Область техники
Настоящее изобретение относится к способу получения 7-хинолинил-3,5-дигидроксигепт-6-еноата.
Более конкретно, оно относится к эффективному и применимому в промышленности способу получения (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата, который может быть полезен в качестве промежуточного соединения в синтезе ингибитора HMG-CoA-редуктазы в качестве средства, понижающего уровень холестерина.
Предшествующий уровень техники
(Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноат представляет собой промежуточное соединение, которое используется в качестве промежуточного соединения для синтеза ингибитора HMG-CoA-редуктазы, и способ его получения, в котором используют боргидрид натрия (NaBH4), раскрыт в JP-A-1-279866, в патенте США 5856336 или в ЕР 0304063 В1:
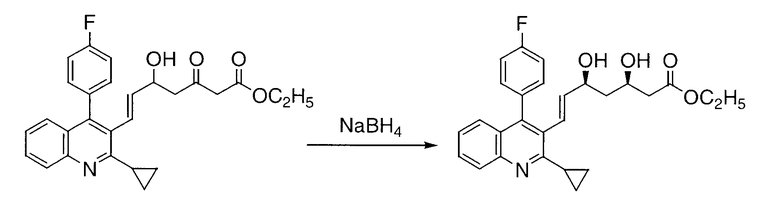
Кроме того, в настоящее время известно, что при получении 1,3-диола восстановлением β-гидроксикетона боргидридом натрия допускается присутствие соединения бора, как и в соответствии с настоящим изобретением, и при этом проходит селективная реакция восстановления, причем в (1) JP-A-61-40243 или в ЕР 0164049 А2 описана реакция, протекающая в присутствии триэтилборана, в (2) Chemistry Letters, 1980, 1415 описана реакция, протекающая в присутствии трибутилборана, а в (3) Chemistry Letters, 1987, 1923 описана реакция, протекающая в присутствии диэтилметоксиборана.
Кроме того, в настоящее время для обработки соединения бора после завершения реакции восстановления согласно (1) и (2) реакционный раствор, содержащий тетрагидрофуран в качестве растворителя, используемого для проведения реакции, непосредственно выливают в 30%-ный водный раствор пероксида водорода, после чего растворитель, который отделяется от воды, добавляют для проведения операции экстракции для получения продукта, а согласно (3) после завершения реакции добавляют метанол, и осуществляют азеотропную отгонку с метанолом, с проведением последующей обычной операции экстракции для получения продукта.
В соответствии с упомянутым ранее, при проведении реакции восстановления β-гидроксикетона в присутствии соединения бора необходимо осуществлять обработку соединения бора после завершения реакции восстановления для эффективного получения продукта.
При использовании способа обработки соединения бора после завершения реакции, которая описана в приведенных выше источниках информации (1), (2) и (3), в лабораторных условиях при проведении реакции на экспериментальном уровне для получения продукта в небольшом количестве особенно серьезных проблем не возникает, но при увеличении масштаба производства способами (1) и (2) присущи экологические проблемы, связанные с обработкой жидких отходов в результате использования большого количества водного раствора пероксида водорода, и способом (3) в том случае, если не использовать значительное количество метанола, обработка соединения бора не может быть осуществлена полностью, и это создает проблему, связанную с недостаточной эффективностью процесса получения продукта. Таким образом, такие способы вряд ли могут быть отнесены к успешно используемым в промышленности способам.
Кроме того, что касается соединения согласно настоящему изобретению, то было установлено, что для того, чтобы выделить желаемое соединение из продукта, получаемого после завершения реакции восстановления, необходимо проводить обработку соединения бора после завершения реакции восстановления, или же, другими словами, желаемое соединение не может быть легко выделено.
В соответствии с этим целью настоящего изобретения является создание простого и пригодного для использования в промышленности способа получения (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата.
Сущность изобретения
Авторы настоящего изобретения провели серьезные исследования для того, чтобы решить указанные проблемы, и в результате этого разработали способ получения, пригодный для использования в промышленности, с точки зрения обработки вышеупомянутых жидких отходов и эффективности производства, что составляет сущность настоящего изобретения.
Таким образом, настоящее изобретение относится к способу получения 7-хинолинил-3,5-дигидроксигепт-6-еноата, представленного формулой (IV):
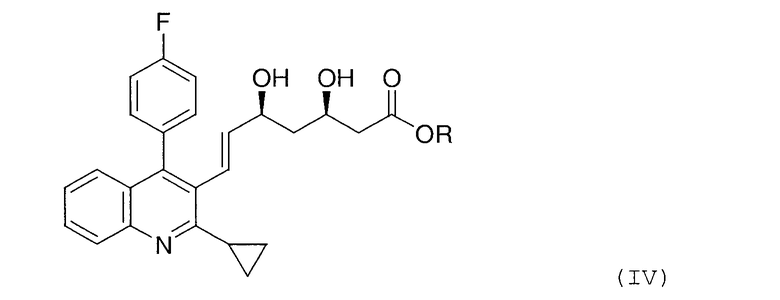
(в которой R представляет собой алкильную группу или арильную группу), отличающемуся тем, что соединение, представленное формулой (I):
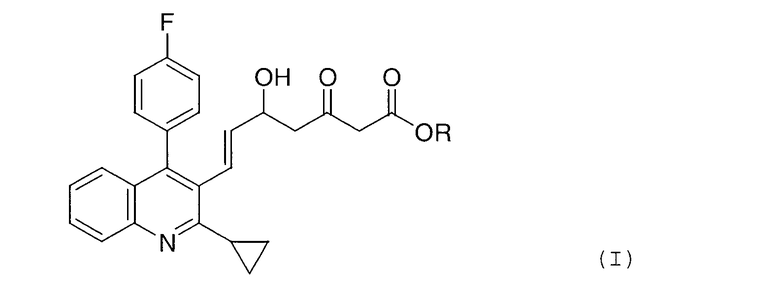
(в которой R является таким, как определено выше), или соединение, представленное формулой (II):
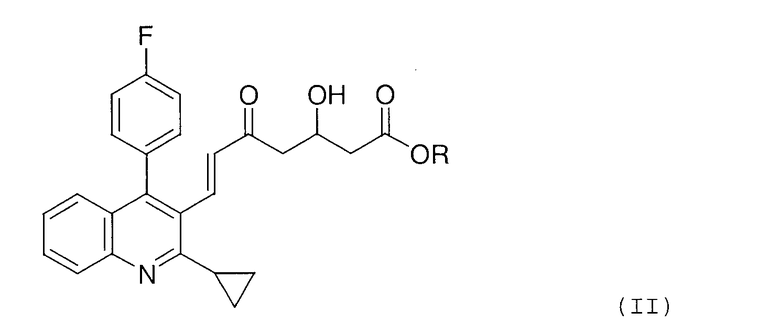
(в которой R является таким, как определено выше), подвергают восстановлению боргидридом натрия в присутствии соединения бора, представленного формулой (III):
R′OBR′′2 (III)
(в которой R' и R" независимо представляют собой алкильную группу), с последующей обработкой полученной реакционной смеси водным раствором пероксида водорода.
Наиболее предпочтительный способ осуществления изобретения
Далее настоящее изобретение будет описано более подробно.
Ниже приведены разъяснения относительно различных терминов, используемых в настоящем описании.
В настоящем описании "н" означает нормальный, "i" - изо, "втор" - вторичный, "трет" - третичный, "с" - цикло, "п" - пара и "о" - орто.
Заместитель R представляет собой алкильную группу или арильную группу.
Алкильная группа представляет собой линейную, разветвленную или циклическую алкильную группу и может представлять собой, например, С1-4 алкильную группу, такую как метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклобутил, 1-метилциклопропил или 2-метилциклопропил.
Арильная группа может представлять собой, например, фенильную группу.
Что касается заместителя R, то предпочтительными являются, например, метил и этил.
Заместители R′ и R′′ независимо представляют собой алкильную группу.
Указанная алкильная группа представляет собой линейную, разветвленную или циклическую алкильную группу и может представлять собой, например, С1-4 алкильную группу, такую как метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклобутил, 1-метилциклопропил или 2-метилциклопропил.
Что касается заместителей R′ и R′′, то в качестве предпочтительных могут быть указаны, например, метил и этил.
Соединение, представленное формулой (I) или (II), используемое в качестве исходного вещества, может быть получено, например, по методике, описанной в JP-A-1-279866, JP-A-8-92217 и JP-A-8-127585.
Настоящее изобретение применяется для осуществления способа получения, в соответствии с которым соединение, представленное формулой (I) или (II) и используемое в качестве исходного вещества, представляет собой оптически активное вещество. В качестве такого оптически активного вещества могут быть указаны 5S-форма соединения (I) или 3R-форма соединения (II).
В качестве соединения бора формулы (III) обычно может быть использован коммерчески доступный продукт. Например, могут быть упомянуты диэтилметоксиборан, дибутилметоксиборан, диэтилэтоксиборан, дибутилэтоксиборан. В качестве предпочтительного соединения может быть указан диэтилметоксиборан.
Количество используемого соединения бора находится в интервале от 0,1 до 5-кратного мольного избытка, предпочтительно в интервале от 0,8 до 3-кратного мольного избытка, особенно предпочтительно, в интервале от 1 до 1,5-кратного мольного избытка, в расчете на субстрат - исходное вещество.
Количество боргидрида натрия, используемого в качестве восстанавливающего агента, находится в интервале от 0,5 до 5-кратного мольного избытка, предпочтительно в интервале от 0,8 до 2,5-кратного мольного избытка, в расчете на субстрат - исходное вещество.
Выбор растворителя, используемого для проведения реакции восстановления, особенно не ограничен, если только он инертен к реакционной системе. Например, могут быть указаны ароматический углеводород, например, такой как бензол, толуол, ксилол, мезителен, хлорбензол, или о-дихлорбензол, алифатический углеводород, например, такой как н-гексан, циклогексан, н-октан или н-декан, галогенированный углеводород, например, такой как дихлорметан, дихлорэтан, хлороформ или четыреххлористый углерод, простой эфир, например, такой как тетрагидрофуран (ТГФ), диэтиловый эфир, трет-бутилметиловый эфир или диметоксиэтан, или спирт, например, такой как метанол, этанол, н-пропанол, изопропанол или н-бутанол. В качестве предпочтительных растворителей могут быть указаны, например, толуол, тетрагидрофуран (ТГФ) или метанол. Более предпочтительным является смешанный растворитель, представляющий собой смесь тетрагидрофурана и метанола.
Количество растворителя, используемого для проведения реакции, находится в интервале от 2-х до 100-кратного избытка по массе, предпочтительно в интервале от 5- до 30-кратного избытка по массе, в расчете на субстрат - исходное вещество.
Температура реакции находится в интервале от -100°С до 0°С, предпочтительно, в интервале от -100°С до -30°С, более предпочтительно, в интервале от -90°С до -60°С.
В соответствии с настоящим изобретением восстановление боргидридом натрия может быть осуществлено либо таким образом, что субстрат формулы (I) или (II) и борсодержащее соединение формулы (III) растворяют в растворителе, после чего, при установленной температуре, добавляют боргидрид натрия, либо таким образом, что борсодержащее соединение формулы (III) и боргидрид натрия сначала помещают в растворитель, а затем по каплям добавляют субстрат формулы (I) или (II).
Настоящее изобретение отличается тем, что после завершения реакции восстановления проводят обработку водным раствором пероксида водорода, для обработки борсодержащего соединения. Однако в том случае, когда в качестве растворителя для проведения реакции используют ароматический углеводород, алифатический углеводород, галогенированный углеводород, нерастворимый в воде простой эфир или подобный им растворитель, предпочтительно, реакционную смесь один раз промыть водой, после чего для проведения обработки добавить водный раствор пероксида водорода.
Кроме того, в том случае, когда в качестве растворителя для проведения реакции используют водорастворимый растворитель, такой как тетрагидрофуран или спирт, предпочтительно добавить растворитель, который отделяется от воды, например толуол, а растворитель, который растворим в воде, используемый для проведения реакции, отогнать, после чего для проведения обработки добавить водный раствор пероксида водорода.
В качестве растворителя для проведения обработки водным раствором пероксида водорода предпочтительным является растворитель, который отделяется от воды. Например, могут быть использованы ароматический углеводород, например, такой как бензол, толуол, ксилол, мезителен, хлорбензол или о-дихлорбензол, алифатический углеводород, например, такой как н-гексан, циклогексан, н-октан или н-декан, или галогенированный углеводород, например, такой как дихлорметан, дихлорэтан, хлороформ или четыреххлористый углерод. В качестве предпочтительного растворителя может быть указан, например, толуол.
Количество используемого растворителя находится в интервале от 2-х до 100-кратного избытка по массе, предпочтительно в интервале от 5- до 30-кратного избытка по массе, в расчете на субстрат - исходное вещество.
Что касается водного раствора пероксида водорода, то его концентрация особенно не ограничена, но с точки зрения эффективности операции и т. п. обычно предпочтителен коммерчески доступный 35%-ный водный раствор пероксида водорода, с которым удобно работать.
Количество водного раствора пероксида водорода, которое следует использовать, может составлять значительный избыток для ускорения реакции, но с точки зрения охраны окружающей среды, целесообразно, чтобы это количество находилось в интервале от эквимольного до 50-кратного мольного избытка, предпочтительно в интервале от эквимольного до 20-кратного мольного избытка, в расчете на субстрат.
Температура находится в интервале от 0°С до 100°С, предпочтительно в интервале от 10°С до 50°С.
Продолжительность обработки может значительно варьироваться в зависимости от используемого растворителя, количества пероксида водорода и температуры, но составляет от 1 до 100 часов.
Кроме того, возможно ускорить реакцию посредством введения неорганического основания во время обработки водным раствором пероксида водорода.
Неорганическое основание может, например, представлять собой гидроксид, например, такой как гидроксид натрия, гидроксид калия, гидроксид магния или гидроксид кальция, или может представлять собой карбонат, например, такой как карбонат натрия, карбонат калия, карбонат магния или карбонат кальция. Предпочтительно использовать карбонат натрия или карбонат калия.
Количество неорганического основания, которое следует использовать, находится в интервале от 0,1 до 20-кратного мольного избытка, предпочтительно в интервале от 0,5 до 5-кратного мольного избытка, в расчете на субстрат.
После завершения реакции водный раствор пероксида водорода отделяют, после чего дополнительно проводят промывку водой, и при необходимости, обработку восстанавливающим агентом, например, таким как сульфит натрия с последующей перекристаллизацией из толуола или смешанного растворителя, представляющего собой смесь толуола с другим растворителем, для выделения (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата.
Далее, при необходимости, осуществляют перекристаллизацию из смешанного растворителя, представляющего собой смесь этилацетата и н-гептана для получения (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата высокой чистоты.
Примеры
Далее настоящее изобретение описано более подробно с помощью примеров, но приведенные примеры никоим образом не ограничивают объем настоящего изобретения.
В соответствии с примерами количественный анализ (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата осуществляли ВЭЖХ (высокоэффективной жидкостной хроматографией) в следующих условиях.
Колонка: L-колонка ODS, (изготовитель Foundation Chemical Evaluation and Research Institute, Япония).
Элюент: смесь этанол/ТГФ/0,01 М ацетат аммония в соотношении 45/3/52.
Температура колонки: 40°С. Скорость потока: 1,0 мл/мин.
Длина волны, при которой проводят детектирование: 254 нм.
Время удерживания: приблизительно 27 минут.
Пример 1
После продувки колбы для проведения реакции азотом, этил- (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-5-гидрокси-3-оксогепт-6-еноат (далее обозначаемый как MOLE) (29,90 г, 66,8 ммоль) растворяют в ТГФ (148,7 г) и метаноле (54,9 г) с последующим охлаждением до -75°С.
После продувки другой колбы для проведения реакции азотом в указанную колбу помещают ТГФ (43,2 г) и диэтилметоксиборан (1,0 М раствор в ТГФ, 80 мл) и далее добавляют боргидрид натрия (3,31 г, 87,5 ммоль). Полученную суспензию охлаждают до -75°С и добавляют по каплям приготовленный заранее раствор MOLE/ТГФ/метанол при температуре от -75°С до -70°С.
После завершения добавления по каплям перемешивание продолжают дополнительно в течение 1 часа при -75°С, после чего реакционный раствор добавляют по каплям в реакционную колбу, содержащую уксусную кислоту (6,5 мл) и толуол (10 г), добавленные для того, чтобы остановить реакцию.
Реакционный раствор нагревают до температуры от 35°С до 40°С, после чего отгоняют ТГФ и метанол при пониженном давлении. После завершения отгонки добавляют толуол (311 г) для растворения, и полученный органический слой дважды промывают водой (230 г).
В результате получают органический слой в количестве 379,6 г.
Полученный органический слой подвергают количественному анализу методом ВЭЖХ, при этом восстановленная форма продукта (включая координированную с бораном форму) содержится в количестве 27,93 г (выход: 93%).
После этого органический слой анализируют методом ЯМР для определения продукта этил-(Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата (далее в настоящем описании обозначаемый как DOLE) и его координированной с бораном формы и при этом устанавливают, что присутствует 20% координированной с бораном формы.
После этого отбирают 17,7 г (с содержанием DOLE, составляющим 1,30 г, 2,9 ммоль) из полученного органического слоя и добавляют к нему безводный карбонат натрия (307 мг, 2,9 ммоль) и 35%-ный водный раствор пероксида водорода (2,8 г, 29 ммоль), с последующим перемешиванием при температуре от 30°С до 35°С в течение 3 часов.
Реакционный раствор анализируют методом ЯМР в отношении содержания координированной с бором формы, при этом в соответствии с полученными результатами содержание координированной с бором формы составляет 0%.
После завершения обработки водным раствором пероксида водорода осуществляют жидкостное разделение и затем добавляют воду (3,8 г) для промывки водой с последующей промывкой 4%-ным водным раствором пиросульфита натрия (4,0 г) и затем дважды промывают водой (3,8 г).
Полученный органический слой количественно исследуют методом ВЭЖХ и при этом устанавливают, что DOLE присутствует в количестве 1,28 г.
Органический слой нагревают до температуры от 40°С до 50°C, после чего отгоняют толуол при пониженном давлении.
После отгонки продукт перекристаллизовывают из этилацетата (2,56 г) и н-гептана (4,39 г), получая 1,22 г DOLE в виде кристаллов.
Пример 2
Из 379,6 г органического слоя, полученного в соответствии с примером 1 и содержащего 27,93 г продукта (включая 20% координированной с бором формы), отбирают 17,7 г (с содержанием DOLE, составляющим 1,30 г, 2,9 ммоль) и добавляют к этому продукту 50%-ный водный раствор карбоната натрия (800 мг, 2,9 ммоль) и 35%-ный водный раствор пероксида водорода (2,8 г, 29 ммоль) с последующим перемешиванием при температуре от 30°С до 35°С в течение 3 часов.
Реакционный раствор анализируют методом ЯМР на присутствие координированной с бораном формы, при этом в соответствии с полученными результатами содержание координированной с бораном формы составляет 0%.
После завершения обработки водным раствором пероксида водорода промывку водой и т.д. проводят таким же образом, как и в примере 1.
Полученный органический слой количественно исследуют методом ВЭЖХ, и при этом устанавливают, что DOLE содержится в количестве 1,20 г.
Органический слой нагревают до температуры от 40°С до 50°C, после чего отгоняют толуол при пониженном давлении.
После отгонки, продукт перекристаллизовывают из этилацетата (2,56 г) и н-гептана (4,39 г), получая 1,14 г DOLE в виде кристаллов.
Ссылочный пример 1
Из 379,6 г органического слоя, полученного в соответствии с примером 1 и содержащего 27,93 г продукта (включая 20% координированной с бораном формы), отбирают 17,7 г (с содержанием DOLE, составляющим 1,30 г, 2,9 ммоль) и непосредственно нагревают при температуре от 40°С до 50°С, не подвергая обработке водным раствором пероксида водорода в соответствии с настоящим изобретением, после чего отгоняют толуол при пониженном давлении.
После отгонки полученный продукт пытаются перекристаллизовать из этилацетата (2,56 г) и н-гептана (4,39 г), но продукт не кристаллизуется, а просто отделяется маслянистый слой, при этом выделить DOLE в виде кристаллов невозможно.
Промышленная применимость
В соответствии с настоящим изобретением предоставляется возможность получения (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата, который может быть полезен в качестве промежуточного соединения для синтеза ингибитора HMG-CoA-редуктазы, с высоким выходом и возможностью использования в промышленности.
Изобретение относится к способу получения 7-хинолинил-3,5-дигидроксигепт-6-еноата, представленного формулой (IV):
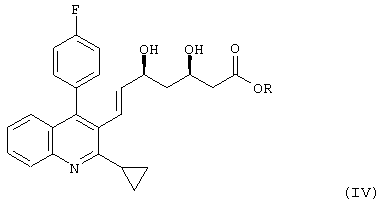
(в которой R представляет собой алкильную группу), заключающемуся в том, что соединение, представленное формулой (I):
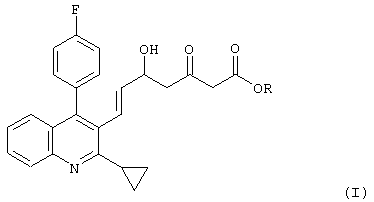
(в которой R является таким, как определено выше), или соединение, представленное формулой (II):
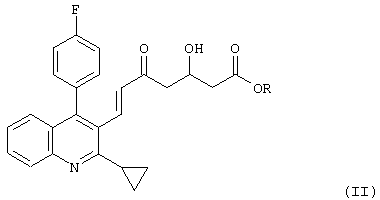
(в которой R является таким, как определено выше), подвергают восстановлению боргидридом натрия в присутствии соединения бора, представленного формулой (III):

(в которой R' и R" независимо представляют собой алкильную группу), с последующей обработкой полученной реакционной смеси водным раствором пероксида водорода в присутствии неорганического основания, которую проводят в двухфазной системе с использованием органического растворителя, который отделяют от воды. 7-Хинолинил-3,5-дигидроксигепт-6-еноат может быть полезен в качестве промежуточного соединения для синтеза фармацевтических средств с высоким выходом и высокой чистоты. Технический результат - разработка простого и пригодного для использования в промышленности способа получения (Е)-7-[2-циклопропил-4-(4-фторфенил)хинолин-3-ил]-3,5-дигидроксигепт-6-еноата. 1 н. и 4 з.п. ф-лы.
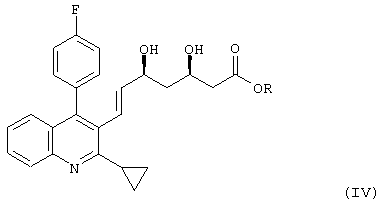
(в которой R представляет собой алкильную группу), отличающийся тем, что соединение, представленное формулой (I)
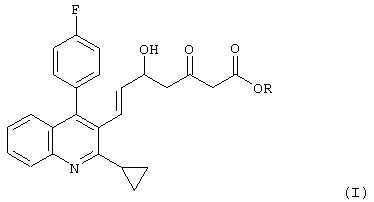
(в которой R является таким, как определено выше), или соединение, представленное формулой (II)
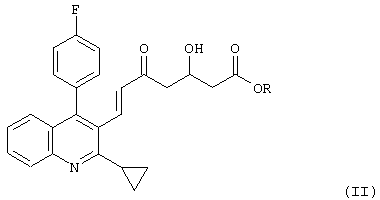
(в которой R является таким, как определено выше), подвергают восстановлению боргидридом натрия в присутствии соединения бора, представленного формулой (III)

(в которой R' и R" независимо представляют собой алкильную группу), с последующей обработкой полученной реакционной смеси водным раствором пероксида водорода в присутствии неорганического основания, которую проводят в двухфазной системе с использованием органического растворителя, который отделяют от воды.
| ТОКАРНЫЙ ПАТРОН | 0 |
|
SU304063A1 |

