Изобретение относится к замещенным пирролам формулы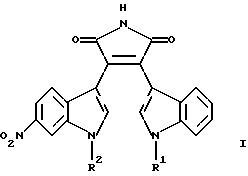
где R1 обозначает водород и R2 обозначает метил или
R1 обозначает метил и R2 обозначает водород, или
R1 обозначает гидроксиметил и R2 обозначает метил,
а также к их фармацевтически приемлемым пролекарствам или фармацевтически приемлемым солям.
Соединения формула I обладают антипролиферативной активностью, более конкретно они ингибируют деление клеток на G2/М-фазе клеточного цикла и в целом их называют ингибиторами "G2/М-фазы клеточного цикла".
Соединения формулы I подпадают под объем соединений, описываемых формулой I US Р 5057614, однако в этом документе они не выделены специально в виде группы или индивидуальных соединений. Кроме того, вышеуказанная активность соединений по настоящему изобретению нигде не описана или не является очевидной из описания US Р 5057614, таким образом, она является неожиданной.
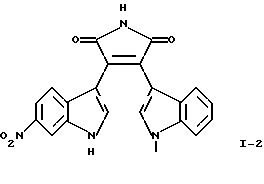
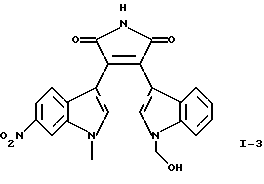
Приведенная выше формула I включает три следующие соединения: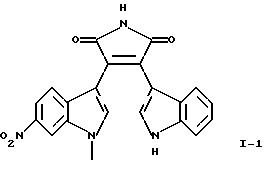
Понятие "фармацевтически приемлемые пролекарства" означает соединения, которые могут быть превращены в физиологических условиях или путем сольволиза в любое из соединений формулы I или в фармацевтически приемлемую соль этих соединений.
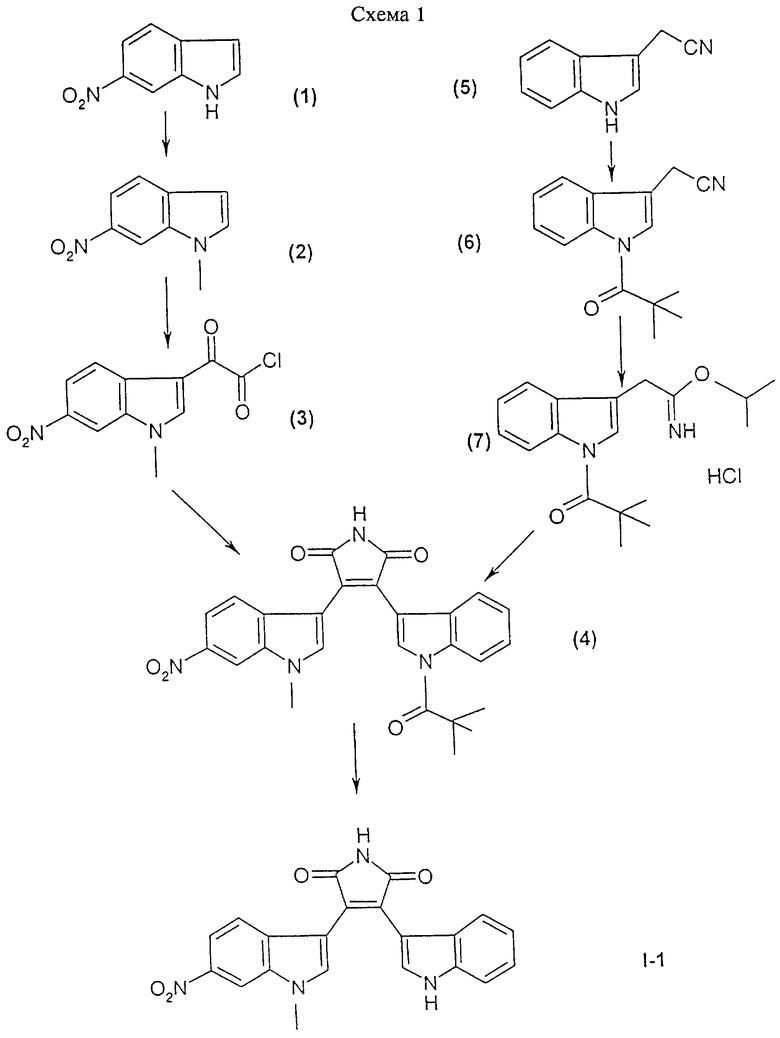
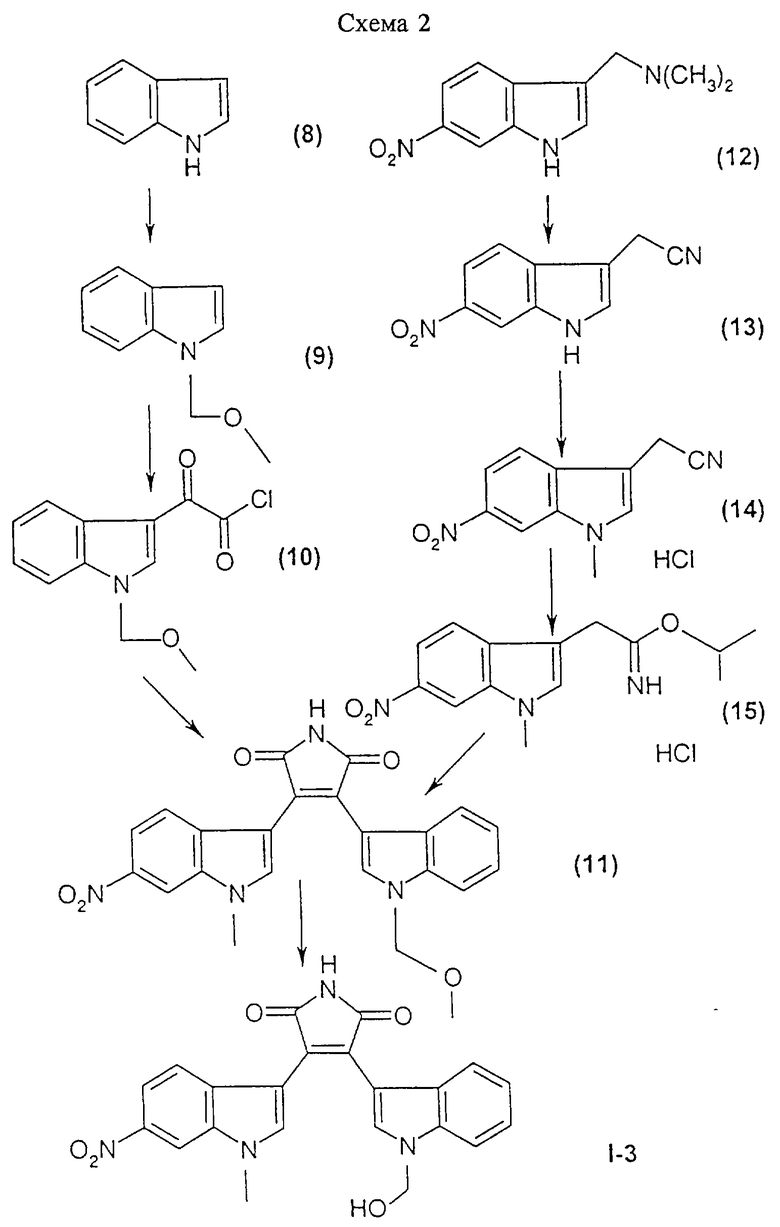
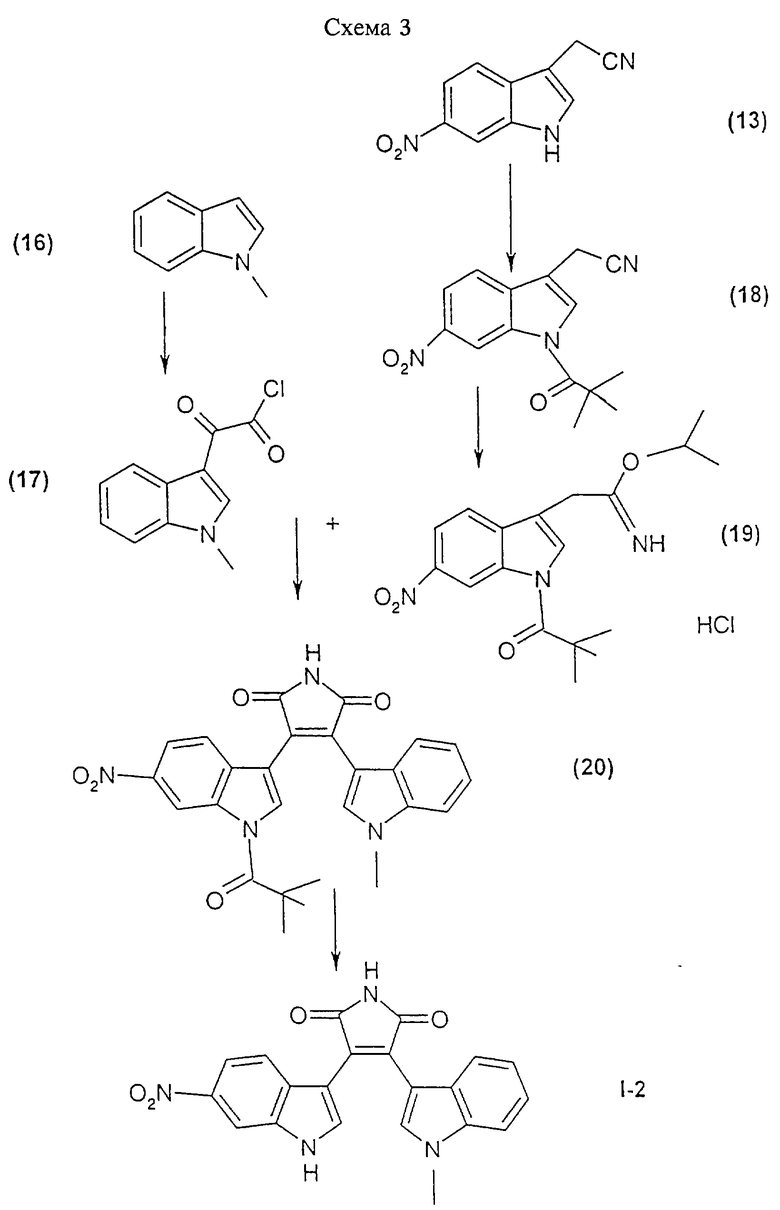
Соединения формулы I, а также фармацевтически приемлемые соли этих соединений получают с помощью реакций, представленных на приведенных схемах 1-3. Синтез каждого из этих соединений также описан в примерах 1-3.
Соединение I-1 может быть получено путем взаимодействия (1-метил-6-нитро-1Н-индол-3-ил)оксоацетилхлорида формулы (3) с гидрохлоридом 1-метилэтилового эфира [1-(2,2-диметилпропионил)-1Н-индол-3-ил]-3-этанимидиновой кислоты формулы (7) и обработки продукта реакции основанием.
Соединение I-2 может быть получено путем взаимодействия (1-метил-1Н-индол-3-ил)оксоацетилхлорида формулы (17) с гидрохлоридом 1-метилэтилового эфира [1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил] -3-этанимидиновой кислоты формулы (19) и обработки продукта реакции основанием.
Соединение I-3 может быть получено путем взаимодействия (1-метоксиметил-1Н-индол-3-ил)оксоацетилхлорида формулы (10) с гидрохлоридом 1-метилэтилового эфира (1-метил-6-нитро-1H-индол-3-ил)-3-этанимидиновой кислоты формулы (15) и обработки продукта реакции кислотой.
Антипролиферативная активность соединений по изобретению продемонстрирована ниже. Эти результаты свидетельствуют о том, что соединения могут применяться для лечения рака, прежде всего твердых опухолей.
Линию клеток карциномы эпителия молочной железы, негативную в отношении рецептора эстрогена (MDA-MB-435), получали из Американской коллекции типовых клеточных культур (АТСС; Роквилл, MD) и выращивали в среде, рекомендованной АТСС. Для анализа влияния тестируемых соединений на рост этих клеток клетки высевали в 96-луночный планшет для культур тканей ("тест-планшет") в количестве 2000 клеток на лунку и инкубировали в течение ночи при 37oС в атмосфере, содержащей 5% СО2. На следующий день тестируемые соединения растворяли в 100%-ном диметилсульфоксиде (ДМСО), получая 10 мМ маточный раствор. Раствор каждого соединения разбавляли стерильной дистиллированной водой до концентрации 1 мМ и затем добавляли в трех повторностях в лунки 96-луночного "планшета-хозяина" ("мастер-планшета"), содержащего среду в количестве, достаточном для получения конечной концентрации 40 мкМ. Соединения серийно разводили в среде в "мастер-планшетах". Одну четверть конечного объема разведенных соединений переносили с дублированием в "тест-планшеты". В ряд лунок с "контрольными клетками" добавляли ДМСО так, чтобы конечная концентрация ДМСО в каждой лунке была равна 0,1%. "Тест-планшеты" вновь помещали в термостат и через 3 дня после добавления тестируемого соединения один "тест-планшет" анализировали как описано ниже. Аналогично этому через 5 дней после добавления тестируемого соединения анализировали как описано ниже второй "тест-планшет".
В каждую лунку добавляли бромид 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-2Н-тетразолия (тиазолиловый синий; МТТ) до получения конечной концентрации 1 мг/мл. Затем планшет инкубировали при 37oС в течение 3 ч. Затем среду, содержащую МТТ, удаляли и в каждую лунку добавляли 50 мкл 100%-ного этанола для растворения образовавшихся метаболитов формазана. Для обеспечения полного растворения планшеты встряхивали в течение 15 мин при комнатной температуре. Абсорбцию регистрировали с помощью ридера для титрационных микропланшетов (фирма Molecular Dynamics) при длине волны 570 нм и эталонной длине волны 650 нм. Процент ингибирования вычисляли путем вычитания фона из результатов, полученных для каждой лунки, затем вычитания из 1,00 величины, полученной делением среднего коэффициента абсорбции, измеренного для каждого тест-варианта по трем повторностям, на средний коэффициент, измеренный для контролей. Ингибирующие концентрации (IC50 и IC90) определяли на основе аппроксимации по методу линейной регрессии зависимости логарифма концентрации от процента ингибирования.
Линию клеток аденокарциномы ободочной кишки SW480 и линию клеток карциномы ободочной кишки НСТ-116 также получали из АТСС и тестировали согласно тому же протоколу, который описан выше, со следующими модификациями. Клетки линии SW480 высевали в количестве 1000 клеток на лунку и анализировали через 6 дней после добавления тестируемого соединения. Клетки линии НСТ-116 высевали в количестве 750 клеток на лунку и анализировали через 4 дня после добавления тестируемого соединения. Для МТТ-анализа планшеты центрифугировали при 1000 об/мин в течение 5 мин перед отсасыванием среды, содержащей МТТ, и для растворения формазана использовали 100 мкл 100%-ного этанола.
Результаты описанных выше тестов in vitro приведены в таблицах I-III.
Для анализа влияния соединений на характеристики прохождения клеточного цикла клетки линии MDA-MB-435 (АТСС; Роквилл, MD) высевали из расчета 1х106 клеток/10 мл на чашки диаметром 10 см в среду для роста, имеющую следующий состав: среда RPMI 1640+10%-ная инактивированная нагреванием фетальная бычья сыворотка, 2 мМ L-глутамин и 50 ед./мл пенициллин-стрептомицина (все продукты от фирмы GIBCO/BRL, Гейтерсбург, MD). Клетки инкубировали в течение ночи при 37oС в атмосфере, содержащей 5% СО2. На следующий день в отдельные чашки добавляли по 10 мкл каждого из тестируемых соединений в 100%-ном растворе ДМСО, получая маточный раствор с 1/1000-кратной конечной концентрацией. Кроме того, в контрольные чашки добавляли 10 мкл 100%-ного ДМСО. Конечная концентрация ДМСО во всех чашках, включая контрольные, составляла 0,1%. Все планшеты вновь помещали в термостат.
После этого в различные моменты времени среду из каждого планшета переносили в центрифужные пробирки объемом 50 мл. Затем оставшийся на чашках слой клеток промывали 5 мл забуференного фосфатом физиологического раствора (ЗФР; фирма GIBCO/BRL). ЗФР удаляли и объединяли со средой, находящейся в соответствующей пробирке. Клетки обрабатывали трипсином в течение 5 мин при 37oС, раствор собирали и объединяли со средой и ЗФР, находящихся в соответствующих пробирках. Затем пробирки центрифугировали в течение 5 мин при 1200 об/мин. Клетки фиксировали путем удаления надосадочной жидкости и потряхивания пробирок для равномерного распределения дебриса и затем добавляли 5 мл холодного 70%-ного этанола при осторожном встряхивании. После этого клетки хранили при -20oС в течение периода времени >24 ч.
Содержащие клетки пробирки вынимали из холодильника и давали нагреться до комнатной температуры в течение 20-30 мин. Пробирки центрифугировали при 3000 об/мин в течение 5 мин. Надосадочную жидкость удаляли, дебрис промывали 5 мл ЗФР и пробирки центрифугировали как описано выше. Затем надосадочную жидкость удаляли и дебрис ресуспендировали в 0,5 мл ЗФР. После этого в каждую пробирку добавляли по 0,5 мл РНКазы А (1 мг/мл в ЗФР) и пробирки инкубировали при 37oС в течение 15 мин. В каждую пробирку добавляли по 100 мкл йодида пропидия (фирма Sigma, Сент-Луис, МО) (1 мг/мл в ЗФР) и затем пробирки инкубировали при комнатной температуре в течение 2-3 мин. Каждый образовавшийся раствор пропускали через пробирку с фильтовальной крышкой (фирма Becton Dickinson, Сан-Джозе, СА, 2235).
Оценки образцов проводили с помощью прибора типа FACSort (фирма Becton Dickinson) с использованием программы фирмы-производителя CellQUEST и результаты анализировали с помощью программного обеспечения фирмы-производителя ModFIT. В результате этих измерений получали оценку процента клеток, находящихся на следующих фазах развития: фаза G0/G1, фаза синтеза ДНК (S) и фаза G2/M.
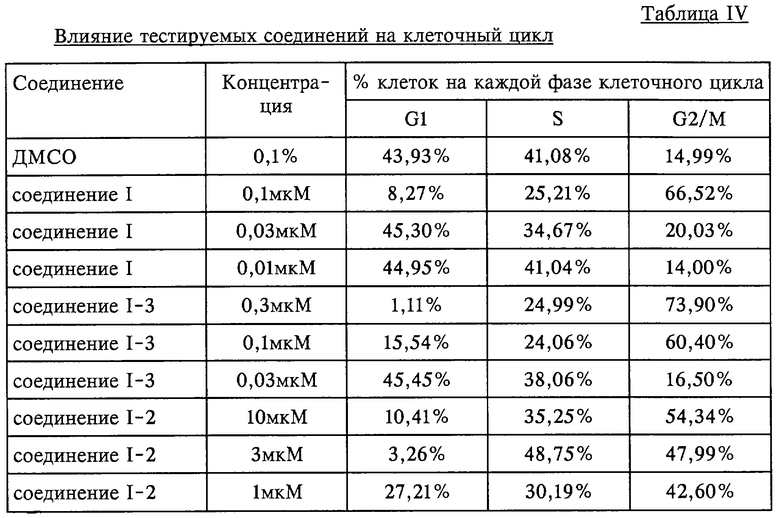
Результаты анализа эксперимента по изучению прохождения клеточного цикла в день 1 после добавления тестируемых соединений I-1, I-2 и I-3 обобщены в таблице IV.
Результаты, обобщенные таблицах I-IV, показывают, что соединения I-1, I-2 и I-3 обладают антипролиферативной активностью; в частности, они вызывают увеличение количества клеток, находящихся в G2/M-фазе клеточного цикла.
Пирролы формулы I и указанные выше их соли могут применяться в качестве лекарственных средств, например, в форме фармацевтических композиций, которые могут вводиться оральным путем, например, в форме таблеток, таблеток с покрытием, драже, желатиновых капсул с твердым или мягким покрытием, растворов, эмульсий или суспензий. Они также могут вводиться ректально, например, в форме суппозиториев или парентерально, например, в форме растворов для инъекций.
Для приготовления фармацевтических композиций эти соединения могут быть включены в препаративную форму вместе с терапевтически инертными неорганическими или органическими носителями. В качестве носителей для таблеток, таблеток с покрытием, драже, желатиновых капсул с твердым покрытием могут использоваться лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли. Пригодными носителями для желатиновых капсул с мягким покрытием являются растительные масла, воски, жиры, полутвердые или жидкие полиолы. Однако, как правило, в зависимости от природы действующего вещества в случае желатиновых капсул с мягким покрытием не требуется никакого носителя. Пригодными носителями для приготовления растворов и сиропов являются вода, полиолы, сахароза, инвертный сахар и глюкоза. Пригодными носителями в случае инъецируемых форм являются вода, спирты, полиолы, глицерин, растительные масла, фосфолипиды и поверхностно-активные вещества, пригодными носителями для суппозиториев являются природные или гидрогенизированные масла, воски, жиры и полутвердые полиолы.
Фармацевтические композиции также могут содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие агенты, эмульгаторы, подслащивающие вещества, красители, корригенты, соли для регулирования осмотического давления, буферы, агенты для нанесения покрытий или антиоксиданты. Они также могут содержать и другие ценные с терапевтической точки зрения вещества.
Как указывалось выше, пирролы формулы I и указанные выше их соли могут применяться для лечения или контроля онкологических болезней. Дозы могут варьироваться в широких пределах и, конечно, их следует регулировать в зависимости от индивидуальных требований в каждом конкретном случае. В целом в случае орального или парентерального введения взрослым людям весом приблизительно 70 кг пригодной может быть суточная доза приблизительно от 10 мг до приблизительно 10000 мг, предпочтительно от приблизительно 200 мг до приблизительно 5000 мг, более предпочтительно до приблизительно 1000 мг, хотя при соответствующих показаниях верхний предел может быть превышен. Суточная доза может вводиться в виде однократной дозы или в виде разделенных доз, или в случае парентерального введения она может вводиться в виде непрерывной инфузии.
Приведенные ниже примеры служат для иллюстрации изобретения.
Пример 1
Получение 3-(1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение I-1)
А. 1-Метил-6-нитро-1H-индол (соединение 2)
К суспензии, содержащей 0,33 г (8,3 ммоля) NaH (60%-ная дисперсия в масле) в 30 мл безводного диметилформамида (ДМФ), добавляли в течение 10 мин при 0-5oС 0,973 г (6,00 ммолей) имеющегося в продаже 6-нитро-1H-индола (соединение 1). После перемешивания при этой температуре в течение 1 ч добавляли 0,75 мл (12,1 ммоля) метилйодида и смесь перемешивали при этой же температуре в течение 30 мин, а затем в течение 1 ч при комнатной температуре, сливали на смесь лед/вода и экстрагировали этилацетатом. Органическую фазу промывали соляным раствором, сушили над MgSO4 и концентрировали, получая 0,814 г (77,5%) 1-метил-6-нитро-1H-индол (соединение 2) в виде твердого вещества желтого цвета. Этот продукт использовали без очистки.
Б. (1-Метил-6-нитро-1Н-индол-3-ил)оксоацетилхлорид (соединение 3)
К раствору, содержащему 1,33 г (7,55 ммоля) 1-метил-6-нитро-lH-индола (соединение 2) в 40 мл простого эфира, добавляли при 0-5oС в атмосфере аргона 1,5 мл (17,2 ммоля) оксалилхлорида. Образовывался осадок. После перемешивания в течение 3 ч образовавшийся твердый продукт фильтровали, промывали небольшим количеством простого эфира и сушили, получая 1,9 г (95%) (1-метил-6-нитро-1Н-индол-3-ил)оксоацетилхлорид (соединение 3) в виде твердого вещества желтого цвета. Этот продукт использовали без очистки.
В. [1-(2,2-Диметилпропионил)-1Н-индол-3-ил]ацетонитрил (соединение 6)
Используя метод, описанный выше в подразделе А, путем реакции N-алкилирования 10,2 г (65 ммолей) имеющегося в продаже (1H-индол-3-ил)ацетонитрила (соединение 5) с 8,7 мл (71 ммоль) триметилацетилхлорида и 3,4 г (85 ммолей) NaH (60%-ная дисперсия в масле) в качестве основания в 115 мл ДМФ получали после хроматографической очистки 6,6 г (38,7%) [1-(2,2-диметилпропионил)-1Н-индол-3-ил]ацетонитрила (соединение 6) в виде масла желтого цвета.
Г. Гидрохлорид 1-метилэтилового эфира [1-(2,2-диметилпропионил)-1Н-индол-3-ил]-3-этанимидовой кислоты (соединение 7)
К суспензии, содержащей 6,6 г (27,5 ммоля) полученного выше на стадии В [1-(2,2-диметилпропионил)-1Н-индол-3-ил]ацетонитрила (соединение 6) в 105 мл 2-пропанола, добавляли при 0-5oС в течение 20 минут по каплям 40 мл (0,563 моля) ацетилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, концентрировали и остаток разбавляли приблизительно 75 мл этилацетата, выдерживали в течение 15 мин в паровой бане и помещали в холодильник. Образовавшийся осадок фильтровали и сушили, получая 6,0 г (65,0%) гидрохлорида 1-метилэтилового эфира [1-(2,2-диметилпропионил)-1Н-индол-3-ил] -3-этанимидовой кислоты (соединение 7) в виде твердого вещества белого цвета.
Д. 3-[1-(2,2-Диметилпропионил)-1Н-индол-3-ил] -4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-дион (соединение 4)
К раствору, содержащему 1,25 г (4,69 ммоля) полученного выше на стадии Б (1-метил-6-нитро-1Н-индол-3-ил)оксоацетилхлорида (соединение 3) и 1,6 г (4,75 ммоля) полученного выше на стадии Г гидрохлорида 1-метилэтилового эфира [1-(2,2-диметилпропионил)-1Н-индол-3-ил] -3-этанимидовой кислоты (соединение 7) в 80 мл метиленхлорида, добавляли при 0oС в атмосфере аргона 2,6 мл (18,65 ммоля) триэтиламина. После перемешивания при этой температуре в течение 30 мин реакционную смесь перемешивали в течение 3,5 ч при комнатной температуре и разбавляли дополнительной порцией метиленхлорида. Органическую фазу промывали водой, 0,5 н. раствором НСl, соляным раствором, сушили над MgSO4 и концентрировали, получая 3,01 г пены. Этот продукт растворяли в 50 мл толуола и обрабатывали при 0oС 987,9 мг (5,19 ммоля) пара-толуолсульфоновой кислоты. После перемешивания в течение 3 ч при комнатной температуре реакционную смесь экстрагировали метиленхлоридом. Органическую фазу промывали насыщенным раствором NаНСО3, соляным раствором, сушили над MgSO4 и концентрировали, получая 3,9 г неочищенного продукта. После хроматографической очистки на колонке с силикагелем получали 1,7 г (77%) 3-[1-(2,2-диметилпропионил)-1Н-индол-3-ил] -4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение 4) в виде твердого вещества оранжевого цвета; tпл>146oC (разл.); МС: (М+), m/z 470.
Е. 3-(1Н-Индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-дион (соединение I-1)
1,7 г (3,61 ммоля) полученного выше на стадии Д 3-[1-(2,2-диметилпропионил)-1Н-индол-3-ил] -4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение 4) в 60 мл метанола обрабатывали 5,6 мл (8,96 ммоля) 1,6 М раствора NaOCH3 в метаноле. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, сливали на смесь 2 н. HCl/лед и экстрагировали этилацетатом. Органические экстракты сушили над безводным MgSO4 и концентрировали, получая после хроматографической очистки 394,7 мг (28%) 3-(1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение I-1) в виде твердого вещества красного цвета; tпл>280oC; МС: (М+), m/z 386.
Пример 2
Получение 3-(1-гидроксиметил-1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение I-3)
А. 1-Метоксиметил-1H-индол (соединение 9)
Используя метод, описанный выше в примере 1, стадия А, путем реакции N-алкилирования 1,17 г (10 ммолей) имеющегося в продаже индола (соединение 8) с 1 мл (13,1 ммоля) простого метилхлорметилового эфира и 0,48 г (12 ммолей) NaH (60%-ная дисперсия в масле) в качестве основания в 22 мл ДМФ получали после хроматографической очистки 1,4 г (86,9%) 1-метоксиметил-1Н-индола (соединение 9).
Б. (1-Метоксиметил-1Н-индол-3-ил)оксоацетилхлорид (соединение 10)
Используя метод, описанный выше в примере 1, стадия Б, путем взаимодействия 0,23 г (1,43 ммоля) полученного выше на стадии А 1-метоксиметил-1H-индола (соединение 9) с 0,25 мл (2,86 ммоля) оксалилхлорида в 3,5 мл простого эфира получали 0,174 г (48,5%) (1-метоксиметил-1Н-индол-3-ил)оксоацетилхлорида (соединение 10) в виде твердого вещества желтого цвета. Этот продукт использовали без очистки.
В. (6-Нитро-1Н-индол-3-ил)ацетонитрил (соединение 13)
К раствору, содержащему 44,27 г (0,204 моля) 6-нитрограмина (соединение 12) [Jackson В. Hester, J. Org. Chem., 29: 1158 (1964)] в 450 мл ацетонитрила, при перемешивании при 0-5oС в течение 1 ч добавляли 44,59 г (0,31 моля) метилйодида. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем добавляли в виде одной порции раствор, содержащий 26,6 г (0,543 моля) цианида натрия в 225 мл воды. Реакционную смесь выдерживали при 32oС в течение ночи, охлаждали до комнатной температуры и продукт экстрагировали трижды, используя всего 800 мл этилацетата и 300 мл воды. Объединенные экстракты промывали водой, 1 н. раствором НСl, насыщенным раствором бикарбоната натрия, сушили над MgSO4 и растворитель выпаривали в вакууме. Остаток оранжево-коричневого цвета (41,3 г) растворяли в 200 мл теплого этилацетата и пропускали через небольшую подушку из силикагеля, получая после выпаривания растворителя 28,9 г (70,4%) (6-нитро-1Н-индол-3-ил)ацетонитрила (соединение 13) в виде твердого вещества желтого цвета.
Г. (1-Метил-6-нитро-1Н-индол-3-ил)ацетонитрил (соединение 14)
К раствору, содержащему 28,9 г (0,143 моля) полученного выше на стадии В (6-нитро-1Н-индол-3-ил)ацетонитрила (соединение 13) в 230 мл диметилформамида, добавляли при комнатной температуре 65,5 г (0,474 моля) порошкообразного карбоната калия. Суспензию перемешивали в течение 40 мин, затем по каплям в течение 65 мин добавляли 25,48 г (0,179 моля) метилйодида. После перемешивания при комнатной температуре в течение ночи реакционную смесь охлаждали и сливали на общее количество 600 мл воды. Осадок фильтровали, промывали небольшим количеством воды и сушили над фосфорным ангидридом до достижения постоянной массы. В результате получали 30,4 г (95,4%) (1-метил-6-нитро-1Н-индол-3-ил)ацетонитрила (соединение 14), который использовали без дальнейшей очистки.
Д. Гидрохлорид 1-метилэтилового эфира (1-метил-6-нитро-1Н-индол-3-ил)-3-этанимидовой кислоты (соединение 15)
Суспензию, содержащую 82 г (0,382 моля) полученного выше на стадии Г (1-метил-6-нитро-1Н-индол-3-ил)ацетонитрила (соединение 14) в 1000 мл 2-пропанола, при 0-10oС при перемешивании барботировали струей газообразного НСl. После введения приблизительно 350 г НСl к реакционной смеси добавляли простой эфир до тех пор, пока не образовывался осадок. Твердый продукт собирали, промывали простым эфиром и сушили в вакууме, получая 102 г (85,7%) гидрохлорида 1-метилэтилового эфира (1-метил-6-нитро-1Н-индол-3-ил)-3-этанимидовой кислоты (соединение 15).
Е. 3-(1-Метоксиметил-1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-дион (11)
Используя метод, описанный выше в примере 1, стадия Д, путем реакции конденсации 1,3 г (5,17 ммоля) полученного выше на стадии Б оксоацетилхлорида (соединение 10) с 1,7 г (5,45 ммоля) полученного выше на стадии Д гидрохлорида 1-метилэтилового эфира (1-метил-6-нитро-1Н-индол-3-ил)-3-этанимидовой кислоты (соединение 15), в 95 мл метиленхлорида получали 1,08 г (48,5%) 3-(1-метоксиметил-1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение 11) в виде твердого вещества оранжевого цвета; tпл>250oC (разл.); МС: (М+), m/z 430.
Ж. 3-(1-Гидроксиметил-1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-дион (соединение I-3)
Раствор, содержащий 727,5 мг полученного выше на стадии Е 3-(1-метоксиметил-1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение 11) в 65 мл ТГФ, обрабатывали приблизительно 40 мл 2 н. НСl. Реакционную смесь кипятили с обратным холодильником в течение 5 ч, охлаждали и продукт экстрагировали этилацетатом. Органическую фазу сушили над MgSO4 и растворитель выпаривали, получая твердое вещество оранжевого цвета. После хроматографической очистки этого продукта получали 123,3 мг 3-(1-гидроксиметил-1Н-индол-3-ил)-4-(1-метил-6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение I-3) в виде твердого вещества красного цвета; tпл 210-213oС; МС: (М+), m/z 416.
Пример 3
Получение 3-(1-метил-1Н-индол-3-ил)-4-(6-нитро-1Н-индол-3-ил)пиррол-2,5-диона (соединение I-2)
А. (1-Метил-1Н-индол-3-ил)оксоацетилхлорид (соединение 17)
Используя метод, описанный выше в примере 1, стадия Б, путем взаимодействия 6 мл (47 ммолей) имеющегося в продаже 1 - метил-1Н-индола (соединение 16) с 8 мл (92 ммоля) оксалилхлорида в 120 мл простого эфира получали 7,6 г (73,2%) (1-метил-1Н-индол-3-ил)оксоацетилхлорида (соединение 17) в виде твердого вещества желтого цвета. Этот продукт использовали без очистки.
Б. [1-(2,2-Диметилпропионил)-6-нитро-1Н-индол-3-ил]ацетонитрил (соединение 18)
Используя метод, описанный выше в примере 1, стадия А, путем N-алкилирования 346,6 мг (1,72 ммоля) полученного в примере 2 на стадии В (6-нитро-1Н-индол-3-ил)ацетонитрила (соединение 13) с использованием 0,3 мл (2,44 ммоля) триметилацетилхлорида и 70,8 мг (1,77 ммоля) NaH (60%-ная дисперсия в масле) в качестве основания в 8 мл ДМФ получали после хроматографической очистки 287,7 мг (43,2%) [1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил] ацетонитрила (соединение 18) в виде масла желтого цвета.
В. Гидрохлорид 1-метилэтилового эфира [1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил]-3-этанимидовой кислоты (соединение 19)
Суспензию, содержащую 1,45 г (5,08 ммоля) полученного выше на стадии Б [1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил]ацетонитрила (соединение 18) в 90 мл 2-пропанола, барботировали при непрерывном перемешивании в течение 3 мин при 0-5oС струей газообразного НСl. Реакционную смесь перемешивали при комнатной температуре в течение 21 ч. Растворитель выпаривали в вакууме, получая 1,95 г (100%) соединения 19 в виде твердого вещества желтого цвета. Этот продукт использовали без очистки.
Г. 3-[1-(2,2-Диметилпропионил)-6-нитро-1Н-индол-3-ил] -4-(1-метил-1Н-индол-3-ил)пиррол-2,5-дион (соединение 20)
Используя метод, описанный выше в примере 1, стадия Г, 1,1 г (4,96 ммоля) полученного выше на стадии А оксоацетилхлорида (соединение 17) подвергали взаимодействию с 1,95 г (5,08 ммоля) полученного выше на стадии В гидрохлорида 1-метилэтилового эфира [1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил] -3-этанимидовой кислоты (соединение 19) и 2,1 мл (17,94 ммоля) триэтиламина в 120 мл метиленхлорида, образовавшийся продукт обрабатывали 1,1 г (5,78 ммоля) моногидрата пара-толуолсульфоновой кислоты в 80 мл толуола, в результате чего получали 1,3 г (62,1%) 3-[1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил] -4-(1-метил-1Н-индол-3-ил)пиррол-2,5-диона (соединение 20) в виде твердого вещества оранжевого цвета; tпл>245oC (разл. ); МС: (М+), m/z 470.
Д. 3-(1-Метил-1Н-индол-3-ил)-4-(6-нитро-1Н-индол-3-ил)пиррол-2.5-дион (соединение I-2)
Используя метод, описанный выше в примере 1, стадия Е, путем взаимодействия 1,3 г (2,76 ммоля) полученного выше на стадии Г 3-[1-(2,2-диметилпропионил)-6-нитро-1Н-индол-3-ил] -4-(1-метил-1Н-индол-3-ил)пиррол-2,5-диона (соединение 20) с 4,3 мл (6,88 ммоля) 1,6 М раствора NaOCH3 в 65 мл метанола для удаления N-защитных групп получали после кристаллизации из этилацетата и гексана 300,6 мг (28,1%) 3-(1-метил-1Н-индол-3-ил)-4-(6-нитро-1Н-индол-3-ил)пиррол-2,5-дионa (соединение I-2) в виде твердого вещества красного цвета; tпл>260oС; МС: (М+), m/z 386.
Пример 4
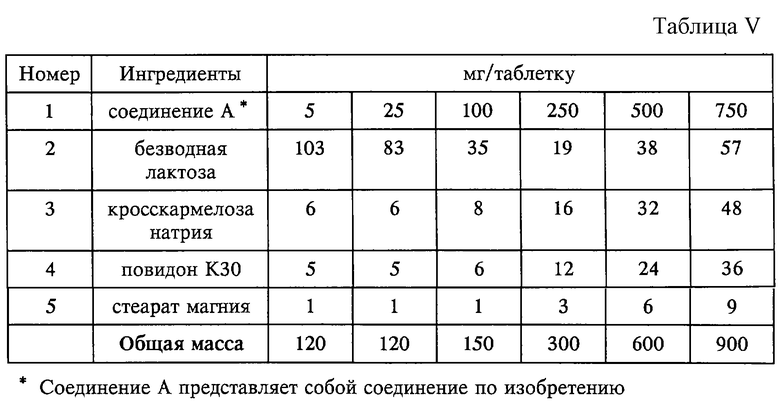
Композиция в форме таблетки (см. табл. V).
Способ приготовления:
1. Смешивают ингредиенты 1, 2 и 3 в пригодном смесителе в течение 15 мин.
2. Гранулируют полученную на стадии 1 порошкообразную смесь вместе с 20%-ным раствором повидона К30 (ингредиент 4).
3. Сушат полученный на стадии 2 гранулят при 50oС.
4. Пропускают полученный на стадии 3 гранулят через пригодное измельчающее устройство.
5. Добавляют ингредиент 5 к полученному на стадии 4 измельченному грануляту и смешивают в течение 3 минут.
6. Прессуют полученный на стадии 5 гранулят с помощью пригодного пресса.
Пример 5
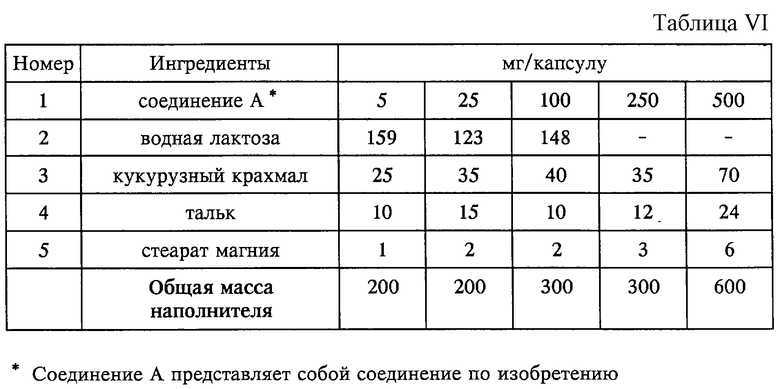
Композиция в форме капсулы (см. табл. VI).
Способ приготовления:
1. Смешивают ингредиенты 1, 2 и 3 в пригодном смесителе в течение 15 мин.
2. Добавляют ингредиенты 4 и 5 и смешивают в течение 3 мин.
3. Заполняют капсулу соответствующего размера.
Пример 6
Композиция в форме раствора/эмульсии для инъекции (см. табл. VII).
Способ приготовления:
1. Растворяют ингредиент 1 в ингредиенте 2.
2. Добавляют ингредиенты 3, 4 и 5 к ингредиенту 6 и смешивают до получения дисперсии, после чего осуществляют гомогенизацию.
3. Добавляют полученный на стадии 1 раствор к полученной на стадии 2 смеси и осуществляют гомогенизацию до тех пор, пока дисперсия не станет прозрачной.
4. Стерилизуют фильтрацией через фильтр с размерами пор 0,2 мкм и заполняют пузырьки.
Пример 7
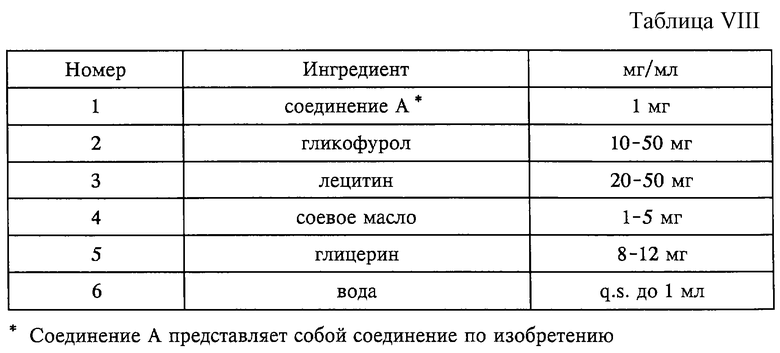
Композиция в форме раствора/эмульсии для инъекции (см. табл. VIII).
Способ приготовления:
1. Растворяют ингредиент 1 в ингредиенте 2.
2. Добавляют ингредиенты 3, 4 и 5 к ингредиенту 6 до получения дисперсии, после чего осуществляют гомогенизацию.
3. Добавляют полученный на стадии 1 раствор к полученной на стадии 2 смеси и осуществляют гомогенизацию до тех пор, пока дисперсия не станет прозрачной.
4. Стерилизуют фильтрацией через фильтр с размерами пор 0,2 мкм и заполняют пузырьки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ | 2000 |
|
RU2261862C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ | 1994 |
|
RU2141960C1 |
| ЗАМЕЩЕННЫЕ БИСИНДОЛИЛИМИДЫ МАЛЕИНОВОЙ КИСЛОТЫ ДЛЯ ИНГИБИРОВАНИЯ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ | 2000 |
|
RU2264398C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛО/4,3-А/ /1,4/ДИАЗЕПИНОВ | 1988 |
|
RU2071962C1 |
| НОВЫЕ ПИРРОЛОКАРБАЗОЛЫ | 1995 |
|
RU2162089C2 |
| НОВЫЕ АЗАИНДОЛТИАЗОЛИНОНЫ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2005 |
|
RU2391342C2 |
| ИМИДАЗОДИАЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2139873C1 |
| Способ получения замещенных пирролов или их фармацевтически приемлемых солей | 1989 |
|
SU1799382A3 |
| ТРИЦИКЛИЧЕСКИЕ ДИКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145606C1 |
| ИМИДАЗО[1,5-а] ПИРИМИДО[5,4-d][1] БЕНЗАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА GABA A | 2002 |
|
RU2287531C2 |




Изобретение относится к новым замещенным бисиндолималеимида формулы (I), а также их фармацевтически приемлемым солям, где R1 обозначает водород и R2 обозначает метил или R1 обозначает метил и R2 обозначает водород, или R1 обозначает гидроксиметил и R2 обозначает метил. Фармацевтическая композиция, обладающая антипролиферативной активностью, включает фармацевтически приемлемый носитель и соединение формулы (I). Технический результат - получение нового соединения, предназначенного для ингибирования пролиферации клеток. 2 с. и 3 з.п.ф-лы, 8 табл.
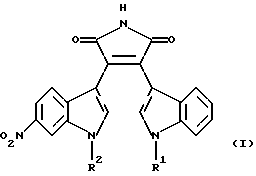
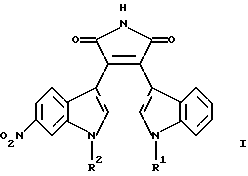
где R1 обозначает водород и R2 обозначает метил, или R1 обозначает метил и R2 обозначает водород, или R1 обозначает гидроксиметил и R2 обозначает метил,
а также их фармацевтически приемлемые соли.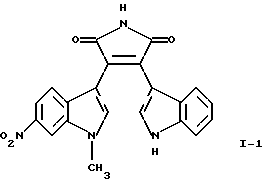
и его фармацевтически приемлемые соли.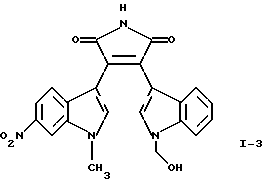
и его фармацевтически приемлемые соли.
| 0 |
|
SU328026A1 | |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| RU 94046306 А1, 27.10.1996. | |||
