Область техники, к которой относится изобретение
Данное изобретение относится к производному пиперидина, обладающему фармакологической активностью, и/или к его соли. Кроме того, данное изобретение относится к лекарственному средству или фармацевтической композиции, содержащей производное пиперидина и/или его соль, описанные выше, в качестве активного ингредиента.
Уровень техники
Доброкачественная гиперплазия простаты (ВРН) представляет собой заболевание, возникающее в основном у мужчин в возрасте старше 50 лет или более и сопровождающееся мочевыми расстройствами, и степень ее распространенности повышается с возрастом. Число пациентов с ВРН в Японии постоянно увеличивалось в последние годы с ростом старения населения. ВРН значительно ухудшает качество жизни пожилых мужчин из-за мочевых расстройств и является серьезной проблемой в плане медицинской экономики, так как представляет собой наиболее часто диагностируемое и подвергаемое лечению заболевание в области урологии.
Было установлено, что два фактора, а именно непосредственное уретральное сдавление из-за гипертрофии простаты (механическая непроходимость) и подъем интрауретрального давления из-за повышенного сокращения гладкой мышцы простаты через симпатический нерв (функциональная непроходимость), одновременно включены в мочевые расстройства, сопровождающие ВРН. Лекарственная терапия может применяться в отношении обоих данных механизмов, при этом в случае механической непроходимости главным образом используются ингибиторы 5α-редуктазы и в случае функциональной непроходимости главным образом используются α1-симпатолитические средства (α1-блокаторы). Ингибиторы 5α-редуктазы вызывают регрессию простаты из-за их антиандрогенного эффекта, основанного на подавлении превращения тестостерона в 5α-дегидротестостерон (DHT), который представляет собой более сильнодействующий андроген, производимый 5α-редуктазой. Регрессирует только эпителий простаты, однако требуется длительный период времени (от нескольких недель до нескольких месяцев), чтобы эффективность лекарственного средства становилась очевидной. C другой стороны, так как α1-блокаторы проявляют свою эффективность как лекарственные средства очень быстро после введения и являются исключительными по безопасности, α1-блокаторы в настоящее время являются первоочередным средством для лечения ВРН. Однако в результате долговременных клинических изучений было установлено, что ингибитор 5α-редуктазы значительно замедлял переход к инвазивной терапии по сравнению с одним применением α1-блокатора и др. (“The New England Journal of Medicine”, 2003, Vol. 349, p. 2387-2398), поэтому применение ингибиторов 5α-редуктазы недавно было рассмотрено снова.
Считалось, что DHT в простате получается под действием 5α-редуктазы из тестостерона, который образуется в яичках и выделяется эндокринологически в простате. Недавно, однако, было опубликовано, что примерно половина DHT и его предшественник, тестостерон, в простате синтезируются из дегидроэпиандростерона (DHEA) - стероида, получаемого из надпочечника, в клетках простаты (“Frontier in Neuroendocrinology”, 2001, Vol. 22, p. 185-212). Данный тип системы продуцирования полового гормона в клетках целевых органов половых гормонов называется интракринологией.
Для ингибиторов 5α-редуктазы трудно ингибировать локальный синтез тестостерона (интракринный синтез тестостерона) в простате. Например, было опубликовано, что концентрация DHT в простате пациентов с BHP была снижена после введения финастерида, ингибитора 5α-редуктазы, приблизительно до 20% концентрации перед введением, в то время как концентрация тестостерона, предшественника, в простате была обратимо повышена в 4 раза (“The Journal of Urology”, 1999, Vol. 161, p. 332-337). Это означает, что хотя ингибитор 5α-редуктазы обладает эффектом по снижению концентрации DHT в простате, он не обладает эффектом по снижению концентрации тестостерона в простате и взамен повышает концентрацию. Так как тестостерон обладает активностью по связыванию андрогенного рецептора, равной примерно половине таковой у DHT, данное локальное повышение концентраций тестостерона в простате, как считают, должно быть частично ответственно за недостаточную лекарственную эффективность финастерида для BPH.
Антиандрогенные терапии с использованием хирургической кастрации и агонистов гонадотропин-высвобождающего гормона также применяются против рака простаты. Сообщается, что антиандрогенные терапии влияют на недостаточный эффект снижения концентраций тестостерона в простате. Например, у пациентов с раком простаты, которые получают антиандрогенную терапию, концентрация тестостерона в крови снижалась до 10% концентрации перед введением, в то время как концентрация DHT в простате оставалась равной примерно 50% (“The Journal of Clinical Endocrinology and Metabolism”, 1995, Vol. 80, p. 1066-1071). Это говорит о том, что концентрация тестостерона в простате также недостаточно снижена. Кроме того, андрогенные рецепторы локализовались в ядрах клеток также при раке простаты, рецидивирующем после антиандрогенной терапии (гормонально-резистентный рак простаты), и не наблюдалось значительной разницы между концентрацией тестостерона в тканях реккурентного рака простаты и концентрацией в нормальной простате (“Clinical Cancer Research”, 2004, Vol. 10, p. 440-448). Данные публикации наводят на мысль, что эффект снижения концентраций тестостерона в простате в существующих терапевтических методах является совершенно недостаточным для лечения реккурентного рака простаты и что подавление механизма синтеза тестостерона в простате, то есть интракринного синтеза тестостерона в простате, может представлять собой новую мишень для терапии рака простаты.
C учетом вышеописанных известных данных, поскольку ингибиторы интракринного синтеза тестостерона в простате обладают эффектом по снижению концентраций тестостерона в простате и не обладают эффектом по снижению концентраций тестостерона в крови, ожидают, что данные ингибиторы будут очень привлекательным средством для лечения ВРН и/или средством для лечения рака простаты, (1) которое может снижать не только концентрацию тестостерона, но также концентрацию DHT в простате и (2) которое может быть лишено вредных эффектов из-за подавления концентрации тестостерона, получаемого из яичек, в крови.
17β-гидроксистероид-дегидрогеназа (17βHSD) является важной для биосинтеза тестостерона. Существует несколько подтипов 17βHSD. 17βHSD типа 5 широко представлен в простате человека и, как было опубликовано, повышает проявление рака простаты и реккурентного рака простаты (“Steroids”, 2004, Vol. 69, p. 795-801; и “Cancer Research”, 2006, Vol. 66, p. 2815-2825). С другой стороны, почти весь тестостерон в крови производится с помощью 17βHSD типа 3 в яичках и экспрессия 17βHSD типа 3 редко наблюдается в других тканях, включая простату (“Nature Genetics”, 1994, Vol. 7, p. 34-39). Таким образом полагают, что 17βHSD типа 5 является ответственным за интракринный синтез тестостерона в простате и селективные ингибиторы 17βHSD типа 5 предполагаются для селективного подавления интракринного синтеза тестостерона в простате. Кроме того, так как вклад 17βHSD типа 5 отмечен также в эстрогензависимых тканях, таких как молочная железа и тому подобное, селективные ингибиторы, как ожидают, будут эффективны для эстрогензависимых заболеваний, таких как рак молочной железы и тому подобное (“Endocrine Reviews”, 2003, Vol. 24, p. 152-182). Кроме того, опубликовано, что AKR1C3 (другое название для 17βHSD типа 5), которая представляет собой подтип альдокеторедуктазы (AKR), метаболизирует полициклический ароматический углеводород (РАН) с образованием реакционноспособных кислородных соединений (ROS) (“The Journal of Biological Chemistry”, 2002, Vol. 277, No. 27, p. 24799-24808), и что единичный нуклеотидный полиморфизм (SNP) AKR1C3 гена, связанного с окислительным стрессом, коррелирует с риском рака легкого (“Carcinogenesis”, 2004, Vol. 25, No. 11, p. 2177-2181). То есть предполагается, что активность AKR1C3 в легких повышает риск рака легкого посредством генерирования ROS от PAH и ожидается, что селективные ингибиторы 17βHSD типа 5 будут эффективны для лечения рака легкого.
В качестве ингибиторов 17βHSD типа 5 известны стероидные производные (патентный документ 1) и NSAID (нестероидные противовоспалительные лекарственные средства), такие как флуфенаминовая кислота, индометацин и тому подобное (непатентный документ 1), производные коричной кислоты (непатентный документ 2) и тому подобное. Хотя механизм действия различный, производное индазола, представляющее собой соединение формулы (А), как известно, эффективно для ВРН (патентный документ 2).
[Хим. 1]
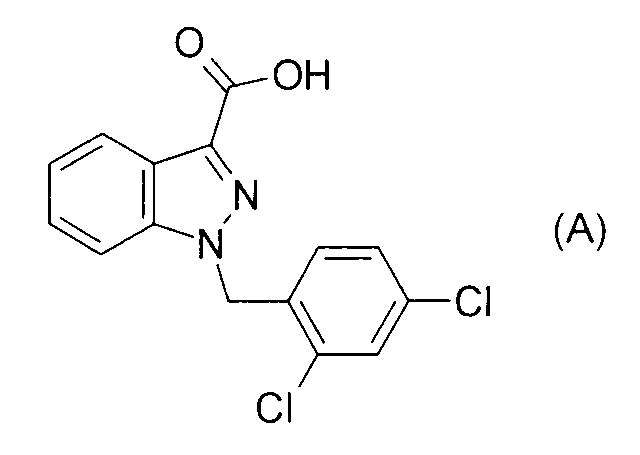
Согласно патентному документу 3 N-замещенные бензимидазоловые производные, включающие соединение формулы (В), обладают ингибирующим действием в отношении c-Kit онкогена и применимы для лечения рака простаты или тому подобного. Однако нет описания индолильной группы и также нет описания ингибирующего действия в отношении 17βHSD типа 5.
[Хим. 2]
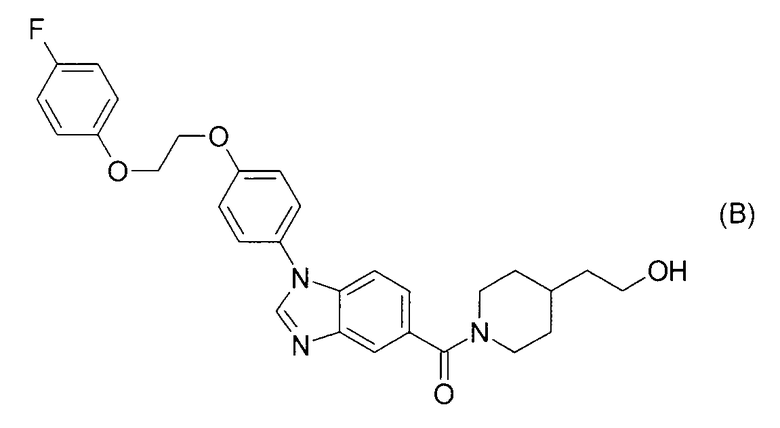
Согласно патентному документу 4 производные бензимидазола, включающие соединение формулы (С), обладают регулирующим действием на тирозинкиназу и применимы для лечения рака простаты или тому подобного. Однако не описана индолильная группа и также нет описания ингибирующего действия в отношении 17βHSD типа 5.
[Хим. 3]
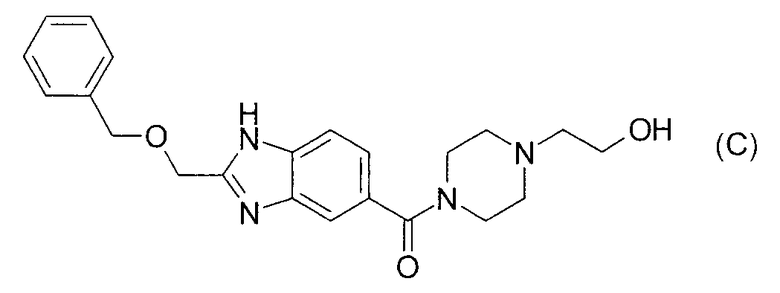
Согласно патентному документу 5 производные индола, включающие соединение формулы (D), обладают гистамин Н4 антагонистическим действием и применимы для лечения воспаления. Однако не описана неосновная (пиперидильная)алканольная структура и также нет описания ингибирующего действия в отношении 17βHSD типа 5 и эффективности в отношении BPH, рака простаты и тому подобного.
[Хим. 4]
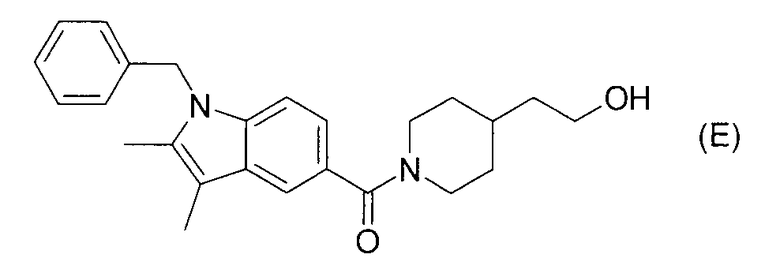
Согласно патентному документу 6 производные индола, включающие соединение формулы (Е), обладают регулирующим действием в отношении каннабиноидного рецептора и применимы для лечения цереброваскулярных нарушений и тому подобного. Однако также нет описания ингибирующего действия в отношении 17βHSD
типа 5 и эффективности в отношении BPH, рака простаты и тому подобного.
[Хим. 5]
[Патентный документ 1] Проспект международной публикации № WO99/046279
[Патентный документ 2] Проспект международной публикации № WO2004/064735
[Патентный документ 3] Проспект международной публикации № WO2005/021531
[Патентный документ 4] Проспект международной публикации № WO2007/056155
[Патентный документ 5] Проспект международной публикации № WO2002/072548
[Патентный документ 6] JP-A-2005-162657
[Непатентный документ 1] Cancer Research, 2004, Vol. 64, p. 1802-1810
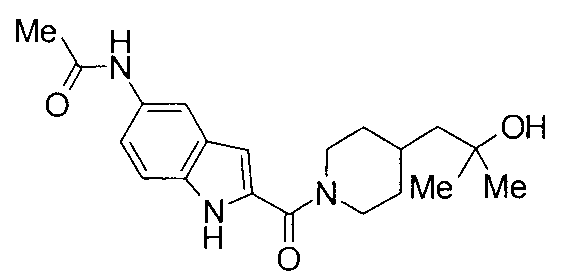
[Непатентный документ 2] Molecular and Cellular Endocrinology, 2006, Vol. 248, p. 233-235
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ПРОБЛЕМА, КОТОРУЮ ИЗОБРЕТЕНИЕ ПРЕДЛАГАЕТ ДЛЯ РЕШЕНИЯ
Задача данного изобретения состоит в том, чтобы предложить соединение, применимое в качестве лекарственного средства, обладающего селективной ингибирующей активностью в отношении 17βHSD типа 5, в частности в качестве средства для лечения доброкачественной гиперплазии простаты и/или рака простаты.
СПОСОБЫ ДЛЯ РЕШЕНИЯ ПРОБЛЕМЫ
В результате интенсивных изучений соединений, обладающих селективной ингибирующей активностью в отношении 17βHSD типа 5, авторы настоящего изобретения установили, что {1-[(индол-2-ил)карбонил]пиперидил}алканол-производное обладает потенциальной селективной ингибирующей активностью в отношении 17βHSD типа 5 и может быть средством для лечения и/или средством для предупреждения заболевания, связанного с 17βHSD типа 5, такого как доброкачественная гиперплазия простаты и рак простаты без сопутствующих вредных эффектов из-за снижения уровня тестостерона. Указанные результаты исследований были положены в основу настоящего изобретения.
То есть данное изобретение относится к соединению формулы (I) или его соли и к фармацевтической композиции, содержащей соединение формулы (I) или его соль и наполнитель.
[Хим. 6]
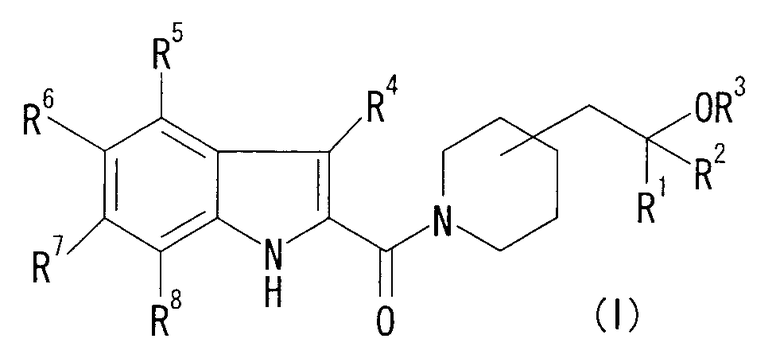
в формуле (I), R1, R2 и R3, которые являются одинаковыми или различными, представляют собой Н или низший алкил;
R4, R5, R6, R7 и R8, которые являются одинаковыми или различными, представляют собой Н, низший алкил, галоген, галоген-низший алкил, нитро, -Х-циклоалкил, который может быть замещенным, -Х-арил, который может быть замещенным, -Х-гетероциклическую группу, которая может быть замещенной, -Х-COOR0, -X-CONR10R11, -X-CN, -X-OR0, -X-SR0, -X-S(O)-низший алкил, -X-S(O)2-низший алкил, -Х-NR10R11, -X-NR0C(O)R10, -X-NR0C(O)OR10, -X-NR0C(O)NR10R11, -X-NR0S(O)2R10, -X-O-галоген-низший алкил, -Х-О-Х-циклоалкил, который может быть замещенным, -Х-О-Х-арил, который может быть замещенным, -Х-О-Х-гетероциклическую группу, которая может быть замещенной, или -Х-О-низший алкилен-OR0; или
R6 и R7 объединены с образованием -О-низший алкилен-О-;
R0, который является одинаковым или различным, представляет собой Н или низший алкил;
R10 и R11, которые являются одинаковыми или различными, представляют собой Н, низший алкил, галоген-низший алкил, -Х-циклоалкил, -Х-арил, или -Х-гетероциклическую группу; или
R10 и R11 вместе с N, к которому они присоединены, образуют насыщенную гетероциклическую группу, которая может быть замещенной; и
Х, который является одинаковым или различным, представляет собой связь или низший алкилен.
В данном описании символы, указанные выше, использованы для того, чтобы, если не оговорено особо, изображать одинаковые значения.
Кроме того, данное изобретение относится к фармацевтической композиции для лечения и/или предупреждения заболевания, связанного с 17βHSD типа 5, причем композиция содержит соединение формулы (I) или его соль, а именно средство для предупреждения и/или средство для лечения заболевания, связанного с 17βHSD типа 5, содержащее соединение формулы (I) или его соль.
Кроме того, данное изобретение относится к применению соединения формулы (I) или его соли для изготовления фармацевтической композиции для лечения и/или предупреждения заболевания, связанного с 17βHSD типа 5.
Кроме того, данное изобретение относится к способу лечения и/или предупреждения заболевания, связанного с 17βHSD типа 5, причем способ включает введение пациенту эффективного количества соединения формулы (I) или его соли.
Кроме того, данное изобретение относится к ингибитору 17βHSD типа 5, содержащему соединение формулы (I) или его соль.
Кроме того, данное изобретение относится к способу приготовления фармацевтической композиции для предупреждения или лечения заболевания, связанного с 17βHSD типа 5, причем способ включает смешивание соединения формулы (I) или его соли и фармацевтически приемлемого носителя, растворителя или эксципиента.
Кроме того, данное изобретение относится к упаковке для продажи, включающей фармацевтическую композицию, содержащую соединение формулы (I) или его соль; и инструкцию, в соответствии с которой соединение формулы (I) или его соль могут быть использованы или должны использоваться для лечения и/или предупреждения заболевания, связанного с 17βHSD типа 5.
ЭФФЕКТ ИЗОБРЕТЕНИЯ
Соединение формулы (I) селективно ингибирует 17βHSD типа 5. В соответствии с этим соединение формулы (I) может быть использовано как средство для предупреждения и/или лечения заболевания, связанного с 17βHSD типа 5. Например, оно может быть использовано как средство для предупреждения и/или лечения заболевания, связанного с андрогеном, так как синтез андрогена подавлен ингибированием 17βHSD типа 5.
НАИЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Ниже в настоящем документе данное изобретение будет описано более подробно.
В определениях данного описания “алкил” и “алкилен” обозначают линейную или разветвленную углеводородную цепь, если, в частности, не оговорено особо.
“Низший алкил” обозначает алкил, содержащий 1-6 атомов углерода (ниже в настоящем описании обозначенный как “С1-6”), и его примеры включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутильную, н-пентильную, н-гексильную группу и тому подобное. В другом варианте осуществления он представляет собой С1-4алкил. В еще другом варианте осуществления он представляет собой метил, этил, н-пропил, изопропил или трет-бутил.
“Низший алкилен” обозначает С1-6алкилен, и его примеры включают метиленовую, этиленовую, триметиленовую, тетраметиленовую, пентаметиленовую, гексаметиленовую, пропиленовую, метилметиленовую, этилэтиленовую, 1,2-диметилэтиленовую, 1,1,2,2-тетраметилэтиленовую группу, и тому подобное. В другом варианте осуществления он представляет собой С1-5алкилен. В еще другом варианте осуществления он представляет собой метилен, этилен, триметилен, тетраметилен или пентаметилен.
“Галоген” обозначает F, Cl, Br или I.
“Галоген-низший алкил” обозначает низший алкил, замещенный одним или несколькими галогенами. В другом варианте осуществления он представляет собой низший алкил, замещенный 1-5 атомами галогена. В еще другом варианте осуществления он представляет собой трифторметил.
“Циклоалкил” представляет собой С3-10 насыщенную углеводородную циклическую группу, в которой может быть мостик. Примеры его включают циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклогептильную, циклооктильную, бицикло[2.2.1]гептильную, адамантильную группу и тому подобное. В другом варианте осуществления он представляет собой С3-8циклоалкил. В еще другом варианте осуществления он представляет собой циклопропил, циклобутил, циклопентил или циклогексил.
“Арил” представляет собой С6-14 от моноциклической до трициклической ароматическую углеводородную циклическую группу, которая содержит циклическую группу, конденсированную с С5-8 циклоалкеном на его участке с двойной связью. Его примеры включают фенильную, нафтильную, тетрагидронафталенильную, инденильную, флуоренильную группу и тому подобное. В другом варианте осуществления он представляет собой фенил или нафтил. В еще другом варианте осуществления он представляет собой фенил.
“Гетероциклическая” группа обозначает циклическую группу, выбираемую из i) 3-8-членной моноциклической, и в другом варианте осуществления из 5-7-членного моноциклического гетероцикла, содержащего 1-4 гетероатома, выбираемого из О, S и N, и из ii) от бициклического до трициклического гетероцикла, содержащего 1-5 гетероатомов, выбираемых из О, S и N, который образован циклической конденсацией моноциклического гетероцикла с одним или двумя циклами, выбираемыми из группы, состоящей из моноциклического гетероцикла, бензольного кольца, С5-8циклоалкана и С5-8циклоалкена. Кольцевой атом, S или N, может быть окислен для образования оксида или диоксида. Кроме того, он может быть соединен мостиком или может образовать спироцикл.
Примеры “гетероциклической” группы включают азиридинил, азетидил, пирролидинил, пиперидил, азепанил, пиперазинил, гомопиперазинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, гомоморфолинил, тиоморфолинил, пирролил, индолил, имидазолил, пиразолил, пиридил, пиримидинил, пиразинил, триазолил, тетразолил, фурил, тиенил, оксазолил, изоксазолил, оксадиазолил, тиазолил, тиадиазолил, бензимидазолил, хинолил, хиназолил, хиноксалинил, бензофуранил, бензотиофенил, бензоксазолил, бензтиазолил, карбазолил, индолинил, тетрагидрохинолинил, тетрагидроизохинолинил, хинуклидинил, дибензофуранилгруппу, и тому подобное.
В другом варианте осуществления она представляет собой моноциклическую или бициклическую 5-10-членную гетероциклическую группу.
В еще другом варианте осуществления она представляет собой пирролидинил, пиперидил, азепанил, пиперазинил, гомопиперазинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, гомоморфолинил, тиоморфолинил, индолил, имидазолил, пиридил, пиримидинил, пиразинил, триазолил, тетразолил, фурил, тиенил, оксазолил, изооксазолил, тиадиазолил, бензимидазолил, тетрагидрохинолинил, тетрагидроизохинолинил или дибензофуранил.
“Насыщенная гетероциклическая” группа обозначает, среди вышеприведенной “гетероциклической” группы, группу, в которой все циклобразующие связи представляют собой одинарные связи.
Примеры “насыщенной гетероциклической” группы включают пирролидинил, пиперидил, азепанил, пиперазинил, гомопиперазинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, гомоморфолинил, тиоморфолинилгруппу и тому подобное.
Выражение “который может быть замещенным” в данном описании обозначает незамещенный или замещенный 1-5 заместителями. Кроме того, если имеются многочисленные заместители, то заместители могут быть одинаковыми или различными.
Заместители для “циклоалкила, который может быть замещенным”, “арила, который может быть замещенным”, или “гетероциклической группы, которая может быть замещенной”, в R4, R5, R6, R7 и R8 включают, например, низший алкил, галоген, галоген-низший алкил, нитро, -X-CN, -X-OR0, -X-SR0, -X-S(O)-низший алкил, -X-S(O)2-низший алкил, -Х-NR10R11, -X-NR0C(O)R10, -X-NR0C(O)OR10, -X-NR0C(O)NR10R11, -X-NR0S(O)2R10, -X-O-галоген-низший алкил или -Х-О-низший алкилен-OR0. В другом варианте осуществления их примеры включают группы, выбираемые из низшего алкила, галогена, галоген-низшего алкила, -CN, и OR0. В еще другом варианте осуществления их примеры включают группы, выбираемые из метила, этила, F, Cl, трифторметила и метокси.
Хотя “циклоалкил”, “фенил”, “циклогексил” и тому подобное описаны как моновалентные группы в данном описании для удобства, они могут представлять собой поливалентные группы, дивалентные или более высокой валентности согласно их структурам. Данное изобретение охватывает эти структуры. Определенные варианты осуществления дивалентных групп соответствуют группам, имеющим суффиксы вышеприведенных кольцевых групп, превращенных в диил в соответствии с номенклатурой органической химии. Например, дивалентная группа, соответствующая фенильной группе, которая является моновалентной группой, представляет собой фенилен.
“Селективный ингибитор 17βHSD типа 5” обозначает ингибитор, в котором ингибирующая активность в отношении 17βHSD типа 3 проявляется 3-кратной или более, предпочтительно 10-кратной или более и более предпочтительно 100-кратной или более величиной относительно ингибирующей активности в отношении человеческого 17βHSD типа 5 (AKK1C3) исходя из величины IC50.
Вариант осуществления соединения формулы (I) данного изобретения будет описан ниже.
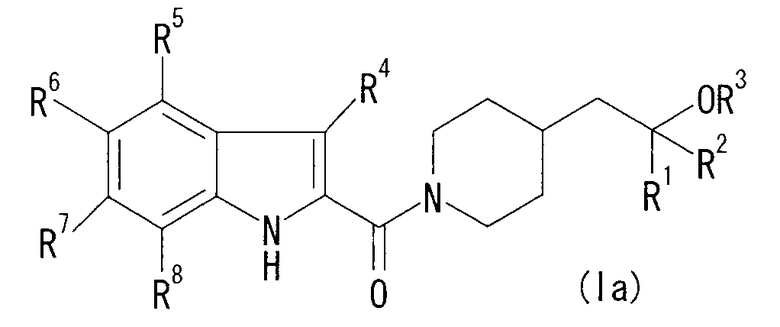
(1) Соединение формулы (Ia)
[Хим.7]
(2) Соединение, в котором R1 означает Н.
(3) Соединение, в котором R1 означает низший алкил.
(4) Соединение, в котором R1 означает метил.
(5) Соединение, в котором R2 означает Н.
(6) Соединение, в котором R2 означает низший алкил.
(7) Соединение, в котором R2 означает метил.
(8) Соединение, в котором R3 означает Н.
(9) Соединение, в котором R3 означает низший алкил.
(10) Соединение, в котором R4 означает Н, низший алкил, галоген или -О-низший алкил.
(11) Соединение, в котором R4 означает Н.
(12) Соединение, в котором R4 означает низший алкил.
(13) Соединение, в котором R4 означает галоген.
(14) Соединение, в котором R4 означает -О-низший алкил.
(15) Соединение, в котором R5 означает Н, низший алкил, галоген или -О-низший алкил.
(16) Соединение, в котором R5 означает Н.
(17) Соединение, в котором R5 означает низший алкил.
(18) Соединение, в котором R5 означает галоген.
(19) Соединение, в котором R5 означает -О-низший алкил.
(20) Соединение, в котором R6 означает Н, низший алкил, галоген, галоген-низший алкил, нитро, циклоалкил, ОН или -О-низший алкил.
(21) Соединение, в котором R6 означает низший алкил, галоген или -О-низший алкил.
(22) Соединение, в котором R6 означает Н.
(23) Соединение, в котором R6 означает низший алкил.
(24) Соединение, в котором R6 означает галоген.
(25) Соединение, в котором R6 означает галоген-низший алкил.
(26) Соединение, в котором R6 означает нитро.
(27) Соединение, в котором R6 означает циклоалкил.
(28) Соединение, в котором R6 означает ОН.
(29) Соединение, в котором R6 означает -О-низший алкил.
(30) Соединение, в котором R7 означает Н, низший алкил, галоген или -О-низший алкил.
(31) Соединение, в котором R7 означает Н.
(32) Соединение, в котором R7 означает низший алкил.
(33) Соединение, в котором R7 означает галоген.
(34) Соединение, в котором R7 означает -О-низший алкил.
(35) Соединение, в котором R8 означает Н, низший алкил, галоген или -О-низший алкил.
(36) Соединение, в котором R8 означает Н.
(37) Соединение, в котором R8 означает низший алкил.
(38) Соединение, в котором R8 означает галоген.
(39) Соединение, в котором R8 означает -О-низший алкил.
(40) Соединение, которое представляет собой комбинацию любых двух или более групп, описанных в вышеприведенных пунктах (1)-(39).
Конкретные примеры соединения вышеприведенного пункта (40) включают следующие соединения.
(41) Соединение, описанное в (1), в котором R1 и R3 означают Н.
(42) Соединение, описанное в (41), где R4, R5, R7 и R8, которые являются одинаковыми или различными, означают Н, низший алкил, галоген или -О-низший алкил, R6 означает Н, низший алкил, галоген, галоген-низший алкил, нитро, циклоалкил, ОН или -О-низший алкил.
(43) Соединение, описанное в (1), в котором R1 и R2 означают низший алкил, и R3 означает Н.
(44) Соединение, описанное в (43), где R4, R5, R7 и R8, которые являются одинаковыми или различными, означают Н, низший алкил, галоген или -О-низший алкил.
(45) Соединение, описанное в (43) или (44), где R6 означает Н, низший алкил, галоген, галоген-низший алкил, нитро, циклоалкил, ОН или -О-низший алкил.
(46) Соединение, описанное в (43)-(45), где R6 означает низший алкил, галоген или -О-низший алкил.
Другой вариант осуществления соединения формулы (I) данного изобретения будет описан ниже.
(47) Соединение, описанное в (1), где R1, R2, R3 и R4, которые являются одинаковыми или различными, означают Н или низший алкил, R5 означает Н, низший алкил, галоген или -О-низший алкил, R6 означает Н, низший алкил, галоген, ОН, -О-низший алкил, -О-низший алкиленфенил, -О-галоген-низший алкил, нитро, амино, -амино-С(О)-низший алкил или пирролил, R7 означает Н, галоген, ОН, или -О-низший алкил, или R6 и R7 объединены, чтобы образовать -О-низший алкилен-О-, и R8 означает Н или галоген.
(48) Соединение, описанное в (47), в котором R1 означает низший алкил.
(49) Соединение, описанное в (48), в котором R1 означает метил.
(50) Соединение, описанное в (47), в котором R2 означает низший алкил.
(51) Соединение, описанное в (50), в котором R2 означает метил.
(52) Соединение, описанное в (47), в котором R3 означает Н.
(53) Соединение, описанное в (47), в котором R4 означает Н.
(54) Соединение, описанное в (47), в котором R5 означает Н, Cl или метил.
(55) Соединение, описанное в (47), в котором R6 означает Н, Cl, метил, метокси или нитро.
(56) Соединение, описанное в (47), в котором R7 означает Н.
(57) Соединение, описанное в (47), в котором R8 означает Н или Cl.
(58) Соединение, которое представляет собой комбинацию из двух или более групп, описанных в вышеприведенных (48)-(57).
Конкретные соединения, охваченные данным изобретением, включают следующие соединения.
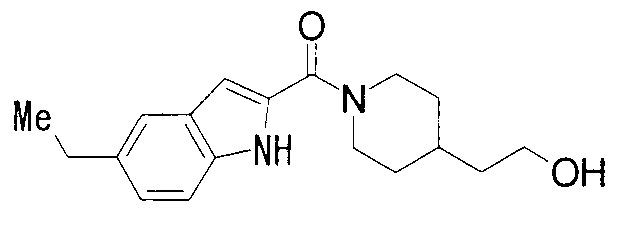
2-{1-[(5-метил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}этанол,
1-[1-(1Н-индол-2-илкарбонил)пиперидин-4-ил]пропан-2-ол,
1-[1-(1Н-индол-2-илкарбонил)пиперидин-4-ил]-2-метилпропан-2-ол,
2-метил-1-{1-[(4-метил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ол,
2-метил-1-{1-[(5-метил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ол,
1-{1-[(3,5-диметил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-трет-бутил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(4-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
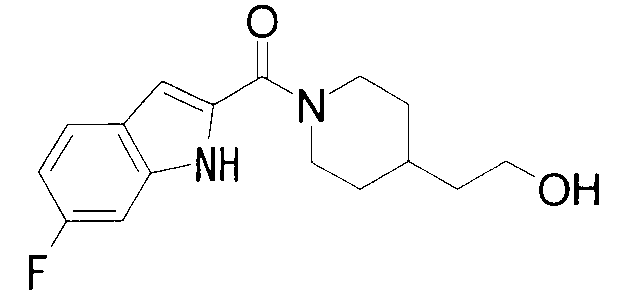
1-{1-[(5-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(4-хлор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-хлор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-бром-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(7-хлор-5-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
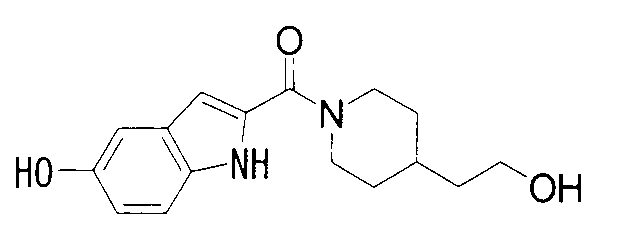
2-{[4-(2-гидрокси-2-метилпропил)пиперидин-1-ил]карбонил}-1Н-индол-5-ол,
1-{1-[(4-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(6-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
2-метил-1-(1-{[(5-(трифторметокси)-1Н-индол-2-ил]карбонил}пиперидин-4-ил)пропан-2-ол, и
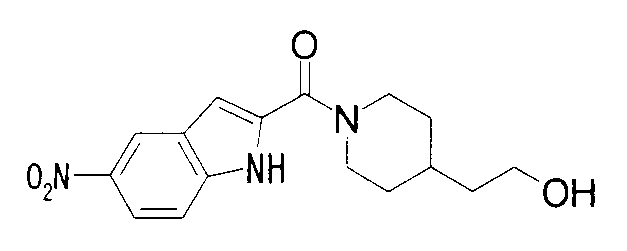
2-метил-1-{1-[(5-нитро-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ол.
Соединение формулы (I) в некоторых случаях может существовать в форме таутомеров или геометрических изомеров в зависимости от вида заместителей. В настоящем описании соединение формулы (I) может быть представлено только в одной форме изомеров, но данное изобретение включает другие изомеры, а также выделенные формы или их смеси.
Кроме того, соединение формулы (I) может иметь асимметрические атомы углерода или аксиальные асимметрии в некоторых случаях и, соответственно, оно может существовать в форме оптических изомеров. Данное изобретение также включает изоляты или смеси оптических изомеров соединения формулы (I).
Кроме того, данное изобретение включает фармацевтически приемлемое пролекарство соединения формулы (I). Фармацевтически приемлемое пролекарство представляет собой соединение, содержащее группу, которую можно превратить в аминогруппу, гидроксильную группу, карбоксильную группу или тому подобное сольволизом или при физиологическом состоянии. Примеры группы, которая образует пролекарство, включает группы, описанные, например, в Prog. Med., 5, 2157-2161 (1985)
или “Pharmaceutical Research and Development” (Hirokawa Publishing Company, 1990), Vol. 7, “Drug Design”, pp. 163-198.
Кроме того, соль соединения формулы (I) представляет собой фармацевтически приемлемую соль соединения формулы (I) и может образовать кислотно-аддитивную соль или соль с основанием в зависимости от типа заместителей. В частности, их примеры включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота, или с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, миндальная кислота, винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, аспарагиновая кислота и глутаминовая кислота, соли с неорганическими основаниями, такие как соли натрия, калия, магния, кальция и алюминия, или с органическими основаниями, такими как соли с метиламином, этиламином, этаноламином, лизином и орнитином, соли с различными аминокислотами и производными аминокислот, такими как соли ацетиллейцина, аммония, и тому подобные.
Кроме того, данное изобретение также включает различные гидраты или сольваты и полиморфные кристаллические вещества соединения формулы (I) и их соли. Кроме того, данное изобретение также включает соединения, меченные различными радиоактивными или нерадиоактивными изотопами.
СПОСОБЫ ПОЛУЧЕНИЯ
Соединение формулы (I) и его соль могут быть получены за счет использования свойств, основанных на типах его основного скелета или заместителей, и применением различных известных синтетических методов. В настоящее время в некоторых случаях исходя из методик получения эффективным является замена функциональной группы соответствующей защитной группой (группой, которая легко может быть превращена в функциональную группу) на стадии исходного продукта до промежуточного в зависимости от типа функциональной группы в процессе получения. Примеры таких функциональных групп включают аминогруппу, гидроксильную группу, карбоксильную группу и тому подобное, и примеры таких защитных групп включают защитные группы, описанные, например, в “Protective Groups in Organic Synthesis (the third edition, 1999)” edited by Greene and Wuts и тому подобном, которые могут быть соответствующим образом выбраны и использованы в зависимости от условий реакции. В данных методах требуемое соединение может быть получено введением защитной группы и выполнением реакции и затем удалением защитной группы, если желательно.
Кроме того, пролекарство соединения формулы (I) может быть получено тем же способом, как и в случае защитных групп, выполнением реакции после введения определенной группы на стадии от исходных продуктов до промежуточных продуктов или применением полученного соединения формулы (I). Реакция может быть осуществлена применением методов, известных специалистам в данной области, таких как этерификация, амидирование, дегидратация и тому подобное.
Далее в настоящем описании будут представлены наглядные способы получения соединения формулы (I). Каждый из способов получения может быть также выполнен с отнесением к ссылкам, прилагаемым к настоящему описанию. Кроме того, способы получения данного изобретения не ограничены примерами, показанными ниже.
[Хим. 8]
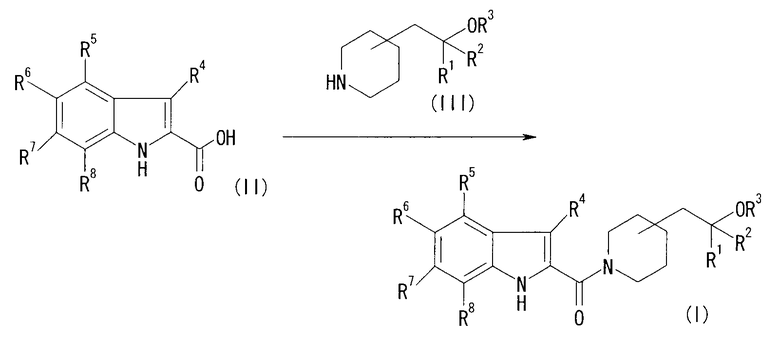
Соединение формулы (I) может быть получено взаимодействием соединения (II) c соединением (III).
Реакцию можно проводить, используя соединение (II) и соединение (III) в эквивалентных количествах или также их в избыточном количестве в присутствии конденсирующего агента в условиях от охлаждения до нагревания, предпочтительно от -20°С до 60°С, обычно с перемешиванием в течение от 0,1 часа до 5 суток, в растворителе, который является инертным для реакции.
В данном случае выбор растворителя не особенно ограничен, но его примеры включают ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; N,N-диметилформамид, диметилсульфоксид, этилацетат, ацетонитрил, вода или их смесь.
Примеры конденсирующего агента включают 1-этил-3-(3-диметиламинопропил)карбодиимид или его гидрохлорид, дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, дифенилфосфорилазид, оксихлорид фосфора и тому подобное, но не ограничены только ими. В некоторых случаях для реакции может быть полезно, например, использовать добавку (например, 1-гидроксибензотриазол или тому подобное). В некоторых случаях может быть полезно для равномерного протекания реакции проводитьреакцию в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, или неорганического основания, такого как карбонат калия, карбонат натрия или гидроксид калия.
Кроме того, может быть также использован метод, в котором соединение карбоновой кислоты (II) превращают в его реакционноспособное производное и затем подвергают взаимодействию с соединением (III). Примеры реакционноспособного производного карбоновой кислоты включают галогенангидрид, полученный взаимодействием с галогенирующим агентом, таким как оксихлорид фосфора или тионилхлорид, смешанный ангидрид кислоты, полученный взаимодействием с изобутилхлорформиатом, или тому подобное, и активный сложный эфир, полученный конденсацией с 1-гидроксибензотриазолом, или тому подобное. Реакция между данными реакционноспособными производными и соединением (III) может быть проведена в условиях от охлаждения до нагревания, предпочтительно от -20°С до 60°С, в растворителе, который является инертным для реакции, таком как галогенированные углеводороды, ароматические углеводороды или простые эфиры.
Кроме того, разные заместители на группах R1-R8 в соединении формулы (I) могут быть легко превращены в другие функциональные группы применением соединения формулы (I) как исходного продукта и применением реакций, описанных в следующих примерах, реакций, очевидных для специалистов в данной области, или их модифицированных методов. Например, такие реакции могут быть осуществлены любой комбинацией способов, которые могут традиционно использоваться специалистами в данной области, например способов О-алкилирования, N-алкилирования, восстановления, гидролиза, амидирования и тому подобного. Их примеры будут описаны ниже.
Соединение формулы (I), содержащее аминогруппу, может быть получено восстановлением нитрогруппы, например, с отсылкой к методу, описанному в “The fourth edition of Courses in Experimental Chemistry (Vol. 26)” edited by the Chemical Society of Japan, Maruzen, 1992.
Соединение формулы (I), содержащее амидогруппу, может быть получено ацилированием аминогруппы, например, с отсылкой к методам, описанным в “The fourth edition of Courses in Experimental Chemistry (Vol. 22)” edited by the Chemical Society of Japan, Maruzen, 1992 и “The fifth edition of Courses in Experimental Chemistry (Vol. 16)” edited by the Chemical Society of Japan, Maruzen, 2005; или “Compendium of Organic Synthetic Methods”, Vols. 1-3.
Соединение формулы (I), содержащее о-дигидроксифенильную группу, может быть получено расщеплением 1,3-диоксоланового кольца, например, с отсылкой к методу, описанному в “Journal of Medicinal Chemisrty”. 2001, 44, 1794-1801.
Соединение формулы (I), содержащее фенильную группу, замещенную пирролильной группой, может быть получено реакцией с образованием пиррольного кольца, например, с отсылкой к методу, описанному в “Tetrahedron Letters”, 1993, 34, 1929-1930.
(Получение исходного соединения)
Исходное соединение (II) может быть получено реакцией Фишера для синтеза индола, например, отсылкой к методу, описанному в “The Fischer Indole Synthesis” edited by Robinson, 1982. Исходное соединение (III), содержащее третичный спирт, может быть получено взаимодействием производного карбоновой кислоты (такого, как сложный эфир) с реактивом Гриньяра, например, с отсылкой к методу, описанному в “Synthesis” 1983, 12, 1030-1031.
[Фармакологический тест]
Отличная селективная ингибирующая активность в отношении человеческого 17βHSD типа 5 соединений данного изобретения была подтверждена тест-методами, описанными в разделах 1-3 ниже. Данный тест может быть выполнен отсылкой к подробностям тест-процедуры, описанной в издании Maniatis, T. et al., Molecular Cloning - A Laboratoty Manual Cold Spring Harbor Laboratory, NY (1982), и тому подобного. Кроме того, гены, кодирующие человеческий 17βHSD типа 5 и типа 3, описанные в разделах 1 и 2 ниже, и человеческий 17βHSD типа 5 и типа 3 могут быть получены методом, описанным в Molecular Endocrinology, 1997, 11(13), 1971-1984.
1. Выделение гена, кодирующего человеческий 17βHSD типа 5, и очистка фермента
кДНК полной длины, кодирующую человеческий 17βHSD тип 5, использованный в фармакологическом тесте данного изобретения, получали PCR методом, используя кДНК, произведенную из клеточной линии, полученной при раке легкого человека, А549 клеток в качестве матрицы. Нуклеотидную последовательность полученной кДНК анализировали методом дидезокситерминаторов и отбирали клон, совместимый с известной последовательностью человеческого 17βHSD типа 5 (номер NM_003739 доступа в GenBank). Escherichia coli BL21 трансформировали с плазмидой, содержащей кДНК, и культивировали в увеличенном масштабе. Белки очищали использованием GSTrapFF колонки (производимой Amersham) и PreScissionProtease (производимой Amersham). Метод очистки выполняли в соответствии с инструкциями, приложенными к GSTrapFF колонке.
2. Выделение гена, кодирующего человеческий 17βHSD типа 3, и очистка фермента
кДНК полной длины, кодирующую человеческий 17βHSD типа 3, использованный в фармакологическом тесте данного изобретения, получали PCR методом, используя кДНК, полученную из человеческих яичек, в качестве матрицы. Нуклеотидную последовательность полученной кДНК анализировали методом дидезокситерминаторов и отбирали клон, совместимый с известной последовательностью человеческого 17βHSD типа 3 (номер ВС034281 доступа в GenBank). Затем клеточную линию, полученную из почки плода человека, 293 клетки, трансформировали с плазмидой, содержащей кДНК, и клетки собирали спустя 24 часа. Собранные клетки затем разрывали в фосфатном буферном растворе, содержащем 5% глицерина (500 мкл на 100-мм планшет фосфатного буферного раствора (рН 7,4, 200 мМ), содержащего 5% глицерина) и центрифугировали (16000 об/мин, 5 мин, 4°С), и супернатант использовали в качестве источника фермента.
3. Измерение ферментативных активностей человеческого 17βHSD типа 5 и типа 3
Активность фермента измеряли, обращаясь к публикации Trevor M. Penning, et al., Biochem. J., 351, 67-77, (2000). В частности, используя 100 мМ буфер фосфата калия (рН 6,0), (1) фермент, очищенный в вышеприведенном разделе 1 в конечной концентрации 10 мкг/мл, (2) андростенедион в конечной концентрации 300 нМ, (3) NADPH в конечной концентрации 200 нМ и (4) тестируемое соединение смешивали для взаимодействия при комнатной температуре в течение 2 часов и затем измеряли количество полученного тестостерона, применяя DELFIA (зарегистрированная торговая марка) тестостероновые реагенты R050-201 (производимые PerkinElmer). Измерения выполняли в соответствии с приложенными инструкциями. Степень снижения образования тестостерона в присутствии соединения получали как относительное значение относительно количества тестостерона в отсутствие системы фермента в 0% и количества тестостерона, полученного в отсутствие системы соединения в 100%. Затем рассчитывали величины IC50 методом логистической регрессии.
Кроме того, в качестве модели in vitro, близкой к живущему организму, активность вышеприведенных ферментов может быть измерена использованием клетки, которая экспрессирует человеческий 17βHSD типа 5 или тому подобное.
Кроме того, LNCaP клетки, экспрессирующие человеческий 17βHSD типа 5, создавали из клеточной линии, происходящей от рака простаты человека, LNCaP клетки, и оценивали активность по ингибированию роста клеток под действием соединения данного изобретения.
4. Создание LNCaP клеток, экспрессирующих человеческий 17βHSD типа 5
LNCaP клетки клеточной линии, происходящей от рака простаты человека, трансформировали с плазмидой, содержащей клон, выбранный в вышеприведенном разделе 1, и затем получали клеточную линию, показывающую стабильную экспрессию.
5. Измерение способности к росту клеток использованием LNCaP клеток, экспрессирующих человеческий 17βHSD типа 5
9000 клетки/лунка из трансформированных клеток, полученных в разделе 4 выше, высевали в 96-луночный планшет и культивировали в продолжении ночи. Затем туда добавляли андростендион в сочетании с тест-соединением в конечной концентрации 10 нМ с последующим культивированием в течение 7 суток. После культивирования считали число клеток, используя CellTiter-Glo (зарегистрированная торговая марка) в люминесцентном анализе жизнеспособности клеток (Promega). CellTiter-Glo (зарегистрированная торговая марка) в люминесцентном анализе жизнеспособности клеток представляет собой реагент, который измеряет число клеток мониторингом уровня внутриклеточной АТФ(АТР) от интенсивности люминесценции люциферазой. Экспериментальное манипулирование проводили согласно приложенным инструкциям. Активность по ингибированию роста клеток в присутствии тест-соединения рассчитывали как относительное значение относительно количества клеток в отсутствие системы андростендиона при пролиферации в 0% и количества клеток в присутствии андростендиона и в отсутствие системы тест-соединения при пролиферации в 100%. Затем рассчитывали величины IC50 методом логистической регрессии.
Таблица 1 показывает величины IC50 по ингибирующей активности в отношении человеческого 17βHSD типа 5 и типа 3 иллюстративных соединений, включенных в соединения данного изобретения, и величины IC50 ингибирующей активности роста клеток с использованием LNCaP клеток, экспрессирующих человеческий 17βHSD типа 5. Аббревиатура “Ex” обозначает номер примера.
Как показано вышеприведенными результатами тестирования, соединения формулы (I) почти не обладают ингибирующей активностью в отношении человеческого 17βHSD типа 3 и обладают ингибирующей активностью, селективной к человеческому 17βHSD типа 5.
В соответствии с этим соединение формулы (I) может быть использовано в качестве средства для предупреждения и/или лечения заболевания, связанного с 17βHSD типа 5. Например, оно может быть использовано в качестве средства для предупреждения и/или лечения заболевания, связанного с андрогеном, так как синтез андрогена подавляется ингибированием 17βHSD типа 5.
Примеры заболеваний, связанных с андрогеном, включают рак простаты, доброкачественную гиперплазию простаты, акне, себоррею, гирсутизм, плешивость, алопецию, раннее половое созревание, гипертрофию надпочечника, синдром поликистозного яичника, рак грудной железы, эндометриоз, лейомиому или тому подобное. Их примеры также включают заболевания, связанные с кислородным стрессом, такие как рак легкого.
Кроме того, поскольку 17βHSD типа 5, как считают, является ответственным для интракринного синтеза андрогена в простате, селективные ингибиторы 17βHSD типа 5 предполагаются для селективного подавления интракринного синтеза андрогена в простате. Поэтому соединение формулы (I) может быть использовано в качестве средства для предупреждения и/или лечения заболевания, связанного с андрогеном, в частности в простате, а именно рака простаты и доброкачественной гиперплазии простаты.
Кроме того, как показано вышеприведенными результатами тестирования, поскольку соединения формулы (I) обладают очень слабой ингибирующей активностью в отношении человеческого 17βHSD типа 3, они предполагаются для селективного подавления интракринного синтеза в простате за счет их селективных ингибирующих эффектов в отношении 17βHSD типа 5 без воздействия на биосинтез тестостерона, производимого из человеческого 17βHSD типа 3 в яичках. Иными словами, так как соединения формулы (I) не влияют на концентрацию тестостерона в крови, они могут быть использованы в качестве средства для лечения и/или предупреждения доброкачественной гиперплазии простаты и рака простаты без вредных эффектов, таких как половая дисфункция из-за снижения концентрации тестостерона в крови, и тому подобное.
Кроме того, как показано вышеприведенными результатами тестирования, поскольку соединения формулы (I) проявляют ингибирующую активность в отношении роста клеток на LNCaP клетках, экспрессирующих человеческий 17βHSD типа 5, они подавляют интракринный синтез тестостерона селективно при раке простаты за счет их селективных ингибирующих свойств в отношении 17βHSD типа 5, таким образом могут быть использованы для лечения и/или предупреждения рака простаты без вредных эффектов.
Кроме того, также полезной является упаковка для продажи, которая содержит вышеупомянутую фармацевтическую композицию и инструкцию, характеризующую вышеназванные эффекты.
Препарат, содержащий один или два, или более видов соединения формулы (I) или его соль в качестве активного ингредиента, может быть приготовлен в соответствии с обычно применяемым методом с использованием фармацевтического носителя, эксципиента или тому подобного, что обычно применяется в данной области.
Введение может быть выполнено в любой форме для перорального введения посредством таблеток, пилюль, капсул, гранул, порошков, жидких препаратов или тому подобного или для парентерального введения посредством инъекций, таких как внутрисуставная инъекция, внутривенная инъекция, внутримышечная инъекция или тому подобное, а также посредством суппозиториев, глазных капель, глазных мазей, жидких препаратов для введения через кожу, мазей, подкожных бляшек, трансмукозальных жидких препаратов, трансмукозальных бляшек, ингаляций и тому подобного.
В качестве твердых композиций для перорального введения согласно данному изобретению использованы таблетки, порошки, гранулы или тому подобное. В такой твердой композиции смешаны один или два, или более типов активных ингредиентов, по меньшей мере, с одним инертным эксципиентом, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон и/или алюминометасиликат магния. Согласно обычному методу композиция может содержать инертные добавки, такие как замасливатель, а именно стеарат магния, дезинтегратор, такой как натрийкарбоксиметилкрахмал, стабилизатор и средство, придающее растворимость. В случае необходимости таблетки или пилюли могут быть покрыты сахаром или пленкой из желудочных или энтеросолюбильных материалов.
Жидкие композиции для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры или тому подобное и содержат обычно применяемый инертный разбавитель, такой как очищенная вода или этанол. В дополнение к инертному разбавителю жидкая композиция может содержать адъювант, такой как средство, придающее растворимость, увлажняющее средство и суспендирующее средство, подсластитель, корригент, ароматизатор и антисептик.
Инъекции для парентерального введения включают стерильные водные и неводные растворы, суспензии или эмульсии. Водный растворитель включает, например, дистиллированную воду для инъекции или физиологический раствор. Примеры неводного растворителя включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, спирты, такие как этанол, полисорбат 80 (японская фармакопея) и тому подобное. Такая композиция может дополнительно содержать тонизирующее средство, антисептик, увлажнитель, эмульгатор, диспергатор, стабилизатор или средство, придающее растворимость. Их стерилизуют, например, фильтрованием через фильтр, задерживающий бактерии, подмешиванием стерилизующего средства или облучением. Кроме того, их также можно использовать приготовлением стерильной твердой композиции или растворением или суспендированием ее в стерильной воде или стерильном растворителе для инъекции перед применением.
Препараты для наружного применения включают мази, пластыри, кремы, желеобразные составы, содержащие лекарственное средство, припарки, спреи, лосьоны, глазные капли, глазные мази и тому подобное. Включены обычно используемые основы мазей, основы лосьонов, водные или неводные жидкости, суспензии, эмульсии и тому подобное. Примеры основ мазей или лосьонов включают полиэтиленгликоль, пропиленгликоль, белый вазелин, обесцвеченный пчелиный воск, полиоксиэтиленгидрированное касторовое масло, глицерилмоностеарат, стеариловый спирт, цетиловый спирт, лауромакрогол, сорбитансесквиолеат и тому подобное.
В качестве трансмукозальных препаратов, таких как препараты для ингаляций и трансназальные препараты, использованы твердые, жидкие или полутвердые формы и могут быть приготовлены в соответствии с традиционно известным методом. Например, к ним может быть добавлен известный эксципиент, а также средство для установления рН, антисептик, поверхностно-активное вещество, замасливатель, стабилизатор, загуститель соответственно или тому подобное. Для их введения может быть использовано соответствующее устройство для ингаляции или продувки. Например, соединение может быть введено само по себе или в виде порошка смеси, приготовленной как препарат, или в виде раствора или суспензии в комбинации с фармацевтически приемлемым носителем с применением обычно известного устройства или опрыскивателя, такого как устройство для ингаляции с дозированным введением. Устройства для ингаляции сухим порошком или тому подобным можно использовать для однократного или многократного введения, и может применяться сухой порошок или капсула, содержащая порошок. В альтернативном случае оно может быть в форме, такой как аэрозоль для опрыскивания под давлением, в котором используется соответствующее вещество-вытеснитель, например подходящий газ, такой как хлорфторалкан, гидрофторалкан или диоксид углерода и тому подобное.
При пероральном введении суточная доза обычно принимает значение от примерно 0,001 до 100 мг/кг, предпочтительно от 0,1 до 30 мг/кг и предпочтительнее от 0,1 до 10 мг/кг массы тела, введенная одной порцией или 2-4 разделенными порциями. В случае внутривенного введения суточная доза подходящим образом принимает значение от примерно 0,0001 до 10 мг/кг массы тела, один раз в сутки или два или более раз в сутки. Кроме того, трансмукозальное средство вводят в дозе от примерно 0,001 до 100 мг/кг массы тела, один раз в сутки или два, или более раз в сутки. Дозу соответственно выбирают в ответ на конкретный случай, принимая во внимание симптомы, возраст, пол и тому подобное.
Соединения формулы (I) могут быть использованы в комбинации c различными средствами для лечения или предупреждения заболеваний, для которых вышеупомянутые соединения формулы (I), как полагают, являются эффективными. Комбинированный состав может быть введен одновременно, или раздельно и непрерывно, или с желаемыми интервалами времени. Составы, предназначенные для совместного введения, могут представлять собой смесь или могут быть приготовлены отдельно.
ПРИМЕРЫ
Способы получения для соединений формулы (I), как активного ингредиента данного изобретения, будут описаны ниже в качестве примеров. Кроме того, способы получения для новых соединений наряду с соединениями, использованными как исходные продукты соединений формулы (I), будут описаны в качестве примеров получения. Способы получения для соединений формулы (I) не ограничены способами получения в конкретных примерах, приведенных ниже, и могут быть выполнены комбинацией данных способов получения или известных способов получения.
Следующие примеры описаны для того, чтобы подробнее объяснить данное изобретение, и данное изобретение не ограничено последующими примерами. Хотя данное изобретение полностью объяснено с помощью примеров, следует подразумевать, что специалисты в данной области примут во внимание, что естественно могут быть произведены различные изменения и модификации. Соответственно, данные изменения и модификации включены в данное изобретение, если они не отступают от области настоящего изобретения.
Следующие сокращения использованы в примерах получения, примерах и таблицах ниже.
Ex: номер примера, REx: номер примера получения, No: номер соединения, mp/т.пл.: температура плавления, data:физико-химические данные (FAB+: FAB-MS (M+H)+, FAB-: FAB-MS (M-H)-, ESI+: ESI-MS (M+H)+, ESI-: ESI-MS (M-H)-, API+: API-ES-MS (M+H)+, EI: EI-MS (M)+, CI: CI-MS (M+H)+, NMR-DMSOd6/ЯМР-ДМСОd6: δ (м.д.) пика(ов) в 1Н ЯМР в DMSO-d6), Str: структурная формула, Syn (REx): номера примеров получения, в которых соответствующие соединения получены при использовании одинакового метода, Syn (Ex): номера примеров, в которых соответствующие соединения получены при использовании одинакового метода, DME: диметоксиэтан, DMF/ДМФА: диметилформамид, DMSO/ДМСО: диметилсульфоксид, THF/ТГФ: тетрагидрофуран, 4M HCl/EtOAc: 4 моль/л раствор хлористоводородная кислота-этилацетат, MeCN: ацетонитрил, МеОН: метанол, tBuOH: трет-бутиловый спирт, RT: время удерживания (минуты) в ВЭЖХ/HPLC.
Пример получения 1
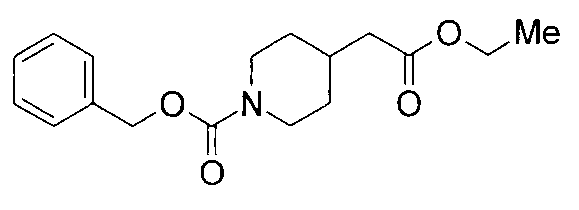
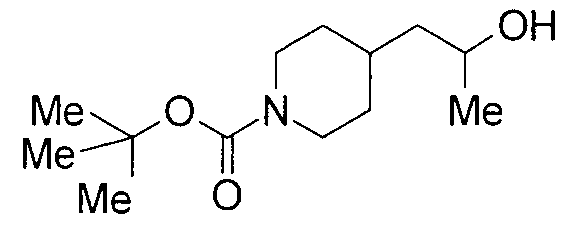
3 г бикарбоната натрия добавляли к раствору 5 г этилпиперидин-4-илацетата в 50 мл диоксана и 50 мл воды при 0°С, и к содержимому добавляли по каплям 4,6 мл бензилхлорформиата с последующим перемешиванием при комнатной температуре в течение 3 суток. Реакционную жидкость концентрировали до половины объема при пониженном давлении с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением 8,9 г бензил 4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилата в виде бесцветного маслянистого вещества.
Пример получения 2
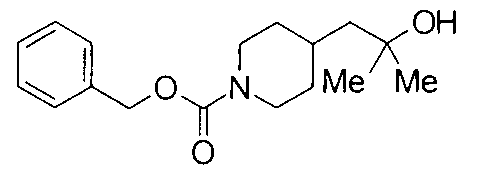
К раствору 8,9 г бензил 4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилата в 100 мл ТГФ добавляли при 0°С 46 мл 1,4 М метилмагнийбромида в толуоле-ТГФ с последующим перемешиванием при комнатной температуре в течение 3 часов. К реакционной жидкости добавляли 1 М водный раствор хлорида аммония с последующей экстракцией этилацетатом. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением 8,5 г бензил 4-(2-гидрокси-2-метилпропил)пиперидин-1-карбоксилата в виде бесцветного маслянистого вещества.
Пример получения 3
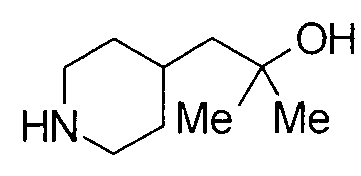
К раствору 8,5 г бензил 4-(2-гидрокси-2-метилпропил)пиперидин-1-карбоксилата в 120 мл метанола добавляли 500 мг 10% палладий-уголь с последующим перемешиванием в атмосфере водорода при комнатной температуре в течение 1 суток. Нерастворимый продукт удаляли фильтрованием через целит и фильтрат концентрировали при пониженном давлении с получением 5,6 г 2-метил-1-(пиперидин-4-ил)-2-пропанола в виде белого вещества.
Пример получения 4
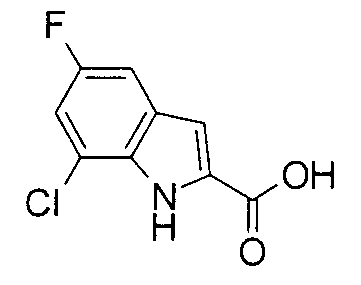
К раствору 250 мг этил 7-хлор-5-фтор-1Н-индол-2-карбоксилата в 2 мл метанола добавляли 5 мл 1 М водного раствора гидроксида натрия с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционную жидкость приводили в подкисленное состояние добавлением 1 М водного раствора хлористоводородной кислоты с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением 210 мг 7-хлор-5-фтор-1Н-индол-2-карбоновой кислоты в виде белого вещества.
Пример получения 5
К раствору 250 мг трет-бутил 4-(2-оксоэтил)пиперидин-1-карбоксилата в 5 мл ТГФ добавляли 1,2 мл 1,4 М метилмагнийбромида в толуоле-ТГФ с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] с получением 236 мг трет-бутил 4-(2-гидроксипропил)пиперидин-1-карбоксилата в виде бесцветного маслянистого вещества.
Пример 1
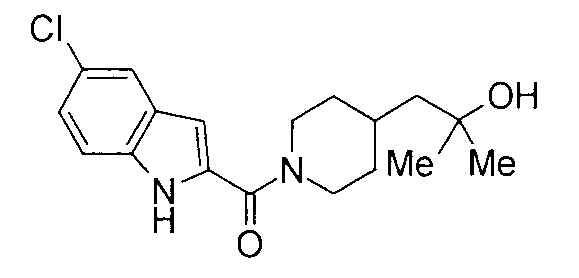
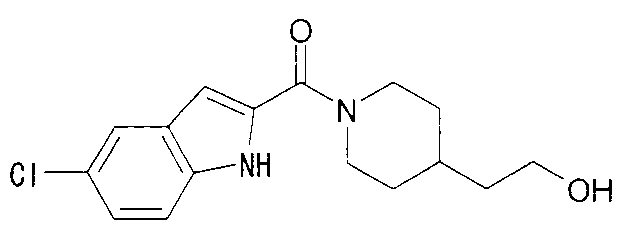
К раствору 370 мг 5-хлориндол-2-карбоновой кислоты и 300 мг 2-метил-1-(пиперидин-4-ил)-2-пропанола в 8 мл ДМФА добавляли 360 мг гидрохлорида 1-этил-3-(диметиламинопропил)карбодиимида и 250 мг 1-гидроксибензотриазола с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли 0,5 М водную хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 0,5 М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем кристаллизовали из диизопропилового эфира с получением 268 мг 1-{1-[(5-хлор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ола в виде белого кристалла.
Пример 24
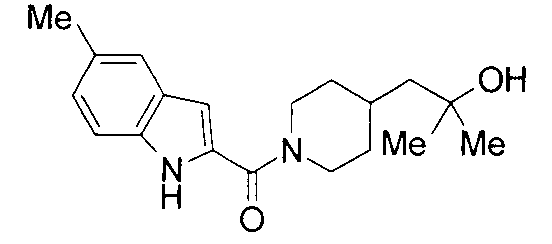
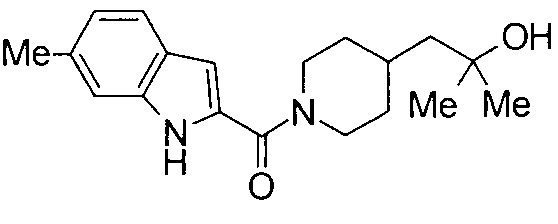
К раствору 1800 мг 5-метилиндол-2-карбоновой кислоты и 1500 мг 2-метил-1-(пиперидин-4-ил)-2-пропанола в 25 мл ДМФА добавляли 2100 мг гидрохлорида 1-этил-3-(диметиламинопропил)карбодиимида и 1500 мг 1-гидроксибензотриазола с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли 0,5 М водную хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 0,5 М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем кристаллизовали из смеси этилацетат/диизопропиловый эфир с получением 2140 мг 2-метил-1-{1-[(5-метил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ола в виде белого кристалла.
Пример 25
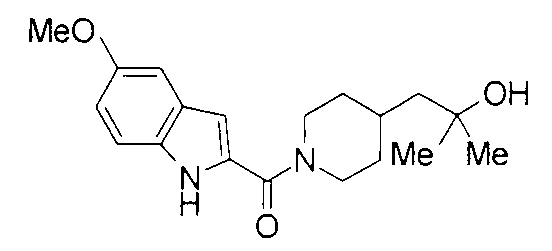
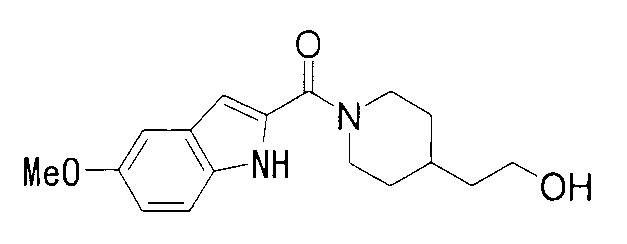
К раствору 478 мг 5-метоксииндол-2-карбоновой кислоты и 432 мг 2-метил-1-(пиперидин-4-ил)-2-пропанола в 8 мл ДМФА добавляли 575 мг гидрохлорида 1-этил-3-(диметиламинопропил)карбодиимида и 169 мг 1-гидроксибензотриазола с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли 0,2 М водную хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 0,2 М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-24:1] и затем кристаллизовали из смеси этилацетат/диизопропиловый эфир с получением 683 мг 1-{1-[(5-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ола в виде белого кристалла.
Пример 26
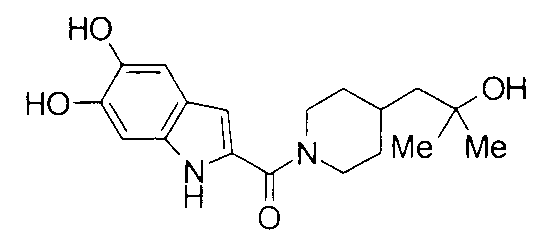
К раствору 150 мг 5Н-[1,3]диоксоло[4,5-f]индол-6-карбоновой кислоты в 3 мл дихлорметана добавляли 1,5 мл 1 М раствора бортрибромида в дихлорметане при 0°C с последующим перемешиванием при комнатной температуре в течение 8 часов. К реакционной жидкости добавляли воду с последующей экстракцией этилацетатом. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. К раствору полученного белого твердого вещества в 3 мл ДМФА добавляли 140 мг 2-метил-1-(пиперидин-4-ил)-2-пропанола, 150 мг гидрохлорида 1-этил-3-(диметиламинопропил)карбодиимида и 105 мг 1-гидроксибензотриазола с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли 0,5 М водную хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 0,5 М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем подвергали затвердеванию, используя этилацетатгексан, с получением 42 мг 2-{[4-(2-гидрокси-2-метилпропил)пиперидин-1-ил]карбонил}-1Н-индол-5,6-диола в виде белого твердого вещества.
Пример 27
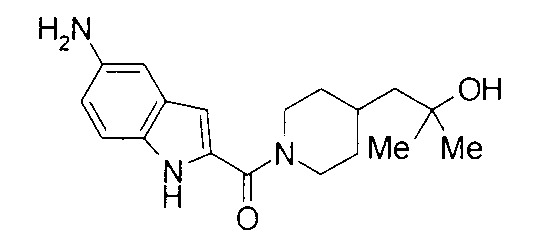
К раствору 838 мг 2-метил-1-{1-[(5-нитро-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ола в 15 мл метанола добавляли 80 мг 10% палладий-уголь с последующим перемешиванием в атмосфере водорода при комнатной температуре в течение 1 суток. Нерастворимый продукт удаляли фильтрованием через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем подвергали затвердеванию с помощью гексана-этилацетата с получением 576 мг 1-{1-[(5-амино-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ола в виде светло-коричневого твердого вещества.
Пример 28
К раствору 105 мг 1-{1-[(5-амино-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ола в 2 мл диоксана и 2 мл ТГФ добавляли 32 мкл уксусного ангидрида с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем подвергали затвердеванию, используя этилацетатгексан, с получением 110 мг N-(2-{[4-(2-гидрокси-2-метилпропил)пиперидин-1-ил]карбонил}-1Н-индол-5-ил)ацетамида в виде белого твердого вещества.
Пример 29
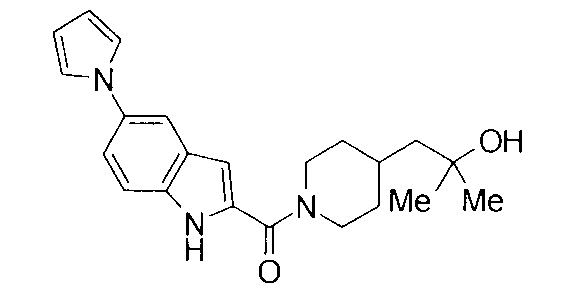
К раствору 127 мг 1-{1-[(5-амино-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ола в 3 мл метанола добавляли 90 мкл 2,5-диметокситетрагидрофурана и 1 мл уксусной кислоты с последующим перемешиванием при 60°С в течение 1 суток. К реакционной жидкости добавляли 1 М хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем подвергали затвердеванию, используя этилацетатгексан, с получением 30 мг 2-метил-1-(1-{[5-(1Н-пиррол-1-ил)-1Н-индол-2-ил]карбонил}пиперидин-4-ил)пропан-2-ола в виде белого твердого вещества.
Пример 30
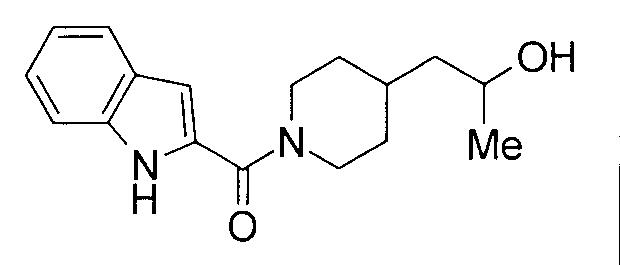
К раствору 233 мг трет-бутил 4-(2-гидроксипропил)пиперидин-1-карбоксилата в 4 мл этилацетата добавляли 3 мл 4 М HCL/EtOAc с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении с получением гидрохлорида 4-(2-гидроксипропил)пиперидина. К раствору гидрохлорида 4-(2-гидроксипропил)пиперидина и 155 мг индол-2-карбоновой кислоты в 5 мл ДМФА добавляли 0,15 мл триэтиламина, 190 мг гидрохлорида 1-этил-3-(диметиламинопропил)карбодиимида и 130 мг 1-гидроксибензотриазола с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли 0,5 М водную хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 0,5 М водным раствором гидроксида натрия и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [хлороформ:метанол=1:0-10:1] и затем подвергали затвердеванию, используя диизопропиловый эфир, с получением 49 мг 1-[1-(1Н-индол-2-илкарбонил)пиперидин-4-ил]пропан-2-ола в виде белого твердого вещества.
Пример 31
К раствору 500 мг индол-2-карбоновой кислоты и 530 мг этилпиперидин-3-илацетата в 8 мл ДМФА добавляли 610 мг гидрохлорида 1-этил-3-(диметиламинопропил)карбодиимида и 430 мг 1-гидроксибензотриазола с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли 0,5 М водную хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 0,5 М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением 519 мг этил [1-(1Н-индол-2-илкарбонил)пиперидин-3-ил]ацетата. К раствору 392 мг полученного этил [1-(1Н-индол-2-илкарбонил)пиперидин-3-ил]ацетата в 7 мл ТГФ добавляли 30 мг боргидрида лития с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [гексан:этилацетат=1:0-2:1] и затем кристаллизовали из диизопропилового эфира с получением 92 мг 2-[1-(1Н-индол-2-илкарбонил)пиперидин-3-ил]этанола в виде белого твердого вещества.
Пример 32
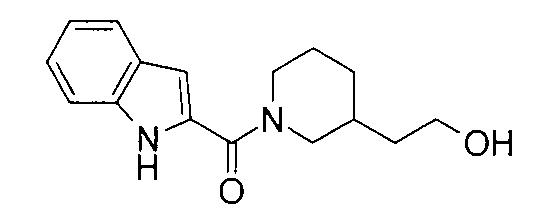
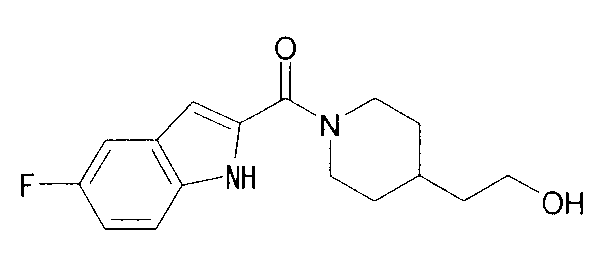
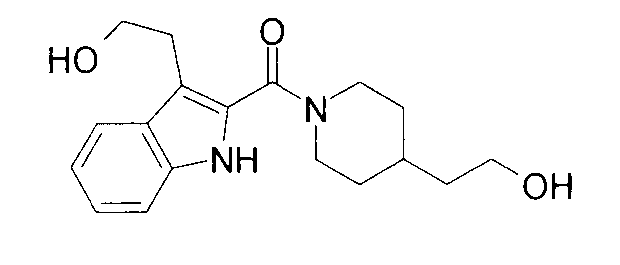
К раствору 4,1 мг 4-пиперидинэтанола, 5,3 мг 5-фтор-1Н-индол-2-карбоновой кислоты и 4,0 мг 1-гидроксибензотриазола в 0,6 мл ДМФА добавляли 100 мг PS-карбодиимида (Argonaut Technologies) при комнатной температуре с последующим перемешиванием в течение ночи. К реакционной жидкости добавляли 50 мг МР-карбоната (Argonaut Technologies) и 50 мг PS-изоцианата (Argonaut Technologies) при комнатной температуре с последующим перемешиванием в течение 2 часов, и нерастворимый продукт фильтровали. Фильтрат концентрировали при пониженном давлении с получением 8,8 мг 2-{1-[(5-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}этанола.
Условия ВЭЖХ, выполненные для определения RT, показаны ниже.
Колонка: Wakosil-II 5C18AR (зарегистрированная торговая марка) (размер частиц: 5 мкм, внутренний диаметр: 2,0 мм и длина: 30 мм).
Подвижная фаза: А раствор, 5 мМ водный раствор трифторуксусной кислоты и В раствор, метанол
Скорость потока: 1,2 мл/мин
Длина волны определения: 254 нм
Температура колонки: 35,0°С
Объем для инъекции: 5 мкл
Соединения примеров вплоть до примера 48, показанные в следующих таблицах, получены способом, аналогичным способам вышеупомянутых примеров. Способы получения и структуры соответствующих соединений примеров показаны в таблицах 4, 5, 8 и 9, и физико-химические данные показаны в таблицах 6, 7 и 10.
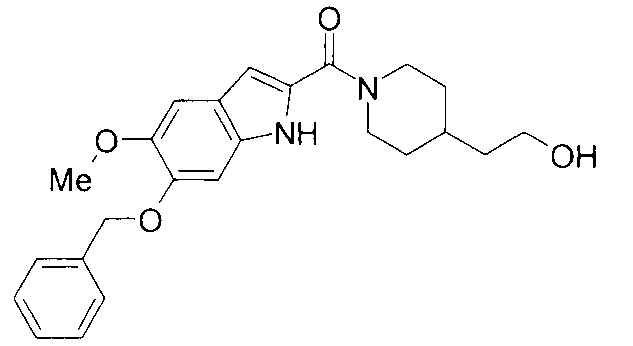
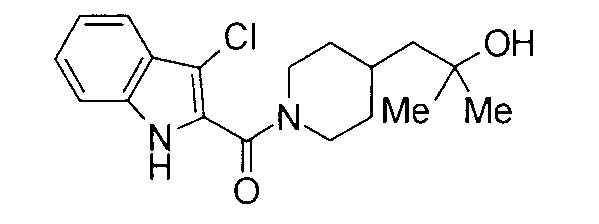
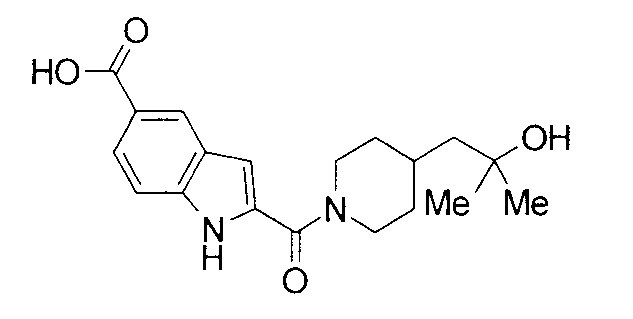
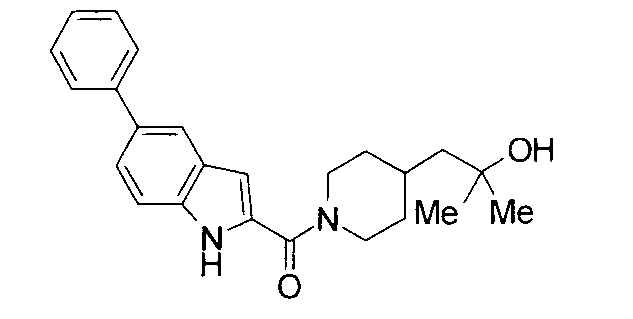
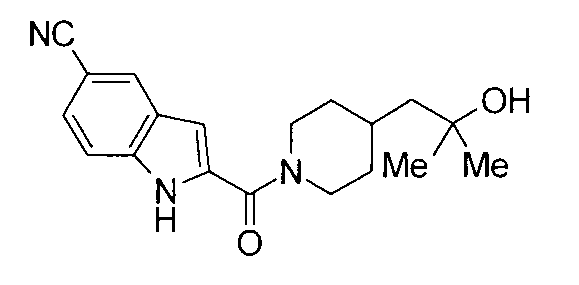
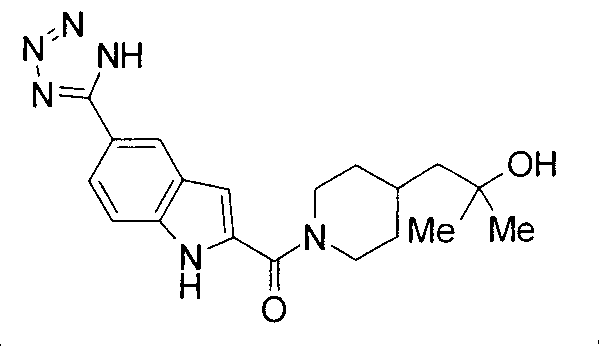
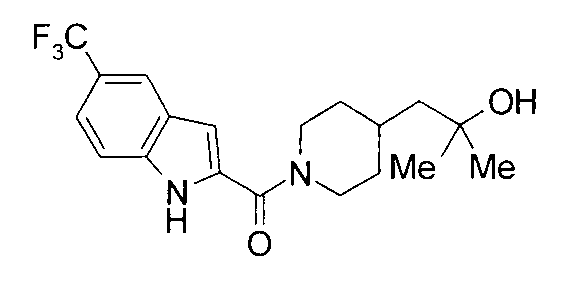
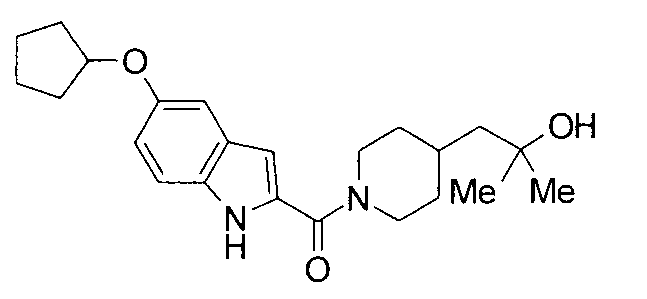
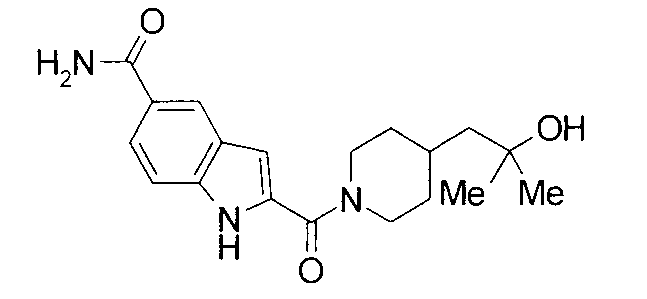
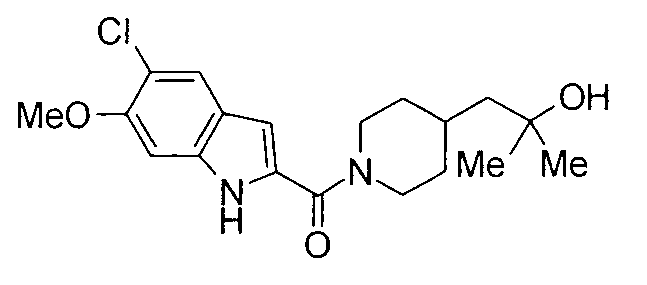
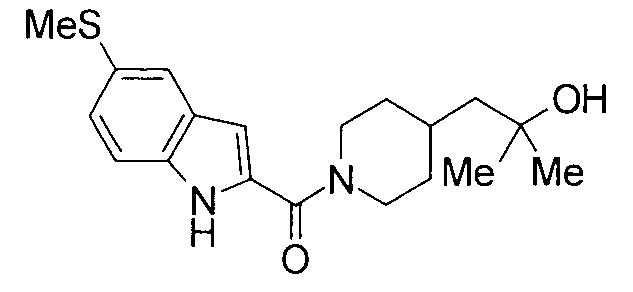
Кроме того, структуры других соединений данного изобретения показаны в таблице 11. Данные соединения легко могут быть получены согласно вышеупомянутым способам получения, методам, описанным в примерах, и методам, очевидным для специалистов в данной области, или их модификациям.
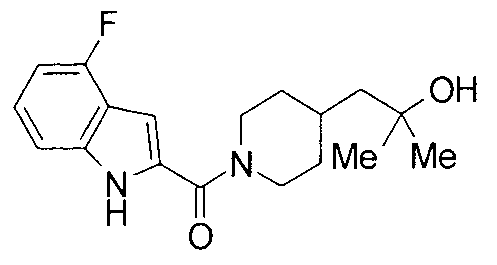
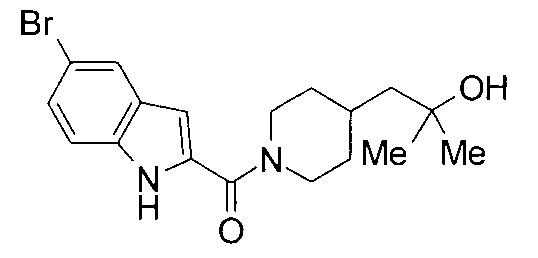
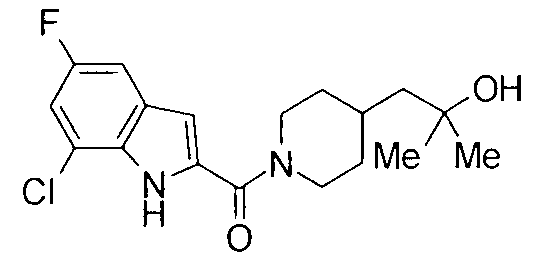
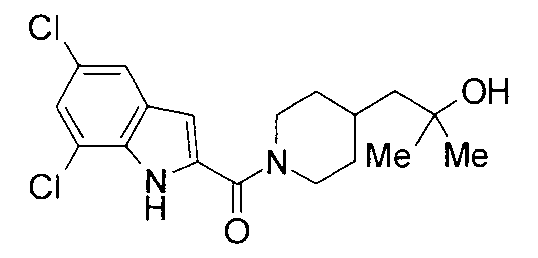
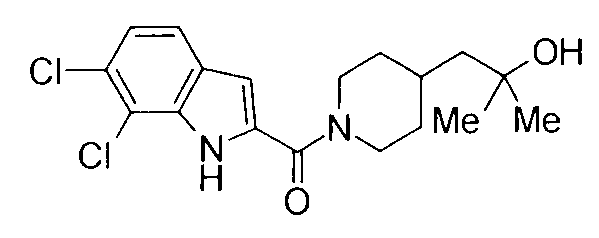
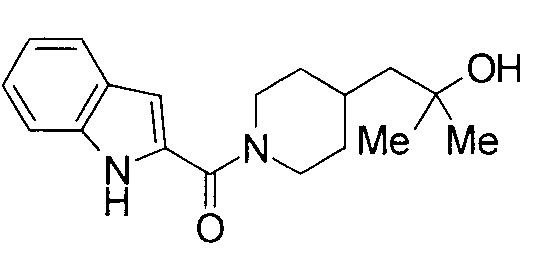
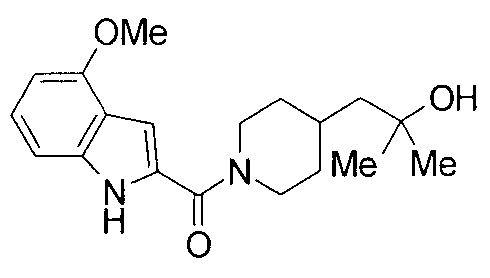
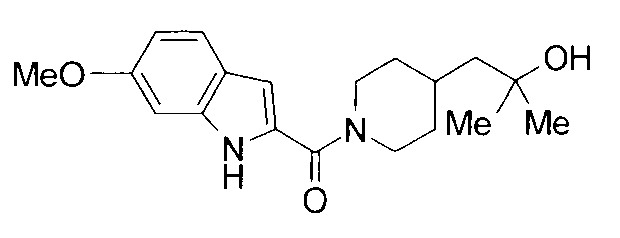
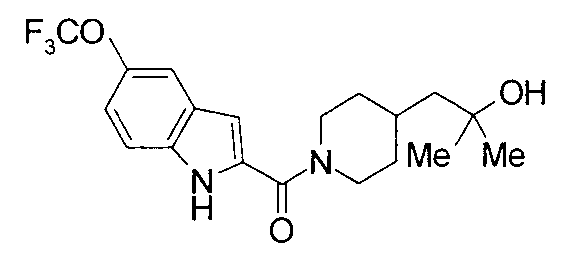
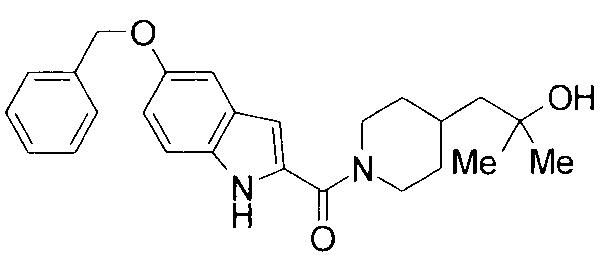
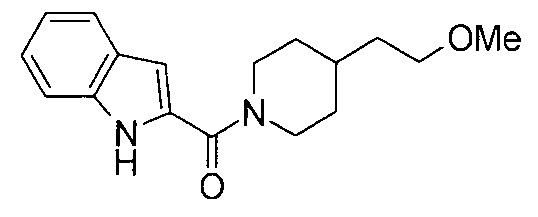
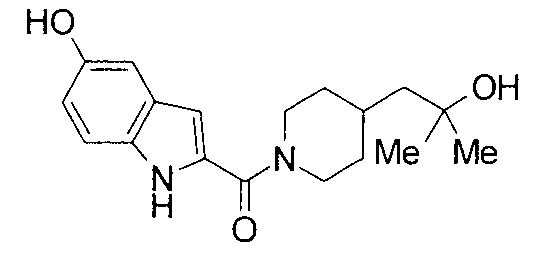
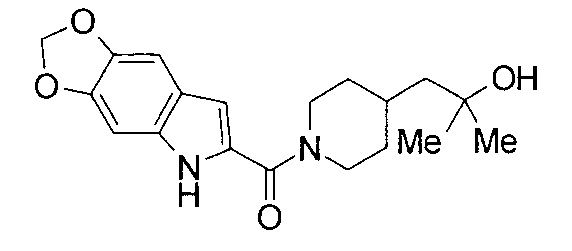
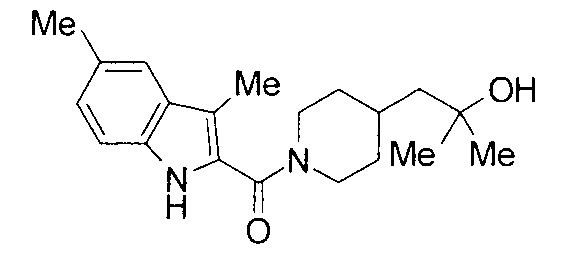
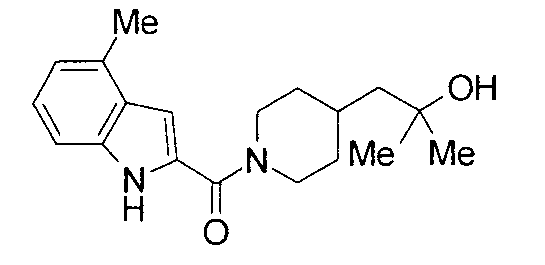
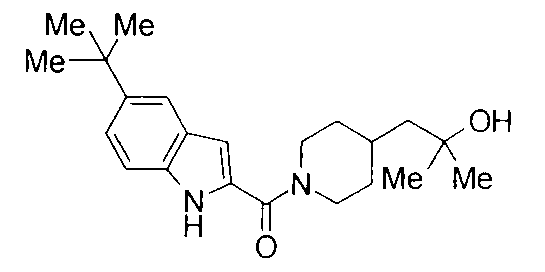
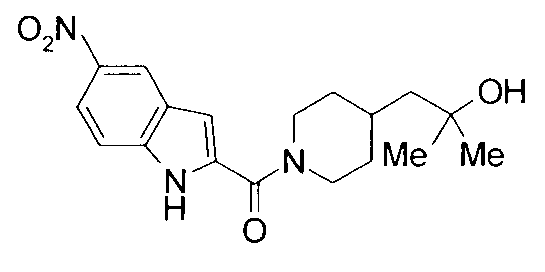
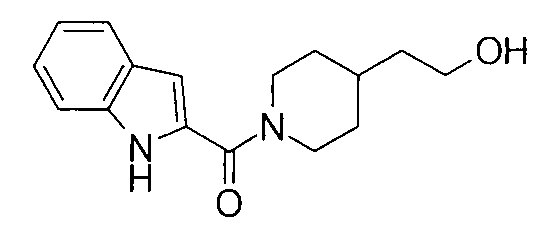
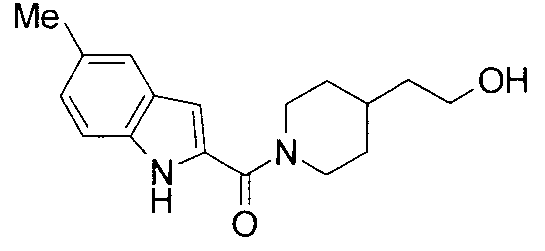
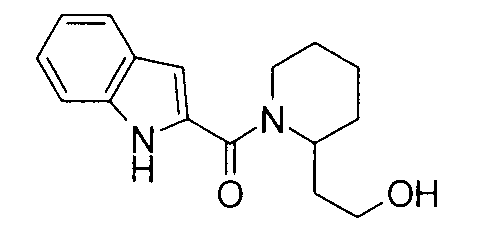
т.пл.: 223-225°C
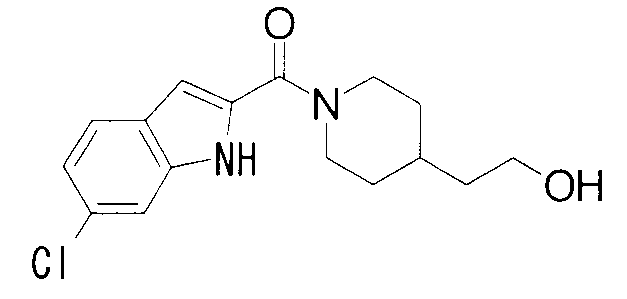
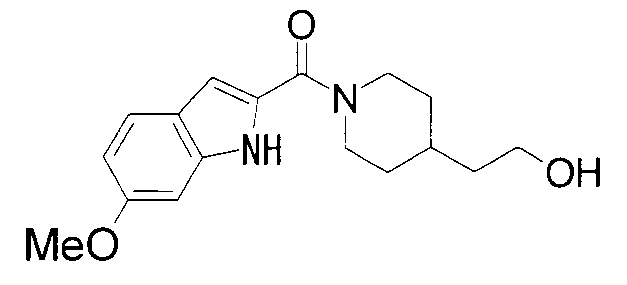
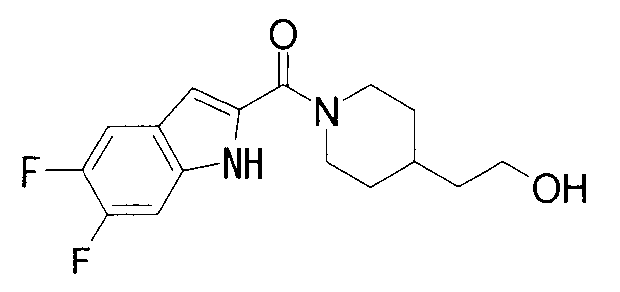
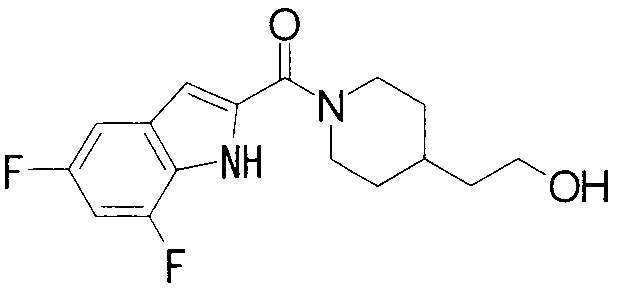
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Поскольку данное соединение, которое представляет собой активный ингредиент лекарственного средства данного изобретения, обладает селективным ингибирующим действием в отношении 17βHDS типа 5 и на основании этого отличным фармакологическим действием, фармацевтическая композиция данного изобретения может быть использована в качестве средства для лечения и/или предупреждения заболеваний, связанных с 17βHDS типа 5, особенно рака простаты, доброкачественной гиперплазии простаты, акне, себорреи, гирсутизма, плешивости, алопеции, раннего полового созревания, гипертрофии надпочечника, синдрома поликистозного яичника, рака грудной железы, рака легкого, эндометриоза, лейомиомы или тому подобного.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 4-АМИНО-ИМИДАЗОХИНОЛИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ АГОНИСТАМИ TLR7 И/ИЛИ TLR8 | 2015 |
|
RU2698902C2 |
| ПРОИЗВОДНЫЕ СПИРОХРОМАНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ АЦЕТИЛ КОЭНЗИМ А КАРБОКСИЛАЗЫ (АСС) | 2006 |
|
RU2422446C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ | 2004 |
|
RU2336273C2 |
| ПИРИДИЛЬНОЕ НЕАРОМАТИЧЕСКОЕ АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛО-1-КАРБОКСИЛАТНОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2408581C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| ПИРИДИЛЬНОЕ НЕАРОМАТИЧЕСКОЕ АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛО-1-КАРБОКСИЛАТНОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2408580C2 |
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ КАССЕТНЫХ ТРАНСПОРТЕРОВ | 2008 |
|
RU2512682C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
Изобретение относится к соединениям формулы (I) или его фармацевтически приемлемым солям:
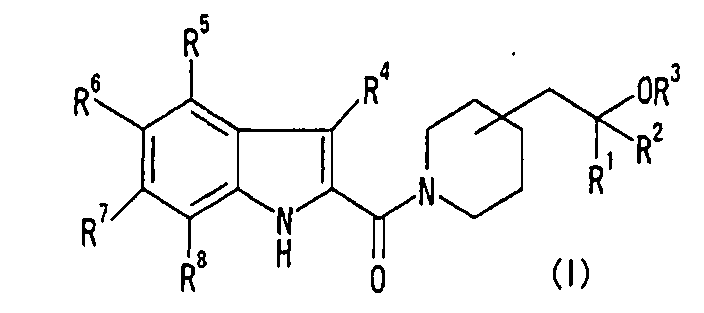
где R1, R2 и R3, которые являются одинаковыми или различными, означают Н, низший алкил; R4, R5, R6, R7 и R8, которые являются одинаковыми или различными, означают Н, низший алкил, галоген, нитро, -X-OR0, -X-NR10R11, -X-NR0C(O)R10, -Х-О-галоген низший алкил, -Х-О-Х-фенил; или R6 и R7 объединены с образованием -0-низший алкилен-О-; R0, который является одинаковым или различным, означает Н, низший алкил; R10, R11, которые являются одинаковыми или различными, означают Н, низший алкил; X, который является одинаковым или различным, означает связь, низший алкилен. Соединения проявляют ингибирующую активность в отношении 17βHSD типа 5, что позволяет использовать их для получения фармацевтической композиции и в способе ингибирования 17βHSD типа 5. 8 н. и 7 з.п. ф-лы, 11 табл., 32 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль: [Хим.9]
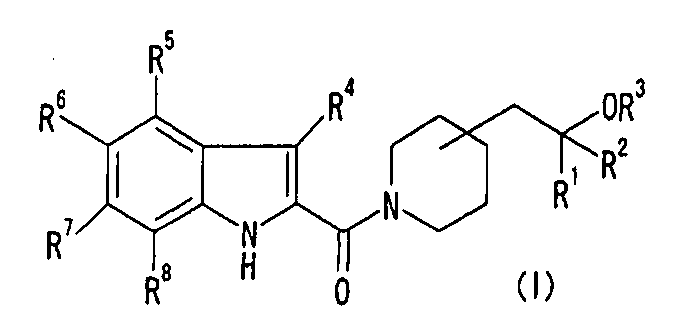
где R1, R2 и R3, которые являются одинаковыми или различными, представляют собой Н или низший алкил;
R4, R5, R6, R7 и R8, которые являются одинаковыми или различными, представляют собой Н, низший алкил, галоген, нитро, -X-OR0, -X-NR10R11, -X-NR0C(O)R10, -Х-О-галоген низший алкил, -Х-О-Х-фенил; или
R6 и R7 объединены с образованием -О-низший алкилен-О-;
R0, который является одинаковым или различным, представляет собой Н
или низший алкил;
R10 и R11, которые являются одинаковыми или различными, представляют собой Н, низший алкил; и
X, который является одинаковым или различным, представляет собой связь или низший алкилен.
2. Соединение формулы (Iа) или его фармацевтически приемлемая соль: [Хим.10]
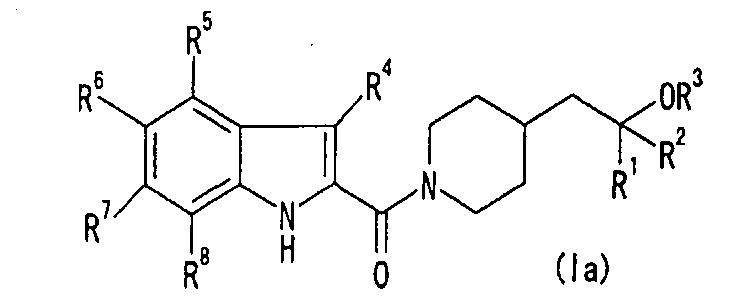
где все символы имеют те же значения, как в п.1.
3. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 и R3 представляют собой Н.
4. Соединение по п.3 или его фармацевтически приемлемая соль, где R4, R5, R7 и R8, которые являются одинаковыми или различными, представляют собой Н, низший алкил, галоген, или -О-низший алкил, и R6 представляет собой Н, низший алкил, галоген, нитро, ОН или -О-низший алкил.
5. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 и R2 представляют собой низший алкил, и R3 представляет собой Н.
6. Соединение по п.5 или его фармацевтически приемлемая соль, где R4, R5, R7 и R8, которые являются одинаковыми или различными, представляют собой Н, низший алкил, галоген, или -О-низший алкил.
7. Соединение по п.5 или 6 или его фармацевтически приемлемая соль, где R6 представляет собой Н, низший алкил, галоген, нитро, ОН, или -O-низший алкил.
8. Соединение по п.7 или его фармацевтически приемлемая соль, где R6 представляет собой низший алкил, галоген, или -О-низший алкил.
9. Соединение по п.5, которое выбрано из группы, включающей
1-[1-(1Н-индол-2-илкарбонил)пиперидин-4-ил]-2-метилпропан-2-ол,
2-метил-1-{1-[(4-метил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ол,
2-метил-1-{1-[5-метил-1H-индол-2-ил)карбонил]иперидин-4-ил}пропан-2-ол,
1-{1-[(5-трет-бутил-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(4-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(4-хлор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-хлор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-бром-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(7-хлор-5-фтор-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
2-{[4-(2-гидрокси-2-метилпропил)пиперидин-1-ил]карбонил}-1Н-индол-5-ол,
1-{1-[(4-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(5-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
1-{1-[(6-метокси-1Н-индол-2-ил)карбонил]пиперидин-4-ил}-2-метилпропан-2-ол,
2-метил-1-(1-{[5-(трифторметокси)-1Н-индол-2-ил]карбонил}пиперидин-4-ил)пропан-2-ол, и
2-метил-1-{1-[(5-нитро-1Н-индол-2-ил)карбонил]пиперидин-4-ил}пропан-2-ол, или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, проявляющая ингибирующую активность в отношении 17β-гидроксистероид-дегидрогеназы (17βHSD) типа 5, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, растворитель или эксципиент.
11. Применение соединения по п.1 или его фармацевтически приемлемой соли для изготовления фармацевтической композиции, проявляющей ингибирующую активность в отношении 17β-гидроксистероид-дегидрогеназы (17βHSD) типа 5.
12. Применение соединения по п.1 или его фармацевтически приемлемой соли в качестве средства, ингибирующего активность 17β-гидроксистероид-дегидрогеназы (17βHSD) типа 5.
13. Способ ингибирования 17β-гидроксистероид-дегидрогеназы (17βHSD) типа 5, включающий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
14. Ингибитор 17βHSD типа 5, содержащий соединение по п.1 или его фармацевтически приемлемую соль.
15. Способ приготовления фармацевтической композиции, проявляющей ингибирующую активность в отношении 17β-гидроксистероид-дегидрогеназы (17βHSD) типа 5, включающий смешивание соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя, растворителя или эксципиента.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2135478C1 |

