Область техники, к которой относится изобретение
Настоящее изобретение относится к новому пиперидиновому производному или его фармацевтически приемлемой соли, а также к медицинской композиции. Более конкретно настоящее изобретение относится к новому пиперидиновому производному или его фармацевтически приемлемой соли, оба они имеют как превосходное действие ингибирования натриевых каналов, так и превосходное анальгетическое действие, а также к медицинской композиции, содержащей указанное выше пиперидиновое производное или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, а более конкретно к фармацевтической композиции, имеющей анальгетическое действие, с уменьшенными побочными воздействиями, в частности, на невропатическую боль, которая действует в качестве ингибитора натриевых каналов.
Уровень техники
Управляемый напряжением натриевый канал представляет собой белок, ответственный за инициирование и распространение потенциалов действий в нейронах. Управляемый напряжением натриевый канал состоит из субъединицы α большего размера с четырьмя доменами, каждый из которых состоит из шести трансмембранных сегментов, в качестве общей структуры, и двух субъединиц β меньшего размера. Главная часть функции канала осуществляется посредством субъединицы α. К настоящему времени известно более чем 10 различных подтипов субъединиц α (Goldin AL, Annals of New York Academy of Sciences 868:38-50, 1999). Каждый подтип управляемого напряжением натриевого канала демонстрирует различные распределения в тканях центральных и периферийных нервов. Эти подтипы регулируют нервную возбудимость и играют важную роль в регулировании физиологических функций в индивидуальных тканях. Предполагается также, что они глубоко связаны с различными патологическими состояниями (Goldin AL, Annual Review of Physiology 63:871-894, 2001).
В последние годы стало ясно, что управляемые напряжением натриевые каналы глубоко вовлечены в невральную болевую трансмиссию, и, как ожидается, ингибиторы натриевых каналов будут превосходными терапевтическими средствами от боли, в частности терапевтическими средствами от невропатической боли (Taylor CP, Current Pharmaceutical Design 2: 375-388, 1996).
Невропатическая боль означает боль, которая возникает в результате дисфункции в центральных или периферийных нейронах, и относится к болезненной диабетической невропатии, боль при раке, тригеминальной невралгии, фантомной боли лимба, постгерпетической невралгии, таламической боли, и тому подобное. Клиническая картина невропатической боли включает в себя стреляющую боль, жгучую боль, гиперальгезию, аллодинию, и тому подобное. В медицинских случаях, для целей снятия боли, используются нестероидные противовоспалительные лекарственные средства, наркотические анальгетики, такие как морфин, и тому подобное. В последнее время антиаритмические лекарственные средства и антиконвульсанты, которые являются ингибиторами натриевых каналов, также начинают использоваться для целей снятия боли.
Нестероидные противовоспалительные лекарственные средства не являются полностью удовлетворительными по анальгетическому воздействию и, кроме того, имеют проблему побочных воздействий (например, желудочно-кишечного расстройства и почечного расстройства). Наркотические анальгетики (например, морфин) являются высоко эффективными в основном для ноцицептивной боли, но имеют проблему больших побочных воздействий на пищеварительную систему, дыхательную систему и центральную нервную систему. Кроме того, эти лекарственные средства имеют, как правило, малое полезное воздействие на невропатическую боль. Обычные ингибиторы натриевых каналов, например антиаритмические лекарственные средства (например, лидокаин и мексилетин) и антиконвульсантные лекарственные средства (например, карбамазепин), начинают использоваться также и для облегчения боли. Однако эти ингибиторы натриевых каналов имеют побочные воздействия на центральную нервную систему (например, конвульсии и сонливость) и побочные воздействия на периферическую нервную систему (например, брахикардию), и по этой причине они имеют проблемы в связи с тем, что введение при достаточно большой дозе является сложным, что делает сложным получение достаточного анальгетического воздействия.
Как описано выше, еще не обнаружено анальгетика, который является эффективным для лечения невралгической боли и при этом обладающего высокой безопасностью. По этой причине является желательным новый ингибитор натриевых каналов, который является высокоэффективным, в частности, для невропатической боли и имеет низкое побочное воздействие. Краткое описание заявки на Международный патент WO 01/53288 (далее упоминается как патентная литература 1) описывает ингибитор натриевых каналов, представленный следующей общей формулой:
Формула 1
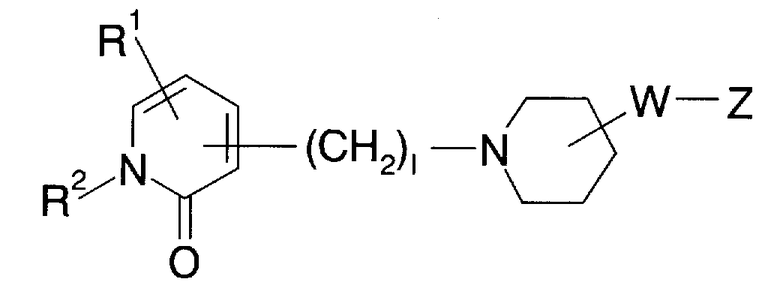
где в указанной выше формуле символ (W) представляет собой C1-6алкиленовую группу, которая может быть замещенной, или что-либо подобное; символ (Z) представляет собой C6-14ароматическую углеводородную кольцевую группу, которая может быть замещенной, или что-либо подобное; символ (l) представляет собой 0 или целое число от 1 до 6; символы (R1) и (R2) представляют собой, каждый, атом водорода или что-либо подобное. Детали этих символов указаны в патентной литературе 1. Соединение, описанное в патентной литературе 1, представляет собой соединение, в котором пиперидиновое кольцо связывается через низшую алкиленовую группу или что-либо подобное [символ (W)] с ароматической углеводородной кольцевой группой или чем-либо подобным [символ (Z)], и 1-положение пиперидинового кольца связывается через низший алкилен с оксодигидропиридиновым кольцом. В то же время соединение по настоящему изобретению отличается от соединения, описанного в патентной литературе 1, по основному скелету, в котором 4-положение пиперидинового кольца связывается через винилен с монозамещенным (-R1) или незамещенным бензольным кольцом и 1-положение пиперидинового кольца имеет ацильную группу [-C(=O)-R4].
Краткое описание заявки на Международный патент WO 01/53288, WO 94/13291 (далее упоминается как патентная литература 2) описывает азотсодержащее гетероциклическое кольцевое производное, имеющее стерильную группу (-CH=CH-бензольное кольцо), представленное следующей общей формулой:
Формула 2
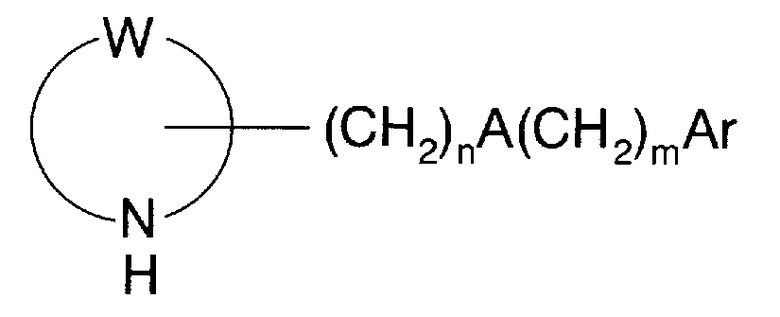
где в указанной выше формуле символ (W) представляет собой -(CH2)4-, -(CH2)5-, -(CH2)2O(CH2)2- или -(CH2)2S(CH2)2-; символ (A)представляет собой связь, -CH=CH-, O, S, NR1 или что-либо подобное; символ (R1) представляет собой атом водорода, C1-3алкил или фенил-C1-3 алкил; символ (Ar) представляет собой арил или гетероарил; символ (n) представляет собой целое число от 0 до 6; и символ (m) представляет собой целое число от 0 до 3. В этой связи детали этих символов приведены в патентной литературе 2.
Краткое описание заявки на Международный патент WO 01/53288 WO 97/19059 (далее упоминается как патентная литература 3) описывает азотсодержащее гетероциклическое кольцевое производное, представленное следующей общей формулой:
Формула 3
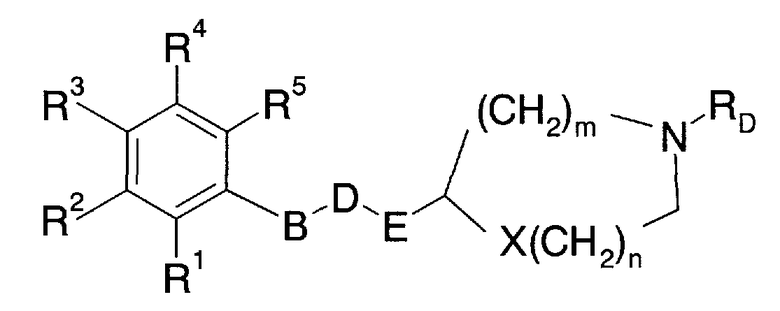
где в указанной выше формуле символ (B) не присутствует или представляет собой низший алкилен, циклоалкилен или что-либо подобное; символ (D) представляет собой -O-, -S-, -C(O)-, -C(O)-O-, -S(O)-, -S(O)2- или что-либо подобное; символ (E) представляет собой низший алкилен или что-либо подобное; символ (X) не присутствует или представляет собой -O-, -S- или что-либо подобное; символы (R1) - (R5) представляют собой, каждый, атом водорода, галогена или что-либо подобное; символ (RD) представляет собой атом водорода, низший алкил или что-либо подобное; символ (n) представляет собой целое число от 0 до 3; и символ (m) представляет собой целое число от 0 до 2. В этой связи детали этих символов приведены в патентной литературе 3.
В патентной литературе 2 и 3, однако, нет ни описания, ни предположения относительно соединения, такого как пиперидиновое производное по настоящему изобретению, в котором 4-положение пиперидинового кольца связывается, через винилен, с монозамещенным (-R1) или незамещенным бензольным кольцом, и 1-положение пиперидинового кольца имеет ацильную группу [-C(=O)-R4]. Кроме того, применение соединений патентной литературы 2 и 3 представляет собой антагонист кальциевого канала (патентная литература 2) и промотор высвобождения ацетилхолина (патентная литература 3), и в этой патентной литературе не имеется ни рассмотрения, ни предположения относительно действия по ингибированию натриевых каналов или анальгетического действия.
Описание изобретения
Настоящее изобретение имеет целью создание нового пиперидинового производного или его фармацевтически приемлемой соли, имеющей превосходное действие ингибирования натриевых каналов и превосходное анальгетическое действие, и медицинской композиции, содержащей новое пиперидиновое производное или его фармацевтически приемлемую соль; в частности, соединения для ингибирования натриевых каналов, демонстрирующего высокое анальгетическое воздействие на невралгическую боль и имеющего низкое побочное воздействие, и медицинской композиции, содержащей соединение в качестве активного ингредиента.
Авторы настоящего изобретения осуществили исследование на азотсодержащих гетероциклических кольцевых производных. В результате было обнаружено, что пиперидиновое производное, в котором 4-положение пиперидинового кольца связывается, через винилен, с монозамещенным (-R1) или незамещенным бензольным кольцом и 1-положение пиперидинового кольца имеет ацильную группу [-C(=O)-R4], или его фармацевтически приемлемая соль, демонстрирует высокую ингибирующее действие (активность) относительно натриевых каналов и, кроме того, демонстрирует хорошее анальгетическое действие на мышей с стрептозотоцин-индуцированным диабетическим нервным расстройством, в качестве животной модели болезненного состоянии. Это открытие приводит к завершению настоящего изобретения. В соответствии с настоящим изобретением, предусматриваются новое соединение и медицинская композиция, содержащая соединение в качестве активного ингредиента, оба описываются ниже.
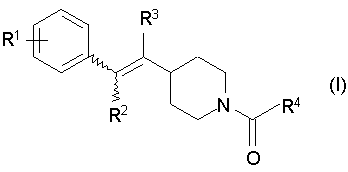
[1] Пиперидиновое производное, представленное следующей далее формулой (I)
Формула 4
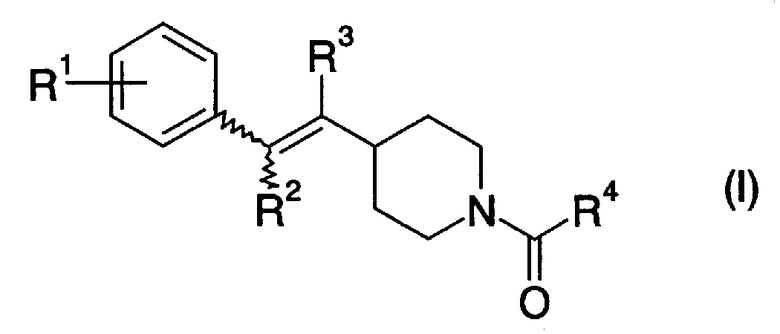
где в указанной выше формуле (I) символы R1 - R4 представляют собой, каждый, любую из одновалентных групп, показанных ниже.
R1 представляет собой атом водорода, атом галогена, низший алкил, который может быть замещенным, -O-низший алкил, который может быть замещенным, -O-арил, арил, циклоалкил, -C(=O)-низший алкил, COOH, -C(=O)-O-низший алкил, -C(=O)-NH2, -C(=O)NH-низший алкил, -C(=O)N-(низший алкил)2,OH, -O-C(=O)-низший алкил, NH2, -NH-низший алкил, -N-(низший алкил)2, -NH-C(=O)-низший алкил, CN или NO2;
R2 и R3 могут быть одинаковыми или отличными друг от друга и представляют собой, каждый, атом водорода, низший алкил или атом галогена; и
R4 представляет собой низший алкил, который может быть замещенным, -O-низший алкил, который может быть замещенным, азотсодержащую гетероциклическую кольцевую группу, которая может быть замещенной, арил, который может быть замещенным, NH2, -NH-низший алкил, который может быть замещенным, или -N-(низший алкил, который может, быть замещенным)2],
или его фармацевтически приемлемая соль.
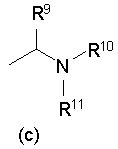
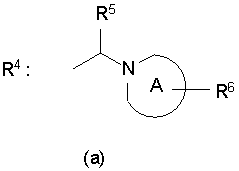
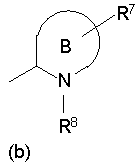
[2] Пиперидиновое производное или его фармацевтически приемлемая соль по п.[1], где, в формуле (I), одновалентная группа, представленная символом R4, представляет собой любую из одновалентных групп (a), (b) и (c), показанных ниже
Формула 5
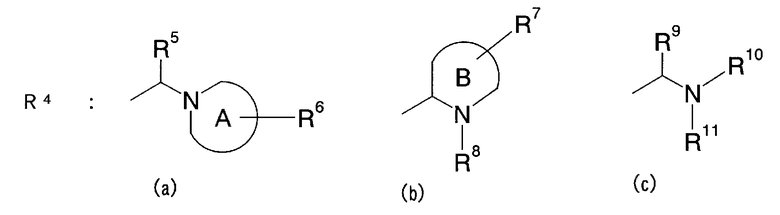
где в указанных выше группах (a), (b) и (c) символы A и B представляют собой, каждый, кольцо, показанное ниже, и символы R5 - R11 представляют собой, каждый, любую из одновалентных групп, показанных ниже. A и B представляют собой, каждый, азотсодержащее гетероциклическое кольцо; R5 и R8 - R11 могут быть одинаковыми или отличными друг от друга и представляют собой, каждый, атом водорода, низший алкил, -C(=O)-O-низший алкил, который может быть замещенным, низший алкилен-O-низший алкил, циклоалкил, или насыщенную или ненасыщенную 5- или 6-членную гетероциклическую кольцевую группу, имеющую 1-3 гетероатома, выбранных из N, S и О;
R6 представляет собой атом водорода, низший алкил, -O-низший алкил, -O-низший алкилен-O-, который связывается с одним атомом углерода из кольца A, с образованием кольца, -C(=O)-O-низший алкил, который может быть замещенным, OH, -низший алкилен-OH или -C(=O)-гетероарил; и
R7 представляет собой атом водорода, низший алкил, -O-низший алкил, -C(=O)-O-низший алкил, OH, -низший алкилен-OH, или -C(=O)-гетероарил.
[3] Пиперидиновое производное или его фармацевтически приемлемая соль по п.[2], где символ R4 представляет собой одновалентную группу (a) и азотсодержащее гетероциклическое кольцо, представленное символом A, представляет собой пирролидиновое, пиперидиновое, морфолиновое, пиперазиновое или оксазепамовое кольцо.
[4] Пиперидиновое производное или его фармацевтически приемлемая соль по п.[2], где символ R4 представляет собой одновалентную группу (b) и азотсодержащее гетероциклическое кольцо, представленное символом B, представляет собой пирролидиновое или пиперидиновое кольцо.
[5] Пиперидиновое производное или его фармацевтически приемлемая соль по п.[2], где символ R4 представляет собой одновалентную группу (c) и R9 - R11 в группе (c) могут быть одинаковыми или отличными друг от друга и представляют собой, каждый, атом водорода, низший алкил, -C(=O)-O-низший алкил, который может быть замещенным, низший алкилен-O-низший алкил, циклоалкил, или насыщенную или ненасыщенную 5- или 6-членную гетероциклическую кольцевую группу, имеющую 1-3 гетероатома, выбранных из N, S и O.
[6] Пиперидиновое производное или его фармацевтически приемлемая соль по п.[5], где, по меньшей мере, один из R9 - R11 в группе (c) представляет собой циклоалкил, а другой (другие) может быть таким же или отличным и представляет собой (представляют собой, каждый) атом водорода, низший алкил, -C(=O)-O-низший алкил, который может быть замещенным, низший алкилен-O-низший алкил, циклоалкил или насыщенную или ненасыщенную 5- или 6-членную гетероциклическую кольцевую группу, имеющую 1-3 гетероатома, выбранных из N, S и O.
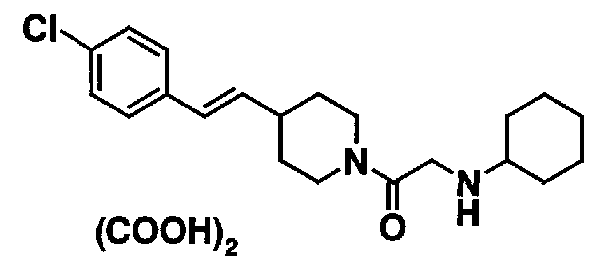
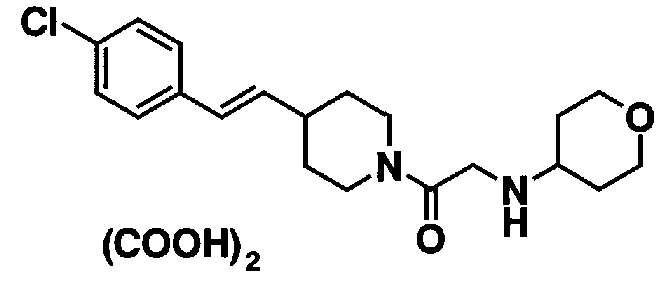
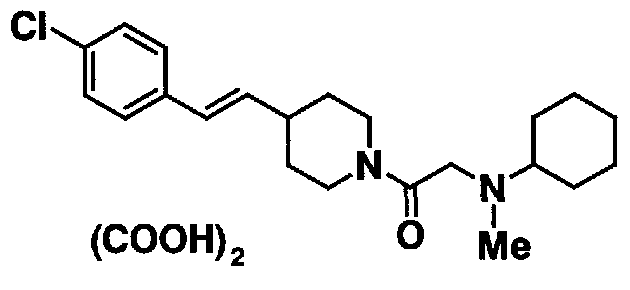
[7] Пиперидиновое производное или его фармацевтически приемлемая соль по любому из пп. [1]-[6], где пиперидиновое производное, представленное формулой (I), представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из 4-(2-{4-[(E)-2-(2-метилфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина, 4-(2-{4-[(E)-2-(4-изопропилфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина, 4-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина, N-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)циклогексанамина и N-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-N-метилциклогексанамина.
[8] Медицинская композиция, содержащая пиперидиновое производное или его фармацевтически приемлемую соль по любому из пп. [1]-[7], и фармацевтически приемлемый носитель.
[9] Медицинская композиция в соответствии с [7], которая представляет собой ингибитор натриевых каналов.
Пиперидиновое производное или его фармацевтически приемлемая соль по настоящему изобретению, как подтверждается, имеет превосходное действие ингибирования натриевых каналов и превосходное анальгетическое действие и демонстрирует высокое анальгетическое воздействие, в частности, на невралгическую боль. По этой причине настоящее соединение является пригодным для использования в качестве ингибитора натриевых каналов с низким побочным воздействием.
Наилучший способ осуществления изобретения
Пиперидиновое производное или его фармацевтически приемлемая соль по настоящему изобретению описывается конкретно. Сначала описывается определение каждого символа, используемого в общей формуле (I) и в одновалентных группах (a), (b) и (c), а также в конкретных примерах каждого символа.
Термин "низший" указывает, если только не указано иного, на углеводородную цепь с прямой цепью или разветвленной цепью, имеющую 1-6 атомов углерода. В качестве "низшего алкила" могут быть рассмотрены, например, C1-6алкилы, такие как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил, изогексил и тому подобное; и предпочтительными являются метил, этил, пропил, бутил и трет-бутил.
В качестве "низшего алкилена" могут быть рассмотрены, например, метилен, этилен, пропилен и изопропилен; и метилен и этилен являются предпочтительными. Термин "циклоалкил" указывает на моно- - трициклическую, алифатическую насыщенную углеводородную кольцевую группу, имеющую 3-14 атомов углерода, и могут быть рассмотрены, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, бициклогептил, бициклооктил, бициклононил, бициклодеканил, трициклононил, трициклодеканил, трициклоундеканил и трициклододеканил, и предпочтительными являются циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Термин "арил" указывает на моно- - трициклическую, ароматическую углеводородную группу, имеющую 6-14 атомов углерода; и могут быть рассмотрены, например, фенил, нафтил, антрил и фенантрил, и фенил и нафтил являются предпочтительными.
Термин "гетероарил" указывает на гетероарил, имеющий 1-3 гетероатома, выбранных из N, S и O, и предпочтительно представляет собой пиридил и пиримидил. Термин "азотсодержащее гетероциклическое кольцо" указывает на моно- или дициклическое, азотсодержащее гетероарильное кольцо, имеющее 5-10 атомов, включая 1-3 атома азота, и может, кроме того, содержать 1-3 атома кислорода или серы, кроме атома (атомов) азота. Могут быть рассмотрены, например, пиррол, имидазол, пиразол, триазол, тетразол, пиридин, пиридазин, пиримидин, пиразин, триазин, индолин, изоиндолин, бензимидазолин, бензопиразолин, пирролопиридин, имидазопиридин, хинолин, изохинолин и хиноксалин. Термин "азотсодержащее гетероциклическое кольцо" также указывает на моно- или дициклический азотсодержащий гетероциклоалкил, имеющий 3-10 атомов, включая 1-3 атома азота, и могут быть рассмотрены, например, азиридин, азетидин, пирролидин, пиперидин, пиперазин, гексагидроазепин, хинуклидин, азабициклооктан (например, азабицикло[3.2.1]октан), диазабициклооктан, азабициклононан и азабициклодекан. Могут включаться, кроме атома (атомов) азота, 1-3 атома кислорода или серы, и могут быть рассмотрены морфолин, оксазепам, оксазол, изооксазол, тиазол, изотиазол, фуразан, и тому подобное. "Азотсодержащее гетероциклическое кольцо" предпочтительно представляет собой пирролидиновое, пиперидиновое, морфолиновое или оксазепамовое кольцо. В качестве "азотсодержащей гетероциклической кольцевой группы" могут быть рассмотрены одновалентные группы из указанных выше "азотсодержащих гетероциклических колец".
В качестве "насыщенной или ненасыщенной 5- или 6-членной гетероциклической кольцевой группы, имеющей 1-3 гетероатома, выбранных из N, S и O" могут быть рассмотрены тетрагидропиранил, фуранил, тиофенил, пирролил и морфолил. Группа включает в себя часть указанных выше "азотсодержащих гетероциклических кольцевых групп" и предпочтительно представляет собой тетрагидропиранил или морфолинил.
В качестве "атома галогена" могут быть рассмотрены фтор, хлор, бром и йод; и фтор и хлор являются предпочтительными.
В термине, выражающем заместитель "низший алкил, который может быть замещенным", "азотсодержащая гетероциклическая кольцевая группа, которая может быть замещенной" или "арил, который может быть замещенным", выражение "который может быть замещенным" указывает на тот, "который может быть замещенным 1-3 одинаковыми или различными заместителями". В качестве примеров азотсодержащей гетероциклической кольцевой группы, которая может быть замещенной, могут быть рассмотрены пиперазинил, морфолинил и имидазолил, все из них могут иметь заместители, такие как OH, низший алкил-O-, NH2, низший алкил-NH-, (низший алкил) 2-N-, арил, низший алкил и тому подобное; однако группа этим не ограничивается. В любом случае в качестве примеров предпочтительных заместителей могут быть рассмотрены фенил, метокси, амино и диметиламино. "Низший алкил, который может быть замещенным" предпочтительно представляет собой замещенную метильную группу, представленную следующей далее формулой
Формула 6
"Азотсодержащая гетероциклическая кольцевая группа, которая может быть замещенной" предпочтительно представляет собой азотсодержащую гетероциклическую кольцевую группу, представленную следующей далее формулой
Формула 7
где в указанной выше формуле каждый символ имеет такое же определение, как дано выше.
В соединении (I) по настоящему изобретению имеются оптические изомеры (например, оптически активные соединения и диастеромеры) или геометрические изомеры, в зависимости от видов заместителей. По этой причине настоящее соединение (I) включает в себя смеси этих оптических изомеров или геометрических изомеров и выделенные соединения.
Также настоящее соединение (I) может образовывать кислотно- или основно-аддитивную соль. В качестве такой соли могут быть рассмотрены, например, аддитивные соли с неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота или что-либо подобное; аддитивные соли с органической кислотой, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота, глютаминовая кислота или что-либо подобное; соли с неорганическим основанием, такие как соли натрия, калия, магния, кальция, алюминия или чего-либо подобного; и соли с органическим основанием, таким как метиламин, этиламин, моноэтаноламин, диэтаноламин, триэтаноламин, циклогексиламин, лизин, орнитин или что-либо подобное. Кроме того, настоящее соединение (I) или его фармацевтически приемлемая соль может образовывать, в некоторых случаях, гидрат, сольват (например, сольват с этанолом) или полиморфное соединение.
Кроме того, соединение (I) по настоящему изобретению включают в себя все соединения (то есть пролекарства), которые могут метаболизироваться и преобразовываться в живом организме в настоящее соединение (I) или его фармацевтически приемлемую соль. В качестве группы, способной образовывать пролекарство по настоящему соединению (I), могут быть рассмотрены, например, группы, описанные в Prog. Med. 5:2157-2161 (1985), и группы, описанные в "Development of Drugs" (Hirokawa Shoten in 1990) Vol. 7 (Molecular Design) 163-198. Эти группы, в частности, представляют собой те, которые могут преобразовываться в первичный амин, вторичный амин, OH, HOC(=O)- или что-либо подобное, по настоящему изобретению, посредством гидролиза или сольволиза, или при физиологических условиях. В качестве пролекарств OH могут быть рассмотрены, например, низший алкил-COO-, который может быть замещенным, арил-C(=O)O-, который может быть замещенным, ROC(=O)-замещенный или незамещенный низший алкилен-C(=O)O-(R представляет собой H- или низший алкил. Это применимо и далее), ROC(=O)-замещенный или незамещенный низший алкенилен-C(=O)O-, ROC(=O)-низший алкилен-O-низший алкилен-C(=O)O-, ROC(=O)-C(=O)O-, ROS(=O)2-замещенный или незамещенный низший алкенилен-C(=O)O-, фталидил-O- и 5-метил-1,3-диоксолен-2-он-4-ил-метилокси.
Далее приводится описание репрезентативных способов получения настоящего соединения (I), синтеза исходных материалов и рецепты.
Способы получения
Настоящее соединение (I) может быть получено посредством различных способов синтеза, использующих характеристики на основе основного скелета или видов заместителей. Здесь описываются два способа получения (первый способ получения и второй способ получения).
Первый способ получения
Первый способ получения представляет собой способ получения настоящего соединения (I) в соответствии с путем реакции, показанным ниже.
Формула 8
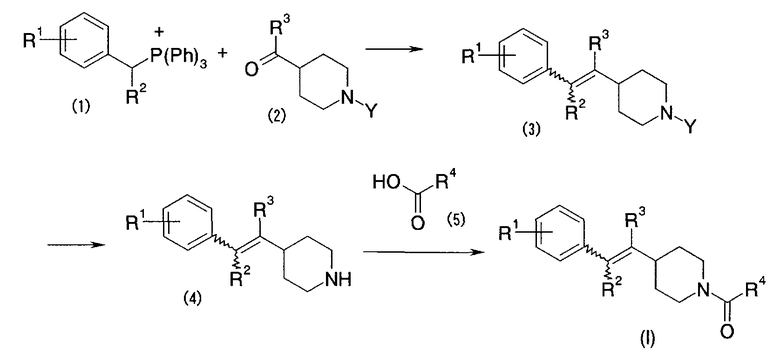
В указанном выше пути реакции символы (R1)-(R4) указывают на указанные выше одновалентные группы. Символ (P) указывает на атом фосфора; символ (Ph) указывает на фенильную группу и символ (Y) указывает на защитную группу для амино. Это же применимо и далее.
В соответствии с первым способом получения, соединение (I) по настоящему изобретению легко может быть получено посредством осуществления, обычным способом, реакции Виттига между фосфониевой солью (1) и альдегидом или кетоном (2) [Org. React., 14, 270-490 (1965); заявка на Международный патент WO 01/53288], с получением соединения (3), удаления аминозащитной группы соединения (3), с получением соединения (4), и осуществления амидирования между соединением (4) и карбоновой кислотой (5). В качестве растворителя для реакции Виттига может использоваться органический растворитель, не участвующий в реакции, такой как тетрагидрофуран, диоксан, диметил сульфоксид, толуол или что-либо подобное. В качестве основания могут использоваться гидрид натрия, трет-бутоксид калия, этоксид натрия, диизопропиламид лития или что-либо подобное. Реакция может осуществляться при температуре от -70°C до температуры дефлегмации. В качестве аминозащитной группы может быть рассмотрена трет-бутоксикарбонильная группа, бензилоксикарбонильная группа и тому подобное. Снятие защиты (удаление защитной группы) может осуществляться посредством обычного снятия защиты (Protective Group in Organic Synthesis, second ed., JOHN WILEY & SONS, INC.). Последующее амидирование может осуществляться посредством обычного способа.
Второй способ получения
Второй способ получения представляет собой способ получения настоящего соединения (I) в соответствии с путем реакции, показанным ниже.
Формула 9
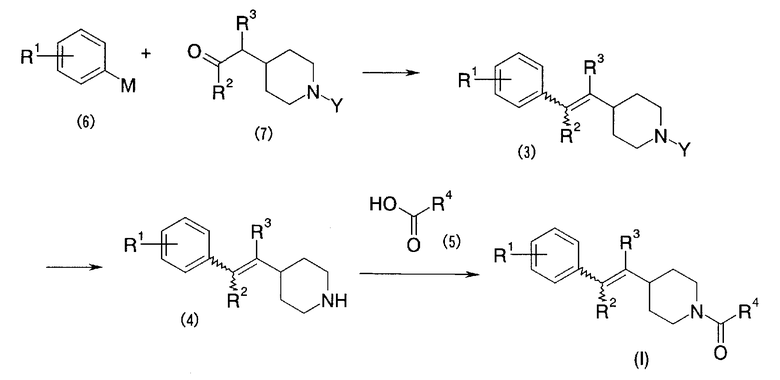
В указанном выше пути реакции символы (R1) - (R4) указывают на указанные выше одновалентные группы; символ (M) указывает на Li, MgCl или что-либо подобное; и символ (Y) указывает на аминозащитную группу. Это же применимо и далее.
В соответствии со вторым способом получения, настоящее соединение (I) может быть получено посредством обычной реакции между арилметаллом (например, ариллитием или арил-Гриньяром) (6) и карбонильным соединением (7) [Org. Synth. III, 200 (1955); Org. React., 6, 339-366 (1964); Org. React., 8, 258-304 (1967)]. В качестве растворителя реакции может использоваться органический растворитель, не участвующий в реакции, такой как простой диэтиловый эфир, тетрагидрофуран, диоксан, диметилсульфоксид, толуол или что-либо подобное. Реакция может осуществляться при температуре от -70°C до температуры дефлегмации. Последующее удаление аминозащитной группы и амидирование могут осуществляться таким же способом, как в первом способе получения.
Настоящее соединение (I) может быть также получено с помощью реакций, иных, чем указанные выше реакции, например реакции Петерсона [Org. React., 38, 1-223 (1990)], и образования тройной связи, с последующим частичным восстановлением [J. Am. Chem. Soc, 77, 3378 (1955); J. Am. Chem. Soc, 99, 2805 (1977); Synthesis, 1973, 457; и Tetrahedron 30, 3817 (1974)].
Синтез исходных материалов
Исходные материалы для настоящего соединения (I) легко могут быть получены в соответствии со способами синтеза, описанными в рассмотренной выше литературе [Org. React., 14, 270-490 (1965); WO 01/53288; Org. React., 16, 1-438 (1968); Org. Synth., III, 200 (1955); Org. React., 6, 339-366 (1964); Org. React., 8, 258-304 (1967)] и Org. Chem. 43, 4099 (1978).
Полученное таким образом соединение (I) по настоящему изобретению выделяется в виде свободной формы или в виде его фармацевтически приемлемой соли. Соль настоящего соединения (I) может быть получена посредством воздействия на настоящее соединение (I) (которое представляет собой свободное основание) обычной реакции образования соли.
Также настоящее соединение (I) или его фармацевтически приемлемая соль выделяется и очищается в виде его гидрата, сольвата или полиморфного соединения. Выделение и очистка осуществляются посредством применения обычных химических операций, таких как экстракция, концентрирование, дистилляция, кристаллизация, фильтрование, перекристаллизация, различные виды хроматографии и тому подобное.
Различные изомеры могут разделяться посредством использования соответствующим образом выбранных исходных материалов или посредством использования различий в физических или химических свойствах между изомерами. Например, оптические изомеры могут очищаться до стереохимически чистых изомеров посредством использования соответствующим образом выбранных исходных материалов или посредством рацемического разрешения рацемических соединений (например, рацемические соединения преобразуются в диастереомерные соли с помощью обычной оптически активной кислоты, с последующим оптическим разрешением).
Рецепты
К настоящему соединению (I) могут применяться различные рецепты, используемые обычно. Репрезентативные рецепты, применимые к настоящему соединению (I), описываются ниже.
Медицинская композиция по настоящему изобретению, содержащая, по меньшей мере, один вид настоящего соединения (I) или его фармацевтически приемлемой соли по настоящему изобретению, может содержать фармацевтически приемлемый носитель. Посредством использования носителя, наполнителя и других добавок все они обычно используются в фармацевтических препаратах, настоящая медицинская композиция получается в форме таблеток, порошка, леденцов, гранул, капсул, пилюль, раствора, препарата для инъекций, суппозитория, мази, пасты или чего-либо подобного и вводится перорально (включая сублингвальное введение) или парентерально.
Клиническая доза настоящего соединения (I) или его фармацевтически приемлемой соли для человека определяется соответствующим образом в каждом индивидуальном случае, принимая во внимание симптом, массу, возраст и пол индивидуального пациента, способ введения, и тому подобное; однако обычно введение осуществляется перорально в общем количестве от 1 мг до 1000 мг, предпочтительно от 10 мг до 200 мг за один или несколько раз в день для взрослого, или внутривенно, в количестве от 1 мг до 500 мг за один или несколько раз в день для взрослого, или вводится внутривенно способом с замедленным высвобождением в течение периода от 1 часа до 24 часов в день. Поскольку доза изменяется в зависимости от различных состояний, как описано выше, количество, меньшее, чем указанная выше доза, может быть достаточным.
В качестве твердой композиции по настоящему изобретению для перорального введения используются таблетки, порошок, гранулы и тому подобное. В такой твердой композиции одно или несколько активных веществ смешивается, по меньшей мере, с одним видом неактивного разбавителя, такого как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, магний метасиликат алюминат или что-либо подобное. Композиция может содержать, в соответствии с обычным способом, добавки, иные, чем неактивные разбавители, например смазывающий материал, такой как стеарат магния, разрыхлитель, такой как крахмал, или целлюлоза кальций гликолят, стабилизатор типа лактозы и солюбилизатор, такой как глютаминовая кислота или аспарагиновая кислота. Таблетки или пилюли могут покрываться сахаром или покрываться пленкой, растворимой в желудке или в кишечнике, с использованием сахарозы, желатина, гидроксипропилцеллюлозы и гидроксипропилметилцеллюлозы фталата и тому подобное.
Жидкая композиция для перорального введения содержит эмульсификатор, растворитель, суспендирующий агент, сироп, эликсир и тому подобное, которые, все, являются фармацевтически приемлемыми, и дополнительно содержит неактивный разбавитель, используемый обычно, такой как очищенная вода, этанол или что-либо подобное. Эта композиция может содержать, наряду с неактивными разбавителями, вспомогательный агент, такой как солюбилизатор, смачивающий агент или суспендирующий агент, подслащивающий агент, ароматизирующий агент, отдушку, консервант и тому подобное.
Препарат для инъекций для парентерального введения содержит солюбилизатор, суспендирующий агент или эмульсификатор, которые, все, являются стерильными и представляют собой водные или неводные растворы. Водный раствор солюбилизатора или суспендирующего агента содержит, например, дистиллированную воду для инъекций и физиологический солевой раствор. Неводный солюбилизатор или суспендирующий агент включают в себя, например, пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, спирт, такой как этанол и Polysolvate 80 (торговое наименование). Такая композиция может дополнительно содержать добавки, такие как изотонический агент, консервант, смачивающий агент, эмульсификатор, диспергирующий агент, стабилизатор, такой как лактоза, вспомогательный агент для солюбилизации и растворения, и тому подобное. Композицию стерилизуют, например, посредством фильтрования через бактериальный фильтр или посредством смешивания со стерилизаторами, или облучения. Альтернативно она может также быть получена в виде асептической твердой композиции, и полученная асептическая композиция предусматривается для использования после растворения, перед использованием, в асептической воде или в асептическом растворителе для инъекций. Настоящее соединение (I) может использоваться в сочетании с терапевтическим препаратом для заболеваний, описанных выше, или с другими лекарственными средствами, пригодными для использования против боли, посредством механизма, иного, чем блокада натриевых каналов. Лекарственное средство, пригодное для использования против боли, которое используется в сочетании, включает в себя наркотические анальгетики, антипиретические анальгетики, нестероидные противовоспалительные лекарственные средства и тому подобное.
Примеры
Настоящее изобретение ниже описывается более подробно посредством примеров [примеров получения настоящего соединения (I)]. Однако настоящее изобретение ни в коем случае не ограничивается этими примерами. Сначала примеры получения исходных материалов, используемых в последующих примерах, описываются как сравнительные примеры.
Сравнительный пример 1
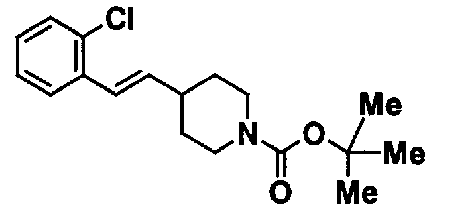
К 15,0 мл N,N-диметилформамидной суспензии, содержащей 2,38 г (2-хлорбензил)(трифенил)фосфонийхлорида, добавляют 0,63 г трет-бутоксида калия. Смесь перемешивают при комнатной температуре в течение 10 минут. К полученной оранжевой суспензии добавляют 1,00 г трет-бутил 4-формилпиперидин-1-карбоксилата, и полученную смесь перемешивают в течение 15 минут для взаимодействия. Полученную реакционную смесь выливают в насыщенный водный раствор хлорида аммония и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (н-гексанэтилацетат), с получением 1,45 г трет-бутил 4-[2-(2-хлорфенил)винил]пиперидин-1-карбоксилата в виде бесцветного масла.
Сравнительные примеры 2-32
Соединения, показанные в таблицах 1-3, получают таким же способом, как в сравнительном примере 1.
Сравнительный пример 33
К 1,45 г трет-бутил 4-[2-(2-хлорфенил)винил]пиперидин-1-карбоксилата добавляют 2,10 г п-толуолсульфоновой кислоты. Смесь перемешивают при 150°C в течение 5 часов для взаимодействия. К полученной реакционной смеси добавляют 10,0 мл воды. Смесь доводят примерно до pH 11 с помощью 10% водного раствора гидроксида натрия и экстрагируют хлороформом. Органический слой промывают водой и насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования отфильтрованный материал концентрируют при пониженном давлении, с получением 0,97 г 4-[(E)-2-(2-хлорфенил)винил]пиперидина в виде желтого масла.
Сравнительные примеры 34-40
Соединения, показанные в таблице 3, получают таким же способом, как в сравнительном примере 33.
Сравнительный пример 41
5,0 мл хлороформового раствора, содержащего 0,55 г 4-[(E)-2-(3-хлорфенил)винил]пиперидина и 0,5 мл триэтиламина, добавляют к 5,0 мл хлороформового раствора, содержащего 0,2 мл хлорацетилхлорида, и полученную смесь перемешивают при комнатной температура в течение 20 минут для взаимодействия. Реакционную смесь выливают в 10% хлористоводородную кислоту. Органический слой отделяют и промывают насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (н-гексанэтилацетат), с получением 0,42 г 1-(хлорацетил)-4-[(E)-2-(3-хлорфенил)винил]пиперидина в виде желтого масла.
Сравнительные примеры 42-45)
Соединения, показанные в таблицах 3 и 4, получают таким же способом, как в сравнительном примере 41.
Пример 1
К 10,0 мл N,N-диметилформамидного раствора, содержащего 0,45 г 4-[(E)-2-(2-хлорфенил)винил]пиперидина, добавляют 0,37 г моногидрохлорид морфолин-4-ил-уксусной кислоты, 0,39 г 1-этил-3-(3-диметиламинопропил)карбодиимида моногидрохлорида, 0,27 г 1-гидроксибензтриазол моногидрата и 0,28 мл триэтиламина. Смесь перемешивают при комнатной температуре в течение 2 дней для взаимодействия. Полученную реакционную смесь выливают в 10% водный раствор карбоната калия и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (этилацетат), с получением 0,56 г 4-(2-{4-[(E)-2-(2-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина в виде бледно-желтых кристаллов. Эти кристаллы получают в растворе 3,0 мл этанола и 1,0 мл тетрагидрофурана. К нему добавляют 2,0 мл этанолового раствора, содержащего 0,14 г щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола, с получением 0,52 г 4-(2-{4-[(E)-2-(2-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина оксалата в виде бесцветных порошкообразных кристаллов.
Примеры 2-54
Соединения, показанные в таблицах 5-13, получают таким же способом, как в примере 1.
Пример 55
К 10,0 мл N,N-диметилформамидного раствора, содержащего 0,80 г 1-(хлорацетил)-4-[(E)-2-(4-хлорфенил)винил]пиперидина, добавляют 0,41 г пиперидин-3-ола и 0,74 г карбоната калия, и полученную смесь перемешивают при комнатной температуре в течение 6 часов для взаимодействия. Полученную реакционную смесь выливают в 10% водный раствор карбоната калия и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (этилацетат-этанол), с получением 0,92 г 1-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)пиперидин-3-ола в виде бесцветного масла. Это масло растворяют в 5,0 мл этанола. К нему добавляют 5,0 мл этанолового раствора, содержащего 0,23 г щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола-этилацетата, с получением 0,96 г 1-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)пиперидин-3-ол оксалата в виде бесцветных порошкообразных кристаллов.
Примеры 56-65
Соединения, показанные в таблицах 13-15, получают таким же способом, как в примере 55.
Пример 66
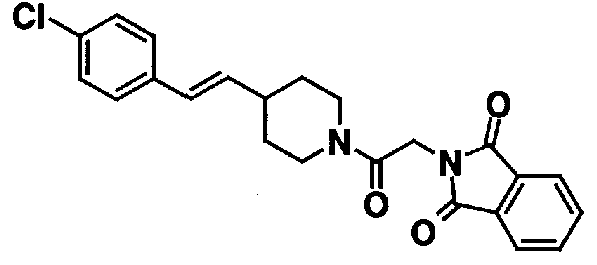
К 20,0 мл N,N-диметилформамидного раствора, содержащего 2,00 г 1-(хлорацетил)-4-[(E)-2-(4-хлорфенил)винил]пиперидина, добавляют 1,86 г фтальимида калия, и полученную смесь перемешивают при 50°C в течение 3 часов для взаимодействия. Полученную реакционную смесь выливают в 10% водный раствор карбоната калия, экстрагируют хлороформом. Органический слой промывают водой и насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (хлороформ-ацетон), с получением 2,46 г 2-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-1H-изоиндол-1,3(2H)диона в виде бесцветных порошкообразных кристаллов.
Пример 67
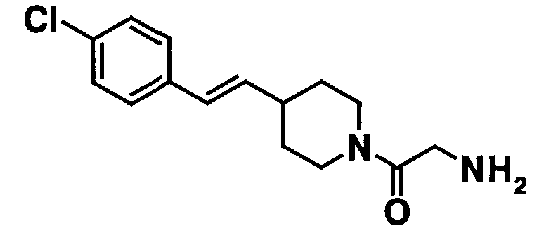
К 50,0 мл метаноловой суспензии, содержащей 2,46 г 2-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-1H-изоиндол-1,3(2H)-диона, добавляют 0,65 мл гидразина моногидрата, и полученную смесь нагревают с обратным холодильником в течение 1,5 часа. Полученную реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении. К остатку добавляют 100,0 мл 5% водного раствора гидроксида натрия и экстрагируют хлороформом. Органический слой промывают водой и насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении, с получением 1,80 г 2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтанамина в виде бесцветного масла.
Пример 68
К 5,0 мл этанолового раствора, содержащего 0,71 г трет-бутил 4-(2-{4-[(E)-2-(3-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)пиперазин-1-карбоксилата, добавляют 10,0 мл 35% раствора хлористоводородной кислоты - этанол. Смесь перемешивают при комнатной температуре в течение 2 часов для взаимодействия. Полученную реакционную смесь концентрируют при пониженном давлении. Остаток три раза азеотропно дистиллируют вместе с 5,0 мл этанола. Полученный остаток перекристаллизуют из этанола, с получением 0,46 г 1-(2-{4-[(E)-2-(3-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)пиперазина дигидрохлорида в виде бесцветных кристаллов.
Примеры 69-74
Соединения, показанные в таблицах 15-16, получают таким же способом, как в примере 68.
Пример 75
К 10,0 мл N,N-диметилформамидной суспензии, содержащей 0,70 г 1-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)пиперазина дигидрохлорида добавляют 0,46 мл триэтиламина и 0,56 г хлорида моногидрохлорида никотиновой кислоты. Смесь перемешивают при комнатной температуре в течение 2 часов для взаимодействия. Полученную реакционную смесь выливают в 10% водный раствор карбоната калия и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли, а затем сушат над сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (н-гексан - этилацетат), с получением 0,73 г 1-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-4-(пиридин-3-илкарбонил)пиперазина в виде бледно-желтого масла. К 3,0 мл этанолового раствора, содержащего его, добавляют 2,0 мл этанолового раствора, содержащего 0,15 г щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола, с получением 0,66 г 1-(2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-4-(пиридин-3-илкарбонил)пиперазина оксалата в виде мутновато-белых порошкообразных кристаллов.
Пример 76
К 20,0 мл метиленхлоридного раствора, содержащего 1,80 г 2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтанамина, добавляют 0,67 мл циклогексанона и 0,35 мл уксусной кислоты, с получением раствора примерно при pH 5. Раствор перемешивают при комнатной температуре в течение 15 минут. К нему добавляют 1,37 г триацетоксиборгидрида натрия, и полученную смесь перемешивают в течение 10 минут для взаимодействия. Полученную реакционную смесь выливают в 10% водный раствор карбоната калия и экстрагируют хлороформом. Органический слой промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (хлороформ-этанол), с получением 1,73 г N-2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)циклогексанамина в виде светло-коричневых кристаллов. 10,0 мл этанолового раствора получают посредством растворения 0,88 г соединения. К раствору добавляют 5,0 мл этанолового раствора, содержащего 0,22 г щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола-этилацетата, с получением 1,0 г N-2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)циклогексанамина оксалата в виде бесцветных порошкообразных кристаллов.
Пример 77
Соединения, показанные в таблице 17, получают таким же способом, как в примере 76.
Пример 78
К 10,0 мл ацетонитрилового раствора, содержащего 0,81 г N-2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)циклогексанамина, при комнатной температуре, добавляют 1,0 мл 37% формальдегидного раствора, и полученную смесь перемешивают в течение 5 минут. К ней добавляют 0,15 мл уксусной кислоты и перемешивают в течение 10 минут для взаимодействия. Добавляют 0,85 г триацетоксиборгидрида натрия, полученную смесь перемешивают в течение 10 минут. Полученную реакционную смесь выливают в 10% водный раствор карбоната калия и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на окиси алюминия (н-гексан-этилацетат), с получением 0,69 г N-2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-N-метилциклогексанамина в виде бесцветного масла. После получения из него 5,0 мл этанолового раствора к нему добавляют 5,0 мл этанолового раствора, содержащего 0,16 г щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола-этилацетата, с получением 0,72 г N-2-{4-[(E)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-N-метилциклогексанамина оксалата в виде бесцветных порошкообразных кристаллов.
Пример 79
Соединения, показанные в таблице 17, получают таким же способом, как в примере 78.
Пример 80
6 мл 20% водного раствора гидроксида натрия добавляют к 30 мл метанолового раствора, содержащего 2,85 г метил 3-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}бензоата. Смесь нагревают с обратным холодильником в течение 1 часа, дистиллируют при пониженном давлении для удаления растворителя. К остатку добавляют для нейтрализации 10% хлористоводородную кислоту. Полученную смесь экстрагируют хлороформом, и экстракт сушат над безводным сульфатом натрия. Высушенный экстракт дистиллируют при пониженном давлении для удаления растворителя. Затем полученные кристаллы суспендируют в этилацетате, полученную суспензию фильтруют, собирая 1,91 г 3-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}бензойной кислоты.
Примеры 81-82
Соединения, показанные в таблице 17, получают таким же способом, как в примере 80.
Пример 83
К 8 мл ацетонитриловой суспензии, содержащей 589 мг 4-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}бензойной кислоты, добавляют 943 мг 1-этил-3-(3-диметиламинопропил)карбодиимида моногидрохлорида, 556 мг 1-гидроксибензтриазола и 1,5 мл 50% водного раствора диметиламина. Смесь нагревают с обратным холодильником в течение 1,5 часа. После охлаждения реакционную смесь концентрируют при пониженном давлении. К остатку добавляют насыщенный водный раствор бикарбоната натрия, и полученную смесь экстрагируют хлороформом. Экстракт промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. Высушенный экстракт дистиллируют при пониженном давлении для удаления растворителя. Остаток очищают с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), с получением 315 мг N,N-диметил-4-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}бензамида в виде бледно-желтого масла. После получения из него 8 мл этилацетатного раствора к нему добавляют 4 мл этанолового раствора, содержащего 74 мг щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола-этилацетата, с получением 290 мг N,N-диметил-4-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}бензамида оксалата.
Примеры 84-88
Соединения, показанные в таблице 18, получают таким же способом, как в примере 83.
Пример 89
6,5 мл 1,2M раствора метиллитий - простой диэтиловый эфир добавляют при -78°C к 20 мл тетрагидрофуранового раствора, содержащего 824 мг метил 4-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}бензоата. Смесь перемешивают при такой же температуре в течение 3 часов. К ней добавляют 20 мл насыщенного водного раствора хлорида аммония, и полученную смесь нагревают до комнатной температуры. Реакционную смесь экстрагируют хлороформом. Экстракт промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. Высушенный экстракт дистиллируют при пониженном давлении для удаления растворителя. Остаток очищают с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), с получением 283 мг 2-4-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}фенил)пропан-2-ола в виде бледно-желтого масла. Затем из него получают 5 мл этилацетатного раствора, к нему добавляют 1 мл этанолового раствора 68 мг щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола, с получением 219 мг 2-4-{(E)-2-[1-(морфолин-4-илацетил)пиперидин-4-ил]винил}фенил)пропан-2-ол оксалата.
Пример 90
К 20 мл метанолового раствора, содержащего 667 мг 4-(2-{4-[(E)-2-(4-нитрофенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина, добавляют 5 мл воды, 0,5 г хлорида аммония и 0,5 г цинкового порошка. Смесь нагревают с обратным холодильником в течение 30 минут. После охлаждения реакционную смесь фильтруют. Фильтрат концентрируют при пониженном давлении. К остатку добавляют воду и экстрагируют этилацетатом. Экстракт промывают насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. Высушенный экстракт дистиллируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), с получением 600 мг 4-{(E)-2-[1-морфолин-4-илацетил]пиперидин-4-ил}винил}анилина в виде бледно-желтого масла. 250 мг соединения получают в растворе в 2 мл этилацетата и 1 мл этанола посредством его растворения. К полученному раствору добавляют 1 мл этанолового раствора, содержащего 68 мг щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола, с получением 231 мг 4-{(E)-2-[1-морфолин-4-илацетил]пиперидин-4-ил}винил}анилина оксалата.
Пример 91
0,5 мл уксусного ангидрида добавляют к 5 мл пиридинового раствора, содержащего 500 мг 4-{(E)-2-[1-морфолин-4-илацетил)пиперидин-4-ил]винил}анилина. Полученную смесь перемешивают при комнатной температуре в течение 6 часов для взаимодействия. Реакционную смесь концентрируют при пониженном давлении. К остатку добавляют 5% водный раствор гидроксида натрия и экстрагируют этилацетатом. Экстракт промывают водой и насыщенным раствором соли, а затем сушат над безводным сульфатом натрия. Высушенный экстракт дистиллируют при пониженном давлении для удаления растворителя. К остатку добавляют этилацетат, и полученную смесь перемешивают. Полученные кристаллы собирают посредством фильтрования, с получением 520 мг N-4-{(E)-2-[1-(2-морфолин-4-илацетил)пиперидин-4-ил]винил}фенил)ацетамида в виде бледно-желтых порошкообразных кристаллов. Затем к нему добавляют раствор 3 мл этилацетата и 3 мл этанола, содержащего 450 мг соединения, 1 мл этанолового раствора, содержащего 110 мг щавелевой кислоты. Полученные кристаллы собирают посредством фильтрования и перекристаллизуют из этанола, с получением 470 мг N-4-{(E)-2-[1-(2-морфолин-4-илацетил)пиперидин-4-ил]винил}фенил)ацетамид оксалата.
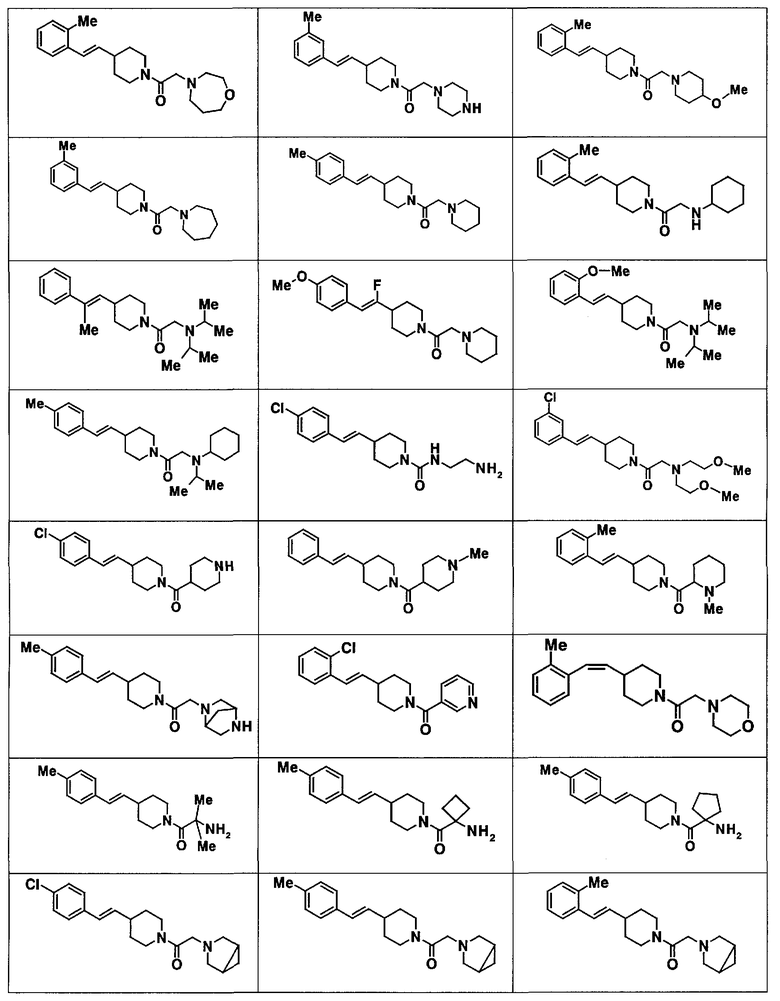
Химические структурные формулы и физические и химические свойства соединений, полученных в приведенных выше сравнительных примерах и примерах, показаны в таблицах 1-19. В дополнение к соединениям, описанным в примерах, соединения, приведенные в таблицах 20, могут быть получены посредством способов, описанных выше, способов, описанных в сравнительных примерах и примерах, способов, известных специалистам в данной области, и их модификаций, без необходимости в каких-либо специальных экспериментах.
Символы в таблицах указывают на следующее.
Rf: № сравнительного примера, EX: № примера, Me: метильная группа, MC: масс-спектр (если не указано иного, FAB или ESI) m/z, ЯМР: спектр ядерного магнитного резонанса (если не указано иного, 400 МГц, 1H-ЯМР, ДМСО-d6, внутренний стандарт TMS) δ (м.д.)
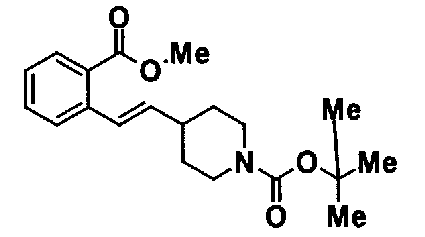
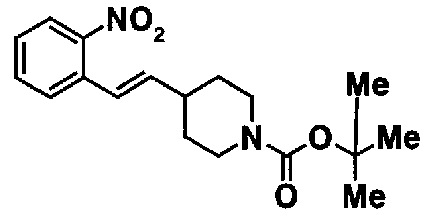
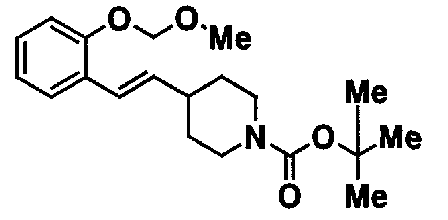
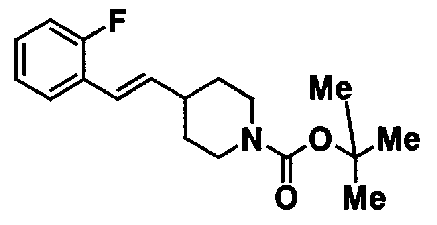
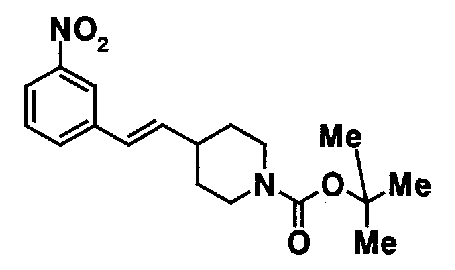
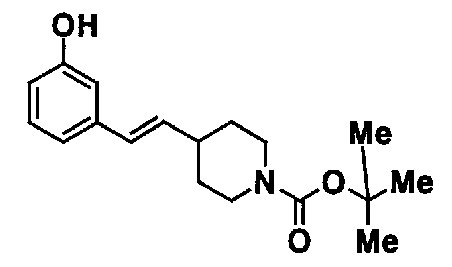
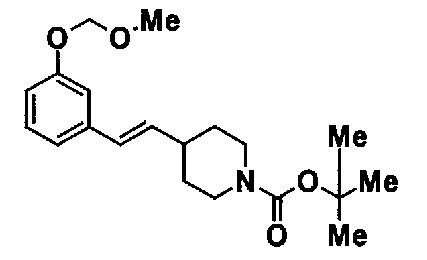
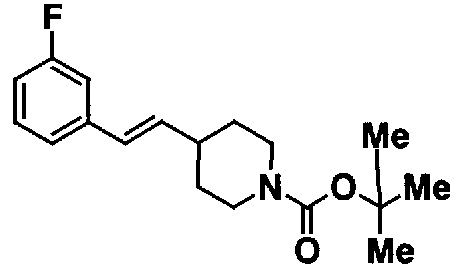
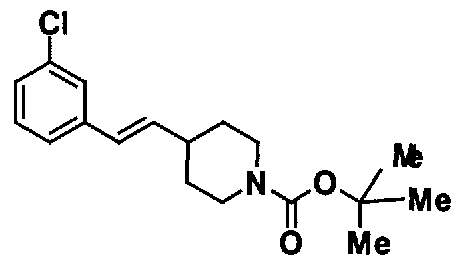
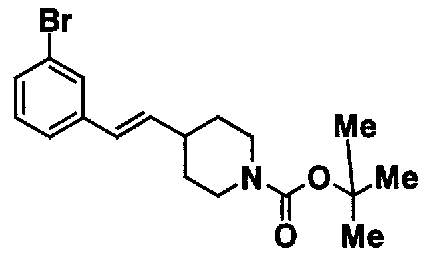
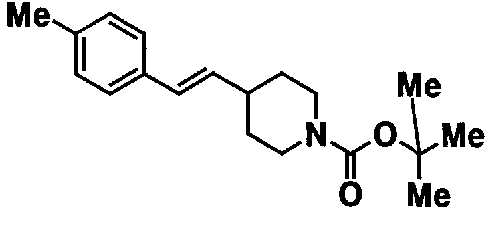
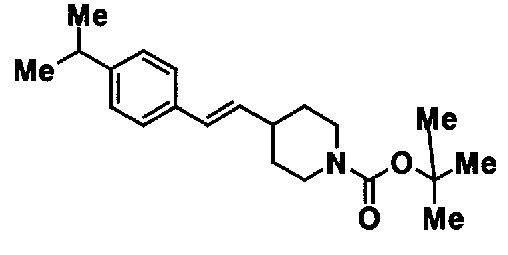
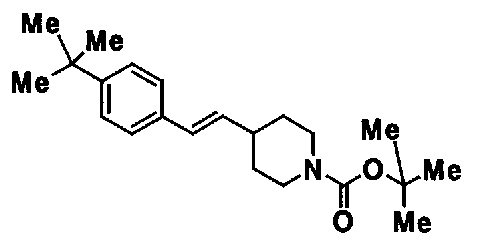
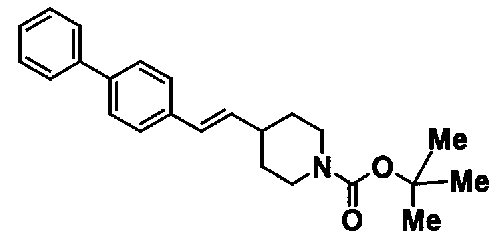
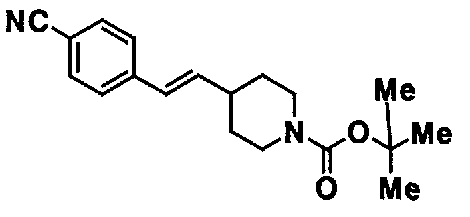
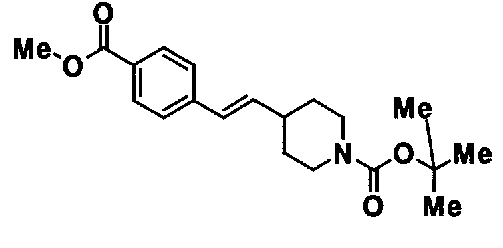
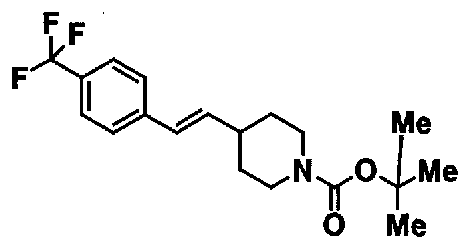
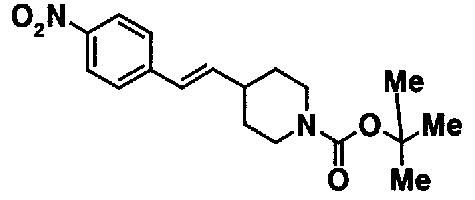
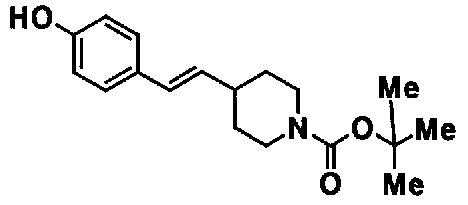
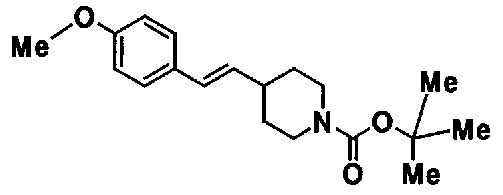
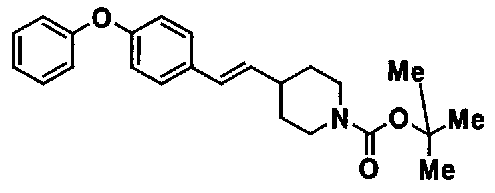
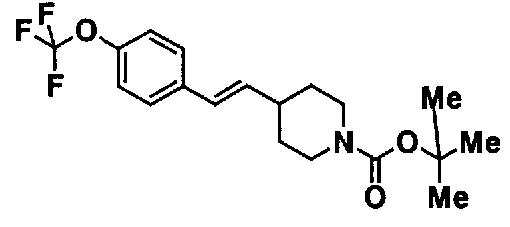
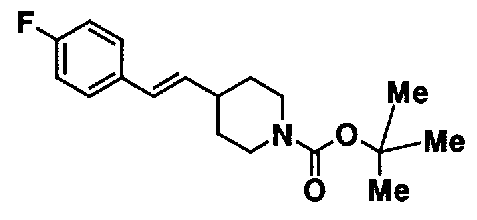
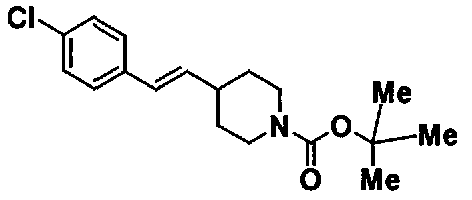
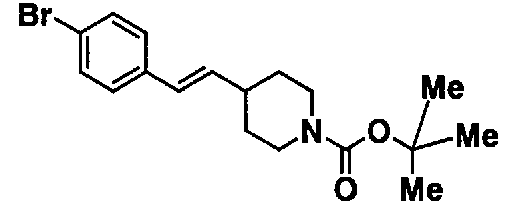
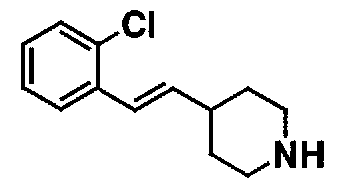
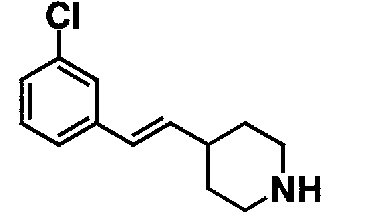
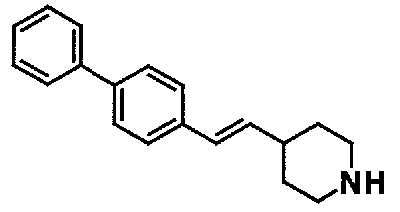
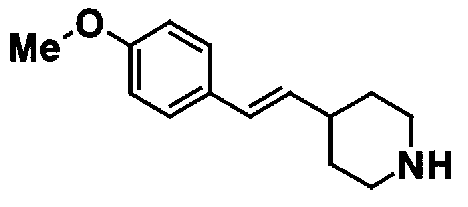
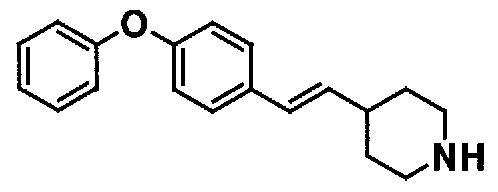
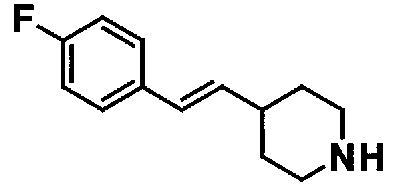
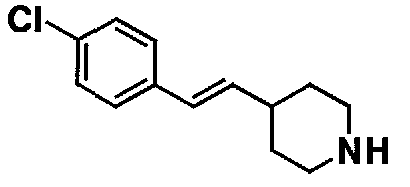
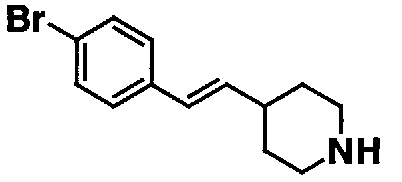
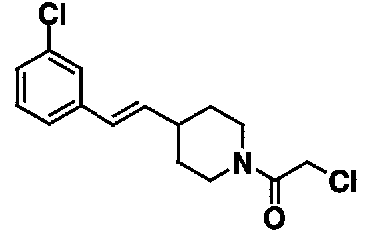
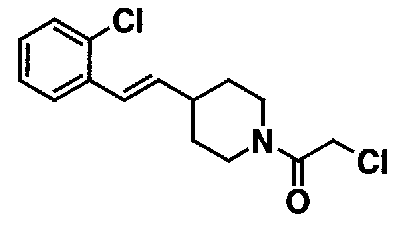
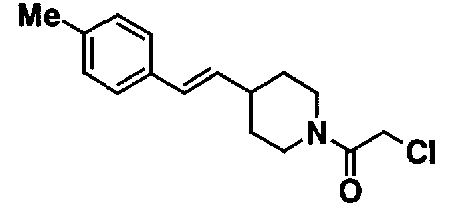
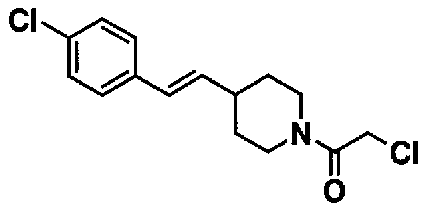
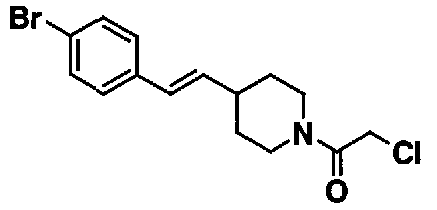
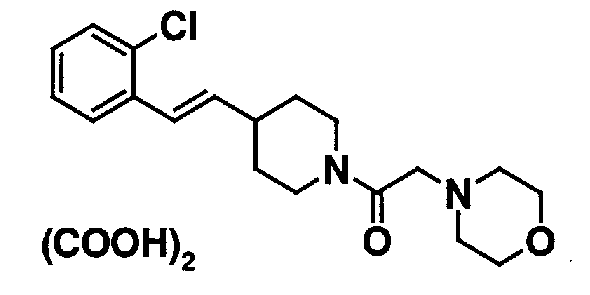
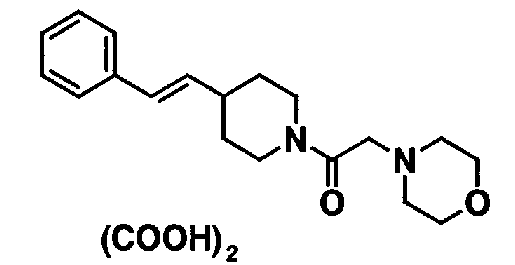
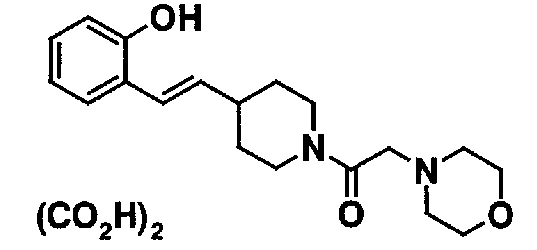
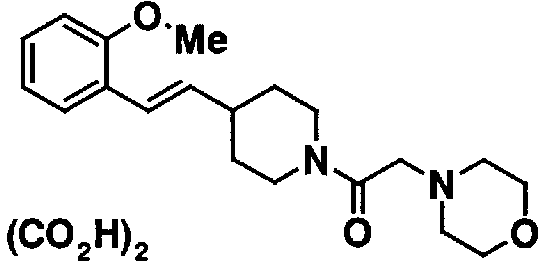
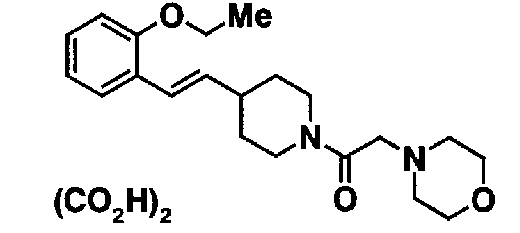
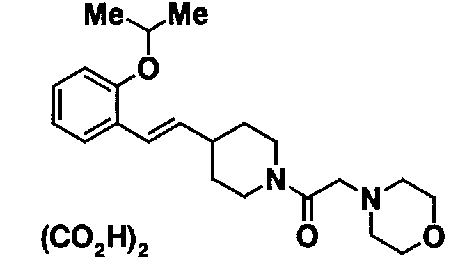
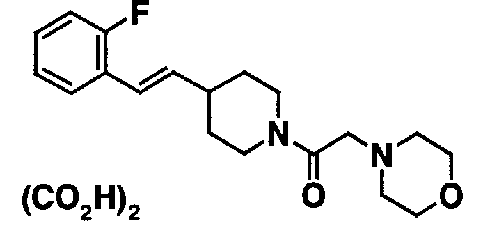
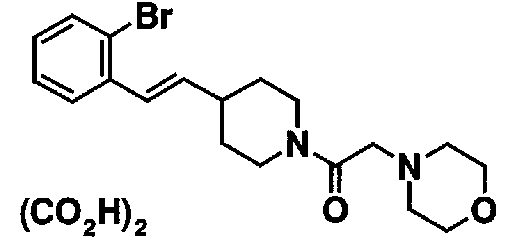
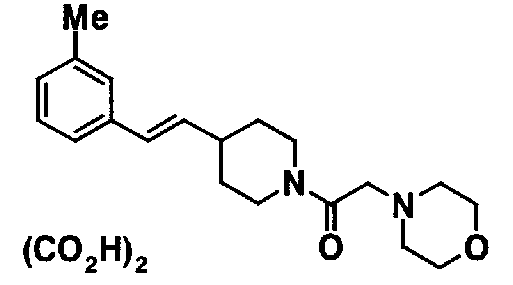
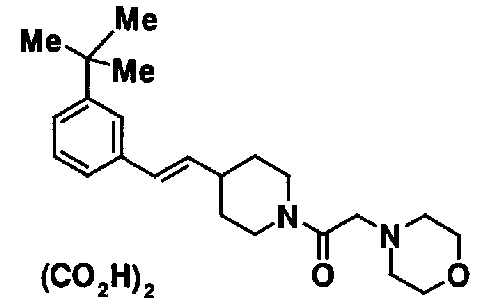
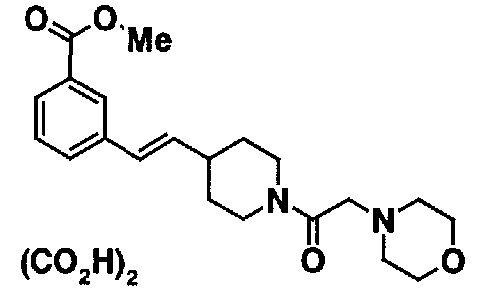
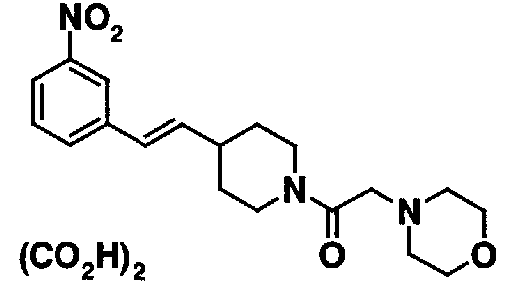
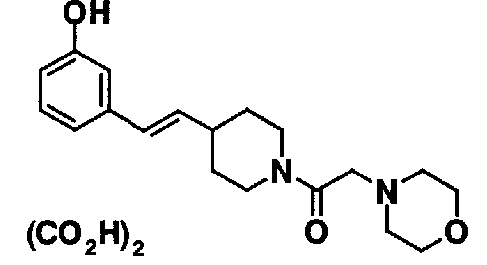
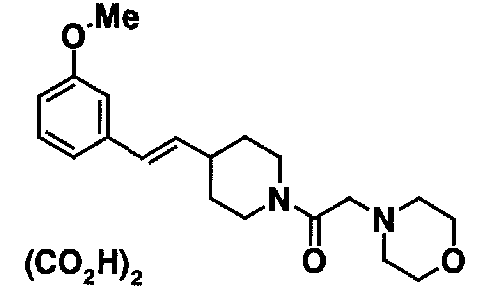
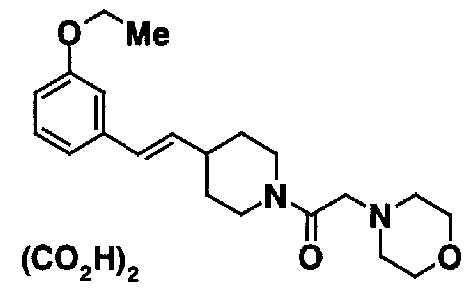
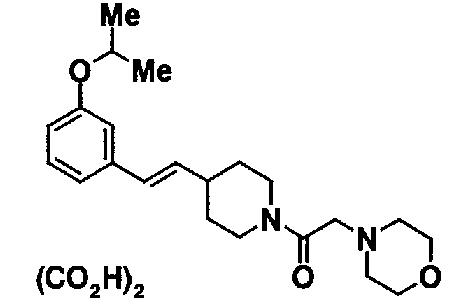
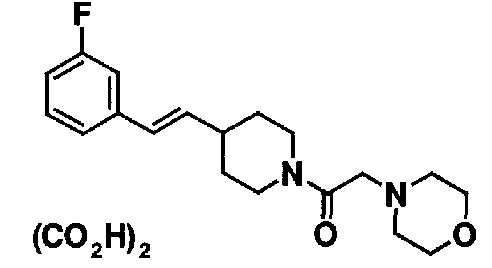
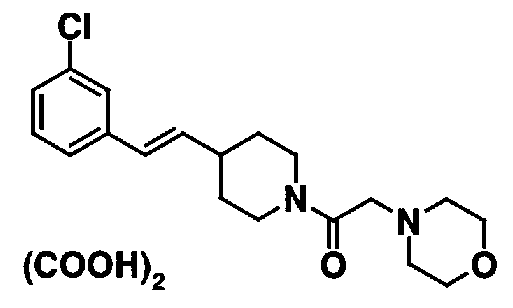
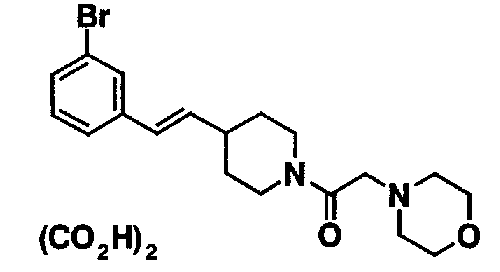
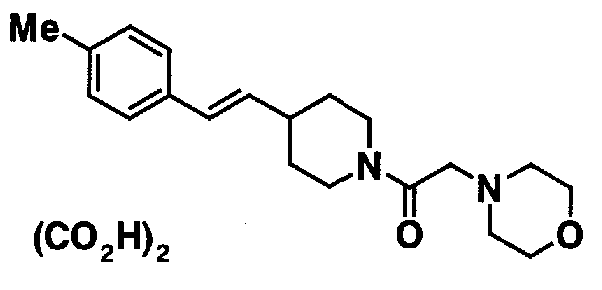
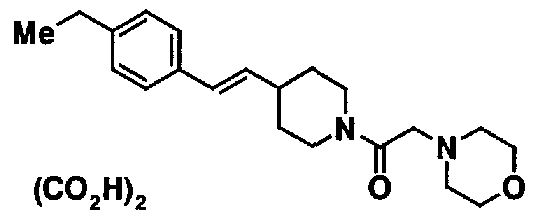
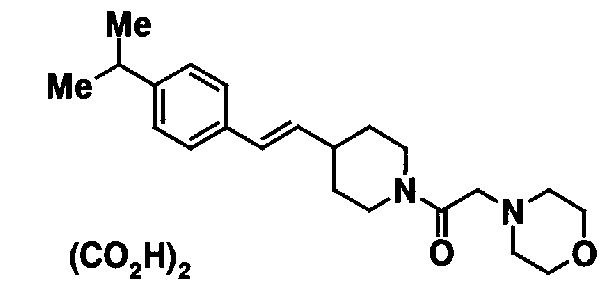
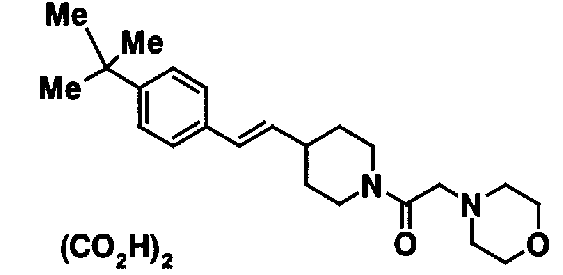
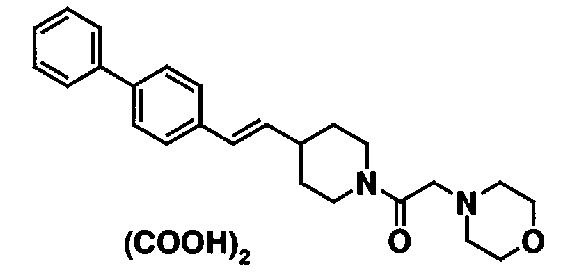
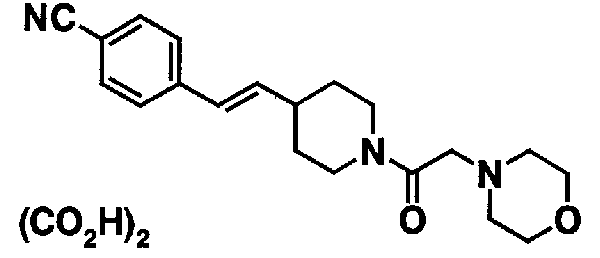
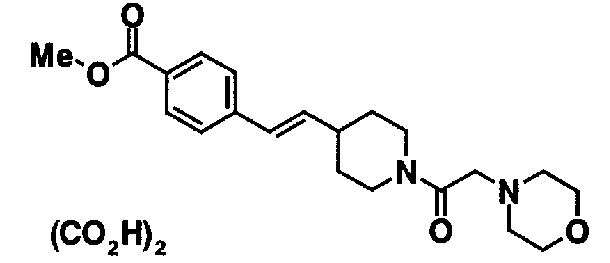
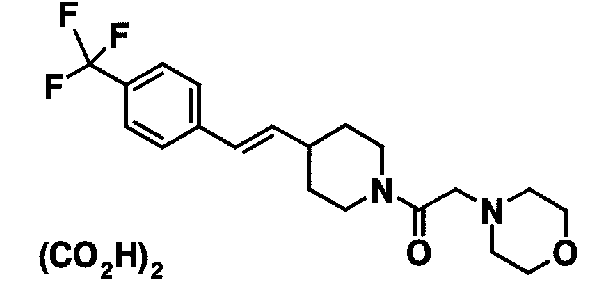
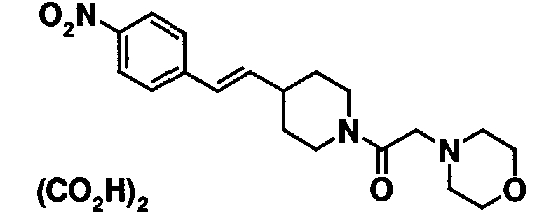
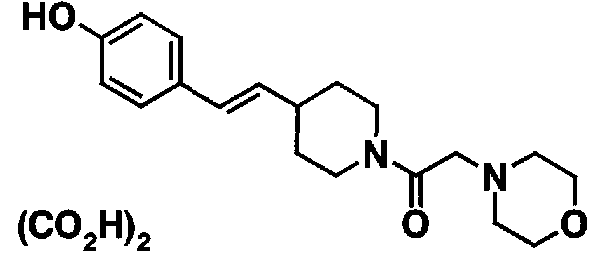
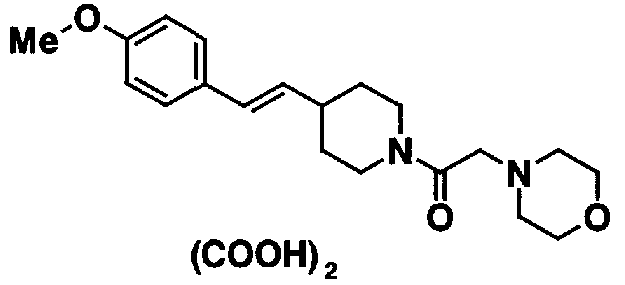
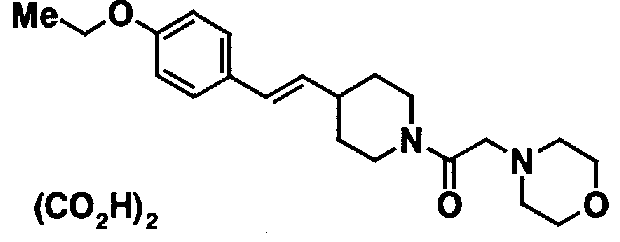
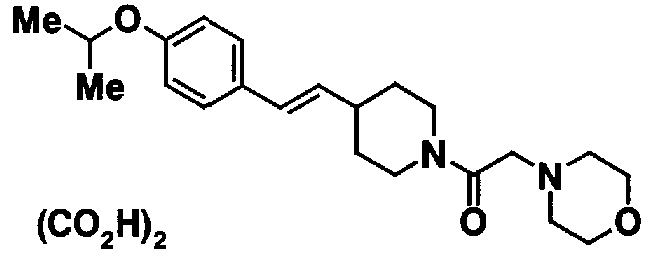
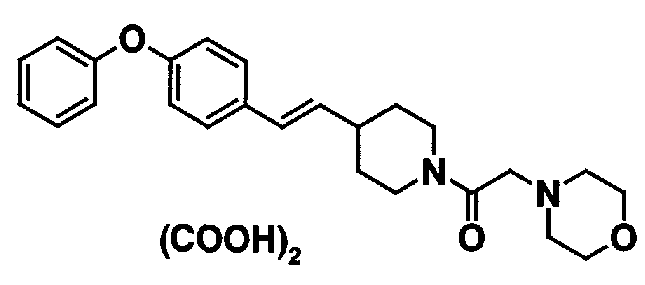
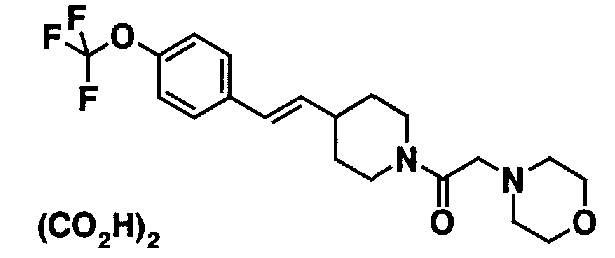
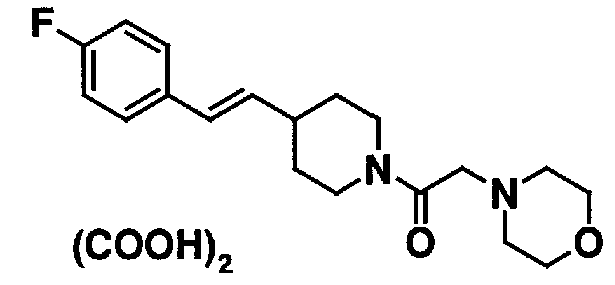
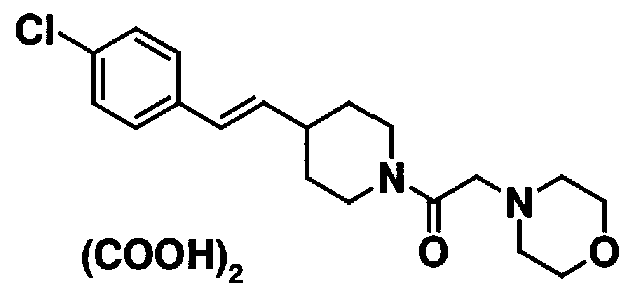
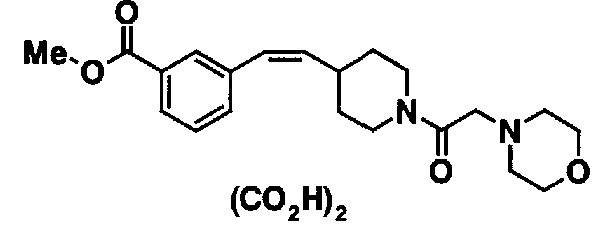

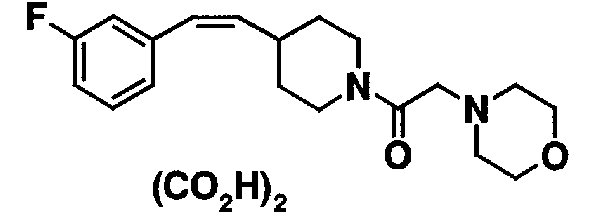
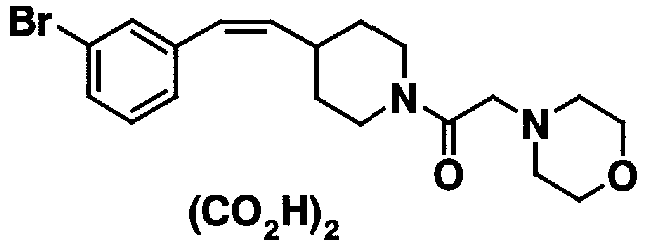
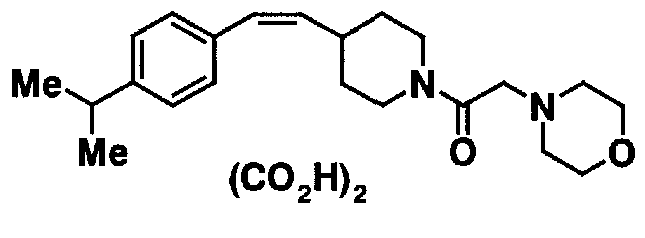
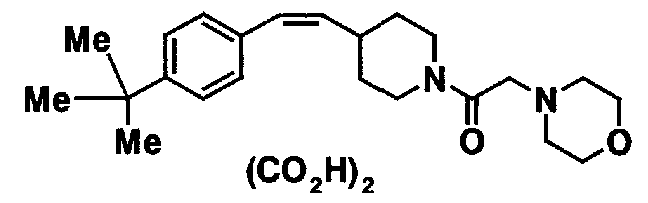
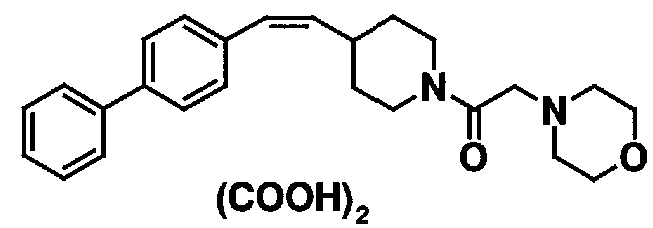
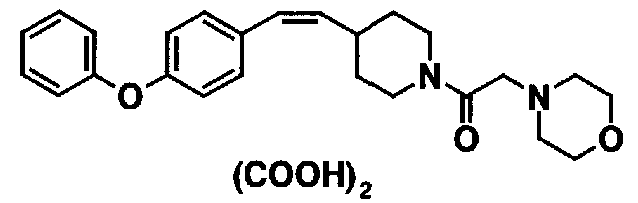
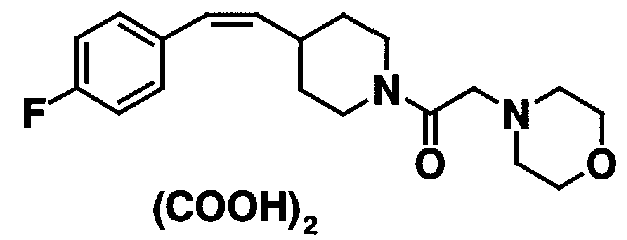
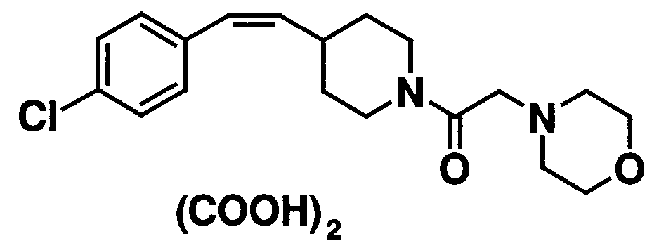
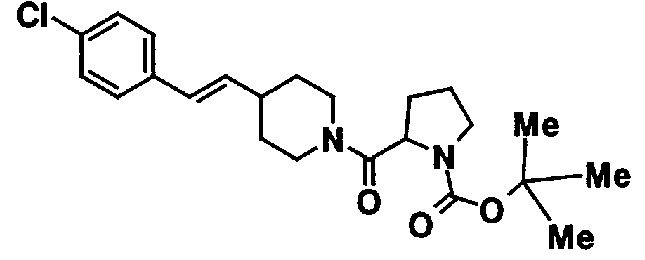
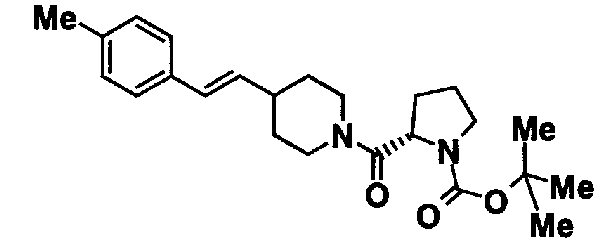
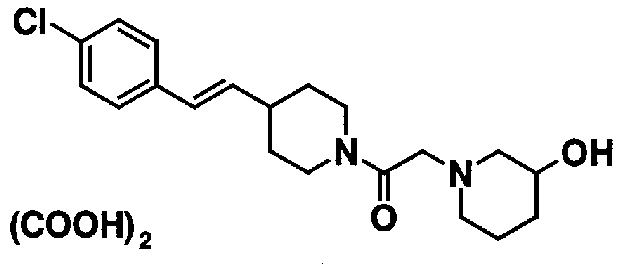
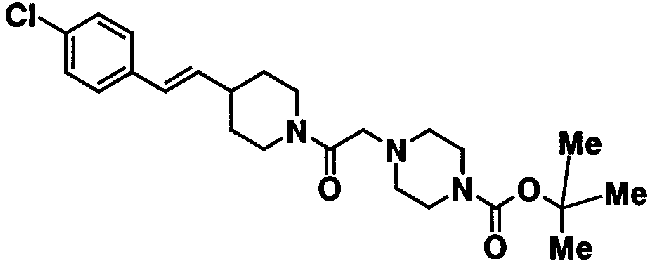
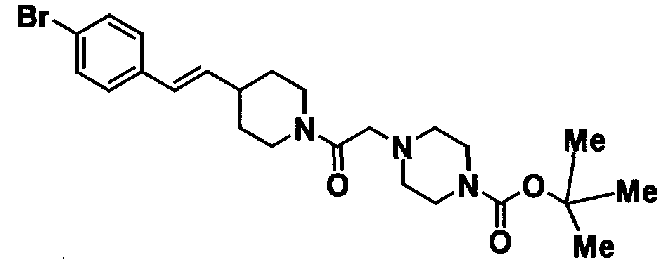
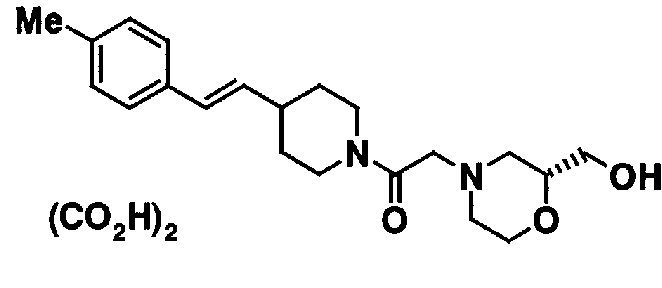
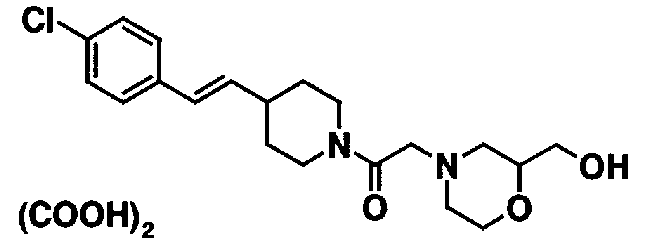
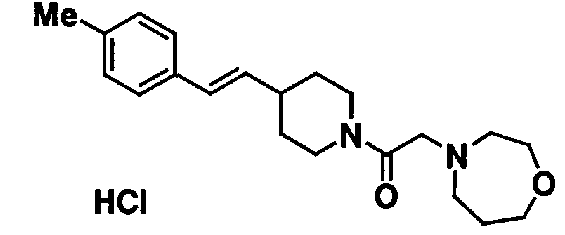
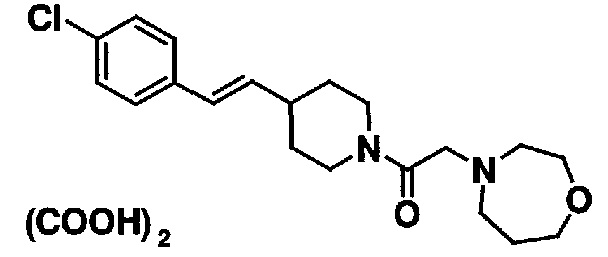
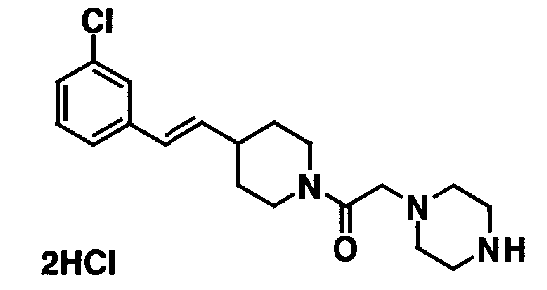
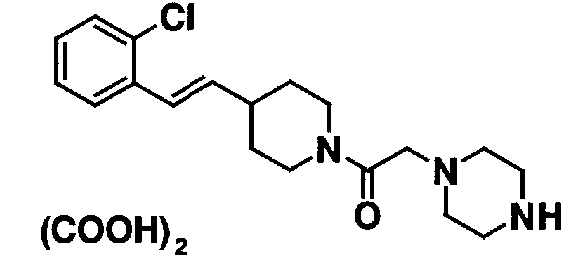
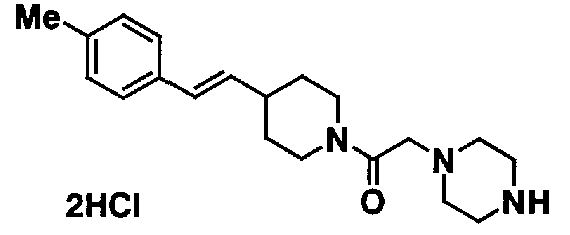
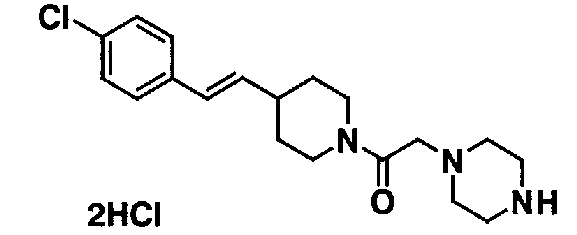
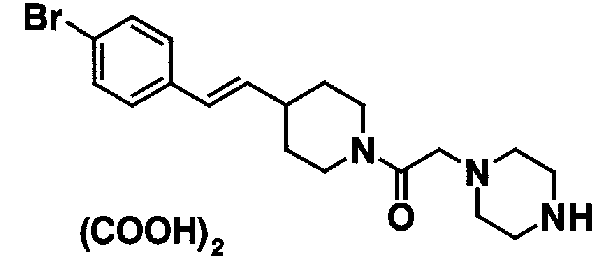
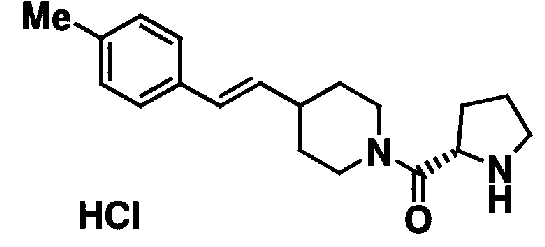
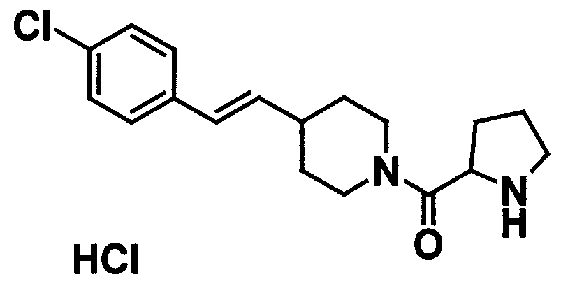
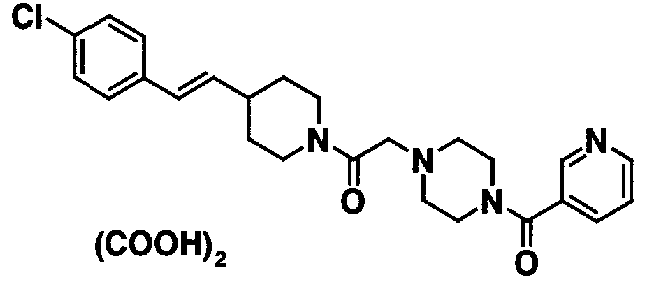
МС: 375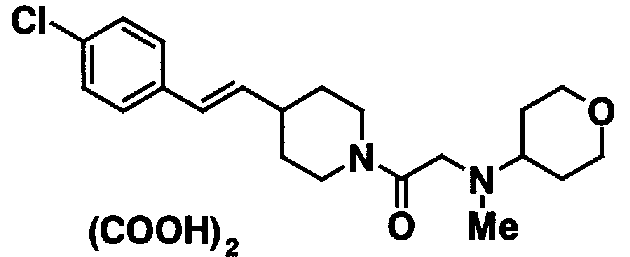
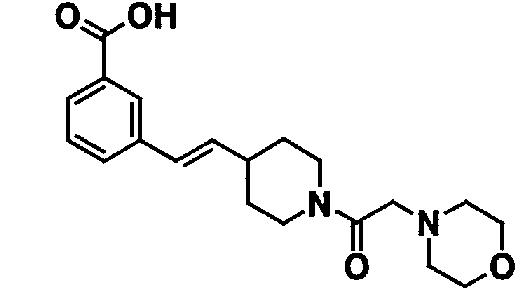
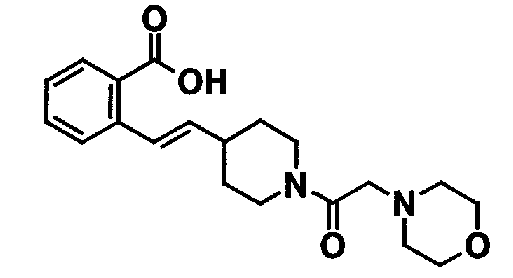
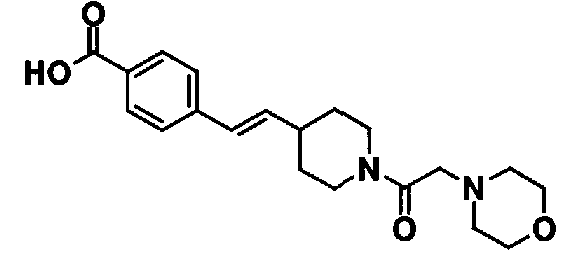
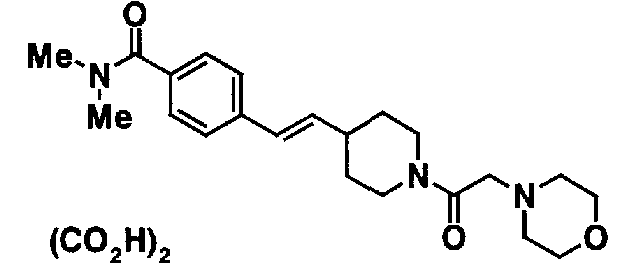
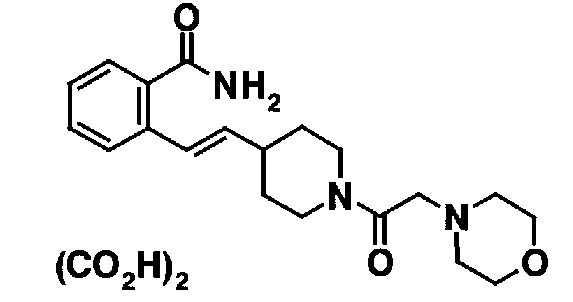
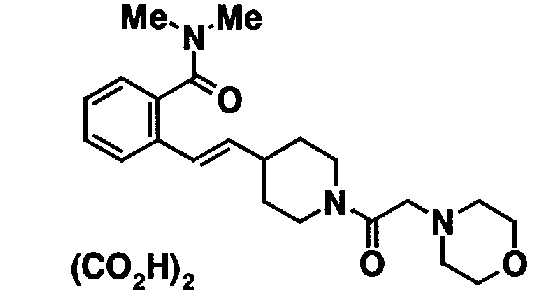
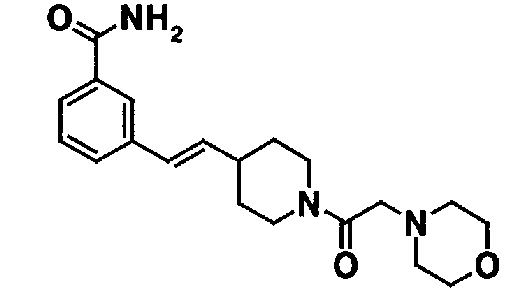
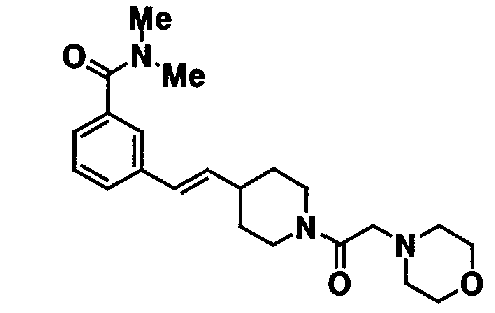
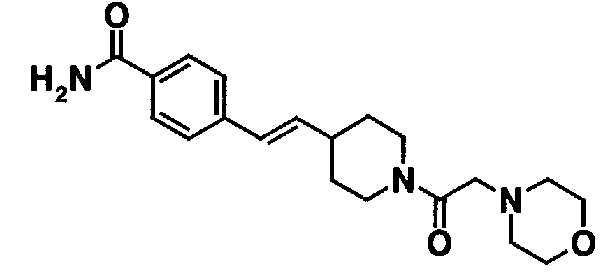
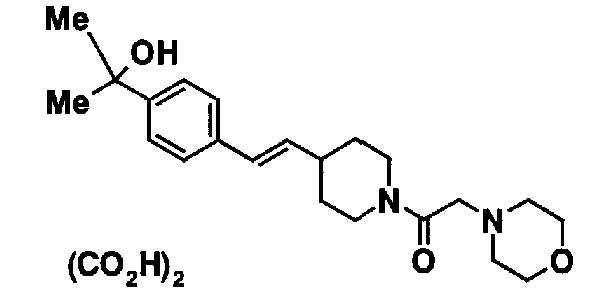
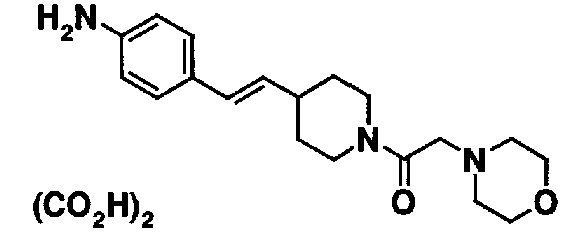
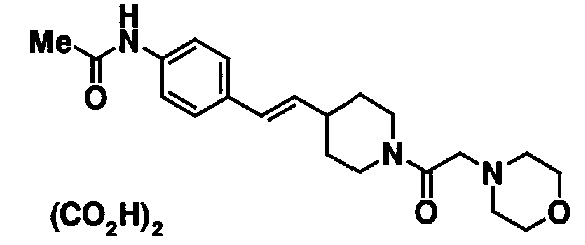
Таблица 20
Фармакологические исследования
Для соединения (I) по настоящему изобретению исследуются действие ингибирования натриевых каналов и анальгетическое действие на животных моделях. Исследования подробно описываются ниже.
Исследование ингибирования натриевых каналов
Действия ингибирования натриевых каналов репрезентативных соединений по настоящему соединению (I) подтверждаются посредством исследования потребления [14C] гуанидина с использованием ткани мозга крысы. Исследования потребления [14C] гуанидина осуществляют посредством модифицикации способа Bonisch et al. (British Journal of Pharmacology 108, 436-442, 1993). [14C] гуанидин используют как трассер для натрия, и измеряется ингибиторная активность на потребление [14C] гуанидина, индуцируемая посредством вератридина, в качестве активатора натриевых каналов, в первичных нейронах церебрального кортекса крысы.
a. Приготовление системы культуры первичных нейронов церебрального кортекса крыс.
Беременных крыс (Wistar, самки, срок беременность = 19 дней) анестезируют простым диэтиловым эфиром и забивают посредством обескровливания с помощью перерезывания сонной артерии. Плоды вырезают у беременной крысы и стерилизуют с помощью этанола для дезинфекции. Затем у плодов отделяют церебральный кортекс. Церебральный кортекс переваривают папаином и диспергируют в культурной среде. Затем отделенные нейроны помещают в 96-луночный белый планшет, покрытый поли-L-лизином, при плотности 2,5×106 клеток/лунку, и культивируют в течение 2 дней в CO2 инкубаторе (37°C, 5% CO2).
b. Оценка исследуемых соединений
Каждую лунку промывают один раз буфером для анализов (135 мМ холин Cl, 5 мМ KCl, 1 мМ MgSO4, 5,5 мМ глюкозы, 1 мг/мл BSA, 10 мМ Hepes-Tris, pH 7,4). Буфер для анализов добавляют в каждую лунку и инкубируют при 25°C в течение 10 минут. Затем буфер для анализов заменяют реакционным раствором (исследуемое соединение, [14C] гуанидин и 100 мкМ вератридина), и инкубируют при 25°C в течение 15 минут. Реакцию останавливают посредством трехкратной промывки холодным промывочным буфером (135 мМ NaCl, 5 мМ KCl, 1 мМ MgSO4, 10 мМ Hepes-Tris, pH 7,4). В каждую лунку добавляют 17 мкл 0,1н NaOH; осуществляют перемешивание; затем добавляют 100 мкл сцинтиллятора и измеряют радиоактивность каждой лунки с использованием жидкостного счетчика сцинтилляций. В каждом исследовании величина потребления [14C] гуанидина, ингибируемая посредством 1 мМ мексилетина, принимается как часть специфичного потребления через натриевый канал. Активность исследуемого соединения на натриевом канале выражается как концентрация 50% ингибирования (IC50) для специфичного потребления.
Как показано в таблице 21, настоящее соединение включает в себя соединения, демонстрирующие значения IC50 примерно 3-30 мкМ и имеют более высокие воздействия, чем мексилетин (примерно 70 мкМ).
Анальгетическое действие на диабетическую невропатию у стрептозотоцин-индуцированных диабетических мышей
Репрезентативные соединения для соединения (I) по настоящему изобретению оцениваются на анальгетическое действие на диабетическую невропатию у стрептозотоцин (STZ)-индуцированных диабетических мышей для подтверждения воздействия на невропатическую боль. Оценку осуществляют с помощью способа Kamei et al. (Pharmacology Biochemistry & Behavior 39, 541-544, 1991) с некоторыми модификациями.
Самцы мышей ICR возрастом 4 недели получают внутрибрюшинную инъекцию массой 200 мг/кг STZ для получения мышей с диабетической невропатией. Анальгетическое действие оценивают посредством исследования придавливания хвоста. Конкретно анальгетическое действие детектируют как пролонгацию времени реакции (в секундах), то есть времени до того, как животное демонстрирует реакцию поворота головы, после того как хвост сдавливают с помощью зажима. В день 14 после инъекции STZ осуществляют предварительное исследование для определения латентности реакции перед введением исследуемого соединения. Только животные, демонстрирующие в предварительном исследовании латентность реакции не более чем 3 секунды, используются для оценки исследуемого соединения на следующий день (день 15 после инъекции STZ). При оценке исследуемого соединения измеряют значения латентности реакции после введения исследуемого соединения. Исследуемое соединение вводят перорально при дозе 30 мг/кг, за 45 минут перед измерением латентности реакции. Анальгетическое действие исследуемого соединения выражается как пролонгация латентности (в секундах), вычисляемая по формуле:
(латентность реакции после введения исследуемого соединения) - (латентность реакции перед введением исследуемого соединения).
Как показано в таблице 22, настоящие исследуемые соединения демонстрируют пролонгацию латентности примерно от 2 до 4 секунд и имеют хорошее анальгетическое действие.
Приведенным выше исследованием подтверждается, что настоящее соединение имеет активность ингибирования натриевых каналов, более высокую, чем у мексилетина. Подтверждается также, что настоящее соединение, когда вводится перорально, показывает хорошее анальгетическое действие на животной модели болезненного состояния, то есть у мышей с диабетической невропатией. Таким образом, подтверждается, что настоящее соединение является эффективным в качестве превосходного ингибитора натриевых каналов для боли, в частности для невропатической боли, связанной с диабетической невропатией, и тому подобное.
Изучение отсутствия побочных воздействий
Для лекарственных средств, используемых в настоящее время с целью лечения невропатической боли, разница между дозой для выраженного анальгетического воздействия и дозой для выраженного побочного воздействия является малой, и по этой причине часто возникает побочное воздействие, делая сложным использование при высокой дозе. Посредством исследования Rotarod, часто используемого в качестве классического способа для детектирования побочного воздействия, подтверждается, что репрезентативные соединения из соединений по настоящему изобретению едва ли вызывают побочное воздействие, даже когда вводятся при дозе, значительно более высокой, чем доза, необходимая для выраженного анальгетического действия. Изучение отличия от дозы побочного воздействия осуществляется посредством частичной модификации способа Christensen et al. (Pain 93, 147-153, 2001). При изучении используют SD-крыс. В каждом опыте каждое исследуемое животное помещают на устройство, которое ускоряется от 4 об/мин до 40 об/мин при постоянном ускорении за 5 минут, и измеряют время (время удерживания, сек) до падения исследуемого животного. В день опыта, сначала, у каждого исследуемого животного измеряют массу тела, и их подвергают воздействию трех опытов перед введением лекарственного средства. Для изучения фармакологического действия выбирают тех исследуемых животных, которые демонстрируют самое продолжительное время удерживания, 90 секунд или больше, в трех опытах. Эти выбранные исследуемые животные разделяются на группы, так что различия в средних временах удерживания перед введением лекарственного средства между группами становится минимальным. Каждое лекарственное средство вводят перорально вместе с растворителем (5 мл/кг). После введения лекарственного средства осуществляют два опыта с таким же временным графиком, как и для исследования с целью измерения воздействия против аллодинии у мышей с лигированными спинальными нервами L5/L6. Например, когда воздействие против аллодинии измеряют через 30 минут после введения лекарственного средства, опыты после введения лекарственного средства осуществляют через 30 минут после введения лекарственного средства, также и в исследовании Rotarod. В качестве времени удерживания после введения лекарственного средства, для каждого исследуемого животного, принимают среднее по двум опытам. Время удерживания каждой группы выражается как среднее значение ± стандартная ошибка. Значимость различия между группой, где вводился растворитель, и группой, где вводилось лекарственное средство, анализируют с использованием теста Дюннета, и уровень p<0,05 оценивается как значимый.
Соединения по настоящему изобретению включают в себя соединения, имеющие такое свойство, что побочное воздействие едва ли вызывается, даже когда они вводятся при дозе, значительно более высокой, чем доза, эффективная при исследовании для измерения воздействия против аллодинии у крыс с лигированием спинального нерва L5/L6.
Воздействие против аллодинии у крыс с лигированием спинального нерва L5/L6
Один из главных симптомов невропатической боли представляет собой значительно пониженный порог реакции на тактильную стимуляцию (аллодиния). Воздействие против аллодинии репрезентативных соединений из соединений по настоящему изобретению подтверждается посредством оценки анальгетического действия на крыс с лигированием спинального нерва L5/L6. Оценки осуществляются посредством способа Kim and Chung (Pain 50, 355-363, 1992) с некоторыми модификациями. Под анестезией с помощью фенобарбитала, самцы SD-крыс возрастом 5 или 6 недель подвергаются хирургической операции для лигирования люмбарных спинальных нервов, левых, как L5, так и L6, плотно, шелковыми нитями. Для оценки анальгетического действия применяется волосковое исследование фон Фрея. То есть заднюю лапу животного колют с помощью волоска и наименьшее усилие на волоске для получения реакции конечности обозначается как порог реакции (логарифм граммов) на механическую стимуляцию. Поскольку в предыдущем исследовании подтверждается, что порог реакции задней лапы животного, нижней лапы на стороне лигирования, явно является низким в течение дней 7-14 после хирургической операции (в состоянии аллодинии), воздействие против аллодинии у исследуемого соединения оценивают в любой день между днями 7 и 14 после операции. В день перед оценкой исследуемого соединения измеряют порог реакции перед введением исследуемого соединения. Животные разделяются на 4-5 групп, так что различия в средних значениях порога реакции перед введением между группами и разброс внутри групп становится малым. При оценке исследуемого соединения измеряют порог реакции после введения исследуемого соединения. Исследуемое соединение вводят перорально за 30 минут до измерения порога реакции. Сильнодействие действия против аллодинии у исследуемого соединения выражают как ЕД50. При вычислении ЕД50 пороги для расположенных на одной стороне и на противоположной стороне, по отношению к лигированию, лап в группе, где вводился растворитель, обозначаются 0% и 100%, соответственно.
Настоящее соединение включает в себя те соединения, которые демонстрируют превосходную ЕД50. В то же время ЕД50 мексилетина составляет примерно 70 мг/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ 2,6-ДИЗАМЕЩЕННЫЙ СТИРИЛ | 2004 |
|
RU2333200C2 |
| ПРОИЗВОДНЫЕ АРИЛ- И ГЕТЕРОАРИЛПИПЕРИДИНКАРБОКСИЛАТОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА FAAH | 2005 |
|
RU2376305C2 |
| ПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2281955C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА | 2008 |
|
RU2470020C2 |
| АЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2493154C2 |
| СОЕДИНЕНИЕ СУЛЬФОНАМИДА ИЛИ ЕГО СОЛЬ | 2007 |
|
RU2425029C2 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДА ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ УКАЗАННОГО ПРОИЗВОДНОГО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2333198C2 |
| ИМИДАЗОДИАЗЕПИНОВОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2712968C2 |
| ПРОИЗВОДНОЕ 4,4-ДИФТОР-1,2,3,4-ТЕТРАГИДРО-5Н-БЕНЗАЗЕПИНА, ЕГО СОЛЬ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2268882C1 |
| НОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ СОДЕРЖАЩИЕ, ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ РАССТРОЙСТВ | 2014 |
|
RU2675818C2 |
Изобретение относится к новым пиперидиновым производным, представленным следующей формулой (I)
Формула 1
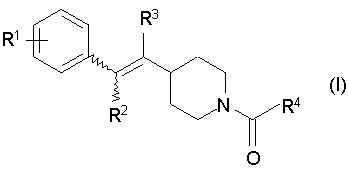
где символы R1-R4 каждый представляет собой любую из одновалентных групп, указанных ниже: R1 представляет собой атом водорода, атом галогена, низший алкил, который может быть замещен атомом галогена или ОН; -O-низший алкил, который может быть замещен атомом галогена; -O-арил, арил, -С(=O)-низший алкил, СООН, -С(=O)-O-низший алкил, -C(=O)-NH2, -С(=O)NH-низший алкил, -С(=O)N-(низший алкил)2, ОН, -O-С(=O)-низший алкил, NH2, -NH-низший алкил, -N-(низший алкил)2, -NH-C(=O)-низший алкил, CN или NO2; R2 и R3 каждый представляет собой атом водорода; и R4 представляет собой любую из одновалентных групп (а), (b) и (с), показанных ниже
Формула 2
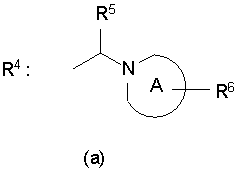
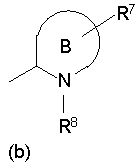
где в указанных выше группах (а), (b) и (с) А представляет собой пирролидиновое, пиперидиновое, морфолиновое, пиперазиновое или оксазепановое кольцо; В представляет собой пирролидиновое или пиперидиновое кольцо; R5 и R8-R11 могут быть одинаковыми или отличными друг от друга и каждый представляет собой атом водорода, -С(=O)-O-низший алкил, циклоалкил или тетрагидропиран; R представляет собой атом водорода, -С(=O)-O-низший алкил, ОН, -низший алкилен-ОН или -С(=O)-пиридин; и R7 представляет собой атом водорода; или к его фармацевтически приемлемым солям.
Изобретение также относится к медицинской композиции. Технический результат - получение новых биологически активных соединений и медицинской композиции на их основе, которая представляет собой ингибитор натриевого канала.
2 н. и 8 з.п. ф-лы, 22 табл.
Формула 1
где символы R1-R4 каждый представляет собой любую из одновалентных групп, указанных ниже
R1 представляет собой атом водорода, атом галогена, низший алкил, который может быть замещен атомом галогена или ОН; -O-низший алкил, который может быть замещен атомом галогена; -O-арил, арил, -С(=O)-низший алкил, СООН, -С(=O)-O-низший алкил, -С(=O)-NH2, -С(=O)NH-низший алкил, -С(=O)N-(низший алкил)2, ОН, -O-С(=O)-низший алкил, NH2, -NH-низший алкил, -N-(низший алкил)2, -NH-С(=O)-низший алкил, CN или NO2;
R2 и R3 каждый представляет собой атом водорода; и
R4 представляет собой любую из одновалентных групп (а), (b) и (с), показанных ниже
Формула 2
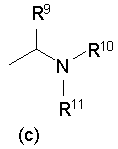
где в указанных выше группах (а), (b) и (с),
А представляет собой пирролидиновое, пиперидиновое, морфолиновое, пиперазиновое или оксазепановое кольцо;
В представляет собой пирролидиновое или пиперидиновое кольцо;
R5 и R8-R11 могут быть одинаковыми или отличными друг от друга и каждый представляет собой атом водорода, -С(=O)-O-низший алкил, циклоалкил или тетрагидропиран;
R6 представляет собой атом водорода, -С(=O)-O-низший алкил, ОН, -низший алкилен-ОН или -С(=O)-пиридин; и
R7 представляет собой атом водорода;
или его фармацевтически приемлемая соль.
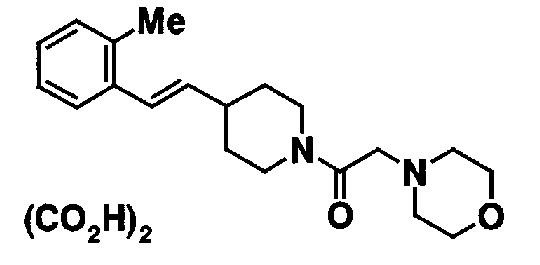
4-(2-{4-[(Е)-2-(2-метилфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина,
4-(2-{4-[(Е)-2-(4-изопропилфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина,
4-(2-{4-[(Е)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)морфолина,
N-(2-{4-[(Е)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)циклогексанамина и
N-(2-{4-[(Е)-2-(4-хлорфенил)винил]пиперидин-1-ил}-2-оксоэтил)-N-метилциклогексанамина.
| WO 00/12074 А2, 09.03.2000 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ТРАНСПОРТИРУЮЩЕЕ УСТРОЙСТВО ДЛЯ СЕКЦИОННЫХПЕЧЕЙ | 0 |
|
SU279681A1 |
| ПРОИЗВОДНЫЕ N-АЛКИЛЕНПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ОПТИЧЕСКИ ЧИСТЫЕ ПРОИЗВОДНЫЕ N-АЛКИЛЕНПИПЕРИДИНА | 1992 |
|
RU2089547C1 |

