ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к N-(2-арилпропионил)амидам, способу их получения и их фармацевтическим композициям, полезным при профилактике и лечении повреждения ткани, обусловленного усиленным пополнением полиморфоядерных нейтрофилов (PMN лейкоцитов) в местах воспаления. В частности, изобретение относится к R-энантиомерам N-(2-арилпропионил)амидов для применения при ингибировании хемотаксиса нейтрофилов, индуцированного ИЛ-8 (IL-8).
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Хемокины образуют семейство цитокинов низкой молекулярной массы, непосредственно вовлеченное в воспалительную ответную реакцию, в перемещение иммунных клеток и в направленную миграцию клеточных элементов. Термин «хемокины», который является сокращением слов хемотактические цитокины, придает большое значение типичной биологической функции этих медиаторов клетки.
Хемокины делят на два подвида, которые отличаются по последовательностям аминокислот СС и СХС, содержащих два цистеиновых остатка, постоянно присутствующих в N-концевой части белка. В одном случае, например в случае моноцитного белка-1-хемоаттрактанта (МСР-1), два цистеиновых остатка являются смежными, в другом случае, например в случае интерлейкина-8 (ИЛ-8) и некоторых его близкородственных аналогов (GRO-α,β,γ, ENA-78, NAP-2, GCP-2), вторая аминокислота расположена между двумя цистеинами.
С функциональной точки зрения хемокины отличаются от других цитокинов клеточной специфичностью их действия: каждый из них регулирует в специфическом пути миграцию и выполняемые функции одного вида клеток. Так, если МСР-1 влияет на движения и управляет движениями моноцитов, ИЛ-8 играет важную роль специфичного для нейтрофилов фактора-хемоаттрактанта. Подтверждение этого обеспечивается присутствием высоких концентраций ИЛ-8 в местах воспаления и в окружающих жидкостях, обнаруженных во время многих острых заболеваний, опосредованных нейтрофилами, а также предотвращением степени тяжести повреждения ткани и пониженной инфильтрацией нейтрофилов, наблюдаемых после введения антител против ИЛ-8 во время экспериментов, проводимых на животных моделях, представляющих нейтрофилзависимые заболевания. Типичными клиническими состояниями являются повреждения, вызванные церебральной реперфузией, и повреждение, вызванное ишемией и реперфузией миокарда.
Указанные наблюдения подтверждают гипотезу, что ИЛ-8 составляет основной медиатор повреждения ткани, вызванного нейтрофилами, в такой степени, что можно предположить, что ИЛ-8 является оптимальной мишенью для терапевтических вмешательств, направленных на устранение острых воспалительных состояний, опосредованных нейтрофилами (N. Mukaida et al., Inflammation Research 47 (Suppl.3) S151, 1998). Для этой цели в качестве альтернативы использованию антител против ИЛ-8 могут представлять большой интерес и клиническую пригодность вещества с низкой молекулярной массой, которые сами, включаясь в межклеточный и внутриклеточный циклы передачи сигнала, могут быть способны ингибировать миграцию нейтрофилов человека, стимулированную ИЛ-8 и его близкородственными аналогами в очень специфическом пути.
Недавно в РСТ/ЕР/9907740 описаны N-ацилсульфонамиды (R)-2-арилпропионовых кислот, оказывающие ингибирующее действие на хемотаксис нейтрофилов, стимулированный ИЛ-8, независимо от воспалительных процессов, связанных с ингибированием циклооксигеназы (СОХ-1 и/или СОХ-2).
С другой стороны, может оказаться, что ингибирование синтеза простагландинов (ПГ), характерное для (S)-энантиомеров 2-арилпропионовых кислот и некоторых их производных, оказывает отрицательное влияние на динамику нейтрофилзависимого процесса воспаления, стимулированного ИЛ-8, так что происходит обострение самого заболевания. В указанных случаях, при ингибировании синтеза ПГ, эндогенный фактор, ПГЕ2, который регулирует синтез фактора-альфа некроза опухоли (TNF-α), исчезает. Следовательно, в соревновании с самим ИЛ-8 TNF-α может содействовать, вместе с цитокинами ИЛ-6 и ИЛ-1 и с молекулами адгезии (Е-селектин, ICAM-1 и С-реактивный белок), обострению степени и тяжести повреждения ткани во время острого инфаркта миокарда (R. Pudil et al., Clin. Chim. Acta, 280, 127, 1999).
Кроме того, оказывается, что известный (R)-2-(4-изобутилфенил)пропионамид (РСТ/ЕР/9907740) является активным при предотвращении и ингибировании хемотаксиса лейкоцитов человека, индуцированного ИЛ-8, свойство, полностью отсутствующее у (S)-энантиомера (таблица 1).
Кроме того, то же самое соединение и соответствующий (R)-N-метил-2-(4-изобутилфенил)пропионамид, хотя и менее сильнодействующий [25+9% ингибирования при концентрации 10-8 М], в качестве ингибитора хемотаксиса лейкоцитов, стимулированного ИЛ-8 (10 нг/мл), характеризуются тем, что они негативно регулируют продуцирование TNF-α (стимулированного в мышиных макрофагах H2O2 и липополисахаридами), а также тем, что они не ингибируют синтез ПГЕ2 в макрофагах после стимуляции липополисахаридами (LPS) при 1 мкг/мл. Вместо этого в таких же экспериментальных условиях S-кетопрофен (взятый в качестве типичного примера (S)-энантиомера 2-арилпропионовых кислот, ингибиторов СОХ) стимулирует в макрофагах амплификацию синтеза TNF-α, индуцируемого LPS с процентным отклонением 300% для синтеза и высвобождения TNF-α; фактически в присутствии контрольных величин цитокина, одного присутствующего в инкубационной среде, меньших, чем поддающийся детектированию минимум (20 пг/мл), величины 10±5 нг/мл обнаружены в присутствии LPS, тогда как величины 39±5 нг/мл обнаружены в присутствии LPS и 10-5 М S-кетопрофена (Ghezzi et al., J. Pharmacol. Exp. Therap., 287, 969-974, 1998). Позднее было обнаружено, что указанное заметное повышение в высвобождении TNF-α является прямым следствием стимуляции мРНК TNF-α S-кетопрофеном (Р.Mascagni et al., Eur. Cytokine Netw., 11:185-192, 2000).
Амиды 2-арилпропионовых кислот с аминоспиртами описаны в ES 500990 и в ES 2007236 для получения N-(α-гидроксиэтил)-d,1-2-(4-изобутил)пропионамидов.
Известны также амиды ибупрофена с L- и D,L-аминокислотами (W. Kwapiszewski et al., Ada Pol. Pharm., 42, 545, 1985) и более широко амиды рацематов и S-энантиомеров 2-арилпропионовых кислот с глицином (Р Singh et al., Indian J. Chem. Sect. В, 29В, 551, 1990) и со следующими аминокислотами: лизином, глутаминовой кислотой и аспарагиновой кислотой [А.Reiner, патент США №4341798]. Более часто указанные соединения были оценены в виде смесей диастереомеров без возможности определить вклад индивидуальных диастереоизомеров.
Амиды энантиомеров 2-арилпропионовых кислот с таурином, глутамином, орнитином, аргинином, глутаминовой кислотой, аспарагиновой кислотой, серином и аланином хорошо известны в качестве мочевых метаболитов этих кислот у различных видов животных (R.I.Jeffrey et al., Xenobiotica, 4, 253, 1978 и цитированные в ней ссылки).
Другие амиды, изученные в качестве пролекарств 2-арилпропионовых кислот, были описаны S. Biniecki et al., PL 114050, H.A.Kguen et al., Arzneim-Forsh, 46, 891, 1986 и G.L.Levitt et al., Russ. J. Org. Chem., 34, 346, 1998. Такие амиды наделены довольно высокой противовоспалительной активностью, связанной с пониженными побочными эффектами и хорошей переносимостью на желудочно-кишечном уровне, которые, как считают, компенсируют потерю эффективности, наблюдаемую по сравнению с их предшественниками.
Потеря всякой остаточной фибринолитической активности была описана для (±)-ибупрофена и других нестероидных противовоспалительных агентов, таких как индометацин, флуфенаминовая кислота и мефенаминовая кислота, после превращения в соответствующие амиды с 2-аминометилпиридином (G.Orzalesi et al., Progress in Fibrinolysis and Thrombolysis, 3, 483, 1978).
В сравнительном исследовании противовоспалительные, аналгезирующие и жаропонижающие свойства, воздействие на поведение и острую токсичность у мышей оценивали для различных амидов ибупрофена, кетопрофена (оба как рацематы) и 3-бензоилфенилуксусной кислоты (R.C.W. Spickett et al., J. Med. Chem. Chim. Ther., 11, 7, 1976). Сравнение проводится с простыми амидами(-CONH2) и их N-этил- и N-диметилпроизводными, соответствующими уреидами и тиоуреидами, а также анилидами и некоторыми циклическими амидами, такими как амиды с 2-аминотиазолидином, 2-аминотиазолом, 2-амино-4-метилпиридином и 1-фенил-2,3-диметил-4-аминопиразолом. Фармакологическое исследование привело к отбору и разработке (R,S)-2-[3-бензоилфенилпропионамидо]-4-метилпиридина, известного также под названием пиркетопрофен (A.Gallardo, патент Великобритании 1436502].
Кроме того, недавно было описано использование R-2-арилпропионовых кислот в качестве лекарств для лечения колоректальных опухолей и муковисцидоза (патент США 5955504 и патент США 5981592).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Теперь обнаружено, что амиды, структурно связанные с (R)-2-(4-изобутилфенил)пропионамидом, характеризующиеся подходящими заместителями, проявляют неожиданно свойства ингибирования хемотаксиса, индуцированного ИЛ-8.
Примерами таких заместителей являются остатки α-аминокислоты, выбранной из группы, состоящей из глицина, L-аланина, D-аланина и L-серина, группы формулы -СН2-СН2ОН, CH2CH2O-CH2-CH2OH или ароматические и гетероароматические радикалы, такие как фенил и пиридил.
Соединения получают взаимодействием (в присутствии подходящего основания) хлорангидрида (R)-2-(4-изобутилфенил) пропионовой кислоты с подходящим амином и с метиловыми эфирами α-аминокислот, ранее указанных.
В последнем случае последующее омыление карбоксиэфиров в нерацемизирующих условиях давало возможность получить свободные кислоты индивидуальных амидов. Амиды изобретения, как таковые или после омыления, имеют хорошие характеристики растворимости.
Было доказано, что свойства ингибирования хемотаксиса, индуцированного ИЛ-8, неожиданно зависят от стереохимии и от стерического, электронного и полярного влияний заместителей на амидном азоте. Фактически было отмечено, например, что амиды с аминокислотами ряда L являются более активными, чем амиды с аминокислотами ряда D. Кроме того, в случае ароматических или гетероароматических амидов присутствие заместителей на ароматическом кольце сильно влияет на активность. Кроме того, полярные взаимодействия внутримолекулярного типа, например, внутримолекулярные водородные связи, имеют иногда доказанную критичность для фармакологической активности.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В следующих абзацах представлены определения известных химических остатков, которые составляют соединения изобретения и предназначаются для использования единообразно по всему описанию и формуле изобретения, если только в противном случае специально представленные определения не предусматривают более широкое определение.
«С1-С4-Алкил», или «С1-С5-алкил», или «C1-С6-алкил» относится к одновалентным алкильным группам, имеющим от 1 до 4, или от 1 до 5, или от 1 до 6 атомов углерода. Примерами этих терминов являются такие группы, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и тому подобное. «Арил» относится к ненасыщенной ароматической карбоциклической группе с атомами углерода от 6 до 14, имеющей одно кольцо (например, фенил) или несколько конденсированных колец (например, нафтил). Предпочтительный арил включает фенил, бифенил, нафтил, фенантренил и тому подобное. «Алкенил» относится к алкенильным группам, предпочтительно имеющим от 2 до 5 атомов углерода и имеющим один или несколько центров алкенильной ненасыщенности. Предпочтительные алкенильные группы включают этенил (-СН=СН2), н-2-пропенил (аллил, -СН2СН=СН2) и тому подобное.
«Замещенный или незамещенный»: если только в противном случае такие термины не ограничены определением отдельного заместителя, выше представленные группы, подобные «алкильной», «алкенильной», «арильной» группам и т.д. могут, необязательно, быть замещены заместителями от 1 до 5, выбранными из группы, состоящей из «С1-С6-алкила», «С1-С6-алкиларила», «С1-С6-алкилгетероарила», «С2-С6-алкенила», первичных, вторичных или третичных аминогрупп или четвертичных аммониевых групп, «ацила», «ацилокси», «ациламино», «аминокарбонила», «алкоксикарбонила», «арила», «гетероарила», карбоксила, циано, галогена, гидрокси, меркапто, нитро, сульфокси, сульфонила, алкокси, тиоалкокси, тригалогенметила и тому подобное. В пределах объема настоящего изобретения указанное «замещение» означает, что оно включает также случаи, когда соседние заместители подвергаются циклизации, особенно, когда включены вицинальные функциональные заместители, таким образом образуя, например, лактамы, лактоны, циклические ангидриды или циклоалканы, а также ацетали, тиоацетали, аминали, образованные циклизацией, например, при необходимости получить защитную группу.
«Фармацевтически приемлемые соли» относятся к солям или комплексам идентифицированных ниже соединений формулы I, которые сохраняют требуемую биологическую активность. Примеры таких солей включают, но не ограничиваются ими, кислотно-аддитивные соли, образованные с неорганическими кислотами (например, хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, азотной кислотой и тому подобное), и соли, образованные с органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, фумаровая кислота, малеиновая кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памоевая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота и полигалактуроновая кислота. Указанные соединения можно также ввести в виде фармацевтически приемлемых четвертичных солей, известных специалисту в данной области. Примеры солей включают также основно-аддитивные соли, образованные с неорганическими основаниями, такими как гидроксид натрия, и с органическими основаниями, такими как трометамин, L-лизин, L-аргинин и тому подобное.
Настоящее изобретение относится к амидам R-энантиомеров 2-арилпропионовых кислот формулы (I)
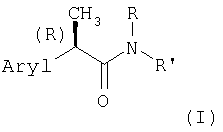
и их фармацевтически приемлемым солям,
где Aryl представляет замещенную или незамещенную арильную группу;
R представляет Н, С1-С4-алкил, аллил, пропаргил, CH2-CO2H
или (СН2)2-CO2Н;
R' представляет:
- аминокислотный остаток, состоящий из неразветвленного или разветвленного С1-С6-алкила, алкенила, циклоалкила, фенилалкила, замещенного одной или несколькими карбоксигруппами CO2Н;
- аминокислотный остаток, состоящий из неразветвленного или разветвленного С1-С6-алкила, алкенила, циклоалкила, фенилалкила, замещенного одной или несколькими карбоксигруппами CO2Н и гетероатомом, выбранным из кислорода или серы;
- остаток формулы -СН2-СН2Х-(CH2-CH2O)nR, где R имеет значения, указанные выше; n равно целому числу от 0 до 5, тогда как Х представляет кислород или серу;
- остаток формулы (R)- или (S)-СН(СН3)-СН2-O-СН2-СН2-ОН;
- остаток формулы OR, где R имеет значения, указанные выше;
- остаток формулы (III)
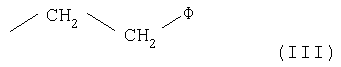
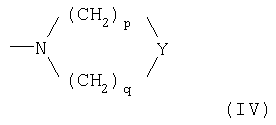
где Ф представляет 2-(1-метилпирролидил), 2-пиридил, 4-пиридил, 1-имидазолил, 4-имидазолил, 1-метил-4-имидазолил, 1-метил-5-имидазолил или группу NRaRb, где каждый из Ra и Rb, которые могут быть одинаковыми или разными, представляет С1-С6-алкил или гидроксиалкил -(СН2)m-ОН, где m равно целому числу 2 или 3, или же Ra и Rb вместе с атомом N, с которым они связаны, образуют гетероцикл из 3-7 членов формулы (IV)
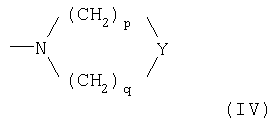
где
- Y представляет прямую связь, СН2, О, S или N-Rc, причем Rc представляет Н, С1-С6-алкил, гидроксиалкил (СН2)m-ОН, остаток -(СН2)m-Ar', где Ar' представляет арил, гетероарил, циклоалифатический и/или гетероциклоалифатический остаток, m' равно нулю или целому числу от 1 до 3, р и q, каждый независимо, представляет целое число от 1 до 3;
- гетероарил, выбранный из группы, состоящей из 2-пиридила или 4-пиридила, 2-пиримидинила или 4-пиримидинила; 2-пиразинила, 5-метил-2-пиразинила; 3-1,2,4-тиазинила; 3-1,2,4-тиазолила, 3-1-бензил-1,2,4-тиазолила; 2-1,3-тиазолидинила, 2-1,3-тиазолила, 1,3-оксазолила, 3-изоксазолила, 4-дигидро-З-оксоизоксазолила, 5-метилизоксазол-4-ила, 2-имидазолила, 4-имидазолил-5-карбоксамида и 2-имидазолил-4,5-дикарбонитрила, 5-инданила, 5-индазолила, 7-азаиндол-3-ила, 2-, 3-, или 4-хинолинила;
для применения в качестве агентов, ингибирующих хемотаксис нейтрофилов, индуцированный интерлейкином-8.
Настоящее изобретение далее относится к новым (R)-энантиомерам 2-арилпропионамидов формулы (Ia)
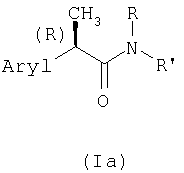
и их фармацевтически приемлемым солям,
где Aryl представляет фенильную группу, замещенную группой, выбранной из изопропила, ацетила, (2'',6''-дихлорфенил)амино, α-гидроксиизопропила, (R,S)-α-гидроксиэтила и его индивидуальных R- и S-изомеров, (R,S)-α-гидроксибензила и его индивидуальных R- и S-изомеров и (R,S)-(α-метилбензила) и его индивидуальных R- и S-изомеров; (R,S)-α-гидрокси-α-метилбензила и его индивидуальных R- и S-изомеров;
R представляет Н, C1-C4-алкил, аллил, пропаргил, CH2-CO2H или (СН2)2-CO2Н;
R' представляет:
- аминокислотный остаток, состоящий из неразветвленного или разветвленного С1-С6-алкила, алкенила, циклоалкила, фенилалкила, замещенного одной или несколькими карбоксигруппами CO2H;
- аминокислотный остаток, состоящий из неразветвленного или разветвленного C1-С6-алкила, алкенила, циклоалкила, фенилалкила, замещенного одной или несколькими карбоксигруппами CO2H и гетероатомом, выбранным из кислорода или серы;
- остаток формулы -СН2-СН2Х-(CH2-CH2O)nR, где R имеет значения, указанные выше; n равно целому числу от 0 до 5, тогда как Х представляет кислород или серу;
- остаток формулы (R)- или (S)-CH(СН3)-CH2-O-CH2-CH2-OH;
- остаток формулы OR, где R имеет значения, указанные выше;
- остаток формулы (III)
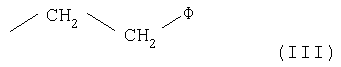
где Ф представляет 2-(1-метилпирролидил), 2-пиридил, 4-пиридил, 1-имидазолил, 4-имидазолил, 1-метил-4-имидазолил, 1-метил-5-имидазолил или группу NRaRb, где каждый из Ra и Rb, которые могут быть одинаковыми или разными, представляет С1-С5-алкил или гидроксиалкил -(CH2)m-OH, где m равно целому числу 2 или 3, или же Ra и Rb вместе с атомом N, с которым они связаны, образуют гетероцикл из 3-7 членов формулы (IV)
где
- Y представляет прямую связь, СН2, О, S или N-Rc, причем Rc представляет Н, С1-С5-алкил, гидроксиалкил (CH2)m-OH, остаток -(СН2)m-Ar', где Ar' представляет арил, ароматический гетероарил, циклоалифатический и/или гетероциклоалифатический остаток, m' равно нулю или целому числу от 1 до 3, каждый р и q, независимо друг от друга, представляет целое число от 1 до 3;
- гетероарил выбран из группы, состоящей из 2-пиридила или 4-пиридила, 2-пиримидинила или 4-пиримидинила; 2-пиразинила, 5-метил-2-пиразинила; 3-1,2,4-тиазинила, 3-1,2,4-тиазолила, 3-1-бензил-1,2,4-тиазолила; 2-1,3-тиазолидинила, 2-1,3-тиазолила, 1,3-оксазолила, 3-изоксазолила, 4-дигидро-3-оксоизоксазолила, 5-метилизоксазол-4-ила, 2-имидазолила, 4-имидазолил-5-карбоксамида и 2-имидазолил-4,5-дикарбонитрила, 5-инданила, 5-индазолила, 7-азаиндол-3-ила, 2-, 3-, или 4-хинолинила.
Примерами арильного остатка Ar' являются фенил, дифенилметил, 4,4'-дифтордифенилметил; примерами гетероарильных ароматических остатков являются пиридил, имидазолил; примерами циклоалифатических или гетероциклоалифатических остатков являются циклогексил, циклопентил, 4-морфолил и 1-пиперидил.
Изобретение далее относится к соединениям, указанным здесь выше, для применения в качестве лекарственных средств.
Термин «арильная группа» предпочтительно означает фенил, необязательно замещенный от одного до трех заместителями, которые являются одинаковыми или разными и выбраны из атомов галогена, С1-С4-алкила, С1-С4-алкокси, гидрокси, С1-С7-ацилокси, циано, нитро, амино, С1-С3-ациламино, галоген-С1-С3-алкила, гидрокси-С1-С3-алкила, галоген-С1-С3-алкокси, гидрокси-С1-С3-арилалкила, бензоила или известных остатков известных противовоспалительных 2-арилпропионовых кислот, таких как ибупрофен, кетопрофен, супрофен, пирпрофен, фенопрофен. Арильная группа более предпочтительно выбрана из группы, состоящей из фенила, 4-метилфенила, 3-изопропилфенила, 4-метоксифенила, 4-ацетоксифенила, 4-бензоилоксифенила, 4-гидроксифенила, 4-изобутилфенила, 4-(2,2-диметил)винилфенила, (СН3)2С=СН-С6Н4-, 4-(2-метил)аллилфенила, 3-бензоилфенила, 3-феноксифенила, 3-бензилфенила, 3-С6Н5-СН(ОН)-фенила, 5-бензоилтиен-2-ила, 4-тиеноилфенила, 1-оксо-2-изоиндолинилфенила, 2-фтор-4-бифенилила, 6-метоксинафтила, 5-бензоил-2-ацетоксифенила, 5-бензоил-2-гидроксифенила, 3-α-метилбензилфенила, 3-гидроксипропилфенила, 3-гидроксиэтилфенила.
Аминокислотный остаток R', имеющий указанные выше значения, предпочтительно представляет остаток L-α-аминокислоты и более предпочтительно выбран из группы, состоящей из аланина, валина, лейцина, изолейцина, норлейцина, фенилаланина, тирозина, гистидина, S-метилцистеина, S-карбоксиметилцистеина, S-2-гидроксиэтилцистеина, метионина, O-метилсерина, O-2-гидроксиэтилсерина, пролина, гидроксипролина, глутаминовой кислоты, аспарагиновой кислоты, глутамина или остатка глицина, фенилглицина, β-аланина, γ-аминомасляной кислоты, δ-аминовалериановой кислоты, цис-4-аминоциклогексанкарбоновой кислоты, транс-4-аминометилциклогексанкарбоновой кислоты, 3-амино-1,5-пентандиовой кислоты или остатка формулы (II)
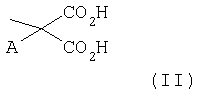
где заместитель А представляет Н, неразветвленный или разветвленный С1-С6-алкил, (CH2)ni-CO2H, где ni равно целому числу между 1 и 3, бензил, п-гидроксибензил, -CH2-O-C2H5, СН2-S-СН3, CH2-S-CH2-CO2H, причем остатки кислот, указанных выше, находятся в форме свободных кислот или солей, как указывается ниже, или в форме их метиловых, этиловых и аллиловых сложных эфиров.
Другой целью настоящего изобретения являются также соли соединений формулы (I) с фармацевтически приемлемыми основаниями или кислотами.
В соединениях формулы (I) R более предпочтительно представляет водород и R' представляет остаток аминокислоты, такой как глицин, цис-4-циклогексанкарбоновая кислота, аминомалоновая кислота, аминометилмалоновая кислота, бензиламиномалоновая кислота, или остаток монокарбоновой или дикарбоновой L-α-аминокислоты или, кроме того, остаток дипептида, выбранного из группы, состоящей из L-аланилглицина, глицил-L-аланина и глицил-D-аланина.
Более предпочтительными являются соединения формулы (I), в которых R представляет водород и R' представляет остаток L-аланина, L-карбоксиметилцистеина, L-фенилаланина, L-лейцина, L-метионина, L-O-метилсерина, L-аланилглицина.
Особенно предпочтительными амидами формулами (I) являются амиды, где R представляет водород и R' является указанной здесь выше группой -CH2-CH2-O-(-CH2-CH2-O)n-R, n равно целому числу от 0 до 2, более предпочтительно целому числу 1.
Предпочтительными амидами формулы (I) являются также амиды, где R представляет водород и R' представляет заместитель формулы (III)
где Ф представляет основный остаток -NRaRb, такой как N,N-диметиламин, N,N-диэтиламин, N,N-диизопропиламин, 4-морфолил, 1-пиперидил, 1-пирролидил, 1-пиперазинил, 1-(4-бензил)пиперазинил, 1-(4-дифенилметил)пиперазинил, 1-(4-[4',4''-дифтордифенил)метил)пиперазинил, 1-(4-этил)пиперазинил, 1-(4-гидроксиэтил)пиперазинил.
Особенно предпочтительными монозамещенными амидами формулы (I) являются амиды, в которых гетероарил R' представляет 2- или 4-пиридил, 2- и 4-пиримидинил, 2-пиразинил, 2-1,3-тиазолил, 1-1,3-тиазолидинил или 2-имидазолидил и более предпочтительно 4-пиридил.
Конкретными примерами соединений изобретения являются:
(R)-(-)-2-(4'-изобутилфенил)-N-метилпропионамид;
(R)-(-)-2-[(4'-изобутил)фенил]-N-карбоксиметилпропионамид;
(R)-(-)-2-[(4'-изобутил)фенил]-N-метоксикарбонилметилпропионамид;
цис-(R)-2-[(4'-изобутил)фенил]-N-(4'-карбоксициклогексил)пропионамид;
транс-(R)-2-[(4'-изобутил)фенил]-N-(4'-карбоксиметилциклогексил)пропионамид;
(R,S')-2-[(4'-изобутил)фенил]-N-(2-метоксикарбонилэтил)пропионамид;
(R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамид;
(R,S')-2-[(4'-метокси)фенил]-N-(2-карбоксиэтил)пропионамид;
(R)-N-[2'-(4''-изобутилфенил)пропаноил]-2-аминоакриловая кислота и ее метиловый эфир;
(R)-(-)-2-[(4'-изобутил)фенил]-N-(2''-гидроксиэтоксиэтил)пропионамид;
(R,S')-2-[(4''-изобутил)фенил]-N-[1'-метил-2'-(2''-гидроксиэтокси)этил]пропионамид;
(R,R')-2-[(4''-изобутил)фенил]-N-[1'-метил-2'-(2''-гидроксиэтокси)этил]пропионамид;
(R)-(-)-2-(4'-изобутилфенил)-N-(2''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-(4'-изобутилфенил)-N-(4''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(3'-бензоил)фенил]-N-(2''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(2'-гидрокси-5'-бензоил)фенил]-N-(2''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(2'-гидрокси-5'-бензоил)фенил]-N-(4''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(2'-гидрокси-5'-бензоил)фенил]-N-карбоксиметилпропионамид;
(R)-(-)-2-(4'-изобутилфенил)-N-(2''-пиразинил)пропионамид и его гидрохлорид;
(R)-(-)-2-(4'-изобутилфенил)-N-(2''-пиримидинил)пропионамид и его гидрохлорид;
(R)-(-)-2-(4'-изобутилфенил)-N-(4''-пиримидинил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(3'-изопропил)фенил]-N-карбоксиметилпропионамид;
(R,S')-(-)-2-[(3'-α-метилбензил)фенил]-N-карбоксиметилпропионамид;
(R,R')-(-)-2-[(3'-α-метилбензил)фенил]-N-карбоксиметилпропионамид.
Для получения амидов изобретения формулы (I) используют известные способы, которые состоят во взаимодействии подходящей активированной формы R-2-арилпропионовой кислоты формулы (V) с амином формулы (VI) в нерацемизирующих условиях реакции в присутствии, если требуется, молярного избытка основания:
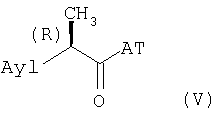
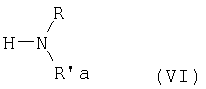
где AT в соединениях формулы (V) представляет остаток, активирующий карбоксигруппу.
Примерами активированных форм 2-арилпропионовых кислот формулы (V) с AT=Н являются соответствующие хлорангидриды (AT=Cl), имидазолиды (AT=1-имидазол), эфиры с фонолами, такими как п-нитрофенол (AT=п-NO2-C6H4O), или активированные формы, полученные взаимодействием в присутствии 1-гидроксибензотриазола (НОВТ) или карбодиимида, такого как дициклогексилкарбодиимид.
Аминами формулы (VI) являются первичные или вторичные амины, где R имеет значения, указанные выше, и R'a представляет:
- остаток эфира L-α-аминокислоты, выбранной из группы, состоящей из аланина, валина, лейцина, изолейцина, норлейцина, фенилаланина, тирозина, гистидина, S-метилцистеина, S-карбоксиметилцистеина, S-2-гидроксиэтилцистеина, метионина, O-метилсерина, O-2-гидроксиэтилсерина, пролина, гидроксипролина;
- остаток эфира глицина, фенилглицина, β-аланина, γ-аминомасляной кислоты, δ-аминовалериановой кислоты, цис-4-аминоциклогексанкарбоновой кислоты, транс-4-аминоциклогексанкарбоновой кислоты, 3-амино-1,5-пентандиовой кислоты;
- остаток малоновой кислоты формулы (II')
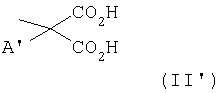
где
- заместитель А' представляет Н, неразветвленный или разветвленный С1-С5-алкил, -(CH2)niCO2-метиловый и/или -этиловый эфир, где ni представляет целое число между 1 и 3, бензил, п-гидроксибенэил, -CH2-O-C2H5, -CH2-S-СН3- и -СН2-S-СН2-СО2-метиловый эфир и/или этиловый эфир;
- остаток формулы -CH2-CH2X-(СН2-СН2О)nR, где R имеет значения, указанные ранее, или остаток формулы (R)- или (S)-СН(СН3)-СН2-O-СН2-СН2-ОН;
- остаток формулы (III)
где Ф имеет значения, указанные ранее;
- гетероарил, который имеет значения, указанные ранее.
Образование амидов формулы (I) взаимодействием активированной формы кислоты формулы (V) со вторичным или первичным амином формулы (VI) обычно проводят при комнатной температуре с использованием общепринятых протонных или апротонных растворителей, предпочтительно обезвоженных на молекулярных ситах, или их смесей. Указанные растворители включают сложные эфиры, такие как этилацетат, метилацетат и этилформиат, нитрилы, такие как ацетонитрил, неразветвленные или циклические простые эфиры, такие как диоксан, тетрагидрофуран, этиловый эфир и сульфолан, амиды, такие как диметилформамид и формамид, галогенированные растворители, такие как дихлорметан, ароматические углеводороды, такие как толуол и хлорбензол, или гетероароматические углеводороды, такие как пиридин и пиколин.
Реакции можно проводить в присутствии основания; предпочтительными неорганическими основаниями являются карбонаты и бикарбонаты щелочных и щелочноземельных металлов, такие как тонкоизмельченные карбонат калия, бикарбонат калия и карбонат магния или карбонат кальция.
Полученный таким образом продукт формулы (Ia):
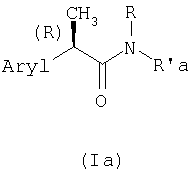
где Aryl, R и R'а имеют значения, описанные ранее, как любое соединение формулы (I), если требуется, можно превратить в другой продукт формулы (I) посредством удаления любых защитных групп, которые могут присутствовать в соединениях формулы (Ia), и/или селективным гидролизом групп сложных эфиров. Особенно предпочтительной сложноэфирной группой, наряду с обычными метильными и этильными группами, является аллильная группа, которую можно удалить в очень селективных и в нерацемизирующих условиях, например, превращением аллильной группы в морфолин, который в присутствии Pd(0), как катализатора, действует в качестве переносчика водорода и в качестве нуклеофильного акцептора, по процедуре, описанной в J. Org. Chem., 54, 751 (1989). При желании соединение формулы (Ia), где R'а представляет остаток α-аминокислоты, β-замещенной свободными или превращенными в простые эфиры тиольными группами, или гидрокси, свободной или этерифицированной алифатической кислотой или сульфоновой (метансульфоновой, бензолсульфоновой, п-толуолсульфоновой) кислотой, можно подвергнуть β-элиминированию указанных заместителей, так чтобы получить, обработкой избытком BBr3, соединения формулы (I), где R' представляет 2,3-дегидроаминокислоту.
Наконец, как указано выше, соединение формулы (Ia) можно превратить в родственный продукт формулы (I) посредством способов образования солей первичных, вторичных или третичных основных групп, присутствующих в соединениях формулы (Ia), с исполазованием для данной цели фармацевтически приемлемых кислот или образованием соли любых карбоксильных или сульфоновых остатков, которые могут присутствовать в соединениях формулы (Ia), с фармацевтически приемлемыми основаниями.
Примерами фармацевтически приемлемых кислот являются одноосновные и многоосновные минеральные кислоты, такие как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота; или одноосновные или многоосновные органические кислоты, такие как уксусная кислота, бензойная кислота, винная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, миндальная кислота, щавелевая кислота и малоновая кислота.
Примерами фармацевтически приемлемых солей являются соли с катионами щелочных или щелочноземельных металлов и, что предпочтительно, натрия и магния, и с органическими основаниями, такими как трометамин, D-глюкозамин, лизин, аргинин, тетраэтиламмоний.
R-Энантиомеры 2-арилпропионовых кислот формулы (Va)
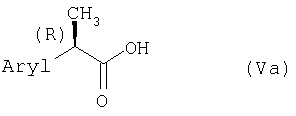
являются известными соединениями, характеризующимися относительно их S-энантиомеров тем, что они иногда являются неэффективными в качестве ингибиторов ферментов циклооксигеназ или их получают по способам, описанным подробно в приведенных ниже примерах. Предпочтительными R-2-арилпропионовыми кислотами формулы (V) являются замещенные R-2-фенилпропионовые кислоты, где замещающей группой на фенильном кольце является 2-(1-оксо-2-изоиндолинил)-, 3-фенокси-, 3-бензоил-, 4-тиеноил-, 4-изобутил, 4-гидрокси-, 4-метокси-, 5-бензоил-2-гидрокси- или где арильной группой является R-2-(5-бензоилокситиен-2-ил)-, 2-(2-фтор-4-бифенил)- и R-2-(6-метоксинафтил).
Особенно предпочтительными R-2-арилпропионовыми кислотами формулы (V) являются кислоты, в которых арильный остаток является одним из R-энантиомеров ибупрофена, кетопрофена, сурпрофена, тиапрофена, напроксена и флурбипрофена. Указанные R-2-арилпропионовые кислоты являются известными соединениями, их можно получить в виде энантиомеров посредством способов оптического разделения соответствующих рацемических 2-арилпропионовых кислот (или (R,S)-2-арилпропионовых кислот). Способы общего и стереоспецифического синтеза индивидуальных 2-арилпропионовых кислот широко описаны. Кроме того, описано превращение (R,S)-2-арилпропионовых кислот в один из энантиомеров через промежуточные 2-арил-2-пропилкетены.
Энантиоселективный синтез 2-арилпропионовых кислот в основном относится к их S-энантиомерам, но может быть модифицирован для получения R-энантиомеров посредством подходящего выбора хирального вспомогательного средства. Для использования арилалкилкетонов в качестве субстратов для синтеза α-арилалкановых кислот, см., например, В.М.Trost and J.H.Rigby, J. Org. Chem., 14, 2936, 1978; для α-арилирования кислот Meldrum см. J.T.Piney and В.A.Rowe, Tetrah. Lett., 21, 965, 1980; для использования винной кислоты в качестве хирального вспомогательного средства см. G.Castaldi et al., J. Org. Chem., 52, 3018, 1987; для использования сложных альфа-гидроксиэфиров в качестве хиральных реагентов см. R.D.Larsen et al., J. Am. Chem. Soc., 111, 7650, 1989 и патент США 4940813 и цитированные в нем ссылки.
Специфический способ получения 2-арилпропионовых кислот, где арилом является 5-бензоил-2-ОН-фенил и его сложные эфиры, описаны в патенте Италии №1283649.
Эффективный способ получения R-энантиомера указанной кислоты состоит в превращении хлорангидрида (R,S)-2-(5-бензоил-2-ацетокси)пропионовой кислоты в 2-(5-бензоил-2-ацетокси)проп-1-кетен обработкой третичным амином, таким как диметилэтиламин, который, в свою очередь, при взаимодействии с R-(-)-пантолактоном дает R-(-)-дигидро-3-гидрокси-4,4-диметил-2(3Н)-фуранон-2-ацетокси-5-бензоилфенилпропионат в виде единственного диастереоизомера (Myers et al., J. Am. Chem. Soc. 119, 6496, 1997 and Larsen R.D. et al., J. Am. Chem. Soc., 111, 7650, 1989). Последующее омыление с применением LiOH дает R-(2-(5-бензоил-2-гидроксифенил)пропионовую кислоту эффективным путем, что позволяет избежать утомительных процедур оптического разделения, например, фракционной кристаллизацией солей правовращающего и/или левовращающего дропропизина.
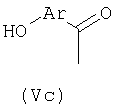
В общей процедуре получения (R)-2-арилпропионовых кислот формулы (Vb) моно- или полизамещенные гидроксиарилкетоны (Vc) подвергают взаимодействию с перфторбутансульфонилфторидом с получением перфторбутансульфонатного эфира (Vd), где n равно целому числу от 1 до 9.
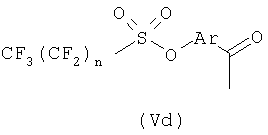
Соединения (Vd) претерпевают перегруппировку Вильгеродта с образованием, после этерификации и альфа-метилирования, производных арилпропионовых кислот (Ve), где n равно целому числу от 1 до 9 и R3 представляет С1-С4-алкил или C2-C4-алкенил.
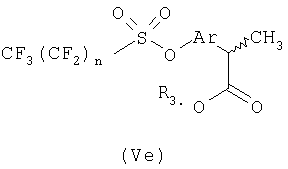
Соединения формулы Ve подвергают взаимодействию с подходящим реагентом трибутилолово-R4, где R4 представляет неразветвленный или разветвленный С1-С6-алкил, С2-С6-алкенил или алкинил, незамещенный или замещенный арильной группой, получая при этом соответствующий (R,S)-2-арилпропионат формулы (Vf).
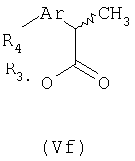
Алкенильные или алкинильные группы можно гидрировать в каталитических условиях с получением соответствующих насыщенных алкильных групп. Соединения формулы (Vf) претерпевают процесс дерацемизации, как описано выше, превращением соответствующих хлорангидридов кислот в кетены, которые взаимодействием с (R)-(-)-пантолактоном и последующим гидролизом образуют чистый R-энантиомер.
Амины формулы (VI) являются известными продуктами, большинство их является коммерчески доступными или их можно получить с использованием известных способов.
Аллиловые эфиры α-аминокислот или ω-аминокислот являются известными продуктами, коммерчески доступными или их можно получить с использованием известных способов; см. публикации Н.Waldmann and H.Kunz Liebigs Ann. Chem., 1712 (1983) или J. Org. Chem., 1989, цитированные ранее.
Для оценки соединений изобретения in vitro использовали полиморфоядерные лейкоциты (далее обозначаемые PMN), выделенные из гепаринизированной крови человека, отобранной у давших согласие здоровых взрослых людей, при помощи седиментации на декстране; мононуклеарные клетки удаляли при помощи Ficoll/Hypaque, тогда как эритроциты удаляли обработкой гипотоническими растворами. Клеточную жизнеспособность PMN вычисляли вытеснением с трипановым голубым, тогда как процент PMN на цитоцентрифугате определяли после окрашивания Diff Quinck по процедуре, описанной W.J.Ming et al., J.Immnunol., 138, 1469, 1987. В каждом из экспериментов, которые будут описаны подробно ниже, предварительные инкубации проводили при температуре 37°С, наблюдая при этом 10-минутные периоды инкубации с исследуемыми соединениями.
В экспериментах хемотаксиса и в экспериментах, предназначенных для измерения уровней иона Са++ цитозоля, использовали рекомбинантный интерлейкин-8 человека (rhIL-8, Pepro Tech); лиофилизованный белок растворяли a HBSS (сбалансированный раствор солей Ханка) при концентрации 100 мкг/мл и затем разбавляли до концентрации 10 нг/мл в экспериментах хемотаксиса, до концентрации 25-50 нг/мл при оценке внутриклеточных модификаций Са++ (т.е. [Са2+]i) и до концентрации 400 нг/мл при оценке активации тирозинкиназы.
Во время анализа хемотаксиса (по W.Falket et al., J.Immunol. Methods, 33, 239, 1980) использовали фильтры без PVP, имеющие пористость 5 мкм, и микрокамеры, изготовленные из плексигласа, подходящие для проведения репликации. Микрокамеру, состоящую из блока плексигласа, содержащего 48 лунок, имеющих емкость 25 мкл, обеспечивали крышкой, в свою очередь, содержащей 48 пор (ячеек), расположенных таким образом, чтобы в микрокамере образовывались верхние отделения, которые имели емкость 50 мкл, как только крышку надевали и завинчивали обратно на нижнюю часть.
Исследуемые соединения добавляли при одной и той же концентрации в лунки верхнего уровня, которые содержали суспензию PMNs, и в лунки нижнего уровня, которые содержали носитель, к которому был добавлен ИЛ-8 (или, при желании, другой стимулирующий агент), или иным образом.
В таблице 2 приводятся результаты оценки in vitro некоторых репрезентативных соединений формулы (I) (10-8 М), по сравнению с (R)-2-(4-изобутилфенил)пропионамидом, в качестве ингибиторов хемотаксиса, индуцированного ИЛ-8.
Результаты показывают неожиданную зависимость активности от ряда факторов, которые являются независимыми друг от друга. Стерический вклад является очевидным, являясь результатом стереохимии аминокислоты, ацилированной R-2-арилпропионовой кислотой (в рассматриваемом случае R-ибупрофеном): после ацилирования D-аланином (5) наблюдается заметное «прокинетическое» парадоксальное действие, которое совершенно отличается от ингибирующего действия на хемотаксис, показанного амидами с глицином (4) и с L-аланином (6).
Кроме того, электронное действие, индуцированное на амидном карбониле заместителями ароматического и гетероароматического типа, значительно влияет на активность: в противоположность хорошей активности 2-пиридиламида и 4-пиридиламида (10, 12), имеется слабая активность в случае анилида (9) и 3-пиридиламида (11).
Наблюдение, что при условии, когда другие заместители являются одинаковыми, присутствие в алкильном остатке R' амидов (3, 7), первичной спиртовой группы в положении γ относительно амидного карбонила сопровождается понижением в биологической активности, которая восстанавливается после ее превращения в простой эфир с остатком -СН2-СН2-ОН (8), указывает на зависимость эффективности биологического действия от участия или не участия амидного карбонила в ван-дер-ваальсовых внутримолекулярных связях.
Строгую зависимость биологического действия от абсолютной конфигурации любых заместителей R', которые могут присутствовать в соединениях формулы (I), кроме того, демонстрируют сравнением активности индивидуальных диастереоизомеров, полученных взаимодействием энантиомеров хлорангидрида 2-(4-изобутилфенил)пропионовой кислоты (ибупрофена) с энантиомерами аланина. Результаты, приведенные в таблице 3, показывают, как каждый из четырех диастереоизомеров ведет себя значимо разным образом, предположительно вследствие взаимодействий рецепторного типа, вплоть до настоящего времени неизвестного, на основе механизма действия указанных соединений.
Из фармакологической оценки энантиомеров амидов ибупрофена и кетопрофена с 4-метил-2-аминопиридином (таблица 4) можно отметить удивительное отсутствие последовательности в биологическом действии в соответствии с присутствием или отсутствием заместителей на кольце пиридина и последующим электронным или стерическим действием на амидный карбонил.
В качестве примера, (R,S')-2-(4-изобутилфенил)-(N-карбоксиэтил)пропионамид ингибирует дозозависимым путем хемотаксис, индуцированный ИЛ-8 (10 нг/мл), при диапазоне концентрации от 10-8 до 10-10 М.
Кроме того, соединения изобретения способны ингибировать повышение внутриклеточной концентрации ионов Са++, индуцированное ИЛ-8, оценку проводили в соответствии с экспериментальной моделью, описанной С.Bizzarri et al., Blood, 86, 2388, 1995. Кроме того, соединения изобретения значительно снижают индуцированную ИЛ-8 активацию тирозинкиназы.
Как ранее обсуждалось, не было обнаружено, что соединения изобретения ингибируют ферменты СОХ при оценке ex vivo по процедуре, описанной Patrignani et al., J. Pharmacol. Exper. Ther., 271, 1705, 1994. Кроме того, почти во всех случаях соединения формулы (I) изобретения не препятствуют продуцированию ПГЕ2 (PGE2), индуцированному в мышиных макрофагах стимуляцией липополисахаридами (1 мкг/мл) при диапазоне концентраций между 10-5 и 10-8 М. Ингибирование продуцирования ПГЕ2, которое, возможно, можно зарегистрировать, находится в большинстве случаев на пределе статистической значимости и часто является более низким, чем 15-20% базовой величины.
Данное незначительное ингибирование синтеза ПГЕ2 позволяет явно отличить соединения изобретения формулы (I) от S-энантиомеров 2-арилпропионовых кислот и от их амидов, которые, наоборот, вследствие заметного ингибирования синтеза ПГЕ2 образуют для самих мышиных макрофагов стимул по отношению к амплификации синтеза TNF-α. Важно, что амплификация в синтезе TNF-α способствует амплифицирующей активации нейтрофилов и поддержанию их хемотаксиса, а также образованию стимула для синтеза ИЛ-8. Для некоторых соединений изобретения формулы (I), кроме того, здесь зарегистрировано ингибирующее действие в отношении синтеза TNF-α, который обычно стимулируется в макрофагах LPS, ингибирующее действие, которое обнаружено также в отношении синтеза самих цитокинов после стимуляции Н2О2. Ввиду вышеуказанного экспериментального доказательства и участия ИЛ-8 и его близкородственных аналогов в качестве самых важных медиаторов и промоторов инфильтрации нейтрофилов при заболеваниях, таких как псориаз (R.J.Nicholoff et al., Am. J.Pathol., 138, 129, 1991), ревматоидный артрит (М.Selz et al., J.Clin. Invest., 87, 463, 1981), неспецифический язвенный колит (Y.R.Manla et al., Clin. Sci., 82, 273, 1992), острая респираторная недостаточность и идиопатический фиброз (E.J.Miller, цитированный ранее, и Р.С.Carre et al., J.Clin. Invest., 88, 1882, 1991), гломерулярный нефрит (Т.Wada et al., J. Exp. Med., 180, 1135, 1994), соединения изобретения формулы (I) используют для лечения указанных заболеваний и для профилактики и лечения повреждений, вызванных ишемией и реперфузией (N.Sekido et al., Nature, 365, 654, 1993).
Соединения изобретения вместе с обычно используемым адъювантом, носителем, разбавителем или эксципиентом можно изготовить в форме фармацевтических композиций и их единичных доз, и в такой форме их можно использовать в виде твердых композиций, таких как таблетки или наполненные капсулы, или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные ими капсулы, все они предназначены для перорального использования или в форме стерильных инъецируемых растворов для парентерального (в том числе для подкожного) использования. Такие фармацевтические композиции и их единичные дозированные формы могут включать ингредиенты в общепринятых пропорциях с добавлением или без добавления дополнительных активных соединений или элементов, и такие единичные дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента, соразмерное с используемым диапазоном назначенных суточных доз.
При использовании в качестве фармацевтических средств амиды данного изобретения обычно вводят в форме фармацевтической композиции. Такие композиции можно получить способом, хорошо известным в фармацевтической области, и они включают, по меньшей мере, одно активное соединение. Соединения данного изобретения обычно вводят в фармацевтически эффективном количестве. Количество соединения, фактически введенного, обычно определяется врачом в свете уместных обстоятельств, включая состояние пациента, которого лечат, выбора пути введения, вводимого фактического соединения, возраста, массы и восприимчивости отдельного пациента и тяжести симптомов пациента и тому подобное.
Фармацевтические композиции изобретения можно вводить различными путями, включая пероральный, ректальный, чрескожный, подкожный, внутривенный, внутримышечный и интраназальный. В зависимости от предназначенного пути доставки соединения предпочтительно изготовляют либо как инъецируемые, либо как пероральные композиции. Композиции для перорального введения могут быть в форме жидких растворов или суспензий в общем объеме или порошков в общей массе. Более обычно, однако, композиции представлены в виде единичной дозированной формы для облегчения точного дозирования. Термин «единичные дозированные формы» относится к физически дискретным единицам, подходящим в качестве унифицированных доз для людей и других млекопитающих, причем каждая единица содержит заданное количество активного материала, рассчитанного для получения требуемого терапевтического действия, в сочетании с подходящим фармацевтическим эксципиентом. Типичные единичные дозированные формы включают предварительно заполненные, предварительно замеренные ампулы или шприцы жидких композиций или пилюли, таблетки, капсулы и тому подобное в случае твердых композиций. В таких композициях амидное соединение обычно является минорным компонентом (приблизительно от 0,1 до приблизительно 50 мас.% или, что предпочтительно, приблизительно от 1 до приблизительно 40 мас.%), причем остальное количество составляют различные наполнители или носители или вспомогательные средства для изготовления, помогающие образованию требуемой дозированной формы.
Жидкие формы, подходящие для перорального введения, могут включать подходящий водный или неводный наполнитель с буферами, суспендирующими и диспергирующими агентами, красителями, ароматизаторами и тому подобное. Жидкие формы, включающие инъецируемые композиции, описанные здесь ниже, всегда хранят в отсутствие света, чтобы избежать любое каталитическое воздействие света, такое как образование гидропероксида или пероксида. Твердые формы могут включать, например, любой из следующих ингредиентов или соединений подобной природы: связующее, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза; дезинтегрирующий агент, такой как альгиновая кислота, примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; вещество, повышающее скольжение, такое как коллоидный диоксид кремния; подслащивающий агент, такой как сахароза или сахарин, или ароматизатор, такой как перечная мята, метилсалицилат или апельсиновый ароматизатор.
Инъецируемые композиции обычно имеют в качестве основы инъецируемый стерильный физиологический раствор или забуференный фосфатом физиологический раствор или другие инъецируемые носители, известные в данной области. Как выше указывалось, амидное производное формулы (I) в таких композициях обычно является минорным компонентом, часто в количестве, составляющим от 0,05 до 10 мас.%, причем остальное количество составляет инъецируемый носитель и тому подобное. Средняя суточная доза будет зависеть от различных факторов, таких как серьезность заболевания и состояния пациента (возраст, пол и масса). Доза обычно варьирует от 1 мг или нескольких мг до 1500 мг соединений формулы (I) в день, ее, необязательно, разделяют на несколько введений. Вследствие низкой токсичности соединений изобретения можно также вводить более высокие дозы на протяжении долгого периода времени.
Вышеописанные компоненты для перорально вводимых или инъецируемых композиций даны только в качестве примеров. Дополнительные материалы, а также способы изготовления композиций и тому подобное изложены в части 8 справочника «Remingtons's Pharmaceutical Sciences Handbook», 18th Edition, 1990, Mack Publishing Company, Easton, Pennsylvania, который включен здесь посредством ссылки.
Соединения данного изобретения можно вводить также в формах пролонгированного действия или из систем доставки с непрерывным высвобождением лекарственного средства. Описание примеров материалов с непрерывным высвобождением лекарственного средства можно найти во включенных материалах в указанном справочнике Ремингтона.
Настоящее изобретение будет далее иллюстрироваться при помощи следующих примеров, которые не должны рассматриваться как ограничивающие объем изобретения.
В описании соединений изобретения формулы (I) были приняты условные обозначения, указывающие абсолютные конфигурации каких-либо хиральных заместителей, которые могут присутствовать в заместителе R' указанных соединений, со штриховыми знаками (например, R', S', S'' и т.д.).
Примерами аббревиатур являются ТГФ для тетрагидрофурана, ДМФ для диметилформамида, НОВТ для 1-гидроксибензотриазола, DCC для дициклогексилкарбодиимида.
ПРИМЕРЫ
Пример 1
(R,S')-2-[(4'-Изобутил)фенил]-N-(2-карбоксиэтил)пропионамид
К раствору R-(-)-ибупрофена (5 г; 24,24 ммоль) в ДМФ (20 мл), охлажденному приблизительно до температуры Т=0°С, при перемешивании добавляют 3 г НОВТ (22,2 ммоль). Спустя 15 минут добавляют смесь гидрохлорида метилового эфира L-аланина (3,2 г; 22,2 ммоль) и триэтиламина (3 мл) в ДМФ (5 мл); наконец, добавляют DCC в виде последовательных порций всего в количестве 5 г (24,24 ммоль). Смесь выдерживают при перемешивании в течение двух часов при температуре Т=0°С и затем в течение ночи при комнатной температуре. После удаления фильтрованием осадка дициклогексилмочевины фильтрат разбавляют этилацетатом (50 мл). Органическую фазу промывают 10% раствором лимонной кислоты (2×20 мл), насыщенным раствором NaHCO3 (2×20 мл) и, наконец, насыщенным раствором NaCl (20 мл). После сушки над Na2SO4 упариванием растворителей при низком давлении получают остаток (3,86 г), который суспендируют в гексане (60 мл) и выдерживают при перемешивании в течение ночи, отделяют белый кристаллический осадок (R,S')-2-[(4'-изобутил)фенил]-N-(2-метоксикарбонилэтил)пропионамида (4,9 г, 16,84 ммоль).
К раствору 2 г (6,87 ммоль) последнего соединения в диоксане (9 мл) добавляют равный объем NaOH (9 мл), и смесь выдерживают при перемешивании при комнатной температуре в течение ночи. После разбавления водой и льдом (130 мл) ее подкисляют концентрированной H2SO4 до слабокислотного значения рН. После исчерпывающей экстракции водной фазы CH2Cl2 (4×20 мл) органические экстракты объединяют, промывают насыщенным раствором NaCl (20 мл), сушат над Na2SO4 и упаривают при низком давлении, получая при этом остаток, который после кристаллизации с использованием этилового эфира (30 мл) дает (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамид (1,81 г, 6,52 ммоль), т. пл. 125-128°С, [α]D=-46 (с=1%; CH3ОН);
1H-ЯМР (CDCl3): δ 7,25-7,1 (м, 4Н), 5,85 (шир.с, CONH), 4,52 (м, 1Н), 3,62 (кв., 1Н, J1=14 Гц, J2=7 Гц), 2,47 (д, 2Н, J=7 Гц), 1,85 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 1,35 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
В альтернативном случае, если так требуется, гидролиз метилового эфира можно проводить также с использованием триметилсилилйодида, например, в хлороформе.
Раствор 1,71 ммоль эфира в CHCl3, к которому добавлен 2,56 ммоль триметилсилилйодида, нагревают в течение нескольких минут до 50°С, затем процесс реакции прерывают охлаждением до комнатной температуры (чтобы минимизировать возможное образование побочного продукта). После упаривания растворителей сырой продукт реакции снова растворяют в этиловом эфире; органическую фазу экстрагируют 1 н NaOH (2×15 мл); основные водные экстракты объединяют, подкисляют и обесцвечивают обработкой тиосульфатом натрия. Водную фазу затем экстрагируют CH2Cl2 (2×15 мл) и органические экстракты, которые объединяют, после обычной обработки (промывание насыщенным раствором NaCl, сушки над Na2SO4), дают требуемый (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамид.
Пример 2
Заменой в процедуре примера 1 L-аланина метиловым эфиром D-аланина и метиловым эфиром глицина получают следующие соединения: (R,R')-2-[(4'-изобутил)фенил]-N-(2''-карбоксиэтил)пропионамид в виде бледно-желтого масла, [α]D=+5 (с=0,5%; СН3ОН);
1H-ЯМР (CDCl3): δ 7,20-7,07 (м, 4Н), 5,97 (шир.с, CONH), 4,45 (м, 1Н), 3,60 (м, 1Н), 2,45 (д, 2Н, J=7 Гц), 1,85 (м, 1Н), 1,53 (м, 3Н), 1,35 (м, 3Н), 0,91 (д, 6Н, J=7 Гц),
(R)-(-)-2-[(4'-изобутил)фенил]-N-карбоксиметилпропионамид, т.пл. 87-90°С.
1H ЯМР (CDCl3): δ 7,23-7,07 (м, 4Н), 5,93 (шир.с, CONH), 4,13-3,93 (м, 2Н), 3,63 (кв., 1Н, J1=8 Гц, J2=15 Гц), 2,45 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Пример 3
(R)-N-[2'-[(4''-Изобутилфенил)пропаноил]-2-аминоакриловая кислота
С использованием этилового эфира L-цистеина в процедуре примера 1 получают (R,R')-2-[(4'-изобутил)фенил]-N-2''-(3''-меркаптокарбоксиэтил)пропионамид. В атмосфере инертного газа к раствору 0,3 г (0,89 ммоль) этого соединения в безводном СН2Cl2 (24 мл), охлажденном до температуры Т=-10°С, по каплям при перемешивании добавляют 1 М раствор BBr3 в CH2Cl2 (6 мл).
Реакционную смесь выдерживают при перемешивании при температуре Т=-10°С в течение одного часа и затем при комнатной температуре в течение шести часов. Смесь затем разбавляют водой (20 мл), две фазы разделяют и водную фазу снова экстрагируют CH2Cl2. Объединенные органические экстракты промывают насыщенным раствором NaHCO3 (3×20 мл). Основную водную фазу затем подкисляют 2 н. HCl до рН 2 и экстрагируют СН2Cl2 (3×10 мл). Объединенные органические экстракты сушат над Na2SO4 и упаривают, получая при этом (R)-N-[2'-[(4''-изобутилфенил)пропаноил]-2-аминоакриловую кислоту (0,080 г, 0,29 ммоль) в виде опалесцентного масла.
1H-ЯМР (CDCl3): δ 7,4-7,2 (м, 4Н), 6,81 (с, 1Н), 6,1 (с, 1Н), 3,80 (м, 1Н), 3,11 (с, 3Н), 3,03 (с, 3Н), 2,60 (м, 2Н), 2,01 (м, 1Н), 1,70 (д, 3Н, J=7 Гц), 1,07 (д, 6Н, J=7 Гц).
Пример 4
Метил-R-N-[2'-[4''-изобутилфенил)пропаноил]-2-аминоакрилат
Продукт получают β-элиминированием в присутствии трет-бутоксида калия (1,1 экв.) в безводном этиловом эфире, исходя из (R,R')-2-[(4'-изобутил)фенил]-N-2''-(3''-меркаптокарбоксиметил) пропионамида (при Т=0°С). После разбавления 1,11 экв. АсОН в этиловом эфире, перераспределения обработкой насыщенным раствором NaH2PO4 в воде, разделения и сушки органической фазы, после упаривания получают метил-(R)-N-[2'-[(4''-изобутилфенил)пропаноил]-2-аминоакрилат в форме бледно-желтого масла;
1H-ЯМР (CDCl3): δ 7,25-7,15 (м, 4Н), 6,57 (с, 1Н), 5,83 (с, 1Н), 3,77 (с, 3Н), 3,63 (м, 1Н), 2,47 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Проведением такой же реакции в присутствии эквивалентного количества воды при 0°С получают свободную кислоту предыдущего примера.
Пример 5
R-(-)-2-[(4'-Изобутил)фенил)-N-(2''-гидроксиэтоксиэтил)пропионамид
Раствор R-(-)-ибупрофена (2 г; 9,69 ммоль) в тионилхлориде (4 мл) нагревают в течение 3 часов при кипячении с обратным холодильником; после охлаждения до комнатной температуры растворитель упаривают при низком давлении, растворяя при этом остаток последовательно два раза в диоксане и упаривая растворители в условиях высокого вакуума для удаления остаточных следов тионилхлорида. Маслянистый желтый остаток (2,16 г; 9,6 ммоль) полученного таким образом R-(-)-ибупрофеноилхлорида растворяют в безводном CH2Cl2 (15 мл). Раствор по каплям при комнатной температуре добавляют к раствору 2-(2-аминоэтокси)этанола (0,97 мл; 9,7 ммоль) и триэтиламина (1,35 мл; 9,7 ммоль) в безводном CH2Cl2 (15 мл). Перемешивание реакционной смеси продолжают в течение ночи при комнатной температуре, затем смесь разбавляют CH2Cl2 (30 мл), органическую фазу промывают 1 н. HCl (2×10 мл) и насыщенным раствором NaCl. После высушивания над Na2SO4 и упаривания растворителя при низком давлении получают остаток, который очищают посредством флэш-хроматографии (элюент CH2Cl2/CH3OH, 98:2), получая при этом в виде прозрачного масла 1,87 г R-(-)-2-[(4'-изобутил)фенил)-N-(2''-гидроксиэтоксиэтил)пропионамида; [α]D=-3,2 (с=3%; EtOH);
1H-ЯМР (CDCl3): δ 7,23 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,77 (шир.с, CONH), 3,75-3,33 (м, 9Н), 2,47 (д, 2Н, J=7 Гц), 1,85 (м, 1Н), 1,63 (шир.с, ОН), 1,53 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Пример 6
С использованием в процедуре предыдущего примера (S)-1-метил-2-(2'-гидроксиэтокси)этиламина получают (R,S')-2-[(4''-изобутил)фенил]-N-[1'-метил-2'-[2'''-гидроксиэтокси)этил]пропионамид; [α]D=-16 (с=1%, СН3ОН);
1H-ЯМР (CDCl3): δ 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,55 (шир.с, CONH), 4,17 (м, 1Н), 3,65 (м, 2Н), 3,55 (м, 4Н), 3,40 (м, 1Н), 2,47 (д, 2Н, J=7 Гц), 2,05 (шир.с, ОН), 1,85 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 1,1 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
С использованием в процедуре предыдущего примера (R)-1-метил-2-(2'-гидроксиэтокси)этиламина получают (R,R')-2-[(4''-изобутил)фенил]-N-[1'-метил-2'-(2'''-гидроксиэтокси)этил]пропионамид.
Пример 7
С использованием в процедуре примера 1 гетероциклического амина, выбранного из группы, состоящей из 2-аминопиридина, 3-аминопиридина и 4-аминопиридина, получают следующие соединения соответственно:
(R)-(-)-2-(4'-изобутил)фенил-N-(2'-пиридил)пропионамид в форме прозрачного масла; [α]D=-56 (с=1%, СН3СН2ОН);
1H-ЯМР (CDCl3): δ 8,25 (м, 2Н), 7,71 (м, 2Н), 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 7,05 (шир.с, CONH), 3,70 (м, 1H), 2,45 (д, 2Н, J=7 Гц), 1,85 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц),
(R)-(-)-2-[(4'-изобутил)фенил]-N-(3''-пиридил)пропионамид в форме воскообразного твердого вещества; [α]D=-96 (с=1%, СН3СН2ОН);
1H-ЯМР (DMSO-d6): δ 8,7 (с, 1Н), 8,22 (д, 1Н, J=5 Гц), 8,03 (м, 1H), 7,13 (м, 3Н), 7,13 (д, 2Н, J=7 Гц), 3,80 (м, 1Н), 2,45 (д, 2Н, J=7 Гц), 1,80 (м, 1H), 1,43 (д, 3Н, J=7 Гц), 0,85 (д, 6Н, J=7 Гц),
(R)-(-)-2-[(4'-изобутил)фенил]-N-(4'-пиридил)пропионамид.
Каждый из этих амидов можно превратить, если это так необходимо, в соответствующие соли по процедурам, которые хорошо известны в данной области, чтобы получить, например, гидрохлорид (R)-(-)-2-[(4'-изобутил)фенил]-N-(4''-пиридил) пропионамида, т.пл. 95-100°С, [α]D=-54 (с=0,2%, СН3ОН);
1H-ЯМР (DMSO-d6): δ 10,91 (с, 1H), 8,87 (д, 2Н, J=7 Гц), 7,83 (д, 2Н, J=7 Гц), 7, 37 (д, 2Н, J=7 Гц), 7,20 (д, 2Н, J=7 Гц), 3,97 (м, 1H), 2,45 (д, 2Н, J=7 Гц), 1,90 (м, 1H), 1,50 (д, 3Н, J=7 Гц), 0,95 (д, 6Н, J=7 Гц).
Подобным же образом ацилированием R-кетопрофена получают следующее соединение: гидрохлорид (R)-(-)-2-[(5'-бензоил)фенил]-N-(2'-пиридил) пропионамида в виде белого порошка; [α]D=-6 (с=1%, СН3СН2ОН);
1H-ЯМР (CDCl3): δ 12,65 (шир.с, NH+), 8,75 (м, 1H), 8,2 (м, 1H), 7,93-7,33 (м, 11Н), 4,20 (м, 1H), 1,67 (д, 3Н, J=7 Гц).
Пример 8
R-(-)-2-(4'-Изобутил)фенил-N-метилпропионамид
Взаимодействием раствора R-ибупрофеноилхлорида в диоксане с водным раствором N-метиламина в условиях реакции Schotten-Baumann R-(-)-2-(4'-изобутил)фенил-N-метилпропионамид получают в форме желтого масла; [α]D=-21 (с=1%, СН3СН2ОН);
1H-ЯМР (CDCl3): δ 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,30 (шир.с, CONH), 3,53 (м, 1Н), 2,73 (д, 3Н, J=7 Гц), 2,45 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Пример 9
С использованием (R)-кетопрофена в процедуре примера 1 получают следующее соединение: (R)-(-)-2-[(5'-бензоил)фенил]-N-карбоксиметилпропионамид в форме вспененного белого твердого вещества; [α]D=-9 (с=1%, СН3ОН);
1H-ЯНР (CDCl3): δ 7,81-7,30 (м, 9Н), 6,17 (шир.с, CONH), 4,1-3,25 (м, 4Н), 1,47 (д, 3Н, J=7 Гц).
Пример 10
С использованием метиловых эфиров цис- и транс-4-аминоциклогексанкарбоновых кислот в процедуре примера 1 получают следующие соединения: цис-(R)-2-[(4'-изобутил)фенил]-N-(4'-карбоксициклогексил) пропионамид и транс-(R)-2-[(4'-изобутил)фенил]-N-(4'-карбоксициклогексил)пропионамид.
Пример 11
К раствору 0,32 г (R)-2-(2-ацетокси-5-бензоил) фенилпропионовой кислоты в 10 мл AcOEt (высушен на молекулярных ситах) при перемешивании добавляют 0,185 г карбонилдиимидазола; затем спустя приблизительно один час добавляют 0,2 г аллилового эфира L-аланиноилглицина. Смесь выдерживают в течение 12 часов при комнатной температуре; реакционную смесь разбавляют AcOEt (5 мл) и промывают несколько раз 2 н H2SO4, водой, 5% NaHCO3 и снова водой до нейтральности и затем упаривают досуха, получая при этом после очистки на колонке с силикагелем 0,41 г аллилового эфира (R)-2-(2-ацетокси-5-бензоил)фенилпропионил-L-аланиноилглицина.
К раствору 0,24 г эфира (0,05 мМ) в ТГФ (10 мл) при перемешивании в атмосфере инертного газа последовательно добавляют 60 мг тетра(трифенилфосфин)палладий(0) и 0,5 мл морфолина. Спустя приблизительно один час растворитель упаривают в условиях вакуума. Остаток растворяют в этилацетате, раствор промывают несколько раз 2 н. Н2SO4 и водой до нейтральности, получая при этом после высушивания над сульфатом натрия, упаривания досуха и фильтрации остатка через колонку кремниевой кислоты, 0,12 г (R)-(2-ацетокси-5-бензоил) фенилпропаноил-L-аланиноилглицина.
«Кремниевая кислота» означает партию SiO2 для колоночной хроматографии, которую после нескольких суспендирований в 6 н HCl промывали до нейтральности и исчезновения следов ионов Cl- в элюате (анализ с AgNO3) и затем реактивировали нагреванием до 120°С в течение, по меньшей мере, 24 часов.
С использоАанием, по той же самой процедуре, (R)-2-(2-фтор-4-бифенил)пропионовой кислоты, (R)-2-(4'-метокси)фенил]пропионовой кислоты, (R)-2-(2-гидрокси-5-бензоил) фенилпропионовой кислоты, (R)-2-(3-феноксифенил)пропионовой кислоты и аллиловых эфиров фенилглицина, глицина и L-аланина, L-фенилаланина, L-аланиноилглицина, глициноил-L-аланина были получены следующие соединения:
R-2-(2-фтор-4-бифенил)пропаноилглицин;
(R)-(2-гидрокси-5-бензоил)фенилпропаноилглицин;
(R)-2-[(4'-метокси)фенил]пропаноил-L-аланин;
(R)-(2-(2-гидрокси-5-бензоил)фенилпропаноилглициноил-L-аланин;
(R)-(2-(2-гидрокси-5-бензоил)фенилпропаноил-L-аланоилглицин;
(R)-(2-(2-гидрокси-5-бензоил)фенилпропаноил-L-фенилаланин;
(R)-(2-(3-феноксифенил)пропаноилфенилглицин;
(R)-(2-(3-феноксифенил)пропаноилглицин.
Пример 12
Взаимодействием R-2-арилпропионовой кислоты, выбранной из группы, состоящей из ибупрофена, супрофена, тиапрофена, флурбипрофена и напроксена с 4-аминопиридином, с карбонилдиимидазолом по процедуре примера 11 получают соответствующие имидазолиды, которые подвергают in situ взаимодействию с 4-аминопиридином и 1-аминэтил-4-(4',4''-дифторфенил)метилпиперазином для получения:
N-[2-[4-(4',4''-дифторфенил)метилпиперазин-1-ил)этил]-R-2-(4-изобутилфенил)пропионамида;
N-(пирид-4-ил)-R-2-(2-фтор-4-бифенил)пропионамида;
N-(пирид-4-ил)-R-2-(6-метоксинафтил)пропионамида;
N-(пирид-4-ил)-R-2-(4-тиеноилфенил)пропионамида;
N-(пирид-4-ил)-R-2-(5-бензоилтиен-2-ил)пропионамида.
Пример 13
Взаимодействием имидазолида R-ибупрофена с аллиловым эфиром N-метилглицина, 3-амино-1,5-пентандиовой кислотой, N-(карбоксиметил)глицином и N-карбоксиэтилглицином по процедуре примера 11 получают следующие соединения соответственно:
N-[R-2-(4-изобутилфенил)пропаноил]-N-метилглицин;
N-[R-2-(4-изобутилфенил)пропаноил]-иминодиуксусную кислоту;
R-3-аза-3-[2-(4-изобутилфенил)пропаноил]-1,6-гександиовую кислоту;
N-3-[2-(4-изобутилфенил)пропаноил]-1,5-пентандиовую кислоту и их аллиловые эфиры.
Взаимодействием метиловых эфиров саркозина, N-аллилглицина и N-пропаргилглицина с R-ибупрофеном по процедуре примера 1 получают следующие соединения:
N-[R-2-(4-изобутилфенил)пропаноил]-N-метилглицин;
N-[R-2-(4-изобутилфенил)пропаноил]-N-аллилглицин;
N-[R-2-(4-изобутилфенил)пропаноил]-N-пропаргилглицин и их метиловые эфиры.
Пример 14
С использованием в примере 11 диаллилового эфира L-S-карбоксиметилцистеина и аллиловых эфиров L-лейцина, L-метионина, L-O-метилсерина и серина взаимодействием с имидазолидами R-ибупрофена, R-кетопрофена и R-индолрофена получают аллиловые эфиры соответствующих амидов, которые обработкой Pd(0)/морфолином превращают в следующие свободные кислоты:
N-[R-2-(4-иэобутилфенил)пропаноил]-L-S-карбоксиметилцистеин;
N-[R-2-(3-бензоилфенил)пропаноил]-L-S-карбоксиметилцистеин;
N-[R-2-(4-изобутилфенил)пропаноил]-L-лейцин;
N-[R-2-(3-бензоилфенил)пропаноил]-L-лецин;
N-[R-2-(1-оксо-2-изоиндолинилфенил)пропаноил]-L-лейцин;
N-[R-2-(4-изобутилфенил)пропаноил]-L-O-метилсерин;
N-[R-2-(3-бензоилфенил)пропаноил]-L-O-метилсерин;
N-[R-2-(1-оксо-2-изоиндолинилфенил)пропаноил]-L-O-метилсерин;
N-[R-2-(4-изобутилфенил)пропаноил]-L-серин;
1H-ЯМР (CDCl3): δ 7,3-7,0 (м, 4Н), 6,45 (шир.с, 1Н), 4,5 (м, 1H), 4,1-4,0 (м, 1Н), 3,9-3,5 (м, 2Н), 2,5-2,3 (м, 3Н), 1,85 (м, 1H), 1,5 (м, 3Н), 0,9 (д, 6Н).
Пример 15 - Получение промежуточных аминов S-1-Метил-2-(2'-гидроксиэтокси)этиламин
Раствор трет-бутилдикарбоната (1,4 г; 6,49 ммоль) в безводном ТГФ (15 мл) добавляют по каплям к раствору S-(+)-2-амино-1-пропанола (0,5 мл; 6,42 ммоль) в безводном ТГФ (15 мл), смесь перемешивают и охлаждают приблизительно до 0°С. Смесь затем выдерживают при перемешивании в течение ночи при комнатной температуре. Растворитель упаривают, остаток растворяют в CH2Cl2 (55 мл); органическую фазу промывают 5% раствором NaH2PO4 (3×10 мл) и сушат над Na2SO4. После упаривания растворителя при низком давлении получают 0,965 г (5,5 ммоль) S-(-)-N-трет-бутоксикарбонил-2-амино-1-пропанол; [α]D=-7,5 (с=1,1%, СН3ОН).
К раствору 0,225 г (1,3 ммоль) данного соединения в безводном ДМФ (7 мл), охлажденному до температуры Т=0°С, добавляют в следующем порядке соединения: NaH (94 мг; 2,34 ммоль, 60% суспензия) и спустя 20 минут 2-(2-бромэтокси) тетрагидро-2Н-пиран (0,24 мл, 1,59 ммоль) и йодид тетра-N-бутиламмония (48 мг, 0,13 ммоль). Реакционную смесь оставляют для нагревания самопроизвольно до комнатной температуры и перемешивание продолжают в течение ночи. Ее затем охлаждают до 0°С перед добавлением по каплям СН3ОН для разложения избыточных реагентов. Ее затем разбавляют водой; водную фазу экстрагируют CH2Cl2 (2×10 мл); органические экстракты объединяют и промывают насыщенным раствором NaCl (2×10 мл), сушат над Na2SO4 и упаривают при низком давлении. Сырой остаток очищают при помощи колоночной хроматографии (элюент: CHCl3/СН3ОН/пиридин, 98:2:1), получая при этом 0,184 г S-(-)-N-трет-бутоксикарбонил-3-(2'-тетрагидропиранилоксиэтокси)-2-пропиламин в форме прозрачного масла; [α]D=-11,7 (с=1%, СН3СН2ОН).
Добавлением трифторуксусной кислоты (0,06 мл) к раствору соединения в безводном CH2Cl3 (10 мл), выдерживанием в течение ночи при комнатной температуре после разбавления водой (5 мл), разделения фаз, подщелачивания водной фазы до рН 10 1 н NaOH, повторной экстракции дихлорметаном и упаривания растворителя получают остаток S-1-метил-2-(2'-гидроксиэтокси)этиламина.
С использованием такой же процедуры, исходя из R-(-)-2-амино-1-пропанола, получают R-1-метил-2-(2'-гидроксиэтокси)этиламин.
Пример 16
Получение соединений, перечисленных в таблицах
А) Получение соединений, перечисленных в таблице 4
С использованием, по процедуре примера 1, индивидуальных энантиомеров 3-ибупрофена, R-ибупрофена, 3-кетопрофена и R-кетопрофена взаимодействием с 4-метил-2-аминопиридином получают следующие соединения:
R-(-)-2-[(4'-изобутил)фенил]-N-(4''-метил-2''-пиридил)пропионамид в форме прозрачного масла; [α]D=-93 (с=1%, СН3СН2OH),
1H-ЯМР (CDCl3): δ 8,13 (с, 1Н), 8,07 (м, 1Н), 7,95 (шир.с, CONH), 7,25 (д, 2Н, J=7 Гц), 7,13 (д, H2, J=7 Гц), 6,83 (д, 1H, J=7 Гц), 3,75, (м, 1Н), 2,45 (д, 3Н, J=7 Гц), 2,35 (с, 3Н), 1,87 (м, 1H), 1,60 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц),
S-(+)-2-[(4'-изобутил)фенил]-N-(4''-метил-2''-пиридил)пропионамид в форме прозрачного масла; [α]D=+98 (с=1,2%, СН3СН2ОН),
1H-ЯМР (CDCl3): δ 8,13 (с, 1H), 8,07 (м, 1H), 7,93 (шир.с, CONH), 7,25 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 6,83 (д, 1H, J=7 Гц), 3,75 (м, 1H), 2,45 (д, 3Н, J=7 Гц), 2,35 (с, ЗН), 1,87 (м, 1H), 1,60 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц),
R-(-)-2-[(5'-бензоил)фенил]-N-(4''-метил-2''-пиридил)пропионамид в форме вспененного белого твердого вещества; [α]D=-83,4 (с=1%, СН3СН2ОН),
1H-ЯМР (CDCl3): δ 8,55 (шир.с, CONH), 8,15 (с, 1H), 8,05 (м, 1H), 8,05 (м, 1H), 7,87-7,43 (м, 9Н), 6,93 (д, 1H, J=7 Гц), 3,85 (м, 1H), 2,40 (с, 3Н), 1,65 (д, 3Н, J=7 Гц),
S-(+)-2-[(5'-бензоил)фенил]-N-(4''-метил-2''-пиридил)пропионамид в форме светло-желтого твердого вещества; [α]D=+87 (с=1%, СН3СН2ОН),
1H-ЯМР (CDCl3): δ 8,88 (шир.с, CONH), 8,2 (с, 1H), 8,05 (м, 1Н), 7,85-7,43 (м, 9Н), 6,93 (д, 1Н, J=7 Гц), 3,90 (м, 1Н), 2,40 (с, 3Н), 1,60 (д, 3Н, J=7 Гц).
В) Получение соединений, перечисленных в таблице 3
(S,R')-2-[(4'-Изобутил)фенил]-N-(2-карбоксиэтил)пропионамид; т.пл. 118-121°С;
[α]D=+39 (с=0,2%; СН3ОН),
1H-ЯМР (CDCl3): δ 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,85 (шир.с, CONH), 4,55 (м, 1Н), 3,60 (м, 1Н), 2,47 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 1,35 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц),
(S,S')-2-[(4'-Изобутил)фенил]-N-(2-карбоксиэтил)пропионамид; т.пл. 85-87°С;
[α]D=-2,8 (с=0,5%; СН3ОН),
1H-ЯМР (CDCl3): δ 7,22-7,10 (м, 4Н), 6,85 (шир.с, CONH), 4,53 (м, 1Н), 3,6 (м, 1Н), 2,47 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,55 (д, 3Н, J=7 Гц), 1,40 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Взаимодействием индивидуальных изомеров ибупрофеноилхлорида с анилином получают следующие соединения:
(S)-(+)-2-[(4'-изобутил)фенил]-N-фенилпропионамид; т.пл. 117-120°С; [α]D=+93 (с=1%; СН3СН2ОН),
1H-ЯМР (CDCl3): δ 7,45-6,97 (м, 10Н), 3,70 (кв., 1Н, J1=15 Гц, J2=7 Гц), 2,45 (д, 3Н, J=7 Гц), 1,87 (м, 1Н), 1,60 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц),
(R)-(-)-2-[(4'-изобутил)фенил]-N-фенилпропионамид; т.пл. 118-120°С; [α]D=-86 (с=1%; СН3СН2ОН),
1H-ЯМР (CDCl3): δ 7,43 (м, 2Н), 7,30 (м, 3Н), 7,17 (м, 2Н), 7,05 (м, 3Н), 3,70 (м, 1Н), 2,45 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,53 (д, 3, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
С) (R)-(-)-2-[(4'-изобутил)фенил]-N-(2'-гидроксиэтил) пропионамид (таблица 2)
К раствору R-ибупрофена (0,25 г, 1,21 ммоль) в безводном этилацетате добавляют 0,11 экв. N,N'-карбонилдиимидазола при комнатной температуре и при перемешивании. После выдерживания смеси 3 часа при комнатной температуре, без выделения промежуточного R-ибупрофеноилимидазолида, добавляют раствор 0,11 экв. 2-аминоэтанола в безводном AcOEt. Перемешивание продолжают в течение 6 часов при комнатной температуре и затем органическую фазу перераспределяют обработкой несколько раз 2 н водным раствором H2SO4. Органические фазы промывают до нейтральности насыщенным раствором NaCl и обезвоживают над Na2SO4. После упаривания растворителя (R)-(-)-2-[(4'-изобутил)фенил]-N-(2'-гидроксиэтил)пропионамид получают в форме бледно-желтого масла;
1H-ЯМР (CDCl3): δ 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,80 (шир.с, CONH), 3,67 (м, 2Н), 3,55 (м, 1Н), 3,35 (м, 2Н), 2,85 (шир.с, ОН), 2,45 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,55 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Е) С использованием процедуры вышеприведенного получения D) и L- и D-аланинола в качестве аминов получают следующие соединения:
(R,R')-2-[(4'-изобутил)фенил]-N-(3''-гидроксипроп-2''-ил)пропионамид; т.пл. 71-74°С; [α]D=+9,2 (с=0,5%; СН3ОН);
1H-ЯМР (CDCl3): δ 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,43 (шир.с, CONH), 4,00 (м, 1Н), 3,6-3,35 (м, 3Н), 2,45 (д, 2Н, J=7 Гц), 1,85 (м, 1Н), 1,47 (м, 4Н), 1,05 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц),
(R,S')-2-[(4'-изобутил)фенил]-N-(3''-гидроксипроп-2-ил)пропионамид; т.пл. 75°С; [α]D=-12 (с=0,5%; СН3ОН),
1H-ЯМР (CDCl3): δ 7,22 (д, 2Н, J=7 Гц), 7,13 (д, 2Н, J=7 Гц), 5,43 (шир.с, CONH), 4,01 (м, 1Н), 3,35 (м, 4Н), 2,45 (д, 2Н, J=7 Гц), 1,87 (м, 1Н), 1,53 (д, 3Н, J=7 Гц), 1,05 (д, 3Н, J=7 Гц), 0,93 (д, 6Н, J=7 Гц).
Пример 17
Общая процедура синтеза 2-арилпропионовых кислот и соответствующих R-энантиомеров
17а - Способ дерацемизации 2-арилпропионовых кислот формулы Va
(R)-2-(2-Гидрокси-5-бензоил)фенилпропионовая кислота и (R)-2-(2-ацетокси-5-бензоил)фенилпропионовая кислота
Суспензию тонкоизмельченного К2СО3 (2,48 г, 18 ммоль) в растворе (R,S)-2-(2-гидрокси-5-бензоилфенил)пропионовой кислоты (2 г; 7,4 ммоль) в безводном ацетоне (35 мл) выдерживают при энергичном перемешивании при комнатной температуре в течение 30 минут; затем по каплям добавляют уксусный ангидрид (2,78 мл; 29,5 ммоль). После окончания прикапывания перемешивание продолжают в течение 12 часов при комнатной температуре. Продукт отфильтровывают от осадка и образовавшийся раствор упаривают досуха при низком давлении.
Раствор остатка в CH2Cl2 промывают неоднократно водой до исчезновения остатков уксусного ангидрида. Органическую фазу сушат над Na2SO4 и упаривают досуха. Раствор остатка в смеси ТГФ: Н2О, 1:1 (30 мл) оставляют при перемешивании на ночь. Последующее упаривание растворителей при низком давлении дает 2-(2-ацетокси-5-бензоилфенил)пропионовую кислоту в форме бледно-желтого масла (1,85 г; 5,92 ммоль);
1H-ЯМР (CDCl3): δ 8,0 (д, 1Н, J=2 Гц), 7,9-7,75 (м, 3Н), 7,67 (м, 1Н), 7,45 (м, 2Н), 7,32 (д, 1Н, J=2 Гц), 4,0 (м, 1Н), 2,35 (с, 3Н), 1,6 (д, 3Н, J=7 Гц).
Раствор 1,5 г (4,8 ммоль) указанной кислоты в безводном толуоле (10 мл), к которому добавлено 2,1 мл оксалилхлорида (24 ммоль), нагревают до температуры Т=60°С до исчезновения исходной кислоты (1,5 час). После охлаждения до комнатной температуры растворитель упаривают сначала в потоке азота и затем в условиях высокого вакуума, получая при этом желтый остаток (1,55 г) хлорангидрида кислоты, который используют как таковой. К раствору соединения в безводном толуоле (15 мл), охлажденному до температуры Т=0°С, добавляют по каплям раствор диметилэтиламина (1,56 мл; 14,4 ммоль) в нескольких мл толуола при перемешивании 3 часа. Реакционную смесь затем охлаждают до температуры Т=-70°С и, наконец, к указанной смеси по каплям добавляют раствор R-(-)-пантолактона (0,656 г; 5,04 ммоль) в безводном толуоле (2 мл). Температуре затем дают возможность подняться до -20°С, и реакционную смесь выдерживают при перемешивании при такой температуре в течение всего 18 часов. Остаток, полученный после упаривания растворителя при низком давлении, очищают при помощи колоночной хроматографии, получая при этом 1,42 г (3,36 ммоль) дигидро-3-гидрокси-4,4-диметил-2(3Н)-фуранон-R-(-)-2-ацетокси-5-бензоилфенилпропионата в форме прозрачного масла и в виде отдельного диастереоизомера;
1H-ЯМР (CDCl3): δ 8,2 (д, 1Н, J=2 Гц), 7,9-7,7 (м, 4Н), 7,32 (м, 2Н), 7,32 (д, 1Н, J=2 Гц), 4,15 (м, 1Н), 4,01 (м, 3Н), 2,35 (с, 3Н), 1,6 (д, 3Н, J=7 Гц), 1,25 (с, 3Н), 1,05 (с, 3Н).
К раствору 1,4 г эфира (3,3 ммоль) в абсолютном этиловом спирте (10 мл), охлажденному до температуры Т=0°С, добавляют 0,37 н. водный раствор гидроксида лития (31,2 мл; 11,55 ммоль). Раствор выдерживают при перемешивании при температуре Т=0°С в течение 2 часов; затем его подкисляют до рН 5,5-6 добавлением по каплям 5% водного раствора лимонной кислоты и, наконец, экстрагируют этилацетатом (3×15 мл). Органические экстракты объединяют и промывают водой (20 мл), сушат над Na2SO4 и упаривают при низком давлении. Последующая очистка остаточного сырого масла при помощи флэш-хроматографии (элюент СН2Cl2/СН3ОН, 95:100), получая при этом R-(-)-2-(2-гидрокси-5-бензоилфенил)пропионовую кислоту в форме белого твердого вещества (0,365 г; 1,35 ммоль); т. пл. 170-172°С;
[α]D=-62 (с=1%; СН3ОН),
1H-ЯМР (CDCl3): δ 9,5 (шир.с, СООН), 8,0 (д, 1Н, J=2 Гц), 7,9-7,75 (м, 3Н), 7,67 (м, 1Н), 7,45 (м, 2Н), 7,32 (д, 1Н, J=2 Гц), 7,05 (с, ОН), 4,0 (м, 1Н), 1,6 (д, 3Н, J=7 Гц).
Последующая этерификация кислоты уксусным ангидридом (0,2 г; 0,74 ммоль) в безводном ацетоне (5 мл) в присутствии тонкоизмельченного К2СО3 (0,25 г; 1,8 ммоль) в виде осадка дает R-(-)-2-(2-ацетокси-5-бензоилфенил)пропионовую кислоту в форме бесцветного масла (0,17 г; 0,545 ммоль): [α]D=-53 (с=1%; СН3ОН),
1H-ЯМР (CDCl3): δ 9,5 (шир.с, СООН), 8,0 (д, 1Н, J=2 Гц), 7,9-7,75 (м, 3Н), 7,67 (м, 1Н), 7,45 (м, 2Н), 7,32 (д, 1Н, J=2 Гц), 4,5 (м, 1Н), 2,37 (с, 3Н), 1,6 (д, 3Н, J=7 Гц).
17b - Общая процедура синтеза 2-арилпропионовых кислот и соответствующих R-энантиомеров формулы Vf (R3=Н)
К перемешиваемому раствору 3-гидроксиацетофенона (80 ммоль) (или, в альтернативном случае, 2-или 4-гидроксиацетофенона) в ацетоне (80 мл) при комнатной температуре добавляют К2СО3 (12,0 г; 8,62 ммоль). После перемешвания 30' при комнатной температуре по каплям добавляют раствор перфторбутансульфонилфторида (15,5 мл; 86,1 ммоль) в ацетоне (30 мл) и образовавшуюся смесь кипятят с обратным холодильником в течение 2 час. После охлаждения до комнатной температуры образовавшееся твердое вещество отфильтровывают и фильтрат упаривают в вакууме, получая при этом сырой остаток, который разбавляют EtOAc (100 мл). Органический раствор перемешивают и промывают насыщенным раствором К2СО3 (20 мл) и затем насыщенным раствором NaCl (20 мл), сушат над Na2SO4 и упаривают в вакууме, получая при этом с количественным выходом перфторбутансульфониловый эфир в виде масла, достаточно чистого для использования на следующей стадии.
Смесь ацетофенонперфторбутансульфонилового эфира (80 ммоль), элементарной серы (2,95 г; 92 ммоль) и морфолина (8,0 мл, 92 ммоль) кипятят с обратным холодильником в течение 6 час. После охлаждения при комнатной температуре смесь осторожно добавляют к перемешиваемой смеси лед/6 н. HCl (40 мл). После разбавления СН2Cl2 (50 мл) две фазы перемешивают и разделяют и водный слой экстрагируют снова CH2Cl2 (2×50 мл). Собранные органические экстракты сушат над Na2SO4 и упаривают в вакууме; образовавшийся сырой желтый маслянистый остаток после очистки флэш-хроматографией (н-гексан/EtOAc, 9:1) дает соответствующий морфолинтиоамид в виде бесцветного масла (выход 73-80%).
К раствору морфолинтиоамида (58 ммоль) и ледяной уксусной кислоты (25 мл) осторожно добавляют 37% HCl (40 мл) и раствор кипятят с обратным холодильником при перемешивании в течение 16 час. После охлаждения до комнатной температуры образованное твердое вещество отфильтровывают и фильтрат, после упаривания, разбавляют водой (50 мл). Водную фазу экстрагируют EtOAc (2×50 мл) и собранные органические экстракты промывают насыщенным раствором NaCl (20 мл), сушат над Na2SO4 и упаривают в вакууме, получая при этом сырой остаток, который при кристаллизации из н-гексана дает (о,м,п)-перфторбутансульфонат-2-фенилуксусную кислоту в виде твердого вещества (выход 90-93%). Последующая обработка конц. H2SO4 в абсол. EtOH при Т=50°С дает соответствующий этиловый эфир с количественным выходом.
Суспензию 60% NaH в минеральном масле (1,6 г; 66,7 ммоль) добавляют небольшими порциями к охлаждаемому льдом и перемешиваемому раствору этил-(о,м,п)-перфторбутансульфонилокси-2-фенилацетата (25 ммоль) в ТГФ (50 мл). Спустя 15' в раствор по каплям добавляют метилйодид (1,88 г; 30,2 ммоль) и образовавшийся темный раствор перемешивают в течение 3,5 час при комнатной температуре. После добавления насыщенного раствора NH4Cl (45 мл) органический растворитель упаривают в вакууме и водную фазу экстрагируют СН2Cl2 (3×50 мл); собранные органические экстракты промывают насыщенным раствором NaCl (20 мл), сушат над Na2SO4 и упаривают в вакууме, получая при этом сырой остаток, который после хроматографирования дает соответствующую 3-перфторбутансульфонилокси-2-фенилпропионовую кислоту в виде бледно-желтого масла (выход 70%).
Исходя из этил-(2-, 3- или 4)-перфторбутансульфонилокси-2-фенилпропионата, рацемические смеси 2-арилпропионовых кислот общей формулы арил-С (СН3)Н-СООН (Va) синтезируют в соответствии с взаимодействием вышеуказанных перфторалкансульфонатов с некоторыми трибутилалкил-, алкенил- или алкинилсоединениями, как описано в Mitchell T.N., Synthesis, 803, 1992 и Ritter K., Synthesis, 735, 1993.
По вышеописанному способу были получены следующие соединения.
17b1 2-[3'-Изопропенилфенил]пропионовая кислота
Этил-3'-перфторбутансульфонилокси-2-фенилпропионат (7,63 ммоль) растворяют в N-метилпирролидоне (30 мл) и обрабатывают сухим LiCl (0,94 г, 22,9 ммоль), трифениларсином (90 мг; 0,3 ммоль) и дипалладийтрибензилиденацетоном (0,193 г; 0,15 ммоль Pd). После 5' выдерживания реакционной смеси при комнатной температуре добавляют трибутилизопропенилолово (2,83 г; 8,55 ммоль) и раствор перемешивают при Т=90°С в течение 5 час. После охлаждения, разбавления насыщенным водным KF и н-гексаном, фильтрования и отделения органической фазы проводят сушку ее над Na2SO4 и упаривание в вакууме. Очистка сырого остатка флэш-хроматографией дает этил-2-[3'-изопропенилфенил]пропионат (1,24 г; 5,3 ммоль). Выход 70%.
К раствору эфира в диоксане (5 мл) добавляют 1 н NaOH (5 мл) и образовавшийся раствор перемешивают в течение ночи при комнатной температуре. После упаривания органического растворителя водную смесь подкисляют до рН 2 2 н. HCl; продукт выделяют фильтрованием в виде белого твердого вещества (1,03 г, 5 ммоль).
1H-ЯМР (CDCl3): δ 10,0 (шир.с, 1Н, СООН), 7,28 (м, 1Н), 7,15 (м, 1Н), 7,05 (м, 2Н), 5,02 (с, 2Н), 3,75 (м, 1Н), 1,45 (д, ЗН, J=7 Гц), 0,78 (с, 3Н).
17b2. 3-[3'-(1''-Стиренил)фенил]пропионовая кислота
Кислоту синтезируют с использованием трибутил-(α-метилстиренил)пропенилолова в качестве реагента, полученного по вышеуказанному способу.
1H-ЯМР (CDCl3): (11,0 (шир.с, 1Н, СООН), 7,38-7,13 (м, 9Н), 3,95 (м, 2Н), 3,81 (м, 1Н), 1,72 (д, 3Н, J=7 Гц).
Вышеуказанные кислоты 17b1 и 17b2 используют также в качестве промежуточных продуктов при синтезе кислот 17b3 и 17b4.
17b3. 2-[3'-(Изопропил)фенил]пропионовая кислота
Смесь этил-2-[3'-(изопропенил)фенил]пропионата (1 г; 4,6 ммоль), полученного по вышеуказанному способу, 95% EtOH и 10% Pd/C (100 мг) гидрируют при комнатной температуре и атмосферном давлении до исчезновения исходного материала (2 час). Катализатор отфильтровывают на целитной панели и после упаривания в вакууме полученное таким образом прозрачное масло (0,99 г; 4,5 ммоль) гидролизуют 1 н. КОН в EtOH (10 мл) при Т=80°С в течение 2 час. После охлаждения при комнатной температуре растворитель упаривают в вакууме и сырой остаток разбавляют EtOAc (20 мл); органическую фазу экстрагируют водой (3×10 мл); водные собранные фазы подкисляют до рН 2 2 н. HCl и экстрагируют обратно EtOAc (2×10 мл); органические собранные экстракты промывают насыщенным раствором NaCl, сушат над Na2SO4 и упаривают в вакууме, получая при этом нужную кислоту (0,75 г; 3,6 ммоль).
1H-ЯМР (CDCl3): δ 10,5 (шир.с, 1Н, СООН), 7,15-7,08 (м, 4Н), 3,55 (м, 1Н), 2,91 (м, 1Н), 1,45 (д, 3Н, J=7 Гц), 1,26 (д, 6Н, J=7 Гц).
Тем же способом и исходя из 3-[3'-(1''-стиренил) фенил]пропионовой кислоты получают следующее соединение.
17b4. (R,S)-2-[3'-(α-Метилбензил)фенил]пропионовая кислота
1H-ЯМР (CDCl3): δ 11,0 (шир.с, 1Н, СООН), 7,38-7,13 (м, 9Н), 4,20 (м, 1Н), 3,78 (м, 1Н), 1,72 (д, 3Н, J=7 Гц), 1,55 (д, 3Н, J=7 Гц).
Каждую из рацемических смесей кислот общей формулы (Vf) превращают в отдельный R-энантиомер посредством стереоспецифического синтеза соответствующих эфиров R-пантолактона (через образование промежуточного кетена) по процедурам, цитированным в Myers A.G. et al., J. Am. Chem. Soc., 119, 6496, 1977 и в Larsen R.D. et al., J. Am. Chem. Soc., 111, 7650, 1989, как описано в примере 17а. Этим путем получены следующие кислоты.
17b5. (R)-2-[(3'-Изопропил)фенил]пропионовая кислота
[α]D=-23 (c=0,5; CH2Cl2)
1H-ЯМР (CDCl3); δ 10,0 (шир.с, 1Н, СООН), 7,15-7,10 (м, 4Н), 3,65 (м, 1Н), 2,90 (м, 1Н), 1,45 (д, 3Н, J=7 Гц), 1,32 (д, 6Н, J=7 Гц).
17b6. (R),(R'S')-2-[3'-(α-Метилбензил)фенил]пропионовая кислота
[α]D=-49 (с=0,5; СН2Cl2)
1H-ЯМР (CDCl3): δ 11,0 (шир.с, 1Н, СООН), 7,38-7,13 (м, 9Н), 4,20 (м, 1Н), 3,78 (м, 1Н), 1,72 (д, 3Н, J=7 Гц), 1,55 (д, 3Н, J=7 Гц).
17b7. (R)-2-[2'-(2,6-Дихлорфениламино)фенил]пропионовая кислота
Получение (R,S)-2-[2'-(2,6-дихлорфениламино)фенил]пропионовой кислоты проводят в соответствии с Geigy, JR; патент Великобритании 1132318 (30.10.1968). Последующее оптическое разделение проводят посредством образования соли с R-(+)-N-метилбензиламином по способу Akguen et al., Arzneim. Forsch. 1996, 46:9, 891-894.
17b8. (R),(R'S')-2-[3'-(α-Гидроксибензил)фенил]пропионовая кислота
К раствору R-(-)-кетопрофена (0,254 г; 1 ммоль) в этиловом спирте (5 мл) добавляют триэтиламин (0,12 г; 1 ммоль) и 5% Pd/C (0,025 г) и смесь затем гидрируют при комнатной температуре и атмосферном давлении в течение 3 час.
После того как катализатор отфильтровывают на целитной панели, фильтрат упаривают и сырой остаток очищают флэш-хроматографией, получая при этом продукт в виде белого порошка (выход 85%).
[α]D=-45,7 (с=1%; CHCl3).
1H-ЯМР (CDCl3): δ 7,41-7,3 (м, 3Н), 7,31-7,14 (м, 6Н), 5,75 (с, 1Н), 4,02 (шир.с, 1Н, ОН), 3,68 (кв., J=7 Гц), 1,4 (д, 3Н, J=7 Гц).
17b9. (R),(R'S')-2-[3'-(α-Гидрокси-α-метилбензил)фенил] пропионовая кислота
К раствору метилового эфира R-(-)-кетопрофена (0,269 г; 1 ммоль) в диэтиловом эфире (10 мл) добавляют 3,0 М метилмагнийбромид (2 ммоль) в диэтиловом эфире и образовавшийся раствор кипятят с обратным холодильником в течение 2 час. После охлаждения при комнатной температуре органическую фазу промывают раствором 5% NaH2PO4 (2×10 мл), сушат над Na2SO4 и упаривают в вакууме, получая при этом сырой остаток, который растворяют в растворе 1:1 1 н. NaOH/MeOH (5 мл). После перемешивания в течение ночи растворитель упаривают в вакууме и водную фазу подкисляют до рН 2; образованный осадок отфильтровывают и промывают водой, получая при этом (R),(R',S')-2-[3'-(α-гидрокси-α-метилбензил)фенил]пропионовую кислоту в виде белого порошка.
[α]D=-45,3 (с=1%; CHCl3).
1H-ЯМР (CDCl3): δ 7,41-7,3 (м, 3Н), 7,31-7,14 (м, 6Н), 4,02 (шир.с, 1Н, ОН), 3,68 (кв., J=7 Гц), 2,12 (с, 3Н), 1,4 (д. 3Н, J=7 Гц).
17b10. (R)-2-[(3'-(α-Гидроксиизопропил)фенил]пропионовая кислота
По той же самой процедуре и исходя из (R)-2-[(3'-ацетил)фенил]пропионовой кислоты, полученной оптическим разделением по способу, указанному выше, из ее рацемической смеси, указанное в заголовке соединение получают в виде белого порошка (выход 70%).
1H-ЯМР (CDCl3): δ 7,31-7,14 (м, 4Н), 4,02 (шир.с, 1Н, ОН), 3,68 (кв., J=7 Гц), 1,85 (с, 6Н), 1,4 (д, 3Н, J=7 Гц).
Пример 18
Общая процедура получения ацилхлоридов 2-арилпропионовых кислот
Раствор (R)-2-[(4'-(изобутил)фенил]пропионовой кислоты (72,8 ммоль) в тионилхлориде (37,5 мл) кипятят с обратным холодильником в течение 3 час; после охлаждения при комнатной температуре растворитель упаривают в вакууме. Сырой маслянистый остаток используют как таковой на следующей стадии.
ИК (пленка) см-1: 1800 (ClC=O)
Пример 19
19а
(R)-2-[(3'-Изопропил)фенил]-N-(карбоксиметил)пропионамид
К охлажденному (Т=0-5°С) раствору (R)-2-[(3'-(изопропил)фенил]пропионовой кислоты (4,75 г; 24,24 ммоль) в ДМФ (20 мл) добавляют гидроксибензотриаэол (НОВТ) (22,2 ммоль) при перемешивании. Спустя 15' добавляют смесь гидрохлорида метилового эфира глицина (2,89 г; 22,2 ммоль) и триэтиламина (3 мл) в ДМФ (5 мл), наконец, по порциям добавляют N,N-дициклогексилкарбодиимид (DCC) (24,24 ммоль). Образовавшуюся смесь перемешивают в течение 2 час при Т=0°С и затем в течение ночи при комнатной температуре. После того, как образованный осадок отфильтровывают, фильтрат разбавляют EtOAc (50 мл); органическую фазу промывают 10% лимонной кислотой (2×20 мл), насыщенным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (20 мл), сушат над Na2SO4 и упаривают в вакууме, получая при этом сырой остаток. После промывания н-гексаном получают чистый эфир в виде белого твердого вещества (5,2 г; 19,4 ммоль).
К раствору эфира (5,2 г; 19,4 ммоль) в диоксане (25 мл) добавляют 1 н. NaOH (25 мл) и образовавшийся раствор перемешивают в течение ночи при комнатной температуре. После упаривания органического растворителя водную смесь подкисляют до рН 2 при помощи 2 н HCl; продукт выделяют фильтрованием в виде белого твердого вещества (4,8 г; 19 ммоль).
[α]D=-53 (с=1%; CHCl3).
1H-ЯМР (CDCl3): δ 10,00 (шир.с, 1Н, СООН), 7,28 (м, 1Н), 7,15 (м, 1Н), 7,05 (м, 2Н), 5,90 (шир.с, 1Н, CONH), 4,12-3,90 (м, 2Н), 3,75 (кв., 1Н, J=7 Гц), 2,34 (м, 1Н), 1,45 (д, 3Н, J=7 Гц), 0,78 (д, 6Н, J=8 Гц).
По такой же процедуре были синтезированы следующие соединения.
19b. (R),(R',S')-2-[3'-(α-Метилбензил)фенил]-N-(карбоксиметил)пропионамид
[α]D=-35 (с=1%; CHCl3).
1H-ЯМР (CDCl3): (10,5 (шир.с, 1Н, СООН), 7,38-7,13 (м, 9Н), 5,85 (шир.с, 1Н, CONH), 4,10-3,95 (м, 2Н), 4,20 (м, 1Н), 3,78 (м,,1Н), 1,72 (д, ЗН, J=7 Гц), 1,55 (д, 3Н, J=7 Гц).
19с. (R),(R',S']-2-[3'-(α-Гидроксибензил)фенил]-N-(карбоксиметил)пропионамид
[α]D=-39,1 (с=1%; CHCl3).
1H-ЯМР (CDCl3): δ 10,05 (шир.с, СООН), 7,41-7,3 (м, 3Н), 7,31-7,14 (м, 6Н), 5,92 (шир.с, 1Н, CONH), 5,75 (с, 1Н), 4,45 (шир.с, 1Н, ОН), 4,12-3,90 (м, 2Н), 3,68 (кв., J=7 Гц), 1,4 (д, 3Н, J=7 Гц).
19d. (R),(R',S')-2-[3'-(α-Гидрокси-α-метилбензил)фенил]-N-(карбоксиметил)пропионамид
[α]D=-41 (с=1%; CHCl3).
1H-ЯМР (CDCl3): δ 9,92 (шир.с, 1Н, СООН), 7,40-7,28 (м, 3Н), 7,25-7,10 (м, 6Н), 5,85 (шир.с, 1Н, CONH), 4,45 (шир.с, 1Н, ОН), 4,10-3,95 (м, 2Н), 3,68 (кв., J=7 Гц), 2,15 (с, ЗН), 1,4 (д, 3Н, J=7 Гц).
Пример 20
(R)-(-)-N-Метокси-2-(4'-(изобутил)фенилпропионамид
К раствору 2-(4'-(изобутилфенил)пропионилхлорида (1 г; 4,34 ммоль) в сухом CH2Cl2 (20 мл) добавляют гидрохлорид O-метилгидроксиламина (0,435 г; 5,201 ммоль) и триэтиламин (1,44 мл; 10,41 ммоль). Образовавшуюся смесь перемешивают при комнатной температуре в течение ночи. Органическую фазу промывают 4 н HCl (2×10 мл), сушат над Na2SO4 и упаривают в вакууме, получая при этом чистый продукт в виде бледно-желтого масла (1 г; 4,2 ммоль).
[α]D=-34 (с=1%; EtOH).
1H-ЯМР (CDCl3): δ 7,8 (шир.с, 1Н, CONH), 7,05 (д, 2Н, J=8 Гц), 6,95 (д, 2Н, J=8 Гц), 3,51 (шир.с, 3Н), 3,45 (м, 1Н), 2,32 (д, 2Н, J=7 Гц), 1,82 (м, 1Н), 1,45 (т, 3Н, J=7 Гц), 0,82 (д, 6Н, J=7 Гц).
Пример 21
R-(-)-2-[(4'-(изобутил)фенил]-N-карбоксиметокси)пропионамид
К охлажденному (Т=0-5°С) раствору (R)-2-[(4'-(изобутил)фенил]пропионовой кислоты (5 г; 24,24 ммоль) в ДМФ (20 мл) при перемешивании добавляют гидроксибензотриазол (НОВТ) (22,2 ммоль). Спустя 15' добавляют смесь полугидрохлорида карбоксиметоксиламина (2,65 г; 12,12 ммоль) и триэтиламина (3 мл) в ДМФ (5 мл); наконец, по порциям добавляют N,N-дициклогексилкарбодиимид (DCC) (24,24 ммоль). Образовавшуюся смесь перемешивают в течение 2 час при Т=0°С и затем в течение ночи при комнатной температуре. После того, как образованный осадок отфильтровывают, фильтрат разбавляют н-гексаном (50 мл); образованный осадок фильтруют и очищают флэш-хроматографией на силикагеле, получая при этом нужный продукт в виде белого порошка (1,69 г; 6 ммоль).
[α]D=-17,6 (с=0,6%; СН3ОН).
1H-ЯМР (CDCl3): δ 9,3 (шир.с, 2Н, CONH+СООН), 7,05 (д, 2Н, J=8 Гц), 6,95 (д, 2Н, J=8 Гц), 4,35 (с, 2Н), 3,45 (м, 1Н), 2,34 (д, 2Н, J=7 Гц), 1,85 (м, 1Н), 1,45 (д, 3Н, J=7 Гц), 0,81 (д, 6Н, J=7 Гц).
Пример 22
R-(-)-2-[(2',6'-Дихлорфенил)амино]фенил-N-[2''-гидрокси-2'''-этоксиэтил)пропионамид
К охлажденному (Т=0-5°С) раствору (R)-2-[2'-(2,6-дихлорфениламино)фенил]пропионовой кислоты (7,51 г; 24,24 ммоль) в ДМФ (20 мл) при перемешивании добавляют гидроксибензотриазол НОВТ (22,2 ммоль). Спустя 15' добавляют 2-аминоэтоксиэтанол (2,33 г; 22,2 ммоль) в ДМФ (5 мл); наконец, по порциям добавляют N,N-дициклогексилкарбодиимид (DCC) (24,24 ммоль). Образовавшуюся смесь перемешивают в течение 2 час при Т=0°С и затем в течение ночи при комнатной температуре. После того, как осадок отфильтровывают, фильтрат упаривают в вакууме; сырой остаток очищают флэш-хроматографией, получая при этом R-(-)-2-[(2',6'-дихлорфенил)амино]фенил-N-(2''-гидрокси-2'''-этоксиэтил)пропионамид в виде белого твердого вещества (6,44 г; 16,7 ммоль).
[α]D=-51 (с=1%; EtOH).
1H-ЯМР (CDCl3): δ 7,35 (д, 2Н, J=8 Гц), 7,20-7,05 (м, 2Н), 7,00-6,85 (м, 2Н), 6,55 (д, 1Н, J=8 Гц), 6,18 (шир.с, 1Н, CONH), 3,85 (м, 1Н), 3,65-3,40 (м, 8Н), 1,45 (д, 3Н, J=7 Гц).
Пример 23
R-(-)-2-[(3'-Ацетил)фенил]-N-(4''-пиримидил)пропионамид
Раствор R-(-)-2-[(3'-ацетил)фенил]пропионилхлорида (0,96 г; 4,27 ммоль) в сухом CH2Cl2 (10 мл) добавляют по каплям к раствору 4-аминопиримидина (1 г; 10 ммоль) в сухом СН2Cl3 (10 мл). Образовавшийся раствор перемешивают при комнатной температуре в течение ночи. Образованный осадок отфильтровывают и фильтрат промывают водой (2×10 мл), насыщенным раствором NaCl, сушат над Na2SO4 и упаривают в вакууме, получая при этом сырой остаток, который очищают кристаллизацией из н-гексана. Чистый продукт получают в виде белого твердого вещества (0,62 г; 2,3 ммоль).
[α]D=-139 (с=0,5%; СН3ОН).
1H-ЯМР (CDCl3): δ 8,80 (с, 1Н), 8,60 (м, 1H), 8,20 (д, 1Н, J=4 Гц), 8,00-7,95 (м, 2Н), 7,81 (шир.с, 1H, CONH), 7,63 (д, 1H, J=7 Гц), 3,80 (кв., 1H, J=7 Гц), 2,6 (с, 3Н), 1,54 (д, 3Н, J=7 Гц).
Пример 24
(R)-2-[(3'-α-Гидроксиизопропил)фенил]-N-(метоксиэтил)пропионамид
К охлажденному (Т=0-5°С) раствору (R)-2-[(3'-α-гидроксиизопропил)фенил]пропионовой кислоты (5,04 г; 24,24 ммоль) в ДМФ (20 мл) при перемешивании добавляют гидроксибензотриазол (НОВТ) (22,2 ммоль). Спустя 15' добавляют O-метилэтаноламин (1,66 г; 22,2 ммоль) в ДМФ (5 мл)); наконец, по порциям добавляют N,N-дициклогексилкарбодиимид (DCC) (24,24 ммоль). Образовавшуюся смесь перемешивают в течение 2 час при Т=0°С и затем в течение ночи при комнатной температуре. После того, как образованный осадок отфильтровывают, фильтрат упаривают в вакууме; сырой остаток очищают флэш-хроматографией, получая при этом (R)-2-[(3'-α-гидроксиизопропил)фенил]-N-(метоксиэтил)пропионамид в виде бесцветного масла (5,3 г; 20 ммоль).
[α]D=-63 (с=0,5%; СН3ОН).
1Н-ЯМР (CDCl3): δ 7,65 (шир.с, 1Н, CONH), 7,31-7,14 (м, 4Н), 4,02 (шир.с, 1Н, ОН), 3,78 (т, 2Н, J=8 Гц)C C,68 (кв., J=7 Гц), 3,4 (т, 2Н, J=8 Гц), 1,85 (с, 6Н), 1,4 (д, 3Н, J=7 Гц).
Экспериментальная модель in vivo инфильтрации полиморфоядерных нейтрофилов сердца, индуцированной реперфузией у ишемической крысы
Эффективность (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамида по предотвращению инфильтрации полиморфоядерных нейтрофилов в сердце крысы, перенесшей ишемию, была подтверждена на модели ишемической крысы.
В указанной экспериментальной модели на крысе ишемию индуцировали 30-минутной окклюзией левой коронарной артерии с последующей 4-часовой реперфузией и являющейся результатом этого инфильтрацией полиморфоядерных нейтрофилов.
Обработка (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамидом (15 мг/кг) значительно снижала инфильтрацию нейтрофилов сердца, индуцированную реперфузией (40% ингибирование), как оценено по активности тканевой миелопероксидазы (МРО) (31±7 и 18±5 мU/мг белка без и с обработкой (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбокси-этил)пропионамидом, соответственно).
Эффективность действия репрезентативных соединений амидов формулы (I) на хемотаксис нейтрофилов человека, индуцированный ИЛ-8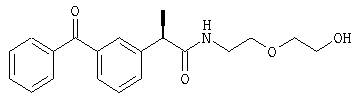
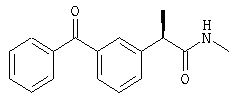
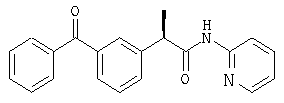
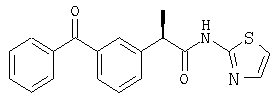
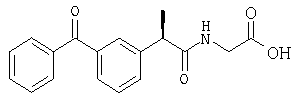
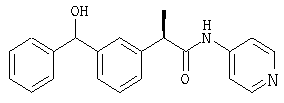
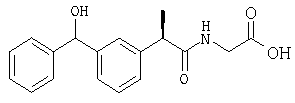
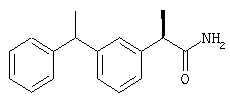
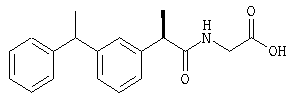
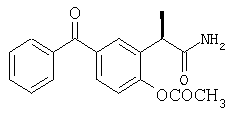
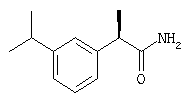
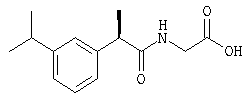
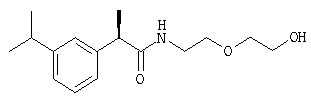
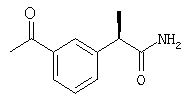
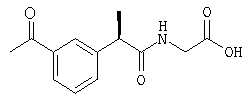
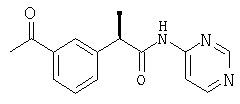
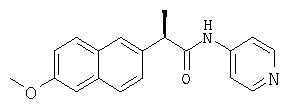
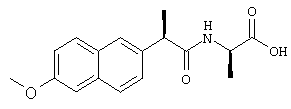
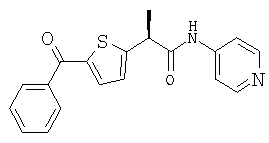
Приложение А
Экспериментальная модель in vivo инфильтрации полиморфоядерных нейтрофилов сердца, индуцированной реперфуэией у ишемической крысы
Эффективность (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил-)пропионамида по предотвращению инфильтрации полиморфоядерных нейтрофилов в сердце крысы, перенесшей ишемию, была подтверждена на модели ишемической крысы.
В указанной экспериментальной модели на крысе ишемию индуцировали 30-минутной окклюзией левой коронарной артерии с последующей 4-часовой реперфузией и являющейся результатом этого инфильтрацией полиморфоядерных нейтрофилов.
Обработка (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамидом (15 мг/кг) значительно снижала инфильтрацию нейтрофилов сердца, индуцированную реперфузией (40% ингибирование), как оценено по активности тканевой миелопероксидазы (МРО) (31±7 и 18±5 мU/мг белка без и с обработкой (R,S')-2-[(4'-изобутил)фенил]-N-(2-карбокси-этил)пропионамидом, соответственно).
Приложение В
Эффективность действия репрезентативных соединений амидов хемотаксис нейтрофилов человека, индуцированный ИЛ-8

| название | год | авторы | номер документа |
|---|---|---|---|
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| ПРИМЕНЕНИЕ ХИРАЛЬНЫХ АРИЛКЕТОНОВ В ЛЕЧЕНИИ НЕЙТРОФИЛ-ЗАВИСИМЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2345759C2 |
| N-(2-АРИЛПРОПИОНИЛ)СУЛЬФОНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1999 |
|
RU2255084C2 |
| ЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ СОЛИ ОМЕГА-АМИНОАЛКИЛАМИДОВ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 2002 |
|
RU2291857C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2005 |
|
RU2375347C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2193029C2 |
| ПРОИЗВОДНЫЕ 2-АРИЛПРОПИОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ ВКЛЮЧАЮЩИЕ | 2005 |
|
RU2410372C2 |
Изобретение относится к (R)-энантиомерам 2-арилпропионамидам формулы (Ia)
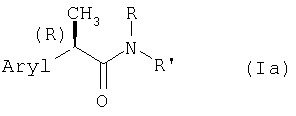
и их фармацевтически приемлемым солям, где Aryl представляет фенильную группу, замещенную группой, выбранной из изопропила, ацетила, (2'',6''-дихлорфенил)амино, α-гидроксиизопропила, (R,S)-α-гидроксибензила и его индивидуального R-изомера, (R,S)-(α-метилбензила) и его индивидуального R-изомера и (R,S)-α-гидрокси-α-метилбензила и его индивидуального R-изомера; R представляет Н или С1-С4-алкил; R' представляет: аминокислотный остаток, состоящий из неразветвленного или разветвленного C1-С6-алкила, замещенного карбоксигруппой CO2H; остаток формулы -СН2-СН2Х-(СН2-СН2O)nR, где R имеет указанные выше значения; n равно целому числу от 0 до 1, тогда как Х представляет собой кислород; гетероарил, выбранный из группы, состоящей из 2-пиримидинила или 4-пиримидинила. Предложена фармацевтическая композиция, ингибирующая хемотаксис нейтрофилов, индуцированный интерлейкином-8, содержащая в качестве активного начала (R)-энантиомеры 2-арилпропионамидов формулы (I) и их фармацевтически приемлемые соли в смеси с его подходящим носителем. Предложен способ получения соединений формулы (Ia). Также предложены (R)-энантиомеры 2-арилпропионовых кислот формулы (Va) и их фармацевтически приемлемые соли. Технический результат - (R)-2-арилпропионамиды, полезные при профилактике и лечении повреждения ткани, обусловленного усиленным пополнением полиморфоядерных нейтрофилов в местах воспаления. 4 н. и 9 з.п. ф-лы, 6 табл.
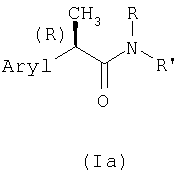
и их фармацевтически приемлемые соли,
где
Aryl представляет фенильную группу, замещенную группой, выбранной из изопропила, ацетила, (2'',6''-дихлорфенил)амино, α-гидроксиизопропила, (R,S)-α-гидроксибензила и его индивидуального R-изомера, (R,S)-(α-метилбензила) и его индивидуального R-изомера и (R,S)-α-гидрокси-α-метилбензила и его индивидуального R-изомера;
R представляет Н или С1-C4-алкил;
R' представляет:
аминокислотный остаток, состоящий из неразветвленного или разветвленного C1-С6-алкила, замещенного карбоксигруппой СО2Н;
остаток формулы -CH2-CH2X-(CH2-CH2O)nR, где R имеет указанные выше значения; n равно целому числу от 0 до 1, тогда как Х представляет кислород;
гетероарил, выбранный из группы, состоящей из 2-пиримидинила или 4-пиримидинила.
(R)-(-)-2-[(3'-изопропил)фенил]-N-карбоксиметилпропионамид;
(R,S')-(-)-2-[(3'-α-метилбензил)фенил]-N-карбоксиметил-пропионамид;
(R,R')-(-)-2-[(3'-(α-метилбензил)фенил]-N-карбоксиметил-пропионамид;
(R)-2-[(3'α-гидроксиизопропил)фенил]-N-(метоксиэтил)-пропионамид;
R(-)-2-[(3'-ацетил)фенил]-N-(4''-пиримидил)пропионамид;
R(-)-2-[(2',6'-дихлорфенил)амино]фенил-N-(2''-гидрокси-2'''-этоксиэтил)пропионамид;
(R),(R',S')-2-[3'-(α-гидроксибензил)фенил]-N-(карбоксиметил)-пропионамид;
(R),(R',S')-2-[3'-(α-гидрокси-α-метилбензил)фенил]-N-(карбоксиметил)пропионамид;
(R)(-)-2-[(3'-ацетил)фенил]-N-карбоксиметилпропионамид;
(R)(-)-2-[(3'-изопропил)фенил]-N-(2''-гидроксиэтокси)-этилпропионамид.
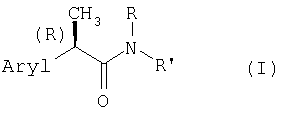
и их фармацевтически приемлемые соли,
где
Aryl представляет фенильную группу, замещенную одной или несколькими группами, независимо выбранными из галогена, С1-С4-алкила, С1-С4-алкокси, гидрокси, С1-С7-ацилокси, циано, нитро, амино, C1-С3-ациламино, галоген-С1-С3-алкила, гидрокси-С1-С3-алкила, галоген-С1-С3-алкокси, гидрокси-С1-С3-арилалкила, бензоила, 3-фенокси, 4-(2-тиенилкарбонил), 3-хлор-4-(3-пирролин-1-ил);
R представляет Н, С1-С4-алкил, аллил, пропаргил, СН2-CO2Н или (СН2)2-CO2Н;
R' представляет
аминокислотный остаток, состоящий из неразветвленного или разветвленного C1-С6-алкила, алкенила, циклоалкила, фенилалкила, замещенного одной или несколькими карбоксигруппами CO2Н и гетероатомом, выбранным из кислорода или серы;
остаток формулы -СН2-СН2Х-(СН2-СН2O)nR, где R имеет значения, указанные выше; n равно целому числу от 0 до 5, тогда как Х представляет кислород или серу;
остаток формулы (R)- или (S)-CH (СН3)-СН2-O-СН2-СН2-ОН;
остаток формулы OR, где R имеет значения, указанные выше;
остаток формулы (III)
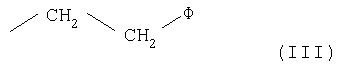
где Ф представляет 2-(1-метилпирролидил), 2-пиридил, 4-пиридил, 1-имидазолил, 4-имидазолил, 1-метил-4-имидазолил, 1-метил-5-имидазолил или группу NRaRb, где каждый из Ra и Rb, которые могут быть одинаковыми или разными, представляет C1-С6-алкил или гидроксиалкил -(СН2)m-ОН, где m равно целому числу 2 или 3, или же Ra и Rb вместе с атомом N, с которым они связаны, образуют гетероцикл из 3-7 членов формулы (IV)
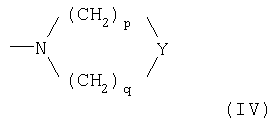
где
Y представляет простую связь, СН2, O, S или N-Rc, причем Rc представляет Н, C1-С6-алкил, гидроксиалкил (CH2)m-OH, остаток -(CH2)m'-Ar', где Ar' представляет арил, гетероарил, циклоалифатический и/или гетероциклоалифатический остаток, m' равно нулю или целому числу от 1 до 3, р и q, каждый независимо, представляет целое число от 1 до 3;
гетероарил, выбранный из группы, состоящей из 2-пиридила или 4-пиридила, 2-пиримидинила или 4-пиримидинила; 2-пиразинила, 5-метил-2-пиразинила; 3-1,2,4-тиазинила; 3-1,2,4-триазолила, 3-1-бензил-1,2,4-триазолила; 2-1,3-тиазолидинила, 2-1,3-тиазолила, 1,3-оксазолила, 3-изоксазолила, 4-дигидро-З-оксоизоксазолила, 5-метилизоксазол-4-ила, 2-имидазолила, 4-имидазолил-5-карбоксамида и 2-имидазолил-4,5-дикарбонитрила, 5-инданила, 5-индазолила, 7-азаиндол-3-ила, 2-, 3-, или 4-хинолинила;
в смеси с его подходящим носителем.
формулы (I) Aryl выбран из фенила, 4-метилфенила, 3-изопропилфенила, 4-метоксифенила, 4-ацетоксифенила, 4-гидроксифенила, 4-изобутилфенила, 3-бензоилфенила, 3-феноксифенила, 3-бензилфенила, 3-С6Н5-СН (ОН)-фенила, 4-тиеноилфенила, 2-фтор-4-бифенилила, 5-бензоил-2-ацетоксифенила, 5-бензоил-2-гидроксифенила, 3-α-метилбензилфенила, 3-гидроксипропилфенила, 3-гидроксиэтилфенила.
(R)-(-)-2-(4'-изобутилфенил)-N-метилпропионамид;
(R)-(-)-2-[(4'-изобутил)фенил]-N-карбоксиметилпропионамид;
(R)-(-)-2-[(4'-изобутил)фенил]-N-метоксикарбонилметилпропионамид;
цис-(R)-2-[(4'-изобутил)фенил]-N-(4'-карбоксициклогексил)пропионамид;
транс-(R)-2-[(4'-изобутил)фенил]-N-(4'-карбоксиметилциклогексил)пропионамид;
(R,S')-2-[(4'-изобутил)фенил]-N-(2-метоксикарбонилэтил)пропионамид;
(R,S')-2-[(4'-изобутил)фенил]-N-(2-карбоксиэтил)пропионамид;
(R,S')-2-[(4'-метокси)фенил]-N-(2-карбоксиэтил)пропионамид;
(R)-N-[2'-(4''-изобутилфенил)пропаноил]-2-аминоакриловая кислота и ее метиловый эфир;
(R)-(-)-2-[(4''-изобутил)фенил]-N-(2''-гидроксиэтоксиэтил)пропионамид;
(R,S')-2-[(4''-изобутил)фенил]-N-[1'-метил-2'-(2''''-гидроксиэтокси)этил]пропионамид;
(R,R')-2-[(4''-изобутил)фенил]-N-[1'-метил-2'-(2''''-гидроксиэтокси)этил]пропионамид;
(R)-(-)-2-(4'-изобутилфенил)-N-(2''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-(4'-изобутилфенил)-N-(4''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(3'-бензоил)фенил]-N-(2''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(2'-гидрокси-5'-бензоил)фенил]-N-(2''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(2'-гидрокси-5'-бензоил)фенил]-N-(4''-пиридил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(2'-гидрокси-5'-бензоил)фенил]-N-карбоксиметилпропионамид;
(R)-(-)-2-(4'-изобутилфенил)-N-(2''-пиразинил)пропионамид и его гидрохлорид;
(R)-(-)-2-(4'-изобутилфенил)-N-(2''-пиримидинил)пропионамид и его гидрохлорид;
(R)-(-)-2-(4'-изобутилфенил)-N-(4''-пиримидинил)пропионамид и его гидрохлорид;
(R)-(-)-2-[(3'-изопропил)фенил]-N-карбоксиметилпропионамид;
(R,S')-(-)-2-[(3'-α-метилбензил)фенил]-N-карбоксиметилпропионамид;
(R,R')-(-)-2-[(3'-α-метилбензил)фенил]-N-карбоксиметилпропионамид;
(R)-2-[(3'-α-гидроксиизопропил)фенил]-N-(метоксиэтил)пропионамид;
R(-)-2-[3'-ацетил)фенил]-N-(4''-пиримидил)пропионамид;
R(-)-2-[(2',6'-дихлорфенил)амино]фенил-N-(2''-гидрокси-2'''-этоксиэтил)пропионамид;
(R),(R',S')-2-[3'-(α-гидроксибензил)фенил]-N-(карбоксиметил)пропионамид;
(R),(R',S')-2-[3'-(α-гидрокси-α-метилбензил)фенил]-N-(карбоксиметил)пропионамид;
(R)(-)-2-[(3'-ацетил)фенил]-N-карбоксиметилпропионамид
(R)(-)-2-[(3'-изопропил)фенил]-N-(2''-гидроксиэтокси)этилпропионамид.
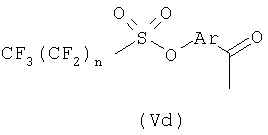
с получением после этерификации и альфа-метилирования, арилпропионовых производных (Ve)
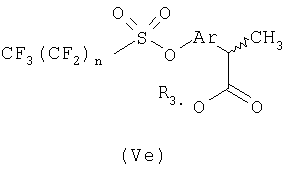
где n равно целому числу от 1 до 9 и R3 представляет С1-С4-алкил или С2-С4-алкенил, и взаимодействие соединений формулы Ve с соединением трибутилолово-R4, где R4 представляет изопропил, ацетил, (2'',6''-дихлорфениламино), α-гидроксибензил, α-метилбензил, α-гидрокси-α-метилбензил с получением соответствующего (R,S)-2-арилпропионата формулы (Vf).
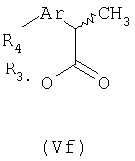
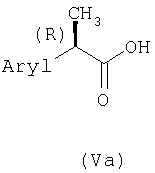
и их фармацевтически приемлемые соли, где Aryl представляет фенильную группу, замещенную группой, выбранной из изопропила, во 2-ом или 3-положении фенильной группы, ацетила, α-гидроксиизопропила, (R,S)-α-гидроксиэтила и его индивидуального R- и S-изомера, (R,S)-α-гидроксибензила и его индивидуального R- и S-изомера, (R,S)-α-метилбензила и его индивидуального R- и S-изомера, (R,S)-α-гидрокси-α-метилбензила и его индивидуального R- и S-изомера.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| ЛЕВИТ Г.Л | |||
| и др | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЖОрХ, 1998, т.34, вып.3, р.378-382 | |||
| WO 00/02903 A1, 20.01.2000 | |||
| MIRANDA, MIGUEL A | |||
| et al | |||
| Enantioselective discrimination in the intramolecular Quenching of an Excited Aromatic Ketone by a Ground-State Phenol | |||
| J | |||
| Am | |||
| Chem | |||
| Soc | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| WO | |||

