Краткое описание изобретения
Настоящее изобретение относится к сульфоновым кислотам, производным указанных кислот и к содержащим их фармацевтическим композициям, которые могут быть использованы для профилактики и лечения повреждения тканей, вызванного усиленным рекрутингом полиморфонуклеарных нейтрофилов (PMN-лейкоцитов) на участках воспаления.
Уровень техники
Отдельные клетки крови (макрофаги, гранулоциты, нейтрофилы, полиморфонуклеарные) отвечают на химические раздражители (когда стимулированы веществами, называемыми хемокинами) путем миграции вдоль градиента концентрации стимулятора посредством процесса, называемого хемотаксис. Основные известные стимуляторы или хемокины представлены продуктами распада комплемента C5a, некоторыми N-формилпептидами, образующимися вследствие лизиса бактериальной поверхности, или пептидами синтетического происхождения, такими как формилметиониллейцилфенилаланин (f-MLP)3, и в основном множеством цитокинов, включающих интерлейкин-8 (IL-8, также называемый CXCL8). Интерлейкин-8 является эндогенным хемотаксическим фактором, продуцируемым большинством нуклеарных клеток, таких как фибробласты и макрофаги.
При некоторых патологических состояниях, характеризующихся усиленным рекрутингом нейтрофилов, большая часть тяжелых повреждений тканей на участке вызвана инфильтрацией нейтрофильных клеток. Недавно было наглядно показано значение активации нейтрофилов в оценке нарушения, связанного с пост-ишемической реперфузией и пульмональной гипероксией.
Биологическая активность IL-8 опосредована взаимодействием интерлейкина с мембранными рецепторами CXCR1 и CXCR2, принадлежащими к семейству из семи трансмембранных рецепторов, экспрессируемых на поверхности нейтрофилов человека и некоторых типов T-клеток (L. Xu et al., J. Leukocyte Biol., 57, 335, 1995). Известны селективные лиганды, которые могут различать CXCR1 и CXCR2: GRO-α служит примером селективного хемотаксического фактора CXCR2.
Хотя известно, что активация CXCR1 играет решающую роль в опосредованном IL-8 хемотаксисе, недавно было выдвинуто предположение о том, что активация CXCR2 может играть патофизиологическую роль в хронических воспалительных заболеваниях, таких как псориаз. Фактически патофизиологическая роль IL-8 в псориазе также подтверждается влияниями IL-8 на функции кератиноцитов.
Действительно, было показано, что IL-8 является эффективным стимулятором пролиферации эпидермальных клеток, а также ангиогенеза, оба указанных процесса являются важными аспектами псориатического патогенеза (A. Tuschil et al. J Invest Dermatol, 99, 294, 1992; Koch AE et al., Science, 258, 1798,1992).
Кроме того, накопленные данные свидетельствуют о том, что патофизиологическая роль IL-8 в развитии меланомы и метастазов может быть опосредована активацией CXCR2 (L.R. Bryan et al., Am J Surg, 174, 507, 1997).
Широко раскрыта потенциальная патогенная роль IL-8 в заболеваниях легких (повреждение легкого, острый респираторный дистресс синдром, астма, хроническое воспаление легких и кистозный фиброз) и в особенности в патогенезе COPD (хронического обструктивного заболевания легких), осуществляемая через путь рецептора CXCR2 (D. WP Hay and H.M. Sarau., Current Opinion in Pharmacology 2001, 1:242-247).
Исследования в отношении вклада сигнала (S)- и (R)-энантиомеров кетопрофена в противовоспалительную активность рацемата и роли указанных энантиомеров в модуляции хемокина показывают (P. Ghezzi et al., J. Exp. Pharm. Ther., 287, 969, 1998), что два энантиомера и соответствующие им соли с хиральными и не хиральными органическими основаниями могут ингибировать зависимым от дозы образом хемотаксис и повышать внутриклеточную концентрацию ионов Ca2+, индуцируемых IL-8 на PMN-лейкоцитах человека (Patent Application US6069172). Впоследствии было показано (C. Bizzarri et al., Biochem. Pharmacol. 61, 1429, 2001), что кетопрофен обладает способностью ингибировать биологическую активность IL-8 в присутствии других молекул, принадлежащих к классу нестероидных противовоспалительных средств (NSAID), таких как флурбипрофен, ибупрофен и индометацин. Ингибирующая активность в отношении фермента циклооксигеназы (COX), характерная для NSAID, ограничивает терапевтическое употребление указанных соединений применительно к лечению нейтрофил-зависимых патологических состояний и воспалительных состояний, таких как псориаз, идиопатический фиброз легких, острая дыхательная недостаточность, нарушения, вызванные реперфузией, и гломерулонефрит. Ингибирование синтеза простагландинов, происходящее под воздействием на ферменты циклооксигеназы, влечет за собой увеличение продуцирования цитокинов, которые, подобно TNF-α, играют роль в усилении нежелательных провоспалительных действий нейтрофилов.
Были обнаружены новые классы эффективных и селективных ингибиторов биологических активностей IL-8, пригодные для введения "in vivo". Амиды и N-ацилсульфонамиды R-2-арилпропионовой кислоты описаны в качестве эффективных ингибиторов IL-8, индуцирующего хемотаксис нейтрофилов и дегрануляцию (WO 01/58852; WO 00/24710). Кроме того, недавно в качестве ингибиторов IL-8 были описаны новые R- и S-2-фенилпропионовые кислоты, полностью лишенные нежелательного СОХ-ингибирующего действия (РСТ/ЕР02/12939).
Подробное описание изобретения
Заявителями установлено, что класс сульфоновых кислот и соответствующие им производные проявляют способность к эффективному ингибированию хемотаксиса нейтрофилов и дегрануляции индуцированного IL-8.
Таким образом, настоящее изобретение относится к применению сульфоновых кислот и соответствующих им производных формулы (I):
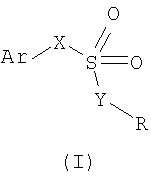
и фармацевтически приемлемых солей указанных соединений,
где
Ar означает фенильную группу, незамещенную или замещенную одним-тремя заместителями, независимо выбранными из группы, включающей галоген, C1-C4-алкил, C1-C4-алкокси, гидрокси, C1-C4-ацилокси, фенокси, циано, нитро, амино, C1-C4-ациламино, галоген-C1-C3-алкил, галоген-C1-C3-алкокси, бензоил, либо Ar означает замещенный или незамещенный 5-6-членный гетероарильный цикл;
X означает либо -CH2-, либо -СН(СН3)-группу или этиленовую группу формулы (II) в E-конфигурации, где R' означает H или CH3;
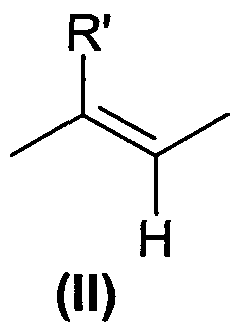
Y выбран из О (кислорода) и NH; и
- когда Y означает О (кислород), R означает H (водород);
- когда Y означает NH, R выбран из группы, включающей:
- H, C1-C5-алкил, C1-C5-циклоалкил, C1-C5-алкенил, C1-C5-ацил;
- остаток формулы -CH2-CH2-Z-(CH2-CH2O)nR", где R" означает H или C1-C5-алкил, n равно целому числу от 0 до 2 и Z означает кислород или серу;
- остаток формулы -(CH2)n-NRaRb, где n равно целому числу от 0 до 5 и каждый из Ra и Rb, которые могут быть одинаковыми или различными, означает C1-C6-алкил, C1-C6-алкенил, либо, альтернативно, Ra и Rb, вместе с атомом азота, к которому они присоединены, образуют гетероцикл из 3-7 членов формулы (III)

где W означает простую связь, CH2, O, S, N-Rc, Rc означает H, C1-C6-алкил или C1-C6-алкилфенил, при получении лекарственного средства для ингибирования IL-8-индуцированного хемотаксиса PMN человека.
Термин "замещенный" в приведенном выше определении означает замещенный группой, выбранной из ряда, включающего C1-C5-алкил, галоген, гидрокси, C1-C5-алкокси, амино, C1-C5-алкиламино, нитро или цианогруппу.
Ar означает замещенную фенильную группу, выбранную из группы, включающей 3'-бензоилфенил, 3'-(4-хлорбензоил)фенил, 3'-(4-метилбензоил)фенил, 3'-ацетилфенил, 3'-пропионилфенил, 3'-изобутаноилфенил, 4'-трифторметансульфонилоксифенил, 4'-бензолсульфонилоксифенил, 4'-трифторметансульфониламинофенил, 4'-бензолсульфониламинофенил, 4'-бензолсульфонилметилфенил, 4'-ацетоксифенил, 4'-пропионилоксифенил, 4'-бензоилоксифенил, 4'-ацетиламинофенил, 4'-пропиониламинофенил, 4'-бензоиламинофенил, или гетероароматический цикл, выбранный из пиридина, пиррола, тиофена, фурана, индола.
Когда Y означает NH, предпочтительными R группами являются
- H, C1-C5-алкил, C1-C5-ацил;
- остаток формулы -CH2-CH2-О-(CH2-CH2O)R", где R" означает H или C1-C5-алкил;
- остаток формулы -(CH2)n-NRaRb, где n равно целому числу от 2 до трех, более предпочтительно 3, и группа NRaRb означает N,N-диметиламин, N,N-диэтиламин, 1-пиперидил, 4-морфолил, 1-пирролидил, 1-пиперазинил, 1-(4-метил)пиперазинил;
Настоящее изобретение также относится к новым сульфоновым кислотам и к соответствующим производным соединений вышеуказанной формулы (I), выбранных из следующих соединений:
1-(4-изобутилфенил)этансульфоновая кислота
1-[4-(1-оксо-2-изоиндолинил)фенил]этансульфоновая кислота
2-(4-фенилсульфонилокси)этансульфоновая кислота
(1-метил-5-ацетилпирролил)-1-метансульфоновая кислота
2-(3-бензоилфенил)этансульфоновая кислота
2-(3-изопропилфенил)этансульфоновая кислота
E-2-(4-изобутилфенил)этенсульфоновая кислота
E-2-(3-бензоилфенил)этенсульфоновая кислота
E-2-(4-метансульфониламинофенил)этенсульфоновая кислота
E-2-(4-трифторметансульфонилоксифенил)этенсульфоновая кислота
E-2-(4-изобутилфенил)этенсульфонамид
E-2-(3-бензоилфенил)этенсульфонамид
E-2-[4-(трифторметансульфонилокси)фенил]этенсульфонамид
E-2-[4-(метансульфониламино)фенил]этенсульфонамид
E-2-(4-изобутилфенил)этен-N-(N,N-диметиламинопропил)сульфонамид
E-2-(3-бензоилфенил)этен-N-(N,N-диметиламинопропил)сульфонамид
E-2-[4-(трифторметансульфонилокси)фенил]этен-N-(N,N-диметиламинопропил)сульфонамид
E-2-[4-(метансульфониламино)фенил]этен-N-(N,N-диметиламинопропил)сульфонамид
E-2-(4-изобутилфенил)этен-N-метилсульфонамид
E-2-(3-бензоилфенил)этен-N-метилсульфонамид
E-2-[4-(трифторметансульфонилокси)фенил]этен-N-метилсульфонамид
E-2-[4-(метансульфониламино)фенил]этен-N-метилсульфонамид
E-2-(4-изобутилфенил)этен-N-(2"-метоксиэтил)сульфонамид
E-2-(3-бензоилфенил)этен-N-(2"-метоксиэтил)сульфонамид
E-2-[4-(трифторметансульфонилокси)фенил]этен-N-(2"-метоксиэтил)сульфонамид
E-2-[4-(метансульфониламино)фенил]этен-N-(2"-метоксиэтил)сульфонамид
(1-метил-5-изобутирилпирролил)-1-метансульфонамид
(1-метил-5-ацетилпирролил)-1-метансульфонамид
1-(4-изобутилфенил)этансульфонамид
1-(3-изопропилфенил)этансульфонамид
1-(4-изобутилфенил)этан-N-(N,N-диметиламинопропил)сульфонамид
1-(3-бензоилфенил)этан-N-(N,N-диметиламинопропил)сульфонамид
1-[4-(трифторметансульфонилокси)фенил]этан-N-(N,N-диметиламинопропил)сульфонамид
1-[4-(метансульфониламино)фенил]этан-N-(N,N-диметиламинопропил)сульфонамид
1-(4-изобутилфенил)этан-N-(2-метоксиэтил)сульфонамид
1-(3-бензоилфенил)этан-N-(2-метоксиэтил)сульфонамид
1-[4-(трифторметансульфонилокси)фенил]этан-N-(2-метоксиэтил)сульфонамид
1-[4-(метансульфониламино)фенил]этан-N-(2-метоксиэтил)сульфонамид
1-(4-изобутилфенил)этан-N-метилсульфонамид
1-(3-бензоилфенил)этан-N-метилсульфонамид
1-[4-(трифторметансульфонилокси)фенил]этан-N-метилсульфонамид
1-[4-(метансульфониламино)фенил]этан-N-метилсульфонамид
1-[4-изобутилфенил]этан-N-ацетилсульфонамид
E-2-(3-бензоилфенил)-2-метилэтенсульфонамид
E-2-(3-изопропилфенил)-2-метилэтенсульфонамид
E-2-(4-изобутилфенил)-2-метилэтенсульфонамид
и фармацевтически приемлемые соли указанных соединений.
Предпочтительно соль представляет собой натриевую соль.
Вышеуказанные этансульфонамиды являются хиральными соединениями, и изобретение касается как рацемической смеси, так и отдельных (+) и (-) энантиомеров.
Соединения по изобретению формулы (I), когда содержат кислотные или основные группы, могут обычно быть выделены в форме соответствующих аддитивных солей как с органическими, так и с неорганическими фармацевтически приемлемыми кислотами или основаниями.
Примерами таких кислот служат хлористоводородная кислота, серная кислота, фосфорная кислота, метансульфоновая кислота, фумаровая кислота, лимонная кислота.
Примерами таких оснований служат гидроксид натрия, гидроксид калия, гидроксид кальция, (D,L)-лизин, L-лизин, трометамин.
Соединения формулы (I), где YR означает OH, получают путем взаимодействия соответствующих соединений формулы (IV), где J означает H или COCH3, с подходящим окислительным агентом, таким как H2O2, HClO и пероксикислоты, предпочтительно м-хлорпербензойная кислота:
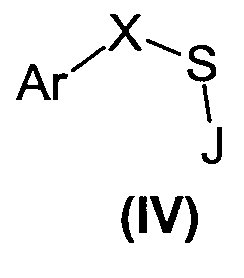
Соединения формулы (I), где Y означает NH и X означает -CH2-, получают путем взаимодействия соответствующих сульфонилгалогенидов, таких как сульфонилхлориды, с одним или двумя эквивалентами амина формулы NH2R в присутствии подходящего органического или неорганического основания, если требуется.
Соединения формулы (I), где Y означает NH и X означает -СН(СН)3-, получают путем взаимодействия соответствующих тиолов формулы (IV) с подходящим N-бромимидом, таким как N-бромфталимид, и последующего окисления атома серы с последующим снятием защиты сульфонамидного производного, как подробно описано в примерах.
Соединения формулы (I), где Y означает NH и X означает группу формулы (II), получают путем взаимодействия соответствующих сульфонилгалогенидов, таких как сульфонилхлориды, с амином формулы NH2R.
Соединения по настоящему изобретению в особенности полезны в качестве ингибиторов IL-8-индуцированного хемотаксиса PMN человека.
Цель настоящего изобретения состоит также в получении новых сульфоновых кислот и вышеуказанных производных соединений для применения в качестве лекарственных средств.
Соединения формулы (I) оценены in vitro на способность ингибировать хемотаксис полиморфонуклеарных лейкоцитов (здесь далее называемых PMN) и моноцитов, индуцируемый фракциями IL-8 и GRO-α. С этой целью для выделения PMN из гепаринизированной крови человека, забранной у здоровых взрослых добровольцев, одноядерные клетки удаляют посредством осаждения на декстране (согласно методике, описанной W.J. Ming et al., J. Immunol., 138, 1469, 1987) и эритроциты с помощью гипотонического раствора. Жизнеспособность клеток определяют путем исключения с применением трипанового синего, тогда как долю циркулирующих полиморфонуклеарных клеток устанавливают по цитоцентрифугату после окрашивания с помощью Diff Quick.
Рекомбинантный IL-8 человека (Pepro Tech) используют в качестве стимуляторов в экспериментах на хемотаксическую активность, получая практически идентичные результаты: лиофилизированный протеин растворяют в объеме HBSS, содержащего 0,2% альбумин сыворотки теленка (BSA), так, чтобы получать основной раствор с концентрацией 10-5 M, который разбавляют HBSS до концентрации 10-9 M, при анализах на хемотаксическую активность.
Во время анализа на хемотаксическую активность (согласно W. Falket et al., J. Immunol. Methods, 33, 239, 1980) используют не содержащие PVP фильтры с пористостью 5 мкм и микрокамеры, пригодные для репликации.
Соединения формулы (I) оценивают в интервале концентраций от 10-6 до 10-10 M; с этой целью указанные соединения добавляют, при одной и той же концентрации, как в верхние поры, так и нижние поры микрокамеры. Оценку способности соединений по изобретению формулы I ингибировать IL-8-индуцируемый хемотаксис моноцитов человека производят согласно методике, описанной Van Damme J. et al. (Eur. J. Immunol., 19, 2367, 1989).
Биологические результаты для некоторых характерных соединений, полученные в испытании на IL-8-индуцируемый хемотаксис PMN, приведены в таблице II (данные по ингибированию, C = 10-8 M).
В особенности предпочтительно использование соединений формулы (I), где Ar-группы означают 3'-бензоилфенил, 3'-(4-хлорбензоил)фенил, 3'-(4-метилбензоил)фенил, 3'-ацетилфенил, 3'-пропионилфенил, 3'-изобутаноилфенил, 4'-трифторметансульфонилоксифенил, 4'-бензолсульфонилоксифенил, 4'-трифторметансульфониламинофенил, 4'-бензолсульфониламинофенил, 4'-бензолсульфонилметилфенил, 4'-ацетоксифенил, 4'-пропионилоксифенил, 4'-бензоилоксифенил, 4'-ацетиламинофенил, 4'-пропиониламинофенил, 4'-бензоиламинофенил, демонстрирующие дополнительную способность к эффективному ингибированию GRO-α-индуцируемого хемотаксиса PMN; такая активность дает возможность терапевтического применения указанных соединений при IL-8-родственных патологиях, где путь CXCR2 участвует специфически или в сочетании с CXCR1-активацией.
Двойные ингибиторы IL-8- и GRO-α-индуцируемых биологических активностей в особенности предпочтительны с точки зрения рассматриваемых терапевтических применений, но описанные соединения, селективно действующие на рецептор IL-8 CXCR1 или рецептор GRO-α/IL-8 CXCR2, могут находить полезные применения в терапии описанных ниже специфических патологий.
Соединения формулы (I), оцененные в крови ex vivo в целом по методике, описанной Patrignani et al., in J. Pharmacol. Exper. Ther., 271, 1705, 1994, признаны абсолютно неэффективными в качестве ингибиторов ферментов циклооксигеназы (COX).
В большинстве случаев соединения формулы (I) не препятствуют продуцированию PGE2, индуцируемого в макрофагах мышей путем стимуляции липополисахаридами (LPS, 1 мкг/мл) при интервале концентраций от 10-5 до 10-7 M. Ингибирование продуцирования PGE2, которое может быть зарегистрировано, в основном находится в пределах статистической ошибки и чаще всего составляет менее 15-20% от исходного уровня. Пониженные эффективности в ингибировании CO создают преимущество для терапевтического применения соединений по изобретению, поскольку ингибирование синтеза простагландинов стимулирует клетки макрофага расширять синтез TNF-α (индуцируемый LPS или пероксидом водорода), являющегося важным медиатором нейтрофильной активации и стимулятором продуцирования цитокина интерлейкина-8.
С учетом обсуждаемых выше экспериментальных данных и роли, выполняемой интерлейкином-8 (IL-8), и родственности этого в процессах, включающих активацию и инфильтрацию нейтрофилов, соединения по изобретению в особенности полезны для лечения заболевания, такого как псориаз (R. J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991). Другие болезни, поддающиеся лечению с помощью соединений по настоящему изобретению, включают кишечные хронические воспалительные патологии, такие как неспецифический язвенный колит (Y. R. Mahida et al., Clin. Sci., 82, 273, 1992) и меланома, хроническое обструктивное заболевание легких (COPD), буллезный пемфигоид, ревматоидный артрит (M. Selz et al., J. Clin. Invest., 87, 463, 1981), идиопатический фиброз (E. J. Miller, цитируемый ранее, и P. C. Carré et al., J. Clin. Invest., 88, 1882, 1991), гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994), и для профилактики и лечения нарушений, вызываемых ишемией и реперфузией.
Ингибиторы активации CXCR1 и CXCR2 находят полезное применение, как подробно описано выше, в особенности в терапии хронических воспалительных патологий (например, псориаза), при которых, как предполагается, активация обоих рецепторов IL-8 играет решающую патофизиологическую роль в развитии заболевания.
Действительно, известно, что активация CXCR1 имеет важное значение для IL-8-опосредованного хемотаксиса PMN (Hammond M et al., J Immunol, 155, 1428, 1995). С другой стороны, считается, что стимуляция активации CXCR2 важна при IL-8-опосредованной пролиферации эпидермальных клеток, а также ангиогенезе у псориатических больных (Kulke R et al., J Invest Dermatol, 110, 90, 1998). Кроме того, селективные антагонисты CXCR2 находят в особенности полезные терапевтические применения в лечении серьезных заболеваний легких, таких как хроническое обструктивное заболевание легких COPD (D. WP Hay and H.M. Sarau., Current Opinion in Pharmacology 2001, 1:242-247).
Кроме того, в дополнение к применению указанных соединений, еще одной целью настоящего изобретения является разработка по использованию соединений формулы (I) в получении лекарственного средства, применяемого для лечения псориаза, неспецифического язвенного колита, меланомы, хронического обструктивного заболевания легких (COPD), буллезного пемфигоида, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и для профилактики и лечения нарушений, вызываемых ишемией и реперфузией. Фармацевтические композиции, содержащие соединение по изобретению и подходящий носитель, также входят в рамки объема настоящего изобретения и приложенных пунктов.
Соединения по изобретению вместе с обычно используемыми вспомогательным средством, носителем, разбавителем или наполнителем могут фактически быть представлены в форме фармацевтических композиций и в соответствующих стандартных дозировках и в такой форме могут быть использованы в виде твердых веществ, таких как таблетки или заполненные капсулы, или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные указанными жидкими формами капсулы, все препараты предназначены для перорального применения или в форме стерильных инъецируемых растворов для парентерального (включая подкожное) применения. Такие фармацевтические композиции и соответствующие им стандартные лекарственные формы могут включать ингредиенты в общепринятых пропорциях с добавкой или без добавки дополнительных активных соединений или действующих начал лекарственных веществ, и указанные стандартные лекарственные формы могут содержать любое подходящее эффективное количество активного ингредиента, соответствующее рекомендуемому для применения интервалу суточных доз.
При использовании в качестве фармацевтических средств кислоты по настоящему изобретению обычно вводят в форме фармацевтической композиции. Такие композиции могут быть получены хорошо известным в фармацевтической области способом и содержат, по меньшей мере, одно активное соединение. Обычно соединения по настоящему изобретению вводят в фармацевтически эффективном количестве. Количество реально вводимого соединения обычно устанавливается лечащим врачом с учетом конкретных обстоятельств, включающих излечиваемое состояние, выбранный способ введения, фактическое вводимое соединение, возраст, массу и реакцию отдельного пациента, тяжесть выраженных симптомов у пациента и тому подобное.
Фармацевтические композиции по изобретению могут быть введены разнообразными путями, включающими пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. В зависимости от выбранного способа доставки соединения предпочтительно формулируют в виде композиций для инъекций или для перорального приема. Композиции для перорального введения могут быть взяты в форме наливных жидких растворов, или суспензий, или нерасфасованных порошков. Однако, чаше всего, композиции выпускаются в виде стандартных лекарственных форм, упрощающих точное дозирование. Термин "стандартные лекарственные формы" относится к физически дискретным единицам, пригодным в качестве однократных дозировок для больных людей и других млекопитающих, каждая единица содержит заранее установленное количество активного материала, рассчитанное на получение требуемого терапевтического эффекта, в сочетании с подходящим фармацевтическим наполнителем. Характерные стандартные лекарственные формы включают предварительно наполненные заранее отмеренным количеством ампулы или шприцы с жидкими композициями либо пилюли, таблетки, капсулы или тому подобное в случае твердых композиций. В таких композициях кислотные соединения представляют собой обычно микрокомпонент (приблизительно от 0,1 до 50 мас.% или предпочтительно приблизительно от 1 до 40 мас.%), оставшуюся часть составляют различные растворители или носители и технологические добавки для формования требуемой лекарственной формы.
Жидкие формы, пригодные для перорального введения, могут включать подходящий водный и неводный растворитель, содержащий буферы, суспендирующие и диспергирующие средства, красители, корригенты и тому подобное. Жидкие формы, включая описанные ниже композиции для инъекций, обычно хранят в отсутствие света, с тем чтобы избежать возможного каталитического действия света, такого как образование гидропероксида или пероксида. Твердые формы могут включать, например, любые из следующих ингредиентов или близкие по природе соединения: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовую камедь или желатин; наполнитель, такой как крахмал или лактоза, дезинтегрирующее средство, такое как альгиновая кислота, Primogel или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; скользящее вещество, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или корригент, такой как мята перечная, метилсалицилат или ароматизирующая добавка со вкусом апельсина.
Композиции для инъекции обычно основаны на стерильном физиологическом растворе для инъекций или фосфатно-забуференном физиологическом растворе либо на других предназначенных для инъекции носителях, известных из уровня техники. Как упомянуто выше, кислотное производное формулы I в таких композициях обычно является микрокомпонентом, часто в пределах от 0,05 до 10 мас.%, при этом оставшуюся часть составляет пригодный для инъекции носитель и тому подобное. Средняя суточная доза зависит от различных факторов, таких как тяжесть заболевания и состояния пациента (возраст, пол и масса). Доза обычно изменяется в пределах от 1 мг или нескольких мг до 1500 мг соединений формулы (I) в день и, необязательно, подразделена в расчете на многократные приемы. Благодаря низкой токсичности соединений по изобретению могут также вводиться большие дозировки в течение длительных периодов времени. Вышеуказанные компоненты композиций для перорального введения или инъекций являются исключительно иллюстративными. Другие материалы, а также способы переработки и тому подобное приведены в Part 8 of "Remington's Pharmaceutical Sciences Handbook", 18th Edition, 1990, Mack Publishing Company, Easton, Pennsylvania, включенном здесь в качестве ссылки.
Соединения по изобретению могут быть также введены в формах для замедленного высвобождения или с помощью систем доставки лекарственных веществ посредством замедленного высвобождения. Описание характерных веществ для замедленного высвобождения также может быть найдено в материалах, включенных в вышеуказанный справочник Ремингтона.
Настоящее изобретение иллюстрируется с помощью следующих примеров, которые не рассматриваются как ограничивающие рамки объема изобретения и приложенных пунктов.
Пример 1
Общая методика синтеза арилметансульфоновых кислот, 1-арилэтансульфоновых кислот формулы R-Ar-C(CH3)H-SO3H и родственных энантиомеров
К охлажденному (T=0-4°C) раствору замещенного бензола (17 ммоль) и ацетилхлорида (18 ммоль) в сухом CH2Cl2 (25 мл) добавляют порциями, при энергичном перемешивании, AlCl3 (18 ммоль). Ледяную баню затем убирают и раствор нагревают при температуре кипения с обратным холодильником до тех пор, пока полное исчезновение исходного материала не станет очевидным (2-3 часа). После охлаждения до комнатной температуры смесь выливают в охлажденную 2N HCl и оставляют перемешиваться на 30'. Кислотный раствор затем переносят в делительную воронку и экстрагируют с помощью CH2Cl2 (3×20 мл).
Собранные органические экстракты промывают насыщенным раствором NaCl (2×25 мл), сушат над Na2SO4 и упаривают в вакууме, получая чистый арилацетофенон (14,45-16,15 ммоль) с высоким выходом (85-95%).
К перемешиваемому раствору арилацетофенона (11,5 ммоль) в метиловом спирте (40 мл) добавляют порциями боргидрид натрия (17,2 ммоль). Смесь нагревают при температуре кипения с обратным холодильником до полного исчезновения исходного материала (3 часа). После охлаждения до комнатной температуры к смеси добавляют 1M HCl и спирт отделяют перегонкой. Водную фазу экстрагируют этилацетатом (3×15 мл) и собранные органические экстракты промывают насыщенным раствором NaCl (2×15 мл), сушат над Na2SO4 и упаривают в вакууме, получая чистый 1-арилэтиловый спирт (выход порядка 75%).
К перемешиваемому раствору 1-арилэтилового спирта (4,5 ммоль) в сухом CHCl3 (10 мл) добавляют тиолуксусную кислоту (5,39 ммоль) и йодид цинка (2,24 ммоль). Реакционную смесь нагревают при температуре кипения с обратным холодильником в течение 3 часов; после охлаждения до комнатной температуры смесь разбавляют водой (15 мл) и переносят в делительную воронку. Две фазы перемешивают встряхиванием и разделяют. Органическую фазу промывают насыщенным раствором NaHCO3 (3×20 мл), затем насыщенным раствором NaCl, сушат над Na2SO4 и упаривают в вакууме, получая чистый 1-арилэтилтиоацетат (выход порядка 80%).
Раствор 1-арилэтилтиоацетата (0,91 ммоль) в ледяной уксусной кислоте (2 мл) перемешивают при 60°C и обрабатывают, добавляя по каплям, 30% H2O2 (4,56 ммоль); полученный раствор перемешивают при 60°C в течение 24 часов, затем уксусную кислоту удаляют азеотропной перегонкой с толуолом.
Остаток разбавляют водой (5 мл), нейтрализуют 1N NaOH, промывают диэтиловым эфиром (2×15 мл) и лиофилизуют, получая натриевую соль 1-арилэтансульфоновой кислоты в виде белого вещества, представляющего собой рацемическую смесь (выход порядка 90%).
Оптическое разделение
Рацемическую натриевую соль 1-арилэтансульфоновой кислоты фильтруют через колонку, заполненную смолой Amberlite IR-120 (H+ форма), элюируют водой и получают продукт в виде пастообразного масла. Разделение двух изомеров осуществляют путем кристаллизации соответствующих (+) или (-) α-фенилэтиламмониевых солей в этанольном растворе, как описано применительно к оптическому разделению арилпропионовых кислот в Akgun H. et al., Arzneim.-Forsch./Drug Res., 46(II), Nr.9, 891-894 (1996). Чистые энантиомеры выделяют в виде натриевых солей.
Согласно приведенному выше способу получены следующие соединения:
натриевая соль (-)-1-(4-изобутилфенил)этансульфоновой кислоты (1)
Соединение синтезируют, исходя из коммерческого изобутилбензола.
[a]D = -35 (c=1; H2O)
1H-ЯМР (ДМСО-d6): δ 7,25 (д, 2H, J=7Гц); 7,05 (д, 2H, J=7Гц); 3,62 (м, 1H); 2,37 (д, 2H, J=7Гц); 1,86 (м, 1H); 1,40 (д, 3H, J=7Гц); 0,91 (д, 6H, J=7Гц).
натриевая соль (+)-1-(4-изобутилфенил)этансульфоновой кислоты (2)
Соединение синтезируют, исходя из коммерческого изобутилбензола.
[a]D = +34,5 (c=1; H2O)
1H-ЯМР (ДМСО-d6): δ 7,25 (д, 2H, J=7Гц); 7,08 (д, 2H, J=7Гц); 3,62 (м, 1H); 2,37 (д, 2H, J=7Гц); 1,86 (м, 1H); 1,42 (д, 3H, J=7Гц); 0,90 (д, 6H, J=7Гц).
натриевая соль (-)-1-[4-(1-оксо-2-изоиндолинил)фенил]этансульфоновой кислоты (3)
Соединение получают по приведенному выше способу, исходя из промежуточного 4-(1-оксо-2-изоиндолинил)ацетофенона. Данное промежуточное соединение получают из промышленно выпускаемых реагентов, фтальальдегида и 4-аминоацетофенона, на основании методики, описанной в Ichiro, T. et al., Heterocycles 43:11, 2343-2346 (1996).
[a]D = -52,4 (c=1; H2O)
1H-ЯМР (ДМСО-d6): δ 7,68 (м, 3H); 7,35 (м, 3H); 7,15 (д, 2H, J=7Гц); 4,68 (с, 2H); 3,65 (кв, 1H, J1=7Гц, J2=3Гц); 1,28 (д, 3H, J=7Гц).
натриевая соль (+)-1-[4-(1-оксо-2-изоиндолинил)фенил]этансульфоновой кислоты (4)
Соединение получают по описанному выше способу, исходя из промежуточного 4-(1-оксо-2-изоиндолинил)ацетофенона. Данное промежуточное соединение получают из промышленно выпускаемых реагентов, фтальальдегида и 4-аминоацетофенона, на основании методики, описанной в Ichiro, T. et al., Heterocycles 43:11, 2343-2346 (1996).
[a]D = +50 (c=1; H2O)
1H-ЯМР (ДМСО-d6): δ 7,708 (м, 3H); 7,35 (м, 3H); 7,18 (д, 2H, J=7Гц); 4,68 (с, 2H); 3,65 (кв, 1H, J1=7Гц, J2=3Гц); 1,30 (д, 3H, J=7Гц).
натриевая соль (-)-2-(4-фенилсульфонилокси)этансульфоновой кислоты (5)
Соединение получают по описанному выше способу, исходя из промежуточного 4-бензолсульфонилоксиацетофенона, полученного из коммерческого 4-гидроксиацетофенона по известным экспериментальным методикам.
[a]D = -47,5 (c=1; H2O)
1H-ЯМР (D2O): δ 7,90 (д, 2H, J=7Гц); 7,70 (т, 1H, J=7Гц); 7,55 (т, 2H, J=7Гц); 7,32 (д, 2H, J=7Гц); 6,95 (д, 2H, J=7Гц); 3,64 (м, 1H); 1,41 (д, 3H, J=7Гц).
натриевая соль (+)-2-(4-фенилсульфонилокси)этансульфоновой кислоты (6)
Соединение получают по приведенному выше способу, исходя из промежуточного 4-бензолсульфонилоксиацетофенона, полученного из коммерческого 4-гидроксиацетофенона по известным экспериментальным методикам.
[a]D = +49 (c=1; H2O)
1H-ЯМР (D2O): δ 7,93 (д, 2H, J=7Гц); 7,70 (т, 1H, J=7Гц); 7,55 (т, 2H, J=7Гц); 7,32 (д, 2H, J=7Гц); 6,91 (д, 2H, J=7Гц); 3,67 (м, 1H); 1,41 (д, 3H, J=7Гц).
натриевая соль (1-метил-5-ацетилпирролил)-1-метансульфоновой кислоты (7)
Синтез (7) осуществляют, исходя из коммерческого реагента метил-1-метил-2-пирролацетата, который при ацилировании по методу Фриделя-Крафтса с ацетилхлоридом дает (1-метил-5-ацетилпирролил)-1-метанацетат. Сложноэфирную группу затем гидролизуют. Согласно экспериментальной методике, описанной в WO 02/0704095, получают родственную натриевую соль (1-метил-5-ацетилпирролил)-1-метансульфоновой кислоты.
1H-ЯМР (ДМСО-d6): δ 7,5 (с, 1H); 6,18 (с, 1H); 3,60 (с, 3H); 3,51 (с, 2H); 2,10 (с, 3H).
натриевая соль (±)-2-(3-бензоилфенил)этансульфоновой кислоты (8)
Синтез (8) осуществляют, исходя из коммерческого реагента, 3-(1-цианоэтил)бензойной кислоты, которая при ацилировании по методу Фриделя-Крафтса в бензоле дает 2-(3'-бензоилфенил)пропионитрил. Следуя экспериментальной методике, описанной в WO 02/0704095, получают родственную натриевую соль 2-(3'-бензоилфенил)этансульфоновой кислоты.
1H-ЯМР (D2O): δ 7,80 (д, 2H, J=7Гц); 7,70 (с, 1H); 7,62 (д, 1H, J=7Гц); 7,51 (м, 2H); 7,30 (м, 3H); 3,62 (м, 1H); 1,40 (д, 3H, J=7Гц).
натриевая соль (±)-2-(3-изопропилфенил)этансульфоновой кислоты (9)
Синтез (9) осуществляют, исходя из доступного реагента, 3-(1-цианоэтил)ацетофенона, который по реакции Виттига и при восстановлении метиленовой группы по хорошо известным методикам дает 2-(3-изопропилфенил)пропионитрил. Следуя экспериментальной методике, описанной в WO 02/0704095, получают родственную натриевую соль 2-(3-изопропилфенил)этансульфоновой кислоты.
1H-ЯМР (D2O): δ 7,30 (м, 2H); 7,10 (м, 2H); 3,92 (м, 1H); 3,63 (м, 1H); 1,42 (д, 3H, J=7Гц); 1,25 (д, 6H, J=8Гц).
Пример 2
Получение E-арилэтенсульфоновых кислот (натриевых солей)
Арилэтансульфоновую кислоту растворяют в тионилхлориде (5 мл) и раствор нагревают при температуре кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры тионилхлорид выпаривают в вакууме и сырой арилэтансульфонилхлорид разбавляют сухим ТГФ (5 мл) и охлаждают до T=0°C на водяной бане со льдом; добавляют 1N водный NaOH (0,64 ммоль) при T=4°C; водяную баню со льдом убирают и реакционную смесь оставляют примерно на 1 час до достижения комнатной температуры, при этом осаждается белое твердое вещество. Органическую соль натрия отделяют фильтрованием в вакууме, промывают ТГФ и сушат в вакууме при 40°C, получая чистую натриевую соль E-арилэтенсульфоновой кислоты (0,32-0,51 ммоль) (выход 50-80%) в виде белого твердого порошка.
Согласно описанной выше методике получены следующие соединения:
натриевая соль E-2-(4-изобутилфенил)этенсульфоновой кислоты (10)
1H-ЯМР (D2O): δ 7,60 (д, 1H, J=8Гц); 7,55-7,32 (м, 4H); 7,05 (д, 1H, J=14Гц); 2,62 (м, 2H); 1,90 (м, 1H); 0,97 (д, 6H, J=7Гц).
натриевая соль E-2-(3-бензоилфенил)этенсульфоновой кислоты (11)
1H-ЯМР (D2O): δ 7,80 (д, 2H, J=7Гц); 7,70 (с, 1H); 7,65 (д, 1H, J=8Гц); 7,62 (д, 1H, J=7Гц); 7,51 (м, 2H); 7,30 (м, 3H); 7,00 (д, 1H, J=14Гц).
натриевая соль E-2-(4-метансульфониламинофенил)этенсульфоновой кислоты (12)
1H-ЯМР (ДМСО-d6): δ 7,60 (д, 1H, J=8Гц); 7,35 (д, 2H, J=8Гц); 7,20 (д, 2H, J=8Гц); 7,07 (д, 1H, J=14Гц); 6,51 (шир.с, 1H, SO2NH); 3,00 (с, 3H).
натриевая соль E-2-(4-трифторметансульфонилоксифенил)этенсульфоновой кислоты (13)
1H-ЯМР (CDCl3): δ 7,62 (д, 1H, J=8Гц); 7,50 (д, 2H, J=7Гц); 7,25 (д, 2H, J=7Гц); 7,05 (д, 1H, J=14Гц).
Пример 3
Общая методика синтеза E-арилэтенсульфонамидов
Раствор арилэтансульфоновой кислоты (0,64 ммоль) растворяют в тионилхлориде (5 мл) и раствор нагревают при температуре кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры тионилхлорид выпаривают в вакууме и сырой арилэтансульфонилхлорид разбавляют сухим ТГФ (5 мл) и охлаждают до T=0°C на водяной бане со льдом; добавляют по каплям выбранный амин (1,28 ммоль). Водяную баню со льдом убирают и реакционную смесь оставляют стоять до достижения комнатной температуры. После полного исчезновения исходного реагента растворители выпаривают в вакууме и к остатку добавляют CHCl3 (10 мл) и воду (10 мл); две фазы перемешивают встряхиванием и разделяют, органическую фазу промывают водой (3×15 мл), сушат над Na2SO4 и упаривают в вакууме, получая сырой продукт, который очищают флэш-хроматографией. Чистые E/Z-арилэтенсульфонамиды (0,32-0,51 ммоль) (выход 50-80%) выделяют в виде бесцветных масел.
Согласно описанной выше методике и при использовании аммиака (0,5 M в 1,4-диоксане) в качестве амина получены следующие соединения:
E-2-(4-изобутилфенил)этенсульфонамид (14)
1H-ЯМР (CDCl3): δ 7,55 (д, 1H, J=14Гц); 7,38 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 6,88 (д, 1H, J=14Гц); 4,75 (шир.с, 2H, SO2NH2); 2,55 (д, 2H, J=7Гц); 1,94 (м, 1H); 1,02 (д, 6H, J=7Гц).
E-2-(3-бензоилфенил)этенсульфонамид (15)
1H-ЯМР (CDCl3): δ 7,80 (д, 2H, J=7Гц); 7,72 (с, 1H); 7,62 (д, 1H, J=8Гц); 7,52 (д, 1H, J=14Гц); 7,50 (м, 2H); 7,30 (м, 3H); 6,88 (д, 1H, J=14Гц); 4,75 (шир.с, 2H, SO2NH2).
E-2-[4-(трифторметансульфонилокси)фенил]этенсульфонамид (16)
1H-ЯМР (CDCl3): δ 7,60 (д, 1H, J=8Гц); 7,52 (д, 2H, J=7Гц); 7,28 (д, 2H, J=7Гц); 7,10 (д, 1H, J=14Гц); 4,85 (шир.с, 2H, SO2NH2).
E-2-[4-(метансульфониламино)фенил]этенсульфонамид (17)
1H-ЯМР (CDCl3): δ 7,55 (д, 1H, J=14Гц); 7,37 (д, 2H, J=8Гц); 7,22 (д, 2H, J=8Гц); 6,90 (д, 1H, J=14Гц); 6,45 (шир.с, 1H, SO2NH); 4,80 (шир.с, 2H, SO2NH2); 2,98 (с, 3H).
Согласно описанной выше методике и при использовании 3-(диметиламино)пропиламина в качестве амина получены следующие соединения:
E-2-(4-изобутилфенил)этен-(N,N-диметиламинопропил)сульфонамид (18)
1H-ЯМР (CDCl3): δ 7,45 (м, 3H); 7,20 (д, 2H, J=7Гц); 6,70 (д, 1H, J=14Гц); 6,40 (шир.с, 1H, SO2NH); 3,18 (м, 2H); 2,55 (м, 4H); 2,30 (с, 6H); 1,92 (м, 1H); 1,75 (м, 2H); 0,97 (д, 6H, J=7Гц).
E-2-(3-бензоилфенил)этен-N-(N,N-диметиламинопропил)сульфонамид (19)
1H-ЯМР (CDCl3): δ 7,82 (д, 2H, J=7Гц); 7,74 (с, 1H); 7,60 (д, 1H, J=8Гц); 7,50 (д, 1H, J=14Гц); 7,45 (м, 2H); 7,26 (м, 3H); 6,70 (д, 1H, J=14Гц); 6,45 (шир.с, 1H, SO2NH); 3,15 (м,, 2H); 2,50 (м, 4H); 2,35 (с, 6H).
E-2-[4-(трифторметансульфонилокси)фенил]этен-(N,N-диметиламинопропил)сульфонамид (20)
1H-ЯМР (CDCl3): δ 7,62 (д, 1H, J=14Гц); 7,48 (д, 2H, J=7Гц); 7,25 (д, 2H, J=7Гц); 7,00 (д, 1H, J=14Гц); 6,50 (шир.с, 1H, SO2NH); 3,17 (м, 2H); 2,48 (м, 4H); 2,35 (с, 6H).
E-2-[4-(метансульфониламино)фенил]этен-(N,N-диметиламинопропил)сульфонамид (21)
1H-ЯМР (CDCl3): δ 7,57 (д, 1H, J=14Гц); 7,37 (д, 2H, J=8Гц); 7,22 (д, 2H, J=8Гц); 6,75 (д, 1H, J=14Гц); 6,50 (шир.с, 2H, SO2NH); 3,15 (м, 2H); 2,98 (с, 3H); 2,50 (м, 4H); 2,40 (с, 6H).
Согласно описанной выше методике и при использовании метиламина (2M в ТГФ) в качестве амина получены следующие соединения:
E-2-(4-изобутилфенил)этен-N-метилсульфонамид (22)
1H-ЯМР (CDCl3): δ 7,55 (д, 1H, J=14Гц); 7,38 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 6,88 (д, 1H, J=14Гц); 4,80 (шир.с, 1H, SO2NH); 2,75 (д, 3H, J=4Гц); 2,55 (д, 2H, J=7Гц); 1,95 (м, 1H); 1,04 (д, 6H, J=7Гц).
E-2-(3-бензоилфенил)этен-N-метилсульфонамид (23)
1H-ЯМР (CDCl3): δ 7,81 (д, 2H, J=7Гц); 7,70 (с, 1H); 7,62 (д, 1H, J=8Гц); 7,55 (д, 1H, J=14Гц); 7,45 (м, 2H); 7,30 (м, 3H); 6,90 (д, 1H, J=14Гц); 4,60 (шир.с, 1H, SO2NH); 2,70 (д, 3H, J=4Гц).
E-2-[4-(трифторметансульфонилокси)фенил]этен-N-метилсульфонамид (24)
1H-ЯМР (CDCl3): δ 7,60 (д, 1H, J=8Гц); 7,52 (д, 2H, J=7Гц); 7,28 (д, 2H, J=7Гц); 7,10 (д, 1H, J=14Гц); 4,85 (шир.с, 1H, SO2NH); 2,70 (д, 3H, J=4Гц).
E-2-[4-(метансульфониламино)фенил]этен-N-метилсульфонамид (25)
1H-ЯМР (CDCl3): δ 7,56 (д, 1H, J=14Гц); 7,35 (д, 2H, J=8Гц); 7,20 (д, 2H, J=8Гц); 6,92 (д, 1H, J=14Гц); 6,50 (шир.с, 1H, SO2NH); 4,70 (шир.с, 1H, SO2NH); 3,00 (с, 3H), 2,75 (д, 3H, J=4Гц).
Согласно описанной выше методике и при использовании 2-метоксиэтиламина в качестве амина получены следующие соединения:
E-2-(4-изобутилфенил)этен-N-(2-метоксиэтил)сульфонамид (26)
1H-ЯМР (CDCl3): δ 7,57 (д, 1H, J=14Гц); 7,38 (д, 2H, J=7Гц); 7,20 (д, 2H, J=7Гц); 6,90 (д, 1H, J=14Гц); 4,80 (шир.с, 1H, SO2NH); 3,74 (м, 2H); 3,55 (м, 2H); 3,45 (с, 3H); 2,52 (д, 2H, J=7Гц); 1,95 (м, 1H); 1,05 (д, 6H, J=7Гц).
E-2-(3-бензоилфенил)этен-N-(2-метоксиэтил)сульфонамид (27)
1H-ЯМР (CDCl3): δ 7,80 (д, 2H, J=7Гц); 7,72 (с, 1H); 7,62 (д, 1H, J=8Гц); 7,55 (д, 1H, J=14Гц); 7,40 (м, 2H); 7,30 (м, 3H); 6,95 (д, 1H, J=14Гц); 4,62 (шир.с, 1H, SO2NH); 3,75 (м, 2H); 3,50 (м, 2H); 3,40 (с, 3H).
E-2-[4-(трифторметансульфонилокси)фенил]этен-N-(2-метоксиэтил)сульфонамид (28)
1H-ЯМР (CDCl3): δ 7,62 (д, 1H, J=8Гц); 7,50 (д, 2H, J=7Гц); 7,30 (д, 2H, J=7Гц); 7,15 (д, 1H, J=14Гц); 4,80 (шир.с, 1H, SO2NH); 3,77 (м, 2H); 3,52 (м, 2H); 3,40 (с, 3H).
E-2-[4-(метансульфониламино)фенил]этен-N-(2-метоксиэтил)сульфонамид (29)
1H-ЯМР (CDCl3): δ 7,58 (д, 1H, J=14Гц); 7,35 (д, 2H, J=8Гц); 7,25 (д, 2H, J=8Гц); 6,90 (д, 1H, J=14Гц); 6,52 (шир.с, 1H, SO2NH); 4,75 (шир.с, 1H, SO2NH); 3,70 (м, 2H); 3,50 (м, 2H); 3,40 (с, 3H); 3,05 (с, 3H).
Пример 4
Общая методика синтеза арилметансульфонамидов
(1-Метил-5-изобутирилпирролил)-1-метансульфонамид (30)
Синтез соединения (30) осуществляют, исходя из коммерческого реагента метил-1-метил-2-пирролацетата, который при ацилировании по методу Фриделя-Крафтса с изобутирилхлоридом дает (1-метил-5-изобутирилпирролил)-1-метанацетат. Сложноэфирную группу затем гидролизуют. Согласно экспериментальной методике, описанной в WO 02/0704095, получают родственную натриевую соль (1-метил-5-изобутирилпирролил)-1-метансульфоновой кислоты.
Раствор натриевой соли (1-метил-5-изобутирилпирролил)-1-метансульфоновой кислоты (0,64 ммоль) растворяют в тионилхлориде (5 мл) и полученный раствор нагревают при температуре кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры тионилхлорид выпаривают в вакууме и сырой (1-метил-5-изобутирилпирролил)-1-метансульфонилхлорид разбавляют сухим ТГФ (5 мл) и охлаждают до T=0°C на водяной бане со льдом; добавляют по каплям раствор аммиака (1,28 ммоль). Водяную баню со льдом убирают и реакционную смесь оставляют стоять до достижения комнатной температуры. После полного исчезновения исходного реагента растворители выпаривают в вакууме и к остатку добавляют CHCl3 (10 мл) и воду (10 мл); две фазы перемешивают встряхиванием и разделяют, органическую фазу промывают водой (3×15 мл), сушат над Na2SO4 и упаривают в вакууме, получая сырой продукт, который очищают флэш-хроматографией. Чистый (1-метил-5-изобутирилпирролил)-1-метансульфонамид (0,60 ммоль) (выход 93%) выделяют в виде желтого масла.
1H-ЯМР (ДМСО-d6): δ 7,5 (с, 1H); 6,18 (с, 1H); 4,65 (шир.с, 2H, SO2NH2); 3,60 (с, 3H); 3,51 (с, 2H); 3,38 (м, 1H); 1,25 (д, 6H, J=8Гц).
Согласно описанной выше методике и при использовании натриевой соли (1-метил-5-ацетилпирролил)-1-метансульфоновой кислоты (7) (полученной по приведенной выше общей методике синтеза арилметансульфоновых кислот) получено следующее соединение:
(1-метил-5-ацетилпирролил)-1-метансульфонамид (31)
1H-ЯМР (ДМСО-d6): δ 7,5 (с, 1H); 6,18 (с, 1H); 4,40 (шир.с, 2H, SO2NH2); 3,60 (с, 3H); 3,51 (с, 2H); 2,10 (с, 3H).
Энантиоселективный синтез (+) и (-) энантиомеров соединений 32 и 33
Энантиоселективный синтез (+) и (-) энантиомеров 1-(4-изобутилфенил)этансульфонамида выполняют, как описано в Davis F.A. et al., J. Org. Chem., 58, 4890-4896, (1993). Методика включает диастереоселективное C-метилирование N-сульфонилкамфоримина, полученного из 4-изобутилбензилсульфонамида (27) и N,N-диизопропил-(1S)-(+)-10-камфорсульфонамида или N,N-диизопропил-(1R)-(-)-10-камфорсульфонамида. Кислотный гидролиз диастереомеров позволяет получать требуемые соединения, оба в виде прозрачных масел.
(-)-1-(4-Изобутилфенил)этансульфонамид (32)
[a]D = -8,5 (c=1,2; CHCl3)
1H-ЯМР (CDCl3): δ 7,30 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 4,25 (м, 1H + шир.с SONH2); 2,45 (д, 2H, J=7Гц); 1,87 (м, 4H); 0,97 (д, 6H, J=7Гц).
(+)-1-(4-Изобутилфенил)этансульфонамид (33)
[a]D = +15 (c=1; CHCl3)
1H-ЯМР (CDCl3): δ 7,30 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 4,25 (м, 1H + шир.с SONH2); 2,45 (д, 2H, J=7Гц); 1,87 (м, 4H); 0,97 (д, 6H, J=7Гц).
Пример 5
Альтернативный синтез арилэтансульфонамидов
Синтез (+)-1-(3-изопропилфенил)этансульфонамида (34)
Указанное в заголовке соединение получают, исходя из коммерческого реагента, 3-(1-цианоэтил)бензойной кислоты, которая согласно экспериментальным методикам, описанным в Kindler K. et al., Chem. Ber., 99, 226 (1966) и Kindler K. et al., Liebigs Ann. Chem., 26, 707 (1967), дает промежуточную 3-изопропилбензойную кислоту. Восстановление до производного бензилового спирта с помощью LiAlH4 и последующая обработка спирта тиолуксусной кислотой приводят к промежуточному этилтиоацетату. Последующий гидролиз производного тиола осуществляют, как описано в Corey E.J. et al., Tet. Lett., 33, 4099 (1992).
К суспензии 3-изопропилбензилтиола (3,85 г; 23,2 ммоль) и трет-бутоксида калия (2,6 г; 23,2 ммоль) в CH2Cl2 (15 мл) добавляют 18-Crown-6 (0,6 г; 2,3 ммоль). После перемешивания в течение 15' при T=0°C-4°C добавляют N-Br-фталимид (5,24 г; 23,2 ммоль). После добавления водяную баню со льдом убирают и раствор оставляют перемешиваться при комнатной температуре на 1 ч; затем органическую фазу промывают водой (3×15 мл), сушат над Na2SO4 и упаривают в вакууме, получая маслянистый остаток, который при очистке флэш-хроматографией дает 3-изопропилбензилтиофталимид (6,05 г; 18,56 ммоль) в виде светло-желтого масла (выход 80%). Последующее метилирование с получением 1-(3-изопропилфенил)этилтиофталимида выполняют, как описано в Davis F.A. et al., J. Org. Chem., 58, 4890-4896, (1993). Конечное соединение, 1-(3-изопропилфенил)этансульфонамид (31), получают окислением с помощью 3-хлорпербензойной кислоты (2 эквивалента) и расщеплением фталимидогруппы с помощью гидразина, согласно хорошо известным из уровня техники методикам.
1H-ЯМР (CDCl3): δ 7,28 (м, 2H); 7,05 (м, 2H); 4,40 (шир.с, 2H, SO2NH2); 3,90 (м, 1H); 3,65 (м, 1H); 1,35 (д, 3H, J=7Гц); 1,20 (д, 6H, J=8Гц).
Алкилирование соответствующих 1-арилэтансульфонамидов (полученных по приведенной выше методике) с помощью 3-диметиламинопропилхлорида в качестве алкилирующего реагента осуществляют в условиях фазового переноса, как описано в Gajda T. et al., Synthesis, 1005 (1981) и Burke P.O. et al., Synthesis, 935 (1985). Получены следующие соединения:
(±)-1-(4-изобутилфенил)этан-N-(N,N-диметиламинопропил)сульфонамид (35)
1H-ЯМР (CDCl3): δ 7,32 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 4,26 (м, 1H); 4,10 (шир.с, 1H, SONH); 3,18 (м, 2H); 2,55 (м, 4H); 2,45 (д, 2H, J=7Гц); 2,40 (с, 6H); 1,85 (м, 4H); 1,00 (д, 6H, J=7Гц).
(±)-1-(3-бензоилфенил)этан-N-(N,N-диметиламинопропил)сульфонамид (36)
1H-ЯМР (CDCl3): δ 7,80 (д, 2H, J=7Гц); 7,70 (с, 1H); 7,62 (д, 1H, J=7Гц); 7,51 (м, 2H); 7,30 (м, 3H); 4,35 (шир.с, 1H, SO2NH); 3,62 (м, 1H); 3,18 (м, 2H); 2,55 (м, 4H); 2,40 (с, 6H); 1,30 (д, 3H, J=7Гц).
(±)-1-[4-(трифторметансульфонилокси)фенил]этан-N-(N,N-диметиламинопропил)сульфонамид (37)
1H-ЯМР (CDCl3): δ 7,50 (д, 2H, J=7Гц); 7,25 (д, 2H, J=7Гц); 4,30 (шир.с, 1H, SO2NH); 3,85 (м, 1H); 3,20 (м, 2H); 2,60 (м, 4H); 2,45 (с, 6H); 1,25 (д, 3H, J=7Гц).
(±)-1-[4-(метансульфониламино)фенил]этан-N-(N,N-диметиламинопропил)сульфонамид (38)
1H-ЯМР (CDCl3): δ 7,37 (д, 2H, J=8Гц); 7,22 (д, 2H, J=8Гц); 6,45 (шир.с, 1H, SO2NH); 4,80 (шир.с, 1H, SO2NH); 3,82 (м, 1H); 3,25 (м, 2H); 2,98 (с, 3H); 2,65 (м, 4H); 2,45 (с, 6H); 1,05 (д, 3H, J=7Гц).
Алкилирование соответствующих 1-арилэтансульфонамидов (полученных по приведенной выше методике) с помощью простого 2-бромэтилметилового эфира в качестве алкилирующего реагента выполняют в условиях фазового переноса, как описано в Gajda T. et al., Synthesis, 1005 (1981) и Burke P.O. et al., Synthesis, 935 (1985). Получены следующие соединения:
(±)-1-(4-изобутилфенил)этан-N-(2-метоксиэтил)сульфонамид (39)
1H-ЯМР (CDCl3): δ 7,30 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 4,25 (м, 1H); 4,80 (шир.с, 1H, SO2NH); 3,74 (м, 2H); 3,55 (м, 2H); 3,45 (с, 3H); 2,45 (д, 2H, J=7Гц); 1,87 (м, 1H); 1,65 (д, 3H, J=7Гц); 0,97 (д, 6H, J=7Гц).
(±)-1-(3-бензоилфенил)этан-N-(2-метоксиэтил)сульфонамид (40)
1H-ЯМР (CDCl3): δ 7,82 (д, 2H, J=7Гц); 7,75 (с, 1H); 7,62 (д, 1H, J=7Гц); 7,55 (м, 2H); 7,30 (м, 3H); 4,25 (шир.с, 1H, SO2NH); 3,75 (м, 2H); 3,60 (м, 1H); 3,55 (м, 2H); 3,48 (с, 3H); 1,55 (д, 3H, J=7Гц).
(±)-1-[4-(трифторметансульфонилокси)фенил]этан-N-(2-метоксиэтил)сульфонамид (41)
1H-ЯМР (CDCl3): δ 7,50 (д, 2H, J=7Гц); 7,25 (д, 2H, J=7Гц); 4,30 (шир.с, 1H, SO2NH); 3,85 (м, 1H); 3,60 (м, 2H); 3,55 (м, 2H); 3,48 (с, 3H); 1,35 (д, 3H, J=7Гц).
(±)-1-[4-(метансульфониламино)фенил]этан-N-(2-метоксиэтил)сульфонамид (42)
1H-ЯМР (CDCl3): δ 7,52 (д, 2H, J=7Гц); 7,28 (д, 2H, J=7Гц); 6,45 (шир.с, 1H, SO2NH); 4,32 (шир.с, 1H, SO2NH); 3,85 (м, 1H); 3,62 (м, 2H); 3,55 (м, 2H); 3,48 (с, 3H); 3,00 (с, 3H); 1,35 (д, 3H, J=7Гц).
Монометилирование соответствующих 1-арилэтансульфонамидов (полученных по приведенной выше методике) с помощью диазометана выполняют, как описано в Muller E. et al., Liebigs Ann. Chem., 623, 34 (1959) и Saegusa T. et al., Tet. Lett., 6131 (1966). Получены следующие соединения:
(±)-1-(4-изобутилфенил)этан-N-метилсульфонамид (43)
1H-ЯМР (CDCl3): δ 7,25 (д, 2H, J=7Гц); 7,18 (д, 2H, J=7Гц); 4,80 (шир.с, 1H, SO2NH); 4,20 (м, 1H); 2,70 (д, 3H, J=4Гц); 2,45 (д, 2H, J=7Гц); 1,87 (м, 1H); 1,65 (д, 3H, J=7Гц); 0,97 (д, 6H, J=7Гц).
(±)-1-(3-бензоилфенил)этан-N-метилсульфонамид (44)
1H-ЯМР (CDCl3): δ 7,82 (д, 2H, J=7Гц); 7,75 (с, 1H); 7,62 (д, 1H, J=7Гц); 7,55 (м, 2H); 7,30 (м, 3H); 4,25 (шир.с, 1H, SO2NH); 4,15 (м, 1H); 2,70 (д, 3H, J=4Гц); 1,55 (д, 3H, J=7Гц).
(±)-1-[4-(трифторметансульфонилокси)фенил]этан-N-метилсульфонамид (45)
1H-ЯМР (CDCl3): δ 7,52 (д, 2H, J=7Гц); 7,28 (д, 2H, J=7Гц); 4,10 (шир.с, 1H, SO2NH); 3,80 (м, 1H); 2,75 (д, 3H, J=4Гц); 1,20 (д, 3H, J=7Гц).
(±)-1-[4-(метансульфониламино)фенил]этан-N-метилсульфонамид (46)
1H-ЯМР (CDCl3): δ 7,50 (д, 2H, J=7Гц); 7,27 (д, 2H, J=7Гц); 6,50 (шир.с, 1H, SO2NH); 4,30 (шир.с, 1H, SO2NH); 3,90 (м, 1H); 3,05 (с, 3H); 2,70 (д, 3H, J=4Гц); 1,32 (д, 3H, J=7Гц).
(±)-1-(4-изобутилфенил)этан-N-ацетилсульфонамид (47)
Соединение синтезируют, как описано выше, путем ацилирования с помощью ацетилхлорида родственного 1-(4-изобутилфенил)этансульфонамида.
1H-ЯМР (CDCl3): δ 7,28 (д, 2H, J=7Гц); 7,20 (д, 2H, J=7Гц); 4,82 (шир.с, 1H, SO2NH); 4,30 (м, 1H); 2,45 (д, 2H, J=7Гц); 1,85 (м, 1H); 1,80 (с, 3H); 1,65 (д, 3H, J=7Гц); 0,97 (д, 6H, J=7Гц).
Пример 6
Общая методика синтеза E/Z-2-арил-2-метилэтенсульфонамидов
Раствор соответствующего арилацетофенона (20 ммоль) (полученного по описанному выше способу общего подхода к синтезу 1-арилэтансульфоновых кислот) в 10 мл трет-бутилового спирта добавляют по каплям в течение 20 мин к коммерческому илиду, йодметилентрифенилфосфорану (25 ммоль), поддерживая реакционную температуру ниже 25°C, и образовавшуюся смесь перемешивают в течение 4 ч при комнатной температуре. По окончании взаимодействия смесь встряхивают с 50 мл пентана и 50 мл воды, фильтруют и слои разделяют. Водный слой экстрагируют 3×50 мл пентана и сушат над сульфатом натрия, получая после очистки флэш-хроматографией чистый 2-(арил)пропениодид (смесь E/Z-изомеров), (выход порядка 70%). Указанное выше олефинирование по Виттигу карбонильного соединения осуществляют, как описано в Sotaro Miyano et al., Bull. Chem. Soc. J., 1197, 52 (1979).
2-(Арил)пропениодид (2 ммоль) растворяют в ацетонитриле (5 мл) и добавляют к раствору тиоацетата калия (4 ммоль) в ацетонитриле (2 мл) при комнатной температуре; реакционную смесь перемешивают в течение 4 часов. Смесь гасят водой и экстрагируют EtOAc; отделенные органические слои сушат, фильтруют и концентрируют, получая 2-арилпропентиоацетат (смесь E/Z-изомеров) (почти количественный выход).
Раствор 2-арил-2-метилэтентиоацетата (1,00 ммоль) в ледяной уксусной кислоте (2 мл) перемешивают при 60°C и обрабатывают по каплям 30% H2O2 (4,56 ммоль); полученный раствор перемешивают при 60°C в течение 24 часов, затем уксусную кислоту удаляют азеотропной перегонкой с толуолом. Остаток разбавляют водой (5 мл), нейтрализуют с помощью 1N NaOH, промывают диэтиловым эфиром (2×15 мл) и лиофилизуют, получая натриевую соль 2-арил-2-метилэтенсульфоновой кислоты в виде белого твердого вещества как смесь E/Z-изомеров (выход порядка 90%).
E/Z-2-арил-2-метилэтенсульфонамиды получают по описанному выше способу общего подхода к синтезу E-арилэтенсульфонамидов, что дает E/Z-2-арил-2-метил-этенсульфонамиды (0,75-0,85 ммоль) (выход 85-95%) в виде бесцветных масел.
Следуя описанной выше методике, синтезируют следующие соединения:
E-2-(3-бензоилфенил)-2-метилэтенсульфонамид (48)
1H-ЯМР (CDCl3): δ 7,75 (м, 3H); 7,62 (м, 2H); 7,53 (м, 4H); 6,15 (д, 1H, J=1,4Гц), 5,96 (д, 1H, J=1,3 Гц); 4,38 (шир.с, 2H, SONH2); 2,10 (д, 3H, J=1,4Гц); 2,0 (д, 3H, J=1,3Гц).
E-2-(3-изопропилфенил)-2-метилэтенсульфонамид (49)
1H-ЯМР (CDCl3): δ 7,28 (м, 1H); 7,15 (м, 1H); 7,05 (м, 2H); 6,15 (д, 1H, J=1,4Гц), 5,96 (д, 1H, J=1,3Гц); 4,38 (шир.с, 2H, SONH2); 3,15 (м, 1H); 2,10 (д, 3H, J=1,4Гц); 2,0 (д, 3H, J=1,3Гц); 1,25 (д, 6H, J=7Гц).
E-2-(4-изобутилфенил)-2-метилэтенсульфонамид (50)
1H-ЯМР (CDCl3): δ 7,32 (д, 2H, J=7Гц); 7,23 (д, 2H, J=7Гц); 6,15 (кв, 1H, J=1,4Гц); 5,96 (кв, 1H, J=1,3Гц); 4,35 (шир.с, 2H, SONH2); 2,45 (д, 2H, J=7Гц); 2,10 (д, 3H, J=1,4Гц); 2,0 (д, 3H, J=1,3Гц); 1,88 (м, 1H); 0,97 (д, 6H, J=7Гц).
Перечень химических названий и структур соединений по примерам 1-6 представлен в таблице I.





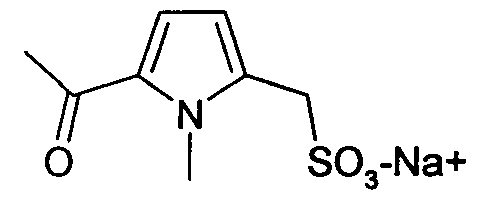


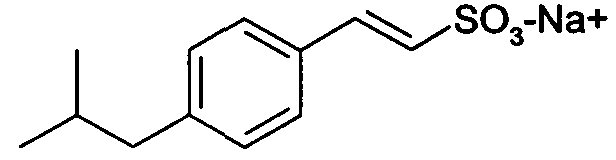
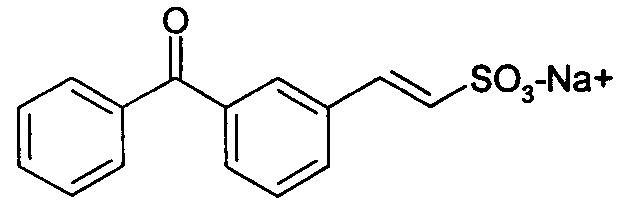

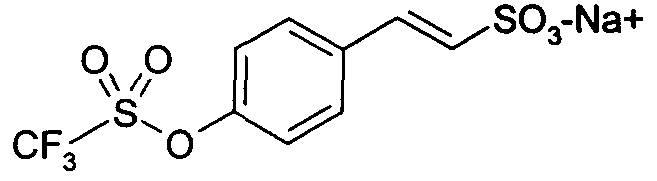
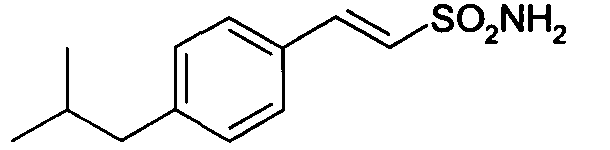

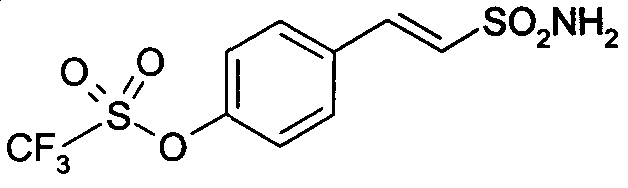











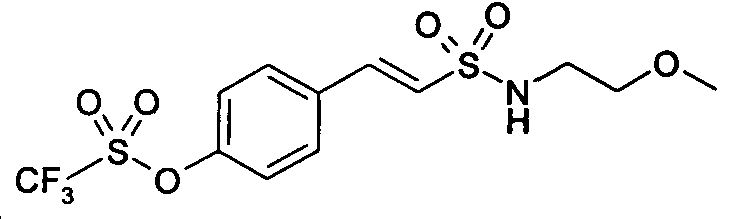

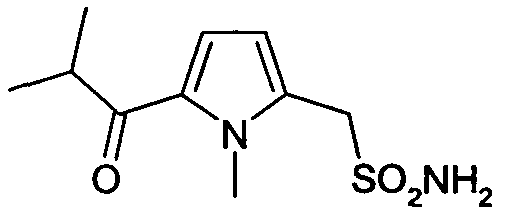
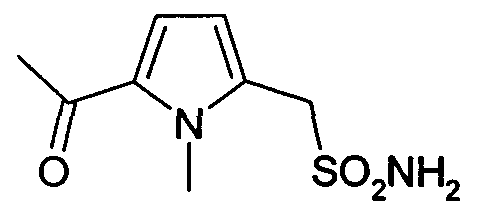






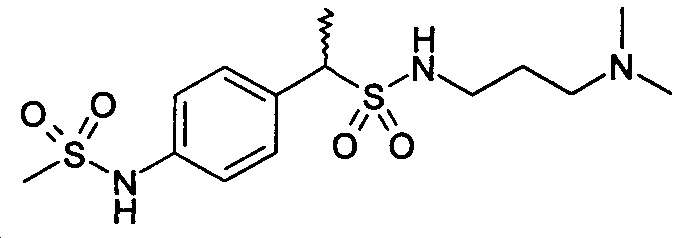









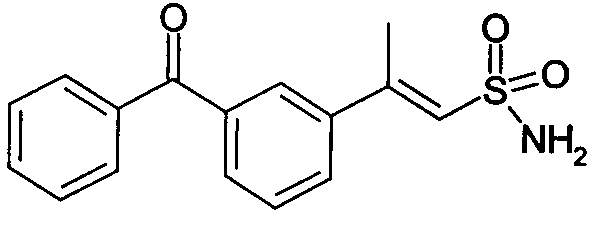

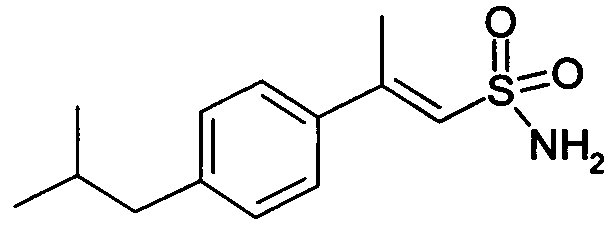
ТАБЛИЦА II
Ингибирование (%) хемотаксиса PMN человека, индуцируемого IL-8 (100 нг/мл)

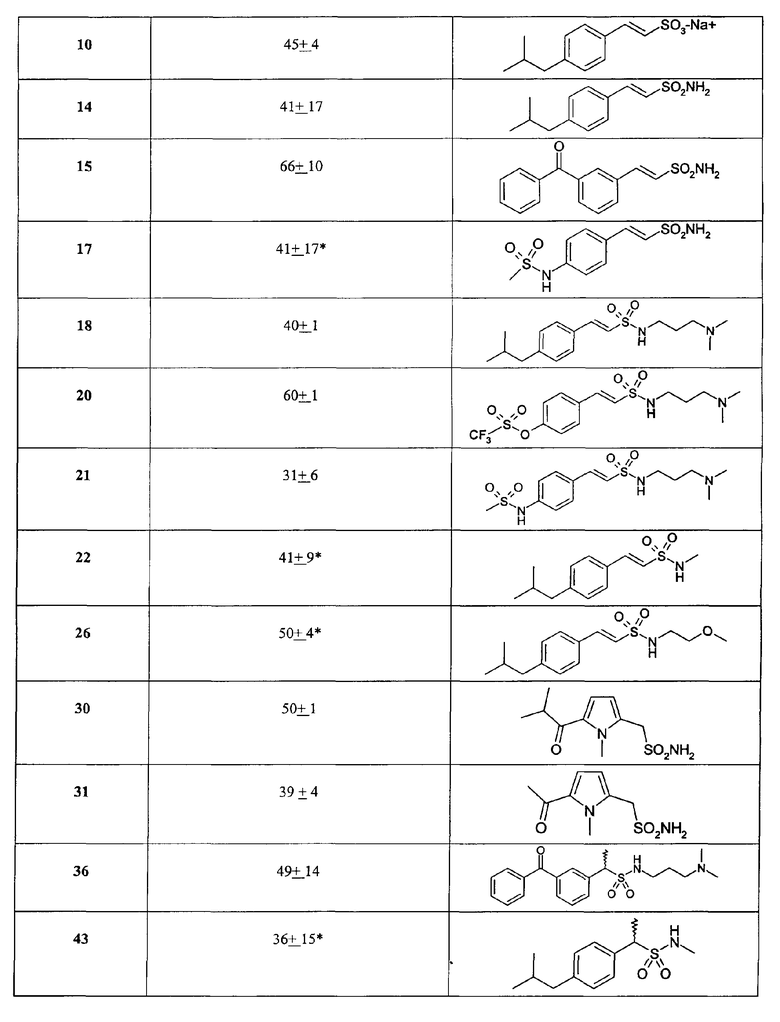
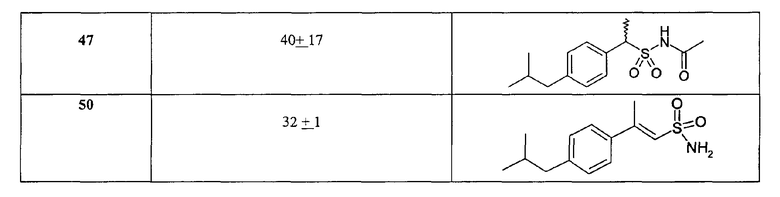
*Соединения исследованы при с = 10-7
| название | год | авторы | номер документа |
|---|---|---|---|
| (R)-АРИЛАЛКИЛАМИНОПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2006 |
|
RU2458051C2 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2317075C2 |
| ЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ СОЛИ ОМЕГА-АМИНОАЛКИЛАМИДОВ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 2002 |
|
RU2291857C2 |
| ПРОИЗВОДНЫЕ 2-ИМИНОПИРРОЛИДИНА | 2002 |
|
RU2270192C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2170737C2 |
| НОВОЕ СОЕДИНЕНИЕ ИНДОЛИНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2003 |
|
RU2318808C2 |
| (R)-2-АРИЛПРОПИОНАМИДЫ, ПОЛЕЗНЫЕ ПРИ ИНГИБИРОВАНИИ ИЛ-8-ИНДУЦИРОВАННОГО ХЕМОТАКСИСА НЕЙТРОФИЛОВ, СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ХЕМОТАКСИС НЕЙТРОФИЛОВ, ИНДУЦИРОВАННЫЙ ИНТЕРЛЕЙКИНОМ-8 | 2001 |
|
RU2273630C2 |
| N-(2-АРИЛПРОПИОНИЛ)СУЛЬФОНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1999 |
|
RU2255084C2 |
Описываются применение сульфоных кислот и их производных формулы (I) и фармацевтически приемлемых солей указанных соединений (значения радикалов см. в формуле изобретения) для получения лекарственного средства для ингибирования хемотаксиса PMN человека, индуцированного IL-8, и сульфоновые кислоты и их производные формулы (I). Описываются три способа получения этих соединений и фармацевтическая композиция на их основе. Соединения формулы (I) обладают ингибирующим действием хемотаксиса PMN человека, индуцируемого IL-8. 6 н. и 5 з.п. ф-лы, 2 табл.


и фармацевтически приемлемых солей указанных соединений,
где Ar означает фенильную группу, замещенную одним заместителем, независимо выбранным из группы, включающей С1-С4-алкил, бензоил, либо Ar означает пирролил, замещенный метилом и ацетилом, либо Ar означает 4'-трифторметансульфонилоксифенил;
Х означает либо -СН2-, либо -СН(СН3)-группу, или этиленовую группу формулы (II)
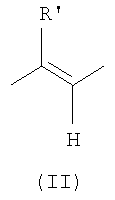
в Е-конфигурации, где R' означает Н или СН3;
Y выбран из О (кислорода) и NH; и
когда Y означает О (кислород), R означает Н (водород);
когда Y означает NH, R выбран из группы, включающей Н, С1-С5-алкил, С1-С5-ацил, остаток формулы -CH2-CH2-Z-(CH2-CH2O)nR", где R" означает С1-С5-алкил, n равно 0, и Z означает кислород или остаток формулы - (СН2)n-NRaRb, где n равно 0-5 и каждый из Ra и Rb, которые могут быть одинаковыми или различными, означают С1-С6-алкил,
при получении лекарственного средства для ингибирования хемотаксиса PMN человека, индуцированного IL-8.
Н, С1-С5-алкил, C1-C5-ацил;
остаток формулы - (CH2)n-NRaRb, где n равно 3, и группа NRaRb означает N,N-диметиламин.
1-{4-изобутилфенил)этансульфоновая кислота,
натриевая соль (-)-1-[4-(2-оксопирролидин-1-ил)фенил]этансульфоновой кислоты,
натриевая соль (+)-1-[4-(2-оксопирролидин-1-ил)фенил]этансульфоновой кислоты,
натриевая соль (+)-1-{4-[(фенилсульфонил)окси]фенил}этансульфоновой кислоты,
натриевая соль (-)-1-{4-[(фенилсульфонил)окси]фенил}этансульфоновой кислоты,
Е-2-(4-изобутилфенил)этенсульфоновая кислота,
Е-2-(3-бензоилфенил)этенсульфоновая кислота,
Е-2-(4-метансульфониламинофенил)этенсульфоновая кислота,
Е-2-(4-трифторметансульфонилоксифенил)этенсульфоновая кислота,
Е-2-(4-изобутилфенил)этенсульфонамид,
Е-2-(3-бензоилфенил)этенсульфонамид,
Е-2-[4-(трифторметансульфонилокси)фенил]этенсульфонамид,
Е-2-[4-(метансульфониламино)фенил]этенсульфонамид,
Е-2-(4-изобутилфенил)этен-N-(N,N-диметиламинопропил)сульфонамид,
Е-2-(3-бензоилфенил)этен-N-(N,N-диметиламинопропил)сульфонамид,
Е-2-[4-(трифторметансульфонилокси)фенил]этен-N-(N,N-диметиламинопропил)сульфонамид,
Е-2-[4-(метансульфониламино)фенил]этен-N-(N,N-диметиламинопропил)сульфонамид,
Е-2-(4-изобутилфенил)этен-N-метилсульфонамид,
Е-2-(3-бензоилфенил)этен-N-метилсульфонамид,
Е-2-[4-(трифторметансульфонилокси)фенил]этен-N-метилсульфонамид,
Е-2-[4-(метансульфониламино)фенил]этен-N-метилсульфонамид,
Е-2-(4-изобутилфенил)этен-N-(2"-метоксиэтил)сульфонамид,
Е-2-(3-бензоилфенил)этен-N-(2"-метоксиэтил)сульфонамид,
Е-2-[4-(трифторметансульфонилокси)фенил]этен-N-(2"-метоксиэтил)сульфонамид,
Е-2-[4-(метансульфониламино)фенил]этен-N-(2"-метоксиэтил)сульфонамид,
(1-метил-5-изобутирилпирролил)-1-метансульфонамид,
(1-метил-5-ацетилпирролил)-1-метансульфонамид,
1-(4-изобутилфенил)этансульфонамид,
1-(3-изопропилфенил)этансульфонамид,
1-(4-изобутилфенил)этан-N-(N,N-диметиламинопропил)сульфонамид,
1-(3-бензоилфенил)этан-N-(N,N-диметиламинопропил)сульфонамид,
1-[4-(трифторметансульфонилокси)фенил]этан-N-(N,N-диметиламинопропил)сульфонамид,
1-[4-(метансульфониламино)фенил]этан-N-(N,N-диметиламинопропил)сульфонамид,
1-(4-изобутилфенил)этан-N-(2-метоксиэтил)сульфонамид,
1-(3-бензоилфенил)этан-N-(2-метоксиэтил)сульфонамид,
1-[4-(трифторметансульфонилокси)фенил]этан-N-(2-метоксиэтил)сульфонамид,
1-[4-(метансульфониламино)фенил]этан-N-(2-метоксиэтил)сульфонамид,
1-(4-изобутилфенил)этан-N-метилсульфонамид,
1-(3-бензоилфенил)этан-N-метилсульфонамид,
1-[4-(трифторметансульфонилокси)фенил]этан-N-метилсульфонамид,
1-[4-(метансульфониламино)фенил] этан-N-метилсульфонамид,
1-[4-изобутилфенил]этан-N-ацетилсульфонамид,
Е-2-(3-бензоилфенил)-2-метилэтенсульфонамид,
Е-2-(3-изопропилфенил)-2-метилэтенсульфонамид,
Е-2-(4-изобутилфенил)-2-метилэтенсульфонамид,
и фармацевтически приемлемые соли указанных соединений.
этансульфонамиды в виде отдельных (-) или (+) энантиомеров.

где J означает Н или СОСН3, с подходящим окислительным агентом, таким как H2O2, HClO или пероксикислота, такая как м-хлорпербензойная кислота.
включающий взаимодействие соответствующего сульфонилгалогенида, такого как сульфонилхлорид, с одним или двумя эквивалентами амина формулы NH2R, где R представляет собой метил, метоксиэтил или диметиламинопропил, или с одним эквивалентом гидроксида натрия с получением соответствующих этенсульфоновых кислот.
где X означает -СН2-, J означает Н, с подходящим N-бромимидом, таким как N-бромфталимид, и последующее метилирование и окисление атома серы с последующим снятием защиты сульфонамидного производного.
| Нагнетательный гидропульсор | 1929 |
|
SU24710A1 |
| Облицовка комнатных печей | 1918 |
|
SU100A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| BULL CHEM SOC JPN, №64, №4, 1991, 1431-1433. | |||

