Область, к которой относится изобретение
Настоящее изобретение относится к основным эфирам жирных кислот и к их применению в качестве противовоспалительных или иммуномодулирующих средств, в частности, для лечения иммунологически опосредованных воспалений, а также в качестве адъювантов для антигенов, участвующих в клеточном и гуморальном ответах.
Сокращения: АА: адъювантный артрит; CFA: полный адъювант Фрейнда; DTH: гиперчувствительность замедленного типа; EAE: экспериментальный аутоиммунный энцефаломиелит; GA: ацетат глатирамера; IFA: неполный адъювант Фрейнда; i.v.: внутривенно; МВР: основный белок миелин; MS: рассеянный склероз; MSCH: гомогенат мышиного спинного мозга; OA: олеиловый спирт; PBS: забуференный фосфатом физиологический раствор; s.c.: подкожно.
Предшествующий уровень техники
1. Воспаление
Воспаление обычно делят на три фазы: острое воспаление, иммунный ответ и хроническое воспаление. Острое воспаление представляет собой начальный ответ на повреждение ткани и опосредуется высвобождением гистамина, серотонина, брадикинина, простагландинов и лейкотриенов. Иммунный ответ, который обычно следует за фазой острого воспаления, вырабатывается в том случае, если иммунокомпетентные клетки активируются в ответ на чужеродные организмы или антигенные вещества, высвобождаемые во время острого или хронического воспалительного ответа. Продуцирование иммунного ответа у хозяина может оказывать благоприятное действие, поскольку оно приводит к фагоцитозу или нейтрализации микроорганизмов. Однако это действие может оказаться неблагоприятным, если оно приводит к хроническому воспалению с непрекращающимся патологическим процессом, как это происходит при ревматоидном артрите.
Лечение пациентов, страдающих воспалениями, приводит к замедлению или прекращению процесса повреждения ткани, а также к ослаблению болей, которые являются ведущим симптомом и главной причиной жалоб пациентов.
Противоспалительные средства обычно подразделяются на стероидные или глюкокортикоидные средства и нестероидные противовоспалительные средства (NSAID). Глюкокортикоиды являются сильнодействующими противовоспалительными средствами, но высокая степень токсичности, ассоциированная с кортикостероидной терапией хронического заболевания, ограничивает их применение за исключением некоторых случаев острых воспалительных состояний. Поэтому предполагается, что главную роль в лечении хронических состояний, таких как ревматоидный артрит, должны играть нестероидные противовоспалительные средства.
Примерами нестероидных противовоспалительных средств являются производные аминоарилкарбоновых кислот, арилуксусных кислот, арилмасляных кислот, арилкарбоновых кислот, арилпропионовых кислот, пиразола, пиразолона, салициловой кислоты и некоторые другие производные различных химических соединений, включая специфические противоартритные/противоревматические средства.
Поэтому необходимость в получении новых нестероидных противовоспалительных средств, которые служили бы альтернативой имеющимся в настоящее время противовоспалительным лекарственным средствам, остается в высокой степени актуальной.
2. Вакцины и адъюванты
Лимфоциты представляют собой центральные клетки иммунной системы, ответственные за приобретенный иммунитет, и являются иммунологическими признаками разнообразия, специфичности, иммунологической памяти и способности распознавания "свое"/"чужое". Зрелые В-клетки отличаются от других лимфоцитов по механизму синтеза и по своей способности представлять мембраносвязанные молекулы иммуноглобулина (антитела), которые служат в качестве рецепторов для антигенов. Взаимодействие между антигеном и мембраносвязанным антителом на зрелых "необученных" В-клетках приводит к активации и дифференцировке В-клеточных клонов с соответствующей специфичностью и к последующему продуцированию В-клеточных клонов, на которых отсутствуют указанные мембраносвязанные антитела, но которые секретируют молекулы антитела с той же самой специфичностью связывания с антигеном.
Т-лимфоциты подобно В-лимфоцитам имеют мембранные рецепторы для антигенов. Однако в отличие от мембраносвязанного антитела на В-клетках Т-клеточный рецептор (TCR) не распознает свободного антигена. Вместо этого TCR распознает только антиген, связанный с аутоантигенной молекулой, кодируемой генами в главном комплексе гистосовместимости (МНС). Для распознавания большинством Т-клеток данный антиген должен быть представлен в ассоциации с МНС-молекулами на поверхности антигенпрезентирующих клеток (АРС) или на вирусинфицированных клетках, раковых клетках и трансплантатах.
Подобно В-клеткам Т-клетки экспрессируют специфические мембранные молекулы. Все субпопуляции Т-клеток экспрессируют TCR, комплекс полипептидов, включающих CD3, и в большинстве случаев, могут распознаваться по присутствию одной или другой из двух мембранных молекул CD4 и CD8. Т-клетки, экспрессирующие мембранную гликопротеиновую молекулу CD4, рестриктированы молекулами МНС класса II по распознаванию антигена, связанного с этими молекулами, тогда как Т-клетки, экспрессирующие CD8, димерный мембранный гликопротеин, рестриктированы МНС-молекулами класса I по распознаванию антигена, связанного с этими молекулами.
В общих чертах экспрессия CD4 и CD8 также определяют две главные субпопуляции Т-лимфоцитов. CD4+-Т-клетки обычно функционируют как Т-хелперные (TН) клетки и рестриктированы молекулами МНС класса II; тогда как CD8+-Т-клетки обычно функционируют как Т-хелперные (TС) клетки и рестриктированы молекулами МНС класса I.
TН-клетки активируются путем распознавания комплекса "антиген-МНС класса II" на антигенпрезентирующих клетках. После активации TН-клетка начинает делиться с образованием клона эффекторных клеток, каждая из которых является специфичной к тому же самому комплексу "антиген-МНС класса II". Эти TН-клетки секретируют различные цитокины, которые играют центральную роль в активации В-клеток, Т-клеток и других клеток, участвующих в иммунном ответе.
Изменения профиля цитокинов, продуцируемых TН-клетками, может приводить к изменению типа иммунного ответа, который вырабатывается другими лейкоцитами. Таким образом, исходя из специфических цитокинов, которые секретируются TН-клетками при их активации, эти TН1-клетки подразделяются на две группы: клетки, продуцирующие TН1-ответ с профилем цитокинов, которые поддерживают воспаление и активируют, главным образом, определенные Т-клетки и макрофаги; и клетки, продуцирующие TН2-ответ, который активирует, главным образом, В-клетки и иммунные антителозависимые ответы. Таким образом, TН1-клетки секретируют IL-2, который индуцирует пролиферацию Т-клеток, и цитокины, такие как IFN-γ, которые опосредуют воспаление ткани. В противоположность этому TН2-клетки секретируют IL-4, который активирует В-клетки, продуцирующие антитела определенных изотипов IgG, и ингибируют продуцирование воспалительных TН1-цитокинов, и IL-10, который ингибирует продуцирование воспалительных цитокинов макрофагами и, таким образом, способствует опосредованному снижению уровня продуцирования цитокинов TН1-клетками и влияет на антигенпрезентирующие клетки путем ингибирования экспрессии МНС класса II.
Аутоиммунный ответ является результатом нерелевантного ответа иммунной системы на аутокомпоненты, приводящего к активации аутореактивных клонов Т- или В-клеток, и генерирования гуморальных или клеточно-опосредованных ответов на эндогенные антигены с последующим повреждением клеток тканей и органов. Иногда такое повреждение вызывается антителами, как, например, в случае аутоиммунных расстройств, таких как болезнь Аддисона, аутоиммунная анемия, например, аутоиммунная гемолитическая анемия и пернициозная анемия, тиреоидит Хашимото и склеродермия.
Многие аутоиммунные расстройства, например инсулинзависимый сахарный диабет (IDDM или диабет типа I), рассеянный склероз, ревматоидный артрит и аутоиммунный тиреоидит, характеризуются деструкцией тканей, опосредованной Т-клетками, активированными эндогенным антигеном. Эти иммунные ответы на аутоантигены поддерживаются путем непрерывной активации аутореактивных Т-лимфоцитов.
Аутоиммунные заболевания могут быть подразделены на орган-специфические аутоиммунные заболевания, при которых иммунный ответ направлен на антиген-мишень, являющийся уникальным для конкретного органа или железы, то есть манифестации такого ответа в значительной степени ограничены этим органом; и системные аутоиммунные заболевания, при которых иммунный ответ направлен на антигены-мишени широкого ряда и которые поражают различные органы и ткани. Примерами орган-специфических иммунных заболеваний являются инсулинзависимый сахарный диабет, рассеянный склероз, ревматоидный артрит, тиреоидит и тяжелая миастения, и примерами системных аутоиммунных заболеваний являются системная красная волчанка и склеродермия.
В патогенезе орган-специфических аутоиммунных заболеваний участвуют TН1-клетки. Так, например, имеются убедительные данные, указывающие на то, что экспериментальный аутоиммунный энцефаломиелит у мышей (ЕАЕ) вызывается CD4+-TН1-клетками, специфичными для иммунизирующего антигена, например основного белка миелина (МВР) или протеолипидного белка (PLP). Это заболевание может передаваться от одного животного к другому посредством Т-клеток животных, иммунизованных либо МВР или PLP, либо посредством клонированных Т-клеточных линий таких животных. Очевидно, что TН1-тип ответов также приводят к развитию заболеваний или состояний, опосредованных другими Т-клеточными линиями, таких как контактный дерматит.
В большинстве случаев орган-специфические аутоиммунные заболевания развиваются вследствие аутореактивности CD4+-Т-клеток. Анализ этих Т-клеток выявил, что TН1/TН2-баланс может так или иначе влиять на развитие аутоиммунного заболевания. TН1-клетки участвуют в развитии аутоиммунного заболевания, тогда как TН2-клетки в некоторых случаях не только обеспечивают защиту от развития такого заболевания, но также и защиту от прогрессирования уже развивающегося заболевания и обеспечивают толерантность и поддержание этой толерантности к аллотрансплантату.
Было разработано несколько способов терапевтического лечения аутоиммунных заболеваний. Идентификация и секвенирование различных аутоантигенов послужили стимулом к разработке новых способов модуляции аутоиммунной Т-клеточной активности. Для лечения аутоиммунных заболеваний было предложено использовать все антигены, участвующие в патогенезе аутоиммунного заболевания, или пептиды, происходящие от последовательностей этих антигенов.
Синтетические пептиды, подходящие для иммуноспецифического лечения аутоиммунного заболевания, представляют собой пептиды, распознаваемые Т-клетками, участвующими в патогенезе аутоиммунного заболевания. Эти пептиды могут иметь последовательность, состоящую из патогенной последовательности, находящейся в последовательности антигена, ответственного за данное заболевание, либо аналог этой последовательности, в которой один или несколько нативных аминокислотных остатков заменены другими аминокислотными остатками, в частности так называемый "модифицированный пептид", который имеет одну аминокислотную замену в эпитопе патогенного нативного аналога (то есть области, контактирующей с TCR), но не имеет каких-либо альтераций в агретопе (то есть в области, контактирующей с МНС).
Каждое аутоиммунное заболевание может быть подвергнуто терапии с использованием идеального пептида, происходящего от последовательности антигена, ассоциированного с данным заболеванием, или модифицированного пептида или другого его аналога. Таким образом, для такого заболевания, как рассеянный склероз (MS), в развитии которого участвуют Т-клетки, реагирующие с аутоантигенами, такими как основный белок миелин (МВР), миелиновый гликопротеин олигодендроцита (MOG) и протеолипидный белок (PLP), требуется терапия с использованием пептида МВР, MOG или PLP или их аналога; для тяжелой миастении может потребоваться терапия с использованием пептида, происходящего от ацетилхолинового рецептора; для тиреоидита может потребоваться терапия с использованием пептида, происходящего от тироглобулина; для диабета типа 1 может оказаться подходящей терапия с использованием пептида, происходящего от фермента глутаминовая кислота-декарбоксилаза (GAD), или пептида, происходящего от последовательности инсулина; для системной красной волчанки может потребоваться терапия с использованием пептида, происходящего от белка Р53, и для синдрома Гийена-Барре может потребоваться терапия с использованием пептида, происходящего от миелинового антигена Р2.
В последние годы для лечения таких заболеваний было предложено использовать пептиды, происходящие от патогенного аутоантигена, ассоциирующегося с аутоиммунным заболеванием, или их аналоги. Так, например, для лечения рассеянного склероза было предложено использовать пептиды, происходящие от последовательности МВР человека (патенты США 5817629 и 6252040) и их аналоги (патенты США 5948764 и 6329499); для лечения диабета было предложено использовать пептидные аналоги 65 кДа-изоформы GAD человека или инсулина (патенты США 5945401 и 6197926 соответственно), и для лечения увеоретинита было предложено использовать аутоантиген или его фрагмент (патент США 5961977). Каждый из этих патентов и все патенты, цитируемые выше, во всей своей полноте введены в настоящее описание посредством ссылки.
Для лечения каждого из указанных различных аутоиммунных заболеваний может оказаться необходимым введение релевантного пептида в адъюванте, который мог бы активировать Т-клетки противовоспалительного TН2-фенотипа. Предполагается, что это может приводить к прекращению аутоиммунного процесса. Существуют также определенные ситуации, не связанные с лечением аутоиммунного заболевания, при которых может оказаться желательной активация специфических Т-клеток с TН2-фенотипом. Однако лечение с участием аутоантигенов должно быть проведено с использованием адъювантов, которые не индуцируют иммунный ответ TН1-типа, способный активировать опасный иммунный TН1-ответ у субъекта, подвергаемого лечению. Таким образом, необходимо идентифицировать адъюванты, способные образовывать комбинации со специфическими антигенами, которые будут индуцировать противовоспалительные Т-клетки TН2-типа.
По своей природе адъюванты представляют собой неспецифические иммуномодуляторы. Адъювант, который может быть использован для указанных выше целей, должен представлять собой неспецифический иммуномодулятор, который может быть использован в комбинации с антигеном или с другой молекулой для терапевтической вакцинации в целях индуцирования активации специфических Т-клеток с нужным противовоспалительным фенотипом.
Было показано, что некоторые пептиды, подходящие для их применения в терапии Т-клеточно-опосредованных заболеваний, расстройств или состояний, таких как аутоиммунные заболевания, являются эффективными при их подкожном (s.c.) введении мышам в масляном носителе, таком как эмульсия минерального масла, известная как неполный адъювант Фрейнда (IFA). Однако IFA, а также полный адъювант Фрейнда (CFA; препарат минерального масла, содержащий различные количества инактивированных микроорганизмов Mycobacterium), не могут быть использованы для введения человеку, поскольку минеральное масло не может разлагаться в организме.
Поэтому необходимость в получении эффективных носителей для пептидной терапии, которые могли бы разлагаться и действовать как адъювант, служащий в качестве носителя, депо-препарата или иммуностимулятора/энхансера, остается актуальной.
3. Материалы известного уровня техники
Некоторые жирные спирты и эфиры жирных кислот были описаны как растворители или эмульгаторы, используемые в фармацевтических композициях. Так, например, цетиловый спирт может быть использован в фармацевтических композициях как эмульгирующий и загущающий агент (The Merck Index, 2001, 13th edition, pp.347-8, #2037), олеиловый спирт может быть использован в лекарственных препаратах в качестве носителя (The Merck Index, 2001, 13th edition, p.1222, #6900) и алкиловые эфиры олеиновой кислоты могут быть использованы в лекарственных препаратах в качестве растворителей (The Merck Index, 2001, 13th edition, p.6899, #6898).
Была описана смесь высших алифатических первичных спиртов, выделенных, главным образом, из пчелиного воска, которая обладает умеренной противовоспалительной активностью. Состав такой смеси не указывается (Rodriguez et al., 1998).
Исследования масс-спектров никотинатов длинноцепочечных спиртов, например октадецил- и (Z)-9-октадецен-1-илникотинатов, выявили структуру длинноцепочечных спиртов (Vetter & Meister, 1981). Эти соединения не обладают какой-либо биологической активностью.
Сложные эфиры 4-аминометилбензойной кислоты (PAMBA) и С6-С16-насыщенных спиртов, например дециловых, ундециловых, тетрадециловых и гексадециловых спиртов, были проанализированы на их антифибринолитическую активность, и было установлено, что они не обладают такой активностью (Markwardt et al., 1966). Было обнаружено, что сложные эфиры РАМВА и короткоцепочечных спиртов способствуют снижению пролиферации in vitro культивированных эндотелиальных клеток, и более эффективным является сложный гексиловый эфир (Beyer & Pilgrim, 1991).
Алкил N,N-дизамещенные аминокислоты, например алкил-N,N-диметиламиноацетат, где алкил представляет собой октил, децил, додецил или тетрадецил, и децил(4-метил-1-пиперазигил)ацетат, были описаны как активаторы чрескожной пенетрации индометацина и, возможно, других лекарственных средств (Wong et al., 1989, патент США 4980378).
Комплексы, используемые в генотерапии и содержащие терапевтически активное вещество и катионный липид, такой как четвертичные соединения пиперазиния, замещенные как в положении 1, так и положении 4 метильным или олеилоксикарбонилметильным радикалами, описаны в патенте США 6291423.
Сложные эфиры N,N-диметиламиноуксусной кислоты и длинноцепочечных алканолов, например тетрадециловых, цетиловых и стеариловых спиртов и алкенолов, используемые в косметических средствах для волос, описаны в JP 2000-302650. Однако конкретного описания олеилового эфира не приводится.
Были получены сложные эфиры бетаина [внутренняя соль гидроксида (карбоксиметил)триметиламмония,] и длинноцепочечных спиртов, таких как дециловый, лауриловый, миристиловый, пальмитиловый, стеариловый и олеиловый спирт, и были исследованы их фармакодинамические свойства (Metayer & Jacob, 1952), либо была протестирована их активность в качестве биоцида при обработке охлаждающей водой (Rucka et al., 1983).
Четвертичные аммониевые соли лауриловых, миристиловых и цетиловых сложных эфиров N-карбоксиметилпиперидиновых, -пиперазиновых и -морфолиновых соединений были описаны как гермициды (Smith et al., 1951).
Сложные стеариловые эфиры аминокислот, например глицина, фенилглицина, аланина, валина, лейцина, лизина, пролина, фенилаланина и тирозина, и сложные стеариловые эфиры пептидов были предложены в качестве адъювантов для бактериальных и вирусных вакцин для введения человеку (Penney et al., 1985, 1993; Nixon-George et al., 1990).
Сложные эфиры DL-ω-фениламинокислот и С4-С10алканолов, такие как октиловый или дециловый эфир DL-2-фенилглицина или октиловый эфир DL-2-(4-диметиламинофенил)глицина, были описаны как антифлогистонные, антигистаминовые, спазмолитические, противоокислительные и противовоспалительные (Schulz et al., 1982; Schewe et al., 1991; Kontogiorgis et al., 2001).
Высшие сложные алкиловые эфиры аминокислот, например лауриловый, миристиловый, цетиловый и стеариловый эфиры глицина, фенилглицина, аланина, валина, норвалина, лейцина, изолейцина, лизина и фенилаланина, и их N-низшие алкильные производные описаны в патенте США 3821403 (Misato et al., 1974) как полезные для борьбы с болезнями растений.
Краткое описание изобретения
В соответствии с настоящим изобретением было обнаружено, что некоторые сложные эфиры длинноцепочечных жирных спиртов и карбоновых кислот, содержащих, по крайней мере, одну основную группу, действуют как противовоспалительные иммуномодуляторы, и поэтому они могут быть использованы для лечения воспаления, в частности, иммунологически опосредованного, а также они могут быть использованы в качестве адъювантов в комбинации со специфическими антигенами, участвующими как в клеточном, так и в гуморальном ответах, где указанный адъювант служит в качестве носителя, депо-препарата или иммунного стимулятора/активатора.
В одном аспекте настоящее изобретение относится к применению соединения общей формулы I:
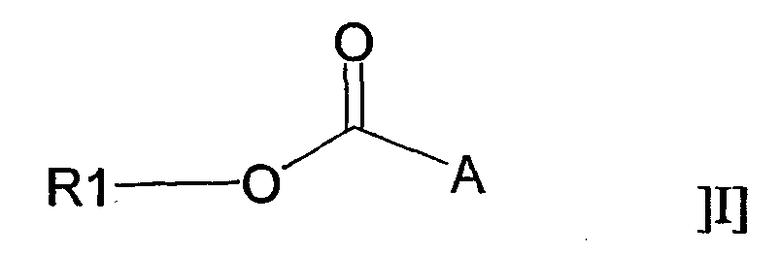
или его фармацевтически приемлемой соли, где R1 представляет собой С12-С24алкил или С10-С24алкенил, и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую или циклическую аминогруппу и/или, по крайней мере, одно гетероароматическое кольцо, содержащее третичный или четвертичный атом азота, для получения фармацевтической композиции для лечения воспаления.
В другом аспекте настоящее изобретение относится к применению адъюванта общей формулы Ia:
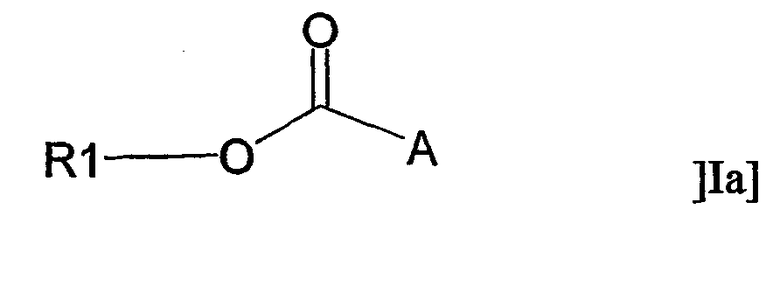
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкил или С10-С24алкенил, и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую или циклическую аминогруппу и/или, по крайней мере, одно гетероароматическое кольцо, содержащее третичный или четвертичный атом азота, за исключением соединений, где R1 представляет собой С18алкил и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую аминогруппу, либо -СО-А представляет собой остаток пролина, для получения терапевтического препарата, дополнительно содержащего антиген.
Антигеном, используемым вместе с адъювантом общей формулы Ia, может быть антиген, участвующий в развитии аутоиммунных заболеваний, рака различных типов, таких как меланома, и инфекционных заболеваний, таких как бактериальные и вирусные инфекции. Терапевтический препарат, содержащий такой антиген и адъювант настоящего изобретения, может быть, в частности, полезен для активации Т-клеток для лечения аутоиммунных заболеваний и для достижения Т-клеточно-опосредованных иммунных эффектов, где предпочтительным является иммунный ответ TН2-типа.
Некоторые из этих соединений, представленных вышеуказанными формулами и определенных ниже, являются новыми и сами по себе представляют другой аспект настоящего изобретения.
Краткое описание чертежей
На Фиг.1 проиллюстрировано влияние октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты (соединения 1) на адъювантный артрит (AA). Соединение 1 внутривенно (i.v.) вводили крысам Lewis в день иммунизации (день 0, треугольники) или на 14-й день после начала развития AA (день +14, круги).
На Фиг.2 проиллюстрировано влияние соединения 1 на экспериментальный аутоиммунный энцефаломиелит (ЕАЕ). Соединение 1 вводили i.v. крысам DA в день иммунизации (день 0, круги).
На Фиг.3 проиллюстрировано влияние соединения 1 на жизнеспособность кожного аллотрансплантата. Соединение 1 вводили i.v. мышам в день трансплантации (день 0, круги).
На Фиг.4 проиллюстрировано ингибирование реакции гиперчувствительности замедленного типа (DTH) у мышей BALB/c после обработки различными концентрациями (1, 2 или 4 мг) тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты (соединения 3).
На Фиг.5 проиллюстрировано влияние различных концентраций (0,5, 1, 2 или 4 мг) соединений 5, 9 и 11 на DTH у мышей. В качестве позитивного контроля использовали дексаметазон (2 мг), и в качестве негативного контроля использовали гидрохлорид этилового эфира (4-метилпиперазин-1-ил)уксусной кислоты (0,5, 1, 2 или 4 мг).
На Фиг.6 проиллюстрировано влияние подкожного (s.c.) введения различных концентраций (10, 50, 100, 200 мг) соединения 3 на AA у крыс. Для сравнения использовали олеиловый спирт (ОА, 100 мг).
На Фиг.7 проиллюстрировано влияние подкожного (s.c.) введения различных концентраций (10, 50, 100, 200 мг) соединения 3 на ЕАЕ у крыс. Для сравнения использовали олеиловый спирт (ОА, 100 мг).
На Фиг.8 проиллюстрировано влияние введения per os различных концентраций (0,2, 1, 2, 10, 20 мг) соединения 3 на EAE у крыс. Для сравнения использовали олеиловый спирт (ОА, 10 мг).
На Фиг.9 проиллюстрировано влияние введения per os битартрат октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты (соединения 9, 100 мг/кг) на ЕАЕ у крыс.
На Фиг.10 показано, что соединение 3 стимулирует иммунологические эффекты ацетата глатирамера (GA) у EAE-индуцированных мышей.
Подробное описание изобретения
В соответствии с одним аспектом настоящее изобретение относится к применению соединения общей формулы I:
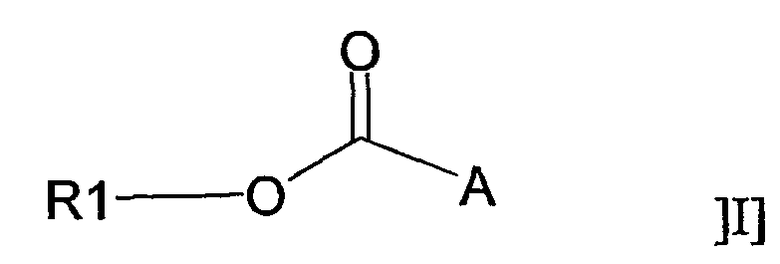
или его фармацевтически приемлемой соли, где R1 представляет собой С12-С24алкил или С10-С24алкенил, и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую или циклическую аминогруппу и/или, по крайней мере, одно гетероароматическое кольцо, содержащее третичный или четвертичный атом азота, для получения фармацевтической композиции для лечения воспаления.
В одном варианте данного аспекта изобретения в указанном противовоспалительном соединении формулы I остаток А выбран из группы, состоящей из:
(i)
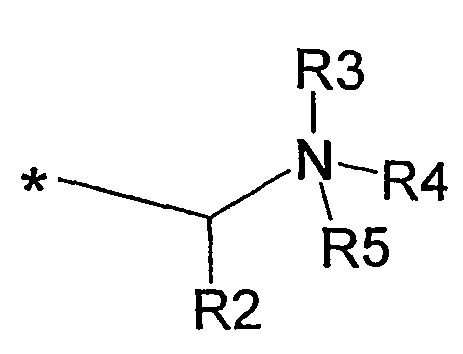
где R2 представляет собой Н, С1-С6алкил, арил или аралкил, где любая арильная группа может быть незамещенной или замещена нитро, циано, галогеном, гидрокси, NR6R7 или СR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(ii) фенила, замещенного NR6R7 или CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил; и
(iii)
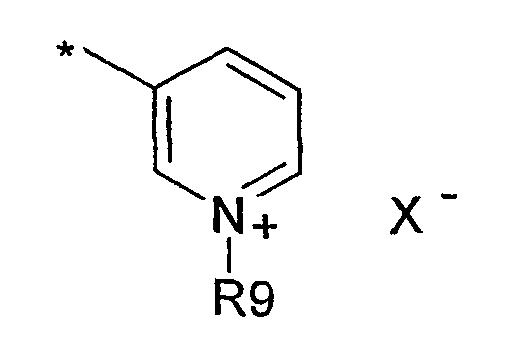
где R9 представляет собой Н, низший алкил или индолил(С1-С4)алкил, и Х- представляет собой противоион, либо R9 представляет собой электронную пару, и Х отсутствует.
В другом аспекте настоящее изобретение относится к применению адъюванта общей формулы Ia:
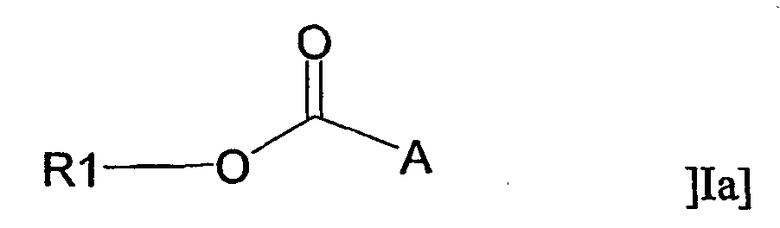
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкил или С10-С24алкенил, и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую или циклическую аминогруппу и/или, по крайней мере, одно гетероароматическое кольцо, содержащее третичный или четвертичный атом азота, за исключением соединений, где R1 представляет собой С18алкил, и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую аминогруппу, либо -СО-А представляет собой остаток пролина, для получения терапевтического препарата, дополнительно содержащего антиген.
В одном варианте данного аспекта изобретения в указанном адъюванте формулы Ia остаток А выбран из группы, состоящей из:
(i)
где R2 представляет собой Н, С1-С6алкил, арил или аралкил, где любая арильная группа может быть незамещенной или замещена нитро, циано, галогеном, гидрокси, NR6R7 или СR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, при условии, что R4 и R5 не являются Н или С1-С6алкилом, если R1 представляет собой октадецил; и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(ii) фенила, замещенного NR6R7 или CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил; и
(iii)
где R9 представляет собой Н, С1-С6алкил или индолил(С1-С4)алкил, и Х- представляет собой противоион, либо R9 представляет собой электронную пару, и Х отсутствует.
В другом аспекте настоящее изобретение относится к новым соединениям общей формулы:
R1-О-СО-А
где:
(i) R1 представляет собой С20-С24алкил или С10-С24алкенил, и А представляет собой остаток формулы:
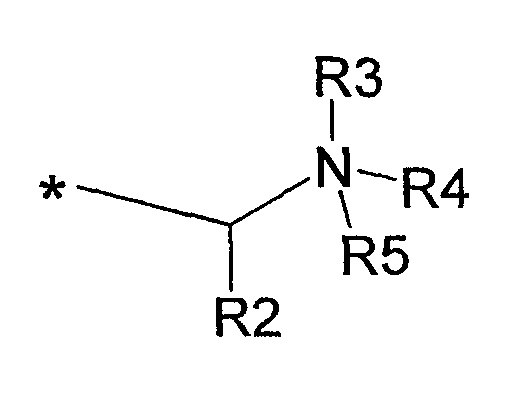
где R2 представляет собой Н, С1-С6алкил, арил или аралкил, где любая арильная группа может быть незамещенной или замещена нитро, циано, галогеном, гидрокси, NR6R7 или СR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(ii) R1 представляет собой С18алкил, и А представляет собой остаток формулы:
где R2 представляет собой Н, R3 представляет собой электронную пару; и R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, или
(iii) R1 представляет собой С12-С16алкил, и А представляет собой остаток формулы:
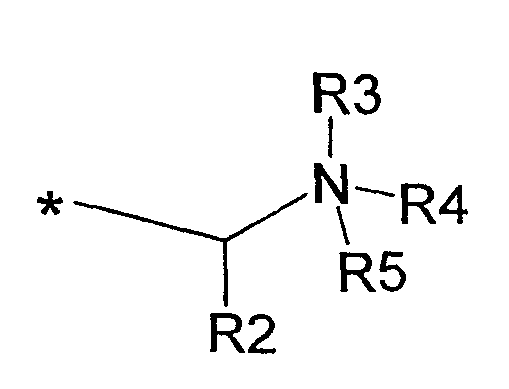
где R2 представляет собой незамещенный арил, либо арил, либо аралкил, где арильная группа замещена нитро, циано, галогеном, гидрокси, NR6R7 или СR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(iv) R1 представляет собой С10алкил, и А представляет собой остаток формулы:
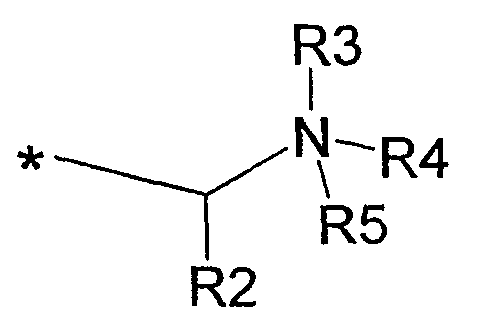
где R2 представляет собой С1-С6алкил, R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил; или
(v) R1 представляет собой С10-С24алкил или С10-С24алкенил, и А представляет собой фенил, замещенный NR6R7 или СR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил, за исключением соединений, где R1 представляет собой С10-С16алкил, и А представляет собой фенил, замещенный -СН2-NH2, или
(vi) R1 представляет собой С10-С24алкил или С10-С24алкенил, и А представляет собой группу формулы:
где R9 представляет собой С1-С6алкил или индолил(С1-С4)алкил, и Х- представляет собой противоион;
и их фармацевтически приемлемых солей.
Для ясности, символ *, используемый во всех формулах, имеющихся в настоящем описании и в формуле изобретения, означает положение, в котором присоединена группа -СО- радикала R1-О-СО-.
В соответствии с настоящим изобретением R1 представляет собой С10-С24алкил или алкенил для адъювантов формулы Ia, либо R1 представляет собой С12-С24алкил или С10-С24алкенил для соединений формулы Ia, используемых для лечения воспаления. В обоих случаях R1 предпочтительно представляет собой С12-С20, более предпочтительно С16-С18 и, наиболее предпочтительно С18-алкил или алкенил. Алкильные группы могут быть прямыми или разветвленными, и предпочтительно выбраны из группы, состоящей из додецила, тетрадецила, гексадецила и октадецила, так, чтобы радикалы R1-О- происходили от насыщенных жирных спиртов, таких как лауриловый спирт, миристиловый спирт, цетиловый спирт (также известный как пальмитиловый спирт) и стеариловый спирт соответственно. Алкенильные группы могут иметь одну или несколько двойных связей и предпочтительно выбраны из группы, состоящей из гексадеценила, октадеценила, октадекадиенила и октадекатриенила, так чтобы радикалы R1-О- происходили от ненасыщенных жирных спиртов, таких как пальмитолеиловый спирт, или предпочтительно от ненасыщенных жирных С18-спиртов, имеющих одну или несколько двойных связей, находящихся в транс-форме либо предпочтительно в цис-форме, таких как, но не ограничивающихся ими, олеиловый спирт (цис-9-октадеценол), линолеиловый спирт (цис-9,12-октадекадиенол), γ-линолениловый спирт (цис-6,9,12-октадекатриенол) и линолениловый спирт (цис-9,12,15-октадекатриенол). В наиболее предпочтительном варианте осуществления изобретения R1 представляет собой цис-9-октадеценильный радикал, также называемый октадец-(Z)-9-енилом или олеилом.
Остаток А может включать первичную, вторичную или третичную аминогруппу или четвертичную аммониевую группу, которая может быть ациклической или циклической. Если указанная аминогруппа является ациклической, то полученные соединения представляют собой сложные эфиры аминокислот.
Таким образом, в одном варианте осуществления изобретения в соединениях, составляющих фармацевтические или терапевтические композиции настоящего изобретения, или в некоторых новых соединениях настоящего изобретения А представляет собой радикал формулы:
где R2 представляет собой Н, С1-С6алкил, арил или аралкил, где любая арильная группа может быть незамещенной или замещена нитро, циано, галогеном, гидрокси, NR6R7 или СR8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил, где ограничения определены выше и в формуле изобретения. Так, например, для адъювантов формулы Ia и для новых соединений R4 и R5 не могут представлять собой Н или С1-С6алкил, если R1 представляет собой октадецил (стеарил); и для новых соединений, если R1 представляет собой С12-С16алкил, то R2 не может представлять собой Н, С1-С6алкил или незамещенный аралкил, либо если R1 представляет собой С10алкил, то R2 не может представлять собой Н, арил или аралкил.
В одном предпочтительном варианте изобретения R2 представляет собой Н. Используемый в данном описании термин "С1-С6алкил" означает прямой или разветвленный алкильный радикал, имеющий 1-6 атомов углерода, такой как, но не ограничивающийся ими, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, пентил и гексил. Используемый термин "арил", взятый отдельно или как часть "аралкильного" радикала, означает ароматический карбоциклический С6-С10радикал, такой как фенил и нафтил. В предпочтительном варианте изобретения арильным радикалом является фенил. "Алкил" в термине "аралкил" означает также С1-6алкильный радикал. В предпочтительном варианте осуществления изобретения аралкильной группой является бензил. В предпочтительных вариантах осуществления изобретения фенильная и бензильная группы являются незамещенными или замещены гидрокси, например п-гидроксибензил.
Некоторые соединения вышеуказанных формул, где R3 представляет собой электронную пару и R4 и R5 являются Н, представляют собой сложные эфиры аминокислот, например эфиры глицина (R2 представляет собой Н), аланина (R2 представляет собой метил), валина (R2 представляет собой изопропил), лейцина (R2 представляет собой изобутил), изолейцина (R2 представляет собой втор-бутил), фенилглицина (R2 представляет собой фенил), фенилаланина (R2 представляет собой бензил) и тирозина (R2 представляет собой п-гидроксибензил).
В соответствии с некоторыми вариантами осуществления изобретения R4 и R5, взятые вместе с атомом азота, с которым они связаны, могут образовывать 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, такое как пирролидин и азипин, предпочтительно 6-членное кольцо и более предпочтительно пиперидин, пиперазин или морфолин. Пиперазиновое кольцо может быть замещено в 4-положении С1-С6алкилом, предпочтительно метилом.
В одном предпочтительном варианте осуществления изобретения R2 представляет собой Н или фенил, R3 представляет собой Н, электронную пару или метил; R4 и R5, каждый представляет собой Н или метил, либо R4 и R5, взятые вместе с атомом N, с которым они связаны, образуют морфолиновое или пиперазиновое кольцо, необязательно замещенное метилом у атома азота. В соответствии с этим вариантом осуществления изобретения примерами соединений, используемых в качестве противовоспалительных средств и в качестве адъювантов, являются новые сложные эфиры, идентифицированные в данном описании как соединения 1, 3, 4, 7, 9, 11:
1. октадец-(Z)-9-ениловый эфир N,N-диметиламиноуксусной кислоты;
3. тартрат октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
4. тартрат октадецилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
7. хлорид 4-метил-4-октадец-(Z)-9-енилоксикарбонилметилморфолин-4-ия;
9. битартрат октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты;
11. HCl-соль октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты.
В другом варианте осуществления изобретения А представляет собой фенил, замещенный NR6R7 или СR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил. В предпочтительном варианте осуществления изобретения А представляет собой фенил, замещенный СR8R8NR6R7, где R8 представляет собой Н, и R6 и R7, каждый представляет собой Н или С1-С6алкил, предпочтительно метил. В соответствии с этим вариантом осуществления изобретения примерами соединений, используемых в качестве противовоспалительных средств и в качестве адъювантов, являются новые сложные эфиры 4-диметиламинометилбензойной кислоты, идентифицированные здесь как соединения 5 и 10:
5. HCl-соль октадец-(Z)-9-енилового эфира 4-диметиламинометилбензойной кислоты;
10. HCl-соль октадец-(Е)-9-енилового эфира 4-диметиламинометилбензойной кислоты.
В еще одном варианте осуществления изобретения А представляет собой пиридильную группу:
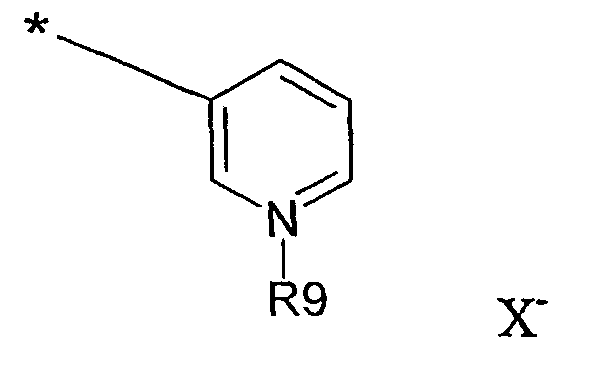
где R9 представляет собой электронную пару, и Х- отсутствует, либо R9 представляет собой метил или индолилэтил, и Х- представляет собой противоион, выбранный из группы, состоящей из хлорида, бромида, иодида и тозилата. Эти соединения представляют собой сложные эфиры никотиновой кислоты. В соответствии с этим вариантом осуществления изобретения примерами соединений, используемых в качестве противовоспалительных средств и в качестве адъювантов, являются соединения, идентифицированные как соединения 2, 6а, 6b, 6с и 8, из которых соединения 6а, 6b, 6с и 8 являются новыми:
2. октадец-(Z)-9-ениловый эфир никотиновой кислоты;
6а. иодид 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
6b. хлорид 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
6с. тозилат 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
8. бромид 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонил-пиридиния.
Сложные эфиры настоящего изобретения представляет собой в основном кристаллические, негигроскопичные и водорастворимые соединения, которые легче поддаются очистке и являются более подходящими для приготовления пероральных и парентеральных препаратов, чем исходные насыщенные или цис-ненасыщенные спирты.
В настоящем изобретении также предусматривается применение фармацевтически приемлемых солей соединений формулы I и Ia, как кислотно-аддитивных солей, так и четвертичных солей ациклических или циклических аммониевых групп или солей пиридиния и противоиона. Фармацевтически приемлемыми кислотно-аддитивными солями указанных соединений являются соли, образованные основанием и неорганической кислотой, такой как хлористоводородная, азотная, фосфорная, серная, бромистоводородная, иодистоводородная и фосфористая кислоты и т.п., и также соли, образованные органическими кислотами, такими как алифатические моно- и дикарбоновые кислоты, фенил-замещенные алкановые кислоты, гидроксиалкановые кислоты, алкандионовые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты и т.п. Например, такими солями являются сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, манделат, бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, п-толуолсульфонат (тозилат), фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат и т.п.
Кислотно-аддитивные соли указанных основных соединений получают стандартным способом путем контактирования свободного основания с достаточным количеством нужной кислоты с получением соли. Форма свободного основания может быть снова получена стандартным способом путем контактирования соли с основанием и выделения свободного основания. Формы свободных оснований отчасти отличаются от соответствующих солевых форм по некоторым физическим свойствам, таким как растворимость в полярных растворителях, но в остальном эти соли эквивалентны их соответствующим свободным основаниям и могут быть использованы для целей настоящего изобретения.
В одном из вариантов осуществления изобретения соединения общей формулы I могут быть получены реакцией длинноцепочечного жирного спирта, такого как олеиловый спирт, с подходящей аминокислотой. Так, например, олеиловый спирт подвергают реакции с N,N-диметилглицином с получением соединения, обозначенного как соединение 1. В другом варианте осуществления изобретения длинноцепочечный жирный спирт подвергают реакции с галогенангидридом никотиновой кислоты, например олеиловый спирт подвергают реакции с никотиноилхлоридом с получением соединения, обозначенного здесь как соединение 2.
В другом варианте осуществления изобретения длинноцепочечный жирный спирт подвергают реакции с хлорангидридом галогенметилуксусной кислоты и затем полученный эфир подвергают реакции с подходящим амином. Так, например, олеиловый спирт подвергают реакции с хлорацетилхлоридом, и полученный олеиловый сложный эфир хлоруксусной кислоты подвергают реакции с пиперазином или с N-метилморфолином, получая свободное основание, которое затем превращают в соль, например в соединение 3 или в соединение 7 соответственно. Аналогичным образом стеариловый спирт подвергают реакции с хлорацетилхлоридом и полученный стеариловый сложный эфир хлоруксусной кислоты подвергают реакции с 4-метилпиперазином, получая свободное основание, которое затем превращают в соль, например в соединение 4.
Другие способы настоящего изобретения описаны в нижеследующих примерах и могут быть применены для получения аналогичных соединений формулы I. Реакции между олеиловым спиртом и активированными аминокислотами могут быть проведены в органических растворителях широкого ряда, как в полярных растворителях, например, ацетонитриле и хлороформе, так и в неполярных растворителях, например, гексане, в широком интервале температур, от комнатной температуры до температуры кипения с обратным холодильником в течение периода времени, составляющего от 1-2 часов до 1-2 дней.
Соли четвертичного пиридиния могут быть получены либо как описано выше, либо алкилированием олеилникотината алкилирующими агентами, известными специалистам в данной области, такими как алкилгалогениды, алкилсульфонаты и галогеналкилиндолы. Олеилгалогенацетаты могут быть подвергнуты реакции с аминами в органических растворителях широкого ряда, как в полярных растворителях, например, в ацетонитриле, так и в неполярных растворителях, например, в гексане, в широком интервале температур, от комнатной температуры до температуры кипения с обратным холодильником в течение периода времени, составляющего от 1-2 часов до 1-2 дней. Маслянистые или низкоплавкие свободные основания, полученные после подходящей обработки и очистки, преобразуют в фармацевтически приемлемые соли. Эти соли обычно представляют собой тугоплавкие и водорастворимые твердые вещества.
Следует отметить, что для получения сложных эфиров настоящего изобретения, где R1 представляет собой цис-алкенильную группу, исходный цис-ненасыщенный спирт, такой как олеиловый спирт, может быть использован в основном в чистой цис-ненасыщенной форме, а именно в виде реагента, который, по крайней мере, на 80% состоит из цис-формы. Так, например, коммерчески доступный олеиловый спирт имеет приблизительно 85%-ную чистоту, и большинство примесей состоит из транс-аналогов (элаидилового спирта).
Иммунная система, как ее природная, так и адаптивная составляющая, участвует в регуляции воспалений любого типа, где воспаление является ключевым фактором в таких процессах, как заживление ран, ремоделирование соединительной ткани, ангиогенез, регенерация органов, нейропротективная реакция, а также в адаптивных иммунных ответах, наблюдаемых при аутоиммунных заболеваниях, аллергиях, реакциях отторжения трансплантата и инфекциях. Поэтому противовоспалительные агенты, которые модулируют воспалительный ответ, такой как ответы, описанные в настоящем изобретении, могут быть использованы для лечения различных состояний.
Воспалительными заболеваниями, расстройствами или состояниями, которые могут быть подвергнуты лечению иммуномодуляторами настоящего изобретения, являются, но не ограничиваются ими, иммунологически опосредованные хронические или острые воспалительные заболевания, расстройства или состояния, выбранные из группы, состоящей из аутоиммунного заболевания, тяжелой аллергии, астмы или воспаления, ассоциированного с заболеванием, расстройством или состоянием, выбранным из группы, состоящей из отторжения трансплантата, хронического дегенеративного заболевания, такого как болезнь Альцгеймера, нейропретективные реакции, регенерация органов, хронические кожные язвы или шизофрения.
Примерами аутоиммунных заболеваний, которые могут быть подвергнуты лечению способами настоящего изобретения, являются рассеянный склероз или артритные состояния у человека, например ревматоидный артрит, реактивный артрит с синдромом Рейтера, анкилозирующий спондилез и другие воспаления суставов, опосредуемых иммунной системой. В нижеследующем описании, касающемся отдельных органов и тканей, будут рассмотрены и другие аутоиммунные заболевания.
Таким образом, настоящее изобретение относится к лечению иммунологически опосредованных воспалительных заболеваний, расстройств или состояний, выбранных из группы, состоящей из тяжелой миастении, синдрома Гийена-Барре или других воспалительных заболеваний нервной системы; псориаза; пузырчатки вульгарной; или других кожных болезней; системной красной волчанки, гломерулонефрита или других почечных заболеваний; атеросклероза или других воспалений кровеносных сосудов; аутоиммунного гепатита; воспалительных заболеваний кишечника, например болезни Крона, панкреатита или других расстройств желудочно-кишечного тракта; сахарного диабета типа 1 (инсулинзависимого сахарного диабета или IDDM), аутоиммунного тиреоидита (тиреоидита Хашимото) или других заболеваний эндокринной системы. В одном из предпочтительных вариантов осуществления изобретения иммунологически опосредованным воспалительным заболеванием, расстройством или состоянием является псориаз.
Одной из моделей, используемых для тестирования противовоспалительной активности агентов настоящего изобретения, является адъювантный артрит (AA), то есть экспериментальное заболевание суставов, индуцируемое у некоторых видов крыс путем иммунизации Mycobacterium tuberculosis в полном адъюванте Фрейнда (CFA). У этих животных развивается артрит, который по своим признакам имитирует ревматоидный артрит человека, и поэтому служит в качестве животных-моделей человеческих артритных состояний, таких как ревматоидный артрит, реактивный артрит при синдроме Рейтера, анкилозирующий спондилит и другие воспаления суставов, которые, как предполагается, опосредуются иммунной системой. Адъювантный артрит также служит в качестве модели иммунологически опосредованного воспаления, включая в основном клеточно-опосредованные аутоиммунные реакции, реакции отторжения трансплантата и аллергические реакции. Так, например, лечение, которое может ослаблять симптомы ревматоидного артрита, предусматривает использование иммуносупрессоров, таких как кортикостероиды, циклоспорин А, азатиоприн и другие иммуносупрессоры, широко применяемые для лечения аутоиммунных заболеваний. Поэтому ослабление симптомов адъювантного артрита указывает на то, что данное средство может быть использовано в качестве противовоспалительных средств широкого применения.
Настоящее изобретение также относится к фармацевтической композиции для лечения воспалений, содержащей фармацевтически приемлемый носитель и описанное выше соединение общей формулы I.
Фармацевтическая композиция настоящего изобретения может быть получена в твердой, полутвердой или в жидкой форме, и, кроме того, она может включать фармацевтически приемлемые наполнители, носители или разбавители и другие инертные ингредиенты и эксципиенты. Такая композиция может быть введена любым подходящими способом, такими как, но, не ограничиваясь ими, пероральное, местное или парентеральное введение, например, путем подкожной, внутривенной, внутримышечной инъекции или любым другим подходящим способом. Поскольку многие соединения являются маслянистыми, то они предпочтительно могут быть введены парентерально и более предпочтительно подкожно. При непрерывном введении каждое соединение настоящего изобретения обычно вводят путем 1-4 инъекций в день или путем непрерывного подкожного вливания, например, с использованием мининасоса. Конкретная доза зависит от состояния пациента и тяжести заболевания и может быть определена лечащим врачом.
Для парентерального введения, соединения могут быть изготовлены смешиванием данного соединения с нужной степенью чистоты в виде разовой лекарственной формы для инъекций (раствора, суспензии или эмульсии), с фармацевтически приемлемым носителем, то есть носителем, который является нетоксичным для реципиентов в используемых дозах и концентрациях и совместимым с другими ингредиентами данного препарата. Обычно указанные препараты получают посредством равномерного и тесного контактирования данного соединения с жидкими носителями или с тонко измельченными твердыми носителями либо с теми и другими. Затем, если это необходимо, полученному продукту придают нужную форму. Предпочтительно используемый носитель представляет собой носитель для парентерального введения и, более предпочтительно раствор, который является изотоничным с кровью реципиента. Примерами таких носителей являются вода, физиологический раствор, раствор Рингера и раствор декстрозы. Могут быть также использованы и безводные носители, такие как жирные масла, а также липосомы. Эти препараты могут быть получены стандартными методами, известными специалистам в данной области, например, методом, описанным в руководстве "Remington's Pharmaceutical Science", А.R. Gennaro, ed., 17th edition, 1985, Mack Publishing Company, Easton, PA, USA.
В другом аспекте настоящее изобретение относится к способу лечения воспаления, в частности иммунологически опосредованного воспаления, где указанный способ предусматривает введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы I, определенного выше.
В другом аспекте настоящее изобретение относится к терапевтическим композициям, содержащим адъювант формулы Ia, определенный выше, и антиген. Указанным антигеном может быть антиген, индуцирующий гуморальный ответ, например токсин, бактериальный или вирусный антиген, либо, что предпочтительно, антиген, индуцирующий клеточный ответ, например, пептид.
Терапевтический препарат может содержать антиген, применяемый для лечения аутоиммунного заболевания, нейродегенеративного заболевания, такого как болезнь Альцгеймера или болезнь Паркинсона, рака, такого как меланома, или инфекционного заболевания, включая как бактериальные, так и вирусные инфекции.
В предпочтительном варианте изобретения указанная терапевтическая композиция предназначена для лечения Т-клеточно-опосредованного заболевания, расстройства или состояния и содержит антиген, распознаваемый воспалительными Т-клетками, ассоциированными с патогенезом указанного Т-клеточно-опосредованного заболевания, расстройства или состояния.
В одном более предпочтительном варианте осуществления изобретения терапевтический препарат содержит антиген, распознаваемый воспалительными Т-клетками, ассоциированными с патогенезом указанного Т-клеточно-опосредованного заболевания, расстройства или состояния, и указанный препарат индуцирует сдвиг Т-клеточного цитокинового ответа TН1-типа в сторону ответа TН2-типа.
Противовоспалительные иммуномодуляторы настоящего изобретения образуют липидные эмульсии, которые при их использовании в качестве вакцинного адъюванта вместе с антигенным веществом, с которым реагируют Т-клетки, участвующие в развитии заболевания, расстройства или состояния, подвергаемого лечению, служат посредниками сдвига Т-клеточного TН1-ответа, продуцируемого перед лечением, в сторону Т-клеточного TН2-ответа, продуцируемого после лечения. Было обнаружено, что такие липидные эмульсии являются биологически активными носителями, сообщающими иммунологическую толерантность, которые могут быть использованы в вакцинах для лечения любого TН1-опосредованного заболевания или состояния. В таких вакцинах антиген обладает иммунологической специфичностью, обеспечивающей терапевтический эффект, в то время как биологически активный носитель настоящего изобретения обеспечивает биологический эффект, то есть сдвиг TН1→TН2. Из-за такого сдвига, опосредованного указанным биологически активным носителем настоящего изобретения, заболевания, расстройства и состояния с соответствующим спектром аутореактивности, могут быть подвергнуты лечению с использованием комбинации одного антигена/носителя, способной индуцировать нужный сдвиг Т-клеточного цитокинового ответа.
В предпочтительном варианте осуществления изобретения указанный терапевтический препарат содержит антиген и противовоспалительный иммуномодуляторный адъювант формулы Ia для лечения орган-специфических аутоиммунных заболеваний, опосредуемых TН1-клетками. Примерами таких заболеваний являются, но не ограничиваются ими, аутоиммунные заболевания, такие как рассеянный склероз, сахарный диабет типа I, ревматоидный артрит и аутоиммунный тиреоидит.
Антигеном, используемым в указанном препарате, является антиген, распознаваемый воспалительными Т-клетками, ассоциированными с патогенезом указанного аутоиммунного заболевания, и который может представлять собой целый белок, участвующий в патологическом процессе; пептид, происходящий от последовательности такого белка; модифицированный пептид, имеющий одну аминокислотную замену в эпитопе патогенного аутоантигенного пептида, или любой другой пептид, распознаваемый воспалительными Т-клетками, ассоциированными с данным заболеванием.
Таким образом, для лечения рассеянного склероза (MS) указанным антигеном может быть МВР, MOG или PLP или пептид, происходящий от последовательности МВР человека, такой как пептиды МВР(75-95), МВР(86-95) и МВР(82-98), описанные в патенте США 5817629, или их аналоги, описанные в патентах США 5948764 и 6239499, либо пептиды MOG или пептиды PLP и их аналоги, где все указанные патенты во всей своей полноте введены в настоящее описание посредством ссылки. Указанным антигеном может быть также ацетат глатирамера, представляющий собой родовое название для ацетатной соли сополимера 1 или Cop1, то есть статистического сополимера, состоящего из четырех аминокислот: тирозина-глутамата-аланина-лизина, который перекрестно реагирует с МВР посредством функциональных групп и способен конкурировать с МВР на МНС класса II за презентацию антигена. В некоторых странах было разрешено применение ацетата глатирамера для лечения MS, выпускаемого под названием COPAXONE® (торговая марка от фирмы Teva Pharmaceuticals Ltd., Petah Tikva, Israel).
Для лечения сахарного диабета типа I (IDDM) пептид может быть получен из декарбоксилазы глутаминовой кислоты (GAD) или из пептидных аналогов GAD, как описано в патенте США 5945401, или из пептидных аналогов инсулина, таких как пептиды, содержащие остатки 9-23 нативной последовательности инсулина В, которая имеет модификации в положениях 12, 13, 15 и/или 16, а также может иметь модификации, описанные в патенте США 6197926, при этом все указанные патенты во всей своей полноте введены в настоящее описание посредством ссылки.
Для лечения других аутоиммунных заболеваний может быть использован пептид, происходящий от последовательности антигена, ассоциированного с данным заболеванием, или его пептидный аналог. Так, например, для лечения аутоиммунного тиреоидита может быть использован пептид, происходящий от последовательности тироглобулина; для лечения ревматоидного артрита может быть использован аутоантиген, происходящий от коллагена II или от микроорганизма Mycobacterium, такого как Mycobacterium tuberculosis, например, белка теплового шока размером 60 кДа, известного как hsp60, который составляет микобактериальный эпитоп, распознаваемый Т-лимфоцитами при адъювантном артрите, или от соответствующего человеческого HSP60 или его пептида; для лечения тяжелой миастении может быть использован пептид, происходящий от последовательности ацетилхолинового рецептора или его аналога, описанного в патенте США 6066621, который во всей своей полноте введен в настоящее описание посредством ссылки; для лечения системной красной волчанки может быть использован пептид, происходящий от последовательности белка Р53; и для лечения синдрома Гийена-Барре может быть использован пептид, происходящий от последовательности миелинового антигена Р2.
Антигеном может быть также непептидный антиген. Примерами непептидных антигенов, которые могут быть использованы в настоящем изобретении, являются, но не ограничиваются ими, фосфолипиды, используемые для лечения фосфолипидного синдрома, холестерин, используемый для лечения атеросклероза, и ДНК-молекулы, используемые для лечения системной красной волчанки.
Используемый антиген необязательно должен быть пептидом. Так, например, TН1-опосредованные аллергические реакции, которые приводят к гиперчувствительности и воспалению кожи, такие как контактный дерматит, могут быть подвергнуты лечению вакциной, содержащей раздражающий антиген и биологически активный носитель/адъювант настоящего изобретения, который индуцирует сдвиг цитокинового ответа TН1-типа в сторону цитокинового ответа TН2-типа. Таким образом, при непрерывном вырабатывании у пациента повышенных уровней антител против данного антигена, воспалительный Т-клеточный ответ, приводящий к раздражению кожи, будет подавляться.
В соответствии с этим биологически активный носитель/адъювант настоящего изобретения, сообщающий иммунологическую толерантность, может быть использован в любое время, желаемое для сообщения толерантности к антигену, который атакуется Т-клетками, то есть в любое время, в течение которого используемая вакцина будет подавлять Т-клеточно-опосредованное состояние, в частности TН1-клеточно-опосредованное состояние. Если будет установлено, что данный антиген активирует реакцию отторжения трансплантата или реакцию "трансплантат против хозяина", то в соответствии с настоящим изобретением предполагается, что введение такого антигена вместе с носителем будет индуцировать сдвиг нежелательного воспалительного TН1-ответа в сторону более желательного TН2-ответа независимо от полного состава набора антигенов, с которыми активно реагируют Т-клетки при данном состоянии.
В одном предпочтительном варианте осуществления изобретения терапевтический препарат способствует ослаблению Т-клеточного цитокинового ответа IL-2-типа или IFN-γ-типа, и усилению Т-клеточного цитокинового ответа IL-4-типа или IL-10-типа.
Для определения секреции цитокинов Т-клетками после их активации пептидами лимфоциты, взятые из периферической крови пациентов, тестируют в анализе на активацию in vitro. Лимфоциты периферической крови выделяют из цельной гипаринизированной крови стандартными методами и культивируют с тестируемым(и) пептидом(ами) в концентрации 5-50 мкг/мл. Супернатанты от культивированных Т-клеток собирают через различные промежутки времени и тестируют на активность различных цитокинов с помощью ELISA или биоанализа(ов).
В соответствии с настоящим изобретением совершенно неожиданно было обнаружено, что некоторые сложные эфиры длинноцепочечных жирных спиртов и карбоновых кислот, содержащих основную группу, могут быть эффективно использованы в качестве адъювантов для активации Т-клеток. Аналогичным образом было совершенно неожиданно обнаружено, что эти препараты являются биологически активными противовоспалительными иммуномодуляторами, сообщающими иммунологическую толерантность.
Преимущество терапевтического препарата настоящего изобретения, содержащего антиген и иммуномодулятор формулы Ia, описываемый в качестве адъюванта, заключается в том, что лечение заболевания может быть ограничено относительно кратковременной обработкой, необходимой для индуцирования протективного иммунного TН2-ответа, то есть специфического TН2-иммунитета, который сам по себе активно подавляет данное заболевание. Введение иммуномодулятора без антигена будет оказывать хроническое или непрерывное подавляющее действие на данное воспалительное заболевание, но после прекращения такого лечения могут наблюдаться рецидивы данного заболевания. При некоторых заболеваниях продолжительное действие непрерывно вводимого отдельного противовоспалительного средства может оказаться нежелательным. Поэтому индуцирование активного и специфического подавления воспалительных процессов может быть преимущественно осуществлено путем объединения иммуномодулятора настоящего изобретения со специфическим антигеном.
Настоящее изобретение также относится к способу лечения Т-клеточно-опосредованного заболевания, расстройства или состояния, где указанный способ предусматривает введение индивидууму, нуждающемуся в этом, эффективного количества терапевтического препарата, содержащего антиген, распознаваемый воспалительными Т-клетками, ассоциированными с патогенезом указанного Т-клеточно-опосредованного заболевания, расстройства или состояния, и адъювант общей формулы Ia, указанной выше.
Настоящее изобретение будет проиллюстрировано следующими неограничивающими примерами.
Примеры
В приводимых примерах следующие соединения будут идентифицированы по их номерам, указанным жирным шрифтом:
1. октадец-(Z)-9-ениловый эфир N,N-диметиламиноуксусной кислоты;
2. октадец-(Z)-9-ениловый эфир никотиновой кислоты;
3. тартрат октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты
4. тартрат октадецилового эфира (4-метилпиперазин-1-ил)уксусной кислоты
5. HCl-соль октадец-(Z)-9-енилового эфира 4-диметиламинометилбензойной кислоты
6а. иодид 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
6b. хлорид 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
6с. тозилат 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
7. хлорид 4-метил-4-октадец-(Z)-9-енилоксикарбонилметилморфолин-4-ия
8. бромид 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
9. битартрат октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты
10. HCl-соль октадец-(Е)-9-енилового эфира 4-N,N-диметиламинометилбензойной кислоты;
11. HCl-соль октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты.
Пример 1
Синтез октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты (соединения 1)
N,N-диметилглицин (1,60 г, 15,4 ммоль) суспендировали в безводном и не содержащем спирта хлороформе (20 мл) и добавляли PCl5 (3,80 г, 17,3 ммоль). Желтую суспензию перемешивали при 55-60°С в течение 1 часа, охлаждали до 30°С и добавляли олеиловый спирт (5,70 мл, 4,85 г, 18,0 ммоль). Желтый раствор нагревали до 63-65°С и перемешивали при этой температуре в течение 3-4 ч. Раствор охлаждали до комнатной температуры и небольшое количество нерастворимых веществ отфильтровывали. К фильтрату добавляли воду и рН этой смеси доводили до 8. Органическую фазу отделяли, промывали 5% NaHCO3 и насыщенным раствором соли, затем сушили и упаривали. Желтый маслянистый остаток (5,07 г) растирали с эфиром и полученную суспензию фильтровали. Фильтрат упаривали досуха и получали 3,18 г неочищенного продукта, который затем очищали колоночной хроматографией (н-гексан, затем этилацетат) с получением указанного в заголовке соединения, называемого соединением 1, в виде желтоватой жидкости (2,6 г, 7,36 ммоль, 41%).
Вычислено: С, 74,73; Н, 12,26; N, 3,96. Найдено: С, 67,88; Н, 11,69; N, 3,13.
1Н-ЯМР (200 МГц, CDCl3) (м.д.) δ: 0,88 (т, 3Н, Ме), 1,27-1,29 (м, 22Н, 11СН2), 1,57-1,67 (м, 2Н, СН2), 2,00 (шир. д, 4Н, 2СН2), 2,37 (с, 6Н, NMe2), 3,18 (с, 2Н, СН2), 4,13 (т, 2Н, СООСН2), 5,31-5,39 (м, 2Н, СН=СН).
ИК: 1755 см-1(СО).
Пример 2
Синтез октадец-(Z)-9-енилового эфира никотиновой кислоты (соединения 2)
Гидрохлорид никотиноилхлорида (17,8 г, 0,1 моль) и олеиловый спирт (26,85 г, 0,1 моль) добавляли в гексан (250 мл). К перемешиваемой смеси в течение 10 минут добавляли пиридин (15,8 г, 0,2 моль) и реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Гидрохлорид пиридина удаляли фильтрованием и промывали гексанами. Объединенный фильтрат экстрагировали водой, сушили над MgSO4 и упаривали досуха. Остаток дважды растворяли в ацетонитриле и образовавшийся клейкий остаток сушили с получением прозрачной подобной воде жидкости, которая после охлаждения превращалась в мылообразное вещество (21,1 г, 56,5 ммоль, 56,5%).
1Н-ЯМР (200 МГц, CDCl3) (м.д.) δ: 0,90 (т, 3Н, Ме), 1,30-1,50 (м, 22Н, 11СН2), 1,75-1,85 (м, 2Н, СН2), 2,00 (шир. д, 4Н, 2СН2), 4,35 (т, 2Н, СООСН2), 5,31-5,39 (м, 2Н, СН=СН), 7,41 (дд, 1Н), 8,30 (дт, 1Н), 8,77 (дд, 1Н), 9,25 (д, 1Н).
Пример 3
Синтез L-тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты (соединения 3)
3а) октадец-(Z)-9-ениловый эфир хлоруксусной кислоты
Олеиловый спирт (147,9 г, 0,55 моль) растворяли в н-гексане (300 мл) и по каплям в течение 10 минут добавляли хлорацетилхлорид (56,5 г, 0,50 моль). Прозрачную реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Следовые количества газообразного HCl удаляли из реакционной смеси, барботируя азот. Этот гексановый раствор неочищенного указанного в заголовке продукта использовали на следующей стадии без дополнительной очистки.
3b) Октадец-(Z)-9-ениловый эфир 4-метилпиперазин-1-илуксусной кислоты (свободное основание)
н-Гексан добавляли к гексановому раствору, полученному, как описано выше в примере 3а (до конечного объема 500 мл), и затем добавляли 1-метилпиперазин (103,6 г, 1,03 моль). Реакционную смесь перемешивали при комнатной температуре в течение 36 ч. Гидрохлорид 1-метилпиперазина удаляли фильтрованием и промывали н-гексаном. Объединенные органические экстракты экстрагировали водой и насыщенным раствором соли, затем сушили и упаривали при пониженном давлении с получением бледно-желтого масла. Полученное масло при его охлаждении до -20°С превращалось в белое твердое вещество.
3с) L-тартратная соль октадец-(Z)-9-енилового эфира 4-метилпиперазин-1-илуксусной кислоты
Свободное основание, полученное, как описано выше в примере 3b, растворяли в метаноле (150 мл) и затем добавляли раствор (L)-(+)-винной кислоты (75 г, 0,5 моль) в метаноле (400 мл). После перемешивания в течение 60 минут при комнатной температуре раствор фильтровали через 20 мкм-фильтр и упаривали досуха. Клейкий остаток сначала гидролизовали ацетонитрилом и затем обрабатывали ацетоном в течение 24 ч. Растворитель декантировали и твердое вещество два раза обрабатывали ацетоном. Полученную суспензию сушили в вакууме (из-за гигроскопичности влажный тартратной соли фильтрацию не проводили). Сухой продукт гомогенизировали и просеивали (500 мкм), получая 197,3 г указанного в заголовке соединения 3 в виде желтоватого порошка (35 ммоль, 64%), т.пл. 69-72°С.
Вычислено: С, 62,34; Н, 9,74; N, 5,01. Найдено: С, 60,33; Н, 9,71; N, 4,91.
1Н-ЯМР (200 МГц, ДМСО-д6) м.д. δ: 0,85 (т, 3Н, Ме), 1,18-1,24 (шир. с, 22Н, 11СН2), 1,54 (шир. с, 2Н, СН2), 1,96 (шир. с, 4Н, 2СН2), 2,56 (с, 3Н, N-Me), 2,69 (с, 4Н, 2СН2), 2,90 (с, 4Н, 2СН2), 3,26 (с, 2Н, N-CH2COO) 4,0 (т, 2Н СН2OCO), 4.18 (c, 1,5Н, тартрат СН), 5,29 (т, 2Н, СН=СН).
ИК(см-1): 1616, 1793(С=О)
Пример 4
Синтез L-тартрата октадецилового эфира (4-метилпиперазин-1-ил)уксусной кислоты (соединения 4)
Стеариловый спирт (27,2 г, 100 ммоль) растворяли в бензоле (150 мл) и по каплям в течение 10 минут добавляли хлорацетилхлорид (8,0 мл, 100 ммоль). Прозрачную реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Следовые количества HCl удаляли, барботируя газообразный азот. Затем добавляли 1-метилпиперазин (28,0 мл, 250 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 36 ч. Гидрохлорид 1-метилпиперазина удаляли фильтрованием и промывали н-гексаном. Фильтрат и промывки объединяли и последовательно экстрагировали водой и насыщенным раствором соли, затем сушили и упаривали досуха, получая свободное основание (38,8 г). Полученное вещество растворяли в этаноле, объединяли с раствором (L)-(+)-винной кислоты (14,18 г) в этаноле (150 мл), перемешивали в течение 60 минут и выдерживали при комнатной температуре в течение 3 дней. Полученное твердое вещество отфильтровывали и сушили с получением 46,5 г (83 ммоль, 83%) указанного в заголовке соединения 4 в виде белого твердого вещества, т.пл. 88-89°С.
Вычислено: С, 62,11; Н, 10,07; N, 5,00. Найдено: С, 61,02; Н, 10,39; N, 5,01.
1Н-ЯМР (200МГц, ДМСО-д6) м.д. δ: 0,85 (т, 3Н, Ме), 1,18-1,30 (шир.с, 30Н, 15СН2), 1,54 (м, 2Н, СН2), 1,96 (шир. с, 4Н, 2СН2), 2,56 (с, 3Н, N-Me), 2,69 (с, 4Н, 2СН2), 2,90 (с, 4Н, 2СН2), 3,26 (с, 2Н, N-CH2COO), 4,02 (т, 2Н СН2OCO), 4,18 (с, 1,5Н, тартрат СН). ИК: 1742 см-1(СО).
Пример 5
Синтез HCl-соли октадец-(Z)-9-енилового эфира 4-диметиламинометилбензойной кислоты (соединения 5)
5а) HCl-соль 4-диметиламинометилбензойной кислоты
Суспензию 4-аминометилбензойной кислоты (92,5 г, 0,61 моль) в 98% муравьиной кислоте (127 мл, 3,36 моль) нагревали при перемешивании до полного растворения и затем добавляли 37% формальдегид (114 мл, 1,53 моль). Раствор нагревали и выдерживали при кипячении с обратным холодильником в течение 10 ч. После охлаждения добавляли 20% HCl (130 мл) и раствор упаривали досуха. Полученный таким образом белый порошок (131,5 г, 0,61 моль, ˜100%) использовали в следующей реакции без дополнительной очистки.
5b) HCl-соль октадец-(Z)-9-енилового эфира 4-диметиламинометилбензойной кислоты
Тионилхлорид (28,6 мл, 0,39 моль) добавляли к перемешиваемой суспензии HCl-соли 4-диметиламинометилбензойной кислоты (65,0 г, 0,3 моль) в ацетонитриле (300 мл), кипятили с обратным холодильником в течение 1 ч и упаривали досуха. Желтоватый твердый остаток суспендировали в ацетонитриле (300 мл), добавляли олеиловый спирт (99,5 мл, 0,31 моль) и смесь кипятили с обратным холодильником в течение 2 ч. Смесь оставляли для охлаждения, неочищенную соль отфильтровывали, промывали ацетонитрилом (500 мл), затем ацетоном (300 мл) и сушили, получая 105 г (0,225 моль, 75%) указанного в заголовке соединения 5 в виде слегка желтоватого твердого вещества, т.пл. 191-193°С.
Вычислено: С, 72,15; Н, 10,38; N, 3,00. Найдено: С, 71,32; Н, 10,68; N, 3,15.
1Н-ЯМР (200МГц, CDCl3) м.д. δ: 0,88 (т, 3Н, Ме), 1,25-1,33 (шир. с, 22Н, 11СН2), 1,75 (м, 2Н, СН2), 2,01 (м, 4Н, 2СН2), 2,83 (с, 6Н, NMe2), 4,30 (т, 2Н, СН2ОСО), 4,39 (с, 2Н, PhCH2), 5,32-5,37 (т, 2H, СН=СН), 7,83 (д, 2Н), 8,10 (д, 2Н)
Пример 6
Синтез соединений 6а, 6b и 6с
6а) Синтез иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния (соединения 6а)
Октадец-(Z)-9-ениловый эфир никотиновой кислоты (3,74 г, 10 ммоль), полученный в соответствии с методикой, описанной в примере 2, и иодметан (3,12 г, 22 ммоль) растворяли в смеси нитрометана (5 мл) и метанола (2,5 мл). Реакционную смесь выдерживали при комнатной температуре в течение 3 дней и упаривали досуха в вакууме. Остаток растворяли в гексане (20 мл), затем гексан декантировали и остаток сушили с получением указанного в заголовке соединения в виде желто-оранжевого клейкого твердого вещества (4,95 г, 9,6 ммоль, 96%).
1Н-ЯМР (200МГц, ДМСО-д6) (м.д.) δ: 0,85 (т, 3Н, Ме), 1,20-1,50 (м, 22Н, 11СН2), 1,70-1,80 (м, 2Н, СН2), 1,96 (шир. д, 4Н, 2СН2), 4,36 (т, 2Н СООСН2), 4,42 (с, 3Н,N-Me), 5,27-5,39 (м, 2Н, СН=СН), 8,25 (дд, 1Н), 8,95 (дт, 1Н), 9,20 (дд, 1Н), 9,55 (д, 1Н).
6b) Синтез хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния (соединения 6b)
Гидрохлорид 1-метилпиридиний-3-карбоксилата (1,74 г, 10 ммоль) добавляли к тионилхлориду (7 мл). Реакционную смесь перемешивали при 68-70°С в течение 1,5 ч и затем упаривали досуха в вакууме. К остатку добавляли гексан (25 мл) и олеиловый спирт (2,69 г, 10 ммоль) и смесь перемешивали при 43°С в течение 3 дней, затем упаривали досуха при пониженном давлении, получая 4,1 г (9,66 ммоль, 96%) клейкого твердого вещества.
1Н-ЯМР (200МГц, ДМСО-д6) (м.д.) δ: 0,85 (т, 3Н, Ме), 1,20-1,50 (м, 22Н, 11СН2), 1,70-1,80 (м, 2Н, СН2), 1,95 (шир. д, 4Н, 2СН2), 4,38 (т, 2Н СООСН2), 4,42 (с, 3Н,N-Me), 5,27-5,40 (м, 2Н, СН=СН), 8,25 (дд, 1Н), 8,95 (дт, 1Н), 9,20 (дд, 1Н), 9,55 (д, 1Н).
6с) Синтез тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния (соединения 6с)
Октадец-(Z)-9-ениловый эфир никотиновой кислоты (3,74 г, 10 ммоль), полученный в соответствии с методикой, описанной в примере 2, растворяли в смеси нитрометана (5 мл) и метанола (2,5 мл). Затем добавляли раствор метил-4-метилбензолсульфоната (1,86 г, 10 ммоль) в смеси нитрометана (2 мл) и метанола (1 мл) и реакционную смесь перемешивали при комнатной температуре в течение 4 дней. Прозрачный раствор упаривали досуха при пониженном давлении с получением 5,2 г (9,3 ммоль, 93%) прозрачного клейкого твердого вещества.
1Н-ЯМР (200МГц, ДМСО-д6) (м.д.) δ: 0,85 (т, 3Н, Ме), 1,20-1,50 (м, 22Н, 11СН2), 1,70-1,80 (м, 2Н, СН2), 1,96 (шир. д, 4Н, 2СН2), 2,25 (с, 3Н толил Me), 4,36 (т, 2Н, СООСН2), 4,42 (с, 3Н, N-Me), 5,27-5,39 (м, 2Н, СН=СН), 7,10 (д, 2Н, Ph), 7,45 (д, 2Н, Ph), 8,25 (дд, 1Н), 8,95 (дт, 1Н), 9,20 (дд, 1Н), 9,55 (д, 1Н).
Пример 7
Синтез хлорида 4-метил-4-октадец-(Z)-9-енилоксикарбонилметил-морфолин-4-ия (соединения 7)
Октадец-(Е)-9-ениловый эфир хлоруксусной кислоты (17,2 г, 50 ммоль) и N-метилморфолин (6,6 мл, 60 ммоль) кипятили в ацетонитриле (100 мл) с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и упаривали досуха. Затем добавляли ацетон, осажденный клейкий остаток отфильтровывали, промывали ацетоном и сушили, получая указанное в заголовке соединение 7 в виде слегка клейкого и гигроскопичного желтоватого твердого вещества, т.пл. 129-131°С (4,11 г, 9,2 ммоль, 18%).
Вычислено: С, 67,31; Н, 10,84; N, 3,14. Найдено: С, 65,58; Н, 11,35; N, 3,48.
1Н-ЯМР (200МГц, CDCl3) (м.д.) δ: 0,88 (т, 3Н, Ме), 1,15-1,50 (м, 22Н, 11СН2), 1,65-1,80 (м, 2Н, СН2), 2,0 (шир. д, 4Н, 2СН2), 3,78 (с, 3Н, N-Me), 4,08-4,36 (м, 10Н, СООСН2, 4СН2 морфолин), 5,28 (с, 2Н, СОСН2N), 5,27-5,39 (м, 2H, СН=СН).
Пример 8
Синтез бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния (соединения 8)
Раствор 3-(2-бромэтил)индола (1,12 г, 5 ммоль) в метаноле (10 мл) добавляли к раствору октадец-(Z)-9-енилового эфира никотиновой кислоты (1,87 г, 5 ммоль) в метаноле (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. и упаривали досуха при пониженном давлении, получая указанное в заголовке соединение 8 в виде клейкого твердого вещества (2,7 г, 4,5 ммоль, 90,3%).
1Н-ЯМР (200МГц, ДМСО-д6) (м.д.) δ: 0,85 (т, 3Н, Ме), 1,20-1,25 (м, 22Н, 11СН2), 1,70-1,80 (м, 2Н, СН2), 1,90-2,05 (шир. д, 4Н, 2СН2), 3,4 (2Н, СН2), 4,31 (т, 2Н, СН2ОСО), 4,95 (т, 2Н, CH2), 5,30-5,39 (м, 2H, СН=СН), 6,30-7,60 (м, 5Н, арил индола), 8,10-9,50 (4Н, пир.), 11,0 (шир. с, 1Н, NH).
Пример 9
Синтез битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты (соединения 9)
К раствору пиперазина (10,77 г, 125 ммоль) в ацетонитриле (200 мл) добавляли раствор неочищенного олеилхлорацетата (полученного из 25 ммоль хлорацетилхлорида) в ацетонитриле (30 мл). Полученную смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и выпаривали досуха. К остатку добавляли воду и этилацетат и фазы разделяли. Органическую фазу промывали равным объемом воды, сушили над сульфатом натрия и выпаривали досуха. Маслянистый остаток растворяли в небольшом объеме метанола и добавляли L-винную кислоту (7,50 г, 2 экв.) в МеОН. Полученный раствор неочищенной соли выпаривали досуха, твердый остаток обрабатывали ацетонитрилом, фильтровали и промывали ацетонитрилом и затем ацетоном. Твердое вещество растворяли в МеОН (400 мл), фильтровали и упаривали досуха. Остаток обрабатывали EtOAc, фильтровали, промывали EtOAc, затем ацетоном и сушили, получая указанное в заголовке соединение 9 в виде белого негигроскопичного порошка, т.пл. 130-132°С (10,45 г, 15 ммоль, 60%).
Вычислено: С, 55,32; Н, 8,42; N, 4,03. Найдено: С, 53,67; Н, 8,66; N, 4,14.
1Н-ЯМР (200МГц, ДМСО-д6) м.д. δ: 0,85 (т, 3Н, Ме), 1,18-1,34 (шир. с, 22Н, 11СН2), 1,54 (шир. с, 2Н, СН2), 1,97 (шир. с, 4Н, 2СН2), 2,73 (с, 4Н, 2СН2), 3,06 (с, 4Н, 2СН2), 3,30 (с, 2Н, N-CH2COO), 4,03 (т, 2Н, СН2ОСО), 4,22 (с, 1,5Н, тартрат CH), 5,33 (т, 2H, СН=СН).
Пример 10
Синтез HCl-соли октадец-(Е)-9-енилового эфира 4-диметиламинометилбензойной кислоты (соединения 10)
Указанное в заголовке соединение 10 (1,0 г, 2,1 ммоль, 46%), т.пл. 194-6°С, получали из гидрохлорида 4-диметиламинометилбензойной кислоты (1,0 г, 4,6 ммоль) в соответствии с методикой, описанной в примере 5b, за исключением того, что вместо олеилового спирта использовали элаидиловый спирт.
Вычислено: С, 72,14; Н, 10,37; N, 3,00. Найдено: С, 71,36; Н, 11,01; N, 3,19.
1Н-ЯМР (200МГц, CDCl3) м.д. δ: 0,88 (т, 3Н, Ме), 1,25-1,33 (шир. с, 22Н, 11СН2), 1,75 (м, 2Н, СН2), 1,96 (м, 4Н, 2СН2), 2,83 (с, 6Н, NMe2), 4,30 (т, 2Н, СН2ОСО), 4,39 (с, 2Н, PhCH2), 5,38 (т, 2H, СН=СН), 7,83 (д, 2Н), 8,10 (д, 2Н).
Пример 11
Синтез HCl-соли октадец-(Z)-9-енилового эфира аминофенилуксусной кислоты (соединения 11)
К раствору N-Вос-фенилглицина (2,51 г, 10 ммоль) в ацетонитриле (50 мл) порциями добавляли 1,1'-карбонилдиимидазол (3,24 г, 20 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа, затем добавляли олеиловый спирт (3,15 мл, 2,68 г, 10 ммоль) и реакционную смесь снова перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали, остаток растворяли в этилацетате (150 мл), затем последовательно промывали 5% NaHCO3, 5% лимонной кислотой и водой, после чего сушили над MgSO4 и упаривали досуха. Остаток растворяли в 1% HCl в этилацетате (100 мл), раствор оставляли на 6 часов при комнатной температуре и упаривали досуха. Полученный таким образом остаток кристаллизовали из смеси эфир/н-гексан с получением не совсем белого твердого вещества (1,6 г, 3,65 ммоль, 36,5%), т.пл. 101-103°С.
Вычислено: С, 71,28; Н, 10,12; N, 3,10. Найдено: С, 70,87; Н, 10,33; N, 3,47.
1Н-ЯМР (200 МГц, ДМСО-д6) (м.д.) δ: 0,85 (т, 3Н, Ме), 1,20-1,50 (м, 22Н, 11СН2), 1,70-1,80 (м, 2Н, СН2), 1,96 (шир. д, 4Н, 2СН2), 4,15 (м, 2Н, СООСН2), 5,25 (с, 1Н, PhCHNH2), 5,27-5,39 (м, 2Н, СН=СН), 7,40-7,60 (м, 5Н, Ph), 9,20 (шир. с, 3Н, N+H3).
Пример 12
Влияние октадец-(Z)-9-енилового эфира диметиламиноуксусной кислоты (соединения 1) на адъювантный артрит (AA)
AA индуцировали путем иммунизации инбредных самок 8-9 недельных крыс Lewis (группы из 8 крыс, полученных от Harlan-Olac) в основание хвоста 0,1 миллилитрами неполного адъюванта Фрейнда (IFA, минеральное масло, содержащее эмульгатор; Sigma), содержащего 1 мг инактивированного микроорганизма Mycobacterium tuberculosis (Difco). Развитие артрита конечностей наблюдали через 12-14 дней и оценивали по 0-16-балльной шкале, и тяжесть воспаления каждой из 4 конечностей оценивали по 0-4-балльной шкале, как описано в литературе (Holoshitz et al., 1983). Пик развития артрита обычно наблюдался примерно на 26-й день после иммунизации.
На день иммунизации одной группе крыс внутривенно (i.v.) вводили 40 мг соединения 1 в 0,5 мг физиологического раствора (треугольники); другой группе крыс i.v. вводили то же самое количество соединения 1 на 14-й день после начала развития AA (круги), и третью группу обрабатывали физиологическим раствором (квадраты). Развитие AA оценивали по 0-16-балльной шкале и степень покраснения и опухания каждой конечности оценивали по 0-4-балльной шкале. Результаты, приведенные на Фиг.1, представлены как среднее±ср.от. Как показано на Фиг.1, соединение 1 может обеспечивать защиту крыс от воспаления, вызываемого AA.
Пример 13
Противовоспалительное защитное действие октадец-(Z)-9-енилового эфира диметиламиноуксусной кислоты (соединения 1), направленное против EAE
EAE представляет собой экспериментальное аутоиммунное заболевание, индуцируемое у некоторых пород крыс путем иммунизации основным белком миелином (МВР) в полном адъюванте Фрейнда (CFA) (у крыс Lewis) или эмульсией спинного мозга крыс в неполном адъюванте Фрейнда (IFA) (у крыс DA). Это экспериментальное заболевание у животных служит моделью аутоиммунного заболевания человека, такого как рассеянный склероз. Это заболевание развивается у животного примерно через 12 дней после иммунизации и характеризуется параличом различной степени, вызываемым воспалением центральной нервной системы. У некоторых пород крыс, таких как крысы Lewis, паралич может длиться вплоть до 6-7 дней, и эти крысы обычно выздоравливают, если только они не погибают в период пика развития острого паралича. У других пород крыс, таких как крысы DA, паралич может перейти в хроническое состояние и может наступать ремиссия.
Для индуцирования и клинической оценки EAE перед иммунизацией спинной мозг, взятый от крыс DA, замораживали, оттаивали и тщательно измельчали шпателем. Крыс иммунизировали одной подкожной инъекцией (непосредственно под кожу) в дорсальное основание хвоста 200 мкл эмульсии, приготовленной из (1:1) IFA (Difco, Detroit, MI, USA) и антигена (объем/масса, то есть 100 мг IFA/100 мг цельного спинного мозга). IFA комплементировали 20 мг/мл штамма 37RA Mycobacterium tuberculosis (Difco)(=полный адъювант Фрейнда, CFA). Эмульсию получали гомогенизацией газонепроницаемым стеклянным шприцем и иглой диаметром 1,2 мм. Крыс регулярно взвешивали и оценивали клинические признаки EAE. Для оценки тяжести заболевания использовали четырехбалльную шкалу: 0 - паралич отсутствует; 1 - слабость хвоста (повисший хвост); 2 - паралич задних лап; 3 - паралич задних и передних лап; 4 - общий тяжелый паралич (Lorentzen et al., 1995).
Группы из 5 или 7 самок 8-9-недельных крыс DA иммунизировали в подушечки задних лап путем инъекции 0,1 мл в подушечку лапы IFA, содержащего 100 мг цельного гомогенизированного спинного мозга, взятого от крыс DA, всего 200 мг на крысу. На день иммунизации крысам внутривенно (i.v.) вводили 40 мг соединения 1 в 0,5 мл физиологического раствора или только физиологический раствор (контроль). Крыс анализировали на тяжесть EAE по 0-4-балльной шкале, как описано выше. Результаты, приведенные на Фиг.2, представлены как среднее±ср.от. Результаты, представленные на Фиг.2, показали, что соединение 1 (круги), по сравнению с необработанным контролем (квадраты) может в значительной степени предотвращать паралич у крыс.
Пример 14
Влияние октадец-(Z)-9-енилового эфира диметиламиноуксусной кислоты (соединения 1) на жизнеспособность кожного трансплантата
Трансплантацию кожи осуществляли в основном, как описано в литературе (Birk et al., 1999). Так, например, мышей выбривали и у умерщвленных доноров с дорсальной стороны, вырезали 1 см2-квадраты кожи, которые затем очищали в забуференном фосфатом физиологическом растворе (PBS). Для получения аллотрансплантата у анестезированных реципиентов (нембутал, 6 мг/мл, 0,25 мл/мышь) вырезали два лоскута кожи из дорсальной области, каждый площадью 1 см2. Затем каждому реципиенту на пораженные участки дорсальной области трансплантировали два донорных аллотрансплантата. На участок вокруг трансплантата наносили гистоакрил (B.Braun Melsungen AG, Melsungen, Germany). Поверх трансплантатов наносили нобекутан (ASTR, Astra Tech, Glos G15, UK).
В данном эксперименте группам из 6 самок 8-недельных мышей BALB/c трансплантировали кожный трансплантат, взятый от самок 8-недельных мышей С57ВL/6, той же толщины и площадью 1 см2. На день трансплантации группу мышей-реципиентов обрабатывали либо физиологическим раствором, либо i.v.-инъекцией 100 мкг физиологического раствора, содержащего 10 мг соединения 1. Затем отмечали день отторжения. Как показано на Фиг.3, кожа, трансплантированная мышам, обработанным соединением 1 (круги), была более жизнеспособной, чем кожа необработанных контрольных мышей (квадраты).
Пример 15
Использование октадец-(Z)-9-енилового эфира диметиламиноуксусной кислоты (соединения 1) в качестве адъюванта для пептида МВР для лечения EAE
Поскольку было показано, что соединение 1 является сильным противовоспалительным иммуномодулятором, то интересно проанализировать его действие в качестве адъюванта, используемого вместе с антигеном, распознаваемым воспалительными Т-клетками, ассоциированными с патогенезом Т-клеточно-опосредованного заболевания, такого как аутоиммунное заболевание, для лечения указанного Т-клеточно-опосредованного заболевания. В качестве модели для MS было выбрано EAE, и в качестве антигена был выбран пептид МВР.
EAE индуцировали у 7-9-недельных самок крыс Lewis инъекцией в подушечку каждой лапы 25 мкг МВР морских свинок, (всего 50 мкг) в CFA (Sigma). Заболевание развивается примерно на 12-й день после иммунизации и характеризуется параличом различной степени, вызываемым воспалением центральной нервной системы. Крыс оценивали на паралич по 0-4-балльной шкале, как описано выше в примере 13.
EAE вызывается Т-клетками, которые распознают определенные детерминанты молекулы МВР. Основная детерминанта МВР у крыс Lewis состоит из пептида с последовательностью 71-90 МВР (называемого далее пептидом р71-90).
Для того чтобы определить, может ли введение энцефалогенного пептида р71-90 МВР вместе с соединением 1, используемым в качестве адъюванта, также ингибировать развитие EAE, группам из 5-8 крыс Lewis подкожно инъецировали пептид р71-90, эмульгированный в 5 мг соединения 1 или в физиологическом растворе, либо только соединение 1 в физиологическом растворе (без пептида), в день иммунизации МВР (день 0) и через пять дней после иммунизации (5-й день), либо на день -14 и день -7 перед иммунизацией МВР. Влияние обработки р71-90/соединением 1 на EAE оценивали, измеряя частоту и тяжесть данного заболевания. Снижение максимальной степени паралича по сравнению со степенью паралича, развивающегося при контрольной обработке пептидом р71-90 в физиологическом растворе или соединением 1 и физиологическим раствором без пептида, указывает на то, что релевантный пептид, такой как пептид р71-90 МВР в эмульсии с соединением 1, обладает способностью модулировать EAE у крыс.
Пример 16
Использование октадец-(Z)-9-енилового эфира диметиламиноуксусной кислоты (соединения 1) в качестве адъюванта для рекомбинантного hsp60 для лечения AA
AA индуцировали у 7-9-недельных самок крыс Lewis иммунизацией в основание хвоста IFA, содержащего 1 мг инактивированного микроорганизма Mycobacterium tuberculosis (Sigma; 0,1 мл 10 мг/мл), как описано выше в примере 12. Степень воспаления сустава оценивали по 0-16-балльной шкале.
Для лечения AA после его индуцирования группы из 5-8 самок 7-9-недельных крыс Lewis обрабатывали 100 мкг рекомбинантного hsp60, то есть микобактериального эпитопа, распознаваемого Т-лимфоцитами при адъювантном артрите в PBS или в эмульсии соединения 1 (5 мг), либо соединением 1 в физиологическом растворе путем подкожной инъекции на день индуцирования AA (день 0) и через 7 дней (день +7), либо на день -14 и день -7. Влияние такой обработки на воспаление сустава анализировали, как описано в примере 12.
Пример 17
Тартрат октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты (соединение 3) ингибирует кожную DTH-реакцию
Гиперчувствительность замедленного типа (DTH), лакализованная воспалительная реакция, индуцированная цитокинами, секретируемыми некоторыми TН-клетками, при их взаимодействии с антигенами некоторых типов, представляет собой установленную экспериментальную модель воспаления кожи.
На первой стадии DTH-реакции индуцировали на коже 8-недельных самок мышей BALB/c. Этих мышей сенсибилизировали местным нанесением на их выбритую брюшину 200 мкл 2% раствора оксазолона (4-этоксиметилен-2-фенил-2-оксазолин-5-она; Sigma, St. Louis, MO) в смеси ацетон/оливковое масло (4:1, об./об.). Через шесть дней после сенсибилизации одно ухо каждой мыши иммунизировали 20 мкл 0,5% раствора оксазолона. Обработку проводили через 30 минут после иммунизации путем нанесения 20 мкл соединения 3 в различных дозах (1, 2 и 4 мг) на сенсибилизированное ухо каждой мыши (контроль: необработанное ухо или ухо, обработанное PBS). Через 24 часа степень воспаления измеряли, проводя тест на опухание уха мыши, и вычисляли процент ингибирования, индуцированного обработкой соединением 3. Результаты представлены на Фиг.4. Обработка 1 мг и 2 мг соединения 3 приводила к ингибированию DTH-реакции (12,5% и 8%, соответственно).
Пример 18
Соединения 5, 9 и 11 ингибируют DTH-реакцию
У мышей индуцировали DTH, как описано в примере 17. Обработку проводили через 30 минут после иммунизации путем нанесения соединений 5, 9 и 11 в дозах 0,5, 1, 2 и 4 мг в 40 мкл. Кроме того, в случае позитивного контроля вводили дексаметазон (2 мг/40 мкл). Группу мышей с индуцированной DTH-реакцией также обрабатывали гидрохлоридом этилового эфира (4-метилпиперазин-1-ил)уксусной кислоты (негативный контроль).
Данные, полученные для тестируемых соединений, а также для дексаметазона указывали на положительный результат в снижении степени воспаления у DTH-модели. Негативный контроль, то есть этиловый эфир не дал никаких положительных результатов. Эти результаты представлены на Фиг.5.
Пример 19
Влияние соединения 3 на адъювантный артрит (AA)
AA индуцировали у 8-недельных самок крыс Lewis, как описано выше в примере 12. В день -7, перед индуцированием заболевания группы крыс подкожно (s.c.) обрабатывали различными количествами соединения 3 (10, 50, 100 или 200 мг). Как показано на Фиг.6, соединение 3 оказывало защитное действие на крыс, направленное против AA у крыс.
Пример 20
Влияние s.c. и per os введения соединения 3 на EAE
ЭАЭ индуцировали у 8-недельных самок крыс Lewis, как описано выше в примере 15, путем введения МВР морских свинок, в CFA.
В одном эксперименте группам крыс (7 животных на группу), за 7 дней до индуцирования заболевания подкожно (s.c.) инъецировали в спину различные количества соединения 3 (10, 50, 100 или 200 мг). Затем крыс исследовали на тяжесть EAE по 0-4-балльной шкале, как описано выше. Результаты представлены на Фиг.7.
В другом эксперименте группам крыс (7 животных на группу) каждый день, начиная со дня индуцирования EAE, перорально (per os) вводили различные количества соединения 3 (0,2, 1, 2, 10 или 20 мг). Затем крыс исследовали на тяжесть EAE по 0-4-балльной шкале, как описано выше. Результаты представлены в таблице 1 и на Фиг.8.
В другом эксперименте соединение 3 перорально вводили через день, начиная со дня индуцирования EAE, и в этом случае указанное соединение было эффективным (данные не приводятся).
Результаты экспериментов показали, что соединение 3 при его подкожном и пероральном введении оказывало протективное действие, направленное против ЭАЭ у крыс.
Пример 21
Влияние перорального введения соединения 9 на EAE
ЭАЭ индуцировали у 8-недельных самок крыс Lewis, как описано выше в примере 15, путем введения МВР морских свинок, в CFA.
Крыс обрабатывали соединением 9 (100 мг/кг) перорально. Затем этих крыс анализировали на тяжесть EAE по 0-4-балльной шкале, как описано выше. Результаты, представленные в таблице 2 и на Фиг.9, показали, что соединение 9 при его пероральном введении оказывало протективное действие, направленное против ЭАЭ у крыс.
Пример 22
Соединение 3 стимулирует иммунологические эффекты ацетата глатирамера (GA)
Ацетат глатирамера (GA) представляет собой иммуномодулятор, используемый для лечения рассеянного склероза (COPAXONE®, торговая марка от фирмы Teva Pharmaceuticals Ltd., Petah Tikva, Israel). GA имеет некоторые хорошо известные иммунологические эффекты, которые могут быть продемонстрированы in vitro и in vivo, а именно образование GA-специфических Т-клеток и снижение тяжести клинических симптомов EAE. Хотя такие эффекты GA и наблюдаются при его введении вместе с некоторыми добавками, однако эти эффекты могут еще более усиливаться. В настоящем примере проиллюстрирован анализ влияния GA-иммунизации и соединения 3, используемого в качестве адъюванта, на исход EAE и на специфический клеточный иммунологический ответ.
Материалы: Ацетат глатирамера (GA) поставлялся Teva Pharmaceuticals Ltd., Petah Tikva, Israel; лиофилизованный гомогенат мышиного спинного мозга (MSCH) от мышей ICR получали в Biological Lab, Netanya, Israel; IFA, М. tuberculosis закупали у Difco; CFA, коклюшный токсин (РТХ), PBS, 2-меркаптоэтанол, конкавалин А (Con A) закупали у Sigma; триптановый голубой закупали у BDH; DССМ1 (ограниченная клеточная культуральная среда) и среда RPMI 1640 поставлялись от Beit-Haemek, Israel; L-глутамин 2 мМ×100, МЕМ (минимальная поддерживающая среда) - заменимые аминокислоты ×100 и пируват натрия 1 мМ×100 (все стерильные) поставлялись от Bio-Lab, Israel.
Экспериментальные животные: EAE тестировали на мышах CSJL/FI, иммунологический ответ оценивали на мышах C57BL/J6. Здоровых и не содержащих каких-либо специфических патогенов самок мышей CSJL/FI и самцов мышей С57BL/J6 получали из Иерусалимского Центра по разведению животных в Израиле (Harlan Animal Breeding Center, Jerusalem, Israel). В начале исследования мыши имели возраст 9 недель и масса 20 г±15%.
А. Протокол теста на EAE:
(i) Мышей разделяли на 4 испытуемые группы, указанные ниже:
(мг/мышь)
(носитель-0,1 мл/мышь)
(ii) Иммунизация: За 11 дней до индуцирования EAE мышей иммунизировали эмульсией GA в IFA (группа 1) или раствором GA и соединения 3 (группа 2), соединением 3 в PBS (группа 3) или только одним PBS (группа 4) путем подкожной инъекции в оба бока в дозе 100 мкл/мышь (в правый и левый бок, 50 мкл/бок).
(iii) Получение эмульсии GA в IFA: Для иммунизации группы 1 раствор GA в PBS (100 мг/мл) разводили 1:2 в IFA до конечной концентрации 50 мг/мл для инъекции 5,0 мг/мышь GA (0,1 мл). Смеси GA+IFA переносили в шприц (2,5 мл), соединенный со вторым шприцем мостиком с люэровским наконечником, и тщательно смешивали путем переноса данного материала из одного шприца в другой в течение примерно 10 минут до тех пор, пока материал не становился в достаточной степени эмульгированным.
(iv) Получение раствора GA с соединением 3: Для иммунизации группы 2 раствор GA в PBS (100 мг/мл) разводили в растворе соединения 3 в PBS (10 мг/мл) до получения конечной концентрации GA 50 мг/мл для иммунизации дозой 5,0 мг/мышь GA. Для иммунизации контрольной группы 3 смешивали равные объемы соединения 3 (10 мг/мл) и PBS (носитель). Конечная концентрация соединения 3 в растворе "соединение 3+GA" для иммунизации группы 2 и соединения 3 для иммунизации контрольной группы 3 составляла 5 мг/мл (уровни доз 0,5 мг/мышь, эквивалентные 25 мг/кг).
(v) Индуцирование EAE: EAE индуцировали путем инъекции энцефалогенного средства, состоящего из MSCH и коммерчески доступного CFA, содержащего Н37Rа Mycobacterium tuberculosis, и коклюшного токсина IV в подушечки лап животных.
(vi) Клинические исследования и оценка: После иммунизации мышей ежедневно наблюдали на аномальные клинические признаки и поведение. Мышей взвешивали перед иммунизацией и затем взвешивали после иммунизации раз в неделю вплоть до окончания исследования. Через десять дней после индуцирования EAE мышей исследовали на клинические признаки EAE, и эти признаки оценивали по соответствующей шкале оценок. Клинические признаки EAE наблюдали и оценивали, начиная с 9-го дня после индуцирования EAE и вплоть до окончания эксперимента по следующей пятибалльной шкале оценок тяжести заболевания: 0 - нормальное поведение; 1 - потеря массы; 2 - слабость хвоста; 3 - гипотония и слабость задних лап; 4 - паралич задних лап; 4 -общий тяжелый паралич; 5 - затрудненное дыхание и/или судороги, и/или полный паралич, или гибель. Все крысы, имеющие оценку 1 и выше, считались больными животными
(vii) Интерпретация результатов:
Вычисление смертности: Определяли число больных и погибших животных в каждой группе, и частоту заболевания и смертность определяли по формуле:
Вычисление процента активности: Процент активности, вычисленный исходя из смертности, определяли следующим образом:
Вычисление средней продолжительности заболевания и начала развития заболевания: Вычисляли среднюю продолжительность заболевания и среднее время до начала развития заболевания в днях.
Вычисление средней максимальной оценки и процента ингибирования: Максимальные оценки для каждой из 10 мышей в тестируемой группе суммировали.
Среднюю максимальную оценку (MMS) для данной группы вычисляли следующим образом:
Σ максимальных оценок для каждой мыши/число мышей в группе.
Процент ингибирования вычисляли следующим образом:
Вычисление средней оценки для группы и процента ингибирования: оценки для каждой из 10 мышей в тестируемой группе суммировали.
Среднюю оценку для данной группы (GMS) вычисляли следующим образом:
Σ оценок для каждой мыши/число мышей в группе.
Процент ингибирования вычисляли следующим образом:
В. Протокол иммунологического теста
(i) Мышей разделяли на 6 тестируемых групп как указано ниже:
(мкг/мышь)
(ii) Иммунизация: За 10 дней до взятия селезенки мышей иммунизировали. Эмульсию GA в CFA и растворы PBS и GA с соединением 3 вводили s.c. в бок мыши в дозе 100 мкл/мышь.
(iii) Получение эмульсии GA в CFA: Раствор GA в PBS (конечная концентрация GA 1,5 мг/мл, для дозы 75 мкг/мышь) разводили в CFA до конечной концентрации 0,75 мг/мл для дозы 75 мкг/мышь GA. Смеси (GA+CFA) переносили в шприц (2,5 мл), соединенный со вторым шприцем мостиком с люэровским наконечником, и тщательно смешивали путем переноса данного материала из одного шприца в другой в течение примерно 10 минут до тех пор, пока материал не становился в достаточной степени эмульгированным.
(iv) Получение раствора GA с соединением 3: Соединение 3 растворяли в PBS до получения 10 мг/мл для дозы 500 мкг/мышь, и разводили 1:10 до получения 1 мг/мл для дозы 50 мкг/мышь. GA (в сосуде объемом 20 мг) растворяли в PBS с получением 20 мг/мл раствора и затем разводили в PBS с получением конечной концентрации GA 1,5 мг/мл для дозы 75 мкг/мышь. Затем равные объемы соединения 3 и GA или PBS для контрольной группы смешивали.
(v) Оценка безопасности: Для тестирования новых композиций на безопасность проводили исследования, в которых мышей ежедневно наблюдали на аномальные клинические признаки и поведение после иммунизации. Мышей оценивали по следующей схеме: 0 - нормальное поведение; 1 - признаки слабой токсичности (пилоэрекция); 2 - умеренные признаки токсичности (неполное закрытие глазного века, сгорбленность, гипоактивность, апатия); 3 - признаки тяжелой токсичности (атаксия, судороги, затрудненное дыхание, паралич, потеря сознания); 4 - гибель. Мышей взвешивали до иммунизации и в день прекращения исследования. Из соображений гуманности исследования завершали после 5-дневного наблюдения, поскольку тяжелое заболевание наблюдалось у 100% мышей во всех группах. Селезенки взвешивали и вычисляли отношение "масса селезенки/масса тела".
(vi) Культивирование клеток: Селезенки мышей для каждой группы собирали и объединяли в стерильной среде RPMI. Клетки экстрагировали, ресуспендировали в обогащенной среде DССМ1 и подсчитывали. Затем клетки культивировали в базальной среде, при 37оС в 5% СО2, при этом в качестве активатора использовали две концентрации GA: 25 мкг/мл и 5 мкг/мл, и в качестве позитивного контроля использовали Con A. Супернатанты собирали и тестировали на уровни секреции цитокинов.
(vii) Детекцию секреции цитокинов в первичных культурах селезенок проводили с помощью ELISA. Клетки активировали in vitro и уровни секреции IL-2 детектировали с использованием коммерчески доступных наборов в соответствии с инструкциями поставщика.
С. Результаты
(i) Результаты для EAE
Краткое описание данных смертности, средних максимальных оценок, средних оценок для группы, среднего периода времени до начала развития заболевания и средней продолжительности заболевания приведены в таблице 3. Подавление признаков EAE наблюдалось в группе, иммунизированной GA (5 мг/мышь)+соединение 3. При этом наблюдалось 60% ингибирование смертности, 16% снижение MMS и 23,7% снижение GMS. Такое подавление признаков EAE в группе, обработанной GA+соединение 3, было выше, чем в группах, обработанных либо соединением 3 без GA, либо GA+IFA (другой добавкой).
(ii) Результаты иммунологических исследований
Результаты анализа на секрецию IL-2 из клеток селезенки иммунизированных мышей представлены в таблице 4 и на Фиг.10. В группах, иммунизированных соединением 3 (без GA) в дозах 50 и 500 мкг/мышь, секреция IL-2 из клеток селезенки не увеличивалась в ответ на GA. Con А (неспецифический активатор) не давал увеличения уровней IL-2 во всех группах. Соединение 3 не влияло на неспецифическую ConA-активацию.
Иммунизация 75 мкг/мышь GA+соединение 3 в дозе 50 мкг/мышь приводила к секреции IL-2 в клетках селезенки в ответ на GA. Уровни IL-2 были выше, чем уровни, наблюдаемые в клетках селезенки, взятых у мышей, иммунизованных одним GA.
Иммунизация 75 мкг/мышь GA+соединение 3 в дозе 500 мкг/мышь также приводила к секреции IL-2 в клетках селезенки в ответ на GA (хотя и в меньшей степени, чем уровни IL-2, наблюдаемые в клетках селезенки, взятых у мышей, иммунизованных одним GA).
В исследованиях на безопасность не наблюдалось какого-либо аномального поведения животных или аномальных клинических признаков. Все оценки были 0.
Соединение 3 и 75 мкг/мышь GA+соединение 3 в обеих дозах не оказывало неблагоприятного воздействия на массу мышей. У всех мышей наблюдалось аналогичное возрастание массы, которое составляло приблизительно 1,5 г/мышь за 10 дней.
Отношения массы селезенки к общей массе тела мышей были аналогичны во всех группах.
D. Выводы
Представленные в данном описании данные показали, что добавление соединения 3 к GA стимулирует его положительное действие на модель EAE (в частности, на выживание) и стимулирует его иммунологический эффект (клеточный ответ клеток селезенки на GA).
Обработка соединением 3 с добавлением или без добавления GA не давала каких-либо признаков токсилогической патологии. Поэтому s.c.-инъекция соединения 3 не приводила к детектируемому острому воспалительному или токсическому эффекту. Кроме того, обработка соединением 3 с добавлением или без добавления GA не давала каких-либо признаков иммуносупрессии.
75 мкг/мышь GA+CFA
75 мкг/мышь GA+CFA
50 мкг/мышь соед.3
75 мкг/мышь GA+50 мкг/мышь соед.3
500 мкг/мышь соед.3
75 мкг/мышь GA+500 мкг/мышь соед.3
Библиография
Beyer S. and Pilgrim H. 1991, Die Hemmung der Endothelzellproliferation durch Ester der 4-Aminomethylbenzoesaure (PAMBA), Pharmazie 46:597-599.
Birk et al., 1999. The 60kDa heat shock protein modulates allograft rejection. Proc. Nat. Acad. Science USA. 96: 5159-63.
Holoshitz, Y. et al., 1983. Lines of Т lymphocytes mediate or vaccinate against autoimmune arthritis, Science 219: 56.
Kontogiorgis C. A., Hadjipavlou-Litinia D.J., and Schulz E., 2001, Antioxidant activity of DLco-phenyl-amino acid octyl esters with anti-inflammatory activity. Arzneim-Forsch./Drug Res,. 51(I):485-488.
Lorentzen J.C. et al., 1995. Protracted, relapsing and demyelinating experimental autoimmune encephalomyelitis in DA rats immunized with syngeneic spinal cord and incomplete Freund's adjuvant, J. Neuroimmunology. 63: 193-205.
Markwardt F., Neuland P., and Klocking H.-P. 1966, Uber die antifibrinolytische Wirkung von Estern der 4-Aminomethylbenzoesaure (PAMBA) Pharmazie 21(6):345-348.
Metayer MM. M. et Jacob J., 1952, Preparation et proprietes pharmacodynamiques de quelques esters de la betaine, Ann. Pharm. Francaise, 10:435-440.
Nixon-George A., Moran Т., Dionne G., Penney C.L., et al. 1990, The adjuvant effect of stearyl tyrosine on a recombinant subunit hepatitis В surface antigen. The Journal of Immunology, 144(12): 4798-4802.
Penney C. L., Shah P., and Landi S., 1985, A simple method for the synthesis of long-chain alkyl esters of amino acids, J. Org. Chem. 50:1457-1459.
Penney С. L., Ethier D., Diorme G., Nixon-George A., Zaghouani H., et al., 1993, Further studies on the adjuvanticity of stearyl tyrosine and ester analogues, Vaccine 11(11):! 129-1134.
Rodriguez, M.D., Gomez, R., Sanchez, M and H. Garcia. 1998, J. Appl. Toxicol 18:313-6.
Rucka M., Oswiecimska M., Witek S. 1983, New biocides for cooling water treatment: Quaternary ammonium salts derivatives of glycine esters. Environment Protection Engineering, 9(3):25-31.
Schewe Ch., Schulz E., Vietinghoff G., Sprung W. D., Kobow M., Loose S., and Schewe Т., 1991, On the mode of action of antiphlogistically active DL-o>-phenyl amino acid esters, Biomed. Biochim. Acta, 50(2): 189-198.
Schulz E., Sprung WD, Kroning G. and Lauge, P. 1982, Pharmacological properties of D,L-2-phenylglycine alkyl esters with special consideration of their anti-inflammatory activity, Agents Actions SuppL, AAS 10:119-128.
Smith D.R., Maienthal M. and Eifert R.L., 1951, Quaternary ammonium salts of heterocyclic bases, JACS, 73:2964-2965.
The Merck Index, 13th Edition, 2001, Merck & Co. Inc., Rahway, N.J., USA.
Vetter W. and Meister W., 1981, Nicotinates as derivatives for the mass spectrometric investigation of long chain alcohols, Organic Mass Spectrometry, 16(3): 118-122.
Wong O., Huntingdon J., Nishihata Т., and Rytting J. H., 1989, New alkyl N,N-dialkyl-substituted amino acetates as transdermal penetration enhancers, Pharmaceutical Research, 6(4):286-295.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ CRTH2 В ТЕРАПИИ | 2004 |
|
RU2412934C2 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СОДЕРЖАЩИЙ ИНГИБИТОР КАРБОКСИПЕПТИДАЗЫ U И ИНГИБИТОР ТРОМБИНА | 2000 |
|
RU2242992C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛИНДИОНА КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2010 |
|
RU2569855C2 |
| СОЕДИНЕНИЯ, СВЯЗЫВАЮЩИЕСЯ С BIR ДОМЕНОМ IAP | 2007 |
|
RU2446170C2 |
| АЗАИНДОЛЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK И ДРУГИХ ПРОТЕИНКИНАЗ | 2005 |
|
RU2403252C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА СФИНГОЗИН-1-ФОСФАТА (S1P) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ МЫШЕЧНОЙ ТКАНИ | 2009 |
|
RU2521286C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2759917C2 |
| МОДУЛЯТОРЫ ROR-ГАММА | 2017 |
|
RU2753490C2 |
| ПРОИЗВОДНЫЕ МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКА, СВЯЗЫВАЮЩЕГО ЖИРНЫЕ КИСЛОТЫ | 2014 |
|
RU2642454C2 |
| НЕКОТОРЫЕ ХИМИЧЕСКИЕ СТРУКТУРЫ, КОМПОЗИЦИИ И СПОСОБЫ | 2009 |
|
RU2537549C2 |
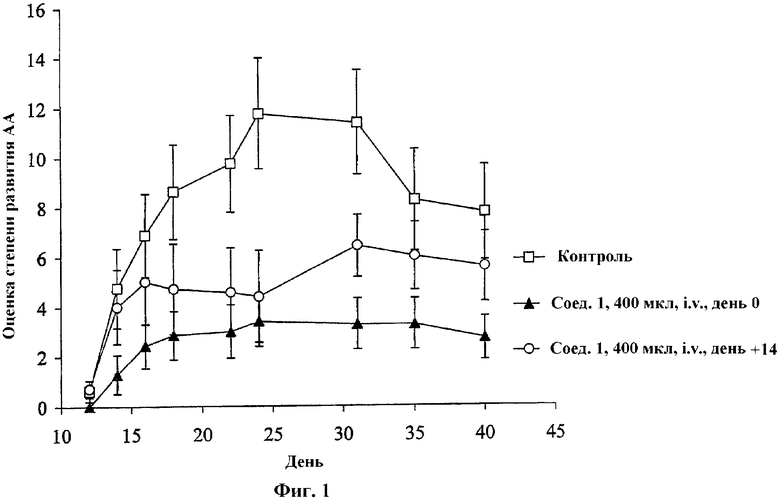
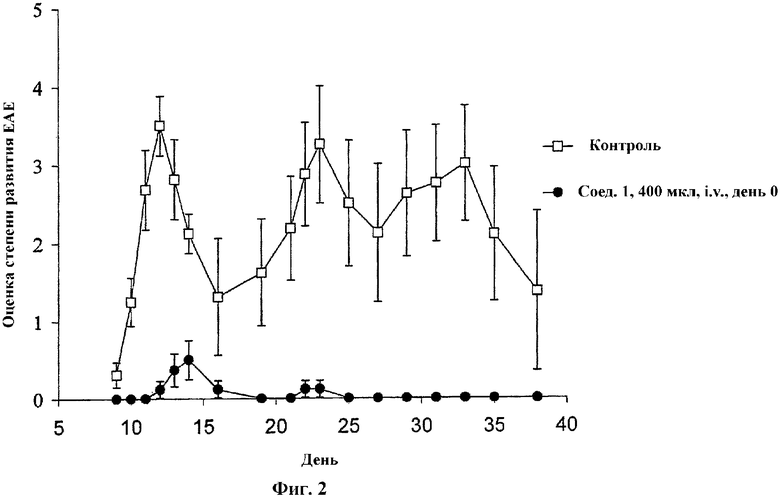
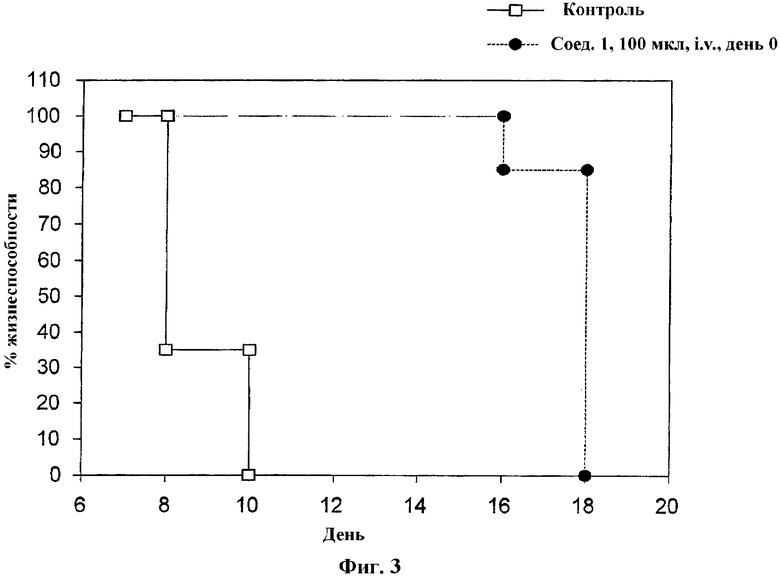
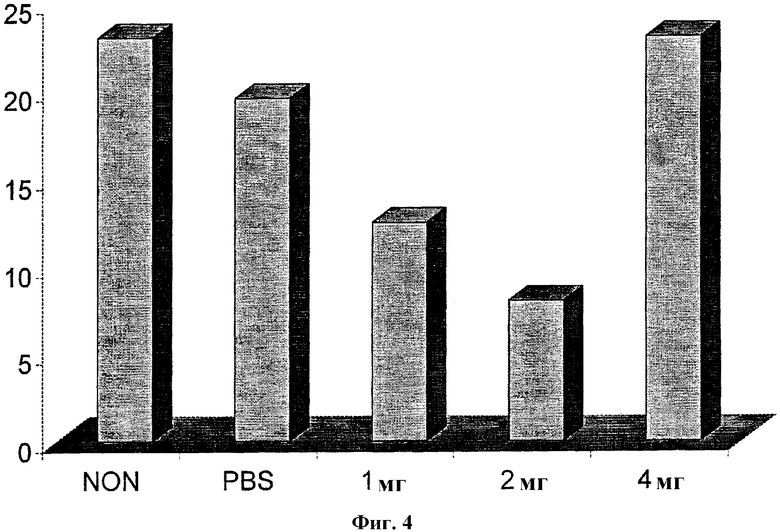
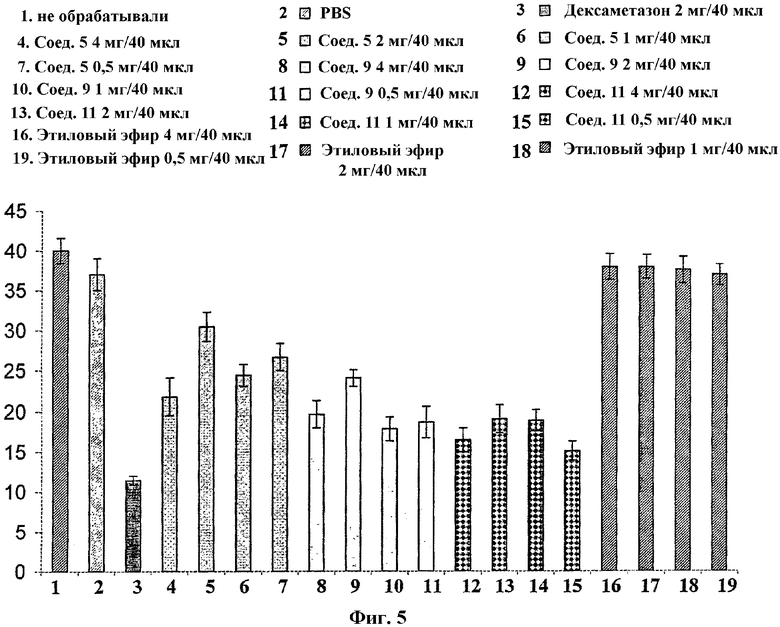
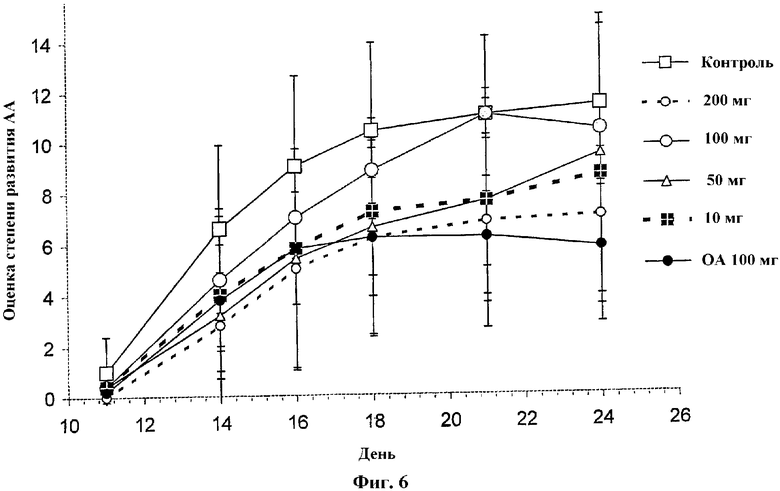
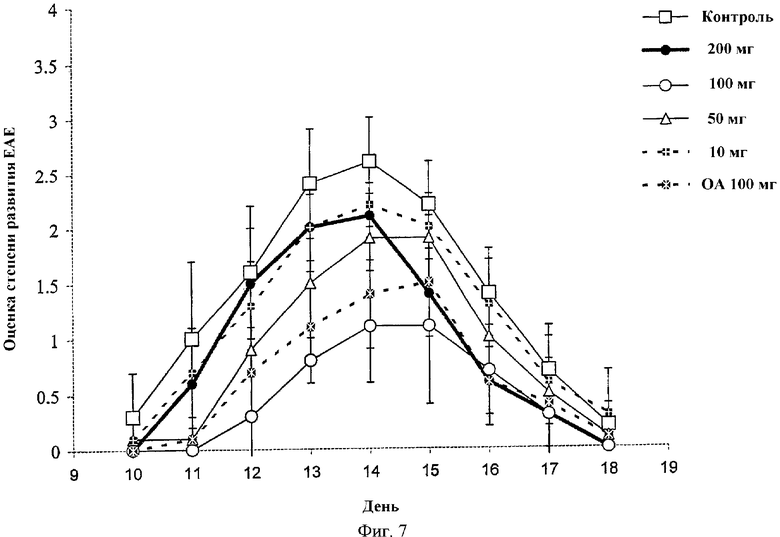
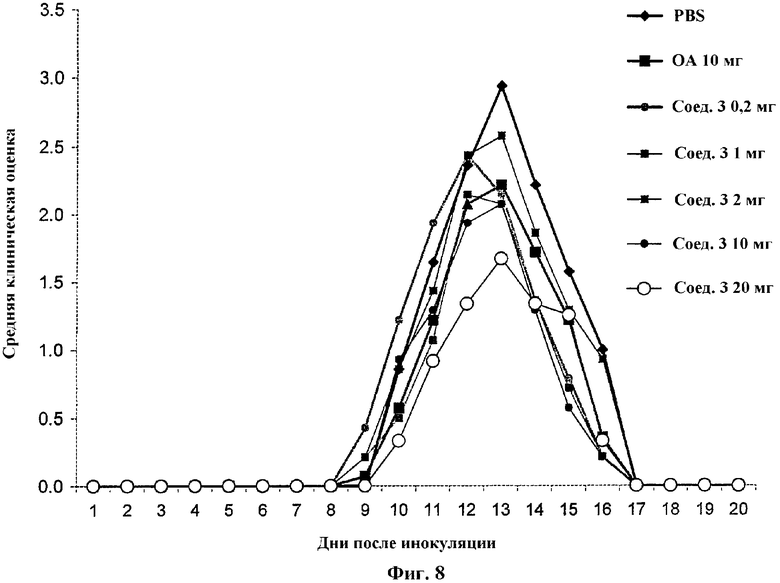
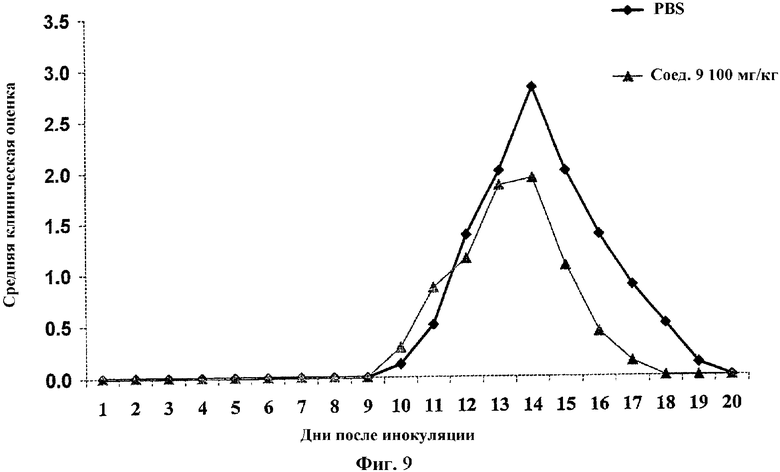
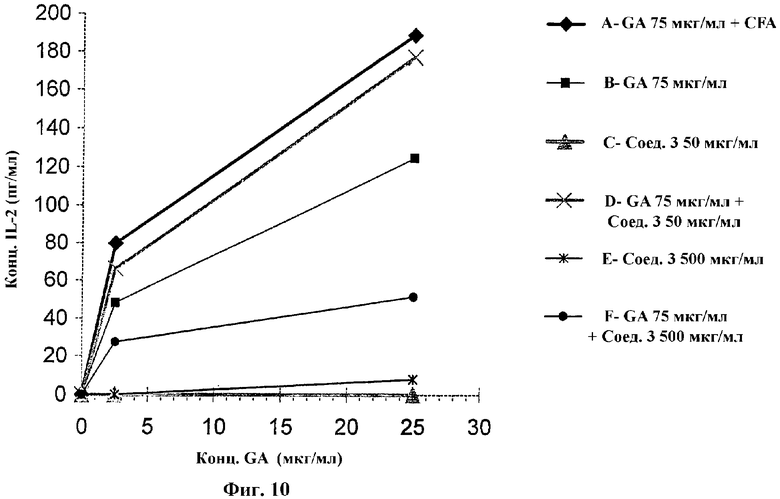
Настоящее изобретение относится к основным эфирам жирных кислот общей формулы R1-О-CO-A или их фармацевтически приемлемым солям, где R1 представляет собой С10-С24алкенил, и А представляет собой остаток, содержащий, по крайней мере, одну ациклическую или циклическую аминогруппу и/или, по крайней мере, одно гетероароматическое кольцо, содержащее третичный или четвертичный атом азота, и к их применению в качестве противовоспалительных или иммуномодулирующих средств, в частности для лечения иммунологически опосредованного воспаления, а также в качестве адъювантов для антигенов, участвующих в клеточных и гуморальных ответах, 10 н. и 170 з.п. ф-лы, 4 табл., 10 ил.
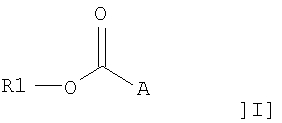
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
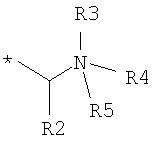
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил; и
(iii)
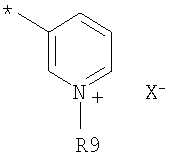
где R9 представляет собой Н, (С1-С6)алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару, и Х отсутствует для получения фармацевтической композиции для лечения иммунологически опосредованного воспаления.
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
тартрата октадецилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец-(Z)-9-енилоксикарбонилметилморфолин-4-ия;
HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты; и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, С1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и
бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
или его фармацевтически приемлемую соль, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил; и
(iii)
где R9 представляет собой Н, (С1-С6)алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару, и Х отсутствует.
(i)
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
тартрата октадецилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец-(Z)-9-енилоксикарбонилметилморфолин-4-ия и HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты; и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, C1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой H или C1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил; и
(iii)
где R9 представляет собой Н, (С1-С6)алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару и Х отсутствует.
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
тартрата октадецилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец(Z)-9-енилоксикарбонилметилморфолин-4-ия;
HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, C1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
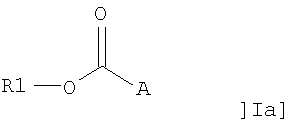
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил; и
(iii)
где R9 представляет собой Н, C1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару и Х отсутствует для получения фармацевтической композиции дополнительно содержащей антиген, где указанный антиген представляет собой антиген, распознаваемый воспалительными Т клетками.
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец(Z)-9-енилоксикарбонилметилморфолин-4-ия;
HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, C1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой H, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил; и
(iii)
где R9 представляет собой Н, (С1-С6)алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару и X отсутствует, причем указанный антиген представляет собой антиген распознаваемый воспалительными Т-клетками.
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец(Z)-9-енилоксикарбонилметилморфолин-4-ия;
HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, C1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или С1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил; и
(iii)
где R9 представляет собой Н, (С1-С6)алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару, и X- отсутствует.
или его фармацевтически приемлемой соли, где R1 представляет собой С10-С24алкенил, и А выбран из
(i)
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или С1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил;
(ii) фенила, замещенного CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил; и
(iii)
где R9 представляет собой Н, (С1-С6)алкил или индолил(С1-С4)алкил, и X- представляет собой противоион или R9 представляет собой электронную пару, и X- отсутствует.
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или C1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец-(Z)-9-енилоксикарбонилметилморфолин-4-ия;
HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, С1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
R1-O-CO-A где:
(i) R1 представляет собой С12-С18алкенил, и А представляет собой остаток формулы:
где R2 представляет собой Н или фенил, необязательно замещенный CR8R8NR6R7; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, и R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил;
(ii) R1 представляет собой С18алкил, и А представляет собой остаток формулы:
где R2 представляет собой Н, R3 представляет собой электронную пару; и R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным С1-С6алкилом,
или
(iii) R1 представляет собой С10-С24алкенил, и А представляет собой фенил, замещенный CR8R8NR6R7, где R6, R7 и R8, каждый независимо представляет собой Н или C1-С6алкил; или
(iv) R1 представляет собой С10-С24алкенил, и А представляет собой остаток формулы:
где R9 представляет собой С1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион;
и их фармацевтически приемлемые соли.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец(Z)-9-енилоксикарбонилметилморфолин-4-ия и битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
иодида 1 -метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
где R2 представляет собой Н или фенил; R3 представляет собой Н, электронную пару или С1-С6алкил; R4 и R5, каждый независимо представляет собой Н или C1-С6алкил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют 5-7-членное насыщенное кольцо, необязательно прерываемое атомом кислорода или атомом азота, необязательно замещенным C1-С6алкилом, где указанное кольцо выбрано из группы, состоящей из пирролидина, пиперидина, морфолина, пиперазина и 4-метилпиперазина.
октадец-(Z)-9-енилового эфира N,N-диметиламиноуксусной кислоты;
тартрата октадец-(Z)-9-енилового эфира (4-метилпиперазин-1-ил)уксусной кислоты;
хлорида 4-метил-4-октадец-(Z)-9-енилоксикарбонилметилморфолин-4-ия;
HCl-соли октадец-(Z)-9-енилового эфира α-амино-α-фенилуксусной кислоты и
битартрата октадец-(Z)-9-енилового эфира пиперазин-1-илуксусной кислоты.
где R9 представляет собой Н, С1-С6алкил или индолил(С1-С4)алкил, и X- представляет собой противоион, либо R9 представляет собой электронную пару, и X- отсутствует.
октадец-(Z)-9-енилового эфира никотиновой кислоты;
иодида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
хлорида 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния;
тозилата 1-метил-3-октадец-(Z)-9-енилоксикарбонилпиридиния и бромида 1-[(2-(1Н-индол-3-ил)этил]-3-октадец-(Z)-9-енилоксикарбонилпиридиния.
| КОНЪЮГАТЫ: ТЕРАПЕВТИЧЕСКОЕ СОЕДИНЕНИЕ - ЖИРНАЯ КИСЛОТА | 1994 |
|
RU2137755C1 |
| US 6118020 A, 21.09.2000 | |||
| Пластика | 1986 |
|
SU1386626A1 |

