Изобретение относится к органическим соединениям, их получению и применению в качестве фармацевтических агентов.
Одним из объектов изобретения являются соединения формулы
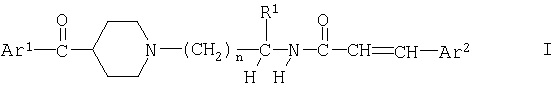
в свободной форме или в форме соли, где
Ar1 обозначает фенил, замещенный одним или несколькими атомами галогена,
Ar2 обозначает фенил или нафтил, который является незамещенным или замещен одним или несколькими заместителя, выбранными из ряда, включающего галоген, циано, гидрокси, нитро, С1-С8алкил, С1-С8галоалкил, С1-С8алкокси или С1-С8алкоксикарбонил,
R1 обозначает водород или С1-С8алкил, необязательно замещенный гидрокси-, С1-С8алкокси-, ацилоксигруппой, -N(R2)R3, галогеном, карбоксигруппой, C1-C8алкоксикарбонилом, -CON(R4)R5 или одновалентной циклической органической группой,
R2 и R3 каждый независимо друг от друга обозначает водород или С1-С8алкил, или R2 обозначает водород и R3 обозначает ацил или - SO2R6, или R2 и R3 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную гетероциклическую группу,
R4 и R5 каждый независимо друг от друга обозначает водород или С1-С8алкил, или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную гетероциклическую группу,
R6 обозначает С1-С8алкил, С1-С8галоалкил или фенил, необязательно замещенный C1-C8-алкилом, и
n обозначает 1, 2, 3 или 4,
при условии, что когда Ar1 обозначает пара-хпорфенил и R1 обозначает водород, то Ar2 не обозначает фенил или пара-нитрофенил.
Понятия, которые применяют в настоящем описании, имеют следующие значения:
"С1-С8алкил" в контексте настоящего описания обозначает С1-С8алкил с прямой или разветвленной цепью, который может представлять собой, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил с прямой или разветвленной цепью, гексил с прямой или разветвленной цепью, гептил с прямой или разветвленной цепью или октил с прямой или разветвленной цепью. Предпочтительно С1-С8алкил обозначает С1-С4алкил.
"С1-С8алкокси" в контексте настоящего описания обозначает C1-С8алкоксигруппу с прямой или разветвленной цепью, которая может обозначать, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси с прямой или разветвленной цепью, гексилокси с прямой или разветвленной цепью, гептилокси с прямой или разветвленной цепью или октилокси с прямой или разветвленной цепью. Предпочтительно С1-С8алкокси обозначает С1-С4алкокси.
"С1-С8галоалкил" в контексте настоящего описания обозначает С1-С8алкил, как он определен выше, замещенный одним или несколькими атомами галогена, предпочтительно одним, двумя или тремя атомами галогена.
"Ацил" в контексте настоящего описания обозначает алкилкарбонил, например С1-С8алкилкарбонил, где С1-С8алкил может представлять собой одну из указанных выше С1-С8алкильных групп, необязательно замещенную одним или несколькими атомами галогена; циклоалкилкарбонил, например С3-С8циклоалкилкарбонил, где С3-С8циклоалкил может представлять собой, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил; 5- или 6-членный гетероциклилкарбонил, несущий в кольце один или два гетероатома, выбранных из группы, включающей азот, кислород и серу, такой как фурилкарбонил или пиридилкарбонил; арилкарбонил, например С6-С10арилкарбонил, такой как бензоил; или аралкилкарбонил, например С6-С10арил-С1-С4алкилкарбонил, такой как бензилкарбонил или фенилэтилкарбонил. Предпочтительно ацил обозначает С1-С4алкилкарбонил.
"Ацилокси" в контексте настоящего описания обозначает алкилкарбонилокси, например С1-С8алкилкарбонилокси, где С1-С8алкил может представлять собой одну из описанных выше С1-С8алкильных групп, необязательно замещенную одним или несколькими атомами галогена; циклоалкилкарбонилокси, например С3-С8циклоалкилкарбонилокси, где С3-С8циклоалкил может обозначать, например, циклопропил, циклобутил, циклопетил, циклогексил, циклогептил или циклооктил; 5- или 6-членную гетероциклилкарбонилоксигруппу, несущую в кольце один или два гетероатома, выбранных из группы, включающей азот, кислород и серу, такую как фурилкарбонилокси или пиридилкарбонилокси; арилкарбонилокси, например С6-С10арилкарбонилоксигруппу, такую как бензоилокси; или аралкилкарбонилоксигруппу, например С6-С10арил-С1-С4алкилкарбонилоксигруппу, такую как бензилкарбонилокси или фенилэтилкарбонилокси. Предпочтительно ацилокси обозначает C1-С4алкилкарбонилокси.
"Галоген" в контексте настоящего описания обозначает фтор, хлор, бром или йод; предпочтительно фтор, хлор или бром.
В Ar1 фенильная группа может быть замещена одним, двумя или тремя, предпочтительно одним или двумя атомами галогена, которые предпочтительно выбирают из атомов фтора и хлора. Когда в качестве заместителя присутствует один атом галогена, он предпочтительно находится в пара-положении относительно указанной карбонильной группы. Когда в качестве заместителей присутствуют два или три атома галогена, то предпочтительно один из них находится в пара-положении относительно указанной карбонильной группы и по меньшей мере один из остальных находится в орто-положении относительно указанной карбонильной группы.
Если Ar2 обозначает замещенный фенил, то он может, например, быть замещен одним, двумя, тремя, четырьмя или пятью, предпочтительно одним, двумя или тремя, указанными выше заместителями. Ar2 может, например, обозначать монозамещенный фенил, заместителем которого предпочтительно является галоген, циано, нитро или С1-С4алкоксигруппа, который предпочтительно находится в орто- или мета-положении относительно указанной -СН=СН-группы. В другом варианте Ar2 может обозначать, например, дизамещенный фенил, заместители которого предпочтительно выбирают из группы, включающей галоген, циано, гидрокси, нитро, С1-С4алкокси, С1-С4алкил и С1-С4галоалкил, особенно предпочтительно заместителями являются два атома галогена (одинаковые или различные атомы галогена), две С1-С4алкоксигруппы, две С1-С4алкильные группы, две С1-С4галоалкильные группы, один галоген и одна цианогруппа, один галоген и одна С1-С4алкоксигруппа, один галоген и одна нитрогруппа, один галоген и одна гидроксигруппа, один галоген и один С1-С4 галоалкил, одна циано- и одна С1-С4алкоксигруппа, одна гидроксигруппа и один С1-С4алкил или одна гидрокси- и одна С1-С4алкоксигруппа. В другом варианте Ar2 может обозначать, например, тризамещенный фенил, заместители которого предпочтительно выбирают из группы, включающей галоген, гидрокси, С1-С4алкокси и С1-С4алкоксикарбонил, особенно предпочтительно заместителями являются три атома галогена (одинаковые атомы или два или три различных атома галогена) или две С1-С4алкоксигруппы и один галоген, гидрокси или С1-С4алкоксикарбонил. Еще в одном варианте Ar2 может обозначать, например, пентазамещенный фенил, заместителями которого предпочтительно являются атомы галогена, прежде всего фтор. Особенно предпочтительными группами Ar2являются цианфенил, прежде всего мета-цианфенил, и дизамещенный фенил, одним из заместителей которого является С1-С4алкоксигруппа, предпочтительно находящаяся в орто-положении относительно -СН=СН-группы, а вторым заместителем, расположенным предпочтительно в пара-положении относительно С1-С4алкоксигруппы, является С1-С4алкокси, галоген, циано или С1-С4алкил.
R1, который обозначает необязательно замещенный С1-С8алкил, предпочтительно представляет собой необязательно замещенный С1-С4алкил, прежде всего С1-С4алкил или замещенный метил или этил. Когда R1 замещен циклической органической группой, то последняя может обозначать карбоциклическую или гетероциклическую группу, например С3-С15карбоциклическую группу или 5-7-членную гетроциклическую группу, несущую один или несколько, предпочтительно один, два или три кольцевых гетероатома, выбранных из группы, включающей азот, кислород и серу. С3-С15карбоциклическая группа может, например, обозначать циклоалифатическую группу, имеющую 3-8 атомов углерода, предпочтительно С5- или С6циклоалкил, такой как циклопентил, метилциклопентил или циклогексил. В другом варианте С3-С15карбоциклическая группа может обозначать, например, С1-С8ароматическую группу, такую как фенил, который может быть незамещенный или замещен С1-С8алкилом, С1-С8алкоксигруппой, галогеном, цианогруппой, -CON(R4)R5, -SO2N(R4)R5 или C1-С8алкилсульфониламиногрупой, где R4 и R5 имеют указанные выше значения. Гетероциклическая группа может нести в кольце один атом азота, кислорода или серы или может нести два атома азота или один атом кислорода и один или два атома азота, или один атом серы и один или два атома азота. Гетероциклическая группа предпочтительно представляет собой гетероциклическую ароматическую группу, прежде всего 5- или 6- членную гетероциклическую группу, такую как фурил, имидазолил, тиазолил или пиридил. В наиболее предпочтительных соединениях R1 обозначает С1-С4алкил, замещенный гидроксигруппой, фенилом, или 5- или 6- членной гетероциклической ароматической группой, несущей один или два кольцевых гетероатома, выбранных из группы, включающей азот, кислород и серу.
Предпочтительными соединениями формулы I в свободной форме или в форме соли являются соединения, в которых
Ar1 обозначает фенил, замещенный фтором или хлором в пара-положении относительно указанной карбонильной группы и необязательно дополнительно замещенный галогеном в орто-положении относительно указанной карбонильной группы,
Ar2 обозначает фенил, монозамещенный заместителем, выбранным из ряда, включающего галоген, циано, нитро и С1-С4алкокси, фенил, замещенный двумя одинаковыми или различными заместителями, выбранными из ряда, включающего галоген, циано, гидрокси, С1-С4алкокси, С1-С4алкил, C1-С4галоалкил и нитро, или фенил, замещенный тремя одинаковыми или различными заместителями, выбранными из ряда, включающего галоген, гидрокси, С1-С4алкокси и С1-С4алкоксикарбонил,
R1 обозначает водород, С1-С4алкил или С1-С4алкил, замещенный гидроксигрупой, С3-С8циклоалкилом, фенилом, фенилом, замещенным С1-С4алкилсульфониламиногруппой, или 5- или 6-членной гетероциклической ароматической группой, несущей один или несколько кольцевых гетероатомов, выбранных из ряда, включающего азот, кислород и серу, и
n обозначает 1 или 2.
Также предпочтительными соединениями формулы I в свободной форме или в форме соли являются соединения, в которых
Ar1 обозначает фенил, замещенный фтором или хлором в пара-положении относительно указанной карбонильной группы,
Ar2 обозначает фенил, замещенный в орто-положении относительно указанной -СН=СН-группы С1-С4алкоксигруппой и замещенный в пара-положении относительно С1-С4 алкоксигруппы цианогруппой, галогеном или С1-С4алкоксигруппой,
R1 обозначает С1-С4алкил, замещенный гидроксигруппой, фенилом, фенилом, замещенным С1-С4алкилсульфониламиногруппой, или 5- или 6-членной гетероциклической ароматической группой, несущей один или два кольцевых гетероатома, выбранных из ряда, включающего азот, кислород и серу, и
n обозначает 1.
Соединения, представленные формулой, обладают способностью образовывать кислотно-аддитивные соли, прежде всего фармацевтически приемлемые кислотно-аддитивные соли. Фармацевтически приемлемые кислотно-аддитивные соли соединения формулы включают соли неорганических кислот, например галогенводородных кислот, таких как фтористоводородная кислота, соляная кислота, бромистоводородная кислота или йодистоводородная кислота, азотной кислоты, серной кислоты, фосфорной кислоты; и органических кислот, например алифатических монокарбоновых кислот, таких как муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота и масляная кислота, алифатических гидроксикислот, таких как молочная кислота, лимонная кислота, винная кислота или яблочная кислота, дикарбоновых кислот, таких как малеиновая кислота или янтарная кислота, ароматических карбоновых кислот, таких как бензойная кислота, пара-хлорбензойная кислота, дифенилуксусная кислота или трифенилуксусная кислота, ароматических гидроксикислот, таких как орто-гидроксибензойная кислота, пара-гидроксибензойная кислота, 1-гидроксинафталин-2-карбоновая кислота или 3-гидроксинафталин-2-карбоновая кислота, и сульфоновых кислот, таких как метансульфоновая кислота или бензолсульфоновая кислота. Эти соли можно получать из соединений формулы с помощью известных процессов солеобразования.
Соединения формулы, которые содержат кислотные, например, карбоксильные группы, также могут образовывать соли с основаниями, прежде всего с фармацевтически приемлемыми основаниями, которые хорошо известны в данной области; такие соли могут включать соли металлов, прежде всего соли щелочных металлов или щелочно-земельных металлов, например соли натрия, калия, магния или кальция, или соли аммиака или фармацевтически приемлемых органических аминов или гетероциклических оснований, таких как этаноламины, бензиламины или пиридины. Эти соли можно получать из соединений формулы I с помощью известных процессов солеобразования.
Когда R1 имеет значение, отличное от водорода, атом углерода, к которому R1 присоединен в формуле, является асимметричным, и в этом случае соединения существуют в виде индивидуальных оптически активных изомерных форм или их смесей, например в виде рацемических или диастереоизомерных смесей. Под объем изобретения подпадают как индивидуальные оптически активные R- и S-изомеры, так и их смеси, например рацемические или диастереоизомерные смеси.
Конкретными предпочтительными соединениями по изобретению являются соединения, описанные в примерах, прежде всего в примерах 4, 9, 10, 15, 18, 19, 20, 21, 23, 24, 25, 28, 29, 30, 37, 38, 40, 42, 43, 44 и 45.
Изобретение относится также к способу получения соединений формулы, предусматривающему
(I) (А) взаимодействие соединения формулы
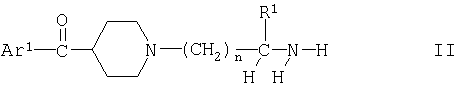
с соединением формулы
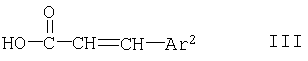
или его образующим амид производным, где Ar1, Ar2, R1 и n имеют указанные выше значения, или
(Б) взаимодействие соединения формулы III или его образующего амид производного с соединением формулы
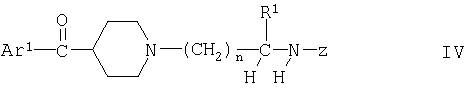
где Ar1, R1 и n имеют указанные выше значения и Z обозначает твердофазный субстрат, химически связанный с указанным атомом азота, и отщепление образовавшего продукта от субстрата путем замены Z на водород; и
(II) выделение продукта в свободной форме или в форме соли.
Согласно варианту (А) способа, оединение формулы II может находиться в свободной форме или в форме соли. Вариант (А) способа можно осуществлять с помощью известных методов, например путем взаимодействия соединения формулы II с галогенангидридом, прежде всего с хлорангидридом кислоты формулы III, с использованием известных процессов образования амидов. Как правило, соединение формулы II в свободной форме или в форме соли подвергают взаимодействию со свободной карбоновой кислотой формулы III, например, с помощью известных процессов, таких как взаимодействие в присутствии третичного амина и связывающего пептид агента, такого как фосфониевая соль тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетрааметилурония или диизопропилкарбодиимид; эту реакцию осуществляют в инертном органическом растворителе, например, в галогенированном углеводороде, таком как дихлорметан; как правило, температура реакции составляет от 0 до 40°С, предпочтительно представляет собой температуру окружающей среды.
Согласно другому пути осуществления варианта (А) способа соединение формулы II, предпочтительно в форме соли, подвергают взаимодействию с образующим амид производным кислоты формулы III, представляющим собой тиоэфир формулы
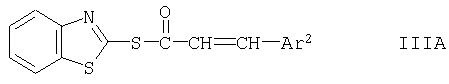
где Ar2 имеет указанные выше значения. Реакцию можно осуществлять с помощью известных процессов или аналогично методам, описанным в примерах. Ее можно осуществлять в присутствии третичного основания, такого как N-метилморфолин. Реакцию, как правило, осуществляют в органическом растворителе, предпочтительно спирте, таком как этанол. Температура реакции может составлять, например, от 30 до 60°С, как правило, от 40 до 50°С.
Вариант (Б) способа можно осуществлять с помощью известных методов, например, путем взаимодействия связанного с субстратом соединения со свободной кислотой в известных условиях пептидного сочетания, например, в присутствии третичного амина и указанного выше связывающего пептид агента. Реакцию можно осуществлять в инертном органическом растворителе, таком как диметилформамид (ДМФ). Приемлемая температура реакции составляет от 0 до 40°С, например, 15-25°С. Продукт можно отщеплять от субстрата известным методом, например обработкой трифторуксусной кислотой в случае, когда атом N связан с СН2 бензильной группы в Z.
Соединения формулы III либо поступают в продажу либо их можно получать известными методами. Соединения формулы III A можно получать взаимодействием кислоты формулы III с 2,2'-дибензотиазолилдисульфидом в присутствии трифенилфосфина и третичного основания, такого как N-метилморфолин, например согласно методам, описанным в примерах.
Соединения формулы II можно получать взаимодействием соединения формулы
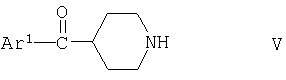
с соединением формулы
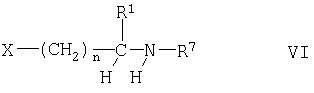
где Ar1, R1 и n имеют указанные выше значения, при условии, что когда R1 содержит реакционноспособную функциональную группу, такую как гидроксигруппа, реакционноспособная группа может присутствовать в защищенной форме, например гидроксигруппа защищена в виде трет-бутоксигруппы, R7 обозначает водород или аминозащитную группу, например трет-бутоксикарбонильную группу, и Х обозначает галоген, и если R7 обозначает защитную группу путем замены R1 в продукте водородом, и если R1 в продукте содержит защищенную функциональную группу путем замены защитной группы водородом. Если R7 обозначает водород, реакцию между соединением формулы V и солью соединения формулы VI можно осуществлять согласно процессам, описанным в патенте US 4559349. Если R обозначает защитную группу, реакцию между соединениями формул V и VI можно осуществлять известными методами, например, в присутствии третичного органического основания, такого как триэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), как правило, в инертном растворителе, например, в полярном растворителе, таком как диметилформамид, реакцию, как правило, проводят при 0-40°С, предпочтительно при температуре окружающей среды. Замену защитной группы R7 водородом можно осуществлять с помощью известных методов; например, если R7 обозначает трет-бутоксикарбонил, обработкой карбоновой кислотой, такой как трифторуксусная кислота. Замену защитной группы в R1 можно осуществлять с помощью известных методов, например, когда R1 содержит гидроксигруппу, защищенную простой эфирной группой, такой как трет-бутоксигруппа, обработкой HBr в карбоновой кислоте, такой как уксусная кислота; когда R7 представляет собой защитную группу, то такая обработка приводит также к замене R7 водородом. Соединения формул V и VI являются известными или их можно получать известными методами.
Если в описании имеется ссылка на защищенные функциональные группы или на защитные группы, то предполагается, что защитные группы можно выбирать в зависимости от природы функциональной группы, например, как описано в "Protective Groups in Organic Synthesis", T.W.Greene и P.G.M.Wuts, John Wiley & Sons Inc, 2-е изд., 1991, где описаны также методы, применяемые для замены защитных групп водородом.
Соединения формулы II можно получать также путем взаимодействия соединения формулы V с соединением формулы
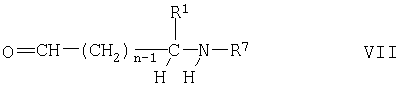
где R1, R7 и n имеют указанные выше значения, и восстановителем, таким как цианборогидрид натрия или триацетоксиборогидрид натрия, например, с использованием известных процессов восстановительного аминирования, как правило, в инертном органическом растворителе, например простом эфире, таком как тетрагидрофуран (ТГФ), температура реакции, как правило, составляет от 0 до 40°С, и если R7 обозначает защитную группу путем замены ее на водород. Соединения формулы VII являются известными или их можно получать с помощью известных процессов.
Соединения формулы II, в которых R1 обозначает гидроксиметил, можно получать также путем взаимодействия соединения формулы V с трет-бутиловым эфиром (R)-4-формил-2,2-диметилоксазолидин-3-карбоновой кислоты формулы
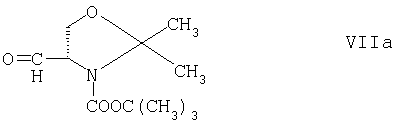
и восстановителем, таким как триацетоксиборогидрид натрия, например, в условиях, описанных выше для взаимодействия соединений формул V и VII, и взаимодействия продукта с реагентом, который может расщеплять оксазолидиновое кольцо, и замены связанной с азотом эфирной группы водородом, например хлористым водородом, в этаноле или диоксане, как это описано ниже в примерах, в этом случае получают соединение формулы II в виде гидрохлорида. Продукт реакции соединений формул V и VIIa, например, для улучшения в случае необходимости энантиомерной чистоты можно перед расщеплением оксазолидинового кольца обрабатывать оптически активной кислотой, такой как ди-О,О-бензоил-L-винная кислота. Соединение формулы VIIa можно получать согласно методу, описанному у A.D.Campbell и др., Synthesis 1707-1709 (1998) или G.Ageno и др., Tetrahedron 51, 8121-8134 (1995).
Соединения формулы II, в которых R обозначает С1-С8 алкоксиметил или ацилоксиметил, можно получать с помощью соответствующей этерификации или ацилирования соединений формулы II, в которых R1 обозначает гидроксиметил.
Соединения формулы IV можно получать взаимодействием соединения формулы V с соединением формулы
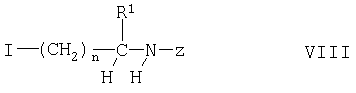
где R1, Z и n имеют указанные выше значения, например, с использованием известных процессов, таких как взаимодействие в инертном органическом растворителе, таком как ДМФ, в присутствии третичного амина, как правило, при температуре от 40 до 60°С. Соединения формулы VIII можно получать взаимодействием соединения формулы
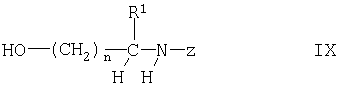
где R1, Z и n имеют указанные выше значения, с йодом, например, с помощью известных процессов, таких как взаимодействие в инертном органическом растворителе, таком как смесь ТГФ и ацетонитрила, в присутствии триарилфосфина и имидазола, как правило, при температуре от 10 до 40°С. Соединения формулы IX можно получать взаимодействием соединения формулы
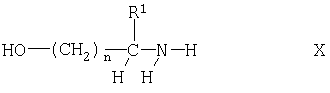
где R1 и n имеют указанные выше значения, с твердофазным субстратом Z, который несет группу, такую как альдегидная группа, вступающую в реакцию с аминогруппой. Такие твердофазные субстраты, включая модифицированные смолы, прежде всего модифицированные полистирольные смолы, имеются в продаже. Соединения формулы Х являются известными или их можно получать известными методами.
Соединения формулы I в свободной форме общепринятыми методами можно превращать в соль, и наоборот, соединения в свободной форме или в форме соли можно получать в форме гидратов или сольватов, содержащих растворитель, который применяют для кристаллизации. Соединения формулы I можно выделять из реакционных смесей и очищать общепринятыми методами. Изомеры, такие как энантиомеры, можно получать общепринятым методом, например, с помощью фракционированной кристаллизации или асимметричного синтеза, из исходных материалов, которые являются соответствующим образом асимметрично замещенными, например, из оптически активных материалов.
Соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли, обозначенные далее так же как агенты по изобретению, можно применять в качестве фармацевтических агентов. Таким образом, изобретение относится также к соединению формулы I в свободной форме или в форме фармацевтически приемлемой соли, предназначенному для применения в качестве фармацевтического агента. Агенты по изобретению действуют в качестве антагонистов CCR-3-рецептора, ингибируя тем самым воспаление и инфильтрацию и активацию воспалительных клеток, прежде всего эозинофилов, и ингибируя аллергическую реакцию. Ингибирующую активность агентов по изобретению можно продемонстрировать с помощью следующего анализа:
Анализ связывания CCR-3
С помощью этого анализа оценивают воздействие агентов по изобретению на связывание человеческого эотаксина с человеческим CCR-3. Рекомбинантные клетки, экспрессирующие человеческий CCR-3, иммобилизуют с использованием покрытых агглютинином из проростков пшеницы (ППА) поливинилтолуиденовых (ПВТ) SPA-гранул (фирма Amersham) посредством специфического связывания между ППА и углеводными остатками гликопротеинов на поверхности клеток. Меченный с помощью [125I] человеческий эотаксин (фирма Amersham) специфично связывается с CCR-3-рецепторами, приводя человеческий [125I]-эотаксин в близкий контакт с SPA-гранулами. Испускаемые человеческим [125I]-эотаксином альфа-частицы возбуждают благодаря их близкому расположению флуорофор в гранулах и вызывают испускание света. Свободный человеческий [125I]-эотаксин в растворе не имеет близкого контакта со сцинтиллятором и поэтому не вызывает испускание света. Следовательно, количество вспышек является мерой того, в какой степени тестируемое соединение ингибирует связывание эотаксина с CCR-3.
Приготовление буфера для анализа: 5,96 г HEPES и 7,0 г хлорида натрия растворяют в дистиллированной воде и добавляют 1 М водный раствор CaCl3 (1 мл) и 1 М водный раствор MgCl2 (5 мл). Значение рН доводят до 7,6 с помощью NaOH и конечный объем раствора доводят до 1 л с помощью дистиллированной воды. Затем в растворе растворяют 5 г бычьего сывороточного альбумина и 0,1 г азида натрия и полученный буфер хранят при 4°С. В день применения в 50 мл буфера добавляют таблетку, содержащую смесь ингибиторов протеазы, типа Complete™ (фирмы Boehringer).
Приготовление буфера для гомогенизации: Трис-основание (2,42 г) растворяют в дистиллированной воде, значение рН раствора доводят до 7,6 с помощью соляной кислоты и раствор разбавляют дистиллированной водой до конечного объема 1 л. Полученный буфер хранят при 4°С. В день применения в 50 мл буфера добавляют таблетку, содержащую смесь ингибиторов протеазы типа Complete™.
Получение мембран: конфлюэнтные клетки крысиного базофильного лейкоза (RBL-2H3), стабильно экспрессирующие CCR-3, выделяют из колб для культуры ткани, используя не содержащий фермент буфер для диссоциации клеток, и ресуспендируют в забуференном фосфатом физиологическом растворе. Клетки центрифугируют (800×g, 5 мин), дебрис ресуспендируют в охлажденном на льду буфере для гомогенизации, используя 1 мл буфера для гомогенизации на 1 г клеток, и инкубируют на льду в течение 30 мин. Клетки гомогенизируют на льду посредством 10 ударов в стеклянной ступке с пестиком. Гомогенат центрифугируют (800 г, 5 мин, 4°С), супернатант еще раз центрифугируют (48000×g, 30 мин, 4°С) и дебрис повторно растворяют в буфере для гомогенизации, содержащем 10 об. % глицерина. Содержание протеина в препарате мембран оценивают методом Брэдфорда (Anal. Biochem., 72:248 (1976)) и аликвоты быстро замораживают и хранят при - 80°С.
Анализ осуществляют в конечном объеме 250 мкл на лунку планшета типа Optiplate (фирма Canberra Packard). В определенные лунки планшета Optiplate добавляют 50 мкл растворов тестируемого соединение в буфере для анализа, содержащем 5% ДМСО (концентрации от 0,01 нМ до 10 мкМ). Для определения общего связывания в определенные другие лунки добавляют 50 мкл буфера для анализа, содержащего 5% ДМСО. Для определения неспецифического связывания в другие выбранные лунки добавляют 50 мкл 100 нМ человеческого эотаксина (фирма R&D Systems) в буфере для анализа, содержащем 5% ДМСО. Во все лунки добавляют по 50 мкл человеческого [125I]-эотаксина (фирма Amersham) в буфере для анализа, содержащем 5% ДМСО в концентрации 250 пМ (получая конечную концентрацию 50 пМ на лунку), по 50 мкл ППА-ПВТ SPA-гранул в буфере для анализа (получая конечную концентрацию 1,0 мг гранул на лунку) и по 100 мкл препарата мембран в концентрации 100 мкг протеина в буфере для анализа (получая конечную концентрацию 10 мкг протеина на лунку). Затем планшет инкубируют в течение 4 ч при комнатной температуре. Планшет запечатывают с помощью TopSeal-S (фирма Canberra Packard) согласно инструкциям производителя. Полученные вспышки подсчитывают с помощью устройства Canberra Packard TopCount, учет каждой лунки осуществляют в течение 1 мин. Из графиков зависимости ингибирования от концентрации обычным образом рассчитывают концентрацию тестируемого соединения, при которой связывание ингибируется на 50% (IC50).
Для соединений из приведенных ниже примеров с помощью вышеописанного анализа установлено, что значения IC50 составляют менее 1 мкМ. Например, для соединений из примеров 1, 2, 4, 7, 9, 13, 20, 23, 25, 28, 30, 38, 40, 43 и 44 значения IC50(нМ) составляют 125, 68, 13, 15, 5, 26, 8, 10, 11, 2, 13, 14, 6, 22 и 25 соответственно.
Для большинства соединений из примеров обнаружено избирательное действие в отношении ингибирования связывания с CCR-3 по сравнению с ингибированием связывания с альфа-1 адренергическим рецептором. Ингибирующее действие агентов по изобретению в отношении связывания с альфа-1 адренергическим рецептором можно продемонстрировать с помощью следующего анализа:
Кору головного мозга самцов крыс Sprague-Dawley (175-200 г) отсекают и гомогенизируют в 10 объемах охлажденного на льду 0,32 М раствора сахарозы (содержащего 1 мМ дигидрат MgCl2 и 1 мМ К2НРО4) в стеклянном/тефлоновом гомогенизаторе. Мембраны центрифугируют при 1000×g в течение 15 мин, дебрис отбрасывают и повторяют центрифугирование. Супернатанты объединяют и центрифугируют при 18000×g в течение 15 мин. Дебрис подвергают осмотическому шоку в 10 объемах воды и выдерживают на льду в течение 30 мин. Суспензию центрифугируют при 39000×g в течение 20 мин, ресуспендируют в буфере Кребса-Хенселейта, рН 7,4 (1,17 мМ MgSO4 (безводный) 4,69 мМ KCl, 0,7 мМ К2HPO4 (безводный), 0,11 М NaCl, 11 мМ D-глюкоза и 25 мМ NaHCO3), содержащем 20 мМ Трис, и выдерживают в течение 2 дней при - 20°С. Затем мембраны подвергают оттаиванию при 20-23°С, трижды промывают буфером Кребса-Хенселейта путем центрифугирования при 18000×g в течение 15 мин, выдерживают в течение ночи при 4°С и вновь трижды промывают. Конечный дебрис ресуспендируют в этом же буфере с помощью стеклянного/тефлонового гомогенизатора из расчета 125 мл/100 мембран. Отбирают образец для определения концентрации протеина (с помощью анализа Брадфорда с использованием гамма-глобулина в качестве стандарта) и оставшиеся аликвоты хранят при - 80°С.
В полученных мембранах анализируют связывания радиолиганда. Анализ проводят в трех повторностях с использованием 96-луночных планшетов, содержащих [125I]-НЕАТ (фирма Amersham) (40 пМ, Kd:58,9+18,7 пМ), немеченое тестируемое соединение и мембрану (57,1 мкг/мл). получая конечный объем 250 мкл (буфер для анализа, содержащий 50 мМ Трис-основание и 0,9% (мас/об) NaCl, рН 7,4). Планшеты инкубируют при 37°С в течение 60 мин, после чего осуществляют быструю вакуумную фильтрацию с использованием 96-луночных фильтрационных планшетов Whatman GF/C. Затем каждый планшет трижды промывают 10 мл охлажденного на льду буфера для анализа с использованием клеточного харвестера фирмы Brandel (Гейтерсбург, штат Мэриленд). После сушки планшетов в течение 3 ч при 50°С в каждую лунку добавляют по 40 мкл сцинтилляциооной жидкости Microscint 20, планшеты инкубируют при комнатной температуре в течение еще 20 мин и оценивают количественно удержанную радиоактивность с помощью сцинтилляционного счетчика типа Packard Topcount NXT.
Маточный растворы тестируемых соединений сначала растворяют в 100% ДМСО и разбавляют буфером для анализа до требуемых концентраций, доводя концентрацию ДМСО до 1 об.%.
Из графиков зависимости ингибирования от концентрации обычным образом рассчитывают концентрацию тестируемого соединения, при которой наблюдается 50%-ное ингибирование (IC50).
С помощью указанного анализа установлено, что для соединений из примеров 1, 2, 4, 7, 9, 13, 20, 23, 25, 28, 30, 38, 40, 43 и 44 значения IC50 (нМ) составляют 210, 221, 94, 48, 58, 53, 89, 131, 387, 72, 121, 1519, 215, 356 и 331.
С учетом их способности ингибировать связывание с CCR-3 агенты по изобретению можно применять для лечения состояний, опосредуемых CCR-3, прежде всего воспалительных или аллергических состояний. Согласно изобретению, лечение может быть симптоматическим или профилактическим.
Таким образом, агенты по изобретению можно применять для лечения воспалительных или обструктивных заболеваний дыхательных путей, что приводит, например, к снижению повреждения ткани, бронхиальной гиперреактивности, к ремоделированию или снижению развития болезни. Воспалительные или обструктивные заболевания дыхательных путей, которые можно лечить согласно настоящему изобретению, включают астму, вне зависимости от ее типа или генеза, в том числе как наследственную (неаллергическую) бронхиальную астму, так и приобретенную (аллергическую) бронхиальную астму, слабую астму, астму средней тяжести, тяжелую астму, бронхиальную астму, вызванную физической нагрузкой астму, профессиональную астму и астму, вызванную бактериальной или вирусной инфекцией. Понятие "лечение астмы" относится также к лечению пациентов возраста менее 4 или 5 лет, которые имеют симптомы стридора и в отношении которых поставлен диагноз или может быть поставлен диагноз "страдающий стридором ребенок", т.е. пациентов, относящихся к имеющей большое медицинское значение категории пациентов, которых в настоящее время часто называют астматиками с начальной или ранней стадией астмы. (Для простоты это конкретное астматическое состояние называют "синдромом страдающего стридором ребенка").
Эффективность профилактического лечения астмы можно выявлять по уменьшению частоты или серьезности симптоматических приступов, например острых приступов астмы или приступов бронхостеноза, улучшению функции легких или снижению повышенной реактивности дыхательных путей. Кроме того, ее можно оценивать по уменьшению частоты применения других средств симптоматического лечения, т.е. лечения, предназначенного для ограничения или устранения симптоматических приступов, если они возникают, например частоты применения противовоспалительных средств (например, кортикостероидов) или бронхолитических средств. Профилактическая эффективность в отношении астмы наиболее наглядно проявляется для пациентов, склонных к так называемому "утреннему погружению". "Утреннее погружение" представляет собой астматический синдром, которым страдает определенный процент астматиков, характеризующийся приступом астмы, например, приблизительно между 4 и 6 часами утра, т.е. в промежутке времени, достаточно отдаленном от времени, когда ранее производилось какое-либо симптоматическое лечение астмы.
Другие воспалительные или обструктивные заболевания или состояния дыхательных путей, для лечения которых можно использовать настоящее изобретение, включают острое поражение легкого (ОЛП), респираторный дистресс-синдром взрослых (РДСВ), хроническое обструктивное заболевание легких или дыхательных путей (ХОЗЛ или ХОЗДП), в том числе хронический бронхит или связанную с ним одышку, эмфизему, а также обострение гиперреактивности дыхательных путей вследствие лечения с использованием других лекарственных средств, в частности лечения с использованием других лекарственных средств, вводимых путем ингаляции. Изобретение можно применять также для лечения бронхита любого типа или генеза, например острого, арахидоидного, катарального, крупозного, хронического или гнойного туберкулезного бронхита. Другие воспалительные или обструктивные заболевания дыхательных путей, для лечения которых можно применять настоящее изобретение, включают пневмокониоз (воспалительное, как правило профессиональное заболевание легких, часто сопровождающееся закупоркой дыхательных путей, которое может быть хроническим или острым и которое связано с постоянным вдыханием пыли) любого типа или генеза, включая, например, алюминоз, антракоз, асбестоз, халикоз, птилоз, сидероз, силикоз, табакоз и биссиноз.
С учетом их противовоспалительной активности, прежде всего в отношении ингибирования активации эозинофилов, агенты по изобретению можно также применять для лечения связанных с эозинофилами заболеваний, например эозинофилии, в частности опосредуемых эозинофилами заболеваний дыхательных путей (например, включая патологическую инфильтрацию эозинофилов легочных тканей), в том числе гиперэозинофилию, поскольку они оказывают воздействие на дыхательные пути и/или легкие, а также, например, для лечения связанных с эозинофилами нарушений заболеваний дыхательных путей, которые являются следствием или которые сопровождают синдром Леффлера, эозинофильную пневмонию, паразитическую (в частности, вызываемую представителями Metazoa) инвазию (включая тропическую эозинофилию), бронхопульмонарный аспергиллез, нодозный полиартериит (включая синдром Чурга-Штрауса), эозинофильную гранулему и связанные с эозинофилами заболевания, поражающие чувствительные к действию лекарств дыхательные пути.
Агенты по изобретению можно применять также для лечения воспалительных или аллергических состояний кожи, например псориаза, контактного дерматита, атопического дерматита, гнездной алопеции, экссудативной эритемы, герпетиформного гепатита, склеродемы, витилиго, гиперчувствительного ангиита, крапивницы, буллезного пемфигоида, системной красной волчанки, пузырьчатки, врожденного буллезного эпидермолиза и других воспалительных или аллергических состояний кожи.
Агенты по изобретению можно применять также для лечения других болезней или состояний, прежде всего болезней или состояний, которые имеют воспалительный компонент, например для лечения болезней и состояний глаз, таких как конъюнктивит, сухой кератоконъюнктивит и весенний конъюнктивит, болезней носа, включая аллергический ренит, например атрофический хронический или сезонный ринит, воспалительных заболеваний желудочно-кишечного тракта, например воспалительного заболевания кишечника, такого как неспецифический язвенный колит и болезнь Крона, заболеваний костной ткани и суставов, включая ревматоидный артрит, псориатический артрит, анкилозирующий спондилоартрит и системный склероз, и других болезней, таких как атеросклероз, рассеянный склероз, диабет (типа I), тяжелая псевдопаралитическая миастения, гипер-IgE-синдром и острое и хроническое отторжение аллотрансплантата, например, после трансплантации сердца, почки, печени, легкого или костного мозга.
Способность агентов по изобретению ингибировать воспалительные состояния, например при воспалительных заболеваниях дыхательных путей, можно продемонстрировать при моделировании на животных, например, мышах или крысах, воспаления дыхательных путей или других воспалительных состояний, например согласно методам, описанным у Szarka и др., J.Immunol. Methods, 202: 49-57 (1997); Renzi и др., Am. Rev. Respir. Dis., 148: 932-939 (1993); Tsuyuki и др., J.Clin. Invest., 96:2924-2931 (1995); и Cernadas и др. Am. J.Respir. CellMol. Biol. 20: 1-8(1999).
Агенты по изобретению можно применять также в качестве вспомогательных терапевтических средств в сочетании с противовоспалительными, бронхолитическими или антигистаминными лекарственными средствами, прежде всего для лечения указанных выше обструктивных или воспалительных заболеваний дыхательных путей, например, в качестве потенцирующих средств для усиления терапевтической активности таких лекарственных средств или в качестве средств для уменьшения требуемых доз или возможных побочных действий таких лекарственных средств. Агент по изобретению можно смешивать с другим лекарственным средством с получением фиксированной фармацевтической композиции или его можно вводить индивидуально до, одновременно или после введения другого лекарственного средства. Такие противовоспалительные лекарственные средства включают стероиды, прежде всего глюкокортикостероиды, такие как будесонид, бекламетазон, флутиказон, циклезонид или мометазон, антагонисты LTB4 (лейкотриен В4), например, описанные в патенте US 5451700, антагонисты LTD, такие как монтелукаст и зафирлукаст, агонисты рецептора допамина, такие как каберголин, бромокриптин, ропинерол и 4-гидрокси-7-[2-[[2-[[3-(2-фенилэтокси)пропил]сульфонил]этил]амино]этил]-2(3Н)-бензотиазолон и его фармацевтически приемлемые соли (например, гидрохлорид, выпускаемый под товарным знаком Viozan®-фирмы AstraZeneca), и ингибиторы PDE4, такие как Ariflo® (фирма GlaxoSmith Kline), рофлумилат (фирма Byk Gulden), V-11294A (фирма Napp), BAY19-8004 (фирма Bayer), SCH-351591 (фирма Schering-Plough) и PD189659 (фирма Parke-Davis). Такие бронхолитические лекарственные средства включают антихолинергические или антимускариновые агенты, в частности бромид ипратропия, бромид окситропия и бромид тиотропия, и агонисты бета-2-адренорецентора, такие как сальбутамол, тербуталин, сальметерол, и прежде всего формотерол, и их фармацевтически приемлемые соли, а также соединения формулы I (в свободной форме или в форме соли или сольвата), описанные в международной заявке на патент РСТ WO 00/75114, которая включена в настоящее описание в качестве ссылки, предпочтительно соединения из приведенных в данной заявке примеров, прежде всего соединение формулы
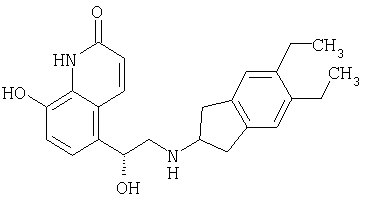
и его фармацевтически приемлемые соли. Антигистаминные лекарственные средства, которые можно использовать для совместной терапии, включают гидрохлорид цетиризина, ацетаминофен, фумарат клемастина, прометазин, лоратидин, деслоратидин, дифенгидрамин и гидрохлорид фексофенадина. Комбинации агентов по изобретению и стероидов, агонистов бета-2, ингибиторов PDE4 или антагонистов LTD4 можно применять, например, для лечения ХОЗЛ или, прежде всего, астмы. Комбинации агентов по изобретению и антихолинергических или антимускариновых агентов, ингибиторов PDE4 или агонистов рецептора допамина можно применять, например, для лечения астмы или, прежде всего, ХОЗЛ.
Другие ценные комбинации агентов по изобретению с противовоспалительными лекарственными средствами представляют собой комбинации с другими антагонистами рецепторов хемокинов, например CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 и CCR-10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, в частности с антагонистами CCR-5, такими как антагонисты фирмы Schering-Plough SC-351125, SCH-55700 и SCH-D, с антагонистами фирмы Takeda, такими как хлорид N-[[4-[[[6,7-дигидро-2-(4-метилфенил)-5Н-бензоциклогептен-8-ил]карбонил]амино]фенил]метил]тетрагидро-N,N-диметил-2Н-пиран-4-аммония (ТАК-770), и с антагонистами CCR-5, описанными в патенте US 6166037 (в частности, в пунктах 18 и 19 формулы изобретения), в WO 00/66558 (в частности, в пункте 8 формулы изобретения) и в WO 00/66559 (в частности, в пункте 9 формулы изобретения).
Согласно вышеизложенному в изобретении, предложен также способ лечения состояния, опосредуемого CCR-3, например воспалительного или аллергического состояния, в частности обструктивного или воспалительного заболевания дыхательных путей, который предусматривает введение пациенту, прежде всего человеку, нуждающемуся в таком лечении, эффективного количества описанного выше соединения формулы I в свободной форме или в форме его фармацевтически приемлемой соли. Другим объектом изобретения является применение описанного выше соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для лечения опосредуемого CCR-3 состояния, например, для лечения, воспалительного или аллергического состояния, прежде всего, обструктивного или воспалительного заболевания дыхательных путей.
Агенты по изобретению можно вводить любым пригодным путем, например перорально, например, в форме таблетки или капсулы; парентерально, например, внутривенно; путем ингаляции, прежде всего при лечении обструктивных или воспалительных заболеваний дыхательных путей; интраназально, например, при лечении аллергического ринита; местно на кожу, например, при лечении атопического дерматита, или ректатально, например, при лечении воспалительного заболевания кишечника.
Еще одним объектом изобретения является фармацевтическая композиция, содержащая соединение формулы I в свободной форме или в форме его фармацевтически приемлемой соли, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем. Композиция может содержать дополнительный терапевтический агент, например описанное выше противоваоспалительное или бронхолитическое лекарственное средство. Такие композиции можно приготовливать с использованием обычных разбавителей или эксципиентов и методов, известных в области приготовления галеновых форм. Так, формы для перорального введения могут представлять собой таблетки и капсулы. Композиции для местного нанесения могут представлять собой кремы, мази, гели или трансдермальные системы введения, например бляшки. Композиции для ингаляции могут представлять собой аэрозоль или другие распыляемые композиции или композиции на основе сухого порошка.
Таким образом, объектом изобретения является (А) агент по изобретению, который можно вводить путем ингаляции, например, в виде аэрозоля или другой распыляемой композиции или пригодных для ингаляции частиц, например, в тонкоизмельченной форме; (Б) предназначенное для ингаляции лекарственное средство, включающее агент по изобретению в форме, пригодной для ингаляции; (В) фармацевтический продукт, включающий такой агент по изобретению в форме, пригодной для ингаляции, в сочетании с устройством для ингаляции; и (Г) устройство для ингаляции, содержащее такой агент по изобретению в форме, пригодной для ингаляции.
Дозы агентов по изобретению, используемые при практическом осуществлении изобретения, естественно должны варьироваться в зависимости, например, от конкретного состояния, подлежащего лечению, ожидаемого действия и пути введения. Как правило, пригодные суточные дозы при введении путем ингаляции составляют приблизительно от 0,01 до 30 мг/кг, а пригодные суточные дозы для перорального введения составляют примерно от 0,01 до 100 мг/кг.
Ниже изобретение проиллюстрировано на примерах.
Примеры 1-47
Соединения формулы I, которые также представлены формулой
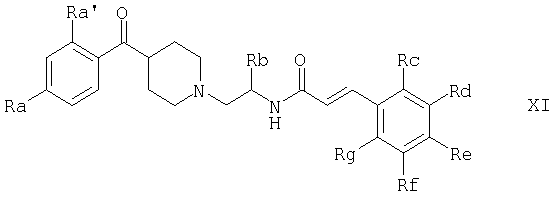
и варианты способов их получения приведены в следующей таблице, причем сами методы описаны ниже. Ra' обозначает H во всех примерах, кроме примера 12, где он обозначает F. В таблице приведены также данные масс-спектрометрии ([МН]+) и, если в примере описано получение соли, указана солеобразующая кислота
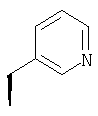
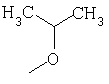
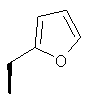
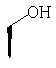
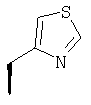
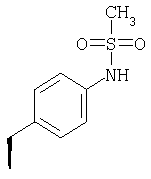
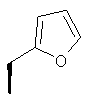
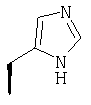
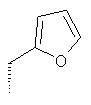
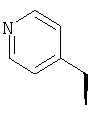
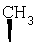
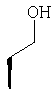
Метод А
Получение трет-бутилового эфира ((R)-2-гидрокси-1-пиридин-3-илметилэтил)карбаминовой кислоты.
К раствору (R)-2-трет-бутоксикарбониламино-3-пиридин-3-илпропионовой кислоты (0,9 г, 3,37 ммоля) в диметоксиэтане (18 мл) добавляют N-метилморфолин (0,44 мл, 4,04 ммоля) и изобутилхлорформиат (0,48 мл, 3,71 ммоля). Реакционную смесь перемешивают при температуре окружающей среды в течение 20 мин и затем фильтруют. Фильтрат обрабатывают водным раствором борогидрида натрия (25 мл, 10,11 ммоля) и реакционную смесь сразу разбавляют водой (200 мл). Перемешивание продолжают в течение 1 ч при температуре окружающей среды. Реакционную смесь распределяют между этилацетатом и водой. Органическую фазу отделяют, сушат над сульфатом магния и выпаривают. Неочищенный продукт очищают экспресс-хроматографией на силикагеле (элюирование EtOAc), получая трет-бутиповъш эфир ((R)-2-гидрокси-1-пиридин-3-илметилэтил)карбаминовой кислоты. [МН]+ 253,5.
Получение трет-бутилового эфира ((R)-2-бром-1-пиридин-3-илметилэтил)карбаминовой кислоты.
К раствору трет-бутилового эфира ((R)-2-гидрокси-1-пиридин-3-илметилэтил)карбаминовой кислоты (0,43 г, 1,70 ммоля) в дихлорметане (10 мл) добавляют четырехбромистый углерод (0,33 г, 2,04 ммоля) и трифенилфосфин (0,23 г, 1,70 ммоля). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 ч, фильтруют и фильтрат распределяют между этилацетатом и соляной кислотой (1 М). Водную фазу отделяют, нейтрализуют насыщенным раствором бикарбоната натрия и экстрагируют дихлорметаном. Дихлорметановый слой сушат на сульфатом магния и выпаривают, получая трет-бутиловый эфир ((R)-2-бром-1 -пиридин-3-илметилэтил)карбаминовой кислоты 1Н-ЯМР (400 МГц, CDCl3) (1,29 (s, 9Н), 3,05 (dd, J 14,3, 9,8, 1H), 3,18 (dd, J 14,3 4,9, 1H), 3,51 (d, J 4,9, 2Н), 4,07-4,16 (m, 1H), 7,84 (dd J 7,9, 5,9, 1H), 8,35 (d, J 7,9, 1H), 8,65 (d, J 5,4, 1H), 8,86 (s, 1H).
Получение трет-бутилового эфира {(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-пиридин-3-илметилэтил}карбаминовой кислоты
(4-Фторфенил)пиперидин-4-илметанон (0,15 г, 0,73 ммоля) добавляют к раствору, содержащему трет-бутиловый эфир ((R)-2-бром-1-пиридин-3-илметилэтил)карбаминовой кислоты (0,21 г, 0,66 ммоля) и 1,8 диазабицикло[5.4.0]ундец-7-ен (0,12 мл, 0,79 ммоля) в диметилформамиде (3 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 24 ч перед распределением между этилацетатом и водой. Этилацетатный слой сушат над сульфатом магния и выпаривают. Неочищенный продукт сушат с помощью экспресс-хроматографии на силикагеле (элюирование смесью 97:3, дихлорметан: метанол), получая трет-бутиловый эфир {(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-пиридин-3-илметилэтил}карбаминовой кислоты. 1Н-ЯМР (400 МГц, CDCl3) δ1,36 (s. 9Н), 1,68-1,85 (br m, 4H), 2,00-2,38 (br m, 4H), 2,78-2,91 (m, 4H), 3,05-3,19 (m 1H), 3,81-3,93 (m, 1H), 7,05 (t, J 8,8, 2Н), 7,12-7,18 (m, 1H), 7,48 (d, J 7,9, 1H), 7,85-7,93 (dd, J 8,8 5,4, 2Н), 8,36 (d, J 1,5, 1H), 8,40 (dd,J 4,9 1,5, 1H).
Получение [1-((R)-2-амино-3-пиридин-3-илпропил)пиперидин-4-ил]-(4-фторфенил)метанона
К раствору трет-бутилового эфира {(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-пиридин-3-илметилэтил}карбаминовой кислоты (0,149 г, 0,34 ммоля) в дихлорметане (2 мл) добавляют трифторуксусную кислоту (0,5 мл) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Реакционную смесь упаривают и остаток растворяют в соляной кислоте (1М), раствор подщелачивают раствором гидроксида натрия (4М) и осадок экстрагируют дихлорметаном. Дихлорметановый слой сушат над сульфатом магния и выпаривают, получая [1-((R)-2-амино-3-пиридин-3-илпропил)пиперидин-4-ил]-(4-фторфенил)метанон. Н-ЯМР (400 МГц, CDCl3) δ1,63-1,85 (m, 4H), 1,88-2,00 (m, 1H), 2,08-2,32 (m, 5H), 2,50 (dd, J 13,5, 7,9, 1H), 2,67 (dd, J 13,5, 4,9, 1H), 2,78-2,98 (m, 2H), 3,04-3,20 (m, 2H), 7,04 (t, J 8,8, 2Н), 7,17 (dd, J 6,9, 4,9, 1H), 7,48 (d, J 7,9, 1H), 7,88 (dd, J 8,8, 5,4, 2H), 8,33-8,45 (m, 2H).
Получение (Е)-3-(3-цианфенил)-N-{(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-пиридин-3-илметилэтил}акриламида.
К раствору (Е)-3-(4-цианфенил)акриловой кислоты (0,022 г, 0,126 ммоля) в дихлорметане (1 мл) добавляют триэтиламин (0,016 мл, 0,126 ммоля) и гексафторфосфат (бензотриазо-1-илокси)трипирролидинфосфония (0,06 г, 0,116 ммоля). Реакционную смесь перемешивают при температуре окружающей среды в течение 5 мин и затем добавляют раствор 1-((R)-2-амино-3-пиридин-3-илпропил)пиперидин-4-ил]-(4-фторфенил)метанона (0,036 г, 0,105 ммоля) в дихлорметане (1 мл). Перемешивание продолжают в течение еще 1,5 ч, затем реакционную смесь фильтруют. Фильтрат упаривают и неочищенный продукт очищают экспресс-хроматографией на силикагеле (дихлорметан: метанол: уксусная кислота, 10:0,5:0,05), получая (Е)-3-(3-цианфенил)-N-{(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-пиридин-3-илметилэтил}акриламид [МН]+ 497,4.
Метод Б
Получение трет-бутилового эфира {(R)-1-бензил-2-[4-(4-фторбензоил)пиперидин-1-ил]этил}карбаминовой кислоты
Раствор, содержащий трет-бутиловый эфир ((R)-1-бензил-2-оксоэтил)карбаминовой кислоты (0,5 г, 2,0 ммоля), (4-фторфенил)пиперидин-4-илметанон (0,414 г, 2,0 ммоля) и триацетоксиборогидрид натрия (0,638 г, 3,0 ммоля) в тетрагидрофуране (20 мл), перемешивают при температуре окружающей среды в течение 24 ч. Растворитель выпаривают и остаток повторно растворяют в дихлорметане и промывают насыщенным раствором бикарбоната натрия. Дихлорметановый слой сушат над сульфатом магния и выпаривают. Неочищенный продукт очищают экспресс-хроматографией на силикагеле (элюирование смесью этилацетат:гексан, 3:1), получая трет-бутиловый эфир [(R)-1-бензил-2-[4-(4-фторбензоил)пиперидин-1-ил]этил}карбаминовой кислоты [МН]+ 441,3.
Получение [1-((R)-2-амино-3-фенилпропил)пиперидин-4-ил](4-фторфенил)метанона.
Раствор трет-бутипового эфира {(R)-1-бензил-2-[4-(4-фторбензоил)пиперидин-1-ил]этил}карбаминовой кислоты (1,12 г, 2,54 ммоля) и трифторуксусной кислоты (3 мл) в дихлорметане (6 мл) перемешивают при температуре окружающей среды в течение 3 ч. Растворитель выпаривают и остаток растворяют в соляной кислоте (2М), промывают этилацетатом и подщелачивают раствором гидроксида натрия (4М) до рН 8-9. Суспензию экстрагируют дихлорметаном, дихлорметановый слой сушат над сульфатом магния и растворитель выпаривают, получая [1-((R)-2-амино-3-фенилпропил)пиперидин-4-ил](4-фторфенил)метанон. [МН]+ 341,7.
Получение (E)-N-{(R)-1-бензил-2-[4-(4-фторбензоил)пиперидин-1-ил] этил} -3-(3-цианфенил)акриламида
К раствору (Е)-3-(4-цианфенил)акриловой кислоты (0,042 г, 0,242 ммоля) в дихлорметане (1 мл) добавляют триэтиламин (0,046 мл, 0,331 ммоля) и гексафторфосфат (бензотриазо-1-илокси)трипирролидинофосфония (0,126 г, 0,242 ммоля). Реакционную смесь перемешивают при температуре окружающей среды в течение 5 мин и затем добавляют раствор [1-((R)-2-амино-3-фенилпропил)пиперидин-4-ил](4-фторфенил)метанона (0,075 г, 0,220 ммоля) в дихлорметане (1 мл). Перемешивание продолжают в течение еще 3 ч, затем реакционную смесь разбавляют дихлорметаном (25 мл) и промывают насыщенным раствором бикарбоната натрия и насыщенным соляным раствором. Дихлорметановый слой сушат над сульфатом магния и растворитель выпаривают. Неочищенный продукт очищают экспресс-хроматографией на силикагеле (элюирование смесью этилацетат: гексан, 5:1), получая (E)-N-{(R)-1-бензил-2-[4-(4-фторбензоил)пиперидин-1-ил]этил}-3-(3-цианфенил)акриламид [MH]+ 496,8.
Метод В
Получение (Е)-3-(5-бром-2-метоксифенил)-N-{2-[4-(4-хлорбензоил)пиперидин-1-ил]этил}акриламида.
К суспензии, содержащей 2-(формил-3-метоксифенокси)этилполистирольную (АМЕВА) смолу (фирма Novabiochem) (6,85 г, 3,33 ммоля) в смеси метанола/дихлорметана (60 мл, 1:1 об/об), добавляют 2-аминоэтанол и триацетоксиборогидрид натрия (4,00 г, 18,85 ммоля) и смесь встряхивают в течение 16 ч при 20°С, затем фильтруют. Смолу промывают метанолом, ДМФ и дихлорметаном, затем сушат под вакуумом. К высушенной смоле добавляют смесь ТГФ/ацетонитрила (50 мл, 1:1 об/об), а затем йод (4,80 г, 18,85 ммоля), имидазол (1,28 г, 18,85 ммоля) и трифенилфосфин (4,90 г, 18,85 ммоля). Полученную суспензию встряхивают в течение 18 ч при 20°С, затем фильтруют. Смолу промывают ТГФ и сушат под вакуумом. К свежеприготовленной смоле (0,50 г, 0,35 ммоля) добавляют раствор гидрохлорида (4-хлорфенил)пиперидин-4-илметанона (0,18 г, 0,70 ммоля), растворенного в ДМФ (2 мл) и диизопропилэтиламине (0,36 г, 2,8 ммоля). Смесь выдерживают при 50°С в течение 16 ч и затем фильтруют. Смолу промывают ДМФ. К промытой смоле добавляют (Е)-3-(5-бром-2-метоксифенил)акриловую кислоту (0,27 г, 1,05 ммоля), тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (0,34 г, 1,05 ммоля), диизопропилэтиламин (0,29 г, 1,05 ммоля) и ДМФ (4 мл) и смесь встряхивают при 20°С в течение 16 ч, затем промывают ДМФ и метанолом, после чего для выделения продукта из смолы обрабатывают смесью трифторуксусная кислота/дихлорметан (6 мл, 1:1 об/об) при 20°С в течение 1 ч. Образовавшуюся смесь фильтруют и фильтрат упаривают под вакуумом, получая требуемый продукт, [MH]+ 506,7.
Метод Г
Получение трет-бутилового эфира {(R)-1-(4-аминобензил)-2-[4-(4-фторбензоил)пиперидин-1 -ил]этил} карбаминовой кислоты
К раствору трет-бутилового эфира [(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-(4-нитробензил)этил]карбаминовой кислоты (1,41 г, 2,90 ммоля) в уксусной кислоте (11 мл), охлажденному до 0°С, добавляют водный раствор хлорида кальция (4 мл, 0,47М) и порошкообразного цинка (3,9 г, 59,6 ммоля). Реакционную смесь перемешивают при 0°С в течение 35 мин и затем фильтруют через пробку из целита. Фильтрат упаривают и остаток растворяют в воде и экстрагируют дихлорметаном. Дихлорметановый слой упаривают и остаток растворяют в воде и подщелачивают водным раствором бикарбоната натрия и экстрагируют дихлорметаном. Дихлорметановый слой сушат над сульфатом магния и упаривают, получая трет-бутиловый эфир {(R)-1-(4-аминобензил)-2-[4-(4-фторбензоил)пиперидин-1-ил]этил}карбаминовой кислоты, [МН]+ 456,5.
Получение трет-бутилового эфира [(R)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-(4-метансульфониламинобензил)этил]карбаминовой кислоты
К раствору трет-бутилового эфира {(R)-1-(4-аминобензил)-2-[4-(4-фторбензоил)пиперидин-1-ил]этил}карбаминовой кислоты (1,19 г, 2,61 ммоля) в дихлорметане (15 мл), охлажденному до 0°С, добавляют триэтиламин (0,37 мл, 2,65 ммоля) и метансульфонилхлорид (0,192 мл, 2,49 ммоля). Реакционной смеси дают нагреться до температуры окружающей среды, перемешивая в течение 1 ч, затем промывают водой и насыщенным соляным раствором, сушат над сульфатом магния и упаривают. Неочищенный продукт очищают экспресс-хроматографией на силикагеле (элюирование в градиенте этилацетата:гексана от 6:4 до 1:0), получая трет-бутиловый эфир [(R)-2-[4-(4-фторбензоил)пиперидин1-ил]-1-(4-метансульфониламинобензил)этил]карбаминовой кислоты, [МН]+ 534,7.
Метод Д
Получение трет-бутилового эфира (S)-4-[4-(4-хлорбензоил)пиперидин-1-илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты
К раствору трет-бутилового эфира (R)-4-формил-2,2-диметилоксазолидин-3-карбоновой кислоты (0,5 г, 2,18 ммоля) в тетрагидрофуране (15 мл) добавляют (4-хлорфенил)пиперидин-4-илметанон (0,49 г, 2,18 ммоля) и триацетоксиборогидрид натрия (0,69 г, 3,27 ммоля) и реакционную смесь перемешивают в течение 3,5 ч при температуре окружающей среды. Растворитель выпаривают и остаток распределяют между этилацетатом (50 мл) и насыщенным раствором бикарбоната натрия (50 мл). Этилацетатный слой сушат над сульфатом магния и упаривают. Неочищенный продукт очищают экспресс-хроматографией на силикагеле (элюирование этилацетатом:гексаном, 1:1), получая трет-бутиловыи эфир (S)-4-[4-(4-хлорбензоил)пиперидин-1-илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты, [МН] 437,2.
Получение гидрохлорида [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил](4-хлорфенил)метанона.
Треот-бутиловый эфир (S)-4-[4-(4-хлорбензоил)пиперидин-1 -илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты (0,68 г, 1,55 ммоля) добавляют к раствору хлористого водорода в этаноле (5 мл, 5,5 М). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч, затем упаривают досуха получая гидрохлорид [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил](4-хлорфенил)метанона, [МН]+ 297,0.
Получение (E)-3-(5-циан-2-метоксифенил)акриловой кислоты
К суспензии ацетата палладия(II) (0,77 г, 3,42 ммоля) в N,N-диметилацетамиде (375 мл) в атмосфере азота добавляют хлорид тетраэтиламмония (19,36 г, 114,5 ммоля), дициклогексилметиламин (35,1 г, 174,5 ммоля) и 3-бром-4-метоксибензонитрил (25,51 г, 118,0 ммоля). Суспензию нагревают до 100-105°С, после чего медленно в течение 45 мин добавляют трет-бутилакрилат (14,82 г, 114,5 ммоля). После перемешивания в течение еще 30-60 мин при 100°С раствор охлаждают до комнатной температуры и разбавляют ТБМЭ (375 мл). Образовавшуюся двухфазную смесь интенсивно перемешивают в течение 10 мин. Верхнюю фазу (ТБМЭ) промывают последовательно водой (100 мл), 10%-ным водным раствором лимонной кислоты (100 мл) и 25%-ным водным раствором NaCl (100 мл). Объединенные водные фазы экстрагируют ТБМЭ (100 мл). После добавления активированного угля (0,4 г) объединенные ТБМЭ-фазы интенсивно перемешивают в течение 10 мин и фильтруют. Добавляют безводный Na2SO4 (10 г) и образовавшуюся суспензию перемешивают в течение еще 10 мин и фильтруют. Фильтрат концентрируют до объема 50-70 мл при пониженном давлении в течение 25-30 мин, добавляют при комнатной температуре к безводной трифторуксусной кислоте (150 мл). Образовавшийся раствор перемешивают при комнатной температуре в течение 60 мин (образовавшийся осадок охлаждают до 0-5°С в ледяной бане и разбавляют этилацетатом (410 мл). После интенсивного перемешивания при 0°С в течение еще 60 мин суспензию фильтруют. Остаток сушат под вакуумом при 45-50°С, получая (Е)-3-(5-циан-2-метоксифенил)акриловую кислоту в виде кристаллического твердого вещества, tпл 252-253°C. MC (ES): [М-Н]- 202.
Получение (Е)-N-{(S)-2-[4-(4-хлорбензоил)пиперидин-1-ил]-1-гидроксиметилэтил}-3-(5-циан-2-метоксифенил)акриламида.
К раствору, содержащему (Е)-3-(5-циан-2-метоксифенил)акриловую кислоту (0,31 г, 1,55 моля), триэтиламин (0,2 мл, 1,55 ммоля) и тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (0,49 г, 1,55 ммоля) в дихлорметане (5 мл), добавляют раствор гидрохлорида [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил](4-хлорфенил)метанона и триэтиламин (0,4 мл, 3,1 ммоля) в дихлорметане (5 мл) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Реакционную смесь разбавляют дихлорметаном (20 мл), последовательно промывают насыщенным раствором бикарбоната натрия (25 мл) и соляным раствором (25 мл), затем сушат над сульфатом магния. Растворитель выпаривают и неочищенный остаток очищают экспресс-хроматографией на силикагеле (метанол:дихлорметан 5:95), получая (Е)-N-{(S)-2-[4-(4-хлорбензоил)пиперидин-1-ил]-1-гидроксиметилэтил}-3-(5-циан-2-метоксифенил)акриламид, [МН]+ 482,2.
Метод Е
Получение трет-бутилового эфира (S)-4-[4-(4-фторбензоил)пиперидин-1-илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты
К раствору (4-фторфенил)пиперидин-4-илметанона (3,5 г, 17 ммолей) в безводном тетрагидрофуране (50 мл) добавляют трет-бутиловый эфир (R)-4-формил-2,2-диметилоксазолидин-3-карбоновой кислоты (3,9 г, 17 ммолей) и триацетоксиборогидрид натрия (5,4 г, 25 ммолей) и реакционную смесь перемешивают в течение 18 ч при температуре окружающей среды. Реакционную смесь фильтруют и растворитель выпаривают, получая твердое вещество белого цвета. Твердое вещество растворяют в дихлорметане (50 мл) и промывают насыщенным раствором бикарбоната натрия (50 мл), водой (2х 50 мл) и соляным раствором (50 мл). Органическую фазу сушат над сульфатом магния и упаривают, получая требуемый продукт, [МН]+ 420,9.
Получение гидрохлорида [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил]-(4-фторфенил)метанона.
К суспензии трет-бутилового эфира (S)-4-[4-(4-фторбензоил)пиперидин-1-илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты (4,9 г, 11,7 ммоля) в этаноле (25 мл) добавляют хлористый водород в диоксане (25 мл, 4 М). Образовавшийся прозрачный раствор перемешивают в течение 4 ч при температуре окружающей среды, при этом образуется осадок белого цвета. Реакционную смесь охлаждают до 0°С и осадок фильтруют, получая требуемый продукт, [MH]+ 281,6.
Получение (Е)-N-{(S)-2-[4-(4-фторбензоил)пиперидин-1-ил]-1-гидроксиметилэтил}-3-(5-циан-2-метоксифенил)акриламида.
К раствору гидрохлорида [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил]-(4-фторфенил)метанона (1,8 г, 5,7 ммоля) и диизопропилэтиламина (2,0 мл, 11,4 ммоля) в дихлорметане (45 мл) добавляют (Е)-3-(5-циан-2-метоксифенил)акриловую кислоту (1,1 г, 5,7 ммоля), а затем тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (1,83 г, 5,7 ммоля). Реакционную смесь перемешивают при температуре окружающей среды в течение 4,5 ч, затем фильтруют и фильтрат промывают водой (50 мл), насыщенным раствором бикарбоната натрия (50 мл), водой (50 мл) и соляным раствором (50 мл). Органическую фазу сушат над сульфатом магния, растворитель выпаривают и остаток очищают экспресс-хроматографией на силикагеле (элюирование в градиенте дихлорметана: метанола от 98:2 до 92:8), получая требуемый продукт, [МН]+ 466,1.
Метод Ж
Получение дибензоил-L-тартрата трет-бутилового эфира (S)-4-[4-(4-хлорбензоил)пиперидин-1-илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты (двухосновного).
К охлажденной (0°С) суспензии борогидрида натрия (2,40 г, 63,55 ммоля) в безводном толуоле (50 мл) в течение 1 ч добавляют в атмосфере инертного газа уксусную кислоту (11,45 г, 189,9 ммоля). Перемешивание продолжают при температуре окружающей среды в течение 5 ч до прекращения выделения водорода (суспензия 1). В другой колбе гидрохлорид 4-(4-хлорбензоил)пиперидина (полученный взаимодействием N-формил-4-(4-хлорбензоил)пиперидина и ацетилхлорида) (5,51 г, 21,18 ммоля) суспендируют в безводном толуоле (20 мл) при комнатной температуре. Добавляют триэтиламин (2,57 г, 25,42 ммоля) и по каплям в течение 45 мин при перемешивании добавляют толуоловый раствор (55 мл) трет-бутиловото эфира (R)-4-формил-2,2-диметилоксазолидин-3-карбоновой кислоты (5,59 г, 24,36 ммоля). Перемешивание продолжают в течение еще 20 мин, затем медленно при перемешивании в течение 60 мин добавляют суспензию 1. Образовавшуюся суспензию перемешивают при температуре окружающей среды до тех пор, пока по данным ТСХ не прекращается поглощение исходных продуктов (14 ч), затем медленно добавляют к раствору NaHCO3 (25 г, 297,6 ммоля) в воде (120 мл). Образовавшуюся эмульсию перемешивают при 20°С в течение 60 мин, отделяют водную фазу смеси и органическую фазу промывают дважды последовательно, используя по 20 мл 10%-ного водного раствора NaHCO3 и воды. После доведения значения рН объединенных водных фаз до 9,5 с помощью твердого Na2CO3 водные фазы экстрагируют толуолом (2×25 мл). К объединенным органическим фазам добавляют целит (0,5 г), после чего их фильтруют и упаривают досуха, получая свободное основание, которое затем растворяют в изопропаноле (35 мл) и выдерживают при температуре дефлегмации. Добавляют по каплям раствор ди-О,О-бензоил-L-винной кислоты (4,0 г, 10,6 ммоля) в изопропаноле (10 мл). После перемешивания при 79-81°С в течение 20 мин смесь охлаждают, разбавляют метил- трет-бутиловым эфиром (ТБМЭ) и продукт кристаллизуют при 0°С, фильтруют, промывают холодной (0°С) смесью ТБМЭ: изопропанола, 1:2 (15 мл) и охлажденным (0°С) ТБМЭ (3×5 мл) и сушат под вакуумом. После перекристаллизации высушенного продукта из изопропанола и последующей сушки под вакуумом получают указанный в заголовке продукт в виде кристаллического твердого вещества tпл 174°C.
МС (ES+): [M}+ 437.
Получение дигидрохлорида [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил](4-хлорфенил)метанона
Дибензоил-L-тартрат трет-бутнлового эфира (S)-4-[4-(4-хлорбензоил)пиперидин-1-илметил]-2,2-диметилоксазолидин-3-карбоновой кислоты (двухосновный) (4,0 г, 3,2 ммоля) суспендируют в н-бутилацетате (40 мл). Добавляют водную (32%) соляную кислоту (2,18 г, 19,2 ммоля) и смесь перемешивают при температуре окружающей среды до тех пор, пока по данным ТСХ не прекращается поглощение исходных продуктов (2 ч). Суспензию перемешивают в ледяной бане в течение еще 3 ч и фильтруют. Твердый продукт промывают холодным (0°С) н-бутилацетатом (2×5 мл) и сушат под вакуумом при 45-50°С, получая указанный в заголовке продукт в виде бесцветных кристаллов tпл 232-237°С. МС (ES+): [МН]+ 297.
Получение S-бензотиазол-2-илового эфира (E)-3-(5-циан-2-метоксифенил)тиоакриловой кислоты
Суспензию, содержащую дисульфид 2,2'-дибензотиазолила (4,0 г, 12,0 ммолей) и трифенилфосфин (3,15 г, 12,0 ммолей) в CH2Cl2 (60 мл), интенсивно перемешивают при 25°С в течение 30 мин. После охлаждения до 0°С в ледяной бане добавляют (Е)-3-(5-циан-2-метоксифенил)акриловую кислоту (полученную согласно методу Д) (2,24 г, 11,0 ммолей), а затем N-метилморфолин (1,21 мл, 11,0 ммолей). Суспензию интенсивно перемешивают и дают нагреться до комнатной температуры в течение ночи. После перемешивания в течение 24 ч при комнатной температуре образовавшийся осадок фильтруют при 0°С и промывают холодным (0°С) CH2Cl2 (10 мл). После сушки под вакуумом при 35°С получают указанный в заголовке продукт в виде кристаллического порошка, tпл 183-185°С. МС(EI): [М]+ 352.
Получение полу-(L)-тартрата (E)-N-{(S)-1-[4-(4-хлорбензоил)пиперидин-1-илметил]-2-гидроксиэтил}-3-(5-циан-2-метоксифенил)акриламида
К суспензии дигидрохлорида [1-((S)-2-амино-3-гидроксипропил)пиперидин-4-ил]-(4-хлорфенил) метанона (9,24 г, 25,0 ммолей) в этаноле (250 мл) добавляют N-метилморфолин (2,53 г, 25,0 ммолей). Суспензию перемешивают при 45°С в течение 30 мин, затем добавляют S-бензотиазол-2-иловый эфир (E)-3-(5-циан-2-метоксифенил)тиоакриловой кислоты (4,40 г, 12,5 ммоля), суспензию разбавляют этанолом (20 мл) и продолжают перемешивание при 45°С в течение 3 ч. Добавляют еще порцию тиоэфира (2,64 г, 7,5 ммоля) и суспензию перемешивают в течение еще 4 ч при 45°С. Добавляют последнюю порцию тиоэфира (1,76 г, 5,0 ммоля) и после перемешивания в течение еще 3 ч добавляют еще одну порцию N-метилморфолина (1,26 г, 12,46 ммоля) и перемешивание продолжают в течение ночи, затем добавляют последнюю порцию N-метилморфолина (1,26 г, 12,46 ммоля). Суспензию сразу фильтруют и фильтрат упаривают досуха при пониженном давлении. Остаток растворяют в СН2Cl2 (250 мл) и промывают последовательно 10%-ным водным Na2СО3 (2×100 мл) и 10%-ным водным NaCl (4×100 мл). Органическую фазу перемешивают с целитом (1 г), фильтруют и упаривают досуха. Остаток сушат под вакуумом и растворяют в этаноле (130 мл). Добавляют при перемешивании при 35°С раствор L-винной кислоты (4,5 г, 30,0 ммолей) в этаноле (100 мл) и образовавшуюся суспензию перемешивают при 50-55°С до получения прозрачного раствора. После помутнения, связанного с образованием кристаллов, суспензию медленно охлаждают до 0°С и перемешивают в ледяной бане в течение еще 45 мин, затем осадок фильтруют, промывают холодным (0°С) этанолом (20 мл) и перекристаллизовывают из этанола, получая указанный в заголовке продукт, tпл 90-120°С (разложение). МС (ES+): [МН]+ 482.
Получение (E)-N-{(S)-1-[4-(4-хлорбензоил)пиперидин-1-илметил]-2-гидроксиэтил}-3-(5-циан-2-метоксифенил)акриламида
10%-ный водный Na2CO3 (100 мл) добавляют при перемешивании при комнатной температуре к суспензии полу-(L)-тартрата (E)-N-{(S)-1-[4-(4-хлорбензоил)пиперидин-1-илметил]-2-гидроксиэтил}-3-(5-циан-2-метоксифенил)акриламида (6,32 г, 10,0 ммолей) в СН2Cl2 (150 мл) и воде (50 мл). После перемешивания при температуре окружающей среды в течение 30 мин фазы разделяют и водную фазу экстрагируют СН2Cl2 (100 мл). Объединенные СН2Cl2-фазы экстрагируют 10%-ным водным NaCl (2×100 мл), перемешивают с целитом (500 мг) и фильтруют после упаривания при пониженном давлении, получая бесцветную пену. После добавления бутилацетата (200 мл) образуется прозрачный раствор, который нагревают до 80°С и дают медленно охладиться до комнатной температуры. После разбавления ТБМЭ (150 мл) суспензию охлаждают до 0°С, осадившиеся кристаллы отфильтровывают, промывают холодной (0°С) смесью бутилацетата/ТБМЭ, 1:1 (50 мл) и сушат под вакуумом при 45-50°С, получая (E)-N-{(S)-1-[4-(4-хлорбензоил)пиперидин-1-илметил]-2-гидроксиэтил}-3-(5-циан-2-метоксифенил)акриламид, tпл 162-163°С. МС (EI): [МН]+ 482.
Изобретение относится к производным пиперидина общей формулы (I)
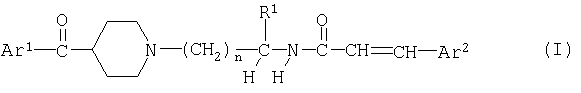
в свободной форме или в форме соли, где
Ar1 обозначает фенил, замещенный одним или несколькими атомами галогена,
Ar2 обозначает фенил или нафтил, который является незамещенным или замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, циано, гидрокси, нитро, С1-С8алкил, С1-С8галоалкил, С1-С8алкокси или С1-С8алкоксикарбонил,
R1 обозначает водород или С1-С8алкил, необязательно замещенный гидрокси-, С1-С8алкокси-, ацилоксигруппой, -N(R2)R3, галогеном, карбоксигруппой, С1-С8алкоксикарбонилом, -CON(R4)R5 или одновалентной циклической органической группой,
R2 и R3 каждый независимо друг от друга обозначает водород или С1-С8алкил, или R2 обозначает водород и R3 обозначает ацил или -SO2R6, или R2 и R3 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную гетероциклическую группу,
R4 и R5 каждый независимо друг от друга обозначает водород или С1-С8алкил, или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную гетероциклическую группу,
R6 обозначает С1-С8алкил, С1-С8галоалкил или фенил, необязательно замещенный С1-С8алкилом, и
n обозначает 1, 2, 3 или 4,
при условии, что когда Ar1 обозначает пара-хлорфенил и R1 обозначает водород, то Ar2 не обозначает фенил или пара-нитрофенил.
Соединения формулы (I) обладают ингибирующей CCR-3 активностью и могут найти применение в медицине. 2 н. и 5 з.п. ф-лы, 1 табл.
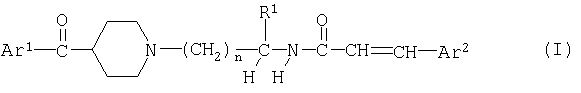
в свободной форме или в форме соли,
где Ar1 обозначает фенил, замещенный одним или несколькими атомами галогена,
Ar2 обозначает фенил или нафтил, который является незамещенным или замещен одним или несколькими заместителями, выбранными из ряда, включающего галоген, циано, гидрокси, нитро, С1-С8алкил, С1-С8галоалкил, С1-С8алкокси или С1-С8алкоксикарбонил,
R1 обозначает водород или С1-С8алкил, необязательно замещенный гидрокси-, С1-С8алкокси-, ацилоксигруппой, -N(R2)R3, галогеном, карбоксигруппой, С1-С8алкоксикарбонилом, -CON(R4)R5 или одновалентной циклической органической группой,
R2 и R3 каждый независимо друг от друга обозначает водород или С1-С8алкил, или R2 обозначает водород и R3 обозначает ацил или -SO2R6, или R2 и R3 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную гетероциклическую группу,
R4 и R5 каждый независимо друг от друга обозначает водород или С1-С8алкил, или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную гетероциклическую группу,
R6 обозначает С1-С8алкил, С1-С8галоалкил или фенил, необязательно замещенный С1-С8алкилом, и
n обозначает 1, 2, 3 или 4,
при условии, что, когда Ar1 обозначает парахлорфенил и R1 обозначает водород, то Ar2 не обозначает фенил или паранитрофенил.
Ar1 обозначает фенил, замещенный фтором или хлором в параположении относительно указанной карбонильной группы и необязательно дополнительно замещенный галогеном в ортоположении относительно указанной карбонильной группы,
Ar2 обозначает фенил, монозамещенный заместителем, выбранным из ряда, включающего галоген, циано, нитро и С1-С4алкокси, фенил, замещенный двумя заместителями, которые могут быть одинаковыми или различными, выбранными из ряда, включающего галоген, циано, гидрокси, С1-С4алкокси, С1-С4алкил, С1-С4галоалкил и нитро, или фенил, замещенный тремя заместителями, которые могут быть одинаковыми или различными, выбранными из ряда, включающего галоген, гидрокси, С1-С4алкокси и С1-С4алкоксикарбонил,
R1 обозначает водород, С1-С4алкил или С1-С4алкил, замещенный гидроксигруппой, С3-С8циклоалкилом, фенилом, фенилом, замещенным С1-С4алкилсульфониламиногруппой, или 5- или 6-членной гетероциклической ароматической группой, несущей один или несколько кольцевых гетероатомов, выбранных из азота, кислорода и серы, и
n обозначает 1 или 2.
Ar1 обозначает фенил, замещенный фтором или хлором в параположении относительно указанной карбонильной группы,
Ar2 обозначает фенил, замещенный в ортоположении относительно указанной -СН=СН-группы С1-С4алкоксигруппой и в параположении относительно С1-С4алкоксигруппы цианогруппой, галогеном или С1-С4алкоксигруппой,
R1 обозначает С1-С4алкил, замещенный гидроксигруппой, фенилом, фенилом, замещенным С1-С4алкилсульфониламиногруппой, или 5- или 6-членной гетероциклической ароматической группой, несущей один или два кольцевых гетероатома, выбранных из азота, кислорода и серы, и
n обозначает 1.
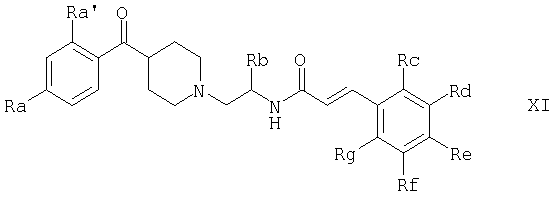
в свободной форме или в форме соли,
где Ra' обозначает водород и Ra, Rb, Rc, Rd, Re, Rg и Rf имеют значения, указанные в следующей таблице
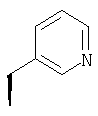
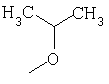
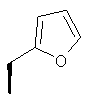
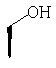
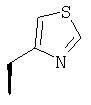
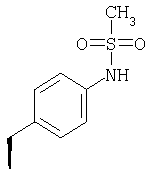
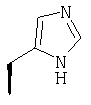
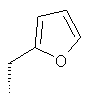
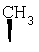
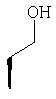
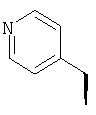
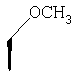
или где Ra и Ra' обозначают фтор, Rb, Rd, Re и Rg обозначают водород, Rc обозначает метокси и Rf обозначает бром.

с соединением формулы
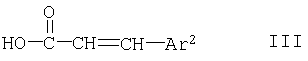
или с его образующим амид производным,
где Ar1, Ar2, R1 и n имеют указанные в п.1 значения,
с последующим выделением целевого продукта в свободной форме или в форме соли.
| RU 2055834 C1, 10.03.1996 | |||
| WO 00/29377 A1, 25.05.2000 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

