Настоящее изобретение касается циклопентиловых соединений, связанных с бензоксазинильной группой через амидофрагмент с использованием кольцевого атома азота бензоксазина. В частности, настоящее изобретение относится к циклопентильным соединениям, связанным с бензоксазинильной группой через амидофрагмент с использованием кольцевого атома азота бензоксазина и дополнительно замещенным гетероциклическим фрагментом, полезным в качестве модуляторов хемокиновых рецепторов.
Хемокины представляют собой семейство небольших (70-120 аминокислот) провоспалительных цитокинов с сильными хемотаксическими активностями. Хемокины являются хемотаксическими цитокинами, которые высвобождаются широким множеством клеток, притягивая различные клетки, такие как моноциты, макрофаги, T-клетки, эозинофилы, базофилы и нейтрофилы, к местам воспаления (рассмотрено в работах Schall, Cytokine, 3, 165-183 (1991), и Murphy, Rev. Immun., 12, 593-633 (1994)). Данные молекулы первоначально были определены по четырем сохранившимся цистеинам и поделены на два подсемейства на основании расположения первой цистеиновой пары. В семействе CXC-хемокинов, которое включает IL-8, GROα, NAP-2 и IP-10, эти два цистеина разделены одной аминокислотой, тогда как в семействе CC-хемокинов, которое включает RANTES, MCP-1, MCP-2, MCP-3, MIP-1α, MIP-1β и эотаксин, эти два остатка являются соседними.
α-Хемокины, такие как интерлейкин-8 (IL-8), нейтрофил-активирующий белок-2 (NAP-2) и белок, стимулирующий активность роста меланомы (MGSA), являются хемотаксическими, главным образом, для нейтрофилов, тогда как β-хемокины, такие как RANTES, MIP-1α, MIP-1β, моноцитарный хемотаксический протеин-1 (MCP-1), MCP-2, MCP-3 и эотаксин, являются хемотаксическими для макрофагов, моноцитов, T-клеток, эозинофилов и базофилов (Deng и др., Nature, 381, 661-666 (1996)).
Хемокины выделяются широким множеством клеточных типов и связываются со специфическими G-белок-ассоциированными рецепторами (GPCR) (рассмотрено в работе Horuk, Trends Pham. Sci., 15, 159-165 (1994)), присутствующими на лейкоцитах и других клетках. Данные хемокиновые рецепторы образуют подсемейство GPCR, которое в настоящее время состоит из пятнадцати охарактеризованных членов, и ряд отдельных веществ. В отличие от рецепторов для разнородных хемоаттрактантов, таких как C5a, fMLP, PAF и LTB4, хемокиновые рецепторы более селективно выражены на подмножествах лейкоцитов. Таким образом, образование специфических хемокинов обеспечивает механизм пополнения подмножеств конкретных лейкоцитов.
При связывании родственных им лигандов хемокиновые рецепторы преобразуют внутриклеточный сигнал, несмотря на ассоциированный тримерный белок G, давая в результате быстрое увеличение внутриклеточной концентрации кальция. Существует, по меньшей мере, семь человеческих хемокиновых рецепторов, которые связываются или реагируют на β-хемокины, со следующими характеристическими примерами: CCR-1 (или "CKR-1" или "CC-CKR-1") [MIP-1α, MIP-1β, MCP-3, RANTES] (Ben-Barruch и др., J. Biol. Chem., 270, 22123-22128 (1995); Beote и др., Cell, 72, 415-425 (1993)); CCR-2A и CCR-2B (или "CKR-2A"/"CKR-2A" или "CC-CKR-2A"/"CC-CKR-2A") [MCP-1, MCP-2, MCP-3, MCP-4]; CCR-3 (или "CKR-3" или "CC-CKR-3") [Эотаксин, Эотаксин 2, RANTES, MCP-2, MCP-3] (Rollins и др., Blood, 90, 908-928 (1997)); CCR-4 (или "CKR-4" или "CC-CKR-4") [MIP-1α, RANTES, MCP-1] (Rollins и др., Blood, 90, 908-928 (1997)); CCR-5 (или "CKR-5" или "CC-CKR-5") [MIP-1α, RANTES, MIP-1β] (Sanson и др., Biochemistry, 35, 3362-3367 (1996)), и антиген группы крови Даффи [RANTES, MCP-1] (Chaudhun и др., J. Biol. Chem., 269, 7835-7838 (1994)). β-Хемокины включают среди других хемокинов эотаксин, MIP ("макрофаговый воспалительный белок"), MCP ("моноцит-хемоаттрактантный белок") и RANTES ("регуляция при активации, нормальный T-экспрессируемый и выделяемый").
Хемокиновые рецепторы, такие как CCR-1, CCR-2, CCR-2A, CCR-2B, CCR-3, CCR-4, CCR-5, CXCR-3, CXCR-4, вовлекаются как важные медиаторы воспалительных и иммунорегуляторных нарушений и заболеваний, включая астму, ринит и аллергические заболевания, а также аутоиммунные патологии, такие как ревматоидный артрит и атеросклероз. Люди, которые являются гомозиготными по удалению 32-пар оснований в гене CCR-5, по-видимому, обладают меньшей восприимчивостью к ревматоидному артриту (Gomez и др., Arthritis & Rheumatism, 42, 989-992 (1999)). Обзор роли эозинофилов в аллергическом воспалении приведен в работе Kita, H. и др., J. Exp. Med., 183, 2421-2426 (1996). Общий обзор роли хемокинов в аллергическом воспалении дан в работе Lustger, A.D., New England J. Med., 338(7), 426-445 (1998).
Подмножество хемокинов представляет сильнодействующие хемоаттрактанты для моноцитов и макрофагов. Среди них лучшими характеристиками обладает MCP-1 (моноцит-хемоаттрактантный протеин-1), основным рецептором которого является CCR2. MCP-1 производится во множестве клеточных типов в ответ на воспалительный стимул у разных видов, включая грызунов и людей, и стимулирует хемотаксис в моноцитах и подмножестве лимфоцитов. В частности, производство MCP-1 коррелирует с инфильтрацией моноцитов и макрофагов на местах воспалений. Удаление MCP-1 или CCR2 посредством гомологической рекомбинации у мышей дает в результате заметное ослабление пополнения моноцитов в ответ на инъекцию тиогликолата и инфицирование Listeria monocytogenes (Lu и др., J. Exp. Med., 187, 601-608 (1998); Kurihara и др. J. Exp. Med., 186, 1757-1762 (1997); Boring и др., J. Clin. Invest., 100, 2552-2561 (1997); Kuziel и др., Proc. Natl. Acad. Sci., 94, 12053-12058 (1997)). Кроме того, данные животные демонстрируют пониженную инфильтрацию моноцитов в грануломатозные поражения, вызванные инъекцией шистосомальных или микобактериальных антигенов (Boring и др., J. Clin. Invest., 100, 2552-2561 (1997); Warmington и др., Am. J. Path., 154, 1407-1416 (1999)). Эти данные предполагают, что MCP-1-индуцированная активация CCR2 играет главную роль в привлечении моноцитов к местам воспалений и что антагонизм данной активности будет продуцировать достаточное подавление иммунной реакции, давая терапевтическую пользу при иммуновоспалительных и аутоиммунных заболеваниях.
Таким образом, агенты, которые модулируют хемокиновые рецепторы, такие как CCR-2 рецептор, были бы полезны при таких нарушениях и заболеваниях.
Кроме того, привлечение моноцитов к воспалительным поражениям в стенках сосудов составляет основной компонент патогенеза атерогенного образования бляшек. MCP-1 продуцируется и выделяется эндотелиальными клетками и интимальными гладкими миоцитами после повреждения стенок сосудов в гиперхолестеринемических условиях. Моноциты, привлекаемые к месту поражения, проникают через стенки сосудов и дифференцируют в пенистые клетки в ответ на высвобождающийся MCP-1. В настоящее время несколько групп исследователей показали, что размер аортального поражения, содержание макрофагов и некроз ослабляются у содержащихся на высокожировой диете MCP-1 -/- или CCR2 -/- мышей, обратноскрещенных с APO-E -/-, LDL-R -/- или Apo B трансгенными мышами (Boring и др. Nature, 394, 894-897 (1998); Gosling и др. J. Clin. Invest., 103, 773-778 (1999)). Таким образом, CCR2-антагонисты могут ингибировать образование атеросклеротических поражений и патологического развития, уменьшая пополнение моноцитов и дифференциацию в артериальных стенках.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
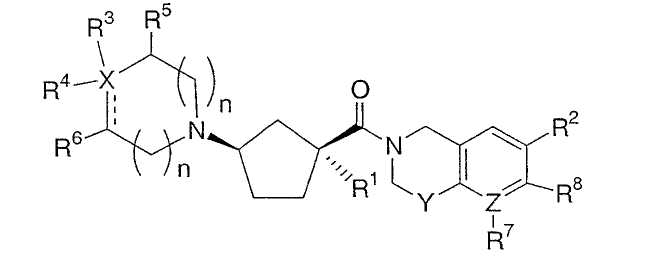
Настоящее изобретение касается циклопентильных соединений, связанных с бензоксазинильной группой через амидофрагмент с использованием кольцевого атома азота бензоксазина и дополнительно замещенных гетероциклическим фрагментом, такие соединения представлены формулой I
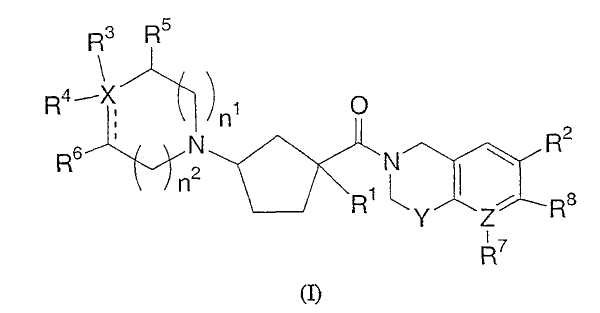
Данные соединения полезны в качестве модуляторов хемокиновых рецепторов CCR-2. Настоящее изобретение также касается соединений, которые являются модуляторами активности хемокиновых рецепторов и полезны при профилактике или лечении некоторых воспалительных и иммунорегуляторных нарушений и заболеваний, аллергических заболеваний, атопических состояний, включая аллергический ринит, дерматит, конъюнктивит и астму, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз. Данное изобретение также касается фармацевтических композиций, содержащих эти соединения, и применения данных соединений и композиций при профилактике или лечении таких заболеваний, в которые вовлечены хемокиновые рецепторы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение касается соединений, представленных формулой I
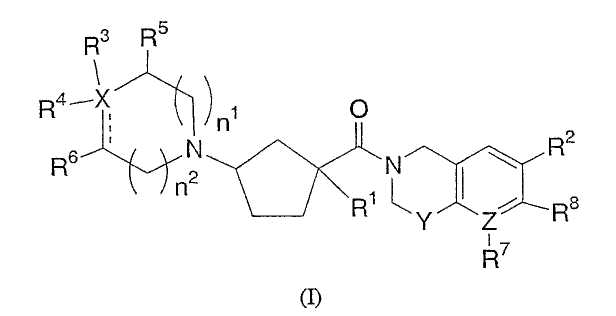
или их фармацевтически приемлемых солей или индивидуальных диастереомеров, в которых
X обозначает C, N, O или S;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил являются необязательно замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, в которых С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает водород, -(С0-6алкил)-фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)-C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил являются необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, -CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикла, и где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, -CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает водород, гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильный или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклический цикл, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген, или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил-C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3, и
пунктирная линия представляет необязательную связь.
Соединения настоящего изобретения включают соединения формулы Ia
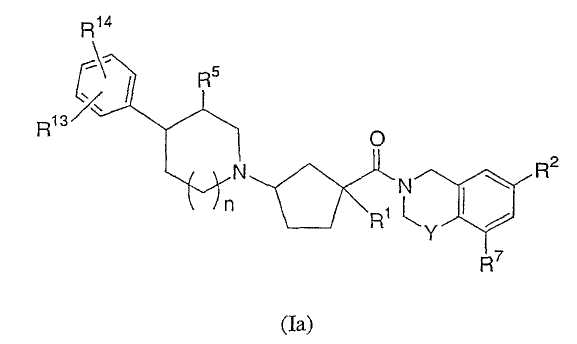
или их фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7 и Y определены, как выше для формулы I,
и где R13 и R14 независимо обозначают водород, галоген, трифторметил, гидрокси, -C1-3алкил, -O-C1-3алкил, -C0-3-CO2H, -C0-3-CO2C1-3алкил, -CN или -C0-3-гетероцикл,
или R13 и R14 объединены вместе, образуя гетероцикл, который сконденсирован с фенильным кольцом и который сам может быть незамещенным или замещенным 1-2 независимыми заместителями из гидрокси, галогена, -CO2R10 или -С1-3алкила, и
n равно 0, 1 или 2.
Соединения настоящего изобретения также включают соединения формулы Ib
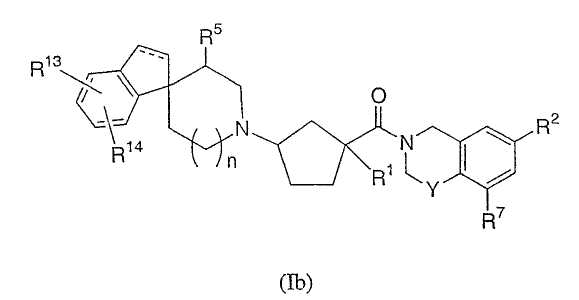
или их фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых пунктирная линия представляет необязательную связь и R1, R2, R5, R7, R13, R14, Y и n определены выше для формулы Ia.
Соединения настоящего изобретения также включают соединения формулы Ic
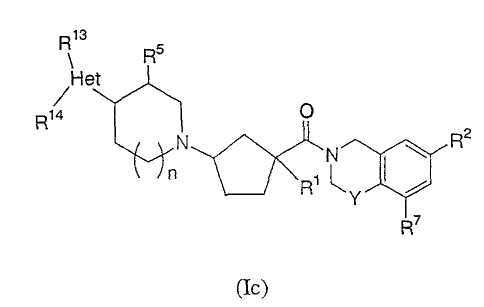
или их фармацевтически приемлемые соли или индивидуальные диастереомеры, в которых R1, R2, R5, R7, R13, R14, Y и n определены выше для Ia и где Het обозначает гетероцикл.
Соединения настоящего изобретения также включают соединения формулы Id
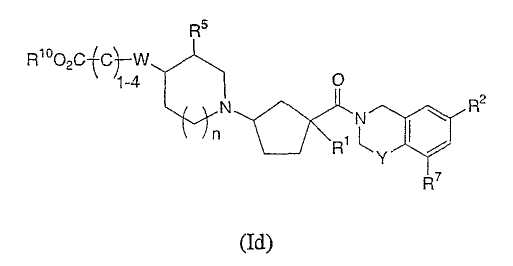
или их фармацевтически приемлемые соли или индивидуальные диастереомеры, в которых R1, R2, R5, R7, R10, Y, W и n определены выше для Ia и где С1-4 углеродная цепь является необязательно замещенной 1-4 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -C0-2алкилфенила,
или где C1-4углеродная цепь является частью C3-7циклоалкильного кольца.
Соединения настоящего изобретения также включают соединения формулы Ie
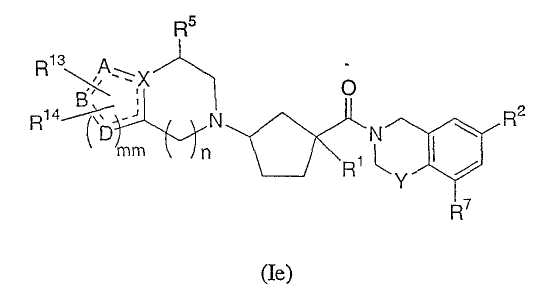
или их фармацевтически приемлемые соли или индивидуальные диастереомеры, в которых R1, R2, R5, R7, R13, R14, X, Y и n определены выше для формулы Ia и
в которых пунктирные линии обозначают необязательную связь,
mm равно 1 или 2 и
A, B, и D каждый независимо представляет собой C, N, O или S или A, B и D в комбинации с mm=2 образуют фенильное кольцо, или в комбинации образуют гетероцикл, если, по меньшей мере, один из X, A, B, D является N, O или S.
Дополнительные соединения настоящего изобретения также включают соединения формулы If
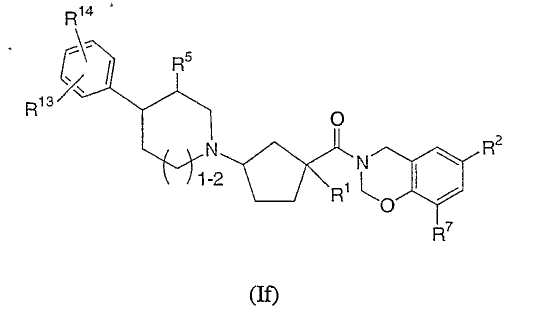
или их фармацевтически приемлемые соли или индивидуальные диастереомеры, в которых R1, R2, R5, R7, R13 и R14 определены выше для Ia
или в которых R13 и R14 объединены вместе, образуя гетероцикл, сконденсированный с фенильным кольцом, и где сам гетероцикл является необязательно замещенным 1-2 независимыми заместителями из гидрокси, галогена, -CO2R10 или -С1-3алкила.
Соединения настоящего изобретения также включают соединения формулы Ig
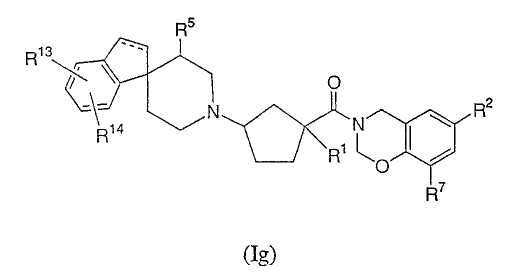
или их фармацевтически приемлемые соли, или индивидуальные диастереомеры, в которых пунктирная линия представляет необязательную связь и R1, R2, R5, R7, R13 и R14 определены выше для Ia.
Соединения настоящего изобретения также включают соединения формулы Ih
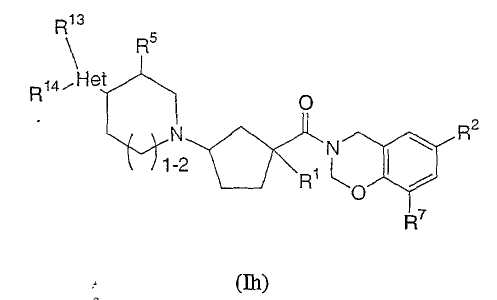
или их фармацевтически приемлемые соли или индивидуальные диастереомеры, в которых R1, R2, R5, R7, R13 и R14 определены выше для Ia и в которых Het обозначает гетероцикл.
Соединения настоящего изобретения также включают соединения формулы Ii
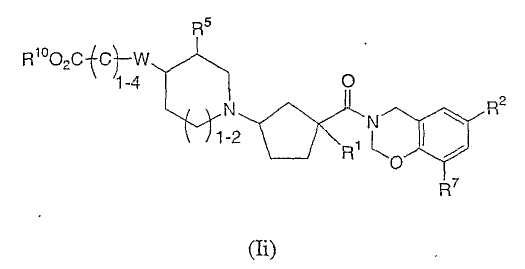
или их фармацевтически приемлемые соли или индивидуальные диастереомеры, в которых R1, R2, R5, R7, R10 и W определены выше для Ia и в которых C1-4 углеродная цепь является необязательно замещенной 1-4 независимыми заместителями из галогена, гидрокси, -C0-6алкила, -O-C1-3алкила, трифторметила или -C0-2алкилфенила.
В некоторых вариантах данного изобретения X обозначает C, Y обозначает -O- и Z обозначает C. Кроме того, в некоторых вариантах данного изобретения R1 обозначает -C1-6алкил, -C0-6алкил-O-С1-6алкил- или -(С0-6алкил)-(C3-7циклоалкил)-(C0-6алкил), в которых алкил и циклоалкил являются необязательно замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, -C1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10.
В другом варианте настоящего изобретения R1 обозначает -C1-6алкил, необязательно замещенный 1-6 независимыми заместителями из галогена, гидрокси, -O-C1-3алкила, трифторметила или -CO2R10, или R1 обозначает -С0-6алкил-O-С1-6алкил, необязательно замещенный 1-6 независимыми заместителями из галогена, трифторметила или -CO2R10, или R1 обозначает -(C3-5циклоалкил)-(С0-6алкил), необязательно замещенный 1-7 независимыми заместителями из галогена, гидрокси, -O-C1-3алкила, трифторметила или -CO2R10.
Еще в одном варианте настоящего изобретения R1 обозначает -C1-6алкил, -C1-6алкилгидрокси или -C1-6алкил, замещенный 1-6 атомами фтора.
Еще в одном варианте настоящего изобретения R1 обозначает изопропил, гидроксиэтил или трифторэтил.
В настоящем изобретении R2 может представлять -С1-6алкил, замещенный 1-6 фтор, -O-С1-6алкил, замещенный 1-6 атомами фтора, хлора, брома или фениламин. Кроме того, R2 может представлять трифторметил, трифторметокси, хлор, бром или фенил. В некоторых вариантах R2 обозначает трифторметил.
В настоящее изобретение включены соединения в которых, если X не является O, R3 обозначает фенил, гетероцикл, C3-7циклоалкил, C1-6алкил, -CO2R10 или -CONH-V-CO2R10, где V обозначает -C1-6алкил- или фенил, и где фенил, гетероцикл, C3-7циклоалкил, и С1-6алкил независимо является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, -C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -гетероцикла или -CONR10R10.
Также в настоящее изобретение включены соединения, в которых, если X не является O, R3 обозначает фенил, гетероцикл, C1-4алкил, -CO2R10 или -CONH-V-CO2R10, где V обозначает -С1-6алкил или фенил, и где фенил, гетероцикл и С1-4алкил каждый независимо необязательно замещен 1-3 независимыми заместителями из галогена, гидрокси, -C1-3алкила, -O-С1-3алкила, -CO2R10 илигетероцикла.
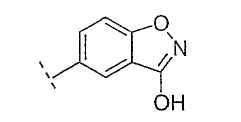
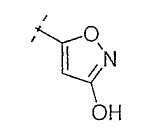
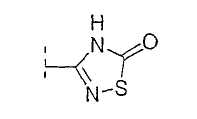
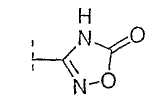
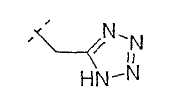
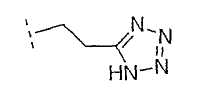
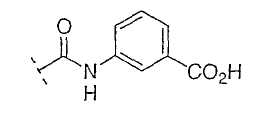
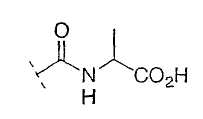
Настоящее изобретение включает также соединения, в которых, если X не является O, R3 выбран из следующей таблицы:
В некоторых вариантах данного изобретения, если Х обозначает C, R4 обозначает водород, гидрокси, -CN или -F.
В настоящем изобретении R3 и R4 могут быть объединены вместе, образуя 1H-инденовый или 2,3-дигидро-1H-инденовый цикл, необязательно замещенный 1-3 независимыми заместителями из галогена, гидрокси, -С1-3алкила, -O-C1-3алкила, -CO2R10 или -гетероциклила.
Кроме того, R5 и R6 могут независимо обозначать водород, гидрокси, -CH3, -O-CH3 или оксо. В настоящем изобретении, если Z не является N, R7 может обозначать H, F или гидрокси. В некоторых вариантах настоящего изобретения R8 обозначает H.
В одном аспекте настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей или индивидуальных диастереомеров, в которых
X обозначает C;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, в которых С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает -ОН, -С0-6алкил, -(С0-6алкил)фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)-C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, -О-SO2-фенил-С0-6алкил, -C(0)-N-(С0-6алкил)(С0-6алкил), -оксазолил-С0-6алкил, -оксазолил-С0-6алкил-O-С0-6алкил, фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил или N-оксидпиридил и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, -CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикла, и где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, -CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген, или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил-C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3, и
пунктирная линия представляет необязательную связь.
В варианте данного одного аспекта настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей или индивидуальных диастереомеров, в которых
X обозначает C;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает -галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, где С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает -ОН, -С0-6алкил, -(С0-6алкил)фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, -O-SO2-фенил-С0-6алкил, -C(O)-N-(С0-6алкил)(С0-6алкил), -оксазолил-С0-6алкил, -оксазолил-С0-6алкил-O-С0-6алкил, фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил или N-оксидпиридил, и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, -CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикла, где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, -CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильный или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген; или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил-C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 2, и
пунктирная линия представляет необязательную связь.
В другом варианте данного одного аспекта настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей или индивидуальных диастереомеров, в которых
X обозначает C;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает -галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, где С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает -ОН, -С0-6алкил, -(С0-6алкил)фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, -O-SO2-фенил-С0-6алкил, -C(O)-N-(С0-6алкил)(С0-6алкил), -оксазолил-С0-6алкил, -оксазолил-С0-6алкил-O-С0-6алкил, фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил или N-оксидпиридил, и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, -CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикла, где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, _CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген; или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил- C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 3, и
пунктирная линия представляет необязательную связь.
Еще в одном варианте данного одного аспекта настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей, или индивидуальных диастереомеров, в которых
X обозначает C;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, где С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает -ОН, -(С0-6алкил), -(С0-6алкил)-фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, -O-SO2-фенил-С0-6алкил, -C(O)-N-(С0-6алкил)(С0-6алкил), -оксазолил-С0-6алкил, -оксазолил-С0-6алкил-O-С0-6алкил, фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил или N-оксидпиридил, и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, -CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикл, где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, -CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильный или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген; или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил- C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 4, и
пунктирная линия представляет необязательную связь.
Во втором аспекте настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей, или индивидуальных диастереомеров, в которых
X обозначает O;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, где С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает -ОН, -С0-6алкил, -(С0-6алкил)-фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, -О-SO2-фенил-С0-6алкил, -C(О)-N-(С0-6алкил)(С0-6алкил), -оксазолил-С0-6алкил, -оксазолил-С0-6алкил-О-С0-6алкил, фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил или N-оксидпиридил, и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, _CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикла, где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, -CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген; или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил- C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3, и
пунктирная линия представляет простую связь.
В третьем аспекте настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей или индивидуальных диастереомеров, в которых
X обозначает N;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, где С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
R3 обозначает -ОН, -(С0-6алкил), -(С0-6алкил)-фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)C3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(C2-6алкенил)-CO2R10, -(С0-6алкил)-SO3H, -(С0-6алкил)-W-С0-4алкил, -(С0-6алкил)-CONR10-фенил, -(С0-6алкил)-CONR12-V-CO2R10, -О-SO2-фенил-С0-6алкил, -C(О)-N-(С0-6алкил)(С0-6алкил), -оксазолил-С0-6алкил, -оксазолил-С0-6алкил-О-С0-6алкил, фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил или N-оксидпиридил, и где R3 отсутствует, если X представляет O, и где С0-6алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила, и где фенил, пиридил, диазолил, тетразолил, тиадиазолонил, оксадиазолонил, тиазолфенил, N-оксидпиридил, гетероцикл, циклоалкил или С0-4алкил является необязательно замещенным 1-5 независимыми заместителями из галогена, трифторметила, гидрокси, С1-3алкила, -O-C1-3алкила, -C0-3-CO2R10, -CN, -(С0-6алкил)-C(O)-(С0-6алкил), -NR10R10, -CONR10R10 или -(C0-3алкил)гетероцикла, где фенил и гетероцикл могут быть сконденсированы с другим гетероциклом, который сам необязательно может быть замещенным 1-2 заместителями, независимо выбранными из гидрокси, галогена, -CO2R10 или -C1-3алкила, и где алкенил является необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, трифторметила, C1-3алкила, фенила или гетероцикла;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
R4 отсутствует, если X представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, С1-6алкилгидрокси, -O-C1-3алкил, -CO2R10, -CONR10R10 или -CN;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген; или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил-C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3, и
пунктирная линия представляет необязательную связь.
В четвертом аспекте настоящее изобретение касается соединений, представленных формулой (I), или их фармацевтически приемлемых солей или индивидуальных диастереомеров, в которых
X обозначает O;
Y обозначает O, S, SO, SO2 или NR9;
Z обозначает C или N;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), -(С0-6алкил)-W-фенил или -(С0-6алкил)-W-гетероцикл, где алкил, фенил, гетероцикл и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями из галогена, гидрокси, -O-С1-3алкила, трифторметила, С1-3алкила, -O-С1-3алкила, -CO2R10, -CN, -NR10R10, -NR10COR10, -NR10SO2R11 или -CONR10R10;
W обозначает простую связь, -O-, -S-, -SO-, -SO2-, -CO-, -CO2-, -CONR10- или -NR9-;
R2 обозначает галоген, -С0-6алкил, С0-6алкил-W-С1-6алкил, С0-6алкил-W-C3-7циклоалкил, С0-6алкил-W-фенил или С0-6алкил-W-гетероцикл, где С1-6алкил, C3-7циклоалкил, фенил и гетероцикл необязательно являются независимо замещенными 1-6 заместителями из галогена, трифторметила, -CN, -C1-6алкила или гидрокси;
V обозначает С1-6алкил или фенил;
R12 обозначает водород, С1-4алкил или R12 присоединен через связь из 1-5 углеродных атомов к одному из атомов углерода V с образованием цикла;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1H-инденил, 2,3-дигидробензофуранил, 1,3-дигидроизобензофуранил, 2,3-дигидробензотиофуранил, 1,3-дигидроизобензотиофуранил, 6H-циклопента[d]изоксазол-3-олил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -C0-3CO2R10, -CN, -NR10R10, -CONR10R10 или -С0-3гетероциклила;
или R3 и R5, или R4 и R6 объединены вместе, образуя фенил или гетероциклическое кольцо, где данный цикл является необязательно замещенным 1-7 независимыми заместителями из галогена, трифторметила, гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10, -CN, -NR10R10 или -CONR10R10;
R5 и R6 независимо обозначают водород, гидрокси, С1-6алкил, С1-6алкил-CO2R10, С1-6алкилгидрокси, -O-C1-3алкил или галоген; или =О, если R5 или R6 присоединен к данному кольцу через двойную связь;
если Z=C, R7 обозначает водород, гидрокси, галоген, С1-6алкил, необязательно замещенный 1-6 атомами фтора, -O-C1-6алкил, необязательно замещенный 1-6 атомами фтора, -NR10R10, -NR10CO2R11, -NR10CONR10R10, -NR10SO2-NR10R10, -NR10-SO2-R11, гетероцикл, -CN, -CONR10R10, -CO2R10, -NO2, -S-R10, -SO-R11, -SO2-R11 или -SO2-NR11R11;
если Z=N, R7 отсутствует или является оксидом (давая в результате N-оксид пиридина);
R8 обозначает водород, С1-6алкил, трифторметил, трифторметокси, хлор, фтор, бром или фенил;
R9 обозначает SO2R11, COR10, CONHR10, CO2R11 или SO2NHR10;
R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-C3-6циклоалкил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
R11 обозначает C1-6алкил, -С0-6алкил- C3-6циклоалкил, бензил или фенил, необязательно замещенный 1-3 независимыми заместителями из галогена, C1-3алкила, C1-3алкокси или трифторметила;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3, и
пунктирная линия представляет необязательную связь.
Типичные соединения настоящего изобретения включают соединения, представленные в примерах, и их фармацевтически приемлемые соли и индивидуальные диастереомеры.
Соединения настоящего изобретения имеют, по меньшей мере, два асимметрических центра в положениях 1 и 3 циклопентильного кольца и один асимметрический центр в положении 4 данного кольца, несущего X. Могут присутствовать дополнительные асимметрические центры в зависимости от природы различных заместителей в молекуле. Каждый такой асимметрический центр будет независимо давать два оптических изомера, и считается, что все возможные оптические изомеры и диастереомеры в смесях и чистых или частично очищенных соединениях включены в область данного изобретения. Абсолютные конфигурации некоторых соединений данной ориентации, где заместители на циклопентильном кольце (амидные и аминные фрагменты) являются цис, изображены на схеме:
Абсолютные конфигурации некоторых соединений данного изобретения представляют соединения указанной ниже ориентации:
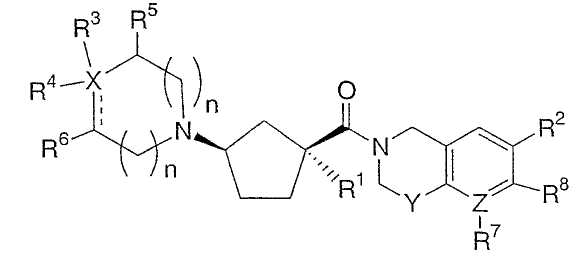
в которых атом углерода, несущий заместитель амин, обозначен как находящийся в (R) абсолютной конфигурации и атом углерода, несущий амидную подгруппу, может быть обозначен как находящийся в (S) или (R) абсолютной конфигурации в зависимости от приоритета для R1. Например, если R обозначает изопропил, то абсолютная стереохимия по атому углерода, несущему амидную подгруппу, будет (S), так как фрагменты амид и амин предпочтительно имеют цис-расположение в циклопентильном кольце.
Необязательные двойные связи изображены пунктирной линией, это означает, что двойная связь может присутствовать или отсутствовать. Специалисту, рассматривающему формулу I, понятно, что, если Х обозначает атом углерода, R4 может оставаться на X, только когда нет двойной связи между Х и атомом углерода, на котором присутствует R6.
Как известно в данной области, можно осуществить независимый синтез диастереомеров и энантиомеров или их хроматографическое разделение при подходящей модификации раскрытой здесь методологии. Их абсолютную стереохимию можно определить методом рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, которые при необходимости получают взаимодействием с реагентом, содержащим асимметрический центр, известной абсолютной конфигурации.
Термин "алкил" обозначает линейные или разветвленные структуры и их комбинации, имеющие указанное количество атомов углерода. Таким образом, например, С1-6алкил включает метил, этил, пропил, 2-пропил, втор- и трет-бутил, бутил, пентил, гексил, 1,1-диметилэтил, циклопропил, циклобутил, циклопентил и циклогексил.
Термин "циклоалкил" обозначает моно-, би- или трициклические структуры, необязательно соединенные с линейными или разветвленными структурами, с указанным количеством атомов углерода. Примеры циклоалкильных групп включают циклопропил, циклопентил, циклогептил, адамантил, циклододецилметил, 2-этил-1-бицикло[4.4.0]децил и подобные.
Термин "замещенный" или "заместитель" в отношении замещения на алкиле, циклоалкиле, фениле, гетероцикле или какой-либо другой химической группе, как предполагается, включает моно- и поли-замещение указанным заместителем в той степени, в которой химически разрешено одинарное или многократное замещение в указанных химических группах. Понятно, что определение заместителя в конкретном положении в молекуле не зависит от его определения в остальных положениях в данной молекуле. Таким образом, например, если R4 определен как -CONR10R10, то каждый R10 независимо выбран из возможных его значений, т.е. каждый R10 может быть таким же или отличаться от любого другого R10.
Термин "необязательно замещенный", как предполагается, включает оба варианта, замещенный и незамещенный. Таким образом, например, необязательно замещенный алкил, где галоген является необязательным заместителем, может представлять пропил или фторпропил.
Как считают специалисты в данной области, используемые здесь термины «гало» или «галоген» включают хлор, фтор, бром и йод. Аналогично C0-8 в C0-8алкиле определен для идентификации группы, которая имеет 0, 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода при линейном или разветвленном расположении, так что C0-8алкил конкретно включает метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, гексил, гептил и октил. Аналогично С0-6 обозначает группу, которая имеет 0, 1, 2, 3, 4, 5 или 6 атомов углерода при линейном или разветвленном расположении, и так далее в отношении других числовых обозначений. C0, как в C0алкиле, представляет непосредственную ковалентную связь в мостиковом положении и атом водорода в терминальном положении. Используемый здесь термин "гетероцикл", как предполагается, включает следующие группы: бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, имидазолил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксетанил, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридазинил, пиридил, пиримидил, пирролил, хиназолинил, хинолил, хиноксалинил, тетрагидропиранил, тетразолил, тетразолопиридил, тиадиазолил, тиазолил, тиенил, триазолил, азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пинолидинил, морфолинил, тиоморфолинил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидрофуранил, дигидроимидазолил, дигидроиндолил, дигидроизоксазолил, дигидроизотиазолил, дигидрооксадиазолил, дигидрооксазолил, дигидропиразинил, дигидропиразолил, дигидропиридинил, дигидропиримидинил, дигидропирролил, дигидрохинолинил, дигидротетразолил, дигидротиадиазолил, дигидротиазолил, дигидротиенил, дигидротриазолил, дигидроазетидинил, метилендиоксибензоил, тетрагидрофуранил, тетрагидротиенил и их N-оксиды.
Используемое здесь выражение "фармацевтически приемлемый" относится к тем соединениям, материалам, композициям и/или дозированным формам, которые входят в область достоверных медицинских заключений, подходят для применения в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмеримо с разумным соотношением польза/риск.
Используемое здесь выражение "фармацевтически приемлемые соли" относится к производным, в которых исходное соединение модифицировано с получением его солей с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничены этим, соли минеральных или органических кислот основных остатков, таких как амины, щелочные или органические соли кислотных остатков, таких как остатки карбоновых кислот, и подобные. Фармацевтически приемлемые соли включают обычные нетоксические соли или четвертичные аммонийные соли исходного соединения, полученного, например, из нетоксических неорганических или органических кислот. Например, такие общеизвестные нетоксические соли включают соединения, производные неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и подобные, и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памоевая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая и подобные.
Фармацевтически приемлемые соли настоящего изобретения можно получить из исходного соединения, которое содержит основный или кислотный фрагмент, по общеизвестным химическим методикам. Обычно такие соли можно получить посредством взаимодействия свободных кислотных или основных форм данных соединений со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или в смеси двух растворителей; обычно применяют неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Подходящие соли обнаружены, например, в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985, p. 1418.
Иллюстрацией данного изобретения является применение соединений, раскрытых здесь в примерах.
Конкретные соединения, входящие в область настоящего изобретения, включают соединения, которые выбраны из группы, состоящей из указанных в заголовке соединений примеров, их фармацевтически приемлемых солей и индивидуальных диастереомеров.
Рассматриваемые соединения полезны в способе модуляции активности хемокиновых рецепторов у пациента, нуждающегося в такой модуляции, включающем введение эффективного количества данного соединения.
Настоящее изобретение касается применения указанных выше соединений в качестве модуляторов активности хемокиновых рецепторов. В частности, данные соединения полезны в качестве модуляторов хемокиновых рецепторов, в частности CCR-2.
Применимость соединений согласно настоящему изобретению в качестве модуляторов активности хемокиновых рецепторов можно продемонстрировать при помощи методологии, известной в данной области, такой как исследование связывания хемокинов, которое раскрыто Van Riper и др., J. Exp. Med., 177, 851-856 (1993), и которое можно легко адаптировать для определения связывания CCR-2.
Сродство к рецепторам в исследовании связывания CCR-2 определяют, измеряя ингибирование связывания 125I-MCP-1 с эндогенным рецептором CCR-2 на различных типах клеток, включая моноциты, клетки THP-1, или после гетерологической экспрессии клонированного рецептора в эукариотических клетках. Клетки суспендируют в связывающем буфере (50 мМ HEPES, pH 7, 2, 5 мМ MgCl2, 1 мМ CaCl2 и 0,50% BSA) и добавляют к исследуемому соединению или ДМСО и 125I-MCP-1 при комнатной температуре в течение 1 час для связывания. Затем клетки собирают на фильтрах GFB, промывают 25 мМ HEPES буфером, содержащим 500 мМ NaCl, и определяют количество связанного с клетками 125I-MCP-1.
При исследовании хемотаксиса осуществляют хемотаксис, используя обедненные T-клетками PBMC, выделенные из цельной венозной крови или крови после лейкофореза и очищенные центрифугированием по Ficoll-Hypaque, с последующим возвращением при помощи обработанных нейраминидазой эритроцитов овец. После выделения клетки промывают HBSS, содержащим 0,1 мг/мл BSA, и суспендируют при 1×107 клеток/мл. В клетки вводят флуоресцентную метку в темноте при помощи 2 мкМ Calcien-AM (молекулярные зонды) в течение 30 мин при 37°C. Меченые клетки промывают дважды и суспендируют при 5x106 клеток/мл в RPMI 1640 с L-глутамином (без фенолового красного), содержащем 0,1 мг/мл BSA. Добавляют разбавленные той же средой MCP-1 (Peprotech) при 10 нг/мл или только среду к ячейкам (27 мкл). Моноциты (150000 клеток) добавляют на верхнюю часть фильтра (30 мкл) через 15 мин предварительной инкубации с ДМСО или разными концентрациями исследуемого соединения. Добавляют к ячейке равные концентрации исследуемого соединения или ДМСО для предотвращения разбавления посредством диффузии. Через 60 мин инкубации при 37°C в 5 % CO2 фильтр удаляют и верхнюю часть промывают HBSS, содержащим 0,1 мг/мл BSA, для удаления клеток, которые не мигрировали в фильтр. Спонтанную миграцию (хемокинез) определяют в отсутствие хемоаттрактанта.
В частности, в указанных выше исследованиях соединения следующих примеров имеют активность в связывании рецептора CCR-2 обычно при значении IC50 менее примерно 1 мкМ. Такие результаты указывают на существенную активность соединений при использовании в качестве модуляторов активности хемокиновых рецепторов.
Хемокиновые рецепторы млекопитающих обеспечивают мишень для взаимодействия или промотирования функции эозинофилов и/или лимфоцитов у млекопитающего, такого как человек. Соединения, которые ингибируют или промотируют функцию хемокиновых рецепторов, особо полезны для модуляции функции эозинофилов и/или лимфоцитов в терапевтических целях. В соответствии с этим соединения, которые ингибируют или промотируют функцию хемокиновых рецепторов, были бы полезны при лечении, профилактике, улучшении, регулировании или снижении риска широкого множества воспалительных и иммунорегуляторных нарушений и заболеваний, аллергических заболеваний, атопических состояний, включая аллергический ринит, дерматит, конъюнктивит и астму, а также аутоиммунные патологии, такие как ревматоидный артрит и атеросклероз.
Например, настоящее соединение, которое ингибирует одну или более функций хемокиновых рецепторов млекопитающих (например, хемокиновых рецепторов человека), можно вводить для ингибирования (т.е. уменьшения или профилактики) воспаления. В результате ингибируется один или более воспалительных процессов, таких как эмиграция лейкоцитов, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождение воспалительных медиаторов. Кроме приматов, таких как люди, можно лечить способом настоящего изобретения различных других млекопитающих. Например, можно лечить млекопитающих, включая (но не ограничиваясь этим) коров, овец, коз, лошадей, собак, кошек, морских свинок, крыс или животных других видов: бычьих, овечьих, лошадиных, псовых, кошачьих, грызунов или мышиных. Однако данный способ можно также применять на других видах, таких как виды птиц (например, цыплята).
Применяя соединения настоящего изобретения, можно лечить заболевания и состояния, связанные с воспалением и инфицированием. В одном варианте заболевание или состояние является таким, в котором для модуляции воспалительной реакции следует ингибировать или промотировать действия лимфоцитов.
Заболевания или состояния людей или других видов, которые можно лечить при помощи ингибиторов функции хемокиновых рецепторов, включают (но не ограничены этим): воспалительные или аллергические заболевания и состояния, включая респираторные аллергические заболевания, такие как астма, особенно бронхиальная астма, аллергический ринит, гиперчувствительные легочные заболевания, гиперчувствительный пневмонит, эозинофильная пневмония (например, синдром Лоффлера (Loeffler), хроническая эозинофильная пневмония), гиперчувствительность замедленного типа, интерстициальные легочные заболевания (ILD) (например, идиопатический легочный фиброз или ILD, связанное с ревматоидным артритом, системная красная волчанка, анкилозирующий спондилит, системный склероз, синдром Шегрена, полимиозит или дерматомиозит); системная анафилаксия или гиперчувствительные реакции, лекарственные аллергии (например, на пенициллин, цефалоспорины), аллергии на укусы насекомых; аутоиммунные заболевания, такие как ревматоидный артрит, псориатический артрит, множественный склероз, системная красная волчанка, астенический бульбарный паралич, юношеский диабет; гломерулонефрит, аутоиммунные тиреодит, болезнь Бехчета; отторжение трансплантата (например, при трансплантации), включая отторжение аллотрансплантата или заболевание трансплантат-относительно-хозяина; воспалительные кишечные заболевания, такие как болезнь Крона и язвенный колит; спондилоартропатия; склеродерма; псориаз (включая псориаз, опосредованный T-клетками) и воспалительный дерматоз, такой как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, крапивница; васкулит (например, некротизированный, кожный и гиперчувствительный васкулит); эозинофильный миозит, эозинофильный фасцит; раковые заболевания кожи или органов с инфильтрацией лейкоцитов. Можно лечить другие заболевания или состояния, при которых следует ингибировать нежелательные воспалительные реакции, включая, но не ограничиваясь этим, реперфузионное поражение, атеросклероз, некоторые гематологические злокачественные заболевания, цитокин-индуцированную токсичность (например, септический шок, эндотоксический шок), полимиозит, дерматомиозит.
Заболевания или состояния людей или других видов, которые можно лечить модуляторами функции хемокиновых рецепторов, включают, но не ограничены этим: иммунодепрессию, такую как иммунодепрессия у людей с синдромом иммунодефицита, например СПИД или другими вирусными инфекциями, у людей, подвергавшихся радиационной терапии, химиотерапии, терапии аутоиммунных заболеваний или лекарственной терапии (например, терапии кортикостероидами), которая вызывает иммунодепрессию; иммунодепрессию вследствие врожденного дефицита функции рецепторов или других причин; инфекционные заболевания, такие как паразитарные заболевания, включая (но не ограничиваясь этим) инфицирование гельминтами, такими как нематоды (круглые черви), (Trichuriasis, Enterobiasis, Ascariasis, Hookworm, Strongyloidiasis, Trichinosis, filariasis), трематоды (плоские черви) (Schistosomiasis, Clonorchiasis), цестоды (ленточные черви) (Echinococcosis, Taeniasis saginata, Cysticercosis), внутренние черви, висцеральные личиночные мигрени (например, Toxocara), эозинофильный гастроэнтерит (например, Anisaki sp., Phocanema sp.) и кожные личиночные мигрени (Ancylostona braziliense, Ancylostoma caninum). Кроме того, лечение указанных выше воспалительных, аллергических и аутоиммунных заболеваний можно также предполагать для промоторов функции хемокиновых рецепторов, если предполагается, что доставка достаточного количества соединения вызывает утрату экспрессии рецепторов на клетках посредством индукции интернализации хемокиновых рецепторов или доставки соединения таким способом, результатом которого является неправильное направление миграции клеток.
Таким образом, соединения настоящего изобретения полезны при лечении, профилактике, улучшении, регулировании или снижении риска широкого множества воспалительных и иммунорегуляторных нарушений и заболеваний, аллергических состояний, атопических состояний, а также аутоиммунных патологий. В специфическом варианте настоящее изобретение касается применения рассматриваемых соединений для лечения, профилактики, улучшения, регулирования или снижения риска аутоиммунных заболеваний, таких как ревматоидный артрит или псориатический артрит.
В другом аспекте настоящее изобретение можно применять для оценки предполагаемых специфических агонистов или антагонистов хемокиновых рецепторов, включая CCR-2. В соответствии с этим настоящее изобретение касается применения данных соединений в подготовке и проведении скрининговых исследований для соединений, которые модулируют активность хемокиновых рецепторов. Например, соединения данного изобретения полезны для выделения рецепторных мутантов, которые являются превосходными инструментами скрининга для более эффективных соединений. Кроме того, соединения данного изобретения полезны при установлении или определении места связывания других соединений с хемокиновыми рецепторами, например, посредством конкурентного ингибирования. Соединения настоящего изобретения также полезны для оценки предполагаемых специфических модуляторов хемокиновых рецепторов, включая CCR-2. Как понимают в данной области, полная оценка специфических агонистов и антагонистов указанных выше хемокиновых рецепторов затруднена наличием недостаточного количества непептидильных (метаболически устойчивых) соединений с высоким связывающим сродством относительно данных рецепторов. Таким образом, соединения данного изобретения являются коммерческими продуктами, которые продаются для данных целей.
Кроме того, настоящее изобретение касается способа производства лекарственного средства для модуляции активности хемокиновых рецепторов у людей и животных, включающего объединение соединения настоящего изобретения с фармацевтическим носителем или разбавителем.
Настоящее изобретение касается также применения настоящих соединений для лечения, профилактики, улучшения, регулирования или снижения риска инфицирования ретровирусом, в частности вирусом герпеса или вирусом иммунодефицита человека (ВИЧ), и лечения и задержки появления последующего патологического состояния, такого как СПИД. Лечение СПИД или профилактику или лечение инфицирования ВИЧ определяют как лечение, включающее (но не ограниченное этим) лечение широкого диапазона состояний ВИЧ-инфицирования: СПИД, ARC (комплекс, связанный со СПИД), оба: симптоматический и асимптоматический, и фактическое или потенциальное воздействие ВИЧ. Например, соединения данного изобретения полезны при лечении инфицирования ВИЧ после подозреваемого воздействия ВИЧ, например после переливания крови, трансплантации органов, замены жидкостей организма, укусов, случайного укола иглой или воздействия крови пациента во время хирургической операции.
В одном аспекте настоящего изобретения рассматриваемые соединения можно применять в способе ингибирования связывания хемокина с хемокиновым рецептором, таким как CCR-2, целевой клетки, который включает контакт целевой клетки с некоторым количеством соединения, которое эффективно при ингибировании связывания хемокина с хемокиновым рецептором.
Пациент, подвергаемый лечению в указанных выше способах, представляет млекопитающее, такое как человек, мужчина или женщина, которое нуждается в модуляции активности хемокиновых рецепторов. Подразумевается, что используемый здесь термин "модуляция" включает антагонизм, агонизм, частичный антагонизм, обратный агонизм и/или частичный агонизм.
Модуляция может относиться к антагонизму активности хемокиновых рецепторов. Термин "терапевтически эффективное количество" обозначает количество рассматриваемого соединения, которое будет вызывать биологическую или медицинскую реакцию ткани, системы, животного или человека, к которому обращается исследователь, ветеринар, врач или другой клиницист.
Подразумевается, что используемый здесь термин "композиция" включает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, который прямо или косвенно получается из комбинации указанных ингредиентов в указанных количествах. Выражение "фармацевтически приемлемый" подразумевает, что носитель, разбавитель или наполнитель должен быть совместим с другими ингредиентами препарата и не вреден для реципиента.
Термины "прием" и/или "введение" соединения следует понимать как обозначающие обеспечение нуждающегося в лечении пациента соединением данного изобретения.
Используемый здесь термин "лечение" касается лечения и предупреждения или профилактической терапии указанных выше состояний. Специалисту в данной области понятно профилактическое введение субтерапевтических доз лекарства.
Комбинированная терапия для модуляции активности хемокиновых рецепторов с целью лечения, профилактики, улучшения, регулирования или снижения риска воспалительных и иммунорегуляторных нарушений и заболеваний, включая астму и аллергические заболевания, а также аутоиммунные патологии, такие как ревматоидный артрит и атеросклероз, и патологии, отмеченные выше, проиллюстрированы при помощи комбинации соединений данного изобретения и других соединений, которые известны для таких применений.
Например, для лечения, профилактики, улучшения, регулирования или снижения риска воспаления, настоящие соединения можно применять в сочетании с противовоспалительным или обезболивающим агентом, таким как опиатный агонист, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, NMDA-антагонист, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидный противовоспалительный агент или цитокин-подавляющий противовоспалительный агент, например, таким соединением как ацетаминофен, аспирин, кодеин, эмбрел, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацитин, пироксикам, стероидный анальгетик, суфентанил, сунлиндак, тенидап и подобные. Аналогично настоящие соединения можно вводить с анальгетиком; потенцирующим средством, таким как кофеин, H2-антагонист, симетикон, гидроксид алюминия или магния; противоотечным средством, таким как фенилэприн, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или леводезокси-эфедрин; средством от кашля, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; диуретиком и седативным или неседативным антигистаминным средством.
Также соединения настоящего изобретения можно применять в комбинации с другими лекарствами, которые используют при лечении/профилактике/депрессии или облегчении заболеваний или состояний, для которых полезны соединения настоящего изобретения. Такие другие лекарства можно вводить обычным способом и в обычном количестве, одновременно или последовательно с соединением настоящего изобретения. Если соединение настоящего изобретения применяют одновременно с одним или несколькими другими лекарствами, то создают фармацевтическую композицию, содержащую другие лекарства в дополнение к соединению настоящего изобретения. В соответствии с этим фармацевтические композиции настоящего изобретения включают соединения, которые также содержат один или более других активных ингредиентов в дополнение к соединению настоящего изобретения.
Примеры других активных ингредиентов, которые можно объединять с соединением настоящего изобретения или вводить отдельно или в тех же фармацевтических композициях, включают (но не ограничены этим): (a) VLA-4 антагонисты, такие как соединения, описанные в патенте США 5510332, WO95/15973, WO96/01644, WO96/06108, WO96/20216, WO96/22966, WO96/31206, WO96/40781, WO97/03094, WO97/02289, WO 98/42656, WO98/53814, WO98/53817, WO98/53818, WO98/54207 и WO98/58902; (b) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, дексаметазон и гидрокортизон; (c) иммуносупрессанты, такие как циклоспорин, такролимус, рапамицин и другие иммуносупрессанты FK-506 типа; (d) антигистаминные агенты (антагонисты H1-гистамина), такие как бромфенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин, пириламин, астемизол, терфенадин, лоратадин, деслоратадин, цетиризин, фексофенадин, дескарбоэтоксилоратадин и подобные; (e) нестероидные антиастматические агенты, такие как β2-агонисты (тербуталин, метапротеренол, фенотерол, изоэтарин, альбутерол, битолтерол и пирбутерол), теофиллин, кромолин натрий, атропин, ипратропийбромид, антагонисты лейкотриена (зафирлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст, SKB-106,203), ингибиторы биосинтеза лейкотриена (зилеутон, BAY-1005); (f) нестероидные противовоспалительные агенты (NSAID), такие как производные пропионовой кислоты (алминопрофен, беноксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (индометацин, ацеметацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозовая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенамовой кислоты (флуфенамовая кислота, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота и толфенамовая кислота), производные бифенилкарбоновой кислоты (дифлунизал и флуфенизал), оксикамы (изоксикам, пироксикам, судоксикам и теноксикан), салицилаты (ацетилсалициловая кислота, сульфасалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (g) ингибиторы циклооксигеназы-2 (COX-2); (h) ингибиторы фосфодиэстеразы типа IV (PDE-IV); (i) другие антагонисты хемокиновых рецепторов, в особенности CCR-1, CCR-2, CCR-3, CXCR-3 и CCR-5; (j) агенты, снижающие холестерин, такие как ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин, розувастатин и другие статины), секвестранты (холестирамин и колестипол), ингибиторы абсорбции холестерина (эзетимиб), никотиновая кислота, производные фенофибровой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (k) антидиабетические агенты, такие как инсулин, сульфонилкарбамиды, бигуаниды (метформин), ингибиторы α-глюкозидазы (акарбоза) и глитазоны (троглитазон и пиоглитазон); (I) препараты интерферона бета (интерферон бета-1α, интерферон бета-1β); (m) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарства, антиметаболиты, такие как азатиоприн и 6-меркаптопурин, и цитотоксические противораковые химиотерапевтические агенты.
Весовое соотношение соединения настоящего изобретения и второго активного ингредиента можно варьировать, и оно зависит от эффективной дозы каждого ингредиента. Обычно применяют эффективную дозу каждого. Таким образом, например, если соединение настоящего изобретения комбинировано с NSAID, весовое соотношение соединения настоящего изобретения и NSAID обычно составляет примерно от 1000:1 до 1:1000, в некоторых случаях примерно от 200:1 до 1:200.
Комбинации соединения настоящего изобретения и других активных ингредиентов обычно также находятся в рамках указанного выше диапазона, но в каждом случае следует применять эффективную дозу каждого активного ингредиента.
В таких комбинациях соединение настоящего изобретения и другие активные агенты можно вводить отдельно или в сопряжении. Кроме того, введение одного элемента может происходить до, одновременно или после введения другого агента(ов).
Соединения настоящего изобретения можно вводить перорально, парентерально (например, посредством внутримышечной, внутрибрюшинной, внутривенной, ICV, внутриполостной инъекции или вливания, подкожной инъекции или имплантата), посредством ингаляционного распыления, носовым, вагинальным, ректальным, подъязычным или локальным способом и можно готовить в виде препаратов, по одному или вместе, в виде подходящих дозированных стандартных препаративных форм, содержащих общеизвестные нетоксические фармацевтически приемлемые носители, адъюванты и наполнители, подходящие для каждого способа введения. Кроме лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки, обезьяны и др., соединения данного изобретения эффективны для применения у людей. Фармацевтические композиции для введения соединений данного изобретения можно удобным образом представить в виде стандартной дозированной формы и приготовить любым способом, хорошо известным в области аптечного дела. Все способы включают стадию объединения активного ингредиента с носителем, который составляет один или более вспомогательных ингредиентов. Вообще, фармацевтические композиции готовят посредством однородного и тщательного объединения активного ингредиента с жидким носителем, или мелко размельченным твердым носителем, или обоими, и затем, если необходимо, придания продукту желательной формы. В фармацевтическую композицию рассматриваемое активное соединение включено в количестве, достаточном для оказания требуемого действия на протекание или состояние заболеваний. Подразумевается, что используемый здесь термин "композиция" включает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, который прямо или косвенно получается из комбинации указанных ингредиентов в указанных количествах.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в виде, удобном для перорального применения, например в виде таблеток, пилюль, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, можно получить в соответствии с любым способом, известным в области производства фармацевтических композиций, и такие композиции могут содержать один или более агентов, выбранных из группы, включающей подслащающие агенты, вкусовые агенты, красители и консерванты, для обеспечения фармацевтически приемлемых и со вкусовыми добавками препаратов. Таблетки содержат активный ингредиент в смеси с нетоксическими фармацевтически приемлемыми наполнителями, которые подходят для производства таблеток. Такими наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например кукурузный крахмал или альгиновая кислота; связывающие агенты, например крахмал, желатин или аравийская камедь, и лубриканты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или на них можно нанести покрытие по известным методикам для замедления разрушения и абсорбции в желудочно-кишечном тракте и обеспечения таким способом продолжительного действия в течение большего периода. Например, можно применять материал для разрушения с задержкой по времени, такой как глицерилмоностеарат или глицерилдистеарат. Можно также нанести на таблетки покрытие по методикам, описанным в патентах США 4256108; 4166452 и 4265874, получая осмотические терапевтические таблетки для регулируемого высвобождения.
Препараты для перорального применения можно также представить в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешивают с водной или масляной средой, например арахисовым маслом, вазелиновым маслом или оливковым маслом.
Водные суспензии содержат активные материалы в смеси с наполнителями, подходящими для производства водных суспензий. Такие наполнители представляют собой суспендирующие агенты, например натрийкарбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь и аравийскую камедь; диспергирующие или смачивающие агенты могут представлять собой встречающиеся в природе фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с алифатическими спиртами с длиной цепью, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными эфирами, производными жирных кислот и гекситола, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации этиленоксида с частичными эфирами, производными жирных кислот и гекситолангидридами, например полиэтиленсорбитанмоноолеат. Водные суспензии также могут содержать один или более консервантов, например этил или н-пропил, пара-гидроксибензоат, один или более красителей, один или более вкусовых агентов и один или более подслащающих агентов, таких как сахароза или сахарин.
Масляные суспензии можно приготовить суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как вазелиновое масло. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Можно добавить подслащающие агенты, такие как указанные выше соединения, и вкусовые агенты для обеспечения вкусного препарата для перорального приема. Такие композиции можно законсервировать, добавляя антиоксидант, такой как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии посредством добавления воды, обеспечивают активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты представлены соединениями, уже упоминавшимися выше. Также могут присутствовать дополнительные наполнители, например подслащающие, вкусовые агенты и красители.
Фармацевтические композиции данного изобретения могут также быть в виде эмульсий «масло в воде». Масляная фаза может представлять собой растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например вазелиновое масло, или их смеси. Подходящими эмульгирующими агентами могут быть встречающиеся в природе смолы, например аравийская камедь или трагакантовая камедь, встречающиеся в природе фосфатиды, например соевый лецитин, и сложные эфиры или частичные сложные эфиры, производные жирных кислот и гекситолангидрида, например сорбитан моноолеат, и продукты конденсации указанных частичных сложных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эмульсии могут также содержать подслащающие и вкусовые агенты.
Сиропы и эликсиры можно приготовить с подслащающими агентами, например глицерином, пропиленгликолем, сорбитом или сахарозой. Такие препараты могут также содержать успокоительные средства, консерванты, вкусовые агенты и красители.
Фармацевтические композиции могут быть в виде стерильных водных или масляных суспензий для инъекций. Данную суспензию можно приготовить в соответствии с известными методиками, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты, которые упоминаются выше. Стерильный препарат для инъекций также может быть стерильным раствором или суспензией для инъекций в нетоксическом парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые можно применять, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, общеизвестно, что в качестве растворителя или суспендирующей среды применяют стерильные нелетучие масла. Для данной цели можно применять любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, в получении препаратов для инъекций находят применение жирные кислоты, такие как олеиновая кислота. Соединения настоящего изобретения можно также применять в виде суппозиториев для ректального введения лекарства. Данные композиции можно получить смешиванием лекарства с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, но жидким при ректальной температуре и, следовательно, плавится в прямой кишке, высвобождая лекарство. Такими материалами являются масло какао и полиэтиленгликоли.
Для локального применения используют кремы, мази, желе, растворы, суспензии и др., содержащие соединения настоящего изобретения. Для целей данной заявки локальное применение включает жидкости для полоскания рта и горла.
Фармацевтическая композиция и способ настоящего изобретения могут также содержать другие терапевтически активные соединения, которые, как здесь отмечено, обычно применяют при лечении указанных выше патологических состояний.
При лечении, профилактике, облегчении, регулировании или снижении риска состояния, которое требует модуляции хемокиновых рецепторов, подходящий уровень дозировки обычно составляет примерно от 0,01 до 500 мг на 1 кг веса тела пациента в день, которые можно вводить в виде единой дозы или многократных доз. Уровень дозировки составляет примерно от 0,1 до 250 мг/кг в день или в некоторых случаях примерно от 0,5 до 100 мг/кг в день. Подходящий уровень дозировки может составлять примерно от 0,01 до 250 мг/кг в день, примерно от 0,05 до 100 мг/кг в день или примерно от 0,1 до 50 мг/кг в день. В рамках данного диапазона дозировка может составлять от 0,05 до 0,5, от 0,5 до 5 или от 5 до 50 мг/кг в день. Для перорального приема предпочтительно получают композиции в виде таблеток, содержащих от 1,0 до 1000 мг активного ингредиента, в некоторых случаях от 2,0 до 500, в других случаях от 3,0 до 200 и еще в других случаях 1, 5, 10, 15, 20, 25, 30, 50, 75, 100, 125, 150, 175, 200, 250, 300, 400, 500, 600, 750, 800, 900 и 1000 мг активного ингредиента для симптоматического регулирования дозировки пациенту, подвергаемому лечению. Соединения можно вводить по схеме от 1 до 4 раз в день, предпочтительно один или два раза в день.
Однако понятно, что конкретный уровень дозы и частоту дозировки для любого конкретного пациента можно варьировать, и они зависят от разнообразных факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия данного соединения, возраст, вес тела, общее состояние здоровья, пол, диету, способ и интервал введения, скорость выделения, лекарственную комбинацию, тяжесть конкретного состояния и организм, принимающий лечение.
Используемые здесь сокращения имеют следующие значения, сгруппированные в таблицу. Сокращения, не приведенные ниже в таблице, имеют свои обычные значения, если специально не указано по-другому.
ОБОЗНАЧЕНИЯ АЛКИЛЬНЫХ ГРУПП
Следующие примеры приведены в качестве иллюстраций определенных вариантов данного изобретения и не предполагают ограничения данного изобретения.
До тех пор, пока не указано по-другому, экспериментальные методики осуществляют в следующих условиях. Все операции проводят при комнатной температуре или температуре окружающей среды, то есть при температуре в диапазоне 18-25°C. Выпаривание растворителя проводят, используя роторный испаритель при пониженном давлении (600-4000 Паскалей: 4,5-30 мм рт.ст.) и температуре бани до 60°C. За ходом реакций следят при помощи тонкослойной хроматографии (ТСХ), и времена взаимодействия приведены только для иллюстрации. Температуры плавления не исправлены, и "d" указывает разложение. Данные температуры плавления получены для материалов, полученных, как описано. В некоторых получениях полиморфизм может приводить к выделению материалов при разных температурах плавления. Структуру и чистоту конечных продуктов оценивают, по меньшей мере, по одной из следующих методик: ТСХ, масс-спектрометрия, спектроскопия ядерного магнитного резонанса (ЯМР) или микроаналитические данные. Выходы, если приведены, даны только для иллюстрации. Данные ЯМР, если приведены, даны в виде величин дельта (δ) для основных диагностических протонов в миллионных долях (м.д.) относительно тетраметилсилана (ТМС) как внутреннего стандарта, определенные при 300 МГц, 400 МГц или 500 МГц с использованием указанного растворителя. Для указания формы сигналов использованы общеизвестные сокращения: c - синглет; д - дублет; т - триплет; м - мультиплет; ш. - широкий и др. Кроме того, Ar обозначает сигнал ароматики. Химические символы имеют свои обычные значения; использованы также следующие сокращения: v (объем), w (масса), т.к. (температура кипения), т.пл. (температура плавления), л (литр(ы)), мл (миллилитры), г (грамм(ы)), мг (миллиграмм(ы)), моль (моли), ммоль (миллимоли), экв. (эквивалент(ы)).
Некоторые способы получения соединения данного изобретения проиллюстрированы на следующих схемах и в примерах. Исходные материалы доступны коммерчески, получены по известным методикам или, как показано здесь.
Один из принципиальных способов, используемых для получения входящих в область настоящего изобретения соединений, который дает структуру 1-9 1,1,3-тризамещенного циклопентана, изображен на схеме 1. Согласно данному способу, кетокислоты 1-1 (получение описано на схеме 2A, схеме 2B, схеме 2C и схеме 2D) взаимодействуют с аминами 1-2 (получение описано на схеме 3). Это можно выполнить различными способами, включающими сначала превращение кислоты в хлорид кислоты при помощи такого реагента, как оксалилхлорид, и затем объединение с амином 1-2 в присутствии основания, такого как триэтиламин. Затем полученный кетоамид 1-3 восстанавливают, например, борогидридом натрия, получая соответствующий спирт, в который далее вводят защиту (Greene, T; Wuts, P.G.М., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, NY 1991), получая его ацетатный сложный эфир 1-4 в стандартных условиях. Затем защитную группу на Y удаляют, применяя условия, подходящие для Y. Например, если PY представляет трет-бутилат, трет-бутилэфирную группу в 1-4 отщепляют в кислотных условиях, таких как безводный 4н. HCl в диоксане, получая 1-5, который в данном случае представляет фенол (YH=OH). При нагревании 1-5 с избытком формальдегида, такого как параформальдегид, в растворителе, таком как толуол, и в присутствии катализатора, такого как TsOH, можно осуществить циклизацию с образованием циклической системы в 1-6 (если Y обозначает кислород, данная циклическая система является бензоксазином). Гидролиз ацетатного эфира в 1-6 можно провести при помощи LiOH или другого основания. Полученный спирт затем окисляют до кетона 1-7. Это можно выполнить, используя разнообразные условия, включая условия окисления по Swern (Mancuso, A. J., Swern, D. Synthesis, (1981), 165). Восстановительное аминирование 1-7 амином 1-8 с применением, например, NaB(OAc)3H или NaBH3CN в качестве восстановителя дает модуляторы хемокиновых рецепторов 1-9. Соединения 1-9, которые можно синтезировать согласно химии, описанной на схеме 1, представляют стереоизомерные смеси (Eliel, E. E., Wilen, S.H., Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York). В частности, соединения 1-9 часто получают в виде смеси цис- и транс-изомеров. Если 1-1 пердставляет отдельный стереоизомер (1-1a), то в результате можно получить только 2 возможных изомера 1-9 (цис и транс); их можно разделить разными способами, включая препаративную ТСХ, флэш-хроматографию, СДЖХ или ВЭЖХ с применением колонки с хиральной стационарной фазой. Если 1-1 является рацемической смесью, можно получить все 4 возможных изомера 1-9. Также их можно разделить методом ВЭЖХ с применением колонки с хиральной стационарной фазой или при комбинации указанных выше способов. Синтез рацемического 1-1 подробно приведен на схеме 2A, тогда как синтез хирального 1-1a описан на схеме 2B и схеме 2C.
Кроме того, соединения 1-9 могут сами модифицироваться, давая новые модуляторы хемокиновых рецепторов 1-9.1. Например, сложноэфирная функциональная группа в соединении 1-9 может гидролизоваться до соответствующей карбоновой кислоты, которая также может быть модулятором хемокиновых рецепторов.
СХЕМА 1
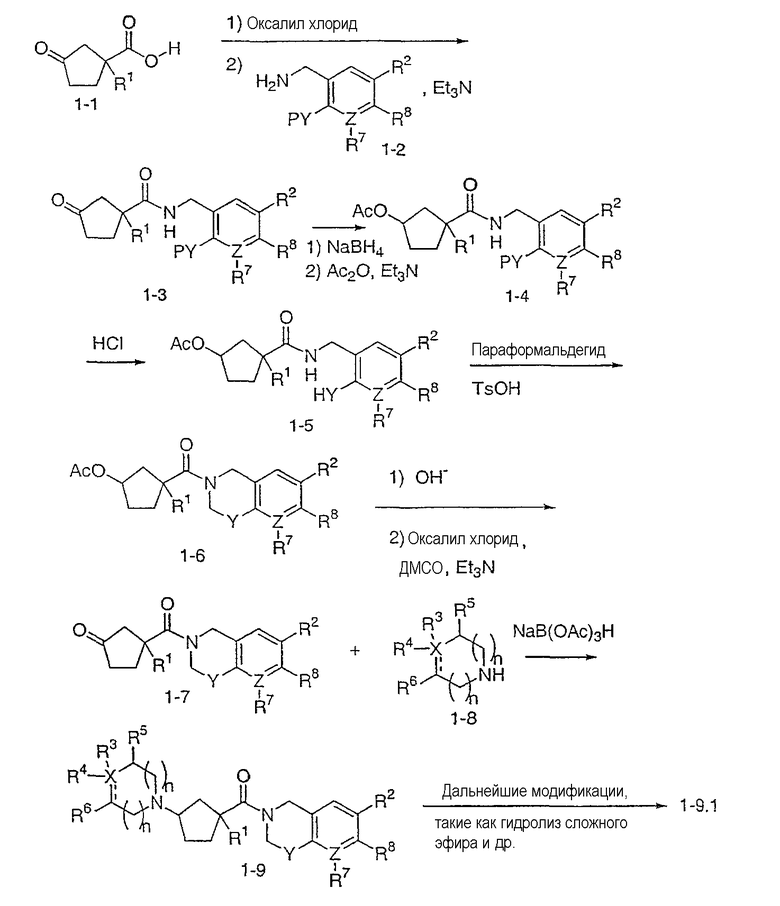
Один из принципиальных способов, применяемых для получения промежуточного продукта 1-1, представлен на схеме 2A. Согласно данному способу 3-оксоциклопентанкарбоновую кислоту (2-1), которую можно синтезировать по известной методике (Stetter, H., Kuhlman, H., Liebigs Ann. Chim., 1979, 944), этерифицируют в стандартных условиях. Если R15 представляет трет-бутильную группу, соответствующий сложный эфир 2-2 можно получить взаимодействием подходящего спирта, в данном случае трет-бутанола, с кислотой 2-1 в присутствии серной кислоты. Защиту оксогруппы в 2-1 можно осуществить рядом способов (Greene, Т., "Wuts, P. G. М., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, NY 1991). Особо подходящую диметилацетальную защитную группу можно ввести с применением триметилортоформиата в качестве реагента в подходящем растворителе, таком как дихлорметан и метиловый спирт, в присутствии кислотного катализатора. По-другому, в случае, когда R15 обозначает метильную группу, кислоту 2-1 можно превратить непосредственно в 2-3, применяя триметилортоформиат и кислотный катализатор, такой как пара-толуолсульфоновая кислота. Алкилирование сложных эфиров 2-3 алкилирующим агентом, таким как алкилхлорид, -бромид или -йодид, в присутствии подходящего основания, такого как литийдиизопропиламид, дает промежуточные продукты 2-4. Сложноэфирную защитную группу, присутствующую в 2-4, можно удалить рядом способов в зависимости природы сложного эфира. Метиловые сложные эфиры (R15=метил) можно гидролизовать в присутствии кислоты или основания при окружающей или повышенной температуре, тогда как трет-бутиловые сложные эфиры (R15=трет-бутил) могут легко расщепляться в кислотных условиях.
СХЕМА 2A
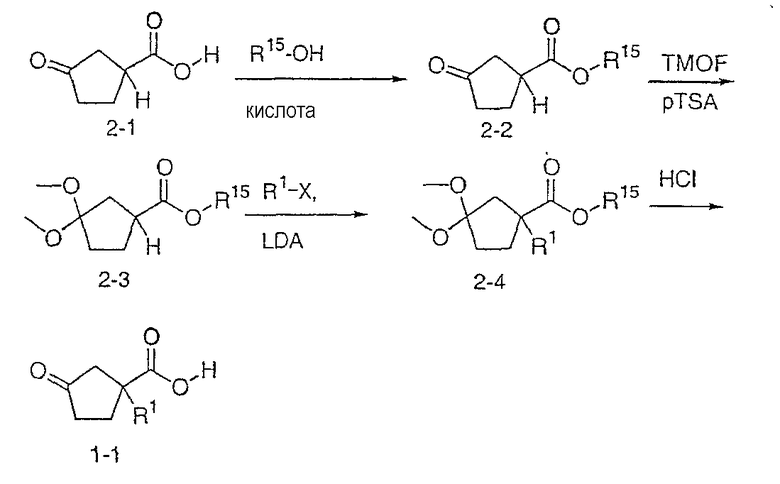
Промежуточный продукт 1-1 можно получить в виде одного стереоизомера (1-1a) различными способами, включая способы, представленные на схеме 2B и схеме 2C. Согласно схеме 2B рацемический 1-1 можно превратить в его бензиловый эфир. Существует много способов проведения данной этерификации, один из которых представляет последовательное превращение в соответствующий хлорид кислоты при помощи, например, оксалилхлорида, затем обработку бензиловым спиртом в присутствии основания, такого как триэтиламин. Затем рацемический бензиловый эфир 2-5 можно разделить методом хиральной препаративной ВЭЖХ с получением 2-5a в виде одного стереоизомера. Удаление бензильной группы с получением хиральной кетокислоты 1-1a можно выполнить несколькими способами. Один удобный способ представляет гидрирование в присутствии катализатора, такого как Pd/C.
СХЕМА 2B
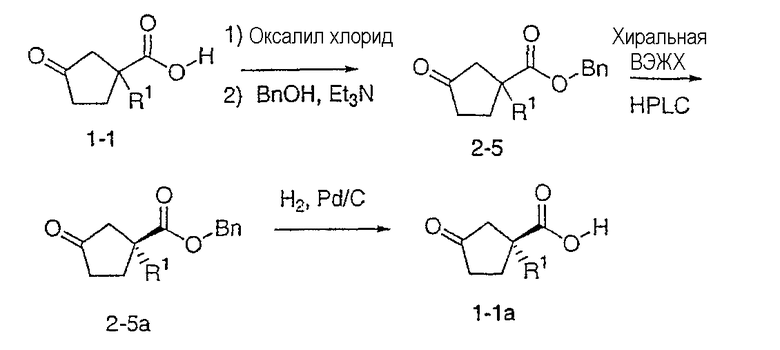
Согласно схеме 2C промежуточный продукт 1-1a хиральную кетокислоту можно получить, исходя из коммерчески доступной оптически чистой аминокислоты 2-6. Защиту карбокислотной группы можно осуществить разнообразными способами. Если R15 обозначает метил, можно выполнить этерификацию посредством обработки метанолом в присутствии кислотного катализатора, такого как HCl. Обработка Boc2O дает защиту аминогруппы 2-7. Стереоселективное алкилирование сложного эфира 2-8 алкилирующим агентом, таким как алкилхлорид, -бромид или -йодид, в присутствии подходящего основания, такого как литий бис(триметилсилил)амид, дает промежуточные продукты 2-9. Гидрирование в присутствии катализатора, такого как Pd/C, дает 2-10. Гидролиз сложного эфира с получением 2-11 можно осуществить в стандартных условиях в зависимости от групп R15. Например, если R13 обозначает метил (метиловый сложный эфир), гидролиз можно выполнить посредством обработки основанием, таким как гидроксид натрия, гидроксид лития или гидроксид калия, при нагревании или без него. Защитную группу Boc можно удалить в стандартных кислотных условиях, таких как HCl, в растворителе, таком как диоксан или TFA. Окисление 2-12 с получением 1-1a (в виде отдельного стереоизомера, если компонент R1 является ахиральным, или смеси стереоизомеров, если компонент R1 имеет хиральный центр) можно осуществить несколькими способами, включающими обработку NBS, с последующей обработкой метилатом натрия.
СХЕМА 2C
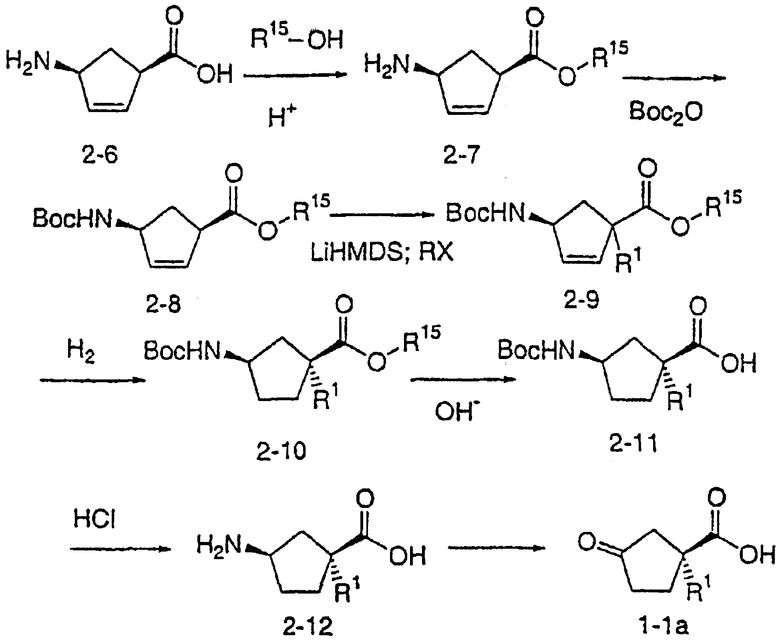
Можно осуществить взаимодействие енолата, полученного из сложного эфира 2-3 (R15 обозначает бензильную или трет-бутильную группу) в присутствии сильного основания, такого как литийдиизопропиламид, с альдегидами (R1аCHO) или кетонами (R1аR2aCO) с получением подходящих гидроксиалкил-замещенных промежуточных продуктов 2-4.1, как указано на схеме 2D. Полученную гидроксигруппу можно защитить различными способами, включающими обработку уксусным ангидридом в присутствии основания, такого как триэтиламин, с получением промежуточных продуктов 2-4.2. Сложноэфирную защитную группу снова удаляют в условиях, подходящих для конкретной защитной группы. В случае трет-бутиловых сложных эфиров (R15 обозначает трет-бутил) удаление защиты проводят в кислотных условиях. Последние обычно вызывают отщепление ацетальной защитной группы, а также данным способом можно получить в одном резервуаре кетокислоты 1-1.1. Их конверсию в конечные модуляторы хемокиновой активности 1-9 можно осуществить, как описано ранее, с модификациией для обеспечения защиты гидроксигруппы в 1-1.1.
СХЕМА 2D
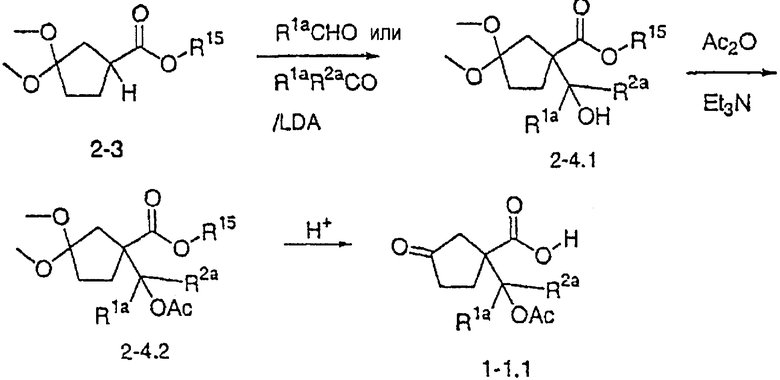
Амины 1-2 можно получить несколькими способами. Один подход показан на схеме 3. Фториды 3-1 (либо коммерчески доступные, либо полученные, как подробно описано в экспериментальном разделе) можно обработать PYH (таким как аммиак, метансульфонамид, трет-бутилтиол, трет-бутиловый спирт) в присутствии основания, такого как NaH, с получением 3-2, образующегося при нуклеофильном ароматическом замещении. Затем можно восстановить нитрильные группы, применяя различные условия, такие как никель Ренея и газообразный водород или боран, получая амины 1-2.
СХЕМА 3
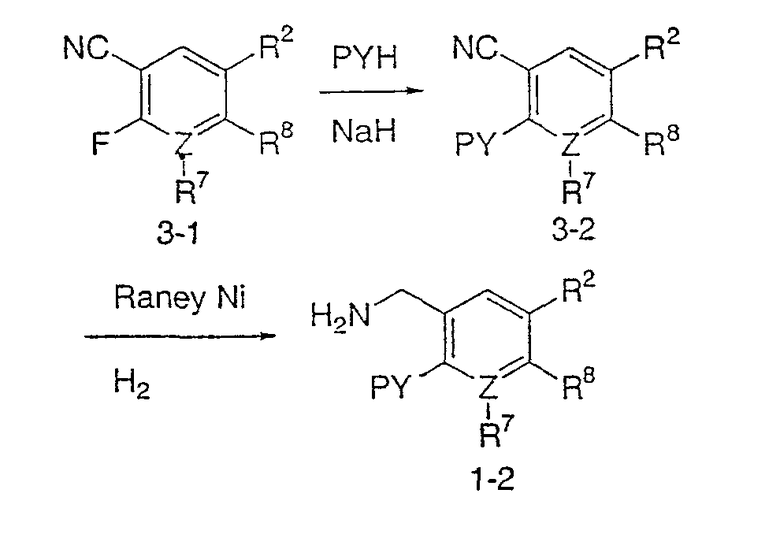
Амины 1-8 получают из разных источников. Некоторые являются коммерчески доступными, некоторые известны из литературы и их можно получить согласно опубликованным методикам, и некоторые получают, как описано здесь. Так как их структуры и способы их получения разнообразны, в данном разделе приведена только одна схема; отдельный синтез аминов 1-8 можно найти ниже. На схеме 4 показан способ синтеза 4-арил-замещенных пиперидинов, а также 4-арил-3-алкилпиперидинов. Можно провести взаимодействие енолтрифлата 4-1 (полученного согласно работе Wustrow, D.J., Wise, L.D., Synthesis (1991), 993-995) с бороновыми кислотами 4-2, как описано Wustrow и Wise. Гидрирование олефина в 4-3 можно осуществить с применением водорода в присутствии катализатора, такого как Pd(OH)2/C. Окисление 4-4 с применением гидрата оксида Ru(IV) и периодата натрия ведет к получению Boc-лактама 4-5. Алкилирование алкилгалогенидом в присутствии основания, такого как LDA, дает 4-6, с преобладанием транс-продукта. Удаление Boc-защитной группы можно осуществить с применением стандартных кислотных условий, таких как HCl в диоксане или TFA/ДХМ. Восстановление лактама 4-7, например, бораном обеспечивает 1-8.2. По-другому, промежуточный продукт 4-4 может сам удалить защиту в кислотных условиях, давая пиперидин 1-8.1.
СХЕМА 4
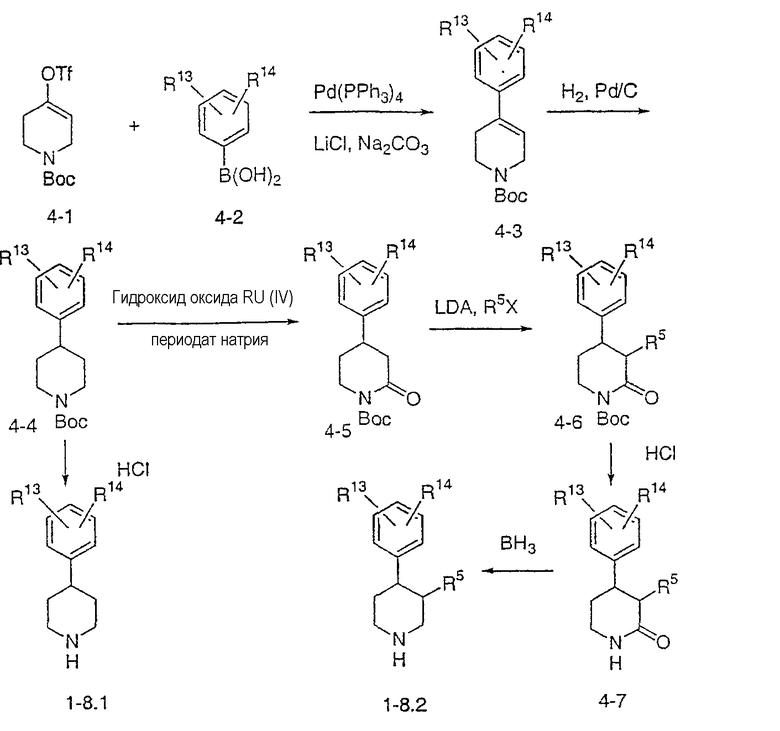
Другие принципиальные способы синтеза модуляторов хемокиновых рецепторов представлены на схеме 5. Согласно данному способу промежуточный продукт 2-11 (описанный на схеме 2C) конденсируют с амином 1-2 (описанным на схеме 1) с применением пептид-связывающего реагента, такого как EDC, с получением 5-1. Защитную группу Boc удаляют в стандартных условиях, таких как HCl в растворителе, таком как диоксан, в случае, когда PY обозначает трет-бутилат, при сопутствующем удалении защитной группы P, получая 5-2. Защиту аминогруппы альтернативной защитной группой, такой как трифторацетат, можно выполнить при помощи трифторуксусного ангидрида в присутствии основания, такого как триэтиламин, с получением 5-3. Циклизацию с получением требуемой конденсированной бициклической кольцевой системы 5-4 осуществляют с применением эквивалента формальдегида, такого как параформальдегид, в присутствии кислотного катализатора, такого как TsOH. Удаление трифторацетатной защитной группы можно выполнить разнообразными способами, включающими обработку K2CO3 в метаноле или другими подходящими основаниями, такими как NaOH или NH3. Обработка полученного амина 5-5 диальдегидом 5-6 в присутствии восстановителя, такого как триацетоксиборогидрид натрия, ведет к двойному восстановительному алкилированию при сопутствующей циклизации с получением 1-9.2. В соответствии со схемой 1 можно осуществить дополнительные модификации, такие как гидролиз сложноэфирной группы, присутствующей в 1-9.2, с получением новых модуляторов хемокиновых рецепторов 1-9.3.
СХЕМА 5
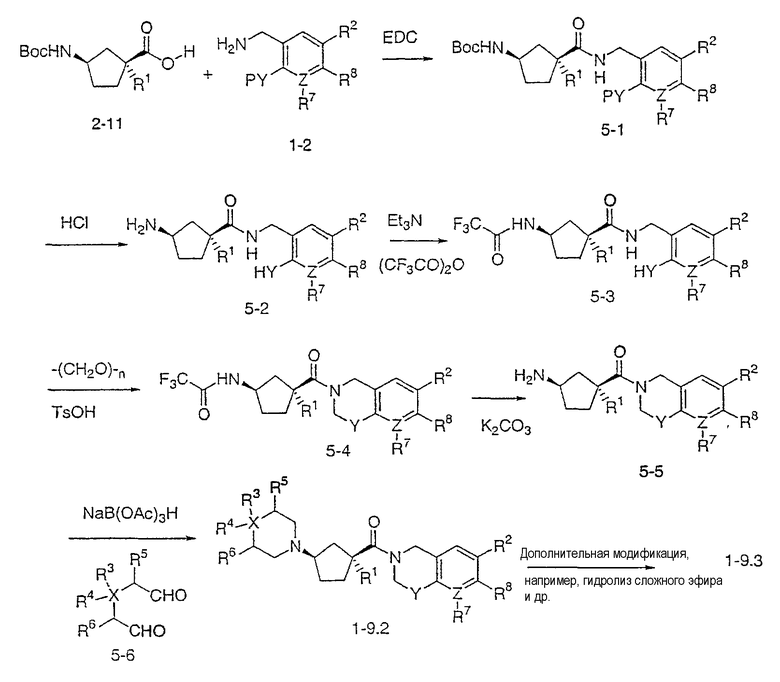
Один путь получения диальдегидов 5-6 представлен на схеме 6. Согласно данному способу производят окислительное отщепление циклоалкена 6-1, например, озоном, а затем диметилсульфидом с получением диальдегида. По-другому, вместо диальдегидов 5-6 можно использовать сами промежуточные продукты озониды 6-2 непосредственно в реакции двойного восстановительного аминирования, ведущей к получению 1-9.2.
СХЕМА 6
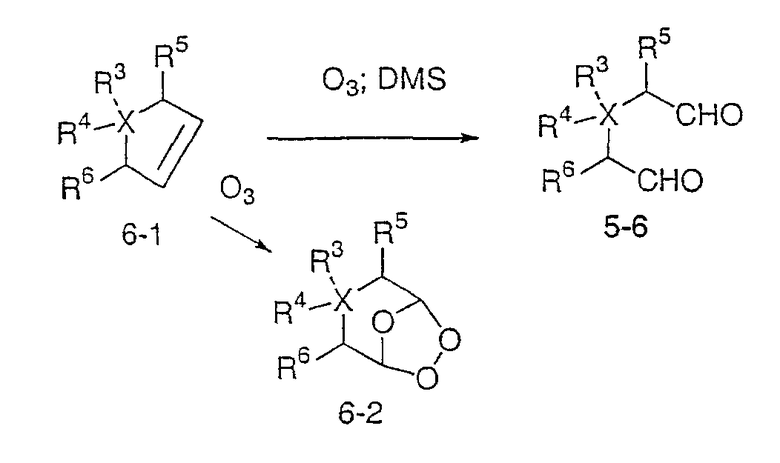
В некоторых случаях порядок проведения вышеупомянутых реакционных схем можно варьировать для содействия взаимодействию или для того, чтобы избежать нежелательных реакционных продуктов. Следующие примеры приведены только для дополнительной иллюстрации и не предполагают ограничений раскрытого изобретения.
Концентрирование растворов обычно проводят на роторном испарителе при пониженном давлении. Флэш-хроматографию выполняют на силикагеле (230-400 меш). СДЖХ обозначает жидкостную хроматографию при среднем давлении, и проводят ее на силикагельной стационарной фазе, если не указано по-другому. ЯМР-спектры получают в растворе CDCl3, если не указано по-другому. Константы взаимодействия (J) даны в герцах (Гц). Сокращения: диэтиловый эфир (эфир), триэтиламин (ТЭА), N,N-диизопропилэтиламин (DIEA), насыщенный водный (насыщ.), комнатная температура (к.т.), час(ы) (час), минута(ы) (мин).
Далее приведены типичные методики получения соединений, применяемых в следующих примерах, или соединений, которыми можно заменить соединения, применяемые в следующих примерах, которые могут не являться коммерчески доступными.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 1
Стадия A:

К охлажденному (0°C) раствору 2-фтор-5-трифторметилбензонитрила (5,23 г, 27,7 ммоль) в 140 мл ТГФ добавляют по капле в быстром темпе суспензию трет-бутилата калия (3,88 г, 34,6 ммоль) в 35 мл ТГФ. Реакционной смеси дают медленно нагреться до к.т. и перемешивают в течение ночи. Реакционную смесь концентрируют при пониженном давлении, затем добавляют эфир и 1 н. раствор HCl и разделяют слои. Эфирный слой промывают насыщенным раствором NaHCO3, затем насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют. Очистка СДЖХ (диоксид кремния, 25% этилацетат/гексан) дает белое кристаллическое твердое вещество. 1H-ЯМР (CDCl3, 500 МГц): δ 7,84 (д, J=2,0 Гц, 1H), 7,73 (дд, J=8,5, 2,0 Гц, 1H), 7,27 (д, J=9,0 Гц), 1,55 (с, 9H).
Стадия B:
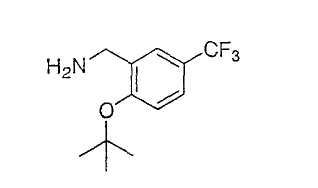
К раствору нитрила, полученного, как описано на стадии A (7,6 г, 31 ммоль), в этаноле (100 мл) добавляют раствор гидроксида аммония (28-30%, 25 мл) и никель Ренея® 2800 (суспензия в воде, ˜3,5 г). Полученную смесь перемешивают при давлении газообразного водорода 50 фунт/дюйм2 (3,515 кг/см2) в течение 24 час с применением аппарата Парра. Затем реакционную смесь фильтруют через целит, промывая этанолом и затем водой. Фильтрат концентрируют досуха при пониженном давлении и полученный таким образом остаток очищают методом флэш-хроматографии [диоксид кремния, градиент от 5 до 10% (1% инкременты) (10% раствора гидроксид аммония (28-30%)/метанол) в ДХМ], получая 1-[2-трет-бутокси-5-(трифторметил)фенил]метанамин в виде бесцветного масла, которое кристаллизуется при хранении в морозильной камере. 1H-ЯМР (500 МГц, CDCl3): δ 7,56 (д, J=2,0 Гц, 1H), 7,44 (дд, J=8,5, 2,0 Гц, 1H), 7,12 (д, 8,5 Гц, 1H), 3,90 (с, 2H), 2,70 (шир.с, 2H), 1,51 (с, 9H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 2
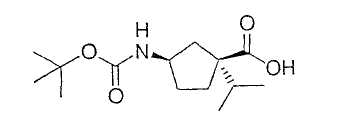
Методика A:
Стадия A:
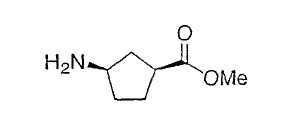
Смесь (1S)-(+)-2-азабицикло[2.2.1]гепт-5-ен-3-она (10,3 г, 94,4 ммоль) в этилацетате (200 мл) и 10% Pd/C (0,5 г) гидрируют при к.т. Через 24 час реакционную смесь фильтруют и выпаривают, оставляя 10,4 г (100%) продукта, который переносят в 250 мл метанола и HCl (12 M, 6 мл). Полученную смесь перемешивают при к.т. до завершения взаимодействия (72 час). Выпаривание метанола с последующей сушкой в высоком вакууме дает указанное в заголовке соединение в виде не совсем белого твердого вещества. 1H-ЯМР (500 МГц, D2O): δ 3,70 (с, 3H), 3,01 (м, 1H), 2,38 (м, 1H), 2,16-1,73 (м, 6H).
Стадия B:
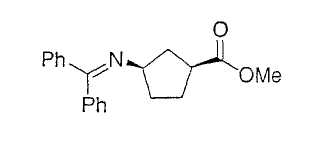
К суспензии промежуточного продукта со стадии A (10,2 г, 56,8 ммоль) в сухом дихлорметане (200 мл) добавляют бензофенонимин (10,2 г, 56,8 ммоль) при к.т. и полученную смесь перемешивают в течение 24 час. Реакционную смесь фильтруют и фильтрат выпаривают, получая желтое масло, которое растирают с эфиром (100 мл), фильтруют и выпаривают. Данную операцию повторяют дважды для гарантии того, что продукт не содержит примесей хлорида аммония. Полученное масло тщательно сушат в вакууме, получая указанное в заголовке соединение, и оно не требует дополнительной очистки. 1H-ЯМР (500 МГц, CDCl3): δ 7,5-7,18 (м, 10H), 3,75 (м, 1H), 3,7 (с, 3H), 2,78 (м, 1H), 2,26-1,71 (м, 6H).
Стадия C:
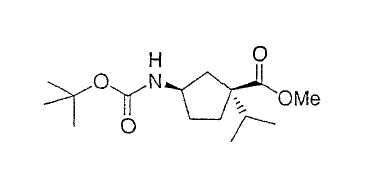
К раствору литийдиизопропиламида (полученного из диизопропиламина (7,7 г, 76 ммоль) и н-бутиллития (30,4 мл, 2,5 M) в гексане, 76 ммоль) в тетрагидрофуране (120 мл) при -78°C добавляют сложный эфир со стадии B (18,0 г, 58,6 ммоль). Полученный красный раствор перемешивают в течение 20 мин, после чего его гасят 2-йодпропаном (14,9 мг, 88 ммоль). Реакционную смесь постепенно нагревают за 3 час до 0°C и данную температуру поддерживают еще в течение 3 час. Реакцию гасят водой и экстрагируют реакционную смесь этилацетатом. Органический слой промывают водой, насыщенным раствором соли, сушат (безводный сульфат магния) и концентрируют, получая масло. К раствору сырого основания Шиффа (20,0 г) в тетрагидрофуране (100 мл) добавляют HCl (5,0 мл, 12 M). Полученную реакционную смесь оставляют перемешиваться при к.т. в течение 3 час. После удаления всех летучих продуктов соль гидрохлорида извлекают в дихлорметан (250 мл), добавляют насыщенный раствор бикарбоната натрия (250 мл) и ди-трет-бутилдикарбонат (26,0 г, 1,4 экв.). Полученную смесь энергично перемешивают в течение ночи при к.т. Органический слой делят и промывают водой, насыщенным раствором соли, сушат (безводный сульфат магния) и концентрируют, получая масло. Очистка методом колоночной флэш-хроматографии (элюент: гексан/этилацетат, 19:1) дает требуемый продукт. 1H-ЯМР (500 МГц, CDCl3): δ 4,79 (широкий, 1H), 4,01 (м, 1H), 3,71 (с, 3H), 2,18-1,60 (м, 6H), 1,44 (с, 9H), 0,87 (д, J=6,9 Гц, 3H), 0,86 (д, J=6,9 Гц, 3H).
Стадия D:
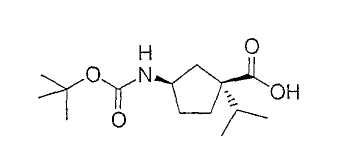
К раствору сложного эфира со стадии С (4,91 г, 17,2 ммоль) в метаноле (100 мл) добавляют раствор LiOH (3,6 г, 85 ммоль) в воде (20 мл) и тетрагидрофуране (10 мл). Полученную смесь нагревают при 80оС до завершения взаимодействия (18 час). Удаляют метанол в вакууме и сырой продукт извлекают смесью вода/этилацетат (200 мл, 1:4) и охлаждают до 0оС. Доводят кислотность смеси до рН 6. Этилацетатный слой отделяют, промывают водой, насыщенным раствором соли, сушат (безводный сульфат магния) и концентрируют, получая масло. Очистка методом колоночной флэш-хроматографии (элюент: гексан/этилацетат (1:1)+2% AcOH) дает (1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновую кислоту. 1H-ЯМР (500 МГц, CDCl3): δ 11,36 (шир., 1H), 6,49 (шир., 1Н), 4,83 (м, 1H), 3,71 (с, 3H), 2,30-1,55 (м, 6H), 1,46 (с, 9H), 0,94 (д, J=6,9 Гц, 3H), 0,933 (д, J=6,9 Гц, 3H).
Методика В:
Стадия А:
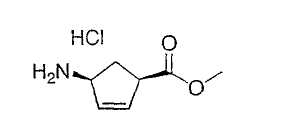
Коммерчески доступную (1R,4S)-4-аминоциклопент-2-ен-1-карбоновую кислоту превращают в гидрохлоридную соль метилового эфира по классической методике.
Стадия B:
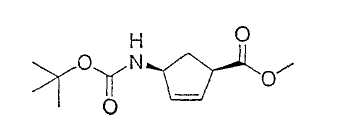
К суспензии амина со стадии A (6,31 г, 35,5 ммоль) в ацетоне (40 мл) и воде (20 мл) добавляют порциями твердый NaHCO3 (6,6 г, 78 ммоль). Через 5 мин добавляют раствор ди-трет-бутилдикарбоната (8,5 г, 39 ммоль) в ацетоне (60 мл) и перемешивают реакционную смесь при к.т. Через 3 час удаляют ацетон в вакууме и остаток распределяют между эфиром (500 мл) и насыщенным водным раствором NaHCO3 (120 мл). Эфирный слой дополнительно промывают водным раствором NaHCO3 (1×100 мл), насыщенным раствором соли (1×100 мл), сушат над безводным Na2SO4, концентрируют и очищают методом флэш-хроматографии (15% этилацетат/гексан), получая продукт.
Стадия C
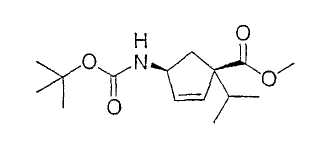
К раствору литийбис(триметилсилил)амида (10,4 г, 62,1 ммоль) в тетрагидрофуране (100 мл) добавляют раствор промежуточного продукта со стадии B (6,71 г, 27,8 ммоль) в тетрагидрофуране (10 мл) за 10 мин при -78°C. Полученный раствор перемешивают при -78°C в течение 30 мин до добавления одной порцией изопропилйодида (3,3 мл, 33 ммоль). Реакционной смеси дают нагреться до -25°C и поддерживают данную температуру в течение ночи. Затем реакцию гасят водным насыщенным раствором NH4Cl (250 мл). Органический слой делят и затем водный слой экстрагируют диэтиловым эфиром (3×100 мл). Комбинированные органические слои далее промывают насыщенным раствором соли (1×100 мл), сушат над безводным Na2SO4, фильтруют, концентрируют и очищают методом флэш-хроматографии (5-10% этилацетат/гексан) с получением продукта в виде прозрачного масла (цис/транс=4,3/1). 1H-ЯМР (500 МГц, CDCl3), цис-изомер: δ 5,79 (с, 2H), 4,75 (м, 1H), 3,72 (с, 3H), 2,28-2,20 (м, 2H), 2,0 (дд, J=15,4 Гц, 1H), 1,45 (с, 9H), 0,85 (д, J=6,6 Гц, 3H), 0,81 (д, J=7 Гц, 3H).
Стадия D:
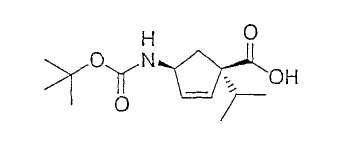
К раствору продукта со стадии C (1,6 г, 5,7 ммоль) в тетрагидрофуране (50 мл), метаноле (50 мл) и воде (10 мл) добавляют моногидрат LiOH (400 мг) и реакционную смесь нагревают при кипении с обратным холодильником в течение ночи до тех пор, пока ТСХ не покажет, что взаимодействие завершено. Органические растворители удаляют в вакууме и водный слой промывают эфиром (1x) и затем медленно подкисляют концентрированной HCl до pH 4. Полученную суспензию экстрагируют CH2Cl2 (3x). Комбинированные органические слои сушат над безводным MgSO4, фильтруют и концентрируют с получением продукта как смеси двух цис/транс изомеров (1,5 г) в виде пенящегося желтого твердого вещества. Данное твердое вещество растворяют в этилацетате (2 мл) при нагревании и разбавляют гексаном (50 мл) с получением прозрачного раствора. Данному раствору дают медленно охладиться до к.т. за 1 час и затем поддерживают при -25°C в морозильной камере в течение ночи.
транс-Изомер кристаллизуется вместе с некоторым количеством требуемого цис-изомера. Маточный раствор собирают и концентрируют с получением указанного в заголовке соединения (только цис-изомер). 1H-ЯМР (500 МГц, CDCl3), цис-изомер: δ 5,80 (м, 2H), 4,80 (м, 1H), 2,40-2,20 (м, 2H), 2,15-2,0 (м, 1H), 1,5 (м, 9H), 1,0-0,8 (м, 3H).
Стадия E:
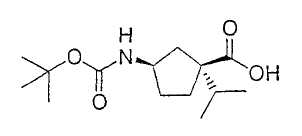
К раствору продукта со стадии D (1 г) в этаноле (30 мл) добавляют 10% Pd/C (100 мг) и полученную смесь перемешивают на аппарате Парра при давлении H2 50 фунтов в течение ночи. Данную смесь фильтруют через целит и концентрируют в вакууме, получая указанное в заголовке соединение (1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновую кислоту. 1H-ЯМР (500 МГц, CDCl3): δ 11,36 (шир., 1H), 6,49 (шир., 1H), 4,83 (м, 1H), 3,71 (с, 3H), 2,30-1,55 (м, 6H), 1,46 (с, 9H), 0,94 (д, J=6,9 Гц, 3H), 0,933 (д, J=6,9 Гц, 3H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 3
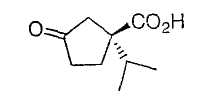
Методика A:
Стадия A:
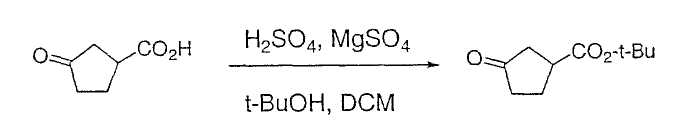
H2SO4 (концентрированная, 15,3 г, 8,30 мл, 156 ммоль) добавляют по капле к энергично перемешиваемой суспензии MgSO4 (75 г, 620 ммоль) в ДХМ (650 мл). Смесь перемешивают в течение 0,5 час, затем добавляют известный циклопентанон-3-карбоксилат (20,0 г, 156 ммоль), а потом трет-бутанол (58 г, 780 ммоль). Реакционный сосуд плотно закупоривают и смесь перемешивают в течение ночи при к.т. На следующее утро добавляют еще 30 мл трет-бутанола. Реакционный сосуд снова плотно закупоривают и перемешивают реакционную смесь в течение выходных дней. Затем реакционную смесь фильтруют через целит. Фильтрат промывают 2 н. NaOH. Водный слой промывают ДХМ методом обратной промывки. Органические слои объединяют, промывают водой, затем насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют, получая трет-бутил 3-оксоциклопентанкарбоксилат. Протекание взаимодействия контролируют при помощи ТСХ с применением смеси 50% этилацетат/гексан и подкрашивания анисальдегидным красителем (SM и полученный краситель красный). 1H-ЯМР (500 МГц, CDCl3): δ 3,02 (p, J=7,8 Гц, 1H), 2,05-2,50 (м, 6H), 1,45 (с, 9H). 13C-ЯМР (125 МГц, CDCl3): δ 217,00, 173,47, 80,99, 41,88, 41,14, 27,94, 26,57.
Стадия B:
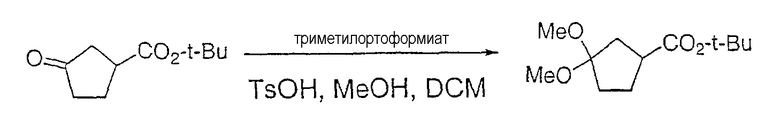
К раствору трет-бутил-3-оксоциклопентанкарбоксилата (19,8 г, 107 ммоль) в смеси (1:1) ДХМ/метанол (150 мл) добавляют триметилортоформиат (46,8 мл, 428 ммоль), а затем TsOH·H2O (˜0,5 г). Реакционную смесь перемешивают при к.т. в течение 2 час. Затем добавляют еще TsOH·H2O (˜0,25 г) и перемешивают реакционную смесь в течение ночи. Реакционную смесь концентрируют при к.т. и полученный остаток растворяют в эфире и промывают насыщенным раствором NaHCO3, затем насыщенным раствором соли. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом флэш-хроматографии (диоксид кремния, 15% этилацетат/гексан) дает трет-бутил-3,3-диметоксициклопентанкарбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 3,21 (с, 3H), 3,20 (с, 3H), 2,80 (м, 1H), 2,10-1,80 (шир.м, 6H), 1,46 (с, 9H). 13C-ЯМР (125 МГц, CDCl3): δ 174,9, 111,2, 80,3, 67,8, 49,2, 42,5, 37,4, 33,8, 28,3, 22,0.
Стадия C:
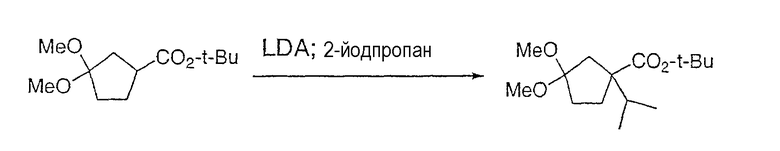
К охлажденному (-78°C) раствору LDA (1,5 M в циклогексане, 41 мл, 61 ммоль) в ТГФ (150 мл) добавляют по капле за 10 мин трет-бутил-3,3-диметоксициклопентанкарбоксилат (9,37 г, 40,7 ммоль) в 25 мл ТГФ. Полученную смесь перемешивают при -78°C в течение 30 мин, затем обрабатывают по капле 2-йодпропаном (16,3 мл, 163 ммоль). После перемешивания в течение дополнительных 10 мин реакционной смеси дают нагреться до к.т. После перемешивания в течение ночи реакционную смесь разбавляют эфиром и промывают насыщенным раствором соли. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. После хранения сырого продукта в вакууме в течение ночи его очищают методом СДЖХ (диоксид кремния, 20% этилацетат/гексан) с получением трет-бутил-1-изопропил-3,3-диметоксициклопентанкарбоксилата. 1H-ЯМР (500 МГц, CDCl3): δ 3,21 (с, 3H), 3,18 (с, 3H), 2,56 (кажущийся д, J=14 Гц, 1H), 2,26 (м, 1H), 1,78-1,89 (м, 3Н).
Стадия D:
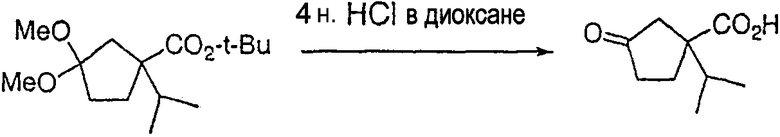
трет-Бутил-1-изопропил-3,3-диметоксициклопентанк
арбоксилат (8,32 г, 30,5 ммоль) растворяют в 4 н. безводном HCl в диоксане (50 мл) и добавляют воду (10 мл). Реакционную смесь перемешивают при к.т. в течение ночи, затем концентрируют. Остаток растворяют в ДХМ, сушат над безводным MgSO4, фильтруют и концентрируют с получением 1-изопропил-3-оксоциклопентанкарбоновой кислоты (используют без очистки). 1H-ЯМР (500 МГц, CDCl3): δ 2,70 (д, J=18,1 Гц, 1H), 2,44-2,39 (м, 1H), 2,30-2,15 (м, 2H), 2,14 (дд, J=18,1, 1,0 Гц, 1H), 2,06 (п, J=6,9 Гц, 1H), 1,98 (м, 1H), 0,98 (дд, J=11,4, 6,9 Гц, 6H).
Стадия E:
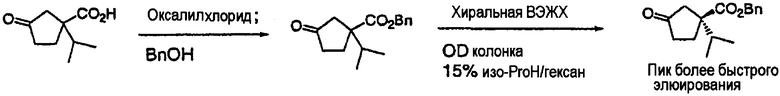
Охлажденный (0°C) раствор 1-изопропил-3-оксоциклопентанкарбоновой кислоты (5,44 г, 32,0 ммоль) в ДХМ (75 мл) обрабатывают оксалилхлоридом (8,36 мл, 95,9 ммоль), а затем 3 каплями ДМФА. Реакционной смеси дают нагреться до к.т. и перемешивают в течение 1,75 час. Затем реакционную смесь концентрируют и хранят в вакууме в течение 30 мин. Полученный хлорангидрид кислоты растворяют в ДХМ (75 мл), охлаждают до 0°C и обрабатывают бензиловым спиртом (8,28 мл, 80,0 ммоль), а затем триэтиламином (8,92 мл, 64,0 ммоль, по капле). Затем добавляют примерно 100 мг ДМАП и реакционную смесь нагревают до к.т. и перемешивают в течение 2 час. Реакционную смесь разбавляют ДХМ и промывают 1 н. раствором HCl, насыщенным раствором NaHCO3 и насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 50% этилацетат/гексан) дает бензил-1-изопропил-3-оксоциклопентанкарбоксилат. 1H-ЯМР (CDCl3, 500 МГц): δ 7,36 (м, 5H), 5,17 (д, J=2,5 Гц, 2H), 2,85 (д, J=18,5 Гц, 1H), 2,48 (м, 1H), 2,29 (дд, J =10,0, 3,0 Гц, 1H), 1,98-2,23 (м, 3H), 1,93 (м, 1H), 0,95 (м, 6H).
Разделение рацемического продукта выполняют методом хиральной ВЭЖХ с применением колонки ChiralCel OD и элюированием смесью 15% 2-пропанол/гексан (100 мг/инъекцию; выполняют с применением программируемой системы ВЭЖХ Gilson). Получают 2,11 г требуемого, быстрее элюируемого изомера, бензил (1S)-1-изопропил-3-оксоциклопентанкарбоксилата.
Стадия F:
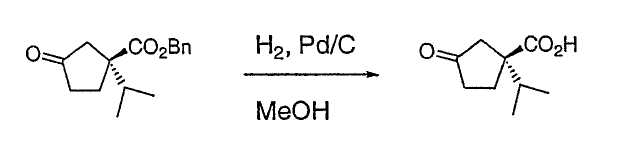
Бензил-(1S)-1-изопропил-3-оксоциклопентанкарбоксилат (1,27 г, 4,88 ммоль) объединяют с Pd/C (10% Degussa, 500 мг) в 20 мл метанола и перемешивают в атмосфере водорода (баллонный) в течение 2 час. Как только взаимодействие проходит часть пути (˜30% конверсии), реакционную смесь фильтруют, добавляют другую порцию Pd/C (500 мг) и перемешивают данную смесь в атмосфере водорода в течение 5 час. Когда взаимодействие подходит к завершению, реакционную смесь фильтруют через целит и концентрируют, получая (1S)-1-изопропил-3-оксоциклопентанкарбоновую кислоту, которая не требует дополнительной очистки. Отметим, что большие количества катализатора применяют потому, что сложный эфир, полученный после хирального разделения, должен быть отравлен примесями. Это уникально для данного конкретного образца. Обычно используют значительно меньшие количества катализатора. 1H-ЯМР спектр идентичен приведенному выше спектру рацемической кислоты (стадия D).
Методика B:
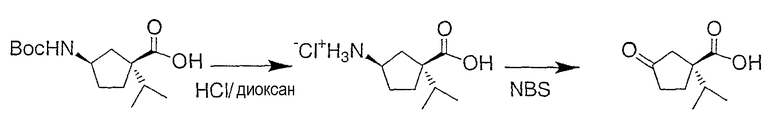
К раствору (1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновой кислоты (7,46 г, 27,5 ммоль) в диоксане (10 мл) добавляют 4 н. HCl в диоксане (30 мл). Реакционную смесь перемешивают при к.т. в течение 2 час, затем концентрируют в вакууме с получением соответствующей соли аминокислоты в виде белого твердого вещества. Данное твердое вещество затем растворяют в CH2Cl2 (100 мл) и добавляют твердый NaHCO3 (7,0 г, 82,5 ммоль). После охдаждения до 0°C медленно добавляют к реакционной смеси раствор NBS (20,0 г, 110 ммоль) в CH2Cl2 (200 мл) за 4 час. После добавления реакционную смесь концентрируют досуха в вакууме и затем растворяют в этаноле (100 мл). К данному этанольному раствору добавляют NaOMe (4,45 г, 82,5 ммоль) и реакционную смесь нагревают при кипении с обратным холодильником. Через 1 час кипения с обратным холодильником реакционную смесь охлаждают до 0°C и добавляют 2 н. водный H2SO4 (50 мл). Смесь перемешивают при к.т. в течение 1 час перед концентрированием в вакууме примерно до объема 60 мл. Оставшуюся смесь распределяют между водой (150 мл) и этилацетатом (100 мл). Водный слой дополнительно дважды экстрагируют этилацетатом. Органические слои объединяют и сушат над безводным MgSO4, концентрируют и очищают методом флэш-хроматографии с получением (1S)-1-изопропил-3-оксоциклопентанкарбоновой кислоты.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 4
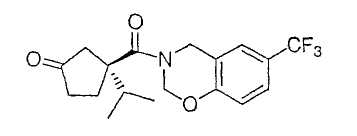
Стадия А:
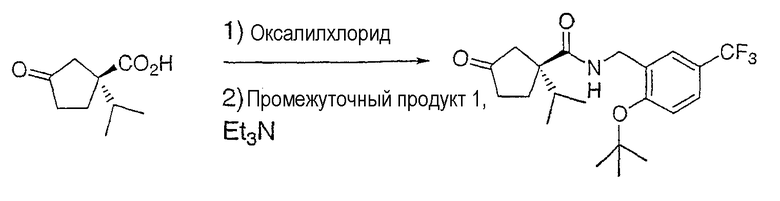
Охлажденный (0°C) раствор (1S)-1-изопропил-3-оксоциклопентанкарбоновой кислоты (588 мг, 3,46 ммоль) в атмосфере азота обрабатывают оксалилхлоридом (1,21 мл, 13,8 ммоль), а затем 1 каплей ДМФА. Реакционной смеси дают нагреться до к.т. (образование пузырьков указывает газообразование) и перемешивают в течение 1,5 час. Затем реакционную смесь концентрируют. Полученный хлорангидрид кислоты повторно растворяют в ДХМ (30 мл), охлаждают до 0°C и обрабатывают промежуточным продуктом 1 (1,28 г, 5,19 ммоль), а затем триэтиламином (0,965 мл, 6,92 ммоль). Полученную реакционную смесь нагревают до к.т. и перемешивают в течение 1,5 час, после чего ее разбавляют и промывают дважды 1 н. раствором HCl, один раз насыщенным раствором NaHCO3 и один раз насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 55% этилацетат/гексан) дает (1S)-N-[2-трет-бутокси-5-(трифторметил)бензил]-1-изопропил-3-оксоциклопентанкарбоксамид. 1H-ЯМР (CDCl3, 500 МГц): δ 7,53 (д, J=2,0 Гц, 1H), 7,48 (дд, J=8,0, 2,0 Гц, 1H), 7,16 (д, J=8,5 Гц, 1H), 6,16 (шир.с, 1H), 4,49 (м, 2H), 2,78 (дд, J=19, 2,0 Гц, 1H), 2,35 (м, 1H), 2,28 (м, 2H), 2,19 (д, J=18 Гц, 1H), 1,92-2,02 (м, 2H), 1,52 (с, 9H), 0,954 (д, J=6,5 Гц, 3H), 0,947 (д, J=7,0 Гц, 3H).
Стадия B:
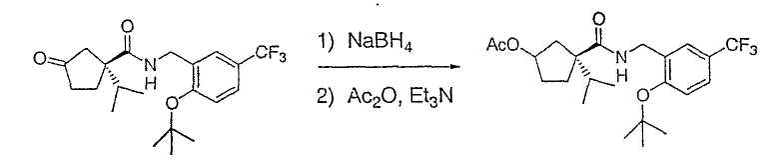
Охлажденный (0°C) раствор (1S)-N-[2-трет-бутокси-5-(трифторметил)бензил]-1-изопропил-3-оксоциклопентанкарбоксамида (1,37 г, 3,43 ммоль) в 10 мл метанола обрабатывают борогидридом натрия (130 мг, 3,43 ммоль). После перемешивания при 0°C в течение 10 мин реакционной смеси дают нагреться до к.т. и перемешивают в течение еще 1 час. Реакционную смесь концентрируют и распределяют остаток между ДХМ и водой. Органический слой промывают насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют, получая смесь цис-/транс-спиртов. Сырую смесь спиртов растворяют в ДХМ (20 мл) и обрабатывают уксусным ангидридом (0,39 мл, 4,1 ммоль), триэтиламином (0,57 мл, 4,1 ммоль) и ДМАП (˜25 мг). Полученную смесь перемешивают при к.т. в течение 2 дней (взаимодействие заканчивается через несколько часов). Реакционную смесь разбавляют ДХМ и промывают 1 н. раствором HCl, насыщенным раствором NaHCO3 и насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Сырой продукт очищают методом СДЖХ (диоксид кремния, 50% этилацетат/гексан) с получением ацетатного продукта в виде смеси двух диастереомеров (цис- и транс-изомеров).
ВЭЖХ-МС. Пик 1: ЭСИ-МС, рассчитано для C23H32F3NO4 443; найдено 466 (M+Na+).
ВЭЖХ-МС. Пик 2: ЭСИ-МС, рассчитано для C23H32F3NO4 443; найдено 466 (M+Na+).
Стадия C:
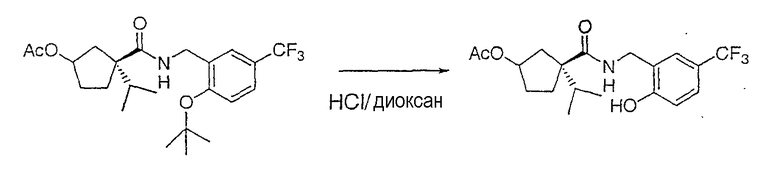
Смесь цис-/транс-ацетатов со стадии B (1,13 г, 2,55 ммоль) растворяют в 4 н. HCl в диоксане (Aldrich, 20 мл). Полученную смесь перемешивают при к.т. в течение 1,25 час, затем концентрируют и хранят в вакууме в течение ночи с получением 1,08 г сырого продукта. ЭСИ-МС: рассчитано для C19H24F3NO4 387; найдено 410 (M+Na+).
Стадия D:
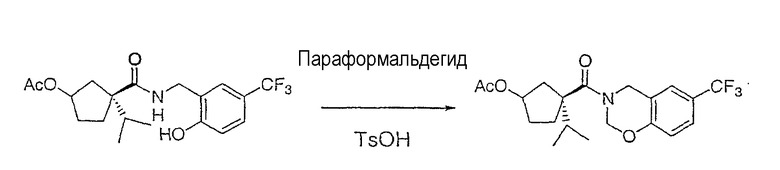
Фенол, полученный, как описано на стадии C (1,16 г, сырой, 2,84 ммоль), объединяют с параформальдегидом (˜1 г) и TsOH·H2O (˜20 мг) в 40 мл толуола. Колбу снабжают ловушкой Дина-Старка и холодильником и перемешивают реакционную смесь при кипении с обратным холодильником в течение 2,5 час. Когда ВЭЖХ-МС анализ показывает, что осталось примерно 20% исходного материала, добавляют дополнительную порцию параформальдегида (˜250 мг) и смесь перемешивают при кипении с обратным холодильником в течение 1 час. Реакционную смесь концентрируют и полученный остаток растворяют в ДХМ и фильтруют, удаляя остатки параформальдегида. Фильтрат концентрируют и очищают методом СДЖХ (диоксид кремния, 50% этилацетат/гексан) с получением (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентилацетата в виде смеси двух изомеров (цис/транс).
ВЭЖХ-МС. Пик 1, ЭСИ-МС: рассчитано для C20H24F3NO4 399; найдено 422 (M+Na+).
ВЭЖХ-МС. Пик 2, ЭСИ-МС: рассчитано для C20H24F3NO4 399; найдено 422 (M+Na+).
Стадия E:
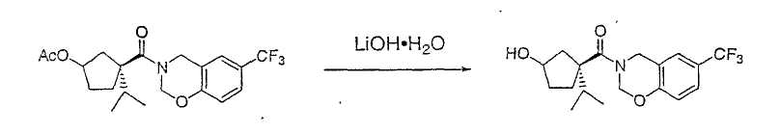
Раствор (3S)-3-изопропил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентилацетата (935 мг, 2,34 ммоль) обрабатывают раствором LiOH·H2O (491 мг, 11,7 ммоль) в деионизированной воде (5 мл). Полученную реакционную смесь перемешивают при к.т. в течение 40 мин, затем разбавляют насыщенным раствором соли и экстрагируют эфиром. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют с получением (3S)-3-изопропил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентанола (смеси двух изомеров), который используют как есть.
ВЭЖХ-МС. Пик 1, ЭСИ-МС: рассчитано для C18H22F3NO3 357; найдено 380 (M+Na+).
ВЭЖХ-МС. Пик 2, ЭСИ-МС: рассчитано для C18H22F3NO3 357; найдено 380 (M+Na+).
Стадия F:
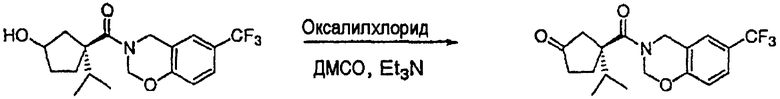
Раствор ДМСО (0,656 мл, 9,24 ммоль) в 7 мл ДХМ добавляют по капле к охлажденному (-78°C) раствору оксалилхлорида (0,403 мл, 4,62 ммоль) в 30 мл ДХМ. Через 5 мин добавляют по капле раствор (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанола (826 мг, 2,31 ммоль) в 8 мл ДХМ. Реакционную смесь перемешивают при -78°C в течение 25 мин, затем обрабатывают по капле чистым триэтиламином (2,58 мл, 18,5 ммоль). После перемешивания при -78°C в течение еще 10 мин реакционной смеси дают нагреться до к.т. и перемешивают в течение 1,5 час. Затем смесь выливают в 1 н. раствор HCl и экстрагируют ДХМ. Органический слой промывают еще раствором 1 н. HCl, затем насыщенным раствором NaHCO3 и насыщенным раствором соли.
Органический слой затем сушат над безводным MgSO4, фильтруют и концентрируют. Сырой продукт очищают методом СДЖХ (диоксид кремния, 75% этилацетат/гексан), получая (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентанон. ЭСИ-МС: рассчитано для C18H20F3NO3 355; найдено 356 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 5
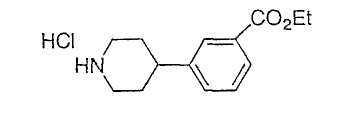
Стадия A:
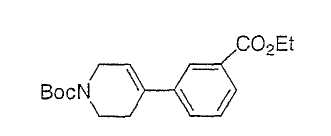
К смеси трет-бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2H)-карбоксилата (полученного согласно Wustrow, D.J., Wise, L.D., Synthesis, (1991), 993-995.; 10,5 г, 31,6 ммоль), 3-(этоксикарбонил)фенилбороновой кислоты (8,59 г, 44,3 ммоль), хлорида лития (3,98 г, 94,8 ммоль) и 2 M раствора Na2CO3 (44 мл) в ДМЭ (107 мл) добавляют Pd(PPh3)4 (1,82 г, 1,58 ммоль) и полученную смесь перемешивают при кипении с обратным холодильником в атмосфере азота в течение 3,5 час. Реакционную смесь охлаждают до к.т., перемешивают в течение ночи, затем частично концентрируют для удаления большей части ДМЭ. К оставшейся водной смеси добавляют ДХМ, 2 M раствор Na2CO3 и ˜10 мл 28% раствора NH4OH. Слои разделяют и органический слой промывают насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом флэш-хроматографии (диоксид кремния, элюент: смесь 10% этилацетат/гексан) дает трет-бутил-4-[3-(этоксикарбонил)фенил]-3,6-дигидропиридин-1(2Н)-карбоксилат. 1H-ЯМР (CDCl3, 500 МГц): δ 8,07 (с, 1H), 7,95 (д, J=7,5 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,43 (т, J=8,0 Гц, 1H), 6,13 (шир.с, 1H), 4,41 (кв., J=7,0 Гц, 2H), 4,12 (шир.с, 2H), 3,68 (т, J=5,5 Гц, 2H), 2,58 (шир.с, 2H), 1,52 (с, 9H), 1,43 (т, J=7,0 Гц, 3H).
Стадия B:
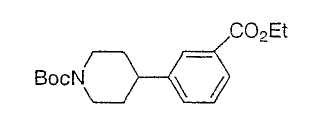
Смесь трет-бутил-4-[3-(этоксикарбонил)фенил]-3,6-дигидропиридин-1(2Н)-карбоксилата (6,48 г, 19,6 ммоль) и Pd(OH)2/C (20% Pd, 1 г) в 50 мл метанола перемешивают в атмосфере водорода (баллонный) в течение 18 час. Затем реакционную смесь фильтруют через набивку из целита и фильтрат концентрируют с получением трет-бутил-4-[3-(этоксикарбонил)фенил]пиперидин-1-карбоксилата, который не требует дополнительной очистки. 1H-ЯМР (CDCl3, 500 МГц): δ 7,91 (м, 2H), 7,40 (м, 2H), 4,40 (кв., J=7,0 Гц, 2H), 4,28 (шир.с, 2H), 2,83 (м, 2H), 2,73 (тт, J=12,5, 4,0 Гц, 1H), 1,85 (шир.д, J=13,0 Гц), 1,67 (д.кв., J=4,0, 12,5 Гц, 2H), 1,51 (с, 9H), 1,42 (т, J=7,0 Гц).
Стадия C:
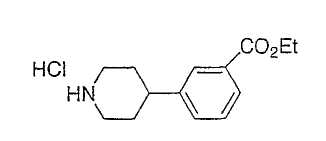
трет-Бутил-4-[3-(этоксикарбонил)фенил]пиперидин-1-карбоксилат (3,24 г, 9,72 ммоль) растворяют в безводном 4 н. HCl в диоксане (около 30 мл) и перемешивают при к.т. в течение 1,5 час. Реакционную смесь концентрируют с получением этил-3-пиперидин-4-илбензоатгидрохлорида в виде светло-желтого твердого вещества, которое не требует дополнительной очистки. ЭСИ-МС: рассчитано для C14H19NO2 233; найдено 234 (M+H).
ПРИМЕР 1
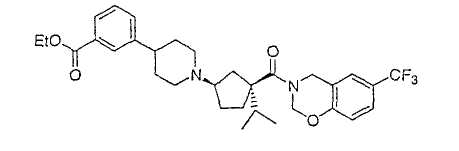
Смесь этил-3-пиперидин-4-илбензоатгидрохлорида (483 мг, 1,79 ммоль), (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентанона (318 мг, 0,896 ммоль), триэтиламина (375 мкл, 2,69 ммоль), 4Å порошкообразных молекулярных сит (˜500 мг) и триацетоксиборогидрида натрия (949 мг, 4,48 ммоль) в 15 мл ДХМ перемешивают при к.т. в течение 5 дней (обычно достаточно 3 дней). Реакционную смесь разбавляют ДХМ и промывают насыщенным раствором NaHCO3, а затем насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом флэш-хроматографии (диоксид кремния, 4% [10% раствора NH4OH (28%)/метанол] в ДХМ) дает продукт в виде смеси цис- и транс-изомеров продукта. Цис (этил 3-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоат) и транс (этил 3-[1-((1S,3S)-3-изопропил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоат) изомеры разделяют методом препаративной ТСХ (диоксид кремния, элюент: 40% ТГФ/гексан) с получением элюируемого в большей степени (цис) изомера и элюируемого в меньшей степени (транс) изомера. Анализ разделенных цис- и транс-изомеров продукта методом хиральной аналитической ВЭЖХ (колонка ChiralCel OD, 10% этанол/гексан) показывает, что каждый является чистым отдельным изомером.
Элюируемый в большей степени (цис) изомер: ЭСИ-МС, рассчитано для C32Н39F3N2O4 572; найдено 573 (M+H).
Элюируемый в меньшей степени (транс) изомер: ЭСИ-МС, рассчитано для C32Н39F3N2O4 572; найдено 573 (M+H).
В некоторых случаях, где амины, применяемые в реакциях восстановительного аминирования, сами собой представляют смеси более чем одного стереоизомера (т.е. если они имеют один или более стереоцентров), обычно можно разделить все возможные изомеры с применением хиральной ВЭЖХ и/или препаративной ТСХ (иногда требуются серии разделений). Типичный пример того, как это выполняют, смотри в ПРИМЕРЕ 3.
ПРИМЕР 2
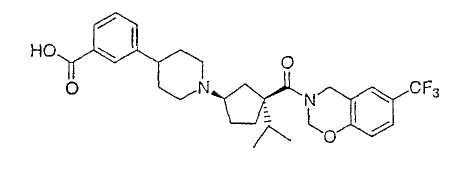
К раствору этил-3-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоата (225 мг, 0,393 ммоль) в этаноле (10 мл) добавляют раствор LiOH·H2O (165 мг, 3,93 ммоль) в 10 мл деионизированной воды. Полученную смесь перемешивают при 50°C в течение 1,5 час, затем частично концентрируют, удаляя большую часть этанола. К полученной водной смеси добавляют насыщенный раствор соли и хлороформа. pH водного слоя доводят до 7 1 M раствором HCl (˜2,5-3 мл). Слои разделяют и водный слой экстрагируют еще два раза хлороформом. Органические слои объединяют и сушат над безводным MgSO4, фильтруют и концентрируют с получением сырого продукта. Очистка ВЭЖХ с обращенной фазой (YMC Pack Pro С18, 100×20 мм внутр. диам.) дает продукт в виде его TFA-соли. Этот продукт превращают в его HCl-соль, растворяя в ДХМ и добавляя избыток 1 н. HCl/эфир, затем концентрируя с получением гидрохлорида 3-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензойной кислоты. ЭСИ-МС: рассчитано для C30H35F3N2O4 544; найдено 545 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 6
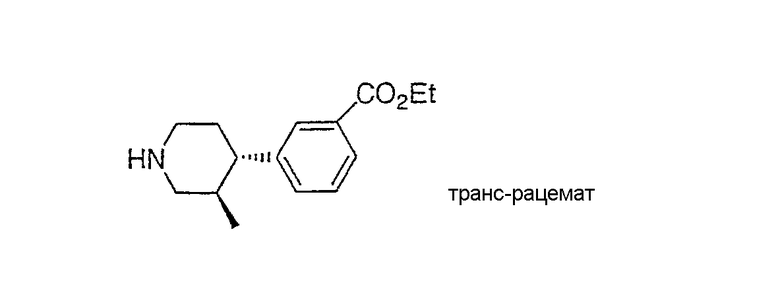
Стадия A:
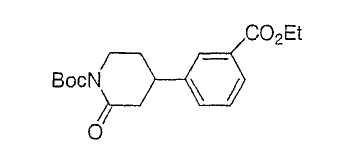
К перемешиваемому раствору трет-бутил-4-[3-(этоксикарбонил)фенил]пиперидин-1-карбоксилата (48 г, 220 ммоль) в хлороформе (900 мл) добавляют гидрат оксида рутения(IV) (6,0 г, 45 ммоль), а затем раствор периодата натрия (150 г, 700 ммоль) в воде (900 мл). Полученную гетерогенную реакционную смесь перемешивают при комнатной температуре в течение 11 дней перед фильтрованием через короткую колонку с целитом. Органический слой удаляют и водный слой экстрагируют дважды ДХМ. Комбинированные органические слои дважды промывают 10% раствором тиосульфата натрия в воде и один раз насыщенным раствором соли. Данный раствор сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Продукт очищают методом флэш-хроматографии (силикагель, 20% ЭА/гексан) с получением 22,5 г (64,8 ммоль) трет-бутил-4-[3-(этоксикарбонил)фенил]-2-оксопиперидин-1-карбоксилат (29%). ЭСИ-МС: рассчитано для C19H25NO5 347,17; найдено 370,1 (M+Na).
Стадия B:
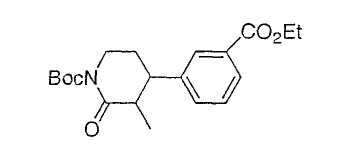
Калий бис(триметилсилил)амид (14 г, 71 ммоль) смешивают с 300 мл ТГФ в высушенной на пламени круглодонной колбе на 1000 мл и полученную смесь охлаждают до -78°C. трет-Бутил-4-[3-(этоксикарбонил)фенил]-2-оксопиперидин-1-карбоксилат (22,5 г, 64,8 ммоль), растворенный в 150 мл ТГФ, медленно добавляют к смеси через делительную воронку и полученную реакционную смесь перемешивают при -78°C в течение 30 мин. Затем добавляют по капле метилйодид (12,1 мл, 195 ммоль) и реакционную смесь перемешивают при -78°C в течение 4 час перед тем, как оставить нагреваться до комнатной температуры в течение ночи. Реакцию гасят насыщенным хлоридом аммония и экстрагируют 3 раза эфиром. Комбинированные эфирные слои промывают насыщенным раствором соли и сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Продукт очищают методом флэш-хроматографии (10-20% ЭА/гексан) с получением 6,1 г транс-рацемата трет-бутил-4-[3-(этоксикарбонил)фенил]-3-метил-2-оксопиперидин-1-карбоксилата (26%). ЭСИ-МС: рассчитано для C20H27NO5: 361,19; найдено 384,25 (M+Na).
Стадия C:
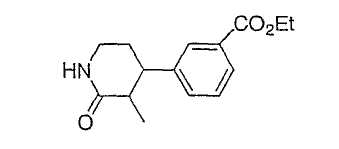
Продукт со стадии B (6,1 г, 17 ммоль) растворяют в 4,0 M HCl в диоксане и перемешивают при комнатной температуре в течение 2 час перед концентрированием при пониженном давлении с получением требуемого продукта в виде оранжевого твердого вещества, которое подается непосредственно на следующую стадию без дополнительной очистки. ЭСИ-МС: рассчитано для C15Н19NO3 261,14; найдено 262,1 (M+H).
Стадия D:
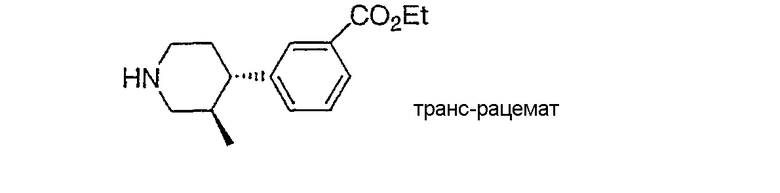
Продукт с предыдущей стадии (общее количество ˜17 ммоль) растворяют в ТГФ (100 мл) и обрабатывают по капле 2,0 M раствором боранметилсульфида в ТГФ (31 мл, 62 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 4 час, перед хранением при 4°C в течение 72 час. Растворитель удаляют при пониженном давлении и полученный остаток растворяют в 0,5 M HCl (водный -38%) в этаноле. Данный раствор нагревают до 50°C и перемешивают в течение 4 час. Растворитель удаляют и методику повторяют снова для гарантии разрушения боранового комплекса. Растворитель удаляют и очищают продукт методом СДЖХ (0-15% (10% NH4OH/MeOH)/ДХМ) с получением требуемого продукта, который имеет чистоту 80%. Данный сырой материал растворяют в ДХМ (100 мл) и обрабатывают ди-трет-бутилдикарбонатом (2,95 г, 13,5 ммоль), диизопропилэтиламином (2,30 мл, 13,5 ммоль) и ДМАП (10 мг). Полученную реакционную смесь перемешивают в течение ночи при комнатной температуре перед разбавлением ДХМ и промывают 1 н. водным насыщенным бикарбонатом натрия и насыщенным раствором соли. Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Промежуточный продукт очищают методом СДЖХ (0-40% EA/гексан). Полученное бесцветное масло растворяют в 4,0 M HCl в диоксане и полученную реакционную смесь перемешивают при комнатной температуре в течение 1,5 час. Реакционную смесь концентрируют досуха с получением 2,13 г (7,52 ммоль) требуемой HCl-соли. ЭСИ-МС: рассчитано для C15H21NO2 247,16; найдено 248,15 (M+H).
ПРИМЕР 3
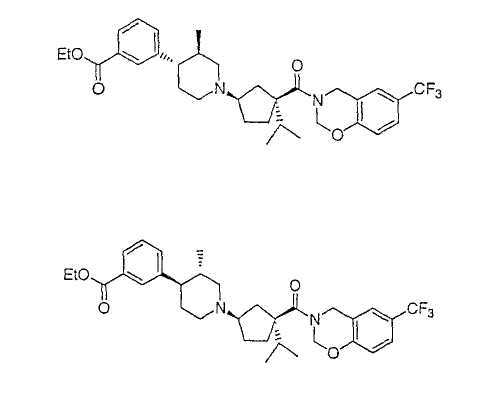
Промежуточный продукт 6 (амин) (830 мг, 2,93 ммоль) объединяют с промежуточным продуктом 17 (990 мг, 2,78 ммоль) и диизопропилэтиламином (501 мкл, 2,93 ммоль) в ДХМ (50 мл). После перемешивания в течение 10 мин данный раствор обрабатывают последовательно порошкообразными 4Å молекулярными ситами (1 г) и триацетоксиборогидридом натрия (2,48 г, 11,7 ммоль). Полученную реакционную смесь оставляют перемешиваться при комнатной температуре в течение 4 дней перед распределением между ДХМ и полунасыщенным водным раствором бикарбоната натрия. Водный слой 3 раза экстрагируют ДХМ методом обратной экстракции. Комбинированные органические слои промывают насыщенным раствором соли и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Полученный сырой продукт очищают методом СДЖХ (0-10% (10% NH4OH/MeOH)/ДХМ) с получением 1,3 г смеси 4 стереоизомеров. Индивидуальные стереоизомеры выделяют методом хиральной ВЭЖХ с применением колонки ChiralCel OD, элюируя смесью 8% этанол/гексан. (Два требуемых цис-изомера (около циклопентана) представлены 3-м и 4-м пиками).
Пик 1: 260 мг изомер - ЭСИ-МС, рассчитано для C33H41F3N2O3 586,30; найдено 587,65 (M+H).
Пик 2: 220 мг изомер - ЭСИ-МС, рассчитано для C33H41F3N2O3 586,30; найдено 587,65 (M+H).
Пик 3: 300 мг изомер - ЭСИ-МС, рассчитано для C33H41F3N2O3 586,30; найдено 587,65 (M+H).
Пик 4: 280 мг изомер - ЭСИ-МС, рассчитано для C33H41F3N2O3 586,30; найдено 587,65 (M+H).
ПРИМЕР 4
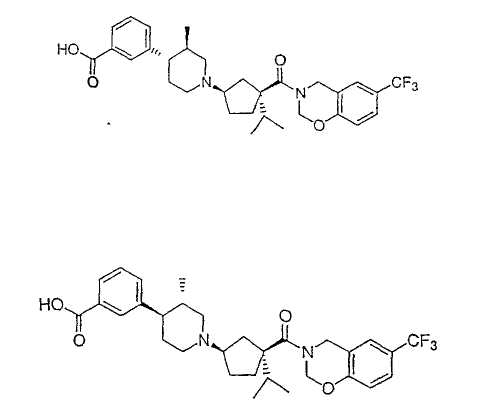
Отдельно растворяют пик 3 (8 мг, 0,01 ммоль) и пик 4 (8 мг, 0,01 ммоль) из примера 3 в этаноле (2 мл) и обрабатывают раствором гидроксида лития (10 мг) в воде (1 мл). Полученные растворы перемешивают при комнатной температуре в течение 22 час перед концентрированием досуха. Полученные сырые продукты очищают методом ВЭЖХ с обращенной фазой (C18, 0-100% MeCN/H2O) и превращают в их HCl-соли, растворяя продукты в ДХМ и обрабатывая полученные растворы 2,0 M HCl в эфире и гексане последовательно. Мутные растворы концентрируют досуха с получением 6,41 мг (из пика 3) и 4,66 мг (из пика 4) требуемых продуктов соответственно.
Из пика 3: ЭСИ-МС: рассчитано для C31H37F3N2O3 558,27; найдено 559,6 (M+H).
Из пика 4: ЭСИ-МС: рассчитано для C31H37F3N2O3 558,27; найдено 559,6 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 7
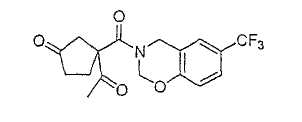
Стадия A:
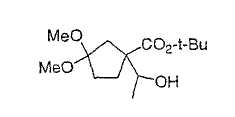
К охлажденному (-78°C) раствору LDA (2 M в смеси ТГФ/гептан, 39 мл, 78 ммоль) в ТГФ (40 мл) добавляют по капле за 15 мин трет-бутил-3,3-диметоксициклопентанкарбоксилат (15,0 г, 65,0 ммоль) в 100 мл ТГФ. Полученную смесь перемешивают при -78°C в течение 40 мин, затем обрабатывают по капле ацетальдегидом (7,27 мл, 130 ммоль). После перемешивания в течение еще 45 мин реакционную смесь выливают в 500 мл 10% раствора лимонной кислоты. Смесь экстрагируют дважды эфиром и объединенные эфирные слои промывают насыщенным раствором соли. Затем эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. ТСХ-анализ показывает, что взаимодействие не проходит до завершения и еще присутствует значительное количество исходного материала. Очистка/разделение методом флэш-хроматографии (диоксид кремния, 30% этилацетат/гексан) дают трет-бутил-1-(1-гидроксиэтил)-3,3-диметоксициклопентанкарбоксилат при соотношении примерно 1:1 трео- и эритро- диастереомерных пар и извлеченного трет-бутил-3,3-диметоксициклопентанкарбоксилата.
Стадия B:
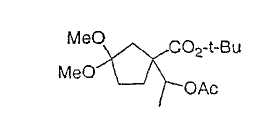
Раствор трет-бутил-1-(1-гидроксиэтил)-3,3-диметоксициклопентанкарбоксилата (8,64 г, 31,5 ммоль) в 100 мл ДХМ обрабатывают триэтиламином (8,78 мл, 63,0 ммоль), а затем уксусным ангидридом (5,94 мл, 63,0 ммоль). Затем добавляют ДМАП (˜200 мг) и полученную смесь перемешивают при к.т. в течение ночи. Реакционную смесь разбавляют ДХМ, промывают дважды 1 н. раствором HCl, один раз насыщенным раствором NaHCO3 и один раз насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом флэш-хроматографии (диоксид кремния, 20% этилацетат/гексан) дает трет-бутил-1-[1-(ацетилокси)этил]-3,3-диметоксициклопентанкарбоксилат.
Стадия C:
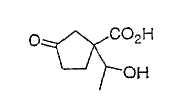
трет-Бутил-1-[1-(ацетилокси)этил]-3,3-диметоксициклопентанкарбоксилат (8,04 г, 25,4 ммоль) растворяют в безводном 4 н. HCl в диоксане (примерно 50 мл) и добавляют 5 мл воды (это была ошибка, которая ведет к неадекватному гидролизу ацетата). Реакционную смесь перемешивают при к.т. в течение 5 час, затем концентрируют. Очистка методом флэш-хроматографии (диоксид кремния, 8% метанол/ДХМ) дает 1-(1-гидроксиэтил)-3-оксоциклопентанкарбоновую кислоту. 1H-ЯМР (CDCl3, 500 МГц): δ 4,09 (кв., J=6,5 Гц, 1H, диастереомер d1), 3,99 (кв., J=6,5 Гц, 1H, диастереомер d2), 2,73-2,78 (м, 2H, d1+d2), 2,37-2,52 (м, примерно 7H, d1+d2), 2,13-2,24 (м, примерно 3H, d1+d2), 1,30 (д, J=6,5 Гц, 3H, изомер d1?), 1,29 (д, J=6,5 Гц, 3H, изомер d2?).
Стадия D:
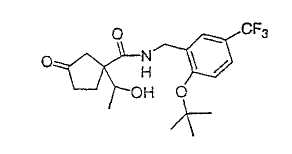
Смесь 1-(1-гидроксиэтил)-3-оксоциклопентанкарбоновой кислоты (3,54 г, 20,6 ммоль), 1-гидрокси-7-азабензотриазола (4,14 г, 30,9 ммоль) и EDC (5,92 г, 30,9 ммоль) в 70 мл ДХМ перемешивают при к.т. в течение 10 мин, затем добавляют 1-[2-трет-бутокси-5-(трифторметил)фенил]метанамин (5,08 г, 20,6 ммоль) в 10 мл ДХМ. Полученную смесь перемешивают при к.т. в течение 50 мин, затем разбавляют ДХМ и промывают водой, а затем насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 85% этилацетат/гексан) дает N-[2-трет-бутокси-5-(трифторметил)бензил]-1-(1-гидроксиэтил)-3-оксоциклопентанкарбоксамид в виде смеси трео- и эритро- энантиомерных пар.
Стадия E:
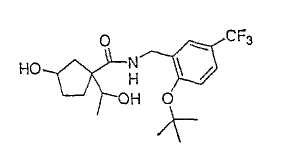
Охлажденный (0°C) раствор N-[2-трет-бутокси-5-(трифторметил)бензил]-1-(1-гидроксиэтил)-3-оксоциклопентанкарбоксамида (6,27 г, 15,6 ммоль) в метаноле обрабатывают борогидридом натрия (591 мг, 15,6 ммоль) и перемешивают в течение 20 мин. Реакционную смесь концентрируют и остаток распределяют между ДХМ и водой. Органический слой промывают насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют с получением N-[2-трет-бутокси-5-(трифторметил)бензил]-3-гидрокси-1-(1-гидроксиэтил)циклопентанкарбоксамида в виде смеси 8 изомеров.
Стадия F:
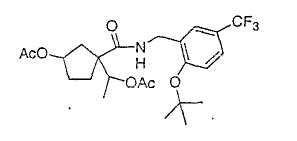
Охлажденный (0°C) раствор N-[2-трет-бутокси-5-(трифторметил)бензил]-3-гидрокси-1-(1-гидроксиэтил)циклопентанкарбоксамида (6,03 г, 14,9 ммоль) в 75 мл ДХМ обрабатывают триэтиламином (6,24 мл, 44,8 ммоль), уксусным ангидридом (4,23 мл, 44,8 ммоль) и ДМАП (примерно 100 мг). Реакционной смеси дают нагреться до к.т. и еще через 2,5 час разбавляют ДХМ и промывают последовательно 1 н. раствором HCl, насыщенным раствором NaHCO3 и насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 60% этилацетат/гексан) дает 1-[3-(ацетилокси)-1-({[2-трет-бутокси-5-(трифторметил)бензил]амино}карбонил)циклопентил]этилацетат в виде смеси 8 диастереомеров. ЭСИ-МС: рассчитано для C24H32F3NO6 478; найдено 510 (M+Na+).
Стадия G:
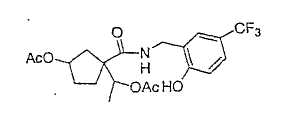
1-[3-(Ацетилокси)-1-({[2-трет-бутокси-5-(трифторметил)бензил]амино}карбонил)циклопентил]этилацетат (7,0 г, 14,4 ммоль) растворяют в безводном 4 н. HCl в диоксане (40 мл) и перемешивают при к.т. в течение 65 мин. Затем реакционную смесь концентрируют с получением 3-[1-(ацетилокси)этил]-3-({[2-гидрокси-5-(трифторметил)бензил]амино}карбонил)циклопентилацетата. ЭСИ-МС: рассчитано для C20H24F3NO6 431; найдено 432 (M+).
Стадия H:
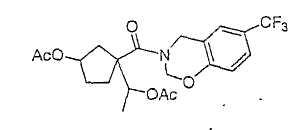
3-[1-(Ацетилокси)этил]-3-({[2-гидрокси-5-(трифторметил)бензил]амино}карбонил)циклопентилацетат объединяют с параформальдегидом (примерно 6 г) и TsOH·H2O (примерно 100 мг) в 130 мл толуола. Колбу снабжают ловушкой Дина Старка и холодильником, реакционную смесь перемешивают при кипении с обратным холодильником в течение 1,5 час. Реакционную смесь фильтруют для удаления оставшегося параформальдегида и фильтрат концентрируют. Очистку полученного остатка выполняют методом СДЖХ (диоксид кремния, 65% этилацетат/гексан), получая 3-[1-(ацетилокси)этил]-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентилацетат в виде смеси 8 диастереомеров. ЭСИ-МС: рассчитывают для C21H24F3NO6 443; найдено 444 (M+).
Стадия I:
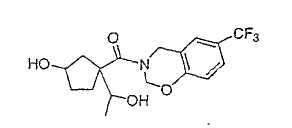
Раствор 3-[1-(ацетилокси)этил]-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентилацетата (5,50 г, 12,4 ммоль) в смеси (1:1) метанол/ТГФ (40 мл) при 0°C обрабатывают по капле раствором LiOH·H2O (520 мг, 12,4 ммоль) в деионизированной воде (20 мл). Полученную смесь перемешивают при температуре от -10 до 0°C в течение 1 час, затем концентрируют при к.т. Остаток распределяют между эфиром и насыщенным раствором соли. Водный слой экстрагируют второй раз эфиром и эфирные слои объединяют и сушат над безводным MgSO4, фильтруют и концентрируют. Анализ методами 1H-ЯМР и ЭСИ-МС показывает, что скорее получают диол, чем требуемый моноацетат, указывая на то, что гидролиз не является селективным. Полученный 3-(1-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанол используют без дополнительной очистки на следующей стадии. ЭСИ-МС: рассчитано для C17H20F3NO4 359; найдено 360 (M+).
Стадия J:

Предварительно охлажденный (-78°C) раствор оксалилхлорида (2,82 мл, 32,4 ммоль) в 150 мл ДХМ обрабатывают по капле раствором ДМСО (4,60 мл, 64,8 ммоль) в 25 мл ДХМ. Еще через 5 мин добавляют по капле раствор 3-(1-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанола (4,33 г, 10,8 ммоль) в 25 мл ДХМ. Реакционную смесь перемешивают еще в течение 30 мин, затем добавляют по капле чистый триэтиламин (18,1 мл, 130 ммоль). Смесь перемешивают еще в течение 10 мин, затем нагревают до к.т. и перемешивают в течение 1,5 час. Реакционную смесь выливают в 1 н. раствор HCl и полученную смесь экстрагируют ДХМ. Органический слой снова промывают 1 н. раствором HCl, затем насыщенным раствором NaHCO3 и в заключение насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 75% этилацетат/гексан) дает 3-ацетил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанон в виде смеси двух энантиомеров. ЭСИ-МС: рассчитано для C17H16F3NO4 355; найдено 356 (M+).
ПРИМЕР 5
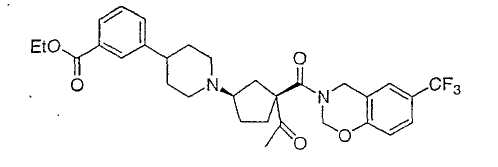
3-Ацетил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентанон (504 мг, 1,42 ммоль) объединяют с этил-3-пиперидин-4-илбензоатгидрохлоридом (670 мг, 2,49 ммоль), триэтиламином (347 мкл, 2,49 ммоль), 4Å порошкообразными молекулярными ситами (примерно 1 г) и триацетоксиборогидридом натрия (1,81 г, 8,52 ммоль) в 20 мл ДХМ. Полученную смесь перемешивают при к.т. в течение 4 дней. Реакционную смесь разбавляют ДХМ и промывают насыщенным раствором NaHCO3, затем насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом флэш-хроматографи (диоксид кремния, 5% метанол/ДХМ) дает требуемый продукт в виде смеси 4 изомеров. Разделение всех четырех изомеров можно осуществить последовательными введениями с применением двух хиральных ВЭЖХ условий (колонка ChiralCel OD, 2 см×25 см, 15% этанол/гексан, затем колонка ChiralPak AD, 2 см×25 см, 60% этанол/гексан; обе колонки от Chiral Technologies, Inc.). Первое разделение дает три пика, где 1-й (132 мг) и 3-й (131 мг) пики представляют отдельные энантиомеры и второй пик (315 мг) представляет смесь двух изомеров. Второе разделение (пика 2) дает два отдельных энантиомера из пиков 2-1 (98 мг) и 2-2 (116 мг). Обнаружено, что пик 3 из первых условий разделения содержит изомер, имеющий большую эффективность и, как предполагают по аналогии, является этил-3-[1-((1R,3S)-3-ацетил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоатом.
Изомер из пика 1 ЭСИ-МС: рассчитано для C31H35F3N2O5 572; найдено 573 (M+).
Изомер из пика 2-1 ЭСИ-МС: рассчитано для C31H35F3N2O5 572; найдено 573 (M+).
Изомер из пика 2-2 ЭСИ-МС: рассчитано для C31H35F3N2O5 572; найдено 573 (M+).
Изомер из пика 3 ЭСИ-МС: рассчитано для C31H35F3N2O5 572; найдено 573 (M+).
ПРИМЕР 6
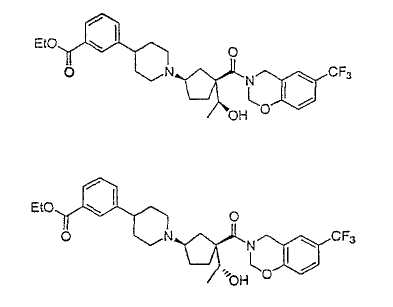
Охлажденный (0°C) раствор этил-3-[1-((1R,3S)-3-ацетил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоата (129 мг, 0,224 ммоль) в метаноле (5 мл) обрабатывают борогидридом натрия (17 мг, 0,45 ммоль), перемешивают в течение 35 мин и концентрируют. К остатку добавляют насыщенный раствор соли и данную смесь экстрагируют дважды ДХМ. Комбинированные органические слои сушат над безводным MgSO4, фильтруют и концентрируют. Разделение двух диастереомерных спиртовых продуктов, этил-3-[1-((1R,3S)-3-(1R-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоата и этил-3-[1-((1R,3S)-3-(1S-гидроксиэтил)-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоата, выполняют методом хиральной ВЭЖХ (колонка ChiralCel OD, 2 см×25 см, 10% этанол/гексан), получая быстрее элюируемый пик (пик 1) и медленнее элюируемый пик (пик 2).
Изомер из пика 1 ЭСИ-МС: рассчитано для C31H37F3N2O5 574; найдено 575 (M+).
Изомер из пика 2 ЭСИ-МС: рассчитано для C31H37F3N2O5 574; найдено 575 (M+).
ПРИМЕР 7
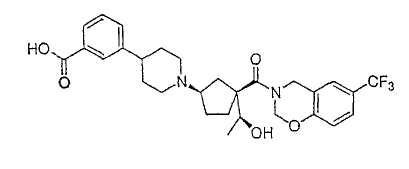
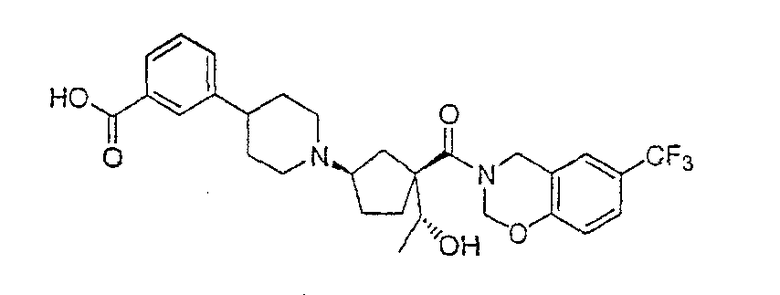
Раствор изомера из пика 1 разделения этил-3-[1-((1R,3S)-3-(1-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоата (12,5 мг, 0,0218 ммоль) в 1 мл этанола обрабатывают раствором LiOH·H2O (9 мг, 0,2 ммоль) в 1 мл деионизированной воды. Полученную смесь перемешивают при 50°C в течение 1 час, затем концентрируют. Очистка ВЭЖХ (YMC Pack Pro C18, 100×20 мм внутр. диам.) дает продукт в виде его TFA-соли. Его превращают в его HCl-соль, растворяя в ДХМ и добавляя избыток 1 н. HCl/эфир, затем концентрируют с получением 3-[1-((1R,3S)-3-(1-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]бензойной кислоты в виде одного изомера с неизвестной стереохимией по 1-гидроксиэтиловому стереоцентру. ЭСИ-МС: рассчитано для C29H33F3N2O5 546; найдено 547 (M+).
Аналогично изомер из пика 2 разделения этил-3-[1-((1R,3S)-3-(1-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]бензоата (20,2 мг, 0,0352 ммоль) превращают в 3-[1-((1R,3S)-3-(1-гидроксиэтил)-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]бензойную кислоту в виде одного изомера с неизвестной (но противоположной стереохимии изомера, полученного выше из пика 1) стереохимией по 1-гидроксиэтиловому стереоцентру. ЭСИ-МС: рассчитано для C29H33F3N2O5 546; найдено 547 (M+).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 8
Стадия A:
К охлажденному (0°C) раствору этаноламина (41,8 г, 0,685 моль) в воде (90 мл) добавляют по капле чистый (R)-пропиленоксид (4,97 г, 85,6 ммоль). Через 1 час при 0°C реакционной смеси дают нагреться до к.т. и перемешивают в течение ночи. Реакционную смесь концентрируют при температуре ˜80°C в вакууме для удаления воды и большей части этаноламина с получением сырого продукта, содержащего некоторое количество остаточного этаноламина. Данный материал используют без дополнительной очистки на стадии B.
Стадия B:
Диол, полученный на стадии A (11,8 г, сырой [˜86% чистоты], примерно 83 ммоль), растворяют в ДХМ (150 мл) и обрабатывают Boc2O (23,4 г, 107 ммоль) в ДХМ (75 мл) за 15 мин. Реакционную смесь перемешивают в течение выходных, концентрируют и очищают методом СДЖХ, элюируя 5% MeOH/EtOAc.
Стадия C:
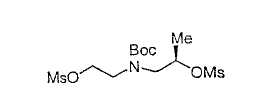
К раствору Boc-защищенного диола, полученного на стадии B (13,2 г, 60,3 ммоль), и триэтиламина (21,0 мл, 15,3 г, 151 ммоль) в ДХМ (150 мл) при 0°C добавляют по капле метансульфонилхлорид (9,56 мл, 14,1 г, 125 ммоль). Реакционную смесь затем перемешивают в течение 1,5 час, еще разбавляют ДХМ (100 мл) и промывают 3 н. HCl (250 мл). Водный слой снова экстрагируют ДХМ (200 мл) и органические слои объединяют и промывают 1 н. HCl (250 мл), насыщенным раствором NaHCO3 (250 мл) и насыщенным раствором соли (250 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют с получением сырого бис-мезилата, который используют немедленно. Если бис-мезилат не использован немедленно, то он претерпевает разложение.
Стадия D:
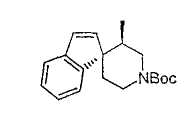
Инден (7,03 мл, 7,00 г, 60,3 ммоль) добавляют по капле за 4 мин к 1,0 M ТГФ-раствору LHMDS (127 мл, 127 ммоль) при 0°C. После перемешивания в течение дополнительных 30 мин, данный раствор переносят при помощи канюли в раствор бис-мезилата (22,6 г, 60,3 ммоль), полученный, как описано выше на стадии C, в ТГФ (75 мл) при 0°C. Смесь перемешивают в течение 2 час, нагревают до к.т. и перемешивают в течение ночи. Реакционную смесь частично концентрируют и затем распределяют между этилацетатом и водой. Водный слой снова экстрагируют этилацетатом и объединяют органические слои. Затем органическую фазу промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют с получением сырого продукта. Очистка методом СДЖХ при элюировании смесью 15% этилацетат/гексан дает пиперидин в виде смеси (˜3:1) транс и цис (определенных по данным 1H-ЯМР). Смесь кристаллизуют из горячего гексана с получением чистого транс-изомера (>20:1 по данным 1H-ЯМР ). 1H-ЯМР (400 МГц, CDCl3): δ 7,29 (дт, J=6,4, 1,6 Гц, 1H), 7,20 (м, 3H), 6,83 (д, J=6,0 Гц, 1H), 6,67 (д, J=5,6 Гц, 1H), 4,20 (шир.с, 2H), 2,97 (шир.т, J=3,2 Гц, 1H), 2,69 (шир.т, J=2,4 Гц, 1H), 2,16 (м, 1H), 2,07 (дт, J=4,4, 13,2 Гц, 1H), 1,49 (с, 9H), 1,25 (м, 1H), 0,31 (д, J=6,8 Гц, 3H).
Стадия E:
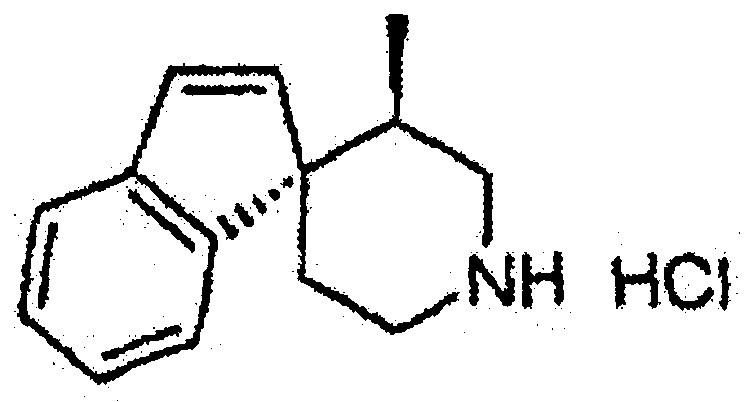
Boc-пиперидин, полученный на стадии D (4,35 г, 14,5 ммоль), растворяют в безводном 4 н. растворе HCl в диоксане и перемешивают при к.т. в течение 1 час. Реакционную смесь затем концентрируют, получая продукт.
EI-МС: рассчитано для C14H17N 199; найдено 200 (M)+.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 9
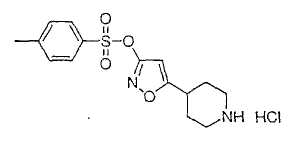
Стадия A:
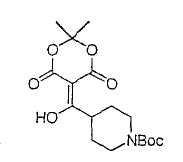
Охлажденный (0°C) раствор Boc-изонипекотовой кислоты (5,97 г, 26,0 ммоль) и кислоты Мелдрума (3,75 г, 26,0 ммоль) в ДМФА (54 мл) обрабатывают по капле диэтилцианофосфонатом (4,34 мл, 28,6 ммоль) и триэтиламином (11,2 мл, 80,7 ммоль). После перемешивания при 0°C в течение дополнительных 30 мин реакционной смеси дают нагреться до к.т. и перемешивают в течение 3 дней. Реакционную смесь концентрируют при пониженном давлении и добавляют эфир и 1 н. раствор HCl. Водный слой снова экстрагируют эфиром и эфирные слои объединяют и промывают 1 н. раствором HCl, дважды водой и в заключение насыщенным раствором соли. Затем сушат неэфирный слой над безводным MgSO4, фильтруют и концентрируют с получением трет-бутил-4-[(2,2-диметил-4,6-диоксо-1,3-диоксан-5-илиден)(гидрокси)метил]пиперидин-1-карбоксилата, который содержит примерно 10% Boc-изонипекотовой кислоты. 1H-ЯМР (CDCl3, 500 МГц): δ 4,24 (шир.м, 2H), 3,97 (тт, J=12, 3,5 Гц, 1H), 2,84 (шир.м, 2H), 1,83 (м, 2H), 1,76 (с, 6H), 1,69-1,75 (м, 2H), 1,48 (с, 9H).
Стадия B:

Раствор трет-бутил-4-[(2,2-диметил-4,6-диоксо-1,3-диоксан-5-илиден)(гидрокси)метил]пиперидин-1-карбоксилата (8,99 г, 25,3 ммоль) и трет-бутил N-(трет-бутоксикарбонилокси)карбамата (5,90 г, 25,3 ммоль) в толуоле (200 мл) перемешивают при 65°C в течение 14 час, затем при к.т. в течение 36 час. Реакционную смесь концентрируют и остаток очищают методом флэш-хроматографии (диоксид кремния, 25% этилацетат/гексан), получая трет-бутил-4-(3-{(трет-бутоксикарбонил)[(трет-бутоксикарбонил)окси]амино}-3-оксопропаноил)пиперидин-1-карбоксилат. Данные 1H-ЯМР-анализа согласуются с продуктом, но сложны из-за наличия ротамеров карбамата.
Стадия C:
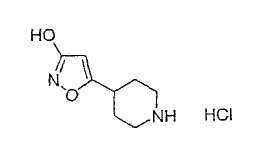
Раствор трет-бутил-4-(3-{(трет-бутоксикарбонил)[(трет-бутоксикарбонил)окси]амино}-3-оксопропаноил)пиперидин-1-карбоксилата (6,2 г, 12,7 ммоль) в 100 мл метанола обрабатывают 4 н. раствором HCl (150 мл) и полученную суспензию перемешивают в течение ночи при к.т. Утром реакционная смесь становится прозрачной. Реакционную смесь концентрируют. Так как сырой продукт нельзя легко очистить, то его используют как есть на следующей стадии. ЭСИ-МС: рассчитано для C8H12N2O2 168; найдено 169 (M+).
Стадия D:
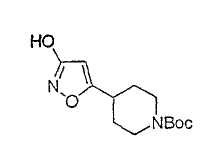
Раствор сырого 5-пиперидин-4-илизоксазол-3-олгидрохлорида (1,77 г, 8,65 ммоль) и 25 NaOH (346 мг, 8,65 ммоль) в смеси воды (15 мл) и диоксана (10 мл) обрабатывают по капле раствором ди-трет-бутилдикарбоната (3,78 г, 17,3 ммоль) в 10 мл диоксана. Полученную смесь перемешивают при к.т. в течение 6 час, затем частично концентрируют, удаляя диоксан. К полученной смеси добавляют 1 н. раствор HCl и эфир. Затем эфирный слой промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют. Требуется следующая серия очисток: СДЖХ (диоксид кремния, постадийный градиент от 75 до 100% этилацетат/гексан), СДЖХ (диоксид кремния, 50% этилацетат/гексан) и в заключение флэш-хроматография (диоксид кремния, 1/30/69 AcOH/этилацетат/гексан). Это дает требуемый трет-бутил-4-(3-гидроксиизоксазол-5-ил)пиперидин-1-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 5,68 (с, 1H), 4,15 (шир.м, 2H), 2,88 (шир.м, 2H), 2,83 (тт, J=11, 3,5 Гц, 1H), 1,99 (м, 2H), 1,62 (д.кв., J=4,0, 13,5 Гц, 2H), 1,49 (с, 9H).
Стадия E:
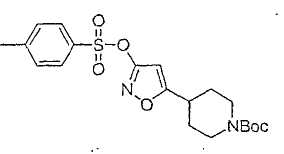
Охлажденный (0°C) раствор трет-бутил-4-(3-гидроксиизоксазол-5-ил)пиперидин-1-карбоксилата (493 мг, 1,84 ммоль) в 5 мл ТГФ обрабатывают толуолсульфонилхлоридом (386 мг, 2,02 ммоль), а затем триэтиламином (295 мкл, 2,12 ммоль). Полученную смесь нагревают до к.т. и перемешивают в течение 2 час. Реакционную смесь фильтруют и фильтрат концентрируют. Остаток очищают методом СДЖХ (диоксид кремния, 50% этилацетат/гексан), получая трет-бутил-4-(3-{[(4-метилфенил)сульфонил]окси}изоксазол-5-ил)пиперидин-1-карбоксилат.
1H-ЯМР (500 МГц, CDCl3): δ 7,87 (д, J=8,5 Гц, 2H), 7,39 (д, J=8,0 Гц, 2H), 6,06 (с, 1H), 4,15 (шир.м, 2H), 2,88 (м, 3H), 2,49 (с, 3H), 2,00 (дд, J=13, 2,5 Гц, 2H), 1,62 (д.кв., J=4,5, 12,5 Гц, 2H), 1,49 (с, 9H).
Стадия F:
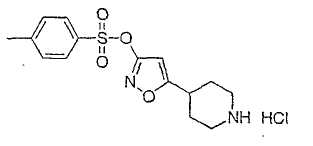
трет-Бутил-4-(3-{[(4-метилфенил)сульфонил]окси}изоксазол-5-ил)пиперидин-1-карбоксилат (719 мг, 1,76 ммоль) растворяют в безводном 4 н. HCl в диоксане (15 мл) и полученный раствор перемешивают при к.т. в течение 1 час. Реакционную смесь концентрируют, получая гидрохлорид 5-пиперидин-4-илизоксазол-3-ил-4-метилбензолсульфоната, который не требует дополнительной очистки. ЭСИ-МС: рассчитано для C15Н18N2O4S 322; найдено 323 (M+H).
ПРИМЕР 8
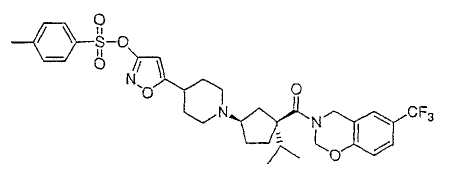
Смесь гидрохлорида 5-пиперидин-4-илизоксазол-3-ил-4-метилбензолсульфоната (182 мг, 0,507 ммоль), (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанона (93,3 мг, 0,263 ммоль), триэтиламина (71 мкл, 0,507 ммоль), 4Å порошкообразных молекулярных сит (˜100 мг) и триацетоксиборогидрида натрия (334 мг, 1,58 ммоль) перемешивают при к.т. в течение 6 дней (обычно достаточно 3 дней). Реакционную смесь разбавляют ДХМ и промывают насыщенным раствором NaHCO3, а затем насыщенным раствором соли. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистку и разделение изомеров цис-5-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]изоксазол-3-ил-4-метилбензолсульфоната и транс-5-[1-((1S,3S)-3-изопропил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]изоксазол-3-ил-4-метилбензолсульфоната выполняют методом препаративной ТСХ (диоксид кремния, элюент: смесь 40% ТГФ/гексан) с получением элюируемого в большей степени цис-изомера и элюируемого в меньшей степени транс-изомера. Анализ разделенных цис- и транс-изомеров продукта методом хиральной аналитической ВЭЖХ (колонка ChiralCel OD) показывает, что каждый является отдельным чистым изомером.
Элюируемый в большей степени цис-изомер: ЭСИ-МС, рассчитано для C33H38F3N3O6S 661; найдено 662 (M+H).
Элюируемый в меньшей степени транс-изомер: ЭСИ-МС, рассчитано для C33H38F3N3O6S 661; найдено 662 (M+H).
ПРИМЕР 9
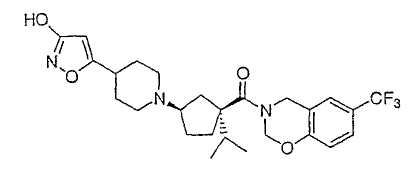
Раствор 5-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]изоксазол-3-ил-4-метилбензолсульфоната (97 мг, 0,15 ммоль) в смеси ТГФ (2 мл) и метанола (2 мл) обрабатывают раствором LiOH·H2O (61,5 мг, 1,47 ммоль) в деионизированной воде (2 мл). Полученную смесь перемешивают при к.т. в течение 1 час, затем концентрируют, добавляют к остатку насыщенный раствор соли и доводят pH до 7 1 н. раствором HCl. Данную смесь экстрагируют три раза хлороформом и объединенные органические слои сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом ВЭЖХ с обращенной фазой (YMC Pack Pro C18, 100 х 20 мм внутр. диам.) дает продукт в виде его TFA-соли. Данный продукт превращают в его HCl-соль, растворяя в ДХМ и добавляя избыток 1 н. HCl/эфир, затем концентрируя с получением гидрохлорида 5-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-ил]изоксазол-3-ола.
ЭСИ-МС: рассчитано для C26H32F3N3O4 507; найдено 508 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 10
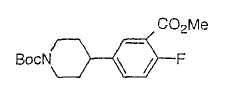
Стадия A:
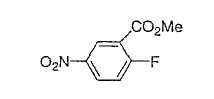
Тионилхлорид (16 мл, 0,22 моль) добавляют по капле к охлажденному (0°C) метанолу (250 мл). После добавления полученный безводный метанольный раствор HCl выливают в колбу, содержащую 2-фтор-5-нитробензойную кислоту (13,77 г, 74,39 ммоль). Полученную таким образом смесь перемешивают в течение ночи при к.т. Реакционную смесь концентрируют и собранный остаток очищают методом флэш-хроматографии с применением градиента [диоксид кремния, 5-30% (700 мл), затем 30-50% (1 л)], получая метил-2-фтор-5-нитробензоат.
Стадия B:
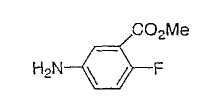
Смесь метил-2-фтор-5-нитробензоата (14,52 г, 72,92 ммоль) и Pd/C (10%, тип Degussa, 726 мг) в метаноле перемешивают при давлении Н2 60 фунт/дюйм2 (4,219 кг/см2), используя аппарат Парра в течение 3 час. Реакционную смесь фильтруют и фильтрат концентрируют с получением метил-5-амино-2-фторбензоата.
Стадия C:
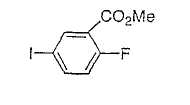
Метил-5-амино-2-фторбензоат (5,21 г, 30,8 ммоль) добавляют к раствору концентрированной серной кислоты (45 мл) в воде (110 мл). Полученную смесь нагревают при 80°C в течение 20 мин, затем охлаждают до 0°C и обрабатывают по капле раствором NaNO2 (2,34 г, 33,9 ммоль) в 10 мл воды. После перемешивания в течение 30 мин добавляют раствор KI (7,16 г, 43,1 ммоль) в 20 мл воды. Затем реакционную смесь нагревают до к.т. и перемешивают в течение ночи в атмосфере азота. Далее реакционную смесь перемешивают при 70°C в течение 15 мин, охлаждают до к.т. и дважды экстрагируют эфиром. Объединенные эфирные слои промывают четыре раза водой, один раз насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом флэш-хроматографии (диоксид кремния, 10% этилацетат/гексан) дает метил-2-фтор-5-йодбензоат.
1H-ЯМР (500 МГц, CDCl3): δ 8,25 (дд, 1H), 7,82 (м, 1H), 6,94 (дд, 1H), 3,95 (с, 3H). ЭСИ-МС: рассчитано для C8H6FIO2 280; найдено 281 (M+H).
Стадия D:
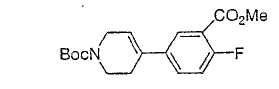
Метил-2-фтор-5-йодбензоат (5,01 г, 17,9 ммоль) объединяют с LiCl (1,52 г, 35,7 ммоль) и трет-бутил-4-(триметилстаннил)-3,6-дигидропиридин-1(2Н)-карбоксилатом (4,28 г, 12,3 ммоль) в 100 мл ДМФА. После перемешивания в течение 20 мин в атмосфере азота добавляют Pd2dba3 (218 мг, 0,238 ммоль), P(2-фурил)3 (276 мг, 1,19 ммоль), реакционную смесь нагревают до 90°C в атмосфере азота и перемешивают в течение 1 час 45 мин. Далее реакционную смесь перемешивают при к.т. в течение ночи. Затем температуру реакции снова доводят до 90°C в течение 1 час. К смеси добавляют насыщенный раствор NaHCO3 и насыщенный раствор соли. Затем полученную водную смесь экстрагируют три раза эфиром. Эфирные слои объединяют и промывают четыре раза водой, сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, градиент 5-25% этилацетат/гексан, при объеме 2 л) дает трет-бутил-4-[4-фтор-3-(метоксикарбонил)фенил]-3,6-дигидропиридин-1(2Н)-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 7,94 (дд, J=7,0, 2,5 Гц, 1H), 7,53 (м, 1H), 7,13 (дд, J=10, 8,5 Гц, 1H), 6,07 (шир.с, 1H), 4,10 (шир.с, 2H), 3,96 (с, 3H), 3,66 (т, 6 Гц, 2H), 2,53 (шир.м, 2H), 1,52 (с, 9H). ЭСИ-МС: рассчитано для C18H22FNO4 335; найдено 236 (M-Boc+H).
Стадия E:
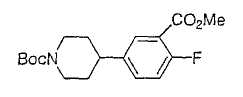
Смесь трет-бутил-4-[4-фтор-3-(метоксикарбонил)фенил]-3,6-дигидропиридин-1(2Н)-карбоксилата (2,11 г, 6,29 ммоль) и 10% Pd/C (525 мг) в этаноле (60 мл) перемешивают в атмосфере водорода с применением баллона, заполненного водородом, в течение 6 час. Реакционную смесь фильтруют и фильтрат концентрируют. 1H-ЯМР-анализ показывает, что остается примерно 5% исходного олефина, поэтому смесь повторно подвергают действию указанных выше условий в течение 4 час. Фильтрование и концентрирование фильтрата дают трет-бутил-4-[4-фтор-3-(метоксикарбонил)фенил]пиперидин-1-карбоксилат, который используют без дополнительной очистки. 1H-ЯМР (500 МГц, CDCl3): δ 7,78 (дд, J=6,5, 2,0 Гц, 1H), 7,37 (м, 1H), 7,10 (дд, J=10,5, 8,0 Гц, 1H), 4,27 (шир.м, 2H), 3,95 (с, 3H), 2,82 (шир.м, 2H), 2,69 (тт, J=12,5, 3,5 Гц, 1H), 1,83 (шир.д, J=13 Гц, 2H), 1,62 (д.кв., J=4,5, 13 Гц, 2H), 1,50 (с, 9H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 11
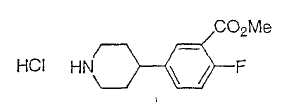
трет-Бутил-4-[4-фтор-3-(метоксикарбонил)фенил]пиперидин-1-карбоксилат (471 мг, 1,40 ммоль) растворяют в безводном 4 н. HCl в диоксане (5 мл) и перемешивают при к.т. в течение 40 мин. Реакционную смесь концентрируют, получая гидрохлорид метил-2-фтор-5-пиперидин-4-илбензоата, который используют без дополнительной очистки. ЭСИ-МС: рассчитано для C13H16FNO2 237; найдено 238 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 12
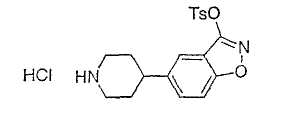
Стадия A:
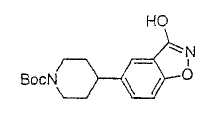
Ацетоксигидроксамовую кислоту (334 мг, 4,45 ммоль) объединяют с трет-бутилатом калия (499 мг, 4,45 ммоль) в 10 мл ДМФА. Полученную суспензию перемешивают при к.т. в течение 43 мин. Затем добавляют раствор трет-бутил-4-[4-фтор-3-(метоксикарбонил)фенил]пиперидин-1-карбоксилата (1,00 г, 2,96 ммоль) в 5 мл ДМФА. Реакционную смесь перемешивают при к.т. в течение ночи. ВЭЖХ-МС анализ показывает, что присутствует небольшое количество продукта и что исходное вещество составляет основную часть материала. Ацетоксигидроксамовую кислоту (670 мг, 8,9 ммоль) объединяют с трет-бутилатом калия (980 мг, 8,9 ммоль) в 10 мл ДМФА. Полученную суспензию перемешивают при к.т. в течение 30 мин. Затем данную смесь добавляют к реакционной смеси и полученную суспензию перемешивают при 50°C в течение 1 час 20 мин. Температуру поднимают до 75°C и перемешивают смесь еще 3 час. Затем температуру поднимают до 90°C и перемешивают смесь в течение 6 час. Затем температуру поднимают до 100°C и перемешивают смесь в течение 6 час. Так как взаимодействие еще не проходит до завершения, ацетоксигидроксамовую кислоту (670 мг, 8,9 ммоль) снова объединяют с трет-бутилатом калия (980 мг, 8,9 ммоль) в 10 мл ДМФА. Полученную суспензия перемешивают при к.т. в течение 30 мин. Затем данную смесь добавляют к реакционной смеси и полученную суспензию перемешивают при 100°C в течение 16 час. Хотя остается примерно 20% исходного материала, реакционную смесь разбавляют эфиром и промывают 1 н. раствором HCl. Проводят обратную экстракцию водного слоя и эфирные слои объединяют и промывают дважды водой и один раз насыщенным раствором соли. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 70% этилацетат/гексан) дает трет-бутил-4-(3-гидрокси-1,2-бензизоксазол-5-ил)пиперидин-1-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 7,62 (д, J=1,5 Гц, 1H), 7,49 (дд, J=8,5, 1,5 Гц, 1H), 7,37 (д, J=9,0 Гц, 1H), 4,30 (шир.м, 2H), 2,85 (шир.м, 2H), 2,80 (тт, J=12,5, 3,5 Гц, 1H), 1,89 (шир.д, J=12,5 Гц, 2H), 1,68 (д.кв., J=4,0, 12,5 Гц, 2H), 1,52 (с, 9H).
Стадия B:
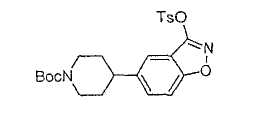
Охлажденный (0°C) раствор трет-бутил-4-(3-гидрокси-1,2-бензизоксазол-5-ил)пиперидин-1-карбоксилата (286 мг, 0,898 ммоль) в ТГФ (5 мл) обрабатывают толуолсульфонилхлоридом (188 мг, 0,988 ммоль), а затем триэтиламином (144 мкл, 1,03 ммоль). Реакционной смеси дают нагреться до к.т. и перемешивают в течение 1,5 час. Реакционную смесь фильтруют и концентрируют фильтрат. Очистка методом СДЖХ (диоксид кремния, 50% этилацетат/гексан) дает трет-бутил-4-(3-{[(4-метилфенил)сульфонил]окси}-1,2-бензизоксазол-5-ил)пиперидин-1-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 7,94 (д, J=8,5 Гц, 2H), 7,47 (с, 3H), 7,41 (д, J=8,0 Гц, 2H), 4,31 (шир.м, 2H), 2,85 (шир.м, 2H), 2,80 (тт, J=12, 3,5 Гц), 2,50 (с, 3H), 1,87 (шир.д, J=13 Гц, 2H), 1,64 (д.кв., J=4,5,13 Гц, 2H), 1,52 (с, 9H). ЭСИ-МС: рассчитано для C24H28N2O6S 472; найдено 473 (M+H).
Стадия C:
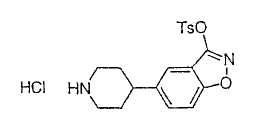
трет-Бутил-4-(3-{[(4-метилфенил)сульфонил]окси}-1,2-бензизоксазол-5-ил)пиперидин-1-карбоксилат (285 мг, 0,603 ммоль) растворяют в безводном 4 н. HCl в диоксане (6 мл) и полученную смесь перемешивают при к.т. в течение 1 час. Реакционную смесь концентрируют, получая гидрохлорид 5-пиперидин-4-ил-1,2-бензизоксазол-3-ил-4-метилбензолсульфоната, который не требует дополнительной очистки. ЭСИ-МС: рассчитано для C19H20N2O4S 372; найдено 373 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 13
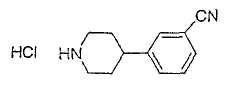
Стадия A:
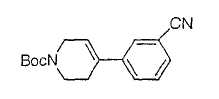
К смеси трет-бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2H)-карбоксилата (полученного согласно работе Wustrow, D.J., Wise, L.D., Synthesis, (1991), 993-995; 3,24 г, 9,77 ммоль), 3-цианофенилбороновой кислоты (2,01 г, 13,7 ммоль), хлорида лития (1,23 г, 29,3 ммоль) и 2 M раствора Na2CO3 (14 мл) в ДМЭ (35 мл) добавляют Pd(PPh3)4 (564 мг, 0,489 ммоль) и полученную смесь перемешивают при кипении с обратным холодильником в атмосфере азота в течение 3,5 час. Реакционную смесь охлаждают до к.т., перемешивают в течение ночи, затем частично концентрируют для удаления большей части ДМЭ. К оставшейся водной смеси добавляют ДХМ, 2 M раствор Na2CO3 и ˜6 мл 28% раствор NH4OH. Слои разделяют и водный слой снова экстрагируют ДХМ. Объединенные органические слои сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, элюент: смесь 45% этилацетат/гексан) дает трет-бутил-4-(3-цианофенил)-3,6-дигидропиридин-1(2Н)-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 7,66 (с, 1H), 7,61 (дд, J=8,0, 1,5 Гц, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,46 (т, J=7,5 Гц, 1H), 6,13 (шир.с, 1H), 4,12 (м, 2H), 3,68 (т, J=5,5 Гц, 2H), 2,53 (шир.с, 2H), 1,52 (с, 9H).
Стадия B:
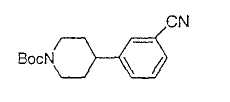
трет-Бутил-4-(3-цианофенил)-3,6-дигидропиридин-1(2Н)-карбоксилат (1,00 г, 3,52 ммоль) объединяют с 20% Pd(OH)2/C (1 г) в 25 мл метанола. Данную смесь перемешивают в атмосфере водорода с применением заполненного водородом баллона в течение 5 час. Реакционную смесь фильтруют через набивку из целита и фильтрат концентрируют. Очистка методом СДЖХ (диоксид кремния, 38% этилацетат/гексан) дает трет-бутил-4-(3-цианофенил)пиперидин-1-карбоксилат. ЭСИ-МС: рассчитано для C17H22N2O2 286; найдено 309 (M+Na+).
Стадия C:
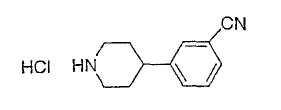
Раствор трет-бутил-4-(3-цианофенил)пиперидин-1-карбоксилата (376 мг, 1,31 ммоль) в 10 мл смеси (1:1) TFA/ДХМ перемешивают при к.т. в течение 1,25 час. Реакционную смесь концентрируют. Очистка методом препаративной ТСХ (диоксид кремния, 1% раствора NH4OH (28%)/9% метанол/ДХМ) дает 3-пиперидин-4-илбензонитрил в виде белого твердого вещества.
ЭСИ-МС: рассчитано для C12Н14N2 186; найдено 187 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 14
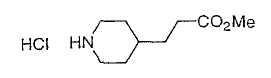
Тионилхлорид (2,23 мл, 30,6 ммоль) добавляют по капле к 25 мл безводного метанола. Полученный безводный метанольный раствор HCl добавляют к коммерчески доступной 3-[1-(трет-бутоксикарбонил)пиперидин-4-ил]пропионовой кислоте (1,97 г, 7,66 ммоль) и полученную смесь перемешивают в течение ночи при к.т. Растворитель удаляют при пониженном давлении, получая гидрохлорид метил-3-пиперидин-4-илпропаноата. ЭСИ-МС: рассчитано для C9H17NO2 171; найдено 172 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 15
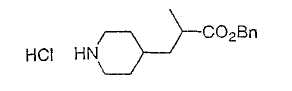
Стадия A:
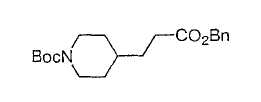
Бензилбромид (2,29 мл, 19,3 ммоль) добавляют по капле к охлажденной (0°C) смеси коммерчески доступной 3-[1-(трет-бутоксикарбонил)пиперидин-4-ил]пропионовой кислоты (4,96 г, 19,3 ммоль) и K2CO3 (6,67 г, 48,3 ммоль) в 40 мл ДМФА. Реакционной смеси дают нагреться до к.т. и перемешивают в течение 3 дней (взаимодействие, по-видимому, завершается только через несколько часов). Реакционную смесь разбавляют эфиром и промывают четыре раза водой, затем один раз насыщенным раствором соли. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 30% этилацетат/гексан) дает трет-бутил-4-[3-(бензилокси)-3-оксопропил]пиперидин-1-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 7,38 (м, 5Н), 5,14 (с, 2H), 4,09 (шир.м, 2H), 2,65 (м, 2H), 2,41 (т, J=7,5 Гц, 2H), 1,63 (м, 4H), 1,47 (с, 9H), 1,40 (м, 1H), 1,10 (д.кв., J=4,0, 12 Гц, 2H).
Стадия B:
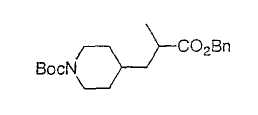
К охлажденному (-78 °C) раствору 1,5 M LDA/циклогексан (6,48 мл, 9,73 ммоль) в 50 мл ТГФ добавляют по капле в атмосфере азота раствор трет-бутил-4-[3-(бензилокси)-3-оксопропил]пиперидин-1-карбоксилата (2,60 г, 7,48 ммоль) в 20 мл ТГФ. Полученную смесь перемешивают при -78°C в течение 45 мин, затем обрабатывают по капле чистым йодметаном (1,40 мл, 22,4 ммоль). После перемешивания при -78°C в течение дополнительных 30 мин реакционной смеси дают нагреться до к.т. и перемешивают в течение ночи. Реакционную смесь разбавляют эфиром и промывают последовательно 1 н. раствором HCl, насыщенным раствором NaHCO3 и насыщенным раствором соли. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 25% этилацетат/гексан) дает трет-бутил-4-[3-(бензилокси)-2-метил-3-оксопропил]пиперидин-1-карбоксилат. 1H-ЯМР (500 МГц, CDCl3): δ 7,38 (м, 5Н), 5,17 (д, J=12 Гц, 1H), 5,12 (д, J=12 Гц, 1H), 4,06 (шир.м, 2H), 2,62 (м, 3H), 1,69 (м, 2H), 1,56 (м, 1H), 1,47 (с, 9H), 1,32 (м, 2H), 1,19 (д, J=7,5 Гц, 3H), 1,06 (м, 2H).
Стадия C:
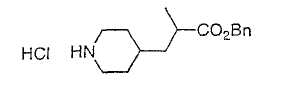
трет-Бутил-4-[3-(бензилокси)-2-метил-3-оксопропил]пиперидин-1-карбоксилат растворяют в 5 мл безводного 4 н. HCl в диоксане и перемешивают при к.т. в течение 1 час. Реакционную смесь концентрируют, получая бензил 2-метил-3-пиперидин-4-илпропаноат. ЭСИ-МС: рассчитано для C16H23NO2 261; найдено 262 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 16
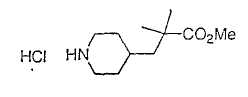
Стадия A:
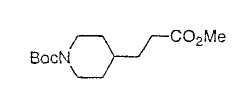
Йодметан (556 мкл, 8,93 ммоль) добавляют по капле к охлажденной (0°C) смеси коммерчески доступной 3-[1-(трет-бутоксикарбонил)пиперидин-4-ил]пропионовой кислоты (2,09 г, 8,12 ммоль) и K2CO3 (2,81 г, 20,3 ммоль) в 20 мл ДМФА. Реакционной смеси дают нагреться до к.т. и перемешивают в течение ночи. Реакционную смесь разбавляют эфиром и промывают четыре раза водой, затем один раз насыщенным раствором соли. Эфирный слой сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 40% этилацетат/гексан) обеспечивает трет-бутил-4-(3-метокси-3-оксопропил)пиперидин-1-карбоксилат.
Стадия B:
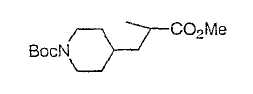
К предварительно охлажденному (-78°C) раствору 1,0 M натрийбис(триметилсилил)амида в ТГФ (13,1 мл, 13,1 ммоль) добавляют по капле в атмосфере азота раствор трет-бутил-4-(3-метокси-3-оксопропил)пиперидин-1-карбоксилата (1,78 г, 6,56 ммоль) в 11 мл ТГФ. После перемешивания еще в течение 25 мин добавляют по капле чистый йодметан (1,23 мл, 19,7 ммоль) и реакционную смесь перемешивают при -78°C в течение еще двух часов. Затем реакционную смесь выливают в 1 н. раствор HCl и полученную смесь экстрагируют дважды эфиром. Комбинированные эфирные слои промывают насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 40% этилацетат/гексан) дает трет-бутил-4-(3-метокси-2-метил-3-оксопропил)пиперидин-1-карбоксилат. ЭСИ-МС: рассчитано для C15H27NO4 285; найдено 186 (M-Boc+H).
Стадия C:
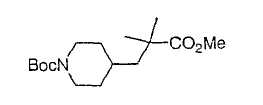
Охлажденный (-78°C) раствор 2,0 M LDA в ТГФ/гексане (Aldrich, 4,72 мл, 9,43 ммоль) в 9 мл ТГФ обрабатывают по капле раствором трет-бутил-4-(3-метокси-2-метил-3-оксопропил)пиперидин-1-карбоксилата (1,35 г, 4,72 ммоль) в 12 мл ТГФ. После перемешивания еще в течение 25 мин добавляют чистый йодметан (1,18 мл, 18,9 ммоль) и реакционную смесь выдерживают при -78°C в течение 2 час. Реакционную смесь выливают на 1 н. раствор HCl и полученную смесь экстрагируют дважды эфиром. Объединенные эфирные слои промывают насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют. Очистка методом СДЖХ (диоксид кремния, 30% этилацетат/гексан) дает трет-бутил-4-(3-метокси-2,2-диметил-3-оксопропил)пиперидин-1-карбоксилат в виде масла, которое кристаллизуется при стоянии в виде светло-желтого твердого вещества. 1H-ЯМР (500 МГц, CDCl3): δ 4,03 (шир.м, 2H), 3,68 (с, 3H), 2,67 (м, 2H), 1,54 (шир.м, 5Н), 1,47 (с, 9H), 1,20 (с, 6H), 1,11 (м, 2H).
Стадия D:
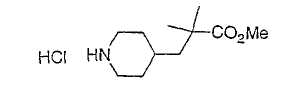
трет-Бутил-4-(3-метокси-2,2-диметил-3-оксопропил)пиперидин-1-карбоксилат (1,40 г, 4,67 ммоль) растворяют в безводном 4 н. HCl в диоксане (10 мл) и полученный раствор перемешивают при к.т. в течение 1 час. Реакционную смесь концентрируют, получая гидрохлорид метил-2,2-диметил-3-пиперидин-4-илпропаноата. ЭСИ-МС: рассчитано для C11H21NO2 199; найдено 200 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 17
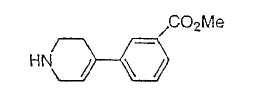
Стадия A:
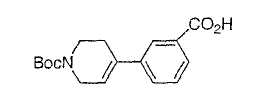
Проводят взаимодействие 3-(дигидроксиборан)бензойной кислоты (4,65 г, 28,0 ммоль) с трет-бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2H)-карбоксилатом (6,65 г, 20,0 ммоль), аналогично описанному ранее синтезу трет-бутил-4-[3-(этоксикарбонил)фенил]-3,6-дигидропиридин-1(2Н)-карбоксилата (промежуточный продукт 5, стадия A), с получением 3-[1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил]бензойной кислоты.
Стадия B:
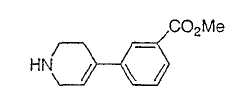
Тионилхлорид (2,39 мл, 32,7 ммоль) добавляют по капле к 15 мл безводного метанола. Полученный безводный метанольный раствор HCl добавляют к 3-[1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил]бензойной кислоте (3,31 г, 10,9 ммоль) и реакционную смесь перемешивают при к.т. в течение 6 час. Растворитель удаляют при пониженном давлении и остаток очищают методом флэш-хроматографии (диоксид кремния, постадийный градиент 5-10% с 1% инкрементами 10% NH4OH (28% раствор)/метанол в ДХМ) с получением метил-3-(1,2,3,6-тетрагидропиридин-4-ил)бензоата. ЭСИ-МС: рассчитано для C13H15NO2 217; найдено 218 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 18
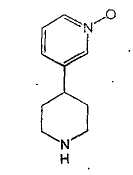
Стадия A:
трет-Бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2H)-карбоксилат (полученный согласно Wustrow, D.J., Wise, L.D., Synthesis, (1991), 993-995; 2,73 г, 8,22 ммоль) растворяют в 80 мл NMP. Затем добавляют в реакционный сосуд трифениларсин (220 мг, 0,66 ммоль), LiCl (1,09 г, 24,7 ммоль) и трис(дибензилиденацетон)дипалладий(0) (156 мг, 0,160 ммоль). Через 10 мин постоянного перемешивания добавляют трибутилстаннилпиридин (4,0 г, 9,7 ммоль) в 10 мл NMP, применяя шприц. Реакционный сосуд повторно откачивают и продувают газообразным N2 (4x). Затем реакционную смесь перемешивают при к.т. в течение 30 мин, потом при 80°C в течение 2,5 час и затем при 65°C в течение 16 час. Потом добавляют 20 мл 1 M раствора KF. Смесь перемешивают в течение 1,5 час и затем разбавляют этилацетатом (100 мл) и фильтруют. Фильтрат дополнительно разбавляют 200 мл 1 M раствора KF и 200 мл этилацетата. Органические и водные слои разделяют и снова экстрагируют водный слой один раз этилацетатом (200 мл). Органические слои объединяют, промывают водой (6×200 мл) и затем насыщенным раствором соли (1×300 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. трет-Бутил-3',6'-дигидро-3,4'-бипиридин-1'(2'H)-карбоксилат получают методом колоночной флэш-хроматографии на силикагеле (1% MeOH в EtOAc). ЭСИ-МС: рассчитано для C15H20N2O2 260,35; найдено 261 (M+H).
Стадия B:
трет-Бутил-3',6'-дигидро-3,4'-бипиридин-1'(2'H)-карбоксилат, полученный на стадии A (1,14 г, 4,38 ммоль), растворяют в 20 мл этанола и добавляют в колбу, содержащую гидроксид палладия на порошкообразном угле (20% Pd). На реакционную смесь воздействуют газообразным Н2 при давлении 50 фунт/дюйм2 (3,515 кг/см2) в течение 6,5 час при энергичном встряхивании. Далее реакционную смесь фильтруют и концентрируют при пониженном давлении. Очистка методом колоночной флэш-хроматографии (силикагель, градиентное элюирование 0-1% MeOH/EtOAc) дает трет-бутил-4-пиридин-3-илпиперидин-1-карбоксилат. ЭСИ-МС: рассчитано для C15H22N2O2 262,37; найдено 263 (M+H).
Стадия C:
m-CPBA (1,12 г, 6,48 ммоль) добавляют к раствору трет-бутил-4-пиридин-3-илпиперидин-1-карбоксилата (850 мг, 3,24 ммоль) в 50 мл CH2Cl2. Реакционную смесь перемешивают при к.т. в атмосфере азота в течение 20 час. Затем реакционную смесь разбавляют дихлорметаном (75 мл) и промывают раствором сульфита натрия (1×100 мл), насыщенным раствором бикарбоната натрия (1×100 мл) и насыщенным раствором соли (1×100 мл). Потом продукт сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Требуемый продукт получают и используют в последующей реакции без дополнительной очистки. ЭСИ-МС: рассчитано для C15H22N2O3 278,35; найдено 223 (M-t-Bu+H).
Стадия D:
N-Оксид, полученный на стадии C (868 мг, 3,12 ммоль), растворяют в 4 н. растворе HCl/диоксан и перемешивают при к.т. в течение 1 час перед концентрированием, получая требуемый амин в виде HCl-соли. ЭСИ-МС: рассчитано для C10H14N2O 178,23; найдено 179 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 19
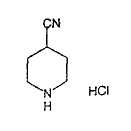
Стадия A:
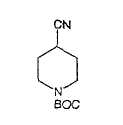
Изонипекотамид (10,2 г, 79,6 ммоль) растворяют в 37,0 мл оксида фосфора. Реакционную смесь нагревают при кипении с обратным холодильником в течение 4 час. После этого реакционную смесь частично концентрируют при пониженном давлении, выливают в колбу, содержащую лед, и обрабатывают 5 M раствором NaOH до pH 12. Добавляют раствор ди-трет-бутилдикарбоната (20,8 г, 95,5 ммоль) и диоксана (40 мл) и реакционную смесь перемешивают при к.т. в атмосфере азота в течение 16 час. Затем реакционную смесь экстрагируют этилацетатом (3×200 мл). Органический слой промывают один раз насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом СДЖХ (силикагель, 0-15% этилацетат/гексан) дает требуемый трет-бутил-4-цианопиперидин-1-карбоксилат в виде белого кристаллического твердого вещества.
Стадия B:
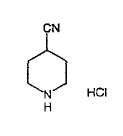
трет-Бутил-4-цианопиперидин-1-карбоксилат (4,87 г, 23,2 ммоль) растворяют в 4 н. растворе HCl/диоксан (30 мл) и перемешивают при к.т. Через 45 мин исходный материал больше не присутствует (по данным ТСХ), и реакционную смесь концентрируют. Продукт получают в виде HCl-соли.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 20
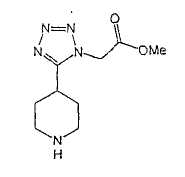
Стадия A:
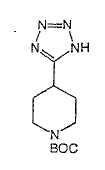
трет-Бутил-4-цианопиперидин-1-карбоксилат (6,98 г, 33,2 ммоль) растворяют в 150 мл толуола и обрабатывают триметилоловоазидом (10,0 г, 48,6 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником и перемешивают в атмосфере азота в течение 4 дней. О завершении взаимодействия судят по ТСХ и затем концентрируют реакционную смесь при пониженном давлении. Смесь растворяют в 200 мл этилацетата и охлаждают до 0°C. Затем барботируют раствор газообразным HCl в течение 10 мин. Еще через 2 час перемешивания при к.т. реакционную смесь концентрируют при пониженном давлении и повторно суспендируют в этилацетате до фильтрования. Отфильтрованные твердые продукты собирают и сушат в вакууме, получая требуемый трет-бутил-4-(1Н-тетраазол-5-ил)пиперидин-1-карбоксилат. ЭСИ-МС: рассчитано для C11H19N5O2 253,30; найдено 254 (M+H).
Стадия B:
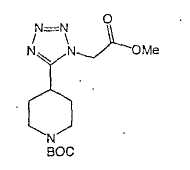
Трифенилфосфин (1,28 г, 4,89 ммоль), DEAD (804 мкл, 4,89 ммоль) и метилгликолат (378 мкл, 4,89 ммоль) добавляют к раствору трет-бутил-4-(1Н-тетраазол-5-ил)пиперидин-1-карбоксилата (619,7 мг, 2,447 ммоль) в дихлорметане (20 мл). Реакционную смесь перемешивают в течение 16 час при к.т. и затем концентрируют при пониженном давлении. Очистка методом СДЖХ (силикагель, 10-50% EtOAc/гексан) дает трет-бутил-4-[1-(2-метокси-2-оксоэтил)-1Н-тетраазол-5-ил]пиперидин-1-карбоксилат. ЭСИ-МС: рассчитано для C14H23N5O4 325,36; найдено 348 (M+Na).
Стадия C:
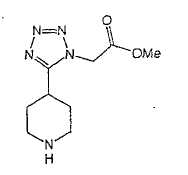
трет-Бутил-4-[1-(2-метокси-2-оксоэтил)-1Н-тетраазол-5-ил]пиперидин-1-карбоксилат (432,7 мг, 1,330 ммоль) растворяют в 4 н. растворе HCl/диоксан (30 мл) и перемешивают при к.т. в течение 4 час перед концентрированием. Продукт получают в виде HCl-соли. ЭСИ-МС: рассчитано для C9H15N5O2 225,25; найдено 226 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 21
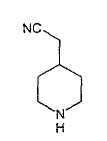
Стадия A:
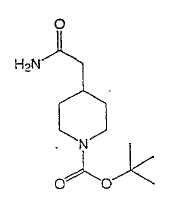
Триэтиламин (3,15 мл, 22,6 ммоль) и изобутилхлорформиат (2,93 мл, 22,6 ммоль) добавляют к охлажденному (0°C) раствору BOC-пиперидинуксусной кислоты (5,0 г, 21 ммоль) в 125 мл дихлорметана. Реакционной смеси дают нагреться до к.т. и перемешивают в течение 15 мин в атмосфере азота. ТСХ показывает полное расходование исходного материала. Непосредственно реакционную смесь барботируют газообразным аммиаком в течение 5 мин. Затем реакционную смесь разбавляют дихлорметаном (100 мл) и промывают раствором бикарбоната натрия (1×100 мл), 1 н. раствором HCl (1×100 мл) и насыщенным раствором соли (1×100 мл). Органические слои объединяют, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом СДЖХ (силикагель, 20-100% EtOAc/гексан) дает требуемый продукт амид. ЭСИ-МС: рассчитано для С12Н22N2O3 242,31; найдено 265 (M+Na).
Стадия B:
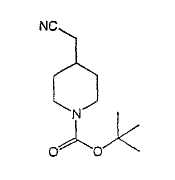
Соединение, полученное на стадии А, растворяют в тетрагидрофуране (50 мл), помещают в атмосферу азота и охлаждают до 0°C. Пиридин (3,9 мл, 48 ммоль) и трифторуксусный ангидрид (13,5 мл, 95,6 ммоль) добавляют по капле к охлажденному раствору. Реакционную смесь нагревают до к.т. и перемешивают в течение 5 час. Затем реакционную смесь разбавляют диэтиловым эфиром (100 мл), промывают 1 н. раствором HCl (1×100 мл), раствором бикарбоната натрия (1×100 мл) и насыщенным раствором соли (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка методом СДЖХ (силикагель, 10-100% EtOAc/гексан) дает требуемый нитрил. ЭСИ-МС: рассчитано для C12Н20N2O2 224,30; найдено 169 (M-t-Bu+H). 1H-ЯМР (500 МГц, CDCl3): δ 4,17 (с, 2H), 2,73 (с, 2H), 2,33 (д, J=6,0 Гц, 2H), 1,81 (д, J 10 =13,0 Гц, 2H), 1,47 (с, 9 H), 1,28 (д, J=12,5, 2H).
Стадия C:
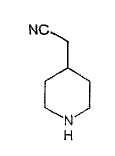
Нитрил, полученный на стадии B (3,12 г, 13,9 ммоль), растворяют в 4 н. растворе HCl/диоксан (100 мл) и перемешивают при к.т. в течение 2 час до концентрирования. Продукт получают в виде HCl-соли. ЭСИ-МС: рассчитано для C7H12N2 124,18; найдено 125 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 22

Стадия A:

Метансульфонилхлорид (4,2 мл, 55 ммоль), триэтиламин (10 мл, 75 ммоль) и 4-диметиламинопиридин (˜10 мг) добавляют к охлажденному (0°C) раствору BOC-защищенного 4-гидроксипиперидина в 200 мл метиленхлорида. Реакционную смесь перемешивают при 0°C в течение 1 час. Затем смесь промывают раствором бикарбоната натрия (2×100 мл) и насыщенным раствором соли (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, получая трет-бутил-4-[(метилсульфонил)окси]пиперидин-1-карбоксилат, который используют без дополнительной очистки. ЭСИ-МС: рассчитано для C11H21NO5S 279,35; найдено 302 (M+Na).
Стадия B:

Добавляют гидрид натрия (519 мг, 21,6 ммоль) к охлажденному (0°C) раствору соединения, полученного на стадии A (4,03 г, 14,4 ммоль), и метил-4-имидазолкарбоксилата (2,0 г, 16 ммоль) в 100 мл ДМФА. Реакционной смеси дают нагреться до к.т. Через 16 час при к.т. не наблюдают требуемого продукта и реакционную смесь нагревают до 50°C в течение 163 час. Хотя остается существенное количество исходного материала (промежуточный мезил), реакционную смесь разбавляют 150 мл диэтилового эфира, промывают водой (5×150 мл) и насыщенным раствором соли (1×150 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом СДЖХ (силикагель, 0-60% EtOAc/гексан) дает требуемый продукт и извлеченный исходный материал. ЭСИ-МС: рассчитано для C15H23N3O4 309,36; найдено 310 (M+H).
Стадия C:
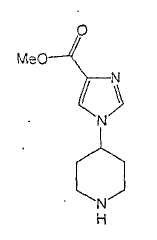
Соединение, полученное на стадии B (174,5 мг, 0,5641 ммоль) растворяют в 4 н. растворе HCl/диоксан (30 мл) и перемешивают при к.т. в течение 2 час до концентрирования. Продукт получают в виде HCl-соли. ЭСИ-МС: рассчитано для С10H15N3O2 209,25, найдено 210 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 23
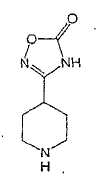
Стадия A:
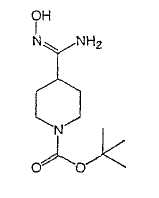
Гидроксиламингидрохлорид (6,0 г, 86 ммоль) и триэтиламин (12 мл, 86 ммоль) растворяют в ДМСО (100 мл), фильтруют и промывают ТГФ (20 мл). Фильтрат частично концентрируют при пониженном давлении, удаляя ТГФ. Нитрил, полученный на стадии A ПРОМЕЖУТОЧНОГО ПРОДУКТА 1 (3,62 г, 17,2 ммоль), растворяют в ДМСО (15 мл) и добавляют к фильтрату. Реакционную смесь нагревают при 75°C в течение 27 час. Затем реакционную смесь разбавляют этилацетатом (100 мл) и промывают водой (1×100 мл). Парционированную водную фазу промывают методом обратной промывки этилацетатом (1×100 мл). Органические порции объединяют, промывают водой (1×100 мл) и затем насыщенным раствором соли (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении.
Требуемый продукт получают и используют на следующих стадиях без дополнительной очистки. ЭСИ-МС: рассчитано для C11H21N3O3 243,30; найдено 188 (M-t-Bu+H).
Стадия B:
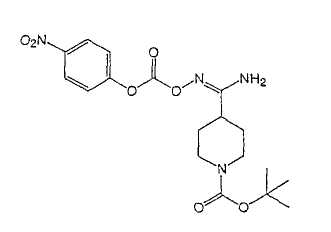
Триэтиламин (2,05 мл, 14,7 ммоль) и пара-нитрофенилхлорформиат (3,23 г, 16,0 ммоль) добавляют к раствору соединения, полученного на стадии A (3,25 г, 13,4 ммоль), в ДХМ (100 мл). Реакционную смесь перемешивают при к.т. в течение 1 час. Затем реакционную смесь промывают 1 н. HCl (1×75 мл), раствором бикарбоната натрия (1×75 мл) и насыщенным раствором соли (1×75 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Требуемый продукт получают и используют на следующих стадиях без дополнительной очистки.
Стадия C:
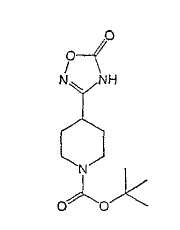
Соединение, полученное на стадии B (4,86 г, 11,9 ммоль), растворяют в бензоле (100 мл) и нагревают при кипении с обратным холодильником в течение 11 час. Реакционную смесь концентрируют при пониженном давлении и очищают методом СДЖХ (силикагель, 0-65% EtOAc/гексан), получая требуемый гетероцикл. ЭСИ-МС: рассчитано для C12Н19N3O4 269,30; найдено 170 (M-BOC+H).
Стадия D:

Соединение, полученное на стадии C (2,19 г, 8,13 ммоль) растворяют в 4 н. растворе HCl/диоксан (50 мл) и перемешивают при к.т. в течение 2 час до концентрирования. Продукт получают в виде HCl-соли. ЭСИ-МС: рассчитано для C7H11N3O2 169,18; найдено 170 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 24
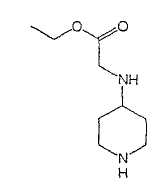
Стадия A:
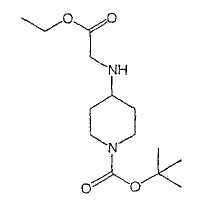
Гидрохлорид этиламиноацетата (4,2 г, 30 ммоль), триэтиламин (4,2 мл, 30 ммоль) и триацетоксиборогидрид натрия (16 г, 75 ммоль) добавляют к раствору BOC-4-пиперидона (3,04 г, 15,1 ммоль) в ДХМ (100 мл). Реакционную смесь перемешивают при к.т. в течение 24 час и определяют завершение взаимодействия по ВЭЖХ/MC. Реакционную смесь разбавляют ДХМ (50 мл), промывают раствором бикарбоната натрия (1×100 мл) и насыщенным раствором соли (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом СДЖХ (силикагель, 10-100% EtOAc/гексан) дает требуемый продукт. ЭСИ-МС: рассчитано для C14H26N2O4 286,37; найдено 231 (M-t-Bu+H).
Стадия B:
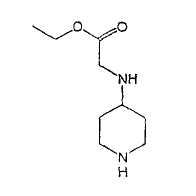
Соединение, полученное на стадии A (2,59 г, 9,04 ммоль), растворяют в 4 н. растворе HCl/диоксан (50 мл) и перемешивают при к.т. в течение 20 мин до концентрирования. Продукт получают в виде HCl-соли. ЭСИ-МС: рассчитано для С9H18N2O2 186,25, найдено 187 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 25
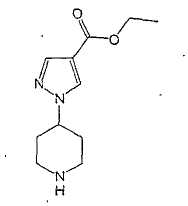
Стадия A:
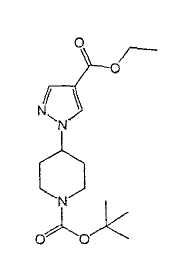
трет-Бутил-4-[(метилсульфонил)окси]пиперидин-1-карбоксилат (3,62 г, 13,0 ммоль) объединяют с этил-4-пиразолкарбоксилатом (2,0 г, 14 ммоль) в 50 мл ДМФА. Смесь охлаждают до 0°C и добавляют гидрид натрия (467 мг, 19,5 ммоль). Затем реакционную смесь нагревают при 50°C в течение 10 час. Потом ее разбавляют диэтиловым эфиром (100 мл), промывают водой (5×100 мл) и насыщенным раствором соли (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка методом СДЖХ (силикагель, 5-100% EtOAc/гексан) дает требуемый продукт. ЭСИ-МС: рассчитано для C16H25N3O4 323,39; найдено 346 (M+Na).
Стадия B:
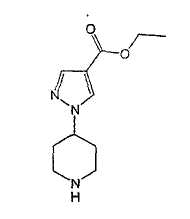
Соединение, полученное на стадии A (1,81 г, 5,60 ммоль) растворяют в 4 н. растворе HCl/диоксан (50 мл) и перемешивают при к.т. в течение 2 час до концентрирования. Продукт получают в виде HCl-соли. ЭСИ-МС: рассчитано для C11H17N3O2 223,27; найдено 224 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 26
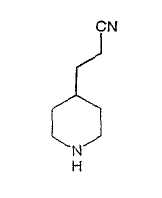
Стадия A:
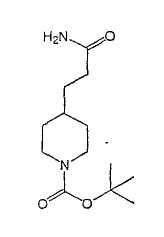
Коммерчески доступную BOC-пиперидинилпропионовую кислоту (2,79 г, 10,8 ммоль) растворяют в 100 мл ДХМ и охлаждают до 0°C. Триэтиламин (1,66 мл, 11,9 ммоль) и изобутилхлорформиат (1,55 мл, 11,9 ммоль) добавляют к охлажденному раствору, реакционную смесь нагревают до к.т. и перемешивают в течение 15 мин. ТСХ показывает полное расходование BOC-пиперидинилпропионовой кислоты. Затем реакционную смесь барботируют газообразным аммиаком в течение 5 мин. Реакционную смесь промывают раствором бикарбоната натрия (1×75 мл), 1 н. HCl (1×75 мл) и насыщенным раствором соли (1×75 мл), сушат над сульфатом магния, фильтруют и концентрируют. Очистка методом СДЖХ (силикагель, 0-100% EtOAc/гексан) дает требуемый продукт. ЭСИ-МС: рассчитано для C13H24N2O3 256,34; найдено 157 (M-BOC+H).
Стадия B:
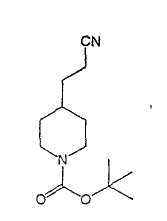
Пиридин (935 мкл, 11,6 ммоль) и трифторуксусный ангидрид (3,3 мл, 23 ммоль) добавляют по капле к перемешиваемому раствору соединения, полученного на стадии A (987,5 мг, 3,852 ммоль), в тетрагидрофуране (20 мл) в атмосфере азота. Реакционную смесь перемешивают при к.т. в атмосфере азота в течение 24 час. Затем реакционную смесь разбавляют диэтиловым эфиром (100 мл), промывают 1 н. HCl (3×100 мл) раствором бикарбоната натрия (2×100 мл) и насыщенным раствором соли (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка методом СДЖХ (силикагель, 10-100% EtOAc/гексан) дает требуемый продукт. ЭСИ-МС: рассчитано для C13H22N2O2 238,33; найдено 183 (M-t-Bu+H).
Стадия C:
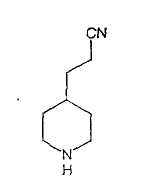
Соединение, полученное на стадии B (709,1 мг, 2,977 ммоль) растворяют в 4 н. растворе HCl/диоксан (50 мл) и перемешивают при к.т. Через 2 час исходный материал больше не присутствует (по данным ТСХ), и реакционную смесь концентрируют. Продукт получают в виде HCl-соли.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 27
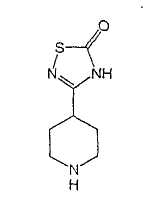
Стадия A:
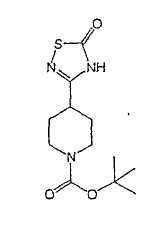
Смесь ранее описанного трет-бутил-4-[(Z)-амино(гидроксиимино)метил]пиперидин-1-карбоксилата (1,26 г, 5,18 моль) и 1,1'-тиокарбонилдиимидазола (1,38 г, 7,77 ммоль) в тетрагидрофуране (20 мл) перемешивают при к.т. в течение 45 мин. Реакционную смесь разбавляют водой (75 мл) и экстрагируют этилацетатом (2×75 мл). Органические порции объединяют, снова промывают водой (1×100 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученное масло растворяют в ТГФ (20 мл). К раствору добавляют бортрифторид диэтилэфират (2,21 г, 15,5 ммоль) и реакционную смесь перемешивают при к.т. в течение 1 час до концентрирования при пониженном давлении. Полученную смесь растворяют в ДХМ (20 мл) и охлаждают до 0°C. Триэтиламин (2,2 мл, 16 ммоль) и ди-трет-бутилдикарбонат (2,3 г, 10 ммоль) добавляют к дихлорметановому раствору и реакционную смесь перемешивают при к.т. в течение 2,5 час до концентрирования. Очистка методом СДЖХ (силикагель, 0-100% этилацетат/гексан) дает требуемый продукт. ЭСИ-МС: рассчитано для C12Н19N3O3S 285,36; найдено 212 (M-O-t-Bu).
Стадия B:
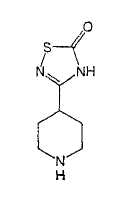
Соединение, полученное на стадии A (411,5 мг, 1,442 ммоль), растворяют в 4 н. растворе HCl/диоксан (20 мл) и перемешивают при к.т. Через 1,5 час реакционную смесь концентрируют и получают требуемый продукт в виде HCl-соли. ЭСИ-МС: рассчитано для C7H11N3OS 185,25, найдено 186 (M+H).
Ряд CCR-2 модуляторов получают способом, аналогичным описанному для примера 1, из (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанона (промежуточный продукт 4) и аминов, доступных из коммерческих источников, известных из научной литературы или описанных в рамках данного документа. цис- и транс-Изомеры, которые получают в результате восстановительного аминирования, разделяют либо методом хиральной ВЭЖХ (с применением колонки ChiralCel OD, 2 см×25 см, или ChiralPak AD, 2 см×25 см, обе доступны от Chiral Technologies, Inc.), либо препаративной ТСХ. В некоторых случаях, где амины, использованные в реакциях восстановительного аминирования, сами представляют смеси более чем одного стереоизомера (т.е. если они имеют один или более стереоцентров), обычно можно разделить все возможные изомеры с применением хиральной ВЭЖХ и/или препаративной ТСХ (иногда требуются серии разделений). В таблице 1 перечислены некоторые типичные примеры этих аналогов (показаны только более биологически активные цис-изомеры).
Аналоги, полученные способом, аналогичным примеру 1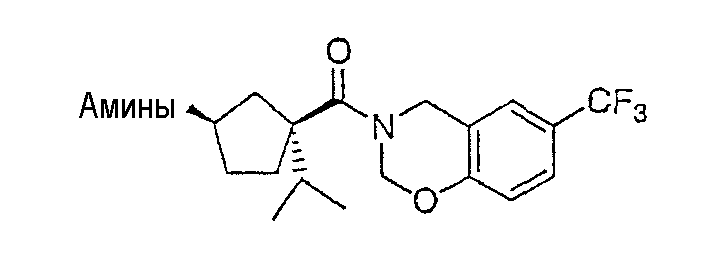

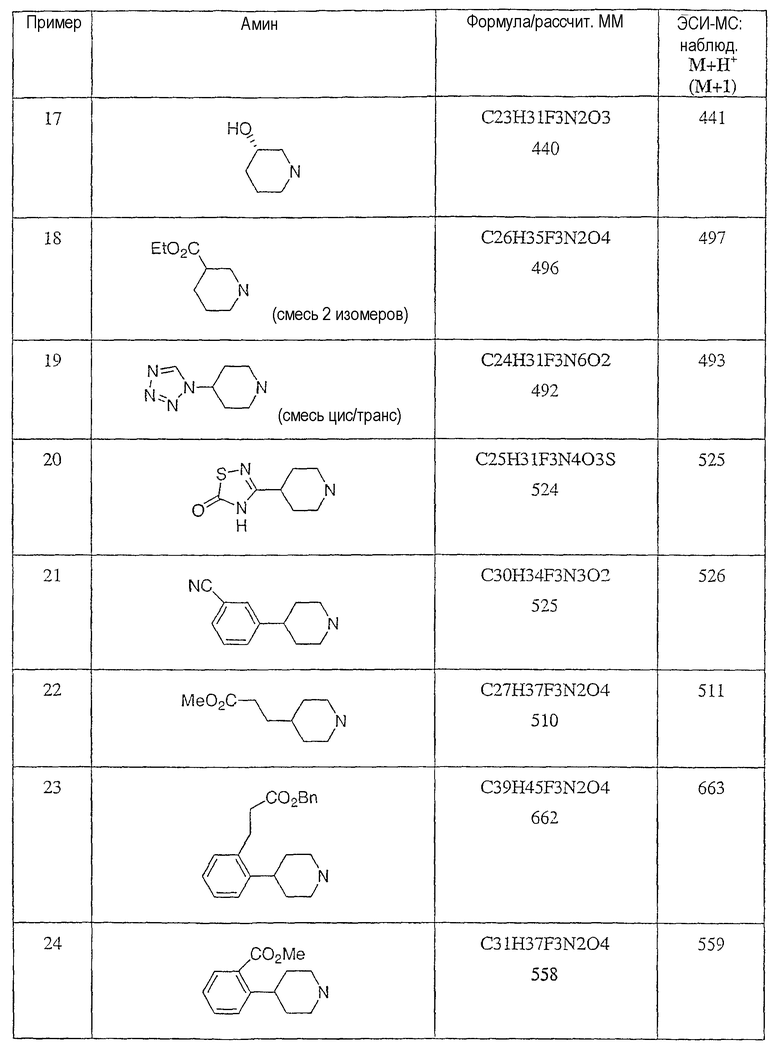
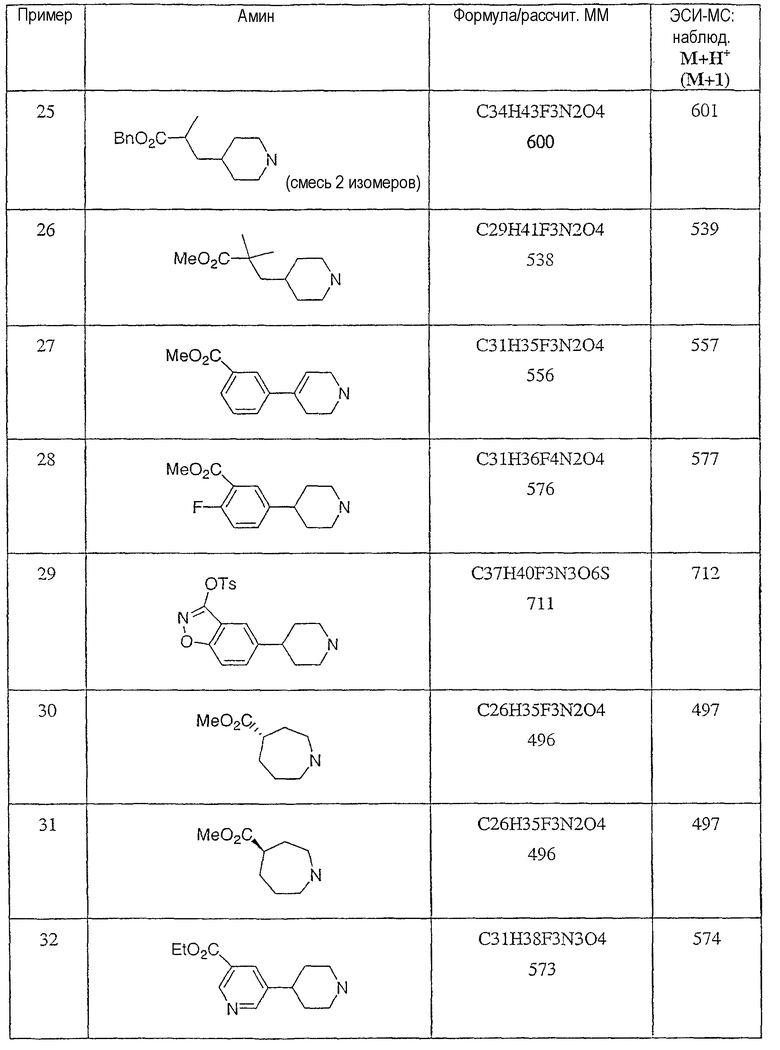
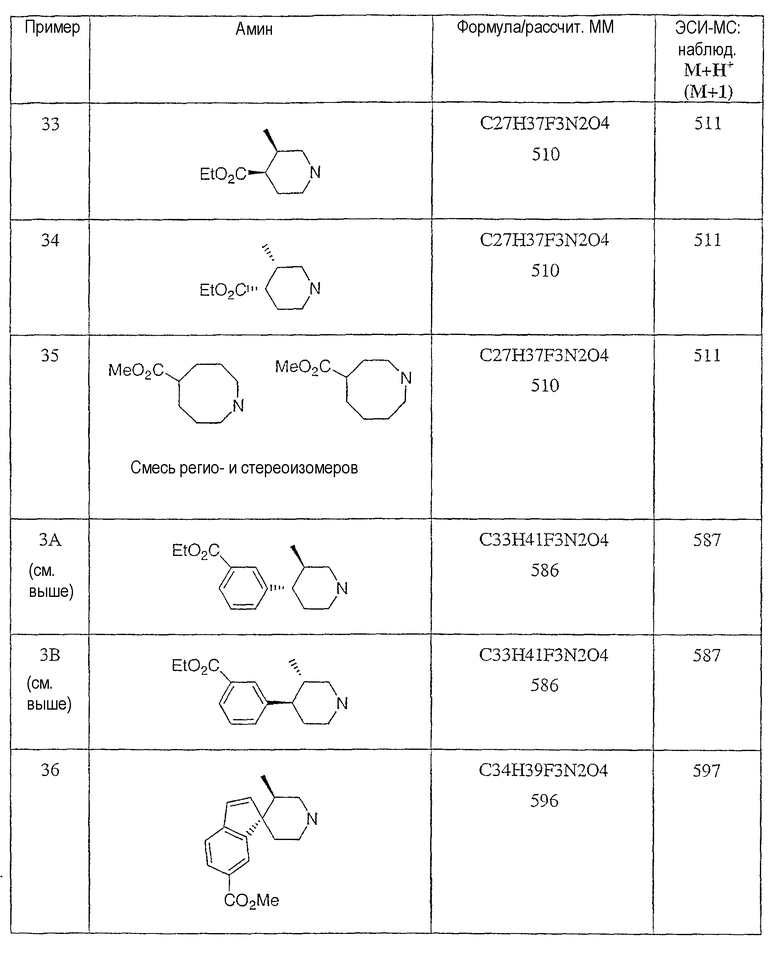
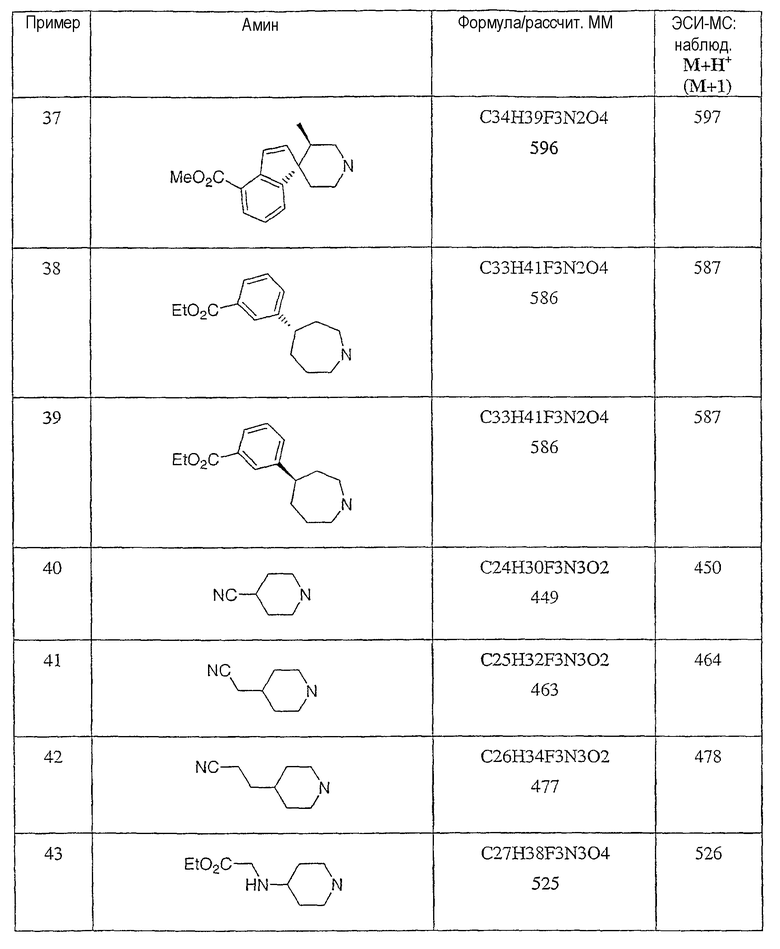
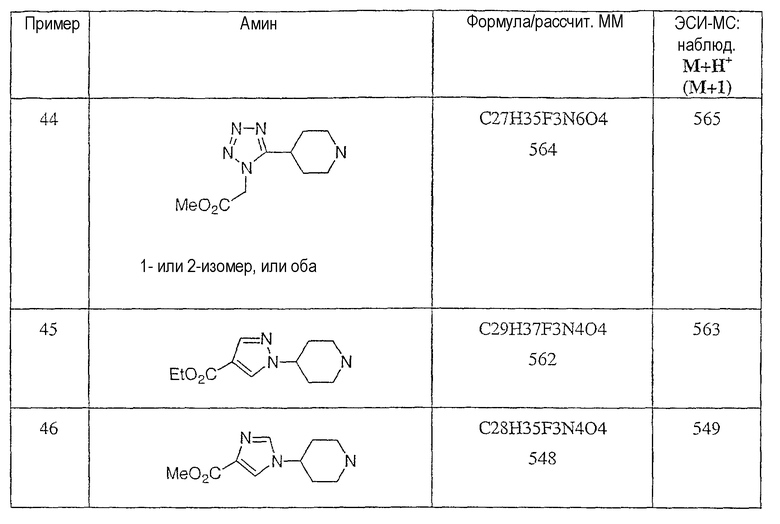
Во многих случаях аналоги, перечисленные в таблице 1, можно далее модифицировать, получая новые целевые модуляторы хемокиновых рецепторов. Например, сложноэфирные группы аналогов, указанных в данной таблице, гидролизуют, получая соответствующие карбоновые кислоты, которые сами являются сильнодействующими модуляторами. Данный гидролиз обычно выполняют в условиях, показанных в примере 2 и примере 4, или с небольшими модификациями этих условий. По-другому, в случае бензиловых сложных эфиров карбоновую кислоту можно получить гидрированием по протоколу, описанному для стадии F промежуточного продукта 3, или с его небольшими модификациями. Список типичных получаемых карбоновых кислот, содержащих модуляторы хемокиновых рецепторов, представлен в таблице 2.
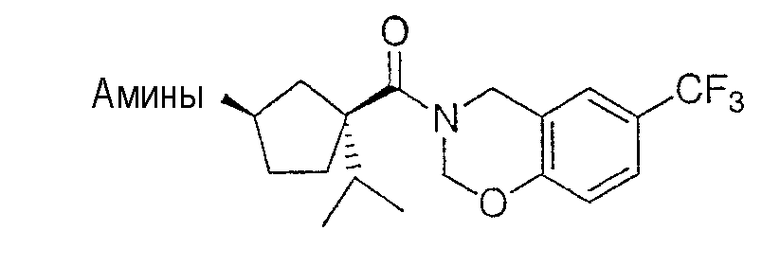
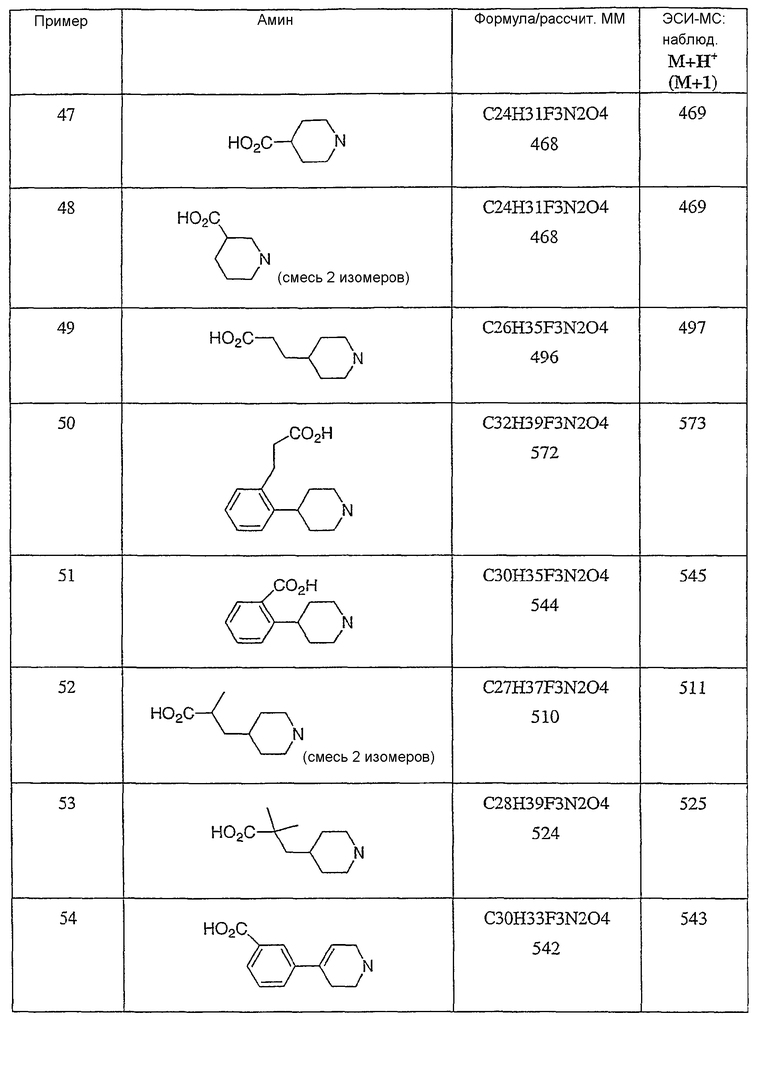
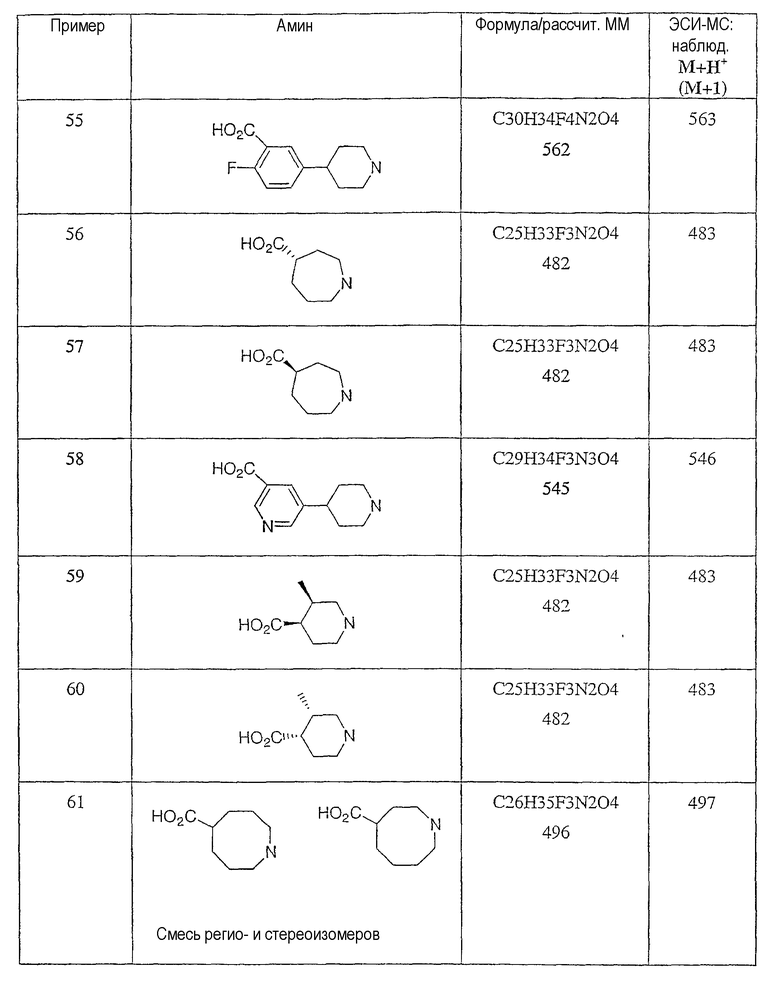
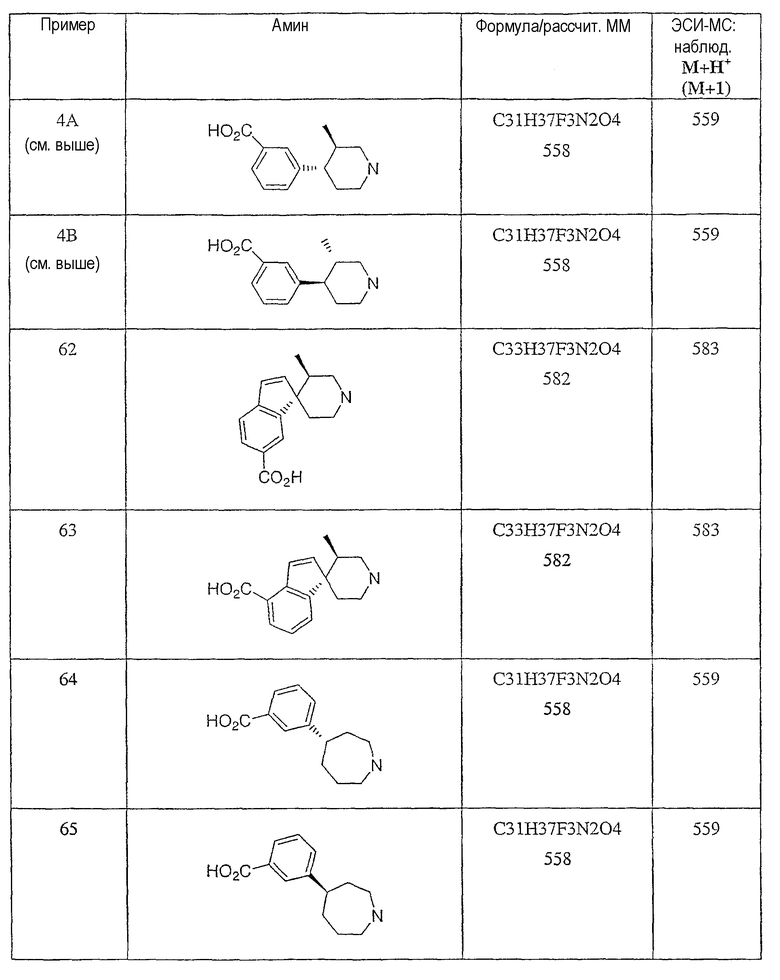
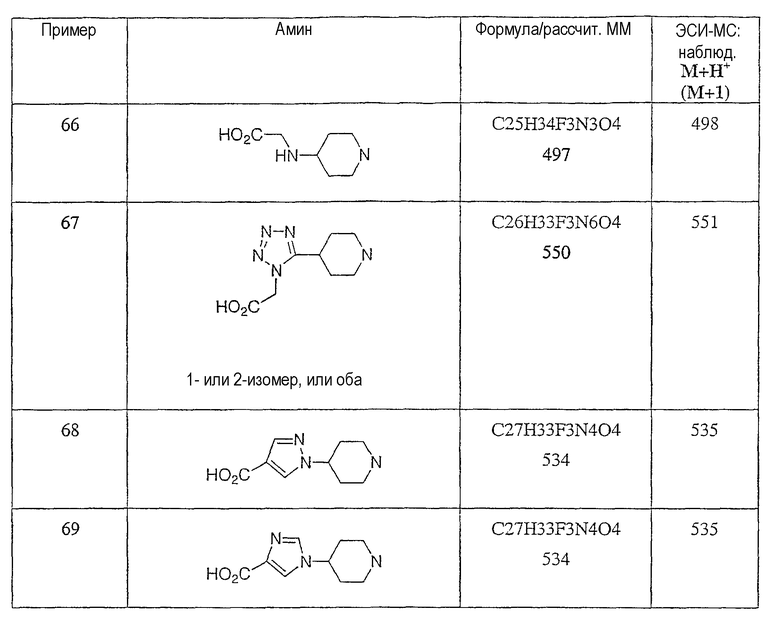
ПРИМЕР 70
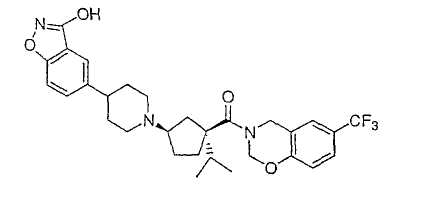
Данное соединение получают, исходя из соединения примера 29, способом, аналогичным показанному в примере 9. ЭСИ-МС: рассчитано для C30H34F3N3O4 557; найдено 558 (M+H).
Другой способ модификации некоторых аналогов, представленных в таблице 1, с получением новых, сильнодействующих модуляторов хемокиновых рецепторов включает преобразование нитрильных групп, обнаруживаемых в некоторых аналогах в таблице 1, в тетразольные группы. Способ осуществления этого описан ниже в примере 71.
ПРИМЕР 71
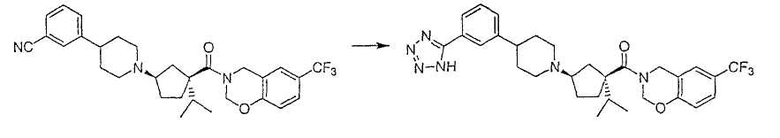
Раствор 3-[1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2H-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ил]бензонитрила (полученного, как описано ранее, 31,7 мг, 0,0603 ммоль) и трибутилоловоазида (50 мкм, 0,18 ммоль) в 10 мл толуола перемешивают в атмосфере азота при кипении с обратным холодильником в течение ночи. Так как взаимодействие не протекает в большой степени, добавляют еще трибутилоловоазид (200 мкл, 0,603 ммоль) и перемешивают реакционную смесь в атмосфере азота при кипении с обратным холодильником в течение 4,5 час. В этот момент добавляют еще трибутилоловоазид (200 мкл, 0,603 ммоль) и перемешивают реакционную смесь в атмосфере азота при кипении с обратным холодильником в течение ночи. Реакционную смесь концентрируют, затем добавляют безводный 1 н. HCl в эфире и смесь перемешивают в течение 15 мин, затем концентрируют. Твердые вещества в колбе отделяют от масла (предположительно Bu3SnCl), отбирая масло пипеткой. К оставшимся твердым веществам добавляют насыщенный раствор соли и ДХМ. Доводят рН водного слоя до 7 и слои разделяют. Водный слой промывают еще дважды ДХМ и затем органические слои объединяют, сушат над безводным MgSO4, фильтруют и концентрируют. После очистки методом препаративной ТСХ (диоксид кремния, 50% этилацетат/гексан для перемещения оставшегося Bu3SnCl, основу собирают) проводят ВЭЖХ с обращенной фазой (YMC Pack Pro C18, 100×20 мМ, вн. диам.) с получением продукта в виде его TFA-соли. Данный продукт превращают в его HCl-соль, растворяя в ДХМ и добавляя избыток 1 н. HCl/эфир, затем концентрируя с получением 3-[((1S,3R)-1-изопропил-3-{4-[3-(1Н-тетраазол-5-ил)фенил]пиперидин-1-ил}циклопентил)карбонил]-6-(трифторметил)-3,4-дигидро-2Н-1,3-бензоксазингидрохлорида. ЭСИ-МС: рассчитано для C30H35F3N6O2 568; найдено 569 (M+H).
Способом, аналогичным описанному непосредственно выше, получают примеры, указанные в таблице 3, посредством конверсии нитрилсодержащих аналогов в соответствующие тетразолсодержащие аналоги.
Тетразолы из нитрилов таблицы 1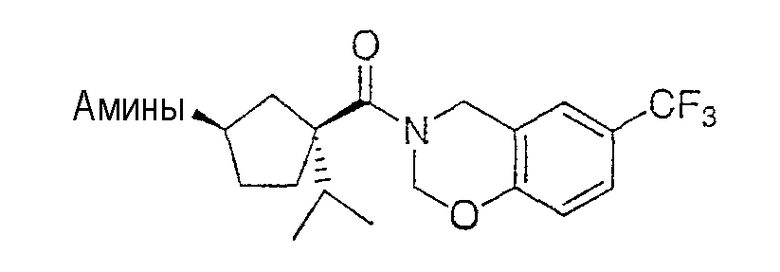
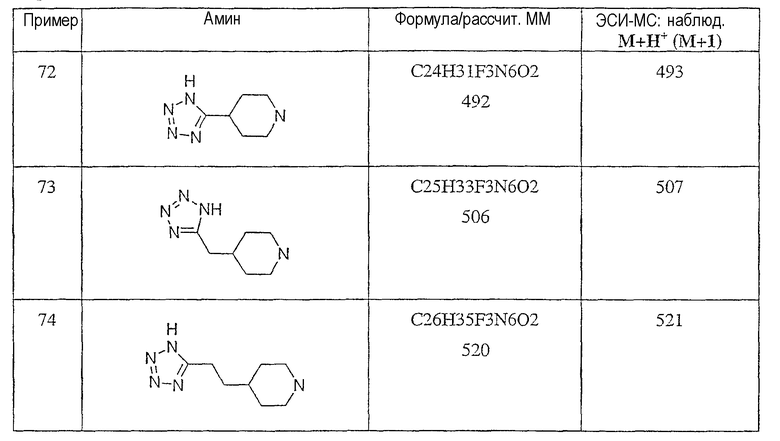
Другой пример модификации модуляторов с получением новых модуляторов описан в двух приведенных ниже примерах.
ПРИМЕР 75
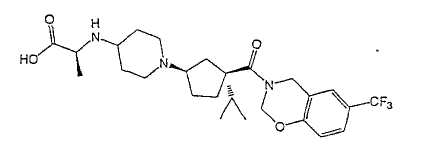
Стадия A:
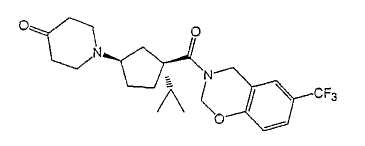
Раствор оксалилхлорида (35,0 мкл, 0,402 ммоль) в 15 мл ДХМ охлаждают до -78°C. К перемешиваемому раствору добавляют по капле ДМСО (57,0 мкл, 0,805 ммоль). Через 5 мин к реакционной смеси добавляют по капле 1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-ол (пример 13, таблица 1, 88 мг) в 5 мл ДХМ за 3 мин. После перемешивания еще в течение 25 мин при -78°C добавляют по капле триэтиламин (224,0 мкл, 1,609 ммоль) в 5 мл ДХМ за 1 мин. Реакционную смесь перемешивают при -78°C в течение 10 мин до удаления бани сухой лед/ацетон. Реакционной смеси дают нагреться до к.т. и перемешивают в течение 1,5 час. Затем реакционную смесь разбавляют ДХМ (50 мл) и промывают 1 н. раствором HCl (1×75 мл), раствором бикарбоната натрия (1×75 мл) и насыщенным раствором соли (1×75 мл). Водные слои объединяют и экстрагируют дихлорметаном (5×100 мл). Органические слои объединяют, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом препаративной ТСХ (силикагель, 0,4% гидроксид аммония, 3,6% MeOH, 96% ДХМ) дает 1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-он. ЭСИ-МС: рассчитано для C23H29F3N2O3 438,48; найдено 457 (гидрат+H).
Стадия B:
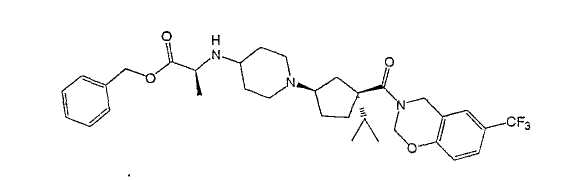
Гидрохлорид L-аланинбензилового эфира (11 мг, 0,062 ммоль), триэтиламин (9,0 мкл, 0,062 ммоль) и триацетоксиборогидрид натрия (22 мг, 0,10 ммоль) добавляют к перемешиваемому раствору 1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4H)-ил]карбонил}циклопентил)пиперидин-4-она (19 мг, 0,021 ммоль) в ДХМ (10 мл). Реакционную смесь перемешивают при к.т. в течение 96 час, однако взаимодействие проходит не до конца. К реакционной смеси добавляют еще 19 мг (0,11 ммоль) гидрохлорида L-аланинбензилового эфира и 15 мкл (0,11 ммоль) триэтиламина. После перемешивания в течение дополнительных 72 час при к.т. реакционную смесь разбавляют дихлорметаном (50 мл), промывают раствором бикарбоната натрия (1×50 мл) и насыщенным раствором соли (1×50 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом препаративной ТСХ (силикагель, 0,25% аммония гидроксид, 2,25% MeOH, 97,5% ДХМ) дает требуемый продукт. ЭСИ-МС: рассчитано для C33H42F3N3O4 601,70; найдено 602 (M+H).
Стадия C:
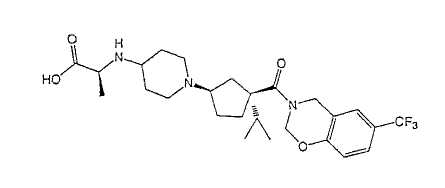
Соединение, полученное на стадии B (5,6 мг, 0,0093 ммоль), растворяют в 5 мл метанола и добавляют в колбу, содержащую палладиевый катализатор (˜3 мг, 10 мас.% на активированном угле). Реакционную колбу повторно откачивают и продувают газообразным водородом (3x). Затем реакционную смесь перемешивают в течение 5 час при к.т. под баллонным водородом. Завершение взаимодействия определяют методом ВЭЖХ/МС, реакционную смесь фильтруют и концентрируют в вакууме. Очистка методом ВЭЖХ с обращенной фазой, затем обработка 1,0 M HCl в диэтиловом эфире дает HCl-соль требуемой кислоты. ЭСИ-МС: рассчитано для C26H36F3N3O4 511,58; найдено 512 (M+H).
ПРИМЕР 76
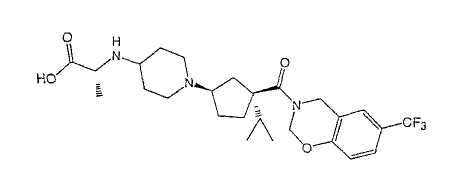
Стадия A:
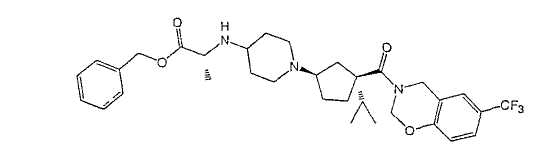
Соль D-аланинбензилового эфира пара-толуолсульфоновой кислоты (22 мг, 0,062 ммоль), триэтиламин (9,0 мкл, 0,062 ммоль) и триацетоксиборогидрид натрия (22 мг, 0,10 ммоль) добавляют к перемешиваемому раствору 1-((1R,3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентил)пиперидин-4-она (19 мг, 0,021 ммоль) в ДХМ (10 мл). Реакционную смесь перемешивают при к.т. в течение 96 час, однако взаимодействие проходит не до конца. К реакционной смеси добавляют еще 39 мг (0,11 ммоль) соли L-аланинбензилового эфира пара-толуолсульфоновой кислоты и 15 мкл (0,11 ммоль) триэтиламина. После перемешивания в течение дополнительных 72 час при к.т. реакционную смесь разбавляют дихлорметаном (50 мл), промывают раствором бикарбоната натрия (1×50 мл) и насыщенным раствором соли (1×50 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Очистка методом препаративной ТСХ (силикагель, 0,25% гидроксид аммония, 2,25% MeOH, 97,5% ДХМ) дает требуемый продукт. ЭСИ-МС: рассчитано для C33H42F3N3O4 601,70; найдено 602 (M+H).
Стадия B:
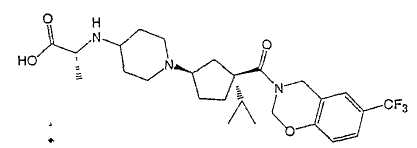
Соединение, полученное на стадии A (5,3 мг, 0,0088 ммоль), растворяют в 5 мл метанола и добавляют в колбу, содержащую палладиевый катализатор (˜3 мг, 10 мас.% на активированном угле).
Реакционную колбу повторно откачивают и продувают газообразным водородом (3x). Затем реакционную смесь перемешивают в течение 5 час при к.т. под баллонным водородом. Завершение взаимодействия определяют методом ВЭЖХ/MS, фильтруют и концентрируют в вакууме. Очистка методом ВЭЖХ с обращенной фазой, затем обработка 1,0 M HCl в диэтиловом эфире дают HCl-соль требуемой кислоты. ЭСИ-МС: рассчитано для C26H36F3N3O4 511,58; найдено 512 (M+H).
В некоторых случаях условия для восстановительного аминирования, описанные в примере 1 и примере 3, не являются оптимальными для получения некоторых аналогов. В частности, это случай, когда аминный компонент в восстановительном аминировании имеет плохую растворимость. В таких случаях применяют альтернативные условия, как описано ниже в примере 77.
ПРИМЕР 77
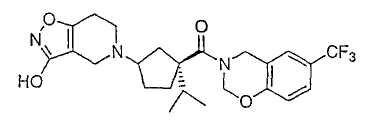
Смесь коммерчески доступного Gaboxadol гидрохлорида (134 мг, 0,760 ммоль), (3S)-3-изопропил-3-{[6-(трифторметил)-2Н-1,3-бензоксазин-3(4Н)-ил]карбонил}циклопентанона (54 мг, 0,15 ммоль), триэтиламина (106 мкл, 0,760 ммоль) и натрия цианборогидрида (57 мг, 0,91 ммоль) в 5 мл метанола перемешивают при к.т. в течение 4 час. Реакционную смесь концентрируют. Очистка методом ВЭЖХ с обращенной фазой (YMC Pack Pro С18, 100 х 20 мМ внутр. диам.) дает продукт в виде его TFA-соли. Данный продукт превращают в его HCl-соль, растворяя в ДХМ и добавляя избыток 1 н. HCl/эфир, затем концентрируя с получением целевого аналога в виде его соли гидрохлорида и в виде смеси цис- и транс-изомеров. ЭСИ-МС: рассчитано для C24H28F3N3O4 479; найдено 480 (M+H).
Другие примеры, полученные с применением условий восстановительного аминирования цианборогидридом натрия, описанных в примере 77, показаны в таблице 4.
Аналоги, полученные с применением NaBH3CN-условий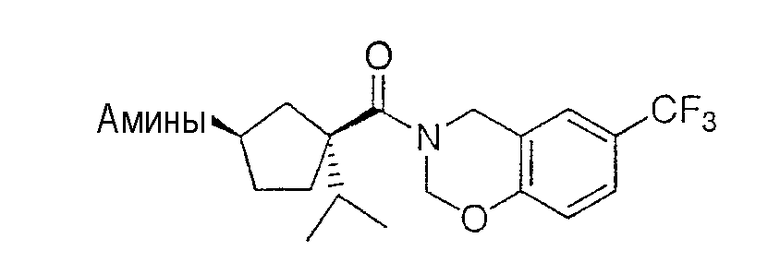
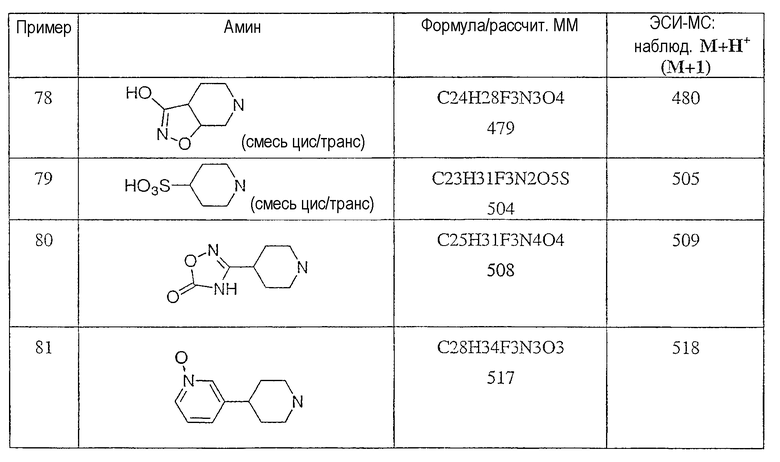
При том, что данное изобретение описано и проиллюстрировано со ссылкой на некоторые конкретные его варианты, специалистам в данной области понятно, что можно делать различные адаптации, изменения, модификации, замещения, устранения или добавления в методиках и протоколах, не отклоняясь от духа и области данного изобретения. Например, можно применять эффективные дозы, отличные от указанных выше конкретных доз, вследствие вариаций в быстроте реагирования млекопитающего, подлежащего по каким-либо показаниям лечению указанными выше соединениями данного изобретения. Также наблюдаемые специфические фармакологические реакции могут меняться в соответствии и в зависимости от конкретных выбранных активных соединений или присутствия фармацевтических носителей, а также типа препарата и используемого способа введения, и такие ожидаемые вариации или различия в результатах предусмотрены в соответствии с целями и инструкциями настоящего изобретения. Таким образом, данное изобретение определено формулой изобретения, которая следует далее, и не ограничено примерами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТРИАЗОЛА КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИДДЕГИДРОГЕНАЗЫ-1 | 2003 |
|
RU2360910C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ДИАМИНО-1,3,5-ТРИАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1997 |
|
RU2186774C2 |
| 1,5-БЕНЗОТИАЗЕПИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИГИПЕРЛИПИДЕМИЧЕСКИХ СРЕДСТВ | 2001 |
|
RU2302414C2 |
| ЛИГАНДЫ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2012 |
|
RU2620375C9 |
| НОВЫЕ СОЕДИНЕНИЯ МИМЕТИКИ ОБРАТНОГО ПОВОРОТА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2457210C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ОПИОИДНЫХ РЕЦЕПТОРОВ | 2003 |
|
RU2332411C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛООКТАНА, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, СОЕДИНЕНИЯ | 2000 |
|
RU2262505C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА НМДА И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2621049C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2571662C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
Изобретение относится к циклопентильным соединениям общей формулы (I)
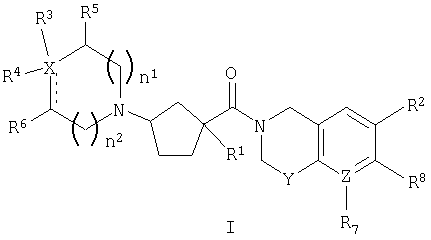
в которой Х обозначает С, N или О; Y обозначает О; Z обозначает С; R1 обозначает водород, -C0-6алкил-W-(C1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), где алкил и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями, выбранными из гидрокси, -O-C1-3алкила, трифторметила, C1-3алкила; W обозначает простую связь, -O-, -СО-, -CO2-, -CONR10- или -NR9-; R2 обозначает -С0-6алкил, С0-6алкил-W-С1-6алкил или С0-6алкил-W-С3-7циклоалкил, где C1-6алкил, С3-7циклоалкил необязательно являются независимо замещенными 1-6 заместителями, выбранными из галогена, трифторметила, -C1-6алкила; R3 обозначает водород, -(С0-6алкил)-фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)-С3-7циклоалкил, -(С0-6алкил)-СО2R10, -(С0-6алкил)-(алкен)-CO2R10, -(С0-6алкил)-SO3Н, -(С0-6алкил)-W-С0-4алкил, -CONR10R10 или -NR10CO2R10, NR10С0-3алкилСО2R10, где фенил и гетероцикл, циклоалкил или С0-6алкил являются необязательно замещенным 1-5 независимыми заместителями, выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -О-С0-3СО2R10, CN, =O, -NR10R10, -CONR10R10, SO3R10 или -(С0-3алкил)гетероцикл, и где фенил может быть сконденсирован с гетероциклом, который сам необязательно может быть замещенным 1-2 гидроксильными группами; R4 отсутствует, если Х представляет О или N, или если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил, CN, -С0-3СО2R10, или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1Н-инденил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из гидрокси, C1-3алкила, -O-C1-3алкила и -С0-3СО2R10, или R3 и R5 или R4 и R6 объединены вместе, образуя фенил или гетероциклильное кольцо, где данное кольцо является необязательно замещенным 1-7 независимыми заместителями выбранными из гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10; R5 и R6 независимо обозначают водород, гидрокси, C1-6алкил или С0-6алкил-СО2R10; если Z=C, R7 обозначает водород, C1-6алкил; R8 обозначает водород; R10 обозначает водород, -C1-6алкил, бензил, фенил или -С0-6алкил-С3-6циклоалкил; n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3; пунктирная линия представляет простую связь или двойную связь, а также к другим соединениям этого ряда; фармацевтической композиции, модулирующей активность хемокиновых рецепторов, способу модуляции активности хемокиновых рецепторов у млекопитающих, а также к способу лечения, улучшения, регулирования или снижения риска воспалительного и иммунорегуляторного нарушения или заболевания. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 12 н. и 9 з.п. ф-лы, 4 табл.
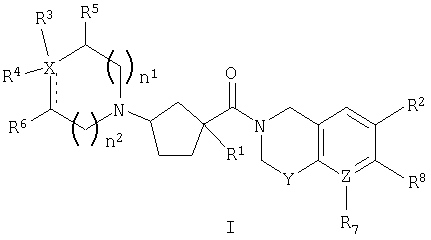
или его фармацевтически приемлемая соль, или его индивидуальный диастереомер, в котором
Х обозначает С, N, или О;
Y обозначает О;
Z обозначает С;
R1 обозначает водород, -С0-6алкил-W-(С1-6алкил)-, -(С0-6алкил)-W-(С0-6алкил)-(С3-7циклоалкил)-(С0-6алкил), где алкил и циклоалкил необязательно являются замещенными 1-7 независимыми заместителями, выбранными из гидрокси, -O-С1-3алкила, трифторметила, C1-3алкила;
W обозначает простую связь, -О-, -СО-, -CO2-, -CONR10- или -NR9-;
R2 обозначает -С0-6алкил, С0-6алкил-W-С0-6алкил или С0-6алкил-W-С3-7циклоалкил, где C1-6алкил, С3-7циклоалкил необязательно являются независимо замещенными 1-6 заместителями, выбранными из галогена, трифторметила, -С1-6алкила;
R3 обозначает водород, -(С0-6алкил)-фенил, -(С0-6алкил)гетероцикл, -(С0-6алкил)-С3-7циклоалкил, -(С0-6алкил)-CO2R10, -(С0-6алкил)-(алкен)-CO2R10, -(С0-6алкил)-SO3Н, -(С0-6алкил)-W-С0-4алкил, -CONR10R10 или -NR10CO2R10, NR10C0-3алкилСО2R10, где фенил и гетероцикл, циклоалкил или С0-6алкил является необязательно замещенным 1-5 независимыми заместителями, выбранными из галогена, трифторметила, гидрокси, C1-3алкила, -О-С0-3CO2R10, CN, =O, -NR10R10, -CONR10R10, SO3 R10 или -(С0-3алкил)гетероцикл, и где фенил может быть сконденсирован с гетероциклом, который сам необязательно может быть замещенным 1-2 гидроксильными группами;
R4 отсутствует, если Х представляет О или N, или, если двойная связь соединяет атомы углерода, к которым присоединены R3 и R6, или R4 обозначает гидрокси, С0-6алкил CN, -С0-3CO2Р10;
или R3 и R4 объединены вместе, образуя 1H-инденил, 2,3-дигидро-1Н-инденил, циклопентанильное или циклогексанильное кольцо, где данный полученный цикл необязательно является замещенным 1-5 заместителями, независимо выбранными из гидрокси, С1-3алкила, -O-C1-3алкила и -С0-3СО2R10;
или R3 и R5 или R4 и R6 объединены вместе, образуя фенил или гетероциклильное кольцо, где данное кольцо является необязательно замещенным 1-7 независимыми заместителями, выбранными из гидрокси, C1-3алкила, -O-C1-3алкила, -CO2R10;
R5 и R6 независимо обозначают водород, гидрокси, C1-6алкил или С0-6алкил-CO2R10;
если Z=C, R7 обозначает водород, C1-6алкил;
R8 обозначает водород;
R10 обозначает водород, -С1-6алкил, бензил, фенил или -С0-6алкил-С3-6циклоалкил;
n1 и n2 независимо равны 0, 1 или 2, причем сумма n1 и n2 равна 0, 1, 2 или 3; и пунктирная линия представляет простую связь или двойную связь.
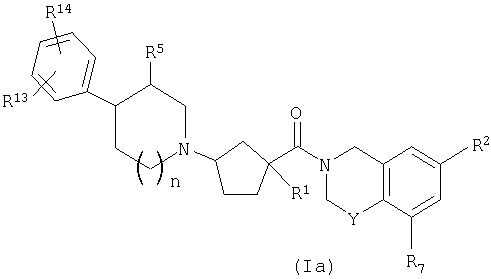
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7 и Y имеют значения, определенные в п.1;
где R13 и R14 независимо обозначают водород, галоген, трифторметил, гидрокси, -С1-3алкил, -O-C1-3алкил, -С0-3-CO2Н, -С0-3-CO2С1-3алкил, -CN или -С0-3-гетероцикл,
или R13 и R14 объединены вместе, образуя гетероцикл, сконденсированный с фенильным кольцом, и который сам может быть незамещенным или замещенным 1-2 независимыми заместителями из гидроксильных групп; и
n равно 0, 1 или 2.
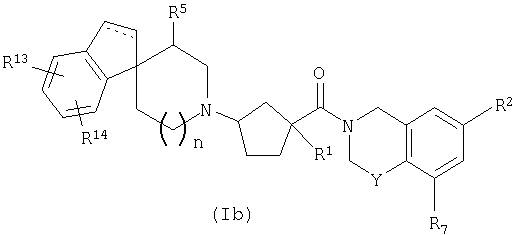
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7 и Y имеют значения, определенные в п.1;
где R13 и R14 независимо обозначают водород, галоген, трифторметил, гидрокси, -C1-3алкил, -O-C1-3алкил, -С0-3-CO2Н, -С0-3-CO2С1-3алкил, -CN или -С0-3-гетероцикл;
или R13 и R14 объединены вместе, образуя гетероцикл, который сконденсирован с фенильным кольцом, и который сам может быть незамещенным или замещенным 1-2 независимыми заместителями из гидроксильных групп; и
n равно 0, 1 или 2.
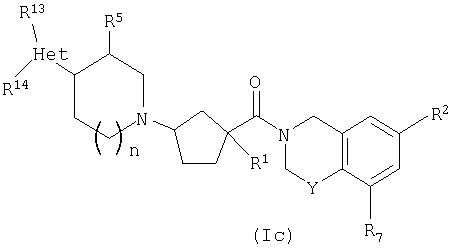
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7 и Y имеют значения, определенные в п.1;
где R13 и R14 независимо обозначают водород, галоген, трифторметил, гидрокси, -С1-3алкил, -O-C1-3алкил, -С0-3-CO2Н, -С0-3-СО2С1-3алкил, -CN или -С0-3-гетероцикл, или R13 и R14 объединены вместе, образуя гетероцикл, который сконденсирован с фенильным кольцом, и который сам может быть незамещенным или замещенным 1-2 независимыми заместителями, выбранными из гидроксильных групп;
n равно 0, 1 или 2; и
Het обозначает гетероцикл.
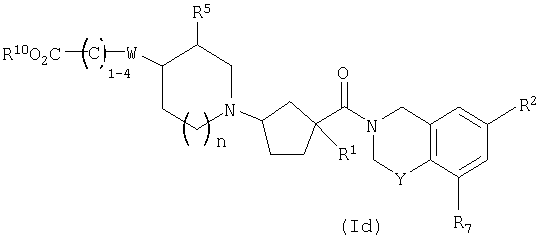
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7, R10, Y и W имеют значения такие, как определенные в п.1;
n равно 0, 1 или 2; и
и C1-4 углеродная цепь является необязательно замещенной 1-4 независимыми заместителями, выбранными из галогена, гидрокси, -С0-6алкила, -О-С1-3алкила, трифторметила или -С0-2алкил-фенила, или где С1-4 углеродная цепь является частью С3-7циклоалкильного кольца.
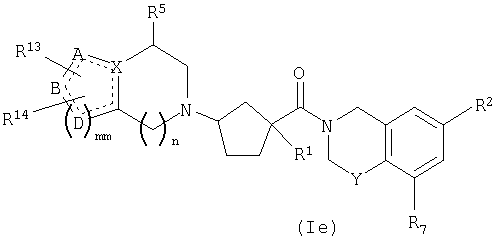
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7, R13, R14, Х и Y имеют значения такие, как определено в п.1;
n равно 0, 1 или 2;
пунктирные линии обозначают необязательную связь,
mm равно 1 или 2; и
А, В и D каждый независимо представляет собой С, N, О или S; или А, В и D в комбинации с mm=2 образуют фенильное кольцо; или в комбинации образуют гетероцикл, если, по меньшей мере, один из X, А, В, D является N, О или S.

или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7, R13 и R14 имеют значения такие, как определено в п.1;
или в которых R13 и R14 объединены вместе, образуя гетероцикл, сконденсированный с фенильным кольцом, и где сам гетероцикл является необязательно замещенным 1-2 независимыми заместителями из гидроксильных групп.
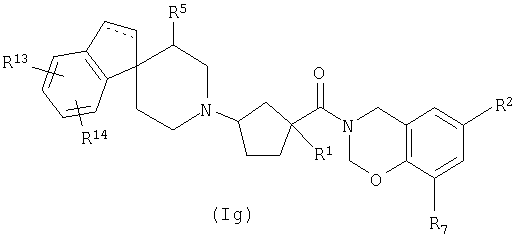
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, где пунктирная линия представляет необязательную связь, в которых R1, R2, R5, R7, R13 и R14 имеют значения такие, как определено в п.1.
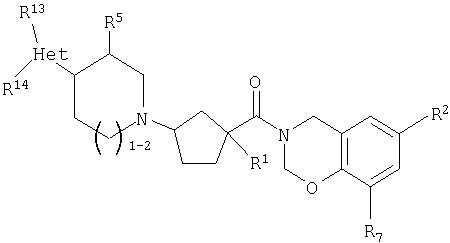
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, R1, R2, R5, R7, R13 и R14 имеют значения такие, как определено в п.1; и Het обозначает гетероцикл.
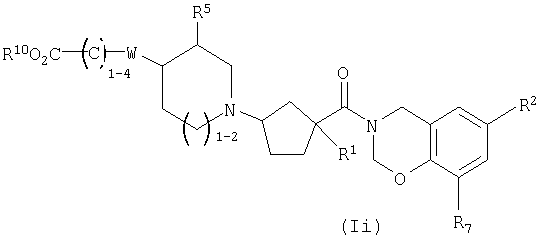
или его фармацевтически приемлемые соли и индивидуальные диастереомеры, в которых R1, R2, R5, R7, R10 и W имеют значения, определенные в п.1; и в которых С1-4 углеродная цепь является необязательно замещенной 1-4 независимыми заместителями, выбранными из галогена, гидрокси, -С0-6алкила, -O-C1-3алкила, трифторметила или -С0-2алкилфенила.
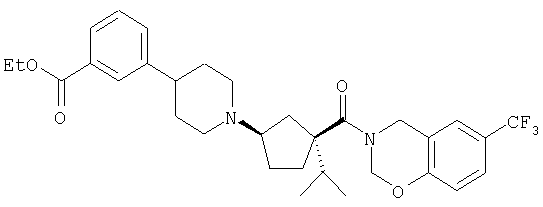
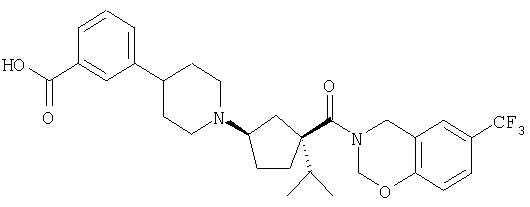
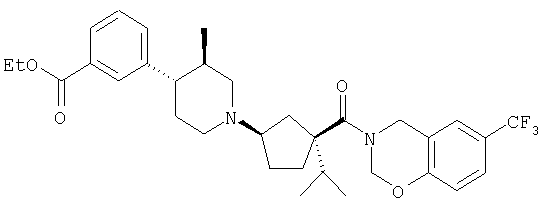
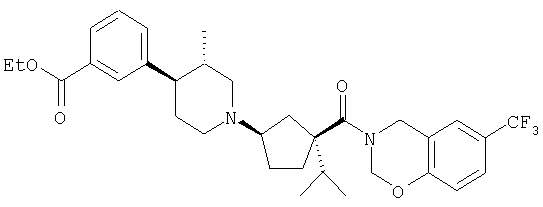
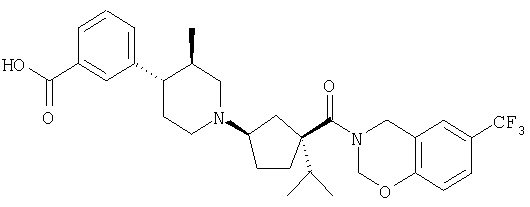
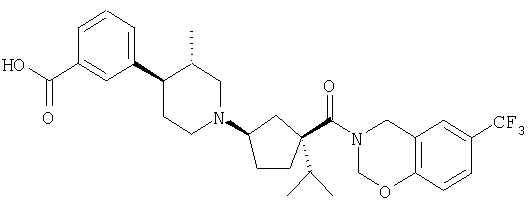
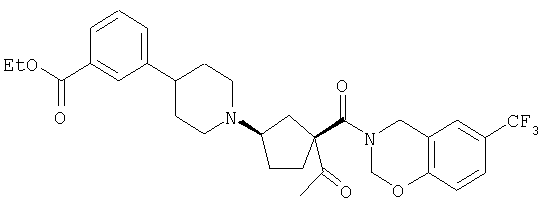
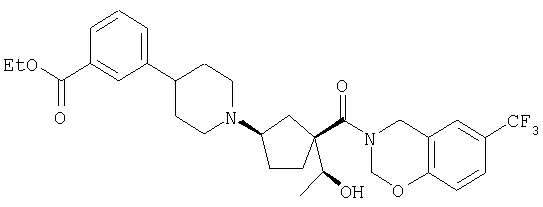

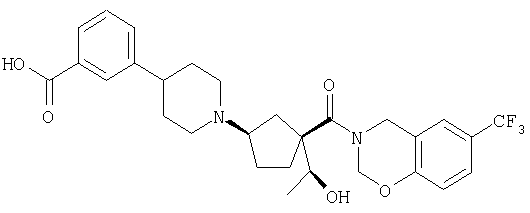
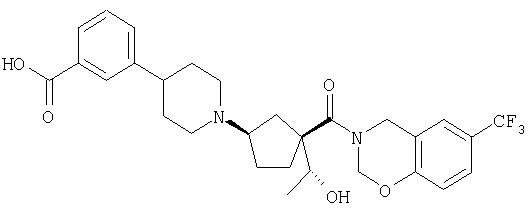
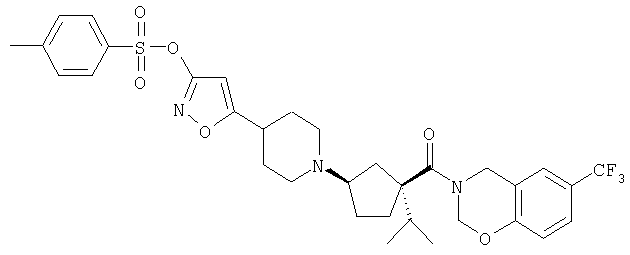
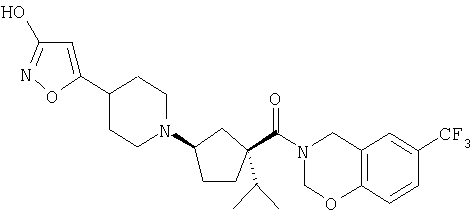
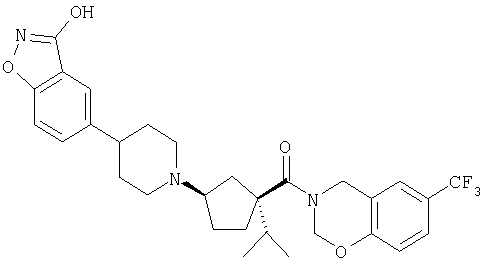
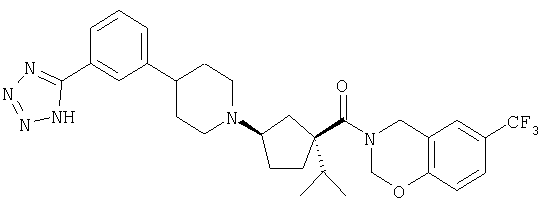
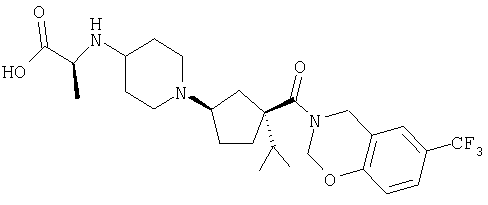
или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
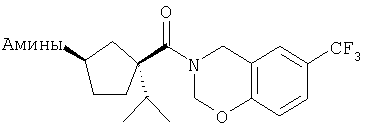
в котором амин является
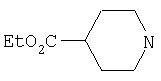
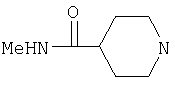
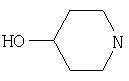
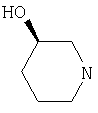
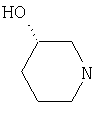
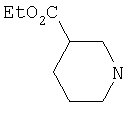
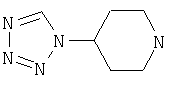
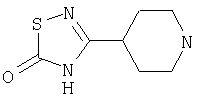
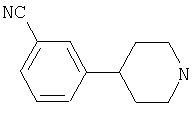
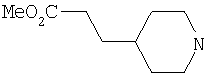
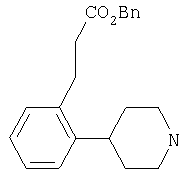
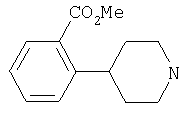
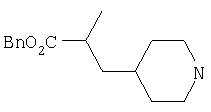

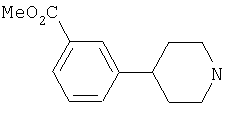
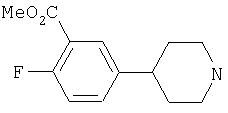
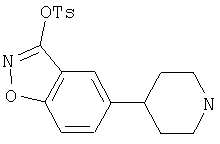
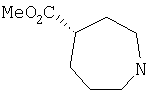
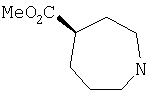
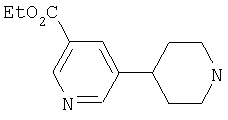
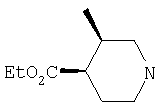
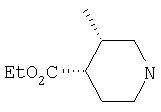
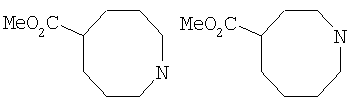
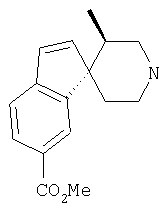

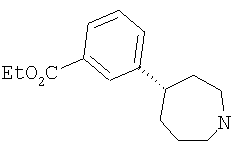
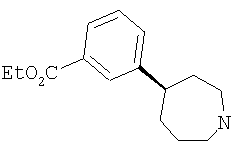
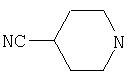
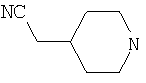
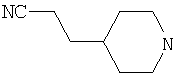
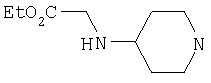
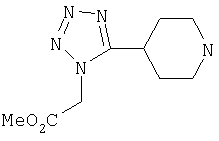
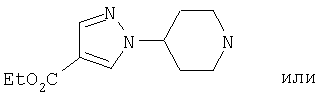
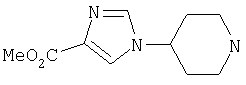
или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
в котором амин является
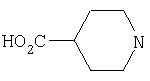

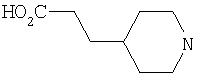
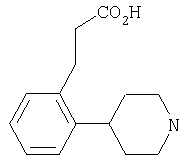

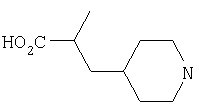

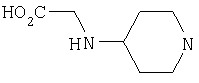
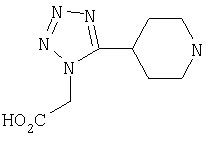
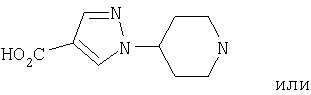
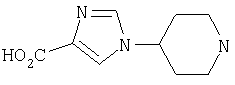
или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
в котором амин является
или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
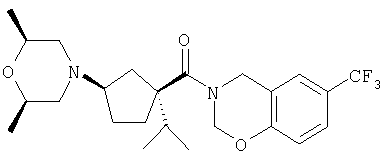
или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
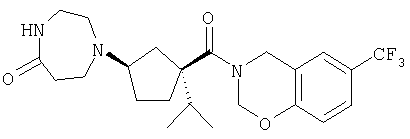
или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
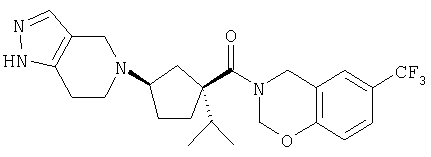
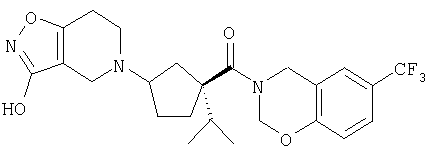

или его фармацевтически приемлемые соли, или индивидуальные диастереомеры.
| ТРИАЗИЛАМИДЫ 4-АРИЛ-2-ГИДРОКСИ-4-ОКСО-3-(2-ОКСО-3,4-ДИГИДРО-2H-1,4-БЕНЗОКСАЗИНИЛ)-2-БУТ ЕНОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНУЮ АКТИВНОСТЬ | 2001 |
|
RU2199537C1 |
| US 5631363 A, 20.05.1997. | |||

