Настоящее изобретение относится к улучшениям химического способа, в частности химического способа получения кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты (1) (изображена ниже), пригодной для изготовления фармацевтического препарата, полезного в лечении в числе прочего гиперхолистеринемии, гиперлипопротеинемии и атеросклероза.
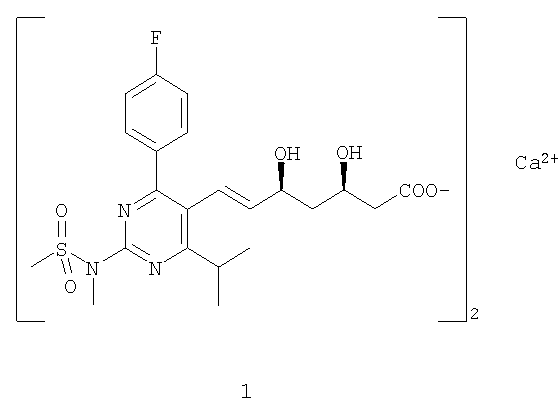
Натриевая соль (2) и кальциевая соль (1) соединения (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновая кислота (упоминаемого ниже как "Агент") раскрыты в европейском патенте 0521471. В данном патенте также описан способ синтеза кальциевой соли (1) через натриевую соль (2), как показано ниже на Схеме 1. Кальциевую соль, образованную таким образом, затем собирают и сушат, и затем ее можно обрабатывать по мере надобности.
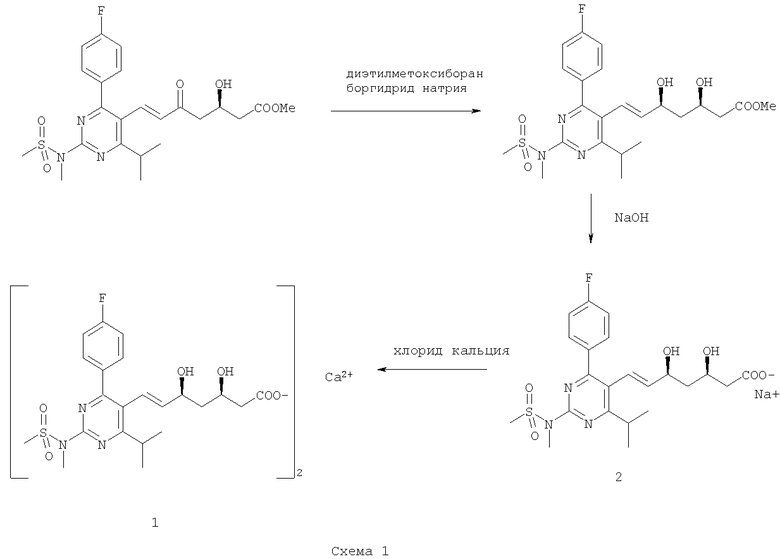
В международной заявке WO 00/49014 (Astrazeneca) описан альтернативный путь получения кальциевой соли (1), также через натриевую соль (2), из соединения ВЕМ (3), который показан ниже на Схеме 2:
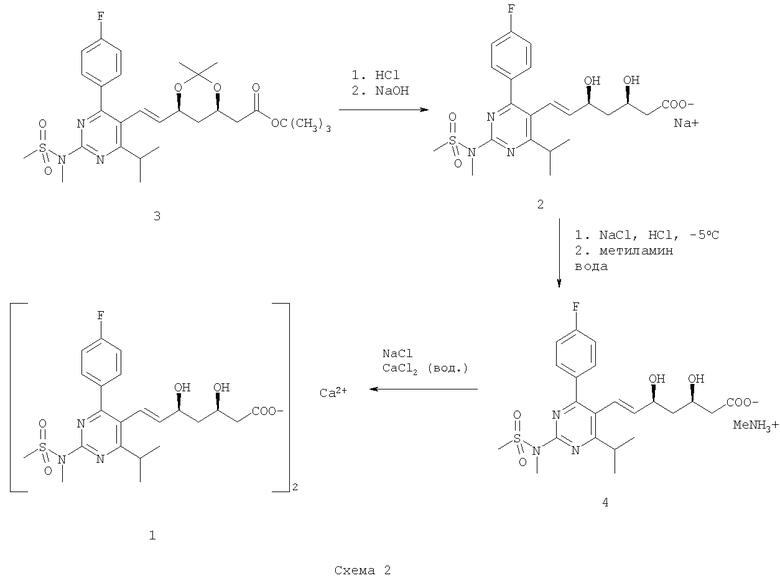
Как описано в WO 00/49014, превращение ВЕМ (3) в кальциевую соль (1) можно осуществить через метиламиновую соль (4), как показано на Схеме 2. Выделение данной промежуточной кристаллической метиламиновой соли позволяет произвести очистку путем перекристаллизации перед окончательным образованием (аморфной) кальциевой соли.
В заявке WO 2004/014872 (Astrazeneca), находящейся на рассмотрении одновременно с настоящей заявкой, описан улучшенный способ выделения кальциевой соли из водорастворимой соли, такой как превращение метиламиновой соли (4) в кальциевую соль (1), показанное на Схеме 2 выше, где улучшение включает регулировку временных и температурных параметров таким образом, чтобы получить оптимальную физическую форму продукта.
Авторы изобретения неожиданно изобрели улучшение способа получения кальциевой соли, результатом которого является улучшение суммарного выхода и уменьшение количества стадий для осуществления превращения ВЕМ (3) в кальциевую соль (1), посредством чего исключается стадия выделения промежуточной соли. Неожиданно, что это не влияло неблагоприятно на качество получаемого в виде кальциевой соли продукта. Способ по настоящему изобретению также является пригодным для алкильных эфиров агента, отличных от трет-бутилового эфира ВЕМ (3).
Согласно настоящему изобретению предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а)-(ж):
а) взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в смешиваемом с водой органическом растворителе с водным раствором кислоты при повышенной температуре;
б) взаимодействие полученного раствора с водным гидроксидом щелочного метала и, возможно, промывание полученного водного раствора соли щелочного металла подходящим органическим растворителем;
в) доведение рН полученного раствора до значения в интервале от рН 6 до рН 11;
г) удаление смешиваемого с водой органического растворителя;
д) возможная фильтрация полученной смеси;
е) добавление водорастворимой кальциевой соли к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, и
ж) выделение продукта со стадии е).
Следует принять во внимание, что в данном способе превращение сложного эфира в кальциевую соль (1) достигается без выделения промежуточной соли указанной кислоты.
Стадия а)
Подходящие для стадии а) растворители в общем случае представляют собой любой смешиваемый с водой органический растворитель, например такие растворители, как ацетонитрил и ацетон. Предпочтительным растворителем является ацетонитрил.
Подходящие водные кислоты представляют собой кислоты, кальциевая соль которых является водорастворимой, так чтобы она не выпадала в осадок на стадии е). В одном воплощении водная кислота представляет собой соляную кислоту. В одном аспекте данного воплощения концентрация водной соляной кислоты составляет приблизительно 0,1 М. В другом аспекте данного воплощения концентрация водной соляной кислоты меньше или равна приблизительно 0,1 М. Обычно концентрация водной соляной кислоты менее 0,05 М, например 0,02 М.
Взаимодействие (1-6С)алкилового эфира Агента с водной кислотой целесообразно проводить при температуре от 30 до 50°С, удобно - при температуре от 35 до 40°С.
Более целесообразно, когда (1-6С)алкиловый эфир Агента, растворенный в ацетонитриле при 35°С, взаимодействует с водной соляной кислотой при 35°С.
Подходящие (1-6С)алкиловые эфиры Агента представляют собой, например, метиловый, этиловый, пропиловый, изопропиловый, н-бутиловый, изобутиловый, пентиловый или гексиловый эфиры. ВЕМ представляет собой предпочтительный пример (1-6С)алкилового эфира (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты.
Исходное вещество ВЕМ (3) можно получить, как описано в WO 00/49014. Аналоги ВЕМ (3) можно получить аналогичными способами, как показано в примерах ниже.
Стадия б)
Стадию б) можно осуществить при температуре от приблизительно 10°С до приблизительно 40°С. Удобно стадию б) проводить при температуре окружающей среды, которая в общем случае предполагается равной 20-25°С, удобно - при приблизительно 25°С.
Пригодный водный гидроксид щелочного металла представляет собой водный гидроксид калия или водный гидроксид натрия.
В одном воплощении водный гидроксид щелочного металла представляет собой гидроксид натрия. В этом воплощении подходящий водный гидроксид натрия имеет концентрацию приблизительно 1 М и его добавляют в количестве, достаточном для образования натриевой соли (2). Следует принять во внимание, что натриевую соль (2) не выделяют и что продукт реакции стадии б) представляет собой водный раствор натриевой соли. Также следует принять во внимание, что данный водный раствор натриевой соли также содержит ацетонитрил.
Водный раствор соли щелочного металла можно промывать толуолом или другим походящим органическим растворителем, для того чтобы перед осуществлением стадии в) удалить непрореагировавший (1-6С)алкиловый эфир Агента, такой как ВЕМ (3), или, если требуется, другие нежелательные минорные компоненты. В общем случае подходящими органическими растворителями для данной стадии промывания являются органические растворители, которые не смешиваются с водой, но смешиваются со смешиваемым с водой органическим растворителем, используемым на стадии а). Если смешиваемый с водой органический растворитель на стадии а) представляет собой ацетонитрил, подходящие органические растворители для стадии промывания представляют собой известные в данной области техники сложноэфирные, эфирные и углеводородные растворители. Примерами таких подходящих растворителей являются ксилол (углеводородный растворитель), метил-трет-бутиловый эфир (МТВЕ) (эфирный растворитель) и этилацетат (сложноэфирный растворитель). Толуол или другой пригодный органический растворитель можно удобно удалять из процесса путем разделения фаз. Любой растворитель, оставшийся после разделения фаз, может быть удален на стадии г). Предпочтительно этот растворитель представляет собой толуол.
В одном воплощении водная соль щелочного металла представляет собой водную натриевую соль. В этом воплощении на стадии б) водный раствор натриевой соли промывают подходящим органическим растворителем. В одном аспекте данного воплощения водный раствор натриевой сопи промывают толуолом, ксилолом, МТВЕ или этилацетатом. В другом аспекте данного воплощения водный раствор натриевой соли промывают толуолом или ксилолом. В другом аспекте данного воплощения водный раствор натриевой соли промывают толуолом. В другом аспекте данного воплощения водный раствор натриевой соли промывают МТВЕ. В другом аспекте данного воплощения водный раствор натриевой соли промывают этилацетатом.
В другом воплощении на стадии б) водный раствор натриевой соли не промывают подходящим органическим растворителем.
В альтернативном воплощении данного изобретения водный гидроксид щелочного металла представляет собой гидроксид калия. Следует принять во внимание, что в данном воплощении в результате образуется калиевая соль, эквивалентная натриевой соли (2). В данном воплощении подходящие температуры, концентрации гидроксида калия и промывающих растворителей те, что описаны выше как пригодные для гидроксида натрия.
Стадия в)
Доводить водный раствор до рН 6-11 удобно путем добавления соляной кислоты, например 0,02 М -1 М водной соляной кислоты. В одном воплощении раствор доводят до рН 8-11. В другом воплощении раствор доводят до рН 9-11, например до приблизительно рН 9-10,5. Целесообразно доводить раствор до приблизительно рН 9-10,5, используя менее чем 0,1 М или 0,1 М соляную кислоту. Более целесообразно доводить раствор до приблизительно рН 10,5, используя приблизительно 0,1 М соляную кислоту. Предпочтительно раствор доводят до приблизительно рН 9, для чего целесообразно использовать 0,02 М водную соляную кислоту. Также могут быть использованы другие неорганические кислоты, известные в данной области техники, при условии что кальциевая соль данной неорганической кислоты является водорастворимой, так чтобы она не выпадала в осадок на стадии е).
Стадия г)
Смешиваемый с водой органический растворитель (и остаточные количества любого органического растворителя, используемого в качестве промывки на стадии б) выше) в общем случае можно удалить посредством дистилляции, которую удобно осуществлять под вакуумом.
Если смешиваемый с водой органический растворитель представляет собой ацетонитрил, дистилляцию целесообразно осуществлять, например, используя вакуум меньший или равный 55 мбар (5,5·103 Па) и температуру, меньшую или равную 45°С. Удобно, когда вакуум составляет приблизительно 52 мбар (5,2·103 Па), а температура составляет приблизительно 33°С. Специалистам в данной области техники необходимо принимать во внимание, что в процессе дистилляции вместе с ацетонитрилом азеотропно может быть удалена вода, и, следовательно, может оказаться желательным добавить дополнительное количество воды в смесь во время процесса дистилляции. Подходящий способ осуществления дистилляции предложен в прилагаемом неограничивающем примере.
Стадия д)
Посредством фильтрация смеси, полученной на стадии г), удаляют любое непрореагировавшее исходное вещество или нерастворимые примеси, которые могли выпасть в осадок во время процесса дистилляции на стадии г). Необходимо принимать во внимание, что для промывки фильтра можно использовать воду. Можно использовать любой фильтр, известный как пригодный в данной области техники. В промышленном масштабе удобно может быть использован GaF-фильтр (например, GaF-фильтр Е6-1825, производимый "Haywood Industrial Products").
Очевидно, что данная фильтрация не всегда является необходимой и может быть пропущена.
Стадия е)
В общем случае подходящая водорастворимая кальциевая соль представляет собой такую любую соль, противоион которой образует с натрием водорастворимую соль так, что она легко удаляется путем промывания продукта после выделения на стадии ж). Подходящие водорастворимые кальциевые соли включают хлорид кальция, бромид кальция и ацетат кальция. Наиболее целесообразно использовать хлорид кальция или бромид кальция.
В одном воплощении водорастворимая кальциевая соль представляет собой хлорид кальция.
В данном воплощении хлорид кальция обычно применяют в виде его дигидратированной формы, которую целесообразно добавлять к фильтрату в виде водного раствора. Можно использовать небольшой избыток хлорида кальция, например 0,6 молярных эквивалентов, по сравнению с Агентом. Хлорид кальция целесообразно добавлять в виде водного раствора с концентрацией 0,1 г/мл. В процессе добавления температуру реакционной смеси целесообразно поддерживать при 32-43°С, более целесообразно при приблизительно 40°С. Скорость добавления хлорида кальция можно регулировать так, чтобы температура реакционной смеси поддерживалась таким образом. Хлорид кальция целесообразно добавлять за 15-20 минут. Смесь можно поддерживать при температуре добавления в течение промежутка времени (здесь именуемого "временем выдерживания") перед выделением кальциевой соли (1). В одном воплощении время выдерживания составляет по меньшей мере 10 минут. В другом воплощении время выдерживания составляет по меньшей мере 20 минут. В еще одном воплощении время выдерживания составляет по меньшей мере 30 минут.
Стадия ж)
Выделение кальциевой соли удобно может быть выполнено путем фильтрации, обычно при температуре приблизительно 20°С (здесь именуемой "температурой фильтрации"). Смесь можно поддерживать при температуре фильтрации в течение промежутка времени до осуществления фильтрации, например в течение 10 до 20 минут, удобно в течение 15 минут. Очевидно, что для промывания фильтрата можно использовать воду.
Согласно настоящему изобретению предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[мeтил(мeтилсульфoнил)aминo]пиpимидин-5-ил](3R,5S)-3,5-дигидpoкcигепт-6-еновой кислоты, включающий следующие стадии (а)-(ж):
а) взаимодействие трет-бутил-(Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)ацетата (ВЕМ) в ацетонитриле с водной соляной кислотой при повышенной температуре;
б) взаимодействие полученного раствора с водным гидроксидом натрия;
в) доведение рН полученного раствора до значения в интервале от рН 6 до рН 11;
г) удаление ацетонитрила;
д) фильтрация полученной смеси;
е) добавление хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-5-еновой кислоты; и
ж) выделение продукта стадии е).
В другом аспекте настоящего изобретения предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий стадии а), б), в), г), е) и ж), как описано выше.
Согласно настоящему изобретению предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при повышенной температуре;
б) взаимодействие полученного раствора с водным гидроксидом натрия;
в) доведение рН полученного раствора до значения интервале от рН 6 до рН 11;
г) удаление ацетонитрила;
д) фильтрация полученной смеси;
е) добавление хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](2R,5S)-3,5-дигидроксигепт-6-еновой кислоты; и
ж) выделение продукта стадии е).
В другом аспекте настоящего изобретения предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий стадии а'), б), в), г), е) и ж), как описано выше.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а)-(ж):
а) взаимодействие трет-бутил-(Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)ацетата (ВЕМ) в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли подходящим органическим растворителем;
в) доведение рН полученного раствора до приблизительно рН 9 путем добавления менее 0,05 М водной соляной кислоты;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,55S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
6) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли подходящим органическим растворителем;
в) доведение рН полученного раствора до приблизительно рН 9 путем добавления менее 0,05 М водной соляной кислоты;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изoпpoпил-2-[мeтил(мeтилcyльфoнил)aминo]пиpимидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[мeтил(мeтилcyльфoнил)aминo]пиpимидин-5-ил](3R,5S)-3,5-дигидpoкcигeпт-6-еновой кислоты, включающий следующие стадии (а)-(ж):
а) взаимодействие трет-бутил-(Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)ацетата (ВЕМ) в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли подходящим органическим растворителем;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли подходящим органическим растворителем;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли подходящим углеводородным, сложноэфирным или эфирным растворителем;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли толуолом, ксилолом, МТВЕ или этилацетатом;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и, возможно, промывание полученного водного раствора натриевой соли толуолом;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и промывание полученного водного раствора натриевой соли подходящим углеводородным, сложноэфирным или эфирным растворителем;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и промывание полученного водного раствора натриевой соли толуолом, ксилолом, МТВЕ или этилацетатом;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий следующие стадии (а')-(ж):
а') взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в ацетонитриле с водной соляной кислотой при температуре 35-40°С;
б) взаимодействие полученного раствора с 1 М гидроксидом натрия при температуре окружающей среды и промывание полученного водного раствора натриевой соли толуолом;
в) доведение рН полученного раствора до приблизительно рН 9-10,5, используя ≤0,1 М водную соляную кислоту;
г) удаление ацетонитрила путем дистилляции при давлении 50-55 мбар (5,0-5,5·103 Па) и температуре 30-35°С;
д) фильтрация полученной смеси;
е) добавление водного раствора дигидрата хлорида кальция к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, при 32-43°С; и
ж) выделение продукта стадии е) путем фильтрации при температуре приблизительно 20°С.
В другом воплощении в настоящем изобретении предложен улучшенный способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий стадии (а')-(ж), как описано в любом аспекте или воплощении выше или ниже, где на стадии б) вместо гидроксида натрия применяют гидроксид калия.
Способ по настоящему изобретению в общем случае приводит к увеличению суммарного процентного выхода продукта (начиная с ВЕМ или другого (1-6С)алкилового эфира) и уменьшению количества стадий по сравнению со способами, известными в данной области техники. Очевидно, что более высокий процентный выход продукта может обеспечить значительный выигрыш по стоимости, когда получение происходит в промышленных масштабах. Уменьшенное количество стадий в способе по настоящему изобретению приводит к меньшему количеству операционных процессов в процессе изготовления, которые могут быть осуществлены в более надежном способе. Уменьшенное количество стадий в способе по настоящему изобретению включает в себя уменьшенную обработку вещества, что может приводить к уменьшенной возможности расщепления и загрязнения продукта. Кроме того, более не требуются некоторые химические реагенты, и уменьшается общее количество отходов и/или сбросов, что является полезным для окружающей среды.
В другом аспекте настоящего изобретения предложено соединение - кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, полученная посредством стадий (а)-(ж) способа, как описано выше.
В другом аспекте настоящего изобретения предложено соединение - кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, полученная посредством стадий (а')-(ж) способа, как описано выше в любом аспекте или воплощении данного изобретения.
Следовательно, в другом аспекте настоящего изобретения предложен продукт, который можно получить способом по настоящему изобретению.
В другом аспекте настоящего изобретения предложен продукт, полученный способом по настоящему изобретению.
Очевидно, что способ по настоящему изобретению можно применять для получения других солей Агента, таких как магниевая соль, путем использования на стадии е) подходящей магниевой соли, такой как хлорид магния. Полученную таким образом соль можно превратить в кальциевую соль (1) способами, известными в данной области техники. Таким образом, в другом аспекте настоящего изобретения предложен способ получения магниевой соли Агента, включающий стадии (а)-(ж), как описано выше, где на стадии (е) вместо водорастворимой кальциевой соли (такой как хлорид кальция) добавляют водорастворимую соль магния (такую как хлорид магния).
Настоящее изобретение далее проиллюстрировано следующими примерами.
Пример 1
ВЕМ (20,0 г) растворяли в ацетонитриле (400 мл) при 40°С, затем охлаждали до 35°С, после чего постепенно добавляли соляную кислоту (0,02 М, 35 мл) при 35°С. Полученный раствор перемешивали при 35°С до завершения реакции, затем охлаждали до 25°С. При 25°С добавляли гидроксид натрия (1,0 М, 38 мл), и полученную смесь перемешивали при этой температуре до завершения реакции. Чтобы довести рН раствора до рН 9, добавляли водную соляную кислоту (1 М). Раствор подвергали дистилляции при пониженном давлении (52 мбар (5,2·103 Па), ≤40°С), пока не было удалено приблизительно 100 мл смеси ацетонитрил/вода. Добавляли воду (100 мл), и дистилляцию продолжали, пока не было удалено еще 100 мл смеси ацетонитрил/вода. Полученную смесь фильтровали через фильтровальную подушку, фильтр промывали водой (30 мл), и фильтраты нагревали до 40°С, затем добавляли раствор дигидрата хлорида кальция (3,07 г) в воде (29,5 мл) на протяжении 20 минут, поддерживая реакционную смесь при 38-41°С.
Реакционную смесь перемешивали в течение еще 15 минут при 40°С, затем охлаждали до 20°С и перемешивали при этой температуре в течение следующих 15 минут. Полученную суспензию фильтровали, промывали водой (3×50 мл) и сушили с получением кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты (15,5 г, 84%-ный выход).
Пример 2
Синтез аналогов ВЕМ показан ниже для изопропилового аналога. Другие аналоги могут быть получены аналогичными методами.
Изопропил-(Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)ацетат
Бис(триметилсилил)амид натрия (80,47 мл, 1,0 М в THF (тетрагидрофуране) по каплям добавляли к охлажденному раствору дифенил-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-илметил]фосфиноксида (40,43 г, 75 мМ) в THF (477,1 мл) при -65°С в течение 30 минут, поддерживая температуру -65°С. К данному раствору на протяжении 35 минут по каплям добавляли изопропил-2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетат в толуоле (21,68 г), поддерживая температуру -65°С. Содержимое сосуда выдерживали при -65°С в течение 15 минут, затем оставляли равномерно нагреваться до 10°С на протяжении 80 минут. Добавляли воду (40,4 мл), затем уксусную кислоту (6,87 г, 114 мМ) с получением двухфазного светло-желтого раствора. Эту партию затем подвергали дистилляции при атмосферном давлении с удалением приблизительно 485 мл продуктов дистилляции. Раствор последовательно промывали водой (84 мл), 7,0%-ным (мас./мас.) бикарбонатом натрия (92,6 г), 1,8%-ным (мас./мас.) бикарбонатом натрия (91,1 г) и водой (63,5 мл). Полученную органическую фазу подвергали дистилляции под вакуумом при 270 мбар (2,7·104 Па), оставляя в перегонной колбе приблизительно 95 мл раствора (удаляя приблизительно 229 мл продуктов дистилляции). В колбу загружали метанол (202 мл) при 50°С, и раствор подвергали дистилляции при атмосферном давлении, удаляя приблизительно 134 мл продуктов дистилляции. К раствору добавляли дополнительную порцию метанола (229 мл) при 50°С, и эту партию охлаждали до 40°С на протяжении 30 минут. Партию охлаждали до 25°С на протяжении 30 минут, до 0-5°С на протяжении 30 минут, затем охлаждали до -8°С на протяжении 20 минут и выдерживали при этой температуре в течение 30 минут. Твердое вещество собирали вакуум-фильтрацией, промывали двумя порциями охлажденного (-8°С) метанола (2×80,6 мл), затем сушили в вакуум-сушильном шкафу при 50°С, 200 мбар (2,0·104 Па), выход=28,9 г (68,3%).
1H NMR δ: 1,15 (q, 1Н) 1,24 (dd, 6H) 1,27 (dd, 6H) 1,40 (s, 3H) 1,49 (s, 3H) 1,55 (dt, 1Н) 2,34 (dd, 1Н) 2,50 (dd, 1Н) 3,38 (spt, 1Н) 3,51 (s, 3H) 3,57 (s, 3H) 4,32 (m, 1Н) 4,43 (m, 1Н) 5,04 (spt, 1Н) 5,47 (dd, 1Н) 6,52 (d, 1Н) 7,08 (t, 2H) 7,65 (dd, 2H)
Изопропил-2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетат
Хлор (газ) (2469,6 мл, 118 мМ) загружали к толуолу (373,3 мл, 16 относительных объемов) при -60°С. Затем на протяжении 30 минут к этому охлажденному раствору по каплям добавляли диметилсульфид (11,67 мл, 121 мМ), поддерживая содержимое при температуре -60°С. После выдерживания в течение 30 минут при этой температуре в сосуд на протяжении 30 минут по каплям добавляли изопропил-2-[(4R,6S)-6-гидроксиметил-2,2-диметил-1,3-диоксан-4-ил]ацетат (24,56 г, 95 мМ) в толуоле (46,7 мл), поддерживая внутреннюю температуру -60°С. Реакционную смесь перемешивали при -60°С в течение 30 минут, после чего на протяжении 30 минут по каплям добавляли триэтиламин (26,36 г, 261 мМ), позволяя внутренней температуре увеличиться до -50°С. Затем реакционную смесь оставляли равномерно нагреваться до 25°С на протяжении 75 минут. Полученную суспензию перемешивали при 25°С в течение 30 минут, затем добавляли воду (77 мл), и данную смесь перемешивали в течение 30 минут. Водный слой отделяли и проверяли рН (значение рН должно находиться в интервале от 7,5 до 8,5). Полученную органическую часть промывали водой (23,3 мл), и органическую часть отделяли для вакуумной дистилляции при 150 мбар (1,5·104 Па). Дистилляцию продолжали, пока не было удалено приблизительно 350 мл толуола. В колбу добавляли толуол (350 мл), и повторяли вакуумную дистилляцию при 150 мбар (1,5·104 Па) с удалением приблизительно 350 мл толуола. Полученный раствор переносили в колбу, содержащую активированные молекулярные сита с размером пор 4 ангстрема, и оставляли на ночь при температуре окружающей среды. Этот раствор использовали непосредственно для стадии сочетания.
Изопропил-2-[(4R,6S)-6-гидроксиметил-2,2-диметил-1,3-диоксан-4-ил]ацетат
Это соединение может быть получено, используя методы, описанные в ЕР 0319847. Аналоги с различными эфирными группами R могут быть получены аналогичным способом.
Дифенил-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-илметил]фосфиноксид
Это соединение может быть получено, как описано в заявке на патент WO 00/49014.
Пример 3: Методика с использованием промывания на стадии (б)
ВЕМ (20,0 г) растворяли в ацетонитриле (140 мл) при 40°С, затем охлаждали до 35°С, после чего постепенно добавляли соляную кислоту (0,02 М, 35 мл) при 35°С. Полученный раствор перемешивали при 35°С до завершения реакции, затем охлаждали до 25°С. Добавляли дополнительное количество ацетонитрила (8 мл), затем добавляли гидроксид натрия (1,0 М, 38 мл) при 25°С, и полученную смесь перемешивали при этой температуре до завершения реакции. Чтобы довести рН данного раствора до приблизительно рН 10,5, добавляли водную соляную кислоту (0,1 М). Воду добавляли так, что общий объем добавленной воды и соляной кислоты (0,1 М) (из предыдущей стадии регулирования рН) составлял 100 мл. Затем добавляли толуол (125 мл), и смесь перемешивали при 40°С в течение 30 минут, затем оставляли отстаиваться в течение 1 часа при 40°С. Затем при 40°С отделяли водную фазу от органической фазы. Водную фазу подвергали дистилляции при пониженном давлении (53 мбар (5,3·103 Па), ≤40°С), пока объем не уменьшался до 135 мл. Полученный водный раствор фильтровали через фильтровальную подушку, фильтр промывали водой, и объединяли с водным раствором реакционной смеси, так что общий объем полученного водного раствора составлял 170 мл. Этот раствор нагревали до 40°С, затем добавляли на протяжении 20 минут раствор дигидрата хлорида кальция (3,05 г) в воде (29,5 мл), поддерживая реакционную смесь при температуре 38-41°С.
Реакционную смесь перемешивали в течение еще 15 минут при 40°С, затем охлаждали до 20°С и перемешивали при этой температуре в течение следующих 15 минут. Полученную суспензию фильтровали, промывали водой (3×53 мл) и сушили с получением кальциевой соли (Е)-7-{4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты (14,7 г при 100%-ной концентрации, 85%-ный выход).
1H NMR δ: 1,21 (d+d, 6Н) 1,32 (dt, 1H) 1,51 (dt, 1H) 2,00 (dd, 1H) 2,14 (dd, 1H) 3,42 (spt, 1H)* 3,45 (s, 3Н) 3,54 (s, 3H) 3,77 (m, 1H) 4,21 (q, 1H) 5,53 (dd, 1H) 6,51 (dd, 1H) 7,27 (t, 2H) 7,71 (dd, 2H)
[1H NMR выполняли для 3%-ного мас./об. раствора в d6-DMSO (где d6-DMSO=2,51δ)].
*частично скрытый.
Изобретение относится к улучшенному способу получения кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]
пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты(кальциевой соли розувастатина), пригодного для изготовления фармацевтического средства, полезного в лечении, в числе прочего гиперхолистеринемии, гиперлипопротеинемии и атеросклероза. Способ включает стадии (а)-(ж): а) взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]
пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в смешиваемом с водой органическом растворителе с водной кислотой при повышенной температуре; б) взаимодействие полученного раствора с водным гидроксидом щелочного метала и, возможно, промывание полученного водного раствора соли щелочного металла подходящим органическим растворителем; в) доведение рН полученного раствора до приблизительно рН 9-10,5 путем добавления водной соляной кислоты; г) удаление смешиваемого с водой органического растворителя; д) возможная фильтрация полученной смеси; е) добавление водорастворимой кальциевой соли к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]
пиримидин-5-ил](1R,5S)-3,5-дигидроксигепт-6-еновой кислоты; и ж) выделение продукта стадии (е). Предпочтительным смешиваемым с водой органическим растворителем представляет собой ацетонитрил, водная кислота на стадии (а) представляет собой соляную кислоту, а стадия (б) включает взаимодействие полученного раствора с водным гидроксидом натрия и промывание полученного водного раствора натриевой соли подходящим углеводородным, сложноэфирным или эфирным растворителем, например толуолом, ксилолом, МТВЕ (метил-трет-бутиловый эфир) или этилацетатом.
1.Способ образования кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты, включающий стадии (а)-(ж):
а) взаимодействие (1-6С)алкилового эфира (Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)уксусной кислоты в смешиваемом с водой органическом растворителе с водной кислотой при повышенной температуре;
б) взаимодействие полученного раствора с водным гидроксидом щелочного металла и, возможно, промывание полученного водного раствора соли щелочного металла подходящим органическим растворителем;
в) доведение рН полученного раствора до приблизительно рН 9-10,5 путем добавления водной соляной кислоты;
г) удаление смешиваемого с водой органического растворителя;
д) возможная фильтрация полученной смеси;
е) добавление водорастворимой кальциевой соли к фильтрату так, чтобы образовалась кальциевая соль (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](1R,5S)-3,5-дигидроксигепт-6-еновой кислоты; и
ж) выделение продукта стадии (е).
2. Способ по п.1, при котором смешиваемый с водой органический растворитель представляет собой ацетонитрил.
3. Способ по п.1, при котором водная кислота на стадии (а) представляет собой соляную кислоту.
4. Способ по п.1, при котором стадия (б) включает взаимодействие полученного раствора с водным гидроксидом натрия и промывание полученного водного раствора натриевой соли подходящим углеводородным, сложноэфирным или эфирным растворителем.
5. Способ по п.1, при котором водный раствор натриевой соли промывают толуолом, ксилолом, МТВЕ (метил-трет-бутиловым эфиром) или этилацетатом.
6. Способ по п.5, при котором водный раствор натриевой соли промывают толуолом.
7. Способ по п.1, при котором (1-6С)алкиловый эфир представляет собой трет-бутиловый эфир.
8. Способ по п.1, при котором стадию (а) осуществляют при 35-40°С.
9. Способ по п.1, при котором стадию (б) осуществляют при температуре окружающей среды.
10. Способ по п.1, при котором на стадии (б) водный гидроксид щелочного металла представляет собой гидроксид калия.
11. Способ по п.1, при котором стадию (г) выполняют при давлении меньшем или равном 55 мбар (5,5·103 Па) и температуре меньшей или равной 45°С.
12. Способ по п.1, при котором на стадии (е) водорастворимая кальциевая соль представляет собой хлорид кальция.
13. Способ по любому из пп.1-12, при котором на стадии (е) кальциевую соль добавляют при 32-43°С.
14. Способ получения кальциевой соли (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновой кислоты посредством осуществления стадий (а)-(ж), указанных в способе по п.1, при котором (1-6С)алкиловый эфир представляет собой трет-бутиловый эфир.
| WO 00/49014 A1, 24.08.2000 | |||
| Система для воспроизведения и передачи расхода жидкости и газа | 1974 |
|
SU521471A1 |
| WO 00/42024 A1, 20.07.2000. | |||

