РОДСТВЕННЫЕ ЗАЯВКИ
По данной заявке испрашивается приоритет предварительной патентной заявки Соединенных Штатов 61/358264, поданной 24 июня 2010 г., содержание которой включено в данное описание путем ссылки на нее.
ИЗВЕСТНЫЙ УРОВЕНЬ ТЕХНИКИ
Гиперлипидемия и гиперхолестеринемия являются состояниями, которые характеризуются хорошо установившейся корреляцией с повышенным риском других состояний, таких как сердечные приступы, атеросклероз и другие ухудшенные нездоровые состояния. Имеются многочисленные агенты, доступные для снижения уровней холестерина и липидов, включая гемфибризол, пробукол и не так давно “статины” (например, ловастатин).
Ниацин (никотиновая кислота), водорастворимый В-комплекс витамина, используется орально для лечения гиперлипидемии. Было показано, что ниацин является эффективным при снижении общего холестерина в плазме (С), липопротеинов низкой плотности LDL-C и триглицеридов липопротеина очень низкой плотности (VLDL-триглицериды), все из которых связаны с рисками для здоровья. Одновременно, ниацин поднимает в сыворотке уровни липопротеинов высокой плотности (HDL-C), которые считаются “здоровым” липопротеином, у пациентов с гиперхолестеринемией типов II, III, IV и V.
Хотя механизм, с помощью которого ниацин изменяет липидограммы, не определен хорошо, было показано, что его механизмы действия включают ингибирование высвобождения свободной жирной кислоты из адипозной ткани (см. Carlson, L. A., Froberg, S.O. and Nye, E.R., Nicotinic acid in the rat. 11. Acute effects of nicotinic acid on plasma, liver, heart, and muscle lipids, Acta Med Scand 180: 571-579, 1966), и повышенную активность липопротеиновой липазы (см. Priego, J.G., Pina, M., Armijo, M., Sunkel, C. and Maroto, M.L., Action of etofibrate, clofibrate and nicotinic acid on the metabolism of lipids in normolipemic rats. Short term effects and method of action, Arch Farmacol Toxicol 5: 29-42, 1979). Более чем 30 миллионов американцев имеют повышенные уровни LDL-C в крови. Ингибиторы HMG-CoA редуктазы (статины) являются наиболее широко используемым классом лекарственных средств для лечения пациентов с повышенными уровнями LDL-C. Ниацин, однако, является единственным лекарственным средством, рекомендуемым American Heart Association для улучшения HDL при первичной профилактике сердечнососудистых заболеваний в дополнение к снижению LDL-C. Ниациновая терапия является не только эффективной по стоимости в качестве монотерапии, но она является также благоприятной в качестве комбинационной терапии, потому что она дополняет действия других классов липид-понижащих лекарственных средств. Однако для изолированной гиперхолестеринемии ниацин является выбором второго или третьего порядка вследствие высокой сферы действия побочных эффектов, связанных с оральной ниациновой терапией. Тем не менее, он имеет терапевтическое преимущество в качестве монотерапии, когда желательным является снижение, как LDL-C, так и триглицеридов, такой как для пациентов с тяжелой комбинированной гиперлипидемией.
Ниацин может использоваться также в сочетании с другими понижающими холестерин агентами, такими как “статины”, для максимизации липид-понижающей активности. Одно исследование показало, что сочетание ниацин/ловастатин является высоко эффективным при снижении LDL-C, триглицеридов и липопротеина (а) (Lp(a)) при сохранении силы ниацина в поднятии HDL-C (Kashyap, M.L., Evans R., Simmons, P.D., Kohler, R.M. and McGoven, M. E., New combination niacin/statin formulation shows pronounced effects on major lipoproteins and well tolerated, J Am Coll Card Suppl, A 35: 326, 2000).
Ниацин широко используется для снижения уровней холестерина в сыворотке, потому что он считается эффективной по стоимости терапией. Ежедневные оральные дозы ниацина 2-3 г у людей снижают уровни общего-С и LDL-C в среднем на 20%-30%, снижают уровни триглицерида на 35%-55%, увеличивают HDL-C на 20%-35%, и снижают Lp(a). Ниацин также снижает общую смертность, также как и смертность от болезни коронарных артерий (см. The Coronary Drug Project Research Group, LAMA 231: 360-381, 1975; и Canner, P.L., Berge, K.G., Wenger, N. К., Stamler, J., Friedman L., Prineas, R.J. and Friedewald, W., Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin, J Am Coll Cardiol 8: 1245-1255, 1986) и он помогает замедлить или отменить прогрессирование атеросклероза (см. Blankenhorn, D.H., Nessim, S. A., Johnson, R. L., Samnarco, M. E., Azen, S.P. and Cashin-Hemphill, L., Beneficial effects of combined colestipol-niacin therapy on coronary atherosclerosis and coronary venous bypass grafts, JAMA 257: 3233-3240, 1987; и Cashin-Hemphill L.; Mack, W. J., Pogoda, J.M., Samnarco, M.T., Azen, S.P. and Blankenhorn, D. H., Beneficial effects of colestipol-niacin on coronary atherosclerosis. A 4-year follow-up, JAMA 264: 3013-3017, 1990).
К сожалению, оральная ниациновая терапия имеет побочные эффекты, которые ограничивают ее полезность. Хотя ниацин является витамином, для снижения холестерина он должен использоваться в терапевтических дозах. При этих дозах, как немедленное высвобождение, так и длительное высвобождение ниацина может иметь некоторые побочные эффекты. Наиболее обычным побочным эффектом ниацина является прилив крови, ощущение тепла на коже, обычно связанное с краснотой и иногда зудом. Прилив крови не является опасным, но большинство пациентов находят его очень некомфортным, что серьезно ограничивает согласие пациента на ниациновую терапию. Вызываемый ниацином прилив крови может в значительной степени смягчаться предварительным лечением ингибиторами циклооксигеназы, полагая, что расширение сосудов вызывается за счет опосредуемого простагландином механизма (см. Carlson, L.А., Nicotinic acid and inhibition of fat mobilizing lipolysis. Present status, of effects on lipid metabolism, Adv Exp Med Biol 109: 225-238, 1978).
Проверка функции печени всегда контролируется у пациентов, принимающих ниацин, поскольку повышение уровней трансаминазы в сыворотке связано с лечением ниацином, а длительное высвобождение ниациновых препаратов связано с более серьезными проблемами печени (см. McKenney, J.M., Proctor, J.D., Harris, S., and Chinchili, V.M., A comparison of the efficacy and toxic effects of substained- vs immediate-release niacin in hypercholesterolemic patients, JAMA 271: 672-777, 1994; и Stafford, R.S., Blumenthal, D. and Pasternak, R.C., Variations in cholesterol management practices of U.S. physicians, J Am Coll Cardiol 29: 139-146, 1997). Другие известные побочные эффекты оральной ниациновой терапии включают активирование пептических язв и ухудшение контроля диабета. Соответственно, безопасность и эффективность оральной ниациновой терапии подрывается потребностью тщательного клинического мониторинга и профилем побочных эффектов соединения.
КРАТКОЕ СОДЕРЖАНИЕ
Один из аспектов настоящего изобретения относится к 6-(морфолиноалкил)-замещенным пиридинам и их фармацевтически приемлемым солям, которые являются активными против ряда болезней млекопитающих. В некоторых воплощениях указанные пиридины или их соли включают функциональную группу, которая является в значительной мере анионной при физиологическом рН.
Еще один аспект изобретения относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; и фармацевтически приемлемый наполнитель. Еще один аспект изобретения относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; ниацин; и фармацевтически приемлемый наполнитель.
Еще один аспект изобретения относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; статин, выбранный из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина; и фармацевтически приемлемый наполнитель. Настоящее изобретение относится также к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; ниацин; статин, выбранный из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина; и фармацевтически приемлемый наполнитель. Дополнительные терапевтические агенты, которые могут вводиться совместно с соединениями изобретения, обсуждаются ниже.
Ниацин или никотиновая кислота имеет установленную эффективность для лечения дислипидемии, но клиническое использование ниацина ограничено кожным приливом крови, как известно, связанным с побочным эффектом. Прилив крови, который оценивается преобладанием такого высокого показателя, как 25%-40%, указан как главная причина для прерывания ниациновой терапии. Ряд исследований установил, что умеренные дозы ингибиторов простагландина снижают ответную реакцию, выражающуюся в кожном приливе крови, из-за введения ниацина. Другие подходы к снижению прилива крови включают в их число регулярное согласованное дозирование, использование составов длительного или продолженного высвобождения, обучение пациентов, дозирование с пищей или в то время, когда ложатся спать, и избежание алкоголя, горячих напитков, острой пищи и горячих ванн или душа близко ко времени или после введения дозы лекарственного средства. В некоторых воплощениях соединения настоящего изобретения могут иметь пониженное число случаев возникновения прилива крови или тяжести прилива крови, когда они вводятся животному, особенно человеку.
Например, соединения настоящего изобретения не вызывают прилив крови у самцов модели мышей C57BL/6, по определению данными лазерной Допплеровской расходо- или стокометрии, при введении в дозах вплоть до 100 мг/кг, и даже более предпочтительно при введении в дозах до 200, 300 или даже 500 мг/кг.
В некоторых воплощениях соединения настоящего изобретения могут характеризоваться вызыванием меньшего прилива крови при введении перорально по сравнению с количеством, эквивалентным молярному количеству NIASPAN® (таблетки длительного высвобождения ниацина, Abbott Laboratories). В некоторых воплощениях соединения настоящего изобретения при введении перорально популяции обычных пациентов показывают снижение числа пациентов с проявлением приливов крови большим, чем или равным 5 по визуальной аналоговой шкале при оральном приеме по сравнению с эквивалентным молярным количеством NIASPAN®.
Настоящее изобретение относится также к способу лечения болезней, расстройств или состояний, выбранных их группы, состоящей из гиперлипидемии, гиперхолестеринемии, гиподистрофии, дислипидемии, атеросклероза и болезни коронарных артерий, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтической композиции настоящего изобретения.
Еще один аспект настоящего изобретения относится к способу лечения заболевания, расстройства или состояния, выбранного из группы, состоящей из метаболического синдрома, ожирения, болезни жировой дистрофии печени и диабета, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтической композиции настоящего изобретения.
Еще один аспект настоящего изобретения относится к способу поднятия уровней липопротеина высокой плотности (HDL) в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтической композиции настоящего изобретения.
Еще один аспект настоящего изобретения относится к способу снижения уровней липопротеина низкой плотности (LDL) или снижения уровней липопротеина (а) в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтической композиции настоящего изобретения.
Еще один аспект настоящего изобретения относится к способу увеличения общих концентраций адипонектина в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтической композиции настоящего изобретения.
Еще один аспект настоящего изобретения относится к способу лечения заболевания, расстройства или состояния, выбранного из группы, состоящей из застойной сердечной недостаточности, сердечнососудистого заболевания, гипертензии, коронарной сердечной болезни, стенокардии, пеллагры, синдрома Хартнупа, карциноидного синдрома, артериальной окклюзивной болезни, гипотироидизма, сужения кровеносных сосудов, остеоартрита, равматоидного артрита, болезни Альцгеймера, расстройств периферической и центральной нервной системы, гематологических болезней, рака, воспаления, респираторных болезней и желудочно-энтерологических болезней, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтической композиции настоящего изобретения.
Дополнительные аспекты, воплощения и преимущества изобретения подробно обсуждаются ниже.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1. Следы масс-спектроскопии жидкостной хроматографии (LC-MS) для ARI-001, полученного от Shanghai SpeedChem (Шанхай, Китай). Для определения чистоты использовали УФ абсорбцию при 215 нм. Область пика при 3,59 мин представляет 98,9% общей области пика следов.
Фигура 2. Относительная стабильность ARI-001 в условиях хранения, как определено в испытании. Вычисляли общую площадь, представляющую пик для ARI-001, а затем нормализовали к общей площади пиков, идентифицированной по следам LC-MS. Это выражалось как % общих пиков. Верхний график, порошковый состав препарата; (■) стандартные условия: 50°С, никакой отмеченной влажности. Нижний график, жидкая форма препарата; (◊) комнатная температура (кт); (■) 4°С; (▲) 20°С.
Фигура 3. Покрытые FPLC следы объединенной плазмы от хомяков, содержащихся или на нормальной еде (жирная линия), или на еде с высоким содержанием жира + высокое содержание сахара (HF/HS) (пунктирная линия).
Фигура 4. FPLC следы объединенной плазмы от хомяков, получающих наполнитель (жирная линия), 1200 мг/кг (mpk) ниацин (пунктирная линия), или 2240 мг/кг ARI-001 (менее жирная линия).
Фигура 5. Изменения липидного параметра как функция дозы ARI-001 (представлено в ммол/кг/d). ARI-001 (■ и соединяющая линия) демонстрирует зависимое от дозы действие на величины липида у HF/HS хомяков. Ниацин (◊) показал значение по отношению к изменениям общего холестерина (ТС), липопротеина высокой плотности (HDL), и только липопротеина низкой плотности (LDL).
Фигуры 6 и 7. Корреляция между параметрами липида и концентрацией ARI-001 в плазме. Каждый параметр липида достигал статистически значимых корреляций с концентрациями ARI-001, за исключением HDL, который был статистически значимым и положительным. Нарисована линия линейной корреляции (жирная линия), а также зона 95% достоверности корреляционной линии (менее жирные кривые). Для каждой серии данных даны коэффициенты корреляции Пирсона, с 95% интервалом достоверности в скобках. Р-величины являются результатами двухвыборочных непарных t-тестов.
Фигура 8 (Левый график). Корреляция между концентрациями в печени и плазме ARI-001 у HF/HS хомяков с дозами в течение 18 дней. Величины для каждой ткани трансформированы с помощью логарифма, и следовательно, оси являются безразмерными. (Правый график) Корреляция между концентрациями в адипозной ткани и плазме у тех же животных. Адипозные концентрации трансформированы с помощью функции “логарифм + 1”, что делает любые величины между -1 и 0 положительными величинами. Это делается для ясности и никоим образом не искажает распределение серии данных.
Фигуры 9-11. Корреляции между концентрациями в печени ARI-001 у HF/HS хомяков и величинами различных параметров липида; также, корреляции между концентрациями в адипозной ткани ARI-001 у HF/HS хомяков и величинами различных липидных параметров. Все величины представляют собой логарифм липидных величин против логарифма концентраций в ткани. Корреляции представлены между концентрацией в ткани и липидными величинами у тех же животных. Адипозные концентрации трансформированы с помощью функции “логарифм + 1”, что делает любые величины между -1 и 0 положительными величинами. Это делается для ясности и никоим образом не искажает распределение серии данных.
Фигуры 12А и В. Параметры испытания функции печени от хомяков, которым давали корм с высоким содержанием жира и дозировали орально наполнитель, ниацин или ARI-001 в течение 18 дней. Изменения в процентах даны относительно наполнителя. Р-величины сообщаются от двухвыборочных непарных t-тестов по сравнению с наполнителем. Фигура 12А, аспартатаминотрансфераза (AST). Фигура 12В, аланинаминотрансфераза (ALT).
Фигура 13. Величины глюкозы, измеренные в плазме от 18-дневного исследования HF/HS хомяков, которым давали ежедневно дозу наполнителя, ниацина или ARI-001. Изменения в процентах сообщаются по сравнению с величинами в группе с наполнителем. Р-величина определяется от двухвыборочного непарного t-теста.
Фигура 14. Концентрации ARI-001 в плазме у мышей, дозированных единственным введением ARI-001 или перорально (РО) или интраперитонеально (внутрибрюшинно) (IP).
Фигуры 15А-15D. Концентрации в плазме ARI-001 у мышей, дозированных множественным ежедневным введением ARI-001 в течение 30 последовательных дней. Использовали четыре различные дозы. Каждая мышь получала одну и ту же указанную дозу ARI-001 каждый день в течение 30 дней.
Фигуры 16А и 16В. Суммарные параметры Смакс и AUC для ARI-001 от 30-дневного исследования с множественным введением мышам дикого типа. Величины даны на графике в виде среднего со стандартной погрешностью. Не было никакого значительного изменения в любом параметре как функции от времени (○) 996 мг/кг/день; (□) 1493 мг/кг/день; (∆) 2240 мг/кг/день; (◊) 3360 мг/кг/день.
Фигура 17. Концентрации ARI-001 в плазме у хомяков Golden Syrian на корме с высоким содержанием жира/высоким содержанием сахара после единственного введения ARI-001 при 5,9 ммол/кг.
Фигура 18. Концентрации ARI-001 в плазме у голодавших обезьян после единственного введения ARI-001 или (♦) 96 мг/кг внутривенно (IV), или (○) 288 мг/кг (mpk) перорально (РО). Y-ось представляет логарифмическую шкалу для ясности. (Вставка): Те же данные, и y-осью в линейном масштабе.
Фигура 19. Концентрации ARI-001 в плазме после единственного введения 288 мг/кг перорально у (♦) кормленых обезьян или (○) голодавших обезьян. Величины являются средними со стандартной погрешностью.
Фигура 20. Концентрации ARI-001 в плазме после повторного ежедневного введения 288 мг/кг перорально у голодавших обезьян. Величины являются средними со стандартной погрешностью. (♦) Пробы показаны на 1 день. (○) Пробы показаны на 7 день.
Фигура 21. ARI-001 не пополняет бета-аррестином клеточную мембрану клеток, экспрессирующих ниациновый рецептор GPR109A. Лиганд относится к ниацину или ARI-001, как указано. RLU, относительно небольшие звенья, по данным измерения при хемилюминесцентном считывании в анализе на активность рецептора в сочетании с G-протеином.
Фигура 22. Ответная реакция дозы на понижение триглицерида с одиночными пероральными дозами ARI-001 у пациентов людей. Процентным изменением является среднее изменение в процентах триглицеридов через 4 часа после введения дозы.
Фигура 23. Концентрация ARI-001 в сыворотке на протяжении времени после одиночного перорального введения указанных количеств ARI-001 людям.
ПОДРОБНОЕ ОПИСАНИЕ
Один аспект изобретения относится к ниациновым аналогам для использования при повышении уровней сывороточного HDL у млекопитающих. В некоторых воплощениях соединения изобретения имеют способность повышения HDL равную или большую, чем ниацин, имея при этом меньшую или никакой склонности вызывать приливы крови, нежелательное побочное действие самого ниацина, когда он используется в дозах, достаточных для поднятия уровней сывороточного HDL. Некоторые не вызывающие приливов крови аналоги ниацина описаны в публикации патентной заявки США № 2009/0312355, которая целиком включена в описание путем ссылки на нее. В некоторых воплощениях ключевые структурные формулы соединений, описанных здесь, явно включают наличие гетероциклилалкильной или гетероаралкильной группы пара к карбоксильной группе в ниацине.
ОПРЕДЕЛЕНИЯ
Для удобства здесь собраны некоторые термины, применяемые в описании, примерах и прилагаемых пунктах формулы изобретения. Все определения, данные и используемые здесь, заменяют словарные определения, определения в документах, включенных путем ссылки на них, и/или обычные значения определяемых терминов.
Неопределенные артикли “a” и “an” используются здесь, чтобы сослаться на один или более чем один (т.е. на, по меньшей мере, один) грамматический объект. Например, английское слово “элемент” (с неопределенным артиклем) означает один элемент или более чем один элемент.
Фраза “и/или”, используемая здесь в описании и в формуле изобретения, должна пониматься как означающая “один из двух или оба” из элементов, в сочетании, т.е. элементов, которые присутствуют в сочетании в некоторых случаях и характерно присутствуют в других случаях. Таким же образом следует толковать множественные элементы, перечисляемые с помощью “и/или”, т.е. таким образом “один или более из элементов” в сочетании. Могут необязательно присутствовать другие элементы, отличные от указанных конкретно с использованием слов “и/или”, относятся ли они или не относятся к элементам, указанным конкретно. Таким образом, как не ограничивающий пример, ссылка на “А и/или В”, используемая в сочетании с не ограничивающим словом, таким как “включающий”, может относиться в одном воплощении только к А (необязательно включая иные элементы, чем В); в еще одном воплощении только к В (необязательно включая иные элементы, чем А); в еще одном воплощении к обоим А и В (необязательно включая другие элементы); и прочее.
Используемое здесь в описании и в формуле изобретения слово “или” следует понимать как имеющее то же значение как в случае “и/или”, определенном выше. Например, когда в перечне указываются раздельные слова или пункты, “или” или “и/или” следует понимать как указывающие включительно, т.е. включение, по меньшей мере, одного, но также включая более чем один, из ряда или перечня элементов, и, необязательно, дополнительных не перечисленных элементов. Только термины, ясно указанные в противоположном смысле, такие как “только один из” или “точно один из”, или, используемые в формуле изобретения, “состоящий из”, будут относиться к включению точно одного элемента из ряда или перечня элементов. В общем, термин “или”, используемый здесь, следует понимать только как включающий исключительные альтернативы (т.е. “один или другой, но не оба”), когда ему предшествуют термины исключительности, такие как “один из двух”, “один из”, “только один из”, или “точно один из”. “Состоящий по существу из”, используемый в формуле изобретения, следует понимать в его обычном значении, используемом в области патентного закона.
Используемую здесь в описании и формуле изобретения фразу “по меньшей мере, один” в ссылке на список одного или более элементов, следует понимать как обозначающую, по меньшей мере, один элемент, выбранный из любого одного или более элементов в перечне элементов, но необязательно включая, по меньшей мере, один из каждого и любого элемента, перечисленного конкретно в списке элементов, и не исключая любые сочетания элементов в перечне элементов. Данное определение также допускает, что могут необязательно присутствовать элементы иные, чем элементы, конкретно указанные в списке элементов, к которым относится фраза “по меньшей мере, один”, относится ли он или не относится к тем конкретно указанным элементам. Таким образом, как не ограничивающий пример “по меньшей мере, один из А и В” (или, эквивалентно, “по меньшей мере, один из А или В”, или эквивалентно “по меньшей мере, один из”) может относиться в одном воплощении к, по меньшей мере одному, необязательно включающему более чем один, А, без никакого присутствующего В (и необязательно включающему иные элементы, чем В); в другом воплощении к, по меньшей мере, одному, необязательно включающему более одного, В, без никакого присутствующего А (и необязательно включающему иные элементы, чем А); в еще одном воплощении к, по меньшей мере, одному, необязательно включающему более, чем один, А, и, по меньшей мере, один, необязательно включающему более, чем один, В (и необязательно включающему другие элементы); и прочее.
Следует понимать также, что если не указывается ясно противоположное, в любых способах, заявленных здесь, которые включают более одной стадии или действия, порядок стадий или действий способа необязательно ограничивается порядком, в котором стадии или действия способа перечисляются.
В пунктах формулы изобретения, также как и в приведенном выше описании, все промежуточные фразы, такие как “включающий” (англ. “comprising” и “including”), “несущий”, “имеющий”, “содержащий”, “вовлекающий или затрагивающий”, “владеющий”, “составленный из”, и аналогичные, следует понимать не в ограничительном смысле, т.е. как означающее включение, но не как ограничение указанным. Только промежуточные фразы “состоящий из” и “состоящий по существу из” должны быть закрытыми или полузакрытыми промежуточными фразами, соответственно, как излагается в Manual of Patent Examining Procedures, Section 2111.03 Патентного ведомства Соединенных Штатов.
Термины “совместное введение” и “совместное назначение” относятся и к одновременному введению (введение двух или более терапевтических агентов в одно и тоже время), и к введению в различное время (введение двух или более терапевтических агентов во время, отличное от времени введения дополнительно терапевтического агента или агентов), пока терапевтические агенты в одно и то же время присутствуют до некоторой степени в организме пациента.
Термин “сольват” относится к фармацевтически приемлемой форме указанного соединения, с одной или более молекулами растворителя, которая сохраняет биологическую эффективность такого соединения. Примеры сольватов включают соединения изобретения в сочетании с такими растворителями, как, например, вода (образуя гидрат), изопропанол, этанол, метанол, диметилсульфоксид, этилацетат, уксусная кислота, этаноламин или ацетон. В них включены также составы сольватных смесей, такие как соединение изобретения в сочетании с двумя или более растворителями.
Имеется в виду, что определение каждого выражения, например, алкил, m, n и аналогичных, когда оно имеет место более чем один раз, в любой структуре, является независимым от определения где-либо еще в той же структуре.
Очевидно понятно, что “замещение” или “замещенный чем-то” включает подразумеваемое условие, что такое замещение находится в соответствии с допустимой валентностью замещенного атома и заместителя, и что замещение приводит в результате к стабильному соединению, например, соединению, которое самопроизвольно не подвергается преобразованию, такому как перегруппировка, циклизация, элиминирование или другая реакция.
Предполагается, что термин “замещенный” включает также все допустимые заместители органических соединений. В широком аспекте, допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Иллюстративные заместители включают, например, заместители, описанные здесь ниже. Допустимые заместители могут быть одним или более и одними и теми же или различными для соответствующих органических соединений. Для целей данного изобретения гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные здесь, которые удовлетворяют валентностям гетероатомов.
Термин “низший” применительно к любой из групп, перечисленных ниже, указывает, что группа содержит менее чем семь атомов углерода (т.е. шесть углеродов или менее). Например, “низший алкил” относится к алкильной группе, содержащей 1-6 атомов углеродов, а “низший алкенил” относится к алкенильной группе, содержащей 2-6 атомов углерода.
Термин “ненасыщенный”, используемый здесь, относится к соединениям и/или группам, которые имеют, по меньшей мере, одну углерод-углеродную двойную связь или углерод-углеродную тройную связь.
Термин “алифатический”, используемый здесь, относится к соединениям и/или группам, которые являются линейными или разветвленными, но не циклическими (называемыми также “ациклическими” или группами “с открытой цепью”).
Термин “циклический”, используемый здесь, относится к соединениям и/или группам, которые имеют одно кольцо, два или более колец (например, спиро, конденсированных, мостиковых). “Моноциклический” относится к соединениям и/или группам с одним кольцом; а “бициклический” относится к соединениям и/или группам с двумя кольцами.
Термин “ароматический” относится к плоской или полициклической структуре, характеризуемой циклически сопряженным молекулярным фрагментом, содержащим 4n+2 электрона, где n представляет собой абсолютную величину целого числа. Ароматические молекулы, содержащие конденсированные или соединенные кольца, называются также бициклическими ароматическими кольцами. Например, бициклические ароматические кольца, содержащие гетероатомы в углеводородной кольцевой структуре, называют также бициклическими гетероарильными кольцами.
Термин “углеводород”, используемый здесь, относится к органическому соединению, состоящему целиком из водорода и углерода.
Для целей данного изобретения химические элементы идентифицируются или определяются в соответствии с Периодической таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 67th Ed., 1986-87, внутренняя крышка переплета.
Термин “гетероатом”, используемый здесь, является признанным в данной области и относится к атому любого элемента, отличному от углерода или водорода. Иллюстративные гетероатомы включают бор, азот, кислород, фосфор, серу и селен.
Термин “алкил” обозначает алифатические или циклический углеводородный радикал, содержащий от 1 до 20, от 1 до 15, или от 1 до 10 атомов углерода. Представительные примеры алкила включают, но не ограничиваются ими, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изо-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, 2-метилциклопентил, 1-(1-этилциклопропил)этил и 1-циклогексилэтил.
Термин “циклоалкил” представляет подгруппу алкила, который относится к циклическому углеводородному радикалу, содержащему от 3 до 15, 3-10 или 3-7 атомов углерода. Представительные примеры циклоалкила включают, но не ограничиваются ими, циклопропил и циклобутил.
Термин “алкенил”, используемый здесь, обозначает углеводородный радикал с прямой или разветвленной цепью, содержащий от 2 до 10 атомов углерода и содержащий, по меньшей мере, одну углерод-углеродную двойную связь, образуемую удалением двух водородов. Представительные примеры алкенила включают, но не ограничиваются ими, этенил, 2-пропенил, 2-метил-2-пропенил, 3-бутенил, 4-пентенил, 5-гексенил, 2-гептенил, 2-метил-1-гептенил и 3-деценил.
Термин “алкинил”, используемый здесь, обозначает углеводородный радикал с прямой или разветвленной цепью, содержащий от 2 до 10 атомов углерода и содержащий, по меньшей мере, одну углерод-углеродную тройную связь. Представительные примеры алкинила включают, но не ограничиваются ими, ацетиленил, 1-пропинил, 2-пропинил, 3-бутинил, 2-пентинил и 1-бутинил.
Термин “алкилен” является признанным в данной области и, как он используется здесь, относится к дирадикалу, получаемому удалением двух атомов водорода алкильной группы, определенной выше.
Термин “карбоциклил”, как здесь используется, обозначает моноциклический или полициклический (например, бициклический, трициклический и др.) углеводородный радикал, содержащий от 3 до 12 атомов углерода, т.е. полностью насыщенный, или имеет одну или более ненасыщенных связей, и во избежание сомнения, степень ненасыщения не является результатом в ароматической кольцевой системе (например, фениле). Примеры карбоциклильных групп включают 1-циклопропил, 1-циклобутил, 2-циклопентил, 1-циклопентенил, 3-циклогексил, 1-циклогексенил и 2-циклопентенилметил.
Термин “гетероциклил”, используемый здесь, относится к радикалу неароматической кольцевой системы, включающей, но не ограниченной ими, моноциклические, бициклические и трициклические кольца, которые могут быть полностью насыщенными или которые могут содержать одно или более звеньев ненасыщения, во избежание сомнения, степень ненасыщения не является результатом в ароматической кольцевой системе, и иметь 3-12 атомов, включающих, по меньшей мере, один гетероатом, такой как атом азота, кислорода или серы. Для целей иллюстрации примерами, которые не следует рассматривать как ограничивающие объем данного изобретения, следующее является примерами гетероциклических колец: азиридинил, азиринил, оксиранил, тииранил, тииренил, диоксиранил, диазиринил, азетил, оксетанил, оксетил, тиетанил, тиэтил, диазетидинил, диоксетанил, диоксетенил, дитиэтанил, дитиэтил, фурил, диоксаланил, пирролил, оксазолил, тиазолил, имидазолил, оксадиазолил, тиадиазолил, триазолил, триазинил, изотиазолил, изоксазолил, тиофенил, пиразолил, тетразолил, пиридил, пиридазинил, пиримидинил, пиразинил, триазинил, тетразинил, хинолинил, изохинолинил, хиноксалинил, хиназолинил, пиридопиразинил, бензоксазолил, бензотиофенил, бензимидазолил, бензотиазолил, бензоксадиазолил, бензтиадиазолил, индолил, бензтриазолил, нафтиридинил, азепины, азетидинил, морфолинил, оксопиперидинил, оксопирролидинил, пиперазинил, пиперидинил, пирролидинил, хиниклудинил, тиоморфолинил, тетрагидропиранил и тетрагидрофуранил. Гетероциклильные группы изобретения замещены 0, 1, 2, 3, 4 или 5 заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, галогена, галогеналкила, фторалкила, гидрокси, алкокси, алкиенилокси, алкинилокси, карбоциклилокси, гетероциклилокси, галогеналкокси, фторалкилокси, сульфгидрила, алкилтио, галогеналкилтио, фторалкилтио, алкиенилтио, алкинилтио, сульфоновой кислоты, алкилсульфонила, галогеналкилсульфонила, фторалкилсульфонила, алкенилсульфонила, алкинилсульфонила, алкоксисульфонила, галогеналкоксисульфонила, фторалкоксисульфонила, алкенилоксисульфонила, алкинилоксисульфонила, аминосульфонила, сульфиновой кислоты, алкилсульфинила, галогеналкилсульфинила, фторалкилсульфинила, алкенилсульфинила, алкинилсульфинила, алкоксисульфинила, галогеналкоксисульфинила, фторалкоксисульфинила, алкенилоксисульфинила, алкинилоксисульфинила, аминосульфинила, формила, алкилкарбонила, галогеналкилкарбонила, фторалкилкарбонила, алкенилкарбонила, алкинилкарбонила, карбоксила, алкоксикарбонила, галогеналкоксикарбонила, фторалкоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, галогеналкилкарбонилокси, фторалкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилсульфонилокси, галогеналкилсульфонилокси, фторалкилсульфонилокси, алкенилсульфонилокси, алкинилсульфонилокси, галогеналкоксисульфонилокси, фторалкоксисульфонилокси, алкенилоксисульфонилокси, алкинилоксисульфонилокси, алкилсульфинилокси, галодиалкилсульфинилокси, фторалкилсульфинилокси, алкенилсульфинилокси, алкинилсульфинилокси, алкоксисульфинилокси, галогеналкоксисульфинилокси, фторалкоксисульфинилокси, алкенилоксисульфинилокси, алкинилоксисульфинилокси, аминосульфинилокси, амино, амидо, аминосульфонила, аминосульфинила, циано, нитро, азидо, фосфинила, фосфорила, силила, силилокси, и любых из названных заместителей, связанных с гетероциклильной группой через алкиленовый фрагмент (например, метиленовый).
Термин “арил”, используемый здесь, обозначает фенильную, нафтильную, фенантренильную или антраценильную группу. Арильные группы настоящего изобретения могут быть необязательно замещенными 1, 2, 3, 4 или 5 заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, галогена, галогеналкила, фторалкила, гидрокси, алкокси, алкиенилокси, алкинилокси, карбоциклилокси, гетероциклилокси, галогеналкокси, фторалкилокси, сульфгидрила, алкилтио, галогеналкилтио, фторалкилтио, алкиенилтио, алкинилтио, сульфоновой кислоты, алкилсульфонила, галогеналкилсульфонила, фторалкилсульфонила, алкенилсульфонила, алкинилсульфонила, алкоксисульфонила, галогеналкоксисульфонила, фторалкоксисульфонила, алкенилоксисульфонила, алкинилоксисульфонила, аминосульфонила, сульфиновой кислоты, алкилсульфинила, галогеналкилсульфинила, фторалкилсульфинила, алкенилсульфинила, алкинилсульфинила, алкоксисульфинила, галогеналкоксисульфинила, фторалкоксисульфинила, алкенилоксисульфинила, алкинилоксисульфинила, аминосульфинила, формила, алкилкарбонила, галогеналкилкарбонила, фторалкилкарбонила, алкенилкарбонила, алкинилкарбонила, карбоксила, алкоксикарбонила, галогеналкилоксикарбонила, фторалкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, галогеналкилкарбонилокси, фторалкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилсульфонилокси, галогеналкилсульфонилокси, фторалкилсульфонилокси, алкенилсульфонилокси, алкинилсульфонилокси, галогеналкоксисульфонилокси, фторалкоксисульфонилокси, алкенилоксисульфонилокси, алкинилоксисульфонилокси, алкилсульфинилокси, галогеналкилсульфинилокси, фторалкилсульфинилокси, алкенилсульфинилокси, алкинилсульфинилокси, алкоксисульфинилокси, галогеналкоксисульфинилокси, фторалкоксисульфинилокси, алкенилоксисульфинилокси, алкинилоксисульфинилокси, аминосульфинилокси, амино, амидо, аминосульфонила, аминосульфинила, циано, нитро, азидо, фосфинила, фосфорила, силила, силилокси, и любых из указанных заместителей, соединенных с гетероциклильной группой через алкиленовый фрагмент (например, метилен).
Термин “арилен” является признанным в данной области и, как он используется здесь, относится к дирадикалу, получаемому удалением двух атомов водорода арильного кольца, определенного выше.
Термин “арилалкил” или “аралкил”, используемый здесь, обозначает арильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через алкильную группу, определенную здесь. Представительные примеры аралкила включают, но не ограничиваются ими, бензил, 2-фенилэтил, 3-фенилпропил и 2-нафт-2-илэтил.
Термин “биарил”, используемый здесь, обозначает арил-замещенный арил, арил-замещенный гетероарил, гетероарил-замещенный арил, или гетероарил-замещенный гетероарил, в которых арил и гетероарил имеют значения, определенные здесь. Представительные примеры включают 4-(фени)фенил и 4-(4-метоксифенил)пиридинил.
Термин “гетероарил”, используемый здесь, включает радикалы ароматических кольцевых систем, включая, но не ограничиваясь ими, моноциклические, бициклические и трициклические кольца, которые имеют 3-12 атомов, включающих, по меньшей мере, один гетероатом, такой как азот, кислород или сера. Для целей иллюстрации примерами, которые не следует рассматривать как ограничивающие объем данного изобретения: аминобензимидазол, бензимидазол, азаиндолил, бензо(b)тиенил, бензимидазолил, бензофуранил, бензоксазолил, бензотиазолил, бензотиадиазолил, бензотриазолил, бензоксадиазолил, фуранил, имидазолил, имидазопиридинил, индолил, индолинил, индазолил, изоиндолинил, изоксазолил, изотиазолил, изохинолинил, оксадиазолил, оксазолил, пуринил, пиранил, пиразинил, пиразолил, пиридинил, пиримидинил, пирролил, пирроло[2,3-d]пиримидинил, пиразоло[3,4-d]пиримидинил, хинолинил, хиназолинил, триазолил, тиазолил, тиофенил, тетрагидроиндолил, тетразолил, тиадиазолил, тиенил, тиоморфолинил, триазолил или тропанил. Гетероарильные группы изобретения замещены 0, 1, 2, 3, 4 или 5 заместителями, независимо выбранными из группы, состоящей из алкила, алкиенила, алкинила, галогена, галогеналкила, фторалкила, гидрокси, алкокси, алкенилокси, алкинилокси, карбоциклилокси, гетероциклилокси, галогеналкокси, фторалкилокси, сульфгидрила, алкилтио, галогеналкилтио, фторалкилтио, алкенилтио, алкинилтио, сульфоновой кислоты, алкилсульфонила, галогеналкилсульфонила, фторалкилсульфонила, алкенилсульфонила, алкинилсульфонила, алкоксисульфонила, галогеналкоксисульфонила, фторалкоксисульфонила, алкенилоксисульфонила, алкинилоксисульфонила, аминосульфонила, сульфиновой кислоты, алкилсульфинила, галогеналкилсульфинила, фторалкилсульфинила, алкенилсульфинила, алкинилсульфинила, алкоксисульфинила, галогеналкоксисульфинила, фторалкоксисульфинила, алкенилоксисульфинила, алкинилоксисульфинила, аминосульфинила, формила, алкилкарбонила, галогеналкилкарбонила, фторалкилкарбонила, алкенилкарбонила, алкинилкарбонила, карбоксила, алкоксикарбонила, галогеналкоксикарбонила, фторалкоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, галогеналкилкарбонилокси, фторалкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилсульфонилокси, галогеналкилсульфонилокси, фторалкилсульфонилокси, алкенилсульфонилокси, алкинилсульфонилокси, галогеналкоксисульфонилокси, фторалкоксисульфонилокси, алкенилоксисульфонилокси, алкинилоксисульфонилокси, алкилсульфинилокси, галогеналкилсульфинилокси, фторалкилсульфинилокси, алкенилсульфинилокси, алкинилсульфинилокси, алкоксисульфинилокси, галогеналкоксисульфинилокси, фторалкоксисульфинилокси, алкенилоксисульфинилокси, алкинилоксисульфинилокси, аминосульфинилокси, амино, амидо, аминосульфонила, аминосульфинила, циано, нитро, азидо, фосфинила, фосфорила, силила, силилокси, и любых из указанных заместителей, соединенных с гетероарильной группой через алкиленовый фрагмент (например, метилен).
Термин “гетероарилен” является признанным в данной области и, как он используется здесь, относится к дирадикалу, получаемому удалением двух атомов водорода гетероарильного кольца, определенного выше.
Термин “гетероарилалкил” или “гетероаралкил”, используемый здесь, обозначает гетероарил, как он определен здесь, соединенный с исходным молекулярным фрагментом через алкильную группу, определенную здесь. Представительные примеры гетероарилалкила включают, но не ограничиваются ими, пиридин-3-илметил и 2-(тиен-2-ил)этил.
Термин “конденсированный бициклил”, используемый здесь, обозначает радикал бициклической кольцевой системы, в которой два кольца орто-конденсированы, и каждое кольцо содержит общее число четыре, пять, шесть или семь атомов (т.е. атомы углерода и гетероатомы), включая два атома слияния, и каждое кольцо может быть полностью насыщенным, может содержать одно или более звеньев ненасыщенности, или может быть полностью ненасыщенным (например, в некоторых случаях ароматическим). Во избежание сомнений, степень ненасыщения в конденсированном бициклиле не дает в результате арильный или гетероарильный фрагмент.
Термин “галоген” обозначает -Cl, -Br, -I или -F.
Термин “галогеналкил” обозначает алкильную группу, определенную здесь, в которой, по меньшей мере, один водород, заменен галогеном, определенным здесь. Представительные примеры галогеналкила включают, но не ограничиваются ими, хлорметил, 2-фторэтил, трифторметил, пентафторэтил и 2-хлор-3-фторпентил.
Термин “фторалкил” обозначает алкильную группу, определенную здесь, в которой несколько или все атомы водорода заменены атомами фтора.
Термин “галогеналкилен”, используемый здесь, относится к дирадикалу, получаемому удалением двух атомов водорода галогеналкильной группы, определенной выше.
Термин “гидрокси”, используемый здесь, обозначает -OH группу.
Термин “алкокси”, используемый здесь, обозначает алкильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через атом кислорода. Представительные примеры алкокси включают, но не ограничиваются ими, метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси, и гексилокси. Термины “алкиенилокси”, “алкинилокси”, “карбоциклилокси” и “гетероциклилокси” определяются аналогично.
Термин “галогеналкокси”, используемый здесь, обозначает алкокси группу, определенную здесь, в которой, по меньшей мере, один водород заменен галогеном, определенным здесь. Представительные примеры галогеналкокси включают, но не ограничиваются ими, хлорметокси, 2-фторэтокси, трифторметокси и пентафторэтокси. Термин “фторалкилокси” имеет определенные значения.
Термин “арилокси”, используемый здесь, обозначает арильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через кислород. Термин “гетероарилокси”, используемый здесь, обозначает гетероарильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через кислород. Термин “гетероарилокси” имеет значения, определенные ранее.
Термин “арилалкокси” или “арилалкилокси”, используемый здесь, обозначает арилалкильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через кислород. Термин “гетероарилалкокси” имеет значения, определенные ранее. Представительные примеры арилокси и гетероарилалкокси включают, но не ограничиваются ими, 2-хлорфенилметокси, 3-трифторметилфенилэтокси и 2,3-диметилпиридинилметокси.
Термин “сульфгидрил” или “тио”, используемый здесь, обозначает -SH группу.
Термин “алкилтио”, используемый здесь, обозначает алкильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через серу. Представительные примеры алкилтио включают, но не ограничиваются ими, метилтио, этилтио, трет-бутилтио и гексилтио. Термины “галогеналкилтио”, “фторалкилтио”, “алкиенилтио”, “алкинилтио”, “карбоциклилтио” и “гетероциклилтио” имеют определенные ранее значения.
Термин “арилтио”, используемый здесь, обозначает арильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через серу. Термин “гетероарилтио” имеет определенные ранее значения.
Термин “арилалкилтио” или “аралкилтио”, используемый здесь, обозначает арилалкильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через серу. Термин “гетероарилалкилтио” имеет значения, определенные аналогичным образом.
Термин “сульфонил”, используемый здесь, относится к -S(=O)2- группе.
Термин “сульфоновая кислота”, используемый здесь, относится к -S(=O)2OH.
Термин “алкилсульфонил”, используемый здесь, обозначает алкильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через сульфонильную группу, определенную здесь. Представительные примеры алкилсульфонила включают, но не ограничиваются ими, метилсульфонил и этилсульфонил. Термины “галогеналкилсульфонил”, “фторалкилсульфонил”, “алкенилсульфонил”, “алкинилсульфонил”, “карбоциклилсульфонил”, “гетероциклилсульфонил”, “арилсульфонил”, “аралкилсульфонил”, “гетероарилсульфонил” и “гетероаралкилсульфонил” имеют значения, определенные аналогично.
Термин “алкоксисульфонил”, используемый здесь, обозначает алкокси группу, определенную здесь, соединенную с исходным молекулярным фрагментом через сульфонильную группу, определенную здесь. Представительные примеры алкоксисульфонила включают, но не ограничиваются ими, метоксисульфонил, этоксисульфонил и пропоксисульфонил. Термины “галогеналкоксисульфонил”, “фторалкоксисульфонил”, “алкенилоксисульфонил”, “алкинилоксисульфонил”, “карбоциклилоксисульфонил”, “гетероциклилоксисульфонил”, “арилоксисульфонил”, “аралкилоксисульфонил”, “гетероарилоксисульфонил” и “гетероаралкилоксисульфонил” имеют определенные аналогично значения.
Термины “трифлил”, “тозил”, “мезил” и “нонафлил” являются признанными в данной области и относятся к трифторметансульфонилу, п-толуолсульфлонилу, метансульфонилу и нонафторбутансульфонильной группам, соответственно. Термины “трифлат”, “тозилат”, “мезилат” и “нонафлат” являются признанными в данной области и относятся к трифторметансульфонатной сложноэфирной, п-толуолсульфонатной, метансульфонатной сложноэфирной и нонафторбутансульфонатной сложноэфирной функциональным группам и молекулам, которые содержат указанные группы, соответственно.
Термин “аминосульфонил”, используемый здесь, обозначает аминогруппу, определенную здесь, соединенную с исходным молекулярным фрагментом через сульфонильную группу.
Термин “сульфинил”, используемый здесь, относится к группе -S(=O)-. Сульфинильные группы имеют значения, определенные выше для сульфонильных групп. Термин “сульфиновая кислота”, используемый здесь, относится к группе -S(=O)OH.
Термин “окси” относится к -О- группе.
Термин “карбонил”, используемый здесь, относится к -С(=О)- группе.
Термин “тиокарбонил”, используемый здесь, относится к -C(=S)- группе.
Термин “формил”, используемый здесь, обозначает -C(=O)H группу.
Термин “ацил”, используемый здесь, относится к любой группе или радикалу формы -C(=O)R, где R является органической группой. Примером ацильной группы является ацетильная группа (-C(=O)CH3).
Термин “алкилкарбонил”, используемый здесь, обозначает алкильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через карбонильную группу, определенную здесь. Представительные примеры алкилкарбонила включают, но не ограничиваются ими, ацетил, 1-оксопропил, 2,2-диметил-1-оксопропил, 1-оксобутил и 1-оксопентил. Термины “галогеналкилкарбонил”, “фторалкилкарбонил”, “алкенилкарбонил”, “алкинилкарбонил”, “карбоциклилкарбонил”, “гетероциклилкарбонил”, “арилкарбонил”, “аралкилкарбонил”, “гетероарилкарбонил” и “гетероаралкилкарбонил” имеют значения, определяемые аналогично.
Термин “карбоксил”, используемый здесь, обозначает -CO2H группу.
“Изостер карбоксильной группы”, используемый здесь, относится к группе, которая является изостерической к карбоксильной группе. Примеры изостеров карбоксильной группы включают тетразолил, оксазолидинонил, 3-изоксазолил, гидроксиизоксазолил, сульфоновую кислоту, сульфиновую кислоту, ацилсульфонамид, фосфоновую кислоту, фосфиновую кислоту, гидантоин, пирролидионил, бороновую кислоту, гидроксамовую кислоту, ацилцианамид и оксадиазолонил.
Термин “алкоксикарбонил”, используемый здесь, обозначает алкокси группу, определенную здесь, соединенную с исходным молекулярным фрагментом через карбонильную группу, определенную здесь. Представительные примеры алкоксикарбонила включают, но не ограничиваются ими, метоксикарбонил, этоксикарбонил и трет-бутоксикарбонил. Термины “галогеналкоксикарбонил”, “фторалкоксикарбонил”, “алкенилоксикарбонил”, “алкинилоксикарбонил, карбоциклилоксикарбонил”, “гетероциклилоксикарбонил”, “арилоксикарбонил”, “аралкилоксикарбонил”, “гетероарилоксикарбонил” и “гетероаралкилоксикарбонил” имеют значения, определяемые аналогично.
Термин “алкилкарбонилокси”, используемый здесь, обозначает алкилкарбонильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через атом кислорода. Представительные примеры алкилкарбонилокси включают, но не ограничиваются ими, ацетилокси, этилкарбонилокси и трет-бутилкарбонилокси. Термины “галогеналкилкарбонилокси”, “фторалкилкарбонилокси”, “алкенилкарбонилокси”, “алкинилкарбонилокси, карбоциклилкарбонилокси”, “гетероциклилкарбонилокси”, “арилкарбонилокси”, “аралкилкарбонилокси”, “гетероарилкарбонилокси” и “гетероаралкилкарбонилокси” имеют значения, определяемые аналогично.
Термин “алкилсульфонилокси”, используемый здесь, обозначает алкилсульфонильную группу, определенную здесь, соединенную с исходным молекулярным фрагментом через атом кислорода. Термины “галогеналкилсульфонилокси”, “фторалкилсульфонилокси”, “алкенилсульфонилокси”, “алкинилсульфонилокси”, карбоциклилсульфонилокси”, “гетероциклилсульфонилокси”, “арилсульфонилокси”, “аралкилсульфонилокси”, “гетероарилсульфонилокси”, “гетероаралкилсульфонилокси”, “галогеналкоксисульфонилокси”, “фторалкоксисульфонилокси”, “алкенилоксисульфонилокси”, “алкинилоксисульфонилокси”, карбоциклилоксисульфонилокси”, “гетероциклилоксисульфонилокси”, “арилоксисульфонилокси”, “аралкилоксисульфонилокси”, “гетероарилоксисульфонилокси” и “гетероаралкилоксисульфонилокси” имеют значения, определяемые аналогично.
Термин “амино” или “амин”, используемый здесь, относится к -NH2 и ее замещенным производным, где один или оба водорода независимо заменены заместителями, выбранными из группы, состоящей из алкила, галогеналкила, фторалкила, алкенила, алкинила, карбоциклила, гетероциклила, арила, аралкила, гетероарила, гетероаралкила, алкилкарбонила, галогеналкилкарбонила, фторалкилкарбонила, алкенилкарбонила, алкинилкарбонила, карбоциклилкарбонила, гетероциклилкарбонила, арилкарбонила, аралкилкарбонила, гетероарилкарбонила, гетероаралкилкарбонила и сульфонильных и сульфинильных групп, определенных выше; или, когда оба атома водорода вместе заменены алкиленовой группой (образуя кольцо, которое содержит азот). Представительные примеры включают, но не ограничиваются ими, метиламино, ацетиламино и диметиламино.
Термин “амидо”, используемый здесь, обозначает аминогруппу, определенную здесь, соединенную с исходным молекулярным фрагментом через карбонил.
Термин “циано”, используемый здесь, обозначает -C≡N группу.
Термин “нитро”, используемый здесь, обозначает -NO2 группу.
Термин “азидо”, используемый здесь, обозначает -N3 группу.
Термин “фосфинил” или “фосфино”, используемый здесь, включает -PH3 и ее замещенные производные, в которых один, два или три атома водорода независимо заменены заместителями, выбранными из группы, состоящей из алкила, галогеналкила, фторалкила, алкенила, алкинила, карбоциклила, гетероциклила, арила, аралкила, гетероарила, гетероаралкила, алкокси, галогеналкокси, фторалкилокси, алкенилокси, алкинилокси, карбоциклилокси, гетероциклилокси, арилокси, аралкилокси, гетероарилокси, гетероаралкилокси и амино.
Термин “фосфорил”, используемый здесь, относится к группе -P(=O)OH2 и ее замещенным производным, где один или оба гидроксила независимо заменены заместителями, выбранными из группы, состоящей из алкила, галогеналкила, фторалкила, алкенила, алкинила, карбоциклила, гетероциклила, арила, аралкила, гетероарила, гетероаралкила, алкокси, галогеналкокси, фторалкилокси, алкенилокси, алкинилокси, карбоциклилокси, гетероциклилокси, арилокси, аралкилокси, гетероарилокси, гетероаралкилокси и амино.
Термин “силил”, используемый здесь, включает H3Si- и ее замещенные производные, в которых один, два или три атома водорода независимо заменены заместителями, выбранными из алкила, галогеналкила, фторалкила, алкенила, алкинила, карбоциклила, гетероциклила, арила, аралкила, гетероарила и гетероаралкила. Представительные примеры включают триметилсилил (TMS), трет-бутилдифенилсилил (TBDPS), трет-бутилдиметилсилил (TBS/TBDMS), триизопропилсилил (TIPS) и [2-(триметилсилил)этокси]метил (SEM).
Термин “силилокси”, используемый здесь, обозначает силильную группу, определенную здесь, соединенную с исходной молекулой атомом кислорода.
Сокращения Me, Et, Ph, Tf, Nf, Ts и Ms представляют метил, этил, фенил, трифторметансульфонил, нонафторбутансульфонил, п-толуолсульфонил и метансульфонил, соответственно. Более обширный перечень сокращений, используемых обычными специалистами в органической химии, есть в первом выпуске каждого тома Journal of Organic Chemistry; данный список символично представлен в таблице, озаглавленной Standard List of Abbreviations.
Термин “лечение”, используемый здесь, охватывает введение и/или применение одного или более описываемых здесь соединений субъекту для профилактики или возможности справляться с состоянием пациента и/или излечения болезненного состояния. “Лечение” для данного изобретения может, но не должно, обеспечить курс лечения; скорее “лечение” может быть в форме умения справляться с состоянием. Когда соединения, описываемые здесь, используются для лечения нежелательных пролиферирующих клеток, включая раковые заболевания, “лечение” включает частичное или полное разрушение нежелательных пролиферирующих клеток с минимальными деструктивными эффектами по отношению к нормальным клеткам. Желаемым механизмом лечения нежелательных быстро пролиферирующих клеток, включающих раковые клетки, на клеточном уровне является апоптоз.
Термин “лечить”, используемый здесь, охватывает таким образом не только исцеление, но также замедление прогрессирования и/или снижение тяжести заболевания, расстройства или состояния. В одном воплощении “лечение” может охватывать “предотвращение” (или профилактику).
Термин “предотвращение”, используемый здесь, включает или профилактику или замедление наступления клинически очевидного развития болезни в целом или предотвращение или замедление наступления очевидной стадии, предшествующей началу заболевания у пациентов с риском заболеть. Это включает профилактическое лечение пациентов с риском развития болезни.
Термин “субъект” для целей лечения включает любого человека или животного, которому поставлен диагноз, или у которого имеются симптомы, или который рискует иметь развитие заболевания. Для способов профилактики субъектом является любой человек или животное.
Термин “необязательно дейтерированный”, используемый здесь, относится к любому радикалу, описанному выше, в котором один или более водородов заменены дейтерием. Примеры дейтерированного алкила включают -CD2H и -CD3.
Термин “полиол”, используемый здесь, относится к малым молекулам и полимерам, которые имеют более чем один гидроксил.
Используемый здесь “карбогидрат” (или, эквивалентно, “сахар”) представляет сахарид (включая моносахариды, олигосахариды и полисахариды) и/или молекулу (включая олигомеры и полимеры), производимую из одного или более моносахаридов, например, восстановлением карбонильных групп, окислением одной или более концевых групп карбоновых кислот, замещением одной или более гидрокси групп атомом водорода, аминогруппой, тиольной группой или сходными гетероароматическими группами, и др. Термин “карбогидрат” включает также производные данных соединений. В некоторых случаях карбогидратом может быть пентоза (т.е. имеющая 5 углеродов) или гексоза (т.е. имеющая 6 углеродов); и в некоторых случаях карбогидратом может быть олигосахарид, включающий пентозное и/или гексозное звенья, например, включая описанные выше.
“Карбогидрат” и “сахар”, используемый здесь, также включает сахаро-миметики и сахароподобные фрагменты. Сахарные миметики хорошо известны специалистам в данной области и включают те, что описаны в “Essentials of Glycobiology” Edited by Varki, A., et al, Cold Spring Harbor Laboratory Press. Cold Spring Harbor, N.Y. 2002. Например, напоминающие сахар группы, охватываемые настоящим изобретением, включают циклиты, такие как циклоалкан, содержащий одну гидроксильную группу на каждом из трех или более кольцевых атомов, как определяется конвенцией IUPAC. В других воплощениях такие цикличные фрагменты включают инозиты, такие как сцилло-инозит. Подходящие подобные сахару фрагменты включают ациклические сахарные группы. Такие группы включают линейные алкиты и эритриты, названы только некоторые из них. Очевидно понятно, что сахарные группы существуют или в циклической или в ациклической форме. Соответственно, ациклические формы сахарных групп охватываются настоящим изобретением как подходящие сахароподобные фрагменты.
Термин “политиол”, используемый здесь, относится к малым молекулам и полимерам, которые имеют более одного тиола.
СОЕДИНЕНИЯ
Ниацин, известный также как никотиновая кислота, имеет структуру
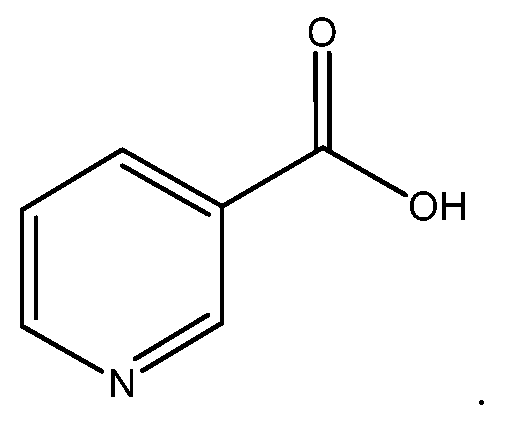
Один аспект изобретения относится к соединениям, представленным структурой I, или их фармацевтически приемлемым солям:
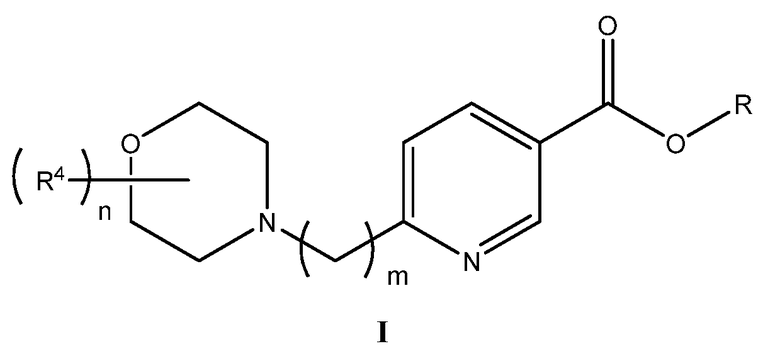
в которой
R представляет собой водород, алкил, галогеналкил, алкенил, алкинил, карбоциклил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, конденсированный бициклил, карбоксиалкил или арилалкениларил;
R4 выбран независимо для каждого случая из группы, состоящей из дейтерия, алкила, алкенила, алкинила, арила, гетероарила, аралкила, гетероаралкила, галогена, нитро, циано, сульфоновой кислоты, алкилсульфоксила, арилсульфоксила, гетероарилсульфоксила, аралкилсульфоксила, гетероаралкилсульфоксила, алкенилсульфоксила, алкинилсульфоксила, алкилсульфонила, арилсульфонила, гетероарилсульфонила, аралкилсульфонила, гетероаралкилсульфонила, алкенилсульфонила, алкинилсульфонила, гидроксила, алкоксила, арилоксила, гетероарилоксила, аралкилокси, гетероаралкилокси, алкенилокси, алкинилокси, тиола, алкилтио, арилтио, аралкилтио, гетероаралкилтио, алкенилтио, алкинилтио, формила, ацила, формилокси, ацилокси, формилтио, ацилтио, амина, алкиламина, ариламина, гетероариламина, аралкиламина, гетероаралкиламина, алкениламина, алкиниламина, формиламина, ациламина, карбоксила, алкилоксикарбонила, арилоксикарбонила, гетероарилоксикарбонила, аралкилоксикарбонила, гетероаралкилоксикарбонила, амидо, алкиламинкарбонила, ариламинкарбонила, гетероариламинкарбонила, аралкиламинкарбонила и гетероаралкиламинкарбонила;
n представляет 0, 1, 2, 3, 4, 5, 6, 7 или 8; и
m представляет собой 1, 2, 3 или 4.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых n представляет собой 0.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых n представляет собой 1.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых n представляет собой 2.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых n представляет собой 3.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых n представляет собой 1, 2 или 3; и R4 замещен заместителем, независимо выбранным из группы, состоящей из низшего алкила, галогена, нитро, циано, сульфоновой кислоты, гидроксила, алкоксила, тиола, алкилтио, формила, ацила, формилокси, ацилокси, формилтио, ацилтио, амина, алкиламина, формиламина, ациламина и карбоксила.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых n представляет собой 1; и R4 выбран из группы, состоящей из низшего алкила, галогена, нитро, циано, сульфоновой кислоты, гидроксила, алкоксила, тиола, алкилтио, формила, ацила, формилокси, ацилокси, формилтио, ацилтио, амина, алкиламина, формиламина, ациламина и карбоксила.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых m представляет собой 1.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых m представляет собой 2.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых m представляет собой 3.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых m представляет собой 4.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых R представляет собой водород.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых R представляет собой низший алкил.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых R представляет собой
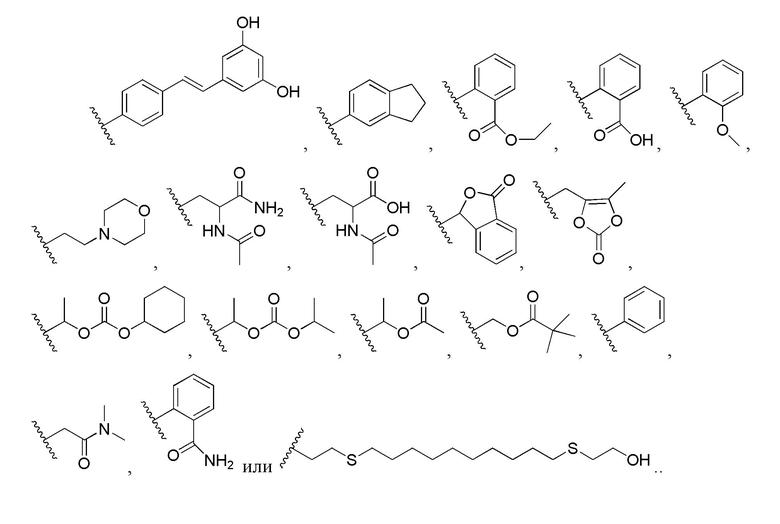
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых R представляет собой алифатическую группу, которая гидролизуется в карбоксил в физиологических условиях.
Еще один аспект изобретения относится к соединению или его фармацевтически приемлемой соли, выбранному из группы, состоящей из
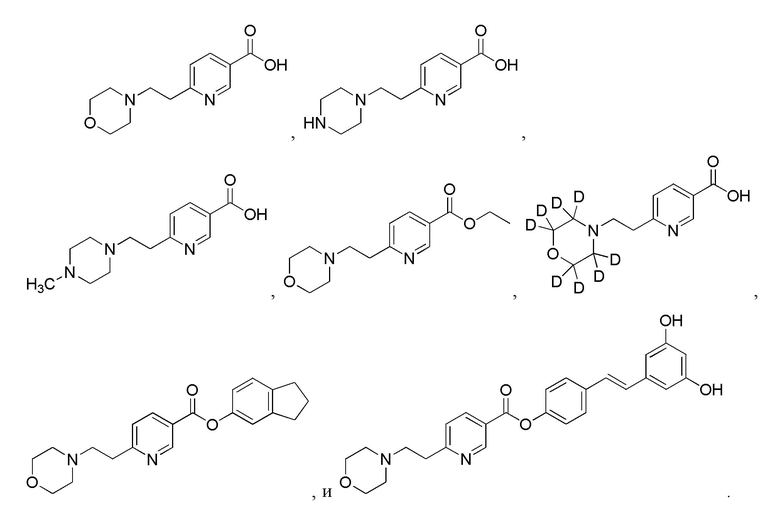
Еще один аспект изобретения относится к соединению
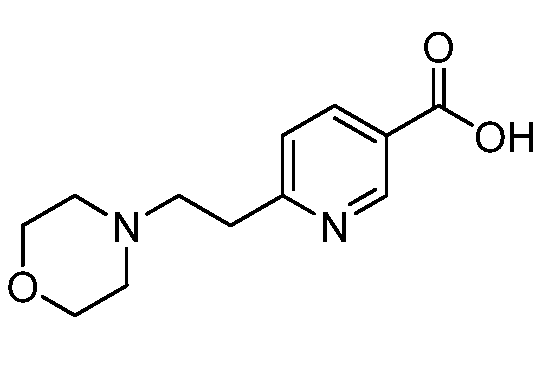
или к его фармацевтически приемлемой соли.
Еще один аспект изобретения относится к соединению, представленному структурой II, или к его фармацевтически приемлемой соли
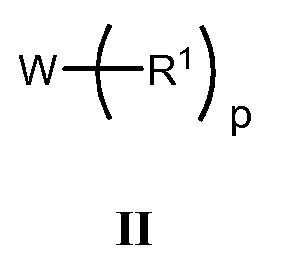
в которой независимо для каждого случая
W представляет собой полиол или политиол;
р представляет собой 2-500 включительно;
R1 представляет собой
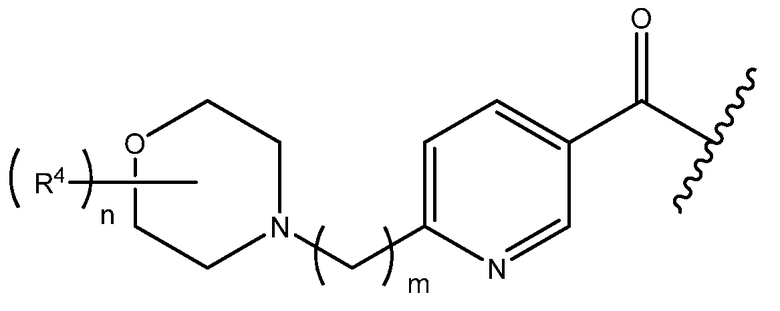
и либо присоединен к полиолу через атом кислорода полиола, либо присоединен к политиолу через атом серы политиола;
R представляет собой водород, алкил, галогеналкил, алкенил, алкинил, карбоциклил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, конденсированный бициклил, карбоксиалкил или арилалкениларил;
R4 выбран независимо для каждого случая из группы, состоящей из дейтерия, алкила, алкенила, алкинила, арила, гетероарила, аралкила, гетероаралкила, галогена, нитро, циано, сульфоновой кислоты, алкилсульфоксила, арилсульфоксила, гетероарилсульфоксила, аралкилсульфоксила, гетероаралкилсульфоксила, алкенилсульфоксила, алкинилсульфоксила, алкилсульфонила, арилсульфонила, гетероарилсульфонила, аралкилсульфонила, гетероаралкилсульфонила, алкенилсульфонила, алкинилсульфонила, гидроксила, алкоксила, арилоксила, гетероарилоксила, аралкилокси, гетероаралкилокси, алкенилокси, алкинилокси, тиола, алкилтио, арилтио, аралкилтио, гетероаралкилтио, алкенилтио, алкинилтио, формила, ацила, формилокси, ацилокси, формилтио, ацилтио, амина, алкиламина, ариламина, гетероариламина, аралкиламина, гетероаралкиламина, алкениламина, алкиниламина, формиламина, ациламина, карбоксила, алкоксикарбонила, арилоксикарбонила, гетероарилоксикарбонила, аралкилоксикарбонила, гетероаралкилоксикарбонила, амидо, алкиламинкарбонила, ариламинкарбонила, гетероариламинкарбонила, аралкиламинкарбонила и гетроаралкиламинкарбонила;
R5 для каждого случая независимо выбран из группы, состоящей из водорода, алкила, алкенила, алкинила, арила, гетероарила, аралкила, гетероаралкила, галогена, нитро, циано, сульфоновой кислоты, алкилсульфоксила, арилсульфоксила, гетероарилсульфоксила, аралкилсульфоксила, гетероаралкилсульфоксила, алкенилсульфоксила, алкинилсульфоксила, алкилсульфонила, арилсульфонила, гетероарилсульфонила, аралкилсульфонила, гетероаралкилсульфонила, алкенилсульфонила, алкинилсульфонила, гидроксила, алкоксила, арилоксила, гетероарилоксила, аралкилокси, гетероаралкилокси, алкенилокси, алкинилокси, тиола, алкилтио, арилтио, аралкилтио, гетероаралкилтио, алкенилтио, алкинилтио, формила, ацила, формилокси, ацилокси, формилтио, ацилтио, амина, алкиламина, ариламина, гетероариламина, аралкиламина, гетероаралкиламина, алкениламина, алкиниламина, формиламина, ациламина, карбоксила, алкилоксикарбонила, арилоксикарбонила, гетероарилоксикарбонила, аралкилоксикарбонила, гетероаралкилоксикарбонила, амидо, алкиламинкарбонила, ариламинкарбонила, гетероариламинкарбонила, аралкиламинкарбонила и гетероаралкиламинкарбонила;
R6 представляет собой водород или низший алкил;
n представляет 0, 1, 2, 3, 4, 5, 6, 7 или 8; и
m представляет собой 1, 2, 3 или 4.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых W представляет собой политиол.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых W представляет собой полиол.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых указанным полиолом является карбогидрат.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых указанным полиолом является мальтит, сорбит, ксилит и изомальт.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых указанным полиолом является сорбит.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых указанным полиолом является инозит.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых указанным полиолом является цис-1,2,3,5-транс-4,6-циклогексангексол.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 2, 3, 4, 5 или 6.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 2-100 включительно. В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 2-50 включительно. В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 2-10 включительно.
В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых указанный р представляет собой 2. В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 3. В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 4. В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 5. В некоторых воплощениях изобретение относится к любому одному из упомянутых выше соединений, в которых р представляет собой 6.
Еще один аспект изобретения относится к соединению или его фармацевтически приемлемой соли, выбранному из группы, состоящей из
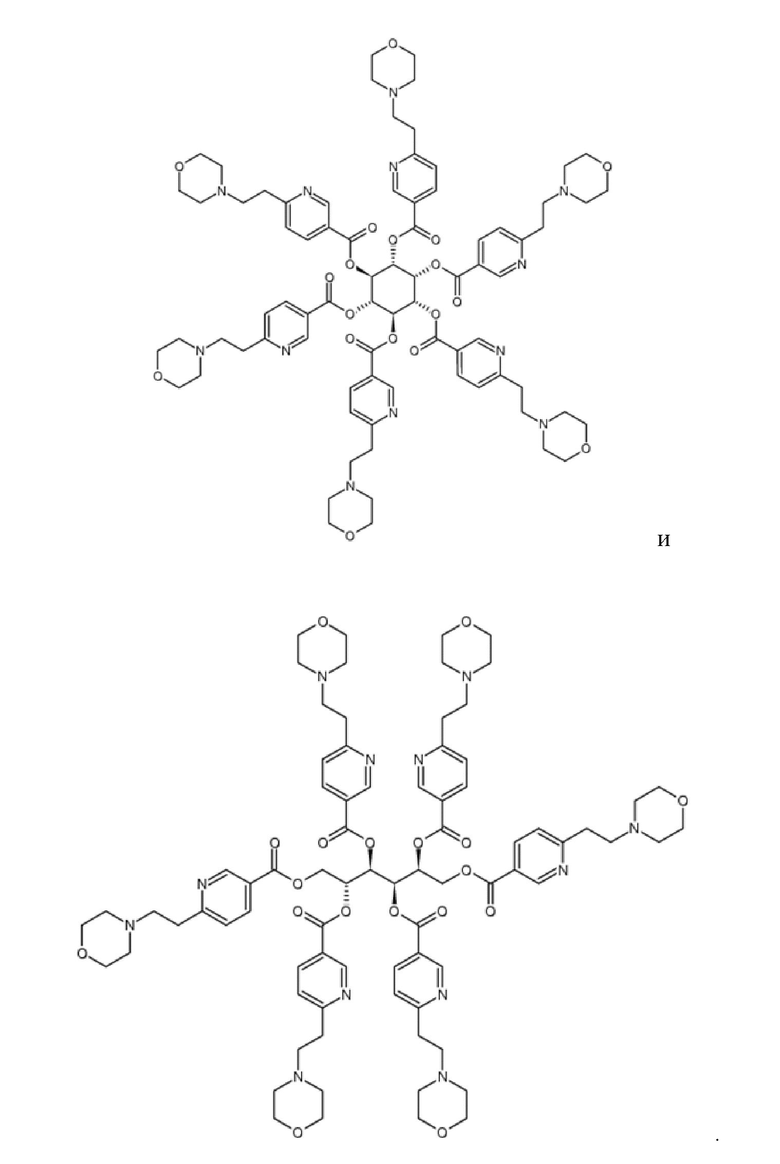
Было обнаружено, что по меньшей мере некоторые из соединений изобретения обладают желательными физиологическими свойствами ниацина, с уменьшенными при этом нежелательными побочными эффектами ниацина. Например, соединения изобретения обладают способностью модулировать, по меньшей мере, один липид желательным образом, без ограничительных побочных эффектов, или без степени ограничения побочных эффектов, характерных для ниацина.
Дополнительно было обнаружено, что, по-видимому, по меньшей мере, некоторые из соединений изобретения не вовлекают рецептор ниацина с высоким сродством GPR109A подобным ниацину образом. GPR109A, называемый также PUMA-G и НМ74А, является членом семейства рецепторов никотиновой кислоты G-протеин-сочетаемых рецепторов (GPCR). Wise A et al. (2003) J Biol Chem 278:99-74; Soga T et al. (2003) Biochem Biophys Res Comm 303:364-9. У GPR109A нокаутированных мышей действия ниацина и на липиды и на приливы крови устраняются. Эффект прилива крови, но не модифицирующие липид эффекты, ниацина приписывают активации с помощью GPR109A ERK ½ МАР киназы, опосредуемой аррестином бета 1 (бета (β)-аррестин). Сообщалось, что у нокаутированных аррестином бета 1 мышей действие ниацина на прилив крови в значительной мере снижается, при этом модифицирующие липид эффекты сохраняются. Walters RW et al. (2009) J Clin Invest 119: 1312-21. Важно, что, по меньшей мере, некоторые из соединений изобретения сильно снижают способность вызывать поступление β-аррестина к мембране клеток, экспрессирующих GPR109A по сравнению с ниацином, сохраняя все же клинически важные липид-модифицирующие эффекты.
Многие из соединений изобретения могут предоставляться в виде солей с фармацевтически приемлемыми противоионами (т.е. в виде фармацевтически приемлемых солей). “Фармацевтически приемлемая соль” обозначает нетоксичную соль, которая после введения реципиенту способна давать или непосредственно, или косвенно соединение или пролекарство соединения данного изобретения. “Фармацевтически приемлемая соль” представляет ионную часть соли, которая не является токсичной, когда высвобождается из соли после введения реципиенту. Фармацевтически приемлемые соли могут образовываться со многими кислотами, включающими, но не ограниченными ими, хлористоводородную, серную, уксусную, молочную, винную, яблочную, янтарную и др. Соли имеют тенденцию быть более растворимыми в водных или других протонных растворителях, чем соответствующие свободные формы оснований.
Кислоты, применяемые обычно для образования фармацевтически приемлемых солей, включают неорганические кислоты, такие как бисульфид водорода, хлористоводородную, бромистоводородную, иодистоводородную, серную и фосфорную кислоты, также как и органические кислоты, такие как пара-толуолсульфоновая, салициловая, винная, битартаровая, аскорбиновая, малеиновая, безиловая, фумаровая, глюконовая, глюкуроновая, муравьиная, глютаминовая, метансульфоновая, этансульфоновая, бензолсульфоновая, молочная, щавелевая, пара-бромфенилсульфоновая, угольная, янтарная, лимонная, бензойная и уксусная кислоты, и родственные им неорганические и органические кислоты. Такие фармацевтически приемлемые соли включают, таким образом, сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моно-кислый фосфат, ди-кислый фосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, субакат, фумарат, малеат, бутин-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, сульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, β-гидроксибутират, гликолят, малеат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и аналогичные соли. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли, образуемые с минеральными кислотами, такими как хлористоводородная и бромистоводородная кислота, и особенно соли, образуемые с органическими кислотами, такими как малеиновая кислота.
Подходящие основания для образования фармацевтически приемлемых солей с кислотными функциональными группами включают, но не ограничиваются ими, гидроокиси щелочных металлов, таких как натрий, калий и литий; гидроокиси щелочноземельных металлов, таких как кальций и магний; гидроокиси других металлов, таких как алюминий и цинк; аммиак и органические амины, такие как незамещенные или гидрокси-замещенные моно, ди- или триалкиламины; дициклогексиламин; трибутиламин; пиридин; N-метил, N-этиламин; диэтиламин; триэтиламин; моно-, бис- или трис-(2-гидрокси-низший алкил амины), такие как моно-, бис- или трис-(2-гидроксиэтил)амин, 2-гидрокси-трет-бутиламин или трис(гидроксиметил)метиламин, N,N-ди-низший алкил-N-(гидрокси низший алкил)амины, такие как N,N-диметил-N-(2-гидроксиэтил)амин или три-(2-гидроксиэтил)амин; N-метил-D-глюкамин; и аминокислоты, такие как аргинин, лизин и аналогичные.
Некоторые соединения изобретения и их соли могут существовать в более чем одной кристаллической форме, и настоящее изобретение включает каждую кристаллическую форму и их смеси.
Некоторые соединения изобретения и их соли могут существовать также в форме сольватов, например, гидратов, и настоящее изобретение включает каждый сольват и их смеси.
Некоторые соединения изобретения могут содержать один или более хиральных центров и существовать в различных оптически активных формах. Когда соединения изобретения содержат один хиральный центр, соединения существуют в двух энантиомерных формах, и настоящее изобретение включает оба энантиомера и смеси энантиомеров, такие как рацемические смеси. Энантиомеры могут разделяться по способам, известным специалистам в данной области, например, путем образования диастереоизомерных солей, которые могут быть разделены, например, с помощью кристаллизации; образования диастереоизомерных производных или комплексов, которые могут разделяться, например с помощью кристаллизации, газо-жидкостной или жидкостной хроматографии; селективной реакции одного энантиомера с энантиомер-специфичным реагентом, например, ферментативной сложной этерификации; или газо-жидкостной или жидкостной хроматографии в хиральной окружающей среде, например, на хиральной подложке, например, двуокиси кремния, со связанным хиральным лигандом или в присутствии хирального растворителя. Очевидно понятно, что когда желаемый энантиомер превращается в какую-либо еще химическую целостность с помощью одной из процедур разделения, описанных выше, может использоваться дальнейшая стадия для освобождения желаемой энантиомерной формы. Альтернативно, конкретные энантиомеры могут синтезироваться с помощью асимметрического синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, или путем превращения одного энантиомера в другой с помощью асимметрического преобразования.
Когда соединение изобретения содержит более одного хирального центра, оно может существовать и в диастереоизомерных формах. Диастереоизомерные соединения могут разделяться с помощью методов, известных специалистам в данной области, например, хроматографии или кристаллизации, и индивидуальные энантиомеры могут отделяться, как описано выше. Настоящее изобретение включает каждый диастереоизомер соединений настоящего изобретения и их смеси.
Некоторые соединения изобретения могут существовать в различных таутомерных формах или в виде различных геометрических изомеров, и настоящее изобретение включает каждый таутомер и/или геометрический изомер соединений изобретения и их смеси.
Некоторые соединения изобретения могут существовать в различных стабильных конформационных формах, которые могут быть разделимыми. Разделение различных конформеров может давать торзионная асимметрия вследствие ограниченного вращения вокруг асимметрической одинарной связи, например, из-за пространственного затруднения или кольцевого напряжения. Настоящее изобретение включает каждый конформационный изомер соединений изобретения и их смеси.
Некоторые соединения изобретения могут существовать в цвиттер-ионной форме, и настоящее изобретение включает цвиттер-ионную форму соединений изобретения и их смеси.
Настоящее изобретение включает также пролекарства. Используемый здесь термин “пролекарства” относится к агенту, который превращается в исходное лекарственное средство in vivo с помощью некоторого физиологического химического процесса (например, пролекарство, доведенное до физиологического показателя рН, превращается в желаемую лекарственную форму). Пролекарства часто являются полезными, в некоторых ситуациях, их может быть легче вводить, чем исходное лекарственное средство. Они могут, например, быть биологически доступными при пероральном введении, тогда как исходное лекарственное средство таким не является. Пролекарство может также иметь улучшенную растворимость в фармацевтических композициях по сравнению с исходным лекарственным средством. Без ограничения, примером пролекарства может быть соединение настоящего изобретения, когда оно вводится в виде сложного эфира (“пролекарства”) для облегчения прохождения через клеточную мембрану, где растворимость в воде не является благоприятной, но затем оно метаболически гидролизуется в карбоновую кислоту сразу внутри клетки, где растворимость в воде является благоприятной. Пролекарства обладают многими полезными свойствами. Например, пролекарство может быть более водорастворимым, чем окончательное лекарственное средство, посредством чего облегчается внутривенное введение лекарственного средства. Пролекарство может также иметь более высокий уровень оральной биодоступности, чем конечное лекарственное средство. После введения пролекарство энзиматически или химически расщепляется, доставляя конечное лекарственное средство в кровь или ткань.
Примерные пролекарства после расщепления высвобождают соответствующую свободную кислоту, и такие гидролизуемые образующие сложный эфир остатки соединений данного изобретения включают, но не ограничиваются ими, карбоновокислотные заместители (например, -С(О)2Н или фрагмент, который содержит карбоновую кислоту), где свободный водород замещен (С1-С4)алкилом, (С2-С12)алканоилоксиметилом, (С4-С9)-1-(алканоилокси)этилом, 1-метил-1-(алканоилокси)этилом, имеющим от 5 до 10 атомов углерода, алкоксикарбонилоксиметилом, имеющим от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этилом, имеющим от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этилом, имеющим от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометилом, имеющим от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этилом, имеющим от 4 до 10 атомов углерода, 3-фталидилом, 4-кротонолактонилом, гамма-бутиролактон-4-илом, ди-N,N-(С1-С2)алкиламино(С2-С3)алкилом (таким, как β-диметиламиноэтил), карбамоил-(С1-С2)алкилом, N,N-ди(С1-С2)алкилкарбамоил-(С1-С2)алкилом и пиперидино-, пирролидино- или морфолино(С2-С3)алкилом.
Другие примерные пролекарства освобождают спирт или амин соединения изобретения, в котором свободный водород гидроксильного или аминового заместителя замещается (С1-С6)алканоилоксиметилом, 1-((С1-С6)алканоилокси)этилом, 1-метил-1-((С1-С6)алканоилокси)этилом, (С1-С6)алкоксикарбонил-оксиметилом, N-(С1-С6)алкоксикарбониламинометилом, сукциноилом, (С1-С6)алканоилом, α-амино(С1-С4)алканоилом, арилактилом и α-аминоацилом, или α-аминоацил-α-аминоацилом, в котором указанными α-аминоалкильными фрагментами независимо являются любые из встречающихся в природе L-аминокислот, найденных в белках, -P(O)(OH)2, -P(O)(O(С1-С6)алкил)2 или гликозил (радикал, получающийся в результате отщепления гидроксила полуацеталя карбогидрата).
Фраза “защитная группа”, используемая здесь, обозначает временные заместители, которые защищают потенциально реакционно-способную функциональную группу от нежелательных химических трансформаций. Примеры таких защитных групп включают эфиры карбоновых кислот, силиловые эфиры спиртов, и ацетали и кетали альдегидов и кетонов, соответственно. Обзор области химии защитных групп дан в (Greene, T.W.; Wuts, P.G.М. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: Нью-Йорк, 1991). Защищенные формы соединений изобретения включены в объем данного изобретения.
Термин “химически защищенная форма”, используемый здесь, относится к соединению, в котором одна или более реакционно-способных функциональных групп защищены от нежелательных химических реакций, то есть находятся в форме защитной или защищающей группы (известной также как маскировочная или маскирующая группа). Удобным или желательным может быть получать, очищать и/или обращаться с активным соединением в химически защищенной форме.
Путем защиты реакционно-способной функциональной группы могут проводиться реакции, вовлекающие другие незащищенные реакционно-способные функциональные группы без затрагивания защищенной группы; защищающая группа может быть удалена, обычно на последующей стадии, не затрагивая по существу остальную часть молекулы. См., например, Protective Groups in Organic Synthesis (T. Green and P. Wuts, Wiley, 1991) и Protective Groups in Organic Synthesis (T. Green and P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Например, гидроксигруппа может защищаться в виде простого эфира (-OR) или сложного эфира (-OC(=O)R), например, как т-бутиловый эфир; бензиловый, бензгидриловый (дифенилметиловый) или тритиловый (трифенилметиловый) эфир; триметилсилиловый или т-бутилдиметилсилиловый эфир; или ацетиловый эфир (-OC(=O)СН3,-OAc).
Например, альдегидная или кетоновая группа может защищаться, соответственно, как ацеталь или кеталь, в котором карбонильная группа (C(=O)) превращается в диэфир (C(OR)2), с помощью реакции, например, с первичным спиртом. Альдегидная или кетоновая группа свободно регенерируется с помощью гидролиза с использованием большого избытка воды в присутствии кислоты.
Например, аминовая группа может защищаться, например, как амид NRC(=O)R) или уретан (-NRC(=O)OR), например, как метиламид (-NHC(=O)CH3); бензилоксиамид (-NHC(=O)OCH2C6H5NH(Cbz); т-бутоксиамид (-NHC=(O=)OC(CH3)3,-NHBoc); 2-бифенил-2-пропоксиамид (-NHC(=O)OC(CH3)2C6H4C6H5NHBoc), как 9-флуоренилметоксиамид (-NHFmoc), как 6-нитровератрилоксиамид (NHNvoc), как 2-триметилсилилэтилоксиамид (-NHTeoc), как 2,2,2-трихлорэтилоксиамид (NHTroc), как аллилоксиамид (=NHAlloc), как 2-(фенилсульфонил)этилоксиамид (=NHPsec; или в подходящих случаях (например, циклических аминах), как нитроксидный радикал.
Например, карбоновокислотная группа может быть защищена как сложный эфир или амид, например, как: бензиловый эфир; т-бутиловый эфир; метиловый эфир или метиламид.
Например, тиольная группа может быть защищена как тиоэфир (-SR), например, как: бензиловый тиоэфир; или ацетамидометиловый эфир (SCH2NHC(=O)CH3).
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Изобретение предоставляет фармацевтические композиции, включающие одно или более соединений, на которые ссылаются выше. В одном аспекте настоящее изобретение предоставляет фармацевтически приемлемые композиции, которые включают терапевтически эффективное количество одного или более из соединений, описанных выше, сформулированных вместе с одним или более фармацевтически приемлемыми носителями (добавками) и/или разбавителями.
Альтернативно или в дополнение, изобретение предоставляет фармацевтические композиции, характеризуемые обладанием одним желаемым терапевтическим эффектом ниацина и уменьшением или отсутствием, по меньшей мере, одного нежелательного побочного эффекта ниацина.
В одном воплощении фармацевтическая композиция формулируется для перорального введения; когда вводится человеку через рот, она снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, выбранного из группы, состоящей из общего холестерина, холестерина липопротеина низкой плотности (LDL), триглицеридов и липопротеина (а); и пероральное введение композиции характеризуется пониженным приливом крови и уменьшением гепатоклеточного повреждения по сравнению с введением эквимолярной оральной дозы ниацина.
Когда вводится человеку перорально, фармацевтическая композиция снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, выбранного из группы, состоящей из общего холестерина, холестерина липопротеина низкой плотности (LDL), триглицеридов и липопротеина (а). Указанная композиция снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, когда такой уровень в сыворотке или плазме уменьшается в измеряемое количество, по сравнению с предварительным лечением, базовой линией или контрольным уровнем. В одном воплощении, данная композиция снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, когда такой уровень в сыворотке или плазме снижается, по меньшей мере, на 5 процентов предварительного лечения, базовой линии или контрольного уровня; то есть уровень в сыворотке или плазме снижается до не более 95 процентов предварительного лечения, базовой линии или контрольного уровня. В одном воплощении, данная композиция снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, когда такой уровень в сыворотке или плазме снижается, по меньшей мере, на 10 процентов предварительного лечения, базовой линии или контрольного уровня. В одном воплощении, данная композиция снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, когда такой уровень в сыворотке или плазме снижается, по меньшей мере, на 15 процентов предварительного лечения, базовой линии или контрольного уровня. В одном воплощении, данная композиция снижает в сыворотке или плазме уровень, по меньшей мере, одного липида, когда такой уровень в сыворотке или плазме снижается на, по меньшей мере, 20 процентов предварительного лечения, базовой линии или контрольного уровня.
Используемый здесь термин “прилив крови” относится к объективному кожному расширению сосудов, часто сопровождаемому краснотой, и/или субъективному опыту ощущения тепла на коже, причем, последнее с или без ощущения зуда. Прилив крови можно объективно измерить с использованием объективных измерений, таких как Допплеровские измерения капиллярного кровяного потока. Альтернативно или дополнительно, прилив крови может измеряться с использованием так называемой визуальной аналоговой шкалы (VAS), которая может быть или на основе наблюдателя, или на основе субъекта. VAS обычно предусматривает отметку признаков или симптомов по шкале от ноля (0) до десяти (10), где ноль соответствует полному отсутствию отмечаемого признака или симптома, а десять соответствует невыносимому или максимальному количеству или степени признака или симптома.
Используемый здесь термин “гепатоклеточное повреждение” относится к токсичному повреждению паренхимальных клеток печени. Гепатоклеточное повреждение может оцениваться с использованием любого подходящего метода. В одном воплощении гепатоклеточное повреждение оценивается путем измерения одного или более ферментов печени в сыворотке. В одном воплощении одним таким ферментом печени является аминотрансфераза (AST, называемая также SGOT). В одном воплощении одним таким ферментом печени является аланинаминотрансфераза (ALT, называемая также SGPT). Уровни AST и ALT в сыворотке измеряются обычно в клинической практике, и нет необходимости описывать здесь методы их измерения. Нормальные уровни обоих из AST и ALT составляют обычно 0-35 ед./л. В противоположность AST, который находится в первую очередь в печени, AST обнаруживается в других тканях, включая сердце, скелетные мышцы, почки и головной мозг, и является таким образом несколько менее специфичным в качестве показателя дисфункции печени. Хотя повышенные уровни AST или ALT могут наблюдаться во множестве непеченочных состояний, включающих инфаркт миокарда, данные состояния обычно свободно отличаются клинически от заболевания печени. При заболевании печени повышения в сыворотке AST и ALT отражают некроз печени, тяжелую форму гепатоклеточного расстройства.
Пероральное введение композиции характеризуется пониженным притоком крови и пониженным повреждением клеток печени по сравнению с введением эквимолярной оральной дозы ниацина. В данном контексте, говорят, что прилив крови снижается, когда он снижается на способное быть измеренным количество или степень по сравнению с соответствующей степенью прилива крови, связанного с введением эквимолярной оральной дозы ниацина. В одном воплощении, говорят, что прилив крови снижается, когда максимум степени или количества прилива крови снижается на измеряемую степень или количество по сравнению с максимальной степенью или количеством прилива крови, ассоциируемого с введением эквимолярной оральной дозы ниацина.
В одном воплощении, указанный прилив крови снижается, когда поток капиллярной крови Допплера снижается, по меньшей мере, на 2 процента по сравнению с потоком капиллярной крови Допплера в соответствующей ткани, связанным с введением эквимолярной оральной дозы ниацина, т.е. поток снижается до не более 98 процентов потока капиллярной крови Допплера в соответствующей ткани, связанного с введением эквимолярной оральной дозы ниацина. В одном воплощении, данный прилив крови снижается, когда поток капиллярной крови Допплера снижается, по меньшей мере, на 5 процентов по сравнению с потоком капиллярной крови Допплера в соответствующей ткани, связанным с введением эквимолярной оральной дозы ниацина. В одном воплощении, данный прилив крови снижается, когда поток капиллярной крови Допплера снижается, по меньшей мере, на 10 процентов по сравнению с потоком капиллярной крови Допплера в соответствующей ткани, связанным с введением эквимолярной оральной дозы ниацина.
В одном воплощении прилив крови снижается, когда отметка VAS для соответствующей ткани снижается, по меньшей мере, на единицу (1) (по шкале от 0 до 10) по сравнению с отметкой VAS для соответствующей ткани, связанной с введением эквимолярной оральной дозы ниацина; т.е. отметка VAS снижается до не более чем на один меньше, чем отметка VAS для соответствующей ткани, связанной с введением эквимолярной оральной дозы ниацина. Например, в одном воплощении, данный прилив крови снижается, когда максимальная отметка VAS составляет 5, тогда как максимальная отметка VAS для соответствующей ткани, связанная с введением эквимолярной оральной дозы ниацина, составляет 6-10.
Конечно, любые из предшествующих сравнений преимущественно проводятся на основе населения. Например, могут сравниваться средние величины отметки AST, ALT или VAS. Аналогично могут сравниваться средние значения максимальной отметки ALT, AST или VAS.
Сравнение проводится по отношению к эквимолярной оральной дозе ниацина. Эквимолярная оральная доза ниацина относится к эквимолярной оральной дозе ниацина в любой форме, включая, например, составы ниацина немедленного, растянутого по времени, длительного и продленного высвобождения. В одном воплощении эквимолярная оральная доза формулируется аналогичным образом, например, в форме таблеток, в которой каждая отдельная таблетка содержит одно и то же или в основном одно и то же молярное количество активного агента. Таким образом, в одном воплощении сравнение может проводиться между оральными дозами, даваемыми в виде одиночных таблеток, каждая из которых содержит 8,2 ммол активного агента (например, 1 г ниацина). В качестве альтернативного примера в еще одном воплощении сравнение может проводиться между оральными дозами, даваемыми в виде двух таблеток, причем каждая таблетка содержит 4,1 ммол активного агента (например, 0,5 г ниацина).
За исключением, возможно отмеченным здесь иным образом, все фармакокинетические сравнения по отношению к “ниацину” являются сравнениями с составом ниацина так называемого немедленного высвобождения.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, в которых пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 40 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 35 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 30 процентов или менее (Смакс) эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 25 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 20 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 15 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 10 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 5 процентов или менее Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1 процент или менее Смакс эквимолярной оральной дозы ниацина. Для каждого из предшествующих воплощений должно быть понятно, что сравнение проводится с составом ниацина немедленного высвобождения. Способы, полезные для измерения концентрации, описаны подробно ниже.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, в которых пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-40 процентов (Смакс) эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-35 процентов Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-30 процентов Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-25 процентов Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-20 процентов Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-15 процентов Смакс эквимолярной оральной дозы ниацина. В одном воплощении пик концентрации в сыворотке или плазме (Смакс) для соединения составляет 1-10 процентов Смакс эквимолярной оральной дозы ниацина. Для каждого из предшествующих воплощений должно быть понятно, что сравнение проводится с составом ниацина немедленного высвобождения. Способы, полезные для измерения концентрации, описаны подробно ниже.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, в которых отношение пика концентрации в сыворотке или плазме (Смакс) к площади под кривой на 24 часа (AUC0-24) (т.е. отношение Смакс/AUC0-24) для соединения составляет 0,35 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,30 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,25 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,20 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,15 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,10 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,05 ч-1 или менее. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,01 ч-1 или менее.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, в которых отношение пика концентрации в сыворотке или плазме (Смакс) к площади под кривой на 24 часа (AUC0-24) (т.е. отношение Смакс/AUC0-24) для соединения составляет 0,10 ч-1 - 0,35 ч-1. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,10 ч-1 - 0,30 ч-1. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,10 ч-1 - 0,25 ч-1. В одном воплощении отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,10 ч-1 - 0,20 ч-1.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, в которых время до пика концентрации в сыворотке или плазме (tмакс) для соединения составляет в интервале 30 минут-5 часов. В одном воплощении время до пика концентрации (tмакс) для соединения составляет в интервале 1-5 часа. В одном воплощении время до пика концентрации (tмакс) для соединения составляет в интервале 1-4 часа. В одном воплощении время до пика концентрации (tмакс) для соединения составляет в интервале 1-3 часа. В одном воплощении время до пика концентрации (tмакс) для соединения составляет в интервале 1-2 часа.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, в которой соединение имеет ЕС50 в отношении функции опосредуемого β-аррестином GPR109A, которая, по меньшей мере, в 10 раз больше, чем ЕС50 ниацина в отношении функции опосредуемого β-аррестином GPR109A. ЕС50 относится к концентрации, при которой особый эффект достигает 50 процентов его максимума. Функция опосредуемого β-аррестином GPR109A описывается здесь далее. В одном воплощении соединение имеет ЕС50 в отношении функции опосредуемого β-аррестином GPR109A, которая, по меньшей мере, в 20 раз больше, чем ЕС50 ниацина в отношении функции опосредуемого β-аррестином GPR109A. В одном воплощении соединение имеет ЕС50 в отношении функции опосредуемого β-аррестином GPR109A, которая, по меньшей мере, в 30 раз больше, чем ЕС50 ниацина в отношении функции опосредуемого β-аррестином GPR109A. В одном воплощении соединение имеет ЕС50 в отношении функции опосредуемого β-аррестином GPR109A, которая, по меньшей мере, в 40 раз больше, чем ЕС50 ниацина в отношении функции опосредуемого β-аррестином GPR109A. В одном воплощении соединение имеет ЕС50 в отношении функции опосредуемого β-аррестином GPR109A, которая, по меньшей мере, в 50 раз больше, чем ЕС50 ниацина в отношении функции опосредуемого β-аррестином GPR109A. В одном воплощении соединение имеет ЕС50 в отношении функции опосредуемого β-аррестином GPR109A, которая, по меньшей мере, в 100 раз больше, чем ЕС50 ниацина в отношении функции опосредуемого β-аррестином GPR109A.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, которая, когда она вводится перорально человеку, увеличивает также уровень в сыворотке или плазме холестерина липопротеина высокой плотности (HDL). Уровень HDL холестерина в сыворотке или плазме увеличивается, по меньшей мере, на измеряемое количество по сравнению с уровнем пред-лечения, базовой линией или контрольным уровнем. Например, в одном воплощении уровень в сыворотке или плазме HDL холестерина увеличивается, по меньшей мере, на 5 процентов по сравнению с уровнем пред-лечения, базовой линией или контрольным уровнем. В одном воплощении уровень HDL холестерина в сыворотке или плазме увеличивается, по меньшей мере, на 10 процентов по сравнению с уровнем пред-лечения, базовой линией или контрольным уровнем. В одном воплощении уровень HDL холестерина в сыворотке или плазме увеличивается, по меньшей мере, на 15 процентов по сравнению с уровнем пред-лечения, базовой линией или контрольным уровнем. В одном воплощении уровень HDL холестерина в сыворотке или плазме увеличивается, по меньшей мере, на 20 процентов по сравнению с уровнем пред-лечения, базовой линией или контрольным уровнем. В одном воплощении уровень HDL холестерина в сыворотке или плазме увеличивается, по меньшей мере, на 25 процентов по сравнению с уровнем пред-лечения, базовой линией или контрольным уровнем.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, которая, когда она вводится перорально человеку, не индуцирует по существу никакого увеличения уровней аспартатаминотрансферазы (AST), аланинаминотрансферазы (ALT), или обеих. В одном воплощении “по существу никакого увеличения” в данном контексте обозначает менее, чем 20 процентное увеличение по сравнению с предлечением, базовой линией или контрольным уровнем. В одном воплощении “по существу никакого увеличения” в данном контексте обозначает менее, чем 10 процентное увеличение по сравнению с предлечением, базовой линией или контрольным уровнем.
В некоторых воплощениях изобретение относится к любой одной из упомянутых выше композиций, которая, когда она вводится перорально человеку, не вызывает по существу никакого увеличения в сыворотке уровней мочевой кислоты, глюкозы или обеих. Нормальные уровни мочевой кислоты в сыворотке составляют 1,5-8,0 мг/дл. В одном воплощении “по существу никакого увеличения уровня мочевой кислоты в сыворотке” обозначает менее, чем 10 процентное увеличение по сравнению с пред-лечением, базовой линией или контрольным уровнем. Нормальные уровни глюкозы в плазме натощак составляют приблизительно 75-115 мг/дл. Нормальные выбранные наугад уровни глюкозы в плазме (через 2 часа после приема пищи) составляют приблизительно <140 мг/дл. В одном воплощении “по существу никакого увеличения уровня глюкозы в сыворотке” обозначает менее, чем 20 процентное увеличение по сравнению с пред-лечением, базовой линией или контрольным уровнем. В одном воплощении “по существу никакого увеличения уровня глюкозы в сыворотке” обозначает менее, чем 15 процентное увеличение по сравнению с пред-лечением, базовой линией или контрольным уровнем. В одном воплощении “по существу никакого увеличения уровня глюкозы в сыворотке” обозначает менее, чем 10 процентное увеличение по сравнению с пред-лечением, базовой линией или контрольным уровнем.
В еще одном аспекте агенты изобретения могут вводиться как таковые, или могут вводиться в смесях с фармацевтически приемлемыми носителями, а могут также вводиться в сочетании с другими агентами. Терапия в сочетании с чем-то включает последовательное, одновременное и раздельное или совместное введение одного или более соединения изобретения, при котором терапевтические эффекты первого вводимого агента не исчезли целиком, когда вводится последующее соединение. Другими словами термин “совместное введение”, используемый здесь, относится и к параллельному введению (введение двух или более терапевтических агентов в одно и то же время) и введению, варьируемому по времени (введение одного или более терапевтических агентов во время, отличающееся от времени введения дополнительно терапевтического агента или агентов), пока терапевтические агенты присутствуют в организме пациента в одно и то же время.
Как описано подробно ниже, фармацевтические композиции настоящего изобретения могут специально формулироваться для введения в твердой или жидкой форме, включая формы, подобранные для следующего: (1) перорального введения, например, жидкие формы (водные или неводные растворы или суспензии), таблетки, например, таблетки для защечного, подъязычного и системного поглощения или всасывания, капсулы, болюсы, порошки, гранулы, пасты для применения на языке; (2) парентерального введения, например, путем внутривенной, внутримышечной, внутриперитонеальной, подкожной или эпидуральной инъекции или инфузии, например, в виде стерильного раствора или суспензии, или состава длительного или непрерывного высвобождения; (3) топического или местного применения, например, в виде крема, мази или кусочков пластыря с регулируемым высвобождением, применяемых на коже; (4) внутриматочного или внутриректального применения, например, в виде пессариев, крема или пены; (5) подъязычно; (6) оккулярно; (7) чрескожно; (8) назально; (9) легочно; или (10) интратекально.
Используемая здесь фраза “терапевтически эффективное количество” обозначает количество соединения, материала или композиции, включающей соединение настоящего изобретения, которое является эффективным для достижения некоторого желаемого эффекта, по меньшей мере, в субпопуляции клеток у животных при приемлемом соотношении польза/риск применительно к любому медицинскому лечению.
Фраза “фармацевтически приемлемый” применяется здесь по отношению к соединениям, материалам, композициям и/или дозировочным формам, которые в объеме здравого медицинского суждения являются подходящими для использования в контакте с тканями людей и животных без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерных с приемлемым соотношением польза/риск.
Фраза “фармацевтически приемлемый носитель”, используемая здесь, обозначает фармацевтически приемлемый материал, композицию или наполнитель, такой как жидкий или твердый наполнитель, разбавитель, эксципиент, производное вспомогательное средство (например, смазка, тальк, стеарат магния, кальция или цинка, или стеариновая кислота) или инкапсулирующий растворитель материал, вовлеченный в перенос или транспортировку соединения у субъекта от одного органа или части организма к другому органу или части организма. Каждый носитель должен быть “приемлемым”, в смысле быть совместимым с другими ингредиентами состава, и безвредным для пациента. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный и картофельный крахмал; (3) целлюлоза и ее производные, такие как натриевая карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) такие наполнители, как масло какао и суппозиторные воски; (9) масла, такие как арахисовое масло, хлопковое масло, подсолнечное, кунжутное, оливковое, кукурузное и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар-агар; (14) буферирующие агенты, такие как гидроокись магния и гидроокись алюминия; (15) альгиновая кислота; (16) свободная от пирогенов вода; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) рН буферные растворы; (21) сложные полиэфиры, поликарбонаты и/или полиангидриды; и (22) другие нетоксичные совместимые вещества, применяемые в фармацевтических составах композиций.
Как отмечалось выше, в некоторых воплощениях настоящие соединения могут содержать основную функциональную группу, такую как амино или алкиламино, и способны таким образом образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми кислотами. Термин “фармацевтически приемлемые соли” в данном отношении, относится к относительно нетоксичным, неорганическим и органическим кислотно-аддитивным солям настоящего изобретения. Данные соли могут получаться in situ при введении наполнителя или в процессе производства дозировочных форм, или отдельно путем реакции очищенного соединения изобретения в форме свободного основания с подходящей органической или неорганической кислотой, и отделения образовавшейся таким образом соли во время последующей очистки. Характерные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептаноат, лактобионат и лаурилсульфонатную соли и аналогичные (см., например, Berge et al. (1977) “Pharmaceutical Salts”, J. Pharm. Sci. 66:1-19).
Фармацевтически приемлемые соли описываемых соединений включают общепринятые нетоксичные соли или четвертичные аммониевые соли соединений, например, из нетоксичных органических или неорганических кислот. Например, такие общепринятые нетоксичные соли включают соли, происходящие из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и аналогичные; и соли, производимые из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, гидроксималеиновая, фенилуксусная, глютаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изотионовая и аналогичные.
В других случаях соединения настоящего изобретения могут содержать одну или более кислотных функциональных групп и, таким образом, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин “фармацевтически приемлемые соли” в данных случаях относится к относительно нетоксичным неорганическим и органическим солям присоединения основания соединений настоящего изобретения. Данные соли аналогичным образом могут быть получены in situ при введении наполнителя или в процессе производства дозировочной формы, или отдельно путем реакции очищенного соединения в свободной форме с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Характерные соли щелочных или щелочноземельных металлов включают соли лития, натрия, калия, кальция, магния и алюминия и аналогичные. Характерные органические амины, пригодные для образования солей присоединения основания, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и аналогичные (см., например, Berge et al., выше).
В композициях могут также присутствовать смачивающие агенты, эмульгаторы и смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также окрашивающие агенты, высвобождающие агенты, агенты покрытий, подсластители, вкусовые и ароматизирующие агенты, консерванты и антиоксиданты.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) растворимые в воде антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и аналогичные; (2) растворимые в масле антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и аналогичные; и (3) хелатирующие металл агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбит, винная кислота, фосфорная кислота и аналогичные.
Составы композиций настоящего изобретения включают формы, подходящие для перорального, назального, местного (включая защечный и подъязычный), ректального, вагинального и/или парентерального введения. Составы могут быть удобно представлены в форме единичных доз и могут быть получены любым методом, хорошо известным в области фармацевтики. Количество активного ингредиента, которое может объединяться с материалом носителя для получения одной дозировочной формы, варьирует в зависимости от субъекта, подлежащего лечению, конкретного способа введения. Количество активного ингредиента, которое может объединяться с материалом носителя для получения одной дозировочной формы, составляет обычно количество соединения, которое дает терапевтический эффект. Обычно, исходя из ста процентов, данное количество будет составлять примерно от 0,1 процента до примерно девяноста девяти процентов активного ингредиента, предпочтительно примерно от 5 до 70 процентов, наиболее предпочтительно примерно от 10 процентов до примерно 30 процентов.
В некоторых воплощениях состав композиции настоящего изобретения включает наполнитель, выбранный из группы, состоящей из циклодекстринов, целлюлоз, липосом, агентов, образующих мицеллы, например, желчных кислот, и полимерных носителей, например, сложных полиэфиров и полиангидридов; и соединение настоящего изобретения. В некоторых воплощениях упомянутый выше состав делает соединение настоящего изобретения орально биодоступным.
Способы получения данных форм или композиций включают стадию объединения соединения настоящего изобретения с носителем и, необязательно, одним или более вспомогательными ингредиентами. Обычно композиции приготавливают с помощью однородного и тщательного смешения соединения настоящего изобретения с жидкими носителями, или тонко измельченными твердыми носителями, или обоими, и, когда необходимо, формования продукта.
Композиции изобретения, подходящие для перорального введения, могут быть в форме капсул, облаток, пилюль, таблеток, пастилок (с использованием основы, обычно сахарозы и акации или трагаканта), порошков, гранул, или в виде раствора или суспензии в водной или неводной жидкости, или в виде эмульсии масло-в-воде или вода-в-масле, или эликсира или сиропа, или в виде пастилок (с использованием инертной основы, такой как желатин и глицерин, или сахарозы и аравийской камеди) и/или в виде полоскания для рта и аналогичных, каждая из которых содержит заданное количество соединения настоящего изобретения в качестве активного ингредиента. Соединение настоящего изобретения может также вводиться в виде болюсов, электуариев или паст.
В твердых дозировочных формах изобретения для перорального введения (капсулы, таблетки, пилюли, драже, порошки, гранулы, пастилки и аналогичные) активный ингредиент смешан с одним или более фармацевтически приемлемыми носителями, такими как цитрат натрия или дикальцийфосфат, и/или любого из следующих: (1) наполнители или разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующие, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; (3) увлажнители, такие как глицерин; (4) дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) задерживающие раствор агенты, такие как парафин; (6) ускорители абсорбции, такие как четвертичные аммониевые соединения и поверхностно-активные вещества, такие как полоксамер и лаурилсульфат натрия; (7) смачивающие агенты, такие как цетиловый спирт, моностеарат глицерина и не-ионные поверхностно-активные вещества; (8) абсорбенты, такие как каолин и бентонитная глина; (9) смазочные агенты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, стеарат цинка, стеарат натрия, стеариновая кислота и их смеси; (10) окрашивающие агенты; и (11) агенты, регулирующие высвобождение, такие как кросповидон или этилцеллюлоза. В случае капсул, таблеток и пилюль фармацевтические композиции могут также включать буферирующие агенты. Твердые композиции похожего типа могут также применяться как наполнители в желатиновые капсулы с мягкой и твердой оболочкой с использованием таких эксципиентов, как лактоза или молоко, также как и полиэтиленгликоли с высоким молекулярным весом, и аналогичные.
Таблетки могут изготавливаться с помощью прессования или формования, необязательно, с одним или более вспомогательными ингредиентами. Прессованные таблетки могут изготавливаться с использованием связующего (например, желатина или гидроксипропилметилцеллюлозы), смазочного агента, инертного разбавителя, консерванта, дезинтегрирующего агента (например, крахмалгликолята натрия или поперечносшитой карбоксиметилцеллюлозы), поверхностно-активного или диспергирующего агента. Формованные таблетки могут производиться формованием в подходящем устройстве смеси порошкообразного соединения, смоченного инертным жидким разбавителем.
Таблетки и другие твердые дозировочные формы фармацевтических композиций настоящего изобретения, такие как драже, капсулы, пилюли и гранулы, могут быть необязательно с метками или с покрытиями и оболочками, такими как энтерические покрытия и другие покрытия, хорошо известные в области изготовления фармацевтических составов. Они могут также быть в виде составов для того, чтобы медленно или контролируемым образом высвобождать активный ингредиент с использованием, например, гидроксипропилметилцеллюлозы в варьируемых пропорциях, чтобы обеспечивать желаемый профиль высвобождения других полимерных матриц, липосом и/или микросфер. Они могут изготавливаться для быстрого высвобождения, например, высушенными замораживанием. Они могут стерилизоваться, например, фильтрованием через задерживающий бактерии фильтр, или включением в них стерилизующих агентов в форме стерильных твердых композиций, которые могут растворяться в воде или какой-либо другой стерильной инъецируемой среде непосредственно перед использованием. Данные композиции могут также содержать необязательно агенты, придающие непрозрачность, а могут быть такими композициями, что они высвобождают активный ингредиент(ы) только, или предпочтительно, в определенной части желудочно-кишечного такта, не обязательно, задерживаемым образом. Примеры композиций, которые могут использоваться, включают полимерные вещества и воски. Активный ингредиент может быть также в микроинкапсулированной форме, если это соответствует, с одним или более из описанных выше наполнителей.
Жидкие дозировочные формы для перорального введения соединений изобретения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активному ингредиенту жидкие дозировочные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солибизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сорбитановые эфиры жирных кислот и их смеси.
Кроме инертных разбавителей, оральные композиции могут также включать вспомогательные агенты, такие как смачивающие, эмульгирующие и суспендирующие агенты, подслащивающие, вкусовые, окрашивающие, ароматизирующие и консервирующие агенты.
Суспензии, в дополнение к активным соединениям, могут содержать суспендирующие агенты как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сорбитановые сложные эфиры, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант, и их смеси.
Составы фармацевтических композиций изобретения для ректального или вагинального введения могут быть представлены в виде медицинских свечей, которые могут быть получены путем смешения одного или более соединений изобретения с одним или более подходящими не-раздражающими наполнителями или носителями, включающими, например, масло какао, полиэтиленгликоль, суппозиторный воск или салицилат, и которые являются твердыми при комнатной температуре, но жидкими при температуре тела и, следовательно, будут размягчаться в прямой кишке или во влагалище и высвобождать активное соединение.
Формы композиций настоящего изобретения, которые являются подходящими для вагинального введения, включают также пессарии, тампоны, кремы, гели, пасты, пены или спреи, содержащие такие носители, которые известны в данной области и соответствуют этой цели.
Дозировочные формы для местного или чрескожного введения соединения данного изобретения включают порошки, спреи, мази, пасты, крема, лосьоны, гели, растворы, пластыри и ингаляторы. Активное соединение может смешиваться в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Мази, пасты, крема и гели могут содержать в дополнение к активному соединению данного изобретения наполнители, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевая кислота, тальк и окись цинка, или их смеси.
Порошки и спреи могут содержать в дополнение к соединению данного изобретения наполнители, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и полиамидный порошок, или смеси данных веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как хлорфторуглеводороды и летучие незамещенные углеводороды, такие как бутан и пропан.
Трансдермальные бляшки имеют дополнительное преимущество обеспечения регулируемого высвобождения соединения настоящего изобретения в организм. Такие дозировочные формы могут получаться растворением и диспергированием соединения в соответствующей среде. Усилители абсорбции могут использоваться также для увеличения движения соединения через кожу. Скорость такого движения может регулироваться или обеспечением скорости регулирования мембраны, или диспергированием соединения в полимерной матрице или геле.
Офтальмические композиции, глазные мази, порошки, растворы и аналогичные также охватываются объемом данного изобретения.
Фармацевтические композиции данного изобретения, подходящие для парентерального введения, включают одно или более соединений изобретения в сочетании с одним или более фармацевтически приемлемых стерильных изотонических водных или неводных растворов, дисперсий, суспензий или эмульсий, или стерильных порошков, которые перед использованием могут преобразовываться в стерильные инъецируемые растворы или дисперсии, которые могут содержать сахара, спирты, антиоксиданты, буферы, бактериостатические агенты, растворенные вещества, которые делают форму композиции изотонической с кровью предполагаемого реципиента, или суспендирующие или загущающие агенты.
Примеры подходящих водных и неводных носителей, которые могут применяться в фармацевтических композициях изобретения, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и аналогичные) и их подходящие смеси, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Надлежащая текучесть может достигаться, например, использованием материалов для покрытия, таких как лецитин, поддержанием требуемого размера частиц в случае дисперсий, и использованием поверхностно-активных веществ.
Данные композиции могут также содержать адъюванты, такие как консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Предотвращение действия микроорганизмов на соединения, о которых идет речь, может гарантироваться путем включения различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенолсорбиновой кислоты и аналогичных. Может быть также желательным включать в композиции изотонические агенты, такие как сахара, хлорид натрия и аналогичные. Дополнительно, пролонгированная абсорбция инъецируемой фармацевтической формы может достигаться включением в нее агентов, которые задерживают абсорбцию, таких как моностеарат алюминия и желатин.
В некоторых случаях, чтобы продлить действие лекарственного средства, желательно замедлить абсорбцию лекарственного средства от подкожной или внутримышечной инъекции. Данный результат может достигаться использованием жидкой суспензии кристаллического или аморфного материала, имеющего плохую растворимость в воде. Скорость абсорбции лекарственного средства зависит затем от скорости растворения, которая в свою очередь может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленная абсорбция вводимой парентерально формы лекарственного средства достигается растворением или суспендированием лекарственного средства в масляном наполнителе.
Инъецируемые запасные формы изготавливаются формированием матриц микрокапсул обсуждаемых соединений в биоразлагаемых полимерах, таких как полиактид-полигликолид. В зависимости от соотношения лекарственного средства и полимера и от природы применяемого конкретного полимера может регулироваться скорость высвобождения лекарственного средства. Примеры других биоразлагаемых полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Запасные инъецируемые формы композиций также получают путем заключения лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканью тела.
Когда соединения настоящего изобретения вводятся людям и животным в качестве фармацевтических средств, они могут использоваться сами по себе или в виде фармацевтической композиции, содержащей, например, 0,1-99% (более предпочтительно, 10-30%) активного ингредиента в сочетании с фармацевтически приемлемым носителем.
Препараты настоящего изобретения могут даваться перорально, парентерально, местно или ректально. Они конечно используются в формах, подходящих для каждого способа введения. Например, они вводятся в форме таблеток или капсул, путем инъекции, ингаляцией, в форме лосьона для глаз, мази, суппозиториев, введением путем инъекции, инфузии или ингаляции, местно в виде лосьона или мази; и ректально, путем суппозиториев.
Фразы “парентеральное введение” и “введенный или вводимый парентерально”, используемые здесь, обозначают способы введения отличные от энтерального (внутрь пищеварительного тракта) и местного введения, обычно инъекцией, и включают без ограничения внутривенную, внутримышечную, внутриартериальную, интратекальную, внутрисуставную, внутриорбитальную, внутрисердечную, внутрикожную, интраперитонеальную, транстрахеальную, подкожную, субкутикулярную, внутрисуставную, субкапсулярную, субарахноидальную, внутриспинальную и внутригрудинную инъекцию и инфузию.
Фразы “системное введение”, “вводимое системно”, “периферийное введение” и “вводимое периферийно”, используемые здесь, обозначают введение соединения, лекарственного средства или другого материала иным путем, чем непосредственно в центральную нервную систему так, что оно входит в систему пациента и, таким образом, подвергается метаболизму и другим аналогичным процессам, например, подкожное введение.
Соединения могут вводиться людям и животным для терапии любым подходящим способом введения, включая через рот, через нос, как, например, в виде спрея, ректально, внутривагинально, парентерально, внутрицистернально и местно, в виде порошков, мазей или капель, включая введение защечно и подъязычно.
Безотносительно выбранного способа введения, соединения настоящего изобретения, которые могут использоваться в подходящей гидратированной форме, и/или фармацевтические композиции настоящего изобретения преобразовываются в фармацевтически приемлемые дозированные формы удобными способами, известные специалистам в данной области.
Фактические дозировочные уровни активных ингредиентов в фармацевтических композициях данного изобретения могут варьироваться так, чтобы получить количество активного ингредиента, которое является эффективным для достижения желаемой терапевтической ответной реакции для конкретного пациента, композиции и способа введения, не являющееся токсичным для пациента.
Выбранный уровень дозы будет зависеть от множества факторов, включая активность применяемого конкретного соединения настоящего изобретения, или его сложного эфира, соли или амида, способ введения, время введения, скорость экскреции или метаболизма конкретно применяемого соединения, скорость и степень абсорбции, длительность лечения, другие лекарственные средства, соединения и/или материалы, используемые в сочетании с применяемым конкретным соединением, возраст, пол, вес, состояние, общее состояния здоровья и медицинскую предысторию подвергаемого лечению пациента и аналогичные факторы хорошо известные в медицинской отрасли техники.
Врач или ветеринар, имеющий обычные знания в данной области, сможет легко определить и назначить эффективное количество требуемой фармацевтической композиции. Например, врач или ветеринар мог бы начать с доз соединений изобретения, применяемых в фармацевтических композициях, при уровнях ниже, чем требуются для того, чтобы достичь желаемого терапевтического эффекта и постепенно увеличивать дозу до тех пор, пока не будет достигнуто желаемое действие.
Обычно подходящей ежедневной дозой соединения изобретения является количество соединения, которое является наименьшей дозой, эффективной для получения терапевтического эффекта. Такая эффективная доза будет зависеть от описанных выше факторов. Обычно пероральная, внутривенная, внутрижелудочковая и подкожная дозы соединений данного изобретения для пациента, используемые для указанных эффектов, будет составлять примерно от 0,0001 до примерно 100 мг на килограмм веса тела в день.
Изобретением в частности рассматривается пероральное введение на людях. Оральная дозировка у взрослых людей составляет обычно порядка 0,05 грамм (50 мг) до 10 грамм в день, даваемая в виде разовой дозы или в виде разделенных доз. В одном воплощении оральная дозировка для взрослых людей составляет 0,5 грамм (500 мг) до 10 грамм в день, даваемая в виде разовой дозы или в разделенных дозах. В одном воплощении оральная дозировка для взрослых людей составляет 0,5-8 грамм в день, даваемая в виде разовой дозы или в разделенных дозах. В одном воплощении оральная дозировка для взрослых людей составляет 0,5-6 грамм в день, даваемая в виде разовой дозы или в разделенных дозах. В одном воплощении оральная дозировка для взрослых людей составляет 0,5-4 грамма в день, даваемая в виде разовой дозы или в разделенных дозах. В одном воплощении оральная дозировка для взрослых людей составляет 0,5-2 грамма в день, даваемая в виде разовой дозы или в разделенных дозах. В одном воплощении оральная дозировка для взрослых людей составляет 0,5-1 грамм в день, даваемая в виде разовой дозы или в разделенных дозах.
При желании эффективная ежедневная доза активного соединения может вводиться в виде двух, трех, четырех, пяти, шести или более поддоз, вводимых отдельно с соответствующими интервалами на протяжении дня, необязательно, в единичных дозированных формах. Примером введения дозировки является одно введение в день.
Хотя соединение настоящего изобретения можно вводить одно, предпочтительно вводить его в виде фармацевтического состава (композиции).
Соединения согласно изобретению могут формулироваться любым удобным образом для использования в человеческой или ветеринарной медицине по аналогии с другими фармацевтическими средствами.
В еще одном аспекте настоящее изобретение предоставляет фармацевтически приемлемые композиции, которые включают терапевтически эффективное количество одного или более обсуждаемых соединений, описанных выше, вместе с одним или более фармацевтически приемлемыми носителями (добавками) и/или разбавителями. Как описано подробно ниже, фармацевтические композиции настоящего изобретения могут конкретно формулироваться для введения в твердой или жидкой форме, включая формы, приспособленные для следующего: (1) пероральное введение, например, в виде доз лекарственного средства для животных (водные или неводные растворы или суспензии), таблетки, болюсы, порошки, гранулы, пасты для применения на языке; (2) парентеральное введение, например, с помощью подкожной, внутримышечной или внутривенной инъекции, как, например, стерильный раствор или суспензия; (3) местное применение, например, в виде крема, мази или спрея, применяемого к коже, легким или слизистым мембранам; или (4) внутривагинально или внутриректально, например, в виде пессариев, крема или пены; (5) подъязычно или защечно; (6) оккулярно; (7) чрескожно; или (8) назально.
Пациентом, получающим данное лечение, является любое нуждающееся в этом животное, включая приматов, особенно людей, и других млекопитающих, таких как лошади, крупный рогатый скот, свиньи и овцы; и в общем домашняя птица и домашние животные.
Соединение изобретения может вводиться как таковое или в смесях с фармацевтически приемлемыми носителями, а может также вводиться в сочетании с, по меньшей мере, одним другим активным соединением. Терапия в сочетании, таким образом, включает последовательное, одновременное или раздельное введение активного соединения таким образом, что терапевтические эффекты первого вводимого соединения не исчезают целиком, когда вводится последующее соединение.
Мицеллы. Технология микроэмульгирования улучшает биодоступность некоторых липофильных (не растворимых в воде) фармацевтических агентов. Примеры включают триметрин (Dordunoo, S.K., et al., Drug Development and Industrial Pharmacy, 17(12), 1685-1713, 1991) и REV 5901 (Sheen, P.C., et al., J Pharm Sci 80(7), 712-714, 1991). Среди прочего, микроэмульгирование представляет увеличенную биодоступность, направляя преимущественно абсорбцию к лимфатической системе вместо циркуляторной системы, которая тем самым обходит печень, и предотвращает разрушение соединений при печеночно-желчной циркуляции.
Хотя рассматриваются все подходящие амфифильные носители, характерными примерами носителей обычно являются те, которые имеют статус Обычно Признаваемых как Безопасные (GRAS), и которые могут и солюбилизировать соединение настоящего изобретения, и микроэмульгировать его на более позднем этапе, когда раствор входит в контакт с комплексной водной фазой (такой как находящаяся в желудочно-кишечном тракте человека). Обычно амфифильные ингредиенты, которые удовлетворяют данным требованиям, имеют показатели HLB (гидрофильно-липофильного баланса) 2-20, и их структуры содержат алифатические радикалы с прямой цепью в пределах С-6 - С-20. Примерами являются полиэтилен-гликозилированные глицериды и полиэтиленгликоли.
Особенно рассматриваются промышленно доступные амфифильные носители, включающие ряд Gelucire, Лабрафил, Лабразол или Лаурогликоль (все производятся и поставляются фирмой Gattefosse Corporation, Saint Priest, Франция), PEG-моноолеат, PEG-диолеат, PEG-монолаурат и дилаурат, лецитин, полисорбат 80, и проч. (производятся и поставляются рядом компаний США и по всему миру).
Полимеры. Гидрофильными полимерами, подходящими для использования в настоящем изобретении, являются полимеры, которые свободно растворимы в воде, могут ковалентно присоединяться к образующему наполнитель липиду, и которые переносятся in vivo без токсичных эффектов (т.е. являются биосовместимыми). Подходящие полимеры включают в их число полиэтиленгликоль (PEG), полимолочную кислоту (называется также полилактид), полигликолевую кислоту (также называется полигликолид), сополимер полимолочной-полигликолевой кислоты и поливиниловый спирт. Характерными примерами полимеров являются полимеры, имеющие молекулярный вес примерно от 100 или 120 дальтон до примерно 5000 или 10000 дальтон, и более предпочтительно примерно от 300 до примерно 5000 дальтон. В одном воплощении полимером является полиэтиленгликоль, имеющий молекулярный вес примерно от 100 до примерно 5000 дальтон, и более предпочтительно примерно от 300 до примерно 5000 дальтон. В одном воплощении полимером является полиэтиленгликоль 750 дальтон (PEG(750)). Полимеры могут также определяться по числу мономеров в них; воплощение настоящего изобретения использует полимеры из, по меньшей мере, примерно трех мономеров, такие PEG полимеры состоят из трех мономеров (приблизительно 150 дальтон).
Другие гидрофильные полимеры, которые могут быть подходящими для использования в настоящем изобретении, включают поливинилпирролидон, полиметоксазолин, полиэтилоксазолин, полигидроксипропилиметакриламид, полиметакриламид, полидиметилакриламид и производные целлюлозы, такие как гидроксиметилцеллюлоза или гидроксиэтилцеллюлоза.
В некоторых воплощениях форма композиции настоящего изобретения включает биосовместимый полимер, выбранный из группы, состоящей из полиамидов, поликарбонатов, полиалкиленов, полимеров акриловых и метакриловых сложных эфиров, поливиниловых полимеров, полигликолидов, полисилоксанов, полиуретанов и их сополимеров, целлюлоз, полипропилена, полиэтиленов, полистирола, полимеров молочной и гликолевой кислоты, полиангидридов, сложных поли(орто)эфиров, поли(масляной кислоты) (англ. poly(butic acid), поли(валериановой кислоты), поли(лактид-со-капролактона), полисахаридов, белков, полигиалуроновых кислот, полицианоакрилатов и смесей или их сополимеров.
Циклодекстрины. Циклодекстринами являются циклические олигосахариды, содержащие 6, 7 или 8 глюкозных звеньев, обозначаемых греческими буквами альфа, бета и гамма, соответственно. Звенья глюкозы связаны альфа-1,4-глюкозидными связями. Как следствие, конформации звеньев сахара, все вторичные гидроксильные группы (при С-2, С-3) расположены на одной стороне кольца, в то время как все первичные гидроксильные группы при С-6 располагаются на другой стороне. В результате, наружные лицевые стороны являются гидрофильными, делая циклодекстрины растворимыми в воде. В противоположность этому, полости циклодекстринов являются гидрофобными, поскольку они выстраиваются по водороду атомов С-3 и С-5, и по эфироподобным кислородам. Данные матрицы позволяют комплексообразование с множеством относительно гидрофобных соединений, включая, например, стероидные соединения, такие как 17-бета-эстрадиол (см., например, Uden et al., Plant Cell Tiss. Org. Cult. 38:1-3-113 (1994)). Комплексообразование имеет место с помощью ванн-дер-ваальсовых взаимодействий и образования водородной связи. Для общего обозрения химии циклодекстринов см. Wenz, Agnew. Chem. Int. Ed. Engl., 33:803-822 (1994).
Физико-химические свойства производных циклодекстрина сильно зависят от вида и степени замещения. Например, их растворимость в воде находится в интервале от нерастворимого (например, триацетил-бета-циклодекстрин) до 147% растворим (вес/объем) (G-2-бета-циклодекстрин). В дополнение к этому они растворимы во многих органических растворителях. Свойства циклодекстринов делают возможным регулирование растворимости разнообразных компонентов составов увеличением или понижением их растворимости.
Описаны многочисленные циклодекстрины и способы их получения. Например, Parmeter (I), et al. (Патент США № 3453259; включенный для ссылки) и Gramera, et al. (патент США № 3459731) описывают электронейтральные циклодекстрины. Другие производные включают циклодекстрины с катионными свойствами (Parmeter (II)), патент США № 3453257, включенный для ссылки), нерастворимые поперечносшитые циклодекстрины (Solms, патент США №3420788, включенный для ссылки), и циклодекстрины с анионными свойствами (Parmeter (III), Патент США №3426011, включенный для ссылки). Среди производных циклодекстрина с анионными свойствами, карбоновые кислоты, фосфористые, фосфиновые, фосфоновые, фосфорные, тиофосфоновые, тиосульфиновые и сульфоновые кислоты присоединены к исходному циклодекстрину (см. Parmeter (III), выше). Кроме того, сульфоалкиловоэфирные производные циклодекстрина описаны Stella et al., (патент США № 5134127, включенный для ссылки).
Липосомы. Липосомы состоят из, по меньшей мере, одной липидной двухслойной мембраны с заключенным водным внутренним отсеком. Липосомы могут характеризоваться по типу мембраны и по размеру. Небольшие однослойные везикулы (SUV) имеют одну мембрану и диаметр обычно между 0,02 и 0,05 мкм; крупные моноламеллярные везикулы (LUVS) обычно крупнее 0,05 мкм. Олиголамеллярные крупные везикулы и многослойные везикулы имеют мультиламеллярные, обычно концентрические, мембранные слои и обычно крупнее 0,1 мкм. Липосомы с несколькими неконцентрическими мембранами, т.е. несколькими более мелкими везикулами, содержащимися в более крупной везикуле, называются мультивезикулярными везикулами.
Один аспект настоящего изобретения относится к составам, включающим липосомы, содержащие соединение настоящего изобретения, в которых липосомная мембрана формируется, обеспечивая липосому с увеличенной несущей полостью. Альтернативно или дополнительно, соединение настоящего изобретения может содержаться внутри, или адсорбироваться на бислое липосомы. Соединение настоящего изобретения может собираться вместе с липидным поверхностно-активным веществом и переноситься во внутреннем пространстве липосомы; в данных случаях липосомная мембрана формируется, чтобы противостоять разрушительным действиям совокупности активный агент/поверхностно-активное вещество.
Согласно одному воплощению настоящего изобретения липидный бислой липосомы содержит липиды, дериватизированные с полиэтиленгликолем (PEG), так что PEG цепи простираются от внутренней поверхности липидного бислоя во внутреннее пространство, инкапсулированное липосомой, и простираются от внешней стороны липидного бислоя в окружающую среду.
Активные агенты, содержащиеся в липосомах настоящего изобретения, находятся в солюбилизированной форме. Совокупности поверхностно-активного вещества и активного агента (такие как эмульсии или мицеллы, содержащие представляющий интерес активный агент) могут улавливаться во внутреннем пространстве липосом согласно настоящему изобретению. Поверхностно-активное вещество действует, чтобы диспергировать и солюбилизировать активный агент, и может быть выбрано из любых алифатических, циклоалифатических или ароматических поверхностно-активных веществ, включая, но не ограничиваясь ими, биосовместимые, лизофосфатидилхолины (LPC) c изменяющейся длиной цепи (например, примерно от С14 до примерно С20). Полимер-дериватизированные липиды, такие как PEG-липиды, могут также использоваться для образования мицелл, так как они действуют, ингибируя слияние мицелла/мембрана, и так как добавление полимера к молекулам поверхностно-активного вещества снижает критическую концентрацию мицелл (СМС) поверхностно-активного вещества и помогает образованию мицелл. Примерами являются поверхностно-активные вещества с СМС в микромолярном интервале; высшие СМС поверхностно-активные вещества могут использоваться для получения мицелл, заключенных внутри липосом настоящего изобретения, однако мономеры поверхностно-активного вещества мицелл могут воздействовать на стабильность липосомного бислоя и являются фактором при проектировании липосомы желаемой стабильности.
Липосомы, пригодные в настоящем изобретении, могут получаться с помощью любых из множества приемов, которые известны в данной области. См., например, патент США № 4235871; опубликованную РСТ заявку WO 96/14057; New RRC, Liposones: A practical approach, IRL Press, Oxford (1990), pp.33-104; Lasic DD, Liposomes from physics to applications, Elsevier Science Publishers BV, Amsterdam, 1993.
Например, липосомы, пригодные в настоящем изобретении, могут получаться с помощью диффундирования липида, дериватизированного с гидрофильным полимером в предварительно образованные липосомы, такого как подвергание предварительно образованных липосом действию мицелл, составленных из сшитых с липидом полимеров, при концентрациях липида, соответствующих конечному мольному проценту дериватизированного липида, который желателен в липосоме. Липосомы, содержащие гидрофильный полимер, могут также получаться с помощью гомогенизации, гидратирования липидной области или экструзионных технологических приемов, которые известны в данной области.
В одном аспекте настоящего изобретения липосомы имеют в основном гомогенные размеры в выбранном размерном интервале. Один эффективный способ сортировки по размеру предусматривает экструдирование водной суспензии липосом через ряд поликарбонатных мембран, имеющих выбранный равномерный размер пор; размер пор мембраны будет грубо соответствовать наиболее крупным размерам липосом, получаемым при экструзии через мембрану. См., например, патент США № 4737323.
Модификаторы высвобождения. Характеристики высвобождения состава настоящего изобретения зависят от инкапсулирующего материала, концентрации инкапсулированного лекарственного средства и от присутствия модификаторов высвобождения. Например, высвобождением можно манипулировать, чтобы оно было зависимым от рН, с использованием чувствительного к рН покрытия, которое обеспечивает высвобождение только при низком показателе рН, как в желудке, или при более высоком рН, как в кишечнике. Энтеросолюбильное покрытие может использоваться для предотвращения высвобождения из окружения не раньше, чем после прохождения через желудок. Многочисленные покрытия или смеси цианамида, инкапсулированного в различные материалы, могут использоваться для достижения начального высвобождения в желудке с последующим более поздним высвобождением в кишечнике. Высвобождением можно также управлять включением солей или агентов, образующих поры, которые могут увеличивать поглощение воды или высвобождение лекарственного средства диффузией из капсулы. Наполнители, которые модифицируют растворимость лекарственного средства, могут также использоваться для регулирования скорости высвобождения. Могут также включаться агенты, которые усиливают разрушение матрикса или высвобождение из матрикса. Они могут добавляться к лекарственному средству в виде отдельной фазы (т.е. в виде частиц), или могут совместно растворяться в полимерной фазе, в зависимости от соединения. Во всех случаях количество должно быть между 0,1 и тридцатью процентами (в/в полимера). Типы усилителей разрушения или разложения включают неорганические соли, такие как сульфат аммония и хлорид аммония, органические кислоты, такие как лимонная кислота, бензойная кислота и аскорбиновая кислота, неорганические основания, такие как карбонат натрия, карбонат калия, карбонат кальция, карбонат цинка и гидроксид цинка, и органические основания, такие как протаминсульфат, спермин, холин, этаноламин, диэтаноламин и триэтаноламин, и поверхностно-активные вещества, такие как Твин® и Плуроник®. Образующие поры агенты, которые добавляют матриксам микроструктуру (т.е. растворимые в воде соединения, такие как неорганические соли и сахара) добавляются в виде частиц. Предел должен быть между одним и тридцатью процентами (в/в полимера).
Поглощением можно также управлять путем изменения времени нахождения частиц в кишечнике. Это может достигаться, например, покрытием частиц, или выбором в качестве инкапсулирующего материала мукозного адгезивного полимера. Примеры включают большинство полимеров со свободными карбоксильными группами, таких как хитозан, целлюлозы и особенно полиакрилаты (используемые здесь полиакрилаты относятся к полимерам, включающим акрилатные группы и модифицированные акрилатные группы, такие как цианоакрилаты и метакрилаты).
Один аспект изобретения относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, где фармацевтический препарат имеет ЕС50 для снижения сывороточного холестерина, LDL и/или триглицеридов, который в среднем у пациентов людей составляет не более 20 процентов половины максимальной концентрации фармацевтического препарата, которая вызывала бы кожное расширение сосудов (прилив крови) у популяции средних пациентов.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, в случае которых ЕС50 для снижения сывороточного холестерина, LDL и/или триглицеридов, составляет не более 1 процента половины максимальной концентрации фармацевтического препарата, которая вызывала бы кожное расширение сосудов (прилив крови) у популяции средних пациентов.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, в случае которых фармацевтический препарат имеет ЕС50 для снижения сывороточного холестерина, LDL и/или триглицеридов, который в среднем у пациентов людей составляет не более 20 процентов половины концентрации фармацевтического препарата, которая вызывала бы увеличение в сыворотке уровней аспартатаминотрансферазы (AST) и аланинаминотрансферазы (ALT), требующего прекращения введения фармацевтического препарата.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, среди которых указанная композиция эффективна для снижения в сыворотке липида, не вызывая при этом ограничивающие лечение (i) гепатотоксичность и (ii) повышение уровней мочевой кислоты, или уровней глюкозы, или того и другого, после введения указанному пациенту, которое потребовало бы, чтобы такое лечение было прервано, когда указанная композиция принимается пациентом один раз в день.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, сформулированных в сочетании со статином.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, в которых статин выбран из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина.
В некоторых воплощениях настоящее изобретение относится к любой одной из упомянутых выше фармацевтических композиций, сформулированных в сочетании, по меньшей мере, с одним дополнительным терапевтическим агентом, выбранным из группы, состоящей из 11β HSD-1 ингибиторов, ингибиторов 5НТ переносчика, 5НТ2с агонистов, 5-LO или FLAP ингибиторов, ингибиторов α-глюкозидазы, АВСА1 усилителей, ACC ингибиторов, ингибиторов ацилСоА:холестерин-О-ацилтрансферазы, ацилэстрогенов, противодиабетических агентов, анти-дислипидемических агентов, анти-гипертензивных агентов, антиоксидантов, Аро А1 миметиков, Аро А1 модуляторов, Аро Е миметиков, ингибиторов белка переноса аполипопротеин-В секреции/микросомального триглицерида (аро-В/МТР), агентов, подавляющих аппетит, аспирина, β3 агонистов, ингибиторов реабсорбции желчной кислоты, веществ, связывающих желчную кислоту, агонистов бомбезина, BRS3 агонистов, СВ1 антагонистов/инверсных агонистов, ССК-А агонистов, ингибиторов абсорбции холестерина, ингибиторов транспортировки холестерина, ингибиторов белка переноса сложного холестерилового эфира (СЕТР), CNTF, CNTF агонистов/модуляторов, сочетания эзетимиба и симвастатина и/или аторвастатина, CSL-111, дегидроэпиандростерона, делипидированного HDL, DGAT антисмысловых олиго агентов, DGAT1 ингибиторов, DGAT2 ингибиторов, ингибиторов переносчика дикарбоксилата, агонистов допамина, антагонистов DP рецепторов, эзетимиба, FAS ингибиторов, ингибиторов связывающего жирную кислоту белка (FABP), ингибиторов переносчика жирной кислоты, ингибиторов белка переносчика жирной кислоты (FATP), ингибиторов прилива крови, модуляторов FXR рецептора, антагонистов рецептора галанина, гемкабена, антагонистов грелина, грелиновых антител, GLP-1 агонистов, агонистов рецептора глюкагон-подобного пептида-1, глюкокортикоидных агонистов/антагонистов, ингибиторов переносчика глюкозы, HDL миметиков, ингибиторных соединений HMG СоА редуктазы, ингибиторов HMG-CoA синтетазы, антагонистов чувствительной к гормонам липазы, агути-родственных белков человека (AGRP), Н3 антагонистов/инверсных агонистов, неорганических секвестрантов холестерина, L-4f, лапаквистата, лептин агонистов/модуляторов, лептинов, ингибиторов липазы, ингибиторов синтеза липопротеина, лорапопранта, индукторов или активаторов рецептора липопротеина низкой плотности, агентов, понижающих Lp(a), агонистов LXR рецептора, ингибитора линкиназы, Mc3r агонистов, Mc4r агонистов, MCH1R антагонистов, MCH2R агонистов/антагонистов, антагонистов концентрирующего меланин гормона, mGluR5 антагонистов, ингибиторов транспортировки микросомального триглицерида, ингибиторов повторного поглощения моноамина, природных растворимых в воде волокон, ингибиторов переносчика NE, агонистов рецептора нейромедина U, антагонистов нейропептида-Y, агонистов ниацина или рецептора ниацина, никотиновой кислоты, норадренергических аноректических агентов, NPY1 антагонистов, NPY2 агонистов, NPY4 агонистов, NPY5 антагонистов, агентов не-стероидных противовоспалительных препаратов (NSAID), омега-3 жирных кислот, опиоидных антагонистов, антагонистов рецептора орексина, PDE ингибиторов, фентермина, ингибиторов фосфатного переносчика, фитофарм соединения 57, растительных станолов и/или сложных жирно-кислотных эфиров растительных станолов, ингибиторов скопления тромбоцитов, PPAR-α агонистов, PPAR-δ агонистов, PPAR-δ частичных агонистов, PPAR-γ агонистов, пробукола, ингибиторов ренинангиотензина, реверсированного-4F, SCD-1 ингибиторов, ингибиторов повторного поглощения сертонина, SGLT2 ингибиторов, ингибиторов скваленэпоксидазы, ингибиторов синтеза сквалена, ингибиторов биосинтеза стерола, симпатомиметических агонистов, агонистов тироидного гормона β, тиромиметических агентов, топирамата, ингибиторов синтеза триглицерида, UCP-1 активаторов, UCP-2 активаторов, UCP-3 активаторов и антагонистов белка, связывающего урокортин.
В некоторых воплощениях настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; ниацин; и фармацевтически приемлемый наполнитель.
В некоторых воплощениях настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; ниацин; статин, выбранный из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина; и фармацевтически приемлемый наполнитель.
В некоторых воплощениях настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; глитазон, выбранный из группы, состоящей из троглитазона, розиглитазона и пиоглитазона; и фармацевтически приемлемый наполнитель.
В некоторых воплощениях настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; ниацин; глитазон, выбранный из группы, состоящей из троглитазона, розиглитазона и пиоглитазона; и фармацевтически приемлемый наполнитель.
В некоторых воплощениях настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; фибрат, выбранный из группы, состоящей из фенофибрата и безафибрата; и фармацевтически приемлемый наполнитель.
В некоторых воплощениях настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения или его фармацевтически приемлемую соль; ниацин; фибрат, выбранный из группы, состоящей из фенофибрата и безафибрата; и фармацевтически приемлемый наполнитель.
СПОСОБЫ
Аспект изобретения относится к способу лечения заболевания, расстройства или состояния, выбранного из группы, состоящей из гиперлипидемии, гиперхолестеринемии, гиподистрофии, дислипидемии, атеросклероза и болезни коронарных артерий, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения болезней, расстройств или состояний, выбранных их группы, состоящей из гиперлипидемии, гиперхолестеринемии, гиподистрофии, дислипидемии, атеросклероза и болезни коронарных артерий, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения болезней, расстройств или состояний, выбранных их группы, состоящей из метаболического синдрома, ожирения, заболевания жирной печени и диабета, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения болезней, расстройств или состояний, выбранных их группы, состоящей из метаболического синдрома, ожирения, заболевания жирной печени и диабета, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу поднятия уровней липопротеина высокой плотности (HDL) в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу поднятия уровней липопротеина высокой плотности (HDL) в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения уровней липопротеина низкой плотности (LDL) в сыворотке или понижения уровней липопротеина (а) в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения уровней липопротеина низкой плотности (LDL) в сыворотке или понижения уровней липопротеина (а) в сыворотке, включающему стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения болезней, расстройств или состояний, выбранных их группы, состоящей из застойной сердечной недостаточности, сердечнососудистой болезни, гипертензии, коронарной болезни сердца, стенокардии, пеллагры, синдрома Хартнупа, карциноидного синдрома, артериальной окклюзивной болезни, гипотироидизма, сужения сосудов, остеоартрита, ревматоидного артрита, болезни Альцгеймера, расстройств периферической и центральной нервной системы, гематологических заболеваний, рака, воспаления, респираторных болезней и гастроэнтерологических заболеваний, включающему стадию введения млекопитающему, нуждающемуся в этом, соединения настоящего изобретения или его фармацевтически приемлемой соли. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения болезней, расстройств или состояний, выбранных их группы, состоящей из застойной сердечной недостаточности, сердечнососудистой болезни, гипертензии, коронарной болезни сердца, стенокардии, пеллагры, синдрома Хартнупа, карциноидного синдрома, артериальной окклюзивной болезни, гипотироидизма, сужения сосудов, остеоартрита, ревматоидного артрита, болезни Альцгеймера, расстройств периферической и центральной нервной системы, гематологических заболеваний, рака, воспаления, респираторных болезней и гастроэнтерологических заболеваний, включающему стадию введения млекопитающему, нуждающемуся в этом, фармацевтической композиции настоящего изобретения. В одном воплощении млекопитающим является человек.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение терапевтически эффективного количества статина. В одном таком воплощении совместным введением является совместное введение перорально.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение терапевтически эффективного количества статина, в котором статин выбран из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном таком воплощении совместным введением является совместное введение перорально.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение терапевтически эффективного количества, по меньшей мере, одного дополнительного терапевтического агента, выбранного из группы, состоящей из 11β HSD-1 ингибиторов, ингибиторов 5НТ переносчика, 5НТ2с агонистов, 5-LO или FLAP ингибиторов, ингибиторов α-глюкозидазы, АВСА1 усилителей, ACC ингибиторов, ингибиторов ацилСоА:холестерин-О-ацилтрансферазы, ацилэстрогенов, противодиабетических агентов, анти-дислипидемических агентов, анти-гипертензивных агентов, антиоксидантов, Аро А1 миметиков, Аро А1 модуляторов, Аро Е миметиков, ингибиторов белка переноса аполипопротеин-В секреции/микросомального триглицерида (аро-В/МТР), агентов, подавляющих аппетит, аспирина, β3 агонистов, ингибиторов реабсорбции желчной кислоты, веществ, связывающих желчную кислоту, агонистов бомбезина, BRS3 агонистов, СВ1 антагонистов/инверсных агонистов, ССК-А агонистов, ингибиторов абсорбции холестерина, ингибиторов транспортировки холестерина, ингибиторов белка переноса сложного холестерилового эфира (СЕТР), CNTF, CNTF агонистов/модуляторов, сочетания эзетимиба и симвастатина и/или аторвастатина, CSL-111, дегидроэпиандростерона, делипидированного HDL, DGAT антисмысловых олиго агентов, DGAT1 ингибиторов, DGAT2 ингибиторов, ингибиторов переносчика дикарбоксилата, агонистов допамина, антагонистов DP рецептора, эзетимиба, FAS ингибиторов, ингибиторов связывающего жирную кислоту белка (FABP), ингибиторов переносчика жирной кислоты, ингибиторов белка переносчика жирной кислоты (FATP), ингибиторов прилива крови, модуляторов FXR рецептора, антагонистов рецептора галанина, гемкабена, антагонистов грелина, грелиновых антител, GLP-1 агонистов, агонистов рецептора глюкагон-подобного пептида-1, глюкокортикоидных агонистов/антагонистов, ингибиторов переносчика глюкозы, HDL миметиков, ингибиторных соединений HMG СоА редуктазы, ингибиторов HMG-CoA синтетазы, антагонистов чувствительной к гормонам липазы, агути-родственных белков человека (AGRP), Н3 антагонистов/инверсных агонистов, неорганических секвестрантов холестерина, L-4f, лапаквистата, агонистов/модуляторов лептина, лептинов, ингибиторов липазы, ингибиторов синтеза липопротеина, лорапопранта, индукторов или активаторов рецептора липопротеина низкой плотности, агентов, понижающих Lp(a), агонистов LXR рецептора, ингибитора линкиназы, Mc3r агонистов, Mc4r агонистов, MCH1R антагонистов, MCH2R агонистов/антагонистов, антагонистов концентрирующего меланин гормона, mGluR5 антагонистов, ингибиторов транспортировки микросомального триглицерида, ингибиторов повторного поглощения моноамина, природных растворимых в воде волокон, ингибиторов переносчика NE, агонистов рецептора нейромедина U, антагонистов нейропептида-Y, агонистов ниацина или рецептора ниацина, никотиновой кислоты, норадренергических аноректических агентов, NPY1 антагонистов, NPY2 агонистов, NPY4 агонистов, NPY5 антагонистов, агентов не-стероидных противовоспалительных препаратов (NSAID), омега-3 жирных кислот, опиоидных антагонистов, антагонистов рецептора орексина, PDE ингибиторов, фентермина, ингибиторов фосфатного переносчика, фитофарм соединения 57, растительных станолов и/или сложных жирно-кислотных эфиров растительных станолов, ингибиторов скопления тромбоцитов, PPAR-α агонистов, PPAR-δ агонистов, PPAR-δ частичных агонистов, PPAR-γ агонистов, пробукола, ингибиторов ренинангиотензина, реверсированного-4F, SCD-1 ингибиторов, ингибиторов повторного поглощения сертонина, SGLT2 ингибиторов, ингибиторов скваленэпоксидазы, ингибиторов синтеза сквалена, ингибиторов биосинтеза стерола, симпатомиметических агонистов, агонистов тироидного гормона β, тиромиметических агентов, топирамата, ингибиторов синтеза триглицерида, UCP-1 активаторов, UCP-2 активаторов, UCP-3 активаторов и антагонистов белка, связывающего урокортин. В одном воплощении совместное введение является пероральным.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение терапевтически эффективного количества, по меньшей мере, одного дополнительного терапевтического агента, выбранного из группы, состоящей из ингибиторов HMG CoA редуктазы, аспирина, ингибиторов белка переноса сложного холестерилового эфира, NSAID, фибратов, протеинконвертазы типа субтилизин/кексин (PCSK9), неорганических секвестрантов холестерина, ингибиторов АцилСоА:холестерин-О-ацилтрансферазы, СЕТР ингибиторов, PPAR α агонистов, PPAR γ агонистов, ингибиторов повторного поглощения желчной кислоты, ингибиторов синтеза триглицерида, активаторов рецептора липопротеина, ингибиторов DGAT1, SCD-1 ингибиторов, ингибиторов липазы, антагонистов DP рецептора, модуляторов аро А1, ингибиторов транспортировки холестерина, метформина, модуляторов рецептора ниацина и ингибиторов DPP-IV. В одном таком воплощении совместное введение является пероральным.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение терапевтически эффективного количества, по меньшей мере, одного дополнительного терапевтического агента, выбранного из группы, состоящей из ингибиторов HMG CoA редуктазы, ингибиторов белка переноса сложного холестерилового эфира, аспирина, NSAID, фибратов, антагонистов DP рецептора, эзетимиба или сочетания эзетимиба и симвастатина. В одном таком воплощении совместное введение является пероральным.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение терапевтически эффективного количества, по меньшей мере, одного ингибитора HMG CoA редуктазы, выбранного из ловастатина, симвастатина, правастатина, аторвастатина, флувастатина, церивастатина, ривастатина, розувастатина кальция и питавастатина. В одном таком воплощении совместное введение является пероральным.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение симвастатина. В одном таком воплощении совместное введение является пероральным.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение ингибитора белка переноса сложного холестерилового эфира. В одном таком воплощении совместное введение является пероральным.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов, дополнительно включающему совместное введение эзетимиба, аспирина, ибупрофена, ацетаминофена или сочетания эзетимиба и симвастатина. В одном таком воплощении совместное введение является пероральным.
Один аспект изобретения относится к способу лечения гиперлипидемии, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения гиперлипидемии, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу повышения в сыворотке уровней липопротеина высокой плотности (HDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу повышения в сыворотке уровней липопротеина высокой плотности (HDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина низкой плотности (LDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина низкой плотности (LDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина (а) (Lp(а)) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина (а) (Lp(а)) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; и терапевтически эффективного количества ниацина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения гиперлипидемии, гиперхолестеринемии, атеросклероза, болезни коронарной артерии, застойной сердечной недостаточности, сердечнососудистого заболевания, гипертензии, коронарной болезни сердца, стенокардии, пеллагры, синдрома Хартнупа, карциноидного синдрома, артериальной окклюзивной болезни, ожирения, гипотироидизма, сужения сосудов, остеоартрита, ревматоидного артрита, диабета, болезни Альцгеймера, гиподистрофии или дислипидемии, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатинга, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения гиперлипидемии, гиперхолестеринемии, атеросклероза, болезни коронарной артерии, застойной сердечной недостаточности, сердечно-сосудистого заболевания, гипертензии, коронарной болезни сердца, стенокардии, пеллагры, синдрома Хартнупа, карциноидного синдрома, артериальной окклюзивной болезни, ожирения, гипотироидизма, сужения сосудов, остеоартрита, ревматоидного артрита, диабета, болезни Альцгеймера, гиподистрофии или дислипидемии, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатинга, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения гиперлипидемии, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения гиперлипидемии, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу повышения в сыворотке уровней липопротеина высокой плотности (HDL) у млекопитающих, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу повышения в сыворотке уровней липопротеина высокой плотности (HDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина низкой плотности (LDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина низкой плотности (LDL) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина (а) (Lp(а)) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу понижения в сыворотке уровней липопротеина (а) (Lp(а)) у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; терапевтически эффективного количества ниацина; и терапевтически эффективного количества статина, выбранного из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, питавастатина, правастатина, розувастатина и симвастатина. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых статином является ловастатин или аторвастатин.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли; и глитазона, выбранного из группы, состоящей из троглитазона, розиглитазона и пиоглитазона. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; и глитазона, выбранного из группы, состоящей из троглитазона, розиглитазона и пиоглитазона. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли; ниацина; и глитазона, выбранного из группы, состоящей из троглитазона, розиглитазона и пиоглитазона. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; ниацина; и глитазона, выбранного из группы, состоящей из троглитазона, розиглитазона и пиоглитазона. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли; и фибрата, выбранного из группы, состоящей из фенофибрата и безафибрата. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; и фибрата, выбранного из группы, состоящей из фенофибрата и безафибрата. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли; ниацина; и фибрата, выбранного из группы, состоящей из фенофибрата и безафибрата. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
Один аспект изобретения относится к способу лечения диабета у млекопитающего, включающему стадию совместного введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции настоящего изобретения; ниацина; и фибрата, выбранного из группы, состоящей из фенофибрата и безафибрата. В одном воплощении совместное введение является пероральным. В одном воплощении млекопитающим является человек.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых млекопитающим являются приматы, бычьи, овечьи, грызуны, лошади, собачьи или кошачьи.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанным млекопитающим является человек.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят перорально.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят внутривенно.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят подъязычно.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят с помощью ингаляции.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят оккулярно.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят чрескожно.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят ректально.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят вагинально.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят место.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят внутримышечно.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят подкожно.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят буккально.
В некоторых воплощениях настоящее изобретение относится к любому из упомянутых выше способов и сопровождающих ограничений, в которых указанное соединение, соединения или фармацевтическую композицию вводят назально.
ПОДТВЕРЖДЕНИЕ ПРИМЕРАМИ
Изобретение теперь описывается в общем, оно будет более легко понятно со ссылкой на следующее описание, которое включено лишь для целей иллюстрации некоторых аспектов и воплощений настоящего изобретения, и не предназначено для ограничения изобретения.
Пример 1: Схемы синтеза
А. Соединение ARI-001
Условия реакции: i. HCHO, Морфолин.HCl, н-PrOH, 100°C; ii. NaOH, затем HCl.
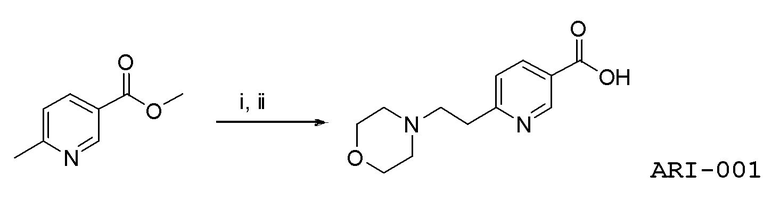
Метиловый эфир 6-метилникотиновой кислоты (4,5 г, 30 ммол), гидрохлорид морфолина (1,85 г, 15 ммол), н-PrOH (18 мл) и раствор формальдегида (в воде 37%)(1,2 г, 15 ммол) добавляли в 50-мл колбу, оснащенную холодильником. Реакционную смесь нагревали с обратным холодильником, с предварительно нагретой до 100°C масляной баней, в течение 2,5 часов под аргоном. Затем смесь оставляли стоять при комнатной температуре на протяжении ночи, и выпадали в осадок желтые игольчатые кристаллы (если осаждение не происходило при комнатной температуре, рекомендовали холодную комнату (4°C) или даже -10°C вымораживание). Выделенные фильтрованием кристаллы и промытые небольшим количеством этилового эфира давали сырой продукт стадии 1 в виде HCl соли (1,5 г, выход 35%; чистота около 95%, побочный продукт двойного присоединения по Манниху, составил около 3%). Данный сырой продукт далее очищали одноразовой перекристаллизацией из н-PrOH и MeOH, получая более чистый продукт стадии 1 в виде не совсем белого или бледно-желтого порошка (1,2 г, выход 28%; чистота была 99,1%, побочный продукт двойного присоединения по Манниху составил 0,9%).
Продукт стадии 1 (1,5 г, 5 ммол) растворяли в MeOH (20 мл) и добавляли воду (10 мл), 1н. LiOH (15 мл) при охлаждении ледяной водой. Полученную в результате смесь перемешивали при комнатной температуре на протяжении ночи, и затем доводили pH до 2-3 с 2н. HCl. После охлаждения в ваакуме остаток дополнительно очищали с помощью препаративной HPLC, элюируя растворителем ацетонитрилом и водой (c добавлением 5 мМ HCl), получая 1,2 г целевого соединения ARI-001 в виде белого порошка (HCl соль, общий выход был около 30% для двух стадий, когда использовали сырой продукт стадии 1).
B. Соединение ARI-002
Условия реакции: i. HCHO, Морфолин.HCl
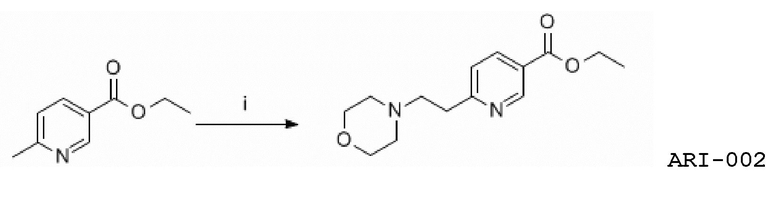
C. Соединение ARI-001D
Условия реакции: i. HCHO, Морфолин-d8.HCl; ii. NaOH, затем HCl.
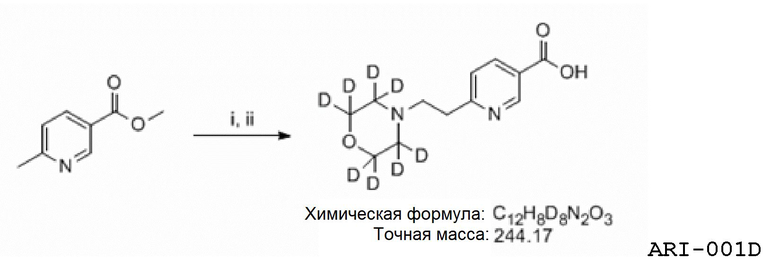
D. Соединение ARI-005
Условия реакции: i. 5-инданол, EDAC, DMAP.
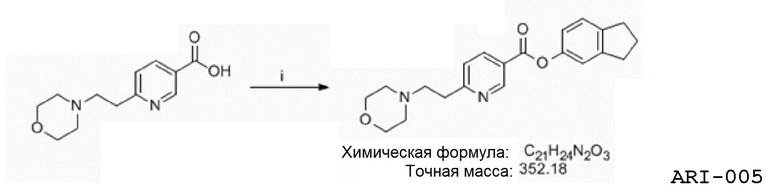
E. Соединение ARI-006 (гекса ARI-001 инозит)
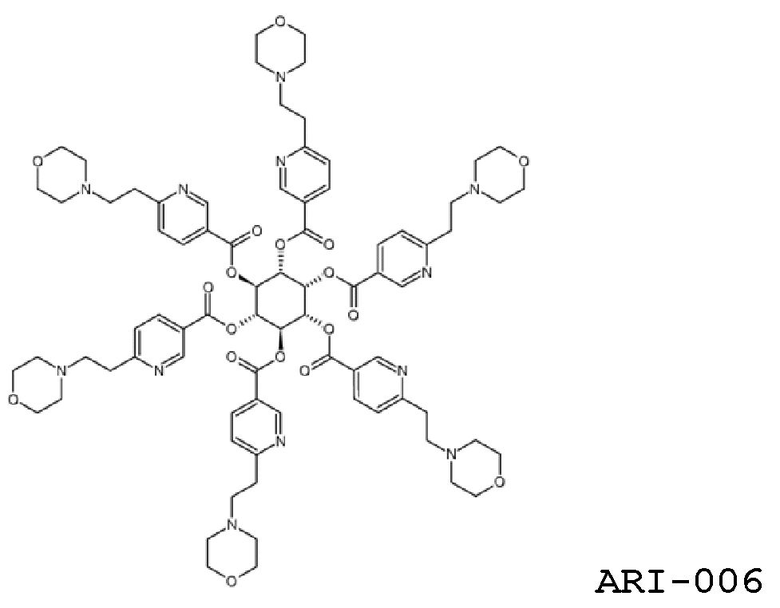
Условия реакции: i. имио-инозит, EDCI, DMAP.
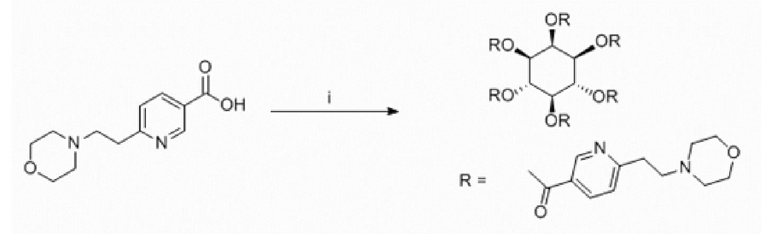
F. Соединение ARI-008
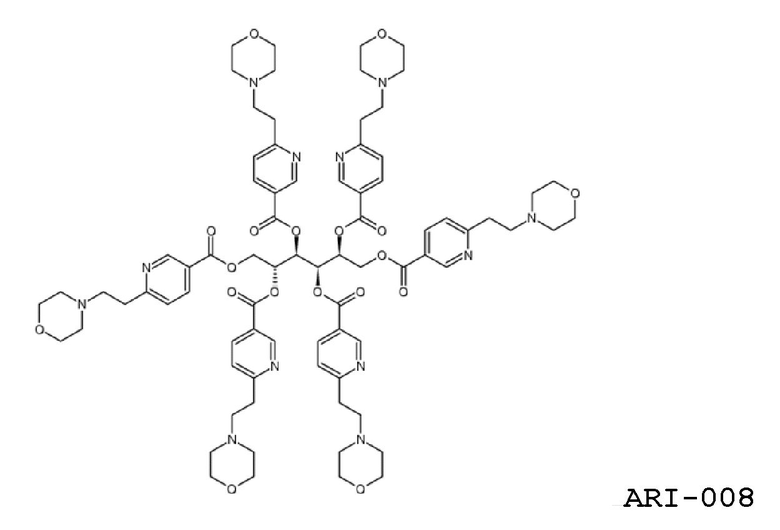
Условия реакции: i. D-сорбит, EDAC, DMAP
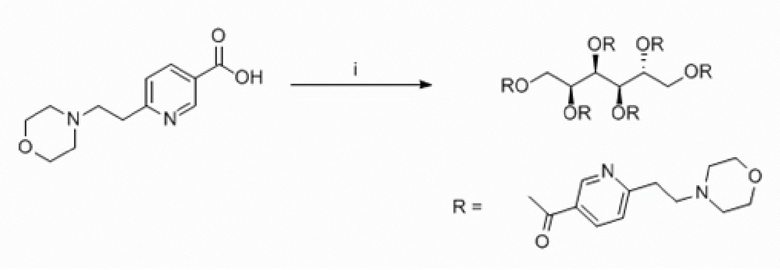
G. Соединение ARI-010
Условия реакции: i. ARI-001, EDAC, DMAP; ii. Bu4NI, BCl3
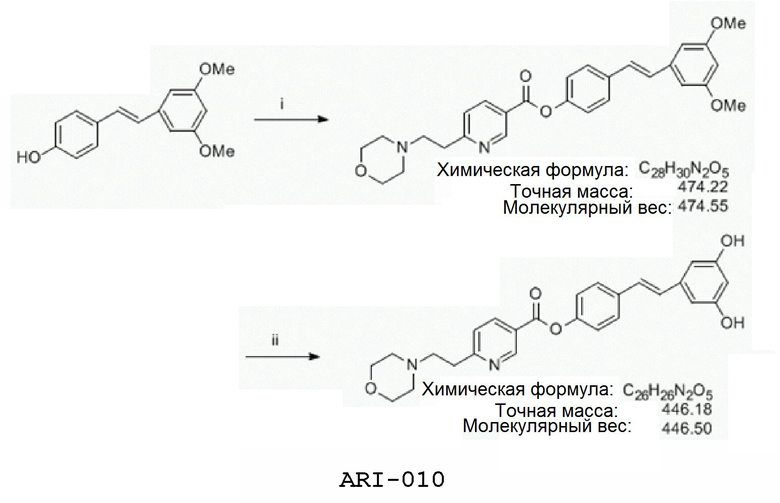
Пример 2: Исследования in vitro
ARI-001 синтезировали, как описано выше. Чистоту определяли независимо перед использованием в экспериментах с помощью жидкостной хроматографии масс-спектроскопии (LC-MS). Образец материала растворяли в воде/ацетонитриле и инжектировали на колонку Discovery C-18 с обращенной фазой. Подвижная фаза начиналась в виде смеси 2%:98% ацетонитрил:вода, которую выдерживали в течение трех минут, после чего начинался линейный градиент, который действовал на протяжении 6 минут, увеличивая процент ацетонитрила до тех пор, пока не достигалось окончательное соотношение 98%:2% ацетонитрил:вода. Данное конечное соотношение выдерживали в течение 3 минут. УФ детекцию проводили при 215 нм, как показано на фигуре 1. Главный пик идентифицировали как ARI-001.
Чистоту определяли по отношению области главного пика (3,59 минут) к сумме областей всех пиков. По данным определения чистота составляла 98,9%. Незначительные пики не идентифицировали.
Стабильность определяли при хранении ARI-001 при 50°С без особых требований к влажности. В различные моменты времени на протяжении 3 месяцев (90 дней) собирали небольшой образец (<5 мг) ARI-001 для анализа с использованием LC-MS в тех же условиях, что использовали для определения чистоты (см. выше). Фигура 2 показывает время стабильности ARI-001 в таких условиях.
В стандартных условиях (50°С, нерегулируемая влажность), ARI-001 оставался на 98,9% чистым в течение периода 65 дней. Только к 90 дням при 50°С ARI-001 начинал показывать какое-либо разложение. Спустя 90 дней, было найдено, что соединение было на 95,9% чистым.
Пример 3: Исследования in vivo
Для исследования действия ARI-001 на модулирование липида разрабатывали модель хомяков и использовали при назначении хронического введения соединения. Данная модель, ее разработка и эффекты ARI-001 описаны здесь.
Действие модификации корма на липидограмму у хомяков. Как и у других грызунов, липидограмму у Golden Syrian хомяков состоит из HDL, с незначительным LDL или VLDL холестерином. Однако на высокожирном корме хомяки испытывают увеличение скопления холестерина, включая триглицериды и свободные жирные кислоты. Добавление в источник питьевой воды сахара, такого как 10% фруктозы, в значительной степени увеличивает скопление триглицерида, включая VLDL. На данном корме хомяки становятся полезной моделью для исследования роли модуляторов на триглицериды и VLDL, и на LDL холестерин. Действительно, литературные источники использовали данную модель для исследования модулирующих триглицериды и свободные жирные кислоты соединений, таких как фенофибрат.
Мы проверяли действие видоизменения корма на липидограмму у самцов Syrian Golden хомяков. Хомяков заказывали у Charles River Labs (Вилмингтон, МА), и просили, чтобы они были весом 111-120 г (соответственно возраст 56-61 день). Хомяков содержали в клетках по 4-5 на клетку, и держали на стандартном световом цикле 12 часов с включенным/12 часов с выключенным светом. Все корма получали у фирмы Dyets, Inc. (Бетлехем, РА). “Нормальным кормом” была стандартная еда грызунов, каталог #5001, производимая в шариках. Вода и данный стандартный корм были доступны данной группе хомяков без ограничений. Корм давали в клетки по мере необходимости, но не менее часто, чем два раза в неделю. “Высокожирным кормом” была та же самая стандартная еда грызунов с добавлением следующего: 11,5% кукурузного масла, 11,5% кокосового масла, 0,5% холестерина и 0,25% дезоксихолата. Это также доступно от Dyets, Inc., как по каталогу #611201. Оба корма заказывали партиями по 10 кг и хранили при 4°С в течение длительности экспериментов (4-8 недель), и при -20°С для более долгого хранения (вплоть до шести месяцев). Вода и данный высокожирный корм были доступны данной группе хомяков без ограничений. Группу “высокожирный + фруктоза” кормили той же едой с добавлением жира как группу с “высокожирным кормом” (#611201), но добавляли в воду фруктозу до конечной концентрации 10%. Фруктозу поставляла Now Foods (по каталогу #6931), а продавала Lucky Vitamine (по каталогу #WB48432). Воду с фруктозой приготавливали добавлением 400 г фруктозы к 4 л воды и перемешиванием при комнатной температуре до растворения. Воду с фруктозой хранили при 4°С до использования. Когда давали хомякам, фруктозную воду держали в бутылках для воды в клетках с хомяками при комнатной температуре, в таких же, как и стандартная вода. Фруктозную воду и высокожирный корм давали данной группе хомяков без ограничений. Все животные оставались на своих соответствующих диетах в течение 21 дня.
Для начала данного эксперимента хомяков произвольно закрепляли за группами, определяемыми указанными выше модификациями диеты. По истечении фиксированного времени собирали у хомяков кровь (N=3-4) для определения содержания липида в плазме. Из-за ограничений в возможности адекватно собрать у хомяков кровь, все образцы крови получали с помощью заключительной сердечной пунктуры, которой предшествовала асфиксация двуокисью углерода. Кровь (приблизительно объемом 2 мл) собирали иглой 22G в 5 мл шприц и переносили в полый цилиндр с K2EDTA. Образцы хранили на льду до центрифугирования (14000 об/мин в течение 10 минут при 4°С) для отделения плазмы. Плазму затем аликвотами помещали в цилиндры для хранения при -80°С до анализа.
Липидные параметры затем определяли с использованием промышленно доступных комплектов от Wako США (Ричмонд, VA) согласно указаниям производителя.
Значения липида от контрольных животных (ноль дней на любую из трех диет, описанных выше) были по существу неразличимыми (см. Таблицу 1). К 21 дню обе модификации диеты оказывали значительное воздействие на липидограммы у данных хомяков при сравнении с животными, содержащимися на диете с обычной едой. Все параметры значительно увеличивались от контрольных (р<0,05 во всех группах), за исключением HDL. Как и предсказывалось, добавление фруктозы к высокожирной диете дополнительно увеличивало концентрации триглицерида и свободной жирной кислоты до большей степени, чем в случае одной высокожирной диеты. HDL был неизменным при сравнении с группой с нормальной диетой, несмотря на модификацию диеты.
Величины липида у хомяков на одной из трех различных диет в течение 21 дня. Хомяков умерщвляли, и их плазму анализировали, как описано выше. Величины представляют собой среднее от измерений каждого параметра (N=3), со стандартным отклонением. ТС: общий холестерин; HDL: холестерин липопротеина высокой плотности; LDL: холестерин липопротеина низкой плотности; TG: триглицериды; FFA: свободные жирные кислоты.
**р<0,01 по сравнению с нормальной диетой в том же параметре
***р<0,001 по сравнению с нормальной диетой в том же параметре
Для разделения различных субпопуляций холестерина от образцов плазмы хомяков использовали быструю жидкостную хроматографию белка (FPLC). Вкратце, плазму от каждого животного внутри заданной группы сливали вместе и наносили на AKTA регулирующую жидкость систему с Superose 6 10/300 GL колонкой (продукт 14-5172-01, GE Life Sciences). 250 мкл образца применяли к инжекционной системе, разбавляли 5 мл буфера (100 мМ Na2HPO4, 100 мМ NaCl, pH 7,5), наносили на колонку и элюировали 23,5 мл буфера со скоростью потока 1,0 мл/мин., фракции размером 0,24 мл. В каждой фракции отдельно измеряли концентрацию холестерина с использованием комплекта на общий холестерин от Wako, как описано выше, со следующим видоизменением: объем образца увеличивали до 30 мкл, а объем реагента снижали до 60 мкл.
Следы FPLC давали непрерывные кривые с тремя явными пиками, представляющими каждую из подгрупп холестерина: VLDL, LDL и HDL. Поскольку колонкой, используемой в данном эксперименте, является колонка с исключением размера, самые крупные частицы появляются первыми, в то время как наиболее мелкие появляются последними. Отсюда, VLDL является первым пиком по следу, с последующим LDL, с HDL, появляющимся последним. Если предположить, что каждый из данных пиков принимает Gaussian распределение, можно развернуть FPLC след в каждый из данных трех компонентов для определения вклада, который делает каждый компонент во всю кривую.
Фигура 3 иллюстрирует FPLC следы плазмы, объединенной от хомяков, содержащихся на двух различных диетах в течение трех недель. VLDL кривая является крайне растянутой на диете высокожирная + фруктоза. LDL пик также является гораздо выше. Интересно, что имеется очень незначительная разница в HDL пике, указывающая на то, что модификация диеты оказывает гораздо более сильное действие на не-HDL холестериновую популяцию. Данная модель затем ведет сама себя к тому, чтобы быть полезным средством, которым можно измерить действие ARI-001 на VLDL и LDL холестерин.
ARI-001 понижает уровни LDL холестерина, триглицеридов и свободной жирной кислоты у HF/HS хомяков. Самцов Golden Syrian хомяков закупали у Charles River Labs (111-120 г, 56-61 день) и акклиматизировали к диете высокожирная + фруктоза (называемая здесь как “HF/HS”), как описано выше, в течение двух недель. Хомяков распределяли на группы по весу тела после двухнедельной акклиматизации к диете. Двенадцать хомяков закрепили за группой наполнителя, 9 - к получению ниацина в дозе 1200 мг/кг, 10 - к получению ARI-001 в дозе 1120 мг/кг, и 10 - к получению ARI-001 в дозе 2240 мг/кг. В конце данного периода акклиматизации (привыкания) к диете хомякам перорально через желудочный зонд вводили 1 мл раствора наполнителя (воды), ниацина, или ARI-001 в одной из двух доз. Хомякам вводили дозу один раз в день всего в течение 18 дней. На протяжении всего периода дозирования все животные оставались на описанной выше HF/HS диете. Дозирующие растворы для каждой группы приготавливали на 7 дней за один раз, хотя приготавливали достаточно, чтобы хватило на 8 дней. Каждый раствор хранили между введениями при комнатной температуре. Группы определяли согласно таблице 2 ниже. Вследствие разницы в молекулярном весе между ниацином и ARI-001 дозы давали и в мг/кг и в ммол/кг. Заметьте, что 1200 мг/кг ниацина эквивалентно 2240 мг/кг ARI-001 на молярной основе.
Распределение на группы для исследования введения дозы в течение 18 дней у хомяков HF/HS
Как показано в Таблице 3, ARI-001, даваемый перорально через желудочный зонд в течение 18 дней по 2240 мг/кг понижал уровни LDL-холестерина и общего холестерина у хомяков HF/HS модели. ARI-001 также понижал уровни триглицеридов и свободной жирной кислоты с очень незначительной разницей между животными, как иллюстрирует небольшое стандартное отклонение. При молярной эквивалентной дозе 1200 мг/кг ниацин не способен был дать данные эффекты на данной модели животных, что говорит о том, что ARI-001, по меньшей мере, в 1,67 раза является более эффективным, чем ниацин, что касается параметра LDL холестерина, и, по меньшей мере, в 1,54 раза более эффективен, чем ниацин, в отношении параметра общего холестерина. Показаны значения триглицеридов и свободных жирных кислот, что указывает на ответ среды всех животных в группе ARI-001, получавшей 2240 мг/кг. То, что имеется очень незначительное стандартное отклонение среди этих данных, отражает впечатляющую скорость реагирования среди данных животных. Соотношение HDL/TC вычисляли делением HDL холестерина каждого отдельного хомяка на уровень его общего холестерина. Доля, которая получилась в результате, является соотношением HDL/ТС. Данный параметр был выше, чем в группе с наполнителем, с впечатляющей статистической значимостью (р<0,001). Это вероятно не представляет сильное увеличение HDL, так как абсолютная HDL величина увеличилась только на 33% в группе с 2240 мг/кг ARI-001 по сравнению с группой с наполнителем. Скорее, очень сильное изменение в соотношении HDL/TC вероятно представляет общесистемное уменьшение холестериновых популяций, за исключением HDL, который не только избавлялся от таких уменьшений, но и возможно увеличивался.
Липидные параметры от HF/HS хомяков, которым вводили перорально дозу наполнителя, ниацина или ARI-001 в течение 18 дней. ТС, общий холестерин. Данные величины представляют собой среднее ± стандартное отклонение. Р-величины сообщаются от двухвыборочных непарных t-тестов в сравнении с обрабатываемыми наполнителем в том же параметре.
(мг/дл)
** р<0,01 по сравнению с наполнителем в том же параметре
*** р<0,001 по сравнению с наполнителем в том же параметре
Измеренные HDL величины были погранично значительными (р=0,06) между группой 2240 мг/кг ARI-001 и наполнителем. Имелось значительное увеличение значения при рассмотрении вычисленных HDL/TC величин. FPLC следы также продемонстрировали значительную разницу в липидограмме между данными двумя группами. FPLC выполняли, как описано выше. Кривые преобразовывали, как описано выше. VLDL и LDL пики не только значительно уменьшались по сравнению с наполнителем, но HDL кривая была заметно больше по ARI-001 следу. См. Фигуру 4.
В отношении изменения уровня липида самцы Golden Syrian хомяков от предыдущего описанного эксперимента продемонстрировали зависимость от дозы ARI-001. 2240 мг/кг является эффективной дозой для 18 дневного ежедневного лечения почти по всем параметрам: общему холестерину, LDL холестерину, триглицеридам и свободным жирным кислотам. Однако 1120 мг/кг демонстрирует только умеренные эффекты по данным параметрам, причем, только результаты по свободным жирным кислотам демонстрируют статистически значимое действие по сравнению с наполнителем. Тем не менее, тенденция ясна между дозами для всех исследованных липидных параметров, как показано на Фигуре 5. Более того, при молярной эквивалентной дозе ARI-001 вызывает более сильную ответную реакцию, чем ниацин, по всем измеренным липидным параметрам.
ARI-001 понижает LDL холестерин, триглицериды и свободные жирные кислоты зависимым от времени образом. Тридцати двум самцам Golden Syrian хомяков давали возможность привыкнуть на протяжении двух недель к HF/HS диете, как описано выше. По истечении двухнедельного индукционного периода, животных распределяли по группам для 18-дневного исследования в отношении действия или наполнителя или ARI-001 (1120 мг/кг), или по группам для 28-дневного исследования в отношении действия или наполнителя или ARI-001 (1120 мг/кг). В каждой из четырех групп было по 8 хомяков. Приготавливали растворы и хранили, как описано выше. Животным давали дозу в объеме 1 мл в день в течение или 18, или 28 последовательных дней, как описано ранее. В конце исследования хомяков умерщвляли, их кровь собирали в пробирки с K2EDTA, плазму отделяли центрифугированием и замораживали до анализа. Анализировали все липиды с использованием промышленно доступного комплекта (Wako USA), как описано выше. В Таблице 4 ниже, животных с вводимым наполнителем, изучаемых и в течение 18 дней, и в течение 28 дней, объединяли в одну группу с наполнителем (N=16).
Липидные параметры от HF/HS хомяков, которым вводили перорально дозу наполнителя или ARI-001 в течение 18 или 28 дней. Приведенные величины представляют собой среднее ± стандартное отклонение. Приводятся Р-величины от двухвыборочных непарных t-тестов в сравнении с обработкой наполнителем в том же параметре.
1120 мг/кг,
18 дней
1120 мг/кг,
28 дней
*** р<0,001 по сравнению с наполнителем в том же параметре
ARI-001 показал благоприятные действия на липиды, когда введение дозы 1120 мг/кг осуществляли в течение до 28 дней. Когда хомякам вводили дозу в течение 28 дней вместо 18 дней, все измеренные липидные параметры достигали статистически значимой разницы по сравнению с наполнителем, за исключением HDL, который не показал никакой разницы по сравнению с наполнителем. Наблюдаемые снижения при введении 1120 мг/кг в течение 28 дней были больше, чем после 18-дневного введения дозы. Однако данные снижения почти не были такими впечатляющими, как наблюдаемые при 18-дневном введении при более высокой дозе 2240 мг/кг.
Корреляция между липидными биомаркерами в плазме и концентрациями ARI-001 в плазме. Плазму от 19 хомяков, которым вводили дозу ARI-001 в описанном выше 18-дневном исследовании, анализировали на концентрации ARI-001. Концентрации определяли для тех образцов, которые собирали через 24 часа после конечного введения дозы. Вкратце, концентрации лекарственного средства в плазме для не-GLP фармакокинетических экспериментов определяли с помощью LC-MS с использованием Applied Biosystems 4000Qtrap спектрометра с электрораспылительной ионизацией. Образцы для анализа приготавливали осаждением белков в плазме холодным метанолом. HPLC образцов выполняли с использованием Agilent Eclipse C18 колонки и градиента метанол/вода, содержащего 0,1% муравьиной кислоты и 5 мМ ацетата аммония. ARI-001 детектировали с использованием множественной реакции мониторинга (MRM) положительным ионным способом. Для количественного анализа измеряли стандартную кривую добавлением известных количеств ARI-001 к плазме от необработанных животных и приготовлением по способу, идентичному образцам от обработанных животных. Все образцы плазмы метили 10 нм/мл обогащенного изотопами ARI-001, который служил в качестве внутреннего стандартам для LC-MS измерений. Все концентрации приводятся в мкМ. Для корреляционного анализа концентрации в плазме ARI-001 у заданного животного соединяли с тем же липидным параметром животного. В анализ включали всех животных из обеих групп с вводимой дозой (1120 мг/кг и 2240 мг/кг). Данные корреляции графически показаны на Фигуре 6 и Фигуре 7. Величины корреляции Пирсона определяли с использованием все данных, как в случае двухвыборочной P-величины для серии данных.
Изменения липидного параметра хорошо коррелировали с уровнями в плазме лекарственного средства, измеренными через 24 часа после последней дозы. Действительно, уровни общего холестерина, HDL холестерина, LDL холестерина, триглицеридов и свободной жирной кислоты все достигали статистически значимых уровней корреляции (р<0,01). Корреляция с триглицеридами была особенно заметной по наличию очень высокой степени статистической значимости, р<0,001. Данные значительные корреляции подтверждают то, что ARI-001 непосредственно ответственен за модулирование липидных величин. Более того, эти данные подтверждают эффекты доза-ответная реакция, показанные на Фигуре 5.
Концентрации ARI-001 в корреляте тканей с уровнями липида в плазме. Образцы ткани собирали от хомяков в описанном выше 18-дневном эксперименте для определения концентрации ARI-001 и в печени и в адипозной ткани. Вкратце, образцы печени и адипозной ткани собирали в конце эксперимента; образцы мгновенно замораживали в жидком азоте, а затем хранили при -80°С до тех пор, пока не использовали для анализа. Для подготовки для анализа образцы печени вырезали из замороженной массы, взвешивали и гомогенизировали измельчителем ткани с последующей ультразвуковой обработкой в буфере. Твердые материалы затем удаляли центрифугированием. Для подготовки для LC-MS анализа гомогенат затем обрабатывали по той же технологии, что описана для подготовки плазмы (см. выше). Образец адипозной ткани вырезали из замороженной массы и переносили в ступку с пестиком, охлаждаемую жидким азотом. Во время измельчения добавляли жидкий азот для того, чтобы образец адипозной ткани оставался твердым. Измельченный образец переносили в предварительно взвешенную пробирку для взвешивания всего образца. Данный измельченный образец экстрагировали метанолом, и данный метанол, содержащий соединение, отделяли от липида охлаждением до -20°С. После сушки метанольный образец растворяли в воде; затем образцы приготавливали для LC-MS анализа таким же образом, как в случае печени и плазмы (см. выше).
Концентрации в печени приводятся в виде нг ARI-001 на мл гомогенизированного образца ткани, экстрагированного буфером. Концентрации в адипозной ткани приводятся в нг ARI-001 на мг ткани, выделенного от процесса измельчения. Вследствие распределения концентраций в ткани и липидных параметров при определении корреляции по всем образцам в печени использовали трансформацию логарифма. В дополнение, концентрации в адипозной ткани трансформировали как логарифм + 1, поскольку трансформация логарифма давала отрицательные величины для данных образцов ткани. Данные трансформации давали возможность показать данные на графике более наглядно, более удобно. Наконец, эти трансформации являются обоснованными, потому что характер коэффициента корреляции Пирсона является безвариантным и для трансформации логарифма и для перестановки.
Фигура 8 графически показывает корреляцию между концентрациями ARI-001 в плазме и в печени (слева), и плазме и адипозной ткани (справа). Обе данные корреляции были высоко статистически значимыми, с р<0,01 для каждой пары параметров. Фигуры 9-11 показывают графически корреляции между концентрациями ARI-001 в каждом образце ткани и липидными параметрами TC, HDL, LDL, TG и FFA. Все измерения концентрации и все липидные параметры трансформировали с помощью логарифма. Коэффициент корреляции Пирсона определяли с использованием всех иллюстрируемых данных. Р-Величину определяли для двухвыборочного t-теста с использованием всех иллюстрируемых данных.
ARI-001 увеличивает АВСА1, ApoAI, SR-BI, CETP и адипонектин мРНК у хомяков HF/HS. Исследование возможного механизма, который мог бы привести к увеличению HDL холестерина, сосредоточенное на изменениях уровней мРНК нескольких генов, связанных с регуляцией HDL. Печень хомяков и адипозную ткань подготавливали по способу, сходному с описанным для определения концентрации ARI-001. Вкратце, для приготовления для анализа из замороженной массы вырезали образец печени, взвешивали и гомогенизировали с помощью измельчителя ткани с последующей ультразвуковой обработкой в буфере. Твердые материалы затем удаляли центрифугированием. Получающийся в результате лизат использовали в анализе qPCR для количественного определения специфичной мРНК, измеряемой (см. выше) с использованием праймеров, предназначенных для представляющих интерес специфических последовательностей. Все количества мРНК нормализировали к обрабатываемым наполнителем животным, и уровни мРНК выражали в виде кратного увеличения или снижения относительно обрабатываемых наполнителем. Адипозную ткань обрабатывали аналогичным печени образом: образец адипозной ткани вырезали из замороженной массы и переносили в ступку с пестиком, охлажденную жидким азотом. Во время измельчения добавляли жидкий азот для обеспечения возможности образцу оставаться твердым. Измельченный образец переносили в предварительно взвешенную пробирку для взвешивания всего образца. Данный измельченный образец обрабатывали буфером и подготавливали для qPCR для определения количеств специфичной мРНК.
Относительные концентрации АВСА1, ApoAI, SR-BI, CETP мРНК на мг ткани печени, и аналогичным образом СЕТР адипозной ткани и адипонектин мРНК на мг адипозной ткани от питавшихся высокожирной едой хомяков в обрабатываемых наполнителем, ниацином или ARI-001 группах. Величины даны в виде кратного изменения по сравнению со средними величинами в группе с наполнителем.
Уровни и АВСА1, и ApoAI мРНК были выше в группе животных, обрабатываемых ARI-001, относительно контрольных животных с наполнителем. Действительно, статистически значимая корреляция была между уровнями HDL и ApoAI мРНК. Это говорит о возможном механизме, по которому ARI-001 может увеличивать HDL у данной модели хомяков.
Корреляции между АВСА1, ApoAI, SR-BI и CETP мРНК на мг ткани печени против уровней HDL, и аналогичным образом между CETP и адипонектин мРНК на мг адипозной ткани против уровней HDL, от хомяков, питавшихся высокожирной едой, в группах, обрабатываемых наполнителем, ниацином или ARI-001.
Пример 4: Фармакологические исследования безопасности (Не- GLP предварительные исследования)
Ввиду известности того, что ниацин связан с токсичностью по отношению к печени и непереносимостью глюкозы при хроническом дозировании, мы проверили, могут ли ARI-001, ниациновый миметик быть связаны аналогичным образом. Для того чтобы изучить это, мы проверяли обычные функциональные ферменты печени AST и ALT в плазме хомяков из эксперимента, описанного в Примере 3. Данным животным вводили дозу исследуемых веществ в течение 18 последовательных дней, причем животные находились на HF/HS диете в течение в общем приблизительно 5 недель. Мы также проверяли уровни глюкозы в плазме данных животных. Для фармакокинетических исследований мы использовали мышей дикого типа для изучения и разового, и повторного введения. Наконец фармакокинетические исследования подкреплялись данными от разового и повторного введения обезьянам.
А. Действие на функцию печени
ARI-001 улучшает функцию печени в испытаниях с хроническим введением дозы хомякам, кормившимся высокожирной едой. Известно, что некоторые составы ниацина вызывают гепатотоксичность у людей. Это привело к исследованию в испытаниях функции печени (AST, ALT) из плазмы хомяков, которым в течение 18 дней вводили дозу ARI-001. AST и ALT измеряли с использованием промышленно доступных наборов (Bio-Quant Diagnostic Kits, Сан Диего, СА). Как и ожидалось, показатели AST и ALT у необработанных хомяков (группа с наполнителем) были очень высокими, так как диета с высоким содержанием жира ведет к гепатомегалии и жирной печени. Данное заболевание отражается в повышенных уровнях AST и ALT. В противоположность этому, уровни AST и ALT у животных, обработанных ARI-001, были значительно ниже, чем в группе с наполнителем. Действительно, AST резко снижалась при дозе 2240 мг/кг, и даже значительно снижалась при дозе 1120 мг/кг. Показатели ALT аналогично снижались зависимым от дозы образом. См. Фигуру 12А и Фигуру 12В.
В. Действия на толерантность к глюкозе
ARI-001 не оказывает никакого действия на уровни глюкозы среди хомяков, кормившихся высокожирной едой. Ниацин известен тем, что оказывает негативное воздействие на уровни глюкозы у пациентов-диабетиков. Модель хомяков, используемых в данном эксперименте, давала популяции с повышенными уровнями глюкозы, как продемонстрировано группой с наполнителем на Фигуре 13. Согласуясь с действиями, показанными на людях, глюкоза у животных, обрабатываемых ниацином, увеличивалась относительно группы с наполнителем. Однако ни одна доза ARI-001 не давала какого-либо значительного изменения уровней глюкозы по сравнению с группой с наполнителем.
Пример 5: Фармакокинетические исследования
А. Исследования на мышах in vivo
Фармакокинетическое исследование на мышах дикого типа с одноразовой дозой. Мышам дикого типа C57BL/6 дозировали одноразовым введением ARI-001 в растворе с помощью или перорального желудочного зонда (PO), или интраперитонеальной инъекции (IP). Затем собирали образцы крови в различные точки времени на протяжении 24-часового периода; затем анализировали плазму на концентрации ARI-001, как описано ранее. ARI-001 вводили в дозе или 2240 мг/кг в виде разового болюса перорально через желудочный зонд, или в дозе 448 мг/кг в виде разового болюса через перитонеальную инъекцию. См. Фигуру 14.
Введение одноразовой дозы ARI-001 перорально через желудочный зонд и с помощью интраперитонеальной инъекции давало фармакокинетические кривые с параметрами, описанными в Таблице 7. К истечению 24 часов временного периода остающаяся концентрация ARI-001 не обнаруживалась.
Фармакокинетические параметры разового введения ARI-001 у мышей данного типа
Фармакокинетическое исследование ARI-001 на мышах дикого типа с множественным введением. Мышам дикого типа C57BL/6 вводили дозу ARI-001 в растворе перорально через желудочный зонд ежедневно в течение 30 последовательных дней. После курса пяти 24-часовых периодов собирали кровь и полученную в результате плазму анализировали на концентрации ARI-001. Использовали четыре дозы: 996 мг/кг, 1493 мг/кг, 2240 мг/кг и 3360 мг/кг. Как и ожидалось, Смакс уровни и общее 24-часовое подвергание воздействию (AUC) были зависимыми от дозы. Однако данные величины были зависимыми от времени, так как не наблюдалось никакой тенденции между данными параметрами и днями введения. См. Фигуры 15A-15D и Фигуры 16А-16В.
В. Дополнительные исследования на мышах in vivo
Фармакокинетические исследования на мышах дикого типа с одноразовой дозой. Мышам дикого типа C57BL/6 дозировали разовым введением ниацин, ARI-001 или соединение 2230С (раскрытое в публикации американской патентной заявки № 2009/0312355 A1, включенной в данное описание путем ссылки), в растворе перорально через желудочный зонд в виде одного болюса (РО). Соединение 2230С имеет структуру
Затем в различные точки времени на протяжении 24-часового периода собирали образцы крови; затем анализировали плазму на концентрации ниацина, ARI-001 и 2230С, как описано ранее. Результаты показаны в Таблице 8.
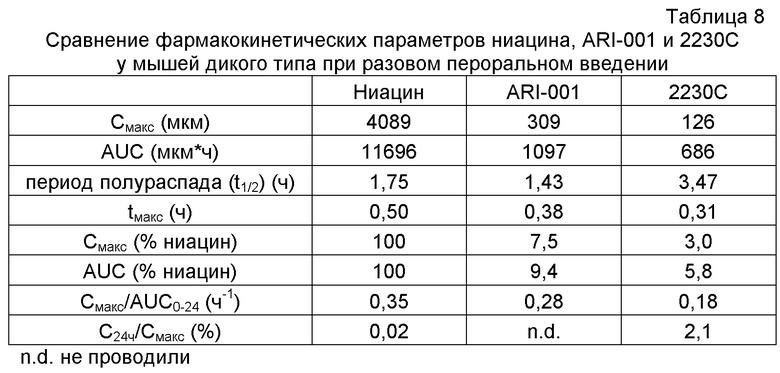
С. Исследования in vivo на хомяках Golden Syrian
Фармакокинетическое исследование ARI-001 с одноразовой дозой на Golden Syrian хомяках. Фармакокинетический профиль ARI-001 оценивали на HF/HS Golden Syrian хомяках, которым давали в виде разовой оральной дозы 5,9 ммол/кг ARI-001. Образцы плазмы собирали с помощью сердечной пунктуры у пяти животных в каждый момент времени для измерения концентраций ARI-001 в плазме на протяжении 24 часов. Результаты показаны на Фигуре 17.
D. Исследования in vivo на обезьянах
Фармакокинетическое исследование ARI-001 на обезьянах макаках с одноразовой дозой. Голодавшим обезьянам давали одноразовым введением ARI-001 в растворе или перорально через желудочный зонд (РО), или через внутривенную инъекцию (IV). В различные временные точки на протяжении 24-часового периода собирали образцы крови; затем анализировали плазму на концентрации ARI-001. ARI-001 вводили или в дозе 288 мг/кг в виде разового болюса перорально через желудочный зонд, или в дозе 96 мг/кг в виде разового болюса с помощью IV инъекции. Результаты показаны на Фигуре 18.
Фармакокинетическое исследование одноразовой дозы ARI-001 на кормившихся или голодавших обезьянах. Кормившихся обезьян кормили и давали им некоторое время для переваривания, прежде чем вводили ARI-001 в виде раствора перорально через желудочный зонд, как описано ранее. Результаты показаны на Фигуре 19.
Фармакокинетика множественного введения ARI-001 на голодавших обезьянах. ARI-001 вводили обезьянам перорально через желудочный зонд один раз в день в течение семи дней. После первого и после последнего введения собирали образцы крови для измерения концентрации в плазме ARI-001. Была очень незначительная разница между концентрациями в плазме в 1-й день против 7-го дня. Наибольшее расхождение было в показателе Смакс, который был выше на 7 день, чем на 1 день. Концентрации через 24 часа или после первой, или после последней доз были по существу идентичными. См. Фигуру 20.
Пример 6: ARI-001 не пополняет β-аррестином клеточную мембрану клеток, экспрессирующих ниациновый рецептор GRP109A высокого сродства
Было продемонстрировано, что индуцируемые ниацином кожные приливы крови опосредуются активацией ниацинового рецептора, GRP109A, зависимым от β-аррестина образом. Walters RW et al. (2009) J Clin Invest 119:1312-21. Готовые для анализа PathHunter eXpress клетки с β-аррестином, экспрессирующие GRP109A, помещали на 96-луночную планшету по 10000 клеток/лунку и стимулировали или ниацином, или ARI-001, каждым в интервале концентраций, в течение 90 минут. Активность G-белок сопряженного рецептора (GPCR) определяли измерением взаимодействия β-аррестина с активированным GPCR с использованием комплементации фрагмента фермента β-галактозадизы. После стимуляции или ниацином, или ARI-001, детектировали сигнал с использованием хемилюминесцентных PathHunter детекционных реагентов. Характерные результаты показаны на Фигуре 21.
В отличие от ниацина, стимуляция GPR109A с помощью ARI-001 при концентрациях вплоть до 10 мМ не пополняла β-аррестином мембрану клеток, экспрессирующих GRP109A. Поскольку известно, что ниацин-индуцируемые приливы крови опосредуются активацией GRP109A β-аррестин-зависимым образом, данная находка согласуется с наблюдением того, что ARI-001 значительно снизил побочный эффект приливов крови по сравнению с ниацином.
Пример 7: Краткое изложение фармакологических исследований ARI-001 на моделях животных
В отношении ARI-001 был проведен ряд исследований фармакокинетики, безопасности и эффективности, на множестве животных, включая мышей, крыс, хомяков Golden Surian, собак и обезьян. Общие результаты данных исследований установили следующее.
ARI-001 снизил уровни в плазме общего холестерина, LDL-C, TG и FFA при увеличении общего уровня HDL-C и HDL-C/TC соотношения. Эффекты изменения липида от разовой дозы в день ARI-001 были более резко выраженными, липидные эффекты, наблюдаемые с почти 2-кратно более высокой дозой ниацина, даваемой один раз в день.
ARI-001, который давали животным ежедневно один раз в течение 28 дней, привел к большим изменениям уровней липида в плазме по сравнению с той же дозой ARI-001, даваемой один раз ежедневно в течение 18 дней.
ARI-001, даваемый один раз день ежедневно, приводил к весьма значительным изменениям липидов в плазме, к большим, чем, или равным эффектам изменения уровня липидов по сравнению с той же общей дозой ARI-001, даваемой дважды в день. В дополнение к сказанному, ARI-001 при разовой дозе в день был более сильным, чем ниацин, в отношении достижения желаемых изменений липидов.
Уровни изменения в плазме TC, HDL-C, TG, LDL-C и FFA коррелировали с концентрациями ARI-001, присутствующего в плазме.
Концентрации ARI-001 в плазме и печени пропорционально связаны.
Концентрация ARI-001 в печени коррелировала с уменьшениями в плазме TC, LDL-C, TG и FFA и увеличениями в плазме HDL-C.
ARI-001 не показывал никаких свидетельств капиллярного сосудорасширения или гиперемии (близко к приливу) в экспериментах на мышах с использованием Допплеровских измерений потока капиллярной крови. Кроме того, у крыс или собак на протяжении 28 дней не наблюдалось клинических симптомов “приливов крови”.
Пример 8: Клиническое испытание ARI-001 на людях
Проводят случайное или беспорядочное, двойное-слепое, плацебо-контролируемое исследование с последовательными повышающимися дозами с наблюдениями в течение 30 часов после введения дозы и обратного осмотра на 8-й день. Субъектами исследования являются здоровые взрослые волонтеры мужского и женского пола, возрастом 18-60 лет, с LDL-C > 130 мг/дл и весом < 85 кг. Субъектов распределяли произвольно для получения изучаемого лекарственного средства или плацебо.
Пять периодов повышения одиночной дозы, с 8 субъектами на группу (6 с лекарственным средством: 2 с плацебо) вовлекало в общем 40 человек. Предоставлялись плацебо, надлежащим образом соотнесенные вслепую. Группа 1 получает 500 мг ARI-001, сформулированного в виде одиночной пероральной таблетки, плюс 11 таблеток плацебо; Группа 2 - 1000 мг, принимаемые в виде двух 500 мг таблеток ARI-001, плюс десять таблеток плацебо; Группа 3 - 2000 мг, принимаемые в виде четырех 500 мг таблеток ARI-001, плюс восемь таблеток плацебо; Группа 4 - 4000 мг, принимаемые в виде восьми 500 мг таблеток ARI-001, плюс четыре таблетки плацебо; Группа 5 - 6000 мг, принимаемые в виде двенадцати 500 мг таблеток ARI-001. Субъекты только с плацебо принимают двенадцать таблеток плацебо. Каждая таблетка представляет собой спрессованную, покрытую пленкой таблетку, подходящую для перорального введения.
Первичными целями исследования является оценка безопасности и способности переносить одиночные дозы ARI-001 здоровыми взрослыми добровольцами при дозах от 500 мг до 6000 мг.
Вторичными целями исследования является установление фармакокинетического профиля ARI-001 в крови после одиночной дозы у здоровых добровольцев; наблюдение изменений в триглицеридах натощак, свободных жирных кислот и других липидных биомаркеров; корреляция уровня дозы и время воздействия лекарственного средства в плазме на протяжении времени с любыми изменениями в триглицеридах натощак, свободных жирных кислотах и других липидных биомаркеров; и установление действия ARI-001 на симптомы приливов крови по визуальной аналоговой шкале (VAS).
Фармакокинетические образцы берут за 0-45 мин до дозы и через 0,5, 1, 1,5, 2, 4, 6, 8, 12, 24, 30 и 168 часов после дозирования. Фактическое время каждого сбора плазмы регистрируют.
При каждом взятии пробы 3 мл крови берут в цилиндр Vacutainer, содержащий EDTA (фиолетовая верхушка), и немедленно охлаждают. В пределах 30 минут сбора отделяют фракцию плазмы с помощью центрифугирования при 2000 об/мин в течение 15 минут при 4°С. Анализ всех проб проводят в центральной лаборатории.
В день 1, до введения дозы, выполняют следующие процедуры:
- Клинические лабораторные испытания, включающие функцию печени (ALT, AST, сывороточный билирубин), CK, гематологию, APTT, PT, анализ мочи и химию липидов (LDL-C, HDL-C, свободные жирные кислоты, триглицериды LPA и ApoA-1)
- 12-электросоединение ECG
- Жизненные признаки
- VAS
- Базисная линия плазмы РК
- Взятие мочи для базисной линии РК между полночью и 0 часами (введение дозы)
После введения исследуемого лекарственного средства выполняют следующие процедуры.
- Клинические лабораторные испытания, включающие функцию печени (ALT, AST, билирубин сыворотки), CK, гематологию и APTT, PT через 6, 12 и 24 часа после дозы
- Химия липидов (LDL-C, HDL-C, свободные жирные кислоты, триглицериды, Lp(a) и ApoA-1) через 4, 12 и 24 часа после дозы
- 12-электросоединение ECG через 1, 2, 4, 6, 8, 12 и 24 часа после дозы
- Анализ мочи через 24 часа после дозы
- Жизненные признаки через 6, 12, 24 и 30 часов после дозы
- VAS через 0,25, 0,5, 1, 2, 4, 8, 12 и 24 часа после дозы
- Физический осмотр через 24 часа после дозы и краткий клинический осмотр через 30 часов после дозы
- Взятие проб крови для РК через 0,5, 1, 1,5, 2, 4, 6, 8, 12, 24 и 30 часов после дозы
- Взятие пробы крови на тропонин через 4 часа после дозы
- Взятие мочи на РК с 6-часовыми интервалами через 0-6, 6-12, 12-18, 18-24 и 24-30 часов после введения дозы
Предварительные результаты данного клинического испытания на людях включают удивительное наблюдение того, что ни один пациент не проявил каких-либо признаков приливов крови при любой дозе ARI-001, вплоть до и включая дозу 6000 мг.
Дополнительные предварительные результаты показаны на Фигуре 22, которая иллюстрирует ответную реакцию на дозу в отношении понижения у людей триглицеридов при одиночных пероральных дозах ARI-001 по данным, измеренным через 4 часа после введения дозы.
Дополнительные предварительные результаты показаны на Фигуре 23, которая иллюстрирует концентрацию ARI-001 в сыворотке, измеряемую на протяжении 24 часов у людей после одиночных пероральных доз, находящуюся в пределах от 500 мг до 6000 мг ARI-001. Показатель Смакс для дозы ARI-001 2000 мг был около 7500 нг/мл (7,5 мг/мл); Смакс для 1500 мг ниацина (грубо эквимолярная доза) был 30000 нг/мл (30 мг/мл).
ВКЛЮЧЕНИЕ В ОПИСАНИЕ С ПОМОЩЬЮ ССЫЛКИ
Все публикации и патенты, упоминаемые здесь, включены в описание целиком путем ссылки на них, как если бы каждая отдельная публикация или патент были конкретно и отдельно включены с помощью ссылки на них. В случае разногласий настоящая заявка, включающая любые определения в ней, послужит для контроля.
ЭКВИВАЛЕНТЫ
Хотя в описании раскрыты конкретные воплощения обсуждаемого изобретения, приведенное выше описание является иллюстративным, а не ограничивающим. Многие видоизменения изобретения станут очевидными специалистам в данной области после ознакомления с данным описанием. Прилагаемыми пунктами формулы изобретения не имеется в виду заявить, что все такие воплощения и видоизменения, и полный объем изобретения должен определяться при обращении к пунктам формулы изобретения наряду с полным объемом эквивалентов и описания вместе с такими вариациями или видоизменениями.
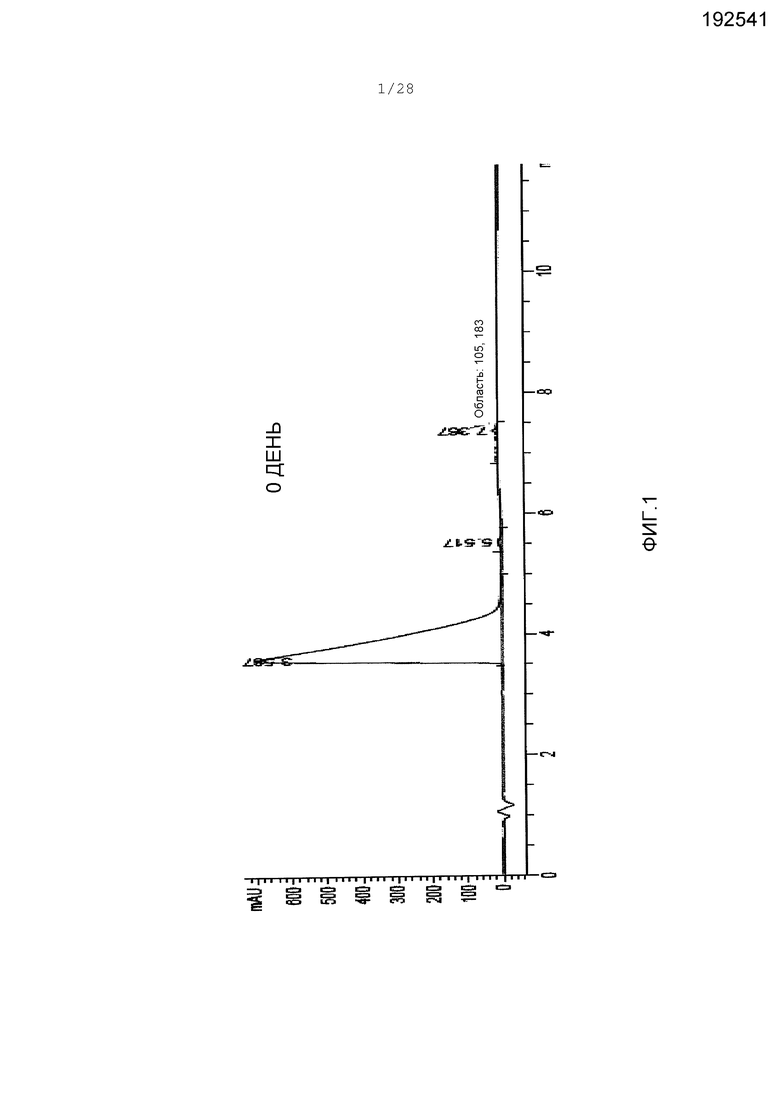
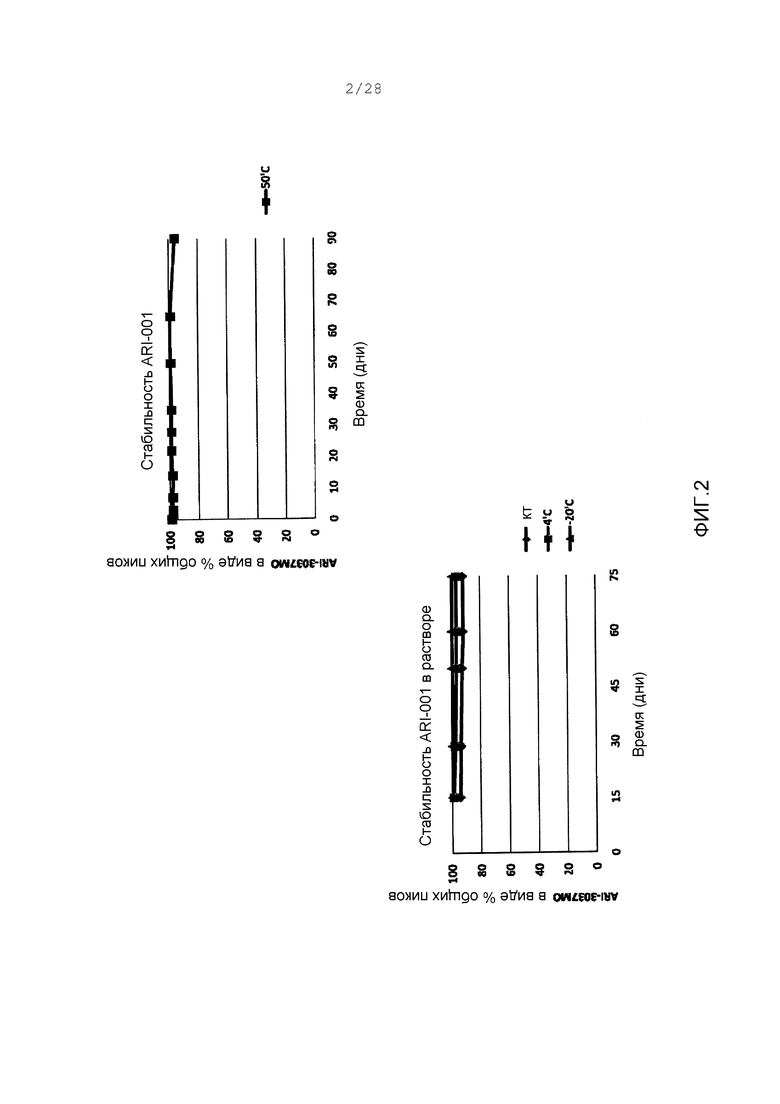
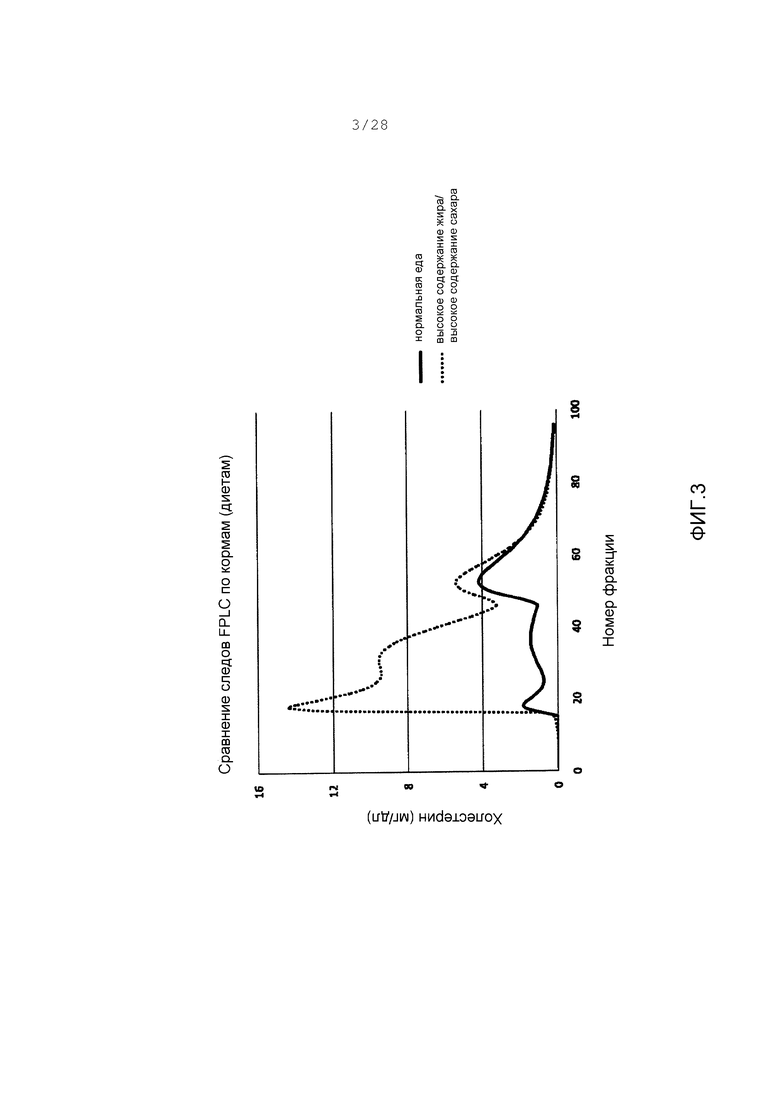
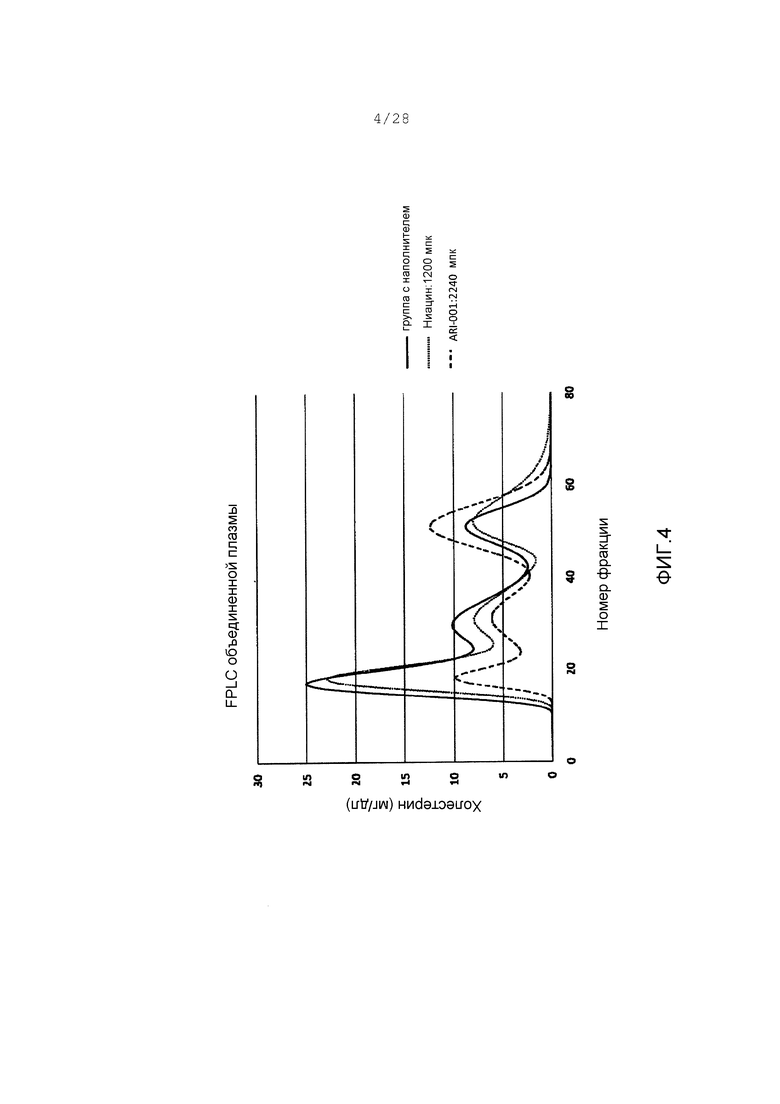
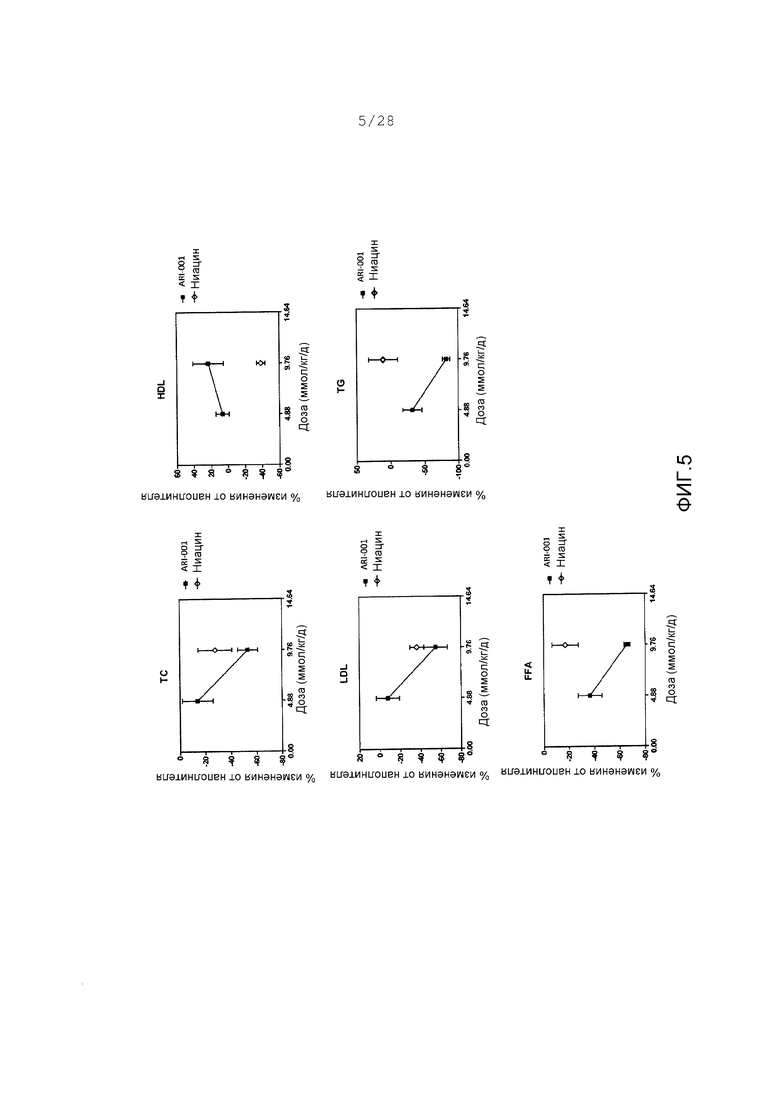
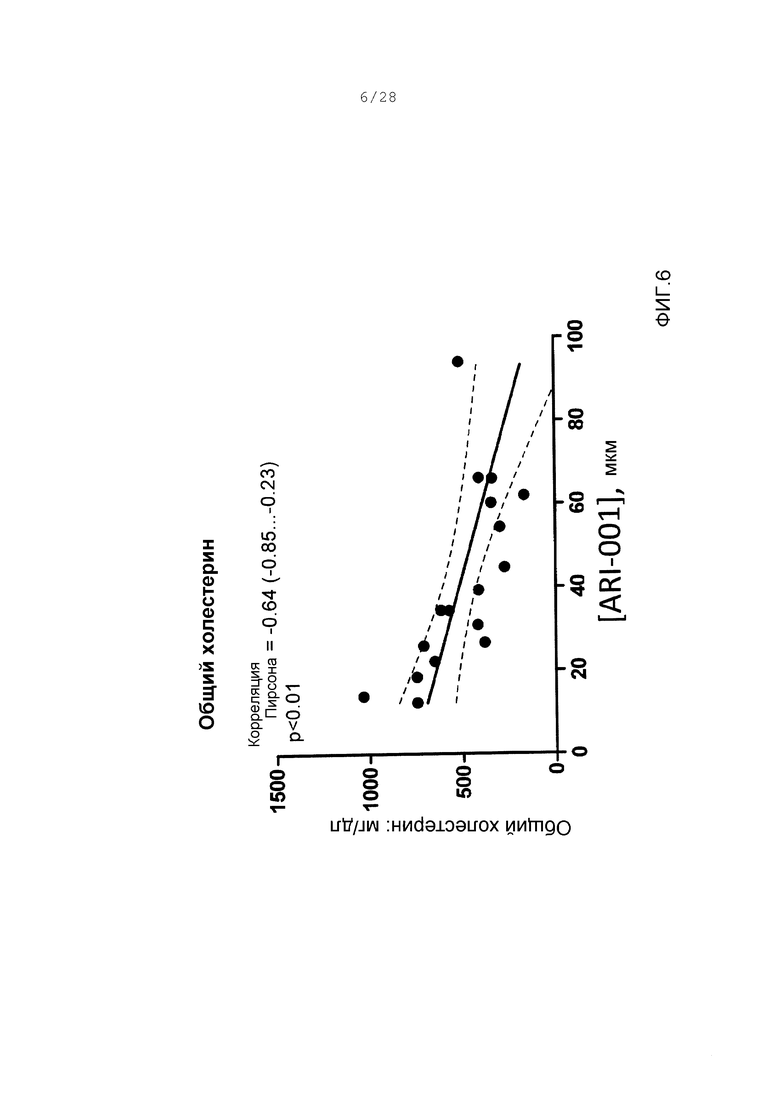
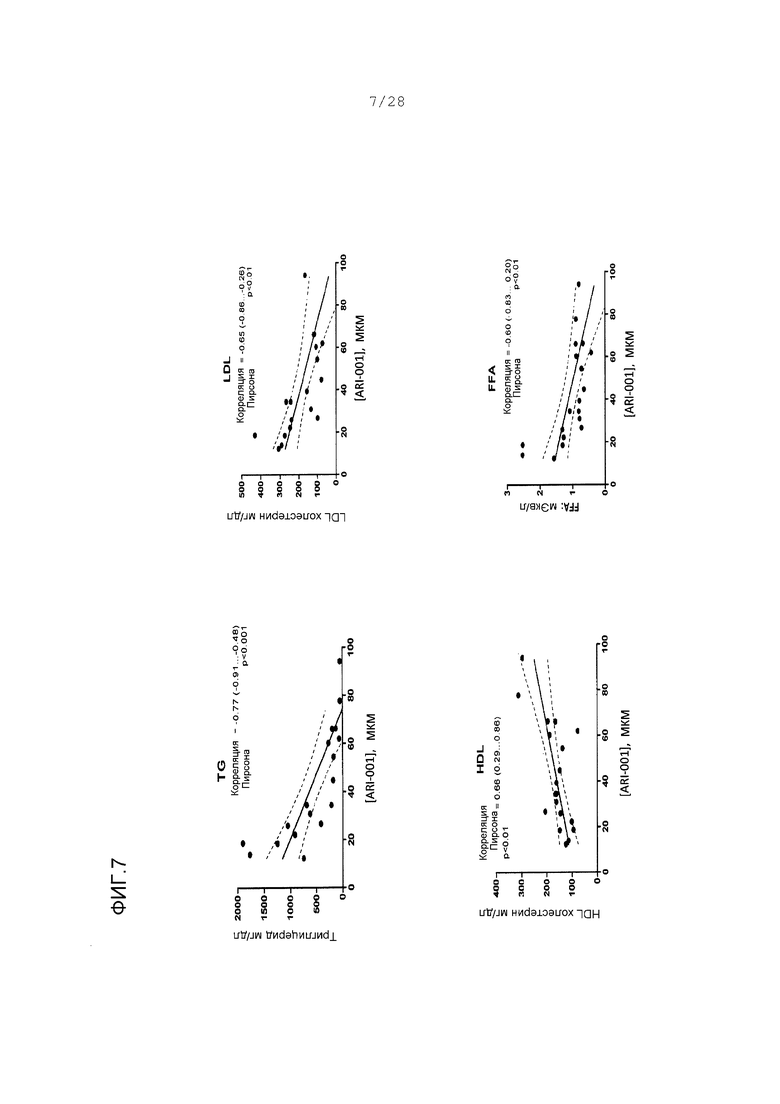
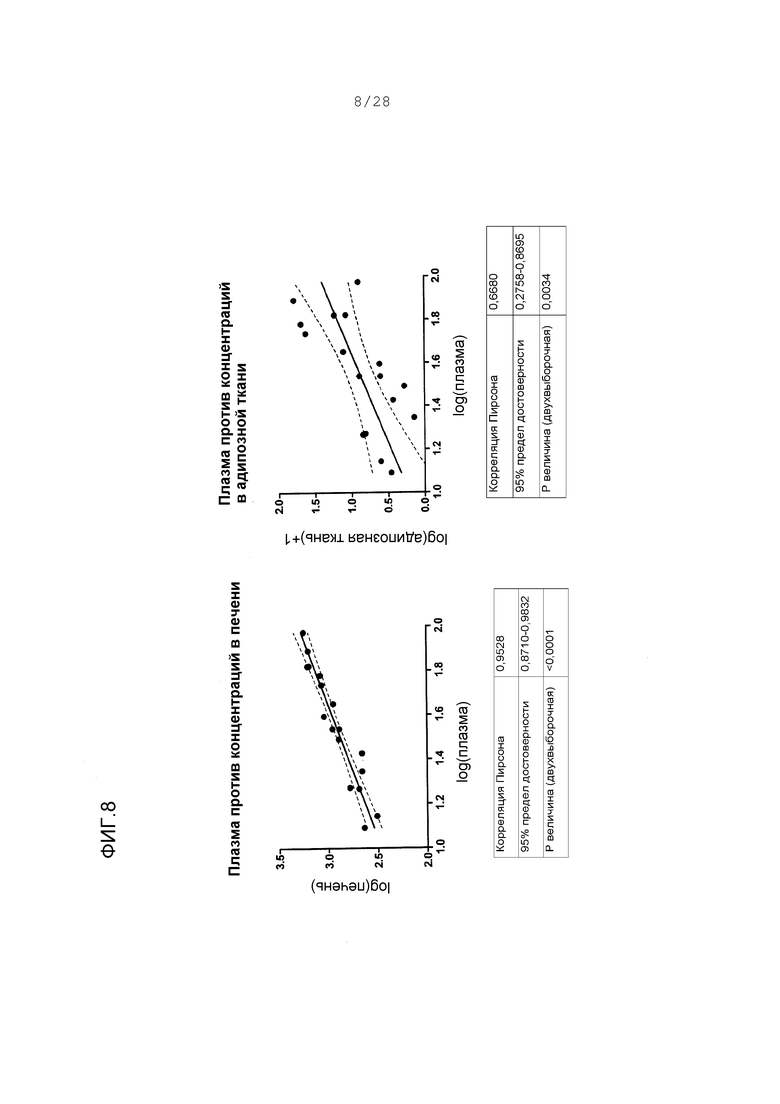
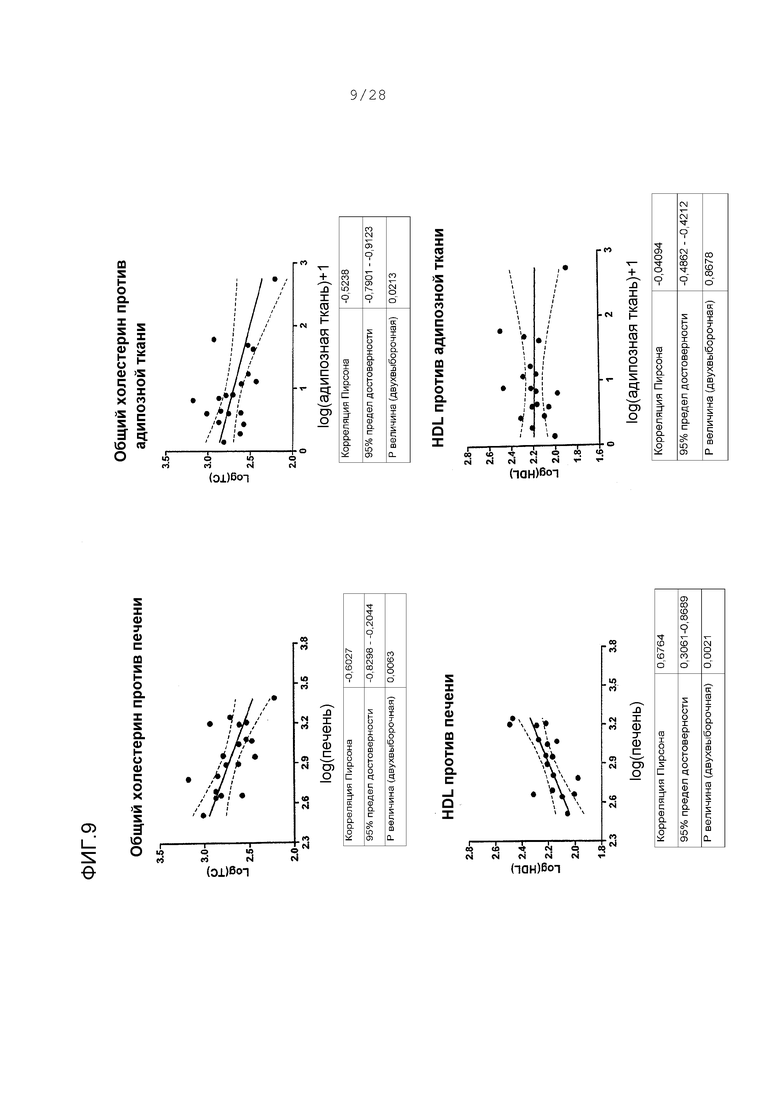
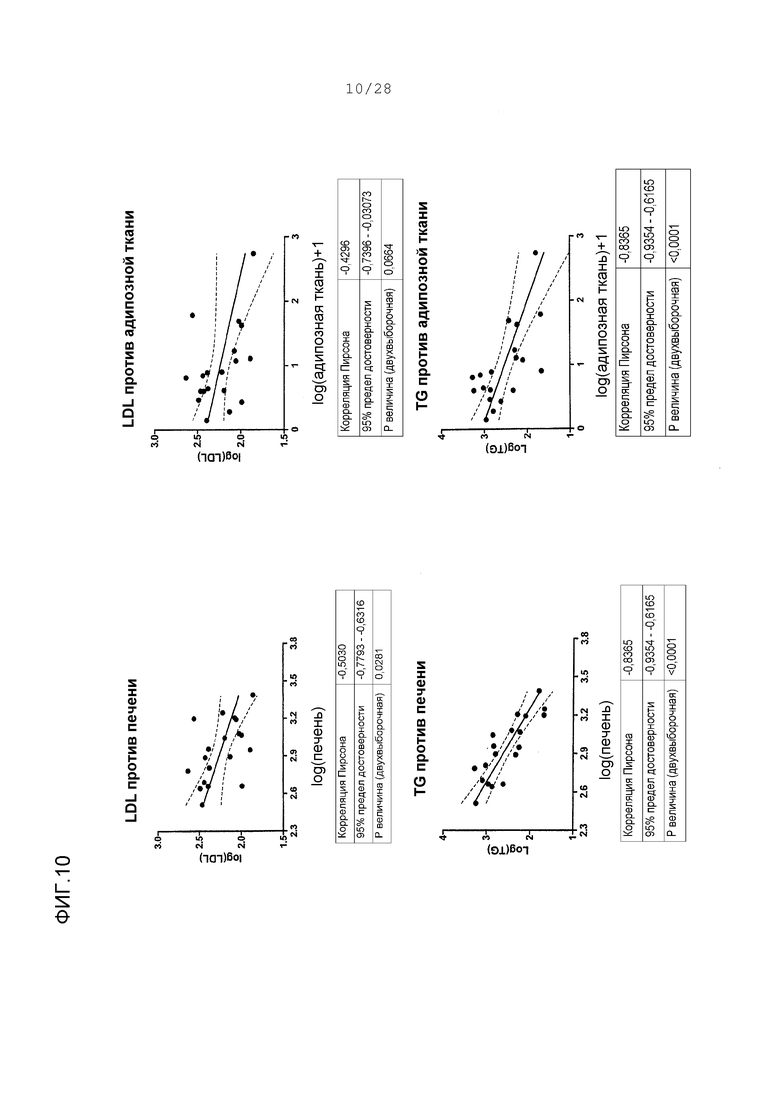
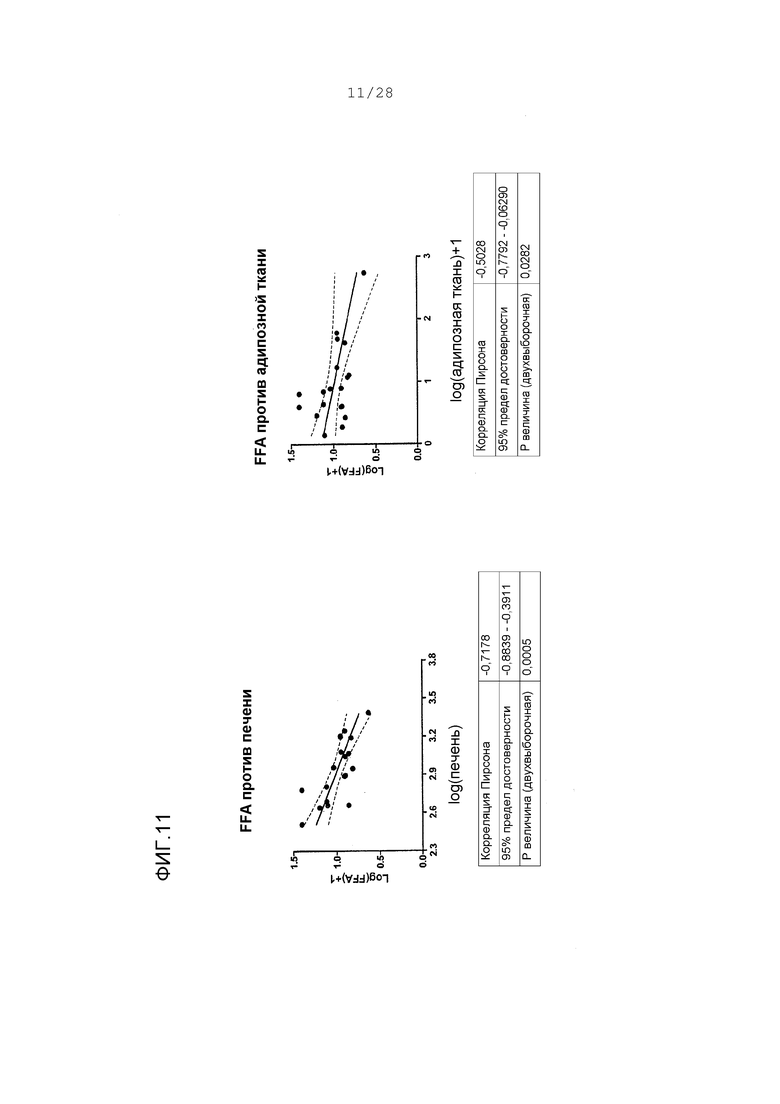
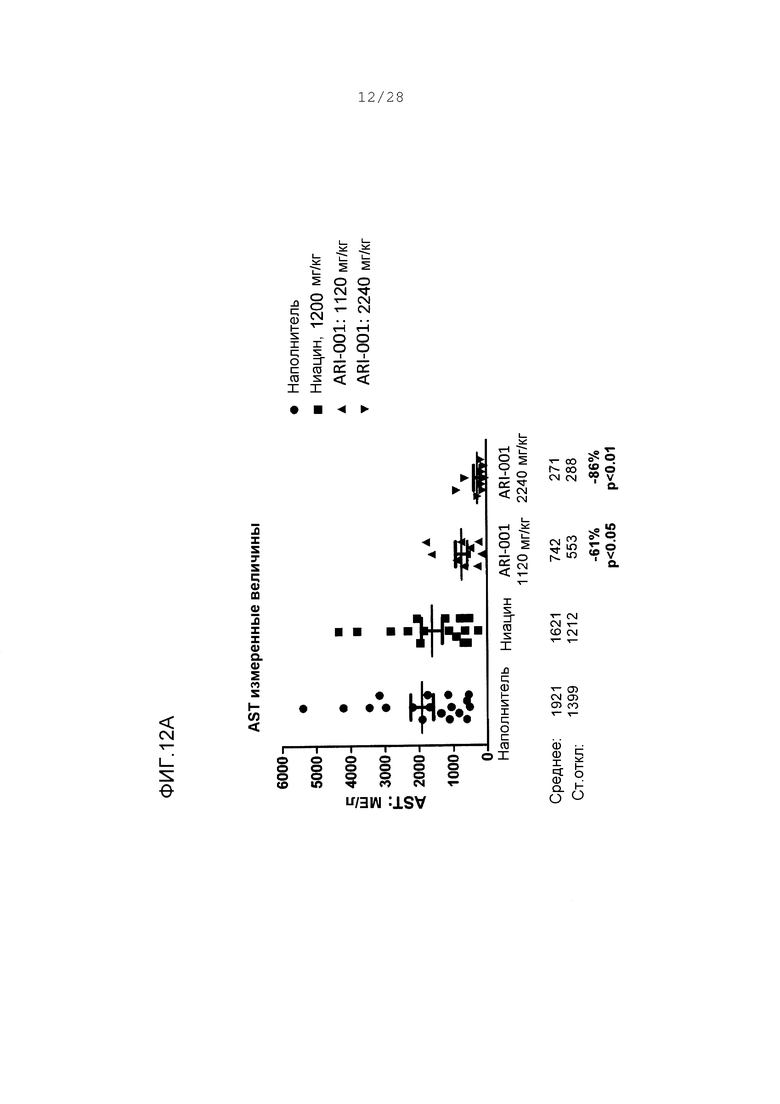
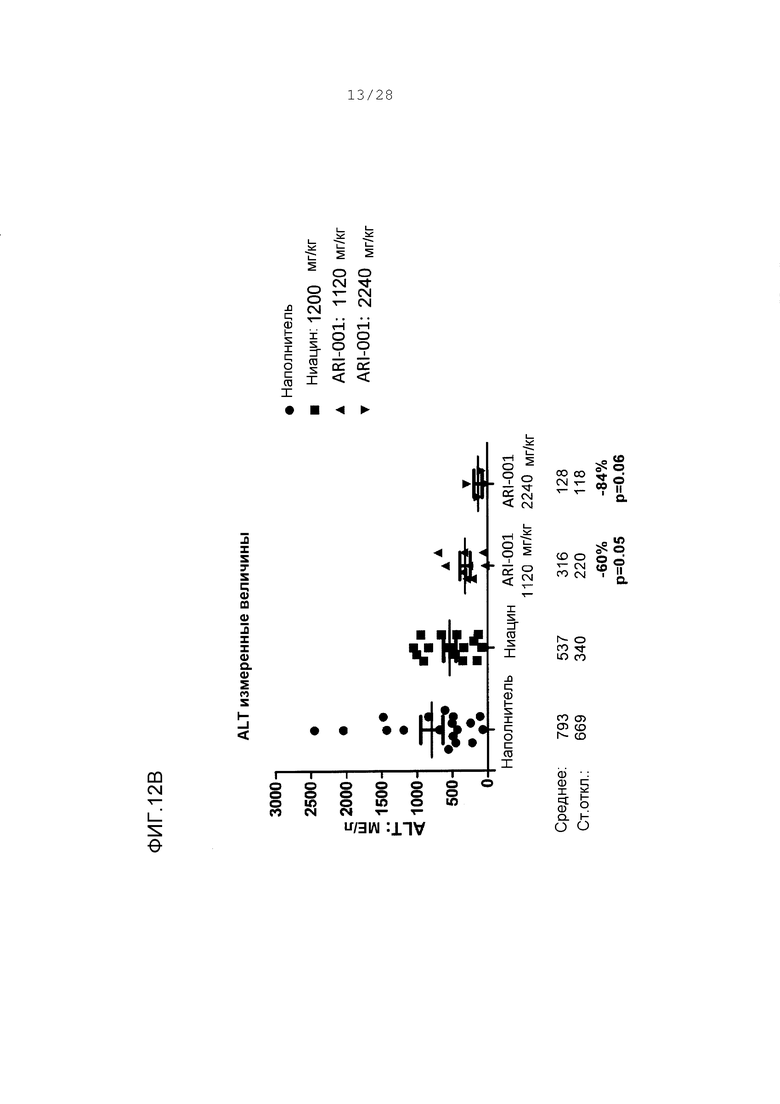
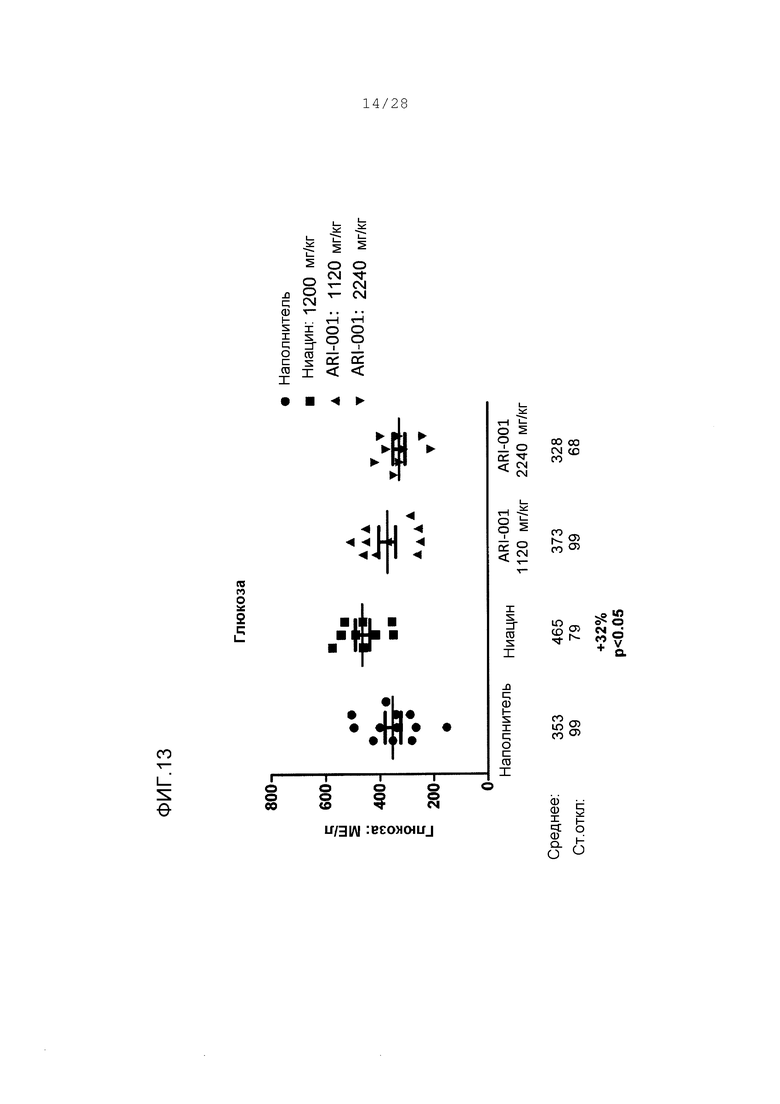
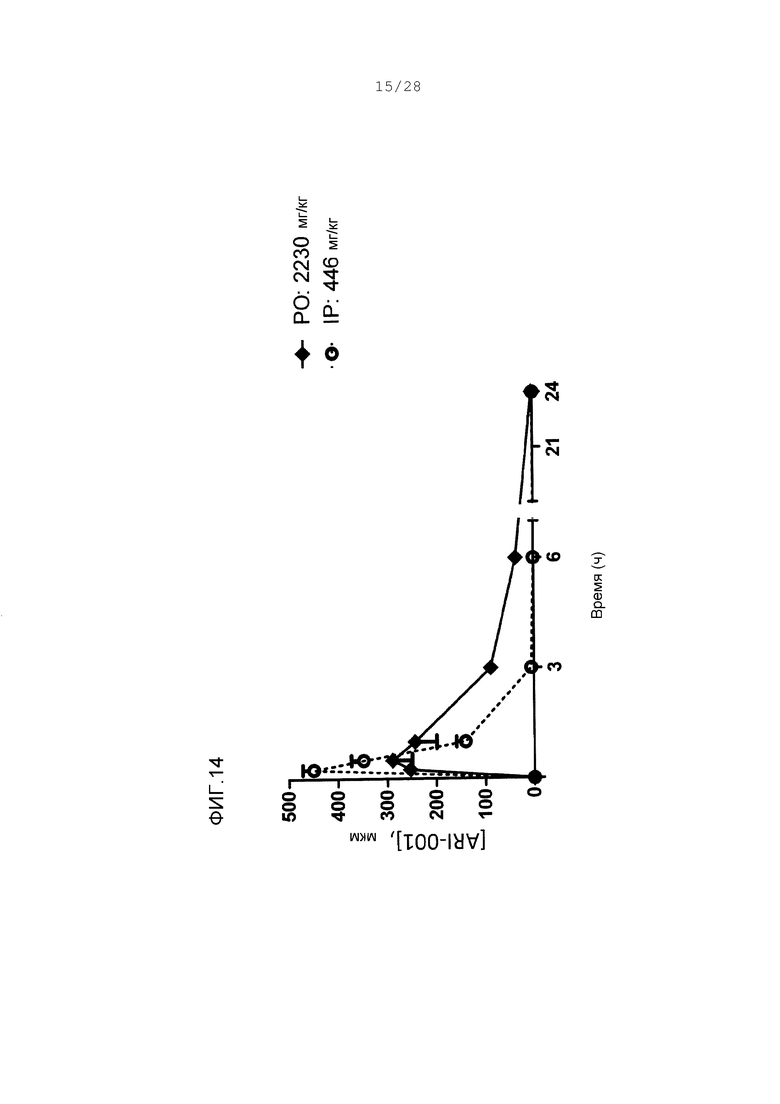
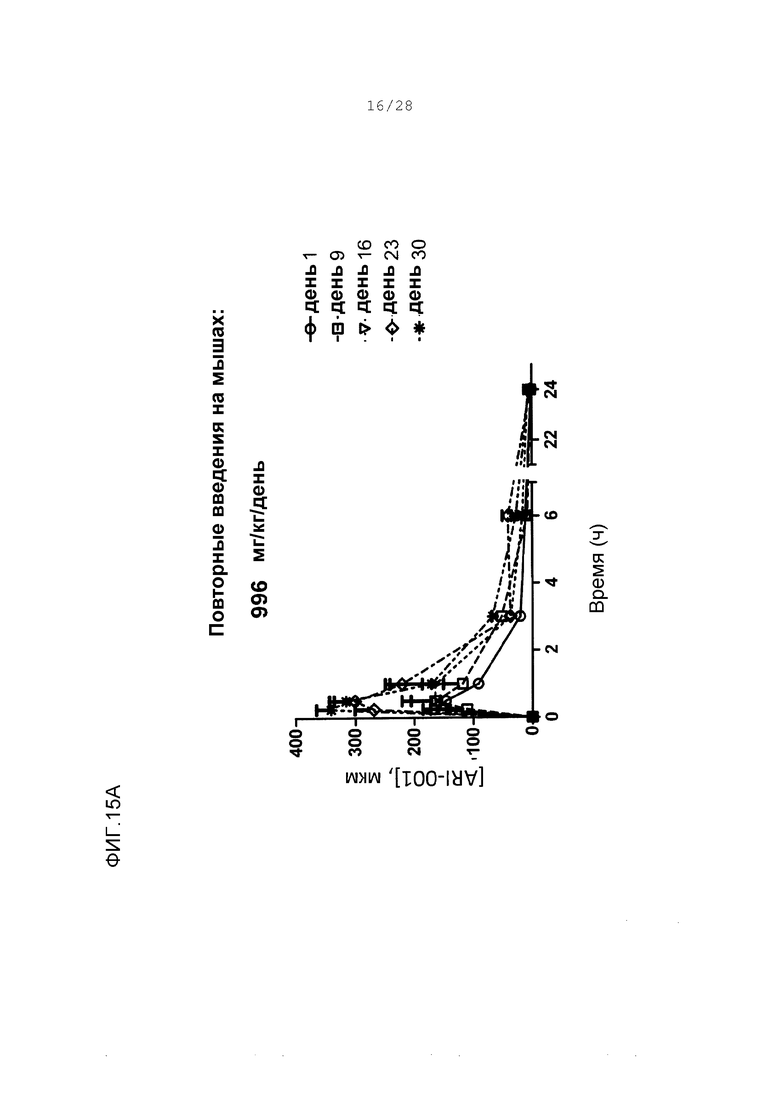
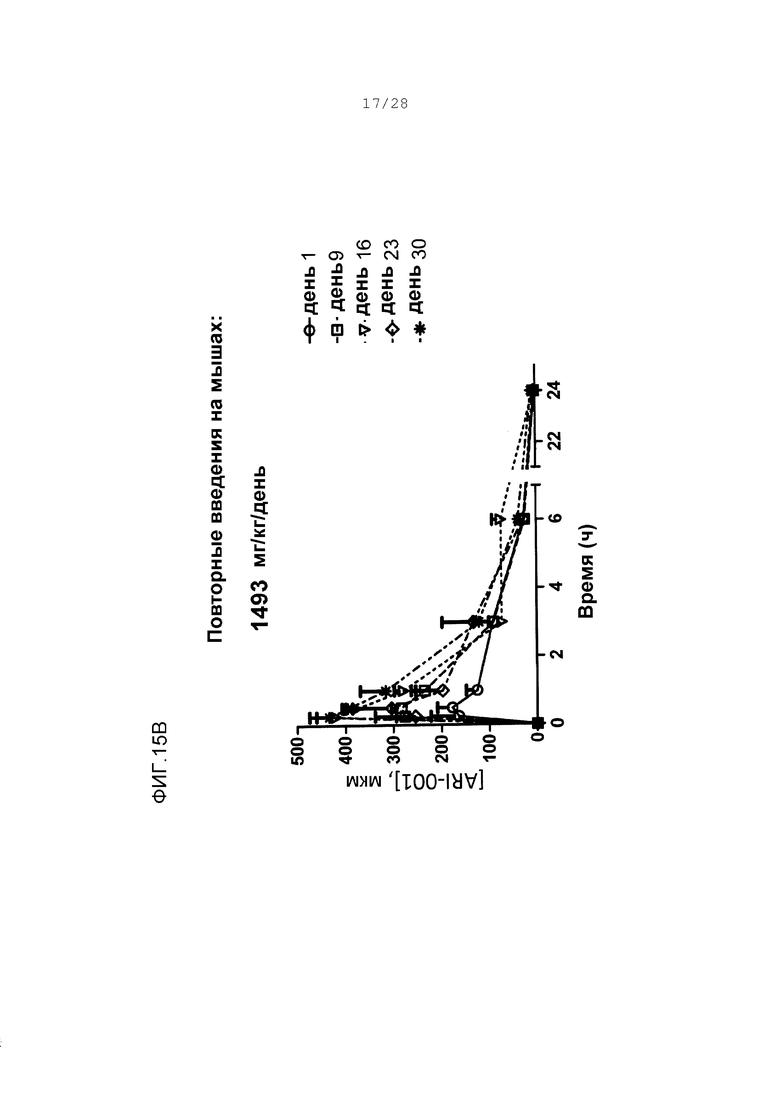
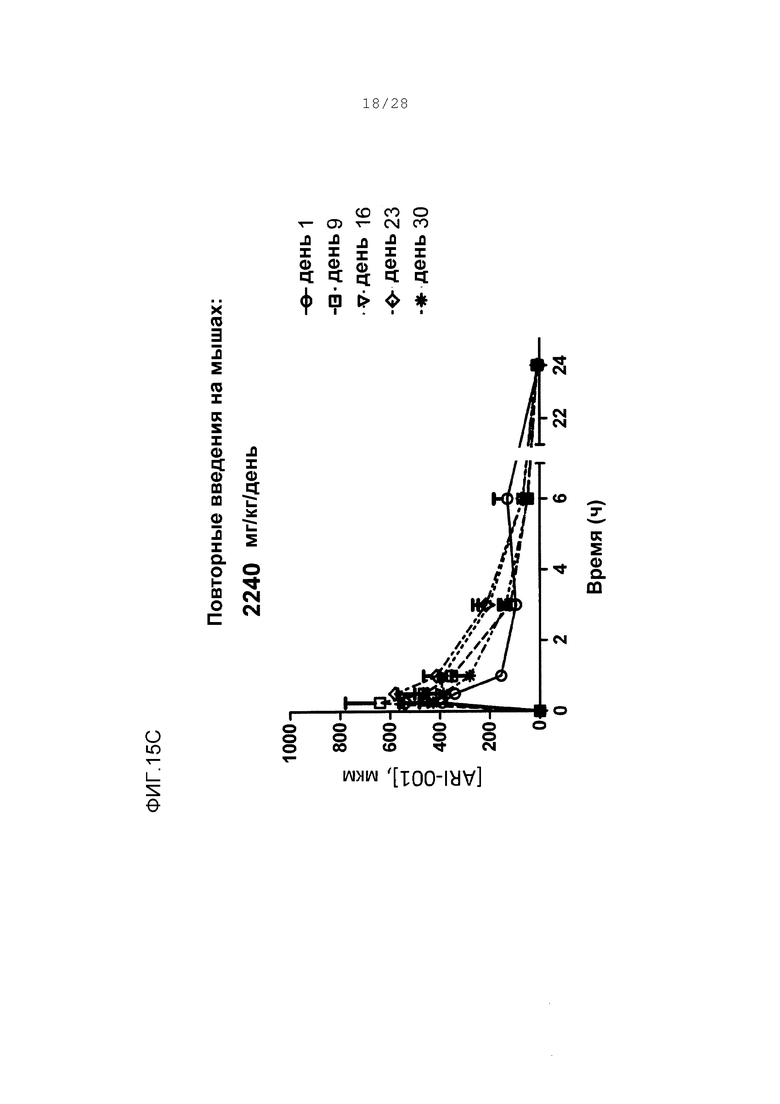
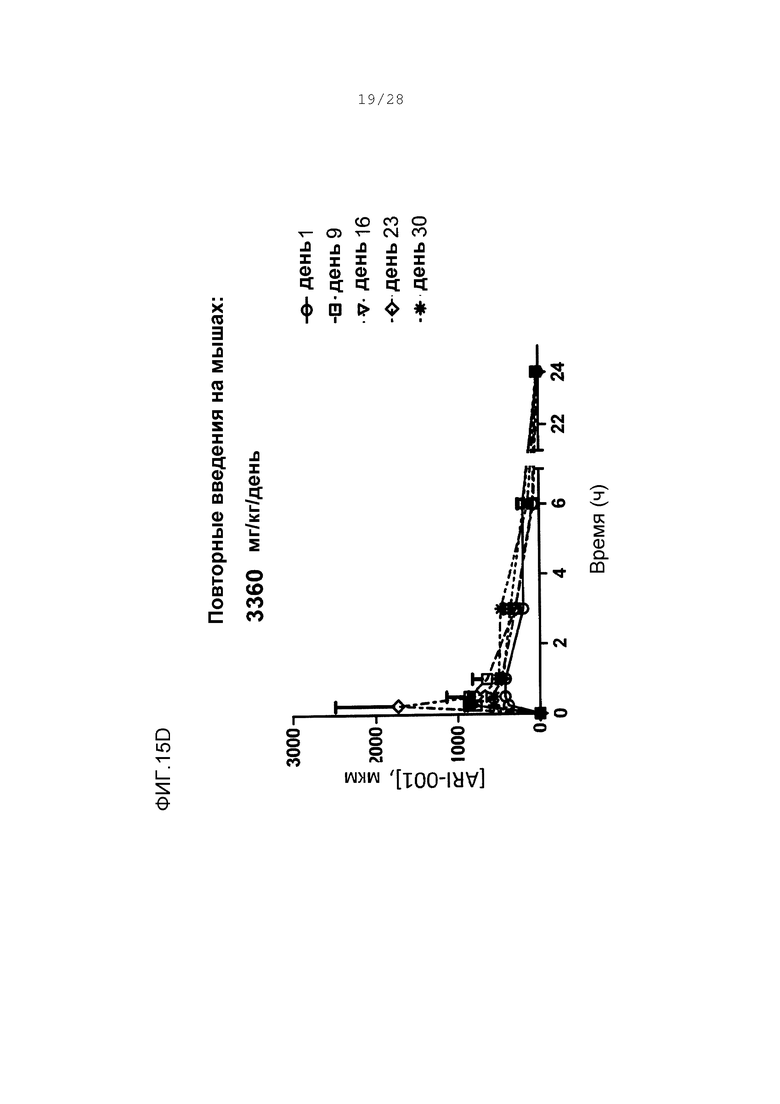
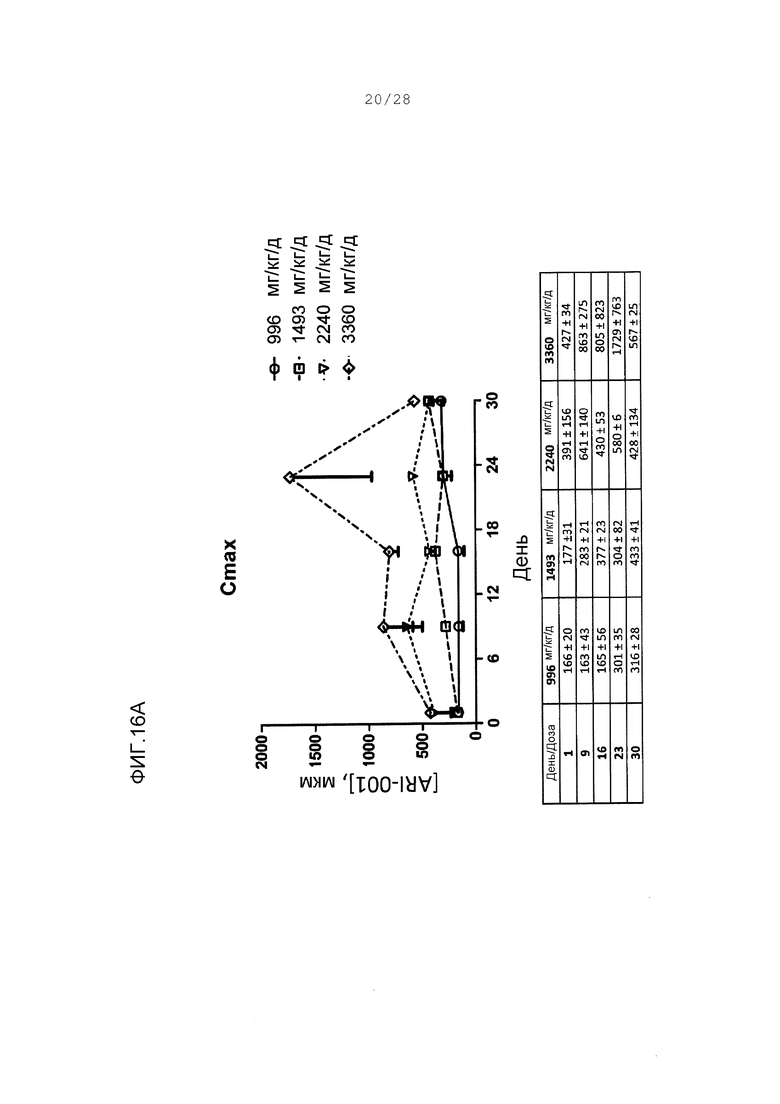
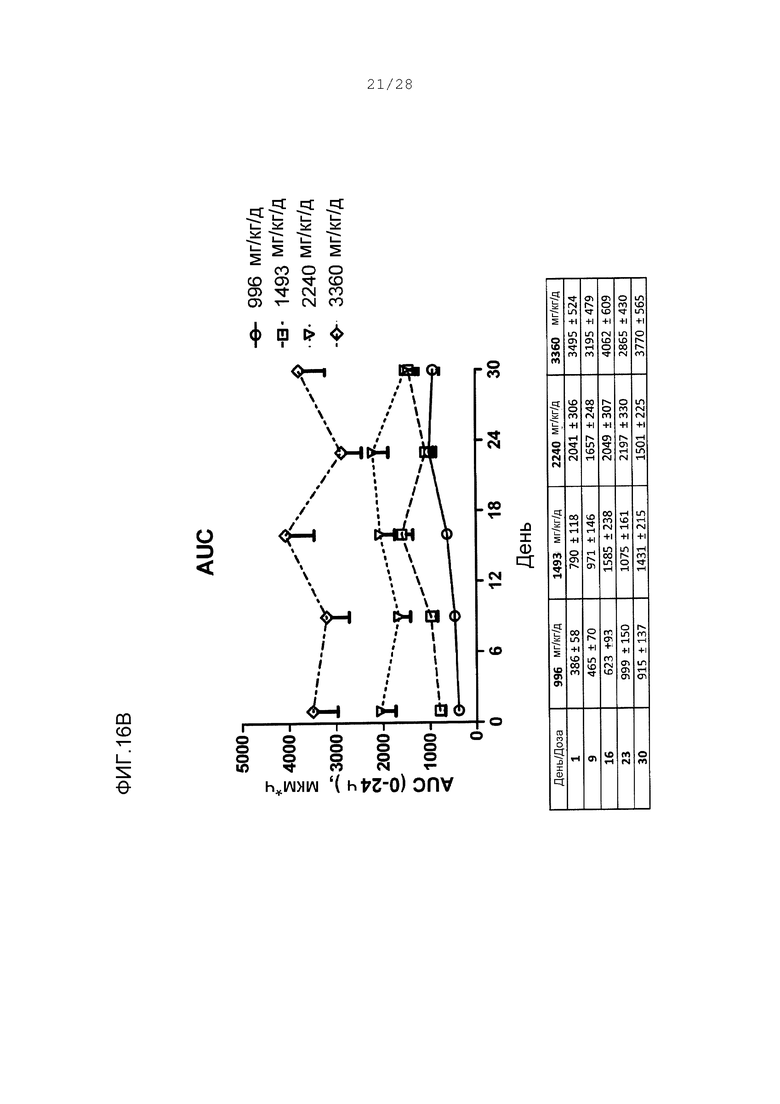
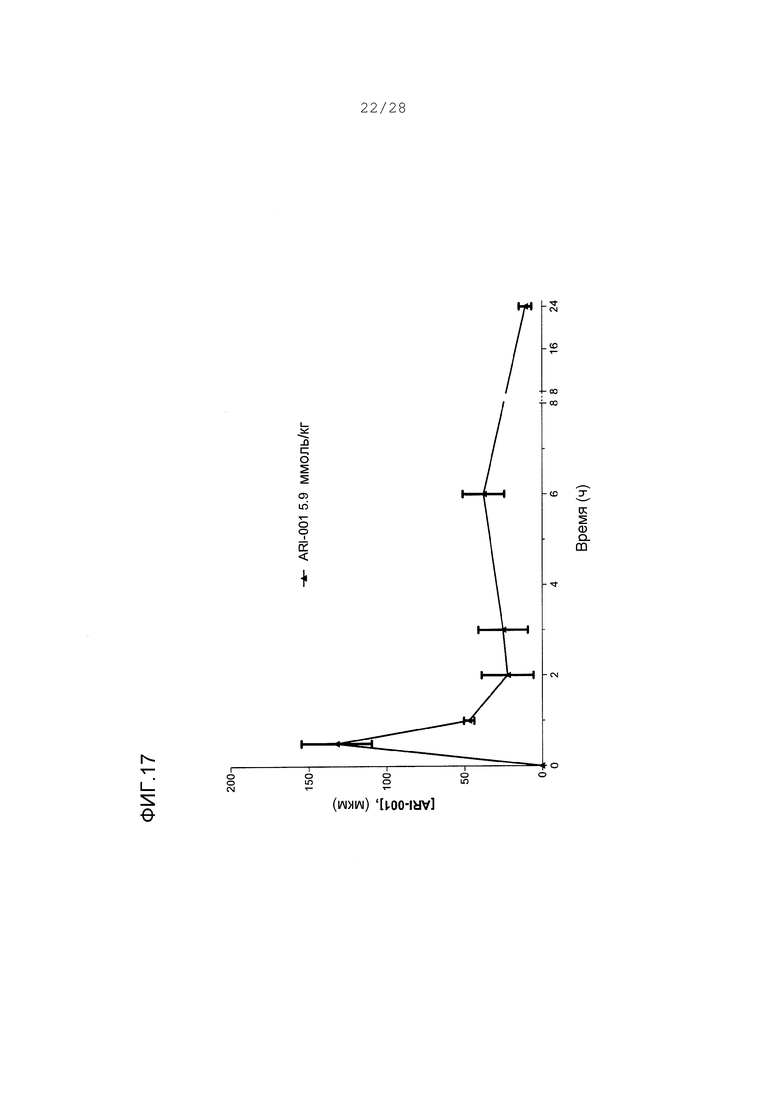
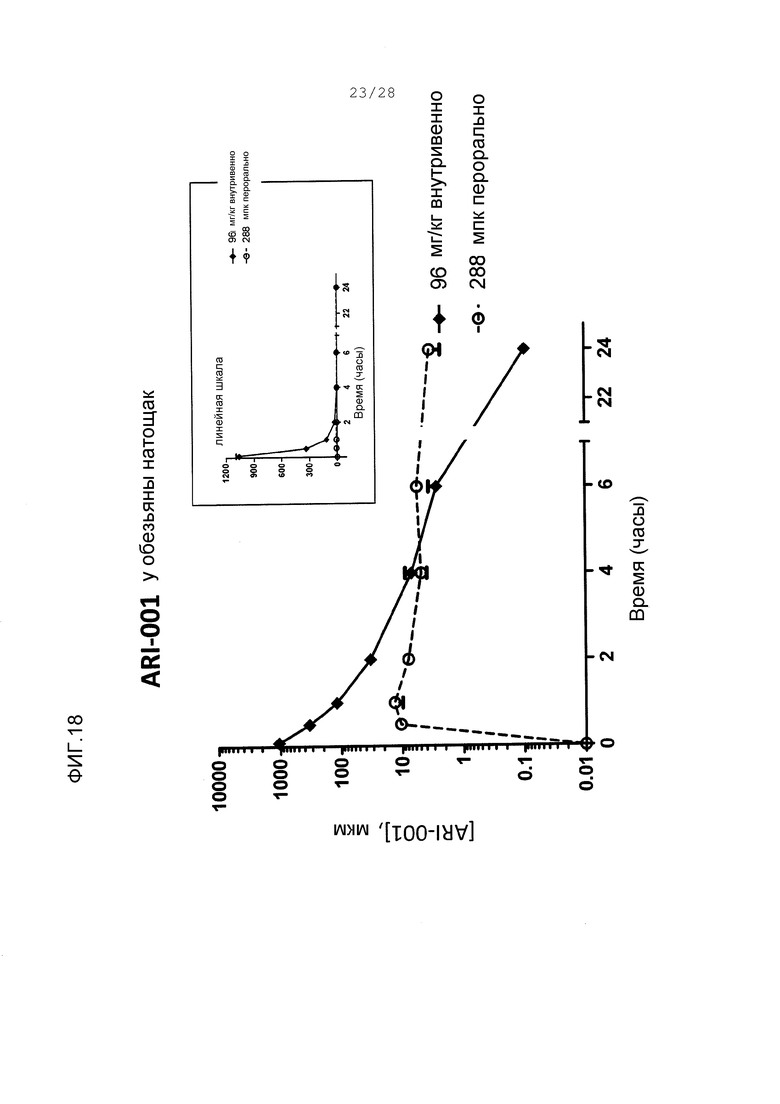
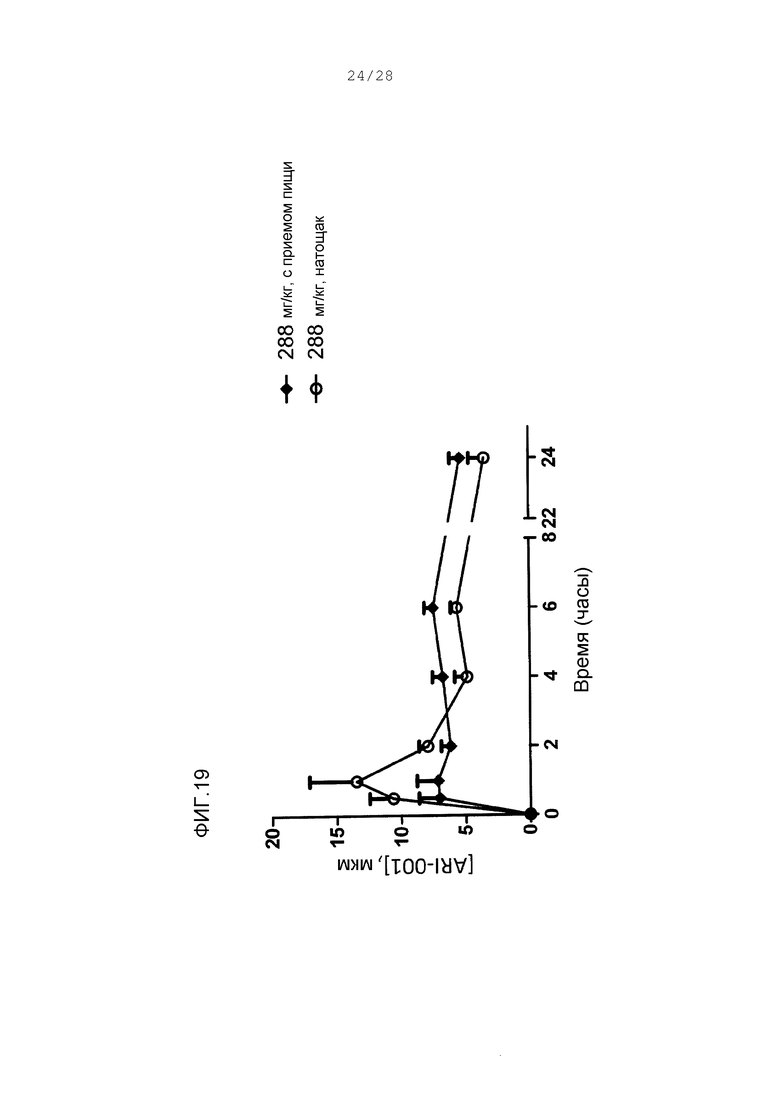
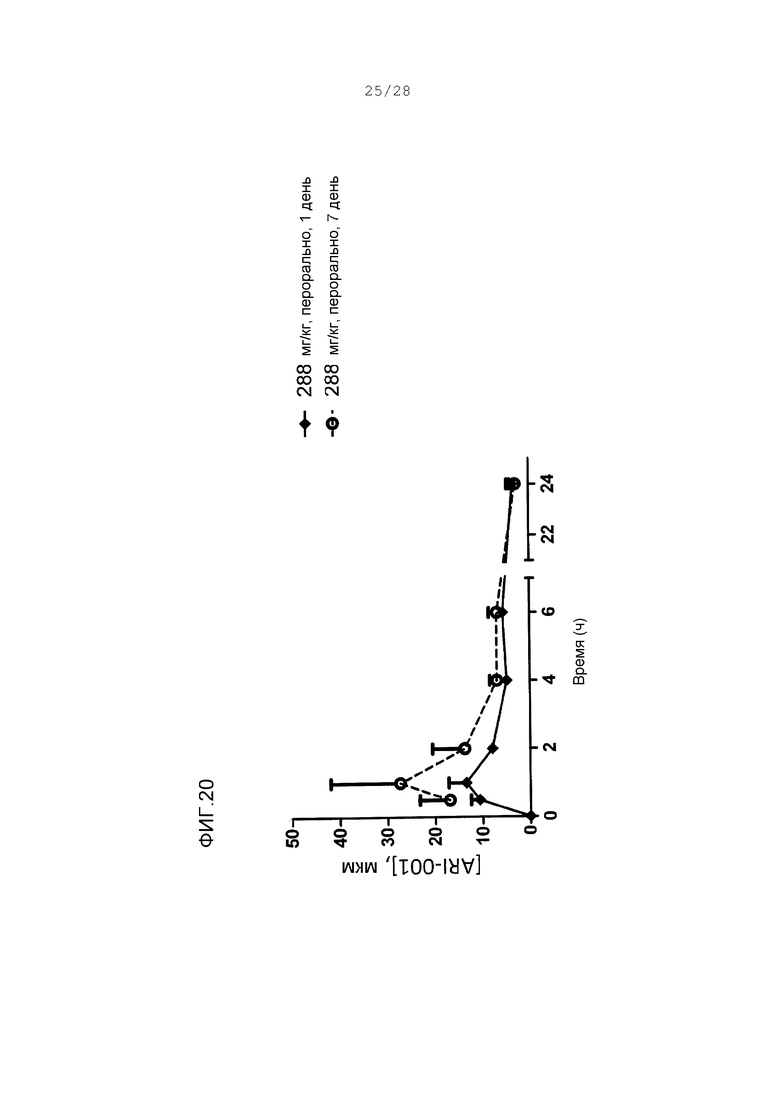

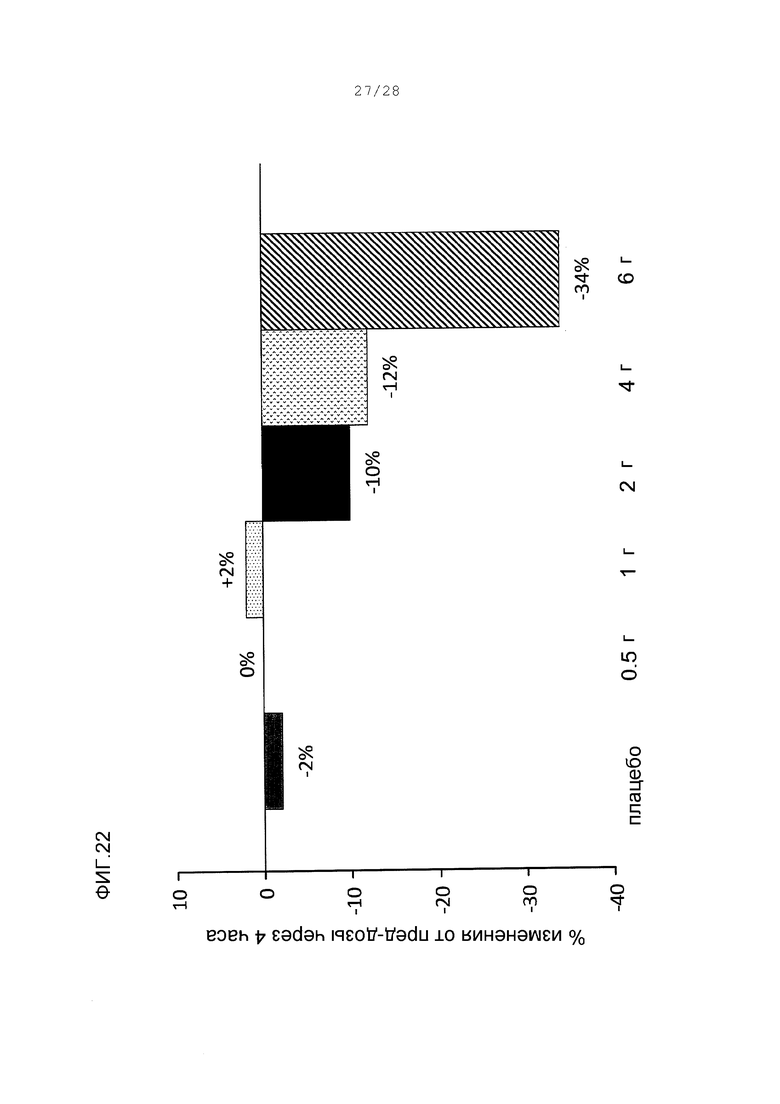
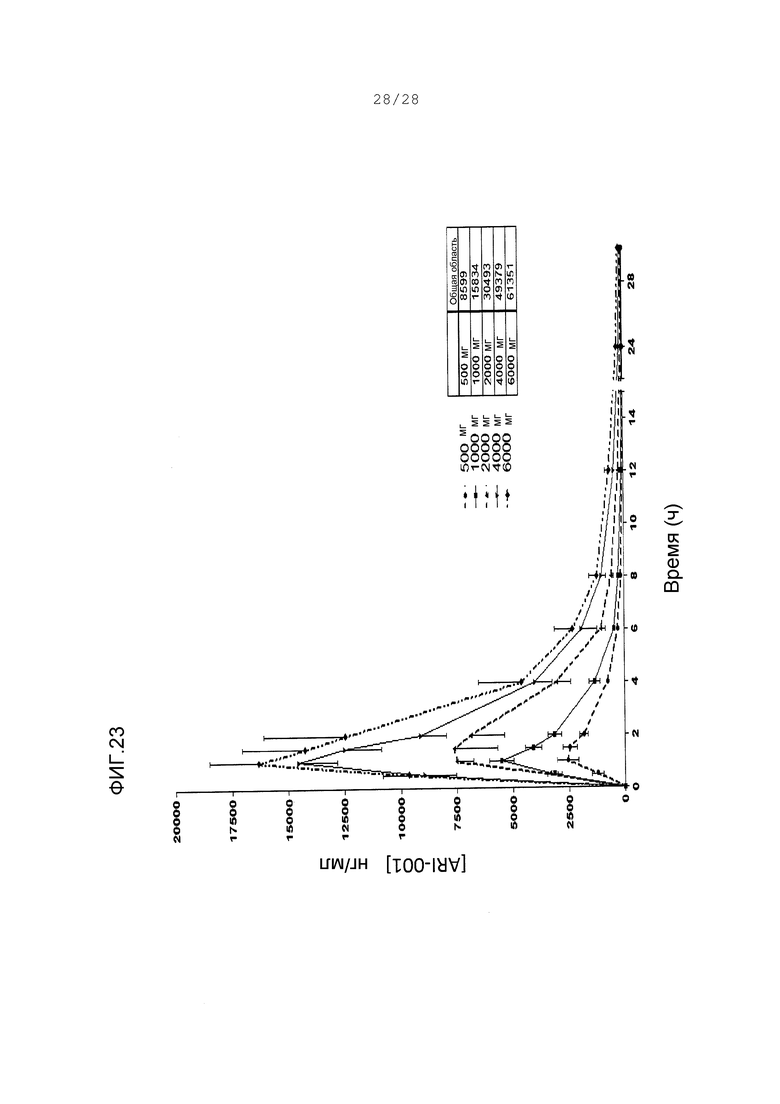
Изобретение относится к соединениям формулы (I), фармацевтической композиции на их основе, способу лечения гиперлипидемии и гиперхолестеринемии, а также к способу повышения в сыворотке уровня липопротеина высокой плотности (HDL) и способу понижения липопротеина низкой плотности (LDL) с их использованием. В общей формуле (I) R представляет собой водород, С1-6алкил, конденсированный бициклил или арилалкениларил. 7 н. и 7 з.п. ф-лы, 23 ил., 8 табл., 8 пр.
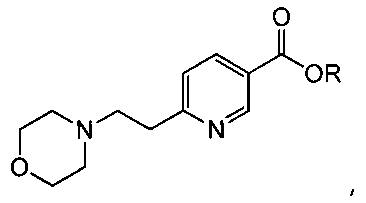 (I)
(I)
1. Соединение, представленное формулой
или его фармацевтически приемлемой солью,
в которой
R представляет собой водород, С1-6алкил, конденсированный бициклил или арилалкениларил.
2. Соединение по п. 1, в котором R представляет собой водород.
3. Соединение по п. 1, в котором R представляет собой С1-6алкил.
4. Соединение по п. 1, в котором R представляет собой конденсированный бициклил.
5. Соединение по п. 1, в котором R представляет собой арилалкениларил.
6. Соединение или его фармацевтически приемлемая соль, выбранное из группы, состоящей из
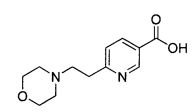 ,
, 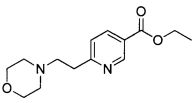 ,
, 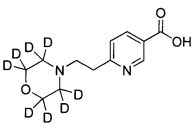 ,
,
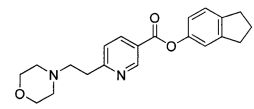 и
и
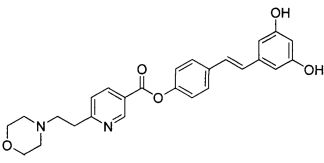 .
.
7. Соединение или его фармацевтически приемлемая соль, выбранное из группы, состоящей из
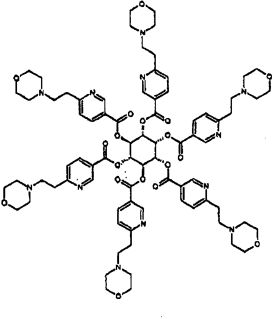 и
и
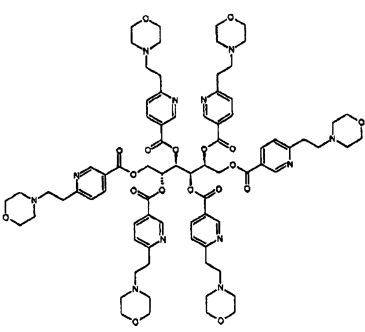 .
.
8. Фармацевтическая композиция для лечения состояния, выбранного из группы, состоящей из гиперлипидемии, гиперхолестеринемии и их комбинации, включающая соединение по любому из пп. 1-6, и фармацевтически приемлемый наполнитель.
9. Фармацевтическая композиция по п. 8, в которой отношение пика концентрации к площади под кривой на 24 часа (Смакс/AUC0-24) для соединения составляет 0,35 ч-1 или менее.
10. Фармацевтическая композиция по п.8 или 9, в которой время до пика концентрации (tмакс) для соединения составляет в интервале 30 минут до 5 часов.
11. Фармацевтическая композиция по любому из пп. 8-10, в которой соединение имеет показатель ЕС50 в отношении функции опосредуемого β-аррестина GPR109A, по меньшей мере, в 10 раз выше, чем показатель ЕС50 ниацина в отношении функции опосредуемого β-аррестина GPR109A.
12. Способ лечения заболевания состояния, выбранного из группы, состоящей из гиперлипидемии, гиперхолестеринемии и их комбинации, включающий стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-6, или фармацевтической композиции по любому из пп. 8-11.
13. Способ повышения в сыворотке уровней липопротеина высокой плотности (HDL), включающий стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-6, или фармацевтической композиции по любому из пп. 8-11.
14. Способ понижения в сыворотке уровней липопротеина низкой плотности (LDL), включающий стадию введения млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-6, или фармацевтической композиции по любому из пп. 8-11.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Swason D.M | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| ВОДЯНЫЕ ЧАСЫ | 1926 |
|
SU4413A1 |
| WO 2004022537 A2, 18.03.2004 | |||
| WO 2003024962 A1, 27.03.2003 | |||
| Производные 1-(морфолинокарбонил)-или 1-(морфолинокарбонилокси)-пиридиний хлорида в качестве дубителей для желатинсодержащих слоев галогенсеребряных фотографических материалов | 1989 |
|
SU1657505A1 |

