Настоящее изобретение относится к металлохелатам с производными, содержащее четыре атома азота макроцикла, конденсированного с пиридиновым циклом, способам их получения и их применению в медицине для получения изображения.
Неоднократно было предложено использование для получения изображения хелатов некоторых производных этого конденсированного макроцикла с парамагнитным или радиоактивным катионом. Можно сослаться на заявки на Европейские патенты ЕР-А-0438206, ЕР-А-0570575 и ЕР-А-0579802, в которых описываются соединения формулы:
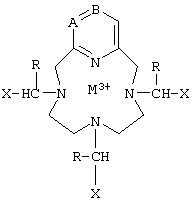
в которой X может означать карбоксилатную или фосфатную группу и R может означать алкил, фенил или один из R означает группу, образующую связь с биологической макромолекулой.
Среди этих соединений соединение, в котором А = В = СН, R = Н, Х = СО2, М = Gd, называемое РСТА, составило объект углубленных исследований, описанных в Inorganic Chemistry, 36 (14), 2992-3000 (1997), и Magn. Reson. Chem., 36, 200-208 (1998); авторы в особенности указывают, что РСТА является замечательным в том, что он обладает особенно высокой продольной релаксируемостью r1, так как она примерно в два раза выше таковой у хелатов гадолиния, используемых в качестве контрастных продуктов для получения изображения путем магнитного резонанса в случае человека.
Известно, что r1 характеризует эффективность парамагнитных продуктов, генерирующих сильный контраст изображений, и особенно представляющим интерес и неожиданным является то, что парамагнитные продукты согласно изобретению обладают релаксируемостью r1 в 10-15 раз выше релаксируемости имеющихся в продаже соединений не только в случае магнитного поля 0,5 Тл, но и также 1 Тл, поля большинства современных приборов для получения изображения, и даже 1,5 Тл, поля наиболее эффективных приборов.
Так как эти новые хелаты, кроме их благоприятных магнитных свойств, являются стабильными in vitro и in vivo, особенно по отношению к возможному разрушению комплекса, обладают незначительной осмоляльностью, хорошим терапевтическим индексом и тем, что в зависимости от природы группы R они могут обладать превосходным сосудистым последействием или специфичностью к органу, их можно преимущественно использовать в случае человека в качестве контрастных продуктов для получения изображения путем магнитного резонанса и в медицинской радиологии, когда ионом металла является радиоактивный элемент.
Настоящее изобретение относится к металлохелатам с соединением формулы (I):

в которой R означает группу формулы (II):

и Z означает связь или группу, выбираемую из групп CH2, CH2-CO-NH или
(CH2)2-NH-CO;
Z' означает связь или группу, выбираемую из групп О, S, NQ, CH2, CO, CO-NQ, NQ-CO, NQ-CO-NQ, CO-NQ-CH2-CO-NQ;
Z" означает связь или группу, выбираемую из групп CO-NQ, NQ-CO, CO-NQ-CH2-CO-NQ;
р и q означают целые числа, сумма которых составляет от 0 до 3;
R1, R2, R3, R4 и R5, независимо друг от друга, выбирают из групп Н, Вr, Cl, I, CO-NQ1Q2 и NQ1-CO-Q2, причем
Q1 и Q2, одинаковые или разные, означают Н или (С1-С6)-алкил, возможно прерываемый атомом или атомами кислорода, и по меньшей мере один из радикалов R1-R5 означает амидогруппу; или R1, R3 и R5, независимо друг от друга, означают Н, Вr, Cl или I;
и R2 и R4 означают группу формулы (III):
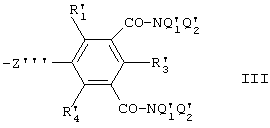
где Z'" означает группу, выбираемую из групп CO-NQ, CONQ-CH2, CO-NQ-CH2-CO-NQ, CO-NQ-(CH2)2-NQ-CO и NQ-CO-NQ и
R'1, R'3 и R'5, одинаковые или разные, означают Н, Вr, Cl или I;
и Q'l и Q'2, одинаковые или разные, означают Н или (С1-С6)-алкил, возможно прерываемый атомом или атомами кислорода;
Q означает Н или (С1-С4)-алкил;
алкильные группы необязательно могут быть моно- или полигидроксилированными.
Ионы металлов могут быть парамагнитными: такие как Gd3+, Fe3+, Tb3+, Мn2+, Dy3+ или Сr3+, или радиоактивными: такие как 99mТc, 67Ga или 111In; ионы, образующие менее стабильные хелаты, которые позволяют протекать реакции переметаллирования, такие как Са2+ или Zn2+, также составляют часть изобретения; парамагнитные хелаты с Gd3+ и Mn2+ особенно пригодны для получения изображения путем магнитного резонанса. Особенно предпочтительными являются парамагнитные хелаты с Gd3+.
Для лучшей гидрофильности и биосовместимости соединений предпочтительно, чтобы группы R1-R5 у одного и того же фенильного ядра вместе включали 6-20 групп ОН или также, чтобы любая присутствующая группа CONQ1Q2 или необязательно группа CONQ'1Q'2 содержала 6-10 групп ОН; еще предпочтительней, чтобы радикалы R2 и R4 были одинаковыми и означали CO-NQ1Q2, каждый из которых включает 6-10 групп ОН, или означали группу формулы (III), в которой каждый CONQ'1Q'2 содержит 6-10 групп ОН; также предпочтительны соединения, в которых R1, R3 и R5 выбирают среди атомов иода или брома, также как и R'1, R'3 и R'5, когда они присутствуют.
Релаксируемость соединений и их фармакокинетика in vivo особенно зависят от числа содержащихся в них фенильных ядер. Например, можно различать соединения, в которых р и q равны 0, особенно когда R2 и R4 означают CO-NQ1Q2, и соединения, в которых сумма р и q составляет 1-3 или лучше 1 или 2 и R2 и R4 означают или нет радикалы формулы (III).
Когда в соединениях формулы (I) R2 и R4 означают CONQ1Q2, то Q1 и Q2 предпочтительно являются (С2-С6)-алкильными группами, возможно прерываемыми атомом кислорода.
С другой стороны, среди соединений формулы (I) предпочтительны соединения, в которых Q означает Н, и среди них предпочтительны таковые, в которых Z означает СН2 или СН2СОNН, Z' означает группу, выбираемую среди CONH, CONHCH2CONH, NHCONH, и Z" означает группу, выбираемую среди CONH и CONHCH2CONH, так же как остаток Z'", когда он присутствует, означает CONH или CONHCH2CONH.
Наконец, другую группу особых соединений составляет группа, в которой р и q равны 1, Z означает СН2 или CH2CONH, Z' и Z" выбирают среди CONH и CONHCH2CONH и R2 и R4 означают CONQ1Q2, с R1, R3, R5 предпочтительно выбираемыми среди Вr и I.
Другими предпочтительными соединениями являются соединения, указанные в п.п.(i) - (ix) ниже:
(i) хелат соединения формулы (I), в которой р и q равны 0 и R2 и R4, являющиеся одинаковыми, означают -CO-NQ1Q2, причем каждая группа -CO-NQ1Q2 включает 6-10 групп ОН;
(ii) хелат соединения формулы (I), в которой р и q равны 0; R1, R3, R5 являются одинаковыми и их выбирают среди Вr и I; и R2 и R4, являющиеся одинаковыми, означают -CO-NQ1Q2, причем каждая группа -CO-NQ1Q2 включает 6-10 групп ОН;
(iii) хелат соединения формулы (I), в которой сумма р + q не равна 0; любая присутствующая группа -CO-NQ1Q2 включает 6-10 групп -ОН; R2 и R4 не отвечают формуле (III). Из этих соединений предпочтительны таковые, в которых сумма р + q составляет 1 или 2;
(iv) хелат соединения формулы (I), в которой сумма р + q не равна 0; R1, R3, R5,являются одинаковыми и их выбирают среди Вr и I; и R2 и R4, являющиеся одинаковыми, означают -CO-NQ1Q2, причем каждый из R2 и R4 включает 6-10 групп -ОН. Из этих соединений предпочтительны таковые, в которых сумма р + q составляет 1 или 2;
(v) хелат соединения формулы (I), в которой Z означает СН2 или CH2-CO-NH; Z' означает группу, выбираемую среди CO-NH, CO-NH-CH2-CO-NH и NH-CONH; Z" означает группу, выбираемую среди CO-NH и CO-NH-CH2-CO-NH; R2 и R4, являющиеся одинаковыми, означают -CO-NQ1Q2, причем каждый из R2 и R4 включает 6-10 групп -ОН; и R1, R3 и R5 являются одинаковыми и их выбирают среди Вr и I. Из этих соединений предпочтительны таковые, в которых сумма р + q составляет 1 или 2;
(vi) хелат соединения формулы (I), в которой Z означает CH2 или CH2-CO-NH; Z' означает группу, выбираемую среди CO-NH, CO-NH-CH2-CO-NH, NH-CONH; Z" означает группу, выбираемую среди CO-NH и CO-NH-CH2-CO-NH; группа Z'", когда она присутствует, означает CO-NH или CO-NH-CH2-CO-NH; и сумма р + q составляет 1 или 2;
(vii) хелат соединения формулы (I), в которой R2 и R4 означают CONQ1Q2 и Q1 и Q2, представляют собой полигидроксилированные, возможно прерываемые атомом кислорода (С2-С6)-алкильные группы;
(viii) хелат соединения формулы (I), в которой Z означает СН2 или CH2-CO-NH;
Z' и Z" выбирают среди CO-NH и CO-NH-CH2-CO-NH, р и q равны 1 и R2 и R4 означают CONQ1Q2. Из этих соединений предпочтительны такие, в которых:
- R2 и R4 вместе включают 6-20 групп -ОН; или
-R1, R3 и R5 являются одинаковыми и их выбирают среди Вr и I; и каждый из R2 и R4 включает 6-10 групп ОН;
(ix) хелат соединения формулы (I), в которой сумма р + q равна 0; R2 и R4 отвечают формуле (III) и R1, R3 и R5 являются одинаковыми и их выбирают среди Вr и I.
Изобретение относится также к способу получения соединений формулы (I), который состоит:
- во введении во взаимодействие конденсированного макроцикла формулы (IV):

с соединением формулы R'OOC-CHX-(CH2)2-COOR', в которой Х означает удаляемую группу, такую как атом галогена, предпочтительно брома, (С1-С3)-алкил-сульфонатную группу, -тозилатную группу или -трифлатную группу, и R' означает Н или (С1-С3)-алкил или бензил, и
- в осуществлении гидролиза или гидрирования сложноэфирных функциональных групп, когда R' отличается от Н,
для получения гексаосновной кислоты формулы (V):

- затем во введении во взаимодействие соли или оксида комплексообразующего металла с гексаосновной кислотой для получения соответствующего хелата или одной из его солей с основанием;
- и, наконец, во введении во взаимодействие с хелатом, в присутствии активирующего карбоксильные функциональные группы агента, амина формулы RNH2, в которой R имеет такое же значение, что и в формуле (I), для получения триамида формулы (I).
Кислота формулы (V) и ее хелаты с металлами, особенно хелат с гадолинием, и их соли с основанием, таким как NaOH, являющиеся промежуточными в синтезе продуктов формулы (I), составляют другой объект изобретения.
Изобретение относится также к содержащим соединение формулы (I) композициям для получения изображения путем магнитного резонанса, когда М означает парамагнитный катион, или для медицинской радиологии, когда М означает радиоактивный элемент, или для рентгенологии, когда М означает катион тяжелого атома, абсорбирующего Х-лучи, причем вышеуказанные композиции могут содержать обычные для введения перорально или парентерально добавки и эксципиенты.
Наконец, изобретение относится к методам получения изображения в медицине, которые состоят во введении пациенту содержащей соединение формулы (I) композиции и наблюдении исследуемой области, получаемой с помощью магнитного резонанса, спинтиграфии или под действием Х-лучей.
Диагностические композиции согласно изобретению вместе с соединением формулы (I) могут содержать добавки, такие как антиоксиданты, буферы, регуляторы осмоляльности, стабилизаторы, соли кальция, магния или цинка, или незначительные количества других хелатов с этими катионами или комплексообразующими соединениями. Примеры приготовления таких композиций находятся в общих руководствах и особенно в Remington's for Pharmaceutical Science, 18-e издание (1990), Mack. Pub. Су.
Разовые дозы зависят от природы контрастного продукта, пути введения, а также от пациента и особенно от природы исследуемого нарушения. Для внутривенной инъекции и наблюдения с помощью магнитного резонанса концентрация раствора составляет 0,001-0,5 моль/л и вводят в количестве 0,001-0,1 ммоль/кг массы пациента.
Контрастные продукты согласно изобретению могут быть использованы для визуализации головного мозга, а также таких органов, как сердце, печень или почки, или всей или части сосудистой системы и для изучения перфузии этих зон и охарактеризовывания аномалий проницаемости, опухолевой, воспалительной или ишемической аномалий.
Различные стадии синтеза соединений согласно изобретению осуществляют в условиях, аналогичных таковым, описанным в литературе для реакций такого же типа.
Макроцикл формулы (IV) может быть получен по способу Richman и Allans, описанному в Inorg. Chem., 32, 5257-5265 (1993).
Замещение атомов азота осуществляют, например, путем воздействия сложного α-бромглутарового эфира в присутствии неорганического или органического основания, такого как NaOH, Na2CO3 или N(C2H5)3, в растворе в полярном растворителе, таком как спирт, или предпочтительно в апротонном растворителе, таком как ацетонитрил или тетрагидрофуран.
Гидролиз сложноэфирных групп осуществляют путем воздействия основания или кислоты в водной или водно-спиртовой среде.
Комплексообразование осуществляют классическим путем, например, как описывается в патенте США 5554748 или в Helv. Chim. Acta, 69,2067-2074 (1986).
Для получения хелата с гадолинием можно вводить во взаимодействие GdCl3 или Gd2О3 с соединением формулы (V) в водном растворе при значении рН, составляющем от 5 до 6,5. Можно также осуществлять обмен катиона комплекса формулы (V) или формулы (I), когда это позволяет относительная стабильность двух комплексов, особенно с помощью ионообменной смолы.
Относительный процент изомеров в полученной смеси, возникающий вследствие наличия трех асимметрических атомов углерода, может быть изменен за счет выдерживания в течение нескольких дней при температуре выше 80°С водного раствора хелата при значении рН, близкого к 3.
Реакция амидирования может быть осуществлена в водной среде, в случае необходимости в присутствии третьего растворителя, такого как диоксан или тетрагидрофуран, с активирующим агентом, таким как растворимый карбодиимид, такой как карбодиимиды, содержащие аминогруппу, описанные в J. Org. Chem., 21,439-441 (1956) и 26, 2525-2528 (1961) или в патенте США 3135748, или четвертичную аммониевую группу, описанные в Org. Synth., V, 555-558, к которым относятся 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI) и 1-циклогексил-3-(2-морфолино-этил)карбодиимидмето-п-толилсульфонат. Реакцию также можно проводить с N-гидроксисульфосукцинимидом, как описывается в Bioconjugate Chem., 5, 565-576 (1994), или 2-сукцинимидо-1,1,3,3-тетраметилуронийтетрафторборатом и аналогичными соединениями, описанными в Tetrahedron Letters, 30.1927-1930 (1989).
Другой способ состоит в получении промежуточного активированного сложного эфира путем введения во взаимодействие, например, N-гидрокси-сульфосукцинимида (NHS) или гидроксибензотриазола (НОВТ) в присутствии карбодиимида, такого как EDCI, с хелатом формулы (V), который может быть солюбилизирован за счет солеобразования с неорганическим катионом, например катионом аммония или натрия.
При использовании 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина (EEDQ) реакцию можно осуществлять в водно-спиртовой среде.
Некоторые из аминов RNH2 представляют собой известные соединения; другие получают аналогичными способами, предпочтительно путем введения фенильных ядер постепенно, исходя из фенила, содержащего группы R1 - R5 и заместитель, пригодный для образования, в зависимости от необходимости, группы Z, Z' или Z".
Можно также сослаться на патенты WO-96/09281 или WO-97/01359 в отношении получения аминов, в которых Z означает CH2CONH, р = q = 0, R1 = R3 = R5 = галоген или Н и R2 = R4 = CONQ1Q2.
В случае некоторых аминоспиртов-предшественников HNQ1Q2, Q1 и/или Q2 означают группу CH2(CHOH)n(CH2OCH2)r(CHOH)tCH2OH, где t = 0, r = 0,1, n = 0 - 4; они могут быть получены
- из аминоспирта или первичного алкиламина, который вводят во взаимодействие с сахаром, после чего осуществляют восстановление полученного имина, как описывается в заявках на Европейские патенты ЕР-А-675105 или ЕР-А-558395;
- или из бензиламина, который вводят во взаимодействие с сахаром, затем (С1-С6)-алкилгалогенидом или -сульфатом, возможно гидроксилированным, после чего удаляют бензильную группу путем каталитического гидрирования.
В зависимости от конфигурации вводимого в реакцию сахара получают различные стереоизомеры.
Аминоспирты в случае, когда r = 1, n = t = 0, получают из 2-аминоэтоксиэтанола, который вводят во взаимодействие с эпоксидом или пригодным гидроксилированным алкилгалогенидом или еще гидроксилированным алифатическим альдегидом, таким как моносахарид, для получения имина, затем восстанавливают каталитическим или химическим путем.Когда r = n = 1, аминоспирты можно получать путем воздействия эпоксида

на пригодный первичный аминоспирт, причем вышеуказанные эпоксиды получают путем окисления соответствующих этиленовых производных с помощью надкислоты или пероксиимидокислоты, как описывается в J. Org. Chem., 26,659-663 (1961) и 48, 888-890 (1983).
Из полученных этими способами аминоспиртов можно указать спирты, в которых Q1 = СН2(СНОН)4СН2OН и Q2 = Q1 или СН2(СНОН)СН2OН;
Q1 = СН2(СНОН)2СН2OН и Q2 = СН2СНОНСН2O(СН2)2OН или (СН2)2OСН2СНОНСН2OН; Q1 = СН2(СНОН)3СН2OН и Q2 = (СН2)2O(СН2)2OН.
В случае, где р и/или q отличны от нуля, образуется мостик Z" между двумя фенильными ядрами, до или после мостика Z'.
Например, соединение формулы (VI):
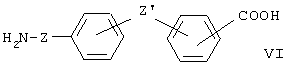
может быть получено, когда Z означает связь из дифенильных производных формулы (VII) или их сложных эфиров:
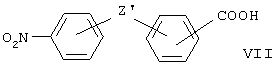
Соединение формулы (VII), в котором Z' означает О, описано в Macromoleculare Chemie, 130. 103-144 (1969); соединение, в котором Z' означает NH, описано в Indian J. Chem., 13, 35-37 (1975); соединение, в котором Z' означает СН2 или СО, описано в J. Pharm. Sci., 55 (3), 295-302 (1966); соединение, в котором Z' означает связь, описано в Synth. Comm., 24 (22), 3307-3313 (1994); и соединение, в котором Z' означает S, описано в II Farmaco, 44 (7-8), 683-684 (1989).
Другие соединения формулы (VII) могут быть получены аналогичными способами: например, когда Z' означает HNCONH, путем воздействия O2NС6Н4NСО на Н2NС6H4СООН в безводной среде, или когда Z' означает NHCO или CONH, путем взаимодействия хлорангидрида ароматической кислоты с пригодным анилином в растворе в апротонном растворителе, таком как CH2Cl2, С6Н5СН3, СН3СОN(СН3)2, или путем взаимодействия ароматической кислоты с анилином в присутствии хлорангидрида сульфокислоты, триэтиламина и диметиламинопиридина, как описывается в Synth. Communications, 25 (18), 2877-2881 (1995).
Восстановление группы NO2 в соединении формулы (VII) до NH2 может быть осуществлено известным образом с помощью водорода в присутствии катализатора или химическим путем.
Когда Z в формуле (VI) означает CH2-CONH, глицин, карбоксильная группа которого активирована и группа NH2 защищена, вводят во взаимодействие с соединением формулы (VI), в котором Z означает связь, или с анилином, содержащим в случае необходимости защищенную группу- предшественник Z'.
Глицин защищен, например, в форме карбамата, в особенности трет-бутилкарбамата (Synthesis, 48 (1986)) и бензидкарбамата (Chem. Ber., 65, 1192 (1932)); в форме фталимида (Tetrahedron Letters, 25, 20, 2093-2096 (1984)); с помощью бензила (Bull. Soc.Chim. Fr., 1012-1015 (1954)), с помощью N-аллила (Tetrahedron Letters, 22, 16, 1483-1486 (1981)) (см. также книгу T.W. Greene "Voir aussi Protective Groups in Organic Synthesis", 315-349 (J. Wiley and Sons, Inc.)). Защитную группу для NH2, фиксированную на Z, обычно удаляют только после введения группы R; классически, фталимидную группу удаляют путем воздействия гидразина, тогда как бензилоксикарбонильную или бензильную группу удаляют путем каталитического гидрирования.
Когда Z = CH2 и Z' = CONH или CONHCH2CONH, 4-аминометилбензойную кислоту, в которой группа NН2 защищена, можно вводить во взаимодействие в форме карбамата или имида, как описывается в J. Org. Chem., 43, 2320-2325 (1978), или в Rec. Trav. Chim. (Pays-Bas), 79, 688 (1960), с замещенной бензойной кислотой, карбоксильная группа которой блокирована путем этерификации.
Когда Z означает (СН2)2NНСО, RNН2, можно получать путем воздействия избытка этилендиамина на пригодный эфир бензойной кислоты, содержащий группу-предшественник Z', возможно защищенную, или более полную часть R.
Соединение формулы (VI) после защиты группы NH2, активированное в форме хлорангидрида кислоты или с помощью агента пептидного связывания, вводят во взаимодействие с группой-предшественником Z", которую содержит концевое фенильное ядро, пригодным образом замещенное с помощью R1 - R5, для получения после удаления защитной группы амина RNH2.
Условия получения аминов RNH2 будут лучше понятны из нижеследующих примеров. Масс-спектры этих продуктов (ионизация электронным распылением) в качестве полученных в примерах продуктов соответствуют ожидаемым структурам.
Соединения А и A': RNH2, где

Соединение А:
(а) 1-Дезокси-1-(2,3-дигидроксипропил)амино-D-галактит из α,D-галактозы:
Растворяют 35 г D-галактозы в 100 мл метанола, содержащих 17 г 3-аминопропандиола, и смесь выдерживают при перемешивании при температуре 25°С в течение 12 ч, после чего добавляют 5 г 10%-ного палладия -на- угле в качестве катализатора и 40 мл воды для осуществления гидрирования имина при температуре 60°С. Катализатор отфильтровывают и среду концентрируют до объема 85 мл. Аминоспирт выделяют путем осаждения во время добавления концентрированного раствора к 30 мл изопропанола при температуре около 35°С.
(b) 5-Амино-2,4,6-трибромизофталевая кислота:
Медленно добавляют 156 г брома к 300 мл водного раствора 50 г 5-аминоизофталевой кислоты и 55 мл 37%-ной соляной кислоты. После перемешивания в течение ночи избыток брома нейтрализуют путем добавления водного раствора бисульфита натрия, после чего осадок выделяют. Выход: 90%.
(с) 5-(Фталимидоацетамидо)-2,4,6-трибромизофталевая кислота:
Медленно добавляют 27 мл тионилхлорида к раствору 69 г N-фталоилглицина в 200 мл диметилацетамида при температуре 10°С, затем после перемешивания в течение 2 ч, при температуре около 15-20°С добавляют 100 г вышеполученной кислоты. После выдерживания в течение ночи при комнатной температуре смесь выливают в 800 мл горячей воды. Таким образом выделяют 140 г конечного продукта.
(d) Хлорангидрид вышеполученной кислоты:
При температуре 18°С к раствору 100 г дикислоты в 300 мл диоксана и 50 мл диметилформамида медленно добавляют 70 мл тионилхлорида. Выпавший после перемешивания в течение трех дней при комнатной температуре осадок желтого цвета отфильтровывают и промывают метил-трет-бутиловым эфиром. Таким образом получают 70 г твердого вещества бежевого цвета.
(е) N',N'-Бис(2,3,4,5,6-пентагидроксигексил)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трибром-5-(фталимидоацетамидо)изофталамид:
Растворяют 125 г аминопроизводного галактита, полученного на стадии (а), в 610 мл N-метилпирролидона при температуре 80°С, после чего при температуре 60°С добавляют 17 г Nа2СО3 и 102 г хлорангидрида дикислоты. После выдерживания при этой температуре в течение 2 ч реакционную среду доводят до комнатной температуры, затем отфильтровывают. Фильтрат вводят в 1,5 л изопропанола; выпавший осадок растворяют в воде и подвергают хроматографии на ионообменной смоле в форме H+ для удаления исходного амина. Таким образом выделяют 136 г твердого продукта.
(f) Соединение А:
125 г Вышеполученного фталимида растворяют в 520 мл N-метилпирролидона и 175 мл воды при температуре 70°С и добавляют 8 мл гидразингидрата, после чего среду выдерживают в течение 2 ч при температуре 90°С. Эту среду затем охлаждают вплоть до температуры 20°С, потом выливают в 1,6 л этанола. Выпавший осадок очищают путем пропускания его водного раствора через ионообменную смолу в форме H+.
Соединение А':
(а') Путем введения во взаимодействие 3-аминопропандиола с D-глюкозой в тех же самых условиях, что и указанные выше на стадии (а) для получения соединения А, получают 1 -дезокси-1 -(2,3 -дигидроксипропил)амино-D-глюцит.
Соединение А' может быть получено путем осуществления стадий b) - f), описанных выше для получения соединения А, исходя из аминопроизводного глюцита, полученного на стадии а'), или же путем осуществления следующей стадии е').
(е') Растворяют 95 г аминопроизводного глюцита, полученного на стадии (а'), в 460 мл диметилацетамида при температуре 90°С, после чего при температуре 65°С добавляют 32 мл триэтиламина и 117 г хлорангидрида дикислоты, описанного на стадии (d) выше при получении соединения А. После выдерживания в течение 4 ч 30 мин при температуре 55-60°С реакционную среду доводят до комнатной температуры и отфильтровывают. Полученный раствор при температуре 50°С медленно выливают в водный раствор гидразина (11 мл в 115 мл воды); после выдерживания в течение 3 ч при температуре 80°С доведенную до комнатной температуры среду подкисляют вплоть до значения рН, равного 1, путем добавления 1н. водного раствора НСl. Осадок отделяют и фильтрат при перемешивании выливают в 3 л этанола. Выпавший осадок высушивают, затем очищают путем диафильтрации для удаления большей части молекул с низкой молекулярной массой и полученный раствор подвергают хроматографии на анионообменных и катионообменных смолах. Выход: 60%.
Конечный амин может быть лиофилизирован. Анализ путем высокоэффективной жидкостной хроматографии (ВЭЖХ) на колонке 2 при использовании элюента 2: СF3СООН в воде, рН 3,3/CH3CN.
Соединение В: RNН2, где
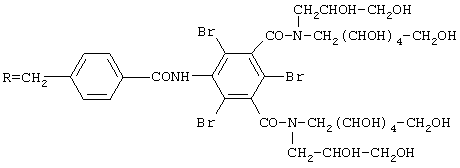
(а) 4-Фталимидометилбензойная кислота:
В течение 72 ч при температуре кипения с обратным холодильником выдерживают смесь 10 г 4-аминометилбензойной кислоты, 14,5 г карбэтоксифталимида, 9,2 мл триэтиламина и 140 мл тетрагидрофурана. Выпавший осадок выделяют при комнатной температуре; после промывки водным раствором кислоты и высушивания получают 14,5 г продукта. Т.пл. = 264°С.
(b) Хлорангидрид вышеполученной кислоты:
К раствору 13,5 г кислоты в 55 мл диоксана добавляют 1 г трикаприлметиламмонийхлорида (aliquat® 336) и 5,3 мл тионилхлорида. После перемешивания в течение 12 ч при температуре 80°С среду концентрируют досуха и оставшееся твердое вещество промывают диизопропиловым эфиром. Масса составляет 14 г.
(с)2,4,6-Трибром-5-(фталимидометилбензамидо)изофталевая кислота
В 50 мл N-метилпирролидона растворяют 14 г хлорангидрида кислоты и 15 г 5-амино-2,4,6-трибромизофталевой кислоты и среду выдерживают при температуре 100°С в течение нескольких часов. Раствор при температуре 20°С выливают в 300 мл воды и выпавший осадок перекристаллизовывают из изопропанола, получая 5,5 г конечного продукта.
ВЭЖХ (высокоэффективная жидкостная хроматография): колонка №1: Symmetry® C18;
100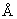 ; 5 мкм; 1 = 25 см; d = 4,6 мм (фирма Waters).
; 5 мкм; 1 = 25 см; d = 4,6 мм (фирма Waters).
Элюент №1: 0,005 М раствор СН3СООNH4 / СН3CN.
Градиент: от 80% до 20% (объем/объем) в течение 15 мин; расход = 1 мл/мин; время удерживания (tr) = 4,5 мин.
(d) Хлорангидрид вышеполученной кислоты:
К раствору 5,5 г кислоты в 40 мл диоксана добавляют 5,6 мл диметилформамида и 9 мл тионилхлорида, поддерживая температуру ниже 5°С. Спустя 30 мин среду выливают в 150 мл воды и выпавший осадок выделяют. Масса = 4,6 г.
(е) Соединение В:
2,3 г Хлорангидрида кислоты вносят в раствор 4 г аминоспирта - 1-дезокси-1-(2,3-дигидроксипропил)амино-D-галактита - в 15 мл N-метилпирролидона при температуре 65°С. Спустя 3 ч 30 мин добавляют 4 мл воды и доводят реакционную среду до температуры 90°С, после чего добавляют 0,3 мл гидразингидрата. После выдерживания в течение 2 ч при температуре 90°С раствор при комнатной температуре выливают в 80 мл этанола. Выделенный осадок растворяют в 10 мл воды и раствор с рН 1 очищают путем хроматографии на анионообменной смоле в форме ОН-типа Амберлит®, затем на катионообменной смоле в форме Н+ типа IМАС® и, наконец, на анионообменной смоле в форме ОН-. Получают 2 г амина.
ВЭЖХ: колонка №2: LiChrospher®; 100 RP18; 5 мкм; l = 25 см; d - 4 мм; (Merck®).
Элюент №2 раствор СF3СООН/СН3СN (рН 33) Градиент: от 98 до 77% (объем/объем) в течение 25 мин; расход = 1 мл/мин.; время удерживания: 18-22 мин.
Соединение С: RNН2, где

К раствору 5,5 г диизобутиламина в 30 мл N-метилпирролидона при температуре 65°С добавляют 2,3 г хлорангидрида кислоты, полученного согласно предыдущей стадии (d). После выдерживания в течение 4 ч при температуре 65°С добавляют 8 мл воды, затем при температуре 90°С добавляют 0,3 мл гидразингидрата; после выдерживания в течение 2 ч при этой температуре реакционную среду при температуре около 20°С выливают в 130 мл воды. Выпавший осадок промывают этанолом, затем снова растворяют в 10 мл воды, раствор доводят до значения рН, равного 1,5, после чего очищают путем хроматографии на анионообменных и катионообменных смолах. Таким образом получают 1,7 г амина.
ВЭЖХ: колонка №2; элюент №2; время удерживания: 18 мин.
Соединение D: RNH2 где

(а) N,N'-[Бис(2,3,4,5,6-пентагидроксигексил)]-2,4,6-трибром-5-(глициламино)изофталамид:
1. Растворяют 15 г дисорбитиламина в 60 мл N-метилпирролидона при температуре 80°С и в среду при температуре 60°С добавляют 1,6 г безводного карбоната натрия, затем 9,6 г хлорангидрида 5-(фталимидоацетамидо)-2,4,6-трибромизофталевой кислоты. После перемешивания в течение 1 ч при этой температуре и 16 ч при комнатной температуре осадок удаляют и раствор выливают в 160 мл изопропанола. Выделенный осадок имеет массу 20 г.
2. Гидразинолиз:
20 г Вышеполученного продукта и 1,7 мл гидразингидрата вносят в 40 мл воды при температуре 70°С. После перемешивания в течение 3 ч подкисляют вплоть до рН, равного 4, путем добавления 6н. соляной кислоты при комнатной температуре. Тогда удаляют выпавший осадок и фильтрат нейтрализуют путем добавления 1н. водного раствора гидроксида натрия. Находящийся в избытке гидразин удаляют путем обратного осмоса. Оставшийся раствор обрабатывают с помощью 1 мл сильной катионообменной смолы, затем с помощью 6,5 мл слабой анионообменной смолы.
Конечный продукт затем экстрагируют из раствора путем фиксации на сильной катионообменной смоле в форме Н+, откуда его элюируют с помощью водного разбавленного (0,1 М) раствора хлорида натрия. Масса = 8 г.
ВЭЖХ: колонка №2; элюент №3;
градиент: СF3СООН в воде (рН 3,4) / СН3СN от 95 до 50% (объем/объем) в течение 50 мин; расход составляет 1мл/мин.; время удерживания: 7 мин.
(b) 4-[4-Нитробензамидо]бензойная кислота:
10 г Хлорангидрида 4-нитробензойной кислоты постепенно добавляют к 7,4 г 4-аминобензойной кислоты и 36 мл диметилацетамида, поддерживая температуру ниже 25°С. После перемешивания в течение 24 ч при температуре 10°С добавляют 50 мл дихлорметана для осаждения искомого продукта. После промывки водой и высушивания выделяют 14,5 г продукта.
(с) 4-[4-Аминобензамидо]бензойная кислота:
Суспензию 13,6 г вышеполученной кислоты в 180 мл воды, к которой добавлено 24 мл 1н. водного раствора гидроксида натрия и 1,4 г 10%-ного палладия-на-угле, подвергают воздействию давления водорода 0,6 МПа в течение 4 ч.
Значение рН конечной суспензии затем доводят до 10, после чего фильтруют через Celite® для удаления катализатора. Осадок, выпавший во время подкисления фильтрата до значения рН, равного 5,3, выделяют и высушивают. Масса = 10,6 г; т.пл. > 260°С.
(d) 4-[4-(Фталимидоацетамидо)бензамидо]бензойная кислота:
К раствору 9 г фталимидоуксусной кислоты в 40 мл диметилацетамида при температуре 10°С добавляют по каплям 3,2 мл тионилхлорида, затем после перемешивания в течение 3 ч, при температуре ниже 20°С добавляют 10,5 г вышеполученной аминокислоты.
После перемешивания в течение 12 ч среду выливают в 400 мл воды и выделенный осадок промывают горячей водой. Масса после высушивания составляет 18 г; т.пл. > 260°С.
(е) Хлорангидрид вышеполученной кислоты:
К 10 г кислоты в виде суспензии в 50 мл диоксана добавляют 2,5 мл тионилхлорида и 1 мл диметилформамида и смесь выдерживают при перемешивании при температуре 50°С в течение 5 часов. После добавления одного объема диизопропилового эфира выделяют 10 г осадка.
Можно также использовать кислоту в виде суспензии в толуоле с трикаприлилметиламмонийхлоридом в качестве катализатора.
(f) N,N'-Бис(2,3,4,5,6-пентагидроксигексил)-2,4,6-трибром-5-(4-[4-(фталимидоацетамидо)бензамидо]бензоилглициламино)изофталамид:
Раствор 2,25 г хлорангидрида кислоты с 5 г N,N'-бис(2,4,5,6-пентагидроксигексил)-2,4,6-трибром-5-(глициламино)изофталамида и 0,7 мл триэтиламина в 25 мл диметилацетамида или N-метилпирролидона выдерживают в течение 12 ч при перемешивании, затем выливают в 60 мл этанола. Таким образом выделяют 6,2 г осадка.
ВЭЖХ: колонка №2; элюент №3; время удерживания = 27-35 мин (смесь изомеров).
(g) Гидразинолиз:
Раствор 0,6 мл гидразингидрата в 10 мл воды добавляют к раствору 10 г вышеполученного фталимида в 40 мл диметилацетамида при температуре 80°С. После перемешивания в течение 3 ч при этой температуре охлажденную смесь выливают в 125 мл этанола. Выделяют 9 г осадка, который очищают путем обработки его водного раствора с помощью сильной анионообменной смолы (ОН-), затем слабой катионообменной смолы (Н+). Масса = 8 г.
Можно также подкислять реакционную среду для отделения осажденного фталилгидразида и удалять растворитель и молекулы с незначительной молекулярной массой путем ультрафильтрации, после чего осуществлять окончательное осаждение в водном этаноле.
ВЭЖХ: колонка №2; элюент №3, но в соотношении 90/10 (объем/объем) при элюировании в изократном режиме при расходе 1 мл/мин; время удерживания = 28-35 мин.
Соединение Е: RNH2, где
(а) 5-(4-Нитробензамидо)-2,4,6-трибромизофталевая кислота:
В течение 18 ч при температуре кипения с обратным холодильником выдерживают 50 г хлорангидрида п-нитробензойной кислоты и 75 г 5-амино-2,4,6-трибромизофталевой кислоты в 400 мл диоксана. После охлаждения осадок отфильтровывают, промывают с помощью 50 мл диоксана и высушивают. Масса = 115 г.
(b) 5-(4-Аминобензамидо)-2,4,6-трибромизофталевая кислота:
Раствор 180 г вышеполученного нитропроизводного в 600 мл воды доводят до рН 6 путем добавления 5н. водного раствора гидроксида натрия и гидрируют при давлении 5×105 Па в присутствии Рt типа 156 (Johnson Matthey) в течение 7 ч. Катализатор отфильтровывают и воду выпаривают при пониженном давлении. Масса = 80 г. ВЭЖХ: колонка №2; элюент №4: СF3СООН в воде (рН 2,8) с метанолом (в соотношении 99/1 по объему); расход: 1 мл/мин.; время удерживания: 3,6 мин (18,8 мин для нитросоединения).
(с)5-(4-[4-Фталимидометил)бензамидо]бензамидо)-2,4,6-трибромизофталевая кислота:
Смесь 10 г 4-аминометилбензойной кислоты, 14,5 г N-карбэтоксифталимида и 9,2 мл триэтиламина в 140 мл тетрагидрофурана выдерживают при ее температуре кипения в течение 72 ч. Осадок, отделенный от реакционной среды путем отфильтровывания при комнатной температуре, промывают диэтиловым эфиром и 1н. водным раствором соляной кислоты. Получают 14,5 г твердого вещества, 12,2 г которого при температуре 10°С растворяют в 90 мл N,N-диметилацетамида и 3,5 мл тионилхлорида; после перемешивания в течение 3 ч в среду вносят 23,4 г полученного на предыдущей стадии анилина и перемешивают в течение одной ночи, после чего смесь выливают в 900 мл воды. Выделенный и промытый водой осадок перекристаллизовывают из 200 мл диоксана. Масса = 30 г.
ВЭЖХ: колонка №2; элюент: 0,1 М раствор СF3СООН в воде/СН3СМ ( в соотношении 90/10 по объему) с градиентом, спустя 20 мин, вплоть до 40/60 в течение 30 мин; расход: 1 мл/мин; время удерживания = 26-29 мин.
(d) Дихлорангидрид кислоты:
30,3 г Полученного на предыдущей стадии производного изофталевой кислоты растворяют в 150 мл диоксана, содержащего 26 мл диметилформамида, и при температуре 5°С добавляют по каплям 42 мл тионилхлорида. После выдерживания при температуре 0°С в течение 30 мин смесь выливают в 550 мл воды и выпавший осадок отфильтровывают, промывают водой и диизопропиловым эфиром. Масса = 26 г после высушивания.
(е)N,N'-Бис(2,3,4,5,6-пентагидроксигексил)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трибром-5-[4-(4-аминометилбензамидо)бензамидо]зофталамид:
1. 10 г Дихлорангидрида кислоты добавляют к раствору 15 г 1-дезокси-1-(2,3-дигидроксипропил)амино-D-галактита в 100 мл N-метилпирролидона при температуре 60°С. После перемешивания в течение 4 ч при этой температуре среду, доведенную до комнатной температуры, выливают в 1 л изопропанола. Выпавший осадок выделяют и высушивают.
ВЭЖХ: колонка №2; элюент №5: 0,01 М раствор СН3СООNH4 в воде/СН3СN; градиент от 85 до 50% (объем/объем) в течение 20 мин; расход: 1 мл/мин.; время удерживания = 16 мин.
2. Удаление фталимидной группы:
20,4 г Вышеполученного твердого вещества при перемешивании и при температуре 80°С вносят в 80 мл N,N'-диметилацетамида, после чего добавляют 1,6 мл гидразингидрата в виде раствора в 20 мл воды. После выдерживания в течение 3 ч при этой температуре реакционную среду при комнатной температуре выливают в 1 л этанола. Выпавший осадок выделяют, высушивают, затем растворяют в 40 мл воды. При температуре О°С добавляют примерно 2 мл 6н. водного раствора HCl для снижения значения рН до 2; среду фильтруют через целит®, затем очищают путем пропускания через ионообменные смолы (анионный Амберлит® и катионный IМАС® ). Получают тогда 6 г искомого продукта. ВЭЖХ: колонка №2; элюент №5; время удерживания = 24-29 мин.
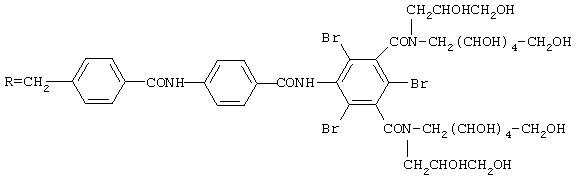
Соединение F: RNH2, где
(а) При температуре 120°С 50 г 1-дезокси-1-(2,3-дигидроксипропил)амино-D-галактита растворяют в 300 мл диметилацетамида, затем при температуре 80°С быстро добавляют 38 г хлорангидрида 5-(фталимидоацетамидо)-2,4,6-трииодоизофталевой кислоты (полученной согласно патенту США 4283381) и 17 мл триэтиламина. После перемешивания в течение 5 ч при температуре 80°С среду фильтруют при комнатной температуре и фильтрат выливают в 800 мл изопропилового спирта и осадок выделяют и высушивают.
Избыток исходного аминоспирта удаляют путем хроматографии водного раствора этого осадка на ионообменной смоле в форме Н+.
Осуществляют гидразинолиз фталимидной группы в водной среде для полученияN,N'-[(2,3,4,5,6-пентагидроксигексил)(2,3-дигидроксипропил)]-2,4,6-трииод-5-глициламиноизофталимида.
(b) К 70 г полученного на стадии (а) амина в виде раствора в 200 мл диметилацетамида добавляют 23 г хлорангидрида 5-(фталимидоацетамидо)-2,4,6-трииодоизофталевой кислоты и 12 мл триэтиламина. После перемешивания в течение 24 ч при комнатной температуре реакционную среду выливают в 1500 мл изопропилового спирта и выделяют выпавший осадок.
Таким образом, полученный сырой фталимид в виде раствора в 160 мл воды с рН 5 хроматографируют на 650 г смолы Амберлит® ХАD 16, элюируя смесью вода/метанол (в соотношении 65/35 по объему). Удаление защитной группы от аминогруппы фталимида осуществляют путем гидразинолиза: 57 г фталимида обрабатывают с помощью 3 мл NН2-NН2 в 200 мл воды при температуре 80°С; спустя 3 ч фталилгидразид осаждают при рН 1 и фильтрат выпаривают, получая масло, которое очищают путем осаждения из этанола и хроматографирования на анионообменной смоле (слабо основной Амберлит®) и катионообменной смоле (IМАС НРIII фирмы Rohm и Нааs).
Аналитическая ВЭЖХ: колонка №2; элюент №8: Н2О/СН3СN; градиент от 95 до 80% в течение 45 мин; время удерживания: 33-49 мин.
В тех же условиях для полученного на стадии (а) амина время удерживания составляет 6-16 мин.
Можно получать другую смесь изомеров соединения F при применении вышеуказанной методики к 1-дезокси-1-(2,3-дигидроксипропил)амино-D-глюциту. Аналитическая ВЭЖХ: колонка №2; элюент №8.
Соответствующий продукт, полученный на стадии (а): время удерживания = 7-24 мин; конечный продукт: время удерживания = 30-40 мин.
Ниже, в качестве иллюстрации описывается получение некоторых хелатов согласно изобретению.
Пример 1
3,6,9,15-Тетраазабицикло[9,3,1]пентадека-1(15), 11,13-триен-3,6,9-три(б-глутаровая) кислота (формула (V)) и комплекс с гадолинием:
1. Растворяют 20 г гетероцикла 3,6,9,15-тетраазабицикло[9,3,1]пентадека-1(15),11,13-триена в 570 мл ацетонитрила и при температуре 80°С добавляют 34 г Nа2СО3, затем постепенно 77 г метил-α-бромглутарата в виде раствора в 110 мл ацетонитрила. После выдерживания среды в течение 24 ч при ее температуре кипения с обратным холодильником ее отфильтровывают при температуре около 20°С и фильтрат выпаривают досуха. Оставшееся твердое вещество очищают путем хроматографии на диоксиде кремния, элюируя смесью этилацетат / гептан (в соотношении 6/4 по объему). Масса = 25 г.
2. Гидролиз
В течение 3 дней при температуре 70°С выдерживают 5,8 г полученного на предыдущей стадии продукта в 15 мл метанола, к которому добавлены 42 мл 5н. водного раствора гидроксида натрия. После добавления 100 мл воды значение рН раствора доводят до 6,5 путем добавления катионообменной смолы (Н+), затем после фильтрации раствор вводят в контакт с анионообменной смолой в форме ОН-. Продукт высвобождают из смолы с помощью смеси уксусная кислота / вода (в соотношении 50/50 по объему).
ВЭЖХ: колонка №2; элюент №6: 0,037н. раствор H2SO4 в воде /СН3СN; градиент от 100% до 20% (объем/объем) в течение 50 мин; расход составляет 1 мл/мин, время удерживания = 12,5 - 13,5 мин (несколько пиков).
3. Комплексообразование:
3 г Кислоты и 1,9 г GdCl3 растворяют в 54 мл воды. Среду доводят до температуры 55°С и значение рН доводят до 5 путем добавления 1н. водного раствора гидроксида натрия. После выдерживания в течение 3 ч при температуре 60°С среду фильтруют при температуре около 20°С, затем выливают в 100 мл этанола. Выделяют натриевую соль комплекса с выходом 70%.
ВЭЖХ: колонка №2; элюент №6; время удерживания = 15-16 мин (несколько пиков).
Пример 2
Соединение формулы (I), где

В 10 мл воды вводят 0,5 г полученного в примере 1 комплекса и 3,15 г амина RNH2. Значение рН доводят до 6 путем добавления 0,1н. водного раствора гидроксида натрия, после добавляют 2 эквивалента гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI). После выдерживания в течение 3 ч при температуре 40°С среду выливают в 100 мл этанола и осадок выделяют.
Очистку осуществляют путем ультрафильтрации водного раствора соединения через мембрану из полиэфиросульфона, с порогом разрыва 3 КДа, с последующей хроматографией на колонке с силанизированным диоксидом кремния LiChroprep® (стандартная смесь RP2 и RР18 (в соотношении 50/50)) фирмы Merck, элюируя смесью вода / СН3СN (градиент от 98 до 95%). Масса = 0,8 г.
ВЭЖХ: колонка №2; элюент №7: H2O / СН3СN; градиент от 98 до 70% (объем/объем) в течение 20 мин; расход составляет 1 мл/мин.; время удерживания = 10 мин.
Пример 3
Соединение формулы (I), где

А) Производное галактозы
8,3 г Соединения А и 1,8 г полученного в примере 1 комплекса растворяют в 100 мл воды. После доведения значения рН до 6 путем добавления 1н. водного раствора НСl добавляют 2,7 г ЕDСI и 150 мг натриевой соли (N-гидроксисукцимидил)-3-сульфокислоты (NHS). После перемешивания в течение 2 ч раствор выливают в 300 мл этанола и выпавший осадок выделяют. Он может быть очищен путем препаративной высокоэффективной жидкостной хроматографии на Lichrospher® С 18-100Е- 12 мкм (фирма Merck) или путем ультрафильтрации. Аналитическая ВЭЖХ: колонка №2; элюент №7; время удерживания = 12 мин.
А') Производное глюкозы:
Путем взаимодействия соединения А' с полученным в примере 1 комплексом, в тех же рабочих условиях, что и в случае А, получают сырое соединение, соответствующее формуле (I). Оно может быть очищено путем хроматографии на колонке Purospher® RP18, элюируя смесью Н2О/СН3СN. Конечный выход: 40%.
Пример 4
Соединение формулы (I), где

2,5 г Полученного согласно примеру 1 комплекса и 11 г соединения В растворяют в 60 мл воды; после добавления 1н. водного раствора НСl вплоть до достижения рН 6 добавляют 45 мл ацетона и 0,5 г гидрата гидроксибензотриазола (НОВТ) и 2,3 г EDCI. Реакционную смесь перемешивают в течение 4 ч при температуре около 20°С, приводя время от времени значение рН к 6 путем добавления 1н. водного раствора гидроксида натрия. Раствор выливают в 500 мл этанола и выделенный осадок высушивают. Масса составляет 11,7 г.
Продукт очищают путем препаративной хроматографии под давлением на колонке Lichrospher® С 18-10 мкм; 1=25 см; d = 50 мм (фирма Merck), элюируя смесью вода / ацетонтрил с градиентом от 95 до 90% в течение 20 мин, затем - 85% в течение 25 мин; расход составляет 80 мл/мин. Выход = 45%.
Аналитическая ВЭЖХ: колонка №2; элюент №8: Н2О/СН3СN; градиент от 95 до 80% в течение 45 мин; время удерживания = 31 мин.
Пример 5
Соединение формулы (I), где

получают согласно способу, описанному в примере 4, но при использовании соединения С.
Аналитическая ВЭЖХ: колонка №2; элюент №8; время удерживания = 25-29 мин.
Пример 6
Соединение формулы (I), где
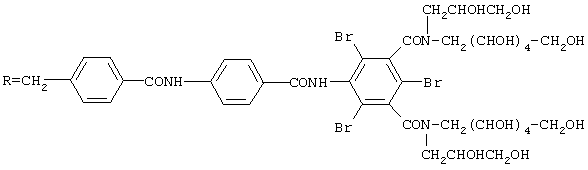
1,6 г Полученного согласно примеру 1 комплекса и 7 г амина Е растворяют в 40 мл воды. При значении рН, равном 7, добавляют 0,3 г НОВТ и 1,5 г EDCI, затем 30 мл ацетона. Среду перемешивают в течение 4 ч, поддерживая значение рН около 6. Осадок, полученный путем выливания раствора в 450 мл этанола, очищают путем препаративной хроматографии высокого давления на колонке Purospher®; L = 250 мм; d = 40 мм; RP18; 10 мкм; 120 (фирма Merck); элюируя смесью вода / ацетонитрил (градиент от 90 до 80%) в течение 5 мин); расход = 80 мл/мин.
(фирма Merck); элюируя смесью вода / ацетонитрил (градиент от 90 до 80%) в течение 5 мин); расход = 80 мл/мин.
Аналитическая ВЭЖХ: колонка №3: Purospher®; RР18 с блокированными остаточными силанильными группами; 5 мкм; L = 250 мм; d = 4 мм; (фирма Merck); элюент №7, но градиент от 85% до 70% в течение 10 мин (объем/объем) по истечении 20 мин; время удерживания = 29 мин.
Пример 7
Соединение формулы (I), где

Продукт получают при применении метода, описанного в предыдущих примерах, при использовании амина D.
Аналитическая ВЭЖХ: колонка №4: Zorbax® 3005В - С18; 1=250 мм, d = 4 мм (фирма Hewlett® Packard; элюент №1, но градиент от 90 до 82% в течение 60 мин; время удерживания = 42-49 мин.
Пример 8
Соединение формулы (I), где

Продукт получают при использовании условий амидирования, аналогичных таковым в предыдущих примерах.
Аналитическая ВЭЖХ: колонка №2; элюент №7, но градиент от 98 до 85% в течение 40 мин; время удерживания = 24 мин.
Пример 9
Соединение формулы (I), где

Аналитическая ВЭЖХ: колонка №2; элюент: H2O/СН3СN в соотношении 95/5 по объему, затем спустя 20 мин градиент для достижения соотношения 85/15 в течение 10 мин; расход = 1 мл/мин; время удерживания = 26 мин.
Пример 10
Соединение формулы (I), где

получают путем введения во взаимодействие соединения F с комплексом примера 1:
1 г Комплекса и 12 г амина Р растворяют в 200 мл воды и значение рН доводят до 6 путем добавления 6н. водного раствора НСl. Тогда добавляют 0,1 г EDCI и 0,25 г НОВТ; среду выдерживают при перемешивании в течение 48 ч, поддерживая значение рН равным 6 путем добавления 2%-ного водного раствора NаНСО3. Конечный сырой продукт выделяют путем введения среды в 10 объемов этанола и очищают путем препаративной хроматографии на колонке Purospher®; RP18, 10 мкм; 120  (фирма Merck), элюируя смесью Н2О/СН3СN. Выход: 30%.
(фирма Merck), элюируя смесью Н2О/СН3СN. Выход: 30%.
Аналитическая ВЭЖХ: колонка №2 - элюент №8; время удерживания = 12 мин.
Изобретение относится к металлохелатам с производными, содержащими четыре атома азота макроцикла, конденсированного с пиридиновым циклом, способам их получения и их применению в медицине для получения изображения. Техническим результатом является получение новых соединений с релаксируемостью r1 в 10-15 раз выше релаксируемости имеющихся в продаже соединений не только в случае магнитного поля 0,5 Тл, но и также 1 Тл, поля большинства современных приборов для получения изображения, и даже 1,5 Тл, поля наиболее эффективных приборов. 4 н. и 11 з.п. ф-лы.

в которой R означает группу формулы (II)

Z означает связь или группу, выбираемую из групп CH2, СН2-CO-NH или (CH2)2-NH-CO;
Z’ означает связь или группу, выбираемую из групп О, S, NQ, CH2, СО, CO-NQ, NQ-CO, NQ-CO-NQ, CO-NQ-CH2-CO-NQ;
Z” означает связь или группу, выбираемую из групп CO-NQ, NQ-CO, CO-NQ-CH2-CO-NQ;
р и q означают целые числа, сумма которых составляет от 0 до 3;
R1, R2, R3, R4 и R5 независимо друг от друга выбирают из групп Н, Br, Cl, I, CO-NQ1Q2 и NQ1-CO-Q2, причем Q1 и Q2, одинаковые или разные, означают Н или (С1-С6)-алкил, возможно прерываемый атомом или атомами кислорода и по меньшей мере один из радикалов R1-R5 означает амидогруппу или R1, R3 и R5 независимо друг от друга означают Н, Br, Cl или I и R2 и R4, одинаковые, означают группу формулы (III)

где Z’” означает группу, выбираемую из групп CO-NQ, CO-NQ-CH2-CO-NQ, CO-NQ-CH2, CO-NQ-(CH2)2-NQ-CO и NQ-CO-NQ;
R’1, R’3 и R’5, одинаковые или разные, означают Н, Br, Cl или I;
Q’1 и Q’2, одинаковые или разные, означают Н или (С1-С6)-алкил, возможно прерываемый атомом или атомами кислорода;
Q означает Н или (С1-С4)-алкил,
при условии, что алкильные группы могут быть при необходимости моно- или полигидроксилированными.
R1=R3=R5=Br; R2=R4=CON(CH2(CHOH)4CH2OH)2 или

CON-CH2(CHOH)4CH2ОH
CH2-CHOH-CH2OH.
Z’=CONH; Z”=CONH-CH2CONH; R1=R3=R5=Br; R2=R4=CON(CH2(CHOH)4CH2OH)2.
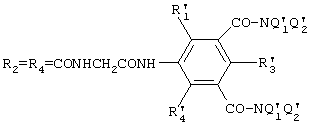
Q’1=Q’2=CH2(CHOH)4CH2OH или Q’1=CH2(CHOH)4CH2OH и Q’2=CH2-CHOH-CH2OH.
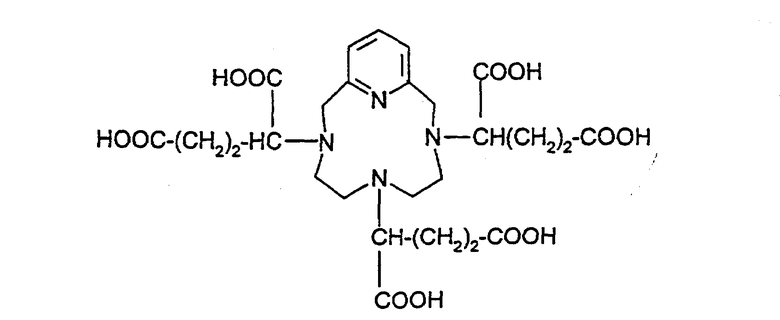
и его соли с неорганическим или органическим основанием.
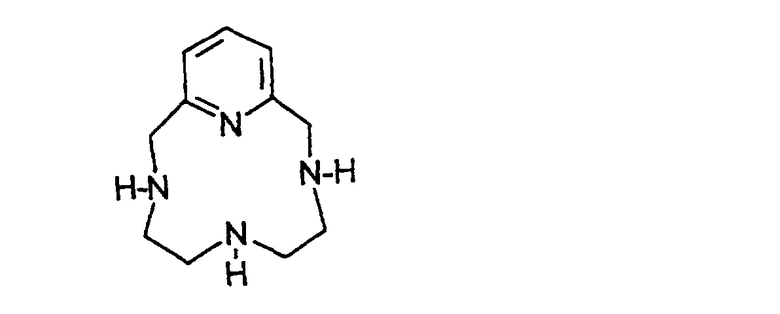
с соединением формулы R’OOC-CHX-(CH2)2-COOR’, в которой Х означает удаляемую группу и R’ означает Н или (С1-С3)-алкил, и в осуществлении гидролиза сложноэфирных функциональных групп, когда R’ отличается от Н, с последующим введением во взаимодействие соли или оксида металла для получения хелата образовавшегося продукта или одной из его солей с основанием и введением во взаимодействие хелата с амином RNH2 в присутствии агента, активирующего карбоксильные группы.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

