ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка является частично продолжающей заявку с серийным №09/325997, зарегистрированную 4 июня 1999 г., которая включена в настоящую заявку на изобретение в качестве ссылки, для всех целей.
ОБЛАСТЬ ТЕХНИКИ
Настоящая заявка касается использования производных (-)(3-тригалометилфенокси)-(4-галофенил)уксусной кислоты и включающих их композиций в лечении резистентности к инсулину, диабета II типа, гиперлипидемии и гиперурикемии.
УРОВЕНЬ ТЕХНИКИ
Сахарный диабет, обычно называемый просто диабетом, обозначает болезненный процесс, происходящий в результате действия многих факторов и характеризующийся повышенным содержанием глюкозы в плазме крови, называемым гипергликемией. См., например, LeRoith, D. et al., (eds.). Diabetes Mellitus (Lippincott-Raven Publishers, PA U.S.A), 1996) и все содержащиеся в этой публикации ссылки. По данным Американской ассоциации диабета, сахарным диабетом больны примерно 6% населения земли. Неконтролируемая гипергликемия связана с повышенной и преждевременной смертностью из-за повышенного риска микрососудистых и макрососудистых болезней, включая нефропатию, невропатию, ретинопатию, гипертензию, заболевание сосудов головного мозга и ишемическую (коронарную) болезнь сердца. В связи с этим контроль гомеостаза глюкозы является решающей важности подходом к лечению диабета.
Существуют две основные формы диабета: диабет I типа (ранее называвшийся инсулин-зависимым диабетом, или IDDM), и диабет II типа (ранее называвшийся инсулин-независимым диабетом, или NIDDM).
Диабет I типа является результатом абсолютной недостаточности инсулина, гормона, регулирующего утилизацию глюкозы. Недостаточность инсулина обычно характеризуется разрушением β-клеток в островках Лангерганса в поджелудочной железе, что, как правило, приводит к абсолютной инсулиновой недостаточности. Диабет I типа имеет две формы: имунно-опосредованный сахарный диабет, возникающий в результате клеточно-опосредованного аутоиммунного разрушения β-клеток поджелудочной железы; и идиопатический сахарный диабет, который относится к формам болезни неизвестной этиологии.
Диабет II типа представляет собой болезнь, которая характеризуется резистентностью к инсулину, сопровождающуюся относительной, а не абсолютной инсулиновой недостаточностью. Диабет II типа может варьировать от преобладающей резистентности к инсулину с относительной инсулиновой недостаточностью до преобладающей инсулиновой недостаточности с некоторой резистентностью к инсулину. Резистентность к инсулину представляет собой уменьшенную возможность инсулина осуществлять свою биологическую функцию в широком диапазоне его концентраций. У индивидуумов с резистентностью к инсулину организм секретирует ненормально высокие количества инсулина, чтобы компенсировать этот дефект. Когда присутствуют неадекватные количества инсулина для компенсации резистентности к инсулину и адекватного контроля содержания глюкозы, то развивается состояние ослабленной толерантности к глюкозе. У значительного числа индивидуумов секреция инсулина далее снижается, а уровни содержания глюкозы в плазме крови повышаются, что приводит к клинической картине диабета. Диабет II типа может являться результатом высокой резистентности к стимулируемым инсулином регуляторным воздействиям на метаболизм глюкозы и липидов в основных чувствительных к инсулину тканях: в тканях мышц, печени и в жировых тканях. Эта резистентность к реакции на инсулин приводит к недостаточной активации инсулином поглощения глюкозы, ее окисления и отложения в мышцах, к неадекватному подавлению инсулином липолиза в жировых тканях, а также продуцирования и выделения глюкозы в печени. При диабете II типа уровни содержания свободных жирных кислот часто повышенные у тучных и у некоторых не страдающих ожирением пациентов, а окисление липидов повышено.
Преждевременное развитие атеросклероза и увеличенная частота сердечно-сосудистых болезней и заболеваний периферических сосудов характерны для пациентов, страдающих диабетом. Гиперлипидемия является важным отягощающим фактором для этих заболеваний. Гиперлипидемия представляет собой состояние, которое характеризуется главным образом ненормальным повышением содержания липидов в сыворотке крови в кровотоке и является важным фактором риска для развития атеросклероза и болезней сердца. Обзор работ по нарушениям метаболизма липидов см., например, в публикации Wilson, J. et al., (ed.). Disorders of Lipid Metabolism, Chapter 23, Textbook of Endocrinology, 9th Edition, (W.B.Sanders Company, Philadelphia, PA U.S.A. 1998); эта публикация и все цитируемые в ней работы включены в настоящую заявку в качестве ссылок. Липопротеины сыворотки крови являются носителями липидов в кровотоке. Их классифицируют на основании их плотности следующим образом: хиломикроны; липопротеины очень низкой плотности (VLDL); липопротеины средней плотности (IDL); липопротеины низкой плотности (LDL); липопротеины высокой плотности (HDL). Гиперлипидемию обычно разделяют на первичную и вторичную гиперлипидемию. Первичная гиперлипидемия обычно вызвана генетическими нарушениями, а вторичную гиперлипидемию, как правило, вызывают другие факторы, такие как различные болезненные состояния, лекарства и факторы, связанные с питанием. В качестве альтернативы, гиперлипидемия может возникнуть в результате сочетания причин, вызывающих как первичную, так и вторичную гиперлипидемию. Повышенные уровни содержания холестерина связаны с рядом болезненных состояний, включая заболевание коронарных артерий, стенокардию, заболевание сонной артерии, параличи, церебральный артериосклероз и ксантому.
Дислипидемия, или ненормальные уровни содержания липопротеинов в плазме крови, является частым нарушением у диабетиков, и доказано, что она является одной из основных причин повышенной встречаемости ишемической болезни сердца и смертности у диабетиков (см., например, Joslin, E. Ann. Chim. Med. (1927) 5: 1061-1079). Проведенные с тех пор эпидемиологические исследования подтвердили наличие этой связи и показали, что смертность от ишемической болезни сердца у диабетиков в несколько раз выше, чем у лиц, не страдающих диабетом (см., например, Garcia, M.J. et al., Diabetes (1974) 23: 105-11 (1974) и Laakso, M. et al., and Lehto, S., Diabetes Reviews (1997) 5(4):294-315). Описаны некоторые отклонения липопротеина от нормы, встречающиеся у больных диабетом (Howard В. et al., Artherosclerosis (1978) 30: 153-162).
Предыдущие исследования 1970-х годов продемонстрировали эффективность рацемического 2-ацетамидоэтил-(4-хлорфенил)-(3-трифторметилфенокси)ацетата (известного также под названием "галофенат") в качестве потенциального терапевтического средства для лечения диабета II типа, гиперлипидемии и гиперурикемии (см., например, Bolhofer, W., U.S. 3,517,050; Jain, A. et al., N. Eng. J. Med. (1975) 293:1283-1286; Kudzma, D. et al., Diabetes (1977) 25:291-95; Kohl, E. et al., Diabetes Care (1984) 7:19-24; McMahon, F.G. et al., Univ. Mich. Med. Center J. (1970) 36:247-248; Simori, С. et al., Lipids (1972) 7: 96-99; Morgan, J.P. et al., Clin. Pharmacol. Therap. (1971) 12:517-524; Aronow, W.S. et al., Clin. Pharmacol. Ther. (1973) 14:358-365 и Fanelli, G.M. et ai., J. Pharm. Experimental Therapeutics (1972) 180:377-396). В этих предшествующих исследованиях влияние рацемического галофената на диабет наблюдали в тех случаях, когда его сочетали с сульфонилмочевинами. Минимальное влияние на глюкозу наблюдали у больных диабетом пациентов, которых лечили одним рацемическим галофенатом. Однако были замечены значительные побочные действия, включая желудочно-кишечные нарушения - кровотечения из желудка и пептические язвы (см., например, Friedberg, S.J. et al., Clin. Res. (1986) Vol. 34, No. 2: 682A).
Кроме того, есть некоторые указания на существование межлекарственных взаимодействий между рацемическим галофенатом и такими агентами, как варфаринсульфат (который называется также 3-(альфа-ацетонилбензил)-4-гидроксикумарин или Кумадин™ (Dupont Pharmaceuticals, E.I. Dupont de Nemours and Co., Inc., Wilmington, DE U.S.A.) (см., например, Vesell, E.S. and Passantanti, G. T., Fed. Proc. (1972) 31(2):538). Кумадин™ представляет собой антикоагулянт, который действует путем ингибирования синтеза зависящих от витамина К факторов свертывания (включающих факторы II, VII, IX и X, а также антикоагулянтные белки С и S). Полагают, что Кумадин™ стереоспецифически метаболизируется печеночными микросомальными ферментами (ферментами цитохром Р450). Изозимы цитохрома Р450, участвующие в метаболизме кумадина, включают 2С9, 2С19, 2С8, 2С18, 1А2 и 3А4. 2С9, по-видимому, является основной формой Р450 печени человека, который модулирует метаболизм нескольких видов лекарств in vivo, включая антикоагулирующую активность Кумадина™ (см., например. Miners, J.O. et al., Bri. J. Clin. Pharmacol. (1998) 45:525-538).
Лекарства, которые ингибируют метаболизм Кумадина™, приводят к дальнейшему уменьшению количества зависящих от витамина К факторов свертывания, в результате чего у пациентов, получающих такое лечение, коагуляция уменьшается в большей степени, чем это желательно (т.е. у пациентов с риском легочной или церебральной эмболии в результате образования сгустков крови в нижних конечностях, сердце или в других участках тела). Просто уменьшить дозу антикоагулянта часто бывает трудно, поскольку пациент нуждается в поддержании соответствующего уровня антикоагуляции, чтобы предотвратить образование сгустков крови. Повышенная антикоагуляция в результате взаимодействия между лекарствами приводит к значительному риску для таких пациентов, с возможностью сильных кровотечений из повреждений мягких тканей, желудочно-кишечных кровотечений (т.е. язв желудка или двенадцатиперстной кишки) и других поражений (например, аневризмы аорты). Кровотечение в результате слишком сильной антикоагуляции требует немедленной медицинской помощи и может привести к смерти, если немедленно не было проведено соответствующее лечение.
Известно, что цитохром Р450 2С9 также участвует в метаболизме некоторых других обычно используемых лекарств, включая дилантин, сульфонилмочевины, такие как толбутамид, а также некоторых нестероидных противовоспалительных средств, таких как ибупрофен. Ингибирование этого фермента может привести к другим неблагоприятным эффектам, связанным с межлекарственными взаимодействиями, кроме тех, которые описаны выше для Кумадина™ (см., например, Pelkonen, О. et al., Xenobiotica (1998) 28:1203-1253; Linn, J.H. и Lu, A.Y., Clin. Pharmacokinet. (1998) 35 (5):361-390).
Прежде чем галофенат станет общепринятым средством для лечения резистентности к инсулину, диабета II типа, гиперлипидемии и гиперурикемии, необходимо решить вышеуказанные проблемы. Настоящее изобретение решает эту и другие задачи путем создания композиций и способов лечения резистентности к инсулину, диабета II типа, гиперлипидемии и гиперурикемии и в то же время обеспечивает улучшение профиля побочных действий.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение касается способа модулирования диабета II типа у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества (-)стереоизомера соединения формулы I,

где R представляет собой радикал, выбранный из группы, состоящей из гидрокси, низшего аралкокси, ди-низшего алкиламино-низшего алкокси, низшего алканамидо низшего алкокси, бензамидо-низшего алкокси, уреидо-низшего алкокси, N'-низшего алкил-уреидо-низшего алкокси, карбамоил-низшего алкокси, галофенокси-замещенного низшего алкокси, карбамоилзамещенного фенокси, карбонил-низшего алкиламино, N,N-ди-низшего алкиламино-низшего алкиламино, галогензамещенного низшего алкиламино, гидрокси-замещенного низшего алкиламино, низшего алканолилоксизамещенного низшего алкиламино, уреидо и низшего алкоксикарбониламино; а Х представляет собой галоген; или его фармацевтически приемлемой соли, причем это соединение по существу не содержит своего (+)стереоизомера.
Некоторые такие способы включают также соединение формулы II:
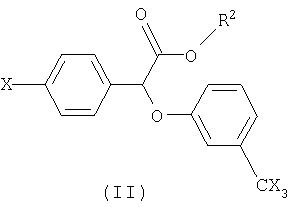
в которой R2 представляет собой радикал, выбранный из группы, состоящей из фенил-низшего алкила, низшего алканамидо-низшего алкила и бензамидо-низшего алкила.
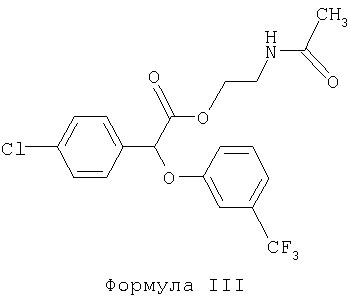
Некоторые такие способы включают также соединение формулы III:
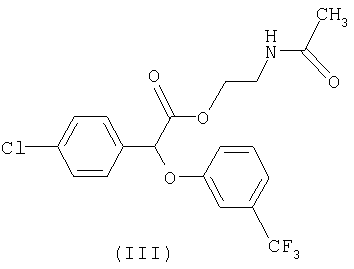
Предпочтительное соединение формулы III известно как "(-)2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетат" или "(-)галофенат".
Настоящее изобретение обеспечивает также способ модулирования резистентности к инсулину у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества (-)стереоизомера соединения формулы I. Некоторые такие способы включают также соединение формулы II. Некоторые такие способы включают также соединение формулы III.
Настоящее изобретение обеспечивает также способ облегчения гиперлипидемии у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества соединения формулы I. Некоторые такие способы включают также соединение формулы II. Некоторые такие способы включают также соединение формулы III.
Настоящее изобретение обеспечивает также способ модулирования гиперурикемии у млекопитающего. Способ включает введение млекопитающему терапевтически эффективного количества соединения формулы I. Некоторые такие способы включают также соединение формулы II. Некоторые такие способы включают также соединение формулы III.
Настоящее изобретение касается также фармацевтических композиций. Фармацевтические композиции включают фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I, II или формулы III.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 показывает ингибирование активности цитохрома Р450 2С9 (CYP2C9) рацемической галофеновой кислотой, (-)галофеновой кислотой и (+)галофеновой кислотой. Гидроксилирование толбутамида измеряли в присутствии увеличивающихся концентраций этих соединений. Рацемическая галофеновая кислота ингибировала активность CYP2C9 с ИК50, составляющей 0,45 мкМ, а (+)галофеновая кислота ингибировала активность CYP2C9 с ИК50, составляющей 0,22 мкМ. В противоположность этому, действие (-)галофеновой кислоты было в 20 раз меньшим с ИК50, составляющей 3,5 мкМ.
Фигура 2 показывает кривую понижения содержания глюкозы во времени после одной пероральной дозы рацемического галофената, (-)энантиомера галофената или (+)энантиомера галофената, в дозе 250 мг/кг у больных диабетом мышей линии ob/ob. Наиболее быстрое наступление действия и наиболее длительное действие продемонстрировал (-)энантиомер. Уменьшение содержания глюкозы было значимым (р<0,05) для (-)энантиомера по сравнению с контролем для всех точек кривой от 3 до 24 часов. Показатели для рацемического галофената и (+)энантиомера были также значимыми (р <0,05) для всех точек от 3 до 24 часов. Содержание глюкозы в плазме через 24 часа составляло 217±16,6 мг/дл у животных, получавших (-)энантиомер, по сравнению с 306±28,5 мг/дл и с 259,3±20,8 мг/дл у животных, получавших (+)энантиомер и рацемат, соответственно. Содержание глюкозы в плазме в контроле, где животные получали только носитель, составляло 408+16,2 мг/дл через 24 часа. Более эффективным был (-)энантиомер, при значительном (р<0,05) отличии от (+)энантиомера как через 3 часа, так и через 24 часа после введения дозы.
Фигура 3 показывает способность рацемического галофената и обоих (-) и (+)энантиомеров галофената понижать содержание глюкозы в плазме у больных диабетом мышей линии ob/ob после ежедневного перорального введения этих соединений. Рацемат давали в дозе 250 мг/кг/день, а энантиомеры давали в дозах 125 мг/г/день и 250 мг/кг/день. Значительное снижение уровней содержания глюкозы по сравнению с контрольными животными наблюдали у животных, которым вводили рацемический галофенат и оба (-) и (+)энантиомера галофената. При низкой дозе введения (125 мг/кг/день) (-) и (+)энантиомеров показатели для (-)энантиомера были значимы через 6, 27 и 30 часов, в то время как показатели для (+)энантиомера были значимы только через 6 и 27 часов после введения дозы.
Фигура 4 показывает уровни содержания инсулина в плазме крови мышей линии ob/ob, получавших рацемический галофенат и оба (-) и (+)энантиомера галофената после ежедневного перорального введения этих соединений. Рацемат давали в дозе 250 мг/кг/день, а энантиомеры давали в дозах 125 мг/г/день и 250 мг/кг/день. По сравнению с контролем, в котором животные получали только носитель, содержание инсулина было ниже у животных, которым вводили рацемический галофенат или любой из энантиомеров галофената. При высокой дозе введения наибольшее понижение содержания инсулина в плазме отмечали через 27 и 30 часов у животных, получавших оба (-) и (+)энантиомера галофената спустя два дня после его введения.
Фигура 5 показывает уровни содержания глюкозы в плазме крови мышей линии ob/ob после голодания в течение ночи, после введения в течение 5 дней носителя, рацемического галофената в дозе 250 мг/кг/день, (-)энантиомера галофената в дозах 125 мг/кг/день и 250 мг/кг/день и (+)энантиомера галофената в дозах 125 мг/кг/день и 250 мг/кг/день. У контрольных животных была гипергликемия, при содержании глюкозы в плазме на уровне 185,4±12,3 мг/дл. У всех животных, получавших галофенат, отмечено значимое (р<0,01) понижение содержания глюкозы. Высокие дозы обоих энантиомеров понижали содержание глюкозы почти до нормальных уровней в 127,3±8,0 мг/дл и 127,2±9,7 мг/дл для животных, получавших (-)энантиомер и (+)энантиомер соответственно.
Фигура 6 показывает уровни содержания инсулина в плазме крови мышей линии ob/ob после голодания в течение ночи, после введения в течение 5 дней носителя, рацемического галофената в дозе 250 мг/кг/день, (-)энантиомера в дозах 125 мг/кг/день и 250 мг/кг/день или (+)энантиомера галофената в дозах 125 мг/кг/день и 250 мг/кг/день. Значительно более низкие уровни содержания инсулина в плазме наблюдали у животных, получавших обе дозы (-)энантиомера. Низкая доза (+)энантиомера не понижала содержание в плазме инсулина, хотя высокая доза (+)энантиомера привела к понижению содержания инсулина в плазме крови.
Фигура 7А показывает уровни содержания глюкозы в плазме крови после перорального контрольного введения глюкозы крысам линии Zucker fatty, которая служит моделью резистентности к инсулину и ослабленной толерантности к глюкозе. Эти животные получали либо носитель (контроль), либо рацемический галофенат, (-)галофенат или (+)галофенат за 5,5 часов до контрольного введения глюкозы. Рацемат давали в дозе 100 мг/кг, а оба энантиомера давали в дозах 50 и 100 мг/кг. У контрольных животных содержание глюкозы повысилось до >250 мг/дл через 30 минут после введения глюкозы, что ясно указывало на ослабленную толерантность к глюкозе. Содержание глюкозы в плазме было пониженным у крыс, получавших рацемический галофенат, особенно между 30 и 60 минутами после контрольного введения глюкозы. Животные, получавшие (-)галофенат в дозе 100 мг/кг, имели самые высокие показатели понижения содержания глюкозы среди всех подопытных животных. Животные, получавшие (-)галофенат, имели более низкие уровни содержания глюкозы, которые сохранялись через 90-120 минут, по сравнению с крысами, получавшими рацемат или (+)галофенат. На фигуре 7В дается сравнение областей приращения под кривой (AUC) для животных каждой группы. Значимые изменения (р<0,05) наблюдали в группах, получавших обе дозы (-)галофената. Хотя AUC была ниже и в других группах, по сравнению с контролем, но эти изменения были незначимыми.
Фигура 8 показывает результаты экспресс-теста на толерантность к инсулину для крыс линии Zucker fatty, которые получали либо носитель (контроль), либо (-)галофенат (50 мг/кг/день) или (+)галофенат (50 мг/кг/день) в течение 5 дней. Этот тест представляет собой измерение чувствительности к инсулину у испытуемых животных, причем наклон кривой понижения содержания глюкозы представляет собой прямое измерение отвечаемости на инсулин. Животные, получавшие (-)галофенат, были существенно более чувствительны к инсулину, чем животные, получавшие носитель (р<0,01) или (+)галофенат (р<0,05).
Фигура 9А показывает уровни содержания холестерина в плазме крови крыс линии Zucker Diabetic Fatty, которые получали в течение 13 дней рацемический галофенат, (-)энантиомер или (+)энантиомер в дозах 50 мг/кг/день, 25 мг/кг/день и 25 мг/кг/день соответственно, по сравнению с животными контрольной группы, которые получали носитель. У животных, получавших как (-)энантиомер, так и рацемат, содержание холестерина в плазме крови понизилось после введения этих соединений. Содержание холестерина у получавших (+)энантиомер животных оставалось относительно постоянным, в то время как у контрольных животных содержание холестерина повышалось. На фигуре 9В приводится сравнение содержание холестерина в плазме животных контрольной группы и групп, получавших испытуемые соединения. Наиболее активным для использованного в испытании вида животных был (-)энантиомер.
Фигура 10А показывает уровни содержания холестерина в плазме крови крыс линии Zucker Diabetic Fatty, которые получали в течение 14 дней либо (-)энантиомер, либо (+)энантиомер галофената, в низкой дозе (12,5 мг/кг/день) или в высокой дозе (37,5 мг/кг/день), по сравнению с животными контрольной группы, которые получали носитель. У животных, получавших высокую дозу, (-)энантиомер привел к самому большому понижению содержания холестерина. На фигуре 10В приводится сравнение различий в содержании холестерина в плазме крови животных контрольной группы и групп, получавших испытуемые соединения. Значимые отличия от контроля наблюдались у животных, получавших (-)энантиомер, через 7 дней при использовании низкой дозы и через 7 и 14 дней при использовании высокой дозы. Для (+)энантиомера значимые отличия получены только через 7 дней после введения высокой дозы.
Фигура 11А показывает уровни содержания триглицерида в плазме крови крыс линии Zucker Diabetic Fatty, которые получали либо (-)энантиомер, либо (+)энантиомер, в низкой дозе (12/5 мг/кг/день) и в высокой дозе (37,5 мг/кг/день), по сравнению с животными контрольной группы, которые получали носитель. Животные, получавшие высокую дозу (-)энантиомера, имели самые низкие уровни содержания триглицерида из всех подопытных групп. На фигуре 11В приводится сравнение различий в содержании триглицерида в плазме крови животных контрольной группы и групп, получавших испытуемые соединения. На 7-й день высокие дозы как (+), так и (-)энантиомеров показали значимое снижение содержания в плазме триглицерида.
Фигура 12 показывает уровни содержания глюкозы в плазме крови крыс линии Zucker Diabetic Fatty, которые получали носитель, (-)галофенат или (+)галофенат, в дни 0, 2 и 3. Введение (-)галофената существенно уменьшило концентрации глюкозы в плазме крови, по сравнению с животными, получавшими носитель.
Фигура 13 показывает концентрации глюкозы в плазме крови у контрольной группы мышей линии C57BL/6J db/db по сравнению с группой, получавшей (-)галофенат. Уровни содержания глюкозы в плазме в контрольной группе прогрессирующе повышались по мере старения животных, в то время как у животных, получавших (-)галофенат, повышение содержания глюкозы в плазме не происходило либо отмечалось его существенное понижение.
Фигура 14 показывает уровни содержания инсулина в плазме крови у контрольной группы мышей линии C57BL/6J db/db по сравнению с группой, получавшей (-)галофенат. Введение (-)галофената поддерживало концентрацию инсулина в плазме, в то время как у животных контрольной группы содержание инсулина в плазме прогрессирующе понижалось.
Фигура 15 показывает процент не больных диабетом животных в контрольной группе мышей линии C57BL/6J db/db, по сравнению с группой, получавшей (-)галофенат. Примерно у 30% мышей группы, получавшей (-)галофенат, диабет не развивался (уровни содержания глюкозы в плазме <250 мг/дл), в то время как у всех животных контрольной группы к возрасту 10 недель развился диабет.
Фигура 16 показывает уровни содержания триглицерида в контрольной группе мышей линии C57BL/6J db/db, по сравнению с группой, получавшей (-)галофенат. Введение (-)галофената привело к понижению гиперлипидемии, в то время как в контрольной группе этого не произошло.
Фигура 17 показывает влияние (-)галофената и (+)галофената на содержание мочевой кислоты в плазме крови крыс, у которых с помощью оксоновой кислоты индуцировали гиперурикемию. Пероральное введение (-)галофената существенно понизило уровни содержания мочевой кислоты в плазме крови. Введение (+)галофената также понижало уровни содержания мочевой кислоты в плазме крови, но это понижение было статистически незначимым.
ОБОЗНАЧЕНИЯ
Термин "млекопитающее" включает, без ограничений, человека, домашних животных (например, собак или кошек), сельскохозяйственных животных (коров, лошадей или свиней), обезьян, кроликов, мышей и лабораторных животных.
Термин "резистентность к инсулину" можно определить в общем виде как нарушение метаболизма глюкозы. Более конкретно, резистентность к инсулину можно определить как уменьшенную способность инсулина проявлять свое биологическое действие в широком диапазоне концентраций, в результате чего его биологический эффект оказывается ниже ожидаемого (см., например, Reaven, G.M., J. Basic & Clin. Phys. & Pharm. (1998) 9:387-406, и Filer, J. Ann. Rev. Med. (1983) 34;145-60). Резистентные к инсулину индивидуумы обладают уменьшенной способностью должным образом утилизировать глюкозу и плохо реагируют или совсем не реагируют на инсулиновую терапию. Проявления резистентности к инсулину включают неэффективную активацию инсулином поглощения, окисления и запасания глюкозы в мышцах, а также неадекватное подавление инсулином липолиза в жировых тканях и продуцирования глюкозы и секретирования ее в печени. Резистентность к инсулину может явиться причиной или одной из причин синдрома поликистоза яичников, ослабленной толерантности к глюкозе (IGT), диабета беременных, гипертензии, ожирения, атеросклероза и ряда других нарушений. В конце концов, индивидуумы, резистентные к инсулину, могут постепенно дойти до состояния диабета. Связь резистентности к инсулину с нетолерантностью к глюкозе, увеличением содержания триглицеридов в плазме крови и уменьшением концентрации холестерина, представляющего собой липопротеин высокой плотности, высоким кровяным давлением, гиперурикемией, пониженной плотностью частиц липопротеинов низкой плотности и с более высокими уровнями циркуляции ингибитора-1 активатора пламиногена получила название "синдрома X" (см., например, Reaven, G.M., Physiol. Rev. (1995) 75:473-486).
Термин "сахарный диабет" или "диабет" обозначает болезнь или состояние, которое, как правило, характеризуется метаболическими дефектами продуцирования и утилизации глюкозы, приводящими к невозможности поддержания необходимых уровней сахара в теле. Результатом этих дефектов является повышенное содержание глюкозы в крови, называемое "гипергликемией". Две основные формы диабета представляют собой диабет I типа и диабет II типа. Как описано выше, диабет I типа, как правило, является результатом абсолютной недостаточности инсулина, гормона, регулирующего утилизацию глюкозы. Диабет II типа часто наблюдается на фоне нормальных или даже повышенных уровней инсулина и может являться результатом неспособности тканей правильно реагировать на инсулин. Большинство пациентов с диабетом II типа являются резистентными к инсулину и имеют относительную недостаточность инсулина, при которой секреция инсулина не может компенсировать резистентность периферических тканей по отношению к инсулину. Кроме того, многие пациенты с диабетом II типа являются тучными. Другие типы нарушений гомеостаза глюкозы включают ослабленную толерантность к глюкозе, которая представляет собой этап метаболизма, промежуточный между нормальным гомеостазом глюкозы и диабетом, и сахарный диабет беременных, который представляет собой не толерантность к глюкозе во время беременности у женщин, у которых в анамнезе ранее не было диабета I типа или II типа.
Термин "вторичный диабет" обозначает диабет, являющийся результатом действия других определенных причин, включающих: генетические дефекты функционирования β-клеток (например, начинающийся при созревании юношеский диабет, называемый "MODY", который представляет собой рано начинающийся диабет II типа с аутосомальной наследуемостью; см., например, Fajans S. et al., Diabet Med. (1996) (9 Suppl. 6): 390-5, и Bell, G. et al., Annu. Rev. Physiol. (1996) 58:171-86; генетические дефекты действия инсулина; болезни экзокринной поджелудочной железы (например, гемохроматоз, панкреатит и муковисцидоз); некоторые эндокринные болезни, при которых избыточное количество гормонов мешает действию инсулина (например, гормон роста при акромегалии и кортизол при синдроме Кушинга); некоторые лекарства, подавляющие секрецию инсулина (например, фенитоин) или ингибирующие действие инсулина (например, эстрогены и глюкокортикоиды); и диабет, вызванный инфекцией (например, краснухой, вирусом Коксаки и CMV (вирусом мозаики цветной капусты); а также другие генетические синдромы.
Рекомендации по диагностике диабета II типа, ослабленной толерантности к глюкозе и диабета беременных предложены Американской ассоциацией по диабету (см., например. The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus, Diabetes Care, (1999) Vol 2 (Suppl 1): S5-19).
Термин "галофеновая кислота" обозначает форму 4-хлорфенил-(3-трифторметилфенокси)уксусной кислоты.
Термин "гиперинсулинемия" обозначает присутствие ненормально повышенного уровня содержания инсулина в крови.
Термин "гиперурикемия" обозначает присутствие ненормально повышенного уровня содержания мочевой кислоты в крови.
Термин "секретор" обозначает вещество или соединение, стимулирующее секрецию. Например, инсулиновый секретор представляет собой вещество или соединение, которое стимулирует секрецию инсулина.
Термин "гемоглобин" или "Hb" обозначает дыхательный пигмент, присутствующий в эритроцитах, который в значительной степени ответственен за транспорт кислорода. Молекула гемоглобина включает четыре полипептидных субъединицы (две системы α-цепей и две системы β-цепей, соответственно). Каждая субъединица образована ассоциацией одного белка глобина и одной молекулы гема, который представляет собой железно-протопорфириновый комплекс. Основным классом гемоглобина, содержащемся в нормальном зрелом гемолизате, является зрелый гемоглобин (называемый "HbA"; а также называемый "HbA0", для того, чтобы отличать его от гликозилированного гемоглобина, называемого "HbA1", описанного ниже), имеющий субъединицы α2β2. В нормальном зрелом гемолизате могут также находиться компоненты, содержащиеся в микроколичествах, такие как HbA2 (α2δ2).
Среди классов зрелого гемоглобина HbAs имеется гликированный гемоглобин (называемый "HbA1" или "гликозилированным гемоглобином"), который в свою очередь можно разделить на фракции HbA1a1, HbA1a2, HbA1b и HbA1c путем фракционирования на ионообменных смолах. Все эти подклассы имеют одинаковую первичную структуру, которая стабилизируется путем образования алдимина (основания Шиффа) аминогруппой N-концевого валина в цепи β-субъединицы нормального гемоглобина HbA и глюкозы (или глюкоза-6-фосфата или фруктозы), с последующим образованием кетоамина путем перегруппировки Амадори.
Термин "гликозилированный гемоглобин" (также называемый "HbA1c", "GHb", "гемоглобин-гликозилированным", "индексом контроля диабета" и "гликогемоглобином", ниже называемый "гемоглобином A1c") обозначает стойкий продукт неферментативного гликозилирования β-цепи гемоглобина глюкозой плазмы. Гемоглобином A1c представлена основная часть гликозилированных гемоглобинов крови. Содержание гликозилированного гемоглобина пропорционально содержанию в крови глюкозы. Вследствие этого, скорость образования гемоглобина A1c прямо пропорционально увеличивается с повышением содержания в крови глюкозы. Поскольку гликозилирование происходит с постоянной скоростью в течение 120 дней продолжительности жизни эритроцита, то измерение уровней содержания гликозилированного гемоглобина отражает среднее содержание в крови глюкозы для данного индивидуума во время предшествующих двух или трех месяцев. Поэтому определение количества гликозилированного гемоглобина HbA1c может служить хорошим указателем при контролировании метаболизма углеводов. В соответствии с этим, уровни содержания в крови глюкозы за последние два месяца можно установить на основании отношения количества HbA1c к общему количеству гемоглобина Hb. Анализ содержания гемоглобина A1c в крови используют в качестве измерения, позволяющего осуществлять долгосрочный контроль за уровнями содержания в крови глюкозы (см., например, Jain, S., et al., Diabetes (1989) 38:1539-1543; Peters A., et al., JAMA (1996) 276:1246-1252).
Термин "симптом" диабета включает, не ограничиваясь нижеперечисленным, полиурию, полидипсию и полифагию, как они используются в настоящей заявке, включая обычное использование этих терминов. Например, "полиурия" обозначает выделение большого объема мочи в течение определенного периода времени; "полидипсия" обозначает хроническую чрезмерную жажду; и "полифагия" обозначает чрезмерное потребление пищи. Другие симптомы диабета включают, например, повышенную восприимчивость к некоторым инфекциям (особенно к грибковым и стафилококковым инфекциям), тошноту и кетоацидоз (повышенное продуцирование кетоновых тел в крови).
Термин "осложнение" диабета включает, не ограничиваясь нижеперечисленным, микрососудистые осложнения и макрососудистые осложнения. Микрососудистые осложнения - это такие осложнения, которые в основном приводят к повреждению мелких кровеносных сосудов. Эти осложнения включают, например, ретинопатию (ослабление или потерю зрения из-за повреждения кровеносных сосудов в глазах); невропатию (повреждение нерва и болезни ног из-за повреждения нервной системы, вызванного кровеносными сосудами); и нефропатию (болезнь почек, вызванная повреждением кровеносных сосудов в почках). Макрососудистые осложнения - это такие осложнения, которые в основном происходят из-за повреждения крупных кровеносных сосудов. Эти осложнения включают, например, сердечно-сосудистые заболевания и заболевания периферической кровеносной системы. К сердечнососудистым заболевания относятся заболевания кровеносных сосудов сердца. См., например, Kaplan, R.M. et al., "Cardiovascular diseases" in "Health and Human Behavior", pp.206-242 (McGraw-Hill, New York 1993). Сердечно-сосудистое заболевание, как правило, представляет собой одну из нескольких форм, например, гипертензию (называемую также повышенным кровяным давлением), коронарную (ишемическую) болезнь сердца, стенокардию и ревматизм сердца. К заболеваниям периферической кровеносной системы относятся заболевания любых кровеносных сосудов вне сердца. Часто они представляют собой сужение кровеносных сосудов, по которым кровь поступает к мышцам ноги или руки.
Термин "атеросклероз" охватывает сосудистые заболевания и состояния, известные и понятные врачам, работающим в соответствующих областях медицины. Атеросклеротическое сердечнососудистое заболевание, коронарная болезнь сердца (известная также под названиями заболевание коронарной артерии или ишемическая болезнь сердца), заболевание сосудов мозга и заболевание периферических кровеносных сосудов - все эти заболевания являются клиническими проявлениями атеросклероза и поэтому охватываются терминами "атеросклероз" и "атеросклеротическое заболевание".
Термин "антигиперлипидемический" относится к понижению избыточных концентраций липидов в крови до желаемых уровней.
Термин "антиуремический" относится к понижению избыточных концентраций мочевой кислоты в крови до желаемых уровней.
Термин "гиперлипидемия" относится к присутствию ненормально повышенной концентрации липидов в крови. Гиперлипидемия может проявляться по меньшей мере в трех формах: (1) гиперхолестеринемия, т.е. повышенное содержание холестерина в крови; (2) гипертриглицеридемия, т.е. повышенное содержание триглицеридов; и (3) комбинированная гиперлипидемия, т.е. комбинация гиперхолестеринемии и гиперглицеридемии.
Термин "модулировать" относится к лечению, профилактике, подавлению, повышению или индуцированию функции или состояния. Например, соединения по настоящему изобретению могут модулировать гипрелипидемию путем понижения содержания холестерина в крови человека, тем самым подавляя гиперлипидемию.
Термин "лечение" обозначает помощь человеку и уход за человеком с целью борьбы с болезнью, состоянием или нарушением, и включает введение соединения по настоящему изобретению, для того, чтобы предотвратить наступление симптомов или осложнений, облегчение симптомов или осложнений или полное устранение болезни, состояния или нарушения.
Термин "профилактика" обозначает помощь человеку и уход за человеком с тем, чтобы не допустить наступления болезни, состояния или нарушения.
Термин "холестерин" обозначает стероидный спирт, который является существенным компонентом клеточных мембран и миелиновых оболочек и, как он используется в настоящей заявке, включает его общепринятое значение. Холестерин также служит предшественником для стероидных гормонов и желчных кислот.
Термин "триглицерид(ы)" ("TGs"), как он используется в настоящей заявке, включает его общепринятое значение. Триглицериды состоят из трех молекул жирной кислоты, эстерифицирующих молекулу глицерина, и служат для запасания жирных кислот, которые используются мышечными клетками для выработки энергии или поглощаются жировыми тканями и откладываются в них в запас.
Поскольку холестерин и триглицериды не растворяются в воде, то они заключены в специальные молекулярные комплексы, известные как "липопротеины", с целью их транспортировки в плазме крови. Липопротеины могут накапливаться в плазме в результате их перепроизводства и/или недостаточного удаления. Существует по меньшей мере пять различных липопротеинов, различающихся по размеру, составу, плотности и функциям. В клетках тонкой кишки поступающие с пищей липиды собираются в большие липопротеиновые комплексы, называемые "хиломикронами", которые имеют высокое содержание триглицеридов и низкое содержание холестерина. В печени сложные эфиры триглицеридов и холестерина уплотняются и выделяются в плазму в виде богатого триглицеридами липопротеина, называемого липопротеином очень низкой плотности ("VLDL"), основная функция которого состоит в эндогенном транспорте триглицеридов, вырабатываемых в печени или выделяемых жировой тканью. В результате ферментативных процессов VLDL могут либо восстанавливаться и поглощаться печенью, либо трансформироваться в липопротеин средней плотности ("IDL"). IDL, в свою очередь, либо поглощается печенью, либо подвергается дальнейшей модификации с образованием липопротеина низкой плотности ("LDL"). LDL либо поглощается и разрушается печенью, либо поглощается внепеченочной тканью. Липопротеин высокой плотности ("HDL") помогает удалять холестерин из периферических тканей посредством процесса, называемого обратным транспортом холестерина.
Термин "дислипидемия" обозначает ненормальное содержание липопротеинов в плазме крови, включая пониженные и/или повышенные уровни содержания липопротеинов (например, повышенные уровни LDL, VLDL и пониженные уровни HDL).
Примеры первичной гиперлипидемии включают, не ограничиваясь нижеперечисленным, следующее:
(1) Семейную гиперхиломикронемию, редкое генетическое нарушение, вызывающее недостаточность фермента LP-липазы, разрушающего молекулы жира. Недостаточность LP-липазы может привести к накоплению больших количеств жира или липопротеинов в крови;
(2) Семейную гиперхолестеринемию, относительно распространенное генетическое нарушение/ возникающее в результате дефекта, вызванного серией мутаций гена рецептора LDL, что приводит к нарушению функционирования рецепторов LDL и/или к отсутствию рецепторов LDL. Это приводит к неэффективному расщеплению LDL рецепторами LDL, в результате чего повышается содержание LDL и общее содержание холестерина в плазме;
(3) Семейную комбинированную гиперлипидемию, известную также как гиперлипидемия многолипопротеинового типа; наследственное нарушение, при которым пациенты и их родственники, пораженные этой болезнью первой степени могут в различные периоды времени иметь высокое содержание холестерина или триглицеридов. Уровни HDL-холестерина при этом часто умеренно понижены;
(4) Семейный дефективный аполипопротеин В-100, представляющий собой относительно распространенное аутосомальное генетическое нарушение. Этот дефект вызван мутацией одного нуклеотида, в результате чего происходит замещение глютамина на аргинин, что может привести к уменьшению сродства частиц LDL к рецептору LDL. Вследствие этого в плазме могут наблюдаться высокие уровни содержания LDL и общего холестерина;
(5) Семейную дисбеталипротеинемию, называемую также гиперлипопротеинемией III типа, представляющую собой необычное наследственное нарушение, приводящее к умеренному или резкому повышению уровней содержания триглицеридов и холестерина в сыворотке крови, с ненормальным функционированием аполипротеина Е. Содержание HDL при этом обычно нормальное; и
(6) Семейную триглицеридемию, обычное наследуемое нарушение, при котором концентрация VLDL в плазме повышена. Это может привести к повышению (от небольшого до умеренного) уровней содержания триглицеридов (обычно не сопровождаемого повышением уровней содержания холестерина) и часто может быть связано с низким содержанием HDL в плазме крови.
Факторы риска для приведенной в качестве примера вторичной гиперлипидемии включают, не ограничиваясь нижеперечисленным, следующее: (1) факторы риска, связанные с болезнью, такие как наличие в анамнезе диабета I типа, диабета II типа, синдрома Кушинга, гипотиреоза и некоторых видов почечной недостаточности; (2) факторы риска, связанные с лекарствами, включающие противозачаточные таблетки; гормоны, такие как эстроген и кортикостероиды; некоторые диуретики; а также различные бетаблокаторы; (3) факторы, риска, связанные с питанием, включающие потребление жира, составляющее более 40% от общего количества калорий; потребление насыщенных жиров, составляющее более 10% от общего количества калорий; потребление холестерина свыше 300 мг в день; привычное и чрезмерное употребление алкоголя; и ожирение.
Термины "тучный" и "ожирение" обозначают, в соответствии с классификацией Всемирной организации здравоохранения, индекс массы тела (BMI), превышающий 27,8 кг/м2 для мужчин и 27,3 кг/м2 для женщин (BMI равен массе(кг)/высоту (м2). Ожирение связано с рядом болезненных состояний, включая диабет и гиперлипидемию. Известно также, что ожирение является фактором риска для развития диабета II типа (см., например, Barrett-Conner, E., Epidemol. Rev. (1989) 11:172-181; и Knowler, et al., Am. J. Clin. Nutr. (1991) 53:1543-1551).
Термин "фармацевтически приемлемые соли" относится к нетоксичным солям щелочных металлов, щелочноземельных металлов и солям аммония, обычно используемым в фармацевтической промышленности, включая соли натрия, калия, лития, кальция, магния, бария, аммония и протаминцинковые соли, получаемые способами, известными в данной области техники. Этот термин включает также нетоксичные кислотно-аддитивные соли, которые, как правило, получают посредством проведения реакции соединений по настоящему изобретению с подходящей органической или неорганической кислотой. Репрезентативные примеры солей включают, не ограничиваясь нижеперечисленным, гидрохлорид, гидробромид, сульфат, бисульфат, ацетат, оксалат, валерат, олеат, лаурат, борат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, напсилат и др.
Термин "фармацевтически приемлемая кислотно-аддитивная соль" относится к солям, сохраняющим биологическую эффективность и свойства свободных оснований и не являющимся нежелательными в биологическом или ином отношении, образованным с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и др., и с органическими кислотами, такими, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, р-толуолсульфоновая кислота, салициловая кислота и др. Описание фармацевтически приемлемых кислотно-аддитивных солей как пролекарств см. в публикации Bundgaard, H., et al., Design of Prodrugs (Elsevier Science Publishers, Amsterdam 1985).
Термин "фармацевтически приемлемый сложный эфир" относится к сложным эфирам, которые сохраняют после гидролиза сложноэфирной связи биологическую эффективность и свойства карбоновой кислоты или спирта и не являются нежелательными в биологическом или ином отношении. Описание фармацевтически приемлемых сложных эфиров в качестве пролекарств см. в публикации Bundgaard, H., цитированной выше. Эти сложные эфиры обычно получают из соответствующей карбоновой кислоты и спирта. Как правило, получение сложных эфиров можно осуществлять с помощью обычных синтетических способов. (См., например, публикацию March, Advanced Organic Chemistry, 3rd Ed., p.1157 (John Wiley & Sons, New York 1985) и цитированные в ней публикации, и Mark et al., Encyclopedia of Chemical Technology, (1980) John Wiley & Sons, New York). Спиртовой компонент сложного эфира обычно включает: (i) С2-С12-алифатический спирт, который может содержать или может не содержать одну или более двойных связей и может содержать или может не содержать разветвленные углеродные цепи; или (ii) С7-С12-ароматические или гетероароматические спирты. Настоящее изобретение предусматривает также использование таких композиций, которые являются сложными эфирами, описанными в настоящей заявке, и в то же время являются их фармацевтически приемлемыми кислотно-аддитивными солями.
Термин "фармацевтически приемлемый амид" относится к амидам, которые сохраняют после гидролиза амидной связи биологическую эффективность и свойства карбоновой кислоты или амина и не являются нежелательными в биологическом или ином отношении. Описание фармацевтически приемлемых амидов в качестве пролекарств см. в публикации Bundgaard, H., ed., цитированной выше. Эти амиды обычно получают из соответствующей карбоновой кислоты и амина. Как правило, получение амидов можно осуществлять с помощью обычных синтетических способов. (См., например, публикацию March et al., Advanced Organic Chemistry, 3rd Ed., p.1152 (John Wiley & Sons, New York 1985) и Mark et al., Encyclopedia of Chemical Technology, John Wiley & Sons, New York 1980). Настоящее изобретение предусматривает также использование таких композиций, которые являются амидами, описанными в настоящей заявке, и в то же время являются их фармацевтически приемлемыми кислотно-аддитивными солями.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
(1) Общие положения
Настоящее изобретение направлено на использование предпочтительно производных (-)(3-тригалометилфенокси)-(4-галофенил)уксусной кислоты, имеющих следующую общую формулу:
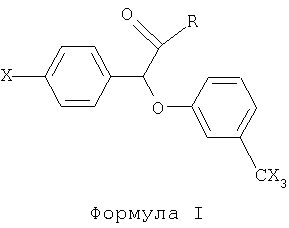
В формуле I R представляет собой функциональную группу, включающую, не ограничиваясь нижеперечисленным, следующее: гидрокси, низший аралкокси, например, фенил-низший алкокси, такой как бензилокси; ди-низший алкиламино-низший алкокси и его нетоксичные фармакологически приемлемые кислотно-аддитивные соли, например, диметиламиноэтокси, диэтиламиноэтокси-гидрохлорид, диэтиламиноэтоксицитрат, диэтиламинопропокси; низший алканамидо низший алкокси, ннапример, формамидоэтокси, ацетамидоэтокси или ацетамидопропокси; бензамидо-низший алкокси, например, бензамидоэтокси или бензамидопропокси; уреидо-низший алкокси, например, уреидоэтокси или 1-метил-2-уреидоэтокси; N'-низший алкил-уреидо-низший алкокси, т.е. R1NH-CONH-CnH2n-O-, где R1 представляет собой низший алкил, а n представляет собой целое число, имеющее значение от 1 до около 5, например, N'-этил-уреидоэтокси или N'-этилуреидопропокси; карбамоил-низший алкокси, например, карбамоилметокси или карбамоилэтокси; галофеноксизамещенный низший алкокси, например, 2-(4-хлорфенокси)этокси или 2-(4-хлорфенокси)-2-метилпропокси; карбамоилзамещенный фенокси, например, 2-карбамоилфенокси; карбокси-низший алкиламино и его нетоксичные фармакологически приемлемые кислотно-аддитивные соли, например, соль карбоксиметиламино и циклогексиламино или карбоксиэтиламин; N,N-ди-низший алкиламино-низший алкиламино и его нетоксичные фармакологически приемлемые кислые соли, например, N,N-диметиламиноэтиламино гидрохлорид, N,N-диэтиламиноэтиламино, N,N-диэтиламиноэтиламиноцитрат или N,N-диметиламинпропиламино-цитрат; гало гензамещенный низший алкиламино, например, 2-хлорэтиламино или 4-хлорбутиламино; гидроксизамещенный низший алкиламино, например, 2-гидроксиэтиламино или 3-гидроксипропиламно; низший алканоилоксизамещенный низший алкиламино, например, ацетоксиэтиламино или ацетоксипропиламино; уреидо; низший алкоксикарбониламино, например, метоксикарбониламино (т.е. -NHCOOCH3) или этоксикарбониламино, (т.е. СНСООС2Н5). В предпочтительном варианте осуществления изобретения R выбирают так, чтобы он представлял собой гидролизируемую группу, такую как сложный эфир или амид, и чтобы после гидролиза сложноэфирной или амидной связи соединение было биологически активным, таким как фармацевтически приемлемые сложные эфиры или амиды, используемые в качестве пролекарств. Х в формуле I представляет собой галоген, например хлор, бром, фтор или йод.
В предпочтительном варианте своего осуществления настоящее изобретение касается использования производных (-)(3-тригалометилфенокси)-(4-галофенил)уксусной кислоты, имеющих следующую общую формулу:
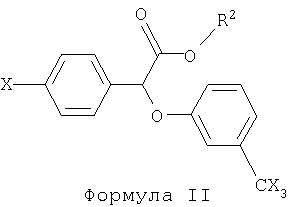
В формуле II R2 представляет собой функциональную группу, включающую, не ограничиваясь нижеперечисленным, следующее: водород, фенил-низший алкил, например, бензил; низший алканамидо-низший алкил, например, ацетамидоэтил; или бензамидо-низший алкил, например, бензамидоэтил. Х в формуле II представляет собой галоген, например хлор, бром, фтор или йод.
В следующем предпочтительном варианте своего осуществления настоящее изобретение касается использования соединения, имеющего формулу:
Соединение формулы III называется "(-)2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетатом" (а также называется "(-)галофенатом").
Изменения в метаболизме лекарств, вызванные ингибированием ферментов цитохром Р450, с очень высокой вероятностью могут привести к существенным неблагоприятным побочным действиям для пациентов. Ранее сообщалось о таких побочных действиях для пациентов, которых лечили рацемическим галофенатом. В настоящих исследованиях выяснилось, что рацемическая галофеновая кислота ингибирует цитохром Р450 2С9, фермент, о котором известно, что он играет важную роль в метаболизме определенных лекарств. Это может привести к возникновению серьезных проблем, связанных со взаимодействием лекарств с антикоагулянтами, противовоспалительными средствами и другими лекарствами, метаболизируемыми этим ферментом. Однако выяснилось, что имеется существенное различие между энантиомерами галофеновой кислоты в отношении их способности ингибировать цитохром Р450 2С9, причем (-)энантиомер примерно в двадцать раз менее активен в этом отношении, чем (+)энантиомер, который был очень активен (см. пример 7). Поэтому использование (-)энантиомеров соединений формулы I, формулы II или формулы III позволяет избежать ингибирования этого фермента и не допустить возникновения неблагоприятных воздействий на метаболизм лекарств, которые ранее наблюдались для рацемического галофената.
Настоящее изобретение охватывает способ модулирования резистентности к инсулину у млекопитающего, включающий: введение млекопитающему терапевтически эффективного количества соединения, имеющего общую структуру формулы I или его фармацевтически приемлемой соли. В предпочтительном варианте осуществления изобретения соединение имеет общую структуру формулы II. Еще в одном предпочтительном варианте осуществления изобретения соединение имеет структуру формулы III. Выяснилось, что способ позволяет избежать неблагоприятных побочных действий, связанных с введением рацемической смеси галофената, путем обеспечения такого количества (-)стереоизомера соединений формулы I, формулы II или формулы III, которое недостаточно для того, чтобы вызвать неблагоприятные побочные действия, связанные с ингибированием цитохрома Р450 2С9.
Настоящее изобретение охватывает также способ модулирования диабета II типа у млекопитающего, включающий: введение млекопитающему терапевтически эффективного количества соединения, имеющего общую структуру формулы I, или его фармацевтически приемлемой соли. В предпочтительном варианте осуществления изобретения соединение имеет общую структуру формулы II. Еще в одном предпочтительном варианте осуществления изобретения соединение имеет структуру формулы III. Выяснилось, что способ позволяет избежать неблагоприятных побочных действий, связанных с введением рацемической смеси галофената, путем обеспечения такого количества (-)стереоизомера соединений формулы I, формулы II или формулы III, которое недостаточно для того, чтобы вызвать неблагоприятные побочные действия, связанные с ингибированием цитохрома Р450 2С9.
Настоящее изобретение охватывает также способ модулирования гиперлипидемии у млекопитающего, включающий: введение млекопитающему терапевтически эффективного количества соединения, имеющего общую структуру формулы I, или его фармацевтически приемлемой соли. В предпочтительном варианте осуществления изобретения соединение имеет общую структуру формулы II. Еще в одном предпочтительном варианте осуществления изобретения соединение имеет структуру формулы III. Выяснилось, что способ позволяет избежать неблагоприятных побочных действий, связанных с введением рацемической смеси галофената, путем обеспечения такого количества (-)стереоизомера соединений формулы I, формулы II или формулы III, которое недостаточно для того, чтобы вызвать неблагоприятные побочные действия, связанные с ингибированием цитохрома Р450 2С9.
Рацемическая смесь галофената (т.е. рацемическая смесь двух энантиомеров в соотношении 1:1) обладает антигиперлипидемическим действием и обеспечивает лечение и снижение гипергликемии, связанной с диабетом, в сочетании с определенными другими лекарствами, обычно используемыми для лечения этой болезни. Однако эта рацемическая смесь, хотя и дает ожидаемый эффект, но вызывает неблагоприятные побочные действия. Термин "неблагоприятные побочные действия" включает, не ограничиваясь нижеперечисленным, тошноту, желудочно-кишечные язвы и желудочно-кишечные кровотечения. Другие побочные действия, о которых сообщалось для рацемического галофената, включают возможность возникновения проблем, связанных с межлекарственными взаимодействиями, особенно включающих трудности с контролированием антикоагуляции посредством Кумадина™. Использование по существу чистых соединений по настоящему изобретению позволяет более четко определить связанную с дозой эффективность, уменьшить неблагоприятные побочные действия и вследствие этого, повысить терапевтический индекс. Таким образом, установлено, что более желательно и предпочтительно вводить (-)энантиомер галофената, вместо рацемического галофената.
Настоящее изобретение охватывает также способ модулирования гиперурикемии у млекопитающего, включающий: введение млекопитающему терапевтически эффективного количества соединения, имеющего общую структуру формулы I, или его фармацевтически приемлемой соли. В предпочтительном варианте осуществления изобретения соединение имеет общую структуру формулы II. Еще в одном предпочтительном варианте осуществления изобретения соединение имеет структуру формулы III. Выяснилось, что способ позволяет избежать неблагоприятных побочных действий, связанных с введением рацемической смеси галофената, путем обеспечения такого количества (-)стереоизомера соединений формулы I, формулы II или формулы III, которое недостаточно для того, чтобы вызвать неблагоприятные побочные действия, связанные с ингибированием цитохрома Р450 2С9.
(2) (-)Энантиомеры формулы I, формулы II и формулы III.
Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость линейно-поляризованного света. При описании оптически активного соединения префиксы R и S используются для обозначения абсолютной конфигурации молекулы вокруг ее хирального центра (ов). Префиксы "d" и "1" или (+) и (-) используются для обозначения знака вращения линейно-поляризованного света данным соединением, причем (-) или 1 обозначают, что соединение является "левовращающим", а (+) или d обозначают, что соединение является "правовращающим". Не существует корреляции между номенклатурой для обозначения абсолютной стереохимии и для вращения энантиомера. Для данной химической структуры эти соединения, называемые "стереоизомерами", являются идентичными, за исключением того, что они являются зеркальными отражениями друг друга. Конкретный стереоизомер может также называться "энантоимером", а смесь таких изомеров часто называют "энантиомерной" или "рацемической" смесью. См., например, Streitwiesser, А. & Heathcock, C.H., Introduction to Organic Chemistry, 2nd Ed., Chapter 7 (MacMillan Publishing Co., U.S.A. 1981).
Химический синтез рацемической смеси галофенатов, производных (-)(3-тригалометилфенокси)-(4-галофенил)уксусной кислоты, можно осуществлять способами, описанными в патенте США №3517050, положения которого включены в настоящую заявку в качестве ссылки. Синтез соединений по настоящему изобретению описан также в нижеприведенных примерах. Отдельные энантоимеры можно получить путем повторного растворения рацемической смеси энантиомеров с помощью обычных способов, известных специалистам в данной области техники и используемых ими. См., например, Jaques, J. et al., в сборнике Enantiomers, Racemates, and Resolutions, John Wiley and Sons, New York (1981). Другие стандартные способы повторного растворения, известные специалистам в данной области техники, включающие, не ограничиваясь нижеперечисленым, простую кристаллизацию и хроматографическое повторное растворение, также можно использовать (см., например, Stereochemistry of Carbon Compounds (1962), E.L.Eliel, McGraw Hill; Lochmuller, J. Chromatography (1975) 13, 283-302). Кроме того, соединения по настоящему изобретению, т.е. оптически чистые изомеры, можно получить из рацемической смеси путем ферментативного биокаталитического повторного растворения. Ферментативное биокаталитическое повторное растворение описано ранее (см., например, патенты США №№5057427 и 5077217, описание которых включены в настоящую заявку в качестве ссылок). Другие способы получения энантиомеров включают стереоспецифический синтез (см., например, Li, A.J. et al., Pharm. Sci. (1997) 86:1073-1077).
Термин "по существу не содержащие своего (+)стереоизомера", как он используется в настоящей заявке, обозначает, что композиции содержат существенно более высокие пропорции (-)изомера галофената по отношению к его (+)изомеру. В предпочтительном варианте осуществления изобретения термин "по существу не содержащая своего (+)стереоизомера", как он используется в настоящей заявке, обозначает, что композиция содержит по меньшей мере 90% масс. (-)изомера и 10% масс. или менее (+)изомера. В более предпочтительном варианте осуществления изобретения термин "по существу не содержащая своего (+)стереоизомера", как он используется в настоящей заявке, обозначает, что композиция содержит по меньшей мере 99% масс. (-)изомера и 1% масс. или менее (+)изомера. В наиболее предпочтительном варианте осуществления изобретения термин "по существу не содержащая своего (+)стереоизомера" обозначает, что композиция содержит более 99% масс. (-)изомера. Эти проценты основаны на общем количестве галофената в композиции. Термины "по существу оптически чистый (1) изомер галофената", "по существу оптически чистый (1) галофенат", "оптически чистый (1) изомер галофената" и "оптически чистый (1) галофената" все относятся к (-)изомеру и охватывают вышеуказанные количества. Кроме того, термины "по существу оптически чистый (d) изомер галофената", "по существу оптически чистый (d) галофенат", "оптически чистый изомер (d) галофената" и "оптически чистый (d) галофенат" все относятся к (+)изомеру и охватывают вышеуказанные количества.
Термин "энантиомерный избыток" или "ее" относится к термину "оптическая чистота" в том смысле, что оба они являются мерами одного и того же феномена. Значение "ее" выражается числами от 0 до 100, причем 0 обозначает рацемический, а 100 - чистый, одинарный энантиомер. Соединение, описываемое как имеющее оптическую чистоту 98%, можно описать, как имеющее 96% "ее".
(3) Комбинированная терапия с применением дополнительных активных агентов
Композиции можно составлять и вводить так, как описано ниже. "Лекарственная форма" определяется как фармацевтический препарат, содержащий смесь различных наполнителей и ключевых компонентов, обеспечивающую относительно стабильную желаемую и полезную форму соединения или лекарства. Для настоящего изобретения, "лекарственная форма" включена в значение термина "композиция". Соединения по настоящему изобретению можно эффективно использовать по отдельности или в сочетании с одним или более дополнительных активных агентов, в зависимости от поставленной цели лечения (см., например. Turner, N. et al., Prog. Drug Res. (1998) 51, 33-94; Haffner, S. Diabetes Care (1998) 21, 160-178; и DeFronzo, R. et al. (eds.). Diabetes Reviews (1997) Vol. 5 No. 4). В ряде работ изучали преимущества комбинированной терапии с использованием пероральных средств (см., например, Mahler, R., J. Clin. Endocrinol. Metab. (1999) 84: 1165-71; United Kingdom Prospective Diabetes Study Group: UKPDS 28, Diabetes Care (1998) 21: 87-92; Bardin, С. W., (ed.), Current Therapy in Endocrinology and Metabolism, 6th Edition (Mosby - Year Book, Inc., St. Louis, МО 1997); Chiasson, J. et al., Ann. Intern. Med. (1994) 121: 928-935; Coniff, R. et al., Clin. Ther. (1997) 19: 16-26; Coniff, R. et al., Am. J. Med. (1995) 98: 443-451; и Iwamoto, Y. et al., Diabet. Med. (1996) 13 365-370; Kwiterovich, P. Am. J. Cardiol (1998) 82(12A): 3U-17U). Эти исследования указывают на то, что модуляцию диабета и гиперлипидемии можно улучшить путем добавления в схему лечения второго агента. Комбинированная терапия включает введение единой лекарственной формы, которая содержит соединение, обладающее общей структурой формулы I (или формулы II или формулы III), и один или более дополнительных активных агентов, а также введение соединения формулы I (или формулы II или формулы III) и каждого активного агента в его собственной отдельной лекарственной форме. Например, соединение формулы I и ингибитор редуктазы HMG-СоА можно вводить человеку вместе, в составе единой композиции для перорального введения, такой как таблетки и капсулы, или каждый агент можно вводить в виде отдельных лекарственных форм. Если используют отдельные лекарственные формы, то соединение формулы I и один или более дополнительных активных агентов можно вводить по существу одновременно (т.е. совместно), либо в отдельные периоды времени, т.е. последовательно. Комбинированная терапия включает все эти схемы.
Примером может служить комбинированная терапия, которая модулирует атеросклероз (т.е. предотвращает наступление симптомов заболевания или связанных с ним осложнений), в которой соединение формулы I вводят в сочетании с одним или более следующих активных агентов: антигиперлипидемический агент; агент, повышающий содержание HDL в плазме крови; антигиперхолестеринемический агент, такой как ингибитор биосинтеза холестерина, например ингибитор гидроксиметилглютарил (HMG)-СоА-редуктазы (который называют также статином, например, такой как ловастатин, симвастатин, правастатин, флувастатин и аторвастатин), ингибитор HMG-CoA-синтазы, ингибитор скваленэпоксидазы, или ингибитор скваленсинтетазы (известный также как ингибитор скваленсинтазы); ингибитор ацил-кофермента ацилтрансферазы холестерина А (АСАТ), такой как мелинамид; пробукол; никотиновая кислота и ее соли и ниацинамид; ингибитор поглощения холестерина, такой как β-ситостерол; анионообменная смола, выводящая желчную кислоту, такая как холестирамин, колестипол или диалкиламиноалкильные производные сшитого декстрана; индуктор рецептора LDL (липопротеина низкой плотности); фиораты, такие как клофибрат, безафибрат, фенофибрат и гемфибризол; витамин В6 (известный также как пиридоксин) и его фармацевтически приемлемые соли, такие соль соляной кислоты; витамин В12 (известный также как цианокобаламин); витамин В3 (известный также как никотиновая кислота и ниацинамид, см. выше); антиоксидантные витамины, такие как витамины С, Е и бета-каротин; бета-блокатор; антагонист ангиотензина II; ингибитор фермента, преобразующего ангиотензин; и ингибитор агрегации тромбоцитов, такой как антагонисты рецепторов фибриногена (т.е. антагонисты рецепторов фибриногена гликопротеина IIb/IIIa) и аспирин. Как указывалось выше, соединение формулы I можно вводить в сочетании с более, чем одним дополнительным активным агентом, например, сочетание соединения формулы I с ингибитором HMG-CoA-редуктазы (например, с ловастатином, симвастатином и правастатином) и аспирином, или соединение формулы I можно вводить с ингибитором HMG-CoA-редуктазы и β-блокатором.
Другим примером комбинированной терапии является лечение ожирения или связанных с ожирением нарушений, при котором соединения формулы I можно эффективно использовать в сочетании, например, с фенилпропаноламином, фентермином, диэтилпропионом, мазиндолом; фенфлурамином, дексфенфлурамином, фентирамином, агонистами β3-адренорецепторов; сибутрамином, ингибиторами желудочно-кишечной липазы (такими, как орлистат) и с лептинами. Другие агенты, используемые при лечении ожирения или связанных с ожирением нарушений, в сочетании с которыми можно эффективно использовать соединения формулы I, включают, например, нейропептид Y, энтеростатин, холецитокинин, бомбезин, амилин, рецепторы гистамина Н3, рецепторы допамина D2, гормон, стимулирующий меланоциты, кортикотропин-рилизинг фактор, галанин и гамма-аминомасляную кислоту (GABA).
Еще одним примером комбинированной терапии является модулирование диабета (или лечение диабета и связанных с ним симптомов, осложнений и нарушений), при котором соединения формулы I можно эффективно использовать в сочетании, например, с сульфонилмочевинами (такими, как хлорпропамид, толбутамид, ацетогексамид, толазамид, глибурид, гликлазид, глиназа, глимепирид и глипизид), бигуанидинами (такими, как метформин), тиазолидиндионами (такими, как циглитазон, пиоглитазон, троглитазон и розиглитазон); дигидроэпиандростероном (называемым также DHEA, или с его конъюгированным сульфатным сложным эфиром DHEA-SO4); антиглюкокортикоидами; ингибиторами TNF-α; ингибиторами α-глюкозидазы (такими, как акарбоза, миглитол и воглибоза), прамлинтидом (синтетическим аналогом человеческого гормона амилина), другими секреторами инсулина (такими, как репаглинид, глихидон и натеглинид), инсулином, а также с вышеописанными активными агентами для лечения атеросклероза.
Следующим примером комбинированной терапии является модулирование гиперлипидемии (лечение гиперлипидемии и связанных с ней осложнений), при котором соединения формулы I можно эффективно использовать в сочетании, например, со статинами (такими, как флувастатин, ловастатин, правастатин или симвастатин), со смолами, связывающими желчную кислоту (такими, как колестипол или холестирамин), с никотиновой кислотой, пробуколом, бетакаротином, витамином Е или витамином С.
В соответствии с настоящим изобретением, терапевтически эффективное количество соединения формулы I (или формулы II, или формулы III) можно использовать для получения фармацевтической композиции, полезной для лечения диабета, лечения гиперлипидемии, лечения гиперурикемии, лечения ожирения, понижения содержания триглицеридов, понижения содержания холестерина, повышения содержания в плазме липопротеина высокой плотности, а также для лечения, профилактики и снижения риска развития атеросклероза.
Кроме того, терапевтически эффективное количество соединения формулы I (или формулы II, или формулы III) и терапевтически эффективное количество одного или более активных агентов, выбранных из нижеперечисленного: антигиперлипидемический агент; агент, повышающий содержание HDL в плазме; антигиперхолестеринемический агент, такой, как ингибитор биосинтеза холестерина, например, ингибитор HMG-CoA-редуктазы, ингибитор HMG-CoA-синтазы, ингибитор скваленэпоксидазы или ингибитор скваленсинтетазы (известный также как ингибитор скваленсинтазы); ингибитор ацил-кофермента ацилтрансферазы холестерина А; пробукол; никотиновая кислота и ее соли; ниацинамид; ингибитор поглощения холестерина; анионообменная смола, выводящая желчную кислоту; индуктор рецептора липопротеина низкой плотности; клофибрат, фенофибрат и гемфиброзил; витамин B6 и его фармацевтически приемлемые соли; витамин B12; антиоксидантный витамин; β-блокатор; антагонист ангиотензина II; ингибитор фермента, преобразующего ангиотензин; ингибитор агрегации тромбоцитов; антагонист рецепторов фибриногена; аспирин; фентирамины, агонисты адренергического рецептора β3; сульфонилмочевины, бигуаниды, ингибиторы α-глюкозидазы, другие секреторы инсулина и инсулин можно использовать вместе для получения фармацевтической композиции, полезной для вышеуказанных видов терапии.
(4) Лекарственные формы и способы введения
В способах по настоящему изобретению соединения формулы I, формулы II или формулы III можно вводить млекопитающему, например человеку, отдельно, в форме его фармацевтической соли или его гидролизируемого предшественника, либо в форме фармацевтической композиции, в которой соединение смешивают с подходящими носителями или наполнителем (наполнителями) в терапевтически эффективном количестве. Под терминами "терапевтически эффективная доза", "терапевтически эффективное количество" или под употребляемыми взамен них терминами "фармакологически приемлемая доза", "фармакологически приемлемое количество" имеют в виду количество соединения по настоящему изобретению или в качестве альтернативы, комбинации, например, соединения по настоящему изобретению, по существу не содержащего своего (+)стереоизомера, и фармацевтически приемлемого носителя, достаточное для достижения желаемого результата, например, излечения симптома или осложнения диабета II типа.
Соединения формулы I, формулы II или формулы III, используемые в способах по настоящему изобретению, можно включать в ряд лекарственных форм для терапевтического использования. Более конкретно, соединения формулы I (или формулы II, или формулы III) можно включать в состав фармацевтических композиций в сочетании с подходящими фармацевтически приемлемыми носителями или разбавителями, и из них можно готовить лекарственные препараты в твердой, полужидкой, жидкой и газообразной форме, такие как таблетки, капсулы, пилюли, порошки, гранулы, драже, гели, взвеси, мази, растворы, суппозитории, растворы для инъекций, ингалянты и аэрозоли. В связи с этим, введение соединений можно осуществлять различными путями, включая пероральное, защечное, ректальное, парентеральное, внутрибрюшинное, внутрикожное, чрескожное, внутритрахеальное введение. Кроме того, соединение можно вводить не системно, а локально, в виде лекарственной формы с замедленным высвобождением активного компонента. Кроме того, соединение можно вводить в липосомах.
Кроме того, соединения формулы I, формулы II или формулы III можно сочетать с обычными наполнителями, разбавителями или носителями и из полученных смесей штамповать таблетки, или готовить их в виде эликсиров или растворов для удобства перорального введения, или для внутримышечного либо внутривенного введения. Соединения можно вводить через кожу или из них можно готовить лекарственные формы с замедленным высвобождением активного компонента и т.п.
Соединения формулы I, формулы II или формулы III можно вводить отдельно или в сочетании друг с другом, или их можно использовать в сочетании с другими известными соединениями (описанными выше). В лекарственных формах соединения можно использовать в виде их фармацевтически приемлемых солей. Они могут содержать гидролизуемые группы. Они также могут применяться отдельно или в походящих сочетаниях, а также в комбинации с другими известными в фармацевтике активными соединениями.
Подходящие лекарственные формы для использования по настоящему изобретению можно найти в публикации Remington's Pharmaceutical Sciences (Mack Publishing Company (1985) Philadelphia, PA, 17th Ed.), которая включена в настоящую заявку в качестве ссылки. Кроме того, краткий обзор способов введения лекарств см. в публикации Langer, Science (1990) 249:1527-1533, которая включена в настоящую заявку в качестве ссылки. Описываемые в настоящей заявке фармацевтические композиции можно изготовить с помощью способов, известных специалистам в данной области техники, например, с помощью обычного смешивания, растворения, гранулирования, дражирования, растирания, эмульгирования, капсулирования, улавливания или лиофилизации. Описываемые ниже способы и наполнители приводятся лишь в качестве примеров и ни в коем случае не ограничивают объем настоящего изобретения.
Для использования в качестве растворов для инъекций из соединений можно приготовить лекарственные формы путем растворения, суспендирования или эмульгирования их в водных и неводных растворителях, таких как растительные или другие подобные масла, синтетические глицериды алифатических кислот, сложные эфиры высших алифатических кислот или пропиленгликоля; по желанию, можно использовать обычные добавки, такие, как солюбилизаторы, изотонические агенты, суспендирующие агенты, эмульгаторы, стабилизаторы и консерванты. Предпочтительно, лекарственные формы из соединений по настоящему изобретению готовят в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнкса, раствор Рингера или буфер на основе физиологического раствора. Для введения через слизистые оболочки в составе лекарственной формы используют пенетранты, проникающие сквозь эти оболочки. Такие пенетранты общеизвестны в данной области техники.
Для перорального введения соединения формулы I, формулы II или формулы III можно легко приготовить в виде лекарственных форм путем соединения их с фармацевтически приемлемыми носителями, хорошо известными в данной области техники. Такие носители позволяют изготовить из соединений таблетки, пилюли, драже, капсулы, эмульсии, липофильные или гидрофильные суспензии, жидкости, гели, сиропы, взвеси, суспензии и т.п., предназначенные для перорального введения пациенту, которому требуется лечение. Фармацевтические препараты для перорального использования можно получить посредством смешивания соединений с твердым носителем, необязательного размалывания полученной смеси и технологической обработки смеси гранул, после добавления, по желанию, подходящих вспомогательных компонентов, с получением таблеток или сердцевины драже. Подходящими наполнителями, в частности, являются сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозные препараты, такие, как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, камедь трагаканта, метилцеллюлоза, гидроксипропилметилцеллюлоза, натриевая карбоксиметилцеллюлоза и/или поливинилпирролидон (PVP). По желанию, можно добавить дезинтеграторы, такие как сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Сердцевины драже покрывают подходящими покрытиями. Для этой цели можно использовать концентрированные растворы сахаров, которые необязательно могут содержать гуммиарабик, тальк, поливинилпирролидон, карбополовый гель, полиэтиленгликоль и/или диоксид титана, лакировочные растворы и подходящие органические растворители или смеси растворителей. В таблетки или покрытия драже можно добавить красители или пигменты с целью идентификации или для того, чтобы различать различные комбинации доз активного соединения.
Фармацевтические препараты, которые можно использовать для перорального введения, включают удобные для глотания капсулы, изготовленные из желатина, а также мягкие запечатанные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Удобные для глотания капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующие вещества, такие как крахмалы, и/или смазывающие вещества, такие как тальк или стеарат магния, и необязательно - стабилизаторы. В мягких капсулах активное вещество может быть растворено или суспендировано в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все лекарственные формы для перорального введения должны быть приготовлены в дозировках, подходящих для такого введения.
Предназначенные для защечного введения композиции могут иметь форму таблеток или лепешек, изготовленных обычным способом.
При введении путем ингаляции соединения для использования по настоящему изобретению удобно вводить в форме аэрозоля, из аэрозольных упаковок или из распылителя, с использованием подходящего газа-вытеснителя, например, дихлоридфторметана, трихлорфторметана, дихлортетрафторэтана, двуокиси углерода или другого подходящего газа, или из порошковых ингаляторов, без использования газов-вытеснителей. В случае аэрозольных упаковок, введение стандартной одноразовой дозы может быть обеспечено с помощью клапана, позволяющего ввести отмеренное количество препарата. Капсулы или картриджи, изготовленные, например, из желатина, для использования в ингаляторе или порошковдувателе, могут содержать порошковую смесь соединения и подходящей порошковой основы, такой как лактоза или крахмал.
Соединения могут использоваться в составе лекарственных форм, предназначенных для парентерального введения путем инъекции, например, инъекции болюсов или непрерывного вливания. Лекарственные формы для инъекции могут быть изготовлены в виде стандартных одноразовых доз, например, в ампулах, или в многодозовых контейнерах, с добавкой консервантов. Композиции могут иметь форму суспензий, растворов или эмульсий в масляных или водных носителях, и могут содержать вспомогательные средства для приготовления лекарственных форм, такие как суспендирующие средства, стабилизаторы и/или диспергаторы.
Лекарственные формы для парентерального введения включают водные растворы активных соединений в воднорастворимой форме. Кроме того, суспензии активных соединений можно приготовить в виде соответствующих масляных суспензий для инъекций. Походящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, повышающие вязкость суспензии, такие как натриевая карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия также может содержать подходящие стабилизаторы или средства, повышающие растворимость соединений, что позволяет приготовить высококонцентрированные растворы. В качестве альтернативы, активный компонент может быть в порошковой форме, для соединения его перед использованием с подходящим носителем, например, со стерильной апирогенной водой.
Лекарственные формы на основе данных соединений могут быть также изготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао, карбовоски, полиэтиленгликоли или другие глицериды, все из которых плавятся при температуре тела, однако отверждаются при комнатной температуре.
Кроме описанных выше лекарственных форм, на основе описываемых соединений могут быть изготовлены длительно действующие препараты. Такие длительно действующие лекарственные формы можно вводить путем имплантации, например, подкожно или внутримышечно) или путем внутримышечной инъекции. Такие лекарственные формы могут быть изготовлены, например, с помощью подходящих полимерных или гидрофобных материалов (например, в виде эмульсии в подходящем масле) или ионообменных смол, либо в виде медленно растворимых производных, например, медленно растворимой соли.
В качестве альтернативы, можно использовать другие системы введения гидрофобных фармацевтических соединений. Хорошо известными примерами носителей для введения гидрофобных лекарств являются липосомы и эмульсии. В предпочтительном варианте осуществления настоящего изобретения можно использовать длительно циркулирующие, т.е. скрытые липосомы. Общее описание таких липосом приводится в патенте США №5013556, Woodle, et al., положения которого включены в настоящую заявку в качестве ссылки. Соединения по настоящему изобретению можно также вводить с использованием способов контролируемого высвобождения и/или с помощью устройств для введения лекарств, таких как описанные в патентах США №№3845770, 3916899, 3536809, 3598123 и 4008719, описания которых включены в настоящую заявку в качестве ссылок.
Могут также использоваться некоторые органические растворители, такие как диметилсульфоксид (DMSO), хотя следует учитывать, что они более токсичны. Кроме того, соединения можно вводить с помощью системы замедленного высвобождения лекарств, такой как полупроницаемая матрица из твердых гидрофобных полимеров, содержащая терапевтическое средство. Разработаны различные типы материалов для замедленного высвобождения лекарств, и они хорошо известны специалистам в данной области техники. Капсулы с замедленным высвобождением активных компонентов могут, в зависимости от их химической природы, высвобождать соединения в течение от нескольких часов до свыше 100 дней.
Фармацевтические композиции могут также включать подходящие твердые или гелеобразные носители или наполнители. Примеры таких носителей и наполнителей включают, не ограничиваясь нижеперечисленным, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли.
Фармацевтические композиции, пригодные для использования по настоящему изобретению, включают композиции, в которых активные компоненты содержатся в терапевтически эффективном количестве. Количество вводимой композиции, конечно, зависит от проходящего лечение субъекта, от массы его тела, тяжести болезни, способа введения и от решения лечащего врача. Эффективное количество вполне в состоянии определить специалист в данной области техники, особенно в свете приведенного здесь подробного описания изобретения.
Для любого соединения, используемого для способа по настоящему изобретению, терапевтически эффективная доза может быть первоначально установлена с помощью анализов в культуре клеток или на моделях животных.
Кроме того, токсичность и терапевтическую эффективность описываемых здесь соединений можно определить с помощью стандартных фармацевтических методов в культуре клеток или на лабораторных животных, например, путем определения ЛД50 (дозы, летальной для 50% популяции) и ЭД50 (дозы, терапевтически эффективной для 50% популяции). Соотношение между токсической и терапевтически эффективной дозами называется терапевтическим индексом и может быть выражено как соотношение между ЛД50 и ЭД50. Соединения, демонстрирующие высокие терапевтические индексы, являются предпочтительными. Данные, полученные из этих анализов в культуре клеток и исследований на животных, можно использовать для расчета диапазона доз, которые нетоксичны для человека. Дозировки таких соединений предпочтительно лежат в пределах диапазона циркулирующих концентраций, включающего ЭД50 с низкой или отсутствующей токсичностью. Доза может варьировать в пределах этого диапазона, в зависимости от используемой лекарственной формы и способа введения. Точную лекарственную форму, способ введения и дозу может выбрать лечащий врач в зависимости от состояния пациента. (См., например, Fingi et al., 1975 In: The Pharmacological Basis of Therapeutics, Ch.1).
Количество активного соединения, которое можно объединить с материалом носителя для получения одноразовой дозы, варьирует в зависимости от болезни, вида млекопитающего и способа введения. Однако, как общее указание, подходящие стандартные дозы для соединений по настоящему изобретению могут, например, предпочтительно содержать между 100 мг и около 3000 мг активного соединения. Предпочтительная стандартная доза составляет между 500 мг и около 1500 мг. Более предпочтительная стандартная доза составляет между 500 мг и около 1000 мг. Такие стандартные дозы можно вводить более одного раза в день, например, 2, 3, 4, 5 или 6 раз в день, но предпочтительно 1 или 2 раза в день, так что общая ежедневная доза для взрослого человека с массой тела в 70 кг находится в интервале от 0,1 до около 250 мг на кг массы тела субъекта на каждое введение. Предпочтительная доза составляет от 5 до около 250 мг на кг массы тела субъекта на каждое введение, и такая терапия может продолжаться в течение ряда недель или месяцев, а в некоторых случаях и лет. Однако следует иметь в виду, что конкретный размер дозы для любого конкретного пациента зависит от ряда факторов, включая активность применяемого соединения; возраст, массу тела, общее состояние здоровья, пол и диету индивидуального пациента; время и способ введения; скорость выделения; другие лекарства, введенные ранее; а также тяжесть болезни, против которой проводится данное лечение, что хорошо понятно специалистам в данной области техники.
Типичная дозировка может представлять собой одну таблетку от 10 до около 1500 мг, принимаемую раз в день или несколько раз в день, или одну капсулу с медленным высвобождением активного компонента или таблетку, принимаемую раз в день и содержащую пропорционально более высокое количество активного компонента. Эффект высвобождения активного компонента с задержкой во времени может быть достигнут с помощью использования материалов капсулы, которые растворяются при разных значениях рН, с помощью капсул, медленно высвобождающих содержащиеся в них компоненты под действием осмотического давления, или с помощью других известных способов контролируемого высвобождения активного компонента.
В некоторых случаях может оказаться необходимым использовать дозировки, находящиеся за пределами указанного интервала, что хорошо понятно специалистам в данной области техники. Кроме того, следует отметить, что клиницисту или лечащему врачу известно, как и когда следует прервать, откорректировать или прекратить терапию в связи с индивидуальной реакцией пациента.
(5) Защитные группы
Для некоторых соединений, имеющих общую структуру формулы I или II, может требоваться использование защитных групп, чтобы осуществить их успешную доработку с получением желаемой структуры. Защитные группы можно выбрать согласно Greene, T.W. et al., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 1991. Защитные группы легко удаляемы, т.е. при желании их можно удалить с помощью способов, не вызывающих расщепление или другое нарушение остальных частей молекулы. Такие способы включают химический и ферментативный гидролиз, обработку химическими восстановителями или окислителями в мягких условиях, обработку ионами фтора, обработку представляющим собой переходный металл катализатором и нуклеофилом, а также каталитическое гидрирование.
Примерами подходящих гидроксильных защитных групп являются: триметилсилил, триэтилсилил, о-нитробензилоксикарбонил, р-нитробензилоксикарбонил, t-бутилдифенилсилил, t-бутилдиметилсилил, бензилоксикарбонил, t-бутилоксикарбонил, 2,2,2-трихлорэтилоксикарбонил и аллилоксикарбонил. Примерами подходящих карбоксильных защитных групп являются бензгидрил, о-нитробензил, р-нитробензил, 2-нафтилметил, аллил, 2-хлораллил, бензил, 2,2,2-трихлорэтил, триметилсилил, t-бутилдиметилсилил, t-бутилдифенилсилил, 2-(триметилсилил)этил, фенацил, р-метоксибензил, ацетонил, р-метоксифенил, 4-пиридилметил и t-бутил.
(6) Способ получения
Способы получения соединений по настоящему изобретению в общем виде изображены на схемах 1 и 2 (и ниже описаны в примерах):
Схема 1:
Схема 2:
В соответствии со схемой 1, замещенный фенилацетонитрил преобразуют в замещенную фенилуксусную кислоту. Замещенную фенилуксусную кислоту преобразуют до активированного производного кислоты (например, хлорангидрида кислоты) с последующей галогенизацией у альфа-углерода и эстерификацией спиртом. Галогенированный сложный эфир обрабатывают замещенным фенолом (например, 3-трифторметилфенолом), получив в результате ариловый эфир, который гидролизуют, получив производное карбоновой кислоты. Производное кислоты преобразуют в активированное производное кислоты, а затем обрабатывают нуклеофилом (например, N-ацетилэтаноламином) с целью получения желаемого продукта.
В соответствии со схемой 2, замещенную фенилуксусную кислоту преобразуют до активированного производного кислоты (например, хлорангидрида кислоты), с последующей галогенизацией у альфа-углерода. Проводят реакцию активированной кислотной части молекулы с нуклеофилом (например, N-ацетилэтаноламином), с получением защищенной кислоты. Галогенированную защищенную кислоту обрабатывают замещенным фенолом (например, 3-трифторметилфенолом), получив в результате желаемый продукт.
Стереоизомеры соединений по настоящему изобретению можно получить с помощью реагентов или катализаторов в их единственной энантиомерной форме, в любом возможном месте описанного процесса или путем повторного растворения смеси стереоизомеров с помощью обычных способов, описанных выше и в примерах. Некоторые из предпочтительных способов включают использование микробного повторного растворения, повторного растворения диастереомерных солей, образованных с хиральными кислотами или хиральными основаниями, и хроматографию с использованием хиральных носителей.
(7) Наборы
Кроме того, настоящее изобретение касается наборов со стандартными дозами соединений формулы I, формулы II или формулы III, в виде дозировок либо для перорального введения, либо для инъекций. Кроме контейнеров, содержащих стандартные дозы, в наборе имеется вкладыш с информационным пакетом, в котором описаны использование и преимущества данных лекарств для лечения симптомов и/или осложнений, связанных с диабетом II типа, а также для лечения гиперлипидемии и гиперурикемии. Предпочтительными соединениями и стандартными дозами являются описанные выше в настоящей заявке.
ПРИМЕРЫ
Соединения формулы I, формулы II или формулы III по настоящему изобретению можно легко получить с помощью способа, представленного выше в схеме 1 и проиллюстрированного в нижеследующих примерах.
ПРИМЕР 1
Этот пример касается получения метилбром-(4-хлорфенил)ацетата.
Исходное соединение, указанное в схеме 1, т.е. 4-хлорфенилуксусную кислоту, легко получить из нескольких коммерческих источников (например, в фирмах Aldrich и Fluka).
5-литровый реактор Morton, оснащенный магнитной мешалкой, устройством для регулирования температуры в резервуаре и дополнительной воронкой, провентилировали с помощью газоочистителя и загрузили в него р-хлорфенилуксусную кислоту (720 г, 4,2 моль) и SOCl2 (390 мл, 630 г, 5,3 моль). Реакционную смесь перемешивали, нагревали и выдерживали при температуре 55°С±5°С в течение 1 часа. Затем в течение 20 минут добавляли бром (220 мл, 670 г, 5,3 моль) и перемешивали при температуре 55°С±5°С в течение 16 часов. Температуру поднимали до 80°С в течение 7 часов, а затем понижали до 9°С на ледяной бане. Затем осторожно добавляли метанол (2,0 л, 1,6 кг, 49,4 моль). Растворитель отгоняли, получив две жидкости весом 1,28 кг. Их растворяли в смеси 0,84 л воды и 2,1 л эфира и разделяли. Органическую фазу однократно промывали 0,78 л 25%-ного (масс.) водного раствора NaCl и сушили над 0,13 кг MgSO4. Полученную смесь фильтровали через фильтровальную бумагу Whatman #1 и отгоняли растворитель, получив 0,985 кг оранжевой жидкости. Анализ методом протонного ЯМР показал, что эта жидкость состояла на 80% из продукта и на 19% из небромированного сложного эфира. Анализ методом ВЭЖХ показал наличие 82% продукта и 18% небромированного сложного эфира. Анализ методом ВЭЖХ проводили на колонке Zorbax S8-C8 при температуре 30°С, при размере колонки 250×4,6 мм и размере частиц 5 мкм. Мобильная фаза представляла собой ацетонитрил: 0,1% H3PO4 в соотношении 60:40 (по объему) при 1,5 мл/мин. Определение проводили при 210 нм. Введенный образец объемом в 1 мкл растворяли в ацетонитриле в концентрации 10 мг/мл. Время удерживания для продукта составило 5,0 мин, а для небромированного сложного эфира - 3,8 мин. Этот сырой продукт очищали путем вакуумной перегонки, получив в результате 96% чистого продукта с выходом 84%. Анализ продукта с помощью протонного ЯМР (CDCl3, 300 МГц) показал сдвиги при 3,79 (s, 3Н), 5,32 (s, 1H) и 7,20-7,55 (m, 4H).
ПРИМЕР 2
Этот пример касается получения метил-(4-хлорфенил)-(3-трифторметилфенокси)ацетата.
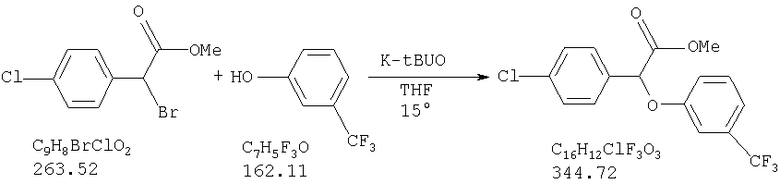
Эта стадия была подобна аналогичной стадии, описанной в патенте США №3517050, за одним исключением, состоящим в том, что вместо натрийметоксида использовали калий-t-бутоксид, чтобы не допустить образования соответствующего метилового эфира. В 5-литровый реактор Morton, оснащенный верхней мешалкой, устройством для замеров температуры в резервуаре и дополнительной воронкой, в атмосфере азота загружали метилбром-(4-хлорфенил) ацетат (830 г, 3,0 моль) и THF (600 мл). Реактор охлаждали до температуры 14°С±3°С на ледяной бане, а затем добавляли подобным же образом охлажденный раствор трифторметил-m-крезола (530 г, 3,3 моль) в 1,0 М калий-1-бутоксиде в THF (3,1 л, 3,1 моль). Реакция проходила экзотермически с типичным подъемом температуры свыше 25°С; добавление реагента контролировали таким образом, чтобы поддерживать температуру на уровне 15°±2°С, и реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. Анализ методом ВЭЖХ проводили на колонке Zorbax S8-C8 при температуре 30°С, при размере колонки 250×4,6 мм и размере частиц 5 мкм. Мобильная фаза представляла собой ацетонитрил: 0,1% Н3PO4 в соотношении 60:40 (по объему) при 1,5 мл/мин. Определение проводили при 210 нм. Введенный образец объемом в 1 мкл растворяли в ацетонитриле в концентрации 10 мг/мл. Время удерживания для продукта составило 9,6 мин, исходный сложный эфир элюировал через 5,0 мин, фенол через 3,0 мин, а небромированный сложный эфир - через 3,8 мин. Растворитель отгоняли с помощью ротационного испарителя, получив желтую вязкую массу, которую растворяли в смеси 4,0 л воды и 12,0 л эфира. Смесь разделяли, органическую фазу однократно промывали 1,6 л 5%-ного (масс.) водного NaOH, a затем 1,6 л воды, и наконец, 1,6 л 25%-ного (масс.) водного NaOH. Органическую фазу сушили над 0,32 кг MgSo4 и фильтровали через фильтровальную бумагу Whatman #1. Отгоняли растворитель, получив 1,0 кг влажных грязно-белых кристаллов. Эти кристаллы подвергали перекристаллизации на ротационном испарителе путем растворения в 1,0 л метилциклогексана при температуре 75°С, с последующим охлаждением до 20°С. Кристаллы фильтровали через фильтровальную бумагу Whatman #1 и промывали тремя порциями по 0,25 л прохладного (15°С) метилциклогексана. Влажный продукт (0,97 кг) сушили до следующего дня, получив в результате 0,81 кг продукта 98%-ной чистоты, что соответствует выходу в 79%. Анализ продукта с помощью протонного ЯМР (CDCl3 300 МГц) показал сдвиги при 3,75 (s, 3Н), 5,63 (s, 1H) и 7,05-7,55 (m, 8H).
ПРИМЕР 3
Этот пример касается получения 4-хлорфенил-(3-трифторметилфенокси)уксусной кислоты.
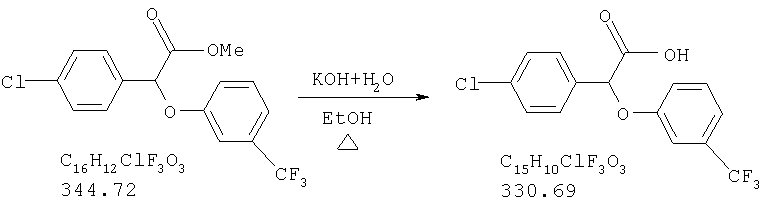
В 12-литровый реактор Morton, оснащенный магнитной мешалкой, устройством для регулирования температуры в резервуаре и парциальным конденсатором горячего орошения, в атмосфере азота загружали метил-4-хлорфенил(3-трифторметилфенокси)ацетат (810 г, 2,3 моль) и абсолютный этанол (5,8 л) и при помешивании нагревали до температуры 57°С, чтобы растворить твердое вещество. Добавляли раствор КОН (520 г, 9,3 моль) в 0,98 л воды. Раствор перегоняли в течение 30 минут, а растворитель отгоняли с помощью ротационного испарителя, получив 2,03 кг смеси двух почти бесцветных жидкостей. Эти жидкости растворяли в воде (16 л) и обрабатывали 16 г нейтрального Norit, затем фильтровали через слой диатомовой земли, оставшийся на фильтровальной бумаге Whatman #1. рН фильтрата понижали от исходного значения 13 до значения от 1 до 2, путем добавления в сумме 2,75 л 3М HCl (8,25 моль). После добавления первых 2,30 л кислоты образовывалось очень клейкое твердое вещество, и в этот момент добавляли эфир (7 л). Разделяли два слоя и органический слой сушили над MgSo4 (230 г) и фильтровали через фильтровальную бумагу Whatman #1. Затем растворитель отгоняли, получив 0,85 кг бесцветного сиропа. Затем материал перекристаллизовали на ротационном испарителе путем добавления метилциклогексана (800 мл) и охлаждения до 18°С при медленном вращении. После этого температуру понижали до 5°С, кристаллы отфильтровали и промывали пять раз порциями по 0,10 л холодного (0°С) метилциклогексана, получив 0,59 кг влажных кристаллов. Влажные кристаллы сушили, получив в результате 0,48 кг продукта (выход 62%), в котором не имелось обнаруживаемых с помощью протонного ЯМР количеств р-хлорфенилуксусной кислоты. Анализ продукта методом протонного ЯМР (CDCl3, 300 МГц) показал сдвиги при 5,65 (s, 1H), 7,02-7,58 (m, 8H) и 10,6 (s, 1H).
ПРИМЕР 4
Этот пример касается получения 4-хлорфенил-(3-трифторметилфенокси)уксусной кислоты.
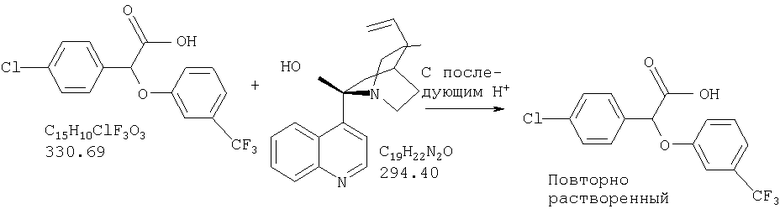
В открытый вверху 12-литровый реактор Morton с верхней мешалкой загружали 4-хлорфенил-(3-трифторметилфенокси)уксусную кислоту (350 г, 1,06 моль) и изопропанол (4,0 л) и нагревали до 65°±3°С. Добавляли взвесь (-)цинхонидина (300 г, 1,02 моль) в изопропаноле (2,0 л), споласкивая все твердые частицы в реактор дополнительной порцией изопропанола (0,8 л). Температура понижалась от 65° до 55°С и сразу образовывался прозрачный оранжевый раствор, после чего полученную смесь выдерживали при 55°±5°С в течение 2 часов. Мелкие кристаллы собирали с помощью фильтрации через фильтровальную бумагу Whatman#1, с однократной промывкой 0,7 л горячего (55°С) изопропанола. Кристаллы сушили в течение 16 часов при температуре окружающей среды в 12,6-литровой вакуумной печи, в потоке азоте в 5 л/мин. Получили 0,37 кг сухого твердого вещества, которое имело 80%-ный избыток ("ее") (+)энантоимера. Избыток энантиомера определяли с помощью ВЭЖХ, с использованием колонки R,R-WhelkO-1 размером 250×4,6 мм, при температуре окружающей среды. Введенные образцы представляли собой аликвоты в 20 мкл растворов образцов в этаноле в концентрации 2 мг/мл. Для элюирования колонки использовали гексан:изопропанол:уксусную кислоту в соотношении 95:5:0,4, при величине расхода 1 мл/мин. Определение осуществляли при 210 нм. (+)Энантиомер элюировал через 7-8 мин, а (-)энантиомер через 11-13 минут. В маточном растворе выпадал вторичный осадок, который почти сразу же фильтровали, промывали и сушили/ получив 0,06 кг соли, которая имела 90% "ее" (-)энантиомера. Подобным же образом получали третий, четвертый и пятый осадки, с массой 0,03 кг, 0,03 кг и 0,7 кг соответственно; при этом избытки (-)энантиомера составляли 88%, 89% и 92% соответственно.
Сырую (+)соль (320 г) перекристаллизовали из смеси этанола (5,9 л) и метанола (1,2 л). Смесь нагревали, перемешивали с помощью верхней мешалки для растворения, охлаждали при температуре окружающей среды в течение 16 часов, фильтровали и дважды промывали 0,20 л смеси этанол:метанол (5:1, по объему). Кристаллы сушили, получив 0,24 кг (+)энантиомера с показателем "ее" в 97%. Это соответствовало 80%-ному выходу этого изомера. Повторно растворенную соль суспендировали в смеси эфира (6,5 л) и воды (4,0 л), перемешивая верхней мешалкой. Величину рН понижали до 0-1, что определяли с помощью полосок индикаторной бумаги, используя раствор концентрированной Н2SO4 (0,13 л) в воде (2,5 л). Фазы разделяли и органическую фазу дважды промывали порциями воды по 6,5 л. Добавляли эфир (1,9 л) и органический слой промывали еще раз водой (6,5 л). После последнего разделения добавляли 0,1 л 25%-го (масс.) водного раствора NaCl, чтобы удалить малейшие следы эмульсии. Продукт сушили над MgSo4 (0,19 кг), фильтровали и удаляли растворитель, получив 0,13 кг бесцветного сиропа, который отверждали. Это соответствовало 97%-ному выходу продукта, который имел 95% "ее" (+)энантиомера, [α]D+5,814° (с. 0,069 в метиловом спирте).
Объединенную сырую (-)соль (200 г) перекристаллизовали из изопропанола (3,1 л). Смесь нагревали, чтобы растворить почти все твердые вещества, и быстро фильтровали, чтобы удалить нерастворимые твердые вещества. Затем смесь охлаждали с перемешиванием при температуре окружающей среды в течение 16 часов, фильтровали и сушили, получив 0,16 кг (-)энантиомера с показателем "ее" в 97%. Это соответствовало выходу этого изомера в 49%. (-)Энантиомер кислоты выделяли так же, как описано выше для (+)кислоты. Растворенную соль суспендировали в эфире и воде, рН понижали с помощью концентрированной Н2SO4 и продукт экстрагировали в органической фазе.
ПРИМЕР 5
А. Получение (-)4-хлорфенил-(3-трифторметилфенокси)-ацетилхлорида.

В 2-литровую испарительную колбу с магнитной мешалкой, переходником Клайссена, термометром для замера температуры в резервуаре и парциальным конденсатором горячего орошения, соединенным с газоочистителем, загружали (-)4-хлорфенил-(3-трифторметилфенокси)уксусную кислоту (143 г, 0,42 моль, в расчете на 97%-ную чистоту) и CHCl3 (170 мл), и нагревали до кипения, чтобы растворить. Добавляли SOCl3 (38 мл, 62,1 г, 0,52 моль). Смесь нагревали до температуры перегонки (конечная температура 68°C) в течение 4,5 часов, а затем отгоняли летучие вещества, получив 151 г желтой мутной жидкости (видимый выход 103%). Полученный материал использовали для следующей стадии без дальнейшей очистки.
В. Получение (+)4-хлорфенил-(3-трифторметилфенокси)-ацетилхлорида.
В 3-литровую испарительную колбу с магнитной мешалкой, переходником Клайссена, термометром для замера температуры в резервуаре и парциальным конденсатором горячего орошения, соединенным с газоочистителем, загружали (+)4-хлорфенил-(3-трифторметилфенокси)уксусную кислоту (131 г, 0,37 моль) и CHCl3 (152 мл) и нагревали до кипения, чтобы растворить. Добавляли SOCl3 (35 мл, 56,5 г, 0,48 моль). Смесь нагревали до температуры перегонки (окончательная температура 70°С) в течение 4 часов, а затем отгоняли летучие вещества, получив 139 г жидкости. Полученный материал использовали для следующей стадии без дальнейшей очистки.
ПРИМЕР 6
А. Получение (-)2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетата.
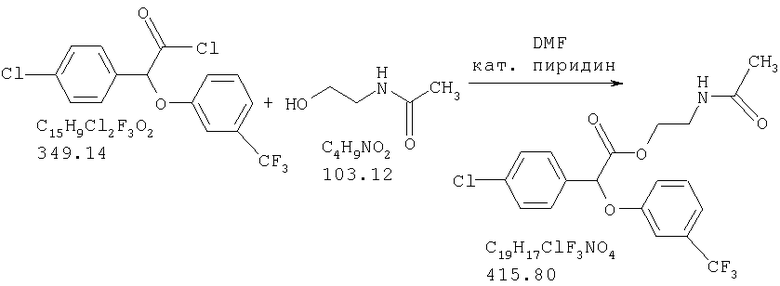
В 3-литровую круглодонную колбу с магнитной мешалкой и термометром для замера температуры в резервуаре в атмосфере азота и на ледяной бане загружали DMF (430 мл), пиридин (37 мл, 36 г, 0,46 моль) и N-ацетоэтаноламин (39 мл, 43 г, 0,42 моль). Смесь охлаждали до температуры от 0° до 5°С и в течение 40 минут добавляли раствор сырого (-)4-хлорфенил-(3-трифторметилфенокси)-ацетилхлорида (151 г, 0,42 моль, в расчете на 100% выход на предыдущем этапе) в эфире (170 мл), так, чтобы поддерживать температуру в резервуаре ниже 13°С. Смесь перемешивали при температуре окружающей среды в течение 16 часов и растворяли путем добавления воды (960 мл), а затем этилацетата (630 мл). Добавление воды приводило к экзотермическому процессу, с подъемом температуры от 24° до 34°С. Добавление этилацетата вызывало падение температуры до 30°С. Слои разделяли и водную фазу однократно экстрагировали этилацетатом (125 мл). Объединенные органические слои один раз экстрагировали 7% (масс.) водным раствором NaHCO3 (125 мл) и пять раз порциями по 60 мл воды, а затем два раза порциями по 60 мл 25% (масс.) водного раствора NaCl. Продукт сушили над MgSo4 (42 г) и фильтровали через фильтровальную бумагу Whatman #1. Растворитель отгоняли с помощью ротационного испарителя, получив 160 г желтого сиропа, что соответствовало 80% выходу, рассчитанному на основе данных протонного ЯМР, которые показали наличие 87% продукта, 8% EtOAc, 4% не бромированного амида и 1% DMF. Этот сироп растворяли в МТВЕ (225 мл) при температуре окружающей среды и охлаждали (до -15°С). Добавляли 85% гексаны (400 мл), при помешивании. Образовались две жидкости, затем кристаллы, а затем смесь превращалась в твердое вещество. Твердую массу соскребали в воронку Бюхнера с фильтровальной бумагой Whatman #1, уплотняли и трижды промывали порциями по 100 мл смеси МТВЕ:гексаны 1:1 (по объему), получив 312 г сырого продукта, вес которого после сушки составил 127 г, что соответствовало выходу в 73%.
В. Получение (+)2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетата.
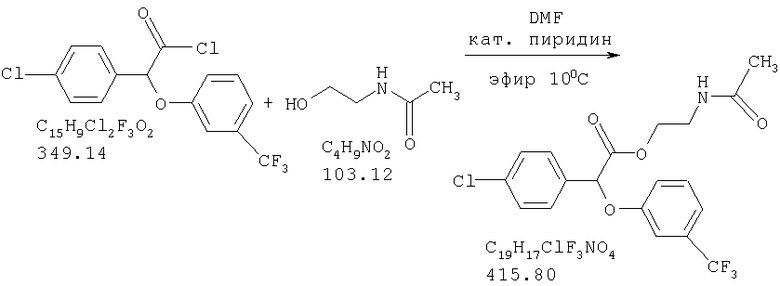
В 3-литровую круглодонную колбу с магнитной мешалкой и термометром для замера температуры в резервуаре, в атмосфере азота и на ледяной бане загружали DMF (365 мл), пиридин (33 мл, 32,3 г, 0,41 моль) и N-ацетоэтаноламин (34 мл, 38,1 г, 0,37 моль). Смесь охлаждали до температуры от 0° до 5°С и в течение 25 минут добавляли раствор сырого (+)4-хлорфенил-(3-трифторметилфенокси)ацетилхлорида (139 г, 0,37 моль, в расчете на 100% выход на предыдущем этапе) в эфире (155 мл), так, чтобы поддерживать температуру в резервуаре ниже 13°С. Смесь перемешивали при температуре окружающей среды в течение 40 часов и растворяли путем добавления воды (850 мл), а затем этилацетата (550 мл). Добавление воды приводило к экзотермическому процессу, с подъемом температуры от 24° до 34°С. Добавление этилацетата привело к падению температуры до 30°С. Слои разделяли и водную фазу однократно экстрагировали этилацетатом (110 мл). Объединенные органические слои два раза промывали порциями по 55 мл воды, а затем пять раз порциями по 55 мл 25% (масс.) водного раствора NaCl, продукт сушили над MgSo4 (30 г) и фильтровали через фильтровальную бумагу Whatman #1. Растворитель отгоняли с помощью ротационного испарителя, получив 168 г желтой жидкости, что соответствовало 86%-ному выходу, рассчитанному на основе данных протонного ЯМР, которые показали наличие 79% продукта, 9% EtOAc, 8% не бромированного амида и 4% DMF. Продукт кристаллизовали в химическом стакане объемом 800 мл путем растворения в МТВЕ (200 мл) при температуре окружающей среды, охлаждения при -15°С в течение 1,4 часа, добавления 85% гексанов (200 мл) и охлаждения в течение 1 часа. Твердую массу соскребали в воронку Бюхнера с фильтровальной бумагой Whatman #1, уплотняли и однократно промывали 100 мл смеси МТВЕ: гексаны 1:1 (по объему), получив 201 г сырого продукта. Продукт сушили в потоке азота и растирали с 85% гексанами (700 мл), используя верхнюю мешалку. Полученный материал фильтровали и сушили, получив 87 г продукта. [α]D+2,769° (с.=0,048 в метиловом спирте). [α]D-2,716° (с.=0,049 в метиловом спирте). Полученные (+) и (-)энантиомеры анализировали также методом ВЭЖХ, используя колонку R,R-WhelkO-1 размером 250×4,6 мм, при температуре окружающей среды. Введенные образцы представляли собой аликвоты по 20 мкл растворов образцов в этаноле с концентрацией 2 мг/мл. Для элюирования колонки использовали изопропанол:гексан в соотношении 60:40, при величине расхода 1 мл/мин. Определение осуществляли при 220 нм. (+)Энантиомер элюировал через 5,0-5,2 мин, а (-)энантиомер - через 5,7-5,9 минут.
ПРИМЕР 7
Данный пример касается ингибирования цитохрома Р450 2С9 (CYP2C9) соединениями по настоящему изобретению.
Активность гидроксилирования толбутамида (100 мкМ 14С-толбутамида; 1 мМ NADPH) анализировали в пуле микросом печени человека (0,6 мг белка/мл) в течение 60 минут при 37°С как с испытуемыми соединениями, так и без них. Испытывали рацемическую галофеновую кислоту, (-)галофеновую кислоту и (+)галофеновую кислоту (в концентрациях от 0,25 мкМ до 40 мкМ). Как показано на фигуре 1, рацемическая галофеновая кислота ингибировала опосредованную CYP2C9 активность гидроксилирования толбутамида в микросомах печени человека, с видимой ИК50 в 0,45 мкМ. Наблюдались существенные различия в способности энантиомеров галофеновой кислоты ингибировать CYP2C9. (+)Галофеновая кислота имела видимую ИК50 в 0,22 мкМ, в то время как (-)галофеновая кислота оказывала почти в 20 раз меньшее действие, с видимой ИК50 в 3,6 мкМ.
ПРИМЕР 8
Данный пример касается зависимости процесса понижения содержания глюкозы от времени для соединений по настоящему изобретению.
А. Материал и методы
Самцов мышей в возрасте 9-10 недель, линии C57BL/6J ob/ob, приобрели в The Jackson laboratory (Bar Harbor, ME, USA). Животных поместили в стандартные лабораторные условия, по 4-5 мышей на клетку, при температуре 22°С и относительной влажности в 50%, и содержали при кормлении кормом для грызунов Пурина и поении водой ad libitum (по желанию). Перед началом эксперимента у каждого животного брали кровь из хвостовой вены. Использовали мышей, у которых содержание глюкозы в плазме крови (без голодания) составляло от 300 до 500 мг/дл. Для каждого варианта использовали группу мышей из 10 особей, которые были распределены по группам таким образом, чтобы средние уровни содержания глюкозы в начале исследования были эквивалентными в каждой группе. Мышам однократно вводили перорально через желудочный зонд одно из нижеследующего: носитель, рацемический галофенат (250 мг/кг), (-)галофенат (250 мг/кг) или (+)галофенат (250 мг/кг). Все соединения вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 2,7% масс./об. метилцеллюлозы. Объем желудочного зонда составлял 10 мл/кг. Образцы крови брали через 1,5, 3, 4,5, 6, 7,5, 9 и 24 часа после введения дозы и анализировали их на содержание глюкозы. Концентрации глюкозы в плазме крови определяли колориметрически с помощью глюкозооксидазного метода (Sigma Chemical Co., St. Louis, МО, USA). Значимость различия между группами (сравнение между группами, получавшими лекарства и носитель, или между группами, получавшими разные варианты лекарства) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Как показано на фигуре 2, рацемический галофенат существенно понижал концентрации глюкозы в плазме крови в большинстве точек времени, с пиком активности в точке 9 часов. (-)Галофенат продемонстрировал понижение концентрации глюкозы в плазме крови уже через 1,5 часа и достиг пика активности в точке 3 часа. Концентрации глюкозы в плазме крови оставались низкими вплоть до 24 часов. (+)Галофенат не продемонстрировал существенной активности до 4,5 часов, а пик его активности был в точке 7,5 часов. После этого концентрация глюкозы в плазме крови начала восстанавливаться. Наблюдались значимые различия между (-) и (+)энантиомерами галофената в точках времени 3 и 24 часа. Активность (-)галофената наступала быстрее и сохранялась более продолжительное время.
ПРИМЕР 9
Данный пример касается активности соединений по настоящему изобретению в отношении понижения содержания глюкозы.
А. Материалы и методы
Самцов мышей в возрасте 8-9 недель, линии C57BL/6J ob/ob, приобрели в The Jackson Laboratory (Bar Harbor, ME, USA). Животных поместили в стандартные лабораторные условия, по 4-5 мышей на клетку, при температуре 22°С и относительной влажности в 50% и содержали при кормлении кормом для грызунов Пурина и поении водой ad libitum (по желанию). Перед началом эксперимента у каждого животного брали кровь из хвостовой вены. Использовали мышей, у которых содержание глюкозы в плазме крови в отсутствие голодания составляло от 300 до 520 мг/дл. Для каждого варианта использовали группу мышей из 10 особей, которые были распределены по группам таким образом, чтобы средние уровни содержания глюкозы в начале исследования были эквивалентными в каждой группе. Мышам один раз в день, в течение 5 дней вводили перорально через желудочный зонд одно из нижеследующего: носитель, рацемический галофенат (250 мг/кг), (-)галофенат (125 и 250 мг/кг) или (+)галофенат (125 и 250 мг/кг). Рацемический галофенат вводили в 2,7% масс./об. метилцеллюлозы, а оба энантиомера - (-) и (+)энантиомеры - вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 2,7% масс./об. метилцеллюлозы. Объем желудочного зонда составлял 10 мл/кг. Образцы крови брали через 3, 6, 27, 30 и 120 часов после введения первой дозы и анализировали их на содержание глюкозы и инсулина. Животные голодали в течение ночи (14 часов) перед забором образцов, осуществлявшимся через 120 часов. Концентрации глюкозы в плазме крови определяли колориметрически с помощью глюкозооксидазного метода (Sigma Chemical Co., St. Louis, МО, USA). Концентрации инсулина в плазме крови определяли колориметрически с помощью набора для определения содержания инсулина у грызунов Rat Insulin RIA Kit из Linco Research Inc. (St. Charles, МО, USA). Значимость различия между группами (при сравнении между группами, получавшими лекарства и получавшими носитель) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Как показано на фигуре 3, (-)галофенат существенно понижал концентрации глюкозы в плазме крови в точках времени 6, 27 и 30 часов. (-)Галофенат в обоих дозах существенно понижал концентрации глюкозы в плазме крови в точках времени 6, 27 и 30 часов. Высокая доза (250 мг/кг) была активна также и в точке 3 часа. (+)Галофенат в дозе 125 мг/кг продемонстрировал понижение концентрации глюкозы в плазме крови в точках времени 6 и 27 часов, в то время как в дозе 250 мг/кг пониженные концентрации глюкозы в плазме крови наблюдались в точках времени 3, 6, 27 и 30 часов. Уровни содержания инсулина в плазме крови показаны на фигуре 4. Рацемический галофенат существенно понижал содержание инсулина в точках 6 и 27 часов. Содержание инсулина было значительно пониженным в группе с введением (-)галофената в точке 27 часов при обеих применявшихся дозах, и было существенно пониженным в точке 30 часов у животных, получавших 250 мг/кг/день. (+)Галофенат существенно понижал содержание инсулина в точках 27 и 30 часов при обеих применявшихся дозах. При дозе 125 мг/кг/день существенное понижение наблюдалось также спустя б часов. После голодания в течение ночи (в точке времени 120 часов) на всех вариантах наблюдались существенно уменьшенные концентрации глюкозы в плазме крови (фигура 5). Содержание инсулина было существенно уменьшенным во всех группах, получавших галофенат, за исключением группы, получавшей (+)галофенат в дозе 125 мг/кг/день (фигура 6).
ПРИМЕР 10
Данный пример касается улучшения показателей резистентности к инсулину и ослабленной толерантности к глюкозе для соединений по настоящему изобретению.
А. Материалы и методы
Самцов крыс в возрасте 8-9 недель, линии Zucker fa/fa (Charles River), поместили в стандартные лабораторные условия (по 2-3 крысы на клетку) при температуре 22°С и относительной влажности в 50% и содержали при кормлении кормом для грызунов Пурина и поении водой ad libitum. Перед началом эксперимента крыс разбили на 6 групп по массе тела. Группа для каждого варианта эксперимента состояла из 8 крыс. Крысам однократно вводили перорально через желудочный зонд одно из нижеследующего: носитель, рацемический галофенат (100 мг/кг), (-)галофенат (50 или 100 мг/кг) или (+)галофенат (50 или 100 мг/кг). Все соединения вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 2,7% масс./об. метилцеллюлозы. Объем желудочного зонда составлял 10 мл/кг. Все крысы получали контрольное пероральное введение глюкозы (1,9 г/кг) через 5,5 часов после введения соединения и через 4 часа после отмены пищи. Образцы крови брали через 0, 15, 30, 60, 90, 120 и 180 минут после контрольного введения глюкозы и анализировали их на содержание глюкозы. Группы, получавшие носитель, (-)галофенат (50 мг/кг) и (+)галофенат (50 мг/кг), подвергали контрольному введению инсулина после ежедневного введения через желудочный зонд соответствующих соединений, продолжавшегося в течение 5 дней. На 5-й день крысам внутривенно вводили инсулин (0,75 Е/кг) через 5,5 часов после последней дозы соединения и через 4 часа после отмены выдачи пищи. Образцы крови брали через 3, 6, 9, 12, 15 и 18 минут после инъекции инсулина и анализировали их на содержание глюкозы в плазме. Концентрации глюкозы в плазме крови определяли колориметрически с помощью глюкозооксидазного метода (Sigma Chemical Co., St. Louis, МО, USA). Значимость различия между группами (при сравнении между группами, получавшими лекарства и получавшими носитель, или между группами, получавшими разные варианты лекарств) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Как показано на фигуре 7А, крысы линии Zucker fatty с ослабленной толерантностью к глюкозе имели пониженные уровни содержания глюкозы в плазме крови после контрольного введения глюкозы вслед за введением галофената. Наиболее эффективным для снижения содержания глюкозы был (-)галофенат, и его эффект сохранялся более продолжительное время, по сравнению с рацематом или (+)энантиомером. Фигура 7В показывает площадь прироста под кривой (AUC) для групп всех вариантов эксперимента. Животные, получавшие (-)галофенат, продемонстрировали существенное уменьшение площади, характеризующей содержание глюкозы, по сравнению с контрольными животными, получавшими носитель. Хотя AUC была уменьшенной и в группах, получавших рацемат или (+)галофенат, но их эффект был не столь велик, как у крыс, получавших (-)галофенат, и наблюдавшиеся отличия были статистически незначимыми.
Изменения в чувствительности к инсулину измеряли путем отслеживания падения содержания глюкозы после внутривенного введения инсулина. Наклон кривой является прямым указанием на чувствительность к инсулину у подопытных животных. Как показано на фигуре 8, чувствительность к инсулину была существенно повышена через 5 дней после лечения (-)галофенатом, по сравнению с контрольными животными, получавшими носитель (р<0,01) и с животными, получавшими (+)галофенат (р<0,05). Лечение (+)галофенатом не оказывало большого влияния на чувствительность к инсулину, которая не имела значимых отличий от этого показателя контрольных животных, получавших носитель (р=0,083). Лечение (-)галофенатом существенно уменьшило резистентность к инсулину у крыс линии Zucker fatty, которая представляет собой принятую модель ослабленной толерантности к инсулину и резистентности к инсулину.
ПРИМЕР 11
Данный пример касается активности соединений по настоящему изобретению в отношении понижения содержания липидов.
А. Материалы и методы
Самцов крыс линии Zucker diabetic fatty (ZDF) получили в GMI Laboratories (Indianopolis, IN) в возрасте 9 недель. Крысам ежедневно перорально через желудочный зонд вводили носитель или энантоимеры галоофената, начиная с возраста 74 дней. Исходные образцы крови для анализа получали за один день до введения соединений и затем в определенные моменты времени, указанные в схеме эксперимента. Кровь анализировали на содержание в плазме триглицеридов и холестерина с помощью стандартных методов.
В. Результаты
В эксперименте I животные получали дозу 25 мг/кг/день. Как показано на фигурах 9А и 9В, значительное понижение содержания в плазме холестерина было отмечено только у животных, получавших (-)галофенат, после 7 и 13 дней лечения. В эксперименте II животные в возрасте 107 дней получали ежедневные дозы по 12,5 мг/кг/день или 37,5 мг/кг/день (-) и (+)энантиомеров галофената. Как показано на фигуре 10А и фигуре 10В, содержание в плазме холестерина было существенно ниже в варианте с высокой дозой после 7 дней, но не после 14 дней лечения (+)галофенатом. В противоположность этому, для (-)галофената в низкой дозе наблюдалось существенное понижение содержания холестерина после 7 дней. При высокой дозе наблюдалось значительно большее понижение содержания в плазме холестерина, как после 7, так и после 14 дней лечения. Как показано на фигуре 11А и фигуре 11В, значительное понижение содержания в плазме триглицеридов также наблюдали через 7 дней после лечения высокой дозой, особенно у животных, получавших (-)энантиомер галофената.
ПРИМЕР 12
Этот пример касается активности аналогов (±)галофената и аналогов (-)галофената в отношении понижения содержания глюкозы.
А. Материалы и методы
Самцов мышей в возрасте 8-9 недель, линии C57BL/6J ob/ob, приобрели в The Jackson Laboratory (Bar Harbor, ME, USA). Животных поместили в стандартные лабораторные условия (по 4-5 мышей на клетку) при температуре 22±3°С и относительной влажности в 50±20% и содержали при кормлении кормом для грызунов Пурина и поении водой ad libitum. Перед началом эксперимента у каждого животного брали кровь из хвостовой вены. Использовали мышей, у которых содержание глюкозы в плазме крови без голодания составляло от 250 до 500 мг/дл. Для каждого варианта использовали группу мышей из 8-10 особей, которые были распределены по группам таким образом, чтобы средние уровни содержания глюкозы в начале исследования были эквивалентными в каждой группе. Мышам один раз в день в течение 1-3 дней вводили перорально через желудочный зонд одно из нижеследующего: носитель, (-)галофеновую кислоту, (±)аналог 14, 29, 33, 34, 35, 36, 37 или 38 в дозе 125 мг/кг или (-)аналог 29, 36, 37 или 38 в дозе 150 мг/кг. Соединения вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 0,9% масс./об. метилцеллюлозы. Объем желудочного зонда составлял 10 мл/кг. Образцы крови брали через 6 часов после введения каждой дозы и анализировали их на содержание глюкозы в плазме. Поглощение пищи и массу тела измеряли ежедневно. Концентрации глюкозы в плазме крови определяли колориметрически с помощью глюкозооксидазного метода (Sigma Chemical Co., St. Louis, МО, USA). Значимость различия между группами (при сравнении между группами, получавшими лекарства и получавшими носитель) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Как показано в таблице 2, соединения оценивали в 5 различных экспериментах. Однократная доза (-)галофеновой кислоты существенно понижала концентрацию глюкозы в плазме крови через 6 часов. Аналог 14 существенно понижал концентрацию глюкозы в плазме крови через 6, 30 и 54 часа. Аналог 33 существенно понижал концентрацию глюкозы в плазме крови через 6 и 54 часа. Аналоги 29 и 38 существенно понижали концентрацию глюкозы в крови через 6, 30 и 54 часа. Аналоги 35 и 36 существенно понижали концентрацию глюкозы в плазме крови через 30 и 54 часа. Аналог 37 существенно понижал концентрацию глюкозы в плазме крови через 54 часа. Однократная доза (-)аналогов 29, 36, 37 и 38 существенно понижала концентрацию глюкозы в плазме крови через 6 часов. Варианты введения различных соединений не оказывали влияния на потребление пищи животными и на массу их тела.
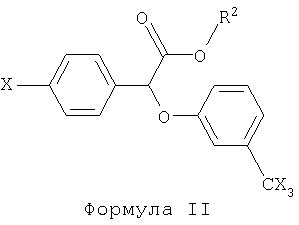
Аналоги (±) и (-)галофената. Соединения описаны со ссылкой на формулу II.
Активность понижения содержания глюкозы аналогами (±)галофената и (-)галофената
ПРИМЕР 13
Этот пример касается сравнения между активностью галофената и (+)галофената.
А. Материалы и методы
Самцов крыс в возрасте 8-9 недель линии ZDF приобрели в фирме Genetic Models, Inc. (Indianapolis, IN). Животных поместили в стандартные лабораторные условия (по 3 крысы на клетку) при температуре 22±3°С и относительной влажности в 50±20% и содержали при кормлении кормом для грызунов Пурина и поении водой ad libitum. Перед началом эксперимента у каждого животного брали кровь из хвостовой вены. Использовали крыс, у которых содержание глюкозы в плазме крови после 4-часового голодания составляло от 200 до 500 мг/дл. Для каждого варианта использовали группу крыс из 8-10 особей, которые были распределены по группам таким образом, чтобы средние уровни содержания глюкозы в начале исследования были эквивалентными в каждой группе. Крысам один раз в день в течение 3 дней вводили перорально через желудочный зонд одно из нижеследующего: носитель, (-)галофенат или (+)галофенат в дозе 50 мг/кг. Соединения вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 0,9% масс./об. метилцеллюлозы. Объем желудочного зонда составлял 5 мл/кг. Образцы крови брали через 5 часов после введения дозы на 2-й и 3-й дни. Концентрации глюкозы в плазме крови определяли колориметрически с помощью глюкозооксидазного метода (Sigma Chemical Co., St. Louis, МО, USA). Значимость различия между группами (при сравнении между группами, получавшими лекарства и получавшими носитель) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Пероральное введение (-)галофената в дозе 50 мг/кг существенно понижало концентрацию глюкозы в плазме крови, в то время, как введение (+)галофената в тех же самых дозах не привело к понижению концентраций глюкозы в плазме крови, по сравнению с животными, получавшими носитель (фигура 12).
ПРИМЕР 14
Этот пример касается фармакокинетического исследования (±)галофената и (-)галофената.
А. Материалы и методы
Самцов крыс массой 225-250 г, линии SD, приобрели в Charles River. Животных поместили в стандартные лабораторные условия (по 3 крысы на клетку) при температуре 22±3°С и относительной влажности в 50±20% и содержали при кормлении кормом для грызунов Пурина и поении водой ad libitum. В левую сонную артерию помещали катетер под обезболиванием с помощью пентобарбитала натрия (50 мг/кг, внутрибрюшинно), после чего животным дали восстановиться в течение 2 дней перед началом эксперимента. Однократную дозу (±)галофената или (-)галофената (50 мг/кг) вводили перорально через желудочный зонд. Соединения вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 0,9% масс./об. метилцеллюлозы. Объем желудочного зонда составлял 5 мл/кг. Образцы крови брали через 1, 2, 4, 6, 8, 12, 24, 48, 72, 96 и 120 часов после введения дозы. Образцы плазмы анализировали на содержание каждой энантиомерной кислоты ((-)галофеновой кислоты и (+)галофеновой кислоты) с помощью хирально-специфического анализа ВЭЖХ, поскольку сложные эфиры являются пролекарствами, предназначенными для преобразования в их соответствующие энантиомерные кислоты in vivo.
В. Результаты
После перорального введения (±)галофената, как (-)галофеновую кислоту, так и (+)галофеновую кислоту обнаружили в образцах плазмы. Как показано в таблице 3, выяснилось, что эти две энантиомерных кислоты имеют разные диспозиционные профили. Элиминация (-)галофеновой кислоты происходила гораздо медленнее, чем (+)галофеновой кислоты. В результате этого AUC для (-)галофеновой кислоты была существенно больше, чем AUC для (+)галофеновой кислоты, а именно, 4708,0 против 758,0 мкг/час/мл, а период полураспада составлял 46,8 часов, против 14,3 часа.
После перорального введения (-)галофената диспозиционный профиль (-)галофеновой кислоты был в основном идентичен введению (±)галофената, поскольку период полураспада был одинаковым (таблица 2). Значения Cmax и AUC (-)галофеновой кислоты были пропорционально выше просто из-за более высокого количества введенного (-)галофената (таблица 3). (+)Галофеновую кислоту также обнаруживали в плазме крови, но ее концентрация была намного ниже, чем (-)галофеновой кислоты. Предполагают, что (+)галофеновая кислота образовалась in vivo, поскольку период полураспада (T1/2) обоих кислот был одинаков.
Эти результаты подтверждают, что использование (-)галофената более желательно, поскольку AUC (-)галофеновой кислоты существенно больше, чем AUC (+)галофеновой кислоты.
Фармакокинетический анализ (-)галофената (-энантиомера) и (+)галофената (+энантиомера)
Концентрации в плазме крови (-)галофеновой кислоты и (+)галофеновой кислоты после однократной дозы (-)галофената
N/A=Нет образца
ПРИМЕР 12
Этот пример касается профилактики развития диабета и лечения гиперглицеридемии с помощью (-)галофената.
А. Материалы и методы
Самцов мышей в возрасте 4 недель, линии C57BL/6J db/db, приобрели в The Jackson Laboratory (Bar Harbor, ME, USA). Животных помещали в стандартные лабораторные условия (по 5 мышей в клетку) при температуре 22±3°С и относительной влажности в 50±20% и содержали при кормлении порошковым кормом Пурина для грызунов (#8640) и поении водой ad libitum. Перед началом эксперимента у каждого животного брали кровь из хвостовой вены для определения содержания глюкозы, инсулина и триглицеридов в плазме крови. Мышей распределяли по группам таким образом, чтобы средние показатели содержания глюкозы и массы тела в начале исследования были эквивалентными в каждой группе. Контрольную группу (20 мышей) кормили порошковым кормом, смешанным с 5% сахарозы, а испытуемую группу (20 мышей) кормили порошковым кормом, смешанным с 5% сахарозы и (-)галофенатом. Количество (-)галофената в корме постоянно корректировали в соответствии с массой тела животного и потреблением им пищи, чтобы добиться получения заданной дозы в 150 мг/кг/день. Образцы крови брали в 8-10 часов утра 1 раз в неделю в течение 9 недель, в условиях без голодания. Потребление пищи и массу тела измеряли каждые 1-3 дня. Концентрации глюкозы и триглицеридов в плазме крови определяли колориметрически с помощью наборов фирмы Sigma Chemical Co. (№315 и №339, St. Louis, МО, USA). Концентрации инсулина в плазме крови определяли с помощью набора для анализов RIA из Linco Research Inc. (St. Charles, МО). Значимость различия между группами (при сравнении между группами, получавшими лекарства и получавшими носитель) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Мыши линии C57BL/6J db/db в возрасте 4 недель находились в преддиабетическом состоянии. Концентрации глюкозы в плазме их крови нормальные, но концентрации инсулина в плазме значительно повышенные. Как показано на фигуре 13, концентрации глюкозы в плазме крови в обеих группах были нормальными в начале эксперимента. После естественного цикла развития диабета уровни содержания глюкозы в плазме в контрольной группе повышались по мере старения животных, в то время, как увеличение содержания глюкозы в плазме крови животных, получавших (-)галофенат, было предотвращено или значительно замедлено. Как показано на фигуре 15, примерно у 30% мышей группы, получавшей (-)галофенат, диабет не развивался, если диабет определять как уровни содержания глюкозы в плазме крови >250 мг/дл. С другой стороны, ни одна из мышей контрольной группы не была не больной диабетом к возрасту 10 недель. Согласуясь с данными изучения содержания глюкозы в плазме, содержание инсулина в плазме крови контрольной группы прогрессивно уменьшалось, что указывает на ослабление способности поджелудочной железы секретировать инсулин. Лечение (-)галофенатом поддерживало концентрацию инсулина в плазме, что указывает на предотвращение ослабления функции поджелудочной железы (фигура 14).
Фигура 16 показывает изменения в содержании триглицеридов в плазме крови мышей линии C57BL/6J db/db в зависимости от их возраста. Введение (-)галофената привело к прекращению увеличения концентрации триглицеридов в плазме во время эксперимента.
ПРИМЕР 16
В этом примере описывается получение (-)2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетата ((-)галофената).
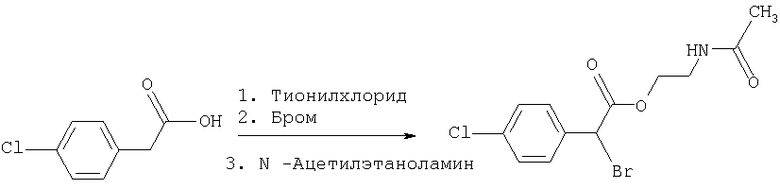
4-Хлорфенилуксусную кислоту соединяли с 1,2-дихлорэтаном и полученный раствор нагревали до 45°С. К реакционной смеси добавляли тионилхлорид и полученную смесь нагревали при 60°С в течение 18 часов. Реакционной смеси дали остыть при комнатной температуре, а затем медленно добавляли ее к раствору N-ацетилэтаноламина в дихлорметане. После продолжавшегося в течение 30 минут перемешивания реакцию гасили водным раствором карбоната калия и тиосульфата натрия. Органический слой промывали водой, сушили над сульфатом магния и фильтровали. После удаления растворителя, проводившегося с помощью ротационного испарителя, получили N-ацетиламиноэтил-2-бром-(4-хлорфенил)ацетат в виде масла.
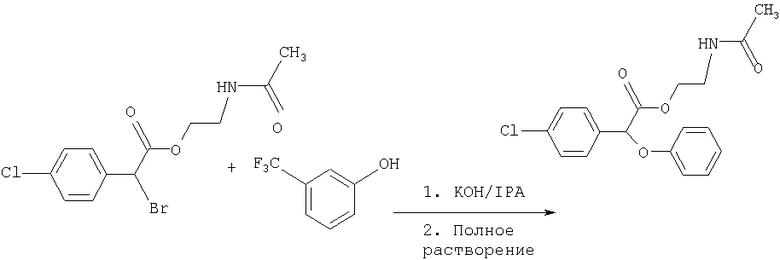
К раствору гидроксида калия в изопропаноле добавляли 3-гидроксибензотрифторид. К раствору изопропанола/феноксида добавляли N-ацетиламиноэтил-2-бром-(4-хлорфенил)ацетат в изопропаноле и перемешивали при комнатной температуре в течение 4 часов. Изопропанол удаляли путем вакуум-фильтрации, а полученную вязкую массу растворяли в этилацетате, дважды промывали водой и один раз - рассолом. После сушки над сульфатом магния и фильтрации растворитель удаляли и получали сырой продукт в виде масла. Сырой продукт растворяли в горячей смеси гексанов/толуола (1:1, об.) и охлаждали до температуры между 0 и 10°С, чтобы кристаллизовать продукт. Осадок на фильтре промывали смесью гексанов/толуола (1:1 об.), а затем сушили в условиях вакуума при 50°С. Выделенное твердое вещество растворяли в горячем растворе изопропанола в гексанах (1:6, об.). После охлаждения образовывался чистый рацемический 2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетат в виде кристаллического твердого вещества. Это твердое вещество собирали с помощью фильтрации, осадок на фильтре промывали горячим раствором изопропанола в гексанах (1:6, об.) и сушили в условиях вакуума при 50°С.
Рацемическое соединение растворяли в растворе, содержащем 20% изопропанола (IPA) и 80% гексана, в концентрации 2,5% масс. Полученный раствор пропускали через колонку Whelk-OR,R Chiral Stationary Phase (CSP) в непрерывном режиме до тех пор, пока не удалили >98% "ее" экстракта. Растворитель выпаривали из экстракта при пониженном давлении, получив в результате (-)2-ацетамидоэтил-4-хлорфенил-(3-трифторметилфенокси)ацетат. (Повторное растворение имитированного движущегося слоя было проведено фирмой Universal Pharm Technologies LLC из 70 Flagship Drive, North Andover, MA 01845).
ПРИМЕР 17
Данный пример касается понижения содержания мочевой кислоты в плазме крови путем введения (-)галофената.
А. Материалы и методы
Самцов крыс линии 3D с массой тела по 275-300 г приобрели в Charles River. Животных помещали (по 3 крысы на клетку) в стандартные лабораторные условия при температуре 22±3°С и относительной влажности в 50±20% и содержали при кормлении порошковым кормом Пурина для грызунов (#8640) и поении водой ad libitum (по желанию). Чтобы добиться гиперуремического состояния, животных содержали на диете, включающей 2,5% масс. оксоновой кислоты (Sigma Chemical Co., St. Louis, МО, USA) в течение всего эксперимента. Оксоновая кислота повышает концентрацию в плазме крови мочевой кислоты за счет ингибирования уриказы. Через 3 дня пребывания на этой диете крыс проанализировали на содержание в плазме мочевой кислоты, и животных, имевших чрезмерные концентрации мочевой кислоты в плазме, исключили из эксперимента. Крыс распределили на три группы так, чтобы средние концентрации мочевой кислоты в каждой группе были эквивалентными. Крысам перорально, через желудочный зонд вводили один раз в день в течение 3 дней одно из нижеследующего: носитель, (-)галофенат или (+)галофенат, в дозе 50 мг/кг. На четвертый день соответствующие крысы получали (-)галофенат или (+)галофенат в дозе 110 мг/кг, и все крысы получали внутрибрюшинную инъекцию оксоновой кислоты (250 мг/кг) через 4 часа после перорального введения лекарства через желудочный зонд. (-)Галофенат и (+)галофенат вводили в жидкой лекарственной форме, содержавшей 5% об. диметилсульфоксида (DMSO), 1% об. твина 80 и 0,9% масс./об. метилцеллюлозы. Оксоновую кислоту вводили в жидкой лекарственной форме, содержавшей 0,9% масс./об. метилцеллюлозы. Объемы желудочного зонда и инъекции составляли по 5 мл/кг. Образцы крови брали через 6 часов после перорального введения дозы через желудочный зонд, на 4-й день. Концентрации мочевой кислоты в плазме крови определяли колориметрически с помощью реагента Infinity Uric Acid Reagent фирмы Sigma Chemical Co. (St. Louis, МО, USA). Значимость различия между группами (при сравнении между группами, получавшими лекарства и получавшими носитель) оценивали с помощью непарного t-критерия Стьюдента.
В. Результаты
Как показано на фигуре 17, пероральное введение (-)галофената существенно понижало концентрации мочевой кислоты в плазме крови. (+)Галофенат также понижал концентрации мочевой кислоты в плазме крови, но это понижение было статистически незначимым.
ПРИМЕР 18
Этот пример касается ингибирования изоформ цитохрома Р450 соединениями по настоящему изобретению.
А. Материалы и методы
Для изучения ингибирующего влияния испытуемых соединений на изоформы цитохрома Р450 1А2, 2А6, 2С9, 2С19, 2D6, 2Е1 и 3А4 использовали нижеследующие пробные субстраты: 100 мкМ фенацетин (CYP1A2), 1 мкМ кумарин (CYP2A6), 150 мкМ толбутамид CYP2C9), 50 мкМ S-мефенитоин (CYP2C19), 16 мкМ декстрометорфан (CYP2D6), 50 мкМ хлорзоказон (CYP2E1) и 80 мкМ тестостерон (CYP3A4). Активность каждой из изоформ определяли на печеночных микросомах человека в присутствии и в отсутствии испытуемого соединения.
Кроме особо оговоренных случаев, инкубацию осуществляли при температуре 37°С. Количество повторов составляло N=3 для всех вариантов эксперимента и для позитивных контролей и N=6 для контролей, в которых использовали только носитель. (-)Галофеновую кислоту (молекулярная масса=330) готовили при комнатной температуре в виде маточных растворов 1000Х в метаноле, затем разбавляли трис-буфером, чтобы получить следующие окончательные концентрации: 0,33, 1,0, 3,3 10 и 33,3 мкМ, в каждом случае содержащие 0,1% метанола. Контроль с носителем (VC), состоящий из микросом и субстрата в трис-буфере, содержащем 0,1% метанола, без испытуемых соединений, включали во все экспериментальные группы. Смеси для позитивных контролей (PC) готовили с использованием нижеследующих известных ингибиторов CYP450; 5 мкМ фурафиллин (CYP1A2), 250 мкМ транилципромин (CYP2A6), 50 мкМ сульфафеназол (CYP2C9), 10 мкМ омепразол (CYP2C19), 1 мкМ хинидин (CYP2D6), 100 мкМ 4-метилпиразол (CYP2E1) и 5 мкМ кетоконазол (CYP3A4). Контроль хроматографической интерференции (CIC) был включен для изучения возможной хроматографической интерференции, вызываемой испытуемым образцом и его метаболитами. Испытуемый образец (в концентрации 33,3 мкг/мл) инкубировали с 1X микросомального белка, 1X NRS и 10 мкл соответствующего органического вещества в течение соответствующего периода времени, как описано ниже.
В этом исследовании использовали стабильные замороженные партии пулов взрослых мужских и женских печеночных микросом, полученных путем дифференциального центрифугирования (см., например, Guengerich, F.P. (1989). Analysis and characterization of enzymes. In: Principles and Methods of Toxicology (A.W. Hayes, Ed.), 777-813. Raven Press, New York). Смеси для инкубации готовили в трис-буфере, содержащем микросомальный белок (1 мг/мл), каждую концентрацию пробных субстратов (в виде маточных растворов 100Х) и испытуемый образец (в каждой концентрации) или PC, соответствующий каждой изоформе. После предварительной инкубации в течение 5 минут при температуре 37°С, добавляли NRS (регенерирующие системы NADPH), чтобы инициировать реакции, и образцы инкубировали при 37°С в течение следующих периодов времени: 30 минут для фенацетина (CYP1A6), 20 минут для кумарина (CYP2A6), 40 минут для толбутамида (CYP2C9), 30 минут для 3-мефенитоина (CYP2C19), 15 минут для декстрометорфана (CYP2D6), 20 минут для хлорзоказона (CYP2E1) и 10 минут для тестостерона (CYP3A4). Реакции инкубации заканчивали в соответствующее время путем добавления равного объема метанола, за исключением инкубации с S-мефенитоином, которую заканчивали путем добавления 100 мкМ перхлорной кислоты. Все субстраты оценивали для их соответствующих концентраций Km, указанных выше.
После каждой инкубации определяли активность изоформ Р450 путем измерения скорости метаболизма для соответствующих пробных субстратов. Для каждого пробного субстрата отслеживали следующие метаболиты: ацетаминофен для CYP1A2; 7-гидроксикумарин для CYP2A6; 4-гидрокситолбутамид для CYP2C9; 4-гидроксимефенитоин для CYP2C19; декстрофан для CYP2D6; 6-гидроксихлорзоказон для CYP2E1; и 6β-гидрокситестостерон для CYP3A4. Активность анализировали с помощью метода ВЭЖХ (In Vitro Technologies, Inc., Baltimore, MD).
Ингибирование рассчитывали с помощью следующего уравнения:
Процент ингибирования = [(контроль с носителем - опытный вариант)/контроль с носителем]×100.
Данные по проценту ингибирования для испытуемых образцов представлены в виде таблиц. Для каждой концентрации испытуемого соединения рассчитывали показатели описательной статистики (среднее и стандартное отклонение), которые затем представляли в таблицах, чтобы проиллюстрировать величину ингибирования. Рассчитывали также значения ИК50 для испытуемых соединений, используя уравнение, соответствующее 4-параметровой кривой, в программе Softmax 2.6.1.
Измерения времени, температуры и концентраций в этом примере были приблизительными.
В. Результаты
Результаты для каждой из 7 изоформ цитохрома Р450, выраженные в виде метаболической активности и процента ингибирования, представлены в таблицах 5-8. (-)Галофеновая кислота ингибировала выработку 4-гидрокситолбутамида (СУР2С9, ИК50=22 мкМ), а также ингибировала выработку 4-гидроксимефенитоина (CYP2C19) в дозах 10 и 33 мкМ. Ингибирования других изоформ CYP450 не наблюдалось. Следует отметить, что ИК50 для CYP2C9 в этом эксперименте была примерно в три раза больше, чем в примере 7 (11 мкМ против 3,6 мкМ). Этот результат скорее всего связан, по меньшей мере частично, с использованием в примере 7 (-)галофеновой кислоты более низкой чистоты (с более низким "ее").
Печеночная микросомальная активность фенацетина (CYP1A2) и кумарина (CYP2A6) в мужских и женских микросомах человека, инкубированных с (-)галофеновой кислотой в дозах 0,33, 1,0, 3,3, 10 и 33,3 мкМ
VC
0,1%
118±2
0
32,0+1,4
0
TRAN
250
NA
NA
0,00±0,00
100
Аббревиатуры: Конц. - концентрация; АС - ацетаминофен;
7 - НС - 7-гидроксикумарин;
CIC - контроль хроматографической интерференции;
VC - контроль - носитель (0,1% метанола); NA - не применимо;
FUR - фурафиллин; TRAN - транилципромин.
Печеночная микросомальная активность толбутамида (CYP2C9) и S-мефенитоина (CYP2C19) в мужских и женских микросомах человека, инкубированных с (-)галофеновой кислотой в дозах 0,33, 1,0, 3,3, 10 и 33,3 мкМ
VC
0,1%
43,0+1,4
0
31,7±0,29
0
SFZ
50
BQL
≈100
NA
NA
4 - ОН ТВ - 4-гиброкситолбутамид; 4-ОН ME - 4-гидроксимефенитоин;
CIC - контроль хроматографической интерференции;
VC - контроль - носитель (0,1% метанола);
NA - не применимо; ОМР - омепразол; SFZ - сульфафеназол;
BQL - ниже определяемого количества.
Печеночная микросомальная активность декстрометорфана (CYP2D6) и хлорзоксазона (CYP2E1) в мужских и женских микросоках человека, инкубированных с (-)галофеновой кислотой в дозах 0,33, 1,0, 3,3, 10 и 33,3 мкМ
VC
0,1%
111±6
0
246±5
0
QIN
1
BQL
≈100
NA
NA
N=3 образцов (VC:N=6).
Аббревиатуры: Конц. - концентрация; DEX - декстрофан;
6 - ОН CZX - 4-гидроксихлорзоксазон;
CIC - контроль хроматографической интерференции;
VC - контроль - носитель (0,1% метанола);
NA - не применимо; 4 - МР - 4-метилпиразол; QUIN - хинидин;
BQL - ниже определяемого количества.
Печеночная микросомальная активность тестостерона (CYP3A4) в мужских и женских микросомах человека, инкубированных с (-)галофеновой кислотой в дозах 0,33, 1,0, 3,3, 10 и 33,3 мкМ
VC
0,1%
1843±9
0
6β-ОНТ - 6β-гидрокситестостерон;
CIC - контроль хроматографической интерференции;
VC - контроль - носитель (0,1% метанола); NA - не применимо;
KTZ - кетоконазол; BQL - ниже определяемого количества.
Хотя настоящее изобретение выше описано в подробностях в целях ясности его понимания, но очевидно, что могут использоваться некоторые его модификации, не выходящие за пределы объема прилагаемой формулы изобретения. Все публикации и патентные документы, цитированные в настоящем описании, включены в него в качестве ссылок во всей их полноте для всех целей в такой же степени, как если бы каждое из них было указано отдельно.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОВЫШЕНИЯ МОЧЕОТДЕЛЕНИЯ У ЧЕЛОВЕКА | 1993 |
|
RU2139053C1 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КАТИОННЫЕ СОЛИ ИЛИ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1992 |
|
RU2079497C1 |
| СПОСОБ ЛЕЧЕНИЯ ДИАБЕТА | 2003 |
|
RU2320343C2 |
| СПОСОБ ЛЕЧЕНИЯ ДИАБЕТА | 2007 |
|
RU2442585C2 |
| N-[2-ГИДРОКСИ-3-(1-ПИПЕРИДИНИЛ)ПРОПОКСИ]ПИРИДИН-1-ОКСИД-3- КАРБОКСИИМИДОИЛХЛОРИД, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2250901C2 |
| Лечение диабета I и II типа | 2012 |
|
RU2646475C2 |
| ПРОТИВОСУДОРОЖНЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ПРОФИЛАКТИКИ РАЗВИТИЯ САХАРНОГО ДИАБЕТА И СИНДРОМА Х | 2001 |
|
RU2268720C2 |
| Способ лечения гипергликемии | 2017 |
|
RU2739255C2 |
| ПРИМЕНЕНИЕ АНАЛОГОВ СИБУТРАМИНА ДЛЯ ПРОФИЛАКТИКИ САХАРНОГО ДИАБЕТА | 1997 |
|
RU2245709C2 |
| НИАЦИНОВЫЕ МИМЕТИКИ И СПОСОБ ИХ ИСПОЛЬЗОВАНИЯ | 2011 |
|
RU2588133C2 |
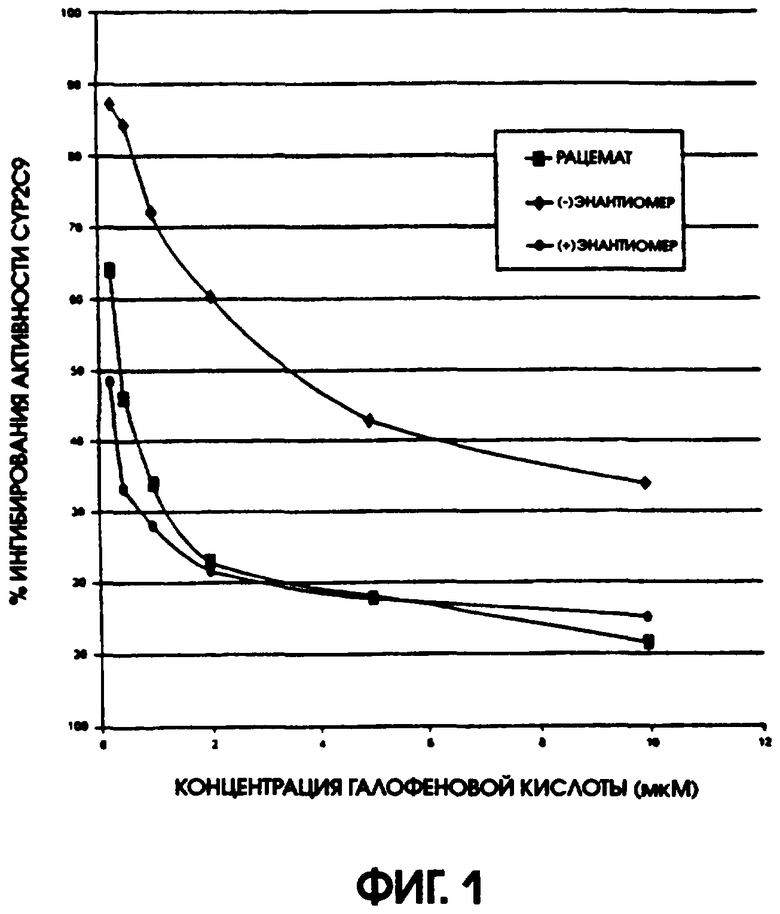
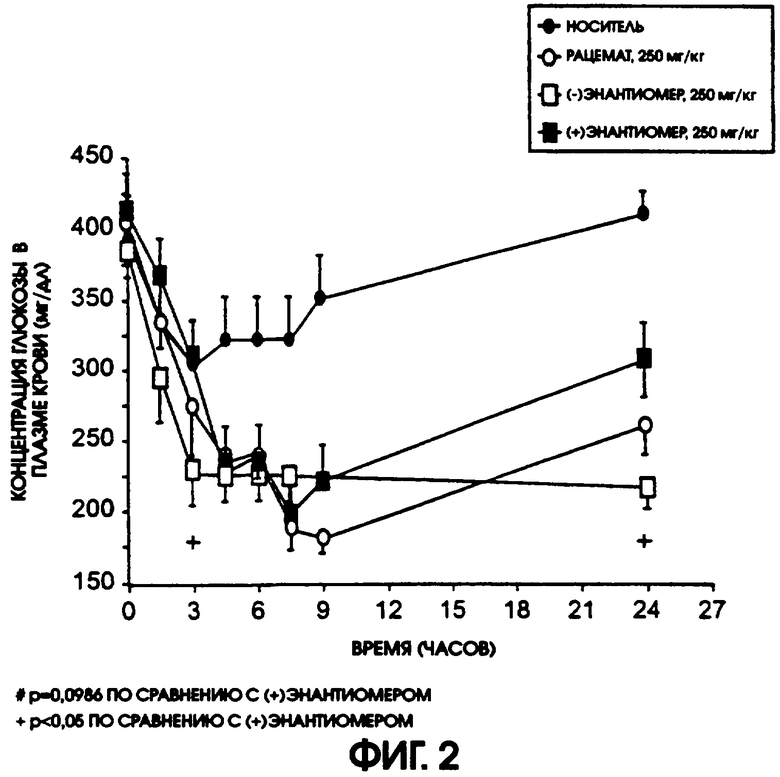
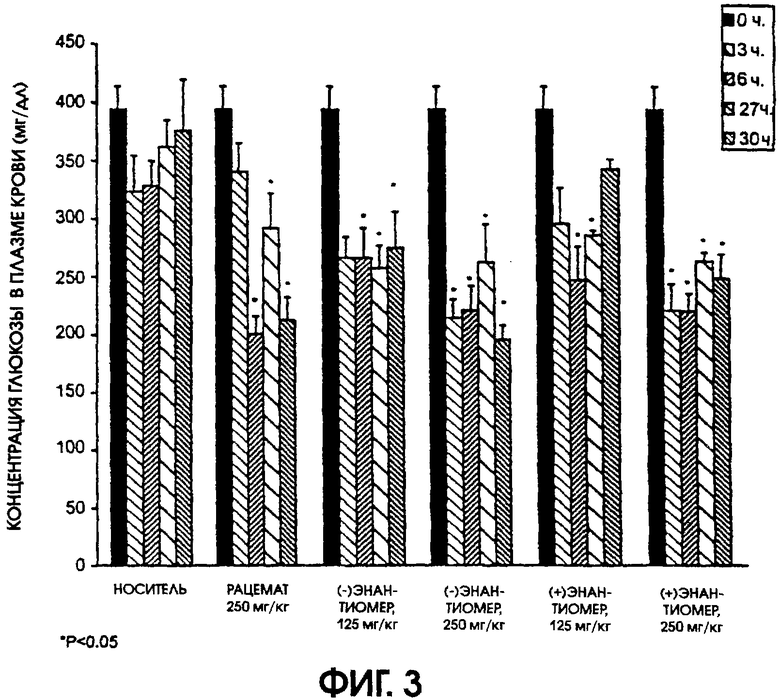
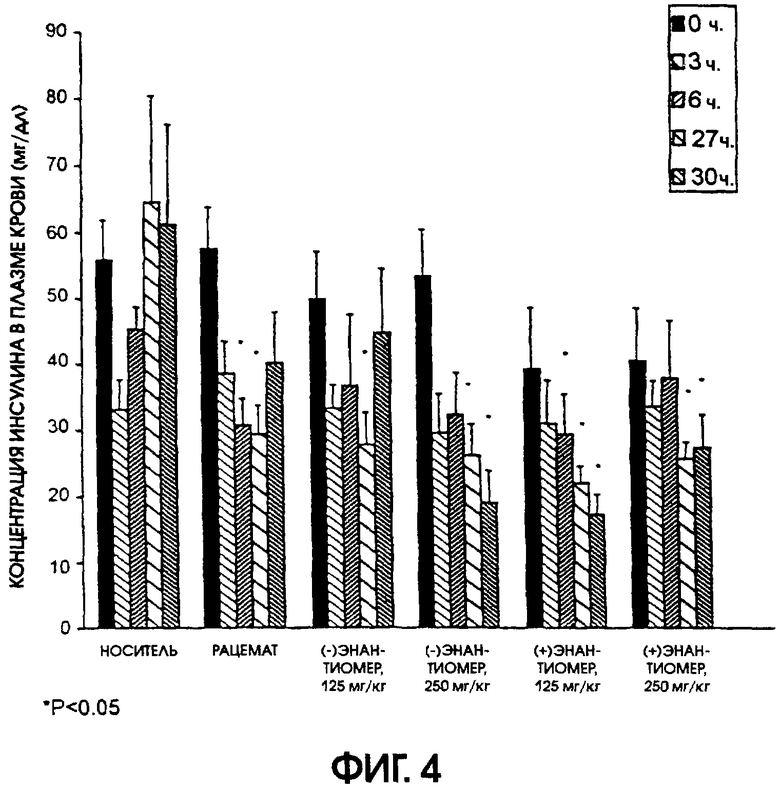
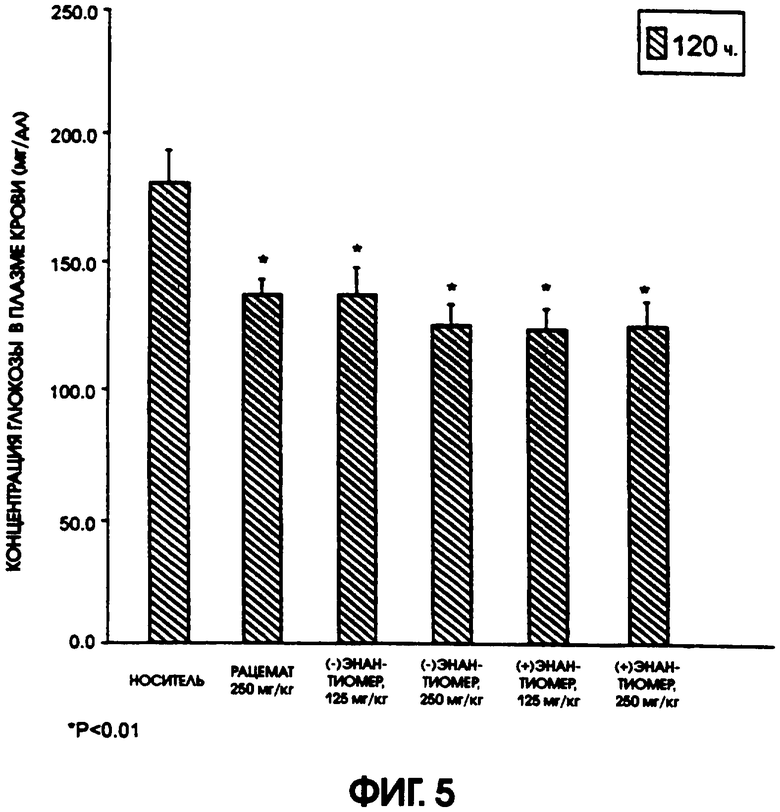
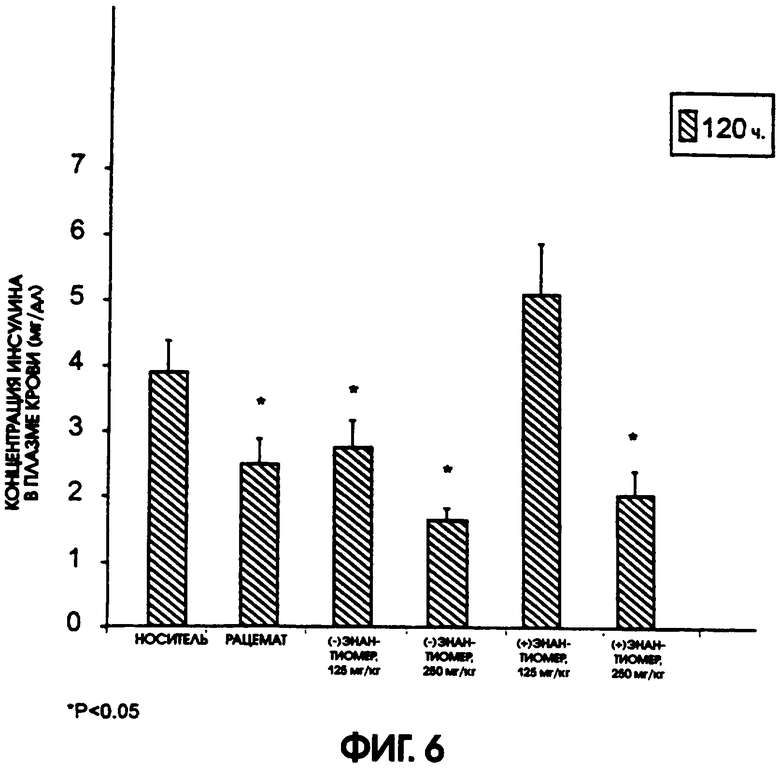

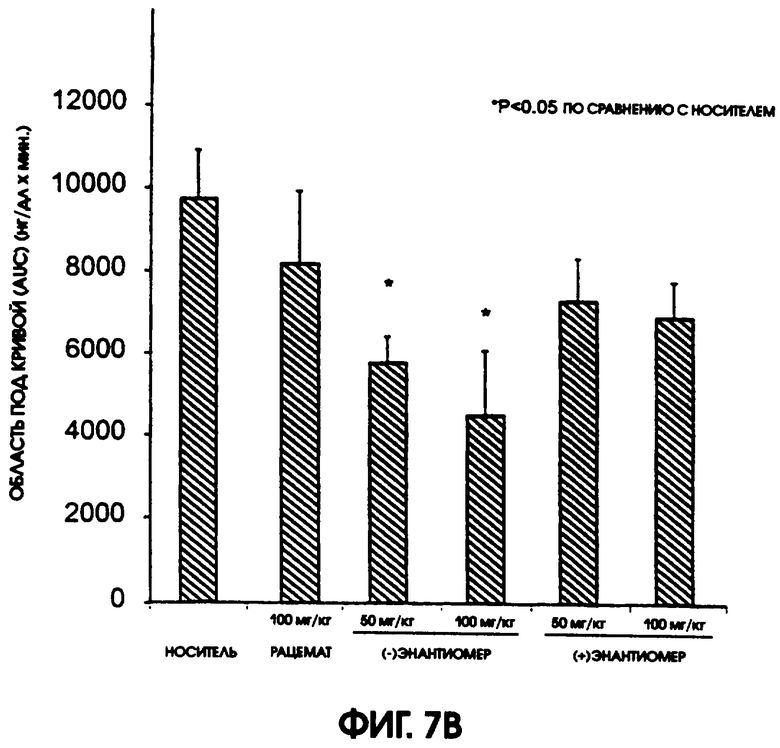
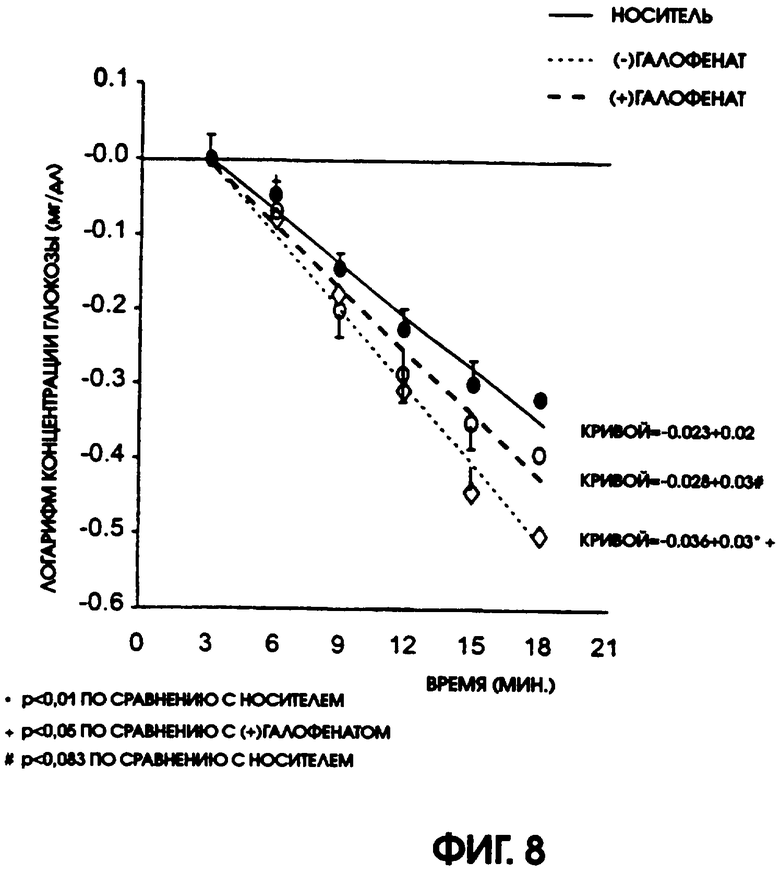
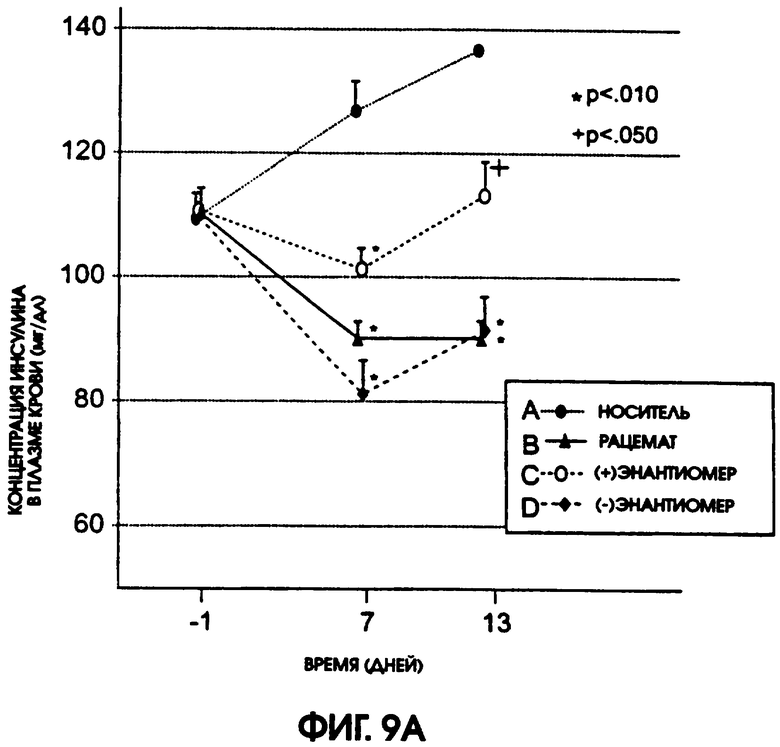
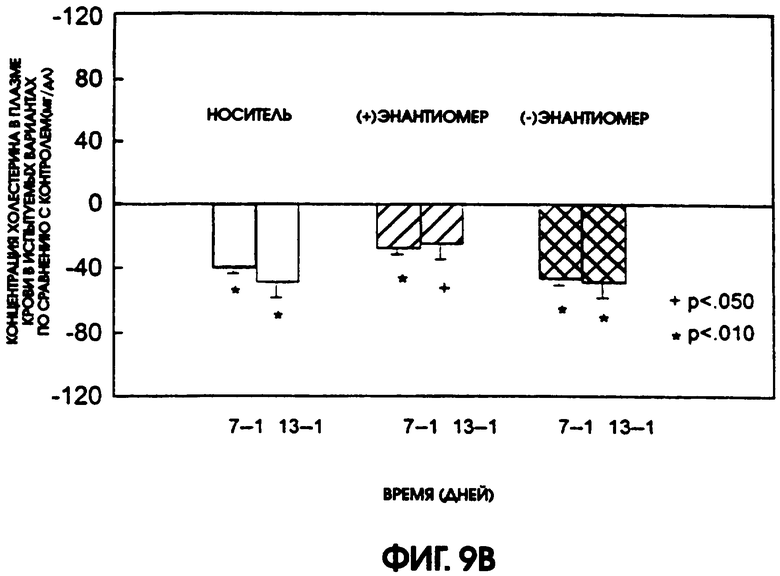
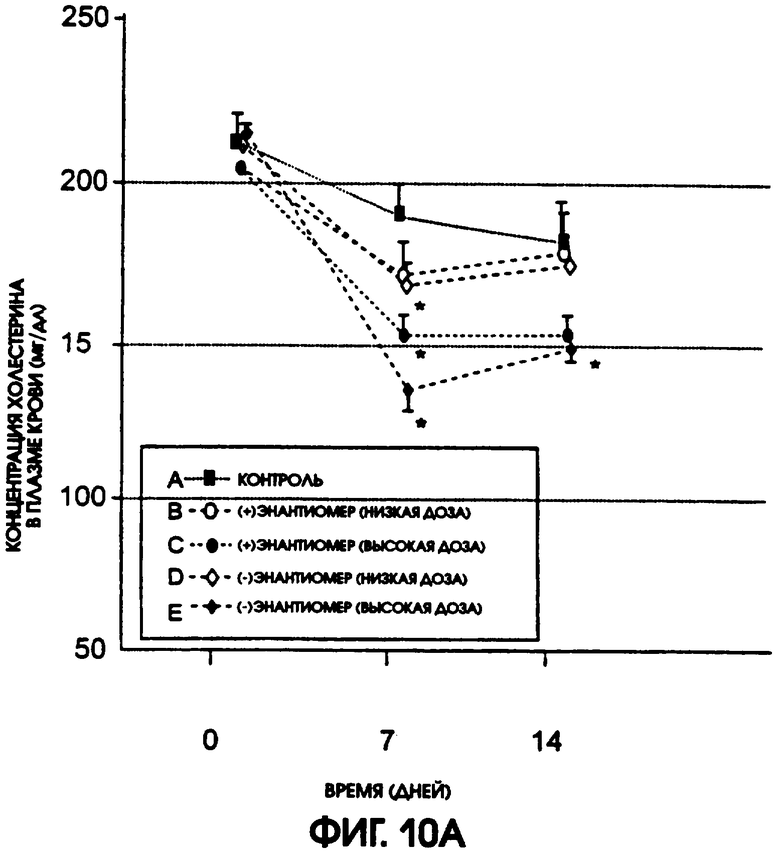
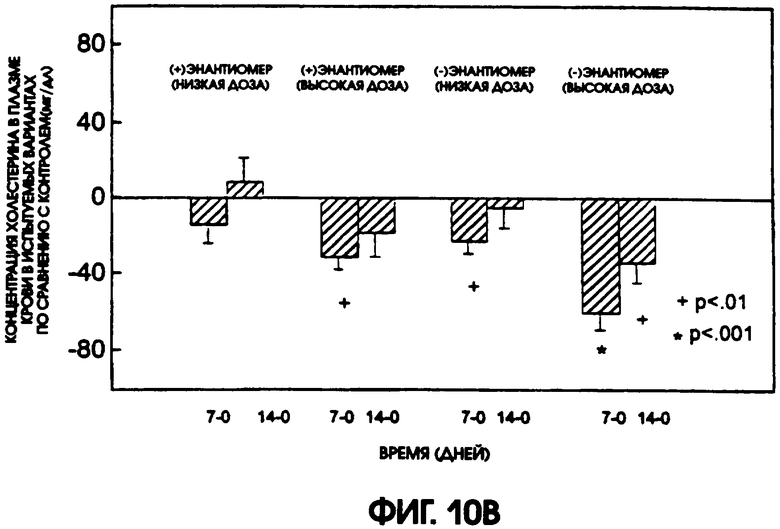
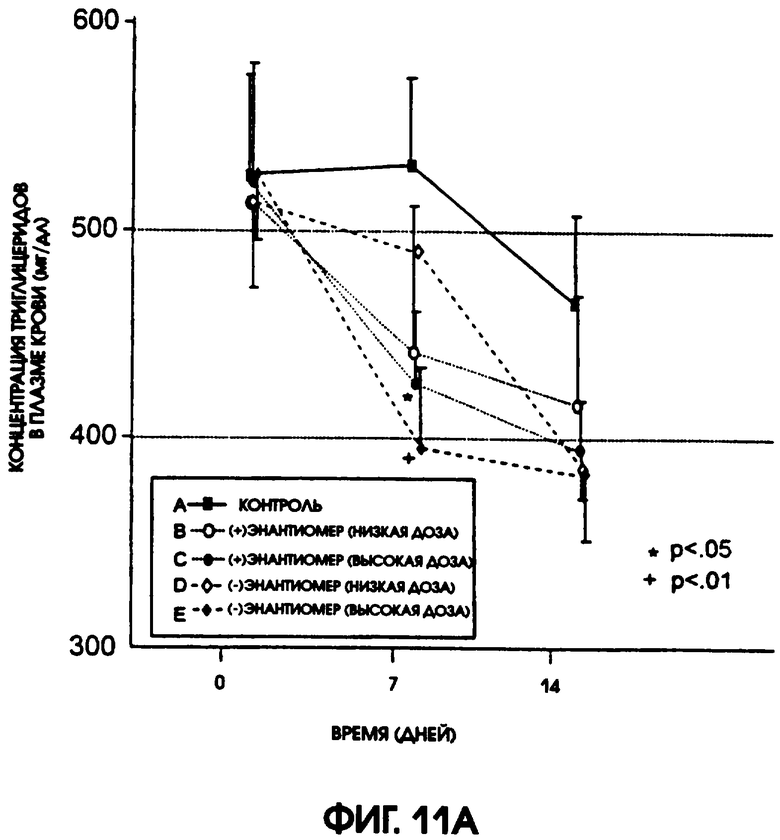
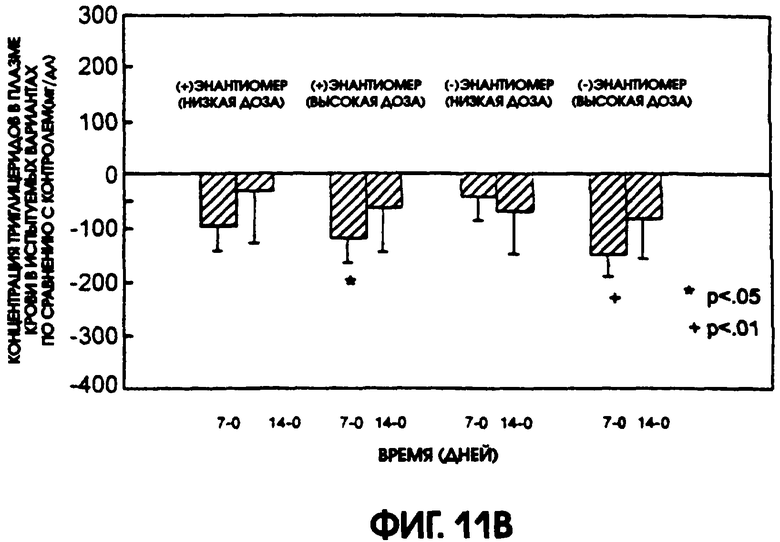
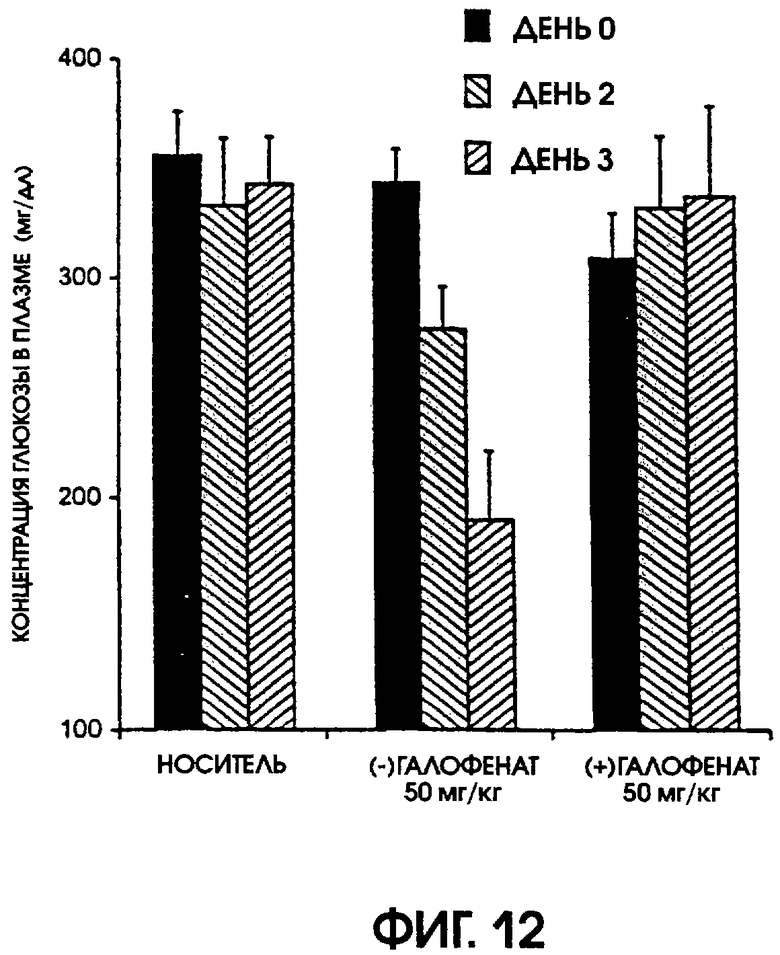
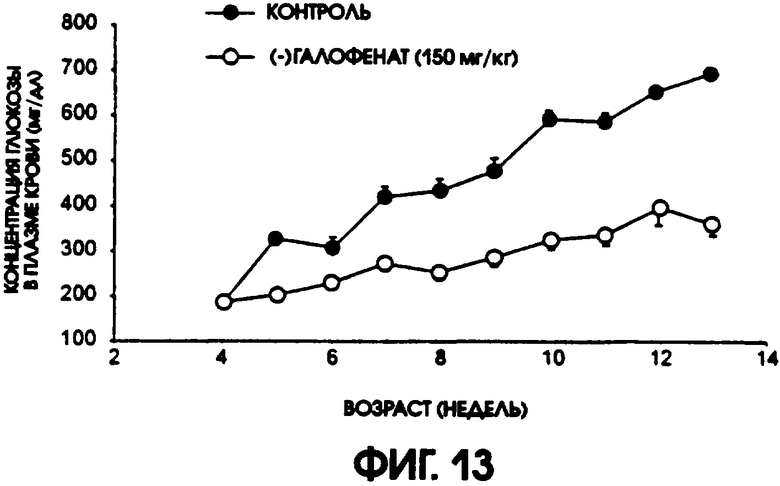
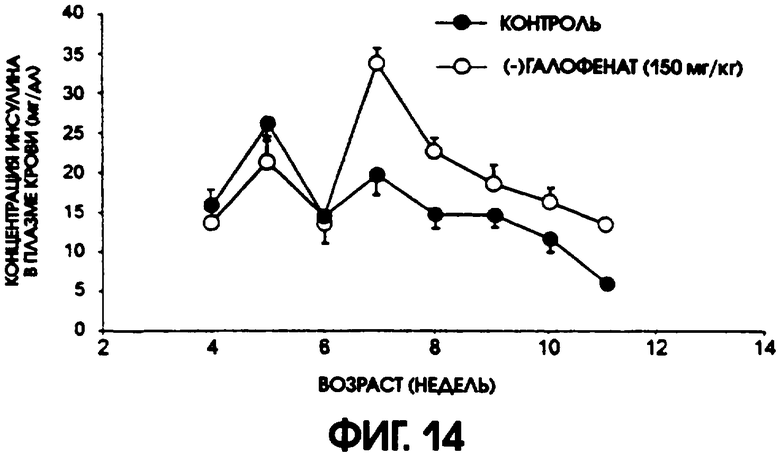
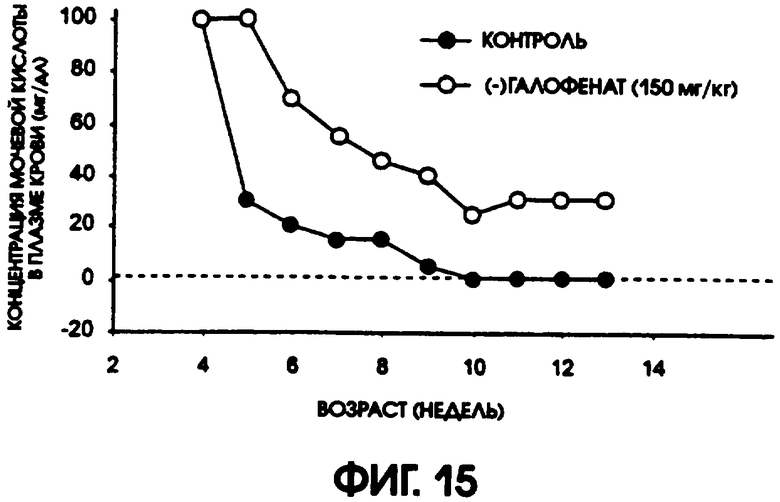

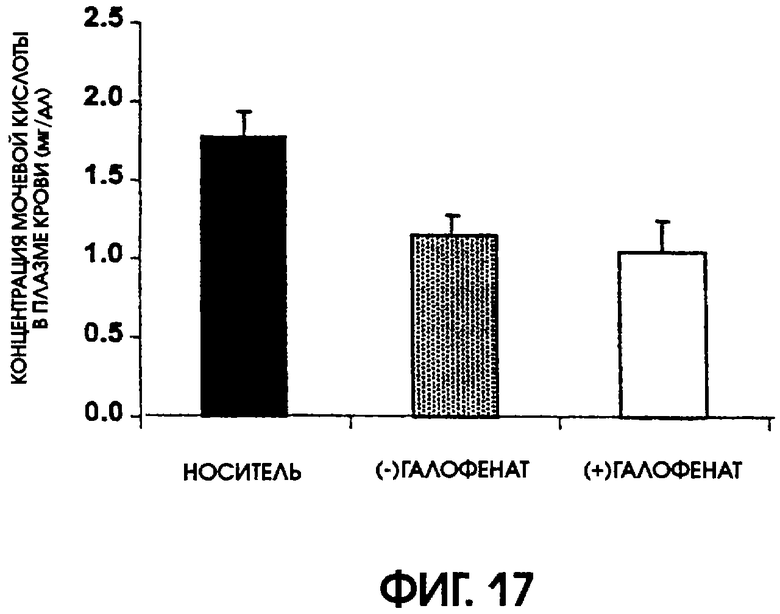
Изобретение относится к усовершенствованным способам модулирования диабета II типа у млекопитающего и модулирования резистентности к инсулину, включающим введение указанному млекопитающему, нуждающемуся в этом, (-)стереоизомера соединения формулы I,

в которой R выбран из группы, состоящей из гидрокси, низшего аралкокси, ди-низшего алкиламино-низшего алкокси, низшего алканамидо низшего алкокси, бензамидо-низшего алкокси, уреидо-низшего алкокси, N'-низшего алкил-уреидо-низшего алкокси, карбамоил-низшего алкокси, галофеноксизамещенного низшего алкокси, карбамоилзамещенного фенокси, или R представляет собой гидролизуемую сложноэфирную группировку; каждый Х независимо представляет собой галоген; или его фармацевтически приемлемой соли, причем (-)стереоизомер по существу не содержит (+)стереоизомера соединения. Изобретение также относится к фармацевтическим композициям, содержащим (-)стереоизомер соединения формулы I и где композиции обладают существенно сниженным ингибирующим эффектом на цитохром Р450 2С9 по сравнению с рацемической композицией, имеющей 0% энантиомерный избыток (-)стереоизомера. 4 н. и 55 з.п. ф-лы, 21 ил., 8 табл.

в которой R выбран из группы, состоящей из гидрокси, низшего аралкокси, динизшего алкиламино-низшего алкокси, низшего алканамидо низшего алкокси, бензамидо-низшего алкокси, уреидо-низшего алкокси, N'-низшего алкил-уреидо-низшего алкокси, карбамоил-низшего алкокси, галофеноксизамещенного низшего алкокси, карбамоилзамещенного фенокси, или R представляет собой гидролизуемую сложноэфирную группировку;
каждый Х независимо представляет собой галоген,
или его фармацевтически приемлемой соли, причем (-)стереоизомер, по существу, не содержит (+)стереоизомера соединения.
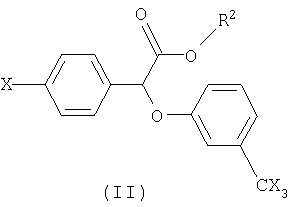
в которой R2 выбран из группы, состоящей из фенил-низшего алкила, низшего алканамидо-низшего алкила и бензамидо-низшего алкила.
в которой R выбран из группы, состоящей из гидрокси, низшего аралкокси, динизшего алкиламино-низшего алкокси, низшего алканамидо низшего алкокси, бензамидо-низшего алкокси, уреидо-низшего алкокси, N'-низшего алкил-уреидо-низшего алкокси, карбамоил-низшего алкокси, галофеноксизамещенного низшего алкокси, карбамоилзамещенного фенокси, или R представляет собой гидролизуемую сложноэфирную группировку;
каждый Х независимо представляет собой галоген,
или его фармацевтически приемлемой соли, причем (-)стереоизомер, по существу, не содержит (+)стереоизомера соединения.
в которой R2 выбран из группы, состоящей из фенил-низшего алкила, низшего алканамидо-низшего алкила и бензамидо-низшего алкила.
в которой R выбран из группы, состоящей из гидрокси, низшего аралкокси, динизшего алкиламино-низшего алкокси, низшего алканамидо низшего алкокси, бензамидо-низшего алкокси, уреидо-низшего алкокси, N'-низшего алкил-уреидо-низшего алкокси, карбамоил-низшего алкокси, галофеноксизамещенного низшего алкокси, карбамоилзамещенного фенокси, или R представляет собой гидролизуемую сложноэфирную группировку; и каждый
Х независимо представляет собой галоген,
или его фармацевтически приемлемой соли, причем это соединение, по существу, не содержит (+)стереоизомера соединения, и где композиция обладает существенно сниженным ингибирующим эффектом на цитохром Р450 2С9 по сравнению с рацемической композицией, имеющей 0%-ный энантиомерный избыток (-)стереоизомера.
в которой R2 выбран из группы, состоящей из фенил-низшего алкила, низшего алканамидо-низшего алкила и бензамидо-низшего алкила.
в которой R выбран из группы, состоящей из гидрокси, низшего аралкокси, динизшего алкиламино-низшего алкокси, низшего алканамидо низшего алкокси, бензамидо-низшего алкокси, уреидо-низшего алкокси, N'-низшего алкил-уреидо-низшего алкокси, карбамоил-низшего алкокси, галофеноксизамещенного низшего алкокси, карбамоилзамещенного фенокси, или R представляет собой гидролизуемую сложноэфирную группировку; и каждый
Х независимо представляет собой галоген,
или его фармацевтически приемлемой соли, причем это соединение, по существу, не содержит (+)стереоизомера соединения, и где композиция обладает существенно сниженным ингибирующим эффектом на цитохром Р450 2С9 по сравнению с рацемической композицией, имеющей 0%-ный энантиомерный избыток (-)стереоизомера.
в которой R2 выбран из группы, состоящей из фенил-низшего алкила, низшего алканамидо-низшего алкила и бензамидо-низшего алкила.
| Feldman E.B., et al | |||
| "Effects of halofenate on glucose tolerance in patients with hyperlipoproteinemia," Journal of Clinical Pharmacology, vol.18, n.5-6, pp.241-248 (1978). |

