ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам получения гиалуронана в рекомбинантной клетке-хозяине.
ОПИСАНИЕ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
Наиболее распространенными гетерополисахаридами в организме являются гликозаминогликаны. Гликозаминогликаны являются неразветвленными углеводными полимерами, состоящими из повторяющихся дисахаридных единиц (только кератансульфат разветвлен в коровой области углевода). Дисахаридные единицы в основном содержат в качестве первой сахаридной единицы один из двух модифицированных сахаров - N-ацетилгалактозамин (GalNAc) или N-ацетилглюкозамин (GlcNAc). Вторая единица обычно представляет собой уроновую кислоту, такую как глюкуроновая кислота (GlcUA) или идуронат.
Гликозаминогликаны являются отрицательно заряженными молекулами и имеют протяженную конформацию, которая придает раствору высокую вязкость. Гликозаминогликаны расположены преимущественно на поверхности клеток или во внеклеточном матриксе. Гликозаминогликаны также обладают низкой сжимаемостью в растворе и в результате идеальны в качестве физиологической смазочной жидкости, например, в суставах. Жесткость гликозаминогликанов придает структурную целостность клеткам и обеспечивает ходы между клетками, позволяя им мигрировать. Гликозаминогликанами с наибольшей физиологической значимостью являются гиалуронан, хондроитинсульфат, гепарин, гепарансульфат, дерматансульфат и кератансульфат. Большинство гликозаминогликанов ковалентно связываются с коровым белком протеогликана посредством специфических олигосахаридных структур. Гиалуронан образует крупные агрегаты с конкретными протеогликанами, но исключением являются свободные углеводные цепи, образующие нековалентные комплексы с протеогликанами.
Определены многочисленные функции гиалуронана в организме (см., Laurent T. C. and Fraser J. R. E., 1992, FASEB J. 6: 2397-2404; and Toole B. P., 1991, "Proteoglycans and hyaluronan in morphogenesis and differentiation." In: Cell Biology of the Extracellular Matrix, pp. 305-341, Hay E. D., ed., Plenum, New York). Гиалуронан присутствует в гиалиновом хряще, синовиальной суставной жидкости и кожной ткани как в дерме, так и в эпидермисе. Гиалуронан также, как предполагают, участвует во многих физиологических функциях, таких как адгезия, развитие, клеточная подвижность, злокачественные опухоли, ангиогенез и заживление ран. Вследствие уникальных физических и биологических свойств гиалуронан используется в глазной и суставной хирургии и разрабатывается для применения в других медицинских процедурах. Продукты гиалуронана также разработаны для применения в ортопедии, ревматологии и дерматологии.
Значимым коммерческим источником гиалуронана являются петушиные гребни. Микроорганизмы являются альтернативным источником. В патенте США № 4801539 описан способ ферментации для получения гиалуроновой кислоты с использованием штамма Streptococcus zooepidemicus с приведенными выходами, составляющими примерно 3,6 г гиалуроновой кислоты на литр. В Европейском патенте № EP0694616 описаны способы ферментации, использующие улучшенный штамм Streptococcus zooepidemicus с приведенными выходами, составляющими примерно 3,5 г гиалуроновой кислоты на литр.
Микроорганизмы, используемые для получения гиалуроновой кислоты путем ферментации, представляют собой штаммы патогенных бактерий, основные из которых представляют собой различные виды Streptococcus. Стрептококки группы A и группы C окружают себя неантигенной капсулой, состоящей из гиалуронана, которая по композиции идентична той, которая обнаружена в соединительной ткани и суставах. Pasteurella multocida, другая патогенная инкапсулированная бактерия, также окружает свои клетки гиалуронаном.
Описаны гиалуронансинтазы позвоночных, бактериальных патогенов и вирусов водорослей (DeAngelis, P. L., 1999, Cell. Mol. Life Sci, 56: 670-682). В WO 99/23227 описана гиалуронатсинтаза группы I Streptococcus equisimilis. В WO 99/51265 и WO 00/27437 описана гиалуронатсинтаза группы II Pasturella multocida. Ferretti et al. описывают оперон гиалуронансинтазы Streptococcus pyogenes, который состоит из трех генов hasA, hasB и hasC, которые кодируют гиалуронатсинтазу, UDP-глюкозодегидрогеназу и UDP-глюкозопирофосфорилазу соответственно (Proc. Natl. Acad. Sci. USA. 98, 4658-4663,2001). В WO 99/51265 описан сегмент нуклеиновой кислоты, имеющий кодирующую область гиалуронансинтазы Streptococcus equisimilis.
Бациллы являются хорошо отработанными системами клеток-хозяев для продукции нативных и рекомбинантных белков. Целью настоящего изобретения является предоставление способов получения гиалуронана в рекомбинантной клетке-хозяине Bacillus.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам получения гиалуроновой кислоты, включающим: (a) культивирование клетки-хозяина Bacillus в условиях, подходящих для продуцирования гиалуроновой кислоты, при этом клетка-хозяин Bacillus содержит конструкцию нуклеиновой кислоты, включающую последовательность, кодирующую гиалуронансинтазу, функционально связанную с промоторной последовательностью, чужеродной в отношении последовательности, кодирующей гиалуронансинтазу; и (b) извлечения гиалуроновой кислоты из среды культивирования.
В предпочтительных осуществлениях конструкция нуклеиновой кислоты дополнительно включает один или более генов, кодирующих ферменты биосинтеза предшественника сахара гиалуроновой кислоты, или клетка-хозяин Bacillus дополнительно включает одну или более вторых конструкций нуклеиновой кислоты, содержащих один или более генов, кодирующих ферменты биосинтеза предшественника сахара.
Еще в одном предпочтительном осуществлении один или более генов, кодирующих предшественник сахара, находятся под контролем того же самого или другого(-их) промотора(-ов), как последовательность, кодирующая последовательность гиалуронансинтазы.
Настоящее изобретение также относится к клеткам-хозяевам Bacillus, включающим конструкцию нуклеиновой кислоты, содержащую последовательность, кодирующую гиалуронансинтазу, функционально связанную с промоторной последовательностью, чужеродной в отношении последовательности, кодирующей гиалуронансинтазу, и к таким конструкциям нуклеиновой кислоты.
Настоящее изобретение также относится к выделенной последовательности нуклеиновой кислоты, кодирующей оперон гиалуронансинтазы, содержащий ген гиалуронансинтазы или его часть и ген UDP-глюкозо-6-дегидрогеназы и, необязательно, один или более генов, выбранных из группы, состоящей из гена UDP-глюкозопирофосфорилазы, гена UDP-N-ацетилглюкозаминпирофосфорилазы и гена глюкозо-6-фосфатизомеразы.
Настоящее изобретение также относится к выделенным последовательностям нуклеиновой кислоты, кодирующим UDP-глюкозо-6-дегидрогеназу, выбранным из группы, состоящей из: (a) последовательности нуклеиновой кислоты, кодирующей полипептид, характеризующийся аминокислотной последовательностью, которая обладает, по меньшей мере, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью по отношению к SEQ ID NO: 41; (b) последовательности нуклеиновой кислоты, которая обладает, по меньшей мере, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% гомологией по отношению к SEQ ID NO: 40; (c) последовательности нуклеиновой кислоты, которая гибридизуется в условиях средней или высокой степени жесткости с (i) последовательностью нуклеиновой кислоты SEQ ID NO: 40, (ii) последовательностью кДНК, содержащейся в SEQ ID NO: 40, или (iii) комплементарной цепью (i) или (ii); и (d) субпоследовательности (a), (b), или (c), которая кодирует полипептидный фрагмент, который имеет активность UDP-глюкозо-6-дегидрогеназы.
Настоящее изобретение также относится к выделенным последовательностям нуклеиновой кислоты, кодирующим UDP-глюкозопирофосфорилазу, выбранным из группы, состоящей из: (a) последовательности нуклеиновой кислоты, кодирующей полипептид, характеризующийся аминокислотной последовательностью, которая обладает, по меньшей мере, примерно 90%, примерно 95% или примерно 97% идентичностью по отношению к SEQ ID NO: 43; (b) последовательности нуклеиновой кислоты, которая обладает, по меньшей мере, примерно 90%, примерно 95% или примерно 97% гомологией по отношению к SEQ ID NO: 42; (c) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с (i) последовательностью нуклеиновой кислоты SEQ ID NO: 42, (ii) последовательностью кДНК, содержащейся в SEQ ID NO: 42, или (iii) с комплементарной цепью (i) или (ii); и (d) субпоследовательности (a), (b), или (c), которая кодирует полипептидный фрагмент, который имеет активность UDP-глюкозопирофосфорилазы.
Настоящее изобретение также относится к выделенным последовательностям нуклеиновой кислоты, кодирующим UDP-N-ацетилглюкозаминпирофосфорилазу, выбранным из группы, состоящей из: (a) последовательности нуклеиновой кислоты, кодирующей полипептид, характеризующийся аминокислотной последовательностью, которая обладает, по меньшей мере, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью по отношению к SEQ ID NO: 45; (b) последовательности нуклеиновой кислоты, которая обладает, по меньшей мере, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% гомологией по отношению к SEQ ID NO: 44; (c) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с (i) последовательностью нуклеиновой кислоты SEQ ID NO: 44, (ii) последовательностью кДНК, содержащейся в SEQ ID NO: 44, или (iii) с комплементарной цепью (i) или (ii); и (d) субпоследовательности (a), (b), или (c), которая кодирует полипептидный фрагмент, который имеет активность UDP-N-ацетилглюкозаминпирофосфорилазы.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фигуре 1 показана химическая структура гиалуронана.
На фигуре 2 показан биосинтетический путь синтеза гиалуронана.
На фигуре 3 показана карта рестрикции pCR2.1-sehasA.
На фигуре 4 показана карта рестрикции pCR2.1-tuaD.
На фигуре 5 показана карта рестрикции pCR2.1-gtaB.
На фигуре 6 показана карта рестрикции pCR2.1-gcaD.
На фигуре 7 показана карта рестрикции pHA1.
На фигуре 8 показана карта рестрикции pHA2.
На фигуре 9 показана карта рестрикции pHA3.
На фигуре 10 показана карта рестрикции pHA4.
На фигуре 11 показана карта рестрикции pHA5.
На фигуре 12 показана карта рестрикции pHA6.
На фигуре 13 показана карта рестрикции pHA7.
На фигуре 14 показана карта рестрикции pMRT106.
На фигуре 15 показана карта рестрикции pHA8.
На фигуре 16 показана карта рестрикции pHA9.
На фигуре 17 показана карта рестрикции pHA10.
На фигуре 18 показана карта рестрикции pRB157.
На фигуре 19 показана карта рестрикции pMRT084.
На фигуре 20 показана карта рестрикции pMRT086.
На фигуре 21 показана карта рестрикции pCJ791.
На фигуре 22 показана карта рестрикции pMRT032.
На фигуре 23 показана карта рестрикции pNNB194neo.
На фигуре 24 показана карта рестрикции pNNB194neo-oriT.
На фигуре 25 показана карта рестрикции pShV3.
На фигуре 26 показана карта рестрикции pShV2.1-amyEΔB.
На фигуре 27 показана карта рестрикции pShV3A.
На фигуре 28 показана карта рестрикции pMRT036.
На фигуре 29 показана карта рестрикции pMRT037.
На фигуре 30 показана карта рестрикции pMRT041.
На фигуре 31 показана карта рестрикции pMRT064.1.
На фигуре 32 показана карта рестрикции pMRT068.
На фигуре 33 показана карта рестрикции pMRT069.
На фигуре 34 показана карта рестрикции pMRT071.
На фигуре 35 показана карта рестрикции pMRT074.
На фигуре 36 показана карта рестрикции pMRT120.
На фигуре 37 показана карта рестрикции pMRT122.
На фигуре 38 показана карта рестрикции pCR2.1-pel5'.
На фигуре 39 показана карта рестрикции pCR2.1-pel3'.
На фигуре 40 показана карта рестрикции pRB161.
На фигуре 41 показана карта рестрикции pRB162.
На фигуре 42 показана карта рестрикции pRB156.
На фигуре 43 показана карта рестрикции pRB164.
На фигуре 44 показана сводка ферментаций различных штаммов Bacillus subtilis, продуцирующих гиалуроновую кислоту, содержащихся в питательной среде примерно с 2 г сахарозы/Lo-ч, при 37°C.
На фигуре 45 показана сводка пиков средних молекулярных масс гиалуроновой кислоты (МДа), полученных в результате ферментаций различных штаммов Bacillus subtilis, продуцирующих гиалуроновую кислоту, содержащихся в питательной среде примерно с 2 г сахарозы/Lo-ч, при 37°C.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам получения гиалуронана, включающим: (a) культивирование клетки-хозяина Bacillus в условиях, подходящих для продукции гиалуроновой кислоты, где клетка-хозяин Bacillus содержит конструкцию нуклеиновой кислоты, включающую последовательность, кодирующую гиалуронансинтазу, функционально связанную с промоторной последовательностью, чужеродной в отношении последовательности, кодирующей гиалуронансинтазу; и (b) извлечения гиалуронана из среды культивирования.
Способы по настоящему изобретению представляют собой улучшение по сравнению с продукцией гиалуронана из патогенных, инкапсулированных бактерий. В инкапсулированных бактериях в капсуле продуцируется большое количество гиалуронана. При переработке и очистке гиалуронана из таких источников вначале необходимо извлечь гиалуронан из капсулы, например, с использованием поверхностно-активного вещества или детергента, такого как SDS. Это приводит к усложнению коммерческого получения гиалуронана, поскольку поверхностно-активное вещество должно добавляться для высвобождения большой порции гиалуронана и впоследствии поверхностно-активное вещество должно быть извлечено перед конечной очисткой.
Настоящее изобретение обеспечивает получение большого количества гиалуронана, которое продуцируется в неинкапсулированной клетке-хозяине в виде свободного гиалуронана. При рассмотрении через микроскоп, с рекомбинантными штаммами Bacillus не ассоциировано видимой капсулы, в то время как патогенные штаммы, традиционно используемые при продукции гиалуронана, содержат капсулу гиалуронана, которая, по меньшей мере, в два раза превышает диаметр самой клетки.
Поскольку гиалуронан рекомбинантной клетки Bacillus экспрессируется непосредственно в культуральную среду, для выделения гиалуронана из культуральной среды может использоваться простой способ. Вначале клетки Bacillus и клеточный дебрис физически удаляют из культуральной среды. Вначале культуральную среду можно развести, если это требуется, чтобы снизить вязкость среды. Для удаления клеток из культуральной среды может использоваться много известных способов, таких как центрифугирование или микрофильтрация. Если требуется, оставшаяся надосадочная жидкость может отфильтровываться, например, путем ультрафильтрации для концентрирования и отделения от гиалуронана низкомолекулярных примесей. После удаления клеток и клеточного дебриса простое осаждение гиалуронана из среды проводят по известным механизмам. Соль, спирт или комбинации соли и спирта могут использоваться для осаждения гиалуронана из фильтрата. Будучи сокращен до осадка, гиалуронан может легко отделяться от раствора физическими средствами. Альтернативно гиалуронан может высушиваться или концентрироваться из раствора фильтрата с использованием способов упаривания, известных в данной области, таких как сушка распылением.
Таким образом, способы настоящего изобретения представляют собой улучшение по сравнению с существующими способами коммерческокого получения гиалуронана путем ферментации в том, что они не требуют применения поверхностно-активного вещества при очистке гиалуронана от клеток в культуре.
ГИАЛУРОНОВАЯ КИСЛОТА
«Гиалуроновая кислота» определяется здесь как несульфатированный гликозаминогликан, состоящий из повторяющихся дисахаридных единиц N-ацетилглюкозамина (GIcNAc) и глюкуроновой кислоты (GIcUA), связанных вместе перемежающимися бета-1,4- и бета-1,3-гликозидными связями (фигура 1). Гиалуроновая кислота также известна как гиалуронан, гиалуронат или HA. Термины гиалуронан и гиалуроновая кислота используются здесь взаимозаменяемо.
В предпочтительном осуществлении гиалуроновая кислота, полученная способами по настоящему изобретению, имеет молекулярную массу, составляющую примерно от 10000 до примерно 10000000 Да. В более предпочтительном осуществлении гиалуроновая кислота, полученная способами по настоящему изобретению, имеет молекулярную массу, составляющую примерно от 25000 до примерно 5000000 Да. В наиболее предпочтительном осуществлении гиалуроновая кислота, полученная способами по настоящему изобретению, имеет молекулярную массу, составляющую примерно от 50000 до примерно 3000000 Да.
Уровень гиалуроновой кислоты, продуцируемой клеткой-хозяином по настоящему изобретению, может определяться модифицированным карбазольным способом (Bitter and Muir, 1962, Anal Biochem. 4: 330-334). Более того, средняя молекулярная масса гиалуроновой кислоты может определяться с использованием стандартных способов, известных в данной области, таких как описанные Ueno et al., 1988, Chem. Pharm. Bull. 36, 4971-4975; Wyatt, 1993, Anal. Chim. Acta 272: 1-40; и Wyatt Technologies, 1999, "Light Scattering University DAWN Course Manual" и "DAWN EOS Manual" Wyatt Technology Corporation, Санта-Барбара, Калифорния.
Гиалуроновая кислота, полученная способами по настоящему изобретению, может подвергаться воздействию различных способов, известных в данной области, для модификации гиалуроновой кислоты, такой как перекрестное связывание, описанное, например, в патентах США № 5616568, 5652347 и 5874417. Более того, молекулярная масса гиалуроновой кислоты может изменяться с использованием известных в данной области способов.
КЛЕТКИ-ХОЗЯЕВА
По способам настоящего изобретения клетка-хозяин Bacillus может представлять собой любую клетку Bacillus, подходящую для рекомбинантного продуцирования гиалуроновой кислоты. Клетка хозяина Bacillus может быть клеткой Bacillus дикого типа или ее мутантом. Клетки Bacillus, подходящие для воплощения настоящего изобретения, включают в качестве неограничивающих примеров клетки Bacillus agaraderhens, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus brevis, Bacillus circulans, Bacillus clausii, Bacillus coagulans, Bacillus firmus, Bacillus lautus, Bacillus lentus, Bacillus licheniformis, Bacillus megaterium, Bacillus pumilus, Bacillus stearothermophilus, Bacillus subtilis, и Bacillus thuringiensis. Мутантные клетки Bacillus subtilis, конкретно адаптированные для рекомбинантной экспрессии, описаны в WO 98/22598. Неинкапсулированные клетки Bacillus являются особенно применимыми по настоящему изобретению.
В предпочтительном осуществлении клетка-хозяин Bacillus представляет собой клетку Bacillus amyloliquefaciens, Bacillus clausii, Bacillus lentus, Bacillus licheniformis, Bacillus stearothermophilus или Bacillus subtilis. В более предпочтительном осуществлении клетка Bacillus представляет собой клетку Bacillus amyloliquefaciens. В другом более предпочтительном осуществлении клетка Bacillus представляет собой клетку Bacillus clausii. В другом более предпочтительном осуществлении клетка Bacillus представляет собой клетку Bacillus lentus. В другом более предпочтительном осуществлении клетка Bacillus представляет собой клетку Bacillus licheniformis. В другом более предпочтительном осуществлении клетка Bacillus представляет собой клетку Bacillus subtilis. В наиболее предпочтительном осуществлении клетка Bacillus представляет собой клетку Bacillus subtilis A164Δ5 (см. патент США № 5891701) или Bacillus subtilis 168Δ4.
Трансформация клетки-хозяина Bacillus конструкцией нуклеиновой кислоты по настоящему изобретению может, например, быть осуществлена путем трансформации протопласта (см., например, Chang and Cohen, 1979, Molecular General Genetics 168: 111-115), путем использования компетентных клеток (см., например, Young and Spizizen, 1961, Journal of Bacteriology 81: 823-829, или Dubnau and Davidoff-Abelson, 1971, Journal of Molecular Biology 56: 209-221), путем электропорации (см., например, Shigekawa and Dower, 1988, Biotechniques 6: 742-751), или путем конъюгации (см., например, Koehler and Thorne, 1987, Journal of Bacteriology 169: 5271-5278).
КОНСТРУКЦИИ НУКЛЕИНОВОЙ КИСЛОТЫ
«Конструкция нуклеиновой кислоты» определяется здесь как молекула нуклеиновой кислоты, одно- или двухцепочечная, которая выделена из встречающегося в природе гена или которая была модифицирована так, что содержит сегменты нуклеиновой кислоты, которые скомбинированы и помещены рядом способом, который не может существовать иначе в природе. Термин «конструкция нуклеиновой кислоты» может быть синонимом термину «экспрессирующая кассета», где конструкция нуклеиновой кислоты содержит все регуляторные последовательности, требуемые для экспрессии кодирующей последовательности. Термин «кодирующая последовательность» определена здесь как последовательность, которая транскрибируется в мРНК и транслируется в интересующий фермент, при помещении под контроль указанных ниже регуляторных последовательностей. Границы кодирующих последовательностей в общем определены участком связывания рибосомы, расположенным непосредственно выше открытой рамки считывания на 5'-конце мРНК, и последовательностью терминации транскрипции, локализованной непосредственно ниже открытой рамки считывания на 3'-конце мРНК. Кодирующая последовательность может включать в качестве неограничивающих примеров ДНК, кДНК и рекомбинантные последовательности нуклеиновой кислоты.
Способы, использованные для выделения или клонирования последовательности нуклеиновой кислоты, кодирующей полипептид, хорошо известны в данной области и охватывают, например, выделение из геномной ДНК, получение из кДНК или их сочетание. Клонирование последовательностей нуклеиновой кислоты из такой геномной ДНК может осуществляться, например, путем использования скрининга антител экспрессирующих библиотек для детекции клонированных фрагментов ДНК с общими структурными свойствами, или хорошо известной полимеразной цепной реакции (ПЦР). См., например, Innis et al, 1990, ПЦР Protocols: A Guide to Methods and Application, Academic Press, New York. Могут использоваться другие процедуры амплификации нуклеиновой кислоты, такие как лигазная цепная реакция, лигированная активированная транскрипция и амплификация, основанная на последовательности нуклеиновой кислоты. Процедуры клонирования могут вовлекать вырезание и выделение требуемого фрагмента нуклеиновой кислоты, содержащего последовательность нуклеиновой кислоты, кодирующую полипептид, вставку фрагмента в векторную молекулу и введение рекомбинантного вектора в клетку Bacillus, где клоны последовательности нуклеиновой кислоты могут реплицироваться. Последовательность нуклеиновой кислоты может быть геномной, кДНК, РНК, полусинтетического, синтетического происхождения, или их сочетанием.
С выделенной последовательностью нуклеиновой кислоты, кодирующей фермент, можно манипулировать различными путями для обеспечения экспрессии фермента. Манипуляция последовательностью нуклеиновой кислоты перед ее встраиванием в конструкцию или вектор может желательно или обязательно зависеть от экспрессирующего вектора или клетки-хозяина Bacillus. Способы модификации последовательностями нуклеиновой кислоты, задействующие способы клонирования, хорошо известны в данной области. Будет понятно, что последовательностью нуклеиновой кислоты можно также манипулировать in vivo в клетке-хозяине с использованием способов, хорошо известных в данной области.
Некоторое количество ферментов участвует в биосинтезе гиалуроновой кислоты. Данные ферменты охватывают гиалуронансинтазу, UDP-глюкозо-6-дегидрогеназу, UDP-глюкозопирофосфорилазу, UDP-N-ацетилгюкозаминпирофосфорилазу, глюкозо-6-фосфатизомеразу, гексокиназу, фосфоглюкомутазу, амидотрансферазу, мутазу и ацетилтрансферазу. Гиалуронансинтаза является ключевым ферментом для получения гиалуроновой кислоты.
«Гиалуронансинтаза» определена здесь как синтаза, которая катализирует удлинение цепи гиалуронана путем добавления сахарных предшественников GlcUA и GlcNAc. Аминокислотные последовательности стрептококковых гиалуронансинтаз, гиалуронансинтаз позвоночных и вирусной гиалуронансинтазы отличаются от гиалуронансинтазы Pasteurella hyaluronan, и, как было предложено, классифицируются как гиалуронансинтазы группы I и группы II, причем гиалуронансинтазы группы I охватывают стрептококковые гиалуронансинтазы (DeAngelis, 1999). Для продукции гиалуронана в клетках хозяина Bacillus менее предпочтительны гиалуронансинтазы эукариотического происхождения, такие как гиалуронансинтазы млекопитающих.
Последовательность, кодирующая гиалуронансинтазу, может представлять собой любую последовательность нуклеиновой кислоты, которая может быть экспрессирована в клетке-хозяине Bacillus. Последовательность нуклеиновой кислоты может быть любого происхождения. Предпочтительные гены гиалуронансинтазы включают любой из группы I или группы II, например гены гиалуронансинтазы группы I из Streptococcus equisimilis, Streptococcus pyogenes, Streptococcus uberis, и Streptococcus equi подвид zooepidemicus, или гены гиалуронансинтазы группы II из Pasturella multocida.
В предпочтительном осуществлении последовательность, кодирующая гиалуронансинтазу, выбрана из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 2, SEQ ID NO: 93, или SEQ ID NO: 103; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 1, SEQ ID NO: 92, или SEQ ID NO: 102; и (c) комплементарной цепи (a) или (b).
В более предпочтительном осуществлении последовательность, кодирующая гиалуронансинтазу, кодирует полипептид, характеризующийся аминокислотной последовательностью SEQ ID NO: 2, SEQ ID NO: 93, или SEQ ID NO: 103; или ее фрагмент, обладающий активностью гиалуронансинтазы.
В другом предпочтительном осуществлении последовательность, кодирующая гиалуронансинтазу, выбрана из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 95; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 94; и (c) комплементарной цепи (a) или (b).
В другом более предпочтительном осуществлении последовательность, кодирующая гиалуронансинтазу, кодирует полипептид, характеризующийся аминокислотной последовательностью SEQ ID NO: 95; или ее фрагмент, обладающий активностью гиалуронансинтазы.
Способы по настоящему изобретению также охватывают конструкции, при помощи которых предшественники сахара гиалуронана поставляются клетке-хозяину или в культуральную среду, или путем кодирования эндогенными генами, неэндогенными генами, или путем комбинации эндогенных и неэндогенных генов в клетке-хозяине Bacillus. Предшественник сахара может представлять собой D-глюкуроновую кислоту или N-ацетилглюкозамин.
По способам настоящего изобретения конструкция нуклеиновой кислоты может дополнительно содержать один или более генов, кодирующих ферменты биосинтеза предшественника сахара гиалуронана. Альтернативно клетка-хозяин Bacillus может дополнительно содержать одну или более вторых конструкций нуклеиновой кислоты, содержащих один или более генов, кодирующих ферменты биосинтеза предшественника сахара. Продукция гиалуронана улучшена путем использования конструкций с последовательностью или последовательностями нуклеиновой кислоты, кодирующими ген или гены, управляющие стадией в пути синтеза предшественника сахара гиалуронана. Под «управлением стадией в пути синтеза предшественника сахара гиалуронана» подразумевается, что экспрессируемый геном белок активен при образовании N-ацетилглюкозамина или D-глюкуроновой кислоты, или сахара, который является предшественником N-ацетилглюкозамина или D-глюкуроновой кислоты (фигура 2).
В предпочтительном способе получения предшественника сахара разработаны конструкции для улучшения продукции гиалуронана в клетке-хозяине, содержащей гиалуронансинтазу, путем культивирования клетки-хозяина, содержащей рекомбинантную конструкцию с гетерологичной промоторной областью, функционально связанной с последовательностью нуклеиновой кислоты, кодирующей ген, управляющий стадией пути синтеза предшественника сахара гиалуронана. В предпочтительном способе клетка-хозяин также содержит рекомбинантную конструкцию, содержащую промоторную область, функционально связанную с гиалуронансинтазой, при этом может использоваться та же или другая промоторная область, что в нуклеиновой кислоте синтазы, участвующей в биосинтезе N-ацетилглюкозамина. В дальнейшем предпочтительном осуществлении клетка-хозяин может содержать рекомбинантную конструкцию с промоторной областью, функционально связанной с различными последовательностями нуклеиновой кислоты, кодирующими второй ген, участвующий в синтезе предшественника сахара гиалуронана.
Таким образом, настоящее изобретение также относится к конструкциям для улучшения продукции гиалуронана путем использования конструкций с последовательностью нуклеиновой кислоты, кодирующей ген, управляющий стадией пути синтеза предшественника сахара гиалуронана. Последовательность нуклеиновой кислоты предшественника сахара может экспрессироваться с того же самого промотора, что и последовательность нуклеиновой кислоты, кодирующая гиалуронансинтазу, или с другого промотора.
Гены, участвующие в биосинтезе предшественников сахара для получения гиалуроновой кислоты, включают ген UDP-глюкозо-6-дегидрогеназы, ген UDP-глюкозопирофосфорилазы, ген UDP-N-ацетилгюкозаминпирофосфорилазы, ген глюкозо-6-фосфатизомеразы, ген гексокиназы, ген фосфоглюкомутазы, ген амидотрансферазы, ген мутазы и ген ацетилтрансферазы.
В клетке, содержащей гиалуронансинтазу, для увеличения пулов сахаров-предшественников, доступных для гиалуронансинтазы, может экспрессироваться любой из двух hasB, hasC и hasD и более, или их гомологов, таких как tuaD, gtaB и gcaD Bacillus subtilis соответственно, а также hasE, или их комбинация. Геном Bacillus описан в Kunst, et al., Nature 390,249-256, "The complete genome sequence of the Gram-positive bacterium Bacillus subtilis" (20 ноября 1997 г.). В некоторых случаях, например, где клетка-хозяин не имеет природной активности гиалуронансинтазы, конструкция может содержать ген hasA.
Последовательность нуклеиновой кислоты, кодирующая ферменты биосинтеза, может быть природной по отношению к клетке-хозяину, тогда как в некоторых случаях может использоваться гетерологичная последовательность. Если экспрессируются два или более гена, они могут являться генами, которые ассоциированы с некоторым другим геном нативного оперона, например с генами оперона HAS Streptococcus equisimilis, который включает hasA, hasB, hasC и hasD. В иных случаях может требоваться применение некоторой комбинации последовательностей генов предшественников без включения в состав оперона его каждого элемента. Использование некоторых генов, природных для клетки-хозяина, и других, экзогенных, может также быть предпочтительным в других случаях. Выбор зависит от доступных наборов сахаров в данной клетке-хозяине, способности клетки приспосабливаться к сверхпродукции без помехи другим функциям клетки-хозяина и от того, регулирует ли клетка экспрессию со своих нативных генов иначе, чем с экзогенных генов.
В качестве одного из примеров в зависимости от требований метаболизма и условий роста клетки и доступных пулов предшественников сахаров может потребоваться увеличить продукцию N-ацетлглюкозамина путем экспрессии последовательности нуклеиновой кислоты, кодирующей UDP-N-ацетилглюкозаминпирофосфорилазы, например гена hasD, гена gcaD Bacillus и их гомологов. Альтернативно предшественник сахара может представлять собой D-глюкуроновую кислоту. В одном из таких осуществлений последовательность нуклеиновой кислоты кодирует UDP-глюкозо-6-дегидрогеназу. Такие последовательности нуклеиновой включают ген tuaD Bacillus, ген hasB Streptococcus и их гомологи. Последовательность нуклеиновой кислоты также может кодировать UDP-глюкозопирофосфорилазу, такую как ген gtaB Bacillus, ген hasC Streptococcus и их гомологи.
По способам настоящего изобретения ген UDP-глюкозо-6-дегидрогеназы может представлять собой ген hasB или tuaD или их гомологи.
В предпочтительном осуществлении ген hasB выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 41, SEQ ID NO: 97 или SEQ ID NO: 105; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 40, SEQ ID NO: 96 или SEQ ID NO: 104; и (c) комплементарной цепи (a) или (b).
В более предпочтительном осуществлении ген hasB кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 41, SEQ ID NO: 97 или SEQ ID NO: 105; или его фрагмент, обладающий активностью UDP-глюкозо-6-дегидрогеназы.
В другом предпочтительном осуществлении ген tuaD выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 12; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 11; и (c) комплементарной цепи (a) или (b).
В другом более предпочтительном осуществлении ген tuaD кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 12, или его фрагмент, обладающий активностью UDP-глюкозо-6-дегидрогеназы.
По способам настоящего изобретения ген UDP-глюкозопирофосфорилазы может представлять собой ген hasC или gtaB или их гомологи.
В предпочтительном осуществлении ген hasC выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 43, SEQ ID NO: 99 или SEQ ID NO: 107; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 42, SEQ ID NO: 98 или SEQ ID NO: 106; и (c) комплементарной цепи (a) или (b).
В более предпочтительном осуществлении ген hasC кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 43, SEQ ID NO: 99 или SEQ ID NO: 107; или его фрагмент, обладающий активностью UDP-глюкозопирофосфорилазы.
В другом предпочтительном осуществлении ген gtaB выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 22; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 21; и (c) комплементарной цепи (a) или (b).
В другом более предпочтительном осуществлении ген gtaB кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 22 или его фрагмент, обладающий активностью UDP-глюкозопирофосфорилазы.
По способам настоящего изобретения ген UDP-N-ацетилглюкозаминпирофосфорилазы может представлять собой ген hasD или gcaD или их гомологи.
В предпочтительном осуществлении ген hasD выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 45; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 44; и (c) комплементарной цепи (a) или (b).
В более предпочтительном осуществлении ген hasD кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 45; или его фрагмент, обладающий активностью UDP-N-ацетилглюкозаминпирофосфорилазы.
В другом предпочтительном осуществлении ген gcaD выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 30; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 29; и (c) комплементарной цепи (a) или (b).
В другом более предпочтительном осуществлении ген gcaD кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 30 или его фрагмент, обладающий активностью UDP-N-ацетилглюкозаминпирофосфорилазы.
По способам настоящего изобретения ген глюкозо-6-фосфатизомеразы может представлять собой ген hasE или его гомолог.
В предпочтительном осуществлении ген hasE выбран из группы, состоящей из (a) последовательности нуклеиновой кислоты, кодирующей полипептид с аминокислотной последовательностью, характеризующейся, по меньшей мере, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90% или примерно 95% идентичностью в отношении SEQ ID NO: 101; (b) последовательности нуклеиновой кислоты, которая гибридизуется в условиях низкой, средней или высокой степени жесткости с SEQ ID NO: 100; и (c) комплементарной цепи (a) или (b).
В более предпочтительном осуществлении ген hasE кодирует полипептид с аминокислотной последовательностью SEQ ID NO: 101 или его фрагмент, обладающий активностью глюкозо-6-фосфатизомеразы.
По способам настоящего изобретения ген гиалуронансинтазы и один или более генов, кодирующих предшественник сахара, находятся под контролем одного и того же промотора. Альтернативно один или более генов, кодирующих предшественник сахара, находятся под контролем одного и того же промотора, но ген гиалуронансинтазы регулируется другим промотором. Дальнейшей альтернативой является то, что ген гиалуронансинтазы и каждый из генов, кодирующих предшественник сахара, находятся под контролем различных промоторов. В предпочтительном осуществлении ген гиалуронансинтазы и один или более генов, кодирующих предшественник сахара, находятся под контролем одного и того же промотора.
Настоящее изобретение также относится к конструкции нуклеиновой кислоты, содержащей выделенную последовательность нуклеиновой кислоты, кодирующую оперон гиалуронансинтазы включающий ген гиалуронансинтазы и ген UDP-глюкозо-6-дегидрогеназы и необязательно один или более генов, выбранных из группы, состоящей из гена UDP-глюкозопирофосфорилазы, гена UDP-N-ацетилглюкозаминпирофосфорилазы, и гена глюкозо-6-фосфатизомеразы. Последовательность нуклеиновой кислоты, кодирующая большую часть оперона гиалуронансинтазы Streptococcus equisimilis находится в SEQ ID NO: 108. Данная последовательность содержит hasB (SEQ ID NO: 40) и hasC (SEQ ID NO: 42) гомологи гена tuaD (SEQ ID NO: 11) и gtaB (SEQ ID NO: 21) Bacillus subtilis соответственно, как и в случае Streptococcus pyogenes, а также гомолог гена gcaD (SEQ ID NO: 29), который обозначен hasD (SEQ ID NO: 44). gcaD Bacillus subtilis кодирует UDP-N-ацетилглюкозаминпирофосфорилазу, которая участвует в синтезе N-ацетилглюкозамина, одного из двух сахаров гиалуронана. Гомолог gcaD из Streptococcus equisimilis, hasD, помещен в Streptococcus equisimilis в оперон гиалуронансинтазы. Последовательность нуклеиновой кислоты также содержит часть гена hasA (последние 1156 п.н. SEQ ID NO: 1).
В некоторых случаях клетка-хозяин будет содержать рекомбинантную конструкцию с гетерологичной промоторной областью, функционально связанной с последовательностью нуклеиновой кислоты, кодирующей ген, управляющий стадией в пути синтеза предшественника сахара гиалуронана, что может сочетаться с экспрессией гиалуронансинтазы с рекомбинантной конструкции. Гиалуронансинтаза может экспрессироваться с той же или другой промоторной области, что и нуклеиновая кислота, кодирующая фермент, участвующий в биосинтезе предшественника. В другом предпочтительном осуществлении клетка-хозяин может содержать рекомбинантную конструкцию с промоторной областью, функционально связанной с другой последовательностью нуклеиновой кислоты, кодирующей второй ген, участвующий в синтезе предшественника сахара гиалуронана.
Последовательностью нуклеиновой кислоты, кодирующая ферменты, участвующие в биосинтезе предшественника(ов) сахара(ов), может экспрессироваться с того же или другого промотора, что и последовательность нуклеиновой кислоты, кодирующая гиалуронансинтазу. В прежнем смысле, конструируют «искусственные опероны», которые могут имитировать оперон Streptococcus equisimilis в том, что будет иметь hasA, hasB, hasC и hasD, или их гомологи, или, альтернативно может задействовать меньше целого комплемента, присутствующего в опероне Streptococcus equisimilis. «Искусственные опероны» могут также содержать ген глюкозо-6-фосфатизомеразы (hasE), а также один или более генов, выбранных из группы, состоящей из гена гексокиназы, гена фосфоглюкомутазы, гена амидотрансферазы, гена мутазы и гена ацетилтрансферазы. В искусственном опероне, по меньшей мере, один из элементов гетерологичен по отношению к одному другому элементу, как, например, промоторная область бывает гетерологична кодирующим последовательностям.
В предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA, tuaD, и gtaB. В другом предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA, tuaD, gtaB, и gcaD. В другом предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA и tuaD. В другом предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA. В другом предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA, tuaD, gtaB, gcaD, и hasE. В другом предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA, hasB, hasC, и hasD. В другом предпочтительном осуществлении конструкция нуклеиновой кислоты содержит hasA, hasB, hasC, hasD, и hasE. На основе указанных выше предпочтительных осуществлений можно заменить указанные гены на их гомологи.
По способам настоящего изобретения конструкции нуклеиновой кислоты содержат последовательность, кодирующую гиалуронансинтазу, функционально связанную с промоторной последовательностью, чужеродной по отношению к последовательности, кодирующей гиалуронансинтазу. Промоторная последовательность может, например, представлять собой одинарный промотор или тандемный промотор.
«Промотор» определяется здесь как последовательность нуклеиновой кислоты, участвующая в связывании РНК-полимеразы для инициации транскрипции гена. «Тандемный промотор» определяется здесь как две или более промоторных последовательностей, каждая из которых функционально связана с кодирующей последовательностью и опосредует транскрипцию кодирующей последовательности в мРНК. «Функционально связанный» определяется здесь как конфигурация, в которой регуляторная последовательность, например промоторная последовательность, подходящим образом помещена в позицию относительно кодирующей последовательности, так что регуляторная последовательность регулирует выработку полипептида, кодируемого кодирующей последовательностью. Как отмечалось ранее, «кодирующая последовательность» определена здесь как последовательность нуклеиновой кислоты, которая транскрибируется в мРНК и транслируется в полипептид при помещении под контроль подходящих регуляторных последовательностей. Границы кодирующей последовательности, в общем, определены участком связывания рибосомы, расположенным непосредственно выше открытой рамки считывания на 5'-конце мРНК, и последовательностью терминации транскрипции, локализованной непосредственно ниже открытой рамки считывания на 3'-конце мРНК. Кодирующая последовательность может включать в качестве неограничивающих примеров ДНК, кДНК, полусинтетические, синтетические и рекомбинантные последовательности нуклеиновой кислоты.
В предпочтительном осуществлении промоторные последовательности могут быть получены из бактериального источника. В более предпочтительном осуществлении промоторные последовательности могут быть получены из грамположительной бактерии, такой как штамм Bacillus, например Bacillus agaradherens, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus brevis, Bacillus circulans, Bacillus clausii, Bacillus coagulans, Bacillus firmus, Bacillus lautus, Bacillus lentus, Bacillus licheniformis, Bacillus megaterium, Bacillus pumilus, Bacillus stearothermophilus, Bacillus subtilis, или Bacillus thuringiensis; или штамм Streptomyces, например, Streptomyces lividans или Streptomyces murinus; или из грамотрицательной бактерии, например E. coli или Pseudomonas sp.
Примерами промоторов, подходящих для управления транскрипцией последовательности нуклеиновой кислоты по способам настоящего изобретения, являются промоторы, полученные из lac-оперона E. coli, гена агаразы (dagA) Streptomyces coelicolor, гена щелочной протеазы (aprH) Bacillus lentus или Bacillus clausii, гена щелочной протеазы (ген субтилизина Карлсберга) Bacillus licheniformis, гена левансахаразы (sacB) Bacillus subtilis, гена альфа-амилазы (amyE) Bacillus subtilis, гена альфа-амилазы (amyL) Bacillus licheniformis, гена мальтогенной амилазы (amyM) Bacillus stearothermophilus, гена альфа-амилазы (amyQ) Bacillus amyloliquefaciens, гена пенициллиназы (penP) Bacillus licheniformis, генов xylA и xylB Bacillus subtilis, гена CryIIIA Bacillus thuringiensis подвида tenebrionis (cryIIIA) или из их частей, из гена прокариотической бета-лактамазы (Villa-Kamaroff et al., 1978, Proceedings of the National Academy of Sciences USA 75: 3727-3731). Другими примерами являются бактериофаговый промотор spo1 и промотор tac (DeBoer et al., 1983, Proceedings of the National Academy of Sciences USA 80: 21-25). Другие промоторы описаны в "Useful proteins from recombinant bacteria" in Scientific American, 1980, 242: 74-94; и в Sambrook, Fritsch, and Maniatus, 1989, Molecular Cloning, A Laboratory Manual, 2d edition, Cold Spring Harbor, New York.
Промотор также может представлять собой «консенсусный» промотор, содержащий последовательность TTGACA в качестве области «-35» и TATAAT в качестве области «-10». Консенсусный промотор может быть получен из любого промотора, который может функционировать в клетке-хозяине Bacillus. Конструирование «консенсусного» промотора может достигаться путем сайт-специфического мутагенеза с образованием промотора, который наиболее совершенно подходит под установленные консенсусные последовательности областей «-10» и «-35» вегетативных промоторов «типа сигма А» Bacillus subtilis (Voskuil et al., 1995, Molecular Microbiology 17: 271-279).
В предпочтительном осуществлении «консенсусный» промотор получают из промотора, полученного из lac-оперона E. coli, гена агаразы (dagA) Streptomyces coelicolor, гена щелочной протеазы (aprH) Bacillus lentus или Bacillus clausii, гена щелочной протеазы (ген субтилизина Карлсберга) Bacillus licheniformis, гена левансахаразы (sacB) Bacillus subtilis, гена альфа-амилазы (amyE) Bacillus subtilis, гена альфа-амилазы (amyL) Bacillus licheniformis, гена мальтогенной амилазы (amyM) Bacillus stearothermophilus, гена альфа-амилазы (amyQ) Bacillus amyloliquefaciens, гена пенициллиназы (penP) Bacillus licheniformis, генов xylA и oxyB Bacillus subtilis, гена CryIIIA Bacillus thuringiensis подвид tenebrionis (cryIIIA) или из их частей, или из гена прокариотической бета-лактамазы, из бактериофагового промотора spo1. В более предпочтительном осуществлении «консенсусный» промотор получают из гена альфа-амилазы (amyQ) Bacillus amyloliquefaciens.
Widner, et al. в патентах США № 6255076 и 5955310 описывают тандемные промоторы, и конструкции и способы использования в клетках Bacillus, включая короткий консенсусный промотор amyQ (также называемый scBAN). Применение стабилизирующей последовательности cryIIIA и конструкции, использующие данную последовательность, для улучшенной продукции в Bacillus также описаны там.
Каждая промоторная последовательность тандемного промотора может представлять собой любую последовательность нуклеиновой кислоты, которая характеризуется транскрипционной активностью в выбранной клетке Bacillus, включая мутантный, укороченный и гибридный промотор, и может быть получена из генов, кодирующих внеклеточные или внутриклеточные полипептиды, гомологичные или гетерологичные для клетки Bacillus. Каждая промоторная последовательность может быть нативной или чужеродной в отношении последовательности нуклеиновой кислоты, кодирующей полипептид, и нативной или чужеродной в отношении клетки Bacillus. Промоторные последовательности могут быть одинаковыми или разными промоторными последовательностями.
Две или более промоторные последовательности тандемного промотора могут одновременно способствовать транскрипции последовательности нуклеиновой кислоты. Альтернативно одна или более промоторных последовательностей тандемного промотора могут способствовать транскрипции последовательности нуклеиновой кислоты на разных стадиях роста клетки Bacillus.
В предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, промотор amyQ гена альфа-амилазы Bacillus amyloliquefaciens. В другом предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, «консенсусный» промотор, имеющий последовательность TTGACA в области «-35» и TATAAT в области «-10». В другом предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, промотор amyL гена альфа-амилазы Bacillus licheniformis. В другом предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, промотор cryIIIA или его части (Agaisse and Lereclus, 1994, Molecular Microbiology 13: 97-107).
В более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, промотор amyL и промотор cryIIIA. В другом более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, промотор amyQ и промотор cryIIIA. В другом более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, «консенсусный» промотор, имеющий последовательность TTGACA в области «-35» и TATAAT в области «-10» и промотор cryIIIA. В другом более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, две копии промотора amyL. В другом более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, две копии промотора amyQ. В другом более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, две копии «консенсусного» промотора, имеющего последовательность TTGACA в области «-35» и TATAAT в области «-10». В другом более предпочтительном осуществлении тандемный промотор содержит, по меньшей мере, две копии промотора cryIIIA.
«Последовательность мРНК процессинга/стабилизации» определяется здесь как последовательность, расположенная ниже одной или более промоторных последовательностей и выше кодирующей последовательности, с которой функционально связаны одна или более промоторных последовательностей, так что мРНК, синтезированные с каждой промоторной последовательности, могут процессироваться с образованием мРНК-транскриптов со стабилизирующей последовательностью на 5'-конце транскрипта. Присутствие такой стабилизирующей последовательности на 5'-конце мРНК-транскриптов повышает их период полужизни (Agaisse and Lereclus, 1994, supra, Hue et al., 1995, Journal of Bacteriology 177: 3465-3471). Последовательность мРНК процессинга/стабилизации комплементарна 3'-концу бактериальной рибосомальной 16S-РНК. В предпочтительном осуществлении последовательность мРНК процессинга/стабилизации приводит к образованию транскриптов, по существу, одного размера со стабилизирующей последовательностью на 5'-конце транскриптов. Последовательность мРНК процессинга/стабилизации предпочтительно комплементарна 3'-концу бактериальной рибосомальной 16S-РНК. См. патенты США № 6255076 и 5955310.
В более предпочтительном осуществлении последовательность процессинга/стабилизации мРНК представляет собой последовательность процессинга/стабилизации мРНК cryIIIA Bacillus thuringiensis, описанную в WO 94/25612 и Agaisse and Lereclus, 1994, supra, или ее части, которые сохраняют функцию процессинга/стабилизации мРНК. В более предпочтительном осуществлении последовательность процессинга/стабилизации мРНК представляет собой последовательность процессинга/стабилизации мРНК SP82 Bacillus subtilis, описанную в Hue et al., 1995, supra, или ее части, которые сохраняют функцию процессинга/стабилизации мРНК.
Когда промотор cryIIIA и его последовательность процессинга/стабилизации мРНК используются в способах настоящего изобретения, может использоваться фрагмент ДНК, содержащий последовательность, описанную в WO 94/25612 и Agaisse and Lereclus, 1994, supra, или ее части, которые сохраняют промоторную функцию и функцию процессинга/стабилизации мРНК. Более того, фрагменты ДНК, содержащие только промотор cryIIIA или только последовательность процессинга/стабилизации мРНК cryIIIA, могут быть получены с использованием способов, хорошо известных в данной области, для конструирования различных тандемных промоторов и комбинаций последовательностей процессинга/стабилизации мРНК. В данном осуществлении промотор cryIIIA и его последовательность процессинга/стабилизации мРНК предпочтительно помещают ниже другой(-их) промоторной(-ых) последовательности(-ей), составляющей(-их) тандемный промотор, и выше кодирующей последовательности интересующего гена.
Выделенная последовательность нуклеиновой кислоты, кодирующая требуемый(-е) фермент(-ы), участвующий(-е) в получении гиалуроновой кислоты, может подвергаться дальнейшим манипуляциям для улучшения экспрессии последовательности нуклеиновой кислоты. Экспрессия понимается так, что она включает любую стадию получения полипептида, включая в качестве неограничивающих примеров транскрипцию, посттранскрипционную модификацию, трансляцию, посттрансляционную модификацию и секрецию. Способы модификации последовательностей нуклеиновой кислоты с использованием способов клонирования хорошо известны в данной области.
Конструкция нуклеиновой кислоты, содержащая последовательность нуклеиновой кислоты, кодирующую фермент, может быть функционально связана с одной или более регуляторными последовательностями, способными управлять экспрессией кодирующей последовательностью в клетке Bacillus, в условиях, совместимых с регуляторными последовательностями.
Термин «регуляторные последовательности» определяется здесь так, что она включает все компоненты, необходимые или предпочтительные для экспрессии кодирующей последовательности, последовательности нуклеиновой кислоты. Каждая регуляторная последовательность может быть нативной или чужеродной в отношении последовательности нуклеиновой кислоты, кодирующей фермент. В дополнение к описанным здесь промоторным последовательностям такие регуляторные последовательности включают в качестве неограничивающих примеров лидерную, сигнальную последовательность и терминатор транскрипции. Как минимум, регуляторные последовательности охватывают промотор и стоп-сигналы транскрипции и трансляции. Регуляторные последовательности могут дополняться линкерами с целью введения конкретных участков рестрикции облегчающих лигирование регуляторных последовательностей с кодирующей областью последовательности нуклеиновой кислоты, кодирующей фермент.
Регуляторная последовательность также может являться подходящей последовательностью терминации транскрипции, последовательностью, распознаваемой клеткой Bacillus для терминации транскрипции. Терминаторная последовательность функционально связана с 3'-концом последовательности нуклеиновой кислоты, кодирующей фермент или последний фермент оперона. Любой терминатор, который функционален в клетке Bacillus по выбору, может использоваться по настоящему изобретению.
Регуляторная последовательность также может являться подходящей лидерной последовательностью, нетранслируемой областью мРНК, которая значима для трансляции клеткой Bacillus. Лидерная последовательность функционально связана с 5'-концом последовательности нуклеиновой кислоты, кодирующей фермент. Любая лидерная последовательность, которая функциональна в клетке Bacillus по выбору, может использоваться по настоящему изобретению.
Регуляторная последовательность может также представлять собой область, кодирующую сигнальный пептид, которая кодирует аминокислотную последовательность, связанную с N-концом полипептида, который может направлять экспрессированный полипептид по секреторному пути клетки. Область, кодирующая сигнальный пептид, может быть природной или может быть получена из чужеродных источников. 5'-конец кодирующей последовательности нуклеиновой кислоты может по сути своей содержать область, кодирующую сигнальный пептид, связанную в природе в рамку считывания трансляции с сегментом кодирующей области, которая кодирует секретируемый полипептид. Альтернативно 5'-конец кодирующей последовательности может содержать область, кодирующую сигнальный пептид, которая чужеродна в отношении таковой части кодирующей последовательности, которая кодирует секретируемый полипептид. Чужеродная область, кодирующая сигнальный пептид, может потребоваться там, где кодирующая последовательность в норме не содержит области, кодирующей сигнальный пептид. Альтернативно область, кодирующая сигнальный пептид, может просто замещать область, кодирующую природный сигнальный пептид, для получения улучшенной секреции полипептида по сравнению с областью, кодирующей природный сигнальный пептид, в норме ассоциированной с данной кодирующей последовательностью. Область, кодирующая сигнальный пептид, может быть получена из гена амилазы или протеазы вида Bacillus. Однако по настоящему изобретению может использоваться любая область, кодирующая сигнальный пептид, способная направлять экспрессированного полипептида в секреторный путь клетки Bacillus по выбору.
Эффективная область, кодирующая сигнальный пептид, для клеток Bacillus представляет собой область, кодирующую сигнальный пептид, полученную из гена мальтогенной амилазы Bacillus NCIB 11837, гена альфа-амилазы Bacillus stearothermophilus, гена субтилизина Bacillus licheniformis, гена бета-лактамазы Bacillus licheniformis, генов нейтральных протеаз Bacillus stearothermophilus (nprT, nprS, nprM), и гена prsA Bacillus subtilis. Другие сигнальные пептиды описаны Simonen and Palva, 1993, Microbiological Reviews 57: 109-137.
Регуляторная последовательность может также представлять собой область, кодирующую пропептид, который кодирует аминокислотную последовательность, расположенную на N-конце полипептида. Полученный в результате полипептид известен как профермент или прополипептид (или, в некоторых случаях, зимоген). Прополипептид, в общем, неактивен и может преобразовываться в зрелый активный полипептид путем каталитического или автокаталитического отщепления пропептида от прополипептида. Кодирующая пропептид область может быть получена из генов щелочной протеазы (aprE) Bacillus subtilis и нейтральной протеазы (nprT) Bacillus subtilis.
Там, где области сигнального пептида и пропептида присутствуют на N-конце полипептида, область пропептида расположена вслед за N-концом полипептида, а область сигнального пептида расположена вслед за N-концом пропептидной области.
Также может потребоваться добавление регуляторных последовательностей, которые обеспечивают регуляцию экспрессии полипептида по мере роста клетки хозяина. Примерами регуляторных систем являются те, что вызывают включение или выключение экспрессии гена в ответ на химический или физический стимул, включая наличие регуляторного соединения. Регуляторные системы в прокариотических системах охватывают операторные системы lac, tac и trp.
ЭКСПРЕССИРУЮЩИЕ ВЕКТОРЫ
По способам настоящего изобретения рекомбинантный экспрессирующий вектор включает последовательность нуклеиновой кислоты, промотор и стоп-сигналы транскрипции и трансляции могут использоваться для рекомбинантной продукции фермента, участвующего в получении гиалуроновой кислоты. Различные последовательности нуклеиновой кислоты и регуляторные последовательности, описанные выше, могут быть объединены вместе для получения рекомбинантного экспрессирующего вектора, который может содержать один или более участков рестрикции, которые позволяют осуществить вставку или замену последовательности нуклеиновой кислоты, кодирующей полипептид или фермент в таких участках. Альтернативно последовательность нуклеиновой кислоты может экспрессироваться путем вставки последовательности нуклеиновой кислоты или конструкции нуклеиновой кислоты, содержащей последовательность, в подходящий вектор для экспрессии. При создании экспрессирующего вектора кодирующая последовательность располагается в векторе так, что кодирующая последовательность функционально связана с подходящими регуляторными последовательностями для экспрессии и, возможно, для секреции.
Рекомбинантный экспрессирующий вектор может представлять собой любой вектор, который подходящим образом может подвергаться процедурам рекомбинантной ДНК, и может осуществлять экспрессию последовательности нуклеиновой кислоты. Выбор вектора обычно зависит от совместимости вектора с клеткой Bacillus, в которую надлежит вводить вектор. Векторы могут быть линейными или замкнутыми кольцевыми плазмидами. Вектор может быть автономно реплицирующим вектором, т.е. вектором, который существует во внехромосомном виде, репликация которого не зависит от хромосомной репликации, например, плазмидой, внехромосомной элементом, минихромосомой или искусственной хромосомой. Вектор может содержать любые средства для обеспечения саморепликации. Альтернативно вектор может быть таким, который при введении в клетку Bacillus интегрируется в геном и реплицируется вместе с хромосомой(-ами), в которую(-ые) он интегрировался. Векторная система может представлять собой единичный вектор или плазмиду или два или более вектора или плазмид, которые вместе содержат общую ДНК, подлежащую введению в геном клетки Bacillus, или может использоваться транспозон.
Векторы по настоящему изобретению предпочтительно содержат элемент(-ы), который(-е) обеспечивают интеграцию вектора в геном клетки хозяина Bacillus или автономную репликацию вектора в клетке независимо от генома.
Для интеграции в геном клетки-хозяина вектор может зависеть от последовательности нуклеиновой кислоты, кодирующей полипептид, или любого другого элемента вектора для интеграции вектора в геном путем гомологичной или негомологичной рекомбинации. Альтернативно вектор может содержать дополнительные последовательности нуклеиновой кислоты для управления интеграцией гомологичной рекомбинации в геном клетки Bacillus. Дополнительные последовательности нуклеиновой кислоты обеспечивают интеграцию вектора в геном клетки Bacillus в точном месте хромосомы. Для увеличения вероятности интеграции в точном месте элементы интеграции должны предпочтительно содержать достаточное число нуклеиновых кислот, например, от 100 до 1500 пар оснований, предпочтительно от 400 до 1500 пар оснований, и наиболее предпочтительно от 800 до 1500 пар оснований, которые высокогомологичны соответствующей последовательности-мишени для усиления вероятности гомологичной рекомбинации. Элементы интеграции могут представлять собой любую последовательность, которая гомологична последовательности-мишени в геноме клетке Bacillus. Более того, элементы интеграции могут относиться к некодирующим или кодирующим последовательностям нуклеиновой кислоты. С другой стороны, вектор может интегрироваться в геном клетки-хозяина путем негомологичной рекомбинации.
Для автономной репликации вектор может дополнительно содержать точку начала репликации, обеспечивающую автономную репликацию вектора в клетке Bacillus. Примерами точек начала репликации являются точки начала репликации плазмид pUB110, pE194, pTA1060, и pAMβ1, обеспечивающие репликацию в Bacillus. Точка начала репликации может иметь мутацию, так что мутация делает ее функцию температурно-чувствительной в клетке Bacillus (см., например, Ehrlich, 1978, Proceedings of the National Academy of Sciences USA 75: 1433).
Векторы предпочтительно содержат один или более подлежащих селекции маркеров, которые обеспечивают простую селекцию трансформированных клеток. Подлежащий селекции маркер представляет собой ген, продукт которого обеспечивает устойчивость к биоциду, устойчивость к тяжелым металлам, от прототрофности до ауксотрофности, и тому подобное. Примерами бактериальных подлежащих селекции маркеров являются гены dal из Bacillus subtilis или Bacillus licheniformis или маркеры, которые обеспечивают устойчивость к антибиотикам, такую как устойчивость к ампициллину, канамицину, хлорамфениколу или тетрациклину. Более того, селекция может осуществляться путем совместной трансформации, например, как описано в WO 91/09129, где подлежащий селекции маркер находится на отдельном векторе.
Более чем одна копия последовательности нуклеиновой кислоты может встраиваться в клетку-хозяин для усиления продукции генного продукта. Увеличение числа копий последовательности нуклеиновой кислоты может быть получено путем интегрирования, по меньшей мере, одной дополнительной копии последовательности в геном клетки-хозяина или путем введения с последовательностью нуклеиновой кислоты амплифицируемого подлежащего селекции маркерного гена, где клетки, содержащие амплифицированные копии подлежащего селекции маркерного гена и за счет этого дополнительные копии последовательности нуклеиновой кислоты, могут отбираться культивированием клеток в присутствии подходящего селективного средства. Подходящий способ достижения амплификации геномных последовательностей ДНК описан в WO 94/14968.
Процедуры, использованные для лигирования описанных выше элементов с целью конструирования рекомбинантных экспрессирующих векторов, хорошо известны специалистам в данной области (см., например, Sambrook et al., 1989, supra).
ПОЛУЧЕНИЕ
По способам настоящего изобретения клетки-хозяева Bacillus культивируют в питательной среде, подходящей для получения гиалуроновой кислоты с использованием способов, известных в данной области. Например, клетки могут культивироваться путем культивирования в флаконах для встряхивания, путем мелко- и крупномасштабной ферментации (включая непрерывную ферментацию, ферментацию одного производственного цикла, подпитываемую ферментацию или ферментацию в твердом состоянии) в лабораторных или промышленных ферментерах, проводимой в подходящей среде и в условиях, обеспечивающих экспрессию ферментов, участвующих в синтезе гиалуроновой кислоты, и выделение гиалуроновой кислоты. Культивирование проходит в подходящей питательной среде, содержащей источники углерода и азота и неорганические соли, с использованием процедур, известных в данной области. Подходящие среды доступны от коммерческих поставщиков или могут быть получены согласно опубликованным композициям (например, в каталогах American Type Culture Collection). Секретированная гиалуроновая кислота может быть получена непосредственно из среды.
Полученная в результате гиалуроновая кислота может быть выделена способами, известными в данной области. Например, гиалуроновая кислота может быть выделена из питательной среды обычными процедурами, включая в качестве неограничивающих примеров центрифугирование, фильтрацию, экстракцию, сушку распылением, упаривание или преципитацию. Выделенную гиалуроновую кислоту затем можно далее очищать путем различных процедур, известных в данной области, включая в качестве неограничивающих примеров хроматографию (например, ионообменную, аффинную, гидрофобную, хроматофокусирование и гель-фильтрацию), электрофоретические процедуры (например, препаративное изоэлектрофокусирование), дифференциальное растворение (например, преципитацию сульфатом аммония) или экстракцию (см., например, Protein Purification, J.-C. Janson and Lars Ryden, editors, VCH Publishers, New York, 1989).
По способам настоящего изобретения клетки-хозяева Bacillus продуцируют более 4 г, предпочтительно более чем примерно 6 г, более предпочтительно более чем примерно 8 г, даже более предпочтительно более чем примерно 10 г, и наиболее предпочтительно более чем примерно 12 г гиалуроновой кислоты на литр.
ДЕЛЕЦИИ/РАЗРУШЕНИЯ
Способы делеции или замещения генов могут использоваться для полного удаления гена подлежащего селекции маркера или другого нежелательного гена. В таких способах делецию подлежащего селекции маркерного гене можно осуществить с использованием плазмиды, которая сконструирована так, что она содержит смежные 5'- и 3'-области, фланкирующие ген подлежащего селекции маркера. Смежные 5'- и 3'-области могут вводиться в клетку Bacillus в температурно-чувствительной плазмиде, например pE194, в ассоциации со вторым подлежащим селекции маркером при пермессивной температуре, чтобы позволить плазмиде установиться в клетке. Затем клетку помещают в непермессивную температуру для отбора клеток, которые содержат плазмиду, интегрированную в хромосому в одной из гомологичных фланкирующих областей. Селекция на предмет интеграции плазмиды осуществляется путем селекции на второй селективный маркер. После интеграции событие рекомбинации во второй гомологичной фланкирующей области стимулируется путем помещения клеток в пермессивную температуру на несколько поколений без селекции. Клетки засевают для получения единичных колоний и колонии анализируют на предмет потери обоих селективных маркеров (см., например, Perego, 1993, In A.L. Sonneshein, J.A. Hoch, and R. Losick, editors, Bacillus subtilis and Other Gram-Positive Bacteria, Chapter 42, American Society of Microbiology, Washington, D.C., 1993).
Подлежащий селекции маркерный ген может также удаляться путем гомологичной рекомбинации путем введения в мутантную клетку фрагмента нуклеиновой кислоты, содержащего 5'- и 3'-области дефектного гена, но лишенного подлежащего селекции маркерного гена, с последующим выбором среды для обратной селекции. Путем гомологичной рекомбинации дефектный ген, содержащий подлежащий селекции маркерный ген, замещают фрагментом нуклеиновой кислоты, лишенным подлежащего селекции маркерного гена. Также могут использоваться другие способы, известные в данной области.
В патенте США № 5891701 описаны способы делеции некоторых генов, включая spoIIAC, aprE, nprE, и amyE.
Другие нежелательные биологические соединения также могут удаляться описанными выше способами, например, красный пигмент, синтезируемый cypX (инвентарный номер BG12580) и/или yvmC (инвентарный номер BG14121).
В предпочтительном осуществлении клетка-хозяин Bacillus не маркирована гетрологичными или экзогенными подлежащими селекции маркерами. В другом предпочтительном осуществлении клетка-хозяин Bacillus не продуцирует какого-либо красного пигмента, синтезируемого cypX и yvmC.
Выделенные последовательности нуклеиновой кислоты, кодирующие полипептиды, характеризующиеся активностью UDP-глюкозо-6-дегидрогеназы, активностью UDP-глюкозопирофосфорилазы или активностью UDP-N-ацетилглюкозаминпирофосфорилазы
Термин «активность UDP-глюкозо-6-дегидрогеназы» определяется здесь как активность UDP-глюкозо:NAD+-6-оксидоредуктазы, которая катализирует преобразование UDP-глюкозы в присутствии 2 NAD+ и воды в UDP-глюкуронат и 2NADH. Для целей настоящего изобретения активность UDP-глюкозо-6-дегидрогеназы определяется по процедуре, описанной Jaenicke and Rudolph, 1986, Biochemistry 25: 7283-7287. Одна единица активности UDP-глюкозо-6-дегидрогеназы определяется как 1,0 мкмоль UDP-глюкуроната, продуцируемого в минуту при 25°C, pH 7.
Термин «активность UDP-глюкозопирофосфорилазы» определяется здесь как активность UTP:  -D-глюкозо-1-фосфатуридилилтрансферазы, которая катализирует преобразование глюкозо-1-фосфата в присутствии UTP в дифосфат и UDP-глюкозу. Для целей настоящего изобретения активность UDP-глюкозопирофосфорилазы определяется по процедуре, описанной Kamogawa et al., 1965, J. Biochem. (Tokyo) 57: 758-765 или Hansen et al., 1966, Method Enzymol. 8: 248-253. Одна единица активности UDP-глюкозопирофосфорилазы определяется как 1,0 мкмоль UDP-глюкозы, продуцируемой за минуту при 25°C, pH 7.
-D-глюкозо-1-фосфатуридилилтрансферазы, которая катализирует преобразование глюкозо-1-фосфата в присутствии UTP в дифосфат и UDP-глюкозу. Для целей настоящего изобретения активность UDP-глюкозопирофосфорилазы определяется по процедуре, описанной Kamogawa et al., 1965, J. Biochem. (Tokyo) 57: 758-765 или Hansen et al., 1966, Method Enzymol. 8: 248-253. Одна единица активности UDP-глюкозопирофосфорилазы определяется как 1,0 мкмоль UDP-глюкозы, продуцируемой за минуту при 25°C, pH 7.
Термин «активность UDP-N-ацетилглюкозаминпирофосфорилазы» определяется здесь как активность UTP:N-ацетил-альфа-D-глюкозамин-1-фосфатуридилтрансферазы, которая катализирует преобразование N-ацетил-альфа-D-глюкозамин-1-фосфата в присутствии UTP в дифосфат и UDP-N-ацетил-альфа-D-глюкозамин. Для целей настоящего изобретения активность UDP-N-ацетилглюкозаминпирофосфорилазы определяется по процедуре, описанной Mangin-Lecreuix et al., 1994, J. Bacteriology 176: 5788-5795. Одна единица активности UDP-N-ацетилглюкозаминпирофосфорилазы определяется как 1,0 мкмоль UDP-N-ацетил-альфа-D-глюкозамина, продуцируемого за минуту при 25°C, pH 7.
Используемый здесь термин «выделенная последовательность нуклеиновой кислоты» относится к последовательности нуклеиновой кислоты, которая, по существу, лишена других последовательностей нуклеиновой кислоты, например, по меньшей мере, очищенная примерно на 20%, предпочтительно по меньшей мере, очищенная примерно, на 40%, более предпочтительно, по меньшей мере, очищенная примерно на 60%, даже более предпочтительно, по меньшей мере, очищенная примерно на 80%, и наиболее предпочтительно, по меньшей мере, очищенная примерно на 90%, что определяют путем электрофореза в агарозном гене. Например, выделенная последовательность нуклеиновой кислоты может быть получена путем стандартных процедур клонирования, используемых в генной инженерии для перемещения последовательности нуклеиновой кислоты из ее природного расположения в другой участок, где он будет репродуцироваться. Процедуры клонирования могут задействовать вырезание и выделение требуемого фрагмента нуклеиновой кислоты, содержащего последовательность нуклеиновой кислоты, кодирующую полипептид, встраивание фрагмента в векторную молекулу, и введение рекомбинантного вектора в клетку-хозяин, где будут реплицироваться множественные копии или клоны последовательности нуклеиновой кислоты. Последовательность нуклеиновой кислоты может быть геномной, кДНК, РНК, полусинтетического, синтетического происхождения или любой их комбинации.
В первом осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, кодирующим полипептиды, характеризующиеся аминокислотной последовательностью, которая обладает степенью идентичности в отношении SEQ ID NO: 41, равной, по меньшей мере, примерно 75%, предпочтительно, по меньшей мере, примерно 80%, более предпочтительно, по меньшей мере, примерно 85%, даже более предпочтительно, по меньшей мере, примерно 90%, наиболее предпочтительно, по меньшей мере, примерно 95%, и даже еще более предпочтительно, по меньшей мере, примерно 97%, которая характеризуется активностью UDP-глюкозо-6-дегидрогеназы (здесь и далее «гомологичные полипептиды»). В предпочтительном осуществлении гомологичные полипептиды характеризуются аминокислотной последовательностью, которая отличается пятью аминокислотами, предпочтительно четырьмя аминокислотами, более предпочтительно тремя аминокислотами, даже более предпочтительно двумя аминокислотами, наиболее предпочтительно одной аминокислотой от SEQ ID NO: 41.
В другом первом осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, кодирующим полипептиды, характеризующиеся аминокислотной последовательностью, которая обладает степенью идентичности в отношении SEQ ID NO: 43, равной, по меньшей мере, примерно 90%, предпочтительно, по меньшей мере, примерно 95%, и более предпочтительно, по меньшей мере, примерно 97%, которая характеризуется активностью UDP-глюкозопирофосфорилазы (здесь и далее «гомологичные полипептиды»). В предпочтительном осуществлении гомологичные полипептиды характеризуются аминокислотной последовательностью, которая отличается пятью аминокислотами, предпочтительно четырьмя аминокислотами, более предпочтительно тремя аминокислотами, даже более предпочтительно двумя аминокислотами, наиболее предпочтительно одной аминокислотой от SEQ ID NO: 43.
В другом первом осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, кодирующим полипептиды, характеризующиеся аминокислотной последовательностью, которая обладает степенью идентичности в отношении SEQ ID NO: 45, равной, по меньшей мере, примерно 75%, предпочтительно, по меньшей мере, примерно 80%, более предпочтительно, по меньшей мере, примерно 85%, даже более предпочтительно, по меньшей мере, примерно 90%, наиболее предпочтительно, по меньшей мере, примерно 95%, и даже еще более предпочтительно, по меньшей мере, примерно 97%, которая характеризуется активностью UDP-N-ацетилглюкозаминпирофосфорилазы (здесь и далее «гомологичные полипептиды»). В предпочтительном осуществлении гомологичные полипептиды характеризуются аминокислотной последовательностью, которая отличается пятью аминокислотами, предпочтительно четырьмя аминокислотами, более предпочтительно тремя аминокислотами, даже более предпочтительно двумя аминокислотами, наиболее предпочтительно одной аминокислотой от SEQ ID NO: 45.
Для целей настоящего изобретения степень идентичности между двумя аминокислотными последовательностями определяется способом Clustal (Higgins, 1989, CABIOS 5: 151- 153) с использованием пакета программного обеспечения Vector NTI AlignX (Informax Inc., Bethesda, MD) со следующими значениями по умолчанию: парное сопоставление, штраф за пробел 10, штраф за изменение 0,1, и матрица счета: blosum62mt2.
Предпочтительно последовательности нуклеиновой кислоты по настоящему изобретению кодируют полипептиды, которые содержат аминокислотную последовательность SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45; или их аллельный вариант; или их фрагмент, который обладает активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы соответственно. В более предпочтительном осуществлении последовательность нуклеиновой кислоты по настоящему изобретению кодирует полипептид, который содержит аминокислотную последовательность SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45. В другом предпочтительном осуществлении последовательность нуклеиновой кислоты по настоящему изобретению кодирует полипептид, который состоит из аминокислотной последовательности SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45; или их аллельного варианта; или их фрагмента, где полипептидный фрагмент обладает активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы соответственно. В другом предпочтительном осуществлении последовательность нуклеиновой кислоты по настоящему изобретению кодирует полипептид, который состоит из аминокислотной последовательности SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45.
Настоящее изобретение также охватывает последовательности нуклеиновой кислоты, которые кодируют полипептид, характеризующийся аминокислотной SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45, которые отличаются от SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44 за счет вырожденности генетического кода. Настоящее изобретение также относится к субпоследовательностям SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44, которые кодируют фрагменты SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45 соответственно, обладающие активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы соответственно.
Субпоследовательность SEQ ID NO: 40 представляет собой последовательность нуклеиновой кислоты, охваченную SEQ ID NO: 40, кроме того, что один или более нуклеотидов с 5'- и/или 3'-концов подвергнуты делеции. Предпочтительно субпоследовательность содержит, по меньшей мере, 1020, более предпочтительно, по меньшей мере, 1080 нуклеотидов, и наиболее предпочтительно, по меньшей мере, 1140 нуклеотидов. Фрагмент SEQ ID NO: 41 представляет собой полипептид, содержащий делецию одной или более аминокислот с N- и/или С-конца данной аминокислотной последовательности. Предпочтительно, фрагмент содержит, по меньшей мере, 340 аминокислотных остатков, более предпочтительно, по меньшей мере, 360 аминокислотных остатков, и наиболее предпочтительно, по меньшей мере, 380 аминокислотных остатков.
Субпоследовательность SEQ ID NO: 42 представляет собой последовательность нуклеиновой кислоты, охваченную SEQ ID NO: 42, кроме того, что один или более нуклеотидов с 5'- и/или 3'-концов подвергнуты делеции. Предпочтительно субпоследовательность содержит, по меньшей мере, 765, более предпочтительно, по меньшей мере, 810 нуклеотидов, и наиболее предпочтительно, по меньшей мере, 855 нуклеотидов. Фрагмент SEQ ID NO: 43 представляет собой полипептид, содержащий делецию одной или более аминокислот с N- и/или С-конца данной аминокислотной последовательности. Предпочтительно фрагмент содержит, по меньшей мере, 255 аминокислотных остатков, более предпочтительно, по меньшей мере, 270 аминокислотных остатков, и наиболее предпочтительно, по меньшей мере, 285 аминокислотных остатков.
Субпоследовательность SEQ ID NO: 44 представляет собой последовательность нуклеиновой кислоты, охваченную SEQ ID NO: 44, кроме того, что один или более нуклеотидов с 5'- и/или 3'-концов подвергнуты делеции. Предпочтительно субпоследовательность содержит, по меньшей мере, 1110, более предпочтительно, по меньшей мере, 1200 нуклеотидов, и наиболее предпочтительно, по меньшей мере, 1290 нуклеотидов. Фрагмент SEQ ID NO: 45 представляет собой полипептид, содержащий делецию одной или более аминокислот с N- и/или С-конца данной аминокислотной последовательности. Предпочтительно фрагмент содержит, по меньшей мере, 370 аминокислотных остатков, более предпочтительно, по меньшей мере, 400 аминокислотных остатков, и наиболее предпочтительно, по меньшей мере, 430 аминокислотных остатков.
Аллельный вариант означает любую из двух или более альтернативных форма гена, занимающих один и тот же хромосомный локус. Аллельный вариант возникает в природе путем мутации и может быть результатом полиморфизма внутри популяции. Генные мутации могут быть молчащими (без изменения в кодируемом полипептиде) или могут кодировать полипептиды, имеющие измененные аминокислотные последовательности. Аллельный вариант полипептида представляет собой полипептид, который кодируется аллельным вариантом гена.
Во втором осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, которые характеризуются уровнем гомологии в отношении SEQ ID NO: 40, равным, по меньшей мере, примерно 75%, предпочтительно, по меньшей мере, примерно 80%, более предпочтительно, по меньшей мере, примерно 85%, даже более предпочтительно, по меньшей мере, примерно 90%, наиболее предпочтительно, по меньшей мере, примерно 95%, и даже еще более предпочтительно, по меньшей мере, примерно 97%.
В другом втором осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, которые характеризуются уровнем гомологии в отношении SEQ ID NO: 42, равным, по меньшей мере, примерно 90%, предпочтительно, по меньшей мере, примерно 95%, и, более предпочтительно, по меньшей мере, примерно 97%.
В другом втором осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, которые характеризуются уровнем гомологии в отношении SEQ ID NO: 44, равным, по меньшей мере, примерно 75%, предпочтительно, по меньшей мере, примерно 80%, более предпочтительно, по меньшей мере, примерно 85%, даже более предпочтительно, по меньшей мере, примерно 90%, наиболее предпочтительно, по меньшей мере, примерно 95%, и даже еще более предпочтительно, по меньшей мере, примерно 97%.
Для целей настоящего изобретения степень гомологии между двумя аминокислотными последовательностями определяется с использованием пакета программного обеспечения Vector NTI AlignX (Informax Inc., Bethesda, MD) со следующими значениями по умолчанию: парное сопоставление, штраф за пробел 15, штраф за удлинение 6,6, и матрица счета: swgapdnamt.
В третьем осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, кодирующим полипептиды, обладающие активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы, которые гибридизуются при условиях очень низкой степени жесткости, предпочтительно при условиях низкой степени жесткости, более предпочтительно при условиях средней степени жесткости, более предпочтительно при условиях средне-высокой степени жесткости, даже более предпочтительно при условиях высокой степени жесткости, и, наиболее предпочтительно, при условиях очень высокой степени жесткости (i) с последовательностью нуклеиновой кислоты SEQ ID NO: 40, SEQ ID NO: 42, или SEQ ID NO: 44, (ii) с последовательностью кДНК, содержащейся в SEQ ID NO: 40, SEQ ID NO: 42, или SEQ ID NO: 44, или (iii) с комплементарной цепью (i) или (ii) (J. Sambrook, E.F. Fritsch, and T. Maniatus, 1989, Molecular Cloning, A Laboratory Manual, 2d edition, Cold Spring Harbor, New York). Субпоследовательность SEQ ID NO: 40, SEQ ID NO: 42, или SEQ ID NO: 44 может, по меньшей мере, составлять 100 нуклеотидов, или предпочтительно, по меньшей мере, 200 нуклеотидов. Более того, соответствующая субпоследовательность может кодировать полипептидный фрагмент, который обладает активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы, или UDP-N-ацетилглюкозаминпирофосфорилазы.
Последовательность нуклеиновой кислоты SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44 или ее субпоследовательности, а также аминокислотная последовательность SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45 или ее фрагмент могут использоваться для конструирования зондов нуклеиновой кислоты для идентификации и клонирования ДНК, кодирующей полипептиды, обладающие активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы соответственно из штаммов различных родов или видов способами, известными в данной области. В частности, такие зонды могут использоваться для гибридизации с геномной или кДНК интересующего рода или вида по стандартным процедурам Саузерн-блоттинга для идентификации и выделения в них соответствующего гена. Такие зонды могут быть заметно короче, чем целая последовательность, но должны составлять в длину, по меньшей мере, 15, предпочтительно, по меньшей мере, 25, и более предпочтительно, по меньшей мере, 35 нуклеотидов. Также могут использоваться более длинные зонды. Могут использоваться зонды на основе ДНК и РНК. Зонды обычно метят для выявления соответствующего гена (например, 32P, 3H, 35S, биотином или авидином). Такие зонды охватываются настоящим изобретением.
Таким образом, библиотека геномной ДНК или кДНК, полученная из таких иных организмов, может подвергаться скринингу на ДНК, которая гибридизуется с описанными выше зондами и которая кодирует полипептид, обладающий активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы. Геномная или другая ДНК таких иных организмов может разделяться путем электрофореза в агарозном или полиакриламидном геле или посредством других способов разделения. ДНК из библиотек или разделенная ДНК может переноситься и иммобилизоваться на нитроцеллюлозе или другом подходящем материале носителя. Для идентификации клона или ДНК, которая гомологична SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44 или их субпоследовательности, материал носителя используют в Саузерн-блоттинге. Для целей настоящего изобретения гибридизация означает, что последовательность нуклеиновой кислоты гибридизуется с меченым зондом нуклеиновой кислоты, соответствующим последовательности нуклеиновой кислоты, показанной в SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44, комплементарной ей цепи или их субпоследовательности при условиях от очень низкой до очень высокой степени жесткости. Молекулы, с которыми гибридизуется при данных условиях зонд нуклеиновой кислоты, выявляют с использованием рентгеновской пленки.
В предпочтительном осуществлении зонд нуклеиновой кислоты представляет собой последовательность нуклеиновой кислоты, которая кодирует полипептид SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45 или их субпоследовательность. В другом предпочтительном осуществлении зонд нуклеиновой кислоты представляет собой SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44. В другом предпочтительном осуществлении зонд нуклеиновой кислоты представляет собой последовательность нуклеиновой кислоты, содержащуюся в плазмиде pMRT106, которая находится в Escherichia coli NRRL B-30536, где последовательность нуклеиновой кислоты кодирует полипептиды, обладающие активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы.
Для длинных зондов, равных, по меньшей мере, 100 нуклеотидов в длину, условия от очень низкой до очень высокой степени жесткости для предгибридизации и гибридизации определяются как 42°C в 5Х SSPE, 0,3% SDS, 200 мкг/мл расщепленной и денатурированной ДНК спермы лосося, и 25% формамида для очень низкой и низкой степени жесткости, 35% формамида для средней и средневысокой степени жесткости или 50% формамида для высокой и очень высокой степени жесткости по стандартным процедурам Саузерн-блоттинга.
Для длинных зондов, равных, по меньшей мере, 100 нуклеотидов в длину, материал носителя в конце отмывают три раза каждый раз в течение 15 минут с использованием 2×SSC, 0,2% SDS, предпочтительно, по меньшей мере, при 45°C (очень низкая степень жесткости), более предпочтительно, по меньшей мере, при 50°C (низкая степень жесткости), более предпочтительно, по меньшей мере, при 55°C (средняя степень жесткости), более предпочтительно, по меньшей мере, при 60°C (средне-высокая степень жесткости), даже более предпочтительно, по меньшей мере, при 65°C (высокая степень жесткости), и наиболее предпочтительно, по меньшей мере, при 70°C (очень высокая степень жесткости).
Для коротких зондов, составляющих примерно от 15 нуклеотидов до примерно 70 нуклеотидов в длину, условия жесткости определяются как предгибридизация, гибридизация, и отмывка после гибридизации при температуре на 5°C - 10°C ниже рассчитанного Tm с использованием расчета по Bolton and McCarthy (1962, Proceedings of the National Academy of Sciences USA 48: 1390) в 0,9 M NaCl, 0,09 M Tris-HCl pH 7,6, 6 мМ EDTA, 0,5% NP-40, 1X раствор Денхардта, 1 мМ пирофосфат натрия, 1 мМ однозамещенный фосфат натрия, 0,1 мМ ATP, и 0,2 мг дрожжевой РНК на мл, по стандартным процедурам Саузерн-блоттинга.
Для коротких зондов, составляющих примерно от 15 нуклеотидов до примерно 70 нуклеотидов в длину, материал носителя отмывают один раз в 6X SCC плюс 0,1% SDS в течение 15 минут, и дважды по 15 минут каждый раз с использованием 6X SSC при температуре, на 5°C - 10°C ниже рассчитанного Tm.
В четвертом осуществлении настоящее изобретение относится к выделенным последовательностям нуклеиновой кислоты, которые кодируют варианты полипептида, характеризующегося аминокислотной последовательностью SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45, содержащие замену, делецию, и/или вставку одной или более аминокислот.
Аминокислотные последовательности полипептидных вариантов могут отличаться от аминокислотной последовательности SEQ ID NO: 41, SEQ ID NO: 43 или SEQ ID NO: 45 вставкой или делецией одного или более аминокислотных остатков и/или заменой одного или более аминокислотных остатков. Предпочтительно аминокислотные замены по природе незначительны, то есть представляют собой консервативные аминокислотные замены, которые значимо не влияют на укладку и/или активность белка; малые делеции, обычно от одной примерно до 30 аминокислот; малые N- или С-концевые удлинения, такие как на N-концевой остаток метионина; малый линкерный пептид примерно до 20-25 остатков; или малое удлинение, которое облегчает очистку путем изменения результирующего заряда или другой функции, такое как полигистидиновый хвост, антигенный эпитоп или связывающий домен.
Примеры консервативных замен лежат в пределах групп основных аминокислот (аргинин, лизин и гистидин), кислых аминокислот (глутаминовая кислота и аспарагиновая кислота), полярных аминокислот (глутамин и аспарагин), гидрофобных аминокислот (лейцин, изолейцин и валин), ароматических аминокислот (фенилаланин, триптофан и тирозин), и малых аминокислот (глицин, аланин, серин, треонин и метионин). Аминокислотные замены, которые, в общем, не изменяют специфичной активности, известны в данной области и описаны, например, H. Neurath and R. L. Hill, 1979, In, The Proteins, Academic Press, New York. Самыми обычно встречающимися заменами являются Ala/Ser, Val/lle, Asp/Glu, Thr/Ser, Ala/Gly, Ala/Thr, Ser/Asn, Ala/Val, Ser/Gly, Tyr/Phe, Ala/Pro, Lys/Arg, Asp/Asn, Leu/lle, Leu/Val, Ala/Glu, и Asp/Gly, а также наоборот.
Модификация последовательности нуклеиновой кислоты по настоящему изобретению может быть необходима для синтеза полипептидов, по существу сходных с данным полипептидом. Термин «по существу сходный» с данным полипептидом относится к не встречающимся в природе формам полипептида. Данные полипептиды могут отличаться по некоторым особенностям конструирования от полипептида, выделенного из своего природного источник, например, варианты, которые отличаются по их специфичной активности, термостабильности, оптимуму рН и тому подобному. Вариант последовательности может конструироваться на основе последовательности нуклеиновой кислоты, представленной в виде полипептида, кодирующего часть SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44, например его субпоследовательности, и/или путем введения нуклеотидных замен, которые не приводят к образованию другой аминокислотной последовательности полипептида, кодируемого последовательностью нуклеиновой кислоты, но которые соответствуют использованию кодонов в организме хозяина, предназначенного для продукции фермента, или путем введения нуклеотидных замен, которые могут приводить к образованию отличной аминокислотной последовательности. Для общего описания нуклеотидной замены см., например, Ford et al., 1991, Protein Expression and Purification 2: 95-107.
Специалистам в данной области понятно, что такие замены могут быть сделаны вне областей, критичных в плане функционирования молекулы и, кроме того, приводить к образованию активного полипептида. Аминокислотные остатки, существенные для активности полипептида, кодируемого выделенной последовательностью нуклеиновой кислоты по изобретению, и поэтому предпочтительно не подлежат замене, могут идентифицироваться по процедурам, известным в данной области, таким как сайт-специфический мутагенез или мутагенез со сканированием аланином (см., например, Cunningham and Wells, 1989, Science 244: 1081-1085). По последнему способу мутации вводятся в каждом положительно заряженном остатке молекулы и полученные в результате мутантные молекулы тестируют на активность фермента для идентификации аминокислотных остатков, критичных для активности молекулы. Участки взаимодействия субстрат-фермент также могут определяться путем анализа трехмерной структуры, что определяется такими способами, как анализ путем ядерного магнитного резонанса, кристаллография или фотоаффинное мечение (см., например, de Vos et al., 1992, Science 255: 306-312; Smith et al., 1992, Journal of Molecular Biology 224: 899-904; Wlodaver et al., 1992, FEBS Letters 309: 59-64).
Полипептиды, кодируемые выделенными последовательностями нуклеиновой кислоты по настоящему изобретению, характеризуются, по меньшей мере, 20%-ной, предпочтительно, по меньшей мере, 40%-ной, более предпочтительно, по меньшей мере, 60%-ной, даже более предпочтительно, по меньшей мере, 80%-ной, даже более предпочтительно, по меньшей мере, 90%-ной, и наиболее предпочтительно, по меньшей мере, 100%-ной активностью UDP-глюкозо-6-дегидрогеназы полипептида SEQ ID NO: 41, активностью UDP-глюкозопирофосфорилазы полипептида SEQ ID NO: 43 или активностью UDP-N-ацетилглюкозаминпирофосфорилазы полипептида SEQ ID NO: 45.
Последовательности нуклеиновой кислоты по настоящему изобретению могут быть получены из микроорганизмов любого рода. Для целей настоящего изобретения используемый здесь в связи с данным источником термин «полученный из» должен означать, что полипептид, кодируемый последовательностью нуклеиновой кислоты, продуцируется источником или клеткой, в которые встроена последовательность нуклеиновой кислоты из источника. В предпочтительном осуществлении полипептид, кодируемый последовательностью нуклеиновой кислоты, секретируется внеклеточно.
Последовательности нуклеиновой кислоты могут быть получены из бактериального источника. Например, данные полипептиды могут быть получены из грамположительной бактерии, такой как штамм Bacillus, например Bacillus agaradherens, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus brevis, Bacillus circulans, Bacillus clausii, Bacillus coagulans, Bacillus lautus, Bacillus lentus, Bacillus licheniformis, Bacillus megaterium, Bacillus stearothermophilus, Bacillus subtilis, или Bacillus thuringiensis ; или Streptomyces, например, Streptomyces lividans или Streptomyces murinus; или из грамотрицательной бактерии, например E coli или Pseudomonas sp.
В предпочтительном осуществлении последовательности нуклеиновой кислоты получают из штамма Streptococcus или Pastuerella.
В более предпочтительном осуществлении последовательности нуклеиновой кислоты получают из штамма Streptococcus equisimilis, Streptococcus pyogenes, Streptococcus uberis, или Streptococcus equi подвид zooepidemicus, или Pasteurella multocida.
В наиболее предпочтительном осуществлении последовательности нуклеиновой кислоты получают из Streptococcus equisimilis, например, последовательности нуклеиновой кислоты, представленные в SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44. В другом наиболее предпочтительном осуществлении последовательность нуклеиновой кислоты представляет собой последовательность, содержащуюся в плазмиде pMRT106, которая содержится Escherichia coli NRRL B-30536. В дальнейшем наиболее предпочтительном осуществлении последовательность нуклеиновой кислоты представляет собой SEQ ID NO: 40, SEQ ID NO: 42 или SEQ ID NO: 44.
Штаммы данных видов общедоступны в некоторых коллекциях культур, таких как American Type Culture Collection (ATCC), Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSM), Centraalbureau Voor Schimmelcultures (CBS), и Agricultural Research Service Patent Culture Collection, Northern Regional Research Center (NRRL).
Более того, такие последовательности нуклеиновой кислоты могут быть идентифицированы и получены из других источников, включая микроорганизмы, выделенные из природы (например, почвы, компостов, воды и т.д.), с использованием указанных выше зондов. Способы выделения микроорганизмов из природных местообитаний хорошо известны в данной области. Затем последовательность нуклеиновой кислоты может выделяться путем сходного скрининга библиотек геномной или кДНК другого микроорганизма. Как только последовательность нуклеиновой кислоты, кодирующая полипептид, детектируется зондом(-ами), данная последовательность может выделяться или клонироваться с использованием способов, которые хорошо известны специалистам в данной области (см., например, выше Sambrook et al., 1989, supra).
Настоящее изобретение также относится к мутантным последовательностям нуклеиновой кислоты, включающим, по меньшей мере, одну мутацию в кодирующей полипептид последовательности SEQ ID NO: 40, SEQ ID NO: 42, и SEQ ID NO: 44, в которой мутантная последовательность нуклеиновой кислоты кодирует полипептид, который состоит из SEQ ID NO: 42, SEQ ID NO: 43 и SEQ ID NO: 45 соответственно.
Способы, использованные для выделения и клонирования последовательности нуклеиновой кислоты, кодирующей полпептид, известны в данной области и охватывают выделение из геномной ДНК, получение из кДНК или их комбинацию. Клонирование последовательностей нуклеиновой кислоты по настоящему изобретению из такой геномной ДНК может осуществляться, например, путем использования хорошо известной полимеразной цепной реакции (ПЦР) или скрининга антителами библиотек экспрессии для детекции клонированных фргаментов ДНК с общими структурными свойствами. См., например, Innis et al., 1990, ПЦР: A Guide to Methods and Application, Academic Press, New York. Могут использоваться другие процедуры амплификации нуклеиновой кислоты, такие как лиганзная цепная реакция (LCR), активированная лигированием транскрипция (LAT) и основанная на последовательности нуклеиновой кислоты амплификация (NASBA). Последовательность нуклеиновой кислоты может клонироваться из штамма Streptococcus, или другого или родственного организма и, таким образом, например, может представлять собой аллельный или видовой вариант кодирующей полипептид области последовательности нуклеиновой кислоты.
Настоящее изобретение также относится к конструкциями нуклеиновой кислоты, содержащим последовательность нуклеиновой кислоты по настоящему изобретению, функционально связанную с одной или более регуляторными последовательностями, которые направляют экспрессию кодирующей последовательности в подходящей клетке-хозяине в условиях, приемлемых для регуляторных последовательностей.
Настоящее изобретение также относится к рекомбинантным экспрессирующим векторам, включающим последовательность нуклеиновой кислоты по настоящему изобретению, промотор и стоп-сигналы транскрипции и трансляции.
Настоящее изобретение также относится к рекомбинантным клеткам-хозяевам, содержащим последовательность нуклеиновой кислоты по изобретению, которые преимущественно используются при рекомбинантной продукции полипептидов.
Настоящее изобретение также относится к способам получения полипептида, обладающего активностью UDP-N-ацетилглюкозаминпирофосфорилазы, включающим (a) культивирование клетки-хозяина в условиях, подходящих для получения полипептида; и (b) извлечения полипептида.
В способах получения по настоящему изобретению клетки культивируют в питательной среде, подходящей для получения полипептида с использованием способов, известных в данной области. Например, клетки могут культивироваться путем культивирования во флаконах для встряхивания, путем мелко- и крупномасштабной ферментации (включая непрерывную ферментацию, ферментацию одного производственного цикла, подпитываемую ферментацию, или ферментацию в твердом состоянии) в лабораторных или промышленных ферментерах, проводимой в подходящей среде и в условиях, обеспечивающих экспрессию и/или выделение полипептида. Культивирование проходит в подходящей питательной среде, содержащей источники углерода и азота и неорганические соли, с использованием процедур, известных в данной области. Подходящие среды доступны от коммерческих поставщиков или могут быть получены согласно опубликованным композициям (например, в каталогах American Type Culture Collection). Если полипептид секретируется в питательную среду, он может быть непосредственно извлечен из среды. Если полипептид не секретируется, он может быть извлечен из клеточных лизатов.
Полипептиды можно выявлять с использованием известных в данной области способов, специфичных для полипептидов. Данные способы выявления могут охватывать применение специфических антител, образование продукта фермента или исчезновение субстрата фермента. Например, ферментный анализ может использоваться для определения активности описанного здесь полипептида.
Полученный в результате полипептид может выделяться способами, известными в данной области. Например, полипептид может выделяться из питательной среды обычными процедурами, включая в качестве неограничивающих примеров центрифугирование, фильтрацию, экстракцию, сушку распылением, упаривание или преципитацию.
Полипептиды затем можно очищать путем различных процедур, известных в данной области, включая в качестве неограничивающих примеров хроматографию (например, ионообменную, аффинную, гидрофобную, хроматофокусирование, и гель-фильтрацию), электрофоретические процедуры (например, препаративное изоэлектрофокусирование), дифференциальное растворение (например, преципитацию сульфатом аммония), SDS-PAGE или экстракцию (см., например, Protein Purification, J.-C. Janson and Lars Ryden, editors, VCH Publishers, New York, 1989).
Настоящее изобретение далее относится к выделенным полипептидам, характеризующимся активностью UDP-глюкозо-6-дегидрогеназы, UDP-глюкозопирофосфорилазы или UDP-N-ацетилглюкозаминпирофосфорилазы, кодируемым описанными выше последовательностями нуклеиновой кислоты.
Настоящее изобретение далее описано следующими примерами, которые следует рассматривать как ограничивающие объем изобретения.
ПРИМЕРЫ
ПРАЙМЕРЫ И ОЛИГОНУКЛЕОТИДЫ
Все праймеры и олигонуклеотиды были приобретены (MWG Biotech Inc., High Point, NC).
Пример 1: ПЦР-амплификация и клонирование гена hasA Streptococcus equisimilis и генов tuaD, gtaB и gcaD Bacillus subtilis
Ген гиалуронансинтазы Streptococcus equisimilis (hasA, инвентарный номер AF023876, SEQ ID NO: 1 [последовательность ДНК] и 2 [выведенная аминокислотная последовательность]) амплифицировали путем ПЦР из плазмиды pKKseD (Weigel, 1997, Journal of Biological Chemistry 272: 32539-32546) с использованием праймеров 1 и 2:
Праймер 1:
5'-GAGCTCTATAAAAATGAGGAGGGAACCGAATGAGAACATTAAAAAACCT-3' (SEQ ID NO: 3)
Праймер 2:
5'-GTTAACGAATTCAGCTATGTAGGTACCTTATAATAATTTTTTACGTGT-3' (SEQ ID NO: 4)
ПЦР-амплификацию проводили в трех параллелях в реакционных смесях объемом 50 мкл, составленных из 1 нг ДНК pKKseD, 0,4 мкМ каждого из праймеров 1 и 2, 200 мкМ каждого dATP, dCTP, dGTP, и dTTP, 1X буфер для ПЦР II (Applied Biosystems, Inc., Foster City, CA) с 2,5 мМ MgCl2, и 2,5 единицы ДНК-полимеразы AmpliTaq Gold™ (Applied Biosystems, Inc., Foster City, CA). Реакции проводили в термоциклере RoboCycler 40 (Stratagene, Inc., La Jolla, CA), запрограммированном на 1 цикл при 95°C в течение 9 минут; 3 цикла, каждый при 95°C в течение 1 минуты, при 52°C в течение 1 минуты и при 72°C в течение 1 минуты; 27 циклов, каждый при 95°C в течение 1 минуты, при 55°C в течение 1 минуты и при 72°C в течение 1 минуты; и 1 цикл при 72°C в течение 5 минут. Продукт ПЦР визуализировали с использованием 0,8%-ного агарозного геля с 44 мМ основания Tris, 44 мМ борной кислоты, 0,5 мМ буфера EDTA (0,5X TBE). Ожидаемый фрагмент составлял примерно 1200 п.н.
ПЦР-фрагмент длиной 1200 п.н. клонировали в pCR2.1 с использованием набора для клонирования TA-TOPO (Stratagene, Inc., La Jolla, СА) и трансформировали в компетентные клетки E. coli OneShot™ по инструкциям производителя (Stratagene, Inc., La Jolla, СА). Трансформантов отбирали при 37°C через 16 часов роста на агаровых чашках с 2X дрожжевым триптоном (YT), дополненным 100 мкг ампициллина на мл. Плазмидную ДНК данных трансформантов очищали с использованием робота QIAGEN (QIAGEN, Valencia, СА) по инструкциям производителя и последовательность ДНК вставок подтверждали путем секвенирования ДНК с использованием прямого праймера M13(-20) и обратного праймера M13 (Invitrogen, Inc, Carlsbad, СА) и следующих внутренних праймеров. Плазмиду, несущую ПЦР-фрагмент длиной 1200 п.н. обозначали pCR2.1-sehasA (фигура 3).
Праймер 3:
5'-GTTGACGATGGAAGTGCTGA-3' (SEQ ID NO: 5)
Праймер 4:
5'-ATCCGTTACAGGTAATATCC-3' (SEQ ID NO: 6)
Праймер 5:
5'-TCCTTTTGTAGCCCTATGGA-3' (SEQ ID NO: 7)
Праймер 6:
5'-TCAGCACTTCCATCGTCAAC-3' (SEQ ID NO: 8)
Праймер 7:
5'-GGATATTACCTGTAACGGAT-3' (SEQ ID NO: 9)
Праймер 8:
5'-TCCATAGGGCTACAAAAGGA-3' (SEQ ID NO: 10)
Ген UDP-глюкозо-6-дегидрогеназы Bacillus subtilis (tuaD, инвентарный номер BG12691, SEQ ID NO: 11 [последовательность ДНК] и 12 [выведеная аминокислотная последовательность]) амплифицировали путем ПЦР из Bacillus subtilis 168 (BGSC 1A1, Bacillus Genetic Stock Center, Columbus, ОН) с использованием праймеров 9 и 10:
Праймер 9:
5'-GGTACCGACACTGCGACCATTATAAA-3' (SEQ ID NO: 13)
Праймер 10:
5'-GTTAACGAATTCCAGCTATGTATCTAGACAGCTTCAACCAAGTAACACT-3' (SEQ ID NO: 14)
ПЦР-амплификацию проводили в трех параллелях в реакционных смесях объемом 30 мкл, составленных из 50 нг хромосомной ДНК Bacillus subtilis 168, 0,3 мкМ каждого из праймеров 9 и 10, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1X буфер для ПЦР II с 2,5 мМ MgCl2, и 2,5 единицы ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 9 минут; 5 циклов, при 95°C в течение 1 минуты, при 50°C в течение 1 минуты и при 72°C в течение 1,5 минут; 32 цикла, каждый при 95°C в течение 1 минуты, при 54°C в течение 1 минуты и при 72°C в течение 1,5 минут; и 1 цикл при 72°C в течение 7 минут. ПЦР-продукт визуализировали с использованием 0,8%-ного агарозного геля с 0,5X буфером TBE. Ожидаемый фрагмент составлял примерно 1400 п.н.
ПЦР-фрагмент длиной 1400 п.н. клонировали в pCR2.1 с использованием набора для клонирования TA-TOPO и трансформировали в компетентные клетки E. coli OneShot™ по инструкциям производителя. Плазмидную ДНК очищали с использованием робота QIAGEN по инструкциям производителя и последовательность ДНК вставок подтверждали путем секвенирования ДНК с использованием прямого праймера M13(-20) и обратного праймера M13 и следующих внутренних праймеров. Плазмиду, несущую ПЦР-фрагмент длиной 1400 п.н. обозначали pCR2.1-tuaD (фигура 4).
Праймер 11:
5'-AGCATCTTAACGGCTACAAA-3' (SEQ ID NO: 15)
Праймер 12:
5'-TGTGAGCGAGTCGGCGCAGA-3' (SEQ ID NO: 16)
Праймер 13:
5'-GGGCGCCCATGTAAAAGCAT-3' (SEQ ID NO: 17)
Праймер 14:
5'-TTTGTAGCCGTTAAGATGCT-3' (SEQ ID NO: 18)
Праймер 15:
5'-TCTGCGCCGACTCGCTCACA-3' (SEQ ID NO: 19)
Праймер 16:
5'-ATGCTTTTACATGGGCGCCC-3' (SEQ ID NO: 20)
Ген UTP-глюкозо-1-фосфоатуридилтрансферазы Bacillus subtilis (gtaB, инвентарный номер BG10402, SEQ ID NO: 21 [последовательность ДНК] и 22 [выведенная аминокислотная последовательность]) ПЦР-амплифицировали из Bacillus subtilis 168 с использованием праймеров 17 и 18:
Праймер 17:
5'-TCTAGATTTTTCGATCATAAGGAAGGT-3' (SEQ ID NO: 23)
Праймер 18:
5'-GTTAACGAATTCCAGCTATGTAGGATCCAATGTCCAATAGCCTTTTTGT-3' (SEQ ID NO: 24)
ПЦР-амплификацию проводили в трех параллелях в реакционных смесях объемом 30 мкл, составленных из 50 нг хромосомной ДНК Bacillus subtilis 168, 0,3 мкМ каждого из праймеров 17 и 18, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1X буфер для ПЦР II с 2,5 мМ MgCl2, и 2,5 единицы ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 9 минут; 5 циклов, при 95°C в течение 1 минуты, при 50°C в течение 1 минуты и при 72°C в течение 1,5 минут; 32 цикла, каждый при 95°C в течение 1 минуты, при 54°C в течение 1 минуты и при 72°C в течение 1,5 минут; и 1 цикл при 72°C в течение 7 минут. ПЦР-продукт визуализировали с использованием 0,8%-ного агарозного геля с 0,5X буфером TBE. Ожидаемый фрагмент составлял примерно 900 п.н.
ПЦР-фрагмент длиной 900 п.н. клонировали в pCR2.1 с использованием набора для клонирования TA-TOPO и трансформировали в компетентные клетки E. coli OneShot™ по инструкциям производителя. Плазмидную ДНК очищали с использованием робота QIAGEN по инструкциям производителя и последовательность ДНК вставок подтверждали путем определения последовательности ДНК с использованием прямого праймера M13(-20) и обратного праймера M13 и следующих внутренних праймеров. Плазмиду, несущую ПЦР-фрагмент длиной 1400 п.н. обозначали pCR2.1-gtaB (фигура 5).
Праймер 19: 5'-AAAAAGGCTTCTAACCTGGC-3' (SEQ ID NO: 25)
Праймер 20: 5'-AAACCGCCTAAAGGCACAGC-3' (SEQ ID NO: 26)
Праймер 21: 5'-GCCAGGTTAGAAGCCTTTTT-3' (SEQ ID NO: 27)
Праймер 22: 5'-GCTGTGCCTTTAGGCGGTTT-3' (SEQ ID NO: 28)
Ген UDP-N-ацетилглюкозаминпирофосфорилазы Bacillus subtilis (gcaD, инвентарный номер BG10113, SEQ ID NO: 29 [последовательность ДНК] и 30 [выведенная аминокислотная последовательность]) ПЦР-амплифицировали из Bacillus subtilis 168 с использованием праймеров 23 и 24:
Праймер 23: 5'-GGATCCTTTCTATGGATAAAAGGGAT-3' (SEQ ID NO: 31)
Праймер 24: 5'-GTTAACAGGATTATTTTTTATGAATATTTTT-3' (SEQ ID NO: 32)
ПЦР-амплификацию проводили в трех параллелях в реакционных смесях объемом 30 мкл, составленных из 50 нг хромосомной ДНК Bacillus subtilis 168, 0,3 мкМ каждого из праймеров 23 и 24, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1X буфер для ПЦР II с 2,5 мМ MgCl2, и 2,5 единицы ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 9 минут; 5 циклов, при 95°C в течение 1 минуты, при 50°C в течение 1 минуты и при 72°C в течение 1,5 минут; 32 цикла, каждый при 95°C в течение 1 минуты, при 54°C в течение 1 минуты и при 72°C в течение 1,5 минут; и 1 цикл при 72°C в течение 7 минут. ПЦР-продукт визуализировали с использованием 0,8%-ного агарозного геля с 0,5X буфером TBE. Ожидаемый фрагмент составлял примерно 1500 п.н.
ПЦР-фрагмент длиной 1500 п.н. клонировали в pCR2.1 с использованием набора для клонирования TA-TOPO и трансформировали в компетентные клетки E. coli OneShot™ по инструкциям производителя. Плазмидную ДНК очищали с использованием робота QIAGEN по инструкциям производителя и последовательность ДНК вставок подтверждали путем секвенирования ДНК с использованием прямого праймера M13(-20) и обратного праймера M13 и следующих внутренних праймеров. Плазмиду, несущую ПЦР-фрагмент длиной 900 п.н. обозначали pCR2.1-gcaD (фигура 6).
Праймер 25:
5'-CAGAGACGATGGAACAGATG-3' (SEQ ID NO: 33)
Праймер 26:
5'-GGAGTTAATGATAGAGTTGC-3' (SEQ ID NO: 34)
Праймер 27:
5'-GAAGATCGGGAATTTTGTAG-3' (SEQ ID NO: 35)
Праймер 28:
5'-CATCTGTTCCATCGTCTCTG-3' (SEQ ID NO: 36)
Праймер 29:
5'-GCAACTCTATCATTAACTCC-3' (SEQ ID NO: 37)
Праймер 30:
5'-CTACAAAATTCCCGATCTTC-3' (SEQ ID NO: 38)
Пример 2: Конструкция оперона hasA/tuaD/gtaB
Плазмиды pDG268Δneo-cryIIIAstab/Sav (патент США № 5955310) и pCR2.1-tuaD (пример 1, фигура 4) расщепляли KpnI и HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент (приблизительно 7700 п.н.) из pDG268Δneo-cryIIIAstab/Sav и меньший фрагмент tuaD (приблизительно 1500 п.н.) из pCR2.1-tuaD очищали в геле с использованием набора для экстракции ДНК QIAquick по инструкциям производителя (QIAGEN, Valencia, СА). Два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 по инструкциям производителя (Roche Applied Science; Indianapolis, IN) и лигированную смесь трансформировали в компетентные клетки E. coli SURE (Stratagene, Inc., La Jolla, СА). Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл.
Плазмидную ДНК очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления KpnI плюс HpaI и на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента KpnI/HpaI tuaD размером примерно 1500 п.н. и обозначали pHA1 (фигура 7).
Плазмиды pHA1 и pCR2.1-gtaB (пример 1, фигура 5) расщепляли XbaI и HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент (приблизительно 9200 п.н.) из pHA1 и меньший фрагмент gtaB (приблизительно 900 п.н.) из pCR2.1-gtaB очищали из 0,8%-ного агарозного геля с 0,5X буферм TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Эти два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и лигированную смесь использовали для трасформации компетентных клеток E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл при 37°С.
Плазмиды очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления XbaI плюс HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента XbaI/HpaI gtaB размером примерно 900 п.н. и обозначали pHA2 (фигура 8).
Плазмиды pHA2 и pCR2.1-sehasA (пример 1, фигура 3) расщепляли SacI плюс KpnI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Более крупный векторный фрагмент (приблизительно 10000 п.н.) из pHA2 и меньший фрагмент hasA (приблизительно 1300 п.н.) из pCR2.1-sehasA очищали из 0,8%-ного агарозного геля с 0,5X буфером TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и лигированную смесь использовали для трансформации компетентных клеток E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, при 37°С. Плазмиды очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления SacI плюс KpnI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента SacI/KpnI hasA размером примерно 1300 п.н. и обозначали pHA3 (фигура 9).
Пример 3: Конструирование оперона hasA/tuaD/gtaB/gcaD
Плазмиды pHA2 (пример 2, фигура 8) и pCR2.1-gcaD (пример 1, фигура 6) расщепляли BamHI и HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент (приблизительно 10000 п.н.) из pHA2 и меньший фрагмент gcaD (приблизительно 1400 п.н.) из pCR2.1-gcaD очищали из 0,8%-ном агарозного геля с 0,5X буфером TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Эти два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и лигированную смесь использовали для трансформации компетентных клеток E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл при 37°С.
Плазмиды очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и расщепляли XbaI плюс HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента BamHI/HpaI gcaD размером примерно 1400 п.н. и обозначали pHA4 (фигура 10).
Плазмиды pHA4 и pCR2.1-sehasA (пример 1, фигура 3) расщепляли SacI и KpnI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Более крупный векторный фрагмент (приблизительно 11000 п.н.) из pHA4 и меньший фрагмент hasA (приблизительно 1300 п.н.) из pCR2.1-sehasA очищали из 0,8%-ном агарозного геля с 0,5X буфером TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и лигированную смесь использовали для трансформации компетентных клеток E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл при 37°С. Плазмиды очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления SacI плюс KpnI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента SacI/KpnI hasA размером примерно 1300 п.н. и обозначали pHA5 (фигура 11).
Пример 4: Конструирование оперона hasA/tuaD/gcaD
Плазмиды pHA1 (пример 2, фигура 7) и pCR2.1-gcaD (пример 1, фигура 6) расщепляли BamHI и HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент (приблизительно 9200 п.н.) из pHA1 и меньший фрагмент gcaD (приблизительно 1400 п.н.) из pCR2.1-gcaD очищали из 0,8%-ного агарозного геля с 0,5X буфером TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Эти два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и лигированную смесь использовали для трансформации компетентных клеток E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл при 37°С.
Плазмиды очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления BamHI плюс HpaI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента BamHI/HpaI gtaB размером примерно 1400 п.н. и обозначали pHA6 (фигура 12).
Плазмиды pHA6 и pCR2.1-sehasA (пример 1, фигура 3) расщепляли SacI и KpnI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Более крупный векторный фрагмент (приблизительно 10200 п.н.) из pHA6 и меньший фрагмент hasA (приблизительно 1300 п.н.) из pCR2.1-sehasA очищали из 0,8%-ном агарозного геля с 0,5X буфером TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Эти два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и лигированную смесь использовали для трансформации компетентных клеток E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл. Плазмиды очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления SacI плюс KpnI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента SacI/KpnI hasA размером примерно 1300 п.н. и обозначали pHA7 (фигура 13).
Пример 5: Конструирование Bacillus subtilis RB161
Плазмиду pDG268MCSΔneo/scBAN/Sav (патент США № 5955310) расщепляли SacI. Расщепленную плазмиду затем очищали с использованием набора для очистки ДНК QIAquick по инструкциям производителя и в конце расщепляли NotI. Более крупный фрагмент плазмиды длиной примерно 6800 п.н. очищали с использованием набора для экстракции ДНК QIAquick из 0,8% агарозного геля с 0,5X TBE по инструкциям производителя (QIAGEN, Valencia, СА). Полученную векторную ДНК затем лигировали с ДНК-вставкой, описанной ниже.
Плазмиду pHA3 (пример 2, фигура 9) расщепляли SacI. Расщепленную плазмиду затем очищали, как описано выше, и в конце расщепляли NotI. Наименьший фрагмент плазмиды длиной примерно 3800 п.н. очищали из геля, как описано выше. Полученный вектор и ДНК-вставку лигировали с использованием набора для быстрого клонирования ДНК (Roche Applied Science; Indianapolis, IN) по инструкциям производителя. Перед трансформацией Bacillus subtilis продукт лигирования, описанный выше, линеаризовали с использованием ScaI для обеспечения интеграции в хромосому путем двойного кроссинговера вместо интеграции в хромосому путем простого кроссинговера. Компетентные клетки Bacillus subtilis 168Δ4 трансформировали продуктами лигирования, расщепленными ScaI. Bacillus subtilis 168Δ4 происходит из типового штамма Bacillus subtilis 168 (BGSC 1A1, Bacillus Genetic Stock Center, Columbus, ОН) и имеет делеции в генах spollAC, aprE, nprE, и amyE. Делецию данных четырех генов проводят, по существу, как описано для Bacillus subtilis A164Δ5, которая подробно описана в патенте США № 5891701.
Устойчивые к хлорамфениколу трансформанты Bacillus subtilis отбирали при 34°C после 16 часов роста на чашках с основой из кровяного агара и триптозы (TBAB), дополненных 5 мкг хромафеникола на мл. Для скрининга интеграции плазмиды путем двойного кроссинговера в локус amyE первичных трансформантов Bacillus subtilis высевали на чашки TBAB, дополненные 6 мкг неомицина на мл, и на чашки TBAB, дополненные 5 мкг хлорамфеникола на мл. Интеграция плазмиды путем двойного кроссинговера в локус amyE не содержала ген устойчивости к неомицину и поэтому делала штамм чувствительным к неомицину. Изоляты также высевали на минимальные чашки для визуализации того, продуцируют они гиалуроновую кислоту или нет. Продуцирующие гиалуроновую кислоту изоляты имеют «влажный» фенотип на минимальных чашках. С использованием скрининга на данных чашках устойчивые к хлорамфениколу и чувствительные к неомицину «влажные» трансформанты (вследствие продукции гиалуроновой кислоты) выделяли при 37°C.
Геномную ДНК выделяли из «влажных» устойчивых к хлорамфениколу и чувствительных к неомицину трансформантов Bacillus subtilis 168Δ4 с использованием колонки QIAGEN tip-20 (QIAGEN, Valencia, СА) по инструкциям производителя. ПЦР-амплификацию проводили на данных трансформантах с использованием синтетических олигонуклеотидов, приведенных ниже, которые основаны на последовательностях генов hasA, tuaD и gtaB, для подтверждения наличия и целостности данных генов в опероне трансформантов Bacillus subtilis.
Реакционные смеси амплификации (25 мкл) состояли примерно из 50 нг геномной ДНК трансформантов Bacillus subtilis 168Δ4, 0,5 мкМ каждого праймера, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфера для ПЦР II, 3 мМ MgCl2, и 0,625 единиц ДНК-полимеразы AmpliTaq Gold™. Реакционные смеси инкубировали в термоциклере RoboCycler 40, запрограммированном на один цикл при 95°C в течение 9 минут; 30 циклов, каждый при 95°C в течение 1 минуты, 55°C в течение 1 минуты и 72°C в течение 2 минут; и на финальный цикл при 72°C в течение 7 минут.
Праймеры 3 и 8 использовали для подтверждения наличия гена hasA, праймеры 3 и 16 для подтверждения наличия гена tuaD, и праймеры 3 и 22 для подтверждения наличия гена gtaB. Интегрант Bacillus subtilis 168Δ4 hasA/tuaD/gtaB обозначали как Bacillus subtilis RB158.
Геномную ДНК выделяли из Bacillus subtilis RB158 с использованием колонки QIAGEN tip-20 по инструкциям производителя и использовали для трансформации компетентной Bacillus subtilis A164Δ5 (делеции генов spollAC, aprE, nprE, amyE и srfC; см. патент США № 5891701). Трансформанты отбирали на чашках TBAB, дополненных 5 мкг хлорамфеникола на мл при 37°C. Интегрант Bacillus subtilis A164Δ5 hasA/tuaD/gtaB идентифицировали по его «влажному» фенотипу и обозначали Bacillus subtilis RB161.
Пример 6: Конструирование Bacillus subtilis RB163
Плазмиду pDG268MCSΔneo/scBAN/Sav (патент США № 5955310) расщепляли SacI. Расщепленную плазмиду затем очищали с использованием набора для очистки ДНК QIAquick по инструкциям производителя и, наконец, расщепляли NotI. Наиболее крупный фрагмент плазмиды длиной примерно 6800 п.н. очищали с использованием набора для экстракции ДНК QIAquick из 0,8% агарозного геля с 0,5X TBE по инструкциям производителя. Полученную векторную ДНК затем лигировали с ДНК-вставкой, описанной ниже.
Плазмиду pHA7 (пример 4, фигура 13) расщепляли SacI. Расщепленную плазмиду затем очищали, как описано выше, и наконец, расщепляли NotI. Наименьший фрагмент плазмиды длиной примерно 4300 п.н. очищали из геля, как описано выше. Полученный вектор и ДНК-вставку лигировали с использованием набора для быстрого клонирования ДНК по инструкциям производителя. Перед трансформацией Bacillus subtilis продукт лигирования, описанный выше, линеаризовали с использованием ScaI для обеспечения интеграции в хромосому путем двойного кроссинговера вместо интеграции в хромосому путем простого кроссинговера. Компетентные клетки Bacillus subtilis 168Δ4 трансформировали продуктами лигирования, расщепленными рестриктазой ScaI.
Устойчивые к хлорамфениколу трансформанты Bacillus subtilis отбирали на чашках TBAB, дополненных 5 мкг хромафеникола на мл, при 37°С. Для скрининга интеграции плазмиды путем двойного кроссинговера в локус amyE первичных трансформантов Bacillus subtilis помещали на чашки TBAB, дополненные 6 мкг неомицина на мл, и на чашки TBAB, дополненные 5 мкг хлорамфеникола на мл, для выделения устойчивых к хлорамфениколу и чувствительных к неомицину «влажных» трансформантов (вследствие продукции гиалуроновой кислоты).
Геномную ДНК выделяли из «влажных», устойчивых к хлорамфениколу и чувствительных к неомицину трансформантов Bacillus subtilis 168Δ4 с использованием колонки QIAGEN tip-20 по инструкциям производителя. Амплификации путем ПЦР проводили на данных трансформантах с использованием праймеров 3, 8, 16, 22 и праймера 30 (пример 1) для подтверждения наличия и целостности данных генов в опероне трансформантов Bacillus subtilis. Реакционные смеси амплификации (25 мкл) состояли примерно из 50 нг геномной ДНК трансформантов Bacillus subtilis 168Δ4, 0,5 мкМ каждого праймера, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфера для ПЦР II, 3 мМ MgCl2, и 0,625 единиц ДНК-полимеразы AmpliTaq Gold™. Реакционные смеси инкубировали в термоциклере RoboCycler 40, запрограммированном на один цикл при 95°C в течение 9 минут; 30 циклов, каждый при 95°C в течение 1 минуты, 55°C в течение 1 минуты, и 72°C в течение 2 минут; и на финальный цикл при 72°C в течение 7 минут.
Праймеры 3 и 8 использовали для подтверждения наличия гена hasA, праймеры 3 и 16 для подтверждения наличия гена tuaD, праймеры 3 и 22 для подтверждения наличия гена gtaB, и праймеры 3 и 30 для подтверждения наличия гена gcaD. Интегрант Bacillus subtilis 168Δ4 hasA/tuaD/gcaD обозначали как Bacillus subtilis RB160.
Геномную ДНК выделяли из Bacillus subtilis RB160 с использованием колонки QIAGEN tip-20 по инструкциям производителя и использовали для трансформации компетентной Bacillus subtilis A164Δ5. Трансформантов отбирали на чашках TBAB, содержащих 5 мкг хлорамфеникола на мл и выращивали 16 часов при 37°C. Интегрант Bacillus subtilis A164Δ5 hasA/tuaD/gcaD идентифицировали по его «влажному» фенотипу и обозначали Bacillus subtilis RB163.
Пример 7: Конструирование Bacillus subtilis TH-1
Оперон гиалуронансинтазы (has) получали из Streptococcus equisimilis с использованием следующей процедуры. Оперон has состоит из генов hasA, hasB, hasC, и hasD. Примерно 20 мкг хромосомной ДНК Streptococcus equisimilis D181 (Kumari and Weigel, 1997, Journal of Biological Chemistry 272: 32539-32546) расщепляли HindIII и разгоняли на 0,8%-ном агарозном геле с 0,5X TBE. ДНК в интервале 3-6 т.п.н. вырезали из геля и очищали с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Полученную вставку ДНК затем лигировали с векторной ДНК, описанной ниже.
Плазмиду pUC18 (2 мкг) расщепляли HindIII и выступающие 5'-концы дефосфорилировали щелочной фосфатазой креветки по инструкциям производителя (Roche Applied Science; Indianapolis, IN). Дефосфорилированный вектор и ДНК-вставку лигировали с использованием набора для быстрого клонирования ДНК по инструкциям производителя. Продукт лигирования использовали для трансформации компетентных клеток E. coli XL10 Gold Kan (Stratagene, Inc., La Jolla, СА). Клетки высевали на чашки с бульоном Лурия (100 мкг/мл ампициллина) и инкубировали в течение ночи при 37°C. Пять чашек, содержащих примерно 500 колоний/чашку, зондировали олигонуклеотидом 952-55-1, показанным ниже, который представляет собой последовательность длиной 54 п.н., идентичную кодирующей цепи вблизи 3'-конца гена hasA Streptococcus equisimilis D181 (нуклеотиды 1098-1151 относительно остатка A стартового кодона трансляции ATG).
Праймер 31:
5'-GTGTCGGAACATTCATTACATGCTTAAGCACCCGCTGTCCTTCTTGTTATCTCC-3' (SEQ ID NO: 39)
Олигонуклеотидный зонд был мечен DIG с использованием набора для мечения DIG 3'-конца олигонуклеотидов по инструкциям производителя (Roche Applied Science; Indianapolis, IN). Гибридизацию и хемилюминесцентную детекцию колоний проводили, как описано в "THE DIG SYSTEM USER'S GUIDE FOR FILTER HYBRIDIZATION", Boehringer Mannheim GmbH.
Идентифицировали семь колоний, гибридизовавшиеся с зондом. Плазмидную ДНК от одного из данных трансформантов очищали с использованием робота QIAGEN (QIAGEN, Valencia, СА) по инструкциям производителя, расщепляли HindIII и разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. ДНК-вставка, как было показано, составляла размером примерно 5 т.п.н. Данную плазмиду обозначали pMRT106 (фигура 14).
Последовательность ДНК клонированного фрагмента определяли с использованием набора для вставки EZ::TN™ <TET-1> по инструкциям производителя (Epicenter Technologies, Madison, Висконсин). Секвенирование выявило, что клонированная ДНК-вставка содержала последние 1156 п.н. гена hasA Streptococcus equisimilis, с последующими тремя другими генами, обозначенными hasB, hasC, и hasD; предположительно все четыре гена содержатся в одном опероне и поэтому транскрибируются совместно. Ген hasB Streptococcus equisimilis содержится в нуклеотидах 1411-2613 (SEQ ID NO: 40 [последовательность ДНК] и 41 [выведенная аминокислотная последовательность]) фрагмента, и ген hasC Streptococcus equisimilis - в нуклеотидах 2666-3565 (SEQ ID NO: 42 [последовательность ДНК] и 43 [выведенная аминокислотная последовательность]) фрагмента, и ген hasD Streptococcus equisimilis - в нуклеотидах 3735-5114 (SEQ ID NO: 44 [последовательность ДНК] и 45 [выведенная аминокислотная последовательность]) фрагмента.
Полипептиды, кодируемые генами hasB и hasC Streptococcus equisimilis, характеризуются некоторой гомологией в отношении генов hasB и hasC соответственно из последовательности оперона has Streptococcus pyogenes (Ferretti et al., 2001, Proc. Natl. Acad. Sci. U.S.A. 98 (8), 4658-4663). Степень идентичности определяли способом Clustal (Higgins, 1989, CABIOS 5: 151-153) с использованием программного обеспечения Vector NTI AlignX (Informax Inc., Bethesda, Мэриленд) со следующими значениями по умолчанию: парное сопоставление, штраф за пробел 10, штраф за удлинение 0,1, и матрица счета: blosum62mt2.
Сравнения аминокислотной последовательности показали, что белок HasB Streptococcus equisimilis характеризуется 70%-ной идентичностью по отношению к белку HasB из Streptococcus uberis (SEQ ID NO: 105); белок HasC Streptococcus equisimilis характеризуется 91%-ной идентичностью по отношению к белку HasC из Streptococcus pyogenes (SEQ ID NO: 99); и белок HasD Streptococcus equisimilis характеризуется 73%-ной идентичностью по отношению к белку GlmU (предполагаемая UDP-N-ацетилглюкозаминпирофосфорилаза) Streptococcus pyogenes (инвентарный № Q8P286). Ген hasD Streptococcus equisimilis кодирует полипептид, который характеризуется 50,7%-ной идентичностью в отношении к ферменту UDP-N-ацетилглюкозаминпирофосфорилазе, кодируемому геном gcaD Bacillus subtilis.
Плазмиду pHA5 (пример 3, фигура 11) расщепляли HpaI и BamHI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент (приблизительно 11000 п.н.) очищали с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Плазмиду pMRT106 расщепляли HindIII, липкие концы заполняли с помощью фрагмента Кленова и расщепляли ДНК BamHI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и меньший фрагмент вставки (приблизительно 1000 п.н., последние 2/3 гена hasD Streptococcus equisimilis) очищали с использованием набора для экстракции ДНК QIAquick по инструкциям производителя.
Два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 по инструкциям производителя и смесью лигирования трансформировали компетентные клетки E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, при 37°С.
Плазмидную ДНК очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления BamHI плюс NotI с разделением на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента BamHI/NotI hasD размером примерно 1100 п.н. и обозначали pHA8 (фигура 15). Плазмиду расщепляли HindIII и лигировали вместе с использованием ДНК-лигазы T4 по инструкциям производителя и лигированную смесь трансформировали в компетентные клетки E. coli SURE. Трансформанты отбирали на чашках агара с 2X YT со 100 мкг ампициллина на мл. Плазмидную ДНК очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления HindIII с разделением на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия единственной полосы размером примерно 9700 п.н. и обозначали pHA9 (фигура 16).
Плазмиду pHA9 расщепляли SacI и NotI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и меньший фрагмент размером приблизительно 2500 п.н. очищали из геля с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Плазмиду pDG268MCSΔneo/scBAN/Sav (патент США № 5955310) расщепляли SacI и NotI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент размером приблизительно 6800 п.н. очищали из геля с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 по инструкциям производителя и лигированную смесь трансформировали в компетентные клетки E. coli SURE (Stratagene, Inc., La Jolla, СА). Трансформантов отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл.
Плазмидную ДНК очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциям производителя и анализировали путем расщепления SaII плюс HindIII с разделением на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали за счет наличия фрагмента SaII/HindIII размером примерно 1600 п.н. и обозначали pHA10 (фигура 17).
Плазмиду pHA10 расщепляли HindIII и BamHI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный векторный фрагмент (приблизительно 8100 п.н.) очищали из геля с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Плазмиду pMRT106 расщепляли HindIII и BamHI. Продукты расщепления разгоняли на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE и более крупный фрагмент вставки размером приблизительно 4100 п.н. очищали из геля с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Два очищенных фрагмента лигировали вместе с использованием ДНК-лигазы T4 и смесь лигирования использовали для трансформации Bacillus subtilis 168Δ4. Трансформанты отбирали на чашках с агаром TBAB, дополненных 5 мкг хлорамфеникола на мл, при 37°C. Примерно 100 трансформантов помещали на TBAB, дополненный хлорамфениколом (5 мкг/мл), и на TBAB, дополненный неомицином (10 мкг/мл), для подсчета устойчивых к хлорафениколу, чувствительных к неомицину колоний; данный фенотип являлся индикатором двойного кроссинговера в локусе amyE. Идентифицировано некбольшое количество таких колоний, и все они проявляли «влажный фенотип», что указывало на продукцию гиалуровной кислоты. Одну из колонии выбирали и обозначали Bacillus subtilis 168Δ4::scBAN/se hasA/hasB/hasC/hasD.
Геномную ДНК выделяли из Bacillus subtilis 168Δ4::scBAN/se hasA/hasB/hasC/hasD с использованием колонки QIAGEN tip-20 по инструкциям производителя, и использовали для трансформации компетентной Bacillus subtilis A164Δ5. Трансформанты отбирали на чашках TBAB, содержащих 5 мкг хлорамфеникола на мл, и выращивали при 37°C в течение 16 часов. Интегрант Bacillus subtilis A164Δ5 hasA/hasB/hasC/hasD идентифицировали по его «влажному» фенотипу и обозначали Bacillus subtilis TH-1.
Пример 8: Конструировнаие Bacillus subtilis RB184
Ген hasA из Streptococcus equisimilis (пример 1) и ген tuaD (гомолог hasB из Bacillus subtilis) (пример 1) клонировали под контролем короткого «консенсусного» промотора amyQ (scBAN) (патент США № 5955310).
Плазмиду pDG268MCSΔneo/scBAN/Sav (патент США 5955310) расщепляли SacI. Затем расщепленную плазмиду очищали с использованием набора для экстракции ДНК QIAquick по инструкциям производителя и, наконец, расщепляли NotI. Наибольший фрагмент плазмиды размером примерно 6800 п.н. очищали из 0,8%-ного агарозного геля с 0,5X буфером TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя. Полученную векторную ДНК затем лигировали с описанной ниже ДНК-вставкой.
Плазмиду pHA5 (пример 3, фигура 11) расщепляли HpaI. Затем расщепленную плазмиду очищали, как описано выше, и наконец, расщепляли XbaI. Затем в дважды расщепленной плазмиде формировали тупые концы вначале путем инактивации XbaI при 85°C в течение 30 минут. Затупление концов проводили путем добавления 0,5 мкл 10 мМ раствора каждого dNTP, 1 мкл 1 ед./мкл ДНК-полимеразы T4 (Roche Applied Science; Indianapolis, IN) и инкубации при 11°C в течение 10 минут. Наконец, полимеразу инактивировали путем инкубации реакционной смеси при 75°C в течение 10 минут. Более крупный фрагмент плазмиды размером примерно 11000 п.н. затем очищали из геля, как описано выше, и повторно лигировали с использованием набора для быстрого клонирования ДНК по инструкуциям производителя. Лигированную смесь трансформировали в компетентные клетки E. coli SURE. Трансформантов отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, при 37°C. Плазмидную ДНК очищали от некоторых трансформантов с использованием робота QIAGEN по инструкциями производителя и анализировали путем расщепления ScaI и разделения на 0,8%-ном агарозном геле с использованием 0,5X буфера TBE. Правильную плазмиду идентифицировали путем наличия фрагмента размером примерно 11 т.п.н. и обозначали pRB157 (фигура 18).
pRB157 расщепляли SacI. Расщепленную плазмиду затем очищали с использованием набора для очистки ДНК QIAquick по инструкциям производителя, и наконец, расщепляли NotI. Наименьший фрагмент плазмиды длиной примерно 2632 п.н. очищали с использованием набора для экстракции ДНК QIAquick из 0,8% агарозного геля с 0,5X TBE по инструкциям производителя. Полученную ДНК-вставку затем лигировали с векторной ДНК, описанной ниже.
Перед трансформацией Bacillus subtilis продукт лигирования, описанный выше, линеаризовали с использованием ScaI для обеспечения интеграции в хромосому путем двойного кроссинговера вместо интеграции в хромосому путем простого кроссинговера. Компетентные клетки Bacillus subtilis 168Δ4 трансформировали продуктами лигирования, расщепленными рестриктазой ScaI.
Устойчивые к хлорамфениколу трансформанты Bacillus subtilis отбирали на чашках TBAB, дополненных 5 мкг хромафеникола на мл. Для скрининга интеграции плазмиды путем двойного кроссинговера в локус amyE первичных трансформантов Bacillus subtilis помещали на чашки TBAB, дополненные 6 мкг неомицина на мл, и на чашки TBAB, дополненные 5 мкг хлорамфеникола на мл, для выделения устойчивых к хлорамфениколу и чувствительных к неомицину «влажных» трансформантов (вследствие продукции гиалуроновой кислоты).
Геномную ДНК выделяли из «влажных», устойчивых к хлорамфениколу и чувствительных к неомицину трансформантов Bacillus subtilis 168Δ4 с использованием колонки QIAGEN tip-20 по инструкциям производителя. Амплификации путем ПЦР проводили на данных трансформантах с использованием праймеров 3, 8, и 16 (пример 1) для подтверждения наличия и целостности hasA и tuaD в опероне трансформантов Bacillus subtilis. Реакционные смеси амплификации (25 мкл) состояли примерно из 50 нг геномной ДНК трансформантов Bacillus subtilis 168Δ4, 0,5 мкМ каждого праймера, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфера для ПЦР II, 3 мМ MgCl2, и 0,625 единиц ДНК-полимеразы AmpliTaq Gold™. Реакционные смеси инкубировали в термоциклере RoboCycler 40, запрограммированном на один цикл при 95°C в течение 9 минут; 30 циклов, каждый при 95°C в течение 1 минуты, 55°C в течение 1 минуты, и 72°C в течение 2 минут; и на финальный цикл при 72°C в течение 7 минут.
Праймеры 3 и 8 использовали для подтверждения наличия гена hasA, праймеры 3 и 16 для подтверждения наличия гена tuaD. Интегрант Bacillus subtilis 168Δ4 hasA/tuaD обозначали как Bacillus subtilis RB183.
Геномную ДНК из Bacillus subtilis RB183 использовали также для трансформации компетентной Bacillus subtilis A164Δ5. Трансформанты отбирали на чашках TBAB, содержащих 5 мкг хлорамфеникола на мл, и выращивали при 37°C в течение 16 ч. Интегрант Bacillus subtilis A164Δ5 hasA/tuaD идентифицировали по его «влажному» фенотипу и обозначали Bacillus subtilis RB184.
Пример 9: Конструирование Bacillus subtilis RB187
Bacillus subtilis RB161 делали компетентным и трансформировали плазмидой pRB115 с делецией cat (Widner et al., 2000, Journal of Industrial Microbiology & Biotechnology 25: 204-212). Отбор по признаку прямой интеграции в хромосому проводили при непермессивной температуре, равной 45°C с использованием отбора эритромицином (5 мкг/мл). При данной температуре точка начала репликации pE194 является неактивной. Клетки способны сохранять устойчивость к эритромицину только путем интеграции плазмиды в ген cat на бактериальной хромосоме. Эти так называемые «интегранты» поддерживались при 45°C для обеспечения роста при такой температуре во время селекции. Для обеспечения потери или «выпячивания» плазмиды, которое будет приводить к делеции из хромосомы большей части гена cat, интегранты выращивали на среде Лурия-Бертани (LB) без селекции при пермессивной температуре, равной 34°C, в течение многих поколений. При данной температуре точка начала репликации pE194 активна и способствует вырезанию плазмиды из генома (Molecular Biological Methods for Bacillus, edited by C. R. Harwood and S. M. Cutting, 1990, John Wiley and Sons Ltd.).
Затем клетки высевают на чашки с неселективным агаром LB, и колонии, содержащие делеции в гене cat и потерю основанного на pE194 репликона, идентифицировали по следующим критериям: (1) чувствительность к хлорамфениколу, указывающая на наличие делеции cat; (2) чувствительность к эритромицину, указывающая на отсутствие гена устойчивости к эритромицину, кодируемого вектором pRB115; и (3) ПЦР, подтверждающая наличие делеции cat в интересующем штамме. ПЦР проводили для подтверждения делеции гена cat в локусе amyE с использованием праймеров 32 и 33:
Праймер 32:
5'-GCGGCCGCGGTACCTGTGTTACACCTGTT-3' (SEQ ID NO: 46)
Праймер 33:
5'-GTCAAGCTTAATTCTCATGTTTGACAGCTTATCATCGG-3' (SEQ ID NO: 47)
Хромосомную ДНК из потенциальных подвергнутых делеции бактерий выделяли с использованием наборов для ПЦР растений REDextract-N-Amp™ (Sigma Chemical Company, St. Louis, МО) следующим образом: единичные колонии Bacillus инокулировали на 100 мкл раствора экстракции (Sigma Chemical Company, St. Louis, МО), инкубировали при 95°C в течение 10 минут и затем разводили в равном объеме раствора разведения (Sigma Chemical Company, St. Louis, МО). ПЦР проводили с использованием 4 мкл экстрагированной ДНК в сочетании с реакционной смесью для ПЦР REDextract-N-Amp и требуемыми праймерами по инструкциями производителя с условиями циклов ПЦР, описанными в примере 5. Продукты реакции ПЦР визуализировали в 0,8%-ном агарозном геле с 0,5X TBE. Подтвержденный штамм обозначали Bacillus subtilis RB187.
Пример 10: Конструирование Bacillus subtilis RB192
Bacillus subtilis RB184 лишали метки путем делеции гена устойчивости к хлорамфениколу (ген cat). Это осуществляли с использованием способа, описанного ранее в примере 9. Полученный в результате штамм обозначали Bacillus subtilis RB192.
Пример 11: Конструирование Bacillus subtilis RB194
Bacillus subtilis RB194 конструировали путем делеции области cypX хромосомы Bacillus subtilis RB187 (пример 9). Область cypX охватывает ген cypX, который кодирует подобный цитохрому P450 фермент, участвующий в синтезе красного пигмента во время ферментации. Для делеции данной области хромосомы конструировали плазмиду pMRT086.
Область хромосомы, несущую опероны cypX-yvmC и yvmB-yvmA, амплифицировали путем ПЦР из Bacillus subtilis BRG-1 в виде одного фрагмента с использованием праймеров 34 и 35. Bacillus subtilis BRG1 по существу является химически мутированным изолятом продуцирующего амилазу штамма Bacillus subtilis, который основан на генетическом фоне Bacillus subtilis A164Δ5, который описан в примере 5. Последовательность данной области идентична опубликованной последовательности типового штамма Bacillus subtilis 168.
Праймер 34: 5'-CATGGGAGAGACCTTTGG-3' (SEQ ID NO: 48)
Праймер 35: 5'-GTCGGTCTTCCATTTGC-3' (SEQ ID NO: 49)
Реакционные смеси амплификации (50 мкл) состояли из 200 нг хромосомной ДНК Bacillus subtilis BRG-1, 0,4 мкМ каждого из праймеров 34 и 35, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфера высококачественного воспроизведения Expand™ (Roche Applied Science; Indianapolis, IN) с 1,5 мМ MgCl2 и 2,6 единицы ферментативной смеси системы ПЦР высококачественного воспроизведения ExpandТМ (Roche Applied Science; Indianapolis, IN). Хромосомную ДНК Bacillus subtilis BRG-1 получали с использованием колонки QIAGEN tip-20 по инструкциям производителя. Реакции амплификации проводили в термоциклере RoboCycler 40 (Stratagene, Inc, La Jolla, СА), запрограммированном на 1 цикл при 95°C в течение 3 минут; 10 циклов, каждый при 95°C в течение 1 минуты, при 58°C в течение 1 минуты и при 68°C в течение 4 минут; 20 циклов, каждый при 95°C в течение 1 минуты, при 58°C в течение 1 минуты, при 68°C в течение 4 минут плюс 20 секунд на цикл с последующим 1 циклом 72°C в течение 7 минут. Продукты реакции анализировали путем электрофореза в агарозном геле с использованием 0,8%-ного агарозного геля с 0,5X буфером TBE.
Полученный в результате фрагмент, содержащий опероны cypX-yvmC и yvmB-yvmA, клонировали в pCR2.1 с использованием набора для клонирования TA-TOPO и трансформировали в клетки E. coli OneShot™ по инструкциям производителя (Invitrogen, Inc., Carlsbad, СА). Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл. Плазмидную ДНК из некоторых трансформантов выделяли с использованием колонок QIAGEN tip-20 по инструкциям производителя и подтверждали путем секвенирования ДНК с использованием прямого праймера M13(-20) и обратного праймера M13 и праймеров с 36 по 51, показанных ниже. Полученную в результате плазмиду обозначали pMRT084 (фигура 19).
Праймер 36: 5'-CGACCACTGTATCTTGG-3' (SEQ ID NO: 50)
Праймер 37: 5'-GAGATGCCAAACAGTGC-3' (SEQ ID NO: 51)
Праймер 38: 5'-CATGTCCATCGTGACG-3' (SEQ ID NO: 52)
Праймер 39: 5'-CAGGAGCATTTGATACG-3' (SEQ ID NO: 53)
Праймер 40: 5'-CCTTCAGATGTGATCC-3' (SEQ ID NO: 54)
Праймер 41: 5'-GTGTTGACGTCAACTGC-3' (SEQ ID NO: 55)
Праймер 42: 5'-GTTCAGCCTTTCCTCTCG-3' (SEQ ID NO: 56)
Праймер 43: 5'-GCTACCTTCTTTCTTAGG-3' (SEQ ID NO: 57)
Праймер 44: 5'-CGTCAATATGATCTGTGC-3' (SEQ ID NO: 58)
Праймер 45: 5'-GGAAAGAAGGTCTGTGC-3' (SEQ ID NO: 59)
Праймер 46: 5'-CAGCTATCAGCTGACAG-3' (SEQ ID NO: 60)
Праймер 47: 5'-GCTCAGCTATGACATATTCC-3' (SEQ ID NO: 61)
Праймер 48: 5'-GATCGTCTTGATTACCG-3' (SEQ ID NO: 62)
Праймер 49: 5'-AGCTTTATCGGTGACG-3' (SEQ ID NO: 63)
Праймер 50: 5'-TGAGCACGATTGCAGG-3' (SEQ ID NO: 64)
Праймер 51: 5'-CATTGCGGAGACATTGC-3' (SEQ ID NO: 65)
Плазмиду pMRT084 расщепляли BsgI для делеции большей части оперонов cypX-yvmC и yvmB-yvmA, оставляя примерно 500 оснований с каждого конца. Расщепленную BsgI ДНК обрабатывали ДНК-полимеразой T4. Плазмиду pECC1 (Youngman et al., 1984, Plasmid 12: 1-9) расщепляли SmaI. Фрагмент размером примерно 5100 п.н. из pMRT084 и фрагмент размером примерно 1600 п.н. из pECC1 выделяли из 0,8%-ного агарозного геля с 0,5X TBE с использованием набора для экстракции ДНК QIAquick по инструкциям производителя, лигировали вместе и трансформировали в клетки E. coli XL1 Blue по инструкциям производителя (Stratagene, Inc., La Jolla, СА). Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл. Трансформанты, несущие правильную плазмиду с делетированной большей частью оперонов cypX-yvmC и yvmB-yvmA, путем ПЦР-амплификации с использованием праймеров 52 и 53. Амплификацию путем ПЦР проводили в реакционных смесях объемом 50 мкл, состоящих из 1 нг плазмидной ДНК, 0,4 мкМ каждого праймера, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфера для ПЦР II с 2,5 мМ MgCl2, и 2,5 единиц ДНК-полимеразы AmpliTaq Gold. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 10 минут; 25 циклов, каждый при 95°C в течение 1 минуты, 55°C в течение 1 минуты и 72°C в течение 1 минуты; и на 1 цикл при 72°C в течение 7 минут. Продукт ПЦР визуализировали с использованием 0,8%-ного агарозного геля с 0,5X TBE. Данную конструкцию обозначали pMRT086 (фигура 20).
Праймер 52: 5'-TAGACAATTGGAAGAGAAAAGAGATA-3' (SEQ ID NO: 66)
Праймер 53: 5'-CCGTCGCTATTGTAACCAGT-3' (SEQ ID NO: 67)
Плазмиду pMRT086 линеаризовали с использованием ScaI и трансформировали в компетентные клетки Bacillus subtilis RB128 в присутствии 0,2 мкг хлорамфеникола на мл. Трансформанты отбирали на чашках TBAB, содержащих 5 мкг хлорамфеникола на мл, после инкубации при 37°C в течение 16 часов. Хромосомную ДНК получали из некоторых трансформантов с использованием колонки QIAGEN tip-20 по инструкциям производителя. Устойчивые к хлорамфениколу колонии подвергали скринингу путем ПЦР на предмет делеции оперонов cypX-yvmC и yvmB-yvmA путем ПЦР с использованием праймеров 36 и 52, 36 и 53, 37 и 52, и 37 и 53. Амплификацию путем ПЦР проводили в реакционных смесях объемом 50 мкл, состоящих из 50 нг хромосомной ДНК, 0,4 мкМ каждого праймера, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфера для ПЦР II с 2,5 мМ MgCl2, и 2,5 единиц ДНК-полимеразы AmpliTaq GoldТМ. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 10 минут; 25 циклов, каждый при 95°C в течение 1 минуты, 55°C в течение 1 минуты и 72°C в течение 1 минуты; и на 1 цикл при 72°C в течение 7 минут. Продукт ПЦР визуализировали с использованием 0,8%-ного агарозного геля с 0,5X TBE. Полученный в результате штамм Bacillus subtilis RB128 с делецией cypX-yvmC и yvmB-yvmA обозначали Bacillus subtilis MaTa17.
Компетентные клетки Bacillus subtilis RB187 (пример 9) трансформировали геномной ДНК Bacillus subtilis MaTa17. Геномную ДНК получали из данного штамма с использованием колонки QIAGEN tip-20 по инструкциям производителей. Трансформанты Bacillus subtilis, устойчивые к хлорамфениколу, отбирали на чашках TBAB, дополненных 5 мкг хлорамфеникола на мл, при 37°C. Первичные трансформанты наносили полосами для выделения отдельных колоний на чашки TBAB, содержащие 5 мкг хлорамфеникола на мл, при 37°C. Полученный в результате штамм с делецией cypX-yvmC и yvmB-yvmA обозначали Bacillus subtilis RB194.
Пример 12: Конструирование Bacillus subtilis RB197
Bacillus subtilis RB197 очень похожа на Bacillus subtilis RB194 c единственным отличием, которое заключается в том, что RB197 содержит меньшую делецию в области cypX: только часть гена cypX делетирована в данном штамме, чтобы получить фенотип cypX-минус. Для того чтобы осуществить данную задачу, плазмиду pMRT122 конструировали, как описано ниже.
Плазмиду pCJ791 (фигура 21) конструировали посредством расщепления плазмиды pSJ2739 (WO 96/23073) при помощи EcoRI/HindIII и лигирования с фрагментом, содержащим подвергнутую делеции форму гена wprA (сериновая протеаза клеточной стенки) из Bacillus subtilis. 5'-область wprA амплифицировали с использованием праймеров 54 и 55, смотри ниже, и 3'-область амплифицировали с использованием праймеров 56 и 57, показанных ниже, из хромомсомной ДНК, полученной из Bacillus subtilis DN1885 (Diderichsen et al., 1990, Journal of Bacteriology 172: 4315-4321). ПЦР-амплификацию проводили в 50 мкл реакционной смеси, состоящей из 1 нг хромомсомной ДНК Bacillus subtilis DN1885, 0,4 мкМ каждого из праймеров 39 и 40, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1X ПЦР-буфера II с 2,5 мМ MgCl2, и 2,5 единиц ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 10 минут; 25 циклов, каждый 95°C в течение 1 минуты, 55°C в течение 1 минуты и при 72°C в течение 1 минуты; и 1 цикл при 72°C в течение 7 минут.
5'- и 3'-фрагменты ПЦР wprA объединяли посредством расщепления при помощи BglII, за которым следовало лигирование, и ПЦР-амплификацию проводили для фрагментов смеси лигирования с использованием праймеров 54 и 57. ПЦР-амплификацию проводили в 50 мкл реакционной смеси, состоящей из 1 нг лигированного фрагмента, 0,4 мкМ каждого праймера, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1X ПЦР-буфера II с 2,5 мМ MgCl2 и 2,5 единиц ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 10 минут; 25 циклов, каждый при 95°C в течение 1 минуты, при 55°C в течение 1 минуты и при 72°C в течение 1 минуты; и 1 цикл при 72°C в течение 7 минут. Продукт ПЦР визуализировали с использованием 0,8% агарозного геля с 0,5Х TBE. Полученный в результате фрагмент ПЦР клонировали в pSJ2739 в качестве EcoRI/HindIII фрагмента, что приводило к получению плазмиды pCJ791 (фигура 21). Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг канамицина на мл после инкубации при температуре 28°C в течение 24-48 часов. Плазмидную ДНК из нескольких трансформантов выделяли с использованием колонок QIAGEN tip-20 согласно инструкциям производителя и подтверждали посредством ПЦР-амплификации с праймерами 54 и 57, используя описанные выше условия.
Праймер 54: 5'-GGAATTCCAAAGCTGCAGCGGCCGGCGCG-3' (SEQ ID NO: 68)
Праймер 55: 5'-GAAGATCTCGTATACTTGGCTTCTGCAGCTGC-3' (SEQ ID NO: 69)
Праймер 56: 5'-GAAGATCTGGTCAACAAGCTGGAAAGCACTC-3' (SEQ ID NO: 70)
Праймер 57: 5'-CCCAAGCTTCGTGACGTACAGCACCGTTCCGGC-3' (SEQ ID NO: 71)
Последовательность выше amyL и 5'-кодирующая область плазмиды pDN1981 (Патент США № 5698415) сливали посредством SOE, используя пары праймеров 58/59 и 60/61, показанных ниже. Полученный в результате фрагмент клонировали в вектор pCR2.1, чтобы получить плазмиду pMRT032, как изложено ниже. ПЦР-амплификации проводили в трех повторах в 50 мкл реакционной смеси, состоящей из 1 нг ДНК pDN1981, 0,4 мкМ каждого из соответствующих праймеров, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1× ПЦР-буфера II с 2,5 мМ MgCl2 и 2,5 единиц ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 9 минут; 3 цикла, каждый при 95°C в течение 1 минуты, при 52°C в течение 1 минуты и при 72°C в течение 1 минуты; 27 циклов, каждый при 95°C в течение 1 минуты, при 55°C в течение 1 минуты и при 72°C в течение 1 минуты; и 1 цикл при 72°C в течение 5 минут. Продукт ПЦР визуализировали с использованием 0,8% агарозного геля с 0,5X TBE. Ожидаемые фрагменты были длиной приблизительно 530 и 466 п.н. соответственно. Окончательный фрагмент SOE получали с использованием пары праймеров 59/60 и клонировали его в вектор pCR2.1, используя набор для клонирования TA-TOPO. Трансформанты отбирали на чашках арага с 2X YT, дополненных 100 мкг/мл ампициллина после инкубации при температуре 37°C в течение 16 часов. Плазмидную ДНК из нескольких трансформантов выделяли с использованием колонок QIAGEN tip-20 согласно инструкциям производителя и подтверждали посредством секвенирования ДНК при помощи прямого праймера M13 (-20) и обратного праймера M13. Плазмиду, несущую слитный фрагмент 5' кодирующей последовательности и последовательности выше amyL, обозначали как pMRT032 (фигура 22).
Праймер 58:
5'-CCTTAAGGGCCGAATATTTATACGGAGCTCCCTGAAACAACAAAAACGGC-3' (SEQ ID NO: 72)
Праймер 59: 5'-GGTGTTCTCTAGAGCGGCCGCGGTTGCGGTCAGC-3' (SEQ ID NO: 73)
Праймер 60: 5'-GTCCTTCTTGGTACCTGGAAGCAGAGC-3' (SEQ ID NO: 74)
Праймер 61: 5'-GTATAAATATTCGGCCCTTAAGGCCAGTACCATTTTCCC-3' (SEQ ID NO: 75)
Плазмиду pNNB194 (pSK+/pE194; патент США № 5958728) расщепляли при помощи NsiI и NotI и плазмиду pBEST501 (Itaya et al. 1989 Nucleic Acids Research 17: 4410) расщепляли при помощи PstI и NotI. Фрагмент вектора длиной 5193 п.н. из pNNB194 и фрагмент длиной 1306 п.н., несущий ген neo из pBEST501 выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя. Выделенные фрагменты лигировали совместно и использовали для трансформации компетентных клеток SURE E. coli согласно инструкциям производителя. Трансформанты, устойчивые к действию ампициллина, отбирали на чашках с 2X YT, дополненных 100 мкг ампициллина на мл. Плазмидную ДНК выделяли из одного такого трансформанта, используя набор для плазмид QIAGEN (QIAGEN Inc., Valencia, CA) и плазмиду подтверждали посредством расщепления NsiI и NotI. Данную плазмиду обозначали как pNNB194neo (фигура 23).
Плазмиду pNNB194neo расщепляли при помощи SacI/NotI и обрабатывали ДНК-полимеразой T4 и dNTP для того, чтобы получить тупые концы с использованием стандартных протоколов. Плазмиду pPL2419 (патент США № 5958728) расщепляли при помощи Ecl136II. Фрагмент вектора длиной 6478 п.н. из pNNB194neo и фрагмент длиной 562 п.н., несущий oriТ из pPL2419 выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя. Очищенные из геля фрагменты лигировали совместно и использовали для трансформации клеток SURE E. coli согласно инструкциям производителя. Трансформанты, устойчивые к действию ампициллина, выбирали на чашках с 2X YT, дополненных 100 мкг ампициллина на мл при температуре 37°C. Плазмидную ДНК выделяли из одного такого трансформанта, используя набор для плазмид QIAGEN и плазмиду подтверждали посредством расщепления NsiI, SacI и BscI. Данную плазмиду обозначали как pNNB194neo-oriT (фигура 24).
Плазмиду pNNB194neo-oriT расщепляли при помощи BamHI и обрабатывали ДНК-полимеразой T4 и dNTP для того, чтобы получить тупые концы с использованием стандартных протоколов. Расщепленную плазмиду очищали из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя. Очищенную плазмиду обрабатывали ДНК-лигазой T4 и использовали для трансформации клеток SURE E coli согласно инструкциям производителя. Трансформанты, устойчивые к действию ампициллина, выбирали на чашках с 2X YT, дополненных 100 мкг ампициллина на мл, при 37°С. Плазмидную ДНК выделяли из одного такого трансформанта, используя набор для плазмид QIAGEN, и разрушения участка BamHI подтверждали посредством расщепления при помощи BamHI и SacI. Полученную плазмиду обозначали как pShV3 (фигура 25).
Плазмиду pShV2.1-amyEΔ (патент США № 5958728) расщепляли при помощи SfiI и NotI и фрагмент вектора длиной 8696 п.н. очищали из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя. Для того чтобы встроить участок BamHI между участками SfiI и NotI pShV2.1-amyEΔ, был сконструирован синтетический линкер, как указано ниже: праймеры 62 и 63 отжигали, смешивая 50 мкМ каждого, прокипятив смесь и позволив смеси медленно остыть.
Праймер 62: 5'-GGGCCGGATCCGC-3' (SEQ ID NO: 76)
Праймер 63: 3'-ATTCCCGGCCTAGGCGCCGG-5' (SEQ ID NO: 77)
Очищенный вектор pShV2.1-amyEΔ и олигонуклеотиды после отжига лигировали совместно и использовали для трансформации компетентных клеток SURE E. coli. Трансформанты, устойчивые к действию хлорамфеникола, отбирали на чашках с LB, дополненных 30 мкг хлорамфеникола на мл при температуре 37°С. Плазмидную ДНК выделяли из одного такого трансформанта с использованием набора для плазмид QIAGEN и вставку участка BamHI подтверждали посредством расщепления при помощи BamHI. Данную плазмиду обозначали как pShV2.1-amyEΔВ (фигура 26).
Плазмиды pShV3 и pShV2.1-amyEΔB расщепляли при помощи SalI/HindIII. Фрагмент вектора длиной 7033 п.н. из pShV3 и фрагмент длиной 1031 п.н., несущий amyEΔ из pShV2.1-amyEΔ, очищали из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя. Очищенные гелем фрагменты лигировали совместно и использовали для трансформации клеток SURE E.coli согласно инструкциям производителя. Трансформанты, устойчивые к действию ампициллина, отбирали на чашках с 2X YT, дополненных 100 мкг ампициллина на мл. Плазмидную ДНК выделяли из одного такого трансформанта с использованием набора для плазмид QIAGEN и плазмиду подтверждали посредством расщепления при помощи SalI и HindIII. Полученную в результате плазмиду обозначали как pShV3А (фигура 27).
Плазмиду pMRT032 расщепляли при помощи KpnI/XbaI, содержащей фрагмент Кленова ДНК-полимеразы в присутствии dNTP, и фрагмент длиной приблизительно 1000 п.н. выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя. Данный фрагмент клонировали в плазмиду pShV3a, расщепленную при помощи EcoRV, и трансформировали в клетки XL1 Blue E. coli согласно инструкциям производителя. Трансформанты отбирали на чашках с агаром и 2X YT, дополненных 100 мкг ампициллина на мл после инкубации при температуре 37°C в течение 16 часов. Плазмидную ДНК из нескольких трансформантов выделяли, используя колонки QIAGEN tip-20 согласно инструкциям производителя, и подтверждали в 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи SacI/SphI. Полученную плазмиду обозначали как pMRT036 (фигура 28).
Плазмиду pMRT036 расщепляли при помощи SalI/HindIII, содержащей фрагмент Кленова ДНК-полимеразы в присутствии dNTP, лигировали и трансформировали в клетки XL1 Blue E coli согласно инструкциям производителя. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг/мл ампициллина после инкубации при температуре 37°C в течение 16 часов. Плазмидную ДНК из нескольких трансформантов выделяли, используя колонки QIAGEN tip-20, согласно инструкциям производителя и подтверждали в 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи SacI/XbaI, PstI и NdeI. Полученную плазмиду обозначали как pMRT037 (фигура 29).
Стабилизирующий фрагмент scBAN/cryIIIA плазмиды pDG268Δneo-cryIIIAstab/Sav (патент США № 5955310) выделяли из 2% агарозного геля с 0,5X TBE как фрагмент SfiI/SacI с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя, лигировали в плазмиду pMRT037, расщепленной при помощи SfiI/SacI, и трансформировали в клетки XL1 Blue E.coli. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, после инкубации при 37°C в течение 16 часов. Плазмидную ДНК из нескольких трансформантов выделяли, используя колонки QIAGEN tip-20 согласно инструкциям производителя, и подтверждали в 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи PstI. Полученную плазмиду обозначали как pMRT041 (фигура 30).
Плазмиды pMRT041 и pCJ791 расщепляли при помощи EcoRI/HindIII. Фрагмент длиной приблизительно 1300 п.н. из pMRT041 и фрагмент длиной приблизительно 4500 п.н. из pCJ791 выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя, лигировали и трансформировали в компетентные клетки Bacillus subtilis 168Δ4. Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл, после инкубации при температуре 30°C в течение 24-48 часов. Плазмидную ДНК из нескольких трансформантов выделяли, используя колонки QIAGEN tip-20 согласно инструкциям производителя, и подтверждали в 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи SacI и EcoRI/HindIII. Полученную плазмиду обозначали как pMRT064.1 (фигура 31).
Участок SacI в положении 2666 в плазмиде pMRT064.1 делетировали посредством SOE с использованием пары праймеров 64 и 65 и пары праймеров 66 и 67, показанных ниже. ПЦР-амплификацию проводили в 50 мкл реакционной смеси, состоящей из 1 нг ДНК pMRT064.1, 0,4 мкМ каждого из праймеров, 200 мкМ каждого из dATP, dCTP, dGTP и dTTP, 1X ПЦР-буфера II с 2,5 мМ MgCl2, и 2,5 единиц ДНК-полимеразы AmpliTaq Gold™. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 10 минут; 25 циклов, каждый при 95°C в течение 1 минуты, при 52°C в течение 1 минуты и при 72°C в течение 1 минуты; и 1 цикл при 72°C в течение 7 минут. Продукт ПЦР визуализировали в 0,8% агарозном геле с 0,5X TBE. Ожидаемые фрагменты имели длину приблизительно 400 и 800 п.н. соответственно. Окончательный фрагмент для клонирования обратно в pMRT064.1 амплифицировали с использованием праймеров 64 и 67. Данный фрагмент клонировали в вектор pCR2.1, используя набор для клонирования TA-TOPO. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг/мл ампициллина, после инкубации при температуре 37°C в течение 16 часов. Трансформанты, несущие правильную плазмиду, подтверждали посредством секвенирования ДНК с использованием прямого и обратного праймеров M13 и праймеров 65, 67 и 68. Данную плазмиду обозначали как pMRT068 (фигура 32) и далее трансформировали в клетки DM1 E.coli (Stratagene, Inc., La Jolla, CA) согласно инструкциям производителя. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл.
Праймер 64: 5'-GGAAATTATCGTGATCAAC-3' (SEQ ID NO: 78)
Праймер 65: 5'-GCACGAGCACTGATAAATATG-3' (SEQ ID NO: 79)
Праймер 66: 5'-CATATTTATCAGTGCTCGTGC-3' (SEQ ID NO: 80)
Праймер 67: 5'-TCGTAGACCTCATATGC-3' (SEQ ID NO: 81)
Праймер 68: 5'-GTCGTTAAACCGTGTGC-3' (SEQ ID NO: 82)
Участок SacI в положении 5463 и 6025 в плазмиде pMRT064.1 делетировали с использованием ПЦР-амплификации при помощи праймеров 69 и 70 и использовали описанные выше условия ПЦР. Полученный фрагмент клонировали в вектор pCR2.1 с использованием набора для клонирования TA-TOPO (Invitrogen, Inc., Carlsbad, CA). Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, после инкубации при температуре 37°C в течение 16 часов. Трансформанты, несущие правильную плазмиду, подтверждали посредством секвенирования ДНК с использованием прямого и обратного праймеров M13. Данную конструкцию обозначали как pMRT069 (фигура 33).
Праймер 69: 5'-CTAGAGGATCCCCGGGTACCGTGCTCTGCCTTTTAGTCC-3' (SEQ ID NO: 83)
Праймер 70: 5'-GTACATCGAATTCGTGCTCATTATTAATCTGTTCAGC-3' (SEQ ID NO: 84)
Плазмиды pMRT068 и pMRT064.1 расщепляли при помощи BclI/AccI. Фрагмент длиной приблизительно 1300 п.н. из pMRT068 и фрагмент длиной приблизительно 3800 п.н. из pMRT064.1 выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя, лигировали и трансформировали в компетентные клетки Bacillus subtilis 168Δ4. Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл после инкубации при температуре 30°C в течение 24-48 часов. Трансформанты, несущие правильную плазмиду, определяли на 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи SacI и EcoRI/AvaI. Полученную конструкцию обозначали как pMRT071 (фигура 34).
Плазмиды pMRT071 и pMRT069 расщепляли при помощи AvaI/EcoRI. Фрагмент длиной 578 п.н. из pMRT069 и фрагмент длиной 4510 п.н. из pMRT071 выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя, лигировали и трансформировали в компетентные клетки Bacillus subtilis 168Δ4. Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл, после инкубации при температуре 30°C в течение 24-48 часов. Трансформанты, несущие правильную плазмиду, определяли на 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи SacI. Полученную конструкцию обозначали как pMRT074 (фигура 35).
Плазмиду pMRT084, описанную в примере 11, расщепляли при помощи SacII/NdeI, обрабатывали ДНК-полимеразой T4, лигировали и трансформировали в клетки XL1 Blue E.coli согласно инструкциям производителя. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, после инкубации при температуре 37°C в течение 16 часов. Трансформанты, несущие правильную плазмиду, определяли на 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи DraI. Полученную плазмиду обозначали как pMRT120 (фигура 36).
Плазмиду pMRT074 расщепляли при помощи HindIII, обрабатывали фрагментом Кленова ДНК-полимеразы и расщепляли при помощи EcoRI. Плазмиду pMRT120 расщепляли при помощи EcoRI/Ecl13611. Фрагмент длиной приблизительно 600 п.н. из pMRT120 и фрагмент длиной приблизительно 4300 п.н. из pMRT074 выделяли из 0,8% агарозного геля с 0,5X TBE, используя набор для очистки ДНК QIAquick согласно инструкциям производителя, лигировали и трансформировали в компетентные клетки Bacillus subtilis 168Δ4. Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл, после инкубации при температуре 30°C в течение 24-48 часов. Трансформанты, несущие правильную плазмиду, определяли на 0,8% агарозном геле с 0,5X TBE посредством рестрикционного анализа при помощи SspI. Полученную конструкцию обозначали как pMRT122 (фигура 37).
Плазмиду pMRT122 трансформировали в компетентные клетки Bacillus subtilis А164Δ5. Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл, после инкубации при температуре 30°C в течение 24-48 часов. Плазмиду вводили в хромосому Bacillus subtilis A164Δ5 посредством гомологичной рекомбинации в локус cypX, инкубируя на чашках со свеженанесенными клетками Bacillus subtilis A164Δ5 (pMRT086) при температуре 45°C в течение 16 часов и выбирая живые растущие колонии. Геномную ДНК выделяли из данного штамма с использованием колонки QIAGEN tip-20 согласно инструкциям производителя и использовали для трансформации Bacillus subtilis RB187 (пример 9). Трансформанты отбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл, после инкубации при температуре 45°C в течение 16 часов. При данной температуре репликон pE194 не способен реплицироваться. Клетки способны поддерживать резистентность к эритромицину, только сохраняя плазмиду в бактериальной хромосоме.
Плазмиду удаляли из хромосомы посредством гомологичной рекомбинации, что приводит к делеции части гена cypX в хромосоме, выращивая трансформанты в среде Лурия-Бертани (LB) без отбора при пермессивной температуре 34°C для множества поколений. При данной температуре такого начала репликация pE194 является активной и фактически содействует вырезанию плазмиды из хромосомы (Molecular Biological Methods for Bacillus, edited by C. R. Harwood and S. M. Cutting, 1990, John Wiley and Sons Ltd.).
После нескольких поколений культивирования клетки помещали на чашки агара с неселективной LB и колонии, без плазмиды и делетированные по cypX и продуцирующие гиалуроновую кислоту, определяли, как следует ниже: (1) клеточный слой был «влажным», когда его помещали на чашку с минимальной средой, указывающей на продукцию гиалуроновой кислоты, (2) чувствительность к эритромицину указывала на потерю основанной на pE194 плазмиды и (3) ПЦР подтвердила наличие делеции 800 п.н. cypX в интересующем штамме посредством использования праймеров 34 и 45.
Хромосомную ДНК из возможных клеток с делецией cypX выделяли с использованием набора для ПЦР растений REDextract-N-Amp™, как следует ниже: единичные колонии Bacillus инокулировали в 100 мкл раствора для экстракции, инкубировали при температуре 95°C в течение 10 минут и затем разбавляли равным объемом раствора для разбавления. ПЦР проводили с использованием 4 мкл экстрагированной ДНК в сочетании с реакционной смесью для ПЦР REDextract-N-Amp™ и необходимых праймеров согласно инструкциям производителя, используя условия циклирования ПЦР, как описано в примере 5. Продукты реакции ПЦР визуализировали с использованием 0,8% агарозного геля с 0,5X TBE. Подтвержденный штамм обозначали как Bacillus subtilis RB197.
Пример 13: Конструирование Bacillus subtilis RB200
Ген cypX Bacillus subtilis RB192 делетировали с использованием тех же самых способов, описанных в примере 9 для Bacillus subtilis RB187. Полученный в результате штамм обозначали как Bacillus subtilis RB200.
Пример 14: Конструирование Bacillus subtilis RB202
Bacillus subtilis A164Δ5ΔcypX конструировали, как следует ниже: плазмиду pMRT122 (пример 12) трансформировали в компетентные клетки Bacillus subtilis A164Δ5. Трансформанты выбирали на чашках агара с TBAB, дополненных 1 мкг эритромицина и 25 мкг линкомицина на мл, после инкубации при температуре 30°C в течение 24-48 часов. Плазмиду вводили в хромосому Bacillus subtilis A164Δ5 посредством гомологичной рекомбинации в локус cypX, инкубируя на чашках со свеженанесенными клетками Bacillus subtilis A164Δ5 (pMRT086) при температуре 45°C в течение 16 часов и выбирая здоровые растущие колонии. Плазмиду удаляли из хромосомы посредством гомологичной рекомбинации, приводящей к делеции части гена cypX в хромосоме, выращивая трансформанты на среде Лурия-Бертани (LB) без селекции при пермессивной температуре 34°C для множества поколений. При данной температуре начало репликации является активным и фактически содействует вырезанию плазмиды из хромосомы (Molecular Biological Methods for Bacillus, edited by C. R. Harwood and S. M. Cutting, 1990, John Wiley and Sons Ltd.). После нескольких поколений культивирования клетки помещали на чашки агара с неселективной LB и колонии без плазмиды и имеющие теперь делецию cypX, определяли, как следует ниже: (1) чувствительность к эритромицину указывала на потерю основанной на pE194 плазмиды и (2) ПЦР подтвердила наличие делеции 800 п.н. cypX в интересующем штамме посредством использования праймеров 34 и 45, как описано выше. Подтвержденный штамм обозначали как Bacillus subtilis A164Δ5ΔcypX.
Bacillus subtilis A164Δ5ΔcypX сделали компетентным и трансформировали геномной ДНК Bacillus subtilis TH1 (пример 7), выделенной с использованием колонки QIAGEN tip-20 согласно инструкциям производителя. Трансформанты отбирали на чашках агара с TBAB, содержащих 5 мкг хлорамфеникола на мл, при температуре 37°C. Интегрант hasA/hasB/hasC/hasD Bacillus subtilis A164Δ5ΔcypX определяли по его «влажному» фенотипу и обозначали как Bacillus subtilis RB201. Ген cat удаляли из Bacillus subtilis RB201, используя те же самые способы, что описаны в примере 9. Полученный в результате штамм обозначали как Bacillus subtilis RB202.
Пример 15: Конструирование Bacillus subtilis MF002 (tuaD/gtaB)
Плазмиду pHA3 (пример 2, фигура 9) расщепляли при помощи Asp718. Затем в расщепленной плазмиде формировали тупые концы при первой инактивации фермента рестрикции при температуре 85°C в течение 30 минут. Формирование тупых концов проводили, добавляя 0,5 мкл 10 мМ каждого dNTP, 1 мкл 1 ед./мкл полимеразы T4 и инкубируя при 11°C в течение 10 минут. В конце полимеразу инактивировали, инкубируя реакционную смесь при 75°C в течение 10 минут. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи NotI. Самый малый фрагмент плазмиды длиной приблизительно 2522 п.н. затем очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК-вставку (tuaD/gtaB) затем лигировали при помощи ДНК вектора, описанной ниже.
Плазмиду pDG268MCSΔneo/scBAN/Sav (патент США № 5955310) расщепляли при помощи Ecl13611. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи NotI. Самый большой фрагмент плазмиды длиной приблизительно 6800 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя.
Восстановленный вектор и ДНК-вставку лигировали с использованием набора для быстрого клонирования ДНК согласно инструкциям производителя. Перед трансформацией в Bacillus subtilis продукт лигирования, описанный выше, линеаризовали с использованием ScaI, чтобы обеспечить интеграцию двойным кроссинговером в хромосоме, а не интеграцию единичным кроссинговером в хромосоме. Компетентные клетки Bacillus subtilis 168Δ4 трансформировали при помощи продукта лигирования, расщепленного при помощи рестрикционного фермента ScaI.
Трансформанты Bacillus subtilis, устойчивые к действию хлорамфеникола, отбирали на чашках с TBAB, дополненных 5 мкг хлорамфеникола на мл. Для скрининга интеграции плазмиды посредством двойного кроссинговера в локусе amyE, первичные трансформанты Bacillus subtilis помещали на чашки с TBAB, дополенные 6 мкг неомицина на мл, и на чашки с TBAB, дополненные 5 мкг хлорамфеникола на мл, для того чтобы выделить трансформанты, устойчивые к действию хлорамфеникола и чувствительные к действию неомицина.
Хромосомную ДНК из устойчивых к действию хлорамфеникола и чувствительных к действию неомицина трансформантов Bacillus subtilis 168Δ4 выделяли с использованием набора для ПЦР растений REDextract-N-AmpТМ (Sigma Chemical Company, St. Louis, Монтана), как следует ниже: единичные колонии Bacillus инокулировали в 100 мкл раствора для экстракции, инкубировали при 95°C в течение 10 минут и затем разбавляли равным объемом раствора для разбавления. ПЦР проводили с использованием 4 мкл экстрагированной ДНК в соединении с реакционной смесью для ПЦР REDextract-N-Amp™ и необходимых праймеров согласно инструкциям производителя, используя условия циклирования ПЦР, как описано в примере 5.
ПЦР-амплификацию проводили с данными трансформантами, используя синетические олигонуклеотиды, описанные выше, для того чтобы подтвердить отсутсвие/наличие и целостность генов hasA, gtaB и tuaD оперона трансформантов Bacillus subtilis. Праймеры 3 и 8 использовали для того, чтобы подтвердить отсутствие гена hasA, праймер 71 и праймер 15 - для подтверждения наличие гена tuaD и праймеры 20 и 71 - для подтверждения наличие гена gtaB. Продукты реакций ПЦР визуализировали в 0,8% агарозном геле с 0,5X TBE. Подтвержденный штамм, интегрант hasA/tuaD/gtaB Bacillus subtilis 168Δ4, обозначали как Bacillus subtilis RB176.
Праймер 71: 5'-AACTATTGCCGATGATAAGC-3' (связывается выше tuaD) (SEQ ID NO: 85)
Геномную ДНК выделяли из устойчивых к действию хлорамфеникола и чувствительных к действию неомицина трансформантов Bacillus subtilis RB176 с использованием колонки QIAGEN tip-20 согласно инструкциям производителя. Геномную ДНК Bacillus subtilis RB176 использовали для трансформации компетентных клеток Bacillus subtilis A164Δ5. Трансформанты выбирали на чашках с TBAB, содержащих 5 мкг хлорамфеникола на мл, и выращивали при температуре 37°C. Интегрант tuaD/gtaB Bacillus subtilis A164Δ5 обозначали как Bacillus subtilis RB177.
Ген cat удаляли из штамма Bacillus subtilis RB177 с использованием способа, описанного в примере 9. Полученный в результате штамм обозначали Bacillus subtilis MF002.
Пример 16: Конструирование плазмиды pRB162 с интеграцией pel
Плазмиду pDG268MCSΔneo/scBAN/Sav (патент США № 5955310) дважды расщепляли при помощи SacI и AatII. Самый большой фрагмент длиной приблизительно 6193 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК вектора затем лигировали со вставкой ДНК, описанной ниже.
5'- и 3'-фрагменты гена пектатлиазы Bacillus subtilis (pel, инвентарный номер BG10840, SEQ ID NO. 86 [последовательность ДНК] и 87 [выведенная аминокислотная последовательность]) ПЦР-амплифицировали из Bacillus subtilis 168 (BGSC 1A1, Bacillus Genetic Stock Center, Columbus, OH) с использованием праймеров 72 (вводит участок рестрикции 5'SpeI) и 73 (вводит участок рестрикции 3'SalI) для фрагмента 5'pel и праймеры 74 (вводит участки рестрикции 5'Sacl/BamHl) и 75 (вводит участки рестрикции 3'NotI/AatII) для фрагмента 3'pel:
Праймер 72:
5'-ACTAGTAATGATGGCTGGGGCGCGTA-3' (SEQ ID NO: 88)
Праймер 73:
5'-GTCGACATGTTGTCGTATTGTGAGTT-3' (SEQ ID NO: 89)
Праймер 74:
5'-GAGCTCTACAACGCTTATGGATCCGCGGCCGCGGCGGCACACACATCTGGAT-3' (SEQ ID NO: 90)
Праймер 75:
5'-GACGTCAGCCCGTTTGCAGCCGATGC-3' (SEQ ID NO: 91)
ПЦР-амплификацию проводили в трех повторах в 30 мкл реакционной смеси, состоящей из 50 нг хромомсомной ДНК Bacillus subtilis, 0,4 мМ каждой из пары праймеров 72/73 для фрагмента 5'pel или пары праймеров 74/75 для фрагмента 3'pel, 200 мкМ каждого из dATP, dCTP, dGTP, и dTTP, 1X буфер II для ПЦР с 2,5 мкМ MgCl2, и 2,5 единиц ДНК-полимеразы AmpliTaq GoldТМ. Реакции проводили в термоциклере RoboCycler 40, запрограммированном на 1 цикл при 95°C в течение 9 минут; 3 цикла, каждый при 95°C в течение 1 минуты, при 52°C в течение 1 минуты и при 72°C в течение 1 минуты; 27 циклов, каждый при 95°C в течение 1 минуты, при 55°C в течение 1 минуты и при 72°C в течение 1 минуты; и 1 цикл при 72°C в течение 5 минут. Продукты ПЦР визуализировали с использованием 0,8% агарозного геля с 0,5X TBE. Ожидаемые фрагменты были длиной приблизительно 530 п.н. для фрагмента 5'pel и 530 п.н. для фрагмента 3'pel.
Фрагменты ПЦР 5'pel длиной 530 п.н. и 3'pel длиной 530 п.н. клонировали в pCR2.1 с использованием набора для клонирования TA-TOPO и трансформировали в компетентные клетки OneShot™ E. coli согласно инструкциям производителя. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, инкубированных при температуре 37°C. Плазмидную ДНК из этих трансформантов очищали с использованием робота QIAGEN согласно инструкциям производителя и последовательность ДНК-вставки подтверждали посредством секвенирования ДНК с использованием праймеров, описанных выше (праймеры 72 и 73 для 5'pel и праймеры 74 и 75 для 3'pel). Плазмиды, содержащие фрагменты ПЦР 530 п.н. и 530 п.н., обозначали как pCR2.1-pel 5' и pCR2.1-pel3' соответственно (фигуры 38 и 39 соответственно).
Плазмиду pCR2.1-pel3' дважды расщепляли при помощи SacI и AatII. Самый маленький фрагмент плазмиды длиной приблизительно 530 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя.
Восстановленный вектор (pDG268MCSΔneo/scBAN) и ДНК-вставки (3'pel) лигировали с использованием набора для быстрого клонирования ДНК согласно инструкциям производителя. Смесь лиригования трансформировали в компетентные клетки SURE E.coli (Stratagene, Inc., La Jolla, CA). Трансформанты выбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, при температуре 37°C.
Плазмидную ДНК из нескольких трансформантов очищали с использованием робота QIAGEN согласно инструкциям производителя и анализировали посредством расщепления при помощи SacI и AatII в 0,8% агарозном геле с использованием 0,5X TBE-буфера. Правильную плазмиду определяли по наличию фрагмента 3'pel SacI/AatII длиной приблизительно 530 п.н. и обозначали ее как pRB161 (фигура 40).
Плазмиду pRB161 дважды расщепляли при помощи SpeI и SalI. Самый большой фрагмент плазмиды длиной приблизительно 5346 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК вектора затем лигировали с ДНК-вставкой, описанной ниже.
Плазмиду pCR2.1-pel5' дважды расщепляли при помощи SpeI и SalI. Самый маленький фрагмент плазмиды длиной приблизительно 530 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick в 0,8% агарозном геле с 0,5X TBE согласно инструкциям производителя.
Восстановленный вектор (pDG268MCSΔneo/scBAN/pel 3') и ДНК-вставку (pel 5') лигировали с использованием набора для быстрого клонирования ДНК согласно инструкциям производителя. Смесь лиригования трансформировали в компетентные клетки SURE E.coli (Stratagene, Inc., La Jolla, СА). Трансформанты выбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл.
Плазмидную ДНК очищали из нескольких трансформантов с использованием робота QIAGEN согласно инструкциям производителя и анализировали посредством расщепления при помощи SpeI и SalI в 0,8% агарозном геле с использованием 0,5X TBE-буфера. Правильную плазмиду определяли по наличию фрагмента pel 5' SpeI/SalI длиной приблизительно 530 п.н. и обозначали ее как pRB162 (фигура 41).
Пример 17: Конструирование pRB156
Плазмиду pHA7 (пример 4, фигура 13) расщепляли при помощи HpaI. Расщепленную плазмиду затем очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи Asp718. В дважды расщепленной плазмиде затем формировали тупые концы при первой инактивации фермента рестрикции при 85°C в течение 30 минут. Формирование тупых концов проводили, добавляя 0,5 мкл 10 мМ каждого dNTP и 1 мкл 1 ед./мкл полимеразы T4 и инкубируя при 11°C в течение 10 минут. Затем полимеразу инактивировали при помощи инкубации реакционной смеси при 75°C в течение 10 минут. Самый большой фрагмент плазмиды длиной приблизительно 8600 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК-вставку (pDG268Δneo-cryIIIAstab/sehasA) затем повторно лигировали с использованием набора для быстрого клонирования ДНК согласно инструкциям производителя.
Смесь лиригования трансформировали в компетентные клетки SURE E.coli (Stratagene, Inc., La Jolla, СА). Трансформанты выбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл, при температуре 37°С. Плазмидную ДНК очищали из нескольких трансформантов с использованием робота QIAGEN согласно инструкциям производителя и анализировали посредством расщепления при помощи SalI в 0,8% агарозном геле с использованием 0,5X TBE-буфера. Правильную плазмиду определяли по наличию фрагмента длиной приблизительно 8755 п.н. и обозначали ее как pRB156 (фигура 42).
Пример 18: Конструирование Bacillus subtilis MF009
Для того чтобы получить Bacillus subtilis MF009, ген hasA под контролем промотора scBAN вводили в локус гена пектатлиазы (pel) Bacillus subtilis MF002.
Плазмиду pRB156 расщепляли при помощи SacI. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи NotI. Самый маленький фрагмент плазмиды длиной приблизительно 1377 п.н. очищали из геля с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК-вставку затем лигировали с ДНК вектора, описанного ниже.
Плазмиду pRB162 (пример 16, фигура 41) расщепляли при помощи NotI. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи SacI. Самый большой фрагмент плазмиды длиной приблизительно 5850 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК вектора затем лигировали с ДНК-вставкой, описанной выше.
Смесь лигирования трансформировали непосредственно в компетентные клетки Bacillus subtilis 168Δ4. Устойчивые к действию хлорамфеникола трансформанты Bacillus subtilis отбирали на чашках с TBAB, дополненных 5 мкг хлорамфеникола на мл, при температуре 37°C. Для выявления интеграции плазмиды посредством двойного кроссинговера в локус pel первичные трансформанты Bacillus subtilis помещали на чашки с TBAB, дополненные 6 мкг неомицина на мл, и на чашки с TBAB, дополненные 5 мкг хлорамфеникола на мл. Интеграция плазмиды посредством двойного кроссинговера в локус pel не включает ген устойчивости к действию неомицина и, следовательно, дает штамм, чувствительный к действию неомицина. Используя данную чашку для скрининга, выделяли трансформанты, устойчивые к действию хлорамфеникола и чувствительные к действию неомицина.
Геномную ДНК выделяли из устойчивых к действию хлорамфеникола и чувствительных к действию неомицина трансформантов Bacillus subtilis 168Δ4 с использованием колонки QIAGEN tip-20 согласно инструкциям производителя. Данную геномную ДНК использовали для трансформации компетентных клеток Bacillus subtilis MF002 (пример 15). Трансформанты выбирали на чашках с TBAB, содержащих 5 мкг хлорамфеникола на мл, и выращивали при температуре 37°C. Интегрант hasA и A164Δ5 tuaD/gtaB Bacillus subtilis определяли по его «влажному» фенотипу и обозначали как Bacillus subtilis MF009.
Пример 19: Конструирование Bacillus subtilis MF010
Плазмиду pDG268MCSΔneo/BAN/Sav (патент США № 5955310) расщепляли при помощи NotI. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи SfiI. Самый маленький фрагмент плазмиды длиной приблизительно 185 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК-вставку затем лигировали с ДНК вектора, описанного ниже.
Плазмиду pRB162 (пример 16, фигура 41) расщепляли при помощи NotI. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи SfiI. Самый большой фрагмент плазмиды длиной приблизительно 5747 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК вектора затем лигировали с ДНК-вставкой, описанной выше.
Восстановленный вектор и ДНК-вставку лигировали с использованием набора для быстрого клонирования ДНК согласно инструкциям производителя. Смесь лигирования трансформировали в компетентные клетки XLI Blue E. coli. Трансформанты отбирали на чашках агара с 2X YT, дополненных 100 мкг ампициллина на мл.
Плазмидную ДНК очищали из нескольких трансформантов с использованием робота QIAGEN согласно инструкциям производителя и анализировали посредством расщепления при помощи BamHI в 0,8% агарозном геле с использованием 0,5X TBE-буфера. Правильную плазмиду определяли посредством линеаризации плазмиды, которая давала фрагмент плазмиды длиной приблизительно 7156 п.н., и обозначали ее как pRB164 (фигура 43).
Плазмиду pRB156 (пример 17, фигура 42) расщепляли при помощи SacI. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи NotI. Самый маленький фрагмент плазмиды длиной приблизительно 1377 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК-вставку затем лигировали с ДНК вектора, описанного ниже.
Плазмиду pRB164 расщепляли при помощи NotI. Затем расщепленную плазмиду очищали с использованием набора для очистки ДНК QIAquick согласно инструкциям производителя и в конце расщепляли при помощи SacI. Самый большой фрагмент плазмиды длиной приблизительно 5922 п.н. очищали с использованием набора для экстракции ДНК из геля QIAquick из 0,8% агарозного геля с 0,5X TBE согласно инструкциям производителя. Восстановленную ДНК вектора затем лигировали с ДНК-вставкой, описанной выше.
Смесь лигирования трансформировали непосредственно в компетентные клетки Bacillus subtilis 168Δ4. Устойчивые к действию хлорамфеникола трансформанты Bacillus subtilis выбирали на чашках с TBAB, дополненных 5 мкг хлорамфеникола на мл, при температуре 37°C. Для выявления интеграции плазмиды посредством двойного кроссинговера в локус amyE первичные трансформанты Bacillus subtilis помещали на чашки с TBAB, дополненные 6 мкг неомицина на мл, и на чашки с TBAB, дополненные 5 мкг хлорамфеникола на мл. Интеграция плазмиды посредством двойного кроссинговера в локус amyE не включает ген устойчивости к действию неомицина и, следовательно, дает штамм, чувствительный к действию неомицина. Используя данную чашку для разделения, выделяли трансформанты, устойчивые к действию хлорамфеникола и чувствительные к действию неомицина.
Геномную ДНК выделяли из устойчивых к действию хлорамфеникола и чувствительных к действию неомицина трансформантов 168Δ4 Bacillus subtilis с использованием колонки QIAGEN tip-20 согласно инструкциям производителя. Данную геномную ДНК использовали для трансформации компетентных клеток Bacillus subtilis MF002 (пример 15). Трансформанты отбирали на чашках с минимальной средой, содержащих 5 мкг хлорамфеникола на мл, и растили при температуре 37°C в течение 16 часов. BAN/hasA Bacillus subtilis A164Δ5 и интегрант scBAN/tuaD/gtaB определяли по его «влажному» фенотипу и обозначали как Bacillus subtilis MF010.
Пример 20: Ферментации
Способность штаммов Bacillus subtilis, перечисленных в таблице 1, продуцировать гиалуроновую кислоту оценивали при различных условиях роста.
Штаммы Bacillus subtilis ферментировали в маленьких ферментерах в среде, состоящей из 6,5 г KH2PO4, 4,5 г Na2HPO4, 3,0 г (NH4)2SO4, 2,0 г Na3-цитрат-2H2O, 3,0 г MgSO4·7H2O, 6,0 мл Mikrosoy-2, 0,15 мг биотина (1 мл этанола в концентрации 0,15 мг/мл), 15,0 г сахарозы, 1,0 мл SB 2066, 2,0 мл P2000, 0,5 г CaCI2·2H2O на литр. Перед автоклавированием среда имела pH от 6,3 до 6,4 (без корректировки). После автоклавирования добавляли CaCI2·2H2O.
Используемой средой для посева являлась среда B-3, то есть агар-3 без агара или среда «S/S-1». Среда агар-3 состояла из 4,0 г питательного бульона, 7,5 г гидролизованного белка, 3,0 г дрожжевого экстракта, 1,0 г глюкозы, и 2% агара. pH не корректировали; перед автоклавированием pH было приблизительно 6,8; после автоклавирования - приблизительно pH 7,7.
Среда для посева во флакон сахароза/соя (S/S-1) состояла из 65 г сахарозы, 35 г соевой муки, 2 г Na3-цитрат·2H2O, 4 г KH2PO4, 5 г Na2HPO4, и 6 мл микроэлементов. pH среды доводили при помощи NaOH приблизительно до 7; после распределения среды по флаконам добавляли 0,2% растительное масло, чтобы подавить образование пены. Микроэлементы состояли из 100 г лимонной кислоты-H2O, 20 г FeSO4·7H2O, 5 г MnSO4·H2О, 2 г CuSO4·5H2O, и 2 г ZnCl2.
Перед инокуляцией рН доводили до 6,8-7,0 при помощи аммиака и впоследствии контролировали при pH 7,0 + 0,2 при помощи аммиака и H3PO4. Температуру поддерживали на 37°C. Перемешивание проводили максимум при 1300 об/мин, используя два 6-лопастных rushton рабочих колеса диаметром 6 см в 3 литровой емкости с первоначальным объемом 1,5 литра. Вентиляция имела максимум 1,5 VVM.
Для питания использовали простой раствор сахарозы. Питание начинали приблизительно через 4 часа после инокуляции, когда концентрация растворенного кислорода (D. O.) все еще продолжала уменьшаться (то есть до истощения сахарозы). Скорость питания линейно возрастала от 0 до приблизительно 6 г сахарозы/L0-ч за промежуток времени 7 часов. В некоторых ферментациях использовали меньшую скорость питания, линейно увеличивающуюся от 0 до приблизительно 2 г сахарозы/L0-ч.
Вязкость становилась заметной приблизительно через 10 часов, и через 24 часа вязкость была очень высокой, приводя к уменьшению концентрации до нижнего предела. Окончательная вязкость достигала 3220 сП. Рост клеточной массы достиг приблизительного максимума (от 12 до 15 г/литр) через 20 часов. Клетки убирали посредством разбавления 1 части культуры 3 частями воды, хорошо перемешивая и центрифугируя приблизительно при 30000 g, для того чтобы получить чистый супернатант и клеточный осадок, который можно промыть и высушить.
Анализ концентрации гиалуроновой кислоты проводили с использованием способа ELISA, основанного на связывающем гиалуронан белке (белок и наборы коммерчески доступны в Seikagaku America, Falmouth, MA).
Bacillus subtilis RB 161 и RB163 культивировали при закрытой ферментации и ферментации с подпитыванием. В процессе подпитывания скорость питания изменяли для культур штаммов Bacillus subtilis RB 161 и RB163. Анализ концентраций гиалуроновой кислоты снова проводили с использованием способа ELISA. Результаты представлены в таблице 2.
Результаты анализа культур для одного и того же штамма при скорости подпитывания 2 г/л сахарозы/L0-ч по сравнению с 6 г/л сахарозы/L0-ч демонстрировали, что более быстрая скорость питания сахарозой незначительно улучшала титры.
Краткое описание цикла штаммов Bacillus при одних и тех же условиях (подпитывание приблизительно 2 г сахарозы/L0-ч при 37°C) показано на фигуре 44. Цифры на фигуре 44 указывают стандартное отклонение данных в многократных циклах при одних и тех же условиях. Данные без цифр представляют единичные циклы. Концентрации гиалуроновой кислоты определяли с использованием модифицированного карбазолового способа (Bitter and Muir, 1962, Anal Biochem. 4: 330-334).
Краткое описание средних молекулярных масс (МДа) пиков массы гиалуроновой кислоты, полученных при ферментации рекомбинантных штаммов Bacillus subtilis при одних и тех же условиях (подпитывание приблизительно 2 г сахарозы/L0-ч, при 37°C), показано на фигуре 45. Молекулярную массу определяли с использованием анализа GPC-MALLS. Данные получали из анализа GPC-MALLS с использованием следующей процедуры. GPC-MALLS (гель проникающая хроматография (или исключение по размеру), сопряженная с многоугольным рассеянием лазерного света), широко используется для характеристики полимеров больших молекулярных масс (MW). Разделение полимеров достигается посредством GPC, основанной на разнице разделения молекул различных MW между элюентом и смолой. Средний молекулярный вес отдельного полимера определяется посредством MALLS, основанного на разнице степени/угла рассеивания для молекул различных MW. Принципы GPC-MALLS и протоколы, подходящие для гиалуроновой кислоты описаны Ueno et al., 1988, Chem. Pharm. Bull. 36,4971-4975; Wyatt, 1993, Anal. Chim. Acta 272: 1-40; и Wyatt Technologies, 1999, «Light Scattering University DAWN Course Manual» и «DAWN EOS Manual» Wyatt Technology Corporation, Santa Barbara, СА). Для GPC использовали HPLC в изократическом режиме на Agilent 1100, колонку Tosoh Biosep G6000 PWxl и для MALLS использовали Wyatt Down EOS. Детектор для рефрактивного индекса Agilent G1362A присоединяли после MALLS для определения концентрации элюата. Различные коммерческие продукты гиалуроновой кислоты с известными молекулярными массами служили в качестве стандартов.
Депонирование биологического материала
Следующий биологический материал депонировали в соответствии с условиями Будапештским Соглашением с Agricultural Research Service Patent Culture Collection, Northern Regional Research Center, 1815 University Street, Peoria, Illinois, 61604 с присвоением следующего инвентарного номера:
Штамм депонировали при условиях, которые обеспечивают во время ожидания решения по данной патентной заявке наличие доступа к культуре лицу, определенному Commissioner of Patents and Trademarks, по заголовкам документов 37 C.F.R. §1.14 и 35 U.S.C. §122. Депонент представляет собой по существу чистую культуру депонированного штамма. Депонент доступен, как это требуется по иностранному патентному законодательству, в странах, где регистрируются параллельные заявки относительно поданной заявки или последующие заявки. Тем не менее, следует понимать, что доступность депонента не является лицензией на осуществление заявленного изобретения, отменяя патентные права, предоставленные государственным актом.
Описанное и заявленное здесь изобретение не ограничивается по объему описанными здесь конкретными осуществлениями, так как данные осуществления предназначены для иллюстрации некоторых аспектов изобретения. Любые равнозначные осуществления подразумеваются как входящие в объем данного изобретения. Несомненно, различные модификации изобретения в дополнение к модификациям, показанным и описанным здесь, очевидны для специалистов в данной области из приведенного выше описания. Такие модификации также, как подразумевается, входят в объем прилагаемой формулы изобретения. В случае конфликта для контроля будет использоваться настоящее описание, содержащее определения.
Здесь приведены различные ссылки, описания которых полностью включены сюда в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| БАКТЕРИЯ РОДА BACILLUS, ПРОДУЦИРУЮЩАЯ ГИАЛУРОНОВУЮ КИСЛОТУ, И СПОСОБ ПОЛУЧЕНИЯ ГИАЛУРОНОВОЙ КИСЛОТЫ С ИСПОЛЬЗОВАНИЕМ УКАЗАННОЙ БАКТЕРИИ | 2019 |
|
RU2719140C1 |
| ТАНДЕМНЫЙ ПРОМОТОР, ФУНКЦИОНИРУЮЩИЙ В БАКТЕРИИ РОДА Bacillus, ТРАНСФОРМИРОВАННАЯ БАКТЕРИЯ РОДА Bacillus - ПРОДУЦЕНТ ЦЕЛЕВОГО ПРОДУКТА, СОДЕРЖАЩАЯ УКАЗАННЫЙ ПРОМОТОР, И СПОСОБ ПОЛУЧЕНИЯ ЦЕЛЕВОГО ПРОДУКТА С ИСПОЛЬЗОВАНИЕМ УКАЗАННОЙ БАКТЕРИИ | 2019 |
|
RU2723722C1 |
| СПОСОБЫ ПОВЫШЕНИЯ УСИЛИВАЮЩЕЙ ЦЕЛЛЮЛОЛИТИЧЕСКОЙ АКТИВНОСТИ ПОЛИПЕПТИДА | 2008 |
|
RU2510417C2 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ ЧЕТЫРЕХУГЛЕРОДНЫХ СПИРТОВ | 2006 |
|
RU2394913C2 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ 1-БУТАНОЛА | 2006 |
|
RU2429295C2 |
| ДНК-конструкция, кодирующая модифицированный вариант протективного антигена Bacillus anthracis | 2015 |
|
RU2622085C2 |
| РЕКОМБИНАНТНАЯ ДНК, КОДИРУЮЩАЯ ФУНКЦИОНАЛЬНО АКТИВНЫЙ ГИБРИДНЫЙ БЕЛОК G17ACA-АЦИЛАЗЫ С ХИТИН-СВЯЗЫВАЮЩИМ ДОМЕНОМ (BrdG17ACA-cbd), РЕКОМБИНАНТНАЯ ПЛАЗМИДА pSVH0108, ОБЕСПЕЧИВАЮЩАЯ ЕГО СИНТЕЗ В КЛЕТКАХ Escherichia coli, И РЕКОМБИНАНТНЫЙ ШТАММ Escherichia coli BL21(DE3)/pSVH0108-ПРОДУЦЕНТ BrdG17ACA-cbd | 2006 |
|
RU2388826C2 |
| ПРОТИВОМИКРОБНЫЕ ПОЛИПЕПТИДЫ ИЗ PSEUDOPLECTANIA NIGRELLA | 2002 |
|
RU2336278C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПУРИНОВЫХ РИБОНУКЛЕОЗИДОВ И РИБОНУКЛЕОТИДОВ | 2008 |
|
RU2422510C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2008 |
|
RU2545699C2 |

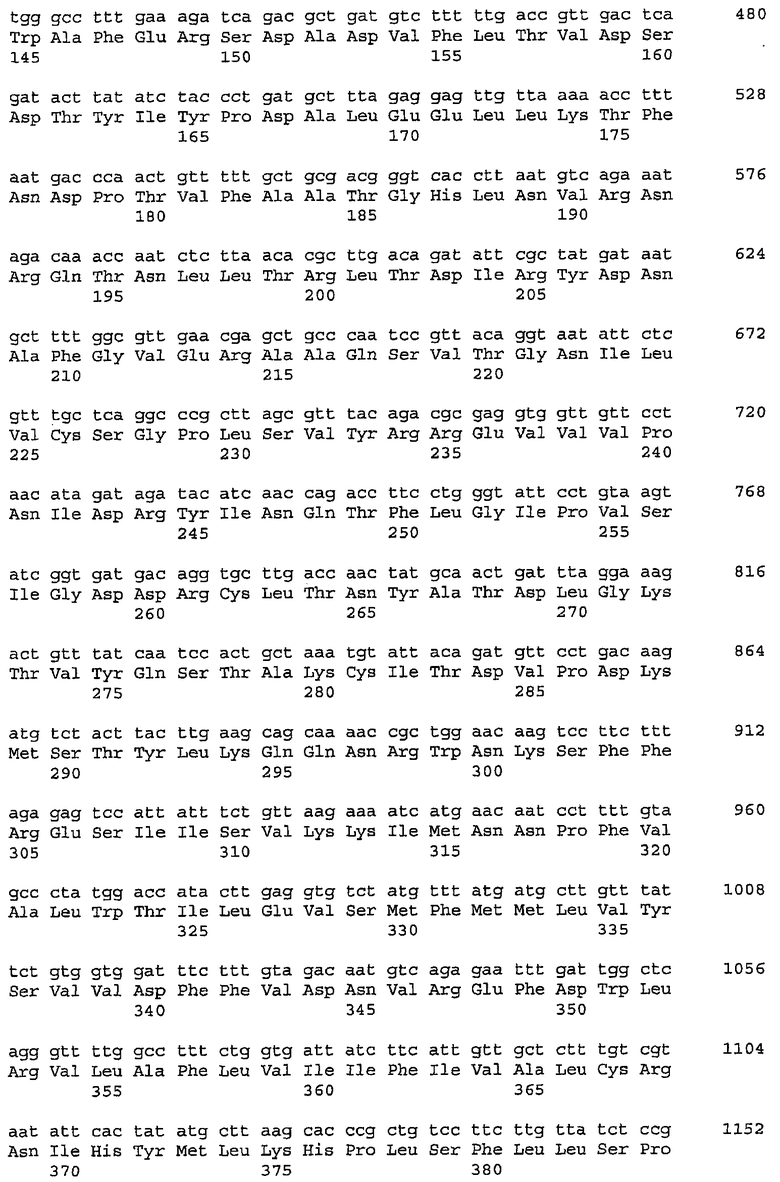




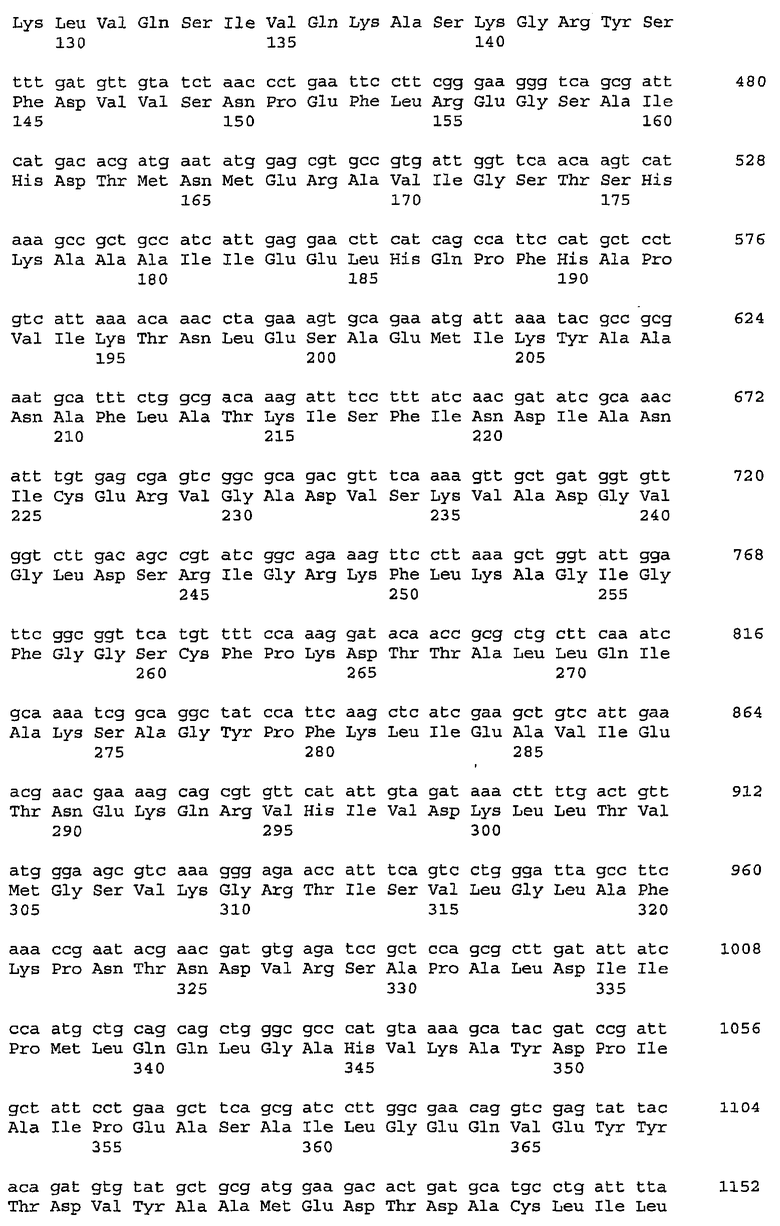




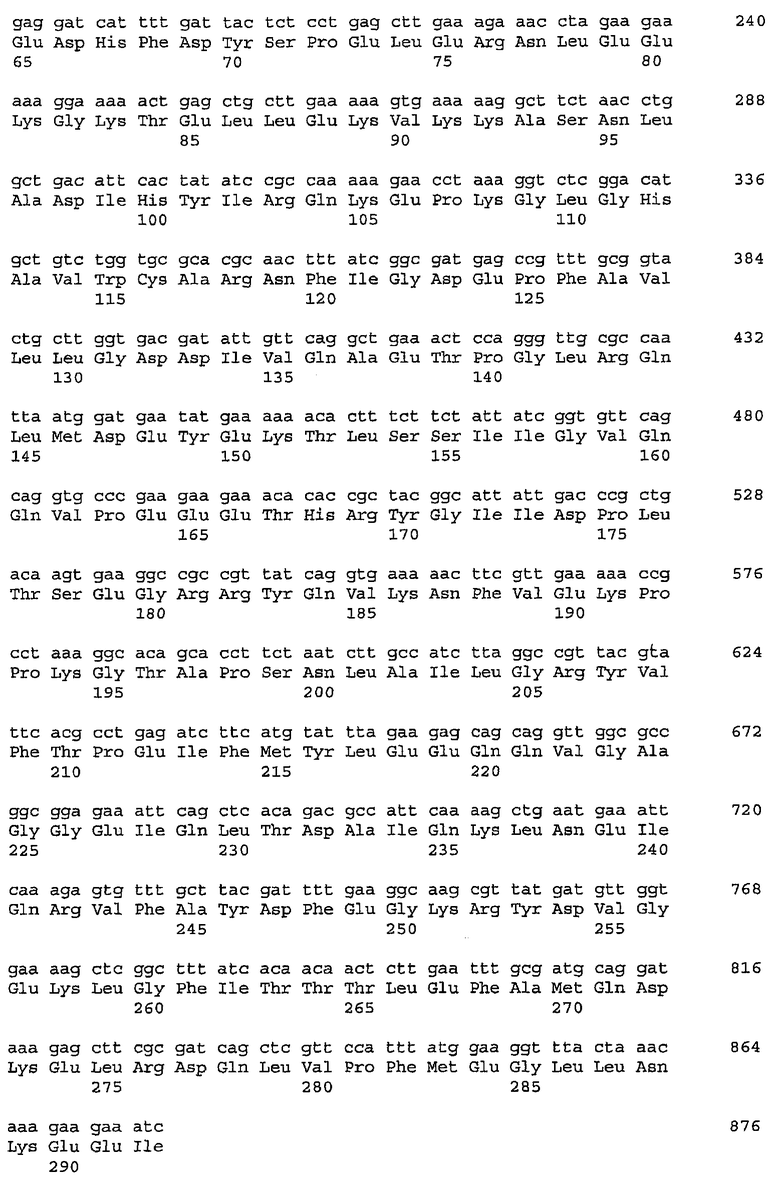



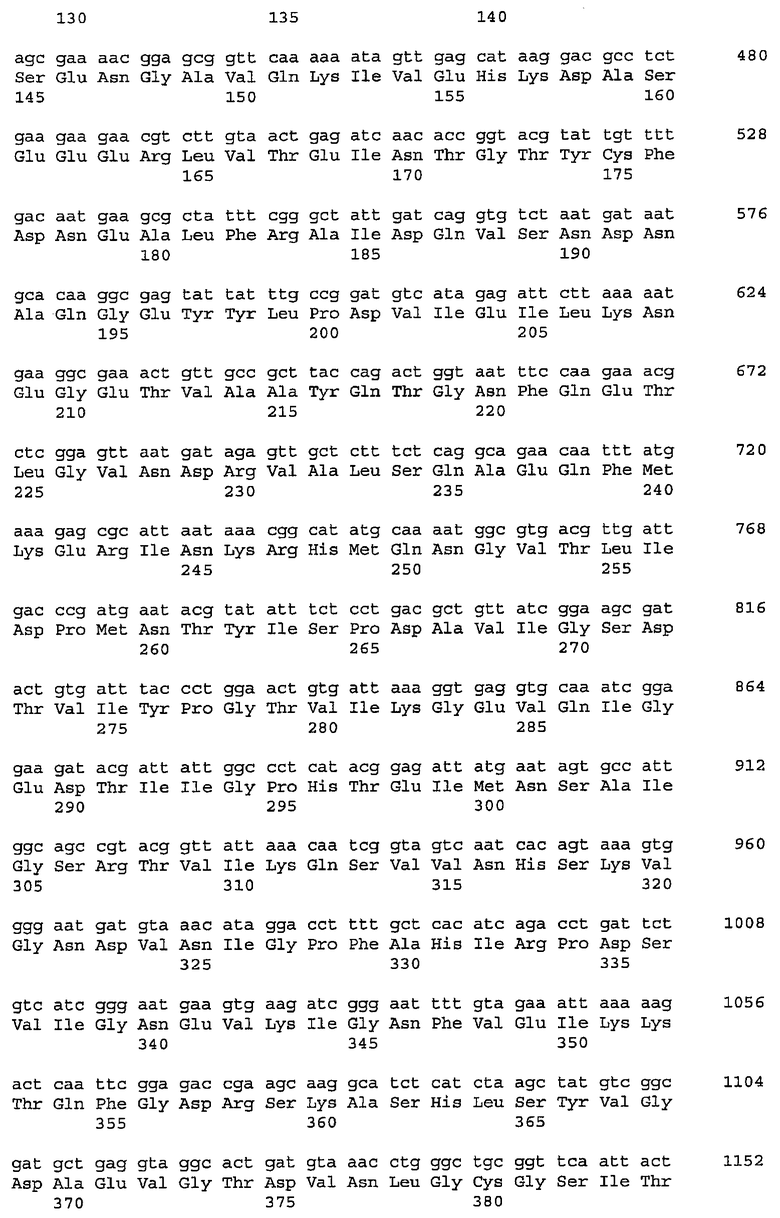












































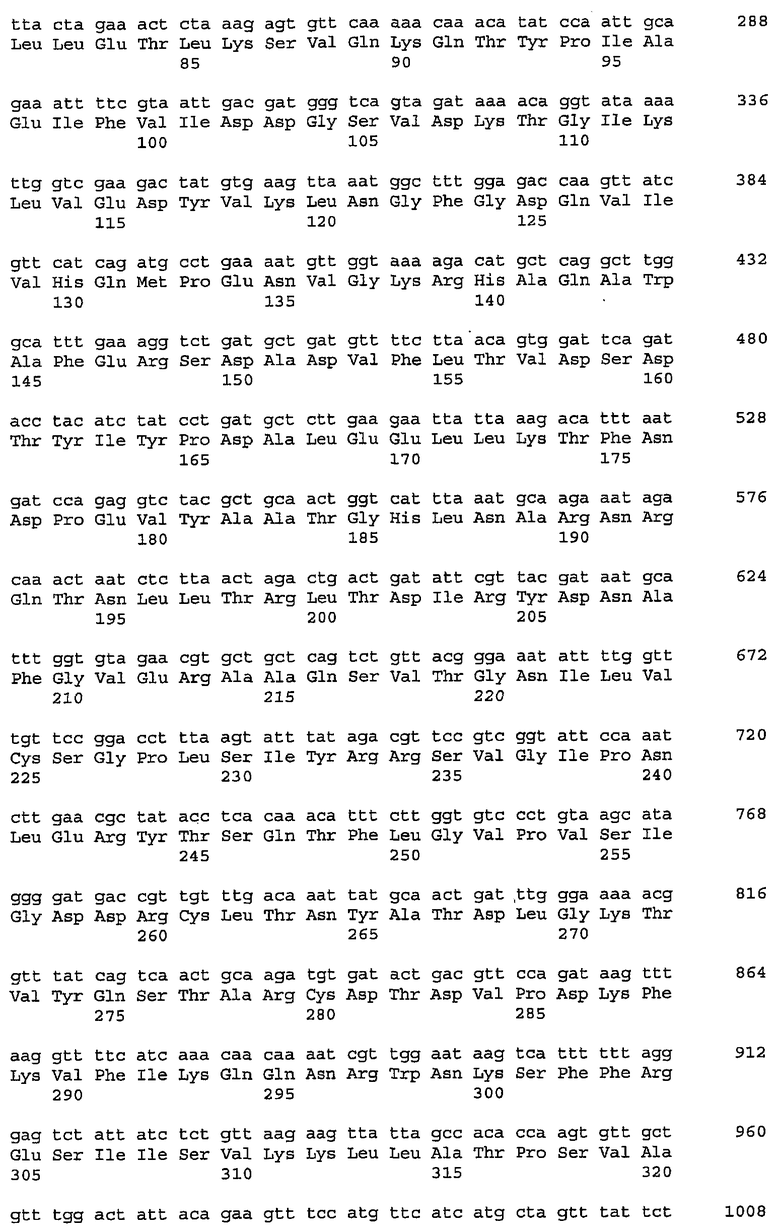












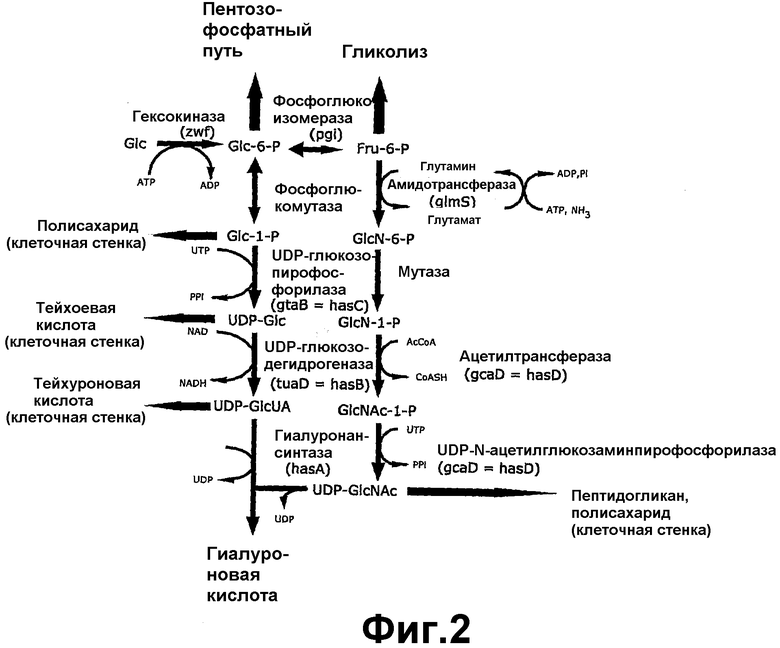


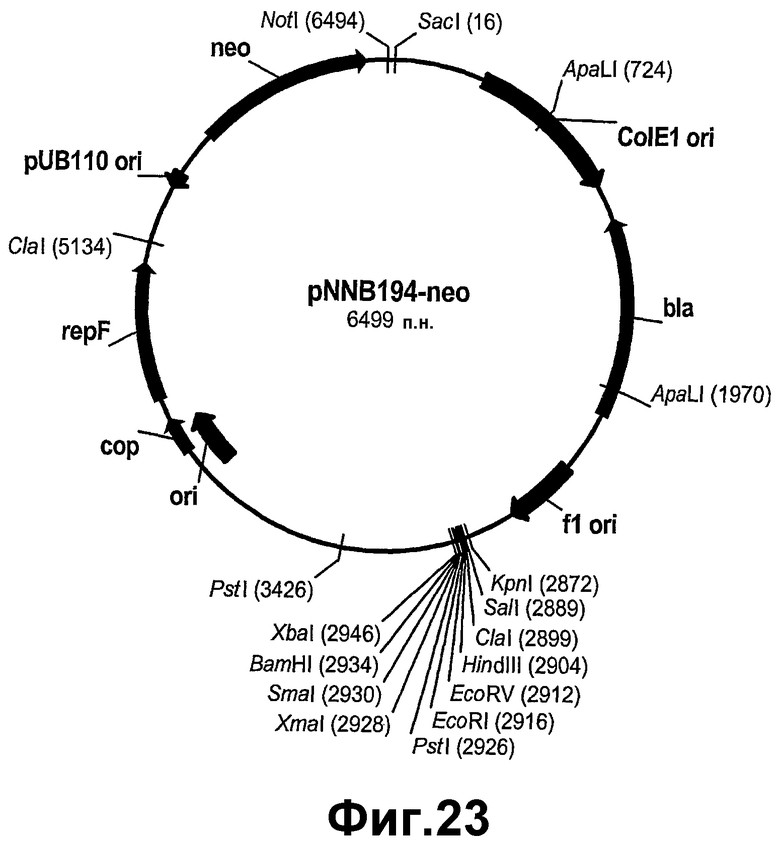
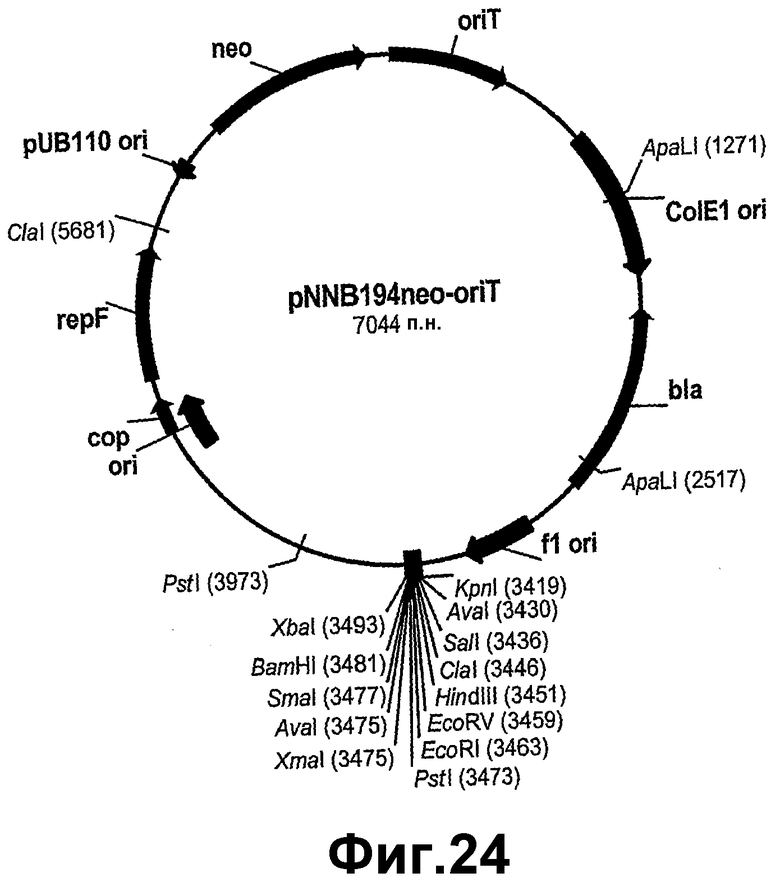

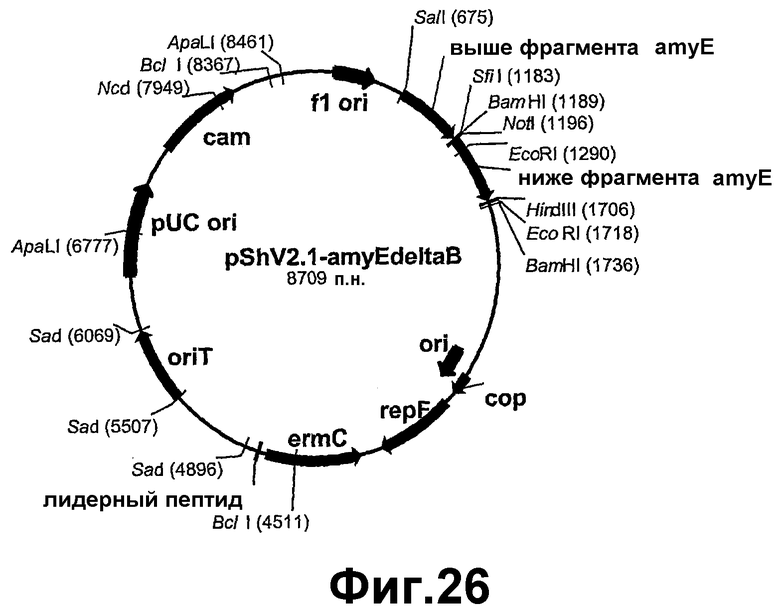
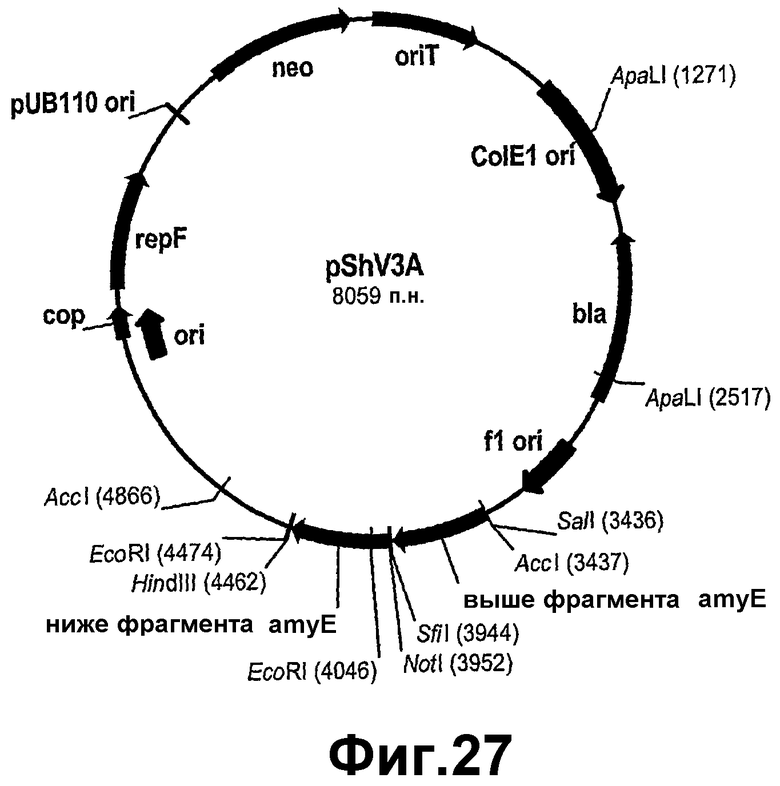

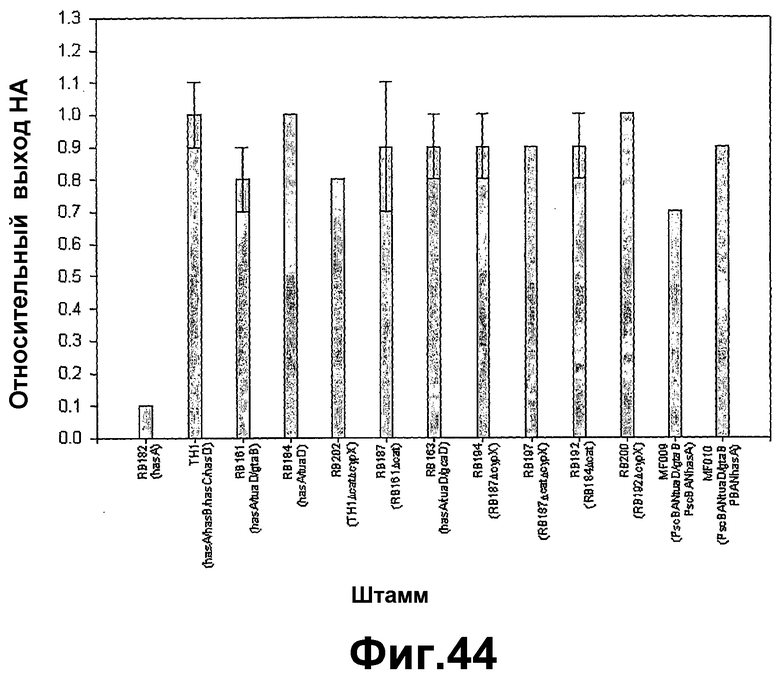
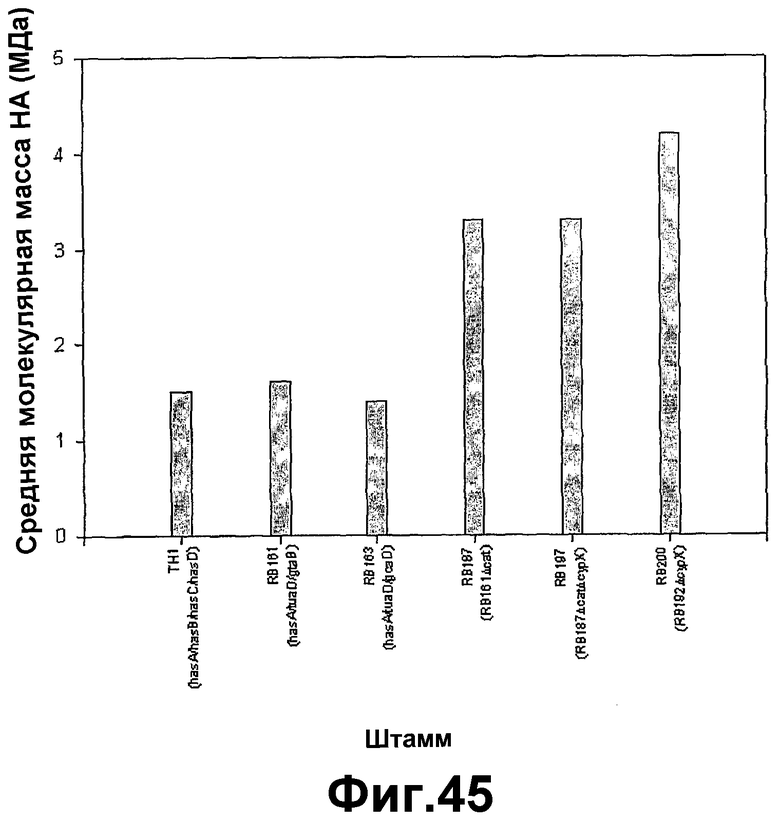
Настоящее изобретение относится к области биотехнологии и может быть использовано в биомедицинской промышленности для получения гиалуроновой кислоты. Предложен способ получения гиалуронана, предусматривающий культивирование клетки-хозяина Bacillus в условиях, подходящих для получения гиалуроновой кислоты, и извлечение целевого продукта из культуральной среды. При этом клетка-хозяин Bacillus содержит генетическую конструкцию, включающую промотор, функционально активный в указанной клетке, и кодирующую область, состоящую из нуклеотидной последовательности, кодирующей стрептококковую гиалуронансинтазу (hasA); последовательности, кодирующей UDP-глюкозо-6-дегидрогеназу Bacillus (tuaD) или аналогичный фермент стрептококкового происхождения (hasB), и последовательность, кодирующую бактериальную или стрептококковую UDP-глюкозопирофосфорилазу (соответственно gtaB и hasC). Применение способа по изобретению обеспечивает возможность получения значительных количеств гиалуронана в хорошо исследованной, непатогенной клеточной системе. 3 и 12 з.п., 45 ил., 2 табл.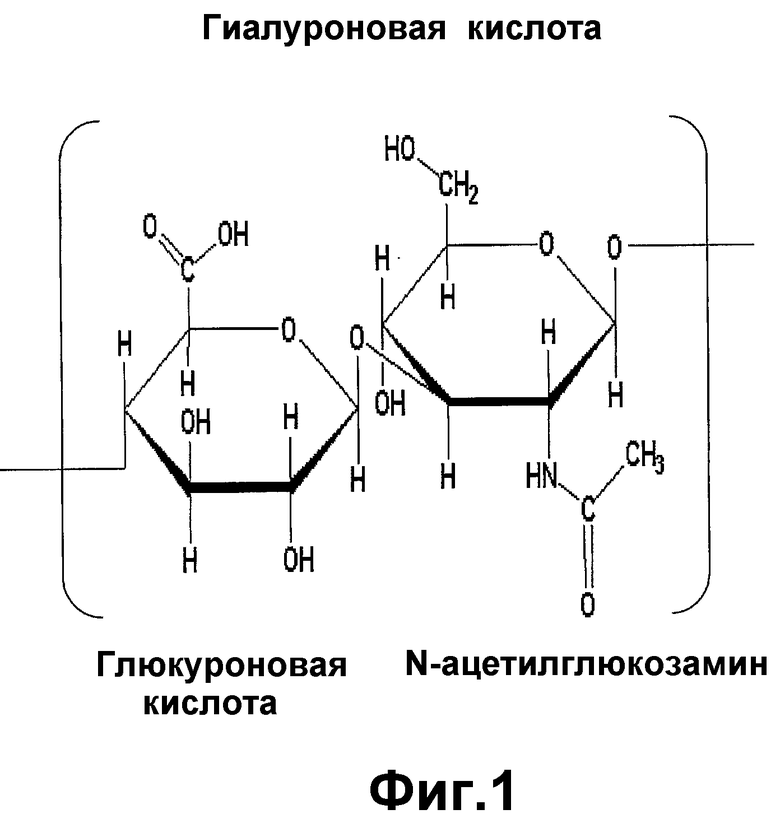
| KUMARI К | |||
| ЕТ AL., J | |||
| Biol | |||
| Chem, 272 (51), 32539-32546, 1997 | |||
| EP 0694616 A, 31.01.1996 | |||
| FERRETTI J | |||
| ЕТ AL., Proc | |||
| Natl | |||
| Acad | |||
| Sci | |||
| USA, 98(8), 46-58, 2001. |

