Изобретение касается аминогуанидинов, применяемых в фармакологии, способов их получения, включающих их фармацевтических композиций и использования их в качестве фармацевтических препаратов.
Более точно, настоящее изобретение относится к новым соединениям формулы (I)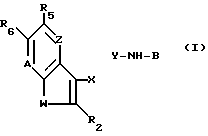
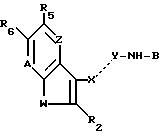
где W S или -NR1, у которого R1 водород, C1-C6алкил или ацил;
R2 водород, галоген или C1-C6алкил;
R5 водород, галоген, C1-C6алкил, гидрокси, нитро, амино, C1-C4алкиламино, ациламино, C2-C6алкоксикарбонил, SO2NRaRb, у которого каждый Ra и Rb независимо водород или C1-C6алкил; циано или триметилсилил;
или, если A -CR7= R5 также C1-C6алкил, замещенный -SO2 -C1-C6алкилом, -SO2NRaRb, -CONRaRb, -NH-SO2-C1-C6алкилом, -N(C1-C6алкил)-SO2-(C1-C6алкил),
-NRaR'b, у которого R'b водород, C1-C6алкил, C3-C7 циклоалкил, C3-C6 алкенил, фенил или фенил-C1-C3 алкил, у которого фенильное кольцо возможно замещено C2-C6 алкоксикарбонил, -PO(C1-C4алкил)2 или гетероциклический радикал; карбокси; CONRaRb; -PO(C1-C4 алкил)2; OCONRcRd, у которого каждый из Rc и Rd независимо являются C1-C6 алкилом; или гетероциклический радикал;
R6 водород или, если R5 OH, то R6 водород или галоген;
Z -CR4= у которого R4 водород, галоген, гидрокси или C1-C6 алкил или, если R5 водород или гидрокси, то Z - также -N=
A -N= или -CR7= у которого R7 водород, галоген, C1-C6 алкил и C1-C6 алкокси,
или R1 и R7 вместе представляют -(CH2)m или -X3(CH2)p-, где m 2, 3 или 4, p 2 или 3 и X3 - кислород, S или -N(C1-C6 алкил)-, причем X3 присоединен к 6-членному кольцу;
X--Y -CR8=N или -CH(R8)-NH-, у которого R8 - водород или C1-C6 алкил; и
B радикал формулы (a) или (b)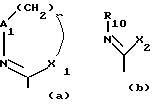
где n 1 или 2;
A1 C=0 или CH2;
X1 S, NR11 или CR12R13, у которого R11 водород или ацил, а каждый из R12 и R13 независимо водород, C1-C4 алкил или C5-C7 циклоалкил;
R10 водород; C1-C12 алкил; C1-C6алкил, замещенный гидрокси, арилом, арилокси, адамантилом, гетероциклическим радикалом, -NR15-CO-R16 или -NH-SO2-арилом; C5-C7циклоалкил; арил; адамантил; ацил или -CONHR14, в котором R14 C1-C10 алкил, C5-C7 циклоалкил, C5-C7 циклоалкил, C5-C7 циклоалкил-C1-C4 алкил, арил, арил-C1-C4 алкил или гетероциклический радикал, R15 - водород или C1-C4 алкил, и R16 C1-C6 алкил, C5-C7 циклоалкил, C5-C7 циклоалкил-C1-C4 алкил, арил или арил-C1-C4 алкил, и
X2 -SR20 или -NR3R'10, у которого R20 C1-C6 алкил, R3 водород или C1-C6 алкил, и R'10 имеет одно из значений, данных выше для R10, или R3 и R'10 вместе с атомом азота, к которому они присоединены, образуют 5-, 6- или 7-членный насыщенный или неароматический ненасыщенный гетероцикл, который может включать дальнейший гетероатом, выбранный из N, S и O, и которые могут быть далее свернуты в бензольное кольцо,
при условии, что:
I) если B радикал формулы (b), только один из R10 и R'10 может быть другим, чем водород, а X2 может быть -SR20, только если R10 водород, и
II) R5 не водород, если B радикал формулы
в которой R'10 4-метилфенил в виде арила, каждый из Z и A являются -CH= W--NH-, R2 и R6 каждый водород, и X --Y это CH=N-,
и его физиологически гидролизуемый или приемлемый сложный или простой эфир, если R5 гидрокси, в свободном виде или в виде соли.
Выражение "физиологически гидролизуемые и приемлемые сложные или простые эфиры" в применении к соединениям формулы I, когда R5 гидрокси, означает эфиры, у которых R5 этерифицирован, и которые могут гидролизоваться при физиологических условиях с получением спирта или кислоты, которые являются физиологически приемлемыми, то есть нетоксичными при желаемых уровнях дозировки.
Примеры эфирной группы в качестве R5 включают, например, C1-C6 алкокси, C1-C6 алкокси, замещенный гидрокси, C1-C4 алкокси, ацилокси, NRaR'b, CONRaRb, у которого Ra, Rb и R'b такие, как определено выше; C2-C6 алкенилокси, Z может быть такжеN-, если R5 эфирная группа.
Примеры сложного эфира качестве R5 включают, например, ацилокси и пиридил-карбонилокси. Если R5 сложный эфир, то он предпочтительно является пиридил-карбонилокси. Если A CR7= то R5 в качестве эфирной группы является предпочтительно ацилокси или пиридил-карбонилокси.
У соединений формулы I алкильные цепи могут быть разветвленными или прямыми. Если R5, R10 или R'10 замещены алкилом, то заместитель предпочтительно размещен на конце алкильной цепи.
Под галогеном предпочтительно понимают фтор или хлор. Если R5 - это гидрокси-замещенный C1-C6 алкокси, то он может также быть алкокси, полизамещенным гидрокси, например, 2,3-дигидрокси-пропокси.
Арил предпочтительно фенил или нафтил, предпочтительно фенил и может быть замещен. Арил-C1-C4 алкил, предпочтительно - фенил-C1-C4 алкил, например бензил или фенэтил, и может быть замещен на фенильном кольце.
Арилокси предпочтительно фенокси, и может быть замещен. Арил-C1-C6 алкокси является, например, бензилокси, и может быть замещен на фенильном кольце. Если арил или арильные группы замещены, то они могут быть моно- или полизамещенными, например, галогеном, C1-C4 алкилом или C1-C6 алкокси. Примерами являются, например, фенил или фенильные группы, моно- или полизамещенные хлором, метилом или метокси.
Ацильные группы или ацилы в составе ацилокси являются предпочтительно RCO, где R -C1-C10 алкил, C2-C10 алкенил, C5-C7 циклоалкил или арил, предпочтительно C1-C10 алкил.
Если каждый из R1 и R11 независимо являются ацилом, то это предпочтительно R'CO, где R' C1-C6 алкил, фенил или фенил-C1-C4алкил, в частности C1-C6 алкил. Если R10 ацил, то это предпочтительно R"CO, где R" это C1-C10 алкил, фенил или фенил-C1-C4 алкил, в частности C1-C10 алкил. Если R5 ацилокси, то это предпочтительно R'-CO-O.
Гетероциклический радикал в виде R5 является, например, радикалом, происходящим от оксазола, тиазола, изоксазола, оксадиазола или тиадиазола. Если R5 является гетероциклическим радикалом или включает такой радикал, предпочтительно радикал формулы (a)
где t 0, 1, 2 или 3;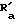 водород или C1-C6 алкил;
водород или C1-C6 алкил;
X' N или CH;
Y O; и
Z' N.
Гетероциклический радикал в виде R10, 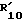 или образованный совместно R3 и
или образованный совместно R3 и  и атом азота, к которому они присоединены, является предпочтительно радикалом, происходящим от 5- или 6-членного насыщенного, ароматического или ненасыщенного гетероцикла, предпочтительно сконденсированного с бензольным кольцом, например пиридин, имидазол, бензимидазол, пирролидин, пирролидон, пиперидин, пиразин или пергидроиндол,
и атом азота, к которому они присоединены, является предпочтительно радикалом, происходящим от 5- или 6-членного насыщенного, ароматического или ненасыщенного гетероцикла, предпочтительно сконденсированного с бензольным кольцом, например пиридин, имидазол, бензимидазол, пирролидин, пирролидон, пиперидин, пиразин или пергидроиндол,
или радикал формулы (c), (d) или (e)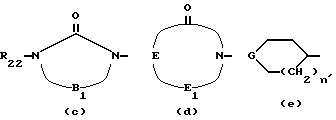
у которых R22 водород или C1-C4 алкил;
B -CH2CH2-, -COCH2- или -(CH2)3-, у которых один или два H могут быть замещены посредством C1-C4 алкила или 1,2-фениленом;
E -CH2CH2-, -CH2N(R17)- или -(CH2)3-, у которых один или два H могут быть замещены C1-C6 алкилом или 1,2-фениленом;
E1 CO или CH2;
R17 водород или CC1-C4 алкил;
G CO, -CHCOOR18, -CHCOR19, 5,5-диметил-1,3-диоксан-2-илиден- или 1,3-диоксолан-2-илиден, где R18 - водород или C1-C6-алкил, а R19 C1-C6 алкил; и n' 0 или 1.
Этот гетероциклический радикал может быть далее замещенным, например, галогеном.
Примерами алкила, замещенного гетероциклическим радикалом, являются, например, 2-(2-пирролидон-1-ил)-этил, 3-бензимидазоллил-пропил.
Если B является радикалом (b), у которого R10 водород, а X2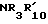 предпочтительно R3 и
предпочтительно R3 и 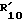 оба не являются водородом.
оба не являются водородом.
У соединений формулы I следующие значения являются предпочтительными либо индивидуально, либо в любом сочетании или под-сочетании:
1. W S, -NH-, -NCH3- или -NC2H5-. Более предпочтительно W -NH.
2. R2 H, CH3, C1 или Br. Более предпочтительно, R2 H.
3. Z CR4=
4. R4 H или C1-C4 алкил, предпочтительно H или метил.
5. Z -N= R5-гидрокси или C1-C4 алкокси, и A - CR7=
6. R5 H; -OH; или C1-C6 алкокси; или, если A - -CR7= то R5 также C1-C6-алкил, замещенный -SO2-C1-C6 алкилом, -SO2NRaRb, -CONRaRb, -NH-SO2-C1-C6 алкилом, -N(C1-C6 алкил)-SO2-C1-C6 алкилом или PO(C1-C4 алкил)2; ацилокси; карбокси; CONRaRb; -PO(C1-C4 алкил)2; или OCONRcRd.
7. A -CR7=
8. R7 H или CH3.
9. X -- Y -CR8=N-.
10. R8 H или CH3.
11. B радикал формулы (a), предпочтительно радикал формулы (a), в которой X1- -NH-.
12. B является радикалом формулы (b).
13. R10 H.
14. X 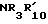 .
.
15. R3 водород или C1-C4 алкил.
16. 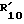 водород, C1-C10 алкил, R"CO, CONHR14, -(CH2)1-5-NH-CO-R16 или C1-C6-алкил, замещенный в ω арилом, радикалом формулы (d) или бензимидазолилом. Более предпочтительно
водород, C1-C10 алкил, R"CO, CONHR14, -(CH2)1-5-NH-CO-R16 или C1-C6-алкил, замещенный в ω арилом, радикалом формулы (d) или бензимидазолилом. Более предпочтительно 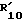 C1-C12 алкил.
C1-C12 алкил.
17. R3 и 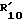 вместе с атомом азота, к которому они присоединены, образуют 5-, 6- или 7-членную, насыщенную или неароматически ненасыщенную гетероциклическую группу, которая может включать далее гетероатом, выбранный из N, S и O, и которая может быть далее сконденсирована с бензольным кольцом. Более предпочтительно R3 и
вместе с атомом азота, к которому они присоединены, образуют 5-, 6- или 7-членную, насыщенную или неароматически ненасыщенную гетероциклическую группу, которая может включать далее гетероатом, выбранный из N, S и O, и которая может быть далее сконденсирована с бензольным кольцом. Более предпочтительно R3 и  вместе с атомом азота, к которому они присоединены, являются пиперидином или пергидроиндолом.
вместе с атомом азота, к которому они присоединены, являются пиперидином или пергидроиндолом.
18. Радикал формулы (d) является
Одна группа соединений по изобретению является группой соединений формулы I, в которой W, R2, A, X -- Y и B такие, как определено выше, Z - CR4= как определено выше, и R5 водород; C1-C6 алкил, гидрокси, C1-C6 алкокси; C1-C6 алкокси, замещенный гидрокси, C1-C4 алкокси, ацилокси, 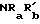 , CONRaR'b или CSNRaRb, где каждый из Ra и Rb независимо являются водородом или C1-C6 алкилом, а
, CONRaR'b или CSNRaRb, где каждый из Ra и Rb независимо являются водородом или C1-C6 алкилом, а 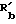 водород, C1-C6 алкил, C3-C7 циклоалкил, C3-C6 алкенил, фенил или фенил-C1-C3 алкил, где фенольное кольцо возможно замещено; C2-C6 алкенилокси; пиридил-карбонилокси; нитро; амино; C1-C4 алкиламино; ациламино; C2-C6 алкоксикарбонил; SO2NRaRb; циано, или триметилсилил;
водород, C1-C6 алкил, C3-C7 циклоалкил, C3-C6 алкенил, фенил или фенил-C1-C3 алкил, где фенольное кольцо возможно замещено; C2-C6 алкенилокси; пиридил-карбонилокси; нитро; амино; C1-C4 алкиламино; ациламино; C2-C6 алкоксикарбонил; SO2NRaRb; циано, или триметилсилил;
или, если A -CR7= R5 также является C1-C6 алкилом, замещенным -SO2-C1-C6 алкилом, SO2NRaRb, CONRaRb, -NH-SO2-C1-C6 алкилом, -N (C1-C6 алкил)-SO2-(C1-C6 алкил), 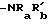 , C2-C6 алкоксикарбонилом или -PO(C1-C4 алкил)2; ацилокси; карбокси; CONRaRb; -PO(C1-C4 алкил)2; или OCONRcRd, где каждый из Rc и Rd независимо является C1-C6 алкилом.
, C2-C6 алкоксикарбонилом или -PO(C1-C4 алкил)2; ацилокси; карбокси; CONRaRb; -PO(C1-C4 алкил)2; или OCONRcRd, где каждый из Rc и Rd независимо является C1-C6 алкилом.
Особенно предпочтительными соединениями формулы I являются такие соединения, у которых W NH, R2 H, Z -CH= или -CCH3= A - -CH= или -CCH3= R5 водород, гидрокси, C1-C6 алкокси, C1-C6 алкил, замещенный -SO2-C1-C6 алкилом, SO2NRaRb-CONRaRb, -NH-SO2-C1-C6 алкилом, N (C1-C6 алкил)-SO2-C1-C6 алкилом, или -PO(C1-C4 алкил)2, ацилокси, карбокси, CONRaRb, -PO(C1-C4 алкил)2 или OCONRcRd.
Также особенно предпочтительными являются соединения формулы I, у которых W NR1; R2 H; Z -N= A -CH= или -CCH3; R -OH или C1-C6 алкокси.
Еще более предпочтительными соединениями формулы I являются те из них, у которых W, R2, Z, A и R5 имеют одно из значений, данных выше, а B является радикалом 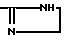 или радикалом формулы (b).
или радикалом формулы (b).
Соединения формулы I могут существовать в свободном виде, в виде соли, а также в виде сольвата или гидрата. Соли могут включать кислотно-аддитивные соли и соли, которые могут быть получены, когда R5 карбокси. Пригодные фармацевтически приемлемые кислотно-аддитивные соли для применения в соответствии с настоящим изобретением, как описано выше, включают, например, хлоргидратные, сульфатные, ацетатные, оксалатные, малеинатные и фумаратные соли. Если R5 карбокси, то подходящими солями являются, например, соли щелочных металлов, таких как натрий или калий, или соли замещенного или незамещенного аммония.
Следует иметь в виду, что соединения формулы I, в которой X -- Y это -CR8=N- и B радикал формулы (b')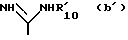
могут существовать как таутомеры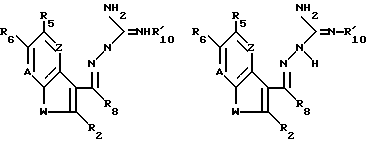
где R2, R5, R6, R8, A, W, Z и 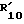 такие, как описано выше.
такие, как описано выше.
Соединения формулы I, в которой Z -N= и R5 гидрокси, также могут существовать как таутомеры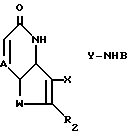
где W, A, R2, B и X -- Y такие, как определено выше.
Следует учитывать, что когда возникают таутомерные формы, то изобретение охватывает все таутомерные формы и их смеси, то есть хотя соединения формулы I определены для удобства со ссылкой только на одну гуанидиновую и одну оксо-форму, следует понимать, что изобретение никоим образом не ограничивается конкретной номенклатурой и использованным графическим изображением. То же касается и исходных материалов, проявляющих гуанидин-таутомеризм, как описано выше.
Кроме того, настоящее изобретение также относится к способу получения соединений формулы I, предусматривающему:
a) для получения соединения формулы I, в которой X -- Y это -CR8=N-, реакцию соединения формулы II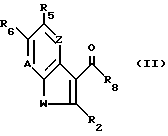
где A, W, Z, R2, R5, R6 и R8 такие, как определено выше с соединением формулы III: H2N-NHB, где B такой, как определено выше, или
b) для получения соединения формулы I, в которой X -- Y это -CHR8=NH-, гидрогенирование соединения формулы I, в которой Y -- X это -CR8=N-;
c) для получения соединения формулы I, в которой B радикал формулы (b'), алкилирование, ацилирование или карбамоилирование соединения формулы Ia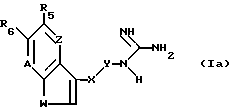
где A, W, Z, R2, R5, R6 и X -- Y такие, как определено выше,
d) для получения соединения формулы I, в которой R5 гидрокси, расщепление по эфирной связи соединения формулы Ib,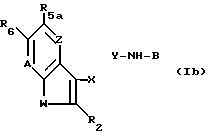
где A, W, Z, R2, R6, X -- Y и B такие, как определено выше, и R5a расщепляемая эфирная группа; или
e) для получения физиологически гидролизуемого и физиологически приемлемого простого или сложного эфира соединения формулы I, в которой R5 гидрокси, этерифицирование или ацилирование соединения формулы I, где R5 гидрокси, и выделение соединений формулы I или их физиологически гидролизуемых и физиологически приемлемых эфиров, полученных таким образом в свободном виде или в виде сольвата или гидрата.
В способе, стадия a) может осуществляться аналогично известным методам, например удобно в присутствии кислоты, например неорганической кислоты, такой как хлорводородная или бромводородная кислота, или органической кислоты, такой как уксусная, p-толуол-сульфоновая или пиридин-p-толуолсульфоновая кислота. Реакция может быть удобно осуществлена в присутствии протонном растворителя, например метанола, этанола или изопропанола. Реакцию преимущественно осуществляют при температуре между комнатной и кипения.
Стадия b) может быть проведена согласно известным методам гидрогенирования. Если R5 бензилокси, то он может быть одновременно присоединен к гидрокси-группе.
Стадия c) может быть проведена путем известных в данной области методов. Алкилирование или ацилирование соединений формулы Ia может быть удобно осуществлено путем реакции с алкилом, циклоалкилом или арил-галогенидом или ацил-гелогенидом или ангидридом соответственно, предпочтительно в присутствии основания, например, триэтиламина или основания Хунига (Hunig). Карбамоилирование может быть удобно проведено путем реакции с изоцианатом, таким как R14NCO, предпочтительно, в присутствии растворителя, например, диметилформамида.
Стадия d) способа может быть проведена аналогично известным в данной области методам эфирного расщепления. Если R5a бензилокси, то она (стадия d) может быть удобно осуществлена посредством гидрогенирования в присутствии катализатора, например палладия на древесном угле. Эта реакция может проводиться в растворителе, например в спирте, при температуре примерно от комнатной до 60oC.
R5a может быть алкокси, замещенный алкокси, алкенилокси или бензилокси.
Стадия e) способа может, например, осуществляться путем реакции соединения формулы I, в которой R5 гидрокси, с ацилгалогенидом, предпочтительно с ацил-хлоридом. Соединения формулы I, в которой R5 - пиридил-карбонилокси, могут быть приготовлены путем реакции соединения формулы I, в которой R5 гидрокси, с галогенидом никотиновой кислоты. Эта реакция может быть удобно осуществлена в растворителе, таком как трифторуксусная кислота или трифторметан-сульфоновая кислота.
Исходные материалы формул II или III либо уже известны или могут быть получены аналогично методам, известным и применяемым в данной области.
Например, соединения формулы II, в которой W -NR1-, могут быть получены согласно следующей схеме реакции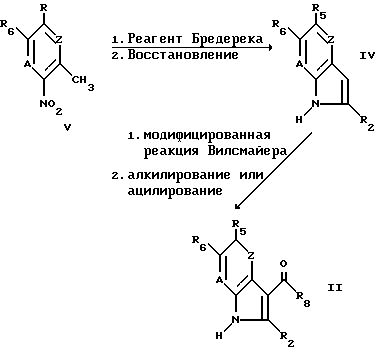
Соединения вышеуказанной формулы IV могут быть удобно получены путем реакции соединения формулы V с реагентом Бредерека, например, (CH3)2NCH(OCH3)2, в отсутствие растворителя или в присутствии растворителя, такого как пирролидин, с последующим восстановлением, например, водородом в присутствии палладиевого катализатора или гидразином в присутствии никеля Ренея (Raney).
Соединения формулы II, в которой W -NR1-, могут быть удобно получены путем подвергания соединения формулы IV модифицированной реакции Вилсмайера, и затем алкилированию или ацилированию.
Модифицированная реакция Вилсмайера может быть осуществлена с использованием диметил-алкиламида в присутствии POCl3, согласно известным в данной области методам. Алкилирование или ацилирование может быть осуществлено известным образом, например, в присутствии основания, например, K2CO3 или C2H5MgBr, в растворе, таком как диметилформамид или тетрагидрофуран.
Соединения формулы III, в которых B радикал формулы (b), в которой X2 не SR20, могут быть удобно получены путем реакции соединения формулы VI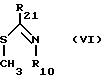
в которой R10 такой, как определено выше, и
R21 либо 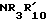 или NHNH2, либо с гидразином, если R21 это
или NHNH2, либо с гидразином, если R21 это 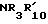 , либо с амином формулы
, либо с амином формулы 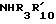 , если R21 это -NHNH2.
, если R21 это -NHNH2.
Реакцию можно преимущественно проводить посредством нагревания при температуре кипения. Ее можно удобно проводить в растворителе, например в спирте, таком как метанол или этанол, вода или диметилформамид, в отсутствии или в присутствии щелочного соединения, например, гидроксида или карбоната калия.
Соединения формулы III, в которой B является радикалом формулы (b), в которой X2 это -SR20, могут быть удобно приготовлены путем алкилирования соединения формулы VII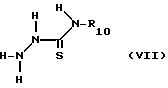
с помощью R20-дел соединения, в соответствии с известными методами.
Поскольку получение исходных материалов не описывается подробно, то следует использовать известные соединения или соединения, которые могут быть получены аналогично известным и используемым в данной области методам, или как раскрыто в следующих примерах.
Эти примеры иллюстрируют изобретение, но не ограничивают его, и все температуры, указанные в них, даны в градусах Цельсия.
Использованы следующие сокращения: ТГФ тетрагидрофуран, ДМФ - диметилформамид, EтOH этиловый спирт, MeOH метиловый спирт, AcOEт - этиловый ацетат, (II) образование пены, (III) образование шлака.
Пример 1. 5-Гидрокси-индол-3-карбоксальдегидамино [3-(2'-пирролидинон-1'-ил)-пропиламино] метиленгидразон.
К раствору 0,9 г 5-гидрокси-индол-3-карбоксальдегида диаминометиленгидразона (4,1 ммоль) в 10 мл ТГФ, содержащего 2 мл ДМФ и 0,9 мл Eт3N (6,2 ммоль) при комнатной температуре добавляют 1,3 г 3-(2'-пирролидинон-1-ил)-1-бромпропана (6,2 ммоль). Смесь перемешивают до утра при 50oC. Здесь смесь охлаждают до комнатной температуры, и растворитель выпаривают. Остаток хроматографируют над SiO2 (элюент: толуол (EтOH) NH3 в соотношении 70:30: 2,5) с получением вышеуказанного в названии соединения в кристаллическом виде. Т. пл. 158oC (II). Масс-спектральный анализ (относительная интенсивность): 343,3 (MH+, 100); 217,2 (20); 168,2 (20); 143,2 (23).
Пример 2. 5-Гидрокси-индол-3-карбоксальдегид амино (N-метил-N-гептиламино)метиленгидразона.
К раствору 0,48 г 5-бензилокси-индол-3-карбоксальдегида амино (N-метил-N-гептиламино)-метилен-гидразона (1,1 ммоль) в EтOH добавляют 0,25 г 10% Pd/C. Суспензию гидрогенируют до утра при 45oC. Затем суспензию фильтруют через силикагель, растворитель выпаривают и остаток хроматографируют на силикагеле (элюент: толуол (EтOH) NH3 в соотношении 85:15:1) с получением целевого соединения. Чистый материал кристаллизуют из CH2Cl2 (гексан в соотношении 2:8). Т. пл. 110o(III). Масс-спектр: m/z: 329 (M+, 40); 128 (40); 111 (60); 73 (50).
Исходные материалы могут быть получены следующим образом:
а) К раствору 3,2 г 5-бензилокси-индол-3-карбоксальдегида (12,7 ммоль) и 5,0 г 1-(N-метил-N-гептил)-3-N'-аминогуанидина, гидроиодида (16,0 ммоль) в 100 мл MeOH добавляют при 5o, раствор MeOH/HCl до pH 3. Через 2 ч, растворитель выпаривают и остаток помещают в AcOEт. Раствор промывают раствором Na2CO3(2N). Органический слой сушат над сульфатом натрия, и растворитель выпаривают. Остаток хроматографируют (элюент: толуол (EтOH) NH3 в соотношении 85:15:0,5) с получением целевого соединения. Масс-спектр m/z (относительная интенсивность): 420 (MH+, 100); 330 (7); 249 (4); 172 (16).
б) 1-(N-метил-N-гептил)-3-N'-аминогуанидина гидроиодид.
Раствор, содержащий 4,7 г 3-метил-изотиополукарбазида гидроиодида (20 ммоль) и 3,7 мл N-метил-N-гептиламина (22 ммоль) в 30 мл метанола подвергают рефлюксу в течение 6 ч. Раствор затем охлаждают до комнатной температуры и растворитель выпаривают с получением 1-(N-метил-N-гептил)-3-N'-аминогуанидина гидроиодида. Полученный сырой материал используют для следующего этапа без дополнительной очистки.
Пример 3. 5-Гидрокси-индол-3-карбоксальдегид амино (N-циклогексилуреидо)метиленгидразона
К раствору 0,8 г 5-гидрокси-индол-3-карбоксальдегида диаминометиленгидразона (3,7 ммоль) в 20 мл ДМФ добавляют в течение 5 мин при 0o к раствору 0,5 мл циклогексил-изоцианата (4,0 ммоль) в 5 мл ДМФ. Раствор перемешивают в течение 4 ч. Растворитель затем выпаривают и остаток хроматографируют (элюент: толуол (EтOH) NH3 85:15:0,5) с получением целевого соединения в виде кристаллов. Т. пл. 135o(II). Масс-спектр: m/z (относительная интенсивность): 343 (MH+, 100); 244 (50); 218 (85); 159 (33).
Пример 4. 5-Гидрокси-6-фтор-индол-3-карбоксальдегид амино(пентиламин)-метилен-гидразона.
Целевое соединение получают по примеру 2. Т. пл. 125o(II).
5-Бензилокси-6-фтор-индол-3-карбоксальдегид, используемый в качестве исходного материала, может быть получен следующим образом:
а) 2-нитро-4-фтор-5-бензилокси-толуол.
К раствору 85,6 г 2-нитро-4-фтор-5-гидрокси-толуола (0,5 моль) в 1300 мл ацетона добавляют при комнатной температуре 138 г K2CO3 (1,0 моль). 72 мл бензил-бромида (0,6 моль) добавляют затем по каплям в течение 1 ч и полученную смесь перемешивают до утра при 60o. Растворитель выпаривают и остаток помещают в AcOEт. Преципитат удаляют фильтрованием, и раствор промывают водой. Органический слой сушат над сульфатом натрия, растворитель выпаривают, и остаток рекристаллизуют из гексана с получением 2-нитро-4-фтор-5-бензилокси-толуола. Т. пл. 95o. Масс-спектр: m/z 261 (M+).
b) 2-[1'-(N, N-диметиламино)-этен-2'-ил] -4-бензилокси-5-фтор-нитробензол.
Раствор 126 г 2-нитро-4-фтор-5-бензилокси-толуола (0,48 моль) в 200 г бис-диметиламино-т-бутокси-метана (1,15 моль) перемешивают до утра при 90o. Затем растворитель выпаривают и остаток кристаллизуют из MeOH с получением b) целевого соединения в виде красных кристаллов. Т. пл. 146o. Масс-спектр: m/z 316 (M+).
c) 5-Бензилокси-6-фтор-индол.
Раствор 9,5 г соединения b) (30,0 ммоль) в 150 мл толуола и 30 мл ТГФ, содержащего 1 г Raney-никеля, гидрогенизируют при комнатной температуре. Через 4 ч суспензию фильтруют через "Ryflo", и растворитель выпаривают. Остаток хроматографируют при среднем давлении (элюент: толуол) с получением целевого соединения, которое кристаллизуют из гексана. Т.пл. 126o. Масс. спектр: m/z 241 (M+).
d) 5-Бензилокси-6-фтор-индол-3-карбоксальдегид.
3,3 мл POCl3 (36,0 ммоль) добавляют по каплям при 0o к 14 мл ДМФ (180 ммоль). Через 15 мин по каплям в течение 10 мин добавляют 7,30 г соединения c) (30 ммоль) в 14 мл ДМФ. Смесь перемешивают 1 ч при комнатной температуре, затем разбавляют холодной водой, а затем добавляют по каплям 7,2 г NaOH в 50 мл воды. Остаток фильтруют и промывают водой. Полученное твердое вещество хроматографируют на SiO2 (элюент: CH2Cl2) и кристаллизуют из эфира с получением целевого (d) соединения. Т. пл. 190o. Масс. спектр: m/z (относительная интенсивность): 269 (M+, 72); 178 (20); 150 (15); 91 (100); 65 (38).
Пример 5. 5-Гидрокси-индол-3-карбоксальдегид амино (бутириламид) метиленгидразона.
К раствору 0,5 г 5-гидрокси-индол-3-карбоксальдегида диаминометиленгидразона (2,3 ммоль) в 5 мл ДМФ добавляют по каплям раствор 0,4 мл бутан-ангидрида (2,5 ммоль) в 5 мл ДМФ. Через 7 ч при комнатной температуре растворитель выпаривают и остаток хроматографируют на SiO2 (элюент: толуол (EтOH) NH3 85: 15:0,3). Таким образом получают целевое соединение, которое осаждают из гексана. Т. пл. 90o (II). Масс. спектр: m/z (относительная интенсивность): 287 (M+, 16); 217 (8); 200 (4); 158 (30); 98 (100); 70 (46).
Пример 6. 5-Гидрокси-индол-3-карбоксальдегид амино (пентил-амино)-метиленгидразона трифторацетат.
Температура плавления 138o.
Пример 7. 5-Гидрокси-индол-3-карбоксальдегида амино (пентиламино)-метиленгидразона трифторацетат.
К раствору 1,0 г 5-гидрокси-индол-3-карбоксальдегид-амино (пентиламино) метиленгидразона (3,5 ммоль) в 10 мл CF3CO2H добавляют 0,72 мл гексаноилхлорида (5,2 ммоль) при 0o. Через 3 ч реакцию гасят 2N раствором Na2CO3, и смесь перемешивают в течение 20 мин. Добавляют AcOEт, и отделяют органический слой, который промывают рассолом и сушат над сульфатом натрия. Растворитель затем выпаривают, и остаток промывают эфиром с получением кристаллического целевого соединения. Т. пл. 205o. Масс. спектр: m/z 385 (М, 20); 160 (30); 158 (25); 69 (100).
Путем описанной выше следующей процедуры, могут быть получены соединения формулы IA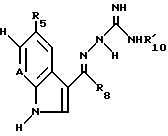
в которой R5, R6 и 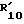 такие, как определено в табл.I.
такие, как определено в табл.I.
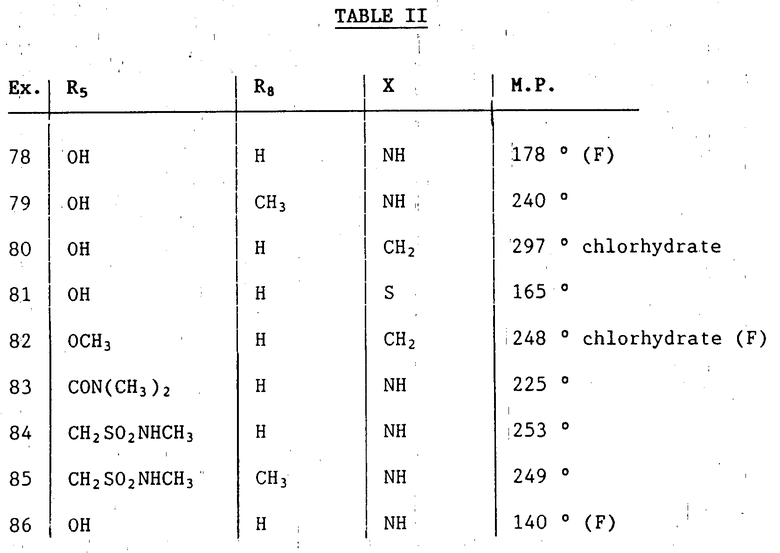
Путем описанной выше процедуры, могут быть получены следующие соединения формулы IB, в которой: R5, R4 и X такие, как определено в табл.II ниже.
Пример 87. 5-Гидрокси-3-[(N'-2'-имидазолин-4'-онил) -гидразометил]-индол.
Температура плавления 110o (II).
Пример 88.

Т. пл. 239o.
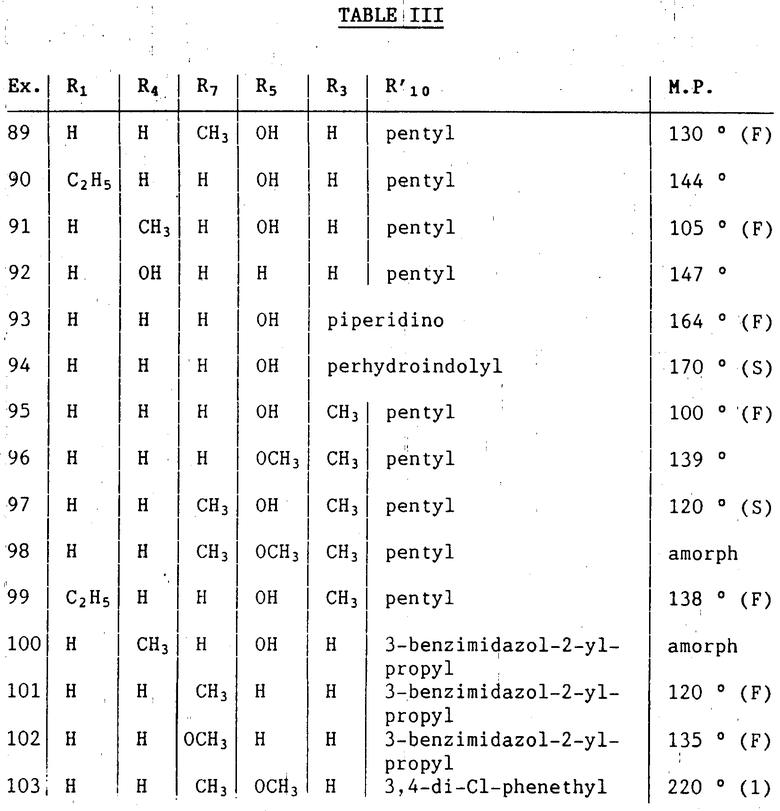
Следующей процедурой, как раскрыто выше, могут быть получены соединения формулы IC, в которой R1, R3, R4, R5, R7 и R'10 такие, как определено в табл.III ниже.
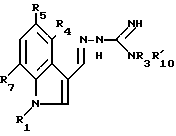
Путем следующей процедуры, раскрытой выше, могут быть получены соединения формулы ID, в которой R5 и 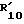 такие, как в табл. IV ниже.
такие, как в табл. IV ниже.
Путем следующей процедуры, как раскрыто выше, могут быть получены следующие соединения формулы IE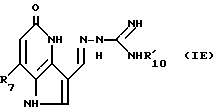
в которой R7 и 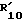 такие, как определено в табл.V ниже.
такие, как определено в табл.V ниже.
Пример 113. (7-Азаиндол)-3-карбоксальдегид амино(пентиламино) метилен-гидразина.
Т. пл. 78o (III).
Пример 114. 5-Гидрокси-6-фтор-индол-3-карбоксальдегид амидингидразона.
Т. пл. 168o (II).
5-(Диметилфосфиноксид)-индол-3-карбоксальдегид, используемый в качестве исходного вещества для получения соединения по примеру 52, может быть приготовлен по примеру 4 d) из индол-5-диметилфосфина оксида.
Индол-5-диметилфосфина оксид может быть приготовлен таким образом:
Пример 115. Индол-5-(диметилфосфина оксид).
Раствор т-бутил-лития в гексане (10 ммоль, 1,7 М) добавляют при -78o к раствору 5-бром-N-бензил-индолина (5 ммоль) в 30 мл эфира. Через 10 мин к смеси добавляют раствор ClPO(Me)2 (10 ммоль) в 10 мл ТГФ. Реакционную смесь оставляют нагреться до комнатной температуры в течение 6 ч. Добавляют воду и AcOEт, органический слой отделяют, и водную фазу экстрагируют AcOEт. Органические фазы соединяют и промывают рассолом, сушат, и растворитель выпаривают. Остаток хроматографируют на SiO2 (элюент: CH2Cl2/MeOH 95:5) с получением целевого a)-соединения. Т. пл. 180o.
b) Индолин-5-(диметилфосфина оксид).
Раствор соединения a) (1,5 ммоль) в 20 мл MeOH, содержащем 0,2 г Pd/C гидрогенизируют в течение 2 ч. Раствор фильтруют через Hyflo, и растворитель выпаривают с получением целевого b)-соединения.
c) Индол-5-(диметилфосфина оксид).
Раствор соединения b) (1,5 ммоль) в 25 мл ксилола, содержащего 100 мг Pd/C подвергают нагреванию с кипячением в течение 3 ч. Раствор фильтруют через Hyflo, и катализатор промывают CH2Cl2. Растворитель выпаривают с получением целевого соединения c). Т. пл. 195o.
5-Гидрокси-бензотиофен-3-карбоксальдегид и 5-метоксибензотиофен-3-карбоксальдегид, используемые в качестве исходных веществ для получения соединений по примерам 104-107; могут быть приготовлены следующим образом:
Пример 116. 5-Метокси-бензотиофен-3-карбоксальдегид.
a) (4-Метокси-бензил)ацетонила сульфид.
0,165 моль гидрида натрия добавляют по частям при 0o к раствору 4-метокси-тиофенола (0,15 моль) в 300 мл ТГФ. Через 1 ч добавляют хлорацетон. Смесь перемешивают до утра при комнатной температуре. Затем выпаривают растворитель, и смесь растворяют в CH2Cl2 и промывают 2N Na2CO3. Органическую фазу сушат, и растворитель выпаривают с получением целевого соединения a), которое используют без дополнительной очистки.
b) 3-Метил-5-метокси-бензотиофен.
0,15 моль соединения a) добавляют в течение 1 ч при кипячении к раствору полифосфорной кислоты (0,17 моль) в 1 л хлорбензола. Этот раствор перемешивают при кипячении до утра. Смесь фильтруют и фильтрат выпаривают. Сырой продукт хроматографируют на SiO2 (элюент: гексан с получением целевого b) соединения.
c) 5-Метокси-3-бромметил-бензотиофен.
0,01 моль NBS и кристалл дибензоилпероксида добавляют к раствору соединения b) (0,01 моль) в 60 мл тетрахлоруглерода. Смесь перемешивают при кипячении в течение 1,5 ч. Суспензию фильтруют и фильтрат выпаривают. Полученное масло кристаллизуется при его охлаждении и далее используется без дополнительной очистки.
d) 5-Метокси-бензотиофен-3-карбоксальдегид.
0,09 моль тетраамина гексаметилена добавляют к раствору соединения c) (0,08 моль) в 150 мл хлороформа. Смесь перемешивают при кипячении в течение 6 ч. После охлаждения суспензии к ней добавляют эфир; и твердое вещество отделяют фильтрованием. Остаток растворяют в 100 мл 50% CH3COOH и смесь нагревают при кипячении в течение 3 ч. Затем добавляют 130 мл воды и 25 мл конц. HCl и смесь кипятят в течение еще 5 мин. Смесь охлаждают на льду. Кристаллизовавшееся целевое соединение d) отделяют фильтрованием и промывают водой. Т. пл. 50o.
Пример 117. 5-Гидрокси-бензотиофен-3-карбоксальдегид.
Раствор BBr3 (0,07 моль) в 15 мл CH2Cl2 добавляют при 0o к раствору 0,014 моль 5-метокси-бензотиофен-3-карбоксальдегида в 80 мл CH2Cl2. После 4 ч стояния добавляют 2N Na2CO3 до pH 7. Органический растворитель выпаривают и суспензию фильтруют. Твердое вещество промывают водой с получением сырого продукта в названии примера, который рекристаллизуют из смеси CH3OH/H2O. Т. пл. 200o.
Соединения формулы I и их фармацевтически приемлемые соли (далее упоминаемые как соединения по изобретению) проявляют фармацевтическую активность и являются поэтому полезными в фармакологии.
В частности, соединения по изобретению обладают стимулирующим действием на работу желудочно-кишечного тракта (перистальтику), что может быть продемонстрировано на стандартных тестовых моделях, например, так.
Однополюсные электроды имплантируют в серозную сторону стенки кишечника (тонкая кишка) собак (Beagle). Из этих электродов сигналы поступают в усилитель и фильтруются с целью регистрации потенциалов низкой и высокой частоты, чтобы отделить медленные сокращения от резких. Количество пиков (резких сокращений) определяют за период в 2 мин. Исходя из этого, получают следующие результаты: длительность фазы I-III, интервал между блоками двух последовательных фаз III, скорость распространения. Один-два цикла записывают перед введением лекарства, которое осуществляют подкожно через 10-15 мин после фазы III, прошедшей большинство дистальных электродов. Контрольные опыты осуществляют обычно посредством введения растворителя. У накормленных собак количество резких сокращений за 30 мин определяли дополнительно. В этом эксперименте соединения по изобретению стимулируют миоэлектрическую активность в дозах порядка примерно от 0,001 до 10 мг/кг s. с. (подкожно).
Далее, стимуляторный эффект на активность работы желудочно-кишечного тракта соединений по изобретению демонстрируется также, например, их действием на перистальтический рефлекс на отделенную ободочную кишку морской свинки.
Мужских особей морских свинок весом 200-400 г глушили и обескровливали. Участки концевого участка ободочной кишки длиной 4-5 см удаляют и суспендируют, как описано Тренделенбургом в "Arch. Exp. Path. Pharmacol." 81, 55-129 (1917), в 20 мл бане при начальной загрузке 1 г. Ткань выдерживают при 37o в модифицированном растворе Кребса (NaCl 118,6; CaCl2 2,7; KCl 4,7; KH2PO4 1,2; MgSO4 0,1; NaHCO3 25,0; глюкоза 5,6 мМ), и продували 5% CO в кислороде. Перистальтику вызывали на 30 с путем увеличения давления в просвете от нуля до 1-4 см с помощью воды. Измерения проводят на реакцию продольной (гладкой) мускулатуры с помощью датчика смещения изотонической силы, а также активность кольцевых мышц с помощью датчика давления. Область под кривой (сокращенно AUC) перистальтических сокращений определяют и кривые реакции в зависимости от концентрации устанавливают с помощью определения AUC, представляющей деятельность продольной и кольцевой мускулатуры. Каждый препарат используют в качестве его собственного контроля, принимая за 100% перистальтическую активность до введения испытываемых соединений. Испытываемые соединения добавляют к серозной стороне и оставляют в контакте с ней в течение 15 мин. В этом эксперименте соединения по изобретению оказывали стимулирующее действие на перистальтическую активность при концентрации порядка примерно от 10-10 до 10-7 М.
Соединения по изобретению являются поэтому полезными для лечения расстройств подвижности желудочно-кишечного тракта, например для нормализации или для улучшения желудочной эвакуации и кишечной проходимости у лиц с расстройством такой активности, например болезнь, характеризующуюся желудочно-пищеводным рефлюксом, пониженной перистальтикой пищевода и/или желудка, и/или тонкого и/или толстого кишечника, или для лечения воспаления пищевода, гастропареза, диспепсии, нежелчной диспепсии псевдонепроходимости (псевдозаворот кишок), непроходимости ободочной кишки, тощей кишки, синдром острого живота, вздутие, боли в области желудка, послеоперационную атонию кишечника, рецидивную тошноту и рвоту, невротическое истощение или дискинезию желчной системы.
Далее, соединения по изобретению также показаны для использования при лечении дискинезии мочевого пузыря, регулирования выделения котизола/альдостерона, или для улучшения памяти и запоминания.
Для вышеуказанных применений, требуемая дозировка будет, конечно, зависеть в значительной степени от пути введения, конкретного состояния подлежащего лечению и требуемого эффекта. Показанные дневные дозы находятся в пределах примерно от 0,01 до 3 мг, например приблизительно от 0,01 до 1 мг для парентерального применения, или примерно от 0,1 до 3 мг для орального применения, предпочтительно вводимые разовой дозой, в разделенных дозах на 2-4 приема в день, или в формах с замедленным высвобождением лекарства. Соответственно, единицы дозировок для орального введения включают примерно 0,0025-1,5 мг активного ингредиента (то есть соединения по изобретению или его фармацевтически приемлемой соли), смешанного с подходящим твердым или жидким, фармацевтически приемлемым разбавителем или носителем.
В соответствии со сказанным выше, настоящее изобретение также обеспечивает:
I) Способ лечения расстройств деятельности желудочно-кишечного тракта, например, путем стимулирования подвижности желудочно-кишечной системы, дискинезии мочевого пузыря, регулирования выделения кортизол/альдостерона или улучшения памяти и запоминания у лица, нуждающегося в таком лечении, предусматривающий введение указанному лицу эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
Далее, было обнаружено, что соединения по изобретению обладают антисеротонинэргическим действием, специфично действующим на рецепторы 5-HT4, как было продемонстрировано на стандартных экспериментальных моделях, например, следующим образом: выделенная из организма морской свинки продольная мускулатура ободочной кишки с прилегающим к ней сплетением мышечного слоя является хорошо испытанной моделью, позволяющей проводить исследования механизма действия нейротрансмиттеров (медиаторов).
Методика. Морских свинок (мужских особей весом 200-400 г) забивали ударом по голове и обескровливали. Длина удаляемого тонкого кишечника составляет примерно 2 см от илео-цекального клапана. Кусочек ободочной кишки растягивают поверх стеклянной палочки и осторожно удаляют мезентерий. Слой продольной мускулатуры отделяют и снимают с нижележащего слоя кольцевой мускулатуры тангенциальными движениями в сторону от мезентериального присоединения (т. е. от брыжейки). Участки продольной мускулатуры длиной 4-5 см помещают в 10 мл бани для органов, содержащей раствор Tyrode при 37o и продувают 5% углекислого газа в кислороде. Раствор Tyrode содержит следующие концентрации веществ, моль/л: NaCl 137,0; CaCl2 1,8; KCl 2,7; MgCl 1,05; NaHCO3 11,9; NaHPO4 0,4; глюкоза 5,6; и метизергид 0,1 μM. Участки кишки поддерживали под постоянным натяжением усилия 500 мг. Сокращения записывали с помощью изотонического качающегося рычага. После уравновешивания в течение 30 мин применяли заданные концентрации карбахола через 10-минутные интервалы, пока не достигалась постоянная реакция.
Получение кривой зависимости концентрации и реакции.
Кривую реакции на некумулятивную концентрацию для 5-HT устанавливают путем добавления возрастающих концентраций агониста к бане органа в интервалы по меньшей мере 15 мин. Предыдущие эксперименты показали, что такие интервалы были достаточно длинными, чтобы избежать тахифилаксиса. Каждую концентрацию оставляют контактировать с тканью в течение 1 мин. Каждый участок кишки используется только однажды для записи двух кривых реакции от концентрации: первая только для одного 5-HT, и вторая для 5-HT в присутствии установленной концентрации антагониста, причем каждый участок служил таким образом своим собственным контролем. Антагонистам позволяют предварительно уравновеситься в течение по меньшей мере 10 мин перед добавлением 5-HT. Концентрации, выражаемые в процентах от максимальной реакции на 5-HT, которые были получены от нескольких препаратов, отмечали как средние значения, чтобы получить логарифмические кривые зависимости реакции от концентрации. Константы ингибирования выражают в виде значений pA2, которые определяют графически по традиционным методам (Arunlakshana и др. 159, McRay, 1978).
В этом эксперименте 5-HT проявляет зависимое от концентрации контрактильное действие. 5-HT вызывает это основное сократительное действие в мускулатуре продольных мышц участка ободочной кишки морской свинки путем высвобождения субстанции P из нервных окончаний в этой ткани. Это действие передает двумя различными рецепторами 5-HT. При низких концентрациях 5-HT активирует нейронный рецептор, который вызывает высвобождение субстанции P. Освобожденная субстанция P активирует нейронные рецепторы субстанции P, и это вызывает высвобождение ацетилхолина, который затем активирует мускариновые рецепторы, размещенные на клетках гладкой мускулатуры, и вызывает их сокращение. При более высоких концентрациях 5-HT активирует второй нейронный рецептор, что приводит к высвобождению субстанции P для активирования рецепторов субстанции P на клетках гладкой мускулатуры, взывая при этом сокращение.
Соединения по изобретению действуют главным образом на блокирование рецепторов высокой аффинности к 5-HT4, ингибируя при этом вызываемое 5-HT сокращение, например, при концентрациях примерно от 10-8 до 10-6 моль/л. Эти соединения проявляют менее антагонистическую активность к рецепторным сайтам низкой аффинности к 5-HT3.
Соединения по изобретению поэтому являются полезными для лечения расстройств подвижности желудочно-кишечного тракта, таких как тахигастрия, затрудненная эвакуация желудка вследствие тахигастрии, синдром острого живота, кишечные спазмы, судороги, запор вследствие повышенного тонуса толстого кишечника, болезнь желудочно-пищеводного рефлекса и дискинезия желчной системы.
Соединения по изобретению также ингибируют образование эрозий желудка, вызываемых некротизирующими агентами, как было продемонстрировано стандартными экспериментами, например, с использованием крыс с вызванными этанолом повреждениями желудка.
Эти эксперименты проводили на мужских особях крыс массой 200-250 г, которых не кормили ночь, но имевших свободный доступ к воде. Испытываемое вещество вводили подкожно или орально через металлическую желудочную трубку. Абсолютный спирт задавали орально через 30 мин после введения испытываемого вещества и животных забивали часом позже. Желудок раскрывали разрезом по большой кривизне и прикалывали в плоском виде. Геморрагические эрозии оценивали двояко: по площади и длине эрозий.
При подкожном введении соединения по изобретению в качестве испытываемого вещества, при дозе примерно от 0,1 μг/кг до 10 мг/кг, наблюдается существенное ингибирование желудочных повреждений, вызываемых этанолом, по сравнению с результатами, полученными с контрольной группой, которая получала плацебо вместо испытываемого вещества.
Соединения по изобретению, соответственно, показаны для использования при лечении или профилактике желудочно-кишечных расстройств, таких как язва желудка и двенадцатиперстной кишки.
Соединения по изобретению далее показаны для лечения диарреи, воспалительных заболеваний желудка и кишечника, например гастрита, дуоденита, включая воспалительные заболевания кишечника, тошноту и рвоту. Далее, они также показаны для лечения аритмии, тахикардии, дискинезии мочевого пузыря, например недержания мочи, для снижения частоты приступов или для смягчения стрессовых реакций.
Для вышеуказанных применений требуемая дозировка, конечно, будет изменяться в зависимости от пути введения, конкретного состояния подлежащего лечению и требуемого эффекта. Как показано, дневная доза находится в пределах примерно от 5μг до 5 мг для парентерального применения и порядка примерно 0,1-100 мг для орального применения, предпочтительно вводимая однажды или в разделенных дозировках 2-4 раза в день, или в формах замедленного высвобождения лекарства. Единичные дозы для орального введения соответственно включают примерно 0,025-50 мг соединения по изобретению, смешанного с соответствующим твердым или жидким фармацевтически приемлемым разбавителем или носителем активного ингредиента.
В соответствии с вышеизложенным настоящее изобретение также обеспечивает:
II) Способ лечения любых из вышеупомянутых расстройств или состояний у лица, которое нуждается в таком лечении, предусматривающий введение указанному лицу эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
Далее, было обнаружено, что соединения по изобретению обладают антагонистическим действием на центральные 5HT-1C рецепторы.
Соединения по изобретению обладают потенциальной связывающей афинностью к центральным 5HT-1C рецепторам, как например было измерено согласно методике, раскрытой Хойером и др. (Hoyer, Eur. J. Pharmacol. 118, 13-23 (1985)).
Соединения по изобретению являются антагонистами пониженной активности (hypolocomotion), что вызывается у крыс введением м-хлорфенил-пиперазина (мCPP), по методике Кеннета и Курзона (Kennet и Curson Br. J. Pharmacol. 94, 137-147 (1988)). В этом эксперименте соединения по изобретению противодействовали вызываемому мCPP эффекту после введения их в дозах примерно от 0,1 до 30 мг/кг живого веса.
Соединения по изобретению поэтому полезны для профилактического лечения мигрени или для лечения психиатрических заболеваний, например возбуждения, навязчивых идей, приступов страха, депрессии, булимии (обжорства), шизофрении, ситуаций с повышенным внутричерепным давлением и приапизма.
Для вышеупомянутых применений требуемая доза будет, конечно, отличаться в зависимости от пути введения, конкретного состояния больного и требуемого эффекта. Показанная дневная доза находится в пределах примерно от 0,5 до 300 мг, предпочтительно применяемая одноразово или в разделенных на 2-4 приема в день дозировках, или в форме медленного высвобождения лекарства.
В соответствии с вышеизложенным, настоящее изобретение также обеспечивает:
III) Способ профилактического лечения мигрени или лечения психиатрических расстройств у лица, нуждающегося в таком лечении, предусматривающий введение указанному лицу эффективного количества соединений по изобретению или его фармацевтически приемлемой соли.
Соединения по изобретению также обладают агонистическим воздействием на 5HT-1Д рецепторы. Их связывающая аффинность к 5HT-1Д рецепторам была определена, например, по методике Вебера и др. (Waeber, Naunynschmiedeburg's Arch. Pharmacol, 337, 595-601 (1988)).
Агонистическое действие далее продемонстрировано следующим экспериментом.
Передние церебральные артерии вырезают из свиных мозгов, полученных от местной скотобойни. Круглые участки длиной 3-4 мм насаживают между двумя I-образными металлическими зубцами и помещают в баню для органов с контролируемой (37oC) температурой, наполненную раствором Кребса, который непрерывно продувают 5% CO2 в кислороде. Агонистически вызванные васкулярные сокращения измеряют изометрически. Для того чтобы измерить только воздействие на 5HT-1Д рецепторы, к раствору в бане добавляют кентасерин (10-7 М), который препятствует сокращениям посредством 5HT-2 рецепторов. Соединения по изобретению вызывают васкулярные сокращения при концентрации от 10-10 до 10-5 М, в частности 10-9 10-7 М.
Соединения по изобретению поэтому полезны для лечения состояний, связанных с головной болью, в частности для лечения мигрени, концентрированной головной боли, хронических пароксизмальных черепных болей и головных болей, связанных с сосудистыми нарушениями, а также для смягчения симптомов, связанных с этими болезнями.
Для вышеупомянутых применений дозировка будет, конечно различаться в зависимости от пути введения, конкретного состояния больного и требуемого эффекта. Показанная дневная доза находится в пределах примерно от 0,5 до 300 мг, предпочтительно вводимая разовым образом, или в разделенных дозировках на 2-4 приема в день, или в форме с замедленным высвобождением лекарства.
В соответствии с вышеизложенным, настоящее изобретение также обеспечивает:
IV) Способ лечения состояний, связанных с болью головного мозга, например таких, как указано выше, предусматривающий введение больному эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
Соединения по изобретению могут быть введены в свободном виде или в виде фармацевтически приемлемой соли. Такие соли могут быть приготовлены традиционными методами и проявляют тот же уровень активности, что и свободное соединение. Пригодные фармацевтически приемлемые соли соединений по изобретению включают, например, гидрохлориды.
Далее, настоящее изобретение также обеспечивает:
V) Соединение по изобретению или его фармацевтически приемлемую соль, для применения в качестве фармацевтического средства, например, в любом из указанных выше способов.
VI) Фармацевтическая композиция, включающая соединение по изобретению, как оно описано выше, или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем или носителем для него. Такая композиция может производиться традиционным образом, например путем смешивания ингредиентов.
Соединения формулы I, в которой R1 H, A -N= или -CH= и Z - -CH= или в которой R1 H, A -CH= Z -N= или -CH= и R5 - гидрокси или C1-C6 алкокси, обладают, например, стимуляторным действием на подвижность желудочно-кишечного тракта и поэтому могут использоваться в способе по изобретению, для лечения расстройств подвижности, например, путем стимулирования перистальтики желудочно-кишечного тракта, как указано выше, для лечения дискинезии мочевого пузыря, модулирования высвобождения кортизола/альдостерона или для улучшения памяти и запоминания. Предпочтительными являются соединения по примерам 13 и 108.
Соединения формулы I, в которой R1 в W и/или R7 в A не являются водородом, обладают, в частности, антисеротонинэргическим действием, оказываемым специфично на 5-HT4 рецепторы, и ингибируют эрозии желудка, вызываемые некротизирующими агентами, и поэтому могут использоваться в качестве противоязвенного или улучшающего подвижность агента в способе по изобретению, для лечения желудочно-кишечных расстройств и для профилактики или для лечения язвы желудка и двенадцатиперстной кишки. Они также показаны для лечения диарреи, воспалений желудка или кишечника, например гастрита, дуоденита, включая воспалительное заболевание кишечника, тошноту и рвоту, аритмии, тахикардии, дискинезию мочевого пузыря, например недержание, для уменьшения частоты приступов, снижения вероятности удара или для смягчения стрессовых реакций. Предпочтительными являются соединения по примерам 89, 90 и 97.
Соединения формулы I, в которой R5 H, -OH, C1-C6 алкокси или нитро; R2 H, Cl, Br или C1-C6 алкил; Z -CR4= где R4 H, C1-C6 алкил, Cl или Br; A - -CR7= где R7 H или C1-C6 алкил; предпочтительно те из них, у которых B является радикалом формулы (b),  - C1-C12 алкил или -C1-C6 алкил, замещенные NH-CO-фенилом или бензимидазолилом, обладают антагонистическим действием на центральные 5HT-1С рецепторы и поэтому могут использоваться при профилактике мигрени и для лечения психиатрических нарушений, таких как возбуждение, навязчивые идеи, приступы страха, депрессия или булимия. Предпочтительным является соединение 38.
- C1-C12 алкил или -C1-C6 алкил, замещенные NH-CO-фенилом или бензимидазолилом, обладают антагонистическим действием на центральные 5HT-1С рецепторы и поэтому могут использоваться при профилактике мигрени и для лечения психиатрических нарушений, таких как возбуждение, навязчивые идеи, приступы страха, депрессия или булимия. Предпочтительным является соединение 38.
Соединения формулы I, в которой R5 H, -OH, C1-C6алкокси, карбокси, C2-C6алкоксикарбонил, CONRaRb, SONH (C1-C6алкил), C1-C6алкил, замещенный SO2 C1-C6 алкилом, или PO(C1-C4алкил), W NH, A CH= Z -CH= и R2 и R6 каждый являются H, особенно те из них, у которых B является радикалом  или радикалом (b), в котором X2 - C1-C12алкил или CONH-C6H11, обладают, в частности, агонистическим действием на 5HT-1Д рецепторы и поэтому могут быть использованы для лечения состояний, связанных с болью головного мозга, например, как указано выше. Особенно предпочтительны соединения по примеру 63.
или радикалом (b), в котором X2 - C1-C12алкил или CONH-C6H11, обладают, в частности, агонистическим действием на 5HT-1Д рецепторы и поэтому могут быть использованы для лечения состояний, связанных с болью головного мозга, например, как указано выше. Особенно предпочтительны соединения по примеру 63.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОНОМЕРНЫЕ ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДНОГО АНТИБИОТИКА | 2005 |
|
RU2424248C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ РЕСПИРАТОРНЫХ ЗАБОЛЕВАНИЙ, ТАКИХ КАК COPD | 2011 |
|
RU2604719C2 |
| КОМПОЗИЦИЯ ДЛЯ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО ПОЛИАМИДА | 1990 |
|
RU2068903C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА | 1993 |
|
RU2098410C1 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830113C2 |
| СРЕДСТВО ДЛЯ ЗАЩИТЫ КУЛЬТУРНЫХ РАСТЕНИЙ ОТ ФИТОТОКСИЧЕСКОГО ПОБОЧНОГО ДЕЙСТВИЯ ГЕРБИЦИДОВ, N-АЦИЛСУЛЬФОНАМИДЫ | 1997 |
|
RU2182423C2 |
| ПРОИЗВОДНЫЕ ГУАНИДИНАЛКИЛ-1,1-БИС-ФОСФОНОВЫХ КИСЛОТ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2114855C1 |
| 5-СУЛЬФАНИЛ-4Н-1,2,4-ТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 2002 |
|
RU2367655C2 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |






Изобретение касается соединений формулы
A-X--Y-NH-B,
где A происходит от возможно замещенного бензотиофена, индола, 4-аза- и 7-аза-бензотиофена или -индола; A несет в своей 5-й позиции водород, галоген, возможно замещенный алкил, гидрокси, нитро, амино, алкиламино, ациламино, алкоксикарбонил, сульфамоил, циано, триметилсилил, карбокси, карбамоил, фосфат, оксикарбамоил, гетероциклический радикал или группу простого или сложного эфира; X--Y - -CR8-N= или CH(R8)-NH, где R8 - H или алкил, и присоединен к A в позиции 3, а B - гетероциклический радикал или остаток формулы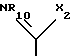
где R10 - H, возможно замещенный алкил, циклоалкил, арил, адамантил, ацил или карбамоил, и X - алкилтио или NR3R10, где R3 - H или алкил, или R3 и R10 вместе с атомом азота, к которому они присоединены, образуют гетероциклический радикал в свободном виде или в виде соли, которые проявляют фармацевтическую активность, например для лечения расстройств желудочно-кишечного тракта. 3 с. и 7 з.п. ф-лы, 5 табл.
где W S или -NR1, где R1 водород, C1 - C6-алкил или ацил;
R2 водород или C1 C6-алкил;
R5 водород, галоген, C1 C6-алкил, гидрокси, C1 C6-алкил, замещенный -SO2-C1 C6-алкил, -SO2NRaRb, -NH-SO2-C1 - C6-алкил или N-RaRb 1, где каждый Ra, Rb и R1 независимо водород или C1 C6-алкил, а также NO2, NH2, CN, триметилсилил, С2 - C6-алкоксикарбонил, SO2NRaRb, -COOH, OCONRcRd, где каждый Rc и Rd независимо - C1 C6-алкил, C1 C6-алкокси, С2 - C6-алкенилокси, бензилокси; C1 C6-алкокси, замещенный ОН, C1 C4-алкокси, ацилокси, -NRaRb 1, -CONRaRb или -CSNRaRb, ацилокси, пиридилкарбонилокси, CONRaRb или RО(C1 - C4-алкил)2;
R6 водород или если R5 ОН, то R6 галоген;
Z -CR4= где R4 водород, гидрокси или C1 - C6-алкил или, если R5 водород или гидрокси, то Z также N
A -CR7 где R7 водород, C1 C6-алкил или C1 C6-алкокси;
B радикал формулы (а)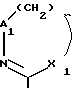
или формулы (b)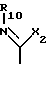
где n 1 или 2;
A1 С=О или СН2;
X1 S, CH2, NR1 1, где R1 1 - водород или ацил;
X2 NR3R'1 0, где R'1 0 представляет собой R1 0, или SR2 0;
R1 0 водород, C1 C12-алкил, С1 - С6-алкил, замещенный гидрокси, фенил, фенил моно- или дизамещенный фтором, хлором, метилом или метоксилом, фенокси или фенокси-, замещенный галогеном, пиридилом, имидазолилом, бензимидазолилом, пирролидинилом, пирролидонилом, пиперидинопиразинилом, пергидроиндолилом, или радикал формы (d)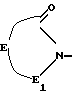
где E -CH2N(R1 7), где R1 7 водород или C1 C4-алкил и один или два водорода, которые могут быть замещены C1 C6-алкилом;
E1 CO или CH2;
R2 0 C1 C6-алкил;
R3 водород или C1 C6-алкил или C1 C6-алкил, замещенный -NR1 5-CO-R1 6 или NH-SO2-арил, где R1 5 водород или C1 - C6-алкил, R1 6 С1 C6-алкил, фенил или фенил-C1 C4-алкил, или CONHR1 4, где R1 4 C1 C10-алкил, С5 - C7-циклоалкил, фенил или фенил-C1 C4-алкил, или
R3 и R'10 вместе с атомом азота, к которому они присоединены, образуют пиперидиновый или пергидроиндолильный радикал;
R2 C1 C6-алкил;
-Х Y- это -CR8=N- или -CH(R8)-NH, где R8 - водород или C1 C6-алкил,
при условии, что I) если B радикал формулы (b), только один из R1 0 и R'1 0 может быть другим, чем водород, а Х2 может быть -SR2 0, если R1 0 водород, и II) R5 не водород, если B радикал формулы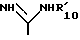
в которой R'1 0 фенил или фенил моно- или дизамещенный фтором, хлором, CH3 или CH3O;
Z и A каждый -CH= W= -NH-;
R2 и R6 каждый водород;
"X Y" это CH=N-,
в свободной форме или в форме соли.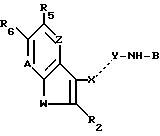
где W, R2, A, "X Y" и B, Z имеют указанные значения;
R5 водород, гидрокси, C1 C6-алкил, C1 - C6-алкил, замещенный -SO2-C1 C6-алкил, -SO2NRaRb, -NH-SO2-C1 C6-алкил или NRaR'b, где каждый из Ra, и R'b независимо - водород или C1 C6-алкил, NO2, NH2, CN, триметилсилил, С2 C6-алкоксикарбонил, -SO2NRaRb, -COOH, -OCONRcRd, где каждый Rc и Rd независимо C1 C6-алкил, C1 - C6-алкокси, С2 C6-алкенилокси, бензилокси, C1 - C6-алкокси, замещенный ОН, C1 C4-алкокси, ацилокси, -NRaR'b, -CONRaRb или -CSNRaRb, ацилокси, пиридилкарбонилокси, CONRaRb или -RO(C1 - C4-алкил)2.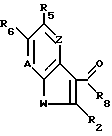
где A, W, Z, R2, R5, R6 и R8 определены в п.1,
с соединением формулы III:
H2N-NHB,
где B указан в п.1,
и, где требуется, проводят гидрогенизацию полученного соединения формулы I, где "X -Y" является -CR8=N-,
и выделяют соединения формулы I в свободной форме или в виде соли, сольватной или гидратной форме.
| EP, патент, 171037, кл.C 07D 209/08, 1985. |
