Настоящее изобретение относится к новым замещенным пирролам общей формулы I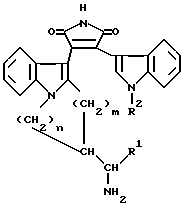
в которой R1 представляет собой низший алкил, низший циклоалкил, арил или низший аралкил, R2 представляет собой водород, арил или низший алкил, m и n равны 1 или 2, а также к фармацевтически приемлемым солям кислотных соединений формулы I с основаниями и основных соединений формулы I с кислотами и медицинским препаратам, например, фармацевтическим композициям на их основе.
В ЕП 0384349 А1, 040490 А1 описаны различные замещенные пирролы, обладающие действием ингибитора протеинкиназы.
Соединения формулы I и их вышеупомянутые соли, предлагаемые согласно настоящему изобретению, показывают более высокую активность по сравнению, например, с описанным аналогом - 3-[8-(аминометил)-6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил]- 4-(1-метил-3-индолил)-1H-пиррол-2,5-дион (соединение A), например, в опытах на противовоспалительную активность на животных.
Таким образом, изобретение относится к соединениям общей формулы I и их вышеупомянутым солям, как самим по себе, так и в качестве терапевтически активных веществ, а также к медицинским препаратам, обладающим действием протеинкиназы, в частности протеинкиназы C.
Вышеупомянутые соединения и их соли можно использовать для лечения или предотвращения заболеваний, в частности, для лечения или предотвращения воспалительных, иммунологических, онкологических, бронхолегочных, дерматологических и сердечно-сосудистых заболеваний, для лечения астмы, СПИДа или диабетических осложнений, или для стимуляции роста волос, или для производства медикаментов против воспалительных, иммунологических, онкологических, бронхолегочных, дерматологических и сердечно-сосудистых расстройств, или против астмы, СПИДа или диабетических осложнений, или для стимуляции роста волос.
В соответствии с настоящим изобретением радикал R2 представляет собой арил или низший алкил, может быть ацилокси, амино, моно(низший алкил)амином, ди(низший алкил)амином, карбокси, низший алкоксикарбонилом или аминокарбонилом. При этом термин "низший алкил" обозначает алкильную группу с линейной или разветвленной цепью, содержащей 1-6 атомов углерода, такую, как метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил и т.п. Термин "низший алкокси", сам по себе или в комбинации, обозначает алкильную группу, которая была определена выше, которая присоединена через атом кислорода; примерами алкоксигрупп являются метокси, этокси, пропокси, изопропокси, бутокси, трет-бутокси и т.п. Термин "низший циклоалкил" обозначает циклоалкильную группу, содержащую 3-6 атомов углерода, а именно, циклопропил, циклобутил, циклопентил и циклогексил. Термин "арил" обозначает незамещенный фенил или фенил, имеющий один или несколько заместителей, выбранных из, например, галогена, низшего алкила и низшего алкокси, такой как п-хлорфенил, п-толил и п-метоксифенил. Термин "низший аралкил" обозначает низшую алкильную группу, которая была определена выше, в которой один атом водорода был замещен арильной группой, которая была определена выше, такую, как бензил, 2-фенилэтил, п-хлорбензил, п-метилбензил и п-метоксибензил. Термин "ацилокси" обозначает ацилоксигруппу, полученную из алкановой кислоты, содержащую до 6 атомов углерода, например, ацетокси, пропионилокси или бутирилокси, или из ароматической карбоновой кислоты, которая может быть замещена, например, галогеном, низшим алкилом и/или низшим алкокси, например, бензоилокси, п-хлорбензоилокси, п-толуоилокси и п-метоксибензоилокси. Термин "галоген" обозначает фтор, хлор, бром или йод.
Соединения формулы I содержат два хиральных атома углерода и могут, таким образом, находиться в рацемической или оптически активной формах. Настоящее изобретение включает в свой объем не только рацемические соединения, но также оптически активные изомеры.
В соединениях формулы I R1 в предпочтительном варианте представляет собой низший алкил, в частности, низший алкил, содержащий 1-3 атома углерода. R2 в предпочтительном варианте представляет собой низший алкил, в частности, метил. В предпочтительном варианте m равно 1, а n равно 2.
Особенно предпочтительными соединениями формулы I являются следующие:
3-[8(S)-[1(R или S)-аминопропил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион; и
3-[8(S)-[1(S)-амино-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион.
Другими предпочтительными соединениями формулы I являются:
3-[8(R или S)-1(R или S)-аминоэтил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион,
3-[8(R или S)-1(R или S)-аминопропил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион,
3-[8(R или S)-1(R или S)-аминобутил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион, и
3-[8(R или S)-1(R или S)-амино-2-метилпропил]-6,7,8,9- тетрагидропиридо[1,2-а]-индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион.
Ниже приведены также предпочтительные соединения, описываемые формулой I:
3-[8(S)-1(R)-амино-2-метилпропил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион,
3-[8(R или S)- [альфа(R или S)-аминобензил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион,
3-[8(S)- [(R или S)-(амино)(циклопентил)метил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион,
3-[2(R или S)-[1(R или S)-амино-2-метилпропил]-2,3-дигидро- 1H-пирроло[1,2-а]индол-9-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5- дион,
3-[8(RS)-[1(RS)-амино-2-метилпропил] -7,8,9,10-тетрагидро-6H- азепино[1,2-а]индол-11-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5- дион,
3-[7(RS)-[1(RS)-амино-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-9-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион, и
3-[8(S)-[1(S)-амино-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-фенил-3-индолил)-1H- пиррол-2,5-дион.
Являющиеся предметом настоящего изобретения соединения формулы I, а также фармацевтически приемлемые соли кислотных соединений формулы I с основаниями и основных соединений формулы I с кислотами, получают отщеплением защитной группы R3 в соединении общей формулы II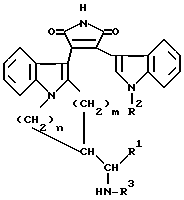
в которой R1, R2, m и n имеют значения, определенные выше, а R3 представляет собой уретанзащитную группу,
и, если необходимо, модифицируют химически активные заместители радикала R2 в соединении формулы I, полученном таким образом, и также, если необходимо, можно осуществить превращение кислотного соединения формулы I в фармацевтически приемлемую соль с основанием или превращение основного соединения формулы I в фармацевтически приемлемую соль с кислотой.
Уретанзащитной группой, обозначаемой R3 в общей формуле II является в предпочтительном варианте низший алкоксикарбонил, в частности, трет-бутоксикарбонил или низший алкоксикарбонил, в частности, бензилоксикарбонил.
Отщепление защитной группы R3 соединения формулы II может быть осуществлено с использованием известных в этой области техники приемов. Например, когда R3 представляет собой низший алкоксикарбонил, отщепление может быть осуществлено обработкой минеральной кислотой, такой, как хлористоводородная кислота, в инертном органическом растворителе, таком, как простой циклический эфир, например, тетрагидрофуран или диоксан, алканоле, например, метаноле или этаноле, сложных эфирах, таких, как этилацетат или галогенированные, в частности, хлорированные углеводороды, например, дихлорметан, или обработкой трифторуксусной кислотой. Когда R3 представляет собой аралкоксикарбонильную группу, отщепление осуществляют гидрогенолизом при помощи известных в этой области техники процедур; например, используя водород в присутствии катализатора, такого, как палладий/древесный уголь.
Функциональная модификация химически активного заместителя, содержащегося в R2 в полученном соединении формулы I, может включать этерификацию карбоксигруппы в низшую алкоксикарбонильную группу, гидролиз ацилоксигруппы в оксигруппу или превращение низшей алкоксикарбонильной группы в карбоксигруппу. Все эти модификации могут быть осуществлены в соответствии с приемами, которые известны в этой области техники.
Превращение кислотного соединения формулы I в фармацевтически приемлемую соль может быть осуществлено обработкой соответствующим основанием при помощи приемов, известных в этой области техники. Соответствующими солями являются соли, которые получают не только из неорганических оснований, таких, как, например, соли натрия, соли калия, соли кальция и т.п., но также и из органических оснований, например, этилендиамин, моноэтаноламин, диэтаноламин и другие подобные соли. Превращение основного соединения формулы I в фармацевтически приемлемую соль может быть осуществлено обработкой соответствующей кислотой при помощи процедур, которые известны в этой области техники. Соответствующими солями являются соли, которые получают не только из неорганических кислот, таких, как, например, хлоргидраты, бромгидраты, фосфаты, сульфаты и т. п., но также из органических кислот, например, ацетаты, цитраты, фумараты, тартраты, малеаты, метансульфонаты, п-толуолсульфонаты и т.п.
Исходные продукты формулы II, указанные выше, являются новыми и также составляют предмет настоящего изобретения. Они могут быть получены, например:
(a) взаимодействием соединения общей формулы III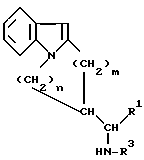
в которой R1, R3, m и n определены выше, с оксалилхлоридом, конденсацией полученного в результате активированного глиоксилата общей формулы IV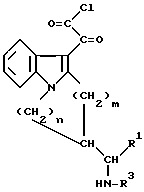
в которой R1, R3, m и n определены ранее, с имидатом общей формулы V
в которой R2 определен ранее, a R4 представляет собой низший алкил, в присутствии сильного основания, и с последующим гидролизом и дегидратацией полученного в результате оксипирролинона общей формулы VI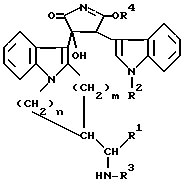
в которой R1, R2, R3, R4, m и n уже были определены ранее, или
(б) взаимодействием активированного глиоксилата формулы IV, приведенной выше, с индолилуксусной кислотой общей формулы VII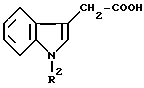
в которой R2 определен ранее,
в присутствии сильного основания и превращения полученного в результате замещенного фурандиона общей формулы VIII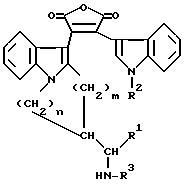
в которой R1, R2, R3, m и n определены ранее, в соответствующий имид формулы II.
Реакция соединения формулы III с оксалилхлоридом обычно осуществляется в присутствии инертного органического растворителя, например, галогенированного алифатического углеводорода, такого, как дихлорметан. Эту реакцию можно также осуществлять при температуре примерно 0oC.
Конденсацию активированного глиоксилата формулы IV с имидатом формулы V, который является известным соединением или аналогом известного соединения, обычно осуществляют в инертном органическом растворителе. Соответствующими основаниями являются, например, третичные амины, такие, как триэтиламин, диизопропилэтиламин, 4-диметиламинопиридин, N-этилморфолин и 1,4-диазабицикло[2.2.2] октан, а также пиридин. Соответствующими растворителями являются, например, галогенированные алифатические углеводороды, такие, как дихлорметан и хлороформ, необязательно галогенированные ароматические углеводороды, такие, как бензол, толуол и хлорбензол, простые циклические эфиры и простые нециклические эфиры, такие, как диметоксиэтан, простой трет-бутилметиловый эфир и тетрагидрофуран, формамиды, такие, как диметилформамид, сложные эфиры, такие, как этилацетат и нитрилы, такие, как ацетонитрил. Конденсацию в предпочтительном варианте осуществляют при температуре примерно от 0oC до 40oC, в частности, при комнатной температуре. Кроме того возможно эту конденсацию осуществлять in situ.
Гидролиз и дегидратацию оксипирролинона формулы VI с целью получения соединения формулы II обычно осуществляют обработкой минеральной кислотой, такой, как хлористоводородная кислота или серная кислота, или органической кислотой, такой, как метансульфоновая кислота или п-толуолсульфокислота, или обработкой ацилирующим реагентом, таким, как трифторуксусный ангидрид, и соответствующим основанием, таким, как пиридин, обычно при примерно комнатной температуре. Оксипирролинон формулы VI в предпочтительном варианте гидролизуют и подвергают дегидратации in situ.
Реакцию активированного глиоксилата формулы IV с индолилуксусной кислотой формулы VII, которая является известным соединением или аналогом известного соединения, обычно осуществляют аналогично приведенной выше конденсацией активированного глиоксилата формулы IV с имидатом формулы V.
Превращение замещенного фурандиона формулы VIII в исходный имид формулы II может быть обычно осуществлено обработкой гексаметилдисилазаном в присутствии алканола, такого, как метанол, в инертном органическом растворителе. Соответствующими растворителями являются, например, галогенированные алифатические углеводороды, такие, как дихлорметан и хлороформ, необязательно галогенированные ароматические углеводороды, такие, как бензол, толуол и хлорбензол, простые циклические эфиры и простые нециклические эфиры, такие, как диметоксиэтан, простой трет-бутилметиловый эфир и тетрагидрофуран, или формамиды, такие, как диметилформамид. Реакцию в предпочтительном варианте осуществляют в интервале примерно от комнатной температуры до 100oC, в частности, при температуре примерно 50oC.
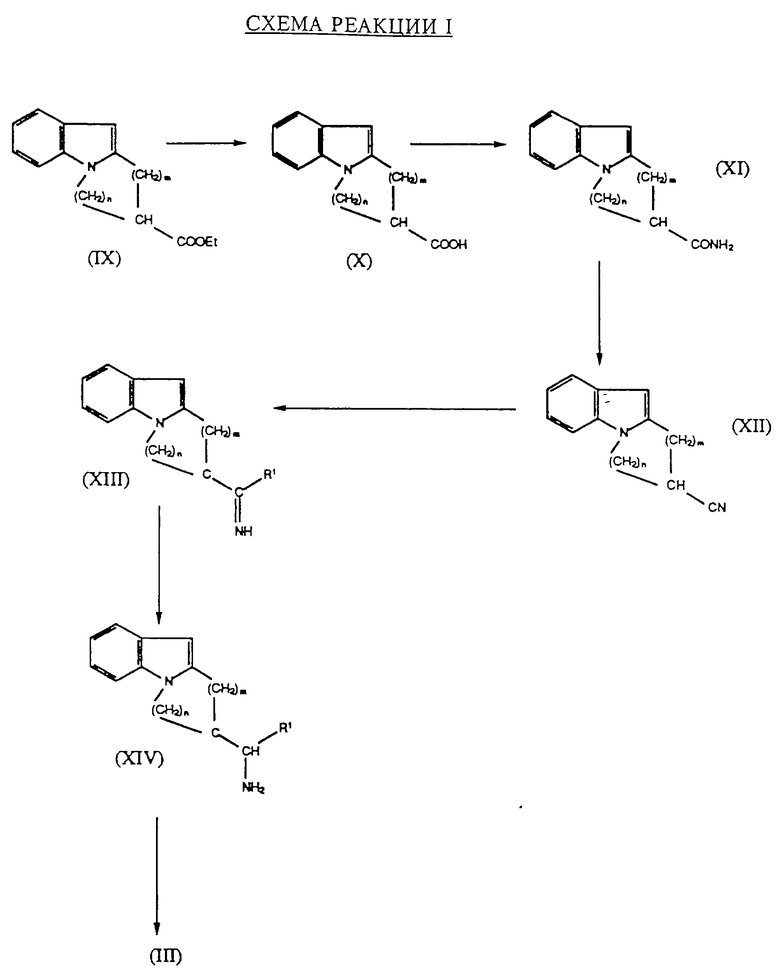
Соединения формулы III, приведенной выше, могут быть получены, например, как это показано на схеме реакции I, в которой R1, m и n определены ранее.
Приведенные на схеме I стадии могут быть осуществлены в соответствии с известными приемами. На первой стадии сложный этиловый эфир формулы IX, который является известным соединением или аналогом известного соединения, подвергают омылению в соответствующую кислоту формулы X, используя, например, раствор гидрата окиси натрия. Затем полученную в результате кислоту амидируют, например, взаимодействием с этилхлорформиатом в присутствии триэтиламина, затем обработкой аммиаком, полученный в результате амид формулы XI превращают в нитрил формулы XII, используя, например, трифторуксусный ангидрид. Затем нитрил формулы XII взаимодействует с реагентом Гриньяра формулы R1-Mg-X, где R1 определен ранее, а X представляет собой галоген, предпочтительно хлор, а полученный в результате имин формулы XIII восстанавливают, используя сложный гидрид металлов, например, алюмогидрид лития, в первичный амин формулы XIV. Такое восстановление предпочтительно осуществляют in situ. Первичный амин формулы XIV превращают в соединение формулы III, например, взаимодействием с хлорформиатом формулы R3Cl или ангидридом формулы R3OR3, в которой R3 имеет значение, которое было определено ранее, в присутствии основания, такого, как триэтиламин.
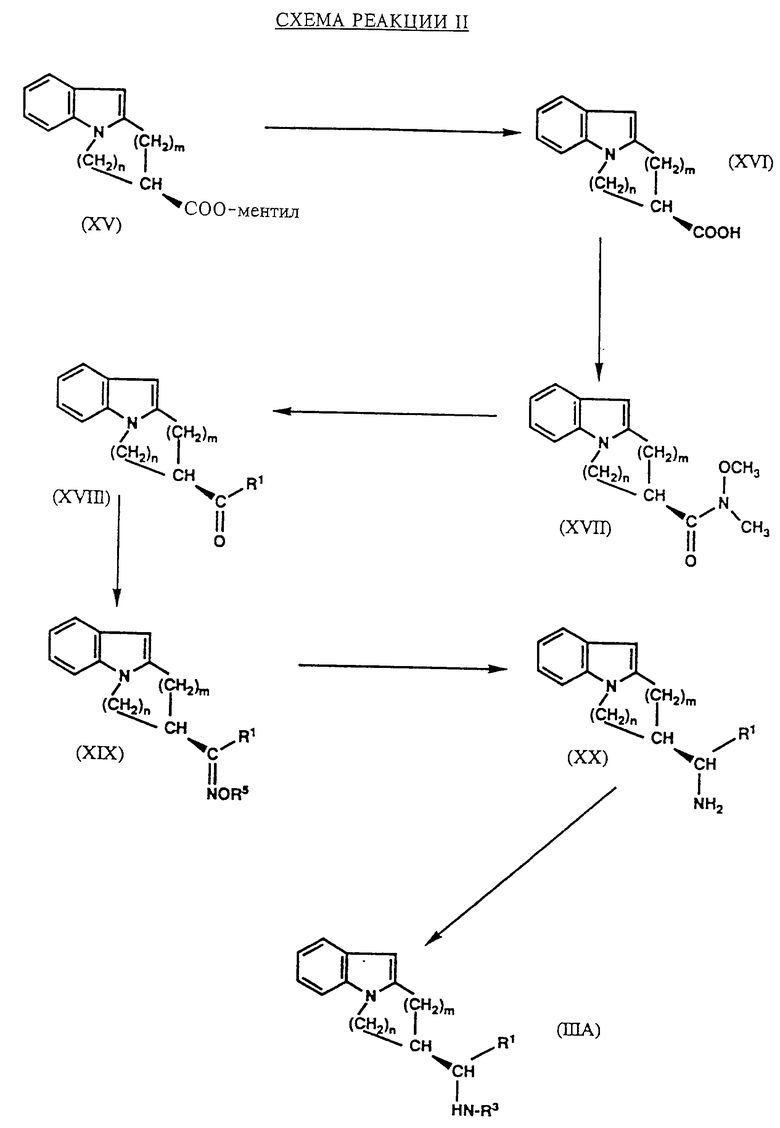
Гомохиральные соединения формулы III, обозначаемые в дальнейшем через IIIA, могут быть получены, например, как это показано на схеме реакции II, в которой R1, m и n уже были определены выше, а R5 представляет собой низший алкил.
Отдельные стадии синтеза, представленного на схеме реакции II, могут быть осуществлены в соответствии с приемами, которые известны в этой области техники. На первой стадии сложный ментиловый эфир формулы XV, который является известным соединением или аналогом известного соединения, превращают с использованием сильной кислоты, например, концентрированной серной кислоты, в соответствующую карбоновую кислоту формулы XVI. Эту кислоту затем конденсируют с N,O-диметилгидроксиламином, а полученный N-метокси-N-метилкарбоксамид формулы XVII взаимодействует с реагентом Гриньяра формулы R1-Mg-X, в которой R1 имеет значение, определенное ранее, а X представляет собой галоген, предпочтительно хлор, чтобы получить кетон формулы XVIII. Реакция этого кетона с гидроксиламином формулы H2N-OR5, в которой R5 был определен выше, дает оксим формулы XIX, который восстанавливают с использованием сложного гидрида металлов, такого, как алюмогидрид лития, в первичный амин формулы XX. Последнее соединение, далее, превращают в гомохиральное исходное соединение формулы IIIA при помощи ацилирования аналогично тому, как это было описано на схеме реакции I для превращения соединения формулы XIV в соединение формулы III.
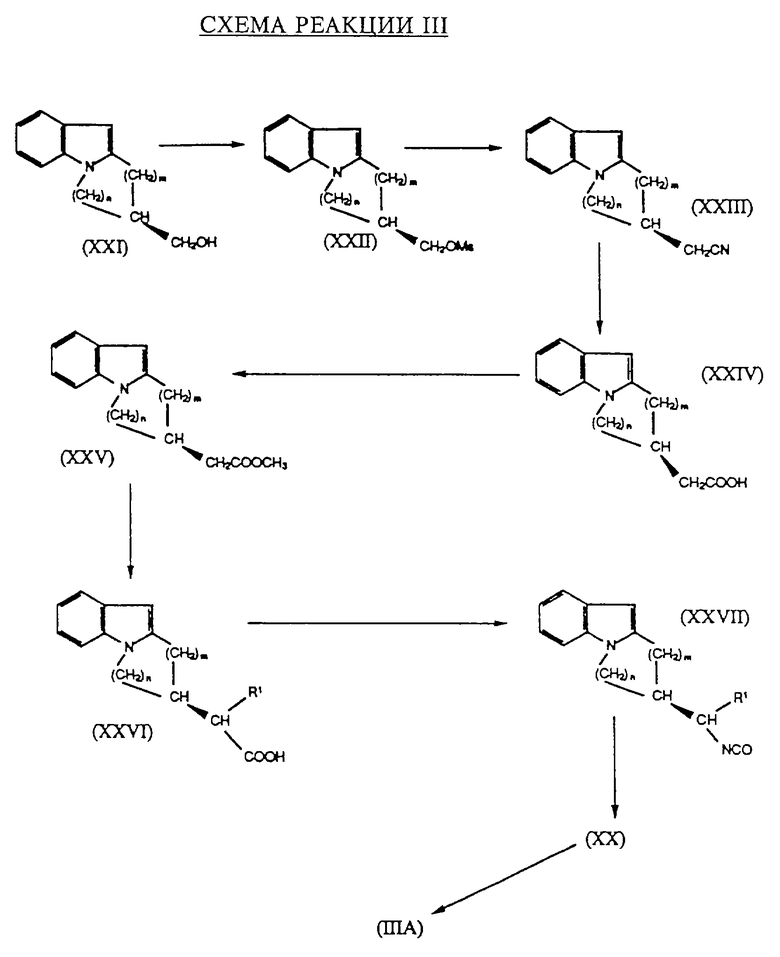
Схема реакции III иллюстрирует альтернативный путь получения гомохиральных исходных соединений формулы IIIA. В этой схеме реакции R1, m и n были определены выше, a Ms представляет собой метансульфонил.
Указанные на схеме III стадии синтеза могут быть осуществлены при помощи известных приемов. Прежде всего спирт формулы XXI подвергают сульфированию, например, с использованием ангидрида метансульфоновой кислоты, а полученный метансульфонат формулы XXII превращают при помощи цианида натрия в нитрил формулы XXIII. Последний подвергают гидролизу, например, используя раствор гидрата окиси натрия, в соответствующую карбоновую кислоту формулы XXIV, которую метилируют, например, используя метанол в присутствии концентрированной серной кислоты в метиловый сложный эфир формулы XXV. Реакция последнего с галидом формулы R1-X, в которой R1 и X были определены выше, в присутствии сильного основания, такого, как диизопропиламид лития, с последующей обработкой раствором гидрата окиси натрия, дает карбоновую кислоту формулы XXVI. Последняя взаимодействует с дифенилфосфорилазидом для получения изоцианата формулы XXVII, который превращают в первичный амин формулы XX при помощи обработки хлористоводородной кислотой. Превращение первичного амина формулы XX в гомохиральное соединение формулы IIIA осуществляют при помощи ацилирования по той же схеме, что была описана в случае схемы реакции I.
Соединения формулы I и их фармацевтически приемлемые соли являются ингибиторами протеинкиназы; они ингибируют клеточные процессы, например, пролиферацию и секретирование, и могут быть использованы для лечения и предотвращения заболеваний, например, для лечения или предотвращения воспалительных расстройств, таких, как артрит, иммунных заболеваний, псориаза, контактного дерматита, при трансплантации органов, а также в онкологии. Они ингибируют инфицирование клеток вирусом иммунодефицита человека или вирусом Эпстейна-Барра и поэтому могут быть использованы при лечении СПИДа и инфекционного мононуклеоза. Соединения и соли настоящего изобретения ингибируют также сокращение гладких мышц и могут быть поэтому использованы при лечении сердечно-сосудистых и бронхолегочных расстройств. Кроме того, их можно также использовать в терапии астмы. Предлагаемые соединения и соли ингибируют также агрегирование тромбоцитов и могут быть использованы для лечения и предотвращения тромбозов. Кроме того, они ингибируют высвобождение медиаторов из активированных нейтрофилов и поэтому могут быть использованы для лечения ишемических заболеваний, например, сердца или головного мозга. Кроме того, они ингибируют нейротоксичность, вызванную повышенными концентрациями глюкозы и поэтому могут быть использованы для лечения диабетических осложнений. Наконец, предлагаемые соединения и соли стимулируют рост волос и поэтому могут быть использованы для предотвращения облысения или восстановления волосяного покрова.
Активность предлагаемых соединений при ингибировании протеинкиназы C может быть проиллюстрирована с использованием in vitro процедурой испытания, которая описана ниже.
Используют систему анализа, описанную Takai et al., BBRC 19, 1218, (1979). Реакционные смеси (100 мкл) содержат 10 мкМ [γ-32P] ATP, 0,2 мг/мл (примерно 15 мкМ) обогащенного лизином гистона, 0,5 мМ CaCl2 и 40 мкг/мл фосфатидилсерина в 25 мМ Трис HCl, 5 мМ MgNO3 (pH 7,5) буфера. Фермент протеинкиназу C выделяют из головного мозга крысы в соответствии с методикой Kikkawa et al., J. Biol. Chem. 257, 13341 (1982).
Реакцию начинают с добавления фермента, осуществляют при температуре 30oC в течение 10 минут, а затем прекращают, используя 1 мл охлажденной льдом трихлоруксусной кислоты. Осажденный кислотой протеин собирали фильтрацией на диски из стекловолокна. Затем эти диски промывают 5%-ной трихлоруксусной кислотой, содержащей 20 мМ пирофосфата натрия (с тем, чтобы удалить непрореагировавшую ATP), затем этанолом. Диски сушат и анализируют на счетчике. Подсчеты на каждом диске, показывали включение 32P из [γ-32P] ATP в гистон. Степень блокировки фермента для каждой концентрации испытываемых соединений рассчитывают по формуле
Импульсы/мин для включения + испыт.соединение + фермент/ Импульсы/мин для включения - испыт.соединение + фермент • 100%
IC50 - значение - это такая концентрация испытываемого соединения, которая снижает на 50% вызванное протеинкиназой включение 32P при условиях анализа, описанных выше.
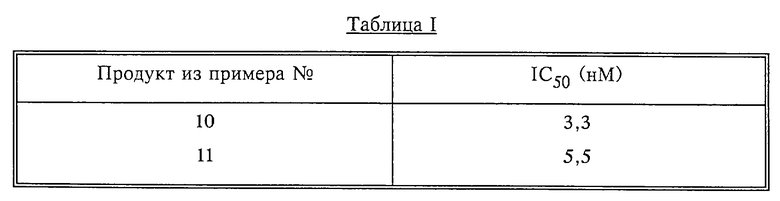
Результаты, полученные в описанном выше испытании для некоторых представителей соединений формулы I, приведены в таб. I. Табл. II показывает результаты испытаний ряда соединений in vivo полученные методом, указанным ниже.
Индукция отека лапы крысы линии AHH/R2, вызываемого 12,13-дибутиратом форбола
Метод:
Женским особям крыс линии AHH/R2 орально вводили (10 мл/кг) испытуемое соединение за один час до индукции отека, вызываемого инъекцией 12,13-дибутирата форбола (ДБуФ) в правую заднюю лапку (0,1 мл 10-3М). Объем отека регистрировали путем плетизмографии через 0,5, 1, 2, 3, 4 и 6 часов после введения.
Тестируемые соединения приготавливают соникацией (разрушением биологического материала) в 10%-ном сукцинилированном желатине в течение 3-х минут с последующим разбавлением требуемым наполнителем. ДБуФ приготавливают растворением 10 мг в 1 мл этанола и 1 мл солевого раствора, получая раствор 10-2М. Этот раствор затем растворяют в солевом растворе до концентрации 10-3M с целью его использования в данном эксперименте.
Соединения формулы I и их вышеупомянутые соли могут быть использованы в качестве медикаментов, например, в виде фармацевтических препаратов. Эти фармацевтические препараты могут быть использованы для орального применения, например, в форме таблеток, таблеток с покрытием, драже, жестких и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако их можно применять ректально (например, в виде суппозиториев) или парентерально (например, в форме растворов для инъекций).
Для получения фармацевтических препаратов из соединений формулы I и их вышеупомянутых солей могут быть использованы терапевтически инертные, неорганические или органические носители. В качестве таких носителей для таблеток, таблеток с покрытием, драже и твердых желатиновых капсул можно использовать, например, лактозу, маисовый крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.п. Соответствующими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.п. В зависимости от природы активного материала носители, однако, как правило, не требуются при использовании мягких желатиновых капсул. Соответствующими носителями для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и т.п. Соответствующими носителями для растворов инъекций являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.п. Соответствующими носителями для суппозиториев являются, например, натуральные или загущенные масла, воски, жиры, полужидкие полиолы и т.п.
Такие фармацевтические препараты могут также содержать консервирующие агенты, солюбилизирующие агенты, стабилизирующие агенты, смачивающие агенты, эмульгирующие агенты, вкусовые агенты, окрашивающие агенты, ароматизирующие агенты, соли для варьирования осмотического давления, буферы, покрывающие агенты или антиоксиданты. Они могут содержать также другие, терапевтически приемлемые вещества. Медикаменты, содержащие соединение формулы I или его соль, которые были определены выше, и терапевтически инертный носитель, а также способ получения таких медикаментов, также являются целями настоящего изобретения. Этот способ позволяет перевести соединения формулы I или его соответствующую соль, в галеновую форму вместе с терапевтически инертным носителем и, если это необходимо, с одним или несколькими другими терапевтически активными веществами.
Как уже упоминалось выше, соединения формулы I и их вышеупомянутые соли могут быть использованы для лечения или предотвращения заболеваний, в частности, для лечения или предотвращения воспалительных, иммунологических, бронхолегочных, дерматологических и сердечно-сосудистых заболеваний, для лечения астмы, СПИДа или диабетических осложнений, или для стимуляции роста волос. Дозировка может варьироваться в широких пределах и будет, естественно, зависеть от индивидуальных требований в каждом конкретном случае. В общем случае при оральном применении для взрослого пациента ежедневная доза изменяется от примерно 5 мг до примерно 500 мг, хотя верхний предел может быть и выше, если это необходимо. Ежедневная доза может быть применена в виде разовой дозы или нескольких доз.
Следующие примеры иллюстрируют настоящее изобретение.
Пример 1
Раствор 1,25 г (2,32 ммоля) 3-[8(R или S)-[1(R или S)-трет- бутоксиформамидоэтил] -6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил]- 4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомера A) в 10 мл ацетата обрабатывали 30 мл насыщенного раствора хлористого водорода в этилацетате и перемешивали при комнатной температуре в течение 18 часов. Полученный твердый материал отделяли фильтрацией и сушили, чтобы получить 1,0 г хлоргидрата 3-[8(R или S)-[1(R или S)-аминоэтил]-6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил]-4-(1- метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 324-325oC.
3-[8(R или S)-[1(R или S)-трет-бутоксиформамидоэтил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-дион (диастереомер A), используемый в качестве исходного продукта, получали следующим образом:
(1) Перемешиваемую суспензию 27 г (126 ммолей) 6,7,8,9-тетрагидропиридо[1,2-а] индол-8-карбоновой кислоты в 30 мл воды и 450 мл ацетона охлаждали до 0oC и обрабатывали последовательно 14,7 г (145 ммолей) триэтиламина и 17,3 г (159 ммолей) этилхлорформиата. Спустя 0,5 часа добавляли 6,3 мл 0,880 аммиака и перемешивание продолжали 1 час. Растворитель удаляли при пониженном давлении и остаток растирали с водным этанолом. Продукт отделяли фильтрацией и сушили, чтобы получить 13 г 6,7,8,9-тетрагидропиридо[1,2-а]индол-8-карбоксамида в виде белого твердого вещества с температурой плавления 165-168oC.
(2) 25,7 г (126 ммолей) трифторуксусного ангидрида добавляли по каплям в перемешиваемую суспензию из 26,5 г (123 ммоля) 6,7,8,9-тетрагидропиридо[1,2-а]индол-8-карбоксамида и 23,4 г (300 ммолей) пиридина в 500 мл сухого диоксана при температуре 10oC. После завершения добавления растворитель удаляли при пониженном давлении и остаток кристаллизовали из метанола, чтобы получить 13 г 6,7,8,9-тетрагидропиридо[1,2-а]индол-8(RS)-карбонитрила в виде светложелтовато-коричневого твердого вещества с температурой плавления 106-109oC.
(3) Раствор 1,4 г (7,2 ммоля) 6,7,8,9-тетрагидропиридо[1,2-а]- индол-8(RS)-карбонитрила в 400 мл сухого толуола обрабатывали 7 мл (21 ммоль) 3M раствора хлорида метилмагния в тетрагидрофуране, а полученный раствор нагревали при кипячении с обратным холодильником в атмосфере азота 0,5 часа. Затем этот раствор добавляли в 17 мл (17 ммолей) 1M раствора алюмогидрида лития в тетрагидрофуране. Полученный раствор нагревали при кипячении с обратным холодильником 15 минут, охлаждали и обрабатывали по каплям примерно 20 мл воды. Осадок отделяли фильтрацией и промывали 100 мл этилацетата, а соединенный фильтрат и промывочные жидкости выпаривали при пониженном давлении, чтобы получить 1,55 г светло-коричневого масла.
Это масло растворяли в 70 мл сухого дихлорметана и обрабатывали 1,5 г (15 ммолей) триэтиламина, затем 1,8 г (7,4 ммоля) дитрет-бутилдикарбоната при 0oC в атмосфере азота. Перемешиваемому раствору давали возможность нагреться до комнатной температуры и перемешивание продолжали 1 час. Растворитель удаляли при пониженном давлении, а продукт подвергали очистке с использованием флэш-хроматографии на силикагеле, используя простой диэтиловый эфир/петролейный эфир (1: 2) для элюирования, чтобы получить 925 мг трет-бутил-[1(R или S)-(6,7,8,9- тетрагидропиридо[1,2-а]индол- 8(R или S)-ил)этил] карбамата в виде смеси диастереомеров в виде белого твердого вещества с температурой плавления 114-117oC.
(4) Перемешиваемый раствор 3,0 г (9,57 ммоля) трет-бутил-[1(R или S)-(6,7,8,9-тетрагидропиридо[1,2-а]индол- 8(R или S)-ил)этил]карбамата в 150 мл дихлорметана по каплям обрабатывали при 0oC 1,28 г (10,3 ммоля) оксалилхлорида. Спустя 5 минут растворитель удаляли при пониженном давлении, а остаток растворяли в 150 мл дихлорметана и обрабатывали 2,94 г (11 ммолей) хлоргидрата изопропил-1-метил-3-индолацетимидата и 4,38 г (43 ммоля) триэтиламина при 0oC в атмосфере азота. После нагревания до комнатной температуры раствор перемешивали 24 часа, промывали водой и сушили над сульфатом магния. Растворитель удаляли выпариванием, а остаток растворяли в 30 мл пиридина. Этот перемешиваемый раствор охлаждали до температуры ледяной бани и обрабатывали по каплям 1,5 мл (10,8 ммолей) трифторуксусного ангидрида. Спустя 15 минут растворитель удаляли при пониженном давлении, а остаток разделяли между 200 мл этилацетата и 200 мл 0,2М раствора хлористоводородной кислоты. Органический слой промывали 50 мл раствора бикарбоната натрия, сушили над сульфатом магния и выпаривали досуха. Остаток подвергали очистке с использованием флэш-хроматографии на силикагеле, применяя этилацетат/петролейный эфир (1:2) для элюирования, чтобы получить 1,35 г 3-[8(R или S)-[1(R или S)- трет-бутоксиформамидоэтил] -6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в форме красного твердого вещества с температурой плавления 255-257oC. Последующее элюирование дает 1,38 г диастереомера B в виде красного твердого вещества с температурой плавления 230-233oC.
Пример 2
По аналогии с процедурой, что была описана в первом абзаце Примера 1, из 1,38 г 3-[8(R или S)-[1(R или S)- трет-бутоксиформамидоэтил]-6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B), полученного так, как это описано в Примере 1(4), получали 930 мг хлоргидрата 3-[8(R или S)-[1(R или S)-аминоэтил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H-пиррол- 2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 254-258oC.
Пример 3
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 180 мг 3-[8(R или S)-[1(R или S)- трет-бутоксиформамидопропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) получали 115 мг хлоргидрата 3-[8(R или S)-[1(R или S)-аминопропил]- 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 321-323oC.
3- [8(R или S)-[1(R или S)-трет-бутоксиформамидопропил]- 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)- 1H-пиррол-2,5-дион (диастереомер A), используемый в качестве исходного продукта, получали следующим образом:
(1) При помощи процедуры, аналогичной той, что описана в Примере 1(3), из 500 мг (2,5 ммоля) 6,7,8,9- тетрагидропиридо[1,2-а]индол-8(RS)-карбонитрила и 2,5 мл (5 ммолей) 2M раствора бромида этилмагния в тетрагидрофуране получали 480 мг трет-бутил-[1(R или S)-6,7,8,9-тетрагидропиридо[1,2-а] индол- 8(R или S)-ил)пропил] карбамата в виде смеси диастереомеров в виде белого твердого вещества с температурой плавления 153-156oC.
(2) При помощи процедуры, аналогичной той, что была описана в Примере 1(4), из 440 мг (1,34 ммоля) трет-бутил-[1(R или S)- (6,7,8,9-тетрагидропиридо[1,2-а]индол-8(R или S)-ил)пропил]карбамата получали 180 мг 3-[8(R или S)-[1(R или S)- трет-бутоксиформамидопропил]-6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в виде красной смолы. Последующее элюирование давало 130 мг диастереомера B в виде красной смолы.
Пример 4
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 130 мг 3-[8(R или S)- 1(R или S)- трет-бутоксиформамидопропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона [диастереомер B, полученный, как это описано в Примере 3(2)] получали 65 мг хлоргидрата 3-[8(R или S)-[1(R или S)-аминопропил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил- 3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 245-249oC.
Пример 5
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 400 мг (0,7 ммоля) 3-[8(R или S)- [1(R или S)-трет-бутоксиформамидобутил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1- метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) получали 310 мг хлоргидрата 3-[8(R или S)-[1(R или S)-аминобутил]-6,7,8,9-тетрагидропиридо- [1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 237-241oC.
3-[8(R или S)-[1(R или S)-трет-бутоксиформамидобутил]- 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)- 1H-пиррол-2,5-дион (диастереомер A), используемый в качестве исходного продукта, получали следующим образом:
При помощи процедуры, аналогичной той, что была описана в Примере 1(3) и (4), из 1,0 г (5 ммолей) 6,7,8,9-тетрагидропиридо- [1,2-а]индол-8(RS)-карбонитрила и 5,0 мл (10 ммолей) 2M раствора хлорида н-пропилмагния в простом диэтиловом эфире получали 470 мг 3-[8(R или S)-[1(R или S)-третбутоксиформамидобутил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 227-229oC. Последующее элюирование давало 285 мг диастереомера B в виде красного твердого вещества с температурой плавления 169-172oC.
Пример 6
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 270 мг (0,48 ммоля) 3-[8(R или S)-[1(R или S)-трет-бутоксиформамидобутил] -6,7,8,9-тетрагидропиридо[1,2-а] - индол-10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B), полученного, как это описано во втором абзаце Примера 5, получали 210 мг хлоргидрата 3-[8(R или S)-[1(R или S)-aминoбутил]- 6,7,8,9-тeтpaгидpoпиpидo[1,2-a]индoл-10- ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 235-238oC.
Пример 7
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 400 мг (0,7 ммоля) 3-[8(R или S)-[1(R или S)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) получали 300 мг хлоргидрата 3- [8 (R или S)-[1(R или S)-амино-2-метилпропил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 254-256oC.
3-[8(R или S)-[1(R или S)-трет-бутоксиформамидо-2-метилпропил] - 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-дион (диастереомер A), используемый в качестве исходного продукта, получали следующим образом:
(1) Раствор 2,0 г (10 ммолей) 6,7,8,9-тетрагидропиридо[1,2-а]- индол-8(RS)-карбонитрила в 150 мл сухого толуола обрабатывали 10 мл (20 ммолей) 2M раствора хлорида изопропилмагния в тетрагидрофуране, а полученный раствор нагревали при кипячении с обратным холодильником 20 минут. Раствор охлаждали и обрабатывали по каплям 24 мл (24 ммоля) 1M раствора алюмогидрида лития в тетрагидрофуране. Полученный раствор перемешивали при комнатной температуре 1,5 часа, охлаждали и по каплям обрабатывали примерно 20 мл воды. Осадок отделяли фильтрацией, промывали 100 мл дихлорметана, а соединенные фильтраты выпаривали при пониженном давлении, чтобы получить 7,2 г бледно-коричневого масла. Это масло растворяли в 100 мл сухого дихлорметана и обрабатывали 2,34 г (23,4 ммоля) триэтиламина, а затем 2,8 г (11,6 ммоля) дитрет-бутилдикарбоната при температуре 0oC в атмосфере азота. Перемешиваемому раствору давали возможность нагреться до комнатной температуры и перемешивание продолжали 70 часов. Растворитель удаляли при пониженном давлении и продукт подвергали очистке с использованием быстрой хроматографии на силикагеле, используя простой диэтиловый эфир/петролейный эфир (1:2) для элюирования, чтобы получить 800 мг трет-бутил-[1(R или S)-[6,7,8,9-тетрагидропиридо[1,2-а]- индол-8(R или S)-ил] -2-метилпропил]карбамата (диастереомер A) в виде белого твердого вещества с температурой плавления 118-119oC. Последующее элюирование давало 420 мг диастереомера B в виде белого твердого вещества с температурой плавления 128-129oC.
(2) Перемешиваемый раствор 770 мг (2,25 ммоля) трет-бутил- [1(R или S)-[6,7,8,9-тетрагидропиридо[1,2-а] индол-8(R или S)- ил]-2-метилпропил]карбамата (диастереомер A) в 30 мл дихлорметана при 0oC обрабатывали по каплям 287 мг (2,4 ммоля) оксалилхлорида. Через 5 минут растворитель удаляли при пониженном давлении, а остаток растворяли в 30 мл дихлорметана и обрабатывали 686 мг (2,57 ммоля) хлоргидрата изопропил-1-метил-3-индолацетимидата и 1,02 г (10 ммолей) триэтиламина при 0oC в атмосфере азота. После нагревания до комнатной температуры раствор перемешивали 18 часов, промывали 50 мл воды и сушили над сульфатом магния. Растворитель удаляли выпариванием, а остаток растворяли в 10 мл пиридина. Перемешиваемый раствор охлаждали до температуры ледяной бани и по каплям обрабатывали 664 мкл (4,8 ммоля) трифторуксусного ангидрида. Спустя 15 минут добавляли 50 мл этилацетата и органическую фазу промывали водой и 2M хлористоводородной кислотой, сушили над сульфатом магния и выпаривали до сухого состояния. Остаток подвергали очистке при помощи флэш-хроматографии на силикагеле, используя этилацетат/петролейный эфир (1: 2) для элюирования, чтобы получить 460 мг 3-[8(R или S)- [1(R или S)-трет-бутоксиформамидо-2-метилпропил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 223-224oC.
Пример 8
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 270 мг (0,48 ммоля) 3-[8(R или S)- [1(R или S)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер B) получали 180 мг хлоргидрата 3-[8(R или S)-[1(R или S)-амино-2-метилпропил]-6,7,8,9- тетрагидропиридо[1.2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол- 2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 248-250oC.
3-[8(R или S)-[1(R или S)-трет-бутоксиформамидо-2- метилпропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1- метил-3-индолил)-1H-пиррол-2,5-дион (диастереомер B), используемый в качестве исходного продукта, получали следующим образом:
При помощи процедуры, аналогичной той, что была описана в Примере 7(2), из 385 мг трет-бутил-[1(R или S)-[6,7,8,9- тетрагидропиридо[1,2-а]индол-8(R или S)-ил] -2-метилпропил] карбамата (диастереомер B), полученного, как это описано в Примере 7(1), получали 270 мг 3-[8(R или S)- [1(R или S)-трет-бутоксиформамидо- 2-метилпропил] -6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил] -4- (1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красной смолы.
Пример 9
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 1,2 г (2,17 ммоля) 3-[8(S)-[1(R или S)-трет-бутоксиформамидопропил] -6,7,8,9-тетрагидропиридо[1,2-а] - индол-10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) получали 850 мг хлоргидрата 3-[8(S)-[1(R или S)- аминопропил]-6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил] -4-(1-метил- 3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 304-308oC.
3-[8(S)-[1(R или S)-тpeт.-бутoкcифopмaмидoпpoпил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H-пиррол- 2,5-дион (диастереомер A), используемый в качестве исходного продукта, получали следующим образом:
(1) Охлажденный льдом раствор 50,0 г (248 ммолей) 8(S)-оксиметил-6,7,8,9-тетрагидропиридо[1,2-а] индола в 500 мл дихлорметана обрабатывали 50 мл (358 ммолей) триэтиламина и 52,0 г (298 ммолей) ангидрида метансульфоновой кислоты в течение 20 минут. Спустя 2 часа добавляли 250 мл воды и органическую фазу промывали последовательно двумя порциями по 250 мл насыщенного раствора бикарбоната натрия и 20 мл воды. Далее органическую фазу сушили над сульфатом магния и выпаривали. Остаточное твердое вещество растирали с простым эфиром, отделяли фильтрацией и сушили в вакууме, чтобы получить 65,4 г [6,7,8,9-тетрагидропиридо- [1,2-а] индол-8(S)-ил]метил-метансульфоната в виде бледно-розового твердого вещества с температурой плавления 114-115oC;
[α]
(2) 18,0 г (367 ммолей) цианида натрия добавляли в раствор 65,0 г (233 ммоля) (6,7,8,9-тетрагидропиридо[1,2-а]индол-8(S)- ил)метил-метансульфоната в 500 мл диметилформамида и смесь нагревали до 70oC 24 часа. Эту смесь разделяли между 1000 мл воды и 600 мл этилацетата. Водную фазу дважды экстрагировали каждый раз 700 мл этилацетата и соединенные экстракты промывали дважды 500 мл воды каждый раз, сушили над сульфатом магния и выпаривали. Полученное коричневое твердое вещество растворяли в этилацетате, а раствор фильтровали через слой из силикагеля. Растворитель удаляли в вакууме, а остаток кристаллизовали из метанола, чтобы получить 25,8 г 6,7,8,9-тетрагидропиридо[1,2-а] - индол-8(S)-ацетонитрила в виде светло-коричневого твердого вещества с температурой плавления 100-101oC;
[α]
(3) Раствор 27,0 г (129 ммолей) 6,7,8,9-тетрагидропиридо- [1,2-а]индол-8(S)-ацетонитрила в 120 мл 2M раствора гидрата окиси натрия в 400 мл 1,2-этандиола нагревали при кипячении с обратным холодильником 4 часа. Добавляли 400 мл этилацетата и органическую фазу промывали последовательно 500 мл воды, 150 мл 2M хлористоводородной кислоты и тремя порциями по 500 мл воды, сушили над сульфатом магния и выпаривали, чтобы получить 29 г 8(S)-(6,7,8,9-тетрагидропиридо[1,2-а]индолил)уксусной кислоты в виде бледно-розового твердого вещества с температурой плавления 118-120oC.
(4) Раствор 29 г (127 ммолей) 8(S)-(6,7,8,9-тетрагидропиридо- [1,2-а] индолил)уксусной кислоты и 5 мл концентрированной серной кислоты в 500 мл метанола нагревали при кипячении с обратным холодильником 1 час. Эту смесь концентрировали при пониженном давлении и продукт отделяли фильтрацией и сушили, чтобы получить 28,4 г метил-[8(S)-6,7,8,9-тетрагидропиридо[1,2-а]индолил]ацетата в виде бледно-розового твердого вещества с температурой плавления 84-87oC.
(5) 10 г (100 ммолей) диизопропиламина в 60 мл сухого тетрагидрофурана обрабатывали при 0oC в атмосфере азота 63 мл (100 ммолей) 1,6М раствора n-бутиллития в гексане и перемешивали 15 минут. Полученный раствор охлаждали до -78oC и раствор 14,9 г (61,3 ммоля) метил-[8(S)-6,7,8,9-тетрагидропиридо[1,2-а] индолил] - ацетата в 60 мл тетрагидрофурана добавляли по каплям. Спустя 30 минут добавляли 15,6 г (100 ммолей) этилиодида и смесь перемешивали 0,5 часа. Затем добавляли еще 8 г этилиодида и смеси давали возможность нагреться до комнатной температуры в течение примерно 1 часа. Эту смесь разделяли между 300 мл простого диэтилового эфира и 20 мл 2M хлористоводородной кислоты. Органическую фазу промывали три раза по 300 мл воды (каждый раз), сушили над сульфатом магния и выпаривали, чтобы получить 17 г бледно-оранжевого масла. Пробу кристаллизовали из метанола, чтобы получить метил-альфа(R или S)-этил-6,7,8,9- тетрагидропиридо [1,2-а]индол-8(S)-ацетат (диастереомеры A+B) в виде бледно-розового твердого вещества с температурой плавления 87-90oC.
(6) Раствор 17 г (63 ммоля) метил-альфа(R или S)-этил- 6,7,8,9-тетрагидропиридо[1,2-а] индол-8(S)-ацетата (диастереомеры A+B) в 150 мл метанола обрабатывали 130 мл 2M раствора гидрата окиси натрия и нагревали при кипячении с обратным холодильником 18 часов. Охлажденный раствор добавляли в 150 мл 4M раствора хлористоводородной кислоты и полученный в результате осадок фильтровали и разделяли между 200 мл дихлорметана и 100 мл воды. Органическую фазу отделяли, сушили над сульфатом магния и выпаривали, чтобы получить 15 г альфа(R или S)-6,7,8,9- тетрагидропиридо[1,2-а]индол-8(S)-уксусной кислоты (диастереомеры A+B) в виде бледно-розового твердого вещества.
(7) Перемешиваемый раствор 15 г (58,4 ммоля) альфа-(R или S)- 6,7,8,9-тетрагидропиридо[1,2-а]индол-8(S)-уксусной кислоты (диастереомеры A+B) в 450 мл толуола обрабатывали 6,5 г (65 ммолей) триэтиламина и 18,6 г (67,5 ммолей) дифенилфосфонилазида при комнатной температуре в атмосфере азота. Спустя 1 час при комнатной температуре смесь нагревали при кипячении с обратным холодильником 0,5 часа, охлаждали и растворитель удаляли при пониженном давлении. Остаток подвергали очистке при помощи флэш-хроматографии на силикагеле, используя простой диэтиловый эфир/петролейный эфир (1:3) для элюирования, чтобы получить 14 г изоцианата в виде бесцветного масла. Это масло растворяли в 400 мл диоксана и 150 мл 2M хлористоводородной кислоты и полученный раствор перемешивали при комнатной температуре 18 часов. Раствор концентрировали и разделяли между 300 мл этилацетата и 2M раствором гидрата окиси натрия. Водный слой экстрагировали 100 мл этилацетата и соединенные органические растворы промывали дважды 400 мл воды (каждый раз), сушили над сульфатом магния и выпаривали до сухого состояния, чтобы получить 7,7 г пены кремового цвета.
Эту пену в 200 мл сухого дихлорметана обрабатывали 7,6 г (75 ммолей) триэтиламина и 10,9 г (50 ммолей) дитрет-бутилдикарбоната и смесь перемешивали при комнатной температуре 3 часа. Растворитель удаляли при пониженном давлении, а остаток подвергали флэш-хроматографии на силикагеле, используя простой диэтиловый эфир/петролейный эфир (1:2) для элюирования, чтобы получить 4,5 г трет-бутил-[1(R или S)-[6,7,8,9-тетрагидропиридо[1,2-а]индол- 8(S)-ил]пропил]карбамата (диастереомеры A+B) в виде белого твердого вещества с температурой плавления 152-158oC.
(8) При помощи процедуры, аналогичной той, что описана в примере 1(4), из 4,3 г (13,1 ммолей) трет-бутил-[1(R или S)- [6,7,8,9-тетрагидропиридо[1,2-а] индол-8(S)-ил] пропил]карбамата (диастереомеры A+B) получали 1,2 г 3-[8(S)-[1(R или S)- трет-бутоксиформамидопропил]-6,7,8,9-тетрагидропиридо[1,2-а]индол- 10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) и 2,0 г диастереомера B, оба в виде красного твердого вещества.
Пример 10
При помощи процедуры, аналогичной той, что описана в первом абзаце примера 1, из 2,0 г (3,62 ммоля) 3-[8(S)-[1(R или S)- трет-бутоксиформамидопропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B), полученного, как это описано в Примере 9, получали 1,28 г хлоргидрата 3-[8(S)-[1(R или S)-аминопропил]-6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 247-253oC.
Пример 11
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 1,46 г 3-[8(S)-[1(S)- трет-бутоксиформамидо-2-метилпропил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона получали 1,29 г хлоргидрата 3-[8(S)-[1(S)- амино-2-метилпропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] - 4-(1-метил-3-индолил)-1H-пиррол-2,5-диона в виде красного твердого вещества с температурой плавления 253-256oC.
3-[8(S)-[1(S)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) К 40 г (114 ммолей) сложного 1-ментилового эфира 6,7,8,9-тетрагидропиридо[1,2-а] индол-8(S)-карбоновой кислоты добавляли 50 мл концентрированной серной кислоты и полученную смесь перемешивали до тех пор, пока весь исходный материал не растворился (примерно 20 минут). Раствор осторожно сливали в 1500 мл смеси лед-вода, а полученный в результате осадок отделяли фильтрацией, промывали петролейным эфиром/толуолом (3:1) и сушили, чтобы получить 24,2 г 6,7,8,9-тетрагидропиридо[1,2-а]индол-8(S)-карбоновой кислоты в виде белого твердого вещества с температурой плавления 251-253oC.
(2) Перемешиваемую суспензию из 24,0 г (111 ммолей) 6,7,8,9-тетрагидропиридо[1,2-а]индол-8(S)-карбоновой кислоты в 500 мл дихлорметана обрабатывали последовательно при 0oC 24 мл (138 ммолей) диизопропилэтиламина, 13,24 г (136 ммолей) хлоргидрата N,O-диметилгидроксиламина, 10 мг диметиламинопиридина и 23,04 г (112 ммолей) дициклогексилкарбодиимида. Полученную смесь перемешивали при комнатной температуре 18 часов и фильтровали, а твердое вещество промывали дважды 100 мл каждый раз дихлорметана. Соединенные фильтраты выпаривали до сухого состояния, а остаток подвергали очистке с использованием флэш-хроматографии на силикагеле, применяя этилацетат/петролейный эфир (1: 3) для элюирования, чтобы получить 22,6 г белого твердого вещества. Пробу растирали с простым эфиром/петролейным эфиром, чтобы получить 6,7,8,9-тетрагидро-N-метокси-N-метилпиридо[1,2-а] индол-8(S)- карбоксамид в виде белого твердого вещества с температурой плавления 78-80oC.
(3) Перемешиваемый раствор 10,0 г (38,7 ммолей) 6,7,8,9-тетрагидро-N-метокси-N-метилпиридо[1,2-а]индол- 8(S)-карбоксамида в 250 мл тетрагидрофурана по каплям обрабатывали при 0oC 60 мл (120 ммолей) 2M раствора изопропилмагнийхлорида в тетрагидрофуране. Эту смесь перемешивали при комнатной температуре 18 часов и сливали в 250 мл насыщенного раствора хлорида аммония. Водную фазу промывали 4 раза 100 мл простого диэтилового эфира каждый раз, а соединенные эфирные экстракты промывали 200 мл соляного раствора, сушили над сульфатом магния и выпаривали до сухого состояния. Продукт подвергали очистке, применяя флэш-хроматографию на силикагеле, используя этилацетат/петролейный эфир (1:3) для элюирования, чтобы получить 4,4 г изопропил- 6,7,8,9-тетрагидропиридо[1,2-а] индол-8(S)-илкетона в виде белого твердого вещества с температурой плавления 78-79oC.
(4) Суспензию из 4,0 г (16,6 ммолей) изопропил-6,7,8,9- тетрагидропиридо[1,2-а] индол-8(S)-илкетона в 120 мл этанола обрабатывали раствором 2,30 г (33 ммоля) хлоргидрата гидроксиламина и 1,0 г (25 ммолей) гидрата окиси натрия в 20 мл воды. Полученную смесь нагревали при кипячении с обратным холодильником 3,5 часа, охлаждали и фильтровали. Полученное твердое вещество сушили, чтобы получить 3,54 г оксима в виде белого твердого вещества.
Этот оксим растворяли в 150 мл сухого тетрагидрофурана и обрабатывали 12,5 мл (12,5 ммоля) 1M раствора алюмогидрида лития в тетрагидрофуране. Полученный раствор нагревали при кипячении с обратным холодильником 3 часа в атмосфере азота, охлаждали и осторожно обрабатывали 150 мл воды. Смесь экстрагировали 200 мл этилацетата, а затем двумя порциями по 150 мл этилацетата и соединенные органические экстракты сушили над сульфатом магния и выпаривали до сухого состояния. Остаток растворяли в 150 мл дихлорметана и полученный раствор обрабатывали 3 мл (21,5 ммоля) триэтиламина и 3,4 г (15,6 ммоля) дитрет-бутилдикарбоната и перемешивали 18 часов. Эту смесь промывали 150 мл насыщенного раствора хлористого аммония, сушили над сульфатом магния и выпаривали при пониженном давлении. После очистки с использованием быстрой хроматографии на силикагеле, применяя простой диэтиловый эфир/петролейный эфир (1: 3) для элюирования, получали 1,4 г трет-бутил-[1(R)-[6,7,8,9-тетрагидропиридо[1,2-а] индол-8(S)- ил] -2-метилпропил]карбамата в виде белого твердого вещества с температурой плавления 122-124oC. Кроме того, после элюирования получали 1,1 г трет-бутил-[1(S)-[6,7,8,9-тетрагидропиридо- [1,2-а] индол-8(S)-ил]-2-метилпропил]карбамата в виде белого твердого вещества с температурой плавления 154-155oC.
(5) При помощи процедуры, аналогичной той, что описана в Примере 1(4), из 1,1 г трет-бутил-[1(S)-[6,7,8,9- тетрагидропиридо[1,2-а]индол-8(S)-ил]-2-метилпропил] карбамата получали 1,46 г 3-[8(S)-[1(8)-трет-бутоксиформамидо-2-метилпропил] - 6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-диона в виде красной пены.
Пример 12
При помощи процедуры, аналогичной той, что описана в первом абзаце Примера 1, из 0,5 г (0,88 ммоля) 3-[8(S)-[1(R)- трет-бутоксиформамидо-2-метилпропил] -6,7,8,9-тетрагидропиридо- [1,2-а] индол-10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона получали 0,41 г хлоргидрата 3-[8(S)-[1(R)-амино-2-метилпропил] - 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-диона в виде красного твердого вещества с температурой плавления 235-242oC.
3-[8(S)-[1(R)-тpeт. -бутoкcифopмaмидo-2-мeтилпpoпил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H-пиррол- 2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
При помощи процедуры, аналогичной той, что описана в Примере 1(4), из 0,94 г трет-бутил-[1(R)-[6,7,8,9-тетрагидропиридо- [1,2-a]индoл-8(S)-ил]-2-метилпропил] карбамата, полученного, как это описано в Примере 11 (1)-(4), получали 1,05 г 3-[8(S)-[1(R)-трет-бутоксиформамидо-2-метилпропил] - 6,7,8,9-тетрагидропиридо-[1,2-а] индол-10-ил]-4-(1-метил-3- индолил)-1H-пиррол-2,5-диона в виде красной пены.
Пример 13
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 100 мг 3-[8(R или S)-[альфа(R или S)- трет-бутоксиформамидобензил] -6,7,8,9-тетрагидропиридо[1,2-а] индол- 10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) получали 50 мг хлоргидрата 3-[8(R или S)-[альфа(R или S)-аминобензил]- 6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил] -4-(1-метил-3-индолил)- 1H-пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 234-237oC.
3-[8(R или S)-[альфа(R или S)-трет-бутоксиформамидобензил]- 6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-дион (диастереомер A), используемый в качестве исходного продукта, получали следующим образом:
(1) При помощи процедуры, аналогичной той, что была описана в Примере 1(3), из 1,0 г (5,1 ммоля) 6,7,8,9-тетрагидропиридо- [1,2-а]индол-8(RS)-карбонитрила и 3,7 мл (11 ммолей) 3M раствора фенилмагнийбромида в тетрагидрофуране получали 0,9 г трет-бутил-[альфа(R или S)-(6,7,8,9-тетрагидропиридо[1,2-а]индол- 8(R или S)-ил)бензил]карбамата в виде смеси диастереомеров в форме белого твердого вещества с температурой плавления 160-165oC.
(2) При помощи процедуры, аналогичной той, что была описана в Примере 1(4), из 800 мг (2,1 ммоля) трет-бутил-[альфа(R или S)- (6,7,8,9-тетрагидропиридо[1,2-а] индол-8(R или S)-ил)бензил]карбамата получали 330 мг 3-[8(R или S)-[альфа(R или S)- трет-бутоксиформамидобензил]-6,7,8,9-тетрагидропиридо[1,2-а]индол- 10-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в виде красной смолы. Дальнейшее элюирование давало 280 мг диастереомера В в виде красной смолы.
Пример 14
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 200 мг 3-[8(R или S)-[альфа(R или S)- трет-бутоксиформамидобензил] - 6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-диона (диастереомер В), полученного, как это описано в Примере 13(2), получали 70 мг хлоргидрата 3-[8(R или S)-[альфа(R или S)-аминобензил] -6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] - 4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 226-233oC.
Пример 15
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 200 мг 3-[8(S)-[(R или S)- (трет-бутоксиформамидо)(циклопентил)метил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) получали 150 мг хлоргидрата 3-[8(S)-[(R или S)-(амино)(циклопентил)метил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 236-241oC.
3-[8(S)-[(R или S)-(трет-бутоксиформамидо)(циклопентил)метил]- 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)- 1H-пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) При помощи процедуры, аналогичной той, что была описана в Примере 11(3), из 2,0 г (7,75 ммоля) 6,7,8,9-тетрагидро-N-метокси- N-метилпиридо[1,2-а]индол-8(S)-карбоксамида и 15 мл (30 ммолей) 2M раствора хлорида циклопентилмагния в простом диэтиловом эфире получали 1,1 г циклопентил-6,7,8,9-тетрагидропиридо[1,2-а] индол- 8(S)-илкетона в виде бледно-желтого твердого вещества с температурой плавления 69oC.
(2) При помощи процедуры, аналогичной той, что была описана в Примере 11(4), из 1,05 г (3,9 ммоля) циклопентил-6,7,8,9- тетрагидропиридо[1,2-а] индол-8(S)-илкетона получали 330 мг 8(S)-[(R или S)-тpeт.-бутoкcифopмaмидо)(циклопентил)метил] -6,7,8,9- тетрагидропиридо[1,2-а]индола (диастереомер A) в виде белого твердого вещества с температурой плавления 140-143oC. В результате дальнейшего элюирования получали 430 мг диастереомера В в виде белого твердого вещества с температурой плавления 58-63oC.
(3) При помощи процедуры, аналогичной той, что была описана в Примере 1(4), из 300 мг 8(S)-[(R или S)- трет-бутоксиформамидо)(циклопентил)метил] -6,7,8,9- тетрагидропиридо[1,2-а] индола (диастереомер A) получали 200 мг 3-[8(S)-[(R или S)-(трет-бутоксиформамидо)(циклопентил)метил]- 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-метил-3- индолил)-1H-пиррол-2,5-диона в виде красной смолы.
Пример 16
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 250 мг 3-[8(S)-[(R или S)- (трет-бутоксиформамидо)(циклопентил)метил]-6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)-1H-пиррол- 2,5-диона (диастереомер B) получали 160 мг хлоргидрата 3-[8(S)- [(R или S)-(амино)(циклопентил)метил]-6,7,8,9-тетрагидропиридо- [1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 241-245oC.
3-[8(S)-[(R или S)-(тpeт. -бутoкcифopмaмидo)(циклопентил)метил] - 6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
При помощи процедуры, аналогичной той, что была описана в Примере 1(4), из 400 мг 8(S)-[(R или S)-(трет-бутоксиформамидо)(циклопентил)метил] -6,7,8,9-тетрагидропиридо[1,2-а] индола (диастереомер B) получали, как это описано в Примере 15 (1)- (2), 250 мг 3-[8(S)-[(R или S)-(трет-бутоксиформамидо)(циклопентил)метил] - 6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-диона в виде красной смолы.
Пример 17
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 40 мг 3-[2(R или S)- [1(R или S)- трет-бутоксиформамидо-2-метилпропил] -2,3-дигидро-1H-пирроло- [1,2-а]индол-9-ил]-4- (1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) получали 20 мг хлоргидрата 3-[2(R или S)-[1(R или S)-амино-2- метилпропил]-2,3-дигидро-1H-пирроло[1,2-а]индол-9-ил] -4-(1- метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер A) в виде красного твердого вещества с температурой плавления 224-230oC.
3-[2(R или S)-[1(R или S)-трет-бутоксиформамидо-2-метилпропил]- 2,3-дигидро-1H-пирроло[1,2-а] индол-9-ил]-4-(1-метил-3-индолил)-1H- пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) Раствор 8,0 г (35 ммолей) этил-2,3-дигидро-1H- пирроло[1,2-а]индол-2(RS)-карбоксилата в 100 мл этанола и 100 мл воды обрабатывали 3,0 г (75 ммолей) гидрата окиси натрия. Эту смесь нагревали при кипячении с обратным холодильником 15 минут, затем охлаждали и подкисляли 60 мл (120 ммолей) 2M хлористоводородной кислоты. Суспензию фильтровали, а твердое вещество промывали 50 мл воды, а затем сушили, чтобы получить 5,9 г 2,3-дигидро-1H- пирроло[1,2-а] индол-2(RS)-карбоновой кислоты в виде белого твердого вещества с температурой плавления 171-173oC.
(2) При помощи процедуры, аналогичной той, что была описана в Примере 11(2), из 4,0 г (20 ммолей) 2,3-дигидро-1H-пирроло[1,2-а]- индол-2(RS)-карбоновой кислоты получали 2,35 г 2,3-дигидро-N- метокси-N-метил-1H-пирроло[1,2-а] индол-2(RS)-карбоксамида в виде белого твердого вещества с температурой плавления 87-88oC.
(3) Суспензию из 840 мг (35 мг атом) магниевой стружки в 60 мл тетрагидрофурана обрабатывали по каплям раствором 4,4 г (37 ммолей) 2-бромпропена в 10 мл тетрагидрофурана. Эту смесь нагревали при кипячении с обратным холодильником еще 30 минут, затем охлаждали до 0oC и добавляли при этой температуре в раствор 2,3 г (9,4 ммоля) 2,3-дигидро-N-метокси-N-метил-1H-пирроло[1,2-а] - индол-2(RS)-карбоксамида в 50 мл тетрагидрофурана. После перемешивания при 0oC в течение 30 минут смесь сливали в 200 мл насыщенного водного раствора хлористого аммония. Раствор экстрагировали 200 мл этилацетата и органическую фазу сушили над сульфатом магния и фильтровали. После добавления петролейного эфира (температура кипения 40-60oC) получали осадок, который отделяли фильтрацией и сушили, чтобы получить 1,6 г белого твердого вещества. Это твердое вещество растворяли в 100 мл этанола и подвергали гидрогенизации над 200 мг 10% палладия/угле при атмосферном давлении в течение 1 часа. Катализатор удаляли фильтрацией, а фильтрат концентрировали при пониженном давлении до тех пор, пока не начнется кристаллизация. Продукт отделяли фильтрацией и сушили, чтобы получить 1,55 г изопропил-2,3-дигидро-1H-пирроло[1,2-а] индол-2(RS)-илкетона в виде белого твердого вещества с температурой плавления 104-105oC.
(4) При помощи процедуры, аналогичной той, что была описана в Примере 11(4), из 1,5 г изопропил-2,3-дигидро-1H-пирроло[1,2-а]индол- 2(RS)-илкетона получали 830 мг трет-бутил-[1(R или S)-[2,3-дигидро- 1H-пирроло[1,2-а]индол-2(R или S)-ил] -2-метилпропил]карбамата в виде смеси диастереомеров. Эту смесь перемешивали в 20 мл этилацетата, насыщенного хлористым водородом, в течение 2 часов. Полученное твердое вещество отделяли фильтрацией и подвергали очистке с использованием быстрой хроматографии на силикагеле, применяя метанол/дихлорметан (1: 10) для элюирования, чтобы получить 150 мг хлоргидрата 2(R или S)-[1(R или S)-амино-2-метилпропил]-2,3-дигидро- 1H-пирроло[1,2-а] индола (диастереомер A) в виде белого твердого вещества. В результате последующего элюирования получали 150 мг диастереомера В в виде белого твердого вещества.
(5) Раствор 100 мг (0,38 ммоля) хлоргидрата 2(R или S)-[1(R или S)-амино-2-метилпропил] -2,3-дигидро-1H-пирроло[1,2-а] индола (диастереомер A) в 30 мл дихлорметана обрабатывали 110 мг (0,5 ммоля) дитрет-бутилдикарбоната и 100 мг (1 ммоль) триэтиламина и перемешивали 72 часа. Этот раствор промывали последовательно 30 мл 1M хлористоводородной кислоты и 30 мл насыщенного водного бикарбоната натрия, а затем сушили над сульфатом магния. После фильтрации, концентрации фильтрата при пониженном давлении и очистки остатка при помощи быстрой хроматографии, используя простой диэтиловый эфир/петролейный эфир (температура кипения 40-60oC) (1:2) для элюирования, получали 100 мг трет-бутил-[1(R или S)- [2,3-дигидро-1H- пирроло[1,2-а]индол-2(R или S)-ил]-2-метилпропил]карбамата (диастереомер A) в виде масла.
(6) При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 55 мг трет-бутил-[I(R или S)- [2,3-дигидро-1H-пирроло[1,2-а] индол-2(R или S)-ил] -2- метилпропил]карбамата (диастереомер A) получали 40 мг 3-[2(R или S)-[1(R или S)-трет-бутоксиформамидо-2-метилпропил] -2,3- дигидро-1H-пирроло[1,2-а] индол-9-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-диона в виде красного масла.
Пример 18
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 80 мг 3-[2(R или S)-[1(R или S)- трет-бутоксиформамидо-2-метилпропил] -2,3-дигидро-1H-пирроло- [1,2-а] индол-9-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) получали 40 мг хлоргидрата 3-[2(R или 8)-амино- 2-метилпропил]-2,3-дигидро-1H-пирроло[1,2-а]индол-9-ил]-4-(1- метил-3-индолил)-1H-пиррол-2,5-диона (диастереомер B) в виде красного твердого вещества с температурой плавления 220-225oC.
3-[2(R или S)-[1(R или S)-трет-бутоксиформамидо-2-метилпропил]- 2,3-дигидро-1H-пирроло[1,2-а] индол-9-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) При помощи процедуры, аналогичной той, что была описана в Примере 17(5), из 90 мг (0,34 ммоля) хлоргидрата 2(R или S)-[1(R или S)-амино-2-метилпропил] -2,3-дигидро-1H-пирроло[1,2-а] индола (диастереомер B), полученного, как это описано в Примере 17(4), получали 100 мг трет-бутил-[1(R или S)-[2,3-дигидро-1H-пирроло- [1,2-а]индол-2(R или S)-ил]-2-метилпропил]карбамата (диастереомер B) в виде масла.
(2) При помощи процедуры, аналогичной той, что описана в Примере 1(4), из 100 мг трет-бутил-[1(R или S)-[2,3-дигидро- 1H-пирроло[1,2-а]индол-2(R или S)-ил] -2-метилпропил] карбамата получали 80 мг 3-[2(R или S)-[1(R или S)-трет-бутоксиформамидо- 2-метилпропил]-2,3-дигидро-1H-пирроло[1,2-а]- индол-9-ил]-4-(1-метил-3-индолил)-1H-пиррол-2,5-диона в виде красного масла.
Пример 19 При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 320 мг 3-[8(RS)-[1(RS)- трет-бутоксиформамидо-2-метилпропил] -7,8,9,10-тетрагидро-6H- азепино[1,2-а] индол-11-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона получали 220 мг хлоргидрата 3-[8(RS)-[1(RS)-амино-2-метилпропил] - 7,8,9,10-тетрагидро-6H-азепино[1,2-а] индол-11-ил] -4-(1-метил-3- индолил)-1H-пиррол-2,5-диона в виде красного твердого вещества с температурой плавления 248-256oC.
3-[8(RS)-[1(RS)-трет-бутоксиформамидо-2-метилпропил] -7,8,9,10- тетрагидро-6H-азепино[1,2-а]индол-11-ил]-4-(1-метил-3-индолил)- 1H-пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) При помощи процедуры, аналогичной той, что была описана в Примере 11(2), из 1,0 г (4,4 ммоля) 7,8,9,10-тетрагидро-6H-азепино- [1,2-а]индол-8(RS)-карбоновой кислоты получали 0,8 г 7,8,9,10-тетрагидро-N-метокси-N-метил-6H-азепино[1,2-а] индол- 8(RS)-карбоксамида в виде белого твердого вещества с температурой плавления 134-135oC.
(2) При помощи процедуры, аналогичной той, что была описана в Примере 17(3), из 0,8 г (2,9 ммоля) 7,8,9,10-тетрагидро-N-метокси- N-метил-6H-азепино[1,2-а] индол-8(RS)-карбоксамида получали 0,56 г изопропил-7,8,9,10-тетрагидро-6H-азепино[1,2-а]индол-8(RS)-илкетона в виде белого твердого вещества с температурой плавления 79-80oC.
(3) При помощи процедуры, аналогичной той, что была описана в Примере 11(4), из 0,56 г (2,2 ммоля) изопропил-7,8,9,10-тетрагидро- 6H-азепино[1,2-а] индол-8(RS)-илкетона получали 330 мг трет-бутил[1(RS)-[7,8,9,10-тетрагидро-6H-азепино[1,2-а] индол- 8(RS)-ил]-2-метилпропил]карбамата в виде смеси диастереомеров в виде белого твердого вещества с температурой плавления 152-153oC.
(4) При помощи процедуры, аналогичной той, что была описана в Примере 1(4), из 300 мг трет-бутил-[1(RS)-[7,8,9,10-тетрагидро- 6H-азепино[1,2-a]- индол-8(RS)-ил] -2-метилпропил] карбамата получали 350 мг 3-[8(RS)-[1(RS)-трет-бутоксиформамидо-2-метилпропил] -7,8,9,10- тетрагидро-6H-азепино[1,2-а] индол-11-ил] -4-(1-мeтил-3- индoлил)-1H-пиppoл-2,5-диoнa в виде красного масла.
Пример 20
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 800 мг 3-[7(RS)-[1(RS)-трет-бутоксиформамидо- 2-метилпропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона получали 480 мг хлоргидрата 3-[7(RS)-[1(RS)-амино-2-метилпропил] -6,7,8,9-тетрагидропиридо- [1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1H-пиррол-2,5-диона в виде красного твердого вещества с температурой плавления 238-244oC.
3-[7(RS)-[1(RS)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-метил-3-индолил)-1Н-пиррол- 2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) При помощи процедуры, аналогичной той, что была описана в Примере 11(2), из 2,0 г (9,3 ммоля) 6,7,8,9-тетрагидропиридо[1,2-а]- индол-7(RS)-карбоновой кислоты получали 1,6 г 6,7,8,9-тетрагидро-N- метокси-N-метилпиридо[1,2-а]индол-7(RS)-карбоксамида в виде бледно-желтого масла.
(2) При помощи процедуры, аналогичной той, что была описана в Примере 17(3), из 1,6 г 6,7,8,9-тетрагидро-N-метокси-N-метилпиридо- [1,2-а]индол-7(RS)-карбоксамида получали 1,05 г изопропил-6,7,8,9- тетрагидропиридо[1,2-а] индол-7(RS)-илкетона в виде коричнево-рыжеватого твердого вещества с температурой плавления 43-44oC.
(3) При помощи процедуры, аналогичной той, что была описана в Примере 11(4), из 1,0 г изопропил-6,7,8,9-тетрагидропиридо- [1,2-а]индол-7(RS)-илкетона получали 800 мг трет-бутил-[1(RS)- [6,7,8,9-тетрагидропиридо[1,2-а] индол-7(RS)-ил] -2- метилпропил]карбамата в виде белого твердого вещества с температурой плавления 55-57oC (в виде смеси диастереомеров).
(4) При помощи процедуры, аналогичной той, что была описана в Примере 1(4), из 700 мг тpeт.-бутил-[1(RS)-[6,7,8,9- тeтpaгидpoпиpидo[1,2-a]индoл- 7(RS)-ил] -2-метилпропил] карбамата получали 800 мг 3-[7(RS)-[1(RS)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил] -4-(1-метил-3-индолил)-1H- пиррол-2,5-диона в виде красной смолы.
Пример 21
При помощи процедуры, аналогичной той, что была описана в первом абзаце Примера 1, из 1,3 г 3-[8(S)-[1(S)- трет-бутоксиформамидо-2-метилпропил]-6,7,8,9-тетрагидропиридо- [1,2-а] индол-10-ил] -4-(1-фенил-3-индолил)-1H-пиррол-2,5-диона получали 1,12 г хлоргидрата 3-[8(S)-[1(S)-амино-2-метилпропил] - 6,7,8,9-тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-фенил-3-индолил)- 1H-пиррол-2,5-диона в виде красного твердого вещества с температурой плавления 235-245oC.
3-[8(S)-[1(S)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил] -4-(1-фенил-3-индолил)-1H- пиррол-2,5-дион, используемый в качестве исходного продукта, получали следующим образом:
(1) Охлажденный льдом раствор из 10 г (51,8 ммоля) 1-фенилиндола в 100 мл безводного простого диэтилового эфира обрабатывали по каплям в течение 5 минут раствором из 6 мл (68,8 ммоля) оксалилхлорида в 20 мл безводного простого диэтилового эфира. Эту смесь перемешивали 3 часа при охлаждении льдом, а затем обрабатывали 25 мл этанола одной порцией. После перемешивания в течение 10 минут растворитель удаляли при пониженном давлении, а остаточное твердое вещество кристаллизовали из 60 мл этанола, чтобы получить 12,38 г этил-1-фенилиндол-3-глиоксилата в виде бледно-желтого твердого вещества с температурой плавления 109-110oC.
(2) Смесь из 10 г (34,1 ммоля) этил-1-фенилиндол-3-глиоксилата и примерно 25 г никеля Ренея в 350 мл этанола и 150 мл воды нагревали при кипячении с обратным холодильником 6 часов. Суспензию фильтровали через фильтровальную бумагу, а твердое вещество промывали четырьмя порциями по 50 мл этилацетата, не давая при этом твердому веществу высохнуть. Фильтрат концентрировали при пониженном давлении, а остаток подвергали очистке с использованием флэш-хроматографии на силикагеле, применяя этилацетат/гексан (1:2) для элюирования. Получали 6,38 г этил-1-фенилиндол-3-ацетата в виде желтого масла.
(3) Раствор из 6,3 г (22,6 ммоля) этил-1-фенилиндол-3-ацетата в 20 мл этанола обрабатывали 20 мл (40 ммолей) 2M раствора гидрата окиси натрия и выдерживали при комнатной температуре 17 часов. Этанол удаляли при пониженном давлении, а водный раствор промывали двумя 20 мл порциями простого диэтилового эфира. Водную фазу подкисляли концентрированной хлористоводородной кислотой, а полученную суспензию хранили при 0oC в течение 2 часов. Суспензию фильтровали, а твердое вещество кристаллизовали из метанола/воды (2:1), чтобы получить 5,6 г 1-фенилиндол-3-уксусной кислоты в виде голубовато-серого твердого вещества с температурой плавления 131-135oC.
(4) Охлажденный льдом раствор из 3 г (8,77 ммоля) трет-бутил-[1(S)-[6,7,8,9-тетрагидропиридо[1,2-а]индол-8(S)-ил]-2- метилпропил]карбамата, полученного, как это описано в Примере 11(4), в 50 мл безводного простого диэтилового эфира обрабатывали по каплям в атмосфере азота в течение 5 минут раствором из 0,85 мл (9,74 ммоля) оксалилхлорида в 5 мл безводного простого диэтилового эфира. Спустя еще 5 минут растворитель удаляли при пониженном давлении, а остаток растворяли в 50 мл сухого дихлорметана. Раствор добавляли по каплям при 0oC в перемешиваемую смесь из 2,2 г (8,77 ммоля) 1-фенилиндол-3-уксусной кислоты и 3,65 мл (26,3 ммоля) триэтиламина в 50 мл сухого дихлорметана. Смесь перемешивали 17 часов, затем растворитель удаляли при пониженном давлении. После очистки при помощи флэш-хроматографии на силикагеле, используя этилацетат/гексан (1:2) для элюирования, последующей кристаллизации из этилацетата/гексана получали 1,6 г 3-[8(S)-[1(S)-трет-бутоксиформамидо-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а] индол-10-ил]-4-(1-фенил-3-индолил)-фуран- 2,5-диона в виде оранжевого твердого вещества с температурой плавления 148-150oC.
(5) Раствор из 1,6 г (2,54 ммоля) 3-[8(S)-[1(S)-тpeт-бутoкcифopмaмидo-2-метилпропил] -6,7,8,9- тетрагидропиридо[1,2-а]индол-10-ил]-4-(1-фенил-3-индолил)-фуран- 2,5-диона в 20 мл сухого N,N-диметилформамида обрабатывали 5,35 мл (25,4 ммоля) гексаметилдисилазана и 0,41 г (12,8 ммоля) метанола. Раствор нагревали до 50oC 3 часа, а затем обрабатывали еще 5,35 мл (24,8 ммоля) гексаметилдисилазана и 0,41 г (12,8 ммоля) метанола. Спустя всего 6 часов растворитель выпаривали при пониженном давлении, а остаток еще раз выпаривали с 20 мл метанола. В результате флэш-хроматографии остатка на силикагеле, используя этилацетат/гексан (1:2) для элюирования, получали 1,35 г 3-[8(S)-[1(S)-трет-бутоксиформамидо-2- метилпропил] -6,7,8,9-тетрагидропиридо[1,2-а] индол-10-ил]-4-(1- фенил-3-индолил)-1H-пиррол-2,5-диона в виде красного твердого вещества с температурой плавления 165-168oC.
Приводимые ниже примеры служат иллюстрацией общих фармацевтических композиций, содержащих соединения, являющиеся предметом настоящего изобретения.
Пример А
Таблетки, содержащие следующие ингредиенты, могут быть получены при помощи известного способа.
Ингредиент - На таблетку
Соединение формулы I - 5,0 мг
Лактоза - 125,0 мг
Маисовый крахмал - 75,0 мг
Тальк - 4,0 мг
Стеарат магния - 1,0 мг
Масса таблетки: 210,0 мг
Пример Б
Капсулы, содержащие следующие ингредиенты, могут быть получены при помощи известного способа.
Ингредиент - На капсулу
Соединение формулы I - 10,0 мг
Лактоза - 165,0 мг
Маисовый крахмал - 20,0 мг
Тальк - 5,0 мг
Масса заполненной капсулы: 200,0 мгу

Описываются новые замещенные пирролы общей формулы I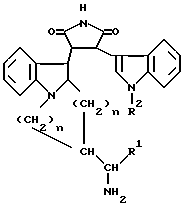
в которой R1 представляет собой низший алкил, низший циклоалкил, арил или низший аралкил; R2 представляет собой водород, арил или низший алкил; m и n равны 1 или 2, а также их фармацевтически приемлемые соли, которые можно использовать для лечения или предотвращения заболеваний, особенно для лечения или предотвращения воспалительных, иммунологических, онкологических, бронхолегочных, дерматологических или сердечно-сосудистых расстройств, для лечения астмы, СПИДа или диабетических осложнений, или для стимуляции роста волос. 2 с. и 7 з.п.ф-лы.
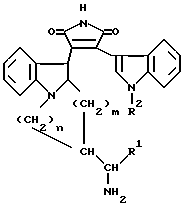
в которой R1 представляет собой низший алкил, низший циклоалкил, арил или низший аралкил;
R2 представляет собой водород, арил или низший алкил;
m и n равны 1 или 2,
а также их фармацевтически приемлемые соли.
Приоритет по пунктам:
10.05.93 по пп.1 - 7 и 9.
| 0 |
|
SU384349A1 | |
| 0 |
|
SU328026A1 | |
| Грузоподъемное устройство | 1973 |
|
SU470490A1 |
| Машковский М.Д | |||
| Лекарственные средства.-М.: Медицина, 1972, ч.2, с.170-172. | |||
