэтил-2,3-диметилфенилкарбоксамида (6,0 г, 29,2 ммоль) в ТГФ (25 мл). Через 15 минут к красному раствору при -78oС добавляли триметилборат (4,1 мл, 35,7 ммоль). Через 5 минут охлаждающую баню убирали и раствор помещали в водяную баню со льдом на 15 минут. Добавляли иод (15,3 г, 60,3 ммоль) и реакционную смесь перемешивали при 0oС в течение 1,5 часа. Реакционную смесь приливали к раствору сульфита натрия (10%, 100 мл). Водный слой экстрагировали этилацетатом (200 мл). Органический слой промывали сульфитом натрия (10%, 100 мл), рассолом (100 мл) и сушили над MgSО4. Удаление растворителя дало смесь N,N-диэтил-[2-(иодметил)-3-метилфенил] -карбоксамида (значительное количество) и N,N-диэтил-[3-(иодметил)-2-метилфенил] -карбоксамида (незначительное количество) в виде масла (8,5 г). Эти изомеры не подвергались разделению с помощью колоночной хроматографии, и поэтому их использовали на следующей стадии без дополнительной очистки.
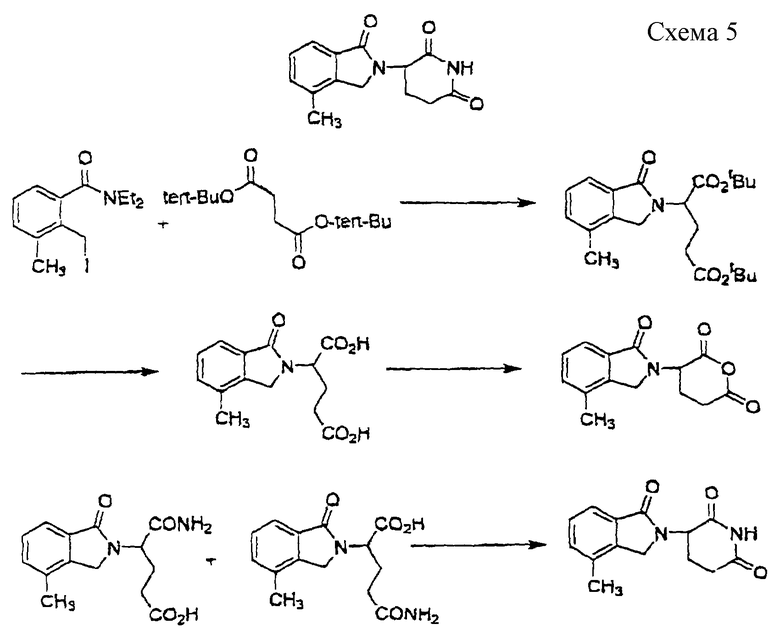
3-(4-Метил-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион (см. схему 5 в конце описания).
Раствор сырого N,N-диэтил-[2-(иодметил)-3-метилфенил] карбоксамида (1,8 г, 5,43 ммоль) и гидрохлорида ди-трет-бутилового эфира глутаминовой кислоты (1,67 г, 5,64 ммоль) и триэтиламин (1,73 мл, 12,4 ммоль) в ацетонитриле (30 мл) нагревали до температуры дефлегмации в течение 2,5 суток. Растворитель удаляли в вакууме. Масло экстрагировали диэтиловым эфиром (50 мл) и водой (40 мл). Водный слой экстрагировали диэтиловым эфиром (50 мл). Объединенные органические слои промывали рассолом (50 мл) и сушили над МgSО4 и углем. Растворитель удаляли в вакууме с получением масла. Это масло очищали с помощью колоночной хроматографии с получением ди-трет-бутил-2-(4-метил-1-оксоизоиндолин-2-ил)пентан-1,5-диоата в виде желтого масла (1,48 г, выход 70%). Это масло (1,48 г, 3,79 ммоль) в муравьиной кислоте (10 мл) нагревали до температуры дефлегмации в течение 18 часов. Растворитель удаляли в вакууме. Это масло суспендировали в метиленхлориде (1 мл) и диэтиловом эфире (50 мл) с получением суспензии. Эту суспензию фильтровали и промывали диэтиловым эфиром с получением 1-(4-метил-1-оксоизоиндолин-2-ил)пропан-1,3-дикарбоновой кислоты в виде твердого вещества белого цвета с желтоватым оттенком (0,78 г, выход 76%). Суспензию вышеуказанной кислоты (0,78 г, 2,86 ммоль) в уксусном ангидриде (20 мл) нагревали до температуры дефлегмации в течение 19 часов. Растворитель удаляли в вакууме. Твердое вещество суспендировали в диэтиловом эфире (10 мл) в течение 30 минут. Эту суспензию фильтровали и промывали диэтиловым эфиром с получением 3-(4-метил-1-оксоизоиндолин-2-ил)-3,4,5-тригидро-2Н-пиран-2,6-диона в виде коричневого твердого вещества (0,6 г, выход 81%). Суспензию вышеуказанного твердого вещества (0,6 г, 2,3 ммоль) и гидроксида аммония (0,4 мл) в тетрагидрофуране (10 мл) перемешивали при комнатной температуре в течение 1 часа. Эту суспензию фильтровали и промывали диэтиловым эфиром с получением смеси 4-карбамоил-2-(4-метил-1-оксоизоиндолин-2-ил)масляной кислоты и 4-карбамоил-4-(4-метил-1-оксоизоиндолин-2-ил)масляной кислоты в виде твердого вещества белого цвета с желтоватым оттенком (640 мг, выход 100%). Суспензию вышеуказанного твердого вещества (640 мг, 2,31 ммоль), карбонилдиимидазола (420 мг, 2,59 ммоль) и диметиламинофенола (ДМАФ) (следовое количество) в ацетонитриле (20 мл) нагревали до температуры дефлегмации в течение 1 суток. Эту суспензию фильтровали и промывали ацетонитрилом с получением твердого вещества белого цвета с желтоватым оттенком. Суспензию этого твердого вещества и угля (120 мг) в ацетоне (100 мл) нагревали до температуры дефлегмации в течение 30 минут. Эту суспензию фильтровали через подушку из целита и промывали ацетоном. Растворитель удаляли в вакууме с получением 3-(4-метил-1-оксоизоиндолин-2-ил)пиперидин-2,6-диона в виде белого твердого вещества (200 мг, выход 34%): температура плавления 271-273oС.
1Н ЯМР (ДМСО (диметилсульфоксид)-d6) δ 1,98-2,04 (m, 1Н, СНН), 2,32 (s, 3Н, СН3), 2,33 2,64 (m, 2H, CH2), 2,86-2,99 (m, 1H, СНН), 4,25 (d, J=17,3 Гц, 1H, CHH), 4,43 (d, J=17,3 Гц, 1H, СНН), 5,14 (dd, J=4,9, 13,1 Гц, 1H, NCH), 7,42-7,44 (m, 2H, Ar), 7,53-7,57 (m, 1H, Ar), 11,01 (s, 1H, NH).
13C ЯМР (AMCO-d6) δ 17,09, 22,55, 31,21, 46,33, 51,51, 120,41, 128,17, 131,37, 132,37, 132,95, 141,03, 168,40, 171,06, 172,89.
Для C14H14N2О3:
Рассчитано: С, 65,11; Н, 5,46; N, 10,85.
Обнаружено: С, 65,12; Н, 5.40; N, 10,56.
Изобретение относится к производным изоиндолина формулы I, где Y обозначает кислород или Н2, один из R1 и R2 представляет собой галоген, алкил, алкокси, алкиламино, диалкиламино, циано или карбамоил, второй из R1 и R2 представляет собой водород, галоген, алкил, алкокси, алкиламино, диалкиламино, циано или карбамоил, R3 представляет собой водород, алкил или бензил. Представленные соединения обладают ингибирующей активностью для понижения нежелательных эффектов TNFα и воспалительных цитокинов и благодаря этому свойству они могут использоваться в качестве активного ингредиента в фармкомпозиции, в способах понижения или ингибирования нежелательных уровней TNFα и воспалительных цитокинов, а также в способах лечения различных заболеваний у млекопитающих. 11 с. и 16 з.п.ф-лы. 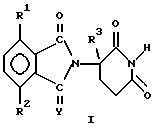
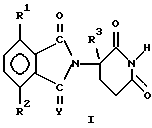
в которой Y представляет собой кислород или H2;
один из R1 и R2 представляет собой галоген, алкил, алкокси, алкиламино, диалкиламино, циано или карбамоил, второй из R1 и R2 независимо представляет собой водород, галоген, алкил, алкокси, алкиламино, диалкиламино, циано или карбамоил;
R3 представляет собой водород, алкил или бензил,
их соли присоединения кислот, которые содержат атом азота, способный к присоединению протона, при условии, что если R1 представляет собой алкиламино, то R2 является иным, чем водород, или если R1 представляет собой хлор, то R2 является иным, чем хлор.
| Способ подавления квадратурных составляющих периодического сигнала | 1981 |
|
SU1075420A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Устройство для распыливания и сжигания мазута | 1978 |
|
SU688771A1 |
| US 5635517 А, 03.06.1997 | |||
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АНГИОТЕНЗИНА, НА ИХ ОСНОВЕ | 1992 |
|
RU2053229C1 |

