Изобретение относится к замещенным 2-(2,6-диоксопиперидин-3-ил)-фталимидам и замещенным 2-(2,6-диоксопиперидин-3-ил)-оксоизоиндолинам, способу снижения уровней фактора некроза опухоли α и лечения воспалительных и аутоиммунных заболеваний у млекопитающего путем введения таких производных, и к фармацевтическим композициям таких производных.
Фактор некроза опухоли α, или ФНОα является цитокином, который продуцируется главным образом мононуклеарными фагоцитами в ответ на различные иммуностимуляторы. При введении животным или людям он вызывает воспаление, лихорадку, сердечно-сосудистые явления, геморрагию, коагуляцию и реакции острой фазы, подобные тем, которые наблюдаются при острых инфекциях и шоковых состояниях. Чрезмерное и неупорядоченное продуцирование ФНОα вовлечено в ряд болезненных состояний. Они включают в себя эндотоксемию и/или синдром токсического шока [Тгасеу et al., Nature 330, 662-664 (1987) и Hinshaw et al. , Circ. Shock 30, 279-292 (1990)], кахексию [Dezube et al., Lancet, 335 (8690), 662 (1990)] и респираторный дистресс-синдром взрослых (РДСВ), при этом концентрацию ФНОα свыше 12000 пг/мл обнаруживали в легочных аспиратах у РДСВ-пациентов [Millar et al., Lancet 2 (8665), 712-714 (1989)]. Системная инфузия рекомбинантного ФНОα в результате приводит также к изменениям, обычно наблюдаемым при РДСВ [Ferrai-Baliviera et al., Arch. Sugr. 124 (12), 1400-1405 (1989)].
По всей вероятности, ФНОα вовлечен в заболевания, связанные с резорбцией кости, в том числе артриты. Лейкоциты, когда они активированы, вызывают резорбцию кости, и имеются данные, позволяющие предполагать, что ФНОα вносит вклад в активность, направленную на резорбцию кости [Bertolini et al., Nature 319, 516-518 (1986) and Johnson et al., Endocrinology 124 (3), 1424-1427 (1989)] . Было показано, что ФНОα стимулирует резорбцию кости и ингибирует остеогенез in vitro и in vivo через стимуляцию образования остеокласта и активацию, связанную с ингибированием функции остеобласта. Хотя ФНОα может быть вовлечен во многие заболевания, связанные с резорбцией кости, в том числе артрит, наиболее тесная связь с заболеванием наблюдается между продуцированием ФНОα тканями опухоли или хозяина и злокачественностью, связанной с гиперкальциемией [Calci. Tissue Int. (US) 46 (Suppl), S3-10 (1990)] . При реакции "трансплантат против хозяина" повышенные уровни ФНОα в сыворотке ассоциированы с главным осложнением, сопровождающим острые аллогенные трансплантаты костного мозга [Holler et al., Blood, 75(4), 1011-1016(1990)].
Церебральная малярия является летальным сверхострым неврологическим синдромом, связанным с высокими уровнями содержания ФНОα в крови, и является наиболее тяжелым осложнением у пациентов, страдающих малярией. Уровни ФНОα в сыворотке соотносятся непосредственно с тяжестью заболевания и позволяют прогнозировать у пациентов острый приступ малярии [Grau et al., N. Engl. J. Med. 320 (24), 1586-1591 (1989)].
Известно, что ангиогенез, индуцированный макрофагами, опосредован ФНОα. Лейбович и др. [Nature, 329, 630-632 (1987)] показали, что ФНОα индуцирует in vivo образование капиллярных кровеносных сосудов в роговице крысы и в хориоаллантойных мембранах цыплят, находящихся в стадии развития, при очень низких дозах, и считается, что ФНОα является кандидатом индуцировать ангиогенез при воспалении, заживлении ран и росте опухоли. Продуцирование ФНОα также связано с условиями возникновения рака, в частности, вызывает образование опухоли [Ching et al., Brit. J. Cancer, (1955) 72, 339-343, и Koch, Progress in Medicinal Chemistry, 22, 166-242 (1985)].
Кроме того, ФНОα играет определенную роль в хронических легочных воспалительных заболеваниях. Отложение частиц диоксида кремния приводит к силикозу, болезни прогрессирующей респираторной недостаточности, вызванной фиброзной реакцией. Антитело к ФНОα полностью блокирует индуцированный диоксидом кремния фиброз легкого у мышей [Pignet et al., Nature, 344:245-247 (1990)]. Высокие уровни продукции ФНОα (в сыворотке и в выделенных макрофагах) были продемонстрированы на животных моделях фиброза, индуцированного диоксидом кремния и асбестом [Bissonnette et al., Inflammation 13 (3), 329-339 (1989)] . Было обнаружено также, что альвеолярные макрофаги от пациентов с саркоидозом легких спонтанно высвобождают избыточное количество ФНОα по сравнению с макрофагами от нормальных доноров [Baughman et al., J.Lab.Clin.Med. 115 (1), 36-42 (1990)].
ФНОα вовлечен также в воспалительную реакцию, которая является следствием реперфузии, называемую реперфузионной травмой, и является основной причиной повреждения ткани в результате утраты кровотока [Vedder et al., PNAS 87, 2643-2646 (1990)] . Кроме того, ФНОα изменяет свойства эндотелиальных клеток и обладает различными прокоагулянтными активностями, такими, как продуцирование увеличения прокоагулянтной активности тканевого фактора и супрессия метаболизма антикоагулянтного белка С, а также ухудшение регулирования экспрессии тромбомодулина [Sherry et al., J.Cell Вiol. 107, 1269-1277 (1988)] . ФНОα обладает провоспалительной активностью, которая наряду с его ранней продукцией (на начальной стадии воспалительного процесса) делает его медиатором повреждения ткани при некоторых важных нарушениях, включая, но не ограничиваясь ими, инфаркт миокарда, инсульт и циркуляторный шок. Особое значение может иметь ФНОα-индуцированная экспрессия факторов адгезии, таких как фактор межклеточной адгезии (ICAM) или фактор адгезии эндотелиальных лейкоцитов (ELAM) на эндотелиальных клетках [Munro et al., Am. J.Path. 135(1), 121-132(1989)].
Было показано, что блокирование ФНОα моноклональными анти-ФНОα антителами полезно при ревматоидном артрите [Elliot et al., Int. J.Pharmac. 1995 17(2), 141-145] и при болезни Крона [von Dullemen et al., Gastroenterology, 1955 109(1), 129-135].
Более того, в настоящее время известно, что ФНОα является сильным активатором репликации ретровирусов, включая активацию ВИЧ-1 [Dun et al., Proc. Nat. Acad. Sci. 86, 5974-5978 (1989); Poll et al., Proc. Nat. Acad. Sci. 87, 782-785 (1990); Monto et al., Blood 79, 2670 (1990); Clouse et al., Immunol. 142, 421-438 (1989); Poll et al., AIDS Res. Hum. Retrovirus, 191-197 (1992)] . СПИД является результатом инфицирования Т-лимфоцитов вирусом иммунодефицита человека (ВИЧ). Идентифицировано по меньшей мере три типа или штамма ВИЧ, то есть ВИЧ-1, ВИЧ-2 и ВИЧ-3. Как следствие ВИЧ-инфекции, Т-клетки, ответственные за иммунитет, ослаблены, и на этом фоне у инфицированных ВИЧ больных наблюдаются оппортунистические инфекции и/или необычные новообразования. Проникновение ВИЧ в Т-лимфоциты требует активации Т-лимфоцитов. Прочие вирусы, такие как ВИЧ-1, ВИЧ-2, инфицируют Т-лимфоциты после активации Т-клеток, и экспрессия и/или репликация таких вирусных белков опосредуется или поддерживается такой активацией Т-клеток. Сразу после активации Т-лимфоциты инфицируются ВИЧ, причем эти Т-лимфоциты должны продолжать оставаться в активированном состоянии для экспрессии гена ВИЧ и/или репликации ВИЧ. Цитокины, особенно ФНОα, вовлечены в экспрессию ВИЧ-белка, опосредованную активированными Т-клетками, и/или репликацию вируса, принимая участие в процессе поддержания активации Т-лимфоцитов. Следовательно, вмешательство в активность цитокинов, такое как предотвращение или ингибирование продуцирования цитокинов, особенно ФНОα, у ВИЧ-инфицированных индивидуумов способствует ограничению поддержания активации Т-лимфоцитов, вызванной ВИЧ-инфекцией.
Моноциты, макрофаги и родственные клетки, такие как клетки Купфера и глиальные клетки, также вовлечены в поддержание ВИЧ-инфекции. Эти клетки подобно Т-клеткам являются мишенями для вирусной репликации, и уровень вирусной репликации зависит от состояния активации клетки [Rosenberg et al., The Immunopathogenesis of HIV Infection, Advances in Immunology, 57 (1989)]. Как было показано, цитокины, такие как ФНОα, активируют репликацию ВИЧ в моноцитах и/или макрофагах [Poll et al., Proc. Natl. Acad. Sci., 87, 782-784 (1990)] , поэтому предотвращение или ингибирование продуцирования или активности цитокинов способствует ограничению продвижения ВИЧ к Т-клеткам. Дополнительные исследования позволили идентифицировать ФНОα как общий фактор активации ВИЧ in vitro и предложить ясный механизм воздействия через нуклеарный регуляторный белок, обнаруженный в цитоплазме клеток [Osborn, et al. , PNAS 86, 2336-2340]. Это доказательство предполагает, что сокращение синтеза ФНОα может иметь антивирусное воздействие при ВИЧ-инфекциях посредством уменьшения транскрипции и, таким образом, продуцирования вируса.
Вирусная репликация латентного ВИЧ при СПИДе в Т-клеточных и макрофаговых линиях может быть вызвана ФНОα [Folks et al., PNAS 86, 2365-2368 (1989)] . Молекулярный механизм для вирус-индуцирующей активности предполагает способность ФНОα активировать ген-регуляторный белок (NFkB), обнаруженный в цитоплазме клеток, который активирует ВИЧ-репликацию посредством связывания с последовательностью вирусного регуляторного гена (LTR (длинные повторы РНК на концах генома)) [Osborn et al. PNAS 86, 2336-2340 (1989)]. При кахексии, связанной со СПИДом, предполагается повышенный уровень ФНОα в сыворотке и высокий уровень спонтанного продуцирования ФНОα в моноцитах периферической крови пациентов [Wright et al., J. Immunol. 141 (1), 99-104 (1988)]. ФНОα, играя разные роли, вовлечен в другие вирусные инфекции, такие как вирус цитомегалии (ВЦМ), вирус гриппа, аденовирус и вирусы семейства герпеса по причинам, подобным тем, которые отмечены выше.
Ядерный фактор kB (NFkB) является плейотропным транскрипционным активатором [Lenardo, et al., Cell 1989, 58, 227-29]. NFkB в качестве транскрипционного активатора вовлечен во множество заболеваний и воспалительных состояний и, как полагают, участвует в регулировании уровня цитокинов, в том числе ФНОα, но не ограничиваясь им, а также является активатором транскрипции ВИЧ [Dbaibo, et al., J. Biol. Chem. 1993, 17762-66; Duh et al., Proc. Natl. Acad. Sci. 1989, 86, 5974-78; Bachelerie et al., Nature 1991, 350, 709-12; Boswas et al. J. Acquired Immune Deficiency Syndrome 1993, 6, 778-786; Suzuki et al. Biochem. And Biophys. Res. Comm. 1993, 193, 277-83; Suzuki et al., Biochem And Biophys. Res. Comm. 1992, 189, 1709-15; Suzuki et al., Biochem. Mol. Bio. Int. 1993, 31 (4), 693-700; Shakhov et al. Proc. Natl. Acad. Sci. USA 1990, 171, 35-47; и Staal et al., Proc. Natl. Acad. Sci. USA 1990, 87, 9943-47). Так, ингибирование связывания NFkB может регулировать транскрипцию гена(ов) цитокина и при посредстве этого модулирования и других механизмов может быть полезным для ингибирования множества болезненных состояний. Соединения по изобретению способны ингибировать действие NFkB в ядрах и поэтому являются полезными при лечении различных заболеваний, включая, но не ограничиваясь ими, ревматоидный артрит, ревматоидный спондилит, остеоартрит, другие артритические состояния, септический шок, сепсис, эндотоксический шок, заболевание "трансплантат против хозяина", истощение, болезнь Крона, язвенный колит, рассеянный склероз, системную красную волчанку, узловатая эритема при лепре, ВИЧ, СПИД и оппортунистические инфекции при СПИДе. Уровни ФНОα и NFkB находятся под влиянием петли взаимной обратной связи. Как отмечено выше, соединения по изобретению воздействуют на уровни как ФНОα, так и NFkB.
Многие клеточные функции опосредованы уровнями аденозин-3',5'-циклического монофосфата (цАМФ). Такие клеточные функции могут вносить вклад в воспалительные состояния и заболевания, включающие в себя астму, воспаление и другие состояния [Lowe and Cheng, Drugs of the Future, 17(9), 799-807, 1992] . Было показано, что повышение уровня цАМФ в воспалительных лейкоцитах ингибирует их активацию и последующее высвобождение воспалительных медиаторов, в том числе ФНОα и NFkB. Повышение уровня цАМФ приводит также к релаксации гладкой мышцы дыхательных путей.
Снижение уровня ФНОα и/или повышение цАМФ, следовательно, вносит значительный вклад в терапевтическую стратегию для лечения многих воспалительных, инфекционных, иммунологических или злокачественных заболеваний. Они включают в себя, не ограничиваясь ими, септический шок, сепсис, эндотоксический шок, гемодинамический шок и синдром сепсиса, постишемическую реперфузионную травму, малярию, микобактериальную инфекцию, менингит, псориаз, конгестивную сердечную недостаточность, фиброзное заболевание, кахексию, отторжение трансплантата, рак, аутоиммунное заболевание, оппортунистические инфекции при СПИДе, ревматоидный артрит, ревматоидный спондилит, остеоартрит, другие артритические состояния, болезнь Крона, язвенный колит, рассеянный склероз, системную красную волчанку, узловатую эритему при лепре, астму, радиационное поражение и гипероксическое альвеолярное повреждение. Ранее предпринятые усилия, направленные на подавление влияния ФНОα, варьируют в пределах от использования стероидов, таких как дексаметазон и преднизолон, до использования как поликлональных, так и моноклональных антител [Beutler et al., Science 234, 470-474 (1985); WO 92/11383].
Подробное описание изобретения
Настоящее изобретение основано на том, что некоторые классы неполипептидных соединений, более полное описание которых приведено в данном описании изобретения, снижают уровни ФНОα.
В частности, изобретение относится к соединениям формулы: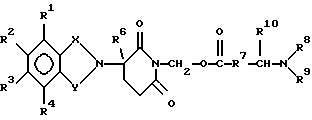
в которой
один из Х и Y представляет собой С=O, а другой из Х и Y представляет собой С=O или СН2;
(1) каждый из R1, R2, R3 и R4 независимо от других представляет собой галогено, алкил, содержащий от 1 до 4 атомов углерода, или алкоксигруппу, содержащую от 1 до 4 атомов углерода, или (2) один из R1, R2, R3 и R4 представляет собой -NHR5, а остальные из R1, R2, R3 и R4 являются водородом;
R5 представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода, или CO-R7-CH(R10)NR8R9;
R6 представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода, бензо, хлоро, или фторо;
R7 представляет собой м-фенилен или п-фенилен или -(СnН2n)-, где n имеет значение от 0 до 4;
каждый из R8 и R9, взятый независимо от другого, представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода, или R8 и R9, взятые вместе, представляют собой тетраметилен, пентаметилен, гексаметилен или -CH2CH2XCH2CH2-, где Х представляет собой -О-, -S- или -NH-;
R10 представляет собой водород, алкил, содержащий от 1 до 8 атомов углерода, или фенил; и
(б) солям присоединения кислоты указанных соединений, которые содержат атом азота, способный протонироваться.
Первую предпочтительную группу соединений составляют соединения формулы I, в которых по меньшей мере один из R1, R2, R3, R4 и R6 является иным чем водород. Среди этих соединений предпочтительной группой являются соединения, в которых каждый из R1, R2 R3 и R4 независимо от других представляет собой галогено, алкил, содержащий от 1 до 4 атомов углерода, или алкоксигруппу, содержащую от 1 до 4 атомов углерода; R6 является водородом, метилом, этилом или пропилом; каждый из R8 и R9, взятый независимо от другого, является водородом или метилом; и R10 является водородом. Из этих соединений одну предпочтительную подгруппу составляют соединения, в которых R7 представляет собой м-фенилен или п-фенилен, тогда как вторую предпочтительную подгруппу составляют соединения, в которых R7 представляет собой -(СnН2n)-, где n имеет значение от 0 до 4.
Следующую предпочтительную группу соединений составляют соединения формулы I, в которой один из R1, R2, R3 и R4 представляет собой -NH2, а остальные из R1, R2, R3 и R4 являются водородом; R6 является водородом, метилом, этилом или пропилом; каждый из R8 и R9, взятый независимо от другого, является водородом или метилом; и R10 является водородом. Из этих соединений, одну предпочтительную подгруппу составляют соединения, в которых R7 представляет собой м-фенилен или п-фенилен, тогда как вторую предпочтительную группу составляют соединения, в которых R7 представляет собой -(CnH2n)-, где n имеет значение от 0 до 4.
Термин "алкил" означает одновалентную насыщенную разветвленную или прямую углеводородную цепь, содержащую от 1 до 8 атомов углерода. Представителями таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Алкоксигруппа относится к алкильной группе, связанной с остатком молекулы через эфирный атом кислорода. Представителями таких алкоксигрупп являются метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втoр-бутокси и трет-бутокси. Предпочтительно, R1, R2, R3 и R4 представляют собой хлоро, фторо, метил или метоксигруппу.
Соединения формулы I используют под наблюдением квалифицированного персонала для ингибирования нежелательных воздействий ФНО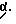 Соединения можно вводить перорально, ректально или парентерально, сами по себе или в комбинации с другими терапевтическими агентами, в том числе антибиотики, стероиды и т.д., млекопитающему, нуждающемуся в лечении.
Соединения можно вводить перорально, ректально или парентерально, сами по себе или в комбинации с другими терапевтическими агентами, в том числе антибиотики, стероиды и т.д., млекопитающему, нуждающемуся в лечении.
Соединения по изобретению также можно применять наружно при лечении или профилактике наружных болезненных состояний, опосредованных или обостренных избыточным продуцированием ФНОα, соответственно, таких как вирусные инфекции, такие как вирусные инфекции, вызванные вирусами герпеса, вирусный конъюнктивит, псориаз, атопический дерматит и т.п.
Соединения по изобретению можно использовать также в ветеринарии для лечения млекопитающих, иных чем люди, нуждающихся в предотвращении или ингибировании продуцирования ФНОα. Опосредованные ФНОα заболевания у животных, предназначенные для терапевтического или профилактического лечения, включают в себя болезненные состояния, такие как отмечено выше, но особенно вирусные инфекции. Примерами являются кошачий вирус иммунодефицита, вирус конской инфекционной анемии, вирус козлиного артрита, вирус висна и вирус маеди, а также другие лентивирусы.
Соединения по изобретению могут быть получены путем взаимодействия вначале формальдегида с промежуточным соединением формулы: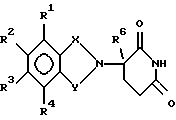
в которой Х и Y являются такими, как определено выше;
(1) каждый из R1, R2 R3 и R4 независимо от других представляет собой галогено, алкил, содержащий от 1 до 4 атомов углерода, или алкоксигруппу, содержащую от 1 до 4 атомов углерода, или (2) один из R1, R2 R3 и R4 представляет собой нитрогруппу или защищенную аминогруппу, а остальные из R1, R2, R3 и R4 являются водородом; и
R6 представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода, бензо, хлоро или фторо.
Полученное в результате N-гидроксиметильное промежуточное соединение формулы II затем подвергают реакции сочетания с производным карбоновой кислоты формулы IV с использованием общеизвестных способов:
где Hal представляет собой реактивный галоген, такой как хлоро, бромо или йодо.
Защитные группы, используемые здесь, означают группы, которые обычно не обнаруживают в конечных терапевтических соединениях, но которые намеренно вводят на какой-нибудь стадии синтеза, для того чтобы защитить группы, которые в противном случае могут видоизменяться в ходе химических превращений. Такие защитные группы удаляют на более поздних стадиях синтеза, и поэтому соединения, несущие такие защитные группы, имеют значение в первую очередь как химические промежуточные соединения (хотя некоторые производные также проявляют биологическую активность). Соответственно, точная структура защитной группы не является важной. Многочисленные реакции образования и удаления таких защитных групп описаны в ряде стандартных работ, в том числе, например, в "Protective Groups In Organic Chemistry", Plenum Press, London and New York, 1973; Green, Th. W. "Protective Groups in Organic Synthesis", Wiley, New York, 1981; "The Peptides", Vol. I, Schroder and Lubke, Academic Press, London and New York, 1965; "Methoden der organischen Chemie", Houben-Weyl. 4th Edition, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974), содержание которых включено в данное описание изобретения ссылкой.
Аминогруппа может быть защищена как амид с использованием селективно удаляемой в мягких условиях ацильной группы, особенно бензилоксикарбонильной, формильной или низшей алканоильной группы, которая является разветвленной в 1- или α-положении к карбонильной группе, в частности третичного алканоила, такого как пивалоил, низшей аканоильной группы, которая замещена в положении 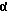 к карбонильной группе, как например трифторацетил.
к карбонильной группе, как например трифторацетил.
Связующие агенты включают в себя такие реагенты, как дициклогексилкарбодиимид и N,N'-карбонилдиимидазол.
После сочетания соединения формулы V могут быть аминированы традиционными способами, например амином в присутствии йодида натрия.
Альтернативно, соединению формулы III дают возможность взаимодействовать с защищенной аминокарбоновой кислотой формулы IV А: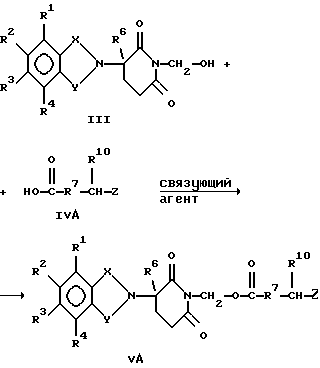
где Z является защищенной аминогруппой.
После этого сочетания защитную группу амина Z удаляют.
В упомянутых выше взаимодействиях, когда один из R1, R2, R3 и R4 представляет собой нитрогруппу, эту нитрогруппу можно превратить в аминогруппу каталитическим гидрированием. Альтернативно, если один из R1, R2, R3 и R4 является защищенной аминогруппой, защитная группа может быть расщеплена с получением соответствующего соединения, в котором один из R1, R2, R3 и R4 является аминогруппой.
В добавление к тому, что они служат в качестве промежуточных соединений, некоторые другие соединения формулы IIА сами являются биологически активными в отношении снижения уровня фактора некроза опухоли α у млекопитающего. К таким соединениям относятся соединения формулы: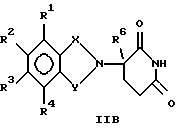
в которой один из Х и Y представляет собой С=O, а другой из Х и Y представляет собой С=O или СН2;
(1) каждый из R1, R2, R3 и R4 независимо от других представляет собой галогено, алкил, содержащий от 1 до 4 атомов углерода, или алкоксигруппу, содержащую от 1 до 4 атомов углерода, или (2) один из R1, R2, R3 и R4 представляет собой -NHR5, а остальные из R1, R2, R3 и R4 являются водородом;
R5 представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода, или CO-R7-CH(R10)NR8R9, где каждый из R7, R8 R9 и R10 является таким, как определено в этом документе; и
R6 представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода, бензо, хлоро или фторо.
Некоторые промежуточные соединения формулы IIА описаны в одновременно рассматриваемых заявках с серийными номерами 08/690258 и 08/701494, содержание которых включено в настоящее описание изобретения ссылкой. Кроме того, алкиловый эфир о-бромметилбензойной кислоты, который соответственно замещен заместителями R1, R2, R3 и R4 и которому дают возможность взаимодействовать с солью α-R6-замещенного α-аминоглутаримида в присутствии акцептора кислоты, такого как триэтиламин, с получением соединений, в которых один из Х и Y представляет собой С=O, а другой из Х и Y представляет собой СН2.
Соединения формулы IIA, в которых Х и Y оба представляют собой С=O, также могут быть получены взаимодействием фталевого ангидрида, который соответственно замещен R1, R2, R3 и R4, с солью α-R6-замещенного α-аминоглутаримида в присутствии уксусной кислоты и ацетата натрия.
Соль α-R6-замещенного α-аминоглутаримида, используемая в вышеупомянутых реакциях, может быть получена циклизацией α-R6-замещенного глутамина, аминогруппа которого защищена. Циклизацию можно проводить, например, N,N'-карбодиимидазолом в присутствии акцептора кислоты, такого как диметиламинопиридин. По окончании реакции защитная группа может быть удалена соответствующим способом. Исключительно в качестве примера: если защитной группой является N-бензилкарбонильная группа, то она может быть удалена каталитическим гидрированием.
В свою очередь, α-R6-замещенные глутамины могут быть получены обработкой ангидрида α-R6-замещенной глутаминовой кислоты, аминогруппа которой защищена аммиаком. Наконец, ангидрид α-R6-замещенной глутаминовой кислоты может быть получен из соответствующей α-R6 -замещенной глутаминовой кислоты с использованием уксусного ангидрида.
Соединения формул I и IIВ обладают центром хиральности и могут существовать в виде оптических изомеров. Как рацематы этих изомеров, так и сами индивидуальные изомеры, а также диастереомеры (при наличии двух хиральных центров) находятся в объеме настоящего изобретения. Рацематы можно использовать как таковые, или их можно разделить на их индивидуальные изомеры механически, например хроматографически с использованием хирального абсорбента. Альтернативно, индивидуальные изомеры могут быть получены в хиральной форме или выделены из смеси химическим путем посредством образования солей с хиральной кислотой, такой как индивидуальные энантиомеры 10-камфорсульфоновой кислоты, камфорной кислоты, α-бромокамфорной кислоты, метоксиуксусной кислоты, винной кислоты, диацетилвинной кислоты, яблочной кислоты, пирролидон-5-карбоновой кислоты и т.п., и последующего высвобождения одного или обоих разделенных оснований, возможно повторяя этот процесс, с тем чтобы получить либо один, либо оба, практически свободные от другого, то есть в форме, имеющей оптическую чистоту >95%.
Настоящее изобретение относится также к физиологически приемлемым нетоксичным солям присоединения кислот соединений формул I и IIВ. Такие соли включают в себя соли, производные от органических и неорганических кислот, таких как, без ограничения, соляная кислота, бромоводородная кислота, фосфорная кислота, серная кислота, метансульфоновая кислота, уксусная кислота, винная кислота, молочная кислота, янтарная кислота, лимонная кислота, яблочная кислота, малеиновая кислота, сорбиновая кислота, аконитовая кислота, салициловая кислота, фталевая кислота, эмбоновая кислота, энантовая кислота и т.п.
Пероральные лекарственные формы включают в себя таблетки, капсулы, драже и сходные формованные, прессованные фармацевтические формы, содержащие от 1 до 100 мг лекарственного средства на стандартную дозу. Для парентерального введения, которое включает в себя внутримышечный, внутриоболочечный, внутривенный и внутриартериальный пути введения, могут быть использованы изотонические физиологические растворы, содержащие от 20 до 100 мг/мл. Ректальное введение может быть осуществлено с использованием суппозиториев, в состав которых входят традиционные носители, такие как масло какао.
Так, фармацевтические композиции содержат одно или более чем одно соединение формул I-IIB, соединенное с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. При приготовлении таких композиций активные ингредиенты обычно смешивают с эксципиентом или разбавляют эксципиентом, либо вводят внутрь такого носителя, который может быть в форме капсулы или саше. Когда эксципиент служит в качестве разбавителя, он может быть твердым, полутвердым или жидким веществом, которое действует как растворитель, носитель или среда для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, эликсиров, суспензий, эмульсий, растворов, сиропов, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных упакованных порошков. Примеры приемлемых эксципиентов включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, аравийскую камедь, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Препараты могут дополнительно включать в себя смазочные агенты, такие как тальк, стеарат магния и минеральное масло, смачивающие агенты, эмульгирующие и суспендирующие агенты, консерванты, такие как метил- и пропилгидроксибензоаты, подсластители или корригенты.
Композиции предпочтительно готовят в виде препарата в стандартной лекарственной форме, подразумевающей физически дискретные единицы, приемлемые в качестве единой дозы или предопределенной доли единой дозы для введения в режиме однократной или многократной дозировки людям или другим млекопитающим, причем каждая единица содержит предопределенное количество активного вещества, рассчитанное на получение желаемого терапевтического эффекта, совместно с приемлемым фармацевтическим эксципиентом. Эти композиции могут быть приготовлены в виде препарата, который будет обеспечивать немедленное, пролонгированное или замедленное высвобождение активного ингредиента после введения пациенту, используя для этого методики, хорошо известные в данной области.
Пероральные лекарственные формы включают в себя таблетки, капсулы, драже и сходные формованные, прессованные фармацевтические формы, содержащие от 1 до 100 мг лекарственного средства на стандартную дозу. Для парентерального введения, которое включает в себя внутримышечный, внутриоболочечный, внутривенный и внутриартериальный пути введения, могут быть использованы изотонические физиологические растворы, содержащие от 20 до 100 мг/мл. Ректальное введение может быть осуществлено с использованием суппозиториев, в состав которых входят традиционные носители, такие как масло какао.
Так, фармацевтические композиции содержат одно или более чем одно соединение формулы I, соединенное с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. При приготовлении таких композиций активные ингредиенты обычно смешивают с эксципиентом или разбавляют эксципиентом, либо вводят внутрь такого носителя, который может быть в форме капсулы или саше. Когда эксципиент служит в качестве разбавителя, он может быть твердым, полутвердым или жидким веществом, которое действует как растворитель, носитель или среда для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, эликсиров, суспензий, эмульсий, растворов, сиропов, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных упакованных порошков. Примеры приемлемых эксципиентов включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, аравийскую камедь, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Препараты могут дополнительно включать в себя смазочные агенты, такие как тальк, стеарат магния и минеральное масло, смачивающие агенты, эмульгирующие и суспендирующие агенты, консерванты, такие как метил- и пропилгидроксибензоаты, подсластители или корригенты.
Композиции предпочтительно готовят в виде препарата в стандартной лекарственной форме, подразумевающей физически дискретные единицы, приемлемые в качестве единой дозы или предопределенной доли единой дозы для введения в режиме однократной или многократной дозировки людям или другим млекопитающим, причем каждая единица содержит предопределенное количество активного вещества, рассчитанное на получение желаемого терапевтического эффекта, совместно с приемлемым фармацевтическим эксципиентом. Эти композиции могут быть приготовлены в виде препарата, который будет обеспечивать немедленное, пролонгированное
или замедленное высвобождение активного ингредиента после введения пациенту, используя для этого методики, хорошо известные в данной области.
Нижеследующие примеры служат дальнейшему раскрытию сущности изобретения, но они не должны быть истолкованы как ограничение его объема, который определен исключительно прилагаемой формулой изобретения.
Пример 1
N-Бензилоксикарбонил-α-метил-глутаминовая кислота
К перемешиваемому раствору α-метил-D,L-глутаминовой кислоты (10 г, 62 ммоль) в 2н. гидроксиде натрия (62 мл) при 0-5oС добавляли бензилхлорформиат (12,7 г, 74,4 ммоль) в течение 30 мин. После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 3 ч. В течение этого времени поддерживали рН около 11 добавлением 2н. гидроксида натрия (33 мл). Затем реакционную смесь экстрагировали эфиром (60 мл). Водный слой охлаждали на ледяной бане, а затем подкисляли 4н. соляной кислотой (34 мл) до рН 1. Полученную в результате смесь экстрагировали этилацетатом (3•100 мл). Объединенные этилацетатные экстракты промывали рассолом (60 мл) и высушивали (МgSО4). Растворитель удаляли в вакууме с получением 15,2 г (83%) N-бензилоксикарбонил-α-метил-глутаминовой кислоты в виде масла: 1H ЯМР (СDСl3) δ 8.73 (m, 5Н), 5.77 (b, 1H), 5.09 (s, 2Н), 2.45-2.27 (m, 4H), 2.0 (s, 3Н).
Аналогичным образом из α-этил-D,L-глутаминовой кислоты и α-пропил-D,L-глутаминовой кислоты получены N-бензилоксикарбонил-α-этилглутаминовая кислота и N-бензилоксикарбонил-α-пропилглутаминовая кислота, соответственно.
Пример 2
N-Бензилоксикарбонил-α-метил-глутаминовый ангидрид
Перемешиваемую смесь N-бензилоксикарбонил-α-метил-глутаминовой кислоты (15 г, 51 ммоль) и уксусного ангидрида (65 мл) нагревали с обратным холодильником в атмосфере азота в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры, а затем концентрировали в вакууме с получением N-бензилоксикарбонил-α-метил-глутаминового ангидрида в виде масла (15,7 г), которое может быть использовано для последующей реакции без дополнительной очистки:
1Н ЯМР (CDCl3) δ 7,44-7.26 (m, 5H), 5.32-5.30 (m, 2H), 5.11 (s, 1H), 2.69-2.61 (m, 2H), 2.40-2.30 (m, 2H), 1.68 (s, 3H).
Аналогичным образом из N-бензилоксикарбонил-α-этилглутаминовой кислоты и N-бензилоксикарбонил-α-пропилглутаминовой кислоты получены N-бензилоксикарбонил-α-этилглутаминовый ангидрид и N-бензилоксикарбонил-α-пропилглутаминовый ангидрид, соответственно.
Пример 3
N-Бензилоксикарбонил-α-амино-α-метилизоглутамин
Перемешиваемый раствор N-бензилоксикарбонил-α-метил-глутаминового ангидрида (14,2 г, 51,5 ммоль) в метиленхлориде (100 мл) охлаждали на ледяной бане. Газообразный аммиак барботировали через охлажденный раствор в течение 2 ч. Реакционную смесь перемешивали при комнатной температуре в течение 17 ч, а затем экстрагировали водой (2•50 мл). Объединенные водные экстракты охлаждали на ледяной бане и подкисляли 4н. соляной кислотой (32 мл) до рН 1. Полученную в результате смесь экстрагировали этилацетатом (3•80 мл). Объединенные этилацетатные экстракты промывали рассолом (60 мл), а затем сушили (МgSO4). Растворитель удаляли в вакууме с получением 11,5 г N-бензилоксикарбонил-α-амино-α-метилизоглутамина:
1Н ЯМР (СDСl3/ДМСО) δ 7,35 (m, 5H), 7,01 (s, 1H), 6,87 (s, 1H), 6.29 (s, 1H), 5,04 (s, 2H), 2.24-1.88 (m, 4H), 1.53 (s, 3H).
Аналогичным образом из N-бензилкарбонил-α-этилглутаминового ангидрида и N-бензилкарбонил-α-пропилглутаминового ангидрида получены N-бензилкарбонил-α-амино-α-этилизоглутамин и N-бензилоксикарбонил-α-амино-α-пропилизоглутамин, соответственно.
Пример 4
N-Бензилоксикарбонил-α-амино-α-метилглутаримид
Перемешиваемую смесь N-бензилоксикарбонил-α-метилизоглутамина (4,60 г, 15,6 ммоль), 1,1'-карбонилдиимидазола (2,80 г, 17,1 ммоль) и 4-диметиламинопиридина (0,05 г) в тетрагидрофуране (15 мл) нагревали с обратным холодильником в атмосфере азота в течение 17 ч. Затем реакционную смесь концентрировали в вакууме до масла. Это масло суспендировали в воде (50 мл) в течение 1 ч. Полученную в результате суспензию фильтровали, и твердое вещество промывали водой и высушивали на воздухе с получением 3,8 г неочищенного продукта в виде белого твердого вещества. Неочищенный продукт очищали флэш-хроматографией (метиленхлорид:этилацетат 8:2) с получением 2,3 г (50%) N-бензилоксикарбонил-α-амино-α-метилглутаримида в виде белого твердого вещества:
Тпл=150,5-152,5oС;
1Н ЯМР (CDCl3) δ 8.21 (s, 1H), 7.34 (s, 5H), 5.59 (s, 1H), 5.08 (s, 2H), 2.74-2,57 (m, 3H), 2.28-2.25 (m, 1H), 1.54 (s, 3H);
13C ЯМР(СDСl3) δ 174.06, 171.56, 154.68, 135.88, 128.06, 127.69, 127.65, 66.15, 54.79, 29.14, 28 70, 21.98;
ЖХВД (жидкостная хроматография высокого давления): колонка Waters Nova-Pak C18, 4 мкм, 3,9•150 мм, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1% Н3РO4 (водн.), 7,56 мин (100%).
Анализ: Вычислено для C14H16N2O4: С, 60.86; Н, 5.84; N, 10.14. Обнаружено: С, 60.88; Н,5.72; N, 10.07.
Аналогичным образом из N-бензилоксикарбонил-α-амино-α-этилизоглутамина и N-бензилоксикарбонил-α-амино-α-пропилизоглутамина получены N-бензилоксикарбонил-α-амино-α-этилглутаримид и N-бензилоксикарбонил-α-амино-α-пропилглутаримид, соответственно.
Пример 5
-α-Амино-α-метилглутаримида гидрохлорид
N-Бензилоксикарбонил-α-амино-α-метилглутаримид (2,3 г, 8,3 ммоль) растворяли в этаноле (200 мл) при осторожном нагревании, и полученному раствору давали возможность охладиться до комнатной температуре. К этому раствору добавляли 4н. соляной кислоты (3 мл) с последующим добавлением 10% Pd/C (0,4 г). Смесь гидрировали в аппарате Парра под давлением водорода 50 фунт./кв. дюйм (350 кПа) в течение 3 ч. К этой смеси добавляли воду для растворения продукта. Эту смесь фильтровали через слой целита, который промывали водой (50 мл). Фильтрат концентрировали в вакууме с получением твердого остатка. Твердое вещество суспендировали в этаноле (20 мл) в течение 30 мин. Суспензию фильтровали с получением 1,38 г (93%) α-амино-α-метилглутаримида гидрохлорида в виде белого твердого вещества:
1H ЯМР (ДМСО-d6) δ 11,25 (s, 1Н), 8.92 (s, 3Н), 2.84-2.51 (m, 2Н), 2.35-2.09 (m, 2Н), 1.53 (s, 3Н);
ЖХВД: колонка Waters Nova-Pak C18, 4 мкм, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1% Н3РO4 (водн.), 1,03 мин (94,6%).
Аналогичным образом из N-бензилоксикарбонил-α-амино-α-этилизоглутаримида и N-бензилоксикарбонил-α-амино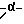 пропилизоглутаримида получены α-амино-α-этилглутаримида гидрохлорид и α-амино-α-пропилглутаримида гидрохлорид, соответственно.
пропилизоглутаримида получены α-амино-α-этилглутаримида гидрохлорид и α-амино-α-пропилглутаримида гидрохлорид, соответственно.
Пример 6
3-(3-Нитрофталимидо)-3-метилпиперидин-2,6-дион
Перемешиваемую смесь α-амино-α-метилглутаримида гидрохлорида (1,2 г, 6,7 ммоль), 3-нитрофталиевого ангидрида (1,3 г, 6,7 ммоль) и ацетата натрия (0,6 г, 7,4 ммоль) в уксусной кислоте (30 мл) нагревали с обратным холодильником в атмосфере азота в течение 6 ч. Затем смесь охлаждали и концентрировали в вакууме. Полученное в результате твердое вещество суспендировали в воде (30 мл) и метиленхлориде (30 мл) в течение 30 мин. Суспензию фильтровали, твердое вещество промывали метиленхлоридом и высушивали в вакууме (60oС, <1 мм) с получением 1,44 г (68%) 3-(3-нитрофталимидо)-3-метилпиперидин-2,6-диона в виде не совсем белого твердого вещества:
Тпл.=265-266,5oС;
1Н ЯМР (ДМСО-d6) δ 11.05 (s, 1H), 8.31 (dd, J=1.1 and 7.9 Гц, 1Н), 8.16-8.03 (m, 2H), 2.67-2.49 (m, 3H), 2.08-2.02 (m, 1H), 1.88 (s, 3H);
13C ЯМР (ДМСО-d6) δ 172,20, 171.71, 165.89, 165.30, 144.19, 136.43, 133.04, 128.49, 126.77, 122.25, 59.22, 28.87, 28.49, 21.04;
ЖХВД: колонка Waters Nova-Pak C18, 4 мкм, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1% Н3РО4 (водн.), 7,38 мин (98%).
Анализ: Вычислено для С14Н11N3О6: С, 53.00; Н, 3.49; N, 13.24. Обнаружено: С, 52.77; Н, 3.29; N, 13.00.
Аналогичным образом из α-амино-α-этилглутаримида гидрохлорида и α-амино-α-пропилглутаримида гидрохлорида получены 3-(3-нитрофталимидо)-3-этилпиперидин-2,6-дион и 3-(3-нитрофталимидо)-3-пропилпиперидин-2.6-дион, соответственно.
Пример 7
3-(3-Аминофталимидо)-3-метил-пиперидин-2,6-дион
3-(3-Нитрофталимидо)-3-метилпиперидин-2,6-дион (0,5 г, 1,57 ммоль) растворяли в ацетоне (250 мл) при осторожном нагревании, а затем охлаждали до комнатной температуры. К этому раствору добавляли 10% Pd/C (0,1 г) в атмосфере азота. Смесь гидрировали в аппарате Парра при давлении водорода 50 фунт. /кв. дюйм (350 кПа) в течение 4 ч. Эту смесь фильтровали через слой целита, который промывали ацетоном (50 мл). Фильтрат концентрировали в вакууме с получением желтого твердого вещества. Твердое вещество суспендировали в этилацетате (10 мл) в течение 30 мин. Затем суспензию фильтровали и высушивали (60oС, <1 мм) с получением 0,37 г (82%) 3-(3-аминофталимидо)-3-метилпиперидин-2,6-диона в виде желтого твердого вещества:
Тпл.=268-269oС;
1Н ЯМР (ДМСО-d6) δ 10.98 (s, 1Н), 7.44 (dd, J=7.1 и 7.3 Гц, 1Н), 6.99 (d, J=8.4 Гц, 1Н), 6.94 (d, J=6,9 Гц, 1Н), 6.52 (s, 2H), 2.71-2.47 (m, 3H), 2.08-1.99 (m, 1H), 1.87 (s, 3H);
13C ЯМР (ДМСО-d6) δ 172.48, 172.18, 169.51, 168.06, 146.55, 135.38, 131.80. 121.51, 110.56, 108.30, 58.29, 29.25, 28.63, 21,00;
ЖХВД: колонка Waters Nova-Pak C18, 4 мкм, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1% Н3РO4 (водн.), 5,62 мин (99,18%).
Анализ: Вычислено для С14Н13N3O4: С, 58.53; Н, 4.56; N, 14.63. Обнаружено: С, 58.60; Н, 4.41; N, 14.36.
Аналогичным образом из 3-(3-нитрофталимидо)-3-этилпиперидин-2,6-диона и 3-(3-нитрофталимидо)-3-пропилпиперидин-2,6-диона получены 3-(3-аминофталимидо)-3-этилпиперидин-2,6-дион и 3-(3-аминофталимидо)-3-пропилпиперидин-2,6-дион, соответственно.
Пример 8
Метиловый эфир 2-бромметил-3-нитробензойной кислоты
Перемешиваемую смесь метилового эфира 2-метил-3-нитробензойной кислоты (17,6 г, 87,1 ммоль) и N-бромсукцинимида (18,9 г, 105 ммоль) в четыреххлористом углероде (243 мл) осторожно нагревали с обратным холодильником с использованием лампы накаливания мощностью 100 Вт, размещенной на расстоянии 2 см над реакционной смесью в течение ночи. Через 18 ч реакционную смесь охлаждали и фильтровали. Фильтрат промывали водой (2•120 мл), рассолом (120 мл) и высушивали (MgSO4). Растворитель удаляли в вакууме с получением желтого твердого вещества. Продукт очищали флэш-хроматографией (гексан:этилацетат 8: 2) с получением 22 г (93%) метилового эфира 2-бромметил-3-нитробензойной кислоты в виде желтого твердого вещества:
Тпл.=69-72oС:
1Н ЯМР (СDСl3) δ 8.13-8.09 (dd, J=1.36 и 7.86 Гц, 1Н), 7.98-7.93 (dd, J= 1.32 и 8.13 Гц, 1Н), 7.57-7.51 (t, J=7.97 Гц, 1Н), 5.16 (s, 2H), 4.0 (s, 3Н);
13С ЯМР (СDСl3) δ 65.84, 150.56, 134.68, 132.64, 132.64, 132.36, 129.09, 53.05, 22.70;
ЖХВД: колонка Waters Nova-Pak C18, 4 мкм, 1 мл/мин, 240 нм, 40/60 СН3СN/0,1% Н3РO4 (водн.), 8,2 мин (99%).
Анализ: Вычислено для C9H8NO4Br: С, 39.44; Н, 2.94; N. 5.11; Вr, 29.15.
Обнаружено: С, 39.51; Н, 2.79; N, 5.02; Вr, 29.32.
Пример 9
3-(1-Oксо-4-нитроизоиндолин-1-ил)-3-метилпиперидин-2,6-дион
К перемешиваемой смеси α-амино-α-метилглутаримида гидрохлорида (2,5 г, 14,0 ммоль) и метилового эфира 2-бромметил-3-нитробензойной кислоты (3,87 г, 14,0 ммоль) в диметилформамиде (40 мл) добавляли триэтиламин (3,14 г, 30,8 ммоль). Полученную в результате смесь нагревали с обратным холодильником в атмосфере азота в течение 6 ч. Смесь охлаждали, а затем концентрировали в вакууме. Полученное в результате твердое вещество
суспендировали в воде (50 мл) и CH2Cl2 в течение 30 мин. Суспензию фильтровали, твердое вещество промывали метиленхлоридом и высушивали в вакууме (60oС, <1 мм) с получением 2,68 г (63%) 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-метилпиперидин-2,6-диона в виде не совсем белого твердого вещества:
Тпл.=233-235oС;
1Н ЯМР (ДМСО-d6) δ 10.95 (s, 1Н), 8.49-8.46 (d, J=8.15 Гц, 1Н), 8.13-8.09 (d, J=7.43 Гц, 1Н), 7.86-7.79 (t, J=7.83 Гц, 1H), 5.22-5.0 (dd, J=19.35 и 34.6 Гц, 2Н), 2.77-2.49 (m, 3H), 2.0-1.94 (m, 1H), 1.74 (s, 3H);
13C ЯМР (ДМСО-d6) δ 173.07, 172.27, 164.95, 143.15, 137.36, 135.19, 130.11, 129.32, 126.93, 57.57, 48.69, 28.9, 27.66, 20,6;
ЖХВД: колонка Waters Nova-Pak C18, 4 мкм, 1 мл/мин, 240 нм, 20/80 СН3СN/0, 1% Н3РO4 (водн.), 4,54 мин (99,6%).
Анализ: Вычислено для С14Н13N3О5: С, 55.45; Н, 4.32; N, 13.86. Обнаружено: С, 52.16; Н, 4.59; N, 12.47.
Заменяя α-амино-α-метилглутаримида гидрохлорид эквивалентными количествами α-амино-α-этилглутаримида гидрохлорида и α-амино-α-пропилглутаримида гидрохлорида, получают соответственно 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-этилпиперидин-2,6-дион и 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-пропилпиперидин-2,6-дион.
Пример 10
3-(1-Oксо-4-аминоизоиндолин-1-ил)-3-метилпиперидин-2,6-дион
3-(1-Оксо-4-нитроизоиндолин-1-ил)-3-метилпиперидин-2,6-дион (1,0 г, 3,3 ммоль) растворяли в метаноле (500 мл) при осторожном нагревании и давали возможность охладиться до комнатной температуры. К этому раствору добавляли 10% Pd/C (0,3 г) в атмосфере азота. Смесь гидрировали в аппарате Парра при давлении водорода 50 фунт. /кв. дюйм (350 кПа) в течение 4 ч. Эту смесь фильтровали через целит, и целит промывали метанолом (50 мл). Фильтрат концентрировали в вакууме с получением не совсем белого твердого вещества. Твердое вещество суспендировали в метиленхлориде (20 мл) в течение 30 мин. Затем суспензию фильтровали, и твердое вещество высушивали (60oС, <1 мм) с получением 0,54 г (60%) 3-(1-оксо-4-аминоизоиндолин-1-ил)-3-метилпиперидин-2,6-диона в виде белого твердого вещества:
Тпл.=268-270oС:
1H ЯМР (ДМСО-d6) δ 10.85 (s, 1Н), 7.19-7.13 (t, J=7.63 Гц, 1Н), 6,83-6.76 (m, 2H), 5.44 (s, 2H), 4.41 (s, 2H), 2.71-2.49 (m, 3H). 1.9-1.8 (m, 1H), 11.67 (s, 3H);
13С ЯМР (ДМСО-d6) δ 173.7, 172.49, 168.0, 143.5, 132.88, 128.78, 125.62, 116.12, 109.92, 56.98, 46.22, 29.04, 27.77, 20,82;
ЖХВД: колонка Waters Nova-Pak C18, 4 мкм, 1 мл/мин, 240 нм, 20/80 СН3СN/0, 1% Н3РO4 (водн.), 1,5 мин (99,6%).
Анализ: Вычислено для С14Н15N3О3: С, 61.53; H, 5.53; N, 15.38. Обнаружено: С, 58.99; H, 5,48; N, 14.29.
Из 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-этилпиперидин-2,6-диона и 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-пропилпиперидин-2,6-диона подобным образом получены 3-(1-оксо-4-аминоизоиндолин-1-ил)-3-этилпиперидин-2,6-дион и 3-(1-оксо-4-аминоизоиндолин-1-ил)-3-пропилпиперидин-2,6-дион, соответственно.
Пример 11
Таблетки, каждая из которых содержит 50 мг 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолина, могут быть приготовлены следующим способом:
Состав (на 1000 таблеток), г:
1-Oксо-2-(2,6-диоксо-3-метилпиперидин-3- ил)-4,5,6,7-тетрафторизоиндолин - 50,0
Лактоза - 50,7
Пшеничный крахмал - 7,5
Полиэтиленгликоль 6000 - 5,0
Тальк - 5,0
Стеарат магния - 1,8
Деминерализованная вода - Сколько требуется
Сначала твердые ингредиенты протирают через сито с размером отверстий 0,6 мм. Затем активный ингредиент, лактозу, тальк, стеарат магния и половину крахмала смешивают. Другую половину крахмала суспендируют в 40 мл воды, и эту суспензию добавляют в кипящий раствор полиэтиленгликоля в 100 мл воды. Полученную в результате пасту добавляют к порошкообразным веществам, и эту смесь гранулируют, с добавлением воды, если необходимо. Гранулят высушивают в течение ночи при 35oС, протирают через сито с размером отверстий 1,2 мм и прессуют с образованием таблеток приблизительно 6 мм в диаметре, которые являются двояковыпуклыми.
Пример 12
Таблетки, каждая из которых содержит 100 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолина, могут быть приготовлены следующим способом:
Состав (на 1000 таблеток), г:
1-Oксо-2-(2,6-диоксопиперидин-3-ил)-4- аминоизоиндолин - 100,0
Лактоза - 100,0
Пшеничный крахмал - 47
Стеарат магния - 3,0
Сначала все твердые ингредиенты протирают через сито с размером отверстий 0,6 мм. Затем активный ингредиент, лактозу, стеарат магния и половину крахмала смешивают. Другую половину крахмала суспендируют в 40 мл воды, и эту суспензию добавляют к 100 мл кипящей воды. Полученную в результате пасту добавляют к порошкообразным веществам, и эту смесь гранулируют, с добавлением воды, если необходимо. Гранулят высушивают в течение ночи при 35oС, протирают через сито с размером отверстий 1,2 мм и прессуют с образованием таблеток приблизительно 6 мм в диаметре, которые являются двояковыпуклыми.
Пример 13
Жевательные таблетки, каждая из которых содержит 75 мг 2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-аминофталимида, могут быть приготовлены следующим способом:
Состав (на 1000 таблеток), г:
2-(2,6-Диоксо-3-метилпиперидин-3-ил)-4- аминофталимид - 75,0
Маннит - 230,0
Лактоза - 150,0
Тальк - 21,0
Глицин - 12,5
Стеариновая кислота - 10,0
Сахарин - 1,5
5% раствор желатина - Сколько требуется
Сначала все твердые ингредиенты протирают через сито с размером отверстий 0,25 мм. Маннит и лактозу смешивают, гранулируют с добавлением раствора желатина, протирают через сито с размером отверстий 2 мм, сушат при 50oС и снова протирают через сито с размером отверстий 1,7 мм. 2-(2-Диоксо-3-метилпиперидин-3-ил)-4-аминофталимид, глицин и сахарин тщательно смешивают, добавляют гранулят маннита и лактозы, стеариновую кислоту и тальк, и все тщательно перемешивают и прессуют с образованием таблеток приблизительно 10 мм в диаметре, которые являются двояковыпуклыми и имеют разделительную канавку на верхней стороне.
Пример 14
Таблетки, каждая из которых содержит 10 мг 2-(2,6-диоксоэтилпиперидин-3-ил)-4-аминофталимида, могут быть приготовлены следующим способом:
Состав (на 1000 таблеток), г:
2-(2,6-Диоксоэтилпиперидин-3-ил)-4- аминофталимид - 10,0
Лактоза - 328,5
Кукурузный крахмал - 17,5
Полиэтиленгликоль 6000 - 5,0
Тальк - 25,0
Стеарат магния - 4,0
Деминерализованная вода - Сколько требуется
Сначала твердые ингредиенты протирают через сито с размером отверстий 0,6 мм. Затем активный имидный ингредиент, лактозу, тальк, стеарат магния и половину крахмала тщательно смешивают. Другую половину крахмала суспендируют в 65 мл воды, и эту суспензию добавляют к кипящему раствору полиэтиленгликоля в 260 мл воды. Полученную в результате пасту добавляют к порошкообразным веществам, и все смешивают и гранулируют, с добавлением воды, если необходимо. Гранулят высушивают в течение ночи при 35oС, протирают через сито с размером отверстий 1,2 мм и прессуют с образованием таблеток приблизительно 10 мм в диаметре, которые являются двояковыпуклыми и имеют разделительную канавку на верхней стороне.
Пример 15
Желатиновые капсулы, заполняемые сухим способом, каждая из которых содержит 100 мг 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолина, могут быть приготовлены следующим способом:
Состав (на 1000 капсул), г:
1-Oксо-2-(2,6-диоксо-3-метилпиперидин-3- ил)-4,5,6,7-тетрафторизоиндолин - 100,0
Микрокристаллическая целлюлоза - 30,0
Лаурилсульфат натрия - 2,0
Стеарат магния - 8,0
Лаурилсульфат натрия просеивают в 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин через сито с размером отверстий 0,2 мм, и эти два компонента тщательно смешивают в течение 10 мин. Затем микрокристаллическую целлюлозу добавляют через сито с размером отверстий 0,9 мм, и все опять тщательно смешивают в течение 10 мин. Наконец, стеарат магния добавляют через сито с размером отверстий 0,8 мм, и после перемешивания в течение дополнительных 3 мин смесь порциями по 140 мг каждая помещают в желатиновые капсулы для сухого способа заполнения размер 0 (удлиненные).
Пример 16
0,2% раствор для инъекций или инфузий может быть приготовлен, например, следующим способом:
1-Oксо-2-(2,6-диоксо-3-метилпиперидин-3- ил)-4,5,6,7-тетрафторизоиндолин - 5,0
Хлорид натрия, г - 22,5
Фосфатный буфер рН 7,4, г - 300,0
Деминерализованная вода, мл - До 2500,0
1-Oксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин растворяют в 1000 мл воды и фильтруют через микрофильтр. Добавляют буферный раствор и доводят объем водой до 2500 мл. Для приготовления форм единицы дозы порции от 1 до 2.5 мл каждая вносят в стеклянные ампулы (каждая содержит соответственно 2 или 5 мг имида).
Пример 17
3-Пентиламино-фталевая кислота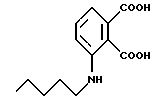
Диметиловый эфир 3-пентиламино-фталевой кислоты (4 ммоль) обрабатывали так же, как описано выше для синтеза 3-(3-хлор-бензиламино)фталевой кислоты. Продукт реакции, который содержал смесь дикислоты и монометиловых эфиров, использовали без дополнительной очистки.
Пример 18
2-(3-Метил-2,6-диоксо-пиперидин-3-ил)-4-пентиламино-изоиндол-1,3-дион
3-Пентиламино-фталевую кислоту (4 ммоль) обрабатывали так же, как описано выше для синтеза 4-(3-хлор-бензиламино)-2-(3-метил-2,6-диоксо-пиперидин-3-ил)-изоиндол-1,3-диона. Остаток суспендировали в диэтиловом эфире (30 мл) в течение ночи и фильтровали с получением 0,63 г (44%) продукта в виде желтого твердого вещества: т.пл. 96-98oС; 1Н ЯМР (ДМСО-d6) δ 11.00 (s, 1Н), 7.55 (t, J=7,8 Гц, 2H), 7.04 (d, J=8,5 Гц, 1Н), 6.94 (d, J=7,0 Гц, 1Н), 6.56 (t, J=5,5 Гц, 1Н), 3.26 (q, J=6,4 Гц, 2H), 2.79-2.48 (m, 3Н), 2.07-1.96 (m, 1H), 1.88 (s, 3Н), 1.61-1.57 (m, 2H), 1.35-1.32 (m, 4H), 0.87 (t, J=5,9 Гц, 3Н); 13С ЯМР (ДМСО-d6) δ 172.46, 172.17, 169.91, 167.98, 146.24, 136.18, 131.99, 116.89, 109.91, 108.72, 58.36, 41.77, 29.26, 28.63, 28.52, 28.35, 21.87, 20.97, 13.90.
Анализ: Вычислено для C19H23N3O4: С, 63.85; Н, 6.49; N, 11.76. Обнаружено: С, 63.63; Н, 6.27; N, 11.68.
Пример 19.
N-[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил]-2-хлорацетамид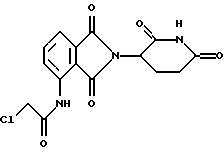
К суспензии 4-амино-2-(2,6-диоксо(3-пиперидил))изоиндолин-1,3-диона (1,37 г, 5 ммоль) в ТГФ (30 мл) добавляли хлорацетилхлорид (0,62 г, 5,5 ммоль). Смесь нагревали с обратным холодильником в течение 30 мин. Растворитель выпаривали в вакууме, и полученное твердое вещество суспендировали в диэтиловом эфире (20 мл) и фильтровали с получением 1,67 г (96%) продукта в виде не совсем белого вещества: 1Н ЯМР (ДМСО-d6) δ 11.18 (s, 1H), 10.31 (s, 1H), 8.54 (d, J=8,4 Гц, 1H), 7.88 (t, J=7,7 Гц, 1H), 7.68 (d, J=7,3 Гц, 1H), 5.17 (dd, J=5,2 и 12.7 Гц, 1H), 4.54 (s, 2Н), 2.90-2.85 (m, 1H), 2.65-2.51 (m, 2H), 2.10-2.06 (m, 1H).
Пример 20.
2-Азидо-N-[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил]-ацетамид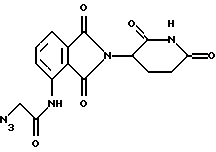
К суспензии N-[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил]-2-хлорацетамида (1,53 г, 4,4 ммоль) в ацетоне (30 мл) добавляли азид натрия (0,43 г, 6,6 ммоль). Смесь нагревали с обратным холодильником в течение 18 ч. Растворитель выпаривали в вакууме с получением 1,49 г (96%) продукта в виде не совсем белого твердого вещества, которое использовали без дополнительной очистки: 1Н ЯМР (ДМСО-d6)  11.19 (s, 1H), 10.20 (s, 1H), 8.49 (d, J= 8,3 Гц, 1H), 7.88 (t, J=7,7 Гц, 1H), 7.68 (d, J=7,3 Гц, 1H), 5.17 (dd, J= 5.1 и 12.7 Гц, 1H), 4.34 (s, 2Н), 2.99-2.84 (m, 1H), 2.65-2.47 (m, 2H), 2.09-2.00 (m, 1H).
11.19 (s, 1H), 10.20 (s, 1H), 8.49 (d, J= 8,3 Гц, 1H), 7.88 (t, J=7,7 Гц, 1H), 7.68 (d, J=7,3 Гц, 1H), 5.17 (dd, J= 5.1 и 12.7 Гц, 1H), 4.34 (s, 2Н), 2.99-2.84 (m, 1H), 2.65-2.47 (m, 2H), 2.09-2.00 (m, 1H).
Пример 21
2-Амино-N-[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил] -ацетамида гидрохлорид (СС-10020)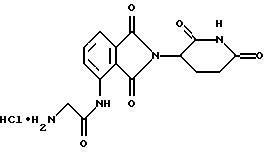
К раствору 2-азидо-N[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил] -2-ацетамида (1,49 г, 4,2 ммоль) в метаноле (50 мл) добавляли 10% Pd-C (0,1 г). Гидрирование при 50 фунт/кв.дюйм (~350 Па) приводило к кристаллизации продукта. Смесь фильтровали с получением в остатке серого твердого вещества, которое перемешивали в Н2О (50 мл). рН водной смеси доводили до 4 путем добавления 3н. HCl. Водную смесь фильтровали через целит для удаления катализатора, и фильтрат перемешивали с 50 мл этилацетата в течение 3 ч. Водный слой упаривали в вакууме с получением твердого вещества, которое суспендировали в этилацетате и фильтровали с получением 0,72 г (45%) продукта в виде не совсем белого твердого вещества. т.пл. 305-307oС; 1Н ЯМР (ДМСО-d6) δ 11.17 (s, 1H), 10.35 (s, 1H), 8.44 (bs, 3Н), 8.32 (d, J=8,2 Гц, 1H), 7.90 (t, J= 8,0 Гц, 1H), 7.71 (d, J=7,2 Гц, 1H), 5.16 (dd, J=5,1 и 12.6 Гц, 1H), 3.97 (s, 2H), 2.99-2.84 (m, 1H), 2.65-2.46 (m, 2H), 2.10-2.06 (m, 1H); 13С ЯМР (ДМСО-d6) 172.81, 169.80, 166.75, 166.56, 166.19, 136.19, 134.91, 131.89, 127.62, 119.47, 118.68, 48.96, 41.13, 30.94, 22.00.
Анализ: Вычислено для С15Н15СlN4О5: С, 48.62; Н, 4.19; N, 15.12. Обнаружено: С, 48.68; Н 4.18; N, 15.05+0,21 Н2О.
Пример 22
N-[2-(2,6-Диоксо-пиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]-2-метиламино-ацетамида гидрохлорид (СС-12048)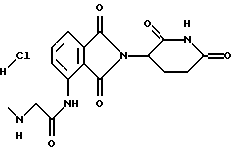
К суспензии N-[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил]-2-хлорацетамида (0,95 г, 2,72 ммоль) в ТГФ (30 мл) добавляли йодид натрия (0,41 г, 2,72 ммоль) и 2М метиламина в ТГФ (4,08 г, 8,15 ммоль). Смесь перемешивали при комнатной температуре в течение 5 ч. Растворитель выпаривали в вакууме с получением в остатке белого твердого вещества. Это твердое вещество суспендировали в этилацетате (200 мл) в течение 2 ч. После этого суспензию промывали водой (3•100 мл), рассолом (100 мл) и сушили (MgSO4). Растворитель выпаривали в вакууме с получением в остатке не совсем белого твердого вещества. Это твердое вещество растворяли в ацетонитриле (20 мл), и к этому раствору добавляли 2М HCl в эфире (2 мл). Смесь перемешивали в течение 1 ч, и растворитель выпаривали в вакууме. Остаток суспендировали в этилацетате в течение 3 ч и фильтровали с получением не совсем белого твердого вещества. Это твердое вещество растворяли в воде (40 мл) и экстрагировали этилацетатом (2•50 мл). рН водной части доводили до 11-12 путем добавления по каплям насыщенного водного карбоната натрия. Водную смесь промывали этилацетатом (3•100 мл). Объединенные этилацетатные экстракты промывали рассолом (100 мл) и сушили (MgSO4). Растворитель выпаривали в вакууме с получением белого твердого вещества. Это твердое вещество растворяли в ацетонитриле (15 мл), и 2М HCl в эфире (2 мл) добавляли к этому раствору. Смесь перемешивали в течение 1 ч, и растворитель выпаривали в вакууме с получением в остатке белого твердого вещества. Это твердое вещество суспендировали в диэтиловом эфире (20 мл) и фильтровали с получением 0,18 г (17%) продукта в виде белого твердого вещества: т.пл. 228-230oС; 1H ЯМР (ДМСО-d6) δ 11.13 (s, 1Н), 10.50 (s, 1H), 9.35 (bs, 2H), 8.28-8.21 (m, 1H), 7.93-7.63 (m, 2H), 5.15 (dd, J=5,1 и 12.6 Гц, 1H), 4.09 (s, 2H), 3.03-2.85 (m, 1H), 2.63-2.47 (m, 5H), 2.10-2.05 (m, 1H); 13C ЯМР (ДМСО-d6) δ 171.99, 169.03, 166.35, 166.07, 164.75, 135.63, 134.46, 131.56, 127.45, 119.14, 118.85, 49.56, 48.82, 32.49, 30.59, 21.69.
Анализ: Вычислено для C16H17ClN4O5: С, 48.99; Н, 4.70; N, 14.28. Обнаружено: С, 48.82; H, 4.72; N, 14.02 + 0.64 H2O.
Пример 23
2-Диметиламино-N-[2-(2,6-диоксо-пиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]-ацетамида гидрохлорид (СС-12057)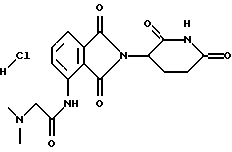
К раствору N-[2-(2,6-диоксо(3-пиперидил))-1,3-диоксоизоиндолин-4-ил]-2-хлорацетамида (0,97 г, 2,76 ммоль) в ТГФ (30 мл) добавляли йодид натрия (0,41 г, 2,76 ммоль) и 2М метиламин в ТГФ (4,14 мл, 8,28 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч. Смесь концентрировали в вакууме с получением маслянистого остатка. Этот остаток растворяли в этилацетате (300 мл) и промывали водой (3•100 мл), рассолом (100 мл) и сушили (MgSO4). Растворитель выпаривали в вакууме с получением в остатке не совсем белого твердого вещества. Это твердое вещество растворяли в ацетонитриле (50 мл), и к этому раствору добавляли 2М HCl в эфире (2 мл). Смесь перемешивали в течение 1 ч, и растворитель выпаривали в вакууме. Остаток суспендировали в этилацетате (35 мл) в течение 3 ч и фильтровали с получением не совсем белого твердого вещества. Это твердое вещество растворяли в воде (70 мл) и промывали этилацетатом (2•50 мл). рН водной части доводили до 11-12 путем добавления по каплям насыщенного водного карбоната натрия. Водную смесь экстрагировали этилацетатом (3•100 мл). Объединенные этилацетатные экстракты промывали рассолом (100 мл) и сушили (MgSO4). Растворитель выпаривали в вакууме с получением в остатке белого твердого вещества. Это твердое вещество растворяли в ацетонитриле (20 мл), и 2М HCl в эфире (2 мл) добавляли к этому раствору. Смесь перемешивали в течение 1 ч, и растворитель выпаривали в вакууме с получением в остатке белого твердого вещества. Это твердое вещество суспендировали в диэтиловом эфире (20 мл) и фильтровали с получением 0,61 г (63%) продукта в виде белого твердого вещества: т.пл. 253-255oС; 1H ЯМР (ДМСО-d6) δ 11.16 (s, 1Н), 10.88 (s, 1H), 10.53 (s, 1H), 8.15 (d, J=7,9 Гц, 1H), 7.91 (t, J=7.4 Гц, 1H), 7.75 (d, J=7.0 Гц, 1Н), 5.15 (dd, J=4.6 и 12.4 Гц, 1H), 4.30 (s, 2H), 2.90 (bs, 7H), 2.65-2.51 (m, 2H), 2.10-2.05 (m, 1H); 13С ЯМР (ДМСО-d6) δ 172.74, 169.72, 166.47, 166.30, 164.16, 136.01, 134.06, 131.93, 128.78, 120.27, 120.03, 57.65, 48.91, 43.14, 30.90, 21.94.
Анализ: Вычислено для C17H19ClN4O5: С, 53.93; Н, 4.95; N, 13.97. Обнаружено: С, 50.73; H,4.91; N, 13.73 + 0.34 Н2O.
Пример 24
N-[2-(2,6-диоксо(3-пиперидил))-1-оксоизоиндолин-4-ил]-хлорацетамид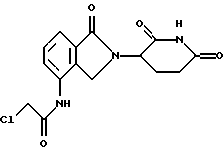
К раствору 3-(4-амино-1-оксоизоиндолин-2-ил)пиперидин-2,6-диона (3,89 г, 15 ммоль) в ТГФ (50 мл) добавляли хлорацетилхлорид (1,86 г, 16,5 ммоль). Смесь нагревали с обратным холодильником в течение 45 мин. К этой реакционной смеси добавляли дополнительное количество хлорацетилхлорида (0,15 г, 0,13 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 30 мин. Растворитель выпаривали в вакууме, и полученное твердое вещество суспендировали в диэтиловом эфире (20 мл) и фильтровали с получением 4,64 г (92%) продукта в виде не совсем белого твердого вещества: 1H ЯМР (ДМСО-d6) δ 11.04 (s, 1H), 10.22 (s, 1H), 7.82 (dd, J=1.6 и 7,2 Гц, 1H), 7.59-7.50 (m, 2H), 5.16 (dd, J=5,1 и 13.2 Гц, 1H), 4.46-4.30 (m, 4H), 3.00-2.85 (m, 1 H), 2.65-2.58 (m, 1H), 2.44-2.28 (m, 1H), 2.06-2.01 (m, 1H).
Пример 25
2-Азидо-N-[2-(2,6-диоксо-(3-пиперидил))-1-оксоизоиндолин-4-ил]-ацетамид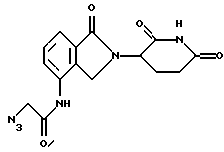
К суспензии N-[2-(2,6-диоксо(3-пиперидил))-1 -оксоизоиндолин-4-ил]-2-хлорацетамида (4,64 г, 13,8 ммоль) в ацетоне (60 мл) добавляли азид натрия (1,35 г, 20,7 ммоль). Смесь нагревали с обратным холодильником в течение 18 ч. К этой реакционной смеси добавляли NaI (2,05 г, 13,8 ммоль) и дополнительное количество азида натрия (0,90 г, 13,8 ммоль). Смесь нагревали с обратным холодильником в течение 18 ч. Растворитель выпаривали в вакууме с получением не совсем белого твердого вещества, которое суспендировали в смеси дихлорметан (50 мл)/Н2O (50 мл) и фильтровали с получением 4,39 г (93%) продукта: 1H ЯМР (ДМСО-d6) δ 11.50-9.52 (bs, 2H), 7.87-7.84 (m, 1H), 7.59-7.50 (m, 2H), 5.17 (dd, J=5.0 и 13.1 Гц, 1Н), 4.44 (d, J=17,6 Гц, 1H), 4.34 (d, J=17,6 Гц, 1H), 4.13 (s, 2H), 3.00-2.86 (m, 1H), 2.65-2.59 (m, 1H), 2.44-2.29 (m, 1H), 2.07-2.02 (m, 1H).
Пример 26
2-Амино-N-[2-(2,6-диоксо(3-пиперидил))-1-оксоизоиндолин-4-ил] -ацетамида гидрохлорид (СС-11028)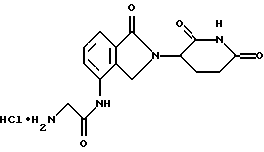
К раствору 2-азидо-N-[2-(2,6-диоксо(3-пиперидил))-1-оксоизоиндолин-4-ил] -2-ацетамида (1,49 г, 4,2 ммоль) в метаноле (50 мл) и 3н. HCl добавляли 10% Pd-C (0,1 г). Гидрирование при 50 фунт/кв.дюйм (~350 Па) приводило к кристаллизации продукта. Смесь фильтровали с получением в остатке серого твердого вещества, которое перемешивали в Н2О (100 мл). Водную смесь фильтровали через целит для удаления катализатора. Водный и метанольный фильтраты объединяли и упаривали с получением белого твердого вещества. Это твердое вещество суспендировали в этилацетате (20 мл), фильтровали, ресуспендировали в метаноле (20 мл) и фильтровали с получением 2,35 г (48%) продукта: т.пл. 293-295oС; 1H ЯМР (ДМСО-d6) δ 11.06 (s, 1H), 10.86 (s, 1H), 8.45 (bs, 2Н), 7.90 (d, J=6,3 Гц, 1H), 7.59-7.51 (m, 2H), 5.17 (dd, J=4,9 и 13.0 Гц, 1H), 4.54 (s, 2H), (d, J=17,8 Гц, 1H), 4.39 (d, J=17,8 Гц, 1H), 3.91 (s, 2H), 3.02-2.88 (m, 1H), 2.68-2.61 (m, 2H), 2.37-2.23 (m, 1H), 2.09-2.05 (m, 1H); 13С ЯМР (ДМСО-d6) δ 172.94, 171.07, 167.74, 165.24, 133.73, 132.86, 128.95, 125.01, 119.65, 51.59, 46.74, 40.90, 31.22, 22.82.
Анализ: Вычислено для C15H17ClN4O4: С, 50.43; Н, 4.94; N, 15.68. Обнаружено: С, 50.08; Н, 4.92; N, 15.53+0,25 Н2O.
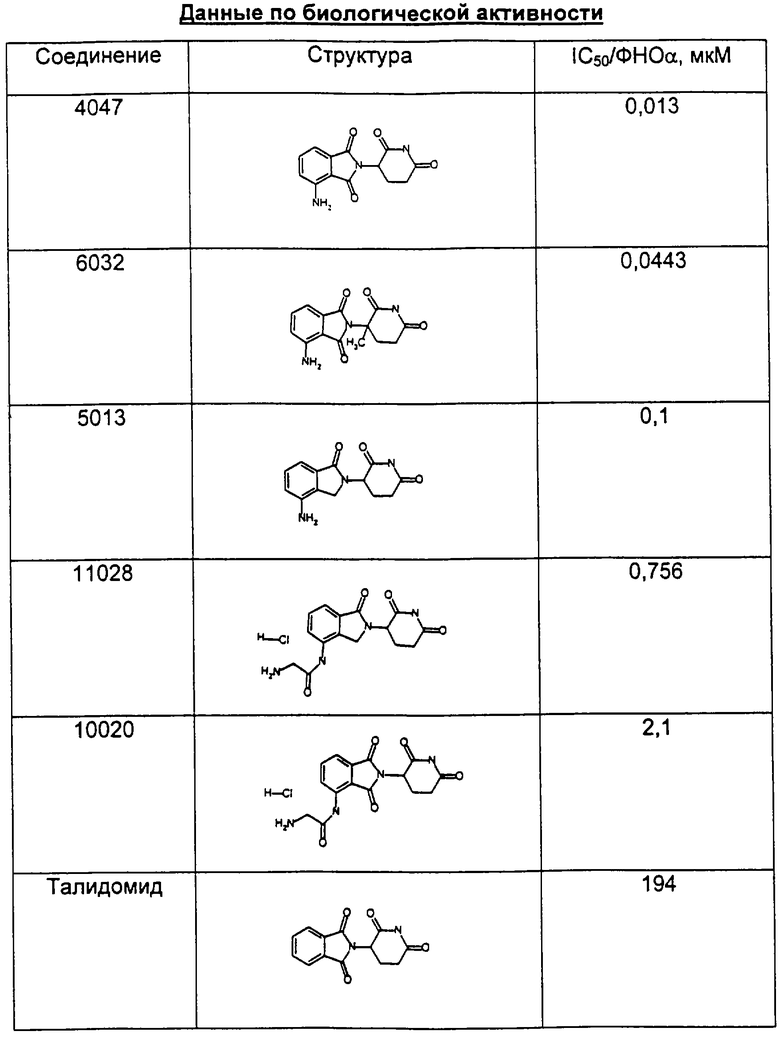
В таблице приведены данные по биологической активности.
Описывается 2,6-диоксопиперидин, состоящий из (а) соединения формулы (1), где один из Х и Y представляет собой С=О, а другой из Х и Y представляет собой С=О или СН2, один из R1, R2, R3 и R4 представляет собой -NHR5, а остальные являются водородом, R5 - водород, С1-8-алкил, СО-R7-СН(R10)NR8R9, R6 - C1-8-алкил, R7-(CnH2n), n=0, каждый из R8 и R9 независимо друг от друга водород, С1-8-алкил, R10 - водород, и (б) соли присоединения кислоты указанных соединений, которые содержат атом азота, способный протонироваться. Описываются также способ снижения нежелательных уровней ФHOα у млекопитающего путем введения эффективного количества указанного соединения и фармацевтическая композиция для снижения нежелательных уровней ФHOα у млекопитающего, содержащая указанное соединение в количестве, достаточном для снижения уровней ФHOα при введении в режиме однократной или многократной дозы, в комбинации с носителем. Технический результат - соединения снижают уровни ФHOα у млекопитающего. 3 с. и 1 з.п. ф-лы, 1 табл.
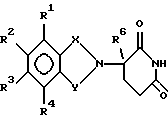
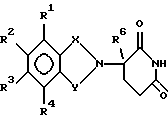
в которой один из Х и Y представляет собой С=O, а другой из Х и Y представляет собой С=O или CH2;
один из R1, R2, R3 и R4 представляет собой -NHR5, а остальные из R1, R2, R3 и R4 являются водородом;
R5 - водород, алкил, содержащий от 1 до 8 атомов углерода, или CO-R7-CH(R10)NR8R9;
R6 - алкил, содержащий от 1 до 8 атомов углерода;
R7 - (CnH2n)-, где n = 0;
каждый из R8 и R9, взятый независимо от другого, представляет собой водород или алкил, содержащий от 1 до 8 атомов углерода;
R10 - водород,
и (б) соли присоединения кислоты указанных соединений, которые содержат атом азота, способный протонироваться.
| DE 4211812, 22.10.1992 | |||
| Устройство для распыливания и сжигания мазута | 1978 |
|
SU688771A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Экономайзер | 0 |
|
SU94A1 |
| RU 99103124 А, 27.01.2001. | |||

