Настоящее изобретение относится к новым N-замещенным азагетероциклическим карбоновым кислотам и их сложным эфирам, в которых замещенная алкильная цепь составляет часть N-заместителя, или их солям к способам их получения, к содержащим их композициям и к их применению для клинического лечения болезненных, гиперальгезических и/или воспалительных состояний, в которых С-волокна играют патофизиологическую роль путем вызывания нейрогенной боли или воспаления. Изобретение также относится к применению настоящих соединений для лечения невосприимчивости к инсулину при инсулин независимом сахарном диабете (ИНСД) или при старении, поскольку известно, что настоящие соединения подавляют активность содержащих нейропептиды С-волокон и, следовательно, ингибируют секрецию и циркуляцию пептидов-антагонистов инсулина, таких как CGRP или амилин.
Предпосылки изобретения.
Нервная система оказывает на воспалительную реакцию значительное влияние. Антидромная стимуляция чувствительных нервов приводит к местной вазодилятации и повышенной сосудистой проницаемости (Janecso et al. Br. J. Pharmacol. 1967, 31, 138-151), и сходная же реакция наблюдается после инъекции пептидов, присутствующих в чувствительных нервах. Из этих и других данных следует, что пептиды, высвобождаемые из чувствительных нервных окончаний, опосредуют многие воспалительные реакции в таких органах, как кожа, сустав, мочевыводящие пути, глаз, мозговые оболочки, желудочно-кишечный тракт и дыхательные пути. Следовательно, ингибирование высвобождения и/или активности пептидов чувствительных нервов может применяться для лечения, например, артрита, дерматита, ринита, бронхиальной астмы, цистита, гингивита, тромбофлебита. Далее, сильное влияние CGRP на активность гликогенсинтазы скелетных мышц и метаболизм глюкозы в мышцах вместе с представлением о том, что данный пептид высвобождается из нервно-мышечного синапса при возбуждении нерва, позволяют предположить, что CGRP играет физиологическую роль в метаболизме глюкозы в скелетных мышцах, направляя фосфорилированную глюкозу от запасания гликогена в сторону гликолитического и окислительного путей (Rossetti et al. Am. J.Physiol. 264, E1-E10, 1993). Данный пептид может представлять собой важный физиологический модулятор внутриклеточного транспорта глюкозы в физиологических таких состояниях, как упражнение, и также могут способствовать снижению активности инсулина и гликогенсинтазы скелетных мышц в таких патофизиологических состояниях, как ИНСД или связанная со старением тучность (Melnyk et al. Obesity Res. 3, 337-344, 1995), при которых уровни содержания циркулирующего в плазме CGRP значительно повышены. Следовательно, ингибирование высвобождения и/или активности нейропептида CGRP может применяться для лечения невосприимчивости к инсулину, связанной с диабетом типа 2 или старением.
В патенте США №4383999 и №4514414 и в ЕР 236342 так же, как и в ЕР 231996, некоторые производные N-(4,4-дизамещенных-3-бутенил)азагетероциклических карбоновых кислот описаны как ингибиторы захвата ГАМК. В ЕР 342635 и ЕР 374801 N-замещенные азагетероциклические карбоновые кислоты, в которых оксимэфирная группа и винилэфирная группа составляют часть N-заместителя, соответственно описаны как ингибиторы захвата ГАМК. Далее, в WO 9107389 и WO 9220658 N-замещенные азациклические карбоновые кислоты описаны как ингибиторы захвата ГАМК.
В дополнение к сделанным выше ссылкам, в патенте США №3074953 этиловый эфир 1-(3-(10,11-дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил)-4-фенил-4-пиперидинкарбоновой кислоты описан как психотропное лекарственное средство. Аналогичные приведенному выше соединению 1-замещенные сложноэфирные производные 4-фенил-4-пиперидинкарбоновой кислоты описаны (J. Med. Chem. 1967, 10, 627-635 и J. Org. Chem. 1962, 27, 230-240) как анальгезирующие, спазмолитические (противосудорожные?) и психотропные средства. В JP 49032544, JP 48040357, FR 2121423, GB 1294550 и DE 2101066 1-замещенные 4-диалкиламино-4-пиперидинкарбоксамиды описаны как психотропные средства для лечения шизофрении и как ингибиторы воспаления.
Описание изобретения.
Настоящее изобретение относится к новым N-замещенным азагетероциклическим карбоновым кислотам и их сложным эфирам формулы I
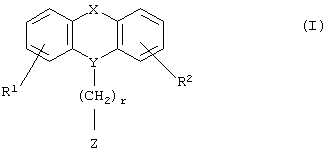
где R1 и R2 независимо представляют собой водород, галоген, трифторметил, NR6R7, гидрокси, C1-6-алкил или С1-6-алкокси; и
Y представляет собой 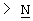 -CH2-,
-CH2-, 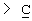 Н-СН2- или
Н-СН2- или 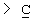 =СН-, где только подчеркнутый атом входит в состав кольцевой системы;
=СН-, где только подчеркнутый атом входит в состав кольцевой системы;
и
Х представляет собой -O-, -S-, -C(R6R7)-, -CH2CH2-, -СН=СН-СН2-, -СН2-СН=СН-, -СН2-(С=O)-, -(С=O)-СН2-, -СН2СН2СН2-, -СН=СН-, -N(R8)-(C=O)-, -(С=O)-N(R8)-, -O-СН2-, -СН2-O-, -S-CH2-, -СН2-S-, -(С=O)-, -N(R9)- или -(S=O)-, где R6, R7, R8 и R9 независимо представляют собой водород или С1-6-алкил; и
r равно 1, 2 или 3; и
Z выбран из
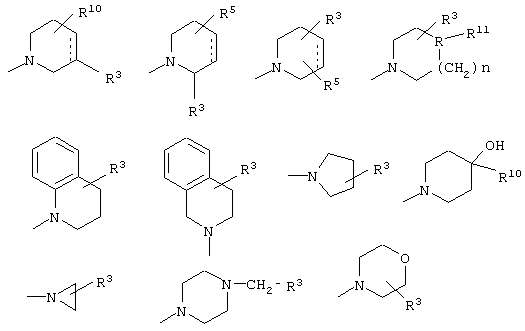
где n равно 1 или 2; и
R3 представляет собой -(СН2)mОН или - (СН2)pCOR4, где m равно 0, 1, 2, 3, 4, 5 или 6, и р равно 0 или 1, и где R4 представляет собой -ОН, -NH2, -NHOH, или С1-6-алкокси; и
R5 представляет собой водород, галоген, трифторметил, гидрокси, С1-6-алкил или C1-6-алкокси; и
R10 представляет собой водород, C1-6-алкил, С1-6-алкокси или фенил, необязательно замещенный галогеном, трифторметилом, гидрокси, С1-6-алкилом или C1-6-алкокси; и
R11 представляет собой водород или С1-6-алкил; и
 представляет собой необязательно одинарную связь или двойную связь;
представляет собой необязательно одинарную связь или двойную связь;
или к их фармацевтически приемлемой соли.
Соединения формулы I могут существовать в виде геометрических и оптических изомеров, и сюда включены все изомеры и их смеси. Изомеры могут быть разделены посредством стандартных методик, таких как хроматографические методы или фракционированная кристаллизация соответствующих солей.
Предпочтительно, соединения формулы I существуют в виде индивидуальных геометрических или оптических изомеров.
Соединения по изобретению могут необязательно существовать в виде фармацевтически приемлемых солей добавления кислот или - когда карбоксильная группа не этерифицирована - в виде фармацевтически приемлемых солей металлов или - необязательно алкилированных - солей аммония.
Примеры таких солей включают в себя неорганические или органические соли добавления кислот, такие как гидрохлорид, гидробромид, сульфат, фосфат, ацетат, фумарат, малеат, цитрат, лактат, тартрат, оксалат или сходные фармацевтически приемлемые соли добавления неорганических или органических если добавления кислот, и включают в себя фармацевтически приемлемые соли, приведенные в Journal of Pharmaceutical Science, 66, 2 (1977), которые включены здесь в качестве ссылки.
Термин "С1-6-алкил", как применяется здесь, отдельно или в сочетании, относится к неразветвленной или разветвленной насыщенной углеводородной цепи, содержащей от 1 до 6 атомов углерода, такой как, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 2-метилбутил, 3-метилбутил, н-гексил, 4-метилпентил, неопентил, н-гексил, 1,2-диметилпропил, 2,2-диметилпропил и 1,2,2-гриметилпропил.
Термин "C1-6-алкокси", как применяется здесь, отдельно или в сочетании, относится к неразветвленному или разветвленному одновалентному заместителю, содержащему C1-6-алкильную группу, присоединенную через кислород простого эфира, свободная связь которого связана с кислородом простого эфира, и содержащему от 1 до 6 атомов углерода, например, метокси, этокси, пропокси, изопропокси, бутокси, пентокси.
Термин "галоген" обозначает фтор, хлор, бром или йод.
Показательные примеры соединений, охватываемых настоящим изобретением, включают:
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинкарбоксамид;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперидинкарбоновая кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинил)метанол;
4-(4-Хлорфенил)-1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинол;
4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперазинкарбоновая кислота;
(23,4R)-1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-гидрокси-2-пирролидинкарбоновая кислота;
4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-морфолинкарбоновая кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-азиридинкарбоновая кислота;
2-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1,2,3,4-тетрагидро-4-изохинолинкарбоновая кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-метил-[1,4]-диазепан-6-карбоновая кислота;
2-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1,2,3,4-тетрагидро-З-изохинолинкарбоновая кислота;
Гидроксамид 1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинкарбоновой кислоты;
(4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)пиперазин-1-ил)уксусная кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинуксусная кислота;
1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновая кислота;
(R)-1-(3-(10,11-Дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил-3-пиперидинкарбоксамид;
(R)-1-(3-(10,11-Дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил-2-пирролидинкарбоновая кислота;
(S)-1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пирролидинкарбоновая кислота;
(R)-1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пиперидинкарбоновая кислота;
1-(3-(10Н-Феноксазин-10-ил)-1-пропил)-4-пиперидинкарбоновая кислота;
1-(3-(3-Хлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинуксусная кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-метил-3-пиперидинкарбоновая кислота;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-хинуклидиний-карбоксилат;
1-(3-(2,8-Дибром-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота;
1-(3-(3,7-Дихлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота;
1-(3-(3-Метил-10,11-дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-4-пиперидинкарбоновая кислота;
1-(3-(3,7-Диметил-10,11-дигидро-5Н-дибенз[b,f]aзeпин-5-ил)-1-пропил-4-пиперидинкарбоновая кислота;
1-(3-(3-Диметиламино-10,11-дигидро-5Н-дибекз[b,f]азетин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота;
или их фармацевтически приемлемая соль.
Как применяется здесь, термин "пациент" включает в себя любое млекопитающее, которому может помочь лечение нейрогенной боли или воспаления или невосприимчивости к инсулину при ИНСД. Термин особенно применим к пациенту-человеку, но не ограничивается им.
Было показано, что новые соединения формулы I подавляют нейрогенное воспаление, которое включает в себя высвобождение нейропептидов из периферических и центральных окончаний чувствительных С-волокон. Это может быть экспериментально продемонстрировано на животных моделях боли или отека лапы, вызванных формалином (Wheeler and Cowan, Agents Actions 1991, 34, 264-269), где новые соединения формулы I проявляют сильный ингибирующий эффект. Соединения формулы I могут быть применены для лечения всех болезненных, гиперальгезических и/или воспалительных состояний, в которых С-волокна играют патофизиологическую роль, вызывая нейрогенную боль или воспаление, т.е.:
Состояния, сопровождающиеся острой болью, как, например, мигрень, послеоперационная боль, ожоги, ушибы, постгерпетическая боль (опоясывающий лишай) и боль, в большинстве случаев связанная с острым воспалением; хронические болезненные и/или воспалительные состояния, как, например, различные типы нейропатии (диабетическая, посттравматическая, токсическая), невралгия, ревматоидный артрит, спондиллит, подагра, воспалительные заболевания кишечника, простатит, боль при злокачественных опухолях, хроническая головная боль, кашель, бронхиальная астма, хронический панкреатит, воспалительные заболевания кожи, включая псориаз и аутоиммунные дерматозы, боль при остеопорозе.
Далее, было продемонстрировано, что соединения общей формулы I увеличивают толерантность к глюкозе у мышей ob/ob, страдающих сахарным диабетом, и что это может быть результатом уменьшения высвобождения CGRP из периферических нервных окончаний. Следовательно, соединения общей формулы I могут быть использованы для лечения ИНСД так же, как и тучности, связанной со старением. Это было экспериментально продемонстрировано с помощью подкожного введения глюкозы мышам ob/ob с или без предварительной пероральной обработки соединением общей формулы I.
Соединения формулы I могут быть получены в соответствии со следующим способом:
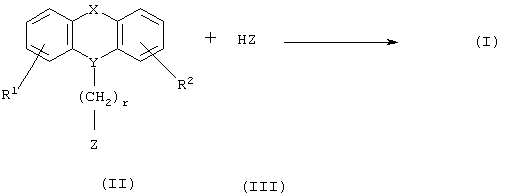
Соединение формулы II, где R1, R2, X, Y и r определены выше, и W представляет собой подходящую уходящую группу, такую как галоген, п-толуолсульфонат или мезилат, могут быть подвергнуты взаимодействию с азагетероциклическим соединением формулы III, где Z определен выше. Данная реакция алкилирования может быть проведена в растворителе, таком как ацетон, дибутиловый эфир, 2-бутанон, метилэтилкетон, этилацетат, тетрагидрофуран (ТГФ) или толуол, в присутствии основания, например, гидрида натрия, и катализатора, например, йодида щелочного металла, при температуре вплоть до температуры кипения используемого растворителя в течение, например, от 1 до 120 ч. Если получают сложные эфиры, в которых R4 представляет собой алкокси, соединения формулы I, где R4 представляет собой ОН, могут быть получены путем гидролиза сложноэфирной группы, предпочтительно, при комнатной температуре в смеси водного раствора гидроксида щелочного металла и спирта, такого как метанол или этанол, например, в течение около от 0,5 до 6 ч.
Соединения формул II и III могут быть легко получены в соответствии со способами, известными специалистам в данной области.
При определенных обстоятельствах может быть необходимо защитить промежуточные продукты, используемые в данных способах, например, соединение формулы III подходящими защитными группами. Например, карбоксильная группа может быть этерифицирована. Введение и удаление таких групп описано в "Protective Groups in Organic Chemistry" J.F.W. McOrnie ed. (New York, 1973).
Фармакологические методы.
Боль или отек лапы, вызванные формалином.
Степень ингибирования боли или отека, вызванных формалином, in vivo для соединений по настоящему изобретений оценивали на мышах, главным образом, по методу Wheeler-Aceto и Cowan (Agents Action 1991, 34, 264-269).
Самкам мышей NMRI массой около 20 г вводили 20 мкл 1% формалина в левую заднюю лапу. Животных затем помещали на нагретый (31° С) стол и оценивали болевую реакцию. Через 1 ч их забивали и кровь спускали. Левую и правую задние лапы удаляли и разницу в массе между лапами использовали в качестве показателя реакции в виде отека лапы, инъецированной формалином.
Уменьшенное высвобождение CGRP.
Самкам мышей ob/ob в возрасте 16 недель подкожно вводили глюкозу (2 г/кг). Через сроки, указанные ниже, по глюкозооксидазному методу в крови из хвостовой вены определяли содержание глюкозы в крови. По завершении исследования животных обезглавливали и туловищную кровь собирали. Содержание иммунореактивного CGRP в плазме определяли с помощью радиоиммунного анализа. Использовали две группы животных. В течение пяти дней перед исследованием одну группу обрабатывали носителем, тогда как другая группа получала соединение формулы I через питьевую воду (100 мг/л).
Значения ингибирования болевой реакции, вызванной формалином, для некоторых показательных соединений приведены в таблице 1.
Что касается приведенных выше показателей, дозировка будет варьировать в зависимости от применяемого соединения формулы I, способа введения и желаемого лечения. Однако, главным образом, удовлетворительные результаты получают, применяя дозировки от около 0,5 мг до около 1000 мг, предпочтительно от около 1 мг до около 500 мг соединений формулы I, которые удобно вводить от 1 до 5 раз ежедневно, необязательно в форме с замедленным высвобождением. Обычно, дозированные формы, подходящие для перорального применения содержат от приблизительно 0,5 мг до приблизительно 1000 мг, предпочтительно, от приблизительно 1 мг до приблизительно 500 мг соединений формулы I, смешанных с фармацевтическим носителем или разбавителем.
Соединения формулы I могут быть введены в виде фармацевтически приемлемой соли добавления кислоты или, где это возможно, в виде соли металла или низшего алкиламмония. Такие солевые формы проявляют приблизительно тот же уровень активности, что и свободные основные формы.
Данное изобретение также относится к фармацевтическим композициям, содержащим соединение формулы I или его фармацевтически приемлемую соль, и, как правило, такие композиции также содержат фармацевтический носитель или разбавитель. Композиции, содержащие соединения по данному изобретению, могут быть получены в соответствии с обычными способами и находятся в традиционных формах, например, в капсулах, таблетках, растворах и суспензиях.
Применяемый фармацевтический носитель может представлять собой широко используемые твердый или жидкий носитель. Примерами твердых носителей являются лактоза, terra alba, сахароза, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния и стеариновая кислота. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло и вода.
Подобным образом носитель или разбавитель могут включать в себя любой материал, увеличивающий время высвобождения, известный в данной области, такой как моностеарат глицерина или дистеарат глицерина, в отдельности или в смеси с воском.
Если для перорального применения используют твердый носитель, препарат может быть таблетирован, помещен в твердую желатиновую капсулу в форме порошка или гранул, или он может быть в виде пастилки или лепешки. Количество твердого носителя будет широко варьировать, но обычно будет находиться в интервале от около 25 мг до около 1 г. Если используют жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы или стерильной жидкости для инъекций, такой как водные или неводные жидкие суспензия и раствор.
Обычно соединения по данному изобретению распределены в виде стандартной лекарственной формы, содержащей 50-200 мг активного ингредиента в или вместе с фармацевтически приемлемым носителем на стандартную лекарственную форму.
Доза соединений по данному изобретению составляет 1-500 мг/сутки, например, около 100 мг на дозу, будучи вводимым пациентам, например, людям, в качестве лекарственного средства.
Типичная таблетка, которая может быть получена обычными методиками таблетирования, содержит
Ядро:
Активный ингредиент (в виде свободного
соединения или его соли) 100 мг
Коллоидный диоксид кремния (Areosil®) 1,5 мг
Целлюлоза, микрокрист. (Avicel®) 70 мг
Модифицированная целлюлозная смола 7,5 мг
(Ac-Di-Sol®)
Стеарат магния
Покрытие:
НРМС, приблиз. 9 мг
*Myvacett® 9-40 Т, приблиз. 0,9 мг
*Ацилированный моноглицерид, применяемый в качестве пластификатора при покрытии пленкой.
Путь введения может представлять собой любой путь, позволяющий эффективно доставлять активное соединение к надлежащему или желаемому месту действия, такой как пероральный или парентеральный, например, ректальный, чрескожный, подкожный, интраназальный, внутримышечный, местный, внутривенный, интрауретральный, глазные капли или мазь, причем пероральный путь введения является предпочтительным.
ПРИМЕРЫ.
Способ получения соединений формулы I и содержащих их препаратов проиллюстрированы далее в следующих примерах, которые, однако, не следует истолковывать как ограничивающие.
Здесь и далее, ТСХ означает тонкослойную хроматографию, CDCl3 означает дейтерированный хлороформ, а ДМСО-d6 означает гексадейтерированный диметилсульфоксид. Структуры соединений подтверждены либо с помощью элементного анализа, либо с помощью ЯМР, где пики, приписываемые характеристическим протонам в указанных в заголовках соединениях, представлены в характерных положениях. Значения сдвигов 1Н ЯМР (δ н) приводятся в миллионных долях (м.д.). Т. пл. означает точку плавления и приводится в ° С и не является точным значением. Колоночную хроматографию проводили, используя метод, описанный W.C. Still et al., J. Org. Chem. (1978), 43, 2923-2925 на силикагеле 60 Merck (Арт. 9385). Соединения, используемые в качестве исходных продуктов, представляли собой либо известные соединения, либо соединения, которые могут быть легко синтезированы способами, известными специалистам.
Пример 1.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты.
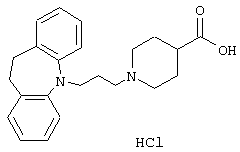
К суспензии 10,11-дигидро-5Н-дибенз[b,f]азепина (15,2 г, 0,078 моль) в толуоле (100 мл) добавляли 3-хлорпропионилхлорид (9,50 мл, 0,099 моль) и полученную смесь нагревали с обратным холодильником в течение 1 ч. Добавляли насыщенный водный бикарбонат натрия (100 мл) и фазы разделяли. Органическую фазу промывали солевым раствором (100 мл), сушили (МgSO4) и концентрировали в вакууме. Это позволяло получить 23,6 г 3-хлор-1-(10,11-дигидро-5Н-дибенз [b,f]азепин-5-ил)-1-пропанона в виде твердого вещества, который использовали на следующей стадии без дополнительной очистки.
Т. пл. 107-108° С.
Вычислено для C17H16ClNO: С 71,45%; Н 5,64%; N 4,90%. Найдено: С 71,45%; Н 5,79%; N 5,01%.
К раствору 3-хлор-1-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанона (14,0 г, 0,044 моль) в тетрагидрофуране (150 мл) при 0° С добавляли боргидрид натрия (6,66 г, 0,176 моль) с последующим добавлением по каплям ледяной уксусной кислоты (10,0 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи и затем нагревали при температуре кипения в течение 2 ч. Добавляли еще боргидрид (6,50 г, 172 ммоль) и затем эфират трифторида бора (20,0 мл, 0,163 моль) и нагревание при температуре кипения продолжали в течение 20 ч. Осторожно добавляли воду (350 мл) и фазы разделяли. Водную фазу экстрагировали толуолом (3× 100 мл). Объединенные органические фазы промывали солевым раствором (3× 100 мл), сушили (МgSO4) и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (100 г), используя градиент гептана и этилацетата (10:0→ 10:2) с получением 4,58 г (38%) 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина в виде масла.
ТСХ: Rf=0,63 (SiO2: этилацетат/гептан = 1:2).
Смесь этилового эфира 4-пиперидинкарбоновой кислоты (2,55 г, 16,2 ммоль), ацетонитрила (13 мл), вышеуказанного хлорида (2,00 г, 0,0074 моль) и йодида калия (1,14 г, 0,0068 моль) нагревали при температуре кипения в течение 4 ч и затем перемешивали при комнатной температуре в течение ночи. Добавляли воду (50 мл) и продукт экстрагировали этилацетатом (3× 20 мл). Объединенные органические экстракты промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя градиент гептана и этилацетата (10:1→ 1:1) с получением 1,6 г (54%) этилового эфира 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,26 (SiO2: этилацетат/гептан = 1:1).
Вычислено для С25Н32N2О2: С 76,50%; Н 8,22%; N 7,14%. Найдено: С 76,34%; Н 8,51%; N 6,88%.
Вышеуказанный эфир (1,01 г, 2,57 ммоль) растворяли в этаноле (10 мл) и добавляли раствор гидроксида натрия (0,59 г, 14,8 ммоль) в воде (1,5 мл). Полученную смесь перемешивали при комнатной температуре в течение 3,5 ч. Добавляли смесь воды (20 мл) и концентрированной соляной кислоты (3,0 мл) и водную фазу экстрагировали дихлорметаном (3× 15 мл). Объединенные органические экстракты промывали солевым раствором (20 мл) и сушили (MgSO4). Выпаривание растворителя давало пену, которую вновь растворяли в смеси метанола (1,0 мл) и этилацетата (5,0 мл). Концентрирование в вакууме позволяло получить твердое вещество, которое суспендировали в этилацетате, нагревали при температуре кипения в течение 1 минуты и давали остыть до комнатной температуры. Твердое вещество отфильтровывали и сушили с получением 0,9 г (88%) указанного в заголовке соединения в виде порошка.
Т. пл. 195-197° С.
Вычислено для C23H28N2O2, Cl: С 68,90%; Н 7,29%; N 6,99%.
Найдено: С 68,90%; Н 7,55%; N 6,72%.
Пример 2.
Гидрохлорид 4-(4-хлорфенил)-1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)пиперидин-4-ола.
К суспензии 10,11-дигидро-5Н-дибенз[b,f]азепина (27,6 г, 0,141 моль) в толуоле (250 мл) добавляли этилмалонилхлорид (25,0 г, 0,166 моль) и полученную смесь нагревали с обратным холодильником в течение 1 ч. Добавляли насыщенный водный бикарбонат натрия (200 мл) и фазы разделяли. Органическую фазу промывали солевым раствором (2× 150 мл), сушили (МgSO4) и концентрировали в вакууме. Это позволяло получить 56,0 г (100%) этилового эфира 3-(10,11-дигидро-5Н-дибенз [b,f]азепин-5-ил)-3-оксопропионовой кислоты в виде масла, которое использовали на следующей стадии без дополнительной очистки.
В круглодонную колбу в атмосфере азота вносили литийалюминийгидрид (20,0 г, 0,527 моль). Добавляли толуол (800 мл) и затем добавляли тетрагидрофуран (80 мл). Полученную суспензию охлаждали до 10-20° С. Полученную смесь перемешивали при комнатной температуре в течение ночи. После охлаждения осторожно добавляли 2 н. гидроксид натрия (200 мл). Добавляли воду (1,0 л), органическую фазу декантировали и водную фазу экстрагировали толуолом (3× 300 мл). Объединенные органические фазы промывали солевым раствором (2× 100 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (175 г), используя градиент гептана и этилацетата (10:0→ 2:1) с получением 21,2 г (59%) 3-(10,11-дигидро-5Н-дибенз[b,f]-азепин-5-ил)-1-пропанола в виде масла.
К перемешиваемому раствору вышеуказанного спирта (1,01 г, 0,004 моль) и триэтиламина (1,02 г, 0,010 моль) в толуоле (25 мл) при 0° С в течение 10 минут по каплям добавляли метансульфонилхлорид (0,6 мл, 0,0077 моль). Полученную смесь перемешивали при 0° С в течение 1,5 ч. Добавляли воду (50 мл) и фазы разделяли. Водную фазу экстрагировали толуолом (50 мл) и объединенные органические фазы промывали солевым раствором (2× 50 мл), сушили (МgSO4) и концентрировали в вакууме. Сырой мезилат перемешивали с 4-(4-хлорфенил)пиперидин-4-олом (0,81 г, 0,008 моль) в ацетонитриле (9 мл) и нагревали при температуре кипения в течение 6 ч. Реакционную смесь оставляли перемешиваться при комнатной температуре на 2 суток. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (3× 15 мл), промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Добавляли воду (50 мл) и смесь подкисляли добавлением концентрированной соляной кислоты (3 мл). Водный раствор экстрагировали дихлорметаном (2× 20 мл), промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученное масло растворяли в смеси этилацетата (15 мл) и метанола (2 мл). Небольшими порциями добавляли гептан до тех пор, пока раствор не становился слегка мутным. Через 4 ч кристаллы отфильтровывали, промывали гептаном и сушили с получением 1,1 г (61%) указанного в заголовке соединения в виде твердого вещества.
Т. пл. 189-191° С.
Вычислено для С28Н30N2О, НСl: С 69,56%; Н 6,67%; N 5,79%. Найдено: С 69,88%; Н 6,92%; N 5,62%.
Пример 3.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинметанола.
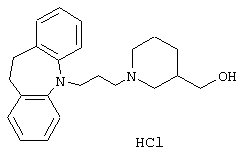
Смесь 3-(гидроксиметил)пиперидина (1,01 г, 0,0088 ммоль), ацетонитрила (9 мл), 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина (0,86 г, 0,003 моль, полученного так же, как описано в примере 1) и йодида калия (0,56 г, 0,003 моль) нагревали при температуре кипения в течение 18 ч. Добавляли воду (20 мл) и продукт экстрагировали этилацетатом (3× 15 мл). Объединенные органические экстракты промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток вновь растворяли в смеси воды (20 мл) и концентрированной соляной кислоты (3,0 мл) и экстрагировали дихлорметаном (3× 10 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили (МgSO4) и концентрировали в вакууме. Остаток упаривали со смесью метанола (0,5 мл) и этилацетата (5 мл) с получением 0,8 г (61%) указанного в заголовке соединения в виде игл.
Т. пл. 145-147° С.
Вычислено для С23N30N2O: С 71,39; Н 8,07; N 7,24. Найдено: С 71,15; Н 8,29; N 7,01.
Пример 4.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинкарбоксамида.
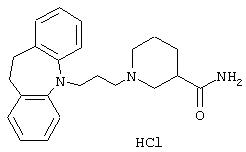
К перемешиваемому раствору 3-(10,11-дигидро-5Н-дибенз [b,f]азепин-5-ил)-1-пропанола (1,44 г, 0,0057 моль, полученного так же, как описано в примере 2) и триэтиламина (1,46 г, 0,014 моль) в толуоле (40 мл) при 0° С в течение 10 минут по каплям добавляли метансульфонилхлорид (0,88 мл, 0,011 моль). Полученную смесь перемешивали при 0° С в течение 1,5 ч. Добавляли толуол (50 мл) и воду (100 мл) и фазы разделяли.
Водную фазу экстрагировали толуолом (50 мл) и объединенные органические фазы промывали солевым раствором (2× 100 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток растворяли в ацетонитриле (20 мл) и добавляли 3-пиперидинкарбоксамид (1,09 г, 0,0085 моль) и карбонат калия (1,76 г, 0,013 моль). Смесь нагревали при температуре кипения в течение 4 ч и перемешивали при комнатной температуре в течение 40 ч. Добавляли воду (20 мл) и продукт экстрагировали этилацетатом (2× 20 мл). Объединенные органические экстракты промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Добавляли воду (20 мл) и концентрированную соляную кислоту (3,0 мл) и смесь экстрагировали дихлорметаном (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток вновь растворяли в смеси теплого этилацетата (10 мл) и метанола (1,0 мл) и после стояния в течение 5 ч при комнатной температуре осадок отфильтровывали и сушили с получением 1,56 г (64%) соединения заголовка.
Время удерживания по данным ЖХВР = 20,44 минут (колонка 4× 250 мм 5 мкм С 18, элюирование градиентом 20-80% 0,1% трифторуксусной кислоты/ацетонитрила и 0,1% трифторуксусной кислоты/воды в течение более 30 минут при 35° С).
Вычислено для C23H29N3O, HCl, 1,5 Н2O: С 64,70%; Н 7,78%; N 9,84%. Найдено: С 65,13%; Н 7,85%; N 9,85%.
Пример 5.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперидинкарбоновой кислоты.
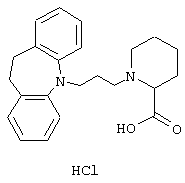
Смесь гидрохлорида этилового эфира 2-пиперидинкарбоновой кислоты (0,60 г, 0,003 моль), ацетонитрила (10 мл), 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз [b,f]азепина (0,60 г, 0,002 моль, полученного так же, как описано в примере 1), йодида калия (0,40 г, 0,002 моль), карбоната калия (1,03 г, 0,008 моль) и NN-диметилформамида (5 мл) нагревали при температуре кипения в течение 85 ч. Добавляли воду (50 мл) и водный раствор экстрагировали этилацетатом (3× 20 мл). Объединенные органические экстракты промывали солевым раствором (2× 50 мл), сушили (МgSO4) и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (15 г), используя градиент гептана и этилацетата (100:0→ 100:25) с получением 0,83 г (97%) этилового эфира 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,58 (SiO2: этилацетат/гептан = 1:1).
Вышеуказанный эфир (0,83 г, 0,002 моль) растворяли в смеси этанола (8 мл), воды (2 мл) и гидроксида натрия (0,58 г, 0,015 моль) в воде (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 50 ч и при 50° С в течение 5 ч. Добавляли воду (50 мл) и концентрированную соляную кислоту (3 мл) и полученную смесь экстрагировали дихлорметаном (3× 10 мл). Объединенные органические экстракты промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток вновь растворяли в метаноле (2 мл) и этилацетате (5 мл) и концентрировали в вакууме. Твердый остаток промывали малым количеством этилацетата и сушили с получением 0,6 г (73%) указанного в заголовке соединения в виде порошка.
Т. пл. 122-126° С.
Вычислено для C23H28N2O2, HCl, 0,25 H2O: С 68,14%; Н 7,33%; N 6,91%. Найдено: С 68,34%;.Н 7,63%; N 6,66%.
Пример 6.
4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперазинкарбоксилат калия.
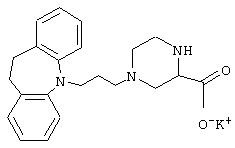
Смесь дигидрохлорида 2-пиперазинкарбоновой кислоты (5,06 г, 0,025 моль), этанола (100 мл) и концентрированной серной кислоты (6,0 мл) нагревали при температуре кипения в течение 6 суток. Добавляли толуол (10 мл) и полученную смесь концентрировали в вакууме до 2/3 от ее исходного объема. Добавляли холодный насыщенный водный карбонат калия (80 мл) и смесь экстрагировали толуолом (3× 100 мл). Объединенные органические экстракты промывали солевым раствором (2× 50 мл), сушили (MgSO4) и концентрировали в вакууме с получением 1,0 г (26%) этилового эфира 2-пиперазинкарбоновой кислоты в виде масла. Масло кристаллизовалось при стоянии при комнатной температуре.
К перемешиваемому раствору 3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанола (1/17 г, 0,0046 моль, полученного так же, как описано в примере 2) и триэтиламина (1,18 г, 0,012 моль) в толуоле (30 мл) при 0° С в течение 10 минут по каплям добавляли метансульфонилхлорид (0,70 мл, 0,009 моль). Полученную смесь перемешивали при 0° С в течение 1 ч. Добавляли толуол (50 мл) и воду (100 мл) и фазы разделяли. Водную фазу экстрагировали толуолом (50 мл) и объединенные органические фазы промывали солевым раствором (2× 100 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток растворяли в ацетонитриле (10 мл) и добавляли этиловый эфир. 2-пиперазинкарбоновой кислоты (1,40 г, 0,0089 моль), карбонат калия (0,67 г, 0,0049 моль) и толуол (5 мл). Полученную смесь нагревали при температуре кипения в течение 18 ч. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (3× 20 мл). Объединенные органические экстракты промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (27 г), используя градиент метанола и этилацетата (5:100→ 20:100) с получением 0,6 г (33%) этилового эфира 4-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперазинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,60 (SiO2: этилацетат/метанол = 1:1).
Смесь вышеуказанного эфира (0,55 г, 0,0014 моль), этанола (5 мл), воды (1 мл) и гидроксида натрия (0,34 г, 0,0085 моль) перемешивали при комнатной температуре в течение 18 ч. Добавляли воду (50 мл) и концентрированную соляную кислоту (3 мл) и раствор промывали дихлорметаном (4× 15 мл). Дихлорметановые экстракты отбрасывали. Водную фазу подщелачивали путем добавления карбоната калия (7,1 г) и смесь экстрагировали дихлорметаном (4× 15 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Это давало 0,6 г масла, которое растирали с ацетонитрилом (2 мл), и затем сушили в вакууме с получением 0,5 г (90%) указанного в заголовке соединения в виде воскообразного твердого вещества.
Т. пл. 151-155° С.
Вычислено для С22Н26N3О3К, 0,5 Н2О: С 64,05%; Н 6,59%; N 10,18%. Найдено: С 64,34%; Н 7,04%; N 10,16%.
Пример 7.
4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1-пиперазинацетат натрия.
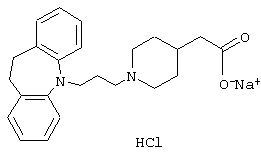
К раствору 3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанола (1,44 г, 0,0057 моль, полученного так же, как описано в примере 2) в дихлорметане (30 мл) при 0° С добавляли триэтиламин (1,73 г, 0,017 моль) с последующим добавлением метансульфонилхлорида (0,9 мл, 0,012 моль). Полученную смесь перемешивали в течение 30 минут при 0° С. Добавляли воду (50 мл), фазы разделяли и органический слой промывали солевым раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток растворяли в ацетонитриле (10 мл), добавляли N-(этоксикарбонилметил) пиперазин (2,44 г, 0,014 моль) и реакционную смесь нагревали при 82° С в течение 5,5 ч. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (3× 20 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (35 г), используя этилацетат в качестве элюента. Это давало 1,5 г (63%) этилового эфира 4-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1-пиперазинуксусной кислоты в виде масла.
ТСХ: Rf=0,18 (SiO2: этилацетат).
Смесь вышеуказанного эфира (0,94 г, 0,0023 моль), этанола (5 мл), воды (1 мл) и гидроксида натрия (0,31 г, 0,0078 моль) перемешивали при комнатной температуре в течение 4,5 ч. Добавляли воду (50 мл) и концентрированную соляную кислоту (3 мл) и раствор промывали дихлорметаном (3× 10 мл). Органические экстракты отбрасывали. Водную фазу подщелачивали путем добавления 4 н. гидроксида натрия (20 мл) и карбоната калия (4 г). Смесь экстрагировали дихлорметаном (5× 20 мл) и объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме с получением 0,9 г пены. Пену растирали и промывали этилацетатом (2 мл) и сушили в вакууме с получением 0,5 г (56%) указанного в заголовке соединения в виде твердого вещества.
Время удержания по данным ЖХВР = 17,81 минут (колонка 4× 250 мм 5 мкм С18, элюция по градиенту 20-80% 0,1% трифторуксусной кислоты/ацетонитрила и 0,1% трифторуксусной кислоты/воды в течение 30 минут при 35° С).
1H ЯМР (400 МГц, СDСl3) δ 1,60 (м, 2Н), 2,10-2,60 (м, 12Н), 3,12 (ушир. С 4Н), 3,68 (м, 2Н), 6,83-7,17 (м, 8Н).
Пример 8.
Гидрохлорид 4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-морфолинкарбоновой кислоты.
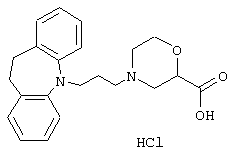
Смесь этилового эфира 2-морфолинкарбоновой кислоты (0,50 г, 0,0031 моль, полученного так же, как описано в Tetrahedron Letters, Том 32, 2281-4, 1991), ацетонитрила (6 мл), карбоната калия (0,50 г, 0,0036 моль), йодида калия (0,54 г, 0,0033 моль), 3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропилового эфира метансульфоновой кислоты (0,36 г, 0,0011 моль, полученного так же, как описано в примере 2) и 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина (0,40 г, 0,0015 моль, полученного так же, как описано в примере 1) нагревали при температуре кипения в течение 22 ч. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (3× 15 мл). Объединенные органические фазы промывали солевым раствором (20 мл), сушили (МgSО4) и концентрировали в вакууме. Продукт очищали колоночной хроматографией на силикагеле (30 г), используя градиент гептана и этилацетата (10:0→ 10:4). Это давало 0,4 г (42%) этилового эфира 4-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-морфолинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,33 (SiО2: этилацетат/гептан = 1:1).
Смесь вышеуказанного эфира (0,40 г, 0,0010 моль) и 4 н. гидроксида натрия (2 мл) перемешивали при комнатной температуре в течение 17 ч. Добавляли воду (50 мл) и концентрированную соляную кислоту (3 мл) и смесь экстрагировали этилацетатом (3× 20 мл). Объединенные органические экстракты промывали солевым раствором (20 мл) и сушили (MqSO4). После фильтрования осушителя из раствора начинало выпадать в осадок твердое вещество. После стояния в течение 4 ч при комнатной температуре осадок отфильтровывали и продукт сушили в вакууме. Это давало 0,2 г (49%) указанного в заголовке соединения в виде твердого вещества.
Т. пл. 196-199° С.
Вычислено для С22Н26N2О3, НСl, 0,25 Н2O: С 64,86%; Н 6,80%; N 6,88%. Найдено: С 65,12%; Н 7,09%; N 6,39%.
Пример 9.
Гидрохлорид 2-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты.
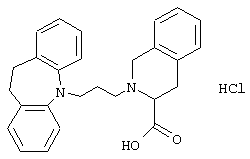
К раствору 3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанола (1,05 г, 0,0041 моль, полученного так же, как описано в примере 2) в дихлорметане (40 мл) при 0° С добавляли триэтиламин (1,28 г, 0,013 моль) с последующим добавлением метансульфонилхлорида (0,9 мл, 0,012 моль). После перемешивания в течение 30 минут при 0° С добавляли воду (50 мл) и фазы разделяли. Органический слой промывали солевым раствором (20 мл), сушили (МgSO4) и концентрировали в вакууме. Остаток растворяли в ацетонитриле (12 мл) и добавляли метиловый эфир 1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты (1,13 г, 0,050 моль), NN-диметилформамид (5 мл), карбонат калия (1,32 г, 0,096 моль) и йодид калия (0,30 г, 0,018 моль). Реакционную смесь нагревали при 82° С в течение 12 ч. Добавляли NN-диметилформамид (5 мл) и нагревание продолжали в течение еще 16 ч. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (3× 20 мл). Объединенные органические экстракты промывали солевым раствором (2× 20 мл), сушили (MgSO4) и концентрировали в вакууме. Продукт очищали колоночной хроматографией на силикагеле (80 г), используя градиент гептана и этилацетата (10:0→ 10:3) с получением 1,4 г (76%) метилового эфира 2-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,61 (SiO2: гептан/этилацетат = 1:1).
Смесь вышеуказанного эфира (0,80 г, 0,0019 моль), этанола (5 мл), тетрагидрофурана (5 мл) и 4 н. гидроксида натрия (4 мл) перемешивали при комнатной температуре в течение 22 ч. Добавляли воду (50 мл) и концентрированную соляную кислоту (2 мл) и смесь экстрагировали этилацетатом (2× 20 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили (MgSO4) и концентрировали в вакууме. Это давало 0,8 г твердого вещества, которое растирали и промывали этилацетатом (2× 5 мл). Сушка в вакууме позволяла получить 0,6 г (75%) указанного в заголовке соединения в виде твердого вещества.
Т. пл. 205-208° С.
Вычислено для C27H28N2O2, HCl, 0,25 Н2O: С 71,51%; Н 6,56%; N 6,18%. Найдено: С 71,34%; Н 6,69%; N 5,90%.
Пример 10.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз [b,f]азепин-5-ил)-1-пропил)-4-пиперидинуксусной кислоты.
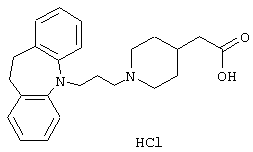
Смесь 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина (1,5 г, 0,0055 моль, полученного так же, как описано в примере 1) и йодида калия (5,4 г, 0,0327 моль) в метилэтилкетоне (100 мл) нагревали при температуре кипения в течение 2,5 ч. Добавляли карбонат калия (1,5 г, 0,109 моль) и этиловый эфир 4-пиперидинуксусной кислоты (1,4 г, 0,0082 моль, полученного так же, как описано в J. Am. Chem. Soc., Том 75, 6249, 1953) и реакционную смесь перемешивали при 75° С в течение ночи. После охлаждения реакционную смесь фильтровали (Hyflo) и растворитель выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (150 г), используя градиент гептана и этилацетата (1:1→ 3:7) с получением 0,6 г этилового эфира 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинуксусной кислоты в виде масла.
ТСХ: Rf=0,10 (SiO2: этилацетат/гептан = 1:1).
К раствору вышеуказанного эфира (0,6 г, 0,0015 моль) в этаноле (5 мл) добавляли 4 н. гидроксид натрия (0,8 мл) и смесь перемешивали при комнатной температуре в течение 2,5 ч и затем оставляли на ночь в холодильнике. Холодной реакционной смеси давали нагреться до комнатной температуры в течение 1 ч и добавляли 4 н. соляную кислоту (1,2 мл) и воду (10 мл). Смесь экстрагировали дихлорметаном (2× 100 мл). Объединенные органические экстракты сушили (MgSO4) и растворитель выпаривали в вакууме. Остаток вновь упаривали со смесью ацетона и изопропилацетата и затем обрабатывали смесью ацетона и изопропилацетата с получением твердого вещества, которое выделяли и сушили в вакууме. Это давало 0,33 г соединения заголовка.
Т. пл. 185-188° С.
Вычислено для С24Н30N2O2, НСl: С 69,47%; Н 7,53%; N 6,75%. Найдено: С 69,21%; Н 7,80%; N 6,45%.
Пример 11.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибензо [а,d]цикло-гептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты.
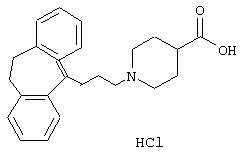
5-(3-Бром-1-пропилиден)-10,11-дигидро-5Н-дибензо[a,d]циклогептен (3,00 г, 0,0096 моль), карбонат калия (8,3 г, 0,060 моль), йодид калия (3,3 г, 0,020 моль) и этиловый эфир 4-пиперидинкарбоновой кислоты (3,1 мл, 0,020 моль) смешивали в метилэтилкетоне (100 мл) и нагревали при температуре кипения в течение 20 ч и перемешивали при комнатной температуре в течение 3 суток. Добавляли воду (100 мл), фазы разделяли и водную фазу экстрагировали этилацетатом. Органическую фазу сушили (МgSO4) и выпаривали в вакууме с получением сырого продукта в почти количественном выходе. Сырой продукт обрабатывали 1 н. соляной кислотой и этилацетатом, выпаривали досуха и упаривали с этилацетатом с получением гидрохлорида этилового эфира 1-(3-(10,11-дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты в виде твердого вещества.
Т. пл. 169-170° С.
Смесь вышеуказнного эфира (0,105 г, 0,27 ммоль), этанола (15 мл) и 1н гидроксида натрия (10 мл) нагревали при температуре кипения в течение 3 часов и затем охлаждали до комнатной температуры. Добавляли воду (75 мл) и смесь подкисляли 5 н. соляной кислотой (3 мл) и экстрагировали дихлор-метаном (3× 75 мл), сушили (MgSO4) и выпаривали в вакууме.
Полученную пену кристаллизовали из ацетона с получением 0,080 г указанного в заголовке соединения в виде кристаллов.
Т. пл. 224-226° С.
Вычислено для C24H27NO2, HCl: С 72,44%; Н 7,09%; N 3,52%. Найдено: С 72,83%; Н 7,38%; N 3,23%.
Пример 12.
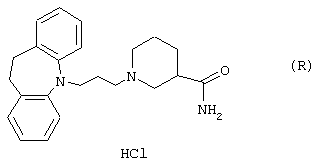
Гидрохлорид (R)-1-(3-(10,11-дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоксамида.
Гидрохлорид (R)-1-(3-(10,11-дигидро-5Н-дибензо[a,d] циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты (4,96 г, 12,5 ммоль, полученного так же, как описано в WO 9518793) растворяли в NN-диметилформамиде (60 мл). Добавляли N-гидроксибензотриазол (1,86 г, 13,8 ммоль) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (2,64 г, 13,8 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 20 минут. Добавляли гидрокарбонат аммония (1,98 г, 25 ммоль) и смесь перемешивали в течение 1 суток при комнатной температуре. Добавляли этилацетат (200 мл) и полученную смесь экстрагировали водой (200 мл), 5% водной лимонной кислоты (200 мл) и насыщенного гидрокарбоната натрия (200 мл). Объединенные водные фазы выпаривали досуха в вакууме и остаток экстрагировали дихлорметаном (200 мл). Полученную дихлорметановую суспензию фильтровали и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (600 мл), используя смесь этилацетата и триэтиламина (95:5) в качестве элюента. Это давало 1,92 г (43%) свободного основания в виде масла, которое затем преобразовывали в гидрохлорид путем растворения в диэтиловом эфире (25 мл) и добавления 1 н. соляной кислоты в диэтиловом эфире (5,9 мл). Фильтрование с последующей сушкой в вакууме позволяло получить 1,53 г (31%) указанного в заголовке соединения в виде твердого вещества.
ТСХ: Rf=0,33 (SiO2: этилацетат/триэтиламин = 95:5).
1Н ЯМР (400 МГц, СDСl3) δ 1,38 (дк, 1Н), 1,75-1,97 (м, 3Н), 2,5-3,7 (м, 13Н), 5,78 (т, 1Н), 7,0-7,3 (м, 8Н), 7,63 (С 1Н), 10,8 (С 1Н).
Т. пл. >250° С.
Вычислено для C24H28N2O, HCl: С 72,62%; Н 7,36%; N 7,06%. Найдено: С 72,24%; Н 7,59%; N 6,87%.
Пример 13.
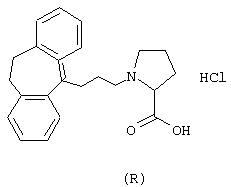
Гидрохлорид (R)-1-(3-(10,11-дигидро-5Н-дибензо[a,d] циклогептен-5-илиден)-1-пропил)-4-пирролидинкарбоновой кислоты.
5-(3-Бром-1-пропилиден)-10,11-дигидро-5Н-дибензо[a,d]циклогептен (3,86 г, 12,3 ммоль, полученного так же, как описано в WO 9518793), карбонат калия (10,2 г, 74 ммоль), йодид калия (4,08 г, 74 ммоль) и гидрохлорид метилового эфира D-пролина (2,45 мл, 14,8 ммоль) смешивали в метилэтилкетоне (40 мл) и нагревали при температуре кипения в течение 20 ч. После охлаждения смесь фильтровали и фильтрат выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (800 мл), используя смесь этилацетата и гептана (1:3) в качестве элюента. Это давало 0,96 г (22%) метилового эфира (R)-1-(3-(10,11-дигидро-5Н-дибензо[а,d] циклогептен-5-илиден)-1-пропил)-2-пирролидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,39 (SiO2: этилацетат/гептан = 1:2).
Вышеуказанный эфир растворяли в 1,4-диоксане (40 мл) и добавляли воду (5 мл). Порциями добавляли 1 н. водный гидроксид натрия (2,15 мл) при комнатной температуре в течение 6 ч. Смесь перемешивали в течение ночи при комнатной температуре. Порциями добавляли 1 н. водный гидроксид натрия (0,84 мл) в течение 20 ч. Добавляли воду (100 мл) и смесь промывали диэтиловым эфиром (2× 100 мл). Водную фазу подкисляли до рН=2 с помощью 1 н. соляной кислоты и экстрагировали дихлорметаном (2× 100 мл). Объединенные органические экстракты сушили (МgSO4) и выпаривали в вакууме с получением 0,29 г (41%) указанного в заголовке соединения в виде аморфного твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 1,95 (ушир. С 2Н), 2,3 (м, 2Н), 2,5-3,4 (м, 11Н), 5,78 (т, 1Н), 7,0-7,3 (м, 8Н).
Вычислено для С23Н25O2, НСl: С 71,96%; Н 6,83%; N 3,65%. Найдено: С 72,15%; Н 7,37%; N 3,40%.
Пример 14.
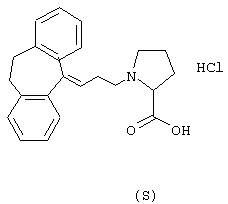
Гидрохлорид (S)-1-(3-(10,11-дигидро-5Н-дибензо[a,d] циклогептен-5-илиден)-1-пропил)-4-пирролидинкарбоновой
5-(3-Бром-1-пропилиден)-10,11-дигидро-5Н-дибензо[a,d]циклогептен (2,00 г, 6,4 ммоль, полученного так же, как описано в WO 9518793), карбонат калия (5,3 г, 38,4 ммоль), йодид калия (2,12 г, 12,8 ммоль) и гидрохлорид метилового эфира L-пролина (1,27 мл, 7,7 ммоль) смешивали в метилэтилкетоне (40 мл) и нагревали при температуре кипения в течение 12 ч. После охлаждения смесь фильтровали и фильтрат выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (800 мл), используя смесь этилацетата и гептана (1:2) в качестве элюента. Это давало 1/64 г (71%) метилового эфира (S)-1-(3-(10,11-дигидро-5Н-дибензо[a,d] циклогептен-5-илиден)-1-пропил)-2-пирролидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,39 (SiO2: этилацетат/гептан = 1:2).
Вышеуказанный эфир (1,3 г, 3,6 ммоль) растворяли в 1,4-диоксане (50 мл) и добавляли воду (5 мл). Порциями добавляли 1 н. водный гидроксид натрия (3,8 мл) при комнатной температуре в течение 6 ч. Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду (100 мл) и смесь промывали диэтиловым эфиром (2× 100 мл). Водную фазу подкисляли до рН=-2 с помощью 1 н. соляной кислоты и экстрагировали дихлорметаном (2× 100 мл). Объединенные органические экстракты сушили (MgSO4) и выпаривали в вакууме с получением 0,80 г (58%) указанного в заголовке соединения в виде аморфного твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 1,95 (ушир. С 2Н), 2,3 (м, 2Н), 2,5-3,4 (м, 11Н), 5,78 (т, 1Н), 7,0-7,3 (м, 8Н).
Вычислено для С23Н25NО2, НСl: С 71,96%; Н 6,83%; N 3,65%. Найдено: С 72,61%; Н 7,30%; N 3,34%.
Пример 15.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибензо [а,d]цикло-гептен-5-илиден)-1-пропил)-2-пиперидинкарбоновой кислоты.
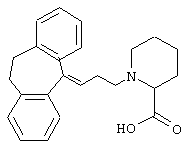
5-(3-Бром-1-пропилиден)-10,11-дигидро-5Н-дибензо[a,d]циклогептен (10,0 г, 22 ммоль, полученной так же, как описано в WO 9518793), карбонат калия (18,24 г, 132 ммоль) и гидрохлорид этилового эфира DL-пипеколиновой кислоты (5,40 г, 26 ммоль) смешивали в этилацетате (50 мл) и нагревали при температуре кипения в течение 16 ч. Добавляли еще карбонат калия (10 г), гидрохлорид этилового эфира DL-пипеколиновой кислоты (2 г) и этилацетат (50 мл) и смесь нагревали при температуре кипения в течение 24 ч. После охлаждения смесь фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (500 мл), элюируя сначала дихлорметаном, а затем этилацетатом. Это позволяло получить 7,92 г (92%) этилового эфира 1-(3-(10/11-дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил)-2-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,54 (SiO2: этилацетат/гептан = 3:7).
К вышеуказанному эфиру (7,7 г, 20 ммоль) добавляли 1 н. соляную кислоту (100 мл) и смесь нагревали в течение 1 ч с одновременной отгонкой воды и этанола (отгоняли 20 мл). Добавляли воду (40 мл) и смесь нагревали в течение 5 ч с отгонкой воды и этанола. После охлаждения к смеси добавляли толуол (100 мл). Осевшие кристаллы выделяли путем фильтрования и промывали 1 н. соляной кислотой с получением сырого продукта (5,3 г). Его часть (2,0 г) подвергали дополнительной очистке путем растворения в воде (100 мл, 75° С) и добавления 37% соляной кислоты (6 мл). Смеси давали охладиться до комнатной температуры. Фильтрование, промывание 1 н. соляной кислотой и сушка в вакууме позволяли получить 1,72 г указанного в заголовке соединения в виде твердого вещества.
Т. пл. 126-127° С.
1H ЯМР (300 МГц, СDСl3) δ 1,5-1,75 (м, 4Н), 2,1 (м, 1Н), 2,5-3,5 (м, ПН), 4,10 (ушир. С 1Н), 5,80 (т, 1Н), 7,05-7,23 (м, 8Н).
Вычислено для C23H26NO2, HCl, 0,5 Н2O: С 70,13%; Н 7,16%; N 3,56%. Найдено: С 69,74%; Н 7,46%; N 3,08%.
Пример 16.
Гидрохлорид 1-(3-(10Н-феноксазин-10-ил)-1-пропил)-4-пиперидинкарбоновой кислоты.
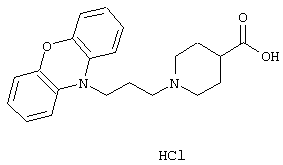
Феноксазин (10,0 г, 54,6 ммоль) растворяли в NN-диметилформамиде (300 мл) в атмосфере азота. Порциями добавляли гидрид натрия (3,27 г, 81,9 ммоль, 60% дисперсия в масле) и полученную смесь перемешивали в течение 20 минут при комнатной температуре. По каплям добавляли 1-бром-3-хлорпропан (21,48 г, 0,136 ммоль). Смесь перемешивали в течение ночи. В течение 4 минут добавляли хлорид аммония (5,5 г, 0,10 моль) и перемешивание продолжали в течение 30 минут. Смесь выливали в воду (800 мл) и экстрагировали дихлорметаном (2× 600 мл). Объединенные органические экстракты сушили (MgSO4) и выпаривали в вакууме. Это позволяло получить 16/0 г сырого 10-(3-хлорпропил)-10Н-феноксазина.
Вышеуказанный сырой хлорид (5,09 г, 17,4 ммоль) растворяли в ацетонитриле (100 мл) и добавляли йодид калия (2,74 г, 16,5 ммоль). Этиловый эфир 4-пиперидинкарбоновой кислоты (6,00 г, 38,2 ммоль) растворяли в ацетонитриле (30 мл) и добавляли к реакционной смеси. Полученную смесь нагревали при температуре кипения в течение 24 ч и оставляли перемешиваться при комнатной температуре в течение 48 ч. Добавляли воду (100 мл) с последующим добавлением этилацетата (100 мл). Водную фазу экстрагировали этилацетатом (3× 100 мл) и объединенные органические фазы промывали солевым раствором (2× 100 мл) и сушили (MgSO4). После выпаривания в вакууме остаток очищали колоночной хроматографией на силикагеле (500 мл), используя смесь гептана и этилацетата (1:1) в качестве элюента. Это давало 5,18 г (77%) этилового эфира 1-(3-(10Н-феноксазин-10-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
ТСХ; Rf=0,2 (SiO2: этилацетат/гептан = 1:1).
Вышеуказанный эфир (1,56 г, 4,04 ммоль) растворяли в смеси 96% этанола (20 мл) и тетрагидрофурана (20 мл). Добавляли раствор гидроксида натрия (0,95 г) в воде (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч. Добавляли 0,1М соляную кислоту (28 мл) и смесь экстрагировали дихлорметаном (3× 30 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили (MgSO4) и выпаривали в вакууме. Дважды добавляли ацетон и раствор выпаривали в вакууме. После третьего добавления ацетона начиналось выпадение осадка и смесь оставляли перемешиваться на 2 ч. После фильтрования твердое вещество вновь суспендировали в ацетоне (25 мл) и оставляли перемешиваться на ночь. Твердое вещество отфильтровывали, промывали ацетоном и сушили. Это позволяло получить 1,30 г (83%) указанного в заголовке соединения в виде твердого вещества.
Т. пл. 196-198° С.
Вычислено для C21H24N2O3, HCl: С 64,86%; Н 6,48%; N 7,20%. Найдено: С 64,82%; Н 6,78%; N 6,77%.
Пример 17.
Гидрохлорид 1-(3-(3-хлор-10,11-дигидро-5Н-дибенз[b,f] азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты.

3-Хлор-10,11-дигидро-5Н-дибенз[b,f]азепин (3,82 г, 16,6 ммоль) растворяли в толуоле (20 мл). По каплям добавляли раствор 3-хлорпропионилхлорида (2,53 г, 19,9 ммоль) в толуоле и полученную смесь нагревали до 95° С и перемешивали при указанной температуре в течение 30 минут. Смесь перемешивали в течение ночи при комнатной температуре. Добавляли еще 3-хлорпропионилхлорид (2,53 г, 19,9 ммоль) и смесь нагревали при 95° С в течение 1,5 ч. После охлаждения добавляли 0,2М гидроксид натрия (10 мл) и фазы разделяли. Органическую фазу разбавляли с помощью дополнительного количества толуола (50 мл) и промывали сначала 0,2М гидроксидом натрия (6× 10 мл), а затем еще 0,2М гидроксидом натрия (3× 20 мл), до тех пор, пока водная фаза не становилась щелочной. Органическую фазу промывали водой (3× 15 мл), солевым раствором (25 мл) и сушили (MgSO4). Выпаривание в вакууме позволяло получить 5,23 г (98%) сырого 3-хлор-1-(3-хлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанона в виде масла. Его подвергали дальнейшей очистке путем добавления гептана и этилацетата (1:1). Это позволяло получить 3,14 г (59%) продукта в виде твердого вещества.
1,0М раствор литийалюминийгидрида в тетрагидрофуране (18,7 мл, 18,7 ммоль) помещали в сухую трехгорлую круглодонную колбу емкостью 250 мл в атмосфере азота. Раствор охлаждали на ледяной бане. Соблюдая меры предосторожности, по каплям добавляли концентрированную серную кислоту (0,5 мл) в течение 10 минут. Для компенсации испарения растворителя добавляли еще сухой тетрагидрофуран (20 мл) и смесь перемешивали в течение 15 минут. Добавляли еще тетрагидрофуран (20 мл) и ледяную баню удаляли. Смесь перемешивали в течение 75 минут при комнатной температуре. Вышеуказанный амид растворяли в сухом тетрагидрофуране (25 мл) и добавляли по каплям в течение 20 минут. Реакционную смесь перемешивали в течение 1 ч. Добавляли воду (0,7 мл) с последующим добавлением 4 н. гидроксида натрия (0,7 мл) и воды (2,1 мл). Перемешивание продолжали в течение 30 минут. Смесь фильтровали (Hyflo) и выпаривали в вакууме с получением 2,70 г (95%) 3-хлор-5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f] азепина в виде масла.
Вышеуказанный хлорид (1,50 г, 4,91 ммоль) растворяли в ацетонитриле (10 мл) и добавляли этиловый эфир 4-пиперидинкарбоновой кислоты (1,70 г, 10,8 ммоль) в ацетонитриле (4 мл) с последующим добавлением йодида калия (0,76 г, 4,6 ммоль). Полученную смесь нагревали при температуре кипения в течение 24 ч. Добавляли воду (50 мл) с последующим добавлением этилацетата (50 мл). Водную фазу экстрагировали этилацетатом (3× 30 мл) и объединенные органические фазы промывали солевым раствором (2× 25 мл) и сушили (MgSO4). После выпаривания в вакууме остаток очищали колоночной хроматографией на силикагеле (150 мл), используя смесь гептана и этилацетата (1:1) в качестве элюента. Это позволяло получить 1,78 г (85%) этилового эфира 1-(3-(3-хлор-10,11-дигидро-5Н-дибенз[b,f] азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,21 (SiO2: этилацетат/гептан = 1:1).
Вышеуказанный эфир (1,70 г, 3,98 ммоль) растворяли в 99% этаноле (20 мл). Добавляли раствор гидроксида натрия (0,92 г) в воде (2,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 1 н. соляную кислоту (23 мл) и смесь экстрагировали дихлорметаном (3× 25 мл). Объединенные органические экстракты промывали солевым раствором (25 мл) и водой (20 мл), сушили (MgSO4) и выпаривали в вакууме. Добавляли дихлорметан и раствор выпаривали повторно. К полученной пене добавляли изопропилацетат и осевшее твердое вещество отфильтровывали и промывали. Это позволяло получить 1,28 г (74%) сырого указанного в заголовке соединения. Продукт повторно растворяли в изопропаноле, раствор декантировали и выпаривали в вакууме. Добавляли дихлорметан и раствор выпаривали в вакууме. К полученной пене добавляли изопропилацетат и осевшее твердое вещество отфильтровывали и промывали. Процедуру повторяли еще раз.
МС(ЕI) 398 (M+-HCl, 18%).
Время удержания по данным ЖХВР = 24,14 минут (колонка 4× 250 мм 5 мкм С18, элюция по градиенту 20-80% 0,1% трифторуксусной кислоты/ацетонитрила и 0,1% трифторуксусной кислоты/воды в течение 30 минут при 35° С).
Пример 18.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинуксусной кислоты.
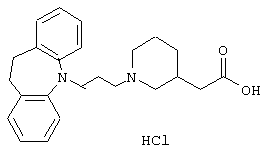
Суспензию 3-пиперидинуксусной кислоты (4,5 г, 0,032 моль, описанной в J. Org. Chem., 28, 602, 1963) в смеси сухого хлороводорода (в избытке) в этаноле перемешивали при температуре окружающей среды. Когда твердое вещество было растворено, раствор перемешивали в течение 2 суток. Растворитель выпаривали в вакууме и остаток повторно упаривали с диэтиловым эфиром (25 мл) и затем перемешивали с диэтиловым эфиром (35 мл) в течение 20 минут. Твердое вещество выделяли путем фильтрования и сушили. Это позволяло получить 6,1 г гидрохлорида этилового эфира 3-пиперидинуксусной кислоты в виде твердого вещества.
Т. пл. 111-113° С.
Смесь йодида калия (19,2 г, 0,12 моль) и метилэтилкетона (180 мл) нагревали при температуре кипения в течение 1 ч. Добавляли раствор 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина (5,2 г, 0,019 моль, полученного так же, как описано в примере 1) в метилэтилкетоне (25 мл) и нагревание при температуре кипения продолжали в течение 3 ч. Добавляли карбонат калия (9,3 г, 0,067 моль) и гидрохлорид этилового эфира 3-пиперидинуксусной кислоты (5,6 г, 0,027 моль) и реакционную смесь нагревали при температуре кипения в течение 2 ч. Температуру снижали до несколько меньшей, чем температура кипения, и смесь оставляли перемешиваться в течение ночи. После охлаждения реакционную смесь фильтровали (Hyflo) и растворитель выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (225 г), используя смесь гептана и этилацетата (1:1) в качестве элюента с получением 5,0 г этилового эфира 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинуксусной кислоты в виде масла.
TCX: Rf=0,19 (SiO2: этилацетат/гептан = 1:1).
К раствору вышеуказанного эфира (2,5 г, 0,0062 моль) в этаноле (10 мл) добавляли 4 н. гидроксид натрия (2,3 мл) и смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли 4 н. соляную кислоту (3,8 мл) и воду (10 мл). Смесь экстрагировали дихлорметаном (2× 250 мл). Объединенные органические экстракты сушили (МgSO4) и растворитель выпаривали в вакууме. Остаток дважды повторно упаривали с ацетоном и некоторое время перемешивали в ацетоне. Твердое вещество выделяли путем фильтрования и сушили. Это позволяло получить 2,4 г указанного в заголовке соединения в виде твердого вещества.
Т. пл. 233-235° С.
Вычислено для С24Н30N2О2, НСl: С 69,47%; Н 7,53%; N 6,75%. Найдено: С 69,59%; Н 7,78%; N 6,50%.
Пример 19.
Гидрохлорид 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-метил-3-пиперидинкарбоновой кислоты.
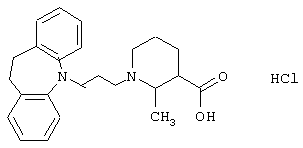
Метиловый эфир 2-метилникотиновой кислоты (4,0 г, 0,026 моль) растворяли в 1 н. соляной кислоте (30 мл) и добавляли 10% палладий на угле (0,8 г). Полученную смесь гидрировали при 200 фунтах на квадратный дюйм (~ 14,065 кг/см2) в течение 10 суток. Реакционную смесь фильтровали и твердое вещество промывали дихлорметаном (100 мл) и водой (50 мл). Объединенные фильтраты выпаривали в вакууме с получением остатка, который повторно упаривали с дихлорметаном (2× 30 мл). Это позволяло получить 5,1 г сырого гидрохлорида метилового эфира 2-метил-3-пиперидинкарбоновой кислоты, который использовали в дальнейшей реакции без очистки.
Смесь йодида калия (17,5 г, 0,11 моль) и метилэтилкетона (180 мл) нагревали при температуре кипения в течение 1 ч. Добавляли раствор 5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина (4,8 г, 0,019 моль, полученного так же, как описано в примере 1) в метилэтилкетоне (25 мл) и нагревание при температуре кипения продолжали в течение 2 ч. Добавляли карбонат калия (8,5 г, 0,061 моль) и гидрохлорид метилового эфира 2-метил-З-пиперидинкарбоновой кислоты (5,1 г, 0,026 моль) и реакционную смесь нагревали при температуре, несколько меньшей, чем температура кипения. После охлаждения реакционную смесь фильтровали (Hyflo) и растворитель выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (275 г), используя смесь гептана и этилацетата (1:1) в качестве элюента с получением 3,4 г метилового эфира 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-метил-3-пиперидинкарбоновой кислоты в виде масла.
К раствору вышеуказанного эфира (3,4 г, 0,0087 моль) в 96% этаноле (15 мл) добавляли 4 н. гидроксид натрия (4,4 мл) и смесь перемешивали при комнатной температуре в течение 4 ч и затем оставляли на ночь в холодильнике. Перемешивание продолжали при комнатной температуре в течение 5 ч и добавляли 4 н. соляную кислоту (6 мл). Растворитель выпаривали, а остаток растирали с ацетоном (30 мл) в течение 10 минут. Твердое вещество выделяли путем фильтрования, промывали ацетоном и сушили в вакууме. Твердое вещество суспендировали в смеси воды (35 мл) и дихлорметана (800 мл). Добавляли насыщенный раствор бикарбоната натрия до тех пор, пока значение рН не становилось равным 8-9, а твердое вещество не растворялось. Фазы разделяли и водную фазу экстрагировали дихлорметаном (250 мл). Объединенные органические экстракты объединяли и объем уменьшали путем выпаривания. Добавляли избыток концентрированной соляной кислоты и смесь выпаривали досуха. Остаток дважды повторно упаривали с дихлорметаном и затем растирали с ацетоном. Твердое вещество выделяли путем фильтрования и сушили в вакууме. Это позволяло получить 2,4 г указанного в заголовке соединения в виде твердого вещества.
Т. пл. 169-170° С.
Вычислено для C24H30N2O2, НСl, Н2О: С 66,59%; Н 7,63%; N 6,47%. Найдено: С 66,64%; Н 7,94%; N 6,23%.
Пример 20.
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-хинуклидинийкарбоксилат.
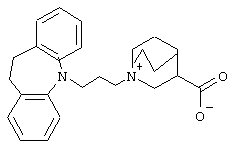
5-(3-Хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепин (1,70 г, 6,25 ммоль, полученный так же, как описано в примере 1) и метиловый эфир хинуклидин-3-карбоновой кислоты (0,85 г, 5,0 ммоль) растворяли в 2-бутаноне (25 мл). Добавляли сухие карбонат калия (4,15 г, 30 ммоль) и йодид натрия (0,75 г, 5 ммоль) и перемешиваемую смесь нагревали при температуре кипения в течение 5 ч. После охлаждения до комнатной температуры добавляли толуол (25 мл) и воду (25 мл). Образовывался маслянистый осадок, который отделяли от растворов путем декантирования и растворяли в дихлорметане (30 мл). Раствор промывали водой (2× 30 мл), 1 н. соляной кислотой, насыщенным раствором гидрокарбоната натрия (30 мл) и водой (30 мл). Органический слой сушили над MgSO4. Выпаривание в вакууме позволяло получить 1,15 г гидроксида 1-(3-(10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-метоксикарбонилхинуклидиния. Сырой продукт сразу использовали на следующей стадии без дополнительной очистки.
Вышеуказанный эфир (1,05 г, 2,50 ммоль) растворяли в этаноле (12,5 мл). Добавляли 2 н. раствор гидроксида натрия (4,1 мл, 8,25 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли воду (10 мл) и этанол выпаривали в вакууме. Оставшийся водный раствор промывали диэтиловым эфиром и экстрагировали 1-бутанолом (3× 20 мл). Бутанольные фазы промывали водой (10 мл) и выпаривали в вакууме. Упаривание остатка в н-гептаном
(3× 20 мл) позволяло получить указанное в заголовке соединение (0,71 г) в виде пены.
Вычислено для C25H30N2O2, 3,5 Н,0: С 66,20%; Н 8,22%; N 6,18%. Найдено: С 66,52%; Н 8,03%; N 5,79%.
Пример 21.
Гидрохлорид 1-(3-(2,8-дибром-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты.
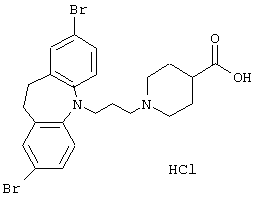
2,8-Дибром-10,11-дигидро-5Н-дибенз[b,f]азепин (1,41 г, 4,0 ммоль, полученный в соответствии с К. Smith et al., Tetrahedron 48, 7479 (1992)) растворяли в толуоле (25 мл). К перемешиваемому раствору при комнатной температуре добавляли триэтиламин (0,60 мл, 4,4 ммоль) и 3-хлорпропионилхлорид (0,50 мл, 5,2 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 ч и при температуре кипения в течение 2,5 ч. Реакционную смесь охлаждали, фильтровали и выпаривали в вакууме. Сырой 3-хлор-1-(2,8-дибром-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)пропан-1-он использовали на следующей стадии без дополнительной очистки.
Раствор литийалюминийгидрида (0,326 г, 8,56 ммоль) в сухом тетрагидрофуране (30 мл) охлаждали на ледяной бане и по каплям добавляли концентрированную серную кислоту (0,428 г, 4,28 ммоль). Раствор перемешивали при комнатной температуре в течение 0,5 ч. Раствор вышеуказанного продукта (1,90 г, 4,28 ммоль) добавляли по каплям и перемешивание продолжали в течение 0,5 ч. Реакцию затем гасили путем осторожного добавления этилацетата (5 мл) с последующим добавлением воды (0,8 мл). Фильтрование смеси и выпаривание фильтрата в вакууме позволяло получить 1,51 г (82%) 2,8-дибром-5-(3-хлорпропил)-10,11-дигидро-5Н-дибенз[b,f]азепина в виде пены, который использовали на следующей стадии без дополнительной очистки.
Вышеуказанный хлорид (1,5 г, 3,5 ммоль) и этилизонипекотат (0,79 г/ 5,0 ммоль) растворяли в 2-бутаноне (40 мл). Добавляли карбонат калия (1,4 г, 10 ммоль) и йодид калия (0,43 г, 2,6 ммоль) и перемешиваемую смесь нагревали при температуре кипения в течение 60 ч. Реакционную смесь фильтровали, фильтрат концентрировали в вакууме и остаток повторно растворяли в диэтиловом эфире (50 мл). При добавлении по каплям 2,6М раствора хлороводорода в диэтиловом эфире (2,0 мл, 5,2 ммоль) продукт выпадал в осадок в виде гидрохлорида. Осадок собирали путем фильтрования и сушили в вакууме с получением 1,3 г (63%) гидрохлорида этилового эфира 1-(3-(2,8-дибром-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде порошка, который использовали на следующей стадии без дополнительной очистки.
Вышеуказанный гидрохлорид сложного эфира (1,30 г, 2,2 ммоль) растворяли в этаноле (15 мл) и добавляли 2 н. гидроксид натрия (4,0 мл). Раствор перемешивали в течение 1,5 ч при комнатной температуре, подкисляли путем добавления 1 н. соляной кислоты до рН 1 и этанол выпаривали в вакууме. Водную суспензию промывали диэтиловым эфиром и фильтровали. Перекристаллизация собранного твердого вещества из этанола позволяла получить 0,25 г (19%) соединения указанного в заголовке.
Т. пл. 165-166° С.
Вычислено для С23H26N2Br2О2, НСl, 0,5 Н2О, 0,5 С2H6O: С 48,79%; Н 5,29%; N 4,74%. Найдено: С 48,64%; Н 5,39%; N 4,58%.
Пример 22.
Гидрохлорид 1-(3-(3,7-дихлор-10,11-дигидро-5Н-дибенз [b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты.
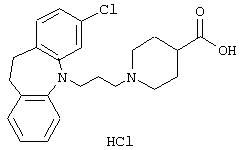
3,7-Дихлор-10,11-дигидро-5Н-дибенз[b,f]азепин (4,6 г, 17 ммоль) растворяли в толуоле (30 мл) и добавляли 3-хлорпропионилхлорид (2,3 мл, 24 ммоль). Полученную смесь нагревали при 95° С в течение 3 ч, перемешивали при комнатной температуре в течение 3 суток и нагревали при 90° С в течение 3 ч. После охлаждения добавляли этилацетат (150 мл) и смесь промывали водой (2× 100 мл). Органическую фазу сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (600 мл), используя смесь этилацетата и гептана (1:4) в качестве элюента. Это позволяло получить 4,7 г (76%) 3-хлор-1-(3,7-дихлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанона в виде масла.
ТСХ: Rf=0,14 (SiO2: этилацетат/гептан = 1:4).
Литийалюминийгидрид (0,44 г, 11,6 ммоль) растворяли в сухом тетрагидрофуране (15 мл) и раствор охлаждали до 0° С. Медленно и соблюдая меры предосторожности, с помощью шприца добавляли концентрированную серную кислоту (0,31 г, 5,8 ммоль). Суспензию перемешивали при комнатной температуре в течение 0,5 ч. Раствор вышеуказанного амида (2,0 г, 5,8 ммоль) в сухом тетрагидрофуране (15 мл) добавляли по каплям к суспензии. Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию гасили путем добавления воды (1,4 мл), 2 н. гидроксида натрия (0,5 мл), воды (4 мл) и карбоната калия (5,0 г). Смесь фильтровали и осадок на фильтре промывали сухим тетрагидрофураном. Объединенные фильтраты выпаривали в вакууме с получением 1,4 г (69%) 5-(3-хлорпропил)-3,7-дихлор-10,11-дигидро-5Н-дибенз[b,f]вазелина в виде масла.
ТСХ: Rf=0,66 (SiO2: этилацетат/гептан = 1:2).
Вышеуказанный хлорид (1,3 г, 3,8 ммоль) растворяли в метилэтилкетоне (20 мл). Добавляли йодид калия (0,63 г, 3,8 ммоль), карбонат калия (3,2 г, 23 ммоль) и этиловый эфир 4-пиперидинкарбоновой кислоты (1/2 г, 7,6 ммоль) и полученную смесь нагревали при температуре кипения в течение 24 ч. После охлаждения добавляли этилацетат (100 мл) и смесь промывали водой (2× 100 мл). Органическую фазу сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (600 мл), используя смесь этилацетата и гептана (1:2) в качестве элюента. Это позволяло получить 0,84 г (48%) этилового эфира 1-(3-(3,7-дихлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидин-карбоновой кислоты в виде масла.
ТСХ: Rf=0,22 (SiO2: этилацетат/гептан = 1:2).
Вышеуказанный эфир (0,8 г, 1,7 ммоль) растворяли в этаноле (20 мл) и добавляли воду (10 мл) и 1 н. гидроксид натрия (1,7 мл). Полученную смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли воду (100 мл) и смесь промывали диэтиловым эфиром (2× 60 мл). рН водной фазы приводили к значению 1 с помощью 5 н. соляной кислоты и водную фазу экстрагировали дихлорметаном (2× 100 мл). Объединенные органические экстракты сушили (MgSO4) и выпаривали в вакууме с получением масла. Добавляли 2-пропанол (15 мл) и полученный осадок отфильтровывали и промывали 2-пропанолом. Сушка в вакууме при 50° С в течение 24 ч позволяла получить 0,33 г (41%) соединения указанного в заголовке.
Т. пл. 237-239° С.
MC(EI) (m/z): 432 (М+, 14%), 303 (43%), 142 (100%).
1Н ЯМР (400 МГц, CDCl3): δ H 1,72-2,02 (м, 5Н), 2,48 (м, 1Н), 2,85 (м, 2Н), 3,05 (м, 2Н), 3,08 (С 4Н), 3,39 (м/ 2Н), 3,80 (т, 2Н), 7,03 (дд, 2Н), 7,17 (д, 2Н), 7,22 (д, 2Н).
Пример 23.
Гидрохлорид 1-(3-(3-метил-10,11-дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты.
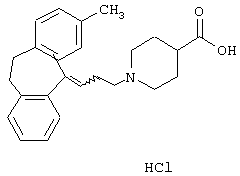
5-(3-Бром-1-пропилиден)-3-метил-10,11-дигидро-5Н-дибензо[а,d]циклогептен (1,05 г, 3,3 ммоль, полученного так же, как описано в WO 9518793) растворяли в метилэтилкетоне (20 мл). Добавляли йодид калия (0,44 г, 6,7 ммоль), карбонат калия (2,76 г, 20 ммоль) и этиловый эфир 4-пиперидинкарбоновой кислоты (0,77 мл, 5,0 ммоль) и полученную смесь нагревали при температуре кипения в течение 1 ч. После охлаждения добавляли этилацетат (60 мл) и смесь промывали водой (2× 60 мл). Органическую фазу сушили (MgSO4) и выпаривали в вакууме. Остаток (1,7 г) очищали колоночной хроматографией на силикагеле (600 мл), используя смесь этилацетата и гептана (1:2) в качестве элюента. Это позволяло получить 0,97 г (73%) этилового эфира 1-(3-(3-метил-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,20 (SiO2: этилацетат/гептан = 1:2).
Вышеуказанный этиловый эфир (0,91 г, 1,7 ммоль) растворяли в этаноле (15 мл) и добавляли воду (5 мл) и 1 н. гидроксид натрия (2,7 мл). Полученную смесь перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь выпаривали в вакууме. Добавляли воду (100 мл) и диэтиловый эфир (70 мл) и фазы разделяли. Значение рН водной фазы устанавливали, равным 2, с помощью 1 н. соляной кислоты и водную фазу экстрагировали дихлорметаном (3× 200 мл). Объединенные органические экстракты сушили (МgSO4) и выпаривали в вакууме с получением 0,12 г (13%) соединения указанного в заголовке. Большая часть продукта выделялась из кислой водной фазы при стоянии. Осадок отфильтровывали, промывали водой и сушили с получением 0,34 г (36%) соединения заголовка.
Т. пл. >250° С.
Вычислено для C25H29NO2, HCl: С 72,89%; Н 7,34%; N 3,40%. Найдено: С 72,65%; Н 7,43%; N 3,26%.
Пример 24.
1-(3-(3,7-Диметил-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота.
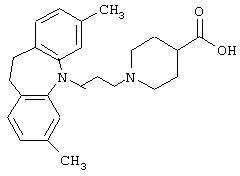
К раствору 3,7-диметил-10,11-дигидро-5Н-дибенз[b,f]азепина (6,45 г, 0,029 моль; полученного так же, как описано в Патенте Великобритании 792615, 1958) и 3-бром-1-пропилтетрагидро-2-пиранилового эфира (8,3 г, 0,037 моль) в сухом бензоле (80 мл) добавляли суспензию амида натрия (3,2 г, 0,041 моль, 50 мас.% суспензия в толуоле). Реакционную смесь нагревали при температуре кипения в течение 20 ч, давали охладиться и добавляли воду (20 мл). Фазы разделяли, из органической фазы выпаривали растворитель и остаток растворяли в смеси метанола (100 мл) и 5 н. НСl (30 мл). Смесь затем нагревали при температуре кипения в течение 15 минут, выпаривали метанол и смесь экстрагировали бензолом (2× 150 мл). Объединенные органические экстракты сушили (К2СО3), фильтровали и растворитель выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (50 мл), используя в качестве элюента сначала бензол для отделения исходного продукта. Затем, используя в качестве элюента хлороформ, выделяли 2,4 г 3-(3,7-диметил-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропанола в виде масла.
Вышеуказанный спирт (2,4 г, 0,0087 моль) растворяли в бензоле (80 мл) и затем добавляли триэтиламин (3,0 мл). После добавления метансульфонилхлорида (1,3 мл, 0,0114 моль) реакционную смесь перемешивали в течение 2 ч. Добавляли воду и фазы разделяли. Органическую фазу сушили (MgSO4) и растворитель выпаривали в вакууме. Остаток растворяли в ацетоне (50 мл). К данному раствору добавляли этиловый эфир 4-пиперидинкарбоновой кислоты (2,0 г, 0,0127 моль) и карбонат калия (3,0 г, 0,0217 моль) и полученную смесь нагревали при температуре кипения в течение 24 ч. Смеси давали охладиться, затем фильтровали и растворитель выпаривали в вакууме с получением остатка, который далее очищали колоночной хроматографией на силикагеле (40 мл), используя хлороформ в качестве элюента. Это позволяло получить 2,6 г (73%) этилового эфира 1-(3-(3,7-диметил-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
Вышеуказанный эфир (2,4 г, 0,057 моль) растворяли в этаноле (50 мл) и добавляли 5 н. NaOH (3 мл). Смесь перемешивали при 40° С в течение 16 ч и этанол выпаривали выпаривали в вакууме с получением остатка, который растворяли в воде (20 мл). К полученному раствору добавляли уксусную кислоту (3 мл) и смесь экстрагировали дихлорметаном (50 мл). Органический экстракт сушили (MgSO4) и растворитель выпаривали в вакууме. К остатку добавляли диэтиловый эфир (50 мл) с получением после фильтрования и сушки 1,85 г (82%) указанного в заголовке соединения в виде твердого вещества.
Т. пл. 207-209° С.
Вычислено для C25H32N2O2, 0,25 Н2O: С 75,63%; Н 8,25%; N 7,06%. Найдено: С 75,58%; Н 8,30%; N 6,89%.
Пример 25.
1-(3-(3-Диметиламино-10,11-дигидро-5Н-дибенз [b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновая кислота.
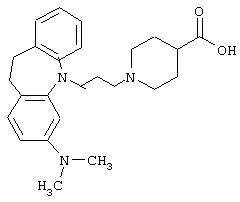
Суспензию амида натрия (2,6 г, 0,033 моль, 50% по массе суспензия в толуоле) добавляли к раствору 3-диметиламино-10,11-дигидро-5Н-дибенз[b,f]азепина (6,1 г, 0,0256 моль; полученного так же, как описано в Патенте Великобритании 1040739, 1966) в сухом бензоле (60 мл). Реакционную смесь нагревали при 70° С в течение 1 ч. Добавляли 3-бром-1-пропилтетрагидро-2-пираниловый эфир (7,35 г, 0,033 моль) и смесь нагревали при температуре кипения в течение 20 ч. К охлажденной реакционной смеси добавляли воду (20 мл) и фазы разделяли. Из органической фазы выпаривали растворитель и остаток растворяли в смеси метанола (100 мл) и 5 н. НСl (30 мл). Смесь нагревали при температуре кипения в течение 15 минут и выпаривали метанол. Добавляли воду (50 мл), рН доводили до значения 8-9 с помощью водного аммиака и смесь экстрагировали бензолом (2× 150 мл). Объединенные органические экстракты сушили (К2СО3), фильтровали и растворитель выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (50 мл), используя в качестве элюента сначала бензол для отделения исходного продукта. Затем, используя в качестве элюента хлороформ, выделяли 3,5 г 3-(3-диметиламино-10,11-дигидро-5Н-дибенз[b,f]aзепин-5-ил)-1-пропанола в виде масла.
Вышеуказанный спирт (3,5 г, 0,0118 моль) растворяли в бензоле (100 мл) и добавляли триэтиламин (4,0 мл) и метан-сульфонилхлорид (1,7 мл, 0,0148 моль). Реакционную смесь перемешивали в течение 2 ч. После добавления воды фазы разделяли. Органическую фазу сушили (MgSO4) и растворитель выпаривали в вакууме с получением остатка, который растворяли в ацетоне (50 мл). К данному раствору добавляли этиловый эфир 4-пиперидинкарбоновой кислоты (2,8 г, 0,0178 моль) и карбонат калия (4,13 г, 0,03 моль) и смесь нагревали при температуре кипения в течение 24 ч. Смеси давали охладиться, затем фильтровали и растворитель выпаривали в вакууме с получением остатка, который далее очищали колоночной хроматографией на силикагеле (50 мл), используя этилацетат в качестве элюента. Это позволяло получить 3,1 г этилового эфира 1-(3-(3-диметиламино-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
Вышеуказанный эфир (1,95 г, 0,0045 моль) растворяли в этаноле (40 мл) и добавляли 5 н. NaOH (3 мл). Смесь перемешивали при 40° С в течение 8 ч и этанол выпаривали выпаривали в вакууме. Остаток растворяли в воде (20 мл). К полученному раствору добавляли уксусную кислоту (3 мл) и смесь экстрагировали дихлорметаном (50 мл). Органический экстракт сушили (MgSO4) и растворитель выпаривали в вакууме. К остатку добавляли диэтиловый эфир (50 мл) с получением после фильтрования и сушки 1,67 г (91%) указанного в заголовке соединения в виде твердого вещества.
Т. пл. 198-202° С.
Вычислено для С25Н33N3О2, 0,25 Н2O: С 72,87%; Н 8,19%; N 10,20%. Найдено: С 72,73%; Н 8,32%; N 10,00%.
Пример 26.
Гидрохлорид (R)-1-(3-(10,11-дигидро-5Н-дибензо[a,d] циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты.
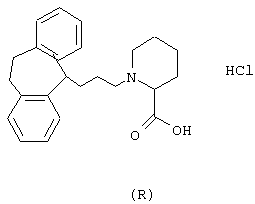
2-Пиперидинкарбоновую кислоту (26 г, 0,201 моль) и (+)-винную кислоту (31,2 г, 0,208 моль) суспендировали в смеси этанола (400 мл) и воды (25 мл). Смесь нагревали до 80° С и раствору давали охладиться до комнатной температуры. Осажденное твердое вещество отфильтровывали, промывали этанолом и сушили с получением 25,4 г (45%) (+)-тартрата (R)-(+)-2-пиперидинкарбоновой кислоты. Маточный раствор выпаривали в вакууме и остаток растворяли в воде (120 мл). Добавляли раствор гидроксида калия (6,5 г, 116 ммоль) в воде (13 мл) и выпавший однозамещенный тартрат калия удаляли путем фильтрования. Выпаривание фильтрата в вакууме позволяло получить 19,5 г сырой (S)-(-)-2-пиперидинкарбоновой кислоты.
Вышеуказанный (+)-тартрат (20 г, 72 ммоль) растворяли в воде (60 мл) и добавляли раствор гидроксида калия (4,0 г, 72 ммоль) в воде (8 мл). Осажденный одно замещенный тартрат калия удаляли путем фильтрования и промывали водой. Маточный раствор выпаривали в вакууме и остаток суспендировали в этаноле (100 мл) и выпаривали в вакууме. Суспендирование в этаноле и выпаривание в вакууме повторяли дважды. Остаток суспендировали в этаноле (200 мл) и по каплям добавляли тионилхлорид (24 мл, 0,28 моль). Полученную смесь нагревали при 70° С в течение 2 ч, охлаждали до комнатной температуры и выпаривали в вакууме. Добавляли диэтиловый эфир (40 мл) и этанол (1 мл) и суспензию перемешивали в течение 30 минут. Твердое вещество отфильтровывали, промывали диэтиловым эфиром и сушили с получением 11,4 г (82%) гидрохлорида этилового эфира (R)-(+)-2-пиперидинкарбоновой кислоты.
[α ]
Газовая хроматография N-ацетильного производного: Rt=46,4 минут. Энантиомерный избыток = 97,9%.
(ГХ проводили на газовом хроматографе Chrompac CP 9000 с F.I.D. определением, используя капиллярную колонку с Chrompac СР-циклодекстрином, длиной 25 м, с внутренним диаметром 0,25 мм, используя скорость потока в щели 40 мл/мин 180 кПа, линейная скорость прохождения газа 27,6 см/С температура на входе и детекторе 200° С и температура в колонке 120° С).
5-(3-Бром-1-пропилиден)-10/11-дигидро-5Н-дибензо[a,d]циклогептен (2,7 г, 8,6 ммоль/ полученный, как описано в WO 9518793), карбонат калия (7,14 г, 52 ммоль), йодид калия (1,4 г, 8,6 ммоль) и гидрохлорид этилового эфира (R)-(+)-2-пиперидинкарбоновой кислоты (3,3 г, 17 ммоль) смешивали в метилэтилкетоне (50 мл) и нагревали при температуре кипения в течение 3 суток. После охлаждения до комнатной температуры добавляли этилацетат (100 мл) и смесь промывали водой (2× 100 мл). Органическую фазу сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (600 мл), используя смесь этилацетата и гептана (1:4) в качестве элюента. Это позволяло получить 2,62 г (78%) этилового эфира (R)-1-(3-(10,11-дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,28 (SiO2: этилацетат/гептан = 1:4).
Вышеуказанный эфир (2,6 г, 6,7 ммоль) растворяли в смеси этанола (25 мл) и 1,4-диоксана (2 мл). Добавляли 1 н. гидроксид натрия (6,7 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли 1 н. гидроксид натрия (1,3 мл) и смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли 1 н. гидроксид натрия (6,9 мл) и этанол (10 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли 1 н. гидроксид натрия (6,9 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли 1 н. гидроксид натрия (6,7 мл) и смесь перемешивали при комнатной температуре в течение 3 суток. Добавляли воду (100 мл) и смесь промывали диэтиловым эфиром (3× 100 мл). рН водной фазы доводили до 1 с помощью 5 н. соляной кислоты и водную фазу экстрагировали дихлорметаном (150 мл). Органический экстракт сушили (МgSO4) и выпаривали в вакууме с получением масла. Добавляли ацетон (50 мл) и раствор выпаривали в вакууме. Обработку ацетоном повторили. Твердое вещество суспендировали в ацетоне и отфильтровывали. Сушка в вакууме при 50° С в течение 24 ч позволяла получить 1,2 г (51%) указанного в заголовке соединения в виде аморфного порошка.
MC(EI) (m/z): 316 (М+, 1%), 142 (100%).
1Н ЯМР (400 МГц, CDCl3): δ H 1,45-1,75 (м, 5Н), 2,1 (м, 1Н), 2,4-2,55 (м, 2Н), 2,75-3,4 (м, 8Н), 3,90 (ушир. С 1Н), 5,80 (т, 1Н), 7,05-7,23 (м, 8Н).
Пример 27.
Гидрохлорид (S)-1-(3-(10,11-дигидро-5Н-дибензо[а,d] циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты.
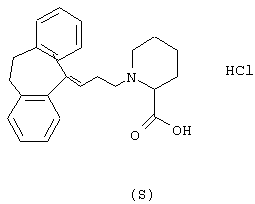
Сырую (S)-(-)-2-пиперидинкарбоновую кислоту (19,5 г, полученную, как описано в примере 26) суспендировали в этаноле (250 мл) и по каплям добавляли тионилхлорид (40 мл, 0,46 моль). После окончания добавления полученную смесь нагревали при температуре кипения в течение 2 ч. Смесь фильтровали горячей, а фильтрат охлаждали до комнатной температуры. Фильтрование и выпаривание в вакууме давали масло, которое кристаллизовали с помощью растирания. Добавляли этанол (10 мл) с последующим медленным добавлением диэтилового эфира (150 мл). Выпавший твердый продукт отфильтровывали, промывали диэтиловым эфиром и сушили путем создания разрежения с получением 13,8 г (выход 35% от расчетного по 2-пиперидинкарбоновой кислоте) гидрохлорида этилового эфира (S)-(-)-2-пиперидинкарбоновой кислоты.
[α ]
Газовая хроматография (осуществляемая, как описано в примере 26) N-ацетильного производного: Rt=47,2 минут. Энантиомерный избыток = 96%.
5-(3-Бром-1-пропилиден)-10,11-дигидро-5Н-дибензо[a,d]циклогептен (2,7 г, 8,6 ммоль, полученный, как описано в WO 9518793), карбонат калия (7,14 г, 52 ммоль), йодид калия (1,4 г, 8,6 ммоль) и гидрохлорид этилового эфира (3)-(-)-2-пиперидинкарбоновой кислоты (3,3 г, 17 ммоль) смешивали в метилэтилкетоне (50 мл) и нагревали при температуре кипения в течение 3 суток. После охлаждения до комнатной температуры добавляли этилацетат (100 мл) и смесь промывали водой (2× 100 мл), сушили (МgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (600 мл), используя смесь этилацетата и гептана (1:4) в качестве элюента. Это позволяло получить 2,3 г (69%) этилового эфира (S)-1-(3-(10,11-дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,22 (SiO2: этилацетат/гептан = 1:4).
Вышеуказанный эфир (2,3 г, 5,9 ммоль) растворяли в смеси этанола (25 мл) и 1,4-диоксана (2 мл). Добавляли 1 н. гидроксид натрия (5,9 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли 1 н. гидроксид натрия (1,18 мл) и смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли 1 н. гидроксид натрия (5,9 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли 1 н. гидроксид натрия (5,9 мл) и смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли 1 н. гидроксид натрия (5,9 мл) и смесь перемешивали при комнатной температуре в течение 3 суток. Добавляли воду (100 мл) и смесь промывали диэтиловым эфиром (3× 100 мл). рН водной фазы доводили до 1 с помощью 5 н. соляной кислоты и водную фазу экстрагировали дихлорметаном (150 мл). Органический экстракт сушили (МgSO4) и выпаривали в вакууме с получением масла. Добавляли ацетон (50 мл) и раствор выпаривали в вакууме. Обработку ацетоном повторили вновь. Твердое вещество суспендировали в ацетоне, отфильтровывали и сушили в вакууме при 50° С в течение 24 ч с получением 0,79 г (37%) указанного в заголовке соединения в виде аморфного порошка.
MC(EI) (m/z): 362 (M+, 1%), 142 (100%).
1Н ЯМР (400 МГц, СDСl3): δ H 1,45-1,75 (м, 5Н), 2,1 (м, 1Н), 2,4-2,55 (м, 2Н), 2,75-3,4 (м, 8Н), 3,80 (ушир. с, 1Н), 5,80 (т, 1Н), 7,05-7,23 (м, 8Н).
Пример 28
Гидрохлорид (1-(2-фенотиазин-10-ил-енил)пиперидин-4-ил-уксусной кислоты
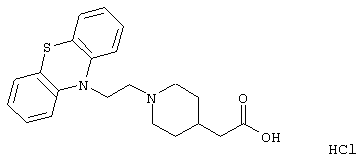
Фенотиазин (7,5 г; 37,6 ммоль) смешивали с хлорацетилхлоридом (30 м ) и перемешивали при комнатной температуре в течение ночи. Смесь выливали в смесь воды со льдом (50 мл) и дихлорметана (50 мл). Медленно добавляли насыщенный раствор бикарбоната натрия (150 мл) и еще дихлорметан (50 мл). Фазы разделяли и водную фазу экстрагировали дихлорметаном (50 мл). Органические фазы промывали водой (75 мл) и насыщенным раствором бикарбоната натрия (50 мл). Для ускорения фазового разделения еще добавляли насыщенный солевой раствор и этанол. Водную фазу промывали дихлорметаном (100 мл) и объединенные органические фазы сушили (MgSO4) и упаривали в вакууме. Оставшийся продукт фильтровали через силикагель с использованием дихлорметана в качестве элюента и упаривали в вакууме. Остаток растворяли в толуоле и добавляли гептан для инициирования осаждения. Добавляли еще толуол и смесь нагревали для растворения. Снова добавляли гептан и смесь оставляли охлаждаться для осаждения. Осадок отфильтровывали и сушили, получая 7,2 г (69%) 2-хлор-1-(фенотиазин-10-ил)этанона.
В сухую трехгорлую колбу в атмосфере азота переносили при помощи шприца 1М раствор алюмогидрида лития в тетрагидрофуране (51,5 мл; 51,5 ммоль). Добавляли безводный тетрагидрофуран (20 мл) и смесь охлаждали на ледяной бане. Концентрированную серную кислоту (1,37 мл; 26 ммоль) смешивали с безводным тетрагидрофураном (10 мл) и добавляли по каплям в течение 10 минут. Полученную смесь перемешивали при комнатной температуре в течение 45 минут. Указанный выше хлорид (7,1 г; 26 ммоль) растворяли в безводном тетрагидрофуране (40 мл) и добавляли по каплям в течение 10 минут. Смесь перемешивали при комнатной температуре в течение 45 минут, затем гасили добавлением воды (5 мл) при охлаждении ледяной баней. Добавляли тетрагидрофуран, а затем воду (10 мл). Добавляли карбонат калия, смесь перемешивали в течение 5 минут, фильтровали и упаривали в вакууме с получением масла. После добавления гептана продукт осаждался, для растворения осадка добавляли толуол и смесь упаривали в вакууме. Остаток растворяли в теплом этаноле и оставляли охлаждаться для осаждения в течение двух дней. Осадок отфильтровывали и сушили, получая 4,62 г (70%) 10-(2-хлорэтил)-10Н-фенотиазина.
Указанный выше хлорид (1,45 г; 5,6 ммоль) растворяли в ДМФ (25 мл) и добавляли этил 4-пиперидилацетат (1,5 г; 7,2 ммоль). иодид калия (0,88 г; 5,5 ммоль) и карбонат калия (2,3 г; 16,7 ммоль). Реакционную смесь нагревали в течение 1 часа при температуре 60° С, оставляли при перемешивании при комнатной температуре в течение ночи, и перемешивали и нагревали, в течение ночи при температуре 60° С. После охлаждения, фильтрования и упаривания добавляли дихлорметан (50 мл) и затем воду (40 мл). Фазы разделяли и органическую фазу сушили (MgSO4) и упаривали в вакууме. Полученный сырой продукт элюировали через двуокись кремния с использованием дихлорметана и метанола (95:5) в качестве элюентов. После упаривания фракций получали 0,67 г этилового эфира (1-(2-(фенотиазин-10-ил)-этил)-4-пиперидинил)уксусной кислоты.
Полученный выше сложный эфир (0,66 г, 1,7 ммоль) растворяли в этаноле (15 мл) и добавляли раствор гидроксида натрия (0,33 г) в воде (2,5 мл). Смесь оставляли для перемешивания в течение 3 часов и рН доводили до ~3 с использованием 1М раствора хлороводородной кислоты (9 мл). Смесь экстрагировали дихлорметаном (35 мл) и органическую фазу сушили (MgSO4) и упаривали. Добавляли ацетон, затем дихлорметан и раствор упаривали. Добавляли ацетон, затем тетрагидрофуран и раствор упаривали. Добавляли ацетон (8 мл), раствор декантировали и упаривали, получая 0,27 г (40%) указанного в заголовке соединения в виде аморфного твердого вещества. Вычислено для C21H24N2O2S· HCl· H2O,%: С 59,63%; Н 6,43%; N 6,62%.
Найдено, %: С 59,66%; Н 6,34%; N 6,37%.
Пример 29
Гидрохлорид 1-(4-(фенотиазин-10-ил)-4-пиперидинкарбоновой кислоты
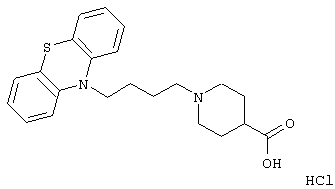
К раствору фенотиазина (4 г, 0,02 моль) в толуоле (40 мл) добавляли 50%-ную суспензию амида натрия в толуоле (2,54 г, 0,03 моль) и реакционную смесь нагревали в атмосфере азота в течение 3 часов при температуре 105° С. После охлаждения смеси до 60° С добавляли 1-бром-4-хлорбутан (6,86 г, 0,04 моль) и реакционную смесь нагревали до температуры кипячения с обратным холодильником в течение 7 часов. Твердое вещество отфильтровывали и фильтрат упаривали в вакууме, получая 8,58 г сырого 10-(4-хлорбутил)-10Н-фенотиазина, который использовали на следующей стадии без дополнительной очистки.
Полученный на предыдущей стадии хлорид (8,58 г) растворяли в метилэтилкетоне (40 мл), добавляли этиловый эфир 4-пиперидинкарбоновой кислоты (5,82 г, 0,037 моль), карбонат калия (13,3 г, 0,089 моль) и иодид натрия (4,44 г, 0,0296 моль), и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 8 часов. Твердое вещество отфильтровывали и фильтрат упаривали в вакууме. Остаток смешивали с бензолом (250 мл) и бензольный раствор отделяли от твердого вещества и фильтровали через целит. После упаривания в вакууме остаток очищали колоночной хроматографией на силикагеле (140 г) с использованием в качестве элюента смеси С 62,40%; Н 6,55%; Cl 8,37%; N 6,61%; S 7,57%. Найдено, %: С 62,34%; Н 6,61%; Cl 8,46%; N 6,47%; S 7,62%.
Пример 30
Гидрохлорид 1-(3-(ксантен-9-илиден)-1-пропил)-4-пиперидинкарбоновой кислоты
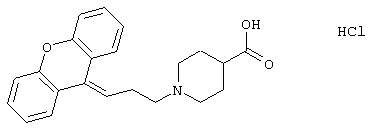
К раствору циклопропилмагнийбромида (полученного из циклопропилбромида (6,05 г, 0,05 моль) и магниевых стружек (1,21 г, 0,05 моль) в безводном тетрагидрофуране (25 мл) в атмосфере сухого азота) по каплям добавляли суспензию ксантона (5,0 г, 0,025 моль) в безводном тетрагидрофуране (30 мл) при температуре 60° С. Реакционную смесь перемешивали в течение 1 часа при температуре 60° С, охлаждали и добавляли к охлажденной на льду смеси насыщенного раствора хлорида аммония (60 мл). Слои разделяли, водный слой дважды экстрагировали эфиром (2× 20 мл), объединенные органические экстракты промывали водой (2× 30 мл), сушили (МgSO4) и упаривали в вакууме. Полученный остаток, 6,27 г, сырого 9-циклопропилксантон-9-ола использовали на следующей стадии без дополнительной очистки.
К раствору полученного выше спирта (6,2 г) в уксусной кислоте (100 мл) при температуре 10-15° С в течение 45 минут добавляли по каплям 15% раствор бромистого водорода в уксусной кислоте (48 мл). Смесь перемешивали при 15° С в течение 30 минут. После фильтрования раствор разбавляли водой (100 мл) и охлаждали в течение ночи. Образовавшийся осадок отфильтровывали, растворяли в эфире (20 мл) и раствор промывали водой (20 мл), сушили (Na2SO4) и упаривали в вакууме.
Масляный остаток (6,2 г) очищали хроматографией на силикагеле с использованием толуола в качестве элюента, и получали 5,34 г (70%) 9-(3-бромопропилиден)ксантона.
ТСХ: Rf=0,88 (н-гексан/ацетон = 2:1).
Смесь полученного, как указано выше, бромида (5,25 г, 0,017 моль), этил 4-пиперидинкарбоксилата (2,74 г, 0,017 моль), безводного карбоната калия (7,23 г, 0,052 моль) и иодида калия (2,9 г, 0,017 моль) в 2-бутаноне (40 мл) нагревали при температуре 60-70° С в течение 5,5 час. После охлаждения неорганические соли отфильтровывали, промывали 2-бутаноном (10 мл) и объединенные фильтраты упаривали в вакууме. Маслянистый остаток (6,6 г) очищали при помощи хроматографии на силикагеле с использованием в качестве элюентов смесей бензола и этилацетата (0-30%) с получением 2,68 г (41%) этилового эфира 1-(3-(9-ксантенилиден)-1-пропил)-4-пиперидинкарбоновой кислоты в виде масла.
Полученный, как указано выше, сложный эфир (2,50 г, 6/6 ммоль) растворяли в этаноле (40 мл), добавляли 20% гидроксид натрия (4,3 мл) и смесь перемешивали при комнатной температуре в течение 6,5 час. Этанол выпаривали в вакууме, смесь разбавляли дихлорметаном (300 мл) и подкисляли уксусной кислотой (рН 7). Добавляли воду (10 мл), фазы разделяли и органический слой сушили (MgSO4) и упаривали в вакууме. Остаток упаривали с ацетоном (40 мл), полученное масло разбавляли эфиром (50 мл), добавляли раствор хлористого водорода в эфире до достижения рН 1, и смесь перемешивали в течение 7 часов. Эфирный раствор декантировали от выпавшего в осадок гидрохлорида, снова добавляли эфир (50 мл) и смесь перемешивали в течение 3 час. Твердый продукт отфильтровывали, промывали эфиром и сушили. В результате получали 1,60 г (63%) указанного в заголовке соединения в виде твердого продукта.
Т. пл. 206-212° С (разл.).
Вычислено для С22Н23NО3·НСl· 1/4СН2Сl2·1/2Н2O, %:
С 64,22%; Н 6,18%; N 3,37%; Cl 12,78%. Найдено, %:
С 64,38%; Н 6,07%; N 3,28%; Cl 12,69%.
Исследование на невосприимчивость к инсулину
Четырем находящимся в сознании нерезистентным 25 граммовым мышам-самцам NMRI интрацервикально вводят гистаминхлорид (90 нмоль) в соответствии с методом Nishibori et al. (J.Pharmacol. Exp. Therap. 241, 582-586, 1987). Содержание глюкозы в крови определяли при времени 0 и через 40 минут после инъекции гистамина. Исследуемые соединения, полученные по примеру 11 настоящей заявки, вводили внутрибрюшинно в дозах 1,0 и 10 мг/кг за 30 минут до инъекции гистамина.
Соединение по примеру 11 показало 52% ингибирования, % ингибирования относится к способности соединения ингибировать рост уровня глюкозы в крови, вызванный гистамином. Этот результат свидетельствует о том, что соединения по настоящему изобретению могут использоваться при лечении невосприимчивости к инсулину.
Описываются N-замещенные азагетероциклические карбоновые кислоты и их сложные эфиры формулы I.
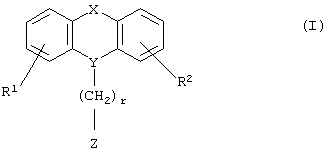
где R1 и R2, независимо, представляют собой водород, галоген, NR6R7 или C1-6-алкил, Y представляет собой 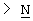 -CH2 или
-CH2 или 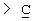 =СН2-, где только подчеркнутый атом входит в состав кольцевой системы, и Х представляет собой -О-, -S-, -СН2СН2-, где R6 и R7, независимо, представляют собой C1-6-алкил, г равно 1, 2 или 3, Z представляет собой гетероцикл, выбранный из (а), (b), (с), (d), (f), (k), (g), (j), способ их получения, фармацевтическая композиция на основе соединения формулы (I).
=СН2-, где только подчеркнутый атом входит в состав кольцевой системы, и Х представляет собой -О-, -S-, -СН2СН2-, где R6 и R7, независимо, представляют собой C1-6-алкил, г равно 1, 2 или 3, Z представляет собой гетероцикл, выбранный из (а), (b), (с), (d), (f), (k), (g), (j), способ их получения, фармацевтическая композиция на основе соединения формулы (I).
Описывается способ ингибирования нейрогенной боли, воспаления и повышения уровня глюкозы в крови у субъекта путем введения субъекту
эффективного количества соединения формулы (I). Технический результат - соединения формулы (I), обладают активностью ингибировать нейрогенную соль, воспаление и повышение уровня глюкозы в крови. 5 с. и 8 з.п. ф-лы, 1 табл.
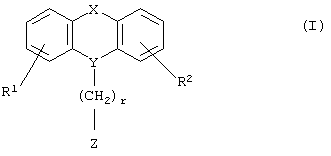
где
R1 и R2, независимо, представляют собой водород, галоген, NR6R7 или C1-6-алкил;
Y представляет собой 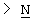 -CH2- или
-CH2- или 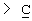 =СН2-, где только подчеркнутый атом входит в состав кольцевой системы; и
=СН2-, где только подчеркнутый атом входит в состав кольцевой системы; и
Х представляет собой -O-, -S-, -СН2СН2-, где R6 и R7, независимо, представляют собой C1-6-алкил;
r равно 1, 2 или 3; и
Z представляет собой (а)
где
R3 представляет собой -(СH2)mОН или -(СH2)рСОR4, где m равно 1, р равно 0 или 1, и где R4 представляет собой -ОН, -NH2 или C1-6-алкокси;
R10 представляет собой водород или С1-6-алкил; и
.... представляет собой одинарную связь;
при условии, что
если R1 и R2 независимо представляют собой водород или галоген, Y представляет собой 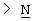 -СН2-, X представляет собой -S- и R10 представляет собой водород, тогда R3 не может быть -(СН2)рСОR4, где р равно 0, и R4 представляет собой –NН2 или С1-6-алкокси; и, далее,
-СН2-, X представляет собой -S- и R10 представляет собой водород, тогда R3 не может быть -(СН2)рСОR4, где р равно 0, и R4 представляет собой –NН2 или С1-6-алкокси; и, далее,
если R1 и R2, независимо, представляют собой водород, галоген или C1-6-алкил; Х представляет собой -O-, -S-, СН2СН2-, и R10 представляет собой водород, тогда R3 не может быть -(CH2)pCOR4, где р равно 0 и R4 представляет собой -ОН или С1-6-алкокси;
или
Z выбран из (b) и (с)
 и
и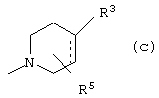
где R3 представляет собой -(CH2)pCOR4, где р равно 0 или 1, и где R4 представляет собой -ОН или C1-6-алкокси;
R5 представляет собой водород;
.... представляет собой одинарную связь;
или
Z выбран из (d), (f) и (k)
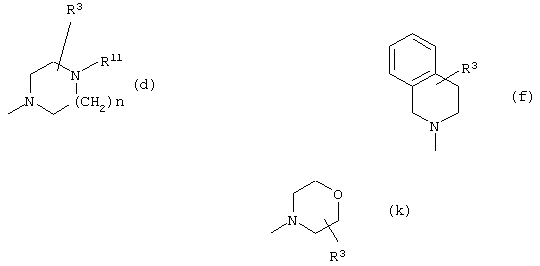
где
n равно 1;
R3 представляет собой -(СН2)рСОR4, где р равно 0 или 1,
и где R4 представляет собой -ОН или C1-6-алкокси;
R11 представляет собой водород;
или
Z представляет собой (g)
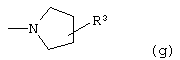
где
R3 представляет собой -(СН2)рСОR4, где р равно 0, и где
R4 представляет собой -ОН или С1-6-алкокси;
или Z представляет собой (j)
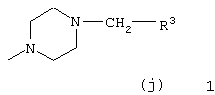
где
R3 представляет собой -(CH2)pCOR4, где р равно 0, и где
R4 представляет собой -ОН или C1-6-алкокси;
или его фармацевтически приемлемая соль.
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинкарбоксамид;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперидинкарбоновую кислоту;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинилметанол;
4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-пиперазинкарбоновую кислоту;
4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-морфолинкарбоновую кислоту;
2-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-1,2,3,4-тетрагидро-4-изохинолинкарбоновую кислоту;
(4-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)пиперазин-1-ил)уксусную кислоту;
1-(3-(10,11-Дигидро-5Н-дибенз[a,d]азепин-5-ил)-1-пропил)-4-пиперидинуксусную кислоту;
1-(3-(10,11-Дигидро-5Н-дибензо[b,f]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновую кислоту;
(R)-1-(3-(10,11-Дигидро-5Н-дибензо[a,d]циклогептен-5-илиден)-1-пропил)-3-пиперидинкарбоксамид;
(R)-1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пирролидинкарбоновую кислоту;
(S)-1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пирролидинкарбоновую кислоту;
1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пиперидинкарбоновую кислоту;
1-(3-(10Н-Феноксазин-10-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(3-Хлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-3-пиперидинуксусную кислоту;
1-(3-(10,11-Дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-2-метил-3-пиперидинкарбоновую кислоту;
1-(3-(2,8-Дибром-10,11-дигидро-5Н-дибенз[a,d]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(3,7-Дихлор-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(3-Метил-10,11-дигидро-5Н-дибенз[a,d]циклогептен-5-илиден)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(3,7-Диметил-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
1-(3-(3-Диметиламино-10,11-дигидро-5Н-дибенз[b,f]азепин-5-ил)-1-пропил)-4-пиперидинкарбоновую кислоту;
(R)-1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пиперидинкарбоновую кислоту;
(S)-1-(3-(10,11-Дигидро-5Н-дибензо[а,d]циклогептен-5-илиден)-1-пропил)-2-пиперидинкарбоновую кислоту;
или их фармацевтически приемлемые соли.
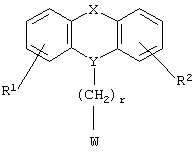
где R1, R2, X, Y и r определены в п.1, и W представляет собой подходящую удаляемую группу, такую как галоген, п-толуолсульфонат или мезилат, с соединением формулы (III)
HZ (III)
где Z определен в п.1, и, при необходимости, полученное соединение формулы (I), где R4 представляет собой C1-6-алкокси, подвергают гидролизу с получением соединения формулы (I), где R4 представляет собой ОН.
Приоритет п.1 (за исключением случая, когда R1 и R2, независимо, обозначают NR6R7), п.2 (за исключением случая, когда R1 и R2 независимо обозначают N(СН3)2), п.3-5, п.6 соединения N 1-4, 6-8, 11-14 - 07.04.1995
Приоритеты по пунктам и признакам:
| GB 830709, 13.03.1960 | |||
| US 4139632, 13.02.1979 | |||
| Машковский М.Д | |||
| Лекарственные средства, Москва, Медицина, 1972, ч.1 стр | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |

