Уровень изобретения
В данном изобретении предлагается региоселективный синтез CCI-779, применимого в качестве противоопухолевого средства.
Сложный 42-эфир рапамицина с 3-гидрокси-2-(гидроксиметил)-2-метилпропионовой кислотой (CCI-779) представляет собой сложный эфир, который показал значительное ингибирующее действие на рост опухоли как на моделях in vitro, так и на моделях in vivo.
CCI-779 может увеличивать срок развития опухолей или время рецидива опухоли, что является более типичным скорее для цитостатических, чем для цитотоксических средств. Считают, что механизм действия CCI-779 подобен механизму действия сиролимуса. CCI-779 связывается и образует комплекс с цитоплазматическим белком FKBP, который ингибирует фермент, mTOR (мишень рапамицина у млекопитающих, также известный как FKBP12-рапамицин связанный белок [FRAP]). Ингибирование активности mTOR-киназы ингибирует многие клеточные процессы сигнальной трансдукции, включая пролиферацию клеток, стимулируемую цитокинами, трансляцию мРНК некоторых ключевых белков, которые регулируют G1 фазу клеточного цикла, и IL-2-индуцируемую транскрипцию, приводящую к развитию клеточного цикла от G1 до S. Механизм действия CCI-779, который приводит к блокированию G1-S фазы, является новым для противораковых лекарственных средств.
Показано, что CCI-779 in vitro ингибирует рост ряда гистологически различных опухолевых клеток. Самыми чувствительными к CCI-779 были рак центральной нервной системы (ЦНС), лейкемия (Т-клетки), рак молочной железы, рак простаты и линии клеток меланомы. Данное соединение подавляло клетки в G1 фазе клеточного цикла.
В опытах in vivo на голых мышах показано, что CCI-779 обладает активностью в отношении ксенотрансплантатов человеческих опухолей различных гистологических типов. Глиомы были особенно чувствительны к CCI-779 и данное соединение было активно в случае модели ортотопической глиомы на голых мышах. Стимуляция клеточной линии глиобластомы человека, вызываемая фактором роста тромбоцитов, значительно подавлялась in vitro под действием CCI-779. Рост некоторых человеческих панкреатических опухолей у мышей, а также одной из двух линий рака молочной железы, исследованный in vivo, также ингибировался под действием CCI-779.
Получение и применение сложных гидроксиэфиров рапамицина, включая CCI-779, раскрыты в патенте США 5362718. Региоспецифический синтез CCI-779 описан в патенте США 6277983.
CCI-779 может быть синтезирован нерегиоспецифическим ацилированием рапамицина, описанным в патенте США 5362718. Данный синтез, однако, усложнен образованием смесей целевого 42-эфира с 31-этерифицированным рапамицином, а также с 31,42-диэтерифицированным рапамицином и непрореагировавшим рапамицином.
CCI-779 может быть также получен ацилированием 31-силилового эфира рапамицина кеталем бис-(гидроксиметил)пропионовой кислоты с последуюшим удалением 31-силилэфирной и кетальной защитной группы бис-(гидроксиметил)пропионовой кислоты, как описано в патенте США 6277983. Однако сырой сложный 42-моноэфир, полученный с помощью данного региоселективного синтеза, требует дальнейшей очистки колоночной хроматографией для удаления остаточных количеств диэфирных побочных продуктов и непрореагировавшего исходного рапамицина.
Сущность изобретения
В данном изобретении предлагается региоселективный синтез 42-эфира рапамицина с 3-гидрокси-2-(гидроксиметил)-2-метилпропионовой кислотой (CCI-779), основанный на химии бороновой кислоты. Данное изобретенеие устраняет сложную и, как правило, трудоемкую очистку сложного 42-моноэфира рапамицина, полученного ранее применявшимися способами.
Другие аспекты и преимущества данного изобретения будут легко понятными из следующего подробного описания изобретения.
Подробное описание изобретения
В данном изобретении предлагается региоселективный синтез сложного 42-эфира рапамицина ацилированием 31-силилового эфира рапамицина соединением формулы
НООС.CR7R8R9
или его смешанным ангидридом, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют Х;
Х представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 каждый независимо представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F;
и
f = 0-6;
с образованием 42-эфирборонат 31-силилового эфира рапамицина.
Далее 42-боронат 31-силилилового эфира рапамицина гидролизуют в умеренно кислых условиях с образованием 42-эфирбороната рапамицина. 42-Эфирборонат рапамицина обрабатывают подходящим диолом. Способ позволяет получить региоспецифический сложный 42-эфир рапамицина.
Получение, выделение и очистка сложного 42-эфира рапамицина от силилового эфира рапамицина в соответствии со способом данного изобретения вызывает реакцию переборонирования, при которой фенилборонатный остаток соединения превращается в диол. Осаждение сложного 42-эфира рапамицина из смеси эфир:гептаны сопровождает данное переборонирование. Синтетический путь данного изобретения имеет несколько отличительных преимуществ по сравнению с синтетической методикой, опубликованной ранее для получения простых и сложных эфиров рапамицина; главным образом, в очистке, в снижении стоимости продуктов, в повышенной безопасности, в повышенной производительности и снижении времени производства. Данный способ изобретения представляет собой новый подход к производству сложных 42-эфиров рапамицина (например, CCI-779). Трудоемкая хроматографическая стадия, ранее используемая во всех больших периодических способах получения представленного CCI-779, была исключена. Большое количество растворителя, требуемое для хроматографии, описанной в патенте США 6277983, стало ненужным, в результате чего снизилась стоимость продуктов. Производственное время в реакторе и ресурсы снизились на 50%. Размер реактора, необходимого для широкомасштабного синтеза CCI-779, уменьшился, при этом увеличилась общая производительность. Новая методика проведения переборонирования, описанная в данном изобретении, уменьшает общее время процесса. Способ очистки, включенный в методику данного изобретения, ликвидирует окончательную очистку с помощью диэтилового эфира в ранее описанных способах синтеза.
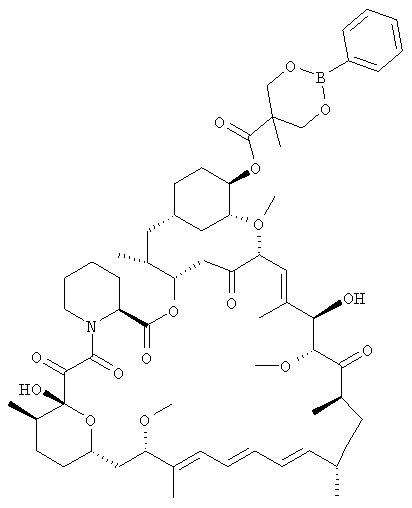
Согласно изобретению 31-силиловый эфир рапамицина ацилируют, используя соединение формулы НООС.CR7R8R9 или его ангидрид, определенное выше, для образования 31-силилэтил-42-эфирбороната. В одном из вариантов осуществления 31-силиловый эфир рапамицина ацилируют, используя 5-метил-2-боронат[1,3-диоксан]-5-карбоновую кислоту (представленную соединением [A] на схеме 1 ниже) или 2,4,6-трихлорбензоильный смешанный ангидрид 5-метил-2-фенил-1,3,2-диоксаборинан-5-карбоновой кислоты.
Один особенно удачный способ получения 31-силиловых эфиров рапамицина представлен в патенте США 6277983. Настоящее изобретение не ограничивается данным способом получения 31-силиловых эфиров. Однако в данной работе предпочтительно, чтобы 31-силиловый эфир рапамицина представлял собой 31-О-триметилсилиловый эфир рапамицина.
В одном из вариантов осуществления 31-силиловый эфир рапамицина отличается тем, что соответствует формуле:
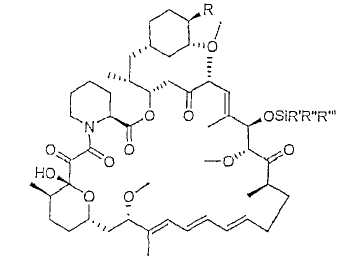
где R выбран из:
-О-С=О.CR7R8R9, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют Х;
Х представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 каждый независимо представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F;
и
f = 0-6;
и где R', R'' и R''' являются одинаковыми или различными и выбраны из алкила с 1-6 атомами углерода, фенила и бензила.
В данном изобретении предлагается соединение [A] как новое соединение, применимое для производства CCI-779 и его аналогов. Получение соединения [A] включает смешивание фенилбороновой кислоты с 2,2-бис(гидроксиметил)пропионовой кислотой при комнатной температуре для получения фенилборинана. Обычно выходы составляют >90%. Реакция может быть проведена в метиленхлориде, но предпочтительный растворитель представляет собой тетрагидрофуран (ТГФ).
Предпочтительно, фенилборинан представляет собой 2-фенил-1,3,2-диоксаборинан-5-карбоновую кислоту, в которой фенил является необязательно замещенным. В другом варианте осуществления фенилборинан представляет собой 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил является необязательно замещенным. Одно особенно желательное замещение в фенильной группе представляет собой алкил, наиболее желателен С1, С2, С3, С4, С5 или С6алкил. Другие арил- (включая фенил-) бороновые кислоты могут быть использованы в данной реакции. Они включают моно, ди и три-замещенные арилбороновые кислоты, в которых заместители являются одинаковыми или различными. Заместители в арильной группе включают галоген, алкил, алкокси, арилокси (например, фенокси), аралкил, нитро, циано, конденсированный фенил, который содержит нафталилбороновая кислота. Термин «алкил», когда используется в случае группы или части группы, такой как алкокси или аралкил, включает алкильные фрагменты, содержащие от 1 до 12 атомов углерода, например, 1-6 атомов углерода. Термин «арил» как группа или часть группы, например, аралкил или арилокси, означает ароматическую группу, содержащую заместители с 6-10 атомами углерода, такие как фенил или нафтил. Предпочтительная арилбороновая кислота представляет собой фенилбороновую кислоту.
Наконец, предпочтительно, чтобы рапамицин бис-силилировался в положениях 31 и 42 триметилсилилхлоридом с последующим десилилированием в положении 42 с помощью разбавленной серной кислоты. Выделенный продукт силилируют в положении 42 ангидридом, полученным из 2-фенилбороновой кислоты. Диметиламинопиридин добавляли в качестве катализатора для доведения реакции до завершения. Для реакции требовалось ˜3 эквивалента смешанного ангидрида, чтобы израсходовать весь 31-триметилсилилрапамицин. После обработки реакционной смеси полученный раствор хранили при температуре от 0 до 10°С, пока он не потребуется для следующей стадии. При стоянии в ацетоновом растворе продукт будет претерпевать диссоциацию до соединения [B]. Данный факт не является проблемой, так как следующая стадия представляет собой гидролиз силильной функциональной группы. Почти полное превращение (< 3%) в [B] было достигнуто через 83 дня в ацетоне при температуре от 0 до 10°С.
Образование смешанного ангидрида может быть проконтролировано системой REACTIR (ASI Applied Systems). Система REACTIR (ASI Applied Systems) представляет собой специально сконструированный прибор для анализа на месте различных химических реакций в реальном времени. При условии, что смешанный ангидрид образуется из карбоновой кислоты и хлорангидрида кислоты, реакция хорошо подходит для контролирования с помощью инфракрасной спектроскопии (ИК). ИК является информативным методом для определения присутствия карбонильных функциональных групп, и в случае REACTIR, для мониторинга появления или исчезновения карбонильных функциональных групп. В обычном REACTIR (ASI Applied Systems) процессе соединение [A] смешивали с диизопропилэтиламином в метиленхлориде и охлаждали до 0-5°С на ледяной бане. Регистрировали ИК-спектр, который служит в качестве начального скана. Затем добавляли 2,4,6-трихлорбензоилхлорид. Снимали второй ИК-спектр, который характеризует смесь при Т=0 мин (т.е. начало реакции). Эксперимент был построен со снятием ИК-спектров каждые 5 мин в течение 5 ч при поддерживании температуры бани от 0 до 5°С. Ключевые характеристические полосы были при 1818 см-1, 1741 см-1 и 1031 см-1. Когда хлорангидрид кислоты был добавлен к смеси соединения [A] и диизопропилэтиламина (Т=0 мин), спектр по существу не показал пикового сигнала. Однако области частот карбонила и ангидрида (С-О-С) со временем увеличивались, указывая на образование смешанного ангидрида.
Реакция со смешанным ангидридом может быть проведена в этилацетате, трет-бутилметиловом эфире, диэтиловом эфире и тетрагидрофуране (ТГФ), но реакции протекают более медленно. Предпочтительный растворитель представляет собой метиленхлорид из-за облегчения завершения реакции. ДМАП является предпочтительным катализатором-основанием для данной реакции. Другие основания, которые могут быть использованы, представляют собой 4-пирролидинопиридин, N-метилимидазол и пиридин.
Смешанный ангидрид представляет собой нестабильное соединение и получается in situ при низких температурах. Он устойчив вплоть до 48 ч при температуре от -5 до 0°С. Он может быть получен при температуре от -50 до 20°С, но предпочтительный температурный интервал составляет от -6 до 5°С. Смешанный ангидрид выдерживают до 8 ч перед реакцией конденсации. Предпочтительное время выдерживания составляет от 4 до 5 ч перед добавлением 31-триметилсилил (TMS) рапамицин - партнера конденсации.
Реакция конденсации может быть проведена при температуре от -20° до 20°С, но предпочтительный температурный интервал составляет от -11° до -5°С. При более высоких температурах реакция замедляется и для завершения требуется дополнительная загрузка смешанного ангидрида. При более низких температурах смешанный ангидрид является более стабильным, тем не менее время реакции увеличивается. Реакция обычно завершается в течение от 12 до 17 ч.
Соединение [B] получали, выделяли и очищали в 3 стадийной последовательности реакции, проводимой в одном реакторе. Основной элемент данной реакции состоял в выборе ацетона в качестве растворителя. Другие растворители, которые могут быть использованы в данном получении, включают диэтиловый эфир, ацетонитрил, этилацетат, ТГФ, трет-бутилметиловый эфир и метиленхлорид. В настоящей работе ацетон является предпочтительным растворителем.
Таким образом, 31-триметилсилил CCI-779 боронат, [D], растворяли в ацетоне с получением концентрата. Однако в некоторых вариантах осуществления гидролиз может быть проведен с использованием однофазной системы водная кислота/органический растворитель.
Гидролиз 31-триметилсилильной группы (для образования [B]) проводят в умеренно кислых условиях. Таким образом, выбранный органический растворитель (например, ацетон) смешивают с разбавленной неорганической кислотой, такой как, например, серная, хлористоводородная или фосфорная кислота. Примеры концентраций подходящей разбавленной кислоты находятся в интервале от примерно 0,1 н. до примерно 3 н., примерно от 0,2 н. до примерно 2 н. или примерно 0,5 н. Обычно данная стадия выполняется при температуре примерно 25°С или ниже, от примерно -5°С до примерно 10°С или примерно от 0°С до примерно 5°С. Желательно, чтобы данная стадия выполнялась при рН от 5 до 6. Необязательно, к смеси добавляют подходящий буфер, например, ацетат натрия, или в присутствии бикарбоната натрия и/или уксусной кислоты, для установления и поддерживания рН в требуемом интервале.
В примерах, приведенных ниже, в реакции гидролиза используется 0,5 н. серная кислота при температуре от 0 до 5°С. Реакция обычно заканчивается за 5-6 часов и соединение [B] выделяли простым фильтрованием. Однако применение реагентов на основе фторидов для удаления 31-триметилсилильной группы нежелательно, так как образуются продукты разложения.
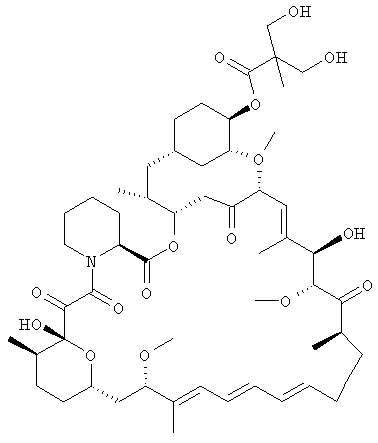
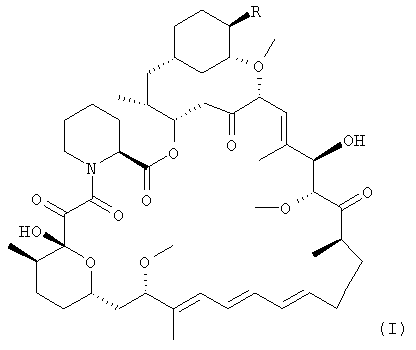
Сложный 42-эфирборонат рапамицина представляет собой новый промежуточный продукт, применимый в способе данного изобретения для получения сложного 42-эфира рапамицина. В одном из вариантов осуществления промежуточный продукт представляет собой сложный 42-эфир рапамицина с 5-метил-2-фенил-1,3,2-диоксаборинан-5-карбоновой кислотой.
В одном из вариантов осуществления в данном изобретении предлагается сложный 42-эфирборонат рапамицина - соединение формулы I:
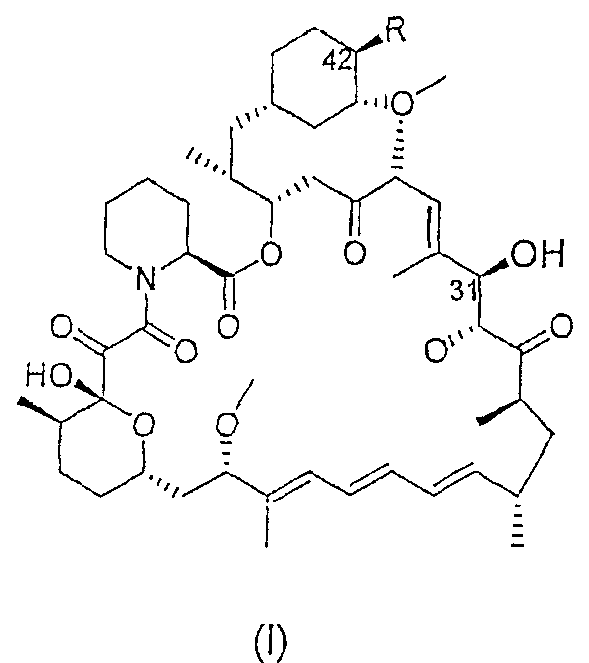
где R выбран из:
-О-С=О.CR7R8R9, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют Х;
Х представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 представляют собой каждый, независимо, водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F;
и
f = 0-6.
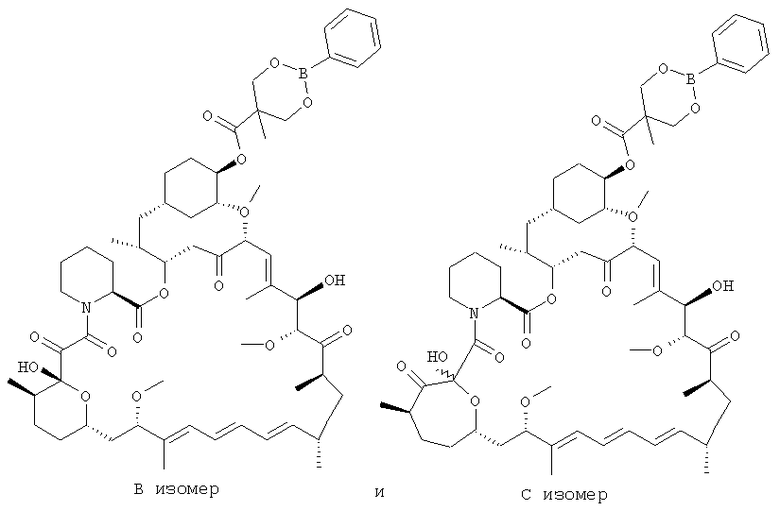
На указанной стадии способа изобретения 42-эфирборонат рапамицина, полученный по способу изобретения, обычно присутствует как В и С изомеры соединения. [Данные два изомера иллюстративного соединения [B] приведены ниже]. На данной стадии отношение изомеров В:С равно обычно <10:1. Авторы изобретения установили, что В изомер является более кристалличным, чем С изомер, и менее растворимым в ацетоне, чем С изомер. Для того, чтобы воспользоваться преимуществом данных свойств, авторы показали, что в натрийацетатном буфере при рН от 5 до 6 отношение изомеров В:С может быть увеличено до примерно 20:1. Путем повышения данного отношения может быть увеличено извлечение соединения [B]. Таким образом, желательно поднять отношение изомеров В:С до, по меньшей мере, 1:1, более желательно, выше 5:1, выше 10:1, выше 15:1, выше 20:1 и, более предпочтительно, до примерно 25:1. Бикарбонат натрия был добавлен для нейтрализации серной кислоты и установления рН до 7-8. Уксусная кислота была добавлена для образования ацетата натрия и снижения рН до 5-6. После выдерживания реакционной смеси в течение 16 ч отношение изомеров составляет ˜25:1. Могут быть использованы другие буферы, такие как ацетат калия и ацетат цинка, но предпочтительный буфер представляет собой ацетат натрия.
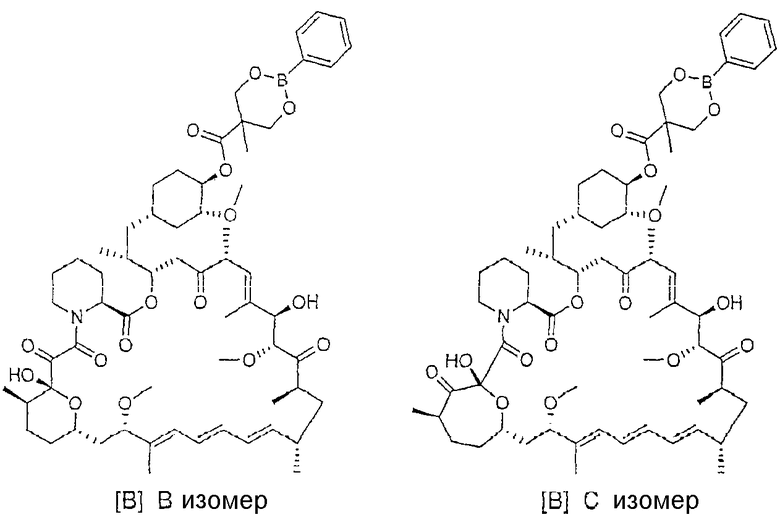
Смесь фильтровали, промывали и сушили для получения неочищенного продукта [B]. Маточные растворы содержат в преобладающем количестве С изомер, бис-сложноэфирные побочные продукты и другие неизвестные примеси, относящиеся к кристаллическому неочищенному исходному продукту рапамицина.
Для облегчения получения чистого продукта важно в данный момент контролировать содержание рапамицина в неочищенном соединении [B]. Содержание рапамицина обычно равно ˜5% (% площади) по данным высокоэффективной жидкостной хроматографии (ВЭЖХ). Перекристаллизации снижают содержание рапамицина до <0,7%. Очистка в подходящих растворителях, таком как ацетон, может снизить уровни рапамицина. Соединение [B], показанное на схеме, представляет собой белый твердый порошок, который является устойчивым при комнатной температуре.
Реакция может быть проведена в растворителе, таком как эфирный растворитель, или предпочтительно ТГФ, при разбавлении реакционной смеси трет-бутилметиловым эфиром или толуолом и применении способа водной экстракции для удаления избытка диола и диолборонатных побочных продуктов. Оба, диол и диолборонат, являются водорастворимыми. Предпочтительный способ устраняет водную экстракцию. Предпочтительный способ включает простую стадию фильтрации. Способ включает растворение соединения [B] в ТГФ, трет-бутилметиловом эфире или ацетонитриле, добавление диола и перемешивание при комнатной температуре в течение 3 ч. Растворитель удаляют дистилляцией до получения реакционной смеси в виде пена/масло. Добавляли эфир и продукт соосаждали гептанами. Способ может быть повторен для получения CCI-779 c выходом от 80% до 90% в расчете на соединение [B].
Первоначальная обработка диолом удаляет основную массу фенилбороновой кислоты в реакционной смеси. Остаточные количества фенилбороновой кислоты, еще присутствующие, легко удаляются дополнительной обработкой диолом. Конечное соединение [C], полученное по данной методике, показывает, что содержание фенилбороновой кислоты является приемлемым. В процессе переборонирования может быть использован избыток диола, но предпочтительное количество составляет от 1 до 5 эквивалентов. Полученный выход в данном переборонировании составил 86%. Общий выход от рапамицина составил от 47 до 50%.
Множество 1,2-, 1,3-, 1,4- и 1,5-диолов может быть использовано для осуществления данного переборонирования. Алкилзамещенные диолы, такие как 2-метил-2,4-пентандиол, являются предпочтительными. Были применены диэтаноламин или полистиролдиэтаноламин на твердой подложке (PS-DEAM). Переборонирование также может быть достигнуто в случае использования реагентов класса карбоновых кислот, таких как щавелевая, малоновая, винная, фталевая и салициловая кислота. 2,2-Бис(гидроксиметил)пропионовая кислота также является эффективной, но не может быть удалена из конечного продукта.
Способ показан на следующей схеме. Данная схема является только иллюстративной и не ограничивает изобретение.
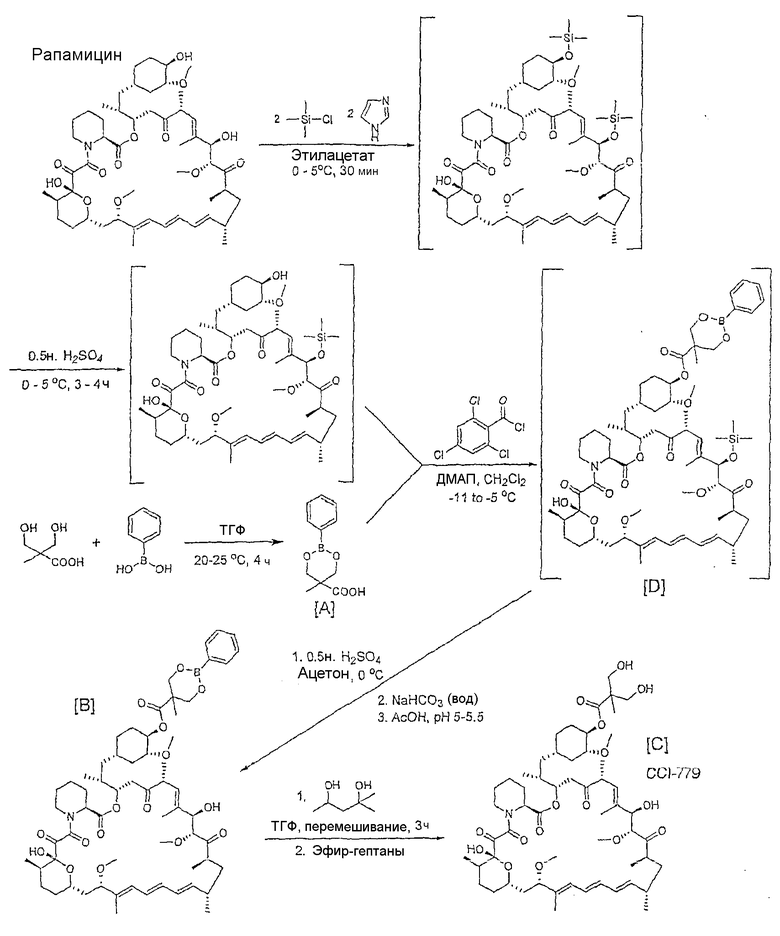
Получение 5-метил-2-фенил-1,3,2-диоксаборинан-5-карбоновой кислоты, [A]
К суспензии 2,2-бис(гидроксиметил)пропионовой кислоты (131 г, 0,98 моль) в тетрагидрофуране (500 мл) добавляли раствор фенилбороновой кислоты (122 г, 1,0 моль) в тетрагидрофуране (500 мл). Смесь перемешивали в течение 3 ч и добавляли толуол (1,0 л). Воду удаляли азеотропной дистилляцией с толуолом. К выпадающему в осадок продукту добавляли гептаны (500 мл), нагревали при кипении с обратным холодильником и охлаждали. Смесь фильтровали и промывали гептанами (2 × 300 мл). Осадок сушили в вакууме при 70-75°С до постоянной массы, получая выход 94%. 1Н ЯМР: δ (ДМСО-d6) 7,65 (д, 2Н, Ar), 7,40 (м, 3Н, Ar), 4,35 (д, 2Н, СН2), 3,92 (д, 2Н, СН2), 1,17 (с, 3Н, СН3).
Получение сложного 42-эфира рапамицина с 5-метил-2-фенил-1,3,2-диоксаборинан-5-карбоновой кислотой, [B]
Как описано в патенте США 6277983 (2001), в 3-литровую колбу помещали рапамицин (100 г, 0,104 моль) и растворяли в этилацетате (1,50 л). Раствор охлаждали до 5-10°С. Добавляли имидазол (30 г, 0,44 моль, 4,23 экв.) и растворяли. В атмосфере азота добавляли триметилсилилхлорид (44 г, 0,405 моль, 4,0 экв.) в течение 30-40 мин, поддерживая температуру при 0-5°С в течение добавления. Смесь выдерживали минимум в течение 0,5 ч. Реакцию контролировали ТСХ (элюент ацетон:гептан 30:70). Реакцию завершали, когда весь рапамицин был израсходован.
Извлекали две-три капли реакционной смеси и сохраняли их в качестве 31,42-бис(триметилсилил)рапамицинового эталонного стандарта. 0,5 н. Серную кислоту (300 мл) добавляли в 3-литровую колбу в течение 0,5 ч, поддерживая температуру 0-5°С. Смесь интенсивно перемешивали и выдерживали в течение 5 ч. Реакцию контролировали тонкослойной хроматографией (ТСХ) (элюент ацетон:гептан 30:70). Реакцию завершали, когда по существу отсутствовал 31,42-бис(триметилсилил)рапамицин. Слои разделяли и нижний водный слой снова экстрагировали этилацетатом (500 мл). Объединенные органические слои промывали насыщенным солевым раствором (500 мл) и насыщенным раствором бикарбонатом натрия (2 × 200 мл) до получения рН 8. Органический слой промывали водой (2 x500 мл) и насыщенным солевым раствором (500 мл) до получения рН от 6 до 7. Раствор сушили над сульфатом магния (100 г) в течение 30 мин, фильтровали в 2-литровую колбу и концентрировали до объема 135 мл. Добавляли этилацетат (500 мл) и массу концентрировали до объема 135 мл. Водную фазу еще раз обрабатывали этилацетатом (500 мл). Добавляли метиленхлорид (300 мл) и раствор выдерживали, пока он не потребовался для следующей стадии.
В 3-литровую колбу, снабженную механической мешалкой, загружали соединение [A] (75 г, 0,341 моль) в метиленхлориде (400 мл). Диизопропилэтиламин (66,1 г, 0,51 моль) добавляли по каплям в течение 20 мин и встряхивалии с метиленхлоридом (25 мл). 2,4,6-Трихлорбензоилхлорид (80 г, 0,328 моль) добавляли и встряхивали с метиленхлоридом (25 мл). Смесь выдерживали в течение 4 ч при 0-5°С и охлаждали до -10±5°С.
Раствор 31-триметилсилилрапамицина добавляли в 3-литровую колбу, содержащую смешанный ангидрид, и встряхивали с метиленхлоридом (25 мл). Получали раствор диметиламинопиридина (48,5 г, 0,397 моль) в метиленхлориде (150 мл), добавляли его в течение 1,5 ч, поддерживая температуру <-8°С, и встряхивали с метиленхлоридом (25 мл). Смесь выдерживали в течение 12 ч при температуре от -11 до -5°С. Реакционную смесь гасили 1 н. серной кислотой (600 мл), поддерживая температуру < 10°С. Смесь перемешивали и выдерживали в течение 30 мин. Значение рН верхнего водного слоя было ≤2. Слои разделяли и нижние органические слои промывали насыщенным солевым раствором (450 мл), насыщенным бикарбонатом натрия (500 мл) до рН ≥8. Органический слой промывали водой (450 мл) до получения рН 6-7. Раствор концентрировали, добавляли ацетон (250 мл) и концентрировали. Процедуру повторяли с другой порцией ацетона (250 мл) и раствор концентрировали.
Раствор разбавляли ацетоном. Добавляли по каплям 0,5 н. серную кислоту (500 мл) в течение 30 мин, поддерживая температуру бани при 0-5°С. Смесь выдерживали минимум в течение 5 ч, за это время продукт осаждался из раствора. Водный бикарбонат натрия (30 г в 375 мл воды) добавляли по каплям в течение 30 мин, поддерживая температуру бани от 0 до 5°С; смесь выдерживали минимум в течение 30 мин. Добавляли уксусную кислоту (25 мл), пока рН не становился равным 5-6, поддерживая температуру бани <10°С. Смесь нагревали до комнатной температуры и выдерживали в течение 16 ч. Твердый продукт фильтровали и промывали водой (2 × 100 мл), затем смесью 1:1 ацетон:вода (2 x 100 мл). Осадок очищали в ацетоне (375 мл), получая 65 г (общий выход 58% относительно рапамицина) продукта [B]. ЖХ/МС: использование электрораспылительной ионизации в режиме регистрации положительных ионов приводило к молекулярному иону [M+Na]=1138,5 атомных единиц массы (а.е.м.).
Получение сложного 42-эфира рапамицина с 2,2-бис(гидроксиметил)пропионовой кислотой, [C]
Соединение [B] (200 г, 0,179 моль) растворяли в тетрагидрофуране (600 мл), к нему добавляли 2-метил-2,4-пентандиол (42,3 г, 0,358 моль, 2,0 экв.) и смесь перемешивали в течение минимум 3 ч. Реакционную смесь концентрировали до состояния пены. Добавляли диэтиловый эфир (1,0 л) и смесь перемешивали в течение 2 ч. Добавляли по каплям гептаны (1,0 л) в течение 1 ч и смесь перемешивали в течение 2 ч. Смесь фильтровали и твердый продукт промывали гептанами (500 мл). Твердые вещества повторно растворяли в ацетоне (400 мл), повторно обрабатывали 2-метил-2,4-пентандиолом (21,1 г, 0,179 моль, 1 экв.) в ацетоне (200 мл), очищали с помощью 0,2-микронного картридж-фильтра и встряхивали с ацетоном (200 мл). Раствор концентрировали до состояния пены, добавляли диэтиловый эфир (1,0 л), предварительно профильтрованный через 0,2-микронный картридж-фильтр, и смесь перемешивали в течение 2 ч. Наблюдалось выпадение осадка при добавлении предварительно профильтрованных гептанов (1,0 л). Выпавшие в осадок твердые вещества фильтровали и промывали эфир:гептаном (2 × 500 мл). Твердые вещества сушили (от 55 до 60°С, 10 мм Hg, минимум 24 ч), получая 159 г (86%) продукта [C]. ЖХ/МС: использование APCI в режиме сканирования положительных ионов приводило к молекулярному иону [M+NH4]=1047,0 а.е.м. 1Н ЯМР продукта (CCI-779) был идентичен продукту, описанному в примере 11 патента США 5362718 (1994).
Все патенты, патентные заявки, статьи и другие документы, относящиеся к данной работе, включены здесь в виде ссылки. Специалисту в данной области будет очевидно, что в конкретные варианты осуществления, описанные здесь, могут быть внесены изменения, не выходящие за пределы объема данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕГИОСПЕЦИФИЧЕСКИЙ СИНТЕЗ ПРОИЗВОДНЫХ 42-ЭФИРА РАПАМИЦИНА | 2005 |
|
RU2387657C2 |
| 42-ОКСИМЫ И ГИДРОКСИЛАМИНЫ РАПАМИЦИНА, СПОСОБ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 1995 |
|
RU2172316C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТЫХ ПОЛИЭФИРКАРБОНАТОВ | 2018 |
|
RU2793322C2 |
| ГИДРОКСИЭФИРЫ РАПАМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ | 1995 |
|
RU2134267C1 |
| КОМБИНАЦИЯ ПИРИМИДИЛАМИНОБЕНЗАМИДА И ИНГИБИТОРА КИНАЗ mTOR | 2006 |
|
RU2443418C2 |
| 4-АМИНО-6-(ГЕТЕРОЦИКЛИЧЕСКИЕ)ПИКОЛИНАТЫ И 6-АМИНО-2-(ГЕТЕРОЦИКЛИЧЕСКИЕ)ПИРИМИДИН-4-КАРБОКСИЛАТЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2014 |
|
RU2672587C2 |
| ПРОТИВООПУХОЛЕВАЯ АКТИВНОСТЬ ССI-779 ПРИ ПАПИЛЛЯРНОМ РАКЕ КЛЕТОК ПОЧЕЧНОГО ЭПИТЕЛИЯ | 2008 |
|
RU2501559C2 |
| ПИРИМИДИНЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПУРИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ИНГИБИРОВАНИЯ ПРОТЕИНКИНАЗ, СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ, ЧУВСТВИТЕЛЬНЫХ К ИНГИБИРОВАНИЮ ПРОТЕИНКИНАЗ, И СПОСОБ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2681081C2 |
| Способ получения полиолов | 2016 |
|
RU2729046C2 |
| КАТАЛИЗАТОРЫ | 2015 |
|
RU2696272C2 |
Изобретение относится к региоселективному синтезу получения сложного 42-эфира рапамицина (CCI-779), который включает: (a) ацилирование 31-силилового эфира рапамицина соединением формулы HOOC.CR7R8R9 или его смешанным ангидридом, где: R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10; R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил; R8 и R9, взятые вместе, образуют X; X представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным; R12 и R13 каждый, независимо, представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F; и f=0-6; для образования 42-эфирбороната 31-силилового эфира рапамицина; (b) селективный гидролиз 42-эфирбороната 31-силилового эфира в умеренно кислых условиях для получения 42-эфирбороната рапамицина; и (c) обработку 42-эфирбороната рапамицина диолом для получения сложного 42-эфира рапамицина. Также заявлены новые промежуточные продукты, применимые в данном способе. Данное соединение применяется в качестве противоопухолевого средства. 9 н. и 39 з.п. ф-лы.
(a) ацилирование 31-силилового эфира рапамицина соединением формулы
HOOC.CR7R8R9
или его смешанным ангидридом,
где R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют X;
X представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 каждый, независимо, представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F; и
f=0-6;
для образования 42-эфирбороната 31-силилового эфира рапамицина;
(b) селективный гидролиз 42-эфирбороната 31-силилового эфира в умеренно кислых условиях для получения 42-эфирбороната рапамицина; и
(с) обработку 42-эфирбороната рапамицина диолом для получения сложного 42-эфира рапамицина.
-OSiR'R"R''',
в которой R', R" и R''' являются одинаковыми или различными и выбраны из алкила с 1-6 атомами углерода, фенила и бензила.
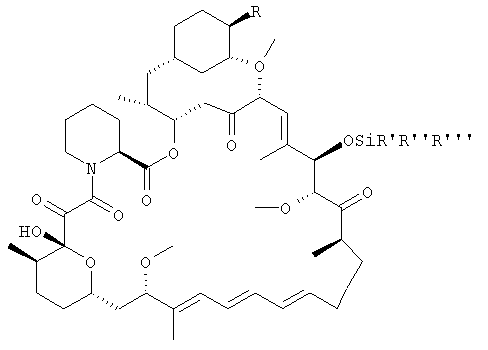
в которой R представляет собой -O-C=O.CR7R8R9, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 иR9, взятые вместе, образуют X;
X представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 каждый, независимо, представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F; и
f=0-6;
R', R" и R''' являются одинаковыми или различными и выбраны из алкила с 1-6 атомами углерода, фенила и бензила.
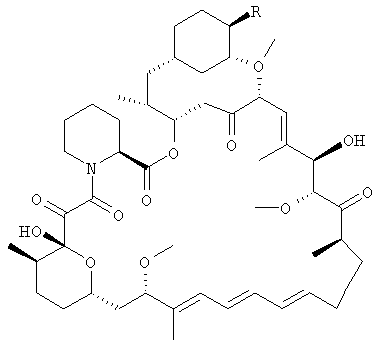
в которой R представляет собой -O-C=O.CR7R8R9, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют X;
X представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 каждый, независимо, представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F; и
f=0-6.
который включает обработку 42-эфирбороната 31-силилового эфира рапамицина подходящим диолом.
в растворе, содержащим изомеры В и С в соотношении < примерно 10:1,
включающий стадию доведения значения рН раствора до рН, равного примерно от 5 до 6.
в которой R выбран из
-O-C=O.CR7R8R9, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют X;
X представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 представляют собой каждый, независимо, водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода,
трифторметил или -F; и
f=0-6.
в которой R выбран из:
-O-C=O.CR7R8R9, где
R7 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, -(CR12R13)fOR10, -CF3, -F или -CO2R10;
R10 представляет собой водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифенилметил, бензил, алкоксиметил с 2-7 атомами углерода, хлорэтил или тетрагидропиранил;
R8 и R9, взятые вместе, образуют X;
X представляет собой 2-фенил-1,3,2-диоксаборинан-5-ил или 2-фенил-1,3,2-диоксаборинан-4-ил, где фенил может быть необязательно замещенным;
R12 и R13 представляют собой каждый, независимо, водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, трифторметил или -F; и
f=0-6;
и где R', R" и R''' являются одинаковыми или различными и выбраны из алкила с 1-6 атомами углерода, фенила и бензила.
HO-C=O.CR7R8R9
в которой R7, R8 и R9 принимают значения, определенные в п.1.
| US 6277983 В1, 21.08.2001 | |||
| US 5362718 А, 08.11.1994 | |||
| Способ получения пироно-рифамицинов | 1973 |
|
SU455545A3 |

