Изобретение относится к замещенным производным 1,4-бензотиазепин-1,1-диоксида, их физиологически совместимым солям и физиологически функциональным производным.
Производные 1,4-бензотиазепин-1,1-диоксида, как и их применение для лечения гиперлипидемии, а также артериосклероза и гиперхолестеринемии уже были описаны [заявка РСТ № PCT/GB 95/01884, публикация № WO 96/05188].
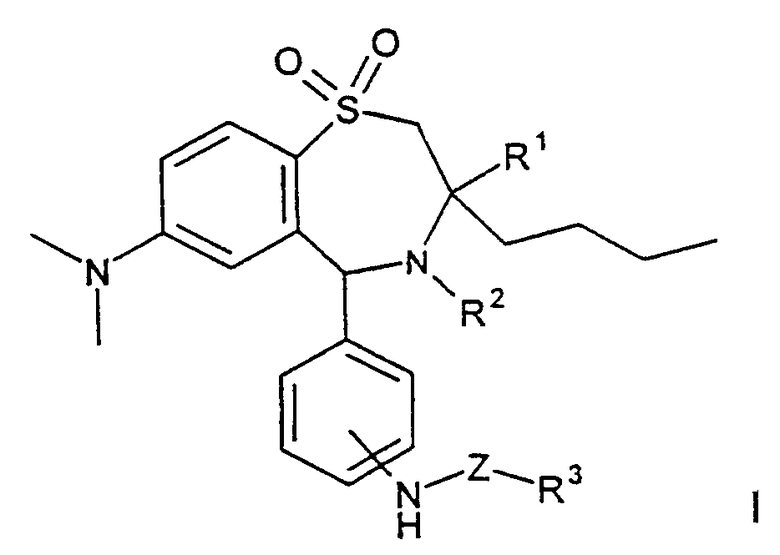
Задачей изобретения является получение терапевтически пригодных соединений, проявляющих гиполипидемическое действие. В особенности задача состояла в поиске новых соединений, которые по сравнению с соединениями, описанными в уровне техники, уже при сниженной дозировке вызывают повышенное выделение желчных кислот. Изобретение касается соединений формулы I
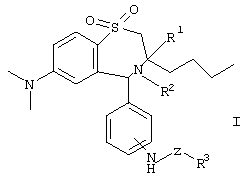
где R1 обозначает метил, этил, пропил, бутил;
R2 обозначает Н, ОН;
R3 обозначает остаток сахарида, остаток дисахарида, остаток трисахарида, остаток тетрасахарида, причем остаток сахарида, остаток дисахарида, остаток трисахарида или остаток тетрасахарида в случае необходимости одно- или многократно замещен сахаридозащитной группой;
Z обозначает -(С=O)n-С0-С16-алкил-, -(С=O)n-С0-С16-алкил-NН-, -(С=O)n-С0-С16-алкил-O-, -(С=O)n-С1-С16-алкил-(С=O)m, ковалентную связь;
n обозначает 0 или 1;
m обозначает 0 или 1;
а также их фармацевтически совместимых солей и физиологически функциональных производных.
Предпочтительными являются соединения формулы I, в которых один или более остатков имеют следующие значения:
R1 - этил, пропил, бутил;
R2 - H, ОН;
R3 - остаток сахарида, остаток дисахарида, причем остаток сахарида или остаток дисахарида в случае необходимости одно- или многократно замещен сахаридозащитной группой;
Z - -(С=O)n-С0-С16-алкил-, -(C=O)n-С0-С16-алкил-NН-, -(С=O)n-С0-С16-алкил-O-, -(С=O)n-С1-С16-алкил-(С=O)m, ковалентная связь;
n - 0 или 1;
m - 0 или 1;
а также их фармацевтически совместимые соли.
Особенно предпочтительными являются соединения формулы I, в которых один или более остатков имеют следующие значения:
R1 - этил, бутил;
R2 - Н, ОН;
R3 - остаток сахарида (моносахарида), причем остаток сахарида в случае необходимости одно- или многократно замещен сахаридозащитной группой;
Z - -(С=O)-С0-С4-алкил-, ковалентная связь;
а также их фармацевтически совместимые соли.
Фармацевтически совместимые соли благодаря их повышенной растворимости в воде по сравнению с исходными или основными соединениями особенно пригодны для медицинских применений. Эти соли должны иметь фармацевтически совместимый анион или катион. Пригодными фармацевтически совместимыми солями присоединения кислот соединений согласно изобретению являются соли неорганических кислот, как соляная кислота, бромистоводородная, фосфорная, метафосфорная, азотная, сульфо- и серная кислоты, а также органических кислот, как, например, уксусная кислота, бензолсульфо-, бензойная, лимонная; этансульфо-, фумаровая, глюконовая, гликолевая, изотионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфо-, янтарная, п-толуолсульфо-, винная и трифторуксусная кислоты. Для медицинских целей особенно предпочтительно применяют хлорид. Пригодными фармацевтически совместимыми основными солями являются соли аммония, соли щелочных металлов (как соли натрия и калия) и соли щелочноземельных металлов (как соли магния и кальция).
Соли с фармацевтически несовместимым анионом также охватываются изобретением в качестве полезных промежуточных продуктов для получения или очистки фармацевтически совместимых солей и/или для использования в нетерапевтических, например, in vitro-применениях.
Используемое здесь понятие "физиологически функциональное производное" означает любое физиологически совместимое производное соединения согласно изобретению, например сложный эфир, который при введении млекопитающему, как, например, человеку, в состоянии образовать (непосредственно или опосредованно) такое соединение или активный метаболит с его участием.
Дальнейшим аспектом этого изобретения являются пролекарства соединений согласно изобретению. Такие пролекарства in vivo могут быть метаболизированы до соединений согласно изобретению. Эти пролекарства сами могут быть или не быть биологически активными.
Соединения согласно изобретению могут также существовать в различных полиморфных формах, например в виде аморфной и кристаллической полиморфных форм. Все полиморфные формы соединений согласно изобретению включены в объем изобретения и являются объектами изобретения.
Последующее касается всех ссылок на "соединение(я) согласно формуле (I)", как описано выше, а также их солей, сольватов и физиологически функциональных производных.
Количество соединения согласно формуле (I), необходимое для достижения желаемого биологического эффекта, зависит от ряда факторов, например выбранного специфического соединения, планируемого применения, вида введения и клинического состояния пациента. В общем, дневная доза находится в области от 0,1 мг до 100 мг (обычно 0,1 мг и 50 мг) в день на килограмм веса тела, например 0,1-10 мг/кг/день. Таблетки или капсулы могут содержать, например, от 0,01 до 100 мг, обычно от 0,02 до 50 мг. В случае фармацевтически совместимых солей вышеназванные количественные данные относятся к весу бензотиазепинового иона соли. Для профилактики или терапии вышеназванных состояний соединения согласно формуле (I) могут применяться непосредственно в виде соединения, однако предпочтительно они находятся в форме фармацевтической композиции с совместимым носителем. Естественно, носитель должен быть совместимым, т.е. совместимым с другими компонентами композиции, и не являться вредным для здоровья пациентов. Носитель может быть твердым веществом, или жидкостью, или обоими и предпочтительно формоваться с соединением в препаративную форму в виде разовой дозы, например в виде таблетки, которая может содержать от 0,05 до 95% биологически активного вещества. Другие фармацевтически активные вещества могут также иметь место, включая другие соединения формулы (I). Фармацевтические композиции согласно изобретению могут быть получены по одному из известных фармацевтических способов, которые, по существу, состоят в том, что компоненты смешиваются с фармакологически совместимыми носителями и/или вспомогательными веществами.
Фармацевтическими композициями согласно изобретению являются такие, которые пригодны для орального и перорального (например, подъязычного) введения, хотя более пригодный способ введения в каждом отдельном случае зависит от вида и тяжести состояния, подвергаемого лечению, и от вида соединения формулы (I). Дражированные препаративные формы и дражированные препаративные формы пролонгированного действия также включены в объем изобретения. Предпочтительными являются кислотоустойчивые и устойчивые к желудочному соку препаративные формы. Пригодные покрытия, устойчивые к желудочному соку, включают ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы и анионные полимеры метакриловой кислоты и метилового эфира метакриловой кислоты.
Пригодные фармацевтические соединения для орального введения могут находиться в отдельных дозированных формах, как, например, капсулы, капсулы с облатками, сосательные таблетки или таблетки, которые по мере надобности содержат определенное количество соединения формулы (I); в виде порошка или гранулятов, в виде раствора или суспензии в водной или неводной жидкости; или в виде эмульсии масло-в-воде или вода-в-масле. Эти композиции могут быть приготовлены, как уже упоминалось, любым пригодным фармацевтическим способом, который включает стадии, на которых приводятся в контакт биологически активное вещество и носитель (который может состоять из одного или нескольких дополнительных компонентов). В общем, композиции получают посредством равномерного и гомогенного смешивания биологически активного вещества с жидким и/или тонко распределенным твердым носителем, после чего продукт в случае необходимости формуют. Так, можно, например, изготовить таблетки, спрессовывая или формуя порошок или гранулят соединения в случае необходимости с одним или несколькими дополнительными компонентами. Прессованные таблетки могут быть изготовлены в пригодной машине посредством таблетирования соединения в свободно текучей форме, как, например, порошок или гранулят, в случае необходимости смешанного со связующим мягчителем, инертным разбавителем и/или одним (несколькими) поверхностно-активным/диспергирующим средством. Формованные таблетки могут быть изготовлены в пригодной машине путем формования порошкообразного соединения, увлажненного инертным жидким разбавителем.
Фармацевтические композиции, пригодные для перорального (подъязычного) приема, представляют собой сосательные таблетки, содержащие соединение согласно формуле (I) с корректирующей вкус добавкой, обычно сахарозой и гуммиарабиком или трагантом, и пастилки, включающие соединение в инертной основе как желатина и глицерин или сахароза и гуммиарабик.
Объектом изобретения являются далее как смеси изомеров формулы I, так и чистые стереоизомеры формулы I, а также смеси диастереоизомеров формулы I и чистые диастереомеры. Разделение смесей происходит хроматографическим путем.
Предпочтительными являются рацемические, а также энантиомерно чистые соединения формулы I со следующей структурой:
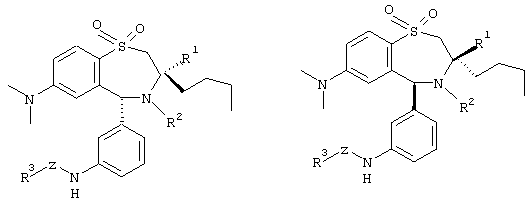
Под остатками моносахаридов понимают соединения, которые образованы от альдоз и кетоз с 3 до 7 атомами углерода, которые могут относиться к D- или L-ряду; к ним относятся также аминосахара, сахарные спирты или сахарные кислоты. В качестве примеров называют глюкозу, маннозу, фруктозу, галактозу, рибозу, эритрозу, глицериновый альдегид, седогептулозу, глюкозамин, галактозамин, глюкуроновую кислоту, галактуроновую кислоту, глюконовую кислоту, галактоновую кислоту, манноновую кислоту, глюкамин, 3-амино-1,2-пропандиол, глюкаровую кислоту и галактаровую кислоту.
Под дисахаридами имеют в виду сахариды, состоящие из двух моносахаридных звеньев. Ди-, три- или тетрасахариды образуются с помощью ацеталеподобной связи из двух или нескольких моносахаридов. При этом связи могут находиться в α- и β-форме. В качестве примеров называют лактозу, мальтозу и целлобиозу.
Если сахар является замещенным, то замещение предпочтительно происходит по атому водорода ОН-группы сахара.
Для гидроксильных групп сахаров в расчет принимаются, в основном, защитные группы, такие как бензильная, ацетильная, бензоильная, пивалоильная, тритильная, трет.-бутилдиметилсилильная, бензилиденовая, циклогексилиденовая или изопропилиденовая защитные группы.
Соединения формулы I и их фармацевтически совместимые соли и физиологически функциональные производные представляют собой идеальное лекарственное средство для лечения нарушений липидного обмена, в особенности гиперлипидемии. Соединения формулы I пригодны также для влияния на уровень холестерина в сыворотке крови, а также для предупреждения и лечения артерио-склеротических симптомов. Соединения могут вводиться в случае необходимости также в комбинации со статинами, как, например, симвастатин, флувастатин, правастатин, церивастатин, ловастатин или аторвастин. Следующие данные подтверждают фармакологическую активность соединений согласно изобретению.
Биологическое испытание соединений согласно изобретению происходило посредством перфузионного теста. В этом испытании исследовали действие соединений согласно изобретению на транспорт желчной кислоты в подвздошной кишке. Испытывались смеси диастереомеров соединений.
Перфузионный тест проводили следующим образом.
Экспериментальная часть
Мужским особям крыс Wistar (диапазон веса 250-350 г) вводили наркотическое средство уретан (1,5 г/кг внутрибрюшинно) и в желчную протоку вводили канюлю с полиэтиленовым зондом. Проксимально к илеоцекальному клапану на расстоянии 8 см выполняли разрез в подвздошной кишке и вшивали силиконовый адаптер для зондов. Второй разрез с имплантацией соответствующего силиконового адаптера выполняли в слепой кишке. Силиконовые зонды присоединяли к адаптеру, для того чтобы ортогонально или открыто (не рециркулирующе) перфундировать подвздошную кишку перфузионным буфером со скоростью перфузии 1 мл/мин.
Перфузионные зонды заполняли перфузионным буфером (137 ммоль NaCl, 0,9 ммоль CaCl2, 0,51 ммоль MgCl2, 8,1 ммоль Na2HPO4, 2,7 ммоль КСl, 1,47 ммоль КН2РO4) (рН 7,4), 1% (об./об.) этанола + 1% ДМСО. Перфузионный буфер содержал тестируемые соединения в указанной концентрации или индифферентную лекарственную основу. Буфер предварительно подогревали до 37°С. Перфузионный буфер содержал 3 ммоля таурохолевой кислоты (ТХК), которая была опять подкреплена с помощью 1000 dpm/мл (число распадов в минуту/мл) 3H ТХК в качестве маркера.
Ход исследования и оценка результатов
Был выбран экспериментальный подход, который позволял определить торможение транспорта желчной кислоты на индивидуальном животном. Желчь собирали с интервалом в 10 минут в течение свыше 90 мин (в случае последующей фазы промывания для тестирования обратимости в течение периода времени до 160 мин). Перфузия буферного раствора, содержащего индифферентную лекарственную основу, в течение свыше 40 мин (первичное тестируемое вещество) следовала перед перфузией перфузионным буфером, который содержал тестируемое соединение в исследуемой концентрации (до 90 мин).
Для расчета торможения в процентном отношении посредством тестируемого соединения получали dpms (распад в мин 3H ТХК) в желчи за 80-90 мин (конец перфузии с тестируемым веществом) при периоде сбора 30-40 мин во время предварительной фазы, если в контрольной фазе выделение 3H ТХК достигало своего максимума и плато. Рассчитывали ЕС50 как эффективную концентрацию среди значений различной концентрации, которая ингибировала максимальное выделение желчной кислоты на 50%.
Результаты приведены в таблице.
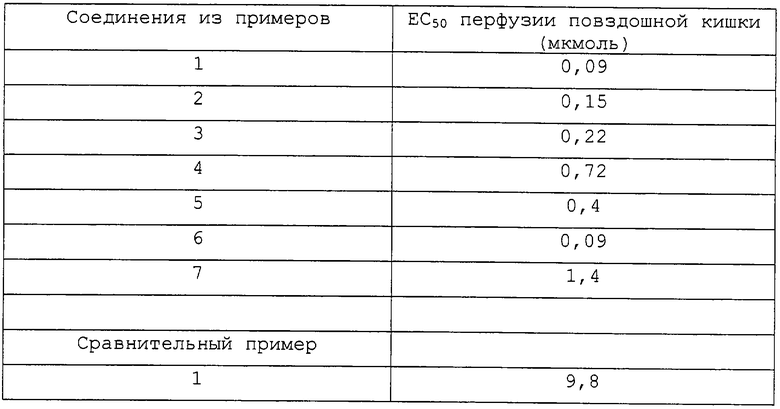
Из данных следует, что соединения формулы I согласно изобретению по отношению к соединениям, описанным в уровне техники, проявляют активность, в 7-100 раз большую.
Следующие примеры более подробно поясняют изобретение, не ограничивая его описанными продуктами и формами выполнения.
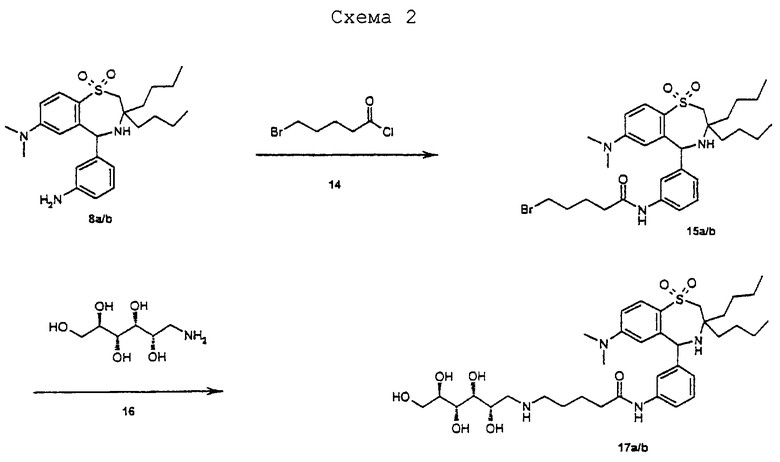
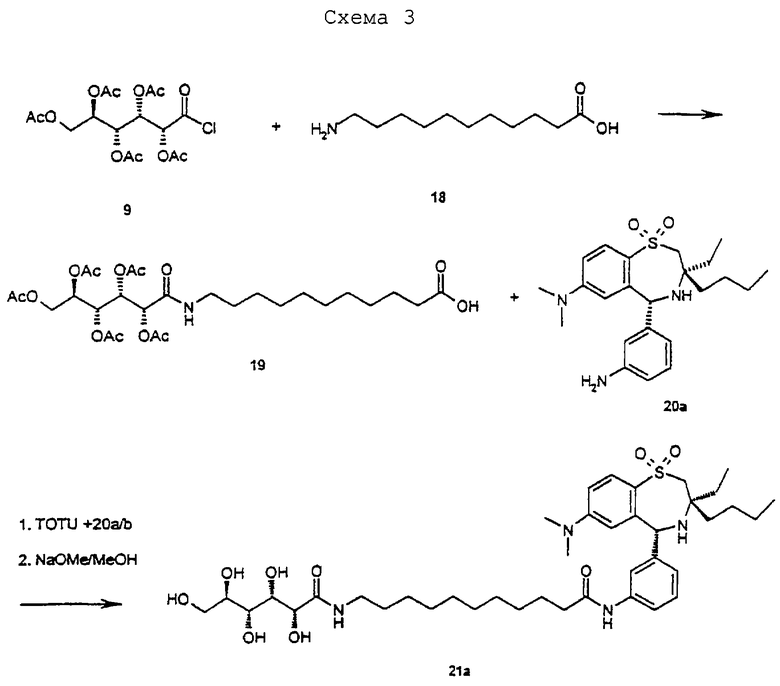
Синтез соединения 2:
20 г (91,6 ммоль) 2,5-дифторбензофенона 1 (Fluka) растворяют в 400 мл ДМСО. Под аргоном добавляют 7,0 г (150 ммоль) сульфида лития. После трех часов при 120°С оставляют охлаждаться до комнатной температуры. Встряхивают с 200 мл 2 М водной НСl и 500 мл этилацетата. Органическую фазу еще дважды промывают раствором NaCl, сушат над MgSO4, фильтруют и концентрируют. Получают 24 г сырого продукта 2 в виде красноватого масла. ТСХ (н-гептан/этилацетат 3:1). Rf=0,3, продукт 1 Rf=0,4. С13Н9FОS (232,28). Масс-спектр (M+H)+=233,1.
Синтез соединения 4:
7 г сырого продукта 2, 2,5 г (16 ммоль) дибутилазиридина 3 (R. Gauthier et al., J.Organomet. Chem. 140 (1977) 245-255) и 300 мг п-толуолсульфокислоты растворяют в 100 мл лутидина. Реакционный раствор кипятят 3 часа в водном сепараторе. Затем концентрируют и осадок очищают флэш-хроматографией. Выход 3,6 г (61%) 4 в виде бесцветного масла. ТСХ (н-гептан/этилацетат 9:1). Rf=0,5. С23Н28FNS (369,55). Масс-спектр (M+H)+=370,3.
Синтез соединения 5:
3,6 г (9,7 ммоль) 4 и 6,0 г NaIО4 суспендируют в 100 мл ацетонитрила, 50 мл метиленхлорида и 30 мл воды. После добавления 200 мг RuСl3 сильно перемешивают 2 часа при комнатной температуре. Раствор разбавляют 200 мл этилацетата и дважды промывают раствором NaCl. После высушивания над MgSO4 концентрируют и очищают флэш-хроматографией. Выход 3,47 г (89%) 5 в виде аморфного твердого вещества. ТСХ (н-гептан/этилацетат 4:1). Rf=0,5, эдукт 4 Rf=0,6. C23H28FNO2S (401,55). Масс-спектр (М+H)+=402,2.
Синтез соединения 6:
3,47 г (8,6 ммоль) 5 растворяют в 24 мл нитрующей смеси (из 14 мл НNО3 и 10 мл H2SO4). Температуру реакции 20°С поддерживают с помощью охлаждения. Через 30 минут выливают раствор в смесь из 700 г льда и 200 мл этилацетата. Водную фазу отделяют и осторожно четырежды промывают 150 мл насыщенного раствора NаНСО3. Затем сушат над MgSO4, концентрируют и очищают флэш-хроматографией. Выход 3,0 г (78%) 6 в виде аморфного твердого вещества. ТСХ (н-гептан/этилацетат 4:1). Rf=0,4. C23H27N2O4SF (446,54). Масс-спектр (М+H)+=447,2.
Синтез соединения 7:
3,0 г (6,7 ммоль) 6 растворяют в 50 мл 33%-ного HNMe2 в этаноле (Fluka) и один час перемешивают при 50°С. Затем оставляют охлаждаться до комнатной температуры и фильтруют образовавшийся продукт. Выход 2,86 г (90%) 7 в виде желтоватых кристаллов с температурой плавления 188°С. ТСХ (н-гептан/этилацетат 2:1). Rf=0,5, эдукт 7 Rf=0,6. C52H33N3O4S (471,62). Масс-спектр (M+H)+=472,3.
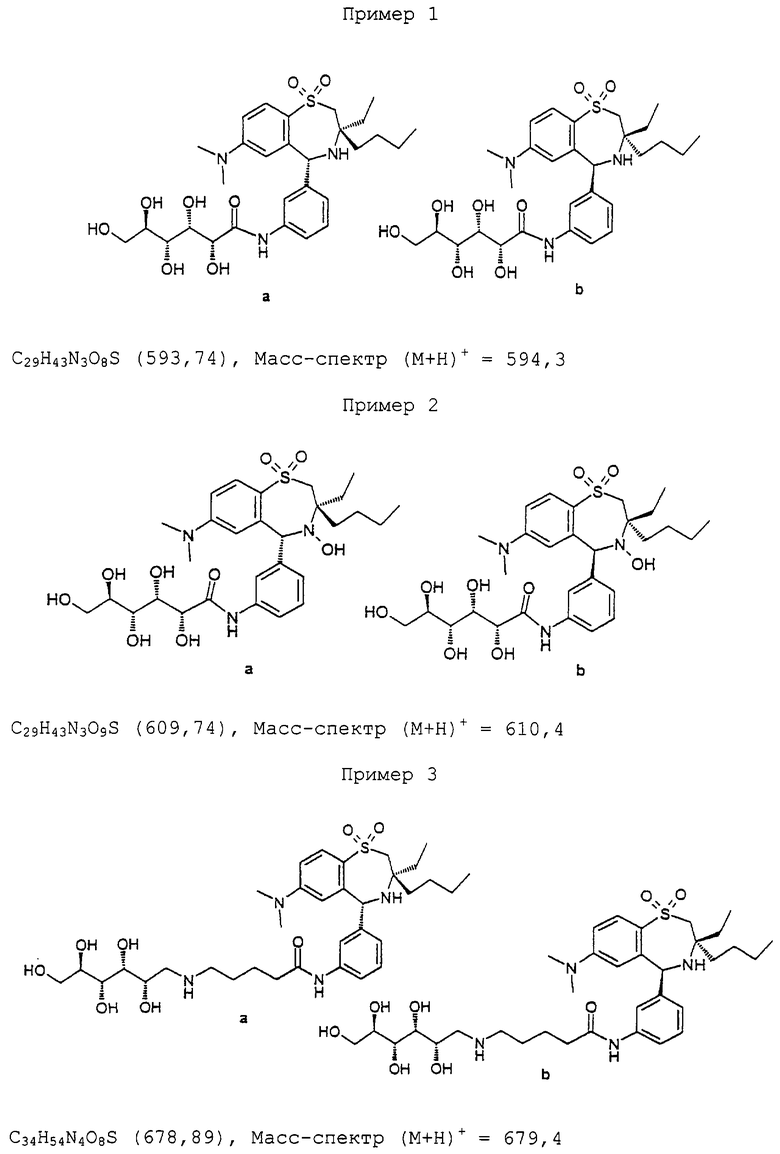
Синтез соединения 8a/b в виде смеси энантиомеров:
1,05 г (2,2 ммоль) 7 суспендируют в 30 мл толуола и добавляют 500 мг платины на активированном угле (10%-ной). 30 часов гидрируют при давлении водорода 150 бар и 100°С в вибрационном автоклаве. Для разделения фильтруют через кизельгель, после промывают 100 мл метанола, концентрируют и осадок очищают флэш-хроматографией. Выход 495 мг (48%) 8a/b в виде аморфного твердого вещества. ТСХ (н-гептан/этилацетат 1:1). Rf=0,3. C25H37N3O2S (443,65). Масс-спектр (М+Н)+=444,3.
Синтез соединения 10а/b в виде смеси диастереомеров:
80 мг (0,18 ммоль) 8а/b и 100 мг (0,24 ммоль) пента-0-ацетил-D-глюконовой кислоты (Org.Synth. Band 5, 887) растворяют в 4 мл ДМФ (диметилформамида). Друг за другом добавляют 100 мг (0,3 ммоль) TOTU (Fluka), 35 мг (0,24 ммоль) оксима (этилового эфира гидроксиимино-циануксусной кислоты; Fluka) и 0,1 мл (0,78 ммоль) NEM (4-этил-морфолина). Через 1 час при комнатной температуре разбавляют 20 мл этилацетата и трижды промывают водой. Органическую фазу сушат над МgSO4, фильтруют и концентрируют. Осадок очищают посредством флэш-хроматографии (этилацетат/н-гептан 2:1) и получают 130 мг (86%) 10а/b в виде аморфного твердого вещества. ТСХ (этилацетат/н-гептан 2:1) Rf=0,3. Продукт 10а/b имел такую же удерживающую способность как продукт 8а/b, но по другому окрашивался 2 М серной кислотой. С41Н57Н3O13S (831,97). Масс-спектр (М+Н)+=832,6.
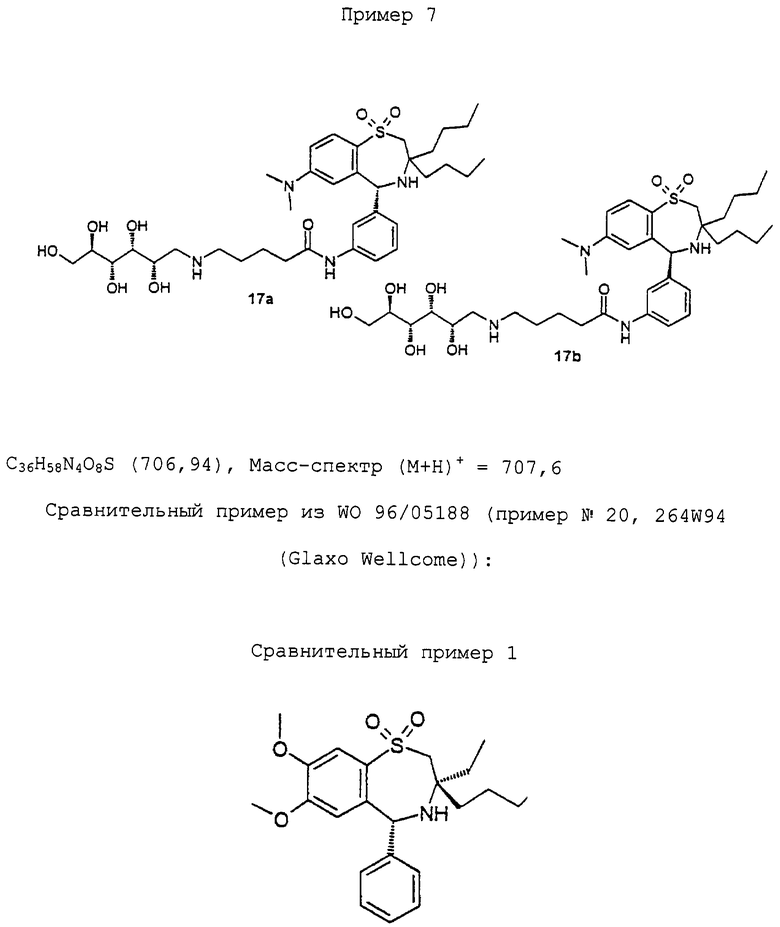
Синтез соединения 11a/b в виде смеси диастереомеров:
130 мг (0,16 ммоль) 10а/b растворяют в 5 мл метанола. После добавления 0,2 мл метанольного 1 М раствора метилата натрия выдерживают один час при комнатной температуре. Затем нейтрализуют метанольным раствором НСl и концентрируют. Осадок очищают флэш-хроматографией (метиленхлорид/метанол/конц.аммиак 30/10/3) и получают 78 мг (80%) 11a/b в виде аморфного твердого вещества. ТСХ (метиленхлорид/метанол/конц. аммиак 30/10/3). Rf=0,4. C31H47N3O8S (621,80). Масс-спектр (М+Н)+=622,4.
Синтез соединения 12а/b в виде смеси диастереомеров:
618 мг (0,74 ммоль) 10а/b растворяют в 30 мл метиленхлорида и добавляют 385 мг (2,23 ммоль) 70%-ной м-хлорпербензойной кислоты (Fluka). Через 30 минут при комнатной температуре разбавляют 100 мл этилацетата и трижды промывают раствором NaHCO3. После сушки МgSO4 концентрируют и получают 700 мг сырого продукта. Этот сырой продукт растворяют в 28 мл 0,05 М раствора TiCl4 в ацетонитриле. После добавления 300 мг твердого NaI оставляют перемешиваться 15 минут. Для разделения смесь разбавляют 150 мл этилацетата и промывают 100 мл 2 М раствора тиосульфата натрия. Органическую фазу сушат над MgSO4, концентрируют и осадок очищают флэш-хроматографией. Выход 550 мг (87% после двух стадий) 12а/b в виде аморфного твердого вещества. ТСХ (н-гептан/этилацетат 1:2). Rf=0,3, эдукт 10а/b Rf=0,35. C41H57N3O14S (847,99). Масс-спектр (М+Н)+=848,5.
Синтез соединения 13а/b в виде смеси диастереомеров:
550 мг (0,65 ммоль) 12а/b растворяют в 20 мл метанола. После добавления 0,3 мл метанольного 1 М раствора метилата натрия выдерживают один час при комнатной температуре. Затем нейтрализуют метанольным раствором НСl и концентрируют. Осадок очищают флэш-хроматографией (метиленхлорид/метанол/конц. аммиак 30/10/3) и получают 370 мг (89%) 13a/b в виде аморфного твердого вещества. ТСХ (метиленхлорид/метанол/конц. аммиак 30/10/3). Rf=0,4. C31H47N3O9S (637,80). Масс-спектр (M+Н)+=638,4.
Синтез соединения 15а/b в виде смеси диастереомеров:
719 мг (1,6 ммоль) 8а/b растворяют в 30 мл метиленхлорида и 2 мл триэтиламина. К этому раствору прикапывают 0,5 мл (3,7 ммоль) 14 и выдерживают 15 минут при комнатной температуре. Затем раствор фильтруют через кизельгель и после этого промывают 100 мл этилацетата. После концентрирования осадок очищают флэш-хроматографией. Выход 950 мг (95%) 15a/b в виде аморфного твердого вещества. ТСХ (н-гептан/этилацетат 1:1). Rf=0,4. С30Н44ВrN3О3S (606,67). Масс-спектр (М+Н)+=607,3.
Синтез соединения 17a/b в виде смеси диастереомеров:
897 мг (1,47 ммоль) 15 a/b растворяют в 20 мл ДМФ. После добавления 1,3 г (7,1 ммоль) 16 (глюкамин, Fluka) нагревают два часа до 80°С. Затем разбавляют 50 мл этилацетата и трижды промывают водой. Органическую фазу сушат над МgSO4, фильтруют и концентрируют. Осадок очищают посредством флэш-хроматографии (метиленхлорид/метанол/конц. аммиак 30/10/3) и получают 700 мг (67%) 17a/b в виде аморфного твердого вещества. ТСХ (метиленхлорид/метанол/конц. аммиак 30/10/3). Rf=0,4. С36Н58N4О8S (706,95). Масс-спектр (М+Н)+=707,4.
Синтез соединения 19:
8,0 г (18,8 ммоль) 9 (хлорида пента-О-ацетил-D-глюконовой кислоты; Org. Synth. Band 5, 887) добавляют к суспензии из 8,0 г (40 ммоль) 18 (Fluka) в 150 мл безводного ДМФ. Эту суспензию сильно перемешивают в течение 20 часов при комнатной температуре. Затем добавляют 500 мл этилацетата и 200 мл воды. Водную фазу еще раз экстрагируют 250 мл этилацетата. Объединенную органическую фазу трижды промывают раствором хлорида натрия, сушат над МgSO4, фильтруют и концентрируют. Выход 9,5 г (86%) 19 в виде бесцветного масла. ТСХ (метиленхлорид/метанол/конц. аммиак 30/10/3). Rf=0,8. C27H43NO13 (589,64). Масс-спектр (М+Н)+=590,4.
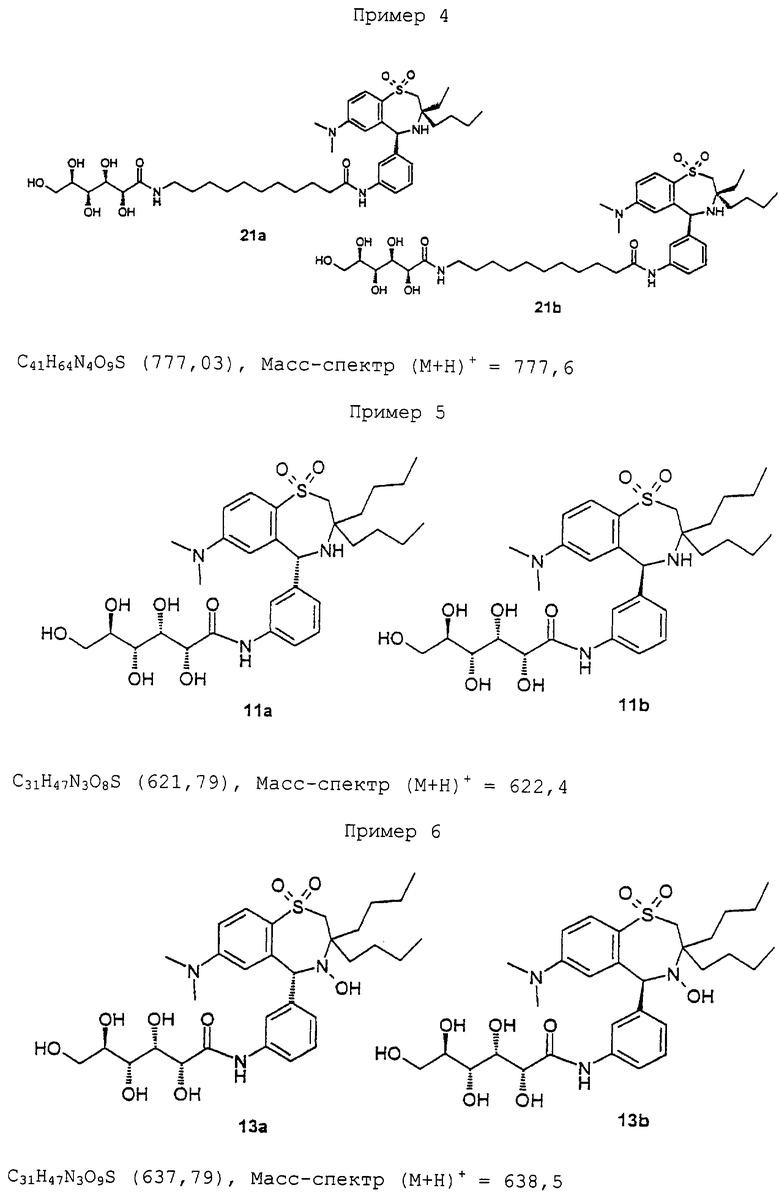
Синтез соединения 21а/b в виде смеси диастереомеров:
200 мг (0,34 ммоль) 19, 70 мг (0,17 ммоль) 20a/b (20a/b является аналогичным 8a/b, проводят последовательность реакцией по схеме 1 с 2-бутил-2-этил-азиридином и 1 (R. Gauthier et al., J. Organomet. Chem. 140 (1977) 245-255), 240 мг TOTU, 80 мг оксима и 0,3 мл NEM подвергают взаимодействию в 4 мл ДМФ аналогично методике соединения 11a/b. После флэш-хроматографии (метиленхлорид/метанол/конц. аммиак 30/5/1) получают 60 мг (46%, после двух стадий) 21 a/b в виде аморфного твердого вещества. ТСХ (метиленхлорид/метанол/конц. аммиак 30/5/1). Rf=0,2. C40H64N4O9S (777,04). Масс-спектр (М+Н)+=777,8.
Изобретение относится к области органической химии. Описываются производные 1,4-бензотиазепин-1,1-диоксида формулы I
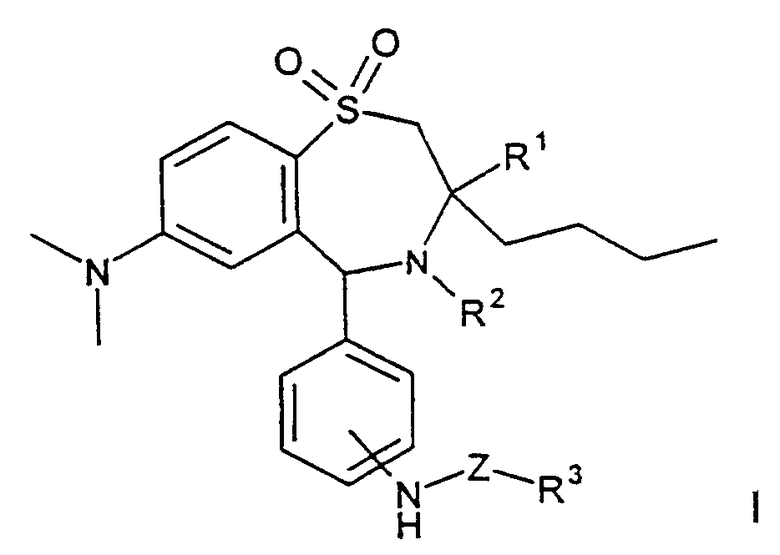
где R1 обозначает метил, этил, пропил, бутил; R2 обозначает Н, ОН;
R3 обозначает остаток сахарида, который в случае необходимости одно- или
многократно замещен сахаридозащитной группой; Z обозначает -(С=О)n-С0-С16 алкил-, -(С=O)n-С0-С16-алкил-NH-, ковалентную связь; n обозначает 0 или 1; m обозначает 0 или 1; а также их фармацевтически совместимые соли и физиологически функциональные производные. Также описываются способ получения соединений формулы (I), лекарственное средство на основе соединений формулы I, обладающие гиполипидемическим действием, и способ получения лекарственного средства. 4 с. и 4 з.п. ф-лы, 1 табл.
где R1 обозначает метил, этил, пропил, бутил;
R2 обозначает Н, ОН;
R3 обозначает остаток сахарида, который, в случае необходимости, одно- или многократно замещен сахаридозащитной группой;
Z обозначает -(С=О)n-С0-С16-алкил-, -(С=O)n-С0-С16-алкил-NН-, ковалентную связь;
n обозначает 0 или 1;
m обозначает 0 или 1,
а также их фармацевтически совместимые соли и физиологически функциональные производные.
R1 - этил, пропил, бутил;
R2 - Н, ОН;
R3 - остаток сахарида, который, в случае необходимости, одно- или многократно замещен сахаридозащитной группой;
Z - -(С=О)n-С0-С16-алкил-, -(C=O)n-C0-C16-алкил-NH-, ковалентная связь;
n - 0 или 1;
m - 0 или 1,
а также их фармацевтически совместимые соли.
R1 - этил, бутил;
R2 - Н, ОН;
R3 - остаток сахарида, который, в случае необходимости, одно- или многократно замещен сахаридозащитной группой;
Z - -(С=О)-С0-С4-алкил-, ковалентная связь;
а также их фармацевтически совместимые соли.
в которой R1, R2 и R3 имеют значения, приведенные для формулы I в п.1, подвергают взаимодействию с соединением формулы III
HO-Z-R3,(III)
в которой R3 и Z имеют значения, приведенные для формулы I в п.1, при отщеплении воды до соединения формулы I, и полученное соединение формулы I, в случае необходимости, переводят в физиологически совместимую соль или физиологически функциональное производное.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Устройство для фазирования синхронных источников импульсов | 1979 |
|
SU864582A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

