ОБЛАСТЬ ИЗОБРЕТЕНИЯ
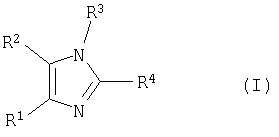
Настоящее изобретение относится к способу получения тетразамещенных производных имидазола общей формулы (I)
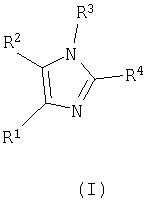
где R1, R2, R3 и R4 определены ниже в описании изобретения.
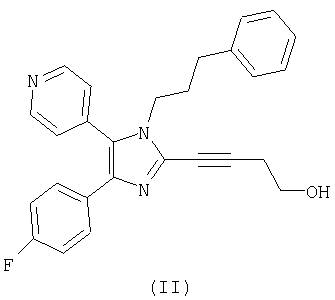
Настоящее изобретение дополнительно относится к способу получения соединения формулы (II)
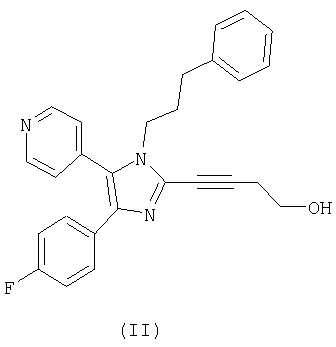
и новым кристаллическим структурам соединения формулы (II).
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения тетразамещенных производных имидазола, представленных формулой (I)
где R1, R2, R3 и R4 определены ниже в описании изобретения.
В наномолярных концентрациях соединения формулы (I) ингибируют активность p38 in vitro. Дополнительно, в наномолярных концентрациях соединения ингибируют секрецию фактора некроза опухоли α (TNF-α) и IL-β in vitro. В экспериментах на животных показано как ингибирование индуцированной липополисахаридами секреции TNF-α, так и ингибирование ревматоидного артрита. Соединения формулы (I) могут применяться при лечении ряда связанных с цитокинами расстройств, включая ревматоидный артрит, воспалительное заболевание кишечника, септический шок, остеопороз, остеоартрит, невропатическую боль, репликацию ВИЧ, связанное с ВИЧ слабоумие, вирусный миокардит, инсулин-зависимый сахарный диабет, инсулин-независимый диабет, заболевание периодонта, рестеноз, гнездную алопецию, T-клеточное истощение при ВИЧ-инфекции или СПИДе, псориаз, острый панкреатит, отторжение аллотрансплантата, аллергическое воспаление легких, атеросклероз, рассеянный склероз, кахексию, болезнь Альцгеймера, удар, болезнь Крона, ишемию, застойную сердечную недостаточность, фиброз легких, гепатит, глиобластому, синдром Гийена-Барре и системную красную волчанку (патент США № 5965583, выданный 12 октября 1999 года).
Настоящее изобретение относится к эффективному способу получения соединений формулы (I). В дополнительном аспекте настоящее изобретение относится к способу получения соединения формулы (II)
Соединение формулы (II) является перорально активным ингибитором p38-киназы. Ингибиторы p38-киназы применяются при супрессии секреции TNF-α моноцитами и, предположительно, могут супрессировать передачу сигнала, инициируемую этим медиатором провоспаления. Поэтому ингибиторы p38-киназы могут применяться как при лечении ряда воспалительных и аутоиммунных расстройств, таких как ревматоидный артрит, сепсис, воспалительное заболевание кишечника, острый респираторный дистресс-синдром, так и при лечении кахексии и резорбции костей (остеопороз и остеоартрит).
Настоящее изобретение дополнительно относится к новым кристаллическим структурам соединения формулы (II), более конкретно к форме A и форме B.
Способ синтеза соединений пиридилимидазола раскрыт в патенте США № 5670527, выданном 23 сентября 1997 года (Adams J.L. et al., правопреемник SmithKline Beecham Corp.), и РСТ заявке WO 96/21452, опубликованной 18 июля 1996 года (Adams, J.L. et al., SmithKline Beecham Corporation).
В патенте США № 5965583 (выдан 12 октября 1999 года), включенном в описание в качестве ссылки, раскрыт способ получения соединений формулы (I). Этот способ включает хроматографическое разделение промежуточных продуктов, что делает его неподходящим для широкомасштабного производства.
Поэтому существует потребность в способе, совместимом с требованиями широкомасштабного производства и который обеспечивает приемлемые уровни чистоты и выхода продукта.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к способу получения соединения формулы (I)
где
R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из C1-C5алкила, галогена или трифторметила) и гетероарила, где гетероарил содержит 5-6 атомов в кольце;
R2 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из C1-C5алкила, галогена или трифторметила) и гетероарила, где гетероарил содержит 5-6 атомов в кольце и необязательно замещен C1-C4алкилом;
R3 выбирают из группы, состоящей из водорода, арилС1-C5алкила, замещенного арилС1-C5алкила (где заместители арила независимо выбирают из одного или более C1-C5алкила, C1-C5алкокси, галогена, амино, C1-C5алкиламино или ди(C1-C5алкил)амино), фталимидоС1-C5алкила, сукцинимидоС1-C5алкила, C1-C5алкилкарбонилС1-C5алкила, арилоксикарбонилС1-C5алкила и гетероарилС1-C5алкила, где
гетероарил содержит 5-6 атомов в кольце;
R4 представляет
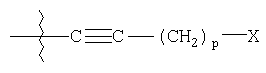
где
p является целым числом от 0 до 9;
X выбирают из группы, состоящей из водорода, гидроксигруппы, винила, замещенного винила (где один или более заместителей выбирают из фтора или хлора), этинила, замещенного этинила (где заместитель выбирают из фтора или хлора), C1-C5алкила, замещенного C1-C5алкила (где заместители алкила выбирают из одной или более C1-C5алкокси, тригалогеналкила, фталамидо или амино), C3-C7циклоалкила, C1-C5алкокси, замещенного C1-C5алкокси (где заместители алкила выбирают из фталимидо или амино), фталимидоокси, фенокси, замещенного фенокси (где заместители фенила выбирают из C1-C5алкила, фтора, хлора или C1-C5алкокси), фенила, замещенного фенила (где заместители фенила выбирают из C1-C5алкила, фтора, хлора или C1-C5алкокси), арилС1-C5-алкила, замещенного арилС1-C5алкила (где заместители арила выбирают из C1-C5алкила, фтора, хлора или C1-C5алкокси), арилгидроксиС1-C5алкиламино, C1-C5алкиламино, ди(C1-C5алкил)амино, нитрила, оксима, бензилоксиимино, C1-C5алкилоксиамино, фталимидо, сукцинимидо, C1-C5алкилкарбонилокси, фенилкарбонилокси, замещенного фенилкарбонилокси (где заместители фенила выбирают из C1-C5алкила, фтора, хлора или C1-C5алкокси), фенилС1-C5алкилкарбонилокси (где заместители фенила выбирают из C1-C5алкила, фтора, хлора или C1-C5алкокси), аминокарбонилокси, C1-C5алкиламинокарбонилокси, ди(C1-C5алкил)аминокарбонилокси, C1-C5алкоксикарбонилокси, замещенного C1-C5алкоксикарбонилокси (где заместители алкила выбирают из группы, состоящей из метила, этила, изопропила и гексила), феноксикарбонилокси, замещенного феноксикарбонилокси (где заместители фенила выбирают из C1-C5алкила, фтора, хлора или C1-C5алкокси), C1-C5алкилтио, замещенного C1-C5алкилтио (где заместители алкила выбирают из гидрокси и фталимидо), C1-C5алкилсульфонила, фенилсульфонила и замещенного фенилсульфонила (где заместители фенила выбирают из фтора, хлора, C1-C5алкокси или трифторметила);
или его фармацевтически приемлемых солей; включающему
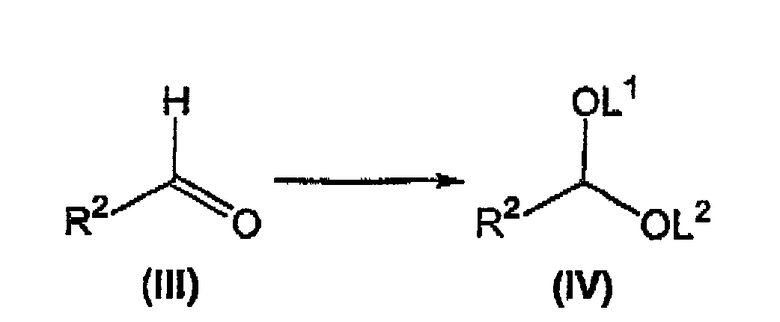
взаимодействие альдегида формулы (III) с получением соответствующего соединения формулы (IV), где L1 и L2 независимо выбирают из группы, состоящей из C1-C4алкила и C1-C4аралкила, или L1 вместе с L2 выбирают из группы, состоящей из -CH2-CH2- (необязательно замещенной 1-4 C1-C3алкилами) и -CH2-CH2-CH2- (необязательно замещенной 1-6 C1-C3алкилами);
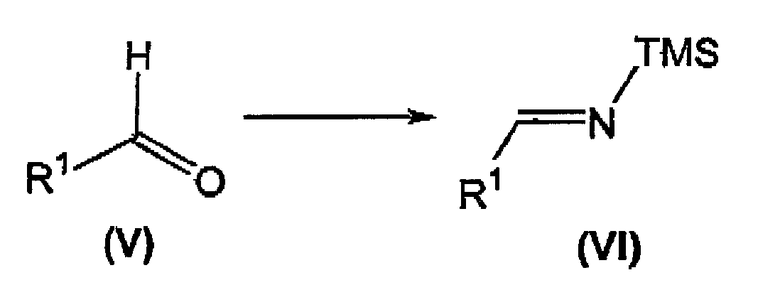
осуществление в отдельном реакционном сосуде взаимодействия альдегида формулы (V) с солью щелочного металла бис(триметилсилил)амида с получением соответствующего триметилсилилзамещенного имина формулы (VI);
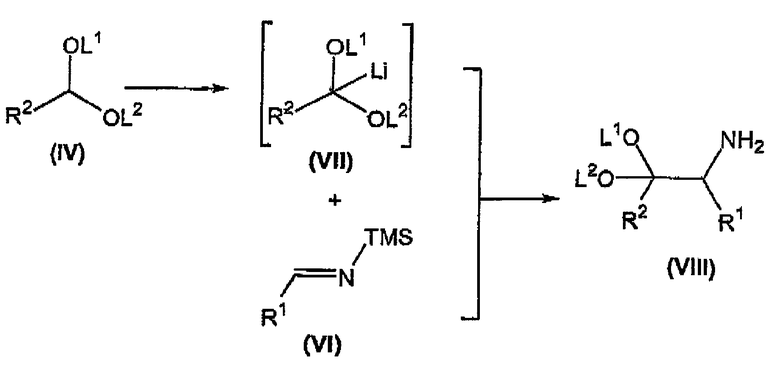
взаимодействие соединения формулы (IV) с алкиллитием с получением соответствующего промежуточного продукта лития формулы (VII);
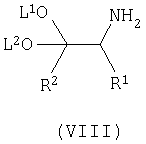
взаимодействие промежуточного продукта лития формулы (VII) с триметилсилилзамещенным имином формулы (VI) с получением соответствующего соединения формулы (VIII);
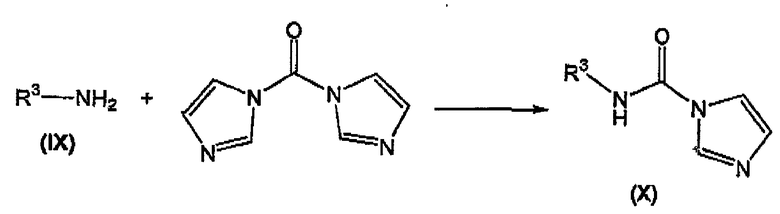
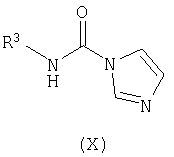
осуществление в отдельном реакционном сосуде взаимодействия замещенного амина формулы (IX) с N,N'-карбонилдиимидазолом с получением соответствующего соединения формулы (X);
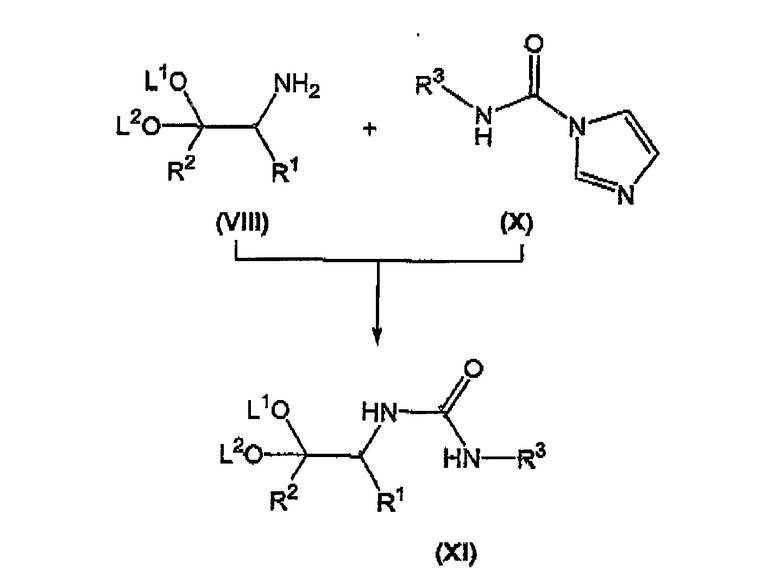
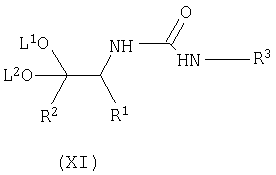
взаимодействие соединения формулы (VIII) с соединением формулы (X) с получением соответствующего соединения формулы (XI);
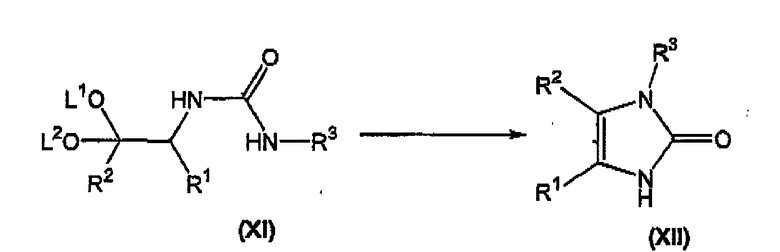
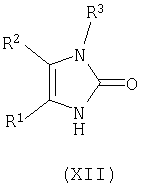
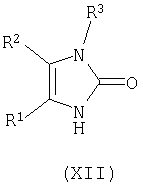
циклизацию соединения формулы (XI) в кислотных условиях при pH менее примерно 7 с получением соответствующего соединения формулы (XII);
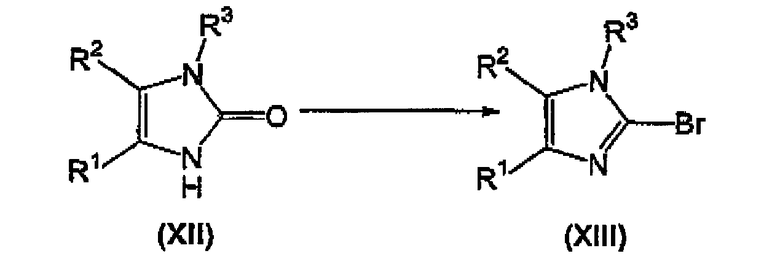
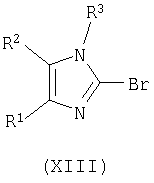
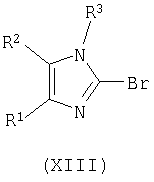
взаимодействие соединения формулы (XII) с POBr3, PBr5, или со смесью PBr3 и Br2, с получением соответствующего соединения формулы (XIII);
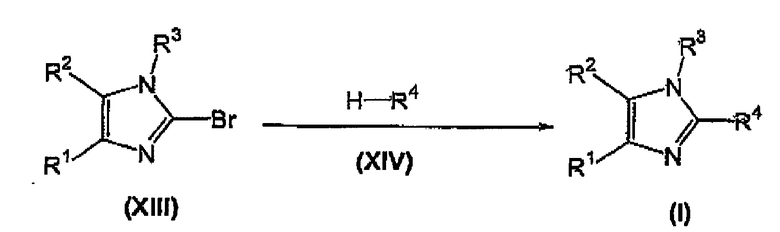
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV) с получением соответствующего соединения формулы (I).
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (II).
В дополнительном аспекте настоящее изобретение относится к промежуточным соединением формулы (XI) и формулы (XII) и способу их получения. В еще одном аспекте настоящее изобретение относится к способу получения промежуточного соединения формулы (XIII).
В дополнительном аспекте настоящее изобретение относится к новым кристаллическим структурам соединения формулы (II), где кристаллические формы, обозначаемые здесь как форма A и форма B, могут быть охарактеризованы по их соответствующим порошковым рентгенограммам.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используемый здесь термин «алкил», применяемый самостоятельно или как часть замещающей группы, включает неразветвленные, разветвленные или циклические алкильные группы. Например, алкильные группы включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, циклопропил, циклобутил, циклопентил, циклогексил и тому подобное.
Если не указано иначе, используемый здесь термин «алкокси» обозначает группу формулы -О-(алкил), например метокси, этокси, н-пропокси, втор-бутокси, трет-бутокси, н-гексилокси и тому подобное.
Если не указано иначе, используемый здесь термин «арил» относится к незамещенным моно- и конденсированным ароматическим кольцам, таким как фенил, нафтил и тому подобное.
Если не указано иначе, используемый здесь термин «гетероарил» обозначает любую пяти- или шестичленную моноциклическую ароматическую кольцевую структуру, содержащую, по крайней мере, один гетероатом, выбираемый из серы, кислорода и азота. В случае пятичленных колец гетероарил будет содержать один атом серы, кислорода или азота и, в дополнение, может содержать до трех дополнительных атомов азота. В случае шестичленных колец гетероарил может содержать до трех атомов азота. Примеры таких гетероарилов включают, но не ограничиваются, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, пиримидин-3-ил, фуран-2-ил, фуран-3-ил, тиофен-2-ил, тиофен-3-ил, пиридазинил, триазинил, тиазолил, оксазолил, пиразолил и тому подобное.
Если не указано иначе, используемый здесь термин «аралкил» обозначает любую C1-C5алкильную группу, замещенную арильной группой, такой как фенил, нафтил и тому подобное. Например, бензил, фенилэтил и тому подобное.
Если не указано иначе, используемый здесь термин «галоген» обозначает хлор, бром, фтор и йод.
Используемый здесь термин «щелочной металл» относится к катиону металла группы I, такому как катион лития, натрия, калия и цезия.
Что касается заместителей, термин «независимо» означает, что в том случае, когда возможно наличие более чем одного из таких заместителей, такие заместители могут быть одинаковыми или могут отличаться друг от друга.
По любому из способов по настоящему изобретению, может быть необходимо и/или желательно защитить чувствительные или реакционно-способные группы в любой из участвующих молекул. Это может быть достигнуто путем применения традиционных защитных групп, таких как описанные в Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; и T.W.Greene & P.G.M.Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991. Защитные группы могут быть удалены на подходящей последующей стадии с применением способов, известных в данной области техники.
Настоящее изобретение относится к способу получения соединений формулы (I), что наиболее полно описывается схемой 1
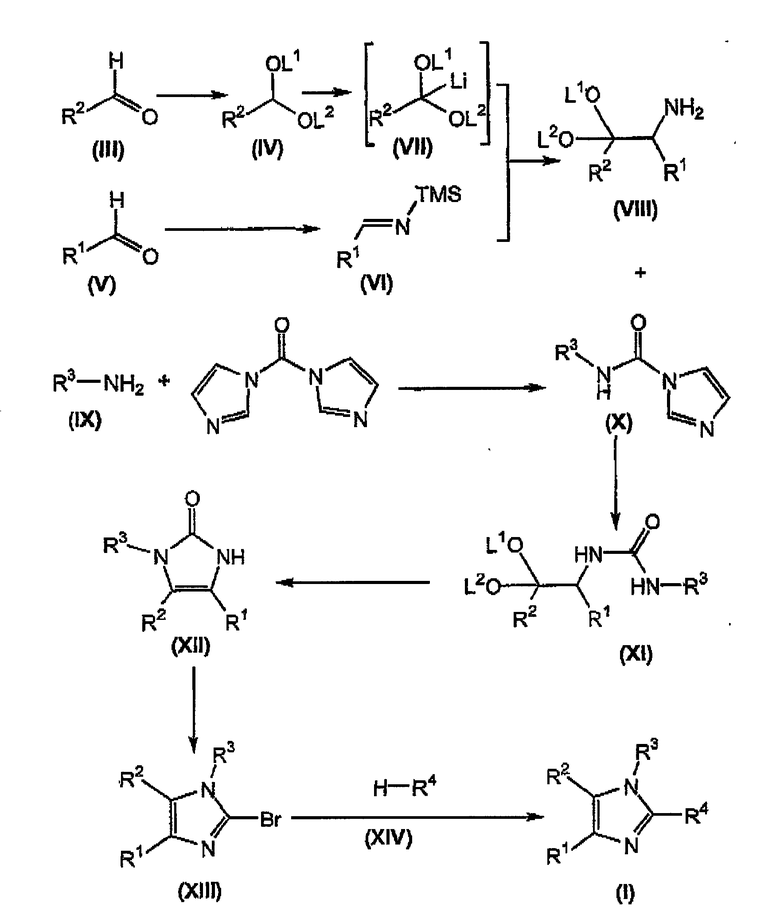
Схема 1
Как изложено выше в схеме 1, альдегид формулы (III), известное соединение или соединение, полученное в соответствии с известной методикой, взаимодействует со спиртом, диолом или триалкоксиметаном, предпочтительно триметоксиметаном, предпочтительно в присутствии метанола, в растворителе, для которого возможно азеотропное удаление воды, таком как бензол, толуол, ксилол и тому подобное, в присутствии кислоты, такой как серная кислота, пара-толуолсульфоновая кислота и тому подобное, предпочтительно серной кислоты, при температуре флегмы с получением соответствующего соединения формулы (IV) (Sheldrake P.W., Synth Commun. (1993) 23(14), 1967-71).
В отдельном реакционном сосуде осуществляют взаимодействие альдегида формулы (V), известного соединения или соединения, полученного в соответствии с известной методикой, с солью щелочного металла бис(триметилсилил)амида, предпочтительно литийбис(триметилсилил)амидом, в органическом растворителе, таком как тетрагидрофуран (ТГФ), диэтиловый эфир, трет-бутилметиловый эфир (MTBE) и тому подобное, предпочтительно ТГФ, при температуре в диапазоне от приблизительно -20°C до приблизительно комнатной температуры, предпочтительно при температуре приблизительно 0°C, с получением соответствующего триметилсилил(TMS)-замещенного имина формулы (VI) (Ojima I. et al., Tetrahedron (1996), 52, 209-224).
Осуществляют взаимодействие соединения формулы (IV) с алкиллитием, таким как метиллитий, этиллитий, н-бутиллитий, и тому подобное, предпочтительно н-бутиллитием, при температуре, которая предотвращает разрушение промежуточного продукта лития формулы (VII), предпочтительно при температуре меньше, чем или равной примерно -20°C, в органическом растворителе, таком как тетрагидрофуран (ТГФ), диэтиловый эфир, трет-бутилметиловый эфир (MTBE), и тому подобное, предпочтительно ТГФ, с получением соответствующего промежуточного продукта лития формулы (VII).
Осуществляют взаимодействие промежуточного продукта лития формулы (VII) с TMS-замещенным имином формулы (VI) в присутствии органического растворителя, такого как тетрагидрофуран (ТГФ), диэтиловый эфир, трет-бутилметиловый эфир (MTBE), и тому подобное, предпочтительно ТГФ, предпочтительно давая возможность реакционной смеси нагреться до примерно комнатной температуры, с получением соответствующего соединения формулы (VIII).
В отдельном реакционном сосуде осуществляют взаимодействие замещенного амина формулы (IX), известного соединения или соединения, полученного в соответствии с известной методикой, с N,N'-карбонилдиимидазолом, известным соединением, в инертном органическом растворителе, таком как тетрагидрофуран (ТГФ), диэтиловый эфир, трет-бутилметиловый эфир (MTBE), толуол, дихлорметан (DCM), и тому подобное, предпочтительно ТГФ, предпочтительно при комнатной температуре, с получением соответствующего соединения формулы (X).
Осуществляют взаимодействие соединения формулы (VIII) с соединением формулы (X), в органическом растворителе, таком как толуол, тетрагидрофуран (ТГФ), диметилформамид (ДМФ), и тому подобное, предпочтительно в толуоле, при температуре в диапазоне примерно 50-150°C, предпочтительно для толуола, при примерно температуре флегмы, с получением соответствующего соединения формулы (XI).
Соединение формулы (XI) циклизуют в кислоте, такой как муравьиная кислота, водная соляная кислота, и тому подобное, предпочтительно в водной соляной кислоте, предпочтительно при температуре в диапазоне примерно 80-150°C, более предпочтительно при температуре в диапазоне примерно 95-100°C, с получением соответствующего соединения формулы (XII).
Осуществляют взаимодействие соединения формулы (XII) с оксибромидом фосфора (POBr3) или пентабромидом фосфора (PBr5), предпочтительно с POBr3, в количестве, равном, по крайней мере, приблизительно 5 эквивалентам, в инертном органическом растворителе, точка кипения которого превышает или равна приблизительно 110°C, таком как тетраметиленсульфон, ксилол, толуол, и тому подобное, предпочтительно в тетраметиленсульфоне, предпочтительно в количестве, равном приблизительно 2 массовым эквивалентам, при температуре, большей или равной приблизительно 110°C, предпочтительно при температуре приблизительно 130°C, с получением соответствующего соединения формулы (XIII).
Альтернативно, осуществляют взаимодействие соединения формулы (XII) со смесью PBr3 и Br2 (дающей in situ PBr5), где отношение PBr3 к Br2 находится в диапазоне от приблизительно 1:2 до приблизительно 2:1, предпочтительно отношение PBr3 к Br2 равно приблизительно 1:1; где количество PBr5, получаемое в смеси PBr3 и Br2, находится в диапазоне примерно 3-3,5 эквивалентов, в растворителе, таком как POCl3, или в инертном органическом растворителе, точка кипения которого превышает или равна приблизительно 110°C, таком как тетраметиленсульфон (сульфолан), ксилол, толуол, и тому подобное, предпочтительно в POCl3, при температуре в диапазоне примерно 10-45°C, предпочтительно при температуре в диапазоне примерно 20-35°C, с получением соответствующего соединения формулы (XIII).
В соединении формулы (XIII) бром замещают взаимодействием с соединением формулы (XIV), известным соединением или соединением, полученным в соответствии с известной методикой, в присутствии катализатора на основе палладия (II), такого как диацетоксибис(трифенилфосфин)палладий (Pd(OAc)2(Ph3P)2), дихлорбис(трифенилфосфин)палладий (PdCl2(Ph3P)2), и тому подобное, или в присутствии катализатора, такого как ацетат палладия (Pd(OAc)2) или хлорид палладия (PdCl2) в присутствии трифенилфосфина, предпочтительно катализатором является диацетоксибис(трифенилфосфин)палладий, предпочтительно в присутствии со-катализатора, такого как йодид меди (I) (CuI), порошковое железо, и тому подобное, предпочтительно CuI, в присутствии органического амина, такого как диизопропиламин, диизопропилэтиламин (DIPEA), триэтиламин (TEA), пиперидин, и тому подобное, или неорганического основания, такого как K2CO3, Cs2CO3, и тому подобное, предпочтительно органического амина, более предпочтительно диизопропиламина, необязательно в инертном органическом растворителе, таком как ТГФ, трет-бутилметиловый эфир (MTBE), диэтиловый эфир, DMF, ацетонитрил, и тому подобное, с нагреванием до температуры в диапазоне примерно 60-100°C, предпочтительно до температуры приблизительно 75°C, с получением соответствующего соединения формулы (I).
Альтернативно, соединение формулы (XI) может быть получено в соответствии со способом, отраженным на схеме 2
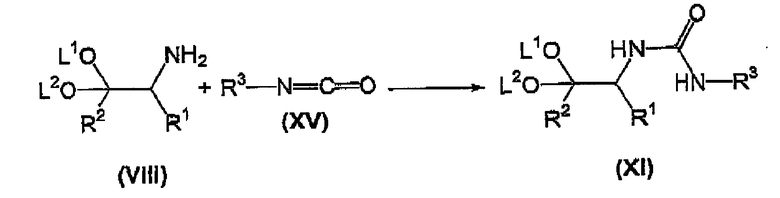
Схема 2
где L1, L2, R1, R2 и R3 имеют значения, определенные выше.
Более конкретно, осуществляют взаимодействие соединения формулы (VIII) с соединением формулы (XV) в инертном органическом растворителе, таком как тетрагидрофуран (ТГФ), диметилформамид (DMF), толуол, и тому подобное, предпочтительно в ТГФ, предпочтительно при комнатной температуре, с получением соответствующего соединения формулы (XI).
В предпочтительном воплощении настоящего изобретения способ применяется для получения соединения формулы (II). Предпочтительно соединение формулы (II) дополнительно очищают при помощи известных способов, таких как перекристаллизация из органического растворителя, такого как толуол, метанол, ацетон, ацетонитрил, и тому подобное, или из смеси органических растворителей, такой как этилацетат/гексан, ТГФ/толуол, этилацетат/толуол, и тому подобное.
Настоящее изобретение дополнительно относится к новым кристаллическим структурам соединения формулы (II). Кристаллические структуры соединения формулы (II) могут быть получены путем перекристаллизации соединения формулы (II) из подходящего органического растворителя, такого как ацетон, ацетонитрил, смесь ТГФ/толуол, и тому подобное.
Как описано выше, перекристаллизация соединения формулы (II) будет приводить к образованию одной из двух новых кристаллических форм, обозначаемых здесь как форма A и форма B. Форму B получают путем перекристаллизации из ацетона или смеси ТГФ:толуол, более предпочтительно из смеси ТГФ:толуол (1:2). Форму A получают путем перекристаллизации из ацетонитрила.
Новые кристаллические формы соединения формулы (II) могут быть охарактеризованы по их соответствующим порошковым рентгенограммам с применением порошкового дифрактометра Siemens D5000T-T, используя CuKα-излучение и следующие системные условия:
a) CuKα-излучение, 35 мА, 40 кВ;
b) оптика
- 1 мм щель, зеркала Гобеля, 0,6 мм щель, и вертикальные солярные (soller) щели между трубкой и образцом,
- LiF монохроматор между образцом и детектором;
c) сканирование от 5 до 35°2Θ при величине шага 0,02 со скоростью 1°2Θ/мин;
d) платформа TTK-450 с изменяемой температурой/влажностью и держатель.
Форма A соединения формулы (II) может быть охарактеризована по своей порошковой рентгенограмме, которая включает основные пики, перечисленные в табл.1.
Пики порошковой рентгенограммы для формы А
Форма В соединения формулы (II) может быть охарактеризована по своей порошковой рентгенограмме, которая включает основные пики, перечисленные в табл.2.
Пики порошковой рентгенограммы для формы В
Следующие примеры более детально описывают настоящее изобретение и предназначены для иллюстрации настоящего изобретения, а никоим образом не для его ограничения.
ПРИМЕР 1
4-(Диметоксиметил)пиридин
К раствору 4-пиридинкарбоксальдегида (100,00 г, 0,93 моль) и триметилортоформиата (159,20 г, 1,50 моль) в метаноле (180 мл) при 0°C в атмосфере азота добавляли концентрированную серную кислоту (41 мл, 0,45 моль). Полученную белую суспензию нагревали с обратным холодильником и перемешивали в течение 24 ч. Реакционный раствор становился прозрачным спустя 2 ч. После охлаждения до комнатной температуры реакционную смесь медленно вливали в 25 мас.% раствор метоксида натрия (360 мл) в метаноле (300 мл). Смесь концентрировали в вакууме до состояния густого светло-коричневого масла. К этому неочищенному маслу добавляли трет-бутилметиловый эфир (500 мл), затем медленно добавляли воду (40 мл) (для превращения неорганических веществ в фильтрируемые твердые вещества). После фильтрации через слой целита фильтрат концентрировали с получением светло-коричневого масла. Неочищенное масло перегоняли в вакууме с получением желаемого продукта в виде бесцветного масла. Выход: 88,91 г (62,4%). Т.кип. 69-71°C при 1 мм рт.ст.
ПРИМЕР 2
2,2-Диметокси-2-(4-пиридил)-1-(4-фторфенил)этанамин
Этап A
К перемешиваемому раствору 1M литийбис(триметилсилил)амида в ТГФ (300 мл, 0,30 моль) в атмосфере азота по каплям при 0оC добавляли 4-фторбензальдегид (37,23 г, 0,30 моль). Полученную смесь перемешивали при комнатной температуре в течение 30 мин с получением раствора.
Этап B
Во втором сосуде 4-диметоксиметилпиридин (38,29 г, 0,25 моль) смешивали с ТГФ (200 мл) и охлаждали до -20°C. К раствору медленно по каплям добавляли 2,5M н-бутиллитий в гексане (120 мл, 0,30 моль) при поддержании температуры реакционного раствора на уровне между -15°С и -20°C. Полученную темно-коричневую реакционную смесь перемешивали при -20°C в течение 15 мин. К реакционной смеси медленно добавляли раствор из описанного выше этапа A. Температуру реакционного раствора поддерживали на уровне ниже -15°C. После добавления темно-коричневую реакционную смесь перемешивали и нагревали до комнатной температуры. Реакционную смесь гасили 2 н. водной HCl (500 мл) до значения pH, равного приблизительно 2, и разделяли полученные слои. Органический слой однократно экстрагировали 1 н. водной HCl (100 мл). Объединенные водные слои промывали этилацетатом (2x150 мл) и затем подщелачивали добавлением 50% водного раствора NaOH до значения pH, равного приблизительно 10. Подщелаченную смесь экстрагировали этилацетатом (400 мл, 2x100 мл). Объединенные этилацетатные экстракты промывали водой (200 мл), солевым раствором (200 мл) и сушили Na2SO4. После концентрирования в вакууме неочищенный продукт получали в виде густого коричневого масла. Выход: 54,70 г (79%).
ПРИМЕР 3
N-(3-Фенилпропил)-1Н-имидазол-1-карбоксамид
К суспензии 1,1'-карбонилдиимидазола (33,0 г, 0,203 моль) в ТГФ (100 мл) при комнатной температуре в атмосфере азота по каплям добавляли 3-фенилпропиламин (25,00 г, 0,185 моль) в ТГФ (50 мл). Реакционная смесь становилась прозрачной во время добавления 3-фенилпропиламина. После окончания добавления прозрачный раствор перемешивали в течение 30 мин при комнатной температуре и затем гасили водой (150 мл) и этилацетатом (200 мл). Слои разделяли и промывали органический слой водой (150 мл), солевым раствором (150 мл) и сушили Na2SO4. Растворители удаляли в вакууме с получением белого воскообразного твердого вещества. Выход: 47,50 г.
ПРИМЕР 4
N-(3-Фенилпропил)-N'-[(2,2-диметокси-2-(4-пиридил)-1-(4-фторфенил)этил)]мочевина
Раствор 2,2-диметокси-2-(4-пиридил)-1-(4-фторфенил)этанамина (51,12 г, 0,185 моль) и N-(3-фенилпропил)-1H-имидазол-1-карбоксамида (42,42 г, 0,185 моль) в толуоле (300 мл) в атмосфере азота перемешивали и нагревали до температуры флегмы в течение 3 ч. Раствор охлаждали до комнатной температуры и разбавляли темно-коричневый раствор этилацетатом (200 мл). Смесь промывали водой (2x200 мл), солевым раствором (200 мл) и сушили Na2SO4. Растворители удаляли в вакууме с получением коричневого твердого вещества, которое перекристаллизовывали из смеси растворителей этилацетата/гексана (1:1) с получением не совсем белого твердого вещества. Выход: 38,00 г (47%).
ПРИМЕР 5
1,3-Дигидро-1-(3-фенилпропил)-4-(4-фторфенил)-5-(4-пиридил)-2H-имидазолин-2-он
N-(3-Фенилпропил)-N'-[(2,2-диметокси-2-(4-пиридил)-1-(4-фторфенил)этил)]мочевину (38,0 г, 86,8 ммоль) растворяли в муравьиной кислоте (100 мл) с получением коричневого раствора. Раствор нагревали до 95-100°C и перемешивали в атмосфере азота в течение 24 ч. Затем раствор охлаждали до комнатной температуры, удаляли муравьиную кислоту на роторном испарителе при пониженном давлении и разбавляли остаток этилацетатом (300 мл). Раствор подщелачивали 6 н. NaOH до значения pH, равного приблизительно 10. В органическом слое медленно образовывалось не совсем белое твердое вещество. Прозрачный водный слой отделяли и экстрагировали этилацетатом (50 мл). Объединенные органические слои разбавляли трет-бутилметиловым эфиром (350 мл) и перемешивали в течение 30 мин. Твердый продукт собирали путем фильтрации, промывали трет-бутилметиловым эфиром (100 мл) и сушили на воздухе в течение 1 ч. Твердый продукт сушили в вакуумной печи при комнатной температуре в течение 24 ч с получением продукта в виде не совсем белого твердого вещества. Выход: 18,11 г (58%). Т.пл.: 198-199,5°C.
ПРИМЕР 6
Гидробромид 2-бром-1-(3-фенилпропил)-4-(4-фторфенил)-5-(4-пиридил)-1H-имидазола
1,3-Дигидро-1-(3-фенилпропил)-4-(4-фторфенил)-5-(4-пиридил)-2H-имидазолин-2-он (5,0 г, 13,4 ммоль) суспендировали в сульфолане (20,0 г) и обрабатывали POBr3 (19,5 г, 68 ммоль). Смесь нагревали до 130°C и перемешивали в атмосфере азота в течение 3-3,5 ч. Реакционный раствор охлаждали до комнатной температуры, разбавляли трет-бутилметиловым эфиром (100 мл) и дополнительно охлаждали до 0оC. Реакционную смесь гасили медленным добавлением 10% раствора NaOH (120 мл) до значения pH, равного приблизительно 10. Слои разделяли и экстрагировали водный слой трет-бутилметиловым эфиром (30x2 мл). Объединенные органические слои промывали водой (50x2 мл), солевым раствором (50 мл) и сушили Na2SO4. Растворитель удаляли в вакууме и растворяли остаток в смеси этилацетата (100 мл) и метанола (5 мл). Раствор обрабатывали 2,88M раствором HBr в этилацетате (9,3 мл, 26,8 ммоль). Полученную желтую суспензию нагревали на паровой бане. К суспензии добавляли метанол (5 мл), что приводило к образованию раствора, и перемешивали раствор в течение ночи при комнатной температуре (приблизительно 18 ч). Затем медленно добавляли этилацетат (50 мл) и перемешивали суспензию в течение дополнительного 1 ч. Осадок собирали путем фильтрации и промывали этилацетатом (50 мл). Твердое вещество сушили в вакуумной печи при комнатной температуре в течение 2 ч с получением продукта в виде желтоватого твердого вещества. Выход: 5,01 г (62%). Т.пл.: 214-216°C (изменение окраски при 205°C).
ПРИМЕР 7
4-(4-Фторфенил)-2-(4-гидрокси-1-бутинил)-1-(3-фенилпропил)-5- (4-пиридил)имидазол
К перемешиваемому раствору 4-(4-фторфенил)-2-йод-1-(3-фенилпропил)-5-(4-пиридил)имидазола (1,42 г, 2,74 ммоль) и 3-бутин-1-ола (0,289 г, 4,1 ммоль) в диизопропиламине (10 мл) добавляли бис(ацетато)бис(трифенилфосфин)палладий (0,102 г, 0,14 ммоль), затем добавляли йодид меди (I) (0,052 г, 0,274 ммоль). Смесь перемешивали при 75°C в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры и гасили водой (100 мл). Смесь экстрагировали этилацетатом (2x50 мл). Объединенный этилацетатный экстракт промывали водой (2x30 мл), солевым раствором (30 мл) и сушили Na2SO4. После удаления растворителей получали неочищенный продукт в виде коричневого твердого вещества. Неочищенный продукт очищали путем перекристаллизации из смеси этилацетата/гексана с получением продукта в виде желтого твердого вещества. Выход: 0,88 г (75%). Т.пл.: 121-122°C.
ПРИМЕР 8
1,3-Дигидро-1-(3-фенилпропил)-4-(4-фторфенил)-5-(4-пиридил)-2H-имидазол-2-он
N-(3-Фенилпропил)-N'-[(2,2-диметокси-2-(4-пиридил)-1-(4-фторфенил)этил)]мочевину (224 г, 0,45 моль) смешивали с 4 н. HCl (800 г) и нагревали с обратным холодильником в течение 4-5 ч (95-100°C). После окончания нагревания реакционную смесь охлаждали до комнатной температуры и доводили до значения pH 13 добавлением 8 н. раствора NaOH (480 г), что приводило к осаждению твердого продукта. В течение 30 мин контролировали значение pH суспензии на уровне pH≥13, добавляя при необходимости гидроксид натрия. Суспензию центрифугировали, водную фазу удаляли и декантировали. Твердое вещество повторно суспендировали в 2 н. растворе NaOH (1000 г), повторно центрифугировали и затем повторно суспендировали в воде (2х1000 г, значение pH водной фазы равно 7). Твердый продукт сушили при 45-50°C в вакууме (в течение приблизительно 4-5 суток) до конечного содержания воды <2% с получением продукта в виде желтовато-коричневого твердого вещества. Выход: 175 г.
ПРИМЕР 9
4-(4-Фторфенил)-2-бром-1-(3-фенилпропил)-5-(4-пиридил)имидазол
1,3-Дигидро-1-(3-фенилпропил)-4-(4-фторфенил)-5-(4-пиридил)-2H-имидазол-2-он (100 г, 0,26 моль) смешивали с POBr3 (268,7 г, 0,93 моль) и сульфоланом (200 г) и нагревали реакционную смесь до температуры 120-125°C в течение 1-2 ч. После окончания нагревания реакционную смесь охлаждали до 40°C. В течение приблизительно 30 мин осторожно добавляли 2 н. раствор NaOH (53 г). Затем в более быстром режиме добавляли дополнительное количество 2 н. раствора NaOH (53 г). Затем реакционную смесь охлаждали до 15-20°C и добавляли 4 н. раствор NaOH (802 г) для доведения pH раствора до значения, равного 7-8. Водную фазу экстрагировали трет-бутилметиловым эфиром (3х143 г) и объединяли органические фазы. К объединенной органической фазе добавляли трет-бутилметиловый эфир (107 г). Раствор промывали водой (2х150 г), что приводило к осаждению твердого осадка, который собирали путем фильтрации.
Гидробромид
Для кристаллизации гидробромида осуществляли замену растворителя трет-бутилметилового эфира на этилацетат.
Фазу трет-бутилметилового эфира концентрировали до 150 г (около половины объема), разбавляли этилацетатом (460 г) и снова концентрировали до 160 г. Полученное масло растворяли в этилацетате (460 г), вводили газообразный HBr (21 г, 0,26 моль) и нагревали раствор с обратным холодильником с получением отдельного желтого масляного слоя. К кипящей смеси (65°C) добавляли метанол (80 г) с получением твердого вещества. Раствор перемешивали и охлаждали до 20-25°C в течение приблизительно 4 ч. Смесь перемешивали в течение ночи и охлаждали до 5°C. Затем к раствору добавляли этилацетат (160 г). Полученный осадок фильтровали на вакуум-фильтре и промывали этилацетатом (10 г) с получением неочищенного продукта в виде желтого твердого вещества.
Выделение и кристаллизация свободного основания
Неочищенный продукт (63 г) растворяли в этилацетате (567 г) и смешивали с насыщенным раствором NaHCO3 (126 г). Смесь перемешивали при 18-25 оC в течение приблизительно 2 ч до тех пор, пока не обнаруживалось дополнительное выделение газа. Значение pH водной фазы поддерживали на уровне 8-9 путем добавления в случае необходимости более насыщенного раствора NaHCO3. Фазы разделяли и концентрировали органическую фазу до приблизительно 1/3 объема. Полученное масло растворяли в этилацетате (100 г) и концентрировали досуха. Масло суспендировали в ацетоне (95 г) и нагревали с обратным холодильником (56±2°C) в течение 1 ч. Смесь охлаждали в течение 3 ч до 36-30°C, выдерживали при этой температуре в течение 2 ч, охлаждали до -10°C и выдерживали при этой температуре в течение 2 ч Полученное твердое вещество фильтровали в вакууме и промывали трет-бутилметиловым эфиром (10 г). Маточный раствор концентрировали, смешивали с ацетоном (41 г), нагревали с обратным холодильником и охлаждали в соответствии с описанной выше процедурой с получением второй порции продукта. Твердые продукты из обеих порций сушили в течение 1-2 ч при 40°С/50 мбар с получением продукта в виде желто-коричневого твердого вещества. Выход: 34 г (30-32%).
ПРИМЕР 10
4-(4-Фторфенил)-2-(4-гидрокси-1-бутинил)-1-(3-фенилпропил)-5- (4-пиридил)имидазол
4-(4-Фторфенил)-2-бром-1-(3-фенилпропил)-5-(4-пиридил)имидазол (30,19 г) смешивали с диизопропиламином (100,56 г). К реакционной смеси по каплям с применением шприца добавляли 3-бутин-1-ол (5,304 г). Затем к реакционной смеси с целью промывки шприца добавляли диизопропиламин (1,810 г), а затем трифенилфосфин (1,805 г), Pd(OAc)2 (0,722 г), порошок железа (0,384 г) и диизопропиламин (78,64 г). В колбе в течение короткого времени обеспечивали азотной подушкой, затем нагревали до 70°C и поддерживали при этой температуре в течение 3 ч.
Описанную выше процедуру повторяли несколько раз. Если по истечении 3 ч оказывалось, что превращение составляло менее 95%, дополнительно добавляли трифенилфосфин (1,805 г) и ацетат палладия (Pd(OAc)2) (0,772 г) и поддерживали температуру до тех пор, пока превращение не достигало 95%.
После окончания реакционную смесь фильтровали для того, чтобы собрать твердый остаток. Отфильтрованный остаток суспендировали в этилацетате (212,17 г) при 40-50°C, фильтровали и выпаривали растворитель досуха. Полученное масло полностью растворяли в первом фильтрате при 70°C. К горячему раствору добавляли воду (148,608 г) и разделяли фазы. Органическую фазу дважды промывали водой (148,608 г) при 70°C. Фазы снова разделяли, промывали органическую фазу насыщенным солевым раствором (148,608 г) и экстрагировали 1 н. HCl (2х146 г). Объединенные фазы HCl повторно экстрагировали этилацетатом (99,07 г). Водную фазу разделяли. К водной фазе по каплям добавляли 25% аммиак (26,948 г) при охлаждении до 5-10°C и при pH 9-10, что приводило к образованию твердого вещества. Суспензию перемешивали в течение приблизительно 45 мин и собирали осадок путем фильтрации. Осадок суспендировали в воде (2х148,61 г) и затем сушили в течение 16 ч при 40°C/50 мбар. Твердое вещество растворяли в смеси этилацетата (412,21 г) и метанола (35,270 г), и смешивали с делоксаном (Deloxan®) (5,00 г). Раствор перемешивали в течение 24 ч при 18-23°C и фильтровали. Отфильтрованный остаток промывали этилацетатом (2х15,34 г). Объединенные маточные растворы и смывы упаривали досуха на роторном испарителе. Остаток растворяли в смеси ТГФ (7,94 г) и толуола (16,0 г) при 70-75°C, медленно охлаждали в течение примерно 2 ч до 18-23°C, что приводило к образованию суспензии. К суспензии добавляли толуол (9,2 г). Твердые вещества суспензии фильтровали на вакуум-фильтре, промывали толуолом (3х1,40 г), а затем гексаном (3х2,02 г). Остаток сушили в течение 16 ч при 50°C/50 мбар с получением продукта в виде не совсем белого твердого вещества. Выход: 20,5 г (70,5%).
Описанную выше процедуру повторяли несколько раз. Перекристаллизация описанного выше остатка приводила к образованию формы B продукта. Перекристаллизация описанного выше остатка из ацетонитрила приводила к образованию формы А продукта.
ПРИМЕР 11
N-(3-Фенилпропил)-N'-[(2,2-диметокси-2-(4-пиридил)-1-(4-фторфенил)этил)]мочевина
К раствору 2,2-диметокси-2-(4-пиридил)-1-(4-фторфенил)этанамина (1,22 г, 4,4 ммоль) в ТГФ (10 мл) добавляли раствор (3-изоцианатопропил)бензола (1,61 г, 10 ммоль) в ТГФ (10 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Реакцию гасили водой (50 мл) и экстрагировали этилацетатом (2х50 мл). Объединенный этилацетатный экстракт промывали водой (50 мл), солевым раствором (50 мл) и сушили над Na2SO4. После удаления растворителя неочищенный продукт очищали колоночной хроматографией с получением продукта в виде светло-коричневого твердого вещества. Выход: 0,83 г (43%). Т.пл.: 162,5-165,5°C.
ПРИМЕР 12
Синтез 4-(4-фторфенил)-2-бром-1-(3-фенилпропил)-5-(4-пиридил)-имидазола
Реакционный сосуд загружали POCl3 (1500,0 г, 9,78 моль). При температуре окружающей среды одной порцией добавляли Br2 (184,9 г, 1,157 моль). Реакционную смесь охлаждали до 10°C и затем в течение 25 мин при энергичном перемешивании добавляли PBr3 (313,0 г, 1,157 моль). Повышали температуру реакционной смеси до 20°C. После добавления продолжали перемешивание в течение дополнительных 1,5 ч, поддерживая температуру в диапазоне 10-20°C. Полученный PBr5 выпадал в осадок в виде желтого твердого вещества. Реакционную смесь нагревали до 25оC и одной порцией добавляли 1,3-дигидро-1-(3-фенилпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол-2-он (150,0 г, 0,386 моль). После добавления реакционную смесь нагревали до приблизительно 30°C и продолжали перемешивание в течение 24 ч. Суспензия становилась темным раствором. POCl3 отгоняли в вакууме при температуре ниже 35°C с получением вязкого масла. Указанное масло добавляли к смеси этилацетата (1000 г) и нашатырного спирта (25 мас.%, 1000 г) в течение приблизительно 1,25 ч при охлаждении. Полученные две фазы разделяли, водную фазу экстрагировали этилацетатом (500 г) и объединенные органические фазы промывали водой (200 г) при 70°C. Органическую фазу концентрировали до приблизительно 30% от начального объема. Затем к нагретой реакционной смеси добавляли триэтиламин (600 г) и удаляли в вакууме дополнительное количество растворителя (приблизительно 150 г), что приводило к кристаллизации желаемого вещества. Реакционную смесь охлаждали до 0°C и перемешивали в течение 12 ч. Продукт отфильтровывали, промывали триэтиламином (50 г) и сушили в вакууме при 40°C с получением неочищенного продукта.
Маточный раствор концентрировали до состояния масла. Затем к маслу добавляли ацетон (25 г), что приводило к выпадению в осадок желаемого вещества. Осадок отфильтровывали, промывали ацетоном (7 г), затем трет-бутилметиловым эфиром (8 г) и сушили в вакууме при 40°C с получением второй порции неочищенного продукта.
Обе порции выделенного продукта суспендировали в смеси триэтиламина (10 г) и ацетона (100 г) при нагревании с обратным холодильником в течение 30 мин, затем охлаждали до 25°C и перемешивали в течение ночи. Осадок отфильтровывали, промывали триэтиламином (25 г), затем ацетоном (10 г) и сушили в вакууме при 40°C с получением указанного в заголовке соединения. Чистота по ВЭЖХ: 99%.
Хотя предшествующее описание раскрывает принципы настоящего изобретения, а примеры предназначены для целей его иллюстрации, необходимо понимать, что осуществление изобретения охватывает все изменения, улучшения и/или модификации, как входящие в объем следующей формулы изобретения и их эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ФУНГИЦИДОВ, И СПОСОБ УНИЧТОЖЕНИЯ ГРИБКОВ | 2000 |
|
RU2234504C2 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ОПОСРЕДУЕМОГО ЦИТОКИНАМИ | 2000 |
|
RU2298008C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2000 |
|
RU2242474C2 |
| СОЕДИНЕНИЯ, ПРЕДСТАВЛЯЮЩИЕ СОБОЙ АЛКИНИЛФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ, ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМИЧЕСКИХ ЗАБОЛЕВАНИЙ И РАССТРОЙСТВ | 2008 |
|
RU2470910C9 |
| 2,6 -ДИГАЛОГЕН-5-АЛКОКСИ-4-ЗАМЕЩЕННЫЕ-ПИРИМИДИНЫ, ПИРИМИДИНКАРБАЛЬДЕГИДЫ И СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2012 |
|
RU2626957C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕРБИЦИДНЫХ ПРОИЗВОДНЫХ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2000 |
|
RU2244715C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ГИДРОКСИ-4-АРИЛ-5-ОКСОПИРАЗОЛИНА С ГЕРБИЦИДНЫМ ДЕЙСТВИЕМ | 1999 |
|
RU2246492C2 |
| ЗАМЕЩЕННЫЕ БИЦИКЛИЧЕСКИЕ ИМИДАЗО-3-ИЛАМИНЫ, ПРИГОДНЫЕ ДЛЯ РЕГУЛЯЦИИ mGluR5-РЕЦЕПТОРА | 2005 |
|
RU2435770C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И ПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ Р38 | 2003 |
|
RU2301233C2 |
| ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 4"-АВЕРМЕКТИНЫ, ОБЛАДАЮЩИЕ ИНСЕКТИЦИДНЫМИ И АКАРИЦИДНЫМИ СВОЙСТВАМИ | 2002 |
|
RU2334755C2 |
Изобретение относится к способу получения тетразамещенных производных имидазола формулы (I)
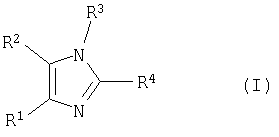
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила); R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот; R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила; R4 представляет
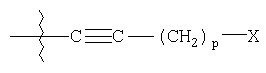
где p является целым числом от 0 до 9; Х представляет гидрокси; или его фармацевтически приемлемых солей, включающий взаимодействие соединения формулы (VIII)
где L1 и L2 независимо выбирают из группы, состоящей из С1-С4алкила, и где R1 и R2 имеют вышеуказанные значения, с соединением формулы (X)
где R3 имеет вышеуказанные значения, с получением соответствующего соединения формулы (XI)
где R1, R2, R3, L1 и L2 имеют вышеуказанные значения, циклизацию соединения формулы (XI) в кислотных условиях при значениях рН менее примерно 7 с получением соответствующего соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения; взаимодействие соединения формулы (XII) с POBr3, PBr5 или со смесью PBr3 и Br2 с получением соответствующего соединения формулы (XIII)
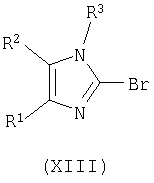
где R1, R2 и R3 имеют вышеуказанные значения; замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV) H-R4 с получением соответствующего соединения формулы (I).
Изобретение также относится к способу получения промежуточных соединений и к кристаллической форме соединения формулы (II)
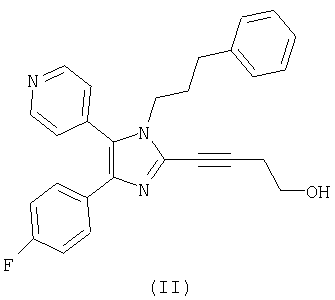
Технический результат - получение нового соединения формулы (II), которое является ингибитором р38-киназы. 14 н. и 6 з.п. ф-лы, 2 табл.
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила;
R4 представляет
где p является целым числом от 0 до 9;
Х представляет гидрокси,
или их фармацевтически приемлемых солей;
включающий взаимодействие соединения формулы (VIII)
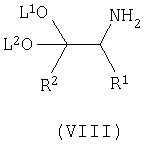
где L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила, и где R1 и R2 имеют вышеуказанные значения,
с соединением формулы (X)
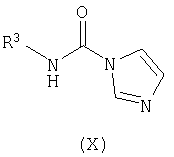
где R3 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (XI)
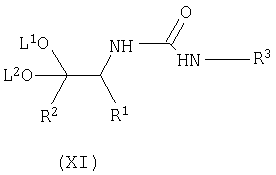
где R1, R2, R3, L1 и L2 имеют вышеуказанные значения,
циклизацию соединения формулы (XI) в кислотных условиях при значениях рН менее примерно 7 с получением соответствующего соединения формулы (XII)
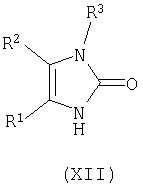
где R1, R2 и R3 имеют вышеуказанные значения;
взаимодействие соединения формулы (XII) с POBr3 или PBr5 с получением соответствующего соединения формулы (XIII)
где R1, R2 и R3 имеют вышеуказанные значения,
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV)

где R4 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (I).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила;
R4 представляет
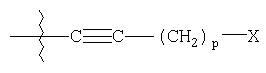
где p является целым числом от 0 до 9;
Х представляет гидрокси;
или их фармацевтически приемлемых солей,
включающий взаимодействие соединения формулы (VIII)
где L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила, и где R1 и R2 имеют вышеуказанные значения,
с соединением формулы (X)
где R3 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (XI):
где R1, R2, R3, L1 и L2 имеют вышеуказанные значения,
циклизацию соединения формулы (XI) в кислотных условиях при значениях рН менее примерно 7 с получением соответствующего соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения,
взаимодействие соединения формулы (XII) со смесью PBr3 и Br2 с получением соответствующего соединения формулы (XIII)
где R1, R2 и R3 имеют вышеуказанные значения,
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV)

где R4 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (I).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот;
R3 выбирают из группы, состоящей из водорода или арилС1- С5алкила;
R4 представляет
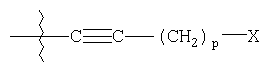
где p является целым числом от 0 до 9;
Х представляет гидрокси,
или их фармацевтически приемлемых солей;
включающий циклизацию соединения формулы (XI)
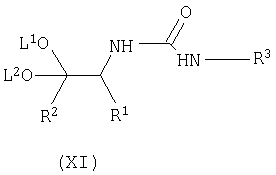
где L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила, и R1, R2 и R3 имеют вышеуказанные значения,
в кислотных условиях при рН менее примерно 7 с получением соответствующего соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения,
взаимодействие соединения формулы (XII) с POBr3 или PBr5 с получением соответствующего соединения формулы (XIII)
где R1, R2 и R3 имеют вышеуказанные значения,
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV)

где R4 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (I).
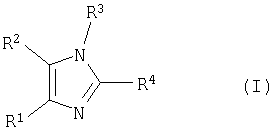
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила;
R4 представляет 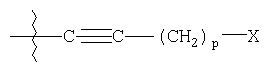
где p является целым числом от 0 до 9;
Х представляет гидрокси,
или их фармацевтически приемлемых солей,
включающий циклизацию соединения формулы (XI)
где L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила, и R1, R2 и R3 имеют вышеуказанные значения, в кислотных условиях при рН менее примерно 7 с получением соответствующего соединения формулы (XII):
где R1, R2 и R3 имеют вышеуказанные значения,
взаимодействие соединения формулы (XII) со смесью PBr3 и Br2 с получением соответствующего соединения формулы (XIII)
где R1, R2 и R3 имеют вышеуказанные значения,
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV)
где R4 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (I).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила;
R4 представляет
где р является целым числом от 0 до 9;
Х представляет гидрокси,
или их фармацевтически приемлемых солей, включающий взаимодействие соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения,
с POBr3 или PBr5 с получением соответствующего соединения формулы (XIII)
где R1, R2 и R3 имеют вышеуказанные значения,
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV)
где R4 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (I).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий азот;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила;
R4 представляет
где р является целым числом от 0 до 9;
Х представляет гидрокси,
или их фармацевтически приемлемых солей, включающий взаимодействие соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения,
со смесью PBr3 и Br2 с получением соответствующего соединения формулы (XIII)
где R1, R2 и R3 имеют вышеуказанные значения,
замещение брома в соединении формулы (XIII) взаимодействием с соединением формулы (XIV)
где R4 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (I).
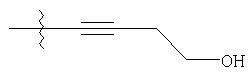
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота;
R3 выбирают из группы, состоящей из водорода или арилС1- С5алкила; и
L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила.
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота; и
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила.
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила; и
L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила,
включающий взаимодействие соединения формулы (VIII)
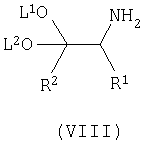
где R1, R2, L1 и L2 имеют вышеуказанные значения,
с соединением формулы (X)
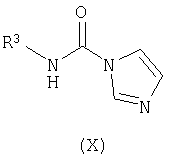
где R3 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (XI).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота; и
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила;
включающий циклизацию соединения формулы (XI)
где L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила, и R1, R2 и R3 имеют вышеуказанные значения,
в кислотных условиях при рН менее примерно 7 с получением соответствующего соединения формулы (XII).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота; и
R3 выбирают из группы, состоящей из водорода или арилС1- С5алкила,
включающий взаимодействие соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения,
с POBr3, PBr5 с получением соответствующего соединения формулы (XIII).
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота; и
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила,
включающий взаимодействие соединения формулы (XII)
где R1, R2 и R3 имеют вышеуказанные значения,
со смесью PBr3 и Br2 с получением соответствующего соединения формулы (XIII).
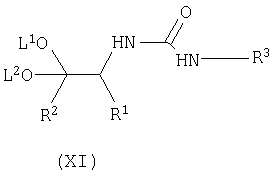
где R1 выбирают из группы, состоящей из фенила, замещенного фенила (где заместители выбирают из галогена или трифторметила);
R2 представляет гетероарил, который обозначает пяти- или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один гетероатом, представляющий атом азота;
R3 выбирают из группы, состоящей из водорода или арилС1-С5алкила; и
L1 и L2 независимо выбирают из группы, состоящей из C1-С4алкила,
включающий взаимодействие соединения формулы (VIII)
где R1, R2, L1 и L2 имеют вышеуказанные значения,
с соединением формулы (XV)
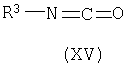
где R3 имеет вышеуказанные значения,
с получением соответствующего соединения формулы (XI).
характеризующаяся следующими пиками порошковой рентгенограммы:
 )
)
| US 5965583 А, 12.10.1999 | |||
| DE 3504677 А, 14.08.1986 | |||
| WO 9931089 А1, 24.06.1999 | |||
| SAKAMOTO Т ЕТ AL, Synthesis, 1992, no.6, р.р.552-554 | |||
| KIM YONG BOON et al, J | |||
| Heterocycl | |||
| Chem., 1994, 31(6), p.p.1653-1656 | |||
| ГОЛОВКО В.В | |||
| и др., Химия гетероциклических соединений, 1986, №10, стр.1339-1342. |

