Изобретение относится к терапевтическим конъюгатам, которые содержат терапевтическое соединение, связанное с от одной до трех ацильных групп жирных кислот, и способам, включающим применение этих соединений. В частности, настоящее изобретение относится к нестероидным противовоспалительным соединениям, соединенным с от одного до трех ацильными производными жирных кислот.
К наиболее широко применяемым терапевтическим средствам относятся нестероидные противовоспалительные лекарственные средства. Их обычно разделяют на три основные группы: карбоновые кислоты, пиразолы и оксикамы.
Особенно интересными по изобретению являются нестероидные противовоспалительные средства, которые содержат группу карбоновой кислоты, такие как салицилаты. Из этих нестероидных противовоспалительных лекарственных средств наиболее обычно применяемыми соединениями являются ацетилсалициловая кислота (аспирин), ибупрофен и индометацин.
Обнаружено, что нестероидные противовоспалительные лекарственные средства и другие терапевтические соединения могут быть связаны с от одной до трех жирных кислот. Предполагают, что эти новые соединения улучшают чрескожную доставку, чрескожное поглощение, период полувыведения и/или другие способы доставки этих нестероидных противовоспалительных и других терапевтических средств. Кроме того, предполагают, что эти новые соединения будут помогать при пероральной, интерназальной и внутриглазной доставке нестероидных противовоспалительных лекарственных средств и других терапевтических средств.
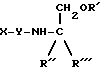
Следовательно, согласно первому аспекту, настоящее изобретение включает соединение следующей формулы: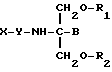
где X представляет собой нестероидное терапевтическое соединение, содержащее карбоксильную группу;
Y - отсутствует или представляет собой одну или две аминокислоты или пептид;
B представляет собой H или CH2O-R3;
R1, R2 и R3 являются одинаковыми или различными и представляют собой водород, метил, этил, гидроксил или ацильную группу, производную жирной кислоты (насыщенной или ненасыщенной), при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой, производной жирной кислоты.
Согласно второму аспекту, настоящее изобретение включает соединение следующей формулы:
X-Y-NH-CH2-CH2O-R4
где X представляет собой нестероидное терапевтическое соединение, содержащее карбоксильную группу;
Y отсутствует или представляет собой одну или две аминокислоты или пептид;
R4 является ацильной группой, производной жирной кислоты.
Согласно третьему аспекту, настоящее изобретение включает способ пролонгирования активности нестероидного противовоспалительного соединения, заключающийся во введении соединения в виде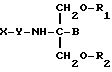
где X представляет собой нестероидное противовоспалительное соединение;
Y отсутствует или представляет собой одну или две аминокислоты или пептид;
B представляет собой H или CH2O-R3;
R1, R2 и R3 являются одинаковыми или различными и представляют собой водород, метил, этил, гидроксил или ацильную группу, образованную из жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой, производной жирной кислоты.
Согласно четвертому аспекту, настоящее изобретение включает способ пролонгирования активности нестероидного противовоспалительного соединения, заключающийся во введении соединения в виде:
X-Y-NH-CH2-CH2O-R4
где X представляет собой нестероидное противовоспалительное соединение;
Y отсутствует или представляет собой одну или две аминокислоты или пептидную или спейсерную группу;
R4 является ацильной группой, производной жирной кислоты.
Согласно пятому аспекту, настоящее изобретение включает способ лечения ожогов, заключающийся во введении соединения формулы
где X представляет собой ибупрофен;
Y отсутствует или представляет собой одну или две аминокислоты или пептид;
B является H или CH2O-R3;
R1, R2 и R3 являются одинаковыми или различными и представляют собой водород, метил, этил, гидроксил или ацильную группу, производную жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой, производной жирной кислоты.
Согласно шестому аспекту, настоящее изобретение включает способ лечения ожогов, заключающийся во введении соединения формулы
X-Y-NH-CH2-CH2O-R4
где X представляет собой ибупрофен,
Y отсутствует или представляет собой одну или две аминокислоты или пептид;
R4 представляет собой ацильную группу, производную жирной кислоты.
Специалистам в данной области понятно, что соединение по первому аспекту настоящего изобретения состоит из терапевтического соединения, присоединенного, необязательно, через аминокислоту(ы), к трометаминовому производному, к которому присоединена ацильная группа, производная жирной кислоты или жирных кислот. Аналогично этому, соединение по второму аспекту может быть признано в качестве терапевтического средства, присоединенного, необязательно, через аминокислоту(ы) к этаноламиновому производному, к которому присоединена ацильная группа, образованная из жирной кислоты.
При предпочтительном осуществлении настоящего изобретения терапевтическое соединение является нестероидным противовоспалительным соединением, которое предпочтительно содержит группу карбоновой кислоты.
Нестероидное противовоспалительное соединение может быть выбрано из широкой области таких известных соединений, включающих салицилат, ацетилсалицилат, салсалат, дифлунизал, фендозал, индометацин, ацеметацин, цинметацин, сулиндак, толметин, зомепирак, диклофенак, изоксепак, ибупрофен, флурбипрофен, напроксен, кетопрофен, фенопрофен, беноксапрофен, индопрофен, пирпрофен, карпрофен, мефенамовую кислоту, флуфенамовую кислоту, меклофенамат, нифлумовую кислоту, толфенамовую кислоту, флуниксин и клониксин. Однако, сейчас предпочтительно, чтобы X был выбран из группы, содержащей ацетилсалицилат, салицилат, индометацин и ибупрофен.
Дальнейшие примеры нестероидных противовоспалительных соединений представлены в работе Weissman, Scientific American, January 1991, 58-64, содержание которой включено здесь в качестве ссылки.
Еще при одном предпочтительном осуществлении изобретения R1 R2 и R3 являются или водородом, или ацильной группой, производной жирной кислоты.
Специалистам в данной области также понятно, что R1 R2 и R3 могут иметь заместители, отличающиеся от метила, этила и гидроксила. Главное требование состоит в том, что, по меньшей мере один из R1 R2 или R3 является ацильной группой, производной жирной кислоты.
При следующем предпочтительном осуществлении по первому аспекту настоящего изобретения каждый из R1 R2 и R3 является ацильной группой, производной жирных кислот, и более конкретно, является одинаковой ацильной группой. Предпочтительно, чтобы жирная кислота имела углеродную цепь из от 3 до 18 атомов углерода, более предпочтительно от 10 до 18 атомов углерода, наиболее предпочтительно 16 атомов углерода. Таким же образом предпочтительно, чтобы R4 представлял собой ацильную группу, производную жирной кислоты, имеющей углеродную цепь из от 3 до 18 атомов углерода, более предпочтительно от 10 до 18 атомов углерода, и наиболее предпочтительно 16 атомов углерода.
По другому предпочтительному осуществлению настоящего изобретения Y отсутствует или является аланином или глицином.
По другому предпочтительному примеру осуществления настоящего изобретения соединение выбрано из группы, содержащей
салицилат-трис-трипальмитат,
салицилат-трис-дипальмитат,
салицилат-трис-монопальмитат,
ибупрофен-аланин-трис-трипальмитат,
ибупрофен-аланин-трис-дипальмитат,
ибупрофен-аланин-трис-монопальмитат,
индометацин-глицин-трис-монопальмитат,
индометацин-глицин-трис-дипальмитат,
индометацин-глицин-трис-трипальмитат,
индометацин-глицин-этаноламин-пальмитат и
индометацин-этаноламин-пальмитат.
Как указано выше, соединения могут, необязательно, содержать линкер. Линкеры, применяемые по настоящему изобретению, хорошо известны и включают:
1. Между соединением с карбоксильной группой и аминогруппой трис
a) линкер с аминогруппой к соединению и карбоксильной группой к трис, такой как аминокислоты и антибиотики;
b) линкер с аминогруппой к соединению и группой сульфоновой кислоты к трис, такой как 2-aминoэтaнcyльфoкиcлoтa (таурин);
c) линкер с аминогруппой к соединению и гидроксильной группой к трис, такой как 2-аминоэтанол (например, через хлоридный или бромидный промежуточный продукт);
d) линкер с гидроксильной группой к соединению и карбоксильной группой к трис, такой как гликолевая кислота, молочная кислота и т.д.;
e) линкер с гидроксильной группой к соединению и группой сульфоновой кислоты к трис, такой как 2-гидроксиэтансульфокислота (изетионовая кислота);
f) линкер с гидроксильной группой к соединению и реакционноспособной галогенидной группой к трис, такой как 2-хлорэтанол;
g) другие примеры потенциально пригодных линкеров между соединением с реакционноспособной карбоксильной группой и аминогруппой трис включают семейства соединений, примерами которых являются п-гидроксибензальдегид, 2-хлоруксусная кислота, 1,2-дибромэтан и этиленоксид.
2. Между соединением с гидроксильной группой и аминогруппой трис
a) линкер с карбоксильной группой к соединению и карбоксильной группой к трис, такой как дикарбоновые кислоты через ангидрид, например янтарный ангидрид, малеиновый ангидрид и т.д.;
b) линкер с карбоксильной группой к соединению и альдегидной группой к трис, такой как глиоксиловая кислота (в присутствии восстановителя, например NaBH4);
c) линкер с карбоксильной группой к соединению и галогенидной группой к трис, такой как хлоруксусная кислота;
d) линкер с карбоксильной группой к соединению и N=C=O-группой к трис, такой как этилизоцианатоацетат.
3. Между соединением с аминогруппой и аминогруппой трис
a) линкер с карбоксильной группой к соединению и карбоксильной группой к трис, такой как дикарбоновые кислоты, через ангидрид, например янтарный ангидрид, малеиновый ангидрид и т.д.;
b) линкер с карбоксильной группой к соединению и альдегидной группой к трис, такой как глиоксиловая кислота (в присутствии восстановителя, например, NaBH4);
c) линкер с карбоксильной группой к соединению и галогенидной группой к трис, такой как хлоруксусная кислота;
d) другие примеры потенциально пригодных линкеров между соединениями с амидогруппой и аминогруппой трис включают семейства соединений, примерами которых являются акролеин (в присутствии восстановителя, например NaBH4) и акриловая кислота.
4. Между соединением с тиольной группой и аминогруппой трис
Примеры потенциально пригодных линкеров между соединением с тиольной группой и аминогруппой трис включают семейства соединений, примерами которых являются хлоруксусная кислота и акролеин (в присутствии восстановителя, например NaBH4).
Для более ясного понимания сущности настоящего изобретения далее описаны его предпочтительные варианты со ссылкой на примеры и чертежи.
Применяются следующие химические аббревиатуры:
ATP1 = аланин-трис-монопальмитат,
ATP2 = аланин-трис-дипальмитат,
ATP3 = аланин-трис-трипальмитат,
CDCl3 = хлороформ-d,
DCC = дициклогексилкарбодиимид,
DCM == дихлорметан,
DCU = дициклогексилкарбамид,
DIEA = диизопропилэтиламин,
DMAP = диметиламинопиридин,
DMF = диметилформамид,
DMSO-d6 = диметилсульфоксид-d6,
GTP1 = глицин-трис-монопальмитат,
GTP2 = глицин-трис-дипальмитат,
GTP3 = глицин-трис-трипальмитат,
HOSu = N-гидроксисукцинимид,
Ибупрофен = α-метил-4-(2-метилпропил)бензолуксусная кислота,
Индометацин = 1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-уксусная кислота,
NSAID = нестероидное противовоспалительное лекарственное средство,
TFA = трифторуксусная кислота,
THF = тетрагидрофуран,
Трис = 2-амино-2-гидроксиметил-1,3-пропандиол,
TSTU = тетрафторборат(O-(N-сукцинимидил)-N,N,N',N'- тетраметилурония,
Z = бензилоксикарбонил.
Материалы и способы
Жидкостная хроматография высокого разрешения (ЖХВР).
Аналитическую ЖХВР проводили на аппаратуре ЖХВР Millipore Waters (Waters Chromatography Division of Millipore, Milford, MA), содержащей систему подачи растворителя серии 6000A с автоматическим регулятором градиента и модулем данных модели 746. Разделение проводили на колонке с обращенной фазой C18 Novapak (100 • 8 мм). Элюирование компонентов, не являющихся ацилпроизводными жирных кислот, проводили при линейном градиенте от 24 до 80% ацетонитрила с 0,1% ТFA в течение 5 минут при объемной скорости потока 2 мл/мин (система A) (Rt A). Определение проводили при 260 нм с применением спектрофотометра Waters Lambda Max, модель 480.
Конъюгаты анализировали на колонке C18 с линейным градиентом от 40% воды /50% ацетонитрила/ 10% THF до 50% ацетонитрила /50 % ТНF, содержащего 0,1% TFA, в течение 5 минут при объемной скорости потока 2 мл/мин (система B) (Rt B).
Препаративная ЖХВР.
Разделение проводили на аппаратуре ЖХВР Millipore Waters Delta Prep 4000, применяя колонки с обращенными фазами C4 и C18 PrepPakR (Prep Nova - Pak HR 40 • 100 мм). Элюирование проводили при линейных градиентах, применяя те же элюентные буферные системы, как и для аналитической ЖХВР, при объемной скорости потока 20 мл/мин.
Ядерный магнитный резонанс (ЯМР).
ЯМР-спектры регистрировали спектрометром Bruker 200 МГц.
Синтезы.
Для синтеза пальмитоилпроизводных индометацина и ибупрофена применяли два альтернативных способа. По одному из них активные сложные эфиры NSAID обрабатывали свежеполученными очищенными GTP1, GTP2, GTP3 или ATP1, ATP2 и ATP3, по другому способу активные эфиры обрабатывали Ala-трис или Gly-трис, продукты очищали и затем пальмитоилировали, получая смесь моно-, ди- и трипальмитатов. Их затем разделяли и очищали, как описано. Производные индометацин-этаноламида и индометацин-глицин-этаноламида синтезировали через активный эфир и пальмитоилировали.
Получение Gly-трис.
Указанное в заголовке соединение получали с количественным выходом гидрогенизацией раствора Z-Gly-трис в этаноле при давлении 40 атм в гидрогенизаторе Парра в присутствии палладия на угле (10%). Удаление бензилоксикарбонильной группы контролировали ЖХВР (система A). Катализатор удаляли фильтрованием и промывали этанолом. После выпаривания растворителя получали указанный в заголовке продукт с 95%-ной чистотой, как показано 1H-ЯМР-спектроскопией. Аналогичным образом получали Ala-трис. Получение Z-Gly-трис и Z-Ala-трис описывается в работе Whittaker, R.G., Hayes, P.J., and Bender, V.J. (1933), Peptide Research 6, 125 и патенте Австралии N 649242.
Получение индометацин-Gly-трис.
В перемешиваемый раствор индометацина (1 г, 2,80 ммоля) в DMF (50 мл) добавляли HOSu (484 мг, 4,2 ммоля). Раствор охлаждали до 0oC и добавляли раствор DCC (618 мг, 3 ммоля) в DCM (5 мл). Перемешивание продолжали при комнатной температуре в течение 2 часов, получая активный эфир с выходом 92% (ЖХВР, система A, Rt 8,38 мин). DCU удаляли фильтрованием и промывали DCM. Объединенные фильтраты добавляли в раствор Gly-трис (1,70 г, 9 ммолей) в DMF (10 мл) и перемешивание продолжали при комнатной температуре в течение 4 часов. Указанное в заголовке соединение получено с выходом 85%, как определено ЖХВР (система A). Растворители удаляли при пониженном давлении, и остаточное масло экстрагировали этилацетатом и сушили (MgSO4), получая указанное в заголовке соединение в виде желтого масла (0,91, 61% от теоретического выхода). ЖХВР (система A) показала один пик, Rt, 5,35.
Получение индометацин-Gly-трис-пальмитатов,
В раствор индометацин-Gly-трис (800 мг, 1,48 ммоля) в DMF (20 мл) при перемешивании при 0oC добавляли пальмитиновую кислоту (570 мг, 2,22 ммоля) в DCM (20 мл), DCC (310 мг, 1,5 ммоля) и DMAP (20 мг). Реакцию контролировали ЖХВР (система B). После выдерживания в этих условиях в течение 16 часов образовались индометацин-Gly-трис-моно, ди- и трипальмитаты в соотношении 66: 30: 4. Растворители выпаривали досуха, остаток снова растворяли в DCM (100 мл) и раствор промывали водой, лимонной кислотой (10%), бикарбонатом натрия (5%) и водой (по 100 мл каждого) и сушили (MgSO4). Моно-, ди- и трипальмитаты разделяли препаративной ЖХВР на колонке C4 (система B), получая продукты, которые дали по одному пику на хроматограмме аналитической ЖХВР, Rt 7,49, 8,93 и 9,80. 1H ЯМР-спектры всех трех продуктов показали, однако, что во время реакции происходило некоторое расщепление молекулы индометацина, наиболее вероятно из-за влияния водной щелочи, поэтому для этих синтезов применяли альтернативный способ.
Получение GTP1, GTP2 и GTP3.
Эти соединения получали гидрогенизацией Z-Gly-трис-моно-, ди- и три-пальмитатов при давлении 40 атм в гидрогенизаторе Парра в присутствии палладия на угле (10%) в этаноле. Удаление бензилоксикарбонильной группы контролировали ЖХВР (система B). После удаления катализатора фильтрованием и выпаривания растворителей получали GTP1, GTP2 и GTP3 с количественными выходами. Синтезы и очистка Z-GTP1, Z-GТР2 и Z-GTP3 и соответствующих аланилсоединений описывается в работе Whittaker, R.G., Hayes Р.J., and Bender, V.J. (1933), Peptide Research 6, 125-128 и патенте Австралии N 649249. Тот же самый источник описывает синтезы моно-, ди- и триформы миристата, лаурата и капрата.
В качестве примеров чистоты приведены следующие данные 1H ЯМР-спектроскопии:
1H ЯМР-спектр GTP2
1H ЯМР-спектр (CDCl3, δ м.д.): 0.87 (6Н, т, пальмитат (CH3), J = 6,4 Гц), 1,19-1,42 (52Н, м, CH2), 1,60 (4Н, м, пальмитат ( β CH3), 2,33 (4Н, т, пальмитат ( α CH2), J = 7,2 Гц), 3,41 (1Н, ш.с, OH), 3,70 (2Н, к, Gly (CH2), J = 7 Гц), 3,80 (2Н, с, трис (CH2)), 4,17 (2Н, д, трис (CH2), J = 10,4 Гц), 4,31 (2Н, д. трис (CH2), J = 11,2 Гц), 7,92 (2Н, ш.с., NH2).
1H ЯМР-спектр Z-GTP2
1H ЯМР-спектр (CDCl3, δ м.д.): 0,89 (6Н, т, пальмитат (CH3), J = 6,4 Гц), 1,19-1,42 (52Н, м, CH2), 1,61 (4Н, м, пальмитат ( β CH2), 2,31 (4Н, т, пальмитат ( α CH2), J = 7,2 Гц), 3,8 (2Н, д, трис(CH2), J = 5,5 Гц), 3,82 (2Н, д, Cly (CH2), J = 8 Гц), 4,21 (2Н, д, трис(CH2OCO), J = 11,6 Гц), 4,39 (2Н, д, трис(CH2OCO), J = 11,4 Гц), 5,14 (2Н, с, Ar-CH2), 5,31 (1Н, т, NH глутамида), J = 5,4 Гц), 6,55 (1Н, с, трис(NН амида), 7,35 (5Н, м Ar(H)).
Получение индометацин-Gly-трис-дипальмитата.
В раствор индометацина (716 мг, 2 ммоля) в сухом DMF (15 мл) добавляли TSTU (Fluka, 85972) (1,20 г, 4 ммоля) в DMF (10 мл). Предполагаемое значение pH раствора 8,3 поддерживали при помощи DIEA и смесь перемешивали при комнатной температуре. Образование активного эфира контролировали обращенно-фазовой ЖХВР на колонке C18 (система A). Реакцию заканчивали за 1 час; Rt индометацина 8,08 мин; индометацин-OSu - 8,38 мин.
Этот раствор добавляли по каплям в раствор Gly-трис-дипальмитата (GTP2, 640 мг, 1 ммоль) в DCM (10 мл). Значение pH поддерживали 8,3 при помощи DIEA и реакцию контролировали ЖХВР на колонке C18 (система B). После подсчета относительной площади, который показал, что реакция прошла на 65-70%, раствор выпаривали досуха при пониженном давлении.
Указанный в заголовке продукт получали в форме белого твердого вещества после хроматографии на силикагеле (Kieselgel S, 0,063-0,2 мм, для колоночной хроматографии, Riedelde Haen A.G.) при элюировании смесью этилацетат/гексан от (1:9) до (6:4). Фракции анализировали ЖХВР (система B). Фракции, содержащие чистый продукт, объединяли, выпаривали досуха и лиофилизировали из трет. бутанола.
Раствор пробы продукта в DCM (10 мг/мл) дает один пик при ЖХВР, Rt B 8,89, и имеет чистоту выше, чем 95%, демонстрируемую 1H ЯМР-спектроскопией.
1H ЯМР-спектр (CDCl3, δ м.д.): 0,86 (6Н, т, пальмитат (CH3), J = 6,4 Гц), 1,02-1,39 (52Н, м, CH2), 1,59 (4Н, м, пальмитат ( β CH2), 2,32 (4Н, т, пальмитат ( α CH2), J = 7,4 Гц), 2,39 (3Н, с, CH3), 3,69 (2Н, с, OCH2), 3,77 (2Н, т, Gly (CH2), J = 5,2 Гц), 3,82 (5Н, с, трис (CH2), OCH3, 4,19 (2Н, д, трис(CH2), J = 12,2 Гц), 4,37 (2Н, д, трис(CH2), J = 11,6 Гц), 6,23 (1Н, т, NH глутамида), J = 5,4 Гц), 6,55 (1Н, с, трис(NH амида), 6,70 (1Н, дд, Ar(CH), J = 2,4; 8,8 Гц), 6,91 (1Н, д, Ar(CH), J = 9,1 Гц), 6,93 (1Н, д, Ar(CH), J = 2,4 Гц), 7,49 (2Н, д, Ar(CH), J = 8,5 Гц), 7,68 (2Н, д, Ar(CH), J = 8,8 Гц).
Получение индометацин-Gly-трис-трипальмитата.
Указанное в заголовке соединение получали способом, аналогичным описанному выше для дипальмитатного производного. Проводили реакцию индометацина (716 мг, 2 ммоля), TSTU (1,2 г, 4 ммоля) и Gly-трис-трипальмитата (950 мг, 1 ммоль), контролируя ее ЖХВР (система B). Указанное в заголовке соединение получали после очистки на силикагеле в виде хроматографически чистого соединения Rt 9,42 (система B).
Получение индометацин-глицина.
В суспензию индометацина (500 мг, 1,40 ммоля) в ацетонитриле (20 мл) добавляли HOSu (177 мг, 1,54 ммоля) и DCC (317 мг, 1,54 ммоля) и раствор перемешивали при комнатной температуре в течение 2 часов. К образованному эфиру индометацина и гидроксисукцинимида добавляли раствор глицина (178 мг, 2,38 ммоля) и триэтиламина (184 мг, 1,82 ммоля) в воде (10 мл) и смесь перемешивали 2 часа, разбавляли водой (60 мл), подкисляли 1 М HCl и экстрагировали этилацетатом (80 мл). После сушки (MgSO4) раствор выпаривали досуха, получая желтое твердое вещество, которое перекристаллизовывали из смеси этилацетат/гексан для получения заглавного продукта (406 мг, 70% от теоретического количества) с чистотой 95%, определенной методом 1H ЯМР-спектроскопии.
Получение индометацин-глицин-этаноламида.
В раствор индометацин-глицина (1,5 г, 3,62 ммоля) в смеси ацетонитрил/DMF (60 мл, 5:1) добавляли HOSu (460 мг, 3,98 ммоль) и DCC (820 мг, 3,98 ммоль) и раствор перемешивали 3 часа до окончания реакции. Добавляли этаноламин (445 мг, 7,3 ммоль) и перемешивание продолжали при комнатной температуре 1 час. Добавляли этилацетат (300 мл) и раствор промывали 1 М HCl (120 мл), водой и насыщенным раствором NaCl (по 120 мл каждого) и сушили (MgSO4). После выпаривания растворителя получали неочищенный указанный в заголовке продукт, который перекристаллизовывали из смеси этилацетат/гексан, получая указанный в заголовке продукт (1,45 г, 88% от теоретического количества) в виде желтого твердого вещества.
Получение индометацин-глицин-этаноламид-пальмитата.
В суспензию индометацин-глицин-этаноламида (410 мг, 0,89 ммоля) в DCM/DMF (25 мл, 9:1) добавляли пальмитиновую, кислоту (240 мг, 0,93 ммоль), DMAP (10 мг) и DCC (192 мг, 0,93 ммоль) и раствор перемешивали 16 часов. Раствор затем разбавляли DCM (50 мл) и промывали 1 М HCl, водой, насыщенным раствором NaCl (по 50 мл каждого) и сушили (MgSO4). После удаления растворителя остаток хроматографировали на диоксиде кремния. Элюировали смесью DCM/этилацетат, получая заглавный продукт в виде желтого твердого вещества. Окончательную очистку проводили перекристаллизацией из этанола (выход 510 мг, 82% от теоретического количества).
Получение индометацин-этаноламида.
В раствор индометацина (1,0 г, 2,80 ммоль) в ацетонитриле (50 мл) добавляли HOSu (335 мг, 2,9 ммоль) и DCC (605 мг, 2,9 ммоль) и раствор перемешивали при комнатной температуре 1 час. Добавляли затем этаноламин (340 мг, 5,5 ммоль) и раствор перемешивали еще 2 часа. DCU удаляли фильтрованием и промывали DCM. Объединенные фильтраты и промывные растворы выпаривали и остаток снова растворяли в DCM (50 мл), промывали 1 М HCl (30 мл) и водой (30 мл) и сушили (MgSO4). После выпаривания растворителя получали заглавное соединение в виде желтого твердого вещества (1,1 г, 98% от теоретического количества). Перекристаллизацией из смеси этилацетат/гексан с несколькими каплями DCM получали чистый продукт в виде слабо-желтого твердого вещества (750 мг, 67% от теоретического количества).
1H ЯМР-спектр (CDCl3, δ м.д.): 2,37 (3Н, с, CH3), 2,78 (1Н, ш.с., OH), 3,36 (2Н, дт, CH2, J = 5,5; 5,5 Гц), 3,65 (4Н, ш.с., CH2CO, CH2OH), 3,82 (3Н, с, OCH3), 6,16 (1Н, ш.т, NH амида, J = 5,5 Гц), 6,68 (1Н, дд, Ar(CH), J = 2,4; 9,1 Гц), 6,87 (1Н, д, Ar(CH), J = 9,1 Гц), 6,90 (1Н, д, Ar(CH), J = 2,4 Гц), 7,46 (2Н, д, Ar(CH), J = 7,9 Гц), 7,64 (2Н, д, Ar(CH), J = 7,9 Гц).
Получение индометацин-этаноламид-пальмитата.
В раствор индометацин-этаноламида (600 мг, 1,50 ммоль) в DCM (10 мл) добавляли пальмитиновую кислоту (405 мг, 1,58 ммоль), DMAP (10 мг) и DCC (320 мг, 1,55 ммоль). Раствор перемешивали при комнатной температуре 16 часов и разбавляли DCM (50 мл), промывали 1 М HCl (50 мл) и водой (50 мл) и сушили (MgSO4). После выпаривания растворителя получали неочищенный продукт в виде слабо-маслянистого твердого вещества, который затем очищали хроматографией на диоксиде кремния. Элюирование проводили смесью DCM/этилацетат с отношением от (9:1) до (8:2), получая заглавное соединение. Перекристаллизация из этилацетата давала 610 мг (66% от теоретического количества) плотного твердого продукта.
1H ЯМР-спектр (CDCl3, δ м.д.): 0,86 (3Н, т, пальмитат (CH3), J = 6,4 Гц), 1,20-1,42 (26H, м), 1,46 (2Н, м, пальмитат ( β CH2), 2,05 (2Н, т, J = 7,9 Гц), 2,38 (3Н, с, CH3), 3,45 (2Н, дт, CONHCH2, J = 5,5; 5,5 Гц), 3,63 (3Н, с, CH2), 3,80 (3Н, с, OCH3), 4,08 (2Н, т, CH2OCO ,J = 5,5 Гц), 5,95 (1Н, ш. т, NH амида, J = 5,5 Гц), 6,67 (1Н, дд, Ar(CH), J = 2,4; 9,1 Гц), 6,84 (1Н, д, Ar(CH), J = 9,1 Гц), 6,85 (1Н, д, Ar(CH), J = 2,4 Гц), 7,47 (2Н, д, Ar(CH), J = 8,5 Гц), 7,70 (2Н, д. Ar(CH), J = 8,5 Гц).
Синтез конъюгатов ибупрофена
Получение ибупрофен-Ala-трис-монопальмитата.
Проводили реакцию ибупрофена (206,3 мг, 1 ммоль) в DMF (5 мл) с TSTU (377 мг, 1,25 ммоль), который был растворен в DMF (5 мл) при pH 8,5 (DIEA). Образование ибупрофен-OSu контролировали ЖХВР (система A) и нашли, что она заканчивается за 10 минут, Rt A 5,70. В эту смесь добавляли раствор ATP1 (536 мг, 1,25 ммоля) в DCM (10 мл) и pH поддерживали 8,5 (DIEA). Образование заглавного продукта контролировали ЖХВР (система B). После завершения реакции растворители выпаривали при пониженном давлении. Препаративной ЖХВР на колонке C4 получали хроматографически чистую пробу, Rt B 7,83.
Ибупрофен-Ala-трис-дипальмитат.
Заглавный продукт получали таким же образом, как описано выше для монопальмитата. Реакцией ибупрофен-OSu с ATP2 (835 мг, 1,25 ммоль) получали хроматографически чистую пробу, Rt B 9,17 минут.
Ибупрофен-Ala-трис-трипальмитат.
Заглавный продукт синтезировали, как описано выше; реакцией ибупрофен-OSu с ATP3 (1,134 г, 1,25 ммоль) получали хроматографически чистый образец, Rt B 10,43.
Молекулярные массы пальмитатов ибупрофена определяли масс-спектрометрией, получая отчетливые пики для масс 619,5, 858,0 и 1096,5, которые хорошо согласуются с рассчитанными молекулярными массами. 1H ЯМР-изучение этих препаратов показало, однако, что этим способом получают продукт в виде рацемической смеси, поэтому применяли альтернативный способ, который включает разделение изомеров кристаллизацией ибупрофен-Ala-трис-соединения.
Получение ибупрофен-Ala-трис.
В растворе ибупрофена (3 г, 14,5 ммоля) в DCM (50 мл) добавляли HOSu (2,49 г, 21,6 ммоля) и DIEA (1,25 мл, 7,25 ммоля) и раствор охлаждали до 0oC. В реакционную смесь добавляли по каплям DCC (3 г, 14,5 ммоля), растворенный в DCM (20 мл). Смесь перемешивали при 0oC в течение 2 часов и затем продолжали перемешивание при комнатной температуре 12 часов, получая активированный эфир (ибупрофен-OSu) с выходом 95% (ЖХВР). Осадок DCU отделяли фильтрованием и в реакционную смесь добавляли Ala-трис (2,77 г, 14,5 ммоля), растворенный в DMF (10 мл) и смесь перемешивали при комнатной температуре в течение 4 часов. Заглавное соединение образовалось с выходом 70% (ЖХВР). Rt A 5,43. Растворители удаляли при пониженном давлении, и маслообразный остаток растворяли в DCM и промывали лимонной кислотой (4%), раствором бикарбоната натрия (5%) и водой. Раствор в DCM сушили (MgSO4) и выпаривали досуха, получая 5,4 г маслообразного соединения. Маслообразный остаток был рацемической смесью 50/50, которую идентифицировали по ее 1H ЯМР-спектру. Дублетные пики метилового протона аланина и ибупрофена диастереоизомеров смещались около на 6 Гц друг от друга.
1H ЯМР-спектр смеси (CDCl2, δ ч. на млн.): 0,89 (6Н, д, Ибу (CH3), J = 6,4 Гц), 1,22, 1,25 (3Н, 2д, Ala (β CH3), J = 5,2 Гц), 1,40, 1,43 (3Н, 2Д, Ибу (β CH3), J = 4,9 Гц), 1,86 (1Н, м, Ибу(CH)), 2,46 (2Н, д, Ибу(CH2), J = 7 Гц), 3,57 (1Н, к, Ибу(α CH), J = 7,9 Гц), 3,67 (6Н, с, трис(CH2), 4,14 (3Н, ш. с. , трис(OH)), 4,28 (1Н, м, Ala(α CH), 5,99, 6,02 (1Н, 2Д, Ala (NH амида), J = 6,7 Гц), 7,09 (1Н, с, трис(NH амида), 7,16 (4Н, м, Ar(H)).
Два различных диастереоизомера удачно разделяли частичной кристаллизацией рацемата в DCM (1,6 г). Аналогичная работа описывалась также M. Bodanzky, "Principles of Peptide Synthesis", volume 16, Springer - Verlag, Berlin 1984. 1H ЯМР-спектры кристаллов и маслообразного остатка подтвердили чистоту диастереоизомеров.
1H ЯМР-спектр кристаллов (CDCl3, δ м.д.): 0,89 (6Н, д, Ибу (CH3), J = 6,4 Гц), 1,3 (3Н, д, Ala ( β CH3), J = 7,6 Гц), 1,48 (3Н, д, Ибу(CH3), J = 7,3 Гц), 1,86 (1Н, м, Ибу(CH)), 2,46 (2Н, д, Ибу (CH2), J = 7 Гц), 3,57 (1Н, к, Ибу ( α CH), J = 7,9 Гц), 3,67 (6Н, с, трис(CH2)), 4,14 (3Н, ш.с., трис(OH)), 4,28 (1Н, м, Ala( α CH), 6,02 (1Н, д, Ala (NH амида), J = 6,4 Гц), 7,09 (1Н, с, трис (NH амида)), 7,16 (4Н, м, Ar(H)).
Ибупрофен-Ala-трис-пальмитаты.
В раствор ибупрофен-Ala-трис (кристаллический продукт, 1,2 г, 3 ммоля) в 50%-ной сухой смеси растворителей DCM/DMF (40 мл) добавляли пальмитиновую кислоту (0,924 г, 3,6 ммоля) и каталитическое количество DMAP (20 мг), и реакционную смесь охлаждали до 0oC. В реакционную смесь добавляли по каплям DCC (0,77 г, 3,6 ммоль), растворенный в DCM (20 мл). Ее перемешивали при 0oC в течение 1 часа и затем при комнатной температуре 20 часов. Содержание моно-, ди- и трипальмитатов было 55, 39 и 6% (определено ЖХВР с системой B).
Растворители выпаривали досуха и остаток снова растворяли в DCM, DCU отделяли фильтрованием и фильтрат промывали раствором бикарбоната натрия (5%) и водой. Разделением этой смеси препаративной ЖХВР на колонке C18 получали с высокой чистотой монопальмитат (520 мг, Rt B 7,81 мин), дипальмитат (720 мг, Rt B 9,24 мин) и трипальмитат (63 мг, Rt B 10,29 мин) (общий выход соединений 56%).
1H ЯМР-спектр монопальмитата (CDCl3, δ м.д.): 0,87 (3Н, т, пальмитат (CH3), J = 6,4 Гц), 0,89 (6Н, д, Ибу (CH3), J = 6,4 Гц), 1,13-1,40 (26Н, м, пальмитат(CH2)), 1,49 (3Н, д, Ибу( β CH3), J = 7 Гц), 1,58 (5Н, м, Ala ( β CH3), пальмитат ( β CH3), 1,86 (1Н, м, Ибу( α CH)), 2,46 (2Н, д, Ибу(CH2), J = 7 Гц), 3,56 (1Н, к, Ибу( α CH), J = 7 Гц), 3,64 (2Н, т, трис (CH2), J = 7,9 Гц), 3,74 (2Н, т, трис(CH2), J = 6,1 Гц), 3,82 (1Н, т, трис(OH), J = 6,4 Гц), 3,99 (1Н, т, трис(OH) , J = 6,7 Гц), 4,26 (2Н, с, трис(CH2)), 4,27 (1Н, м, Ala( α CH)), 5,77 (1Н, д, Ala (NH амида), J = 6,7 Гц), 6,87 (1Н, с, трис-NH), 7,16 (4Н, м Ar(H)).
Получение салицилат-трис.
Метилсалицилат (2,3 г, 15,1 ммоля) обрабатывали трис (14,8 г, 120,8 ммоля) в DMF (182 мл) и водой (121 мл) при pH 9,0 и 60oC в течение 48 часов. Образование указанного в заголовке продукта (Rt 3,95) из метилсалицилата (Rt 7,66) контролировали ЖХВР (система A) до прекращения реакции при 75% превращения. Смесь затем выпаривали досуха при пониженном давлении и пробу растворяли в воде. pH устанавливали 4,5 и заглавный продукт очищали препаративной ЖХВР и затем ионообменной хроматографией (смола Amberlite 1R 63), получая хроматографически чистый салицилат-трис (3,66 г, 65% от теоретического количества).
Салицилат-трис теперь готов для реакции с 1-3 жирными кислотами.
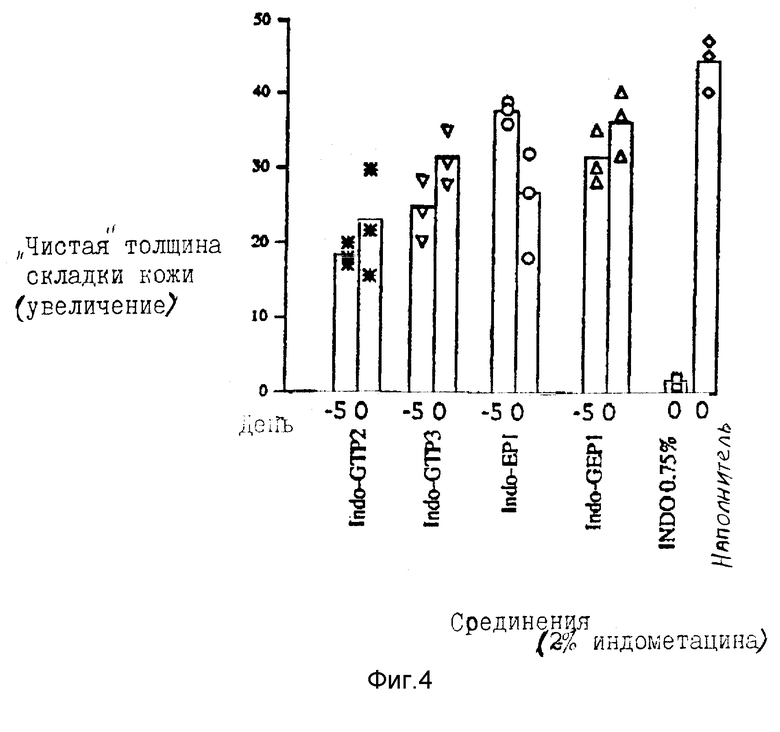
Противовоспалительное действие модифицированных нестероидных противовоспалительных лекарственных средств после индуцированного острым воздействием УФВ-лучей фотоповреждения у безволосых мышей SKH-1
Введение.
Постоянное воздействие субэритемных доз ультрафиолетовых (УФ) лучей на безволосых мышей-альбиносов (SKH-1) индуцирует как видимые, так и гистологические изменения в коже (Bissett et al., 1990). Острое воздействие УФВ-лучей индуцирует солнечную эритему и отек. Оба эти результата временные и исчезают в течение нескольких дней (Reeve et al., 1991).
Kaidbey и Kurban в 1976 г. и Snyder исследовали предел, до которого индометацин при местном применении мог подавлять солнечную эритему у людей и морских свинок. Было найдено, что в этих экспериментах индометацин оказывает глубокое воздействие на эритему. Оказалось, что наиболее эффективна концентрация индометацина 2,5% в наполнителе пропиленгликоль : этанол : диметилацетамид (19:19:2). Субъекты, получившие 3 MED (средние эритемные дозы) УФ-лучей и лечение индометацином с такой концентрацией, проявили общее защитное действие индометацина.
Метод.
Группы из 3 самок мышей подвергали действию 3 MED УФВ-лучей на их дорсальную поверхность и затем сразу смазывали (путем ровного нанесения местного раствора на дорсальную поверхность) 60 мкл или 100 мкл наполнителя или наполнителя, содержащего повышаемые концентрации одного или нескольких NSAID или конъюгатов NSAID с жирными кислотами. Применяемый наполнитель был модификацией ранее известных наполнителей и состоял из этанола и DMSO соотношении 5:1. Испытываемые NSAID и их конъюгаты использовали после УФВ-облучения для снижения возможности их солнцезащитного действия (0 день) или за 1-5 дней до УФВ-облучения. Отек затем определяли измерением толщины кожной складки (SFT) сложенного средне-дорсального кусочка кожи при помощи ручного микрометра. Измерения проводили в 0 часов, непосредственно перед облучением УФВ-лучами, и через 24 часа после облучения УФВ-лучами. Контрольную группу, смазанную 60 или 100 мкл наполнителя, применяли для определения усиления отека кожи из-за воспалительной реакции. Было найдено, что точка времени 24 часа соответствует максимальному отеку кожи. Величину SFТ до УФВ-облучения вычитали из величины SFT после 24 часов для определения увеличения результирующей (чистой) толщины складки кожи (NSFT).
Результаты.
Защитное действие индометацина от отека
В качестве предварительного изучения определяли способность индометацина защищать мышей skh-1 от отека. Индометацин в пределах концентраций от 0,1% до 2,5% наносили после облучения мышей УФВ-лучами; применяли также необлученных контрольных мышей. Сначала нашли, что при концентрациях 2% и выше индометацин вызывает летальный исход после 36 часов, индуцируя типичное желудочно-кишечное кровотечение. Это привело к тому, что концентрацию индометацина 1% установили как высшую концентрацию в дальнейших экспериментах. Было найдено, что молодые мыши даже более восприимчивы к желудочно-кишечному поражению и предел концентрации был установлен 0,75%, Данные измерения NSFT для этого эксперимента представлены на фиг. 1 ( NSFT для индометацина через 24 часа).
NSFT для индометацина через 24 часа).
Мыши, которые не были подвергнуты действию УФВ-лучей и были смазаны наполнителем или наполнителем плюс индометацином, не обнаружили признака эритемы или отека, показав тем самым, что ни наполнитель, ни способ применения, ни индометацин не индуцировали воспалительную реакцию. Мыши, которых подвергали действию УФВ-лучей и обрабатывали индометацином, показали снижение эритемы и отека с повышением концентраций индометацина. Концентрации индометацина 0,1% имели среднее противовоспалительное действие, максимальное защитное действие имело место при концентрации индометацина 1% и выше.
Результаты испытания конъюгатов индометацин-ацил жирной кислоты
Конъюгаты индометацин-ацил жирной кислоты исследовали с применением необлученной группы самок мышей Skh-1. Ранее было определено, что неочищенные конъюгаты индометацин-ацил жирной кислоты не обладали защитным действием при концентрациях ниже 1% (считая на индометацин, мас./об.%) (данные не приведены). Концентрации, испытанные в следующих экспериментах, были 2% (считая на индометацин, мас./об.%), конъюгаты наносили на кожу после действия на нее УФВ-лучей или за 5 дней, 3 дня или 1 день до воздействия УФВ-лучей.
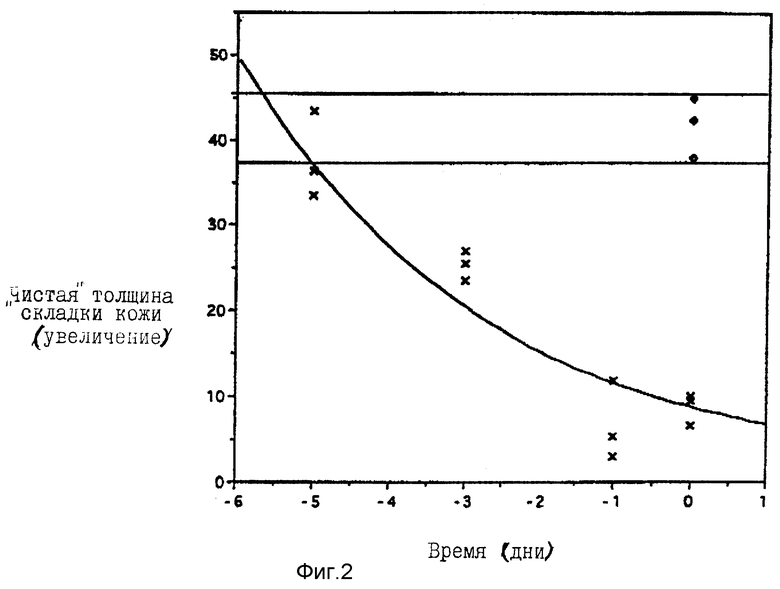
Аликвоту 100 мкл 1%-ного (мас./об.%) раствора индометацина в наполнителе этанол/ DMSO (5: 1) наносили смазыванием на дорсальную поверхность самок безволосых мышей (Skh-1) во временные интервалы до их обработки УФВ-лучами, в дни от -5 до -1, и после обработки УФВ-лучами (день 0). Их NSFT затем подсчитывали для времени 24 часа после облучения, эти данные представлены на фиг. 2 ( наполнитель, x 1%-ный индометацин). Логарифмические выражения этих данных указывают, что 1%-ный индометацин теряет свое защитное действие на индуцированный УФВ-лучами отек с повышением интервала времени применения до облучения.
наполнитель, x 1%-ный индометацин). Логарифмические выражения этих данных указывают, что 1%-ный индометацин теряет свое защитное действие на индуцированный УФВ-лучами отек с повышением интервала времени применения до облучения.
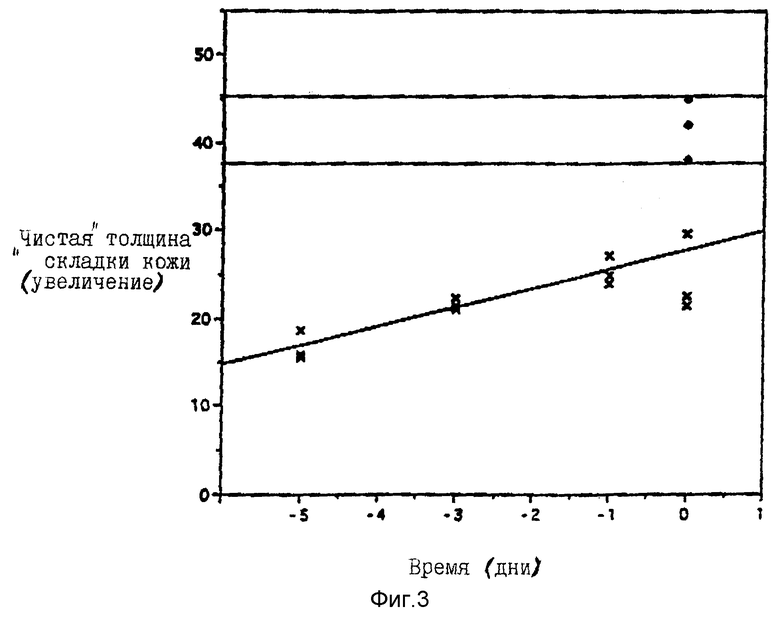
На фиг. 3 группы мышей Skh-1 смазывали 100 мкл 2% неочищенного конъюгата индометацин-глицин-трис-дипальмитат (загрязнен 30% GTP2, не содержит несвязанный индометацин) в те же интервалы времени до облучения УФВ-лучами, как для свободного индометацина на фигуре 2 ( наполнитель, x 2%-ный индометацин-GTP2). Линейное выражение этих данных дает профиль, отличающийся от профиля, наблюдаемого при применении немодифицированного индометацина. Фотозащита от УФВ-индуцированного отека наиболее явно выражена при проведении ее за 5 дней до облучения УФВ-лучами. Это указывает на то, что имела место пролонгированная защита от УФВ-индуцированного отека.
наполнитель, x 2%-ный индометацин-GTP2). Линейное выражение этих данных дает профиль, отличающийся от профиля, наблюдаемого при применении немодифицированного индометацина. Фотозащита от УФВ-индуцированного отека наиболее явно выражена при проведении ее за 5 дней до облучения УФВ-лучами. Это указывает на то, что имела место пролонгированная защита от УФВ-индуцированного отека.
Было найдено, что индометацин защищает приблизительно на 50% при применении его за 3 дня до обработки УФВ-лучами, но такого защитного действия не было при применении его за 5 дней до облучения (фиг. 2). Было найдено, что индо-GTP2 проявляет наибольшее защитное действие при применении за 5 дней до облучения УФВ-лучами.
На фиг. 4 группы из 3 мышей были смазаны 2%-ными растворами (эквивалент индометацина) очень чистого индометацин-глицин-трис-дипальмитата (индо-GTP2), индометацин-глицин-трис-трипальмитата (индо-GТР3), индометацин-этаноламин-моно-пальмитата (индо-ЕР1) или индометацин-глицин-этаноламин-моно-пальмитата (индо-GEP1) и испытаны на пролонгированную защитную активность путем смазывания их за 5 дней до УФВ-облучения или на немедленную активность смазыванием их сразу после УФВ-облучения, день 0. Контрольных мышей обрабатывали 0,75% индометацина и наполнителем (этанол: DMSO, 5:1). Было показано, что индометацин-GTP2 обладает наиболее высокими уровнями пролонгированной и немедленной активности. Это подтверждает результаты, представленные на фиг. 3 для неочищенного препарата.
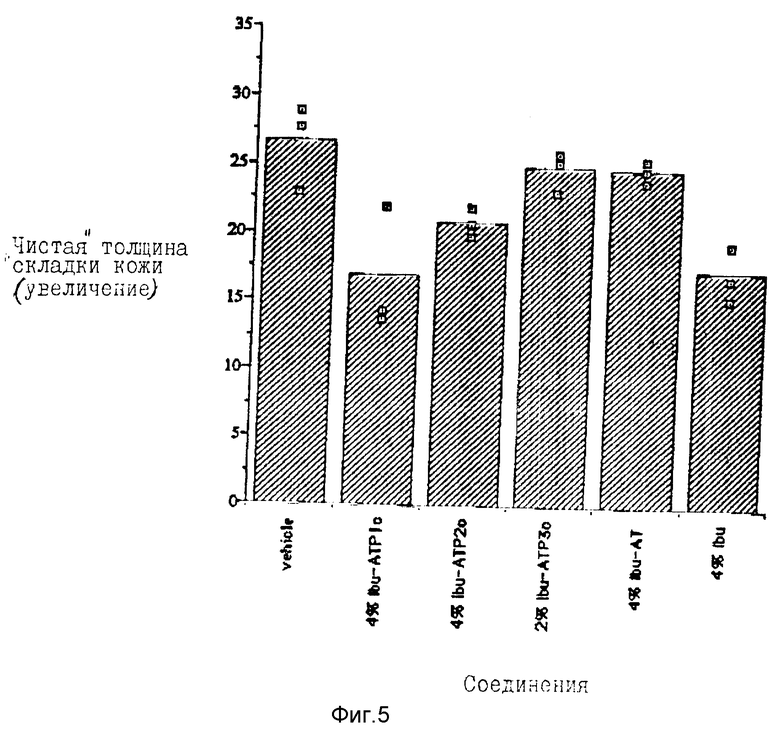
Результаты испытания ибупрофен-аланин-трис-ацил жирной кислоты
Эксперимент, исследующий противовоспалительные действия конъюгатов ибупрофен-ацил жирной кислоты, показал, что они активны в защите против УФВ-индуцированного отека. Фиг. 5 показывает результаты применения 2 и 4% конъюгатов ибупрофен-жирная кислота.
Группы из 3 мышей облучали 3 MED УФВ-лучей и затем смазывали 4%-ными (эквивалент ибупрофена) растворами ибупрофен-аланин-трис-монопальмитата (Ibu-ATP1c), ибупрофен-аланин-трис-дипальмитата (Ibu-ATP2c), ибупрофен-аланин-трис-трипальмитата (Ibu-ATP3c при 2%) или ибупрофен-аланин-трис (Ibu-AT). Контрольным мышам наносили 4%-ный ибупрофен (Ibu) или только наполнитель.
Результаты немедленной активности (от высшей к низшей): 4% Ibu = 4% Ibu - ATP > 4% Ibu - ATP2 > 4% Ibu-AT = 2% Ibu - ATP3 (с = кристаллическая форма Ibu-аланин-трис, применяемая в синтезе).
Влияние конъюгата ибупрофен-трис-ацил жирной кислоты на ожоги
Следующим открытием при испытании конъюгатов ибупрофен-жирная кислота была их неожиданная способность ослаблять УФВ-ожоги. Результаты ряда экспериментов с ибупрофен-аланин-трис-монопальмитатом показаны в таблице, где индивидуальные баллы представлены для каждой мыши, т.е. результаты, обозначенные как 5, 4, 3, интерпретируются как первая мышь, имеющая сильный ожог, вторая мышь, имеющая ожог от умеренного до сильного, и третья мышь, имеющая умеренный ожог (ожог второй степени).
Результаты этих экспериментов показывают, что ибупрофен-аланин-трис-монопальмитат (Ibu-ATP1) обладает защитным действием на УФВ-индуцированный ожог в значительно большей степени, чем немодифицированный ибупрофен. Это было видно по первоначальному снижению ожоговой реакции и затем более скорому разрешению ожога, приводящему к обычному эпидермису. Этот результат, по-видимому, не зависит от снижения отека, так как 2%-ный ибупрофен (как конъюгат или свободное лекарственное средство) обладает слабым действием на отек при этой концентрации. Кроме того, уровни индометацина, которые предотвращают образование отека, по-видимому, обладают слабым действием или не оказывают влияния на заживление ожога.
Заключение.
Конъюгаты NSAID-жирная кислота изменяют биологическую доступность индометацина и ибупрофена при местном их применении. Это изменение биологической доступности, возможно, обусловлено медленной высвобождающей способностью, которая допускает пролонгированную защиту от УФВ-индуцированного отека. Конъюгаты ибупрофен-ацил жирной кислоты оказывают повышенное защитное действие на ожог путем ускоренного лечения ожога после острого УФВ-облучения.
Авторы настоящего изобретения показали, что компонент соединений настоящего изобретения, являющийся нестероидным противовоспалительным соединением, сохраняет свою активность в конъюгатах и что эта активность поддерживается и пролонгируется. Так как в научной литературе допускается, что нестероидные противовоспалительные соединения могут обладать противоопухолевой активностью, считают, что соединения по настоящему изобретению могут также найти применение в этой области.
Специалистам в данной области науки должно быть понятно, что можно сделать различные вариации и/или модификации настоящего изобретения, как показано на конкретных примерах осуществления настоящего изобретения, без отступления от существа и объема данного изобретения, как широко описанного. Настоящие примеры осуществления изобретения, следовательно, следует рассматривать во всех аспектах как иллюстративные и не ограничивающие.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДОСТАВКА НУКЛЕИНОВЫХ КИСЛОТ | 1995 |
|
RU2160278C2 |
| КОНЪЮГАТЫ ТЕРАПЕВТИЧЕСКОГО СОЕДИНЕНИЯ С ЖИРНОЙ КИСЛОТОЙ | 1996 |
|
RU2166512C2 |
| СПОСОБ НАНЕСЕНИЯ КРАСИТЕЛЯ НА КЕРАТИНОВЫЕ ВОЛОКНА (ВАРИАНТЫ) | 1993 |
|
RU2128257C1 |
| СПОСОБ МОДИФИКАЦИИ, ПО МЕНЬШЕЙ МЕРЕ, ЧАСТИ ПОВЕРХНОСТИ ПОЛИМЕРА | 1996 |
|
RU2163246C2 |
| СПОСОБ ФУМИГАЦИИ ПРОДУКТОВ ФОСФИНОМ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1990 |
|
RU2088097C1 |
| УСОВЕРШЕНСТВОВАННАЯ ФУМИГАЦИЯ МЕЛКИХ СЫПУЧИХ ПРОДУКТОВ | 1994 |
|
RU2135046C1 |
| МАГНИЕВОЕ ЛИТЬЕ ПОД ДАВЛЕНИЕМ | 1998 |
|
RU2212980C2 |
| ФУМИГАНТ, ФУМИГАНТНАЯ КОМПОЗИЦИЯ И СПОСОБ ОКУРИВАНИЯ | 1993 |
|
RU2141762C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФИНА, УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СИСТЕМА БЕЗОПАСНОСТИ ДЛЯ ПРОИЗВОДСТВА ФОСФИНА | 1991 |
|
RU2087415C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ СМЕШИВАНИЯ | 1998 |
|
RU2216393C2 |

Изобретение относится к терапевтическим конъюгатам, которые содержат терапевтическое соединение, соединенное с одной, двумя или тремя ацильными группами, производными жирных кислот. Терапевтические соединения предпочтительно являются нестероидными противовоспалительными средствами, которые включают карбоксильную кислотную группу. Соединения включают использование производного трометамина или этаноламина для связывания ацильных групп, производных жирных кислот, с терапевтическим соединением. Описываются новые конъюгаты: терапевтическое соединение - жирная кислота общей формулы I, где Х - представляет собой нестероидное противовоспалительное соединение, содержащее карбоксильную группу, Y отсутствует или представляет собой одну или две аминокислоты или пептид, В - водород или СН2ОR3, R4 - ацильная группа, производное жирной кислоты, R1, R2, и R3 - одинаковые или различные и представляют собой либо водород, метил, этил, гидроксил, либо ацильную группу, производную жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой, производной жирной кислоты. 35 з.п. ф-лы, 5 ил. 1 табл.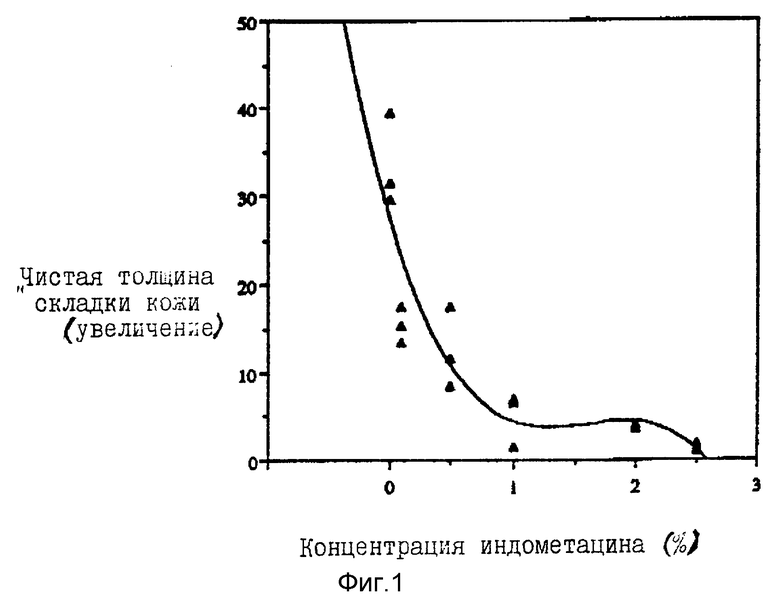
где
а) R' = R1, R'' = В, R''' = CН2ОR2, б) R' = R4, R'' = R''' = H;
Х - представляет собой нестероидное противоспалительное соединение, содержащее карбоксильную группу;
Y отсутствует или представляет собой одну или две аминокислоты или пептид;
В - водород или СН2ОR3;
R4 = ациальная группа, производное жирной кислоты;
R1, R2, R3 - одинаковые или различные и представляют собой либо водород, метил, этил, гидроксил, либо ацильную группу, производную жирной кислоты, при условии, что по меньшей мере один из R1, R2, R3 является ациальной группой, производной жирной кислоты.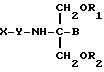
значения радикалов указаны в п.1.
X-Y-NH-СН2-СН2ОR4
где Х - нестероидное противоспалительное соединение, содержащее карбоксильную группу;
Y - отсутствует или представляет собой одну или две аминокислоты или пептид;
R4 - ацильная группа, производное жирной кислоты.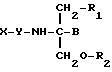
где Х представляет собой нестероидное противоспалительное соединение;
Y отсутствует или представляет собой одну или две аминокислоты или пептид;
В представляет собой Н или СН2ОR3;
R1, R2, R3, одинаковые или разные, представляют собой либо водород, метил, этил, гидроксил, либо ацильную группу, производную жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой производной жирной кислоты.
X-Y-NH-СН2-СН2О-R4,
где значения радикалов указаны в п.10.
| Способ получения сложных эфиров аминокислот или их физиологически совместимых солей | 1989 |
|
SU1836380A3 |
| Огнетушитель | 0 |
|
SU91A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| US 3686238 A, 1972 | |||
| Whittaker R.G | |||
| et | |||
| al | |||
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| Williams L.P | |||
| et | |||
| al | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |

