Изобретение относится к способу получения энантиомерно чистых ингибиторов HMG-CoA редуктазы, стадиям способа и к новым промежуточным соединениям.
Ингибиторы HMG-CoA редуктазы (также называемые ингибиторами β-гидрокси-β-метилглутарил-коэнзим-А редуктазы и также называемые статинами) являются активными агентами, которые могут предпочтительно использоваться для понижения в крови уровня липидов, включая холестерин, и могут использоваться, например, для профилактики или лечения гиперлипидемии и атеросклероза.
Класс ингибиторов HMG-Co-редуктазы включает соединения, имеющие различные структуры. Например, могут быть упомянуты соединения, выбранные из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, питавастатина (ранее итавастатин), правастатина, росувастатина и симвастатина или, в каждом случае, из их фармацевтически приемлемых солей.
Предпочтительными ингибиторами HMG-Co-редуктазы являются агенты, которые присутствуют на рынке, наиболее предпочтительными являются флувастатин, аторвастатин, питавастатин, особенно его кальциевая соль, или симвастатин или его фармацевтически приемлемая соль.
Аторвастатин формулы
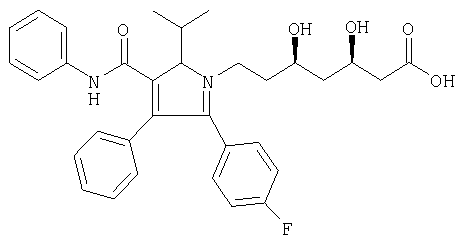
описан и заявлен в US 5273995.
Церивастатин формулы
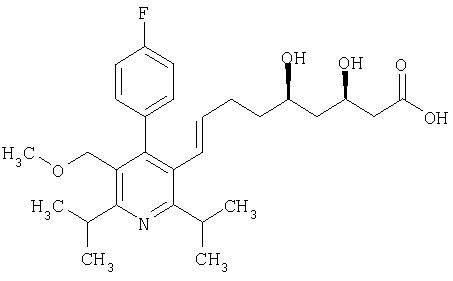
описан и заявлен в US 5177080.
Рацемический флувастатин с син-конфигурацией гидроксигрупп в формуле
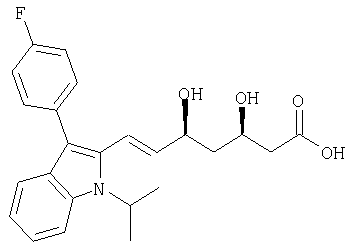
описан и заявлен в US 5345772.
Ловастатин формулы
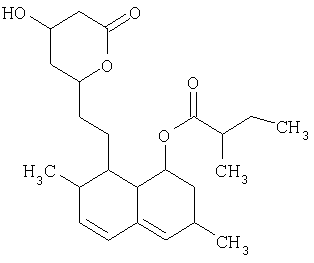
описан и заявлен в US 4231938.
Питавастатин формулы
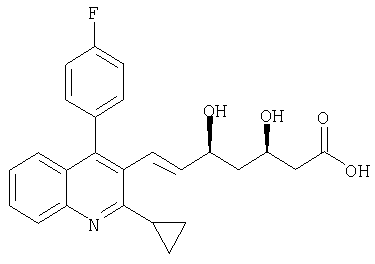
описан и заявлен в US 5856336.
Правастатин формулы
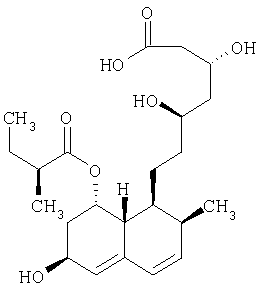
описан и заявлен в US 4410629.
Росувастатин формулы
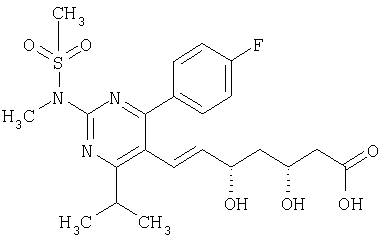
описан и заявлен в US 5260440.
Симвастатин формулы
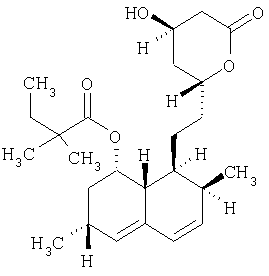
описан и заявлен в US 4444784.
Структуры активных агентов, определенные выше или далее по их названиям генериков или товарных знаков, могут быть обнаружены в текущем издании стандартного сборника "The Merck Index" или из баз данных, например, Patents International или LifeCycle Patents International, соответственно (например, IMS World Publications). Их соответствующее содержание, таким образом, включено в качестве ссылки. Любой специалист в данной области техники может полностью идентифицировать активные агенты и, основываясь на этих ссылках, аналогично может получить и протестировать фармацевтические показания и свойства на стандартных испытательных моделях in vitro и in vivo.
Кислотные представители ингибиторов HMG-Co-редуктазы разрабатываются в виде солей, например, флувастатин в виде натриевой соли и питавастатин в виде кальциевой соли.
Соответствующие активные ингредиенты или их фармацевтически приемлемые соли могут также использоваться в форме сольвата, такого как гидрат, или включая другие растворители, используемые для кристаллизации.
По существу, статины включают циклический основной элемент и элемент боковой цепи формулы
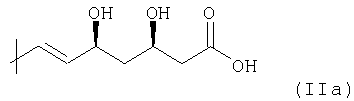
(группа 3,5-дигидроксигепт-6-еновой кислоты), который может образовывать 5 соответствующую группу лактона структурной формулы
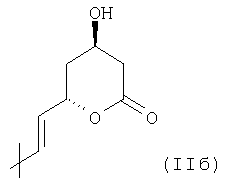 или
или
или 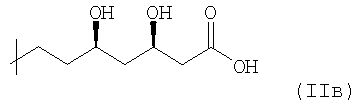
(производное 3,5-дигидроксигептановой кислоты), которая может образовывать соответствующую группу лактона структурной формулы 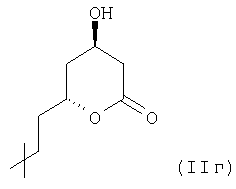 .
.
В указанных элементах боковой цепи (IIa) или (IIв), соответственно, 3,5-син структура диола и R-конфигурация в С-3 являются существенными особенностями, так как соответствующие статины с этим конкретным элементом проявляют самую высокую биологическую активность.
Цель настоящего изобретения состоит в обеспечении энантиоселективного синтеза соединений формулы (I) с высокими выходами и, кроме того, с гарантированным уменьшением экологического загрязнения окружающей среды, что является экономически выгодным, например, благодаря меньшему количеству стадий реакции в последовательности реакции получения соединений формулы I, и приводящим в значительной степени к энантиомерно чистым целевым продуктам и к хорошо кристаллизуемым продуктам. Кроме того, другая цель настоящего изобретения состоит в обеспечении способа, который может быть осуществлен в крупном масштабе и может таким образом использоваться для соответствующего способа производства. Кроме того, существует потребность в устранении любого разделения и расщепления стереоизомеров.
Неожиданно было обнаружено, что способ настоящего изобретения достигает вышеупомянутые цели. Способ относится к энантиоселективному синтезу, использующему по существу так называемую конденсацию Виттига-Водсворта-Эммонса (Виттига-Хорнера) или Виттига посредством химической десимметризации. Например, может быть достигнут энантиомерный избыток (ее) соединения формулы (I) или его соли ≥95%, предпочтительно ≥98% и наиболее предпочтительно ≥99%. Кроме того, может быть легко получен ее ≥99,5%. Кроме того, в соответствии с настоящим изобретением может быть легко достигнут диастереомерный избыток (de)≥95%, предпочтительно ≥98% и наиболее предпочтительно ≥99% для соединения формулы (I) или его соли, полученных в соответствии с настоящим изобретением.
Изобретение относится к способу получения энантиомерно чистой формы ингибитора HMG-CoA редуктазы производного мевалоновой кислоты формулы (I)
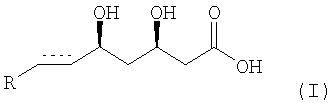
или соли, особенно его фармацевтически приемлемой соли с основанием или его лактона, где
элемент  представляет собой -СН2-СН2- или -СН=СН-, и R представляет собой циклический остаток.
представляет собой -СН2-СН2- или -СН=СН-, и R представляет собой циклический остаток.
Солью соединения формулы (I) является, например, соль с основанием, предпочтительно, его соответствующая фармацевтически приемлемая соль.
Лактон соединения формулы (I) представлен формулой (Ia)
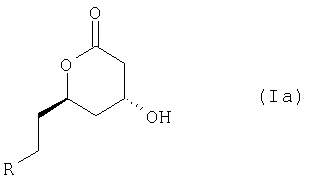 или
или 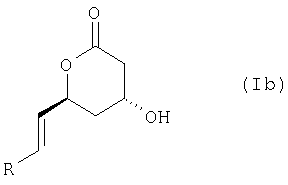
Соответствующий циклический остаток R включает циклический остаток, выбранный из группы, состоящей из
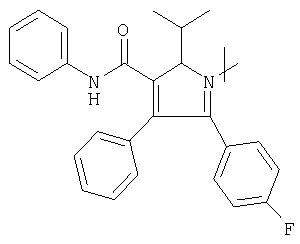 ;
; 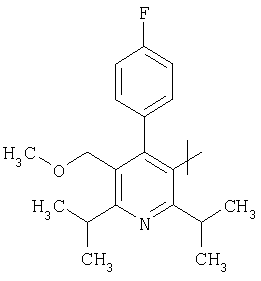 ;
;
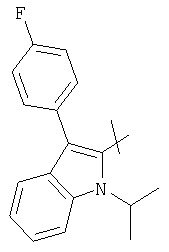 ;
;
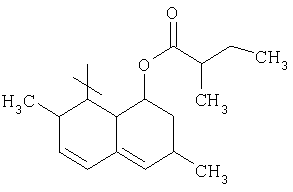 ;
; 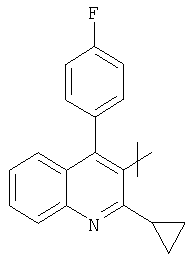 ;
;
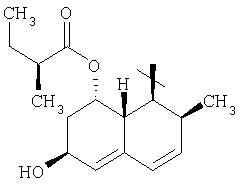 ;
; 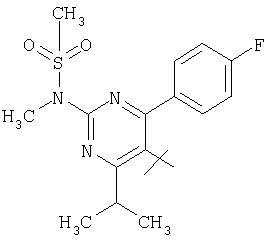 ;
;
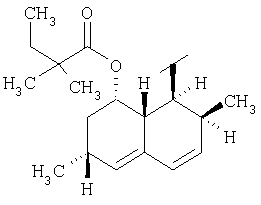 .
.
Многочисленные экспериментальные испытания неожиданно привели к последовательности способа получения, который отвечает вышеупомянутым критериям, проявляя указанные преимущества.
По сравнению со способом, раскрытым в J.Org. Chem. 1991, 56, 3744-3747, было неожиданно обнаружено, что способ в соответствии с настоящим изобретением может быть упрощен, опуская (i) стадию окисления с N2O4 для получения соответствующего N-нитрозамина, (и) гидролиз для получения свободной кислоты и (iii) метилирование для получения метилового эфира.
Было доказано, что последовательность реакции в соответствии с настоящим изобретением, особенно при использовании соответствующего исходного материала и промежуточных соединений, имеющих амидную группу, когда амидный элемент представлен формулой
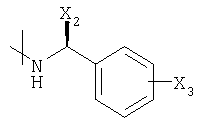 , особенно
, особенно 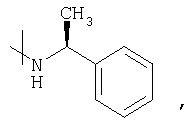
например, вместо применения соответствующего эфира, имеющего спиртовой элемент формулы
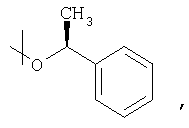
приводит к вышеупомянутым преимуществам.
Кроме того, в недавней публикации [Bioorg. Med. Chem. Lett. 9 (1999) 2977-2982)] был раскрыт улучшенный способ получения питавастатина путем постадийного образования боковой цепи формулы (IIa), что приводит к образованию смеси диастереомеров, которые нужно разделять. Следовательно, одна половина диастереомера не может использоваться. В противоположность указанной процедуре, способ настоящего изобретения явно более экономичный.
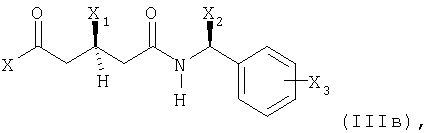
Способ получения энантиомерно чистой формы соединения формулы
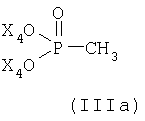
или его соли в соответствии с настоящим изобретениям характеризуется (а) реакцией соединений (IIIa) или (IIIб)
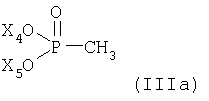 или
или 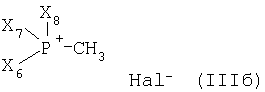
где Х4 и Х5, независимо друг от друга, представляют собой С1-С7-алкил или фенил-С1-С7-алкил;
Х6, Х7 и Х8, независимо друг от друга, представляют собой фенил, который является незамещенным или замещен одним или несколькими заместителями, выбранными из группы, состоящей из С1-С7алкила, гидрокси, С1-С7алкокси, С2-С8алканоилокси, галогена, нитро, циано и CF3; и Hal- представляет собой галогеновый анион;
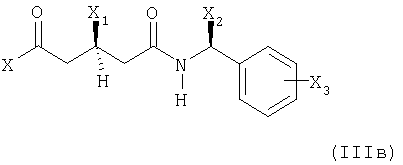
с металлированным алканом с образованием соответствующего илида и затем реакцией полученного илидного промежуточного соединения с соединением формулы
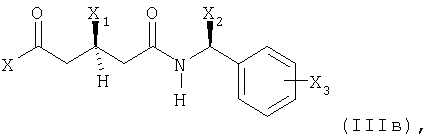
где
X представляет собой этерифицированный гидрокси, этерифицированный гидрокси или незамещенный или моно- или дизамещенный амино;
X1 представляет собой защищенный гидрокси;
Х2 представляет собой С1-С7алкил и
Х3 представляет собой водород или один или несколько заместителей, например, выбранных из группы, состоящей из С1-С7алкила, гидрокси, С1-С7алкокси, С2-С8алканоилокси, галогена, нитро, циано и CF3;
(б) необязательно, при необходимости, преобразованием полученного соединения формулы (IIIг)
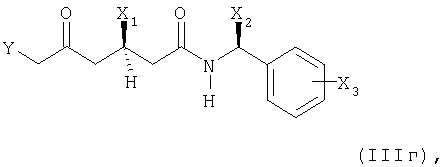
в котором X1, X2 и Х3 имеют вышеупомянутые значения и Y представляет собой группу формулы (Х4O)(Х5O)Р(=O)- или (X6)(X7)(X8)P+Hal- и Х4, Х5, Х6, Х7, X8 и Hal- имеют вышеупомянутые значения;
в соединение формулы (IIIд)
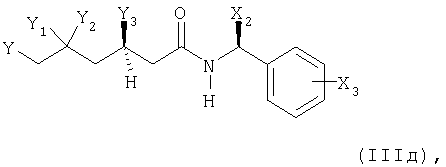
в котором Х2, Х3 и Y имеют вышеупомянутые значения и где Y1 представляет собой гидрокси или защищенный гидрокси, и Y2 представляет собой водород, и Y3 представляет собой гидрокси или защищенный гидрокси, и Y1 и Y3 образуют син-диольную конфигурацию; или в котором
Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk, являющийся C1-С7алкилиденом; и Y2 представляет собой водород, и Y1 и Y3 образуют син-диольную конфигурацию;
(в) реакцией соединения формулы (IIIд),
в которой Х2, Х3 и Y имеют вышеупомянутые значения и в которой
Y1 представляет собой гидрокси или защищенный гидрокси, и Y2 представляет собой водород, и Y3 представляет собой гидрокси или защищенный гидрокси, и Y1 и Y3 образуют син-диольную конфигурацию; или в которой
Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk, являющийся C1-С7алкилиденом; и Y2 представляет собой водород, и Y1 и Y3 образуют син-диольную конфигурацию; или в которой
Y1 и Y3 вместе представляют собой оксогруппу и Y3 представляет собой защищенный гидроксил (соответствующий соединениям формулы (IIг));
с альдегидом формулы (IIIe) R-CH(=O), приводя к получению соединения формулы (IIIж)
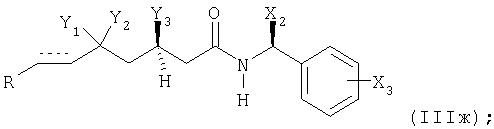
в которой R, Х2, Х3, Y1, Y2 и Y3 и элемент имеют вышеупомянутые значения;
при необходимости, восстанавливая соответствующие соединения формулы (IIIж), в которой элемент представляет собой -СН=СН-, с получением соединения, в котором указанный элемент представляет собой -СН2-СН2-; и
(г) если получают соединение формулы (IIIж), в котором один из Y1 и Y3 представляет собой защищенный гидрокси и другой представляет собой гидрокси, или оба Y1 и Y3 представляют собой защищенные гидрокси, и в каждом случае Y2 представляет собой водород; и Y1 и Y3 образуют син-конфигурацию; или
Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk, являющийся C1-С7алкилиденом, и Y1 и Y3 образуют син-конфигурацию; и Y2 представляет собой водород; или
удалением защиты гидроксигруппы(групп) с получением соединения формулы
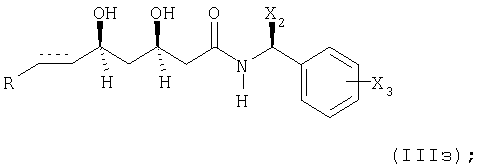 или
или
при необходимости, восстановлением соответствующего соединения формулы (IIIз), в котором элемент представляет собой -СН=СН-, с получением соединения, в котором указанный элемент представляет собой -СН2-СН2-;
(д) если получают соединение формулы (IIIж), в котором Y1 и Y2 вместе образуют оксогруппу =O; и Y3 представляет собой защищенный гидрокси (X1); преобразованием указанного соединения формулы (IIIж) в соединение формулы (IIIи)
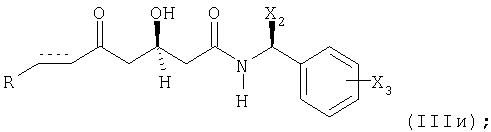
удалением защитной гидроксигруппы;
в которой R, X2, Х3 и элемент имеют вышеупомянутые значения; и последующим восстановлением указанного соединения формулы (IIIи) в соединение формулы (IIIз);
(е) гидролизом соединения формулы (IIIз) в соединение формулы (I) или его соли и
(ж) выделением полученного соединения формулы (I) или его соли;
и, при необходимости, преобразованием полученной свободной кислоты формулы (I) в его соль или в лактон формулы (Ia) или (Iб), соответственно, или преобразованием полученного лактона формулы (Ia) или (Iб) в кислоту формулы (I) или ее соль.
Наиболее предпочтительно, в соединениях формул (IIIв), (IIIг), (IIIд), (IIIж), (IIIз) и (IIIи), в каждом случае X2 представляет собой метил и Х3 представляет собой водород.
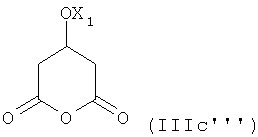
В соответствии с настоящим способом, описанным выше и далее, может быть получен любой из энантиомеров, например, используя соединение формулы (IIIc"") или ее энантиомер для стадии десимметризации. Кроме того, используя рацемическую смесь соединения формулы (IIIc""), могут быть получены рацемические формы упомянутых ингибиторов HMG-CoA редуктазы.
Используемые выше и далее общие термины имеют следующие значения, если не определено иначе.
Этерифицированный гидрокси представляет собой, например, С1-С7алкокси, ар-С1-С7алкокси, С3-С8пиклоалкокси, С3-С8-циклоалкил-С1-С7алкокси.
Этерифицированный гидрокси представляет собой, например, R-CO-O, ароилокси, С2-С8-алканоилокси или ар-С2-С8-алканоилокси.
В моно- или дизамещенном амине аминогруппа является монозамещенной или, независимо друг от друга, дизамещенной заместителем, выбранным из группы, состоящей из С1-С7алкокси, С1-С7алкила, ар-С1-С7алкила, С3-C8-циклоалкила, С3-С8-циклоалкил-С1-С7алкила.
С1-С7Алкокси представляет собой, например, метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, изобутилокси, втор-бутилокси, трет-бутилокси или соответствующий пентилокси, гексилокси, или гептилокси остаток. С1-С4алкокси является предпочтительным. Метокси является особенно предпочтительным.
С1-С7Алкил представляет собой, например, остаток метила, этила, н-пропила, изопропила, н-бутила, изобутила, втор-бутила, трет-бутила или соответствующего пентила, гексила или гептила. С1-С4алкил, особенно метил, является предпочтительным.
С3-С8циклоалкокси и С3-С8циклоалкокси в С3-С8циклоалкокси-С1-С7алкиле являются предпочтительно С3-С8циклоалкокси, например, циклопропокси, циклопентокси или циклогексилокси.
С3-С8циклоалкил представляет собой, в частности, С3-С8циклоалкил, такой как циклопропил, циклобутил, циклопентил или циклогексил. Циклопропил является особенно предпочтительным.
С3-С8алканоил в С3-С8алканоилокси представляет собой, в частности, ацетил, пропионил, бутирил, изобутирил или пивалоил. С3-С8алканоил является предпочтительным.
Галоген представляет собой, в частности, галоген с атомным номером включительно до 35, то есть фтор, хлор или бром, и в более широком смысле включают йод. Фтор или хлор являются предпочтительными.
Фенил-С1-С7алкил представляет собой, в частности, фенил-С1-С4алкил, такой как бензил или 1- или 2-фенэтил.
С1-С7Алкилиден представляет собой, в частности, метилен, этилиден, 1,1-или 2,2-пропилиден, а также 1,1- или 2,2-бутилиден или 1,1-, 2,2- или 3,3-пентилиден. С2-С5алкилиден является предпочтительным.
Защищенный гидрокси (Х2 и/или Х4) представляет собой силилокси, этерифицированный гидрокси, тетрагидропиранилокси. Силилокси представляет собой, например, три-С1-С7алкилсилилокси, особенно трет-бутилдиметилсилилокси.
С1-С7Алкилен представляет собой предпочтительно С1-С4алкилен, например, метилен, 1,2-этилен, 1,2- или 1,3-пропилен, а также включает С2-С7алкилиден, предпочтительно С2-С4алкилен, например, 1,1-этилен, 1,1- или 2,2-пропилиден. Наиболее предпочтительным является 2,2-пропилиден.
Арильный остаток (ар) представляет собой предпочтительно карбоциклический арил, такой как фенил, бифенилил или нафтил, или гетероциклический арил, такой как пиридил. Соответственно может быть незамещенным или замещенным одним или несколькими, например, двумя или тремя, остатками, например, выбранными из группы, состоящей из С1-С7алкила, гидрокси, С1-С7алкокси, С2-С8алканоилокси, галогена, нитро, циано и CF3.
Реакции, описанные в вариантах выше и ниже, проводятся, например, в отсутствии или обычно в присутствии подходящего растворителя или разбавителя или их смеси, реакция, при необходимости, проводится с охлаждением, при комнатной температуре или при нагревании, например, в температурном диапазоне от -80°С до температуры кипения реакционной среды, предпочтительно приблизительно от -10° до +200°С, и, при необходимости, в закрытом сосуде, под давлением, в атмосфере инертного газа и/или при безводных условиях.
Стадия (а):
В реакции стадии (а) реакцию соединения формулы (IIIa) или (IIIб), соответственно, с металлированным алканом проводят в инертном растворителе, таком как эфир, предпочтительно тетрагидрофуран, и при низких температурах, например, от -78° до 0°С, предпочтительно при -78°С. Последующее добавление соединения формулы (IIIв) производят при тех же условиях реакции, предпочтительно в растворителе тетрагидрофуране и при -78°С.
Предпочтительный Х представляет собой С1-С7алкокси, особенно метокси или этокси, или N-С1-С7алкил-Н-С1-С7алкоксиамино, наиболее предпочтительно N-метил-N-метоксиамино. Соответствующие N-C1-C7алкил-N-C1-С7алкоксиаминопроизводные являются новыми.
Соответственно, настоящее изобретение также относится к соединению формулы (IIIв), в котором Х представляет собой N-C1-C7алкил-N-C1-С7алкоксиамино, наиболее предпочтительно N-метил-N-метоксиамино.
Метилированными алканами являются, например, алканы щелочных металлов, такие как бутиллитий или гексиллитий и т.д. Предпочтительным металлированным алканом является бутиллитий.
Стадия (б)
Х4 и Х5, в каждом случае, предпочтительно представляют собой C1-С7алкил, особенно С1-С4алкил, наиболее предпочтительно метил или этил.
Х6, Х7 и Х8, в каждом случае представляют собой, предпочтительно, фенил.
Галид Hal- является предпочтительно хлоридом, а также бромидом и йодидом.
Предпочтительной является реакция с соединением формулы (IIIa).
Для получения соединения формулы (IIIд), в котором Х2, Х3 и Y имеют вышеупомянутые значения, и в котором Y1 представляет собой гидрокси или защищенный гидрокси, и Y2 представляет собой водород, и Y3 представляет собой гидрокси или защищенный гидрокси, и Y1 и Y3 образуют син-диольную конфигурацию; соответствующее соединение формулы (IIIд), в котором Y1 и Y2 вместе образуют оксогруппу, восстанавливают подходящим восстановительным агентом.
Восстановление осуществляют соответствующим восстановительным агентом, например, каталитическим гидрированием в присутствии катализатора гидрирования, например катализатора рутения, такого как (Ru(cod)(nu-3-(2-метилалли))2, восстановлением гидридом, например гидридом, который, при необходимости, может быть комплексным, таким как гидрид, образованный из элемента 1-ой и 3-ей основной группы периодической таблицы элементов, например, боргидрид или алюмогидрид, например, литийборгидрид, литийалюмогидрид, диизобутилалюмогидрид (может быть необходима дополнительная стадия восстановления, используя цианоборгидрид щелочного металла, такой как цианоборгидрид натрия), а также биборан.
Предпочтительным восстановительным агентом является, например, гидрид, например боргидрид щелочного металла, особенно натрий боргидрид, предпочтительно в присутствии ди-С1-С7алкил-С1-С7алкоксиборана, наиболее предпочтительно диэтилметоксиборана.
Восстановление осуществляют в инертном растворителе, таком как эфир, предпочтительно тетрагидрофуран, и при низких температурах, например от -78° до 0°С, предпочтительно при -78°С. Для расщепления соответствующего борного эфира реакционную смесь затем окисляют окислительным агентом, таким как пероксид, особенно, перекись водорода. Окисление осуществляют в инертном растворителе, таком как нитрил, предпочтительно ацетонитрил, и в температурном диапазоне, например, от 0°С до температуры кипения растворителя, предпочтительно в диапазоне от 20° до 50°С.
При необходимости в полученном соединении формулы (IIIд), в котором Y1 представляет собой гидрокси и Y2 представляет собой водород, гидроксигруппа Y1 является защищенной, например, реакцией с галидом, например, галидом формулы Y1-Hal и Hal является галогеном, особенно, хлорид, бромид или йодид.
При необходимости, в полученном соединении формулы (IIIд), в котором Y1 представляет собой гидрокси и Y2 представляет собой водород, защищенную гидроксигруппу X1 удаляют, например, обработкой сильной кислотой, такой как минеральная кислота, например Н3PO4. Предпочтительно, этерифицированную гидроксигруппу или силилоксигруппу отщепляют обработкой кислотой.
Для получения соединения формулы (IIIд), в котором Х2, Х3 и Y имеют определенное выше значение и в котором Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk является С1-С7алкилиденом; и Y2 представляет собой водород, и Y1 и Y3 образуют син-диольную конфигурацию; соответствующее соединение формулы (IIIд), в котором Y1 и Y3, каждый, представляет собой гидрокси и Y2 представляет собой водород, и Y1 и Y3 образуют син-диольную конфигурацию; этерифицируют обработкой, например, соединением формулы Hal-Alk-Hal, например, в присутствии основания.
Стадия (в):
Стадию (в) осуществляют в присутствии основания, такого как алкан щелочного металла, особенно бутиллития, или гидрида, например, гидрида натрия или карбоната щелочного металла, особенно К2СО3 или Cs2СО3, или объемного амина, такого как 1,5-диазабицикло[4,3,0]нон5-ен (DBN) и 1,8-диазабицикло[5,4,0]ундек7-ен (DBU), особенно DBU в присутствии хлорида лития или гидроксида щелочного металла, особенно КОН. Образование соединения формулы (IIIж) осуществляют в инертном растворителе, таком как тетрагидрофуран, или в протонном растворителе, таком как спирт, предпочтительно изопропанол или этанол, и в температурном диапазоне, например, от -78°С до температуры кипения растворителя, предпочтительно от комнатной температуры до 45°С, в зависимости от используемого основания и растворителя.
Предпочтительными соединениями формулы (IIIг) являются соединения, в которых Y представляет собой группу формулы (Х4O)(Х5O)Р(=O)- и Х4 и Х5, в каждом случае особенно представляют собой, С1-С4алкил, предпочтительно метил или этил.
Стадия (г):
Для получения соединения формулы (IIIз) соединение формулы (IIIж), в котором один из Y1 и Y3 является защищенным гидрокси и другой представляет собой гидрокси или оба Y1 и Y3 являются защищенными гидрокси и в каждом случае Y2 представляет собой водород; и Y1 и Y3 образуют син-конфигурацию; или
Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk является C1-С7алкилиденом и Y1 и Y3 образуют син-конфигурацию; и Y2 представляет собой водород; используется в качестве исходного материала, и защиту гидроксигруппы (групп) удаляют, или -O-Alk-O-группу расщепляют обработкой, например сильной кислотой, такой как минеральная кислота, например гидрогалогеновая кислота, особенно HCl, или фосфорная кислота, особенно Н3PO4.
Если защищенным гидрокси является соответствующая силилоксигруппа, ее можно расщепить фторидной солью, например, тетрабутиламмонийфторидом, или кислотой, такой как минеральная кислота, например, гидрогалогеновая кислота или фосфорная кислота, особенно Н3PO4. Гидроксизащитная группа расщепляется в инертном растворителе, таком как нитрил, предпочтительно ацетонитрил, и в температурном диапазоне, например, от -78°С до температуры кипения растворителя, предпочтительно в диапазоне от 0° до 50°С.
Предпочтительной гидроксизащитной группой является трет-бутилдиметилсиланилоксигруппа, которая расщепляется с помощью минеральной кислоты, например H2SO4, HF, Н3PO4, особенно HCl.
Стадия (д):
Для получения соединения формулы (IIIз) соединение формулы (IIIж), в котором Y1 и Y2 вместе образуют оксогруппу и Y3 представляет собой защищенный гидрокси; используется в качестве исходного материала и защитную группу Y3 удаляют, например, обработкой сильной кислотой, такой как минеральная кислота, например, гидрогалогеновая кислота или фосфорная кислота, особенно Н3PO4.
Если защищенный гидрокси представляет собой соответствующую силилоксигруппу, ее можно расщепить фторидной солью, например тетрабутиламмонийфторидом или кислотой, такой как минеральная кислота, например гидрогалогеновая кислота или фосфорная кислота, особенно Н3PO4. Гидроксизащищенная группа расщепляется в инертном растворителе, таком как нитрил, предпочтительно ацетонитрил, и в температурном диапазоне, например, от -78°С до температуры кипения растворителя, предпочтительно в диапазоне от 0° до 50°С.
Предпочтительная гидроксизащитная группа представляет собой трет-бутилдиметилсиланилоксигруппу, которая расщепляется с помощью минеральной кислоты, например H2SO4, HF, Н3PO4, особенно HCl.
Полученное соединение формулы (IIIж), в котором Y1 и Y3 вместе образуют оксогруппу, и Y3 представляет собой гидрокси, восстанавливают подходящим восстановительным агентом, например, каталитическим гидрированием в присутствии катализатора гидрирования, например, катализатора рутения, такого как (Ru(cod)(nu-3-(2-метилалли))2, восстановлением гидридом, например, гидридом, который, при необходимости, может быть комплексным, таким как гидрид, образованный из элемента 1-ой и 3-ой основной группы периодической таблицы элементов, например боргидрид или алюмогидрид, например литийборгидрид, литийалюмогидрид, диизобутилалюмогидрид (может быть необходима дополнительная стадия восстановления, используя цианоборгидрид щелочного металла, такой как цианоборгидрид натрия), а также диборан.
Предпочтительным восстановительным агентом является, например, гидрид, например боргидрид щелочного металла, особенно натрий боргидрид, предпочтительно в присутствии ди-С1-С7алкил-С1-С7алкоксиборана, наиболее предпочтительно диэтилметоксиборана.
Восстановление осуществляют в инертном растворителе, таком как эфир, предпочтительно тетрагидрофуран, и при низких температурах, например, от -78° до 0°С, предпочтительно при -78°С. Для расщепления соответствующего борного эфира реакционную смесь затем окисляют окислительным агентом, таким как пероксид, особенно, перекись водорода. Окисление осуществляют в инертном растворителе, таком как нитрил, предпочтительно ацетонитрил, и в температурном диапазоне, например, от 0°С до температуры кипения растворителя, предпочтительно в диапазоне от 20° до 50°С.
Стадия (е):
Стадию гидролиза (е) осуществляли, например, обработкой амида формулы (IIIж) сильным основанием, таким как гидроксид щелочного металла, предпочтительно NaOH или Ca(OH)2, и подкисляя полученную реакционную смесь.
Стадия (ж):
Стадию выделения (ж) соединения формулы (I) осуществляли в соответствии с обычными методами выделения, такими как кристаллизация полученного соединения формулы (I) из реакционной смеси или хроматографией реакционной смеси.
Исходный материал формулы (IIIв) может быть получен, например, этерификацией или амидированием соединения формулы
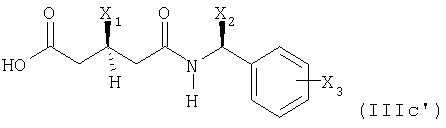
известным способом.
Кроме того, настоящее изобретение относится к способу получения соединения формулы
в котором Х представляет собой N-С1-С7алкил-N-С1-С7алкоксиамино, предпочтительно, N-метил-N-метоксиамино,
особенно формулы
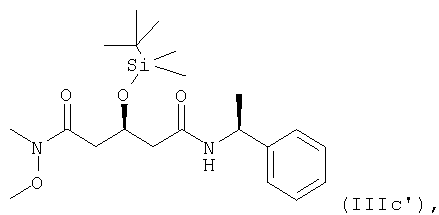
где указанный способ включает реакцию соединения формулы
в котором X1 представляет собой защищенный гидрокси, особенно трет-бутилдиметилсилилокси,
особенно 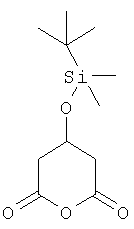 (Соединение (1)),
(Соединение (1)),
с соединением формулы
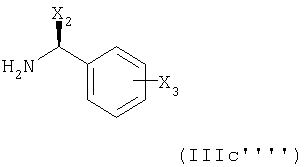
в котором Х2 и Х3 имеют указанные выше значения, в присутствии стерически затрудняющего амина или предпочтительно в присутствии по крайней мере двух эквивалентов указанного выше 1-фенетиламина, и последующее амидирование N-С1-С7алкил-N-С1-С7-алкоксиамином, особенно N-метилом-N-метоксиамином.
Стерически затрудняющий амин представляет собой, например, N-C1-С7алкил-N-С1-С7-алкоксиамино, особенно N-этилдиизопропиламин.
Способ получения исходного материала формулы (IIIв), особенно соединения формулы (IIIв), в котором Х представляет собой N-С1-С7алкил-N-С1-С7-алкоксиамино, особенно N-метил-N-метоксиамино, является аналогичным объекту настоящего изобретения. Может быть достигнута существенно более высокая селективность.
Соединение формулы (IIIв), в котором Х представляет собой N-C1-С7алкил-N-С1-С7-алкоксиамино, особенно N-метил-N-метоксиамино, является аналогичным объекту настоящего изобретения.
Кроме того, настоящее изобретение также относится к стадии реакции (а), особенно при использовании соединения формулы (IIIв), в котором Х предпочтительно представляет собой N-С1-С7алкил-N-С1-С7-алкоксиамино, наиболее предпочтительно N-метил-N-метоксиамино. При применении этого метода не наблюдается никакого существенного β-отщепления защищенной гидроксигруппы X1, особенно ввиду присутствия фенилэтиламида, который стабилизирует защищенную гидроксигруппу, предотвращая β-отщепление и приводя к высокой селективности для реакции замещения.
При использовании соответствующих диэфиров из уровня техники известно, что β-отщепление происходит как побочная реакция и селективность значительно снижается.
Кроме того, настоящее изобретение также относится к стадии реакции (б), особенно при использовании соединения формулы (IIIв), в котором Х предпочтительно представляет собой N-С1-С7алкил-N-С1-С7-алкоксиамино, наиболее предпочтительно N-метил-N-метоксиамино. Здесь аналогично не наблюдалось никакого β-отщепления и может быть достигнута значительная селективность реакции.
Настоящее изобретение, кроме того, относится к соответствующим соединениям формулы (IIIг), особенно соединениям, в которых X1 представляет собой силилокси, предпочтительно трет-бутилдиметилсилилокси.
Настоящее изобретение, кроме того, относится к соответствующим соединениям формулы (IIIд), особенно соединениям, в которых Y3 представляет собой силилокси, предпочтительно трет-бутилдиметилсилилокси.
Кроме того, настоящее изобретение также относится к стадии реакции (в), особенно при использовании соединения формулы (IIIд), в котором Y3 представляет собой силилокси, предпочтительно трет-бутилдиметилсилилокси. Здесь аналогично не наблюдалось никакого β-отщепления и может быть достигнута значительная селективность реакции.
Соединение формулы (IIIд) предпочтительно представлено следующими формулами
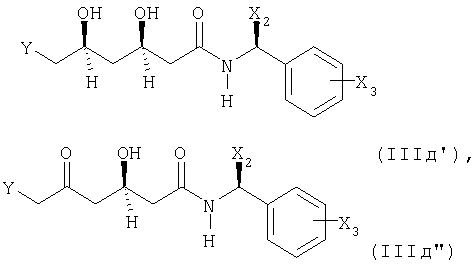
или
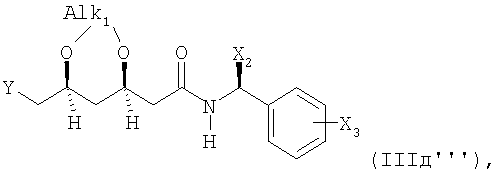
где Alk1 представляет собой предпочтительно 1-метил-1,1-этилиден.
Настоящее изобретение аналогично относится к соединению формул (IIIд'), (IIIд'') и (IIIд''') и к их получению.
Настоящее изобретение, кроме того, относится к соответствующим соединениям формулы (IIIж), особенно соединениям, в которых Y3 представляет собой силилокси, предпочтительно трет-бутилдиметилсилилокси.
Настоящее изобретение аналогично относится к новому соединению, как описано в части рабочих примеров.
Настоящее изобретение аналогично относится к конкретным продуктам, непосредственно полученным последовательностью способа или отдельным стадиям способа, особенно соответствующим продуктам, которые находятся по существу в энантиомерно чистой форме.
Преобразование кислоты формулы (I) в соль осуществляли известным способом. Таким образом, например, соль соединений формулы I с основанием получали обработкой основанием. Соли могут быть преобразованы в свободные соединения общепринятыми способами, и соли с основанием могут быть преобразованы, например, обработкой подходящим кислотным агентом с получением свободной кислоты.
Преобразование кислоты формулы (I) в соответствующий лактон формулы (Ia) или (Iб), соответственно, осуществляли в присутствии кислоты, предпочтительно минеральной кислоты, в подходящем растворителе, например, протонном или апротонном, таком как этанол или ацетонитрил. В зависимости от кислоты, преобразование осуществляли в температурном диапазоне, например, от -78° до температуры кипения растворителя. Наиболее предпочтительно используется Н3PO4 в ацетонитриле при 60°С.
Преобразование лактона формулы (Ia) или (Iб), соответственно, в соль кислоты формулы (I) осуществляли, например, в смеси протонного растворителя, например, этанола, и воды, используя гидроксид щелочного металла, такой как LiOH, NaOH или Са(ОН)3. Альтернативно, лактон может быть гидролизован, используя гидроксид щелочного металла, такой как LiOH, NaOH, и полученная соль может быть преобразована в кальциевую соль кислоты питавастатина добавлением водного раствора CaCl2 в воде.
Вариант способа в соответствии с настоящим изобретением включает прямое образование соединения лактона формулы (I). Образование указанного лактона может быть осуществлено обработкой соединения формулы (IIIз) кислотой, такой как минеральная кислота, предпочтительно Н3PO4.
Способ получения соединений формулы (I) и их солей может быть, например, иллюстрирован с помощью следующей схемы реакции получения питавастатина:
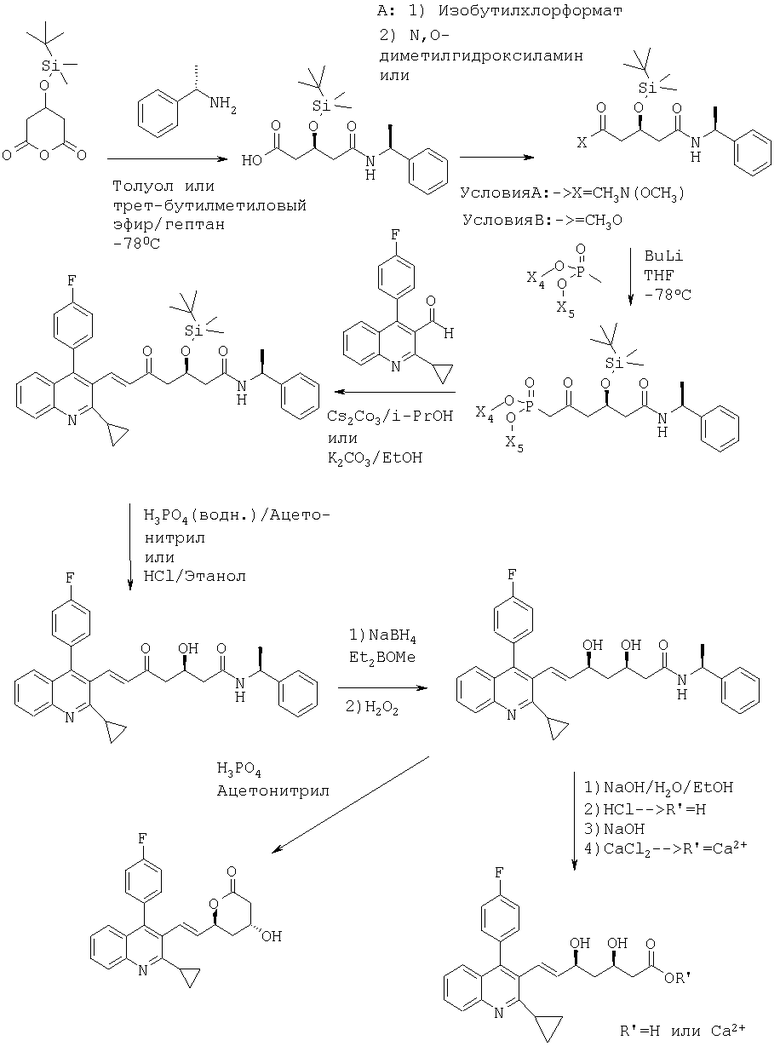
Рабочие примеры:
Получение исходных соединений
Соединение(1)
3-(трет-Бутилдиметилсилилокси)глутаровый ангидрид может быть приобретен от Aldrich.
3-(трет-Бутилдиметилсилилокси)глутаровый ангидрид может быть получен следующим образом:
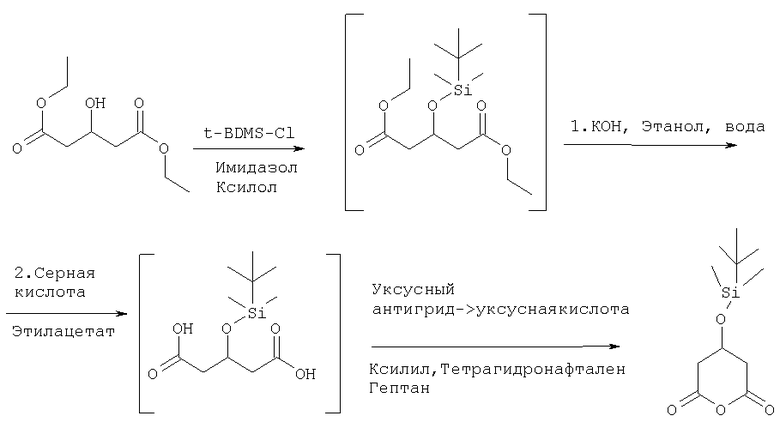
4-Горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником наполняли диэтиловым эфиром 3-гидроксиглутаровой кислоты (107,5 г, 0,5 моль), имидазолом (44,2 г 0,6 моль) и ксилолом (200 мл). Полученную смесь нагревали до 70-80°С и по каплям добавляли трет-бутилдиметилхлорсилан (84,1 г, 0,55 моль), растворенный в ксилоле (100 мл), в течение 30-40 минут. Перемешивание затем продолжали в течение от 3 до 6 часов при 70-80°С. Впоследствии эмульсию охлаждали до комнатной температуры и добавляли воду (200 мл). Находящуюся внизу водную фазу удаляли и оставшуюся органическую фазу снова промывали водой (100 мл). Затем к органической фазе добавляли этанол (95%, 200 мл) и раствор гидроксида калия (119,8 г 49,2%, 1,05 моль). Перемешивание продолжали в течение приблизительно 10-20 часов при 15-25°С. Затем к жидкому раствору добавляли воду (1200 мл) и этилацетат (300 мл). Полученную эмульсию охлаждали до 0-15°С и медленно при 0-15°С добавляли серную кислоту (приблизительно 230 мл 20% раствора в воде), для получения значения рН 3,0-3,5 (подкисление должно проводиться без прерывания, так как промежуточная монокалиевая соль очень нестабильна и может разлагаться). Эмульсию затем нагревали до 15-20°С и удаляли низший водный слой. Оставшуюся органическую фазу промывали водой (200 мл). Добавляли тетрагидронафтален (100 мл) и воду (200 мл). После небольшого перемешивания водную фазу снова удаляли и оставшуюся органическую фазу упаривали в вакууме на 1/2 (200-60 мбар) при 60-70°С. Затем добавляли уксусный ангидрид (103,5 г, 1,0 моль) в течение 20-40 минут при 55-65°С и раствор перемешивали в течение 2-4 часов. Раствор снова упаривали в вакууме (80°С, 100-20 мбар). Затем медленно добавляли гептан (600 мл) при 30-40°С для кристаллизации продукта. Жидкий раствор охлаждали до -15°С, перемешивали в течение приблизительно 1 часа и кристаллы собирали, промывали гептаном (100 мл) и высушивали в вакууме (60°С, 10-20 мбар). Розовые подобные песку кристаллы, tпл 58-68°С (частично), затем 80-82°С, использовали на следующей стадии.
Соединение (2)
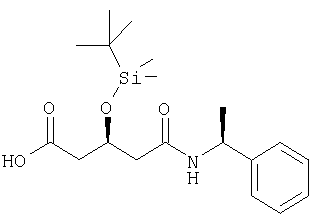
(3S,1'S)-3-[(трет-Бутилдиметилсилил)окси]-5-[(1-фенилэтил)амино]-5-оксопентановая кислота может быть получена в соответствии с процедурой, описанной в Donald. S.Karanewsky, Mary F.Malley и Jack Z.Gougoutas, J.Org.Chem. 1991, 56, 3744-3747, соответствующий способ при этом включен в настоящее описание в качестве ссылки.
Процедура Karanewsky и др. может быть улучшена для достижения более высокой диастереоселективности и более высоких выходов желаемого диастереомера путем замены триэтиламина на более стерически затрудняющее основание, особенно N-этилдиизопропиламин. Указанная замена приводит к лучшим соотношениям и выходу желаемого диастереомера по сравнению с ненужным. Когда триэтиламин заменен на (S)-(-)-1-фенэтиламин в качестве основания, желаемый диастереомер получали с более высоким выходом по сравнению с описанным способом Karanewsky и др. Это означает, что использовали по крайней мере 2 моля (S)-(-)-1-фенэтиламина, один моль в качестве субстрата и по крайней мере один моль в качестве основания. Лучшие соотношения и выхода желаемого диастереомера достигались, если второй мольный эквивалент (S)-(-)-1-фенэтиламина использовали в качестве основания вместо третичного амина. (3S,1'S)-3-[(трет-бутилдиметилсилил)окси]-5-[(1-фенилэтил)амино]-5-оксопентановая кислота, полученная в соответствии с этими процедурами, имеет обычно >98% de и >98% ее.
Улучшенный метод описан ниже.
4-Горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником наполняли ангидридом (соединение 1) (30 г, 0,123 моль), трет-бутилметиловым эфиром (210 мл) и гептаном (120 мл). Чистый раствор охлаждали до -78°С, получая мутный раствор. Медленно добавляли (S)-(-)-1-фенэтиламин (31,3 г, 0,258 моль, 99,6% ее) в гептане (120 мл) в течение 60-90 минут при -78/-75°С. Чистый вязкий раствор перемешивали в течение приблизительно 2 часов. Затем температуру поднимали до 20-25°С. Добавляли воду (100 мл) и фосфорную кислоту (приблизительно 100 мл 20% раствора в воде), таким способом поддерживая температуру между 30-35°С и доводя значение рН до 2,5-3,5. Смесь затем кипятили с обратным холодильником (70°С) и перемешивали в течение приблизительно 30 минут. Затем смесь охлаждали до 0-5°С и отфильтровывали для получения белых кристаллов, которые промывали смесью гептан/вода 1:1 (80 мл), с последующим растворением в этаноле (20% в воде, 100 мл). Полученные кристаллы высушивали в вакууме (50-60°С, 10-20 мбар). tпл 170-172°С. (3S,1'S)-3-[(трет-бутилдиметилсилил)окси]-5-[(1-фенилэтил)амино]-5-оксопентановая кислота, полученная в соответствии с этой процедурой, имеет обычно >98% de и >98% ее.
Соединение (3)
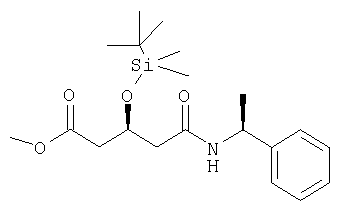
Метиловый эфир (3S,1'S)-3-[(трет-бутилдиметилсилил)окси]-5-[(1-фенилэтил)амино]-5-оксопентановой кислоты может быть получен в соответствии с процедурой Karanewsky и др., указанной выше.
Соединение (4)
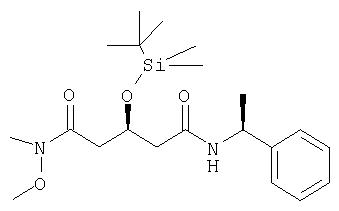
Метоксиметиламид-((S)-1-фенилэтил)амид (S)-3-(трет-бутилдиметилсиланилокси)-пентандионовой кислоты может быть получен следующим образом: 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником наполняли кислотой (соединение 2) (10 г, 27,36 ммоль) и дихлорметаном (200 мл). При перемешивании при комнатной температуре добавляли N-метилморфолин (6,03 мл, 54,71 ммоль), получая чистый раствор. Реакционную смесь охлаждали до -20°С. К реакционной смеси добавляли изобутилхлорформат (3,76 мл, приблизительно 95 вес.% чистоты, 27,36 ммоль) при от -15 до -20°С. Смесь перемешивали в течение 15 мин при от -15 до -20°С и обрабатывали гидрохлоридом N,O-диметилгидроксиламина (2,695 г, 27,36 ммоль). Перемешивание продолжали в течение 1 часа и реакционную смесь нагревали до комнатной температуры. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и обрабатывали водой (200 мл) с получением двухфазного раствора. Слои разделяли и водный слой извлекали дихлорметаном (2×200 мл). Органический слой промывали соляным раствором (200 мл), высушивали над безводным MgSO4 и растворитель упаривали при пониженном давлении с получением сырого метоксиметиламид-((S)-1-фенилэтил)амида(S)-3-(трет-бутилдиметилсиланилокси)пентандионовой кислоты. Сырой продукт повторно перекристаллизовывали из гексана с получением чистого метоксиметиламид-((S)-1-фенилэтил)амида (S)-3-(трет-бутилдиметилсиланилокси)пентандионовой кислоты. tпл 69,6-70,1°С; [α]D 25=-27°(CHCl3, с=1); MS (ES+, m/z): 431 ([M+Na]+, 100%); ИК (KBr): Сильные поглощения при 3304, 1666, 1630, 1541 и 831 см-1.
Пример 1:
a)
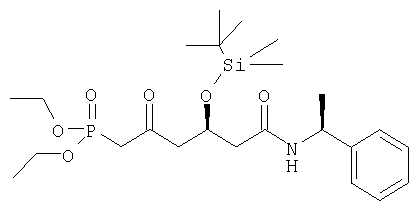
Диэтиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((8)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты может быть получен следующим образом: 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником наполняли диэтиловым эфиром метанфосфоновой кислоты (4,65 г, 30,59 ммоль) и тетрагидрофураном (11 мл). Раствор охлаждали до -78°С и добавляли бутиллитий (15,3 мл 1,6 М раствора в гексане, 24,47 ммоль). После перемешивания в течение дополнительных 60 минут при -78°С к реакционной смеси добавляли раствор метоксиметиламид-((S)-1-фенилэтил)амида (S)-3-(трет-бутилдиметилсиланилокси)пентандионовой кислоты (2,5 г) в тетрагидрофуране (10 мл) при поддержании температуры при -78°С. Перемешивание продолжали в течение 1 часа при этой температуре и реакцию останавливали медленным добавлением раствора уксусной кислоты (1,84 г) в тетрагидрофуране (1,25 мл) при -78°С. Реакционную смесь затем нагревали до комнатной температуры и выливали в этилацетат (125 мл) и соляной раствор (125 мл). Двухфазную смесь перемешивали в течение 10 минут и органический слой отделяли. Водный слой извлекали этилацетатом. Органические слои объединяли, промывали водой, высушивали над безводным сульфатом магния и растворитель упаривали в вакууме с получением сырого диэтилового эфира [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты в виде вязкого масла. Сырой продукт очищали колоночной хроматографией с этилацетатом/гексаном (1:1) в качестве элюента, с получением диэтилового эфира [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты в виде высоко вязкого масла. [α]D 20=-40,2°(CHCl3, с=1); MS (ES+, m/z): 522 ([M+Na]+, 100%); ИК (Пленка): сильные поглощения при 3297, 2930, 1717, 1652, 1542, 1254, 1026, 837 см-1.
Альтернативно, диэтиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5 ((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты также может быть получен с помощью метилового эфира (3S,1'S)-3-[(трет-бутилдиметилсилил)окси]-5-[(1-фенилэтил)амино]-5-оксопентановой кислоты в качестве исходного материала вместо метоксиметиламид-((S)-1-фенилэтил)амида(S)-3-(трет-бутилдиметилсиланилокси)пентандионовой кислоты, применяя вышеупомянутый описанный метод.
б)
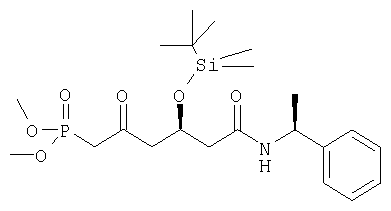
Диметиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты может быть получен следующим образом: диметиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил-пентил]фосфоновой кислоты получали с помощью процедуры, как описано для диэтилового эфира [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты.
Диметиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты получали в виде высоковязкого масла, которое затвердевало при хранении в холодильнике. [α]D 20=-37,7°(CHCl3, с=1); MS (ES+, m/z): 494 ([M+Na]+, 100%); ИК (Пленка): сильные поглощения при 3299, 2955, 2929, 2855, 1717, 1651, 1542, 1256, 1035, 838, 779, 701 см-1.
в)
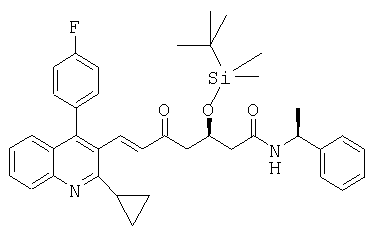
((S)-1-фенилэтил)амид (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-оксогепт-6-еновой кислоты может быть получен следующим образом: Диэтиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5 ((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты (3 г, 6 ммоль) помещали в 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником. Добавляли изопропанол (12 мл) с получением раствора. Карбонат цезия (1,96 г, 6 ммоль) добавляли, далее добавляли 2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-карбальдегид (1,75 г, 6 ммоль). Реакционную смесь растворяли изопропанолом (6 мл) и перемешивали в течение ночи при комнатной температуре. Затем реакцию останавливали добавлением водного раствора лимонной кислоты (60 мл) и водную смесь экстрагировали дважды трет-бутилметиловым эфиром (2×120 мл). Органические слои объединяли, промывали водой (120 мл), высушивали над безводным сульфатом магния и растворитель упаривали при пониженном давлении с получением сырого продукта в виде коричневато-желтой пены. Сырой продукт очищали колоночной хроматографией на силикагеле с гексаном/этилацетатом (7:3) в качестве элюента, с получением ((S)-1-фенилэтил)амида (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-оксо-гепт-6-еновой кислоты в виде светло-желтой твердой пены. [α]D 20=-28,8°(CHCl3, с=1); MS (ES+, m/z): 659 ([M+Na]+, 100%); ИК (KBr): Сильные поглощения при 1647, 1605, 1540, 1513, 1253, 1223, 1094, 1066, 837, 778, 763, 6999 см-1.
Альтернативно, ((S)-1-фенилэтил)амид (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-оксогепт-6-еновой кислоты может быть получен из диметилового эфира [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((8)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты, например, используя следующую процедуру.
Диметиловый эфир [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты (424,9 г, 910 ммоль) помещали в 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником. Добавляли этанол (950 мл) с получением раствора. Раствор охлаждали до 0-4°С в ледяной бане. Добавляли тонко измельченный карбонат калия (119,8 г, 858,1 ммоль), далее добавляя 2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-карбальдегид (250 г, 858,1 ммоль). Реакционную смесь растворяли в этаноле (950 мл) и нагревали до комнатной температуры. После перемешивания в течение 30 минут при комнатной температуре реакционную смесь нагревали до 40-45°С и перемешивали при этой температуре в течение 48 часов. Реакцию затем останавливали выливанием в раствор 5% водной лимонной кислоты (1800 мл). Реакционную смесь переносили в разделительный сосуд и извлекали трет-бутилметиловым эфиром (3500 мл). Слои разделяли и органический слой промывали водой (3500 мл) и соляным раствором (3500 мл). Водные слои экстрагировали снова трет-бутилметиловым эфиром (5500 мл) и органические слои объединяли. Органический слой высушивали над сульфатом магния (70 г) и растворитель упаривали в вакууме с получением коричневого тягучего сырого продукта, которое может быть очищено колоночной хроматографией, как описано выше, с получением чистого ((S)-1-фенилэтил)амида (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-оксогепт-6-еновой кислоты. Альтернативно, сырой продукт может использоваться для следующей стадии без дальнейшей очистки.
г)
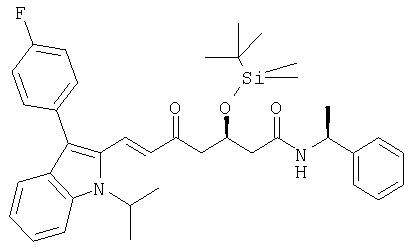
((S)-1-Фенилэтил)амид (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-5-оксогепт-6-еновой кислоты может быть получен из 3-(4-фторфенил)-1-изопропил-1H-индол-2-карбальдегида и диметилового эфира [(R)-4-(трет-бутилдиметилсиланилокси)-2-оксо-5-((S)-1-фенилэтилкарбамоил)пентил]фосфоновой кислоты в соответствии с описанной выше процедурой. После хроматографии на силикагеле с н-гексаном/этилацетатом (7:3) в качестве элюента получали ((S)-1-фенилэтил)амид (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-5-оксогепт-6-еновой кислоты в виде желтой пены. Rf=0,65 (ТСХ на силикагеле с этилацетатом/гексаном 9:1 в качестве элюента). [α]D 20=-40,1° (МеОН, с=1); MS (ES+, m/z): 649 (100%, M+Na+) ИК (KBr): характерные сигналы в 3309 (широкий), 3062, 2954, 2929, 2884, 2855, 1652, 1591, 1540, 1496, 1453, 1371, 1339, 1251, 1222, 1156, 1138, 1093, 1015, 970, 837, 778, 743, 699, 565 см-1; Микроанализ: вычисленный (найденный) для С38Н47FN2O3Si: 72,81 (72,69)% С, 7,56 (7,73)% Н, 4.47 (4,61)% N, 3,03 (2,99)% F, 4,48 (4,36)% Si.
д)
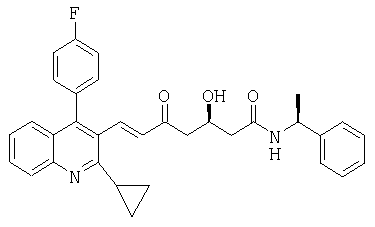
((S)-1-Фенилэтил)амид (Е)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксо-гепт-6-еновой кислоты может быть получен следующим образом: ((S)-1-фенилэтил)амид (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-оксогепт-6-еновой кислоты (2,0 г, 3,14 ммоль) помещали в 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником. Добавляли ацетонитрил (30 мл) и смесь перемешивали с получением раствора. Добавляли предварительно полученный раствор фосфорной кислоты (10 мл 1 М раствора в воде) и реакционную смесь нагревали до 45°С. Перемешивание продолжали в течение 29 часов при этой температуре. Затем реакционную смесь выливали в водный насыщенный раствор NaHCO3 (65 мл) и смесь извлекали этилацетатом (2×70 мл). Органические слои промывали соляным раствором, высушивали над безводным сульфатом магния и растворитель упаривали при пониженном давлении с получением твердой пены. Сырой продукт использовали без дальнейшей очистки на следующей стадии.
Аналитический образец сырого продукта очищали колоночной хроматографией на силикагеле, используя этилацетат в качестве элюента, и впоследствии перекристаллизовывали из трет-бутилметилового эфира/гексана, получая чистый ((S)-1-фенилэтил)амид (Е)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты. tпл 150-151°С; [α]D 20=-26,9°(CHCl3, с=1); MS (ES+, m/z): 545 ([M+Na+), 523 (MH+, 100%); ИК (KBr): Сильные поглощения в 3344, 1693, 1631, 1603, 1549, 1513, 1488, 1409, 1344, 1219, 1055, 1030, 768, 697 см-1; Микроанализ: вычисленный (найденный) для С33Н31FH2O3: 75,84 (75,74)% С, 5,98 (6,13)% Н, 5,36 (5,39)% N, 3,64 (3,63)% F.
Альтернативно, ((S)-1-фенилэтил)амид (Е)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты может быть получен, используя соляную кислоту для снятия защиты в соответствии со следующей процедурой: Сырой ((S)-1-фенилэтил)амид (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-оксогепт-6-еновой кислоты (655,1 г) из примера в) помещали в 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником. Добавляли этанол (2400 мл) с получением раствора. Раствор охлаждали до 0-4°С и по каплям добавляли соляную кислоту (2 М, водный раствор, 657,1 г, 1276 ммоль) при этой температуре. Реакционную смесь нагревали до 25°С и перемешивали при этой температуре в течение дополнительных 4 часов для завершения снятия защиты. Реакционную смесь затем выливали в водный раствор бикарбоната натрия (7400 мл 2% раствора) и экстрагировали этилацетатом (5000 мл). Органический слой промывали соляным раствором (1800 мл), водные слои объединяли и экстрагировали этилацетатом (2000 мл). Органические слои объединяли, высушивали над сульфатом магния и растворитель упаривали в вакууме с получением 561,2 г коричневого тягучего сырого продукта. Сырой продукт растворяли в толуоле (860 мл) и добавляли н-гексан (1075 мл). Смесь нагревали при 60°С в течение 2 часов и при 50°С в течение 1 часа. Добавляли н-гексан (1075 мл) при 50°С и суспензию охлаждали до комнатной температуры. Перемешивание продолжали в течение ночи при комнатной температуре и в течение дополнительных 3 часов при 0°С. Твердый продукт выделяли фильтрацией, фильтр промывали ледяным н-гексаном (502 мл) и высушивали в вакууме с получением ((S)-1-фенилэтил)амида (E)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты как желтого порошка.
е)
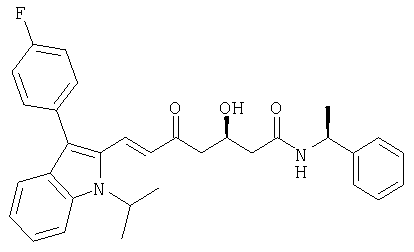
((S)-1-Фенилэтил)амид (Е)-(R)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты может быть получен из ((S)-1-фенилэтил)амида (Е)-(R)-3-(трет-бутилдиметилсиланилокси)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-5-оксогепт-6-еновой кислоты снятием защиты с помощью HCl в этаноле в соответствии с описанной выше процедурой для получения ((S)-1-фенилэтил)амида (Е)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты. Хроматография на силикагеле с этилацетатом/н-гексаном (9:1) в качестве элюента приводит к получению ((S)-1-фенилэтил)амида (E)-(R)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты в виде желтой пены. Rf=0,30 (ТСХ на силикагеле, этилацетат/гексан 9:1 в качестве элюента). [α]D 20=-35,5° (СН3ОН, с=1) MS (ES+, m/z): 535 ([MNa]+, 100%); ИК (KBr): 3307 (широкий), 3061, 2973, 2932, 1645, 1590, 1539, 1495, 1452, 1421, 1371, 1339, 1273, 1221, 1155, 1138, 1105, 1095, 1048, 1016, 973, 910, 839, 815, 744, 719, 700 см-1. Микроанализ: вычисленный (найденный) для С32Н33FN2O3: 74,98 (74,20)% С, 6,49 (6,49)% Н, 5,46 (5,49)% N, 3,71 (3,58)% F.
ж)
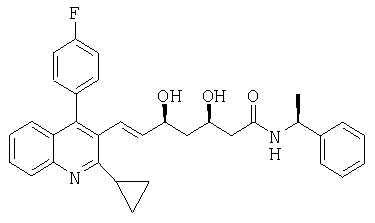
((S)-1-Фенилэтил)амид (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты может быть получен следующим образом:
Сухую 3-горлую круглодонную колбу, оборудованную мешалкой, цифровым термометром и входным отверстием для аргона наполняли сухим тетрагидрофураном (7,5 мл) и охлаждали до -78°С. В потоке аргона добавляли боргидрид натрия (172,5 мг, 4,56 ммоль), сопровождая введением диэтилметоксиборана (0,694 г 50% раствора в тетрагидрофуране 3,47 ммоль) при -78°С. Смесь перемешивали в течение 5 минут при этой температуре. Затем медленно добавляли раствор сырого ((S)-1-фенилэтил)амида (E)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты (1,84 г, 3,52 ммоль) в сухом тетрагидрофуране (1,8 мл) и сухом метаноле (2,2 мл) в течение приблизительно 30 минут при -78°С и перемешивание продолжали еще в течение 1 часа при этой температуре. Для обработки реакционную смесь выливали в раствор бикарбоната натрия (0,383 г) в воде (12 мл) и добавляли изопропилацетат (30 мл). Двухфазный раствор перемешивали в течение 30 минут при комнатной температуре, пока не прекращалось выделение газа. Слои разделяли и органический слой промывали соляным раствором (2×30 мл). Растворитель упаривали в вакууме с получением сухого желтого твердого вещества. Твердое вещество растворяли в изопропилацетате (15 мл) и раствор нагревали до 45-50°С. Медленно добавляли перекись водорода (0,994 г 35% водного раствора, 10,23 ммоль) при 45-50°С и перемешивание продолжали в течение дополнительных 2 часов. Реакцию останавливали добавлением соляного раствора (15 мл) при 45-50°С. Двухфазную смесь перемешивали в течение 20 минут при 45-50°С и слои разделяли. Органический слой обрабатывали водным раствором Na2SO3 (0,658 г в 15 мл воды) при 45-50°С, смесь перемешивали в течение 5 минут и слои разделяли. Наконец, органический слой промывали водным, полунасыщенным раствором NaCl (15 мл), высушивали над сульфатом магния и растворитель упаривали в вакууме с получением сырого продукта в виде желтоватой твердой пены. Очистка сырого продукта колоночной хроматографией на силикагеле с этилацетатом/гексаном (9:1) в качестве элюента приводила к получению ((S)-1-фенилэтил)амида(Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты, которая может быть перекристаллизована из трет-бутилметилового эфира следующим образом: ((S)-1-фенилэтил)амид (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты (1,197 г) растворяли в трет-бутилметиловом эфире (10 мл) и по каплям добавляли фракцию гексана (4 мл) при перемешивании. Чистый раствор обрабатывали суспензией маленькой части затравочных кристаллов и перемешивание продолжали в течение дополнительного часа при комнатной температуре. Полученную суспензию охлаждали до 0-5°С и перемешивали в течение 2 часов при этой температуре. Продукт выделяли фильтрацией, промывали ледяной фракцией гексана (5 мл) и высушивали в вакууме при 40°С с получением прозрачного ((S)-1-фенилэтил)амида (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты. 1Н-ЯМР и микроанализ показал присутствие приблизительно 1:1 формы сольвата кристаллов с трет-бутилметиловым эфиром, tпл: диапазон плавления 57-76°С; [α]D 20=-27,0°(CHCl3, с=1); MS (ES+, m/z): 547 ([M+Na]+), 525 (МН+, 100%); ИК (KBr): Сильные поглощения в 3479, 3296, 2977, 1642, 1557, 1513, 1490, 1215, 1116, 1068, 763, 698 см-1; Микроанализ: вычисленный (найденный) для С33Н33FN2O3+С5Н12О: 74,48 (74,47)% С, 7,40 (7,30)% Н, 4,57 (4,64)% N, 3,10 (3,13)%F.
Альтернативно, ((S)-1-фенилэтил)амид (Е)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты, осажденный из толуола, может быть преобразован в ((S)-1-фенилэтил)амид (E)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты также в соответствии со следующей процедурой:
Натрия боргидрид (30,26 г) помещали в сухую 4-горлую круглодонную колбу, оборудованную механической мешалкой, цифровым термометром, входным отверстием для азота и обратным холодильником. Добавляли высушенный тетрагидрофуран (1877 мл) и суспензию охлаждали до -78°С. Добавляли раствор диэтилметоксиборана (54,85 г, 548,4 ммоль) в тетрагидрофуране (54,85 г) в течение 15 мин при -78°С и реакционную смесь перемешивали в течение дополнительных 5 мин при этой температуре. К реакционной смеси медленно добавляли раствор ((S)-1-фенилэтил)амида (E)-(R)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты (290,7 г, 556 ммоль) в сухом тетрагидрофуране (488 мл) и метаноле (581 мл) в течение 2,5 часов, поддерживая температуру реакции при -78°С. После перемешивания в течение дополнительного одного часа при -78°С реакционную смесь выливали в ледяной раствор бикарбоната натрия (92,15 г) в воде (4610 мл). Добавляли изопропилацетат (7346 мл) и двухфазную смесь перемешивали до образования двух ясных стадий. Органический слой отделяли и водный слой экстрагировали изопропилацетатом (2×3150 мл). Органические слои объединяли, промывали соляным раствором (2×3150 мл) и растворитель упаривали в вакууме с получением твердого вещества в виде желтой пены. Твердое вещество растворяли в изопропилацетате (630 мл) и раствор нагревали до 50°С. Добавляли перекись водорода (174,02 г 35% водного раствора, соответствуя 60,907 г пероксиду водорода, 1790 ммоль) в течение 45 мин при 50°С, и реакционную смесь перемешивали в течение дополнительного часа при этой температуре. Реакцию останавливали добавлением соляного раствора (3980 мл) при 45-50°С и разбавляли изопропилацетатом (2390 мл). Двухфазную смесь перемешивали в течение 20 минут при 45-50°С и слои разделяли. Органический слой обрабатывали водным раствором Na2SO3 (118 г в 2850 мл воды) при 45-50°С, смесь перемешивали в течение 5 минут и слои разделяли. Наконец, органический слой промывали водным полунасыщенным раствором NaCl (2×2390 мл), высушивали над сульфатом магния и растворитель упаривали в вакууме с получением сырого продукта (349 г) в виде желтой твердой пены. Сырой продукт растворяли в трет-бутилметиловом эфире (706 мл) и раствор охлаждали до 0°С. После перемешивания в течение 30 мин при 0°С полученную суспензию нагревали до 30°С и перемешивали в течение 15 мин при этой температуре. Суспензию охлаждали до 25°С и перемешивали в течение ночи при этой температуре. Наконец, суспензию перемешивали в течение 2 часов при 0°С и в течение 3 часов при -20°С. Продукт выделяли фильтрацией, промывали н-гептаном (2×75 мл) и высушивали в вакууме при 40°С, получая прозрачный ((S)-1-фенилэтил)амид (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты в виде сольвата с трет-бутилметиловым эфиром. Диапазон плавления 58-69°С. В соответствии с ВЭЖХ, кристаллы включали 99,89% желаемого син-(3R,5S)-продукта и 0,11% анти-(3R,5R)-эпимера.
з)
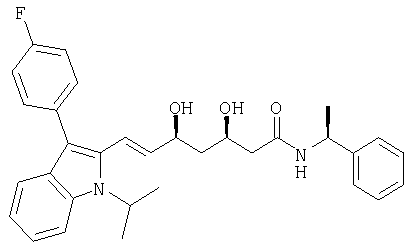
((S)-1-Фенилэтил)амид (E)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты может быть получен из ((S)-1-фенилэтил)амида (E)-(R)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3-гидрокси-5-оксогепт-6-еновой кислоты в соответствии с процедурой, описанной выше для получения ((S)-1-фенилэтил)амида (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты. Хроматография на силикагеле с этилацетатом/гексаном (9:1) в качестве элюента приводит к получению ((S)-1-фенилэтил)амида (Е)-(3R,5S)-7-[3-(4-фторфенил)-, 1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты в виде легкой желтой пены. Rf=0,175 (ТСХ на силикагеле, этилацетат/гексан 9:1 в качестве элюента). [α]D 20=-16,7°(СН3ОН, с=1). MS (ES+, m/z): 537 ([M+Na]+, 100%); ИК (KBr): 3310 (широкий), 3049, 2974, 2934, 2875, 1644, 1604, 1545, 1501, 1457, 1420, 1371, 1346, 1219, 1155, 1104, 1065, 1045, 1018, 970, 944, 837, 814, 742, 718, 700, 565.
и)
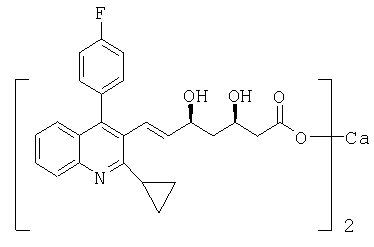
Кальциевая соль (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты может быть получена следующим образом: ((S)-1-фенилэтил)амид (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты (0,50 г, 0,95 ммоль) растворяли в этаноле (12,5 мл). Добавляли воду (12,5 мл) и гидроксид натрия (5 мл 1 М раствора, 5 ммоль) и смесь нагревали приблизительно до 80°С в масляной бане. Перемешивание продолжали в течение 18 часов приблизительно при 80°С. Растворитель упаривали при пониженном давлении и остаток растворяли в воде (50 мл). Водный раствор экстрагировали дважды трет-бутилметиловым эфиром (2×50 мл). Водный слой упаривали в вакууме до конечного объема приблизительно 25 мл. Добавляли воду (25 мл), далее добавляли хлорид кальция (0,049 г, 0,044 ммоль) при перемешивании при 30-35°С. Происходит осаждение. Суспензию охлаждали до комнатной температуры. Суспензию перемешивали в течение 2 часов при комнатной температуре и в течение дополнительных 2,5 часов при 15°С. Остаток отделяли фильтрацией и фильтр промывали водой (5 мл). Продукт высушивали в вакууме с получением кальциевой соли питавастатина в виде белого порошка. Кислотный компонент соли идентичен кислотному компоненту подлинного образца кальциевой соли (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты, (соответствующей кальциевой соли питавастатина) в ИК и ВЭЖХ.
Альтернативно, (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновая кислота и ее кальциевая соль соответственно могут быть получены следующим образом: ((S)-1-фенилэтил)амид (E)-(3R, 5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты (4,0 г, 6,53 ммоль) растворяли в этаноле (40 мл). Добавляли воду (40 мл) и порошок гидроксида натрия (2,64 г, 66 ммоль) и смесь нагревали при 50-55°С в течение 26 часов до контроля в процессе (ВЭЖХ), показавшее полное преобразование. Медленно добавляли соляную кислоту (59 мл 1 М раствора, 59 ммоль) в течение 15 минут. Растворитель упаривали при пониженном давлении и остаток растворяли в воде (80 мл). Водный раствор экстрагировали трет-бутилметиловым эфиром (3×80 мл) и органическую фазу удаляли. Водную фазу упаривали при пониженном давлении и остаток повторно растворяли в воде (176 мл). Добавляли соляную кислоту (6,53 мл 1 М раствора, 6,53 ммоль) для осаждения кислоты, далее добавляли этилацетат (176 мл). Смесь перемешивали в течение 15 мин и слои разделяли. Органический слой промывали водой (90 мл). К органическому слою добавляли древесный уголь (0,5 г) и смесь перемешивали при 30-35°С в течение нескольких часов. Добавляли фильтровальное вспомогательное вещество (Cellflock, 1,0 г), и перемешивание продолжали в течение дополнительных 30 мин. Древесный уголь удаляли фильтрацией со вспомогательным веществом, получая чистый раствор, и растворитель упаривали при 30-35°С при пониженном давлении, получая (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновую кислоту в виде белого твердого вещества. Для образования кальциевой соли кислоту (2,55 г, 6,05 ммоль) суспендировали в воде (40,5 мл) и добавляли гидроксид натрия (0,260 г, 6,5 ммоль) с получением чистого раствора соответствующей соли натрия. К раствору соли натрия по каплям добавляли раствор хлорида кальция (0,399 г, 3,49 ммоль) в воде (2 мл). Суспензию получали немедленно после добавления хлорида кальция. Суспензию перемешивали в течение 4 часов при 20-25°С и в течение 2 часов при 15-17°С. Продукт выделяли фильтрацией, фильтр промывали холодной водой и высушивали в вакууме при 20-25°С с получением кальциевой соли (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты, в виде белого прозрачного порошка, включая 10,6% (по весу) воды. [α]D 20=+22,92°(1:1 ацетонитрил/вода, с=1). Рентгеновский анализ показал присутствие кристаллической модификации А. Соотношение энантиомера, имеющего (3S,5R) конфигурацию, было ниже предела обнаружения 0,05% в соответствии с колоночным электрофорезом. Продукт имел чистоту более 99,7% в соответствии с ВЭЖХ и включал 0,09% соответствующих эпимеров (сумма (3S,5S) и (3R,5R) эпимеров, которые не были отделены в ВЭЖХ). Соответствующий лактон не мог быть обнаружен в пределе обнаружения 0,05%.
к)
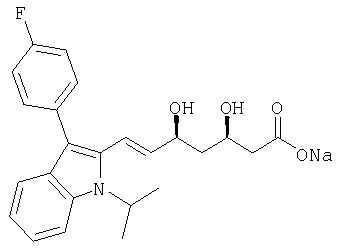
Натриевая соль (Е)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты может быть получена из ((S)-1-фенилэтил)амида (Е)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1H-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты гидролизом с гидроксидом натрия в соответствии с описанной выше процедурой для получения (E)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты. После завершения гидролиза избыток гидроксида натрия нейтрализовали добавлением соляной кислоты и растворитель упаривали при пониженном давлении. Как вариант обработки, остаток растворяли в воде при 60-70°С и раствор охлаждали до комнатной температуры. Происходит кристаллизация. Полученную суспензию охлаждали до 5°С, перемешивали в течение нескольких часов при этой температуре для окончания кристаллизации и продукт выделяли фильтрацией. Получали натриевую соль (E)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты в виде бежево-желтого прозрачного продукта. Прозрачный продукт может быть растворен в воде и лиофилизован с получением лиофилизата, который является растворимым в органических растворителях, например, в дихлорметане. [α]D 20=-20° (с=0,8 в дихлорметане). Кислотный компонент соли идентичен кислотному компоненту Флувастатина ® в нехиральном ВЭЖХ. ИК и MS спектры продукта подтверждали предложенную структуру и соответствовали опубликованным данным для натриевой соли (Е)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты (см. Tempkin и др., Tetrahedron 1997, 53, 10659-10670).
Альтернативно, натриевая соль (Е)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты может быть получена гидролизом (4R,6S)-6-{(Е)-2-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]винил}-4-гидрокситетрагидропиран-2-она с NaOH.
л)
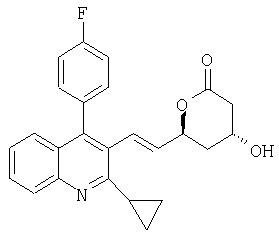
(4R,6S)-6-{(Е)-2-[2-Циклопропил-4-(4-фторфенил)хинолин-3-ил]винил}-4-гидрокситетрагидропиран-2-он может быть получен следующим образом: ((S)-1-фенилэтил)амид (Е)-(3R,5S)-7-[2-циклопропил-4-(4-фторфенилхинолин-3-ил]-3,5-дигидроксигепт-6-еновой кислоты (0,2 г, 0,381 ммоль) помещали в 3-горлую круглодонную колбу и добавляли ацетонитрил (10 мл). Раствор обрабатывали ортофосфорной кислотой (0,113 г, 1,14 ммоль) и нагревали до 65-70°С. Реакционную смесь перемешивали в течение 18 часов при 65-70°С. Для обработки смесь растворяли в трет-бутилметиловом эфире (30 мл) и раствор продукта промывали три раза водой (3×30 мл). Органический слой высушивали над безводным сульфатом магния и растворитель упаривали в вакууме с получением 135,2 мг сырого продукта в виде бесцветной твердой пены. Сырой продукт очищали колоночной хроматографией на силикагеле с этилацетатом/гексаном (7:3) в качестве элюента. MS (ES+, m/z): 404 (МН, 100%); ИК (KBr): Сильные поглощения в 3410, 1736, 1709, 1513, 1490, 1216, 1159, 1062, 1038, 970, 766 см-1. 1H-ЯМР подтверждал предполагаемую структуру.
м)
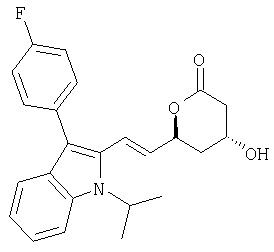
(4R,6S)-6-{(Е)-2-[3-(4-Фторфенил)-1-изопропил-1Н-индол-2-ил]винил}-4-гидрокситетрагидропиран-2-он может быть получен из (E)-(3R,5S)-7-[3-(4-Фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновой кислоты или ее производного в соответствии с известными процедурами, смотри, например. F.G.Kathawala, международный патент WO 84/02131 (1984).
(н)
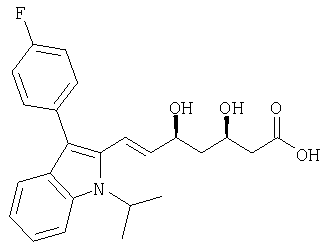
(Е)-(3R,5S)-7-[3-(4-фторфенил)-1-изопропил-1Н-индол-2-ил]-3,5-дигидроксигепт-6-еновая кислота может быть получена из соответствующей натриевой соли [раздел (к)] нейтрализацией кислотой, например HCl, и экстракцией этилацетатом, который можно удалить в вакууме.
Изобретение относится к способу получения энантиомерно чистых ингибиторов HMG-CoA редуктазы. Описывается способ получения соединения формулы
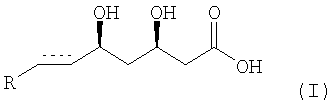
или его соли, особенно фармацевтически приемлемой соли с основанием, или его лактона, где элемент  представляет собой -СН2-СН2- или -СН=СН- и R представляет собой циклический остаток. Технический результат: обеспечение энантиоселективного синтеза соединений формулы (I), с высокими выходами, уменьшение экологического загрязнения окружающей среды, возможность крупномаштабного производства. 4 з.п. ф-лы.
представляет собой -СН2-СН2- или -СН=СН- и R представляет собой циклический остаток. Технический результат: обеспечение энантиоселективного синтеза соединений формулы (I), с высокими выходами, уменьшение экологического загрязнения окружающей среды, возможность крупномаштабного производства. 4 з.п. ф-лы.
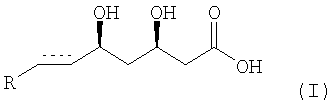
или его соли, особенно фармацевтически приемлемой соли с основанием, или его лактона, где элемент  представляет собой -СН2-СН2- или -СН=СН- и R представляет собой циклический остаток, включающий
представляет собой -СН2-СН2- или -СН=СН- и R представляет собой циклический остаток, включающий
(а) реакцию соединений (IIIa) или (IIIб)
 или
или 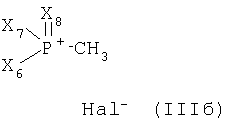
где Х4 и Х5, независимо друг от друга, представляют собой С1-С7-алкил или фенил-С1-С7-алкил;
Х6, Х7 и X8, независимо друг от друга, представляют собой фенил, который является незамещенным или замещен одним или несколькими заместителями, выбранными из группы, состоящей из С1-С7алкила, гидрокси, С1-С7алкокси, С2-С8алканоилокси, галогена, нитро, циано и CF3;
и HaI- представляет собой галогеновый анион;
с металлированным алканом с образованием соответствующего илида и затем реакцию полученного илидного промежуточного соединения с соединением формулы
где Х представляет собой этерифицированный гидрокси, этерифицированный гидрокси или незамещенный или моно- или дизамещенный амино;
Х1 представляет собой защищенный гидрокси;
Х2 представляет собой С1-С7алкил и
Х3 представляет собой водород или один или несколько заместителей, например, выбранных из группы, состоящей из С1-С7алкила, гидрокси, С1-С7алкокси, С2-С8алканоилокси, галогена, нитро, циано и CF3;
(б) необязательно, при необходимости, преобразование полученного соединения формулы (IIIг)
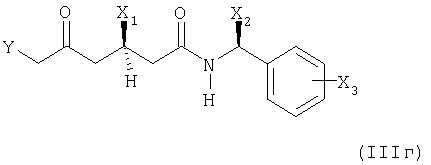
в котором Х1, X2 и Х3 имеют вышеупомянутые значения и Y представляет собой группу формулы (Х4O)(Х5O)Р(=O)- или (X6)(X7)(X8)P+Hal- и Х4, Х5, Х6, Х7, X8 и HaI- имеют вышеупомянутые значения;
в соединение формулы (IIIд)
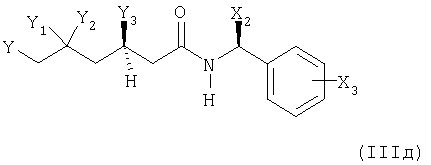
в котором Х2, Х3 и Y имеют вышеупомянутые значения и где Y1 представляет собой гидрокси или защищенный гидрокси, и Y2 представляет собой водород, и Y3 представляет собой гидрокси или защищенный гидрокси, и Y1 и Y3 образуют син-диольную конфигурацию; или в котором
Y1 и Y2 вместе представляют собой -O-Alk-O- и Alk, являющийся
С1-С7алкилиденом; и Y3 представляет собой водород, и Y1 и Y3 образуют синдиольную конфигурацию;
(в) реакцию соединения формулы (IIIд),
в которой Х2, Х3 и Y имеют вышеупомянутые значения и в которой Y1 представляет собой гидрокси или защищенный гидрокси, и Y2 представляет собой водород, и Y3 представляет собой гидрокси или защищенный гидрокси, и Y1 и Y3 образуют син-диольную конфигурацию; или в которой Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk, являющийся С1-С7алкилиденом; и Y2 представляет собой водород, и Y1 и Y3 образуют син-диольную конфигурацию;
или в которой Y1 и Y2 вместе представляют собой оксогруппу и Y3 представляет собой защищенный гидроксил (соответствующий соединениям формулы (IIг));
с альдегидом формулы (IIIe) R-CH(=O), приводя к получению соединения формулы (IIIж)
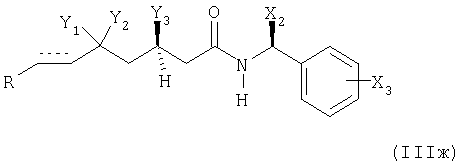
в которой R, Х2, Х3, Y1, Y2 и Y3 и элемент имеют вышеупомянутые значения;
при необходимости, восстанавливая соответствующие соединения формулы (IIIж), в которой элемент представляет собой -СН=СН-, с получением соединения, в котором указанный элемент представляет собой -СН2-СН2-; и
(г) если получают соединение формулы (IIIж), в котором один из Y1 и Y3 представляет собой защищенный гидрокси и другой представляет собой гидрокси, или оба Y1 и Y3 представляют собой защищенные гидрокси, и в каждом случае Y2 представляет собой водород; и Y1 и Y3 образуют син-конфигурацию; или
Y1 и Y3 вместе представляют собой -O-Alk-O- и Alk, являющийся
С1-С7алкилиденом, и Y1 и Y3 образуют син-конфигурацию; и Y2 представляет собой водород; или
удаление защиты гидроксигруппы (групп) с получением соединения формулы
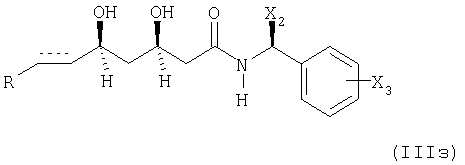
или при необходимости, восстановлением соответствующего соединения формулы (IIIз), в котором элемент представляет собой -СН=СН-, с получением соединения, в котором указанный элемент представляет собой -СН2-СН2-;
(д) если получают соединение формулы (IIIж), в котором Y1 и Y2 вместе образуют оксогруппу =O; и Y3 представляет собой защищенный гидрокси (Х1); преобразование указанного соединения формулы (IIIж) в соединение формулы (IIIи)
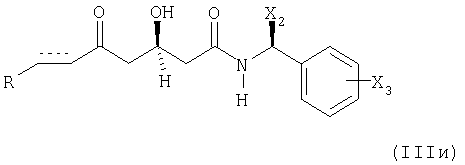
удалением защитной гидроксигруппы;
в которой R, X2, Х3 и элемент имеют вышеупомянутые значения; и последующее восстановление указанного соединения формулы (IIIи) в соединение формулы (IIIз);
(е) гидролиз соединения формулы (IIIз) в соединение формулы (I) или его соль и
(ж) выделение полученного соединения формулы (I) или его соли;
и, при необходимости, преобразование полученной свободной кислоты формулы (I) в его соль или в лактон формулы (Ia) или (Iб), соответственно, или преобразование полученного лактона формулы (Ia) или (Iб) в кислоту формулы (I) или ее соль.
представляет собой -СН=СН- иR представляет собой циклический остаток формулы
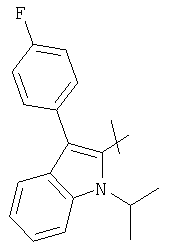 или
или 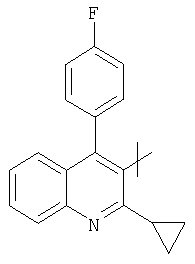
| US 5049577 А, 17.09.1991 | |||
| P.D | |||
| THEISEN ET AL: J.ORG.CHEM., Vol.53, no | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| ФИЛЬТР КОЛЬЦЕВОГО ТИПА | 0 |
|
SU340007A1 |
| US 5034399 A, 23.07.1991 | |||
| US 5354772 A, 11.10.1994 | |||
| US 5856336 A, 05.01.1999 | |||
| N-АЦИЛИРОВАННОЕ СОЕДИНЕНИЕ АРИЛПИРРОЛА, СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ, СПОСОБ ПОЛУЧЕНИЯ N-АЦИЛИРОВАННЫХ СОЕДИНЕНИЙ АРИЛПИРРОЛА | 1991 |
|
RU2099326C1 |

