Область техники
Изобретение относится к азольному соединению, которое можно применять в качестве активного ингредиента фармацевтической композиции, в частности, фармацевтической композиции для лечения нейропатической боли.
Предшествующий уровень техники
Известно, что гидролаза амидов жирных кислот (FAAH) гидролизует эндоканнабиноид для его инактивации (непатентный документ 1). «Эндоканнабиноид» является родовым термином для обозначения биомолекул, которые действуют на каннабиноидные рецепторы для проявления их физиологической активности. В качестве типичных эндоканнабиноидов известны анандамид, пальмитоилэтаноламид, олеамид и 2-арахидоноилглицерин. Кроме того, известно, что дельта-9-тетрагидроканнабинол, который считается активным ингредиентом гашиша (марихуаны), инактивирует каннабиноидный рецептор (непатентный документ 2).
У млекопитающих до настоящего времени было известно наличие двух типов каннабиноидных рецепторов - CB1 и CB2. CB1 экспрессирован в центральной и периферической нервной системе, и при активации он проявляет психологическое действие, анальгетическое действие или тому подобные. CB2 экспрессирован в иммунной системе и при активации он проявляет противовоспалительное действие, анальгетическое (противовоспалительное) действие или тому подобные.
Известно, что нестероидные противовоспалительные лекарственные средства и наркотические анальгетические лекарственные средства, такие как морфин и тому подобные, которые представляют собой обычные анальгетики, слабо эффективны для лечения нейропатической боли. В области медицины для облегчения боли применяются антиэпилептические лекарственные средства, такие как прегабалин и тому подобные, и антидепрессантные лекарственные средства, такие как дулоксетин и тому подобные, применяются для облегчения боли, но их анальгетические эффекты недостаточны, и имеются проблемы с центральными побочными эффектами, такими как сонливость, головокружение и тому подобные.
Агонист каннабиноидных рецепторов проявляет эффективность у пациентов с нейропатической болью, но его применение в значительной степени ограничено вследствие его психологического действия (непатентный документ 3).
С другой стороны, когда ингибитор FAAH вводится животному, то он проявляет анальгетический эффект против нейропатической боли и воспалительной боли, но побочные эффекты, отмечаемые, когда агонист каннабиноидных рецепторов вводится животному, такие как седативное действие, сниженная температура тела, каталепсия и тому подобные, не наблюдаются (непатентные документы 4 и 5), и, таким образом, ожидается, что ингибитор FAAH будет превосходным фармацевтическим средством для лечения боли, в частности, фармацевтическим средством для лечения нейропатической боли.
В качестве соединений, обладающих ингибирующей активностью в отношении FAAH, известны соединения, которые способны действовать в качестве анальгетического лекарственного средства, анксиолитического лекарственного средства, антиэпилептического лекарственного средства, противорвотного средства, средства для лечения сердечнососудистых заболеваний или средства против глаукомы.
Например, в патентом документе 1 описано соединение, представленное следующей формулой (А) в качестве соединения, обладающего ингибирующей активностью в отношении FAAH.
Формула 1
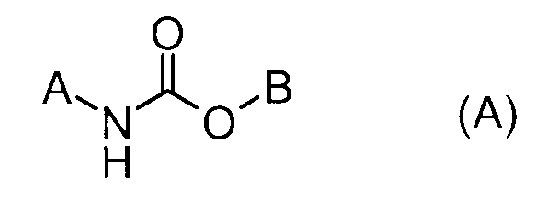
(В формуле В представляет разнообразные кольцевые группы, которые могут быть замещены, и тому подобные, а A представляет фенил, который может быть замещен, фенилалкил, который может быть замещен, дибензофуранил, дибензотиенил, нафтоил, индолил, флуоренил или карбазолил. Детали можно найти в данной публикации).
Кроме того, в патентном документе 2 описано соединение, представленное следующей формулой (B) в качестве соединения, обладающего ингибирующей активностью в отношении FAAH.
Формула 2
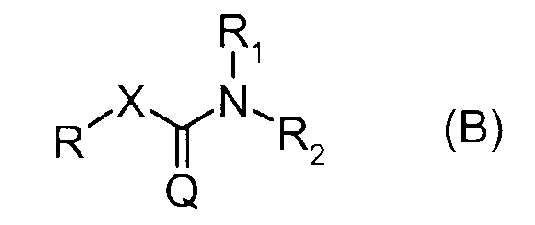
(В формуле R представляет разнообразные кольцевые группы, которые могут быть замещены, или тому подобные, а X и Q являются одинаковыми или отличаются друг от друга и представляют соответственно O и S. Кроме того, R1 и R2 могут комбинироваться с атомом N, с которым они могут быть соединены для образования замещенного или незамещенного кольца. Детали можно найти в данной публикации).
Кроме того, в патентном документе 3 описано соединение, представленное следующей формулой (C) в качестве соединения, обладающего ингибирующей активностью в отношении FAAH.
Формула 3
(Детали можно найти в данной публикации).
Все соединения, раскрытые в этом документе, имеют структуры, отличающиеся от соединения формулы (I) по настоящему изобретению.
Кроме того, в патентных документах 4 и 5 раскрывается соединение мочевины, представленное следующей формулой (D), в качестве ингибитора FAAH.
Формула 4
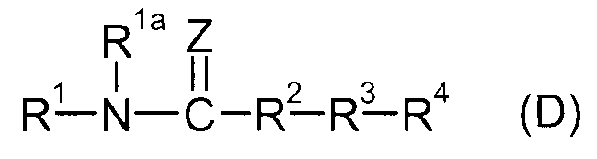
(В этой формуле Z представляет O или S, а R2 представляет пиперидин-1,4-диил или пиперазин-1,4-диил, каждый из которых может быть замещенным. Детали можно найти в указанных публикациях).
Список документов
Патентные документы
[Патентный документ 1] Описание Международной заявки № WO2003/065989
[Патентный документ 2] Описание Международной заявки № WO2004/033422
[Патентный документ 3] Описание Международной заявки № WO2006/088075
[Патентный документ 4] Описание Международной заявки № WO2006/054652
[Патентный документ 5] Описание Международной заявки № WO2007/020888
Не патентные документы
[Не патентный документ 1] "Annual review o f biochemistry", (USA), 2005, Vol. 74, p. 411-432
[Не патентный документ 2] "Current Medicinal Chemistry", (USA), 1999, Vol. 6, p. 635-664
[Не патентный документ 3] "Expert opinion on p harmacotherapy", (UK), 2006, Vol. 7, p. 607-615
[Не патентный документ 4] "British Journal of Ph armacology", (UK), 2007, Vol. 152, p. 624-32
[Не патентный документ 5] "Nature Medicine", (UK), 2003, Vol. 9, p. 76-81
Краткое описание сущности изобретения
Проблема, которую предстоит разрешить изобретением
Предоставляется соединение, которое может применяться в качестве активного ингредиента фармацевтической композиции, в частности, фармацевтической композиции для лечения боли, причем при использовании указанного соединения отсутствуют или снижаются опасения, связанные с побочными эффектами и зависимостью, подобными тем, которые наблюдаются при применении гашиша.
Средства для решения проблемы
Заявители провели обширные исследования соединений, обладающих ингибирующей активностью в отношении FAAH, и, в результате, обнаружили, что соединение формулы (I) проявляет превосходную ингибирующую активность в отношении FAAH, посредством чего и было создано настоящее изобретение.
То есть настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли и к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, и фармацевтически приемлемый эксципиент.
Формула 5
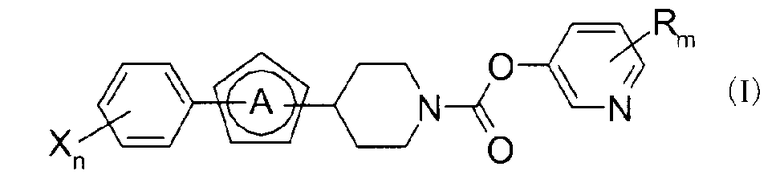
где
кольцо A представляет азольное кольцо,
R является одинаковым или отличается друг от друга и представляет H или низший алкил,
X является одинаковым или отличается друг от друга и представляет H, галоген или галоген-низший алкил,
n и m являются одинаковыми или отличаются друг от друга и представляют 1 или 2.
Кроме того, настоящее изобретение относится к фармацевтической композиции для лечения нейропатической боли, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, то есть средство для лечения нейропатической боли, содержащее соединение формулы (I) или его фармацевтически приемлемую соль.
Кроме того, настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для получения фармацевтической композиции для лечения нейропатической боли, и к способу для лечения нейропатической боли, включающему введение пациенту эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Эффекты изобретения
Соединение формулы (I), или его фармацевтически приемлемая соль, обладает ингибирующей активностью в отношении FAAH и может применяться в качестве средства для предотвращения и/или лечения заболеваний, связанных с FAAH, в частности, нейропатической боли.
Наилучший вариант осуществления изобретения
Далее настоящее изобретение будет описано в деталях.
«Низший алкил» представляет собой линейный или разветвленный алкил, имеющий 1-6 атомов углерода (который далее просто обозначен в виде C1-6), например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-гексильную группу или тому подобные. В другом варианте осуществления он представляет собой метил или этил, и в еще одном варианте осуществления - метил.
«Азольное кольцо», среди моноциклических, конъюгированных, ненасыщенных 5-членных колец, означает кольцо, имеющее 2-4 гетероатома, выбранных из O, S и N, в качестве атомов, составляющих кольцо, в котором по меньшей мере один из гетероатомов представляет N. В соответствии с расположением гетероатомов в кольце, его примеры включают 1,2-азол, 1,3-азол, 1,2,4-азол, 1,2,3,4-азол и тому подобные; примеры 1,2-азола включают пиразол, изоксазол и изотиазол; примеры 1,3-азола включают имидазол, оксазол и тиазол; примеры 1,2,4-азола включают 1,2,4-триазол, 1,2,4-оксадиазол и 1,2,4-оксатиазол; и примеры 1,2,3,4-азола включают тетразол и тому подобные соединения. Когда «азольное кольцо» представляет собой двухвалентную кольцевую группу, то она представляет собой двухвалентную группу, образованную удалением атомом водорода в любом положении.
«Галоген» означает F, Cl, Br или I.
«Галоген-низший алкил» означает линейный или разветвленный алкил, имеющий 1-6 атомов углерода (C1-6алкил), замещенный 1-5 атомами галогена.
«Нейропатическая боль» означает боль, вызванную дисфункцией периферической или центральной нервной системы, и ее примеры включают диабетическую нейропатическую боль, постгерпетическую боль, нейропатию, вызванную ВИЧ, нейропатию, вызванную противораковыми средствами, боль после повреждения спинного мозга или боль, сопровождающую рассеянный склероз, и тому подобные. Основные клинические симптомы нейропатической боли включают сжимающую боль, жгучую боль, гиперальгезию, аллодинию и тому подобные.
Ниже будут представлены варианты осуществления соединения формулы (I).
(1) Соединение, в котором кольцо А представляет 1,2-азол, 1,3-азол 1,2,4-азол или 1,3,4-азол; в другом варианте осуществления кольцо А представляет 1,2,4-оксадиазол, 1,2,4-триазол, 1,3-оксазол или пиразол, или в еще одном варианте осуществления кольцо А представляет собой одно из колец, представленных следующими формулами (II)-(VI):
Формулы 6
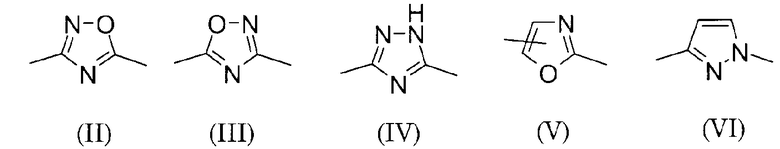
В другом варианте осуществления кольцо А представляет собой одно из колец, представленных формулами (IV)-(VI). В еще одном варианте осуществления соединение содержит кольцо А, которое представляет собой кольцо, представленное формулой (IV). В еще одном варианте осуществления соединение содержит кольцо А, которое представляет собой кольцо, представленное формулой (V). В еще одном варианте осуществления соединение содержит кольцо А, которое представляет собой кольцо, представленное формулой (VI).
(2) Соединение, в котором Rm представляет H, 2-метил, 6-метил или 2,6-диметил, а в другом варианте осуществления Rm представляет H, 2-метил или 6-метил.
(3) Соединение, в котором Xn представляет H, 2-фтор-, 3-фтор-, 4-фтор- или 3,4-дифтор-.
(4) Соединение, которое представляет комбинацию двух или более из групп, как описано выше в пунктах (1)-(3).
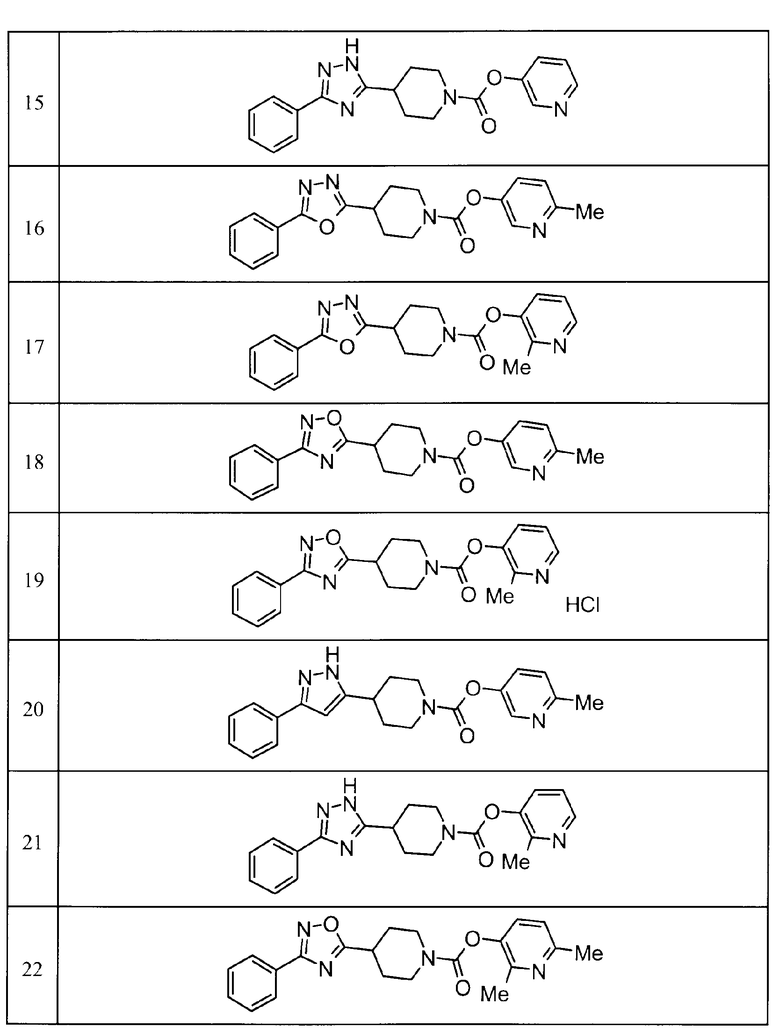
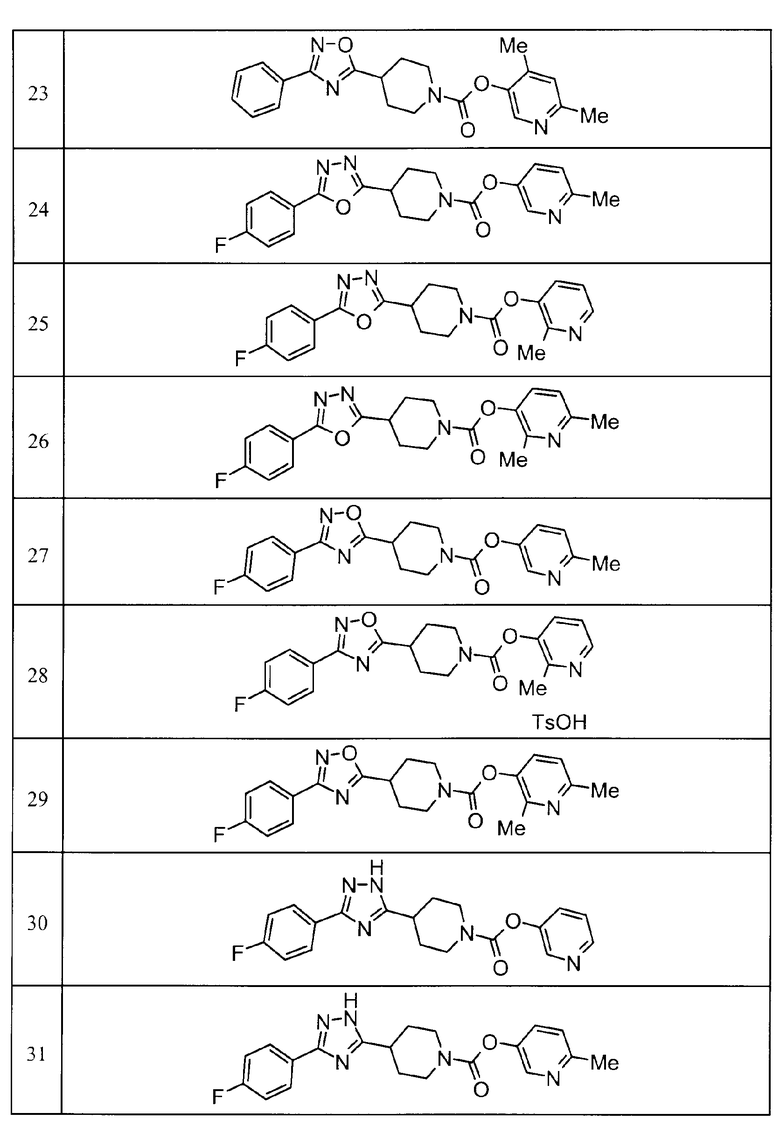
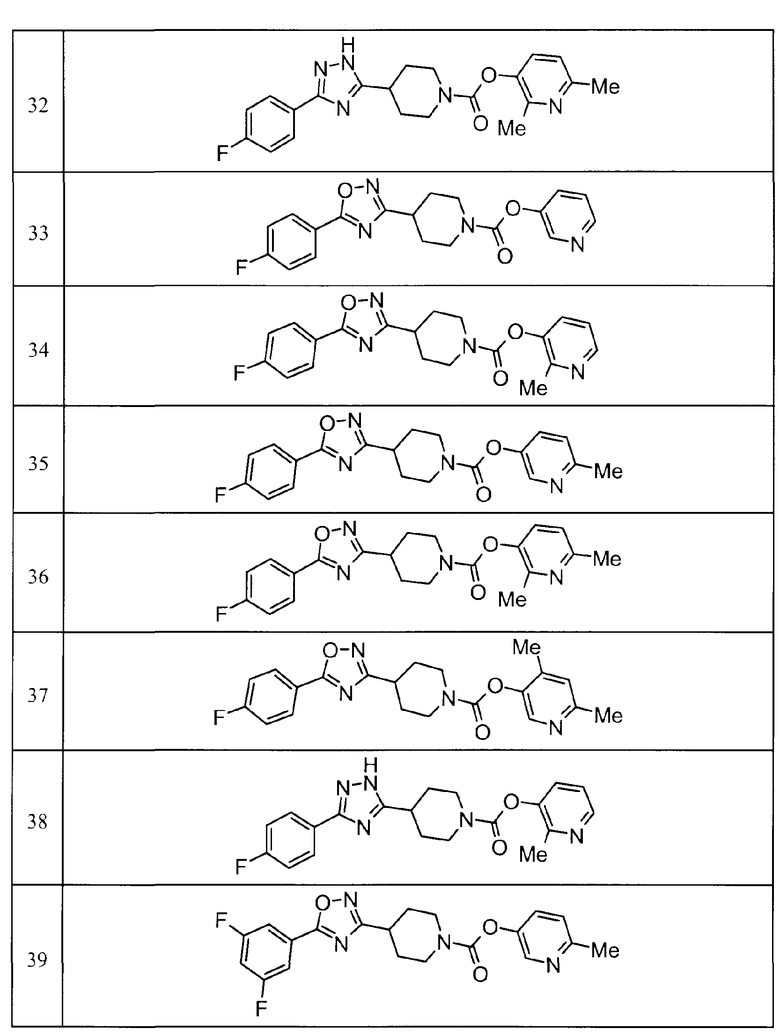
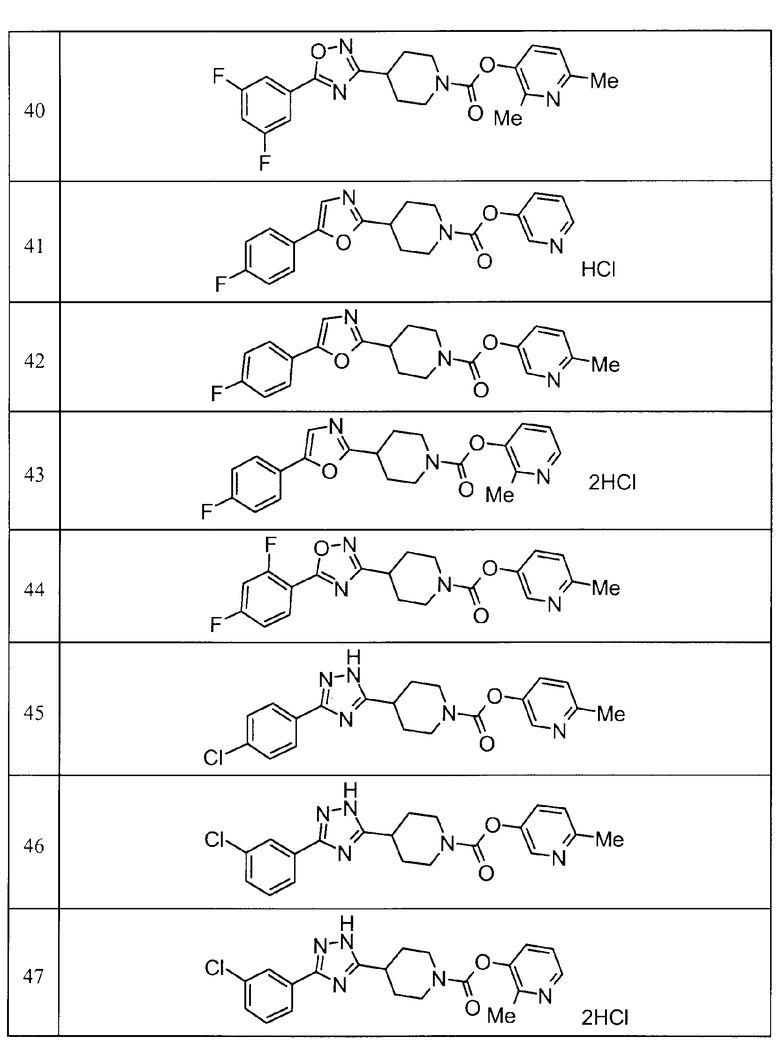
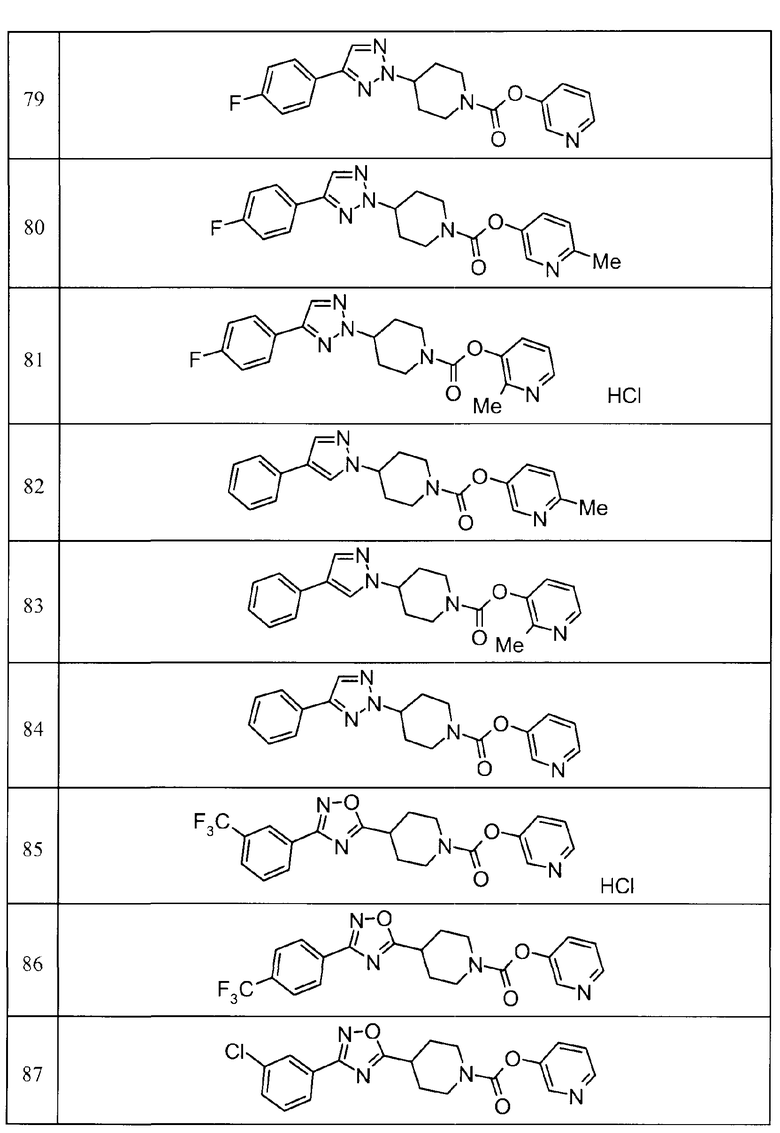
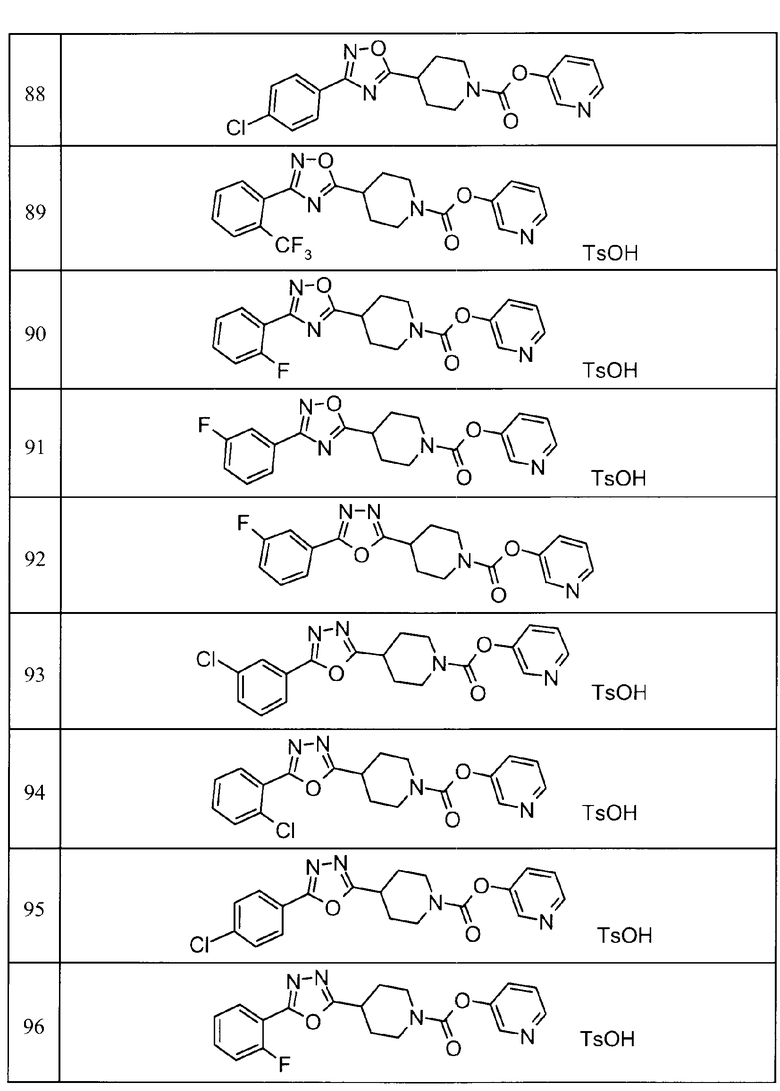
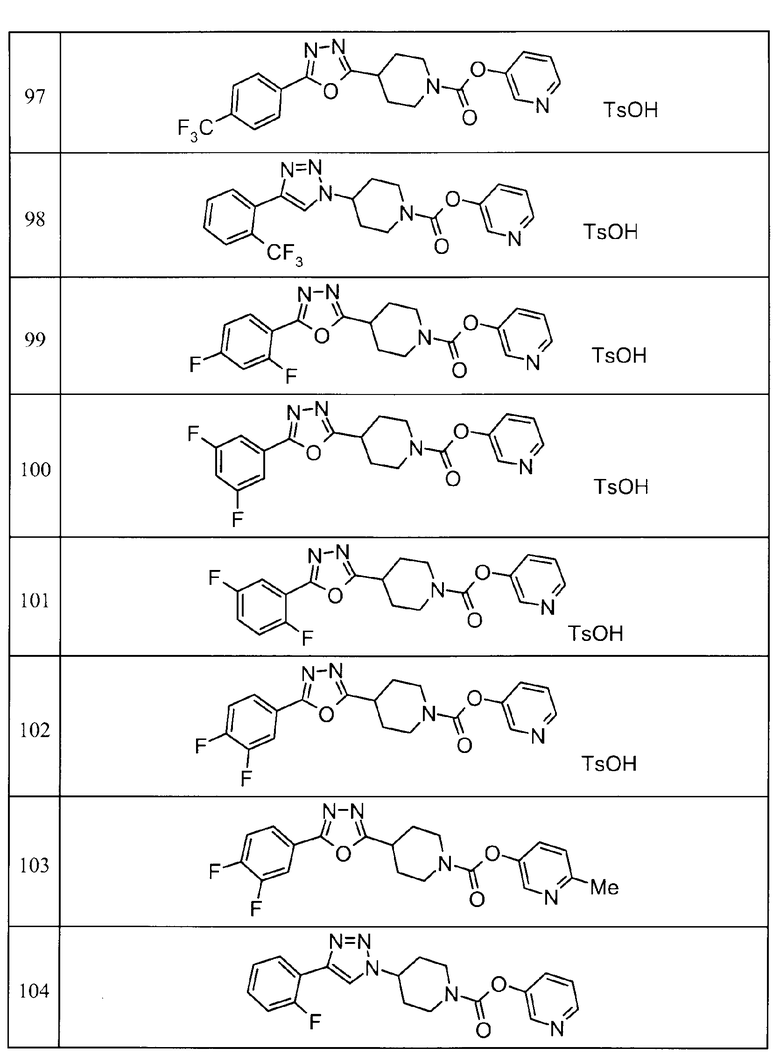
Примеры определенных соединений, охватываемых настоящим изобретением, включают соединения, представленные в пунктах (5) или (6) ниже, или их фармацевтически приемлемые соли:
(5) пиридин-3-ил 4-[3-(4-фторфенил)-1,2,4-оксадиазол-5-ил]пиперидин-1-карбоксилат,
пиридин-3-ил 4-(3-фенил-1H-1,2,4-триазол-5-ил)пиперидин-1-карбоксилат,
6-метилпиридин-3-ил 4-[3-(4-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат,
6-метилпиридин-3-ил 4-[5-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат,
2-метилпиридин-3-ил 4-[5-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат,
2,6-диметилпиридин-3-ил 4-[5-(3,4-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидин-1-карбоксилат,
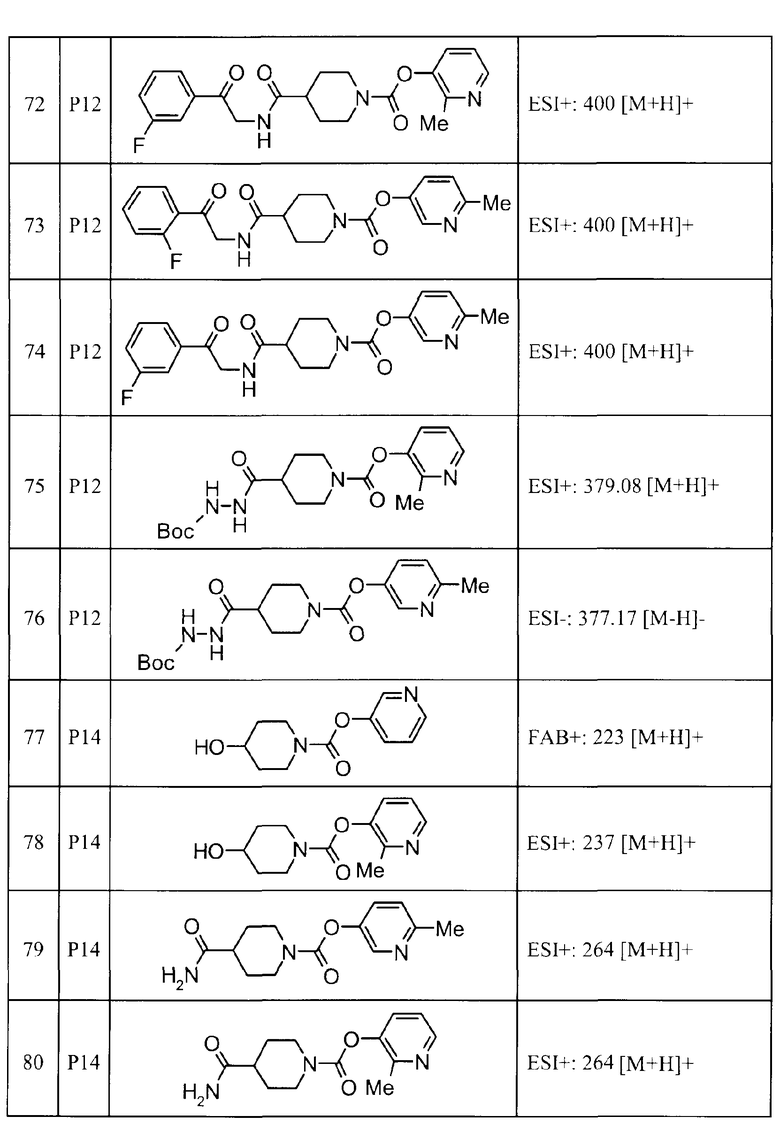
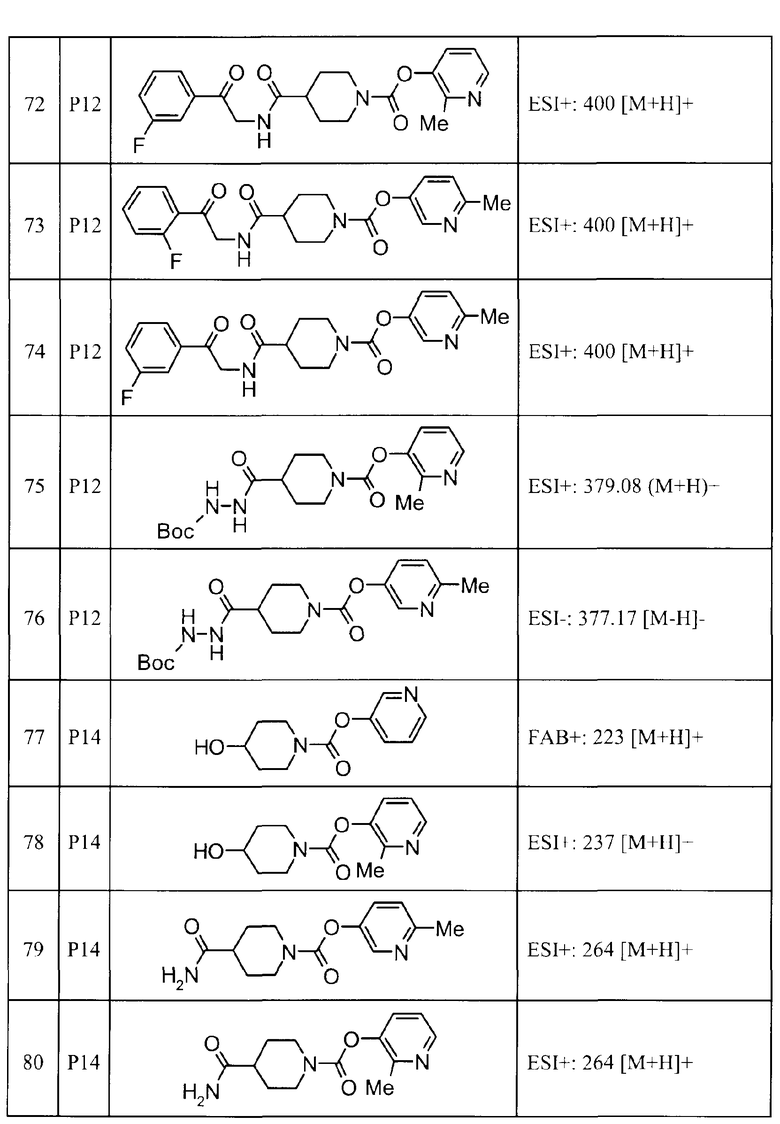
2-метилпиридин-3-ил 4-[3-(2-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат,
6-метилпиридин-3-ил 4-(3-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилат,
2-метилпиридин-3-ил 4-[5-(3-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат, и
6-метилпиридин-3-ил 4-[4-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат.
(6) пиридин-3-ил 4-(3-фенил-1H-1,2,4-триазол-5-ил)пиперидин-1-карбоксилат,
6-метилпиридин-3-ил 4-[3-(4-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат,
6-метилпиридин-3-ил 4-[5-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат,
2,6-диметилпиридин-3-ил 4-[5-(3,4-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидин-1-карбоксилат,
2-метилпиридин-3-ил 4-[3-(2-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат,
6-метилпиридин-3-ил 4-(3-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилат,
2-метилпиридин-3-ил 4-[5-(3-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат и
6-метилпиридин-3-ил 4-[4-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат.
В некоторых случаях, в зависимости от вида заместителей, соединение формулы (I) может иметь таутомеры или геометрические изомеры. В настоящем описании соединение формулы (I) будет описано только в одной форме изомеров, тем не менее настоящее изобретение включает другие изомеры, изолированные формы изомеров или их смесь.
Кроме того, в некоторых случаях, соединение формулы (I) может иметь асимметричный(е) атом(ы) углерода или ось асимметрии, и, соответственно, оно может существовать в форме оптических изомеров. Настоящее изобретение включает и изолированную форму этих оптических изомеров соединения формулы (I), и их смесь.
Кроме того, фармацевтически приемлемые пролекарства соединения, представленного формулой (I), также включены в настоящее изобретение. Фармацевтически приемлемое пролекарство относится к соединению, имеющему группу, которая может быть превращена в аминогруппу, гидроксильную группу, карбоксильную группу или тому подобные, путем сольволиза или в физиологических условиях. Примеры групп для образования пролекарства включают группы, описанные в публикации Prog. Med., 5, 2157-2161 (1985) or "Iyakuhin no Kaihatsu (Pharmaceutical Research and DevE1opment)" (Hirokawa Publishing Company, 1990), vol. 7, Bunshi Sekkei (Drug Design), 163-198.
Кроме того, соединение формулы (I) может образовывать кислотно-аддитивную соль или соль с основанием, в зависимости от вида замещения, и эти соли включены в настоящее изобретение, пока они представляют собой фармацевтически приемлемые соли. В частности, их примеры включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная, иодистоводородная, серная, азотная, фосфорная и другими, и с органическими кислотами, такими как муравьиная, уксусная, пропионовая, щавелевая, малоновая, сукциновая, фумаровая, алеиновая, молочная, яблочная, миндальная, винная, дибензолвинная, дитолилвинная, лимонная, метансульфокислота, этансульфокислота, бензолсульфокислота, п-толуолсульфокислота, аспартановая, глутаминовая и другие, солями калия, магния, кальция, алюминия и тому подобными, или слои с органическими основаниями, такие как метиламин, этиламин, этаноламин, лизин, орнитин и тому подобные, соли с различными аминокислотами и производными аминокислот, такие как ацетиллейцин и тому подобные, соли аммония и другие.
Кроме того, настоящее изобретение также включает различные гидраты или сольваты и любые из кристаллических полиморфов соединения формулы (I) и их фармацевтически приемлемые соли. Настоящее изобретение также включает соединения, меченные различными радиоактивными или нерадиоактивными изотопами.
Способы получения
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть получены применением различных известных способов синтеза с использованием характеристик, основанных на их основных скелетах или видах заместителей. В то же время, в зависимости от типа функциональных групп, с точки зрения методик получения, в некоторых случаях эффективно замещение функциональных групп соответствующей защитной группой (группой, которая способна легко превращаться в функциональную группу), на стадии получения исходного материала или промежуточного соединения. Примеры защитной группы включают защитные группы, описанные в руководстве “Protective Groups in Organic Synthesis (4th edition, 2007)”, написанном Greene и Wuts, и тому подобные, которые могут быть соответствующим образом выбраны и использованы, в зависимости от условий реакции. В этих способах желательное соединение может быть получено введением защитной группы для поведения реакции, а затем, при желании, удалением защитной группы.
Кроме того, пролекарства соединения формулы (I) могут быть получены введением определенной группы на стадии получения исходного материала или промежуточного соединения таким же образом, как для указанных выше защитных групп, или проведением реакции с использованием полученного соединения формулы (I). Реакция может проводиться путем применения способа, известного специалисту в данной области, такого как обычная этерификация, амидирование, дегидратация и тому подобные.
Ниже будут описаны репрезентативные способы получения соединения формулы (I). Каждый из способов получения может также проводиться с обращением к ссылкам, прилагаемым к объяснению. Кроме того, способы получения по настоящему изобретению не ограничиваются приведенными ниже примерами.
Способ получения 1
Схема 7
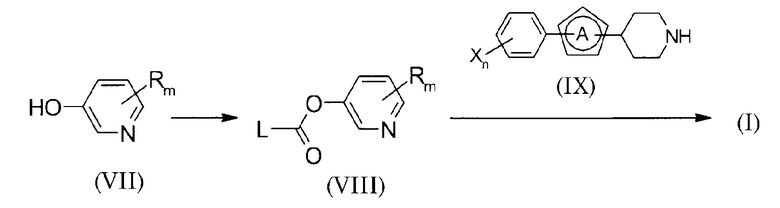
(В схеме L представляет уходящую группу).
Соединение формулы (I) может быть получено превращением соединения (VII) в производное сложного эфира карбоновой кислоты (VIII), которое затем взаимодействует с аминным соединением (IX).
Приведенные в настоящем описании примеры уходящих групп включают Cl, имидазолил, фенокси и 4-нитрофенокси группу.
Первая стадия проводится взаимодействием соединения (VII) с эквивалентным количеством или избытком карбонилирующего реагента в присутствии основания в условиях от охлаждения до нагревания, предпочтительно, при температуре от -20°С до 80°С в инертном растворителе, обычно в течение примерно от 0,1 часа до суток. На следующей стадии, без обработки реакционной смеси, полученной на первой стадии, эквивалентное количество или избыточное количество аминного соединения (IX) добавляется к реакционной смеси, и смесь подвергается реакции в условиях от охлаждения до нагревания, предпочтительно, при температуре от -20°С до 80°С в инертном растворителе, обычно в течение примерно от 0,1 часа до суток. Растворитель, используемый при этой реакции, конкретно не ограничивается, но его примеры включают галоидированные углеводороды, такие как дихлорметан (DCM), 1,2-дихлорэтан (DCE), хлороформ и тому подобные, ароматические углеводороды, такие как бензол, толуол, ксилол и тому подобные, простые эфиры, такие как простой диэтиловый эфир, тетрагидрофуран (THF), диоксан, диметоксиэтан (DME) и тому подобные, N,N-диметилформамид (DMF), диметилсульфоксид (DMSO), этилацетат, ацетонитрил или их смесь. Примеры карбонилирующего реагента включают дифосген, трифосген, 1,1'-карбонилдиимидазол (CDI), 4-нитрофенил хлорформиат, фенил хлорформиат и тому подобные. Когда производное сложного эфира карбоновой кислоты (VIII), которое представляет собой промежуточное соединение, устойчиво, то оно может быть сначала выделено, а затем подвергнуто следующей реакции. Далее, для реакции, используемой в этом способе получения, можно сослаться на следующую публикацию: “Organic Functional Group Preparations”, написанную S.R. Sandler и W. Karo, 2nd Edition< Viol. 2, Academic Press Inc., 1991.
Способ получения 2
Схема 8
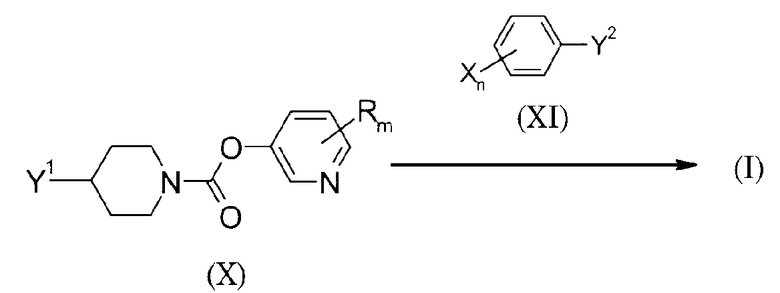
(В схеме одна из Y1 и Y2 представляет, например, группу, выбранную из -CO2H, -CONH2, -CONH-NH2, -N3, -OH и тому подобных, и, таким образом, другая представляет группу, выбранную из -C(=N-OH)-NH2, -C(=O)NH-NH2, -C(=O)-CH2-Br, этинильной группы и тому подобных. То же относится к описанному ниже).
Настоящий способ получения представляет собой способ, в котором соединение (X) взаимодействует с соединением (XI) с получением кольца А, посредством этого получая соединение формулы (I). Соединение формулы (I) может быть получено выбором подходящего вида Y1 и Y2 в соответствии с желательным кольцом А с использованием способа синтеза азольного кольца. Например, когда осуществляется получение соединения формулы (I), в котором кольцо А представляет собой 1,2,4-оксадиазол-3,5-диил, то могут использоваться соединение (X) и соединение (XI), в которых Y1 представляет -CO2H, а Y2 представляет -C(=N-OH)-NH2. Далее, когда кольцо А представляет собой 1,3,4-оксадиазол-2,5-диил, то в качестве Y1 и Y2 могут использоваться соответственно -CO2H и -C(=N-OH)-NH2. Далее, когда кольцо А представляет собой 1,2,3-триазол-1,4-диил, то в качестве Y1 и Y2 могут использоваться соответственно -N3 и этинильная группа. Далее, когда кольцо А представляет собой 1,3-оксазол-2,4-диил, то в качестве Y1 и Y2 могут использоваться соответственно -CONH-NH2 и -C(=O)-CH2-Br. Далее, когда кольцо А представляет собой тетразол-2,5-диил, то в качестве Y1 и Y2 могут использоваться соответственно тетразол-3-ил и -OH. Кроме того, ссылку на различные способы синтеза азольного кольца можно найти в следующей публикации: @Yetrocyclic Compounds, New Edition, Applications, написанной Hiroshi Yamanaka, Tohru Hino, Masako Nakagawa Takao Sakamoto, опубликованной Kodansha Ltd., Scientific, 2004.
Способ получения 3
Схема 9
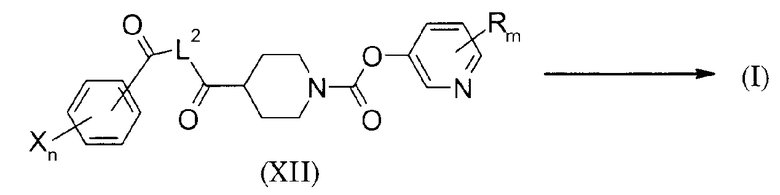
(В схеме L2 представляет двухвалентную связывающую цепь, имеющую 1-2 атома, релевантных длине цепи, и представляет собой, например, -HN-NH-, -CH2-NH- или метилен. То же относится к описанному ниже).
Настоящий способ получения представляет собой способ, в котором кольцо А синтезируется внутримолекулярной замыкающей кольцо реакцией соединения (XII) и, таким образом, получается соединение формулы (I). Например, соединение формулы (I), в котором кольцо А представляет собой пиразол-3,5-диил, может быть получено взаимодействием соединения (XII), в котором L2 представляет метилен, в присутствии гидразина моногидрата. Далее, соединение формулы (I), в котором кольцо А представляет собой 1,3,4-оксадиазол-2,5-диил, может быть получено взаимодействием соединения (XII), в котором L2 представляет собой -HN-NH-, в основных условиях с использованием тозилхлорида. Далее, соединение формулы (I), в котором кольцо А представляет собой 1,3-оксазол-2,5-диил, может быть получено взаимодействием соединения (XII), в котором L2 представляет собой -CH2-NH- в присутствии фосфорного оксихлорида. В этой связи, для различных способов синтеза азольного кольца, можно сослаться на следующую публикацию: Heterocyclic Compounds, New Edition, Appplicastions, написанную Hiroshi Yamanaka, Torhu Hino, Masako Nakagawa и Takao Sakammoto, опубликованную Kodansha, Ltd., Scientific, 2004.
Способы синтеза исходного материала
Способ 1 получения исходного материала
(Схема 10)
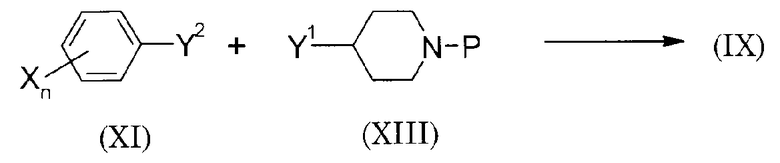
(В схеме P представляет защитную группу аминогруппы. И она представляет собой, например, трет-бутоксикарбонильную группу. То же относится к описанному ниже).
Аминное соединение (IX) может быть получено взаимодействием соединения (XI) с соединением (XIII) для образования кольца A и удалением защитной группы аминогруппы. В качестве Y1 и Y2 может использоваться такая же группа, как в описанном выше способе получения 2 в соответствии с видом целевого кольца A.
Способ 2 получения исходного материала
(Схема 11)
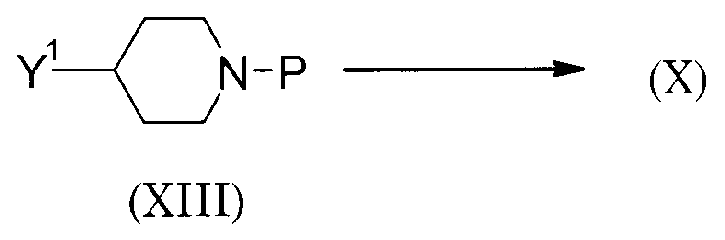
Соединение (X) может быть получено удалением защитной группы аминогруппы соединения (XIII), которое затем взаимодействует с описанным выше соединением (VIII). Реакция может проводиться таким же образом, как в описанном выше способе получения 1.
Способ 3 получения исходного материала
(Схема 12)
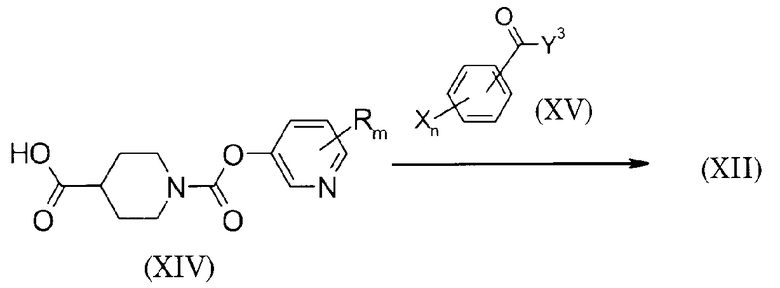
Соединение (XII) может быть получено взаимодействием соединения (XIV) с соединением (XV). В качестве Y3 подходящий заместитель выбирается в соответствии с видом L2 соединения (XII). Например, когда L2 соединения (XII) представляет собой метилен, то метил может использоваться в качестве Y3; когда L2 представляет -HN-NH-, то -HN-NH2 может использоваться в качестве Y3; и когда L2 представляет -CH2-NH-, то -CH2-NH2 может использоваться в качестве Y3.
Соединение формулы (I) может быть выделено и очищено в виде его свободного соединения, фармацевтически приемлемой соли, гидрата, сольвата или кристаллического полиморфного соединения. Фармацевтически приемлемая соль соединения формулы (I) может быть получена посредством обычной реакции образования соли.
Выделение и очистка могут проводиться использованием общих химических операций, таких как экстракция, фракционная кристаллизация, различные типы фракционной хроматографии и тому подобные.
Различные изомеры могут отделяться выбором соответствующего исходного соединения или использованием разности физико-химических свойств между изомерами. Например, оптические изомеры могут быть превращены в каждый стереохимически чистый изомер посредством общих способов оптического разрешения (например, фракционной кристаллизацией, превращающей соединение в диастереомерные соли с оптически активными основаниями или кислотами, хроматографией с использованием хиральной колонки или тому подобными способами). Далее, они могут также быть получены из соответствующего оптически активного исходного соединения.
Фармакологическая активность соединения по настоящему изобретению была подтверждена следующими тестами.
Пример тестирования 1
Скрининг для выявления вещества, ингибирующего активность FAAH с использованием клеток, полученных из раковой опухоли мочевого пузыря человека:
(1) Скрининг для выявления вещества, ингибирующего активность FAAH:
Полученные из раковой опухоли мочевого пузыря человека клетки клеточной линии 5637 (HTB-9; ATCC) высевали на 48-луночный культуральный планшет в количестве 1×105 клеток/лунку с использованием среды RPMII640 (Invitrogen), содержащей 10% фетальной телячьей сыворотки (HyClone). После культивирования при 37°С в течение 12 часов или дольше клетки промывали 400 мкг/лунку буфером (сбалансированный солевой раствор Хэнка, 20 мМ Hepes-NaOH (pH 7,4). Тестируемое вещество, растворенное в DMSO (диметилсульфоксиде), добавляли к раствору субстрата (указанный выше буфер, содержащий 3 мкКи/мл радиоактивно меченый анандамид (Анандамид [этаноламин 1-3H]) и 10 мкМ анандамида) с тем, чтобы концентрация составляла от 0,003 нМ до 30 нМ. В качестве контроля добавляли один DMSO. К указанным выше клеткам добавляли 100 мкл/лунку раствора субстрата и инкубировали в СО2 инкубаторе при 37°С в течение 30 минут. Затем, планшет для культуры клеток переносили на лед; раствор субстрата удаляли отсасыванием; и к нему добавляли ледяной раствор для лизиса клеток (указанный выше буфер, содержащий 0,5% Triton X-100 и 100 мкМ соединения, обладающего ингибирующей FAAH активностью, сложный 3'-карбамоилбифенил-3-иловый эфир циклогексилкарбаминовой кислоты (URB597; Cayman chemical; Kathuria et al., Nature Med., Vol. 9, pp. 76-81, 2003)). Полученный в результате клеточный лизат отдельно переносили в пробирки для образцов емкостью 1,5 мл, к которым добавляли 150 мкл раствора хлороформа/метанола в соотношении 1:1 (по объему) с последующим перемешиванием. После центрифугирования (15000 об/мин, 2 минуты) разрушенный продукт этаноламин (этаноламин 1-3H) отделялся в верхнем слое (слое воды/метанола), и не вступивший в реакцию радиоактивно меченый анадамид отделялся в нижнем слое (слое хлороформа). 25 мкл верхнего слоя переносили в 96-луночный, устойчивый к органическому растворителю микроплашет (PicoPlate-96; Perkin E1mer), к нему добавляли 150 мкл Microscint-20 (Perkin E1mer) и проводили измерение сцинтилляционным счетчиком микропланшет (TopCountTM; Beckman). Вещество, которое, по сравнению с контролем, давало уменьшенную величину измерения, выбирали в качестве вещества, ингибирующего активность FAAH.
(2) Измерение величины IC50 вещества, ингибирующего активность FAAH:
Соединение, растворенное в DMSO до концентрации 10 мМ, добавляли к раствору субстрата с тем, чтобы концентрация составляла от 0,003 нМ до 30 нМ. В соответствии со способом, описанным выше, соединение анализировали для выявления их влияния на активность FAAH. В качестве отрицательного контроля использовали DMSO, а в качестве положительного контроля к раствору субстрата добавляли URB597 до концентрации 10 мкМ. При измеренной величине положительного контроля, установленной на 0%, и измеренной величине отрицательного контроля, установленной на 100%, получали величины IC50 тестируемых веществ.
Пример тестирования 2
Скрининг для выявления вещества, ингибирующего активность FAAH с использованием гомогената ткани крыс, которым вводили тестируемое вещество:
(1) Введение крысам и получение гомогената ткани:
Тестируемое вещество, суспендированное в 0,5% растворе метилцеллюлозы (MC), перорально вводили 6-недельным самцам крыс SD (Japan SLC) в дозе 1 мг/кг. В качестве контроля двум крысам перорально вводили 0,5% раствор MC. Через 60 минут крыс умерщвляли декапитацией под эфирным наркозом, и затем у них брали правое полушарие.
К взятой ткани мозга крыс добавляли 2 мл ледяного буфера (50 мМ Tris-HCl (pH 8,0), 1 мМ EDTA (этилендиаминтетрауксусной кислоты)), и мозговую ткань гомогенизировали гомогенизатором на льду для получения однородного раствора. Далее, с использованием генератора ультразвуковых волн (UR-20P (Регулятор мощности 4), Tommy Seiko) раствор гомогената подвергали ультразвуковой фрагментации в течение 5 секунд. Концентрацию белка в полученных гомогенатах измеряли в соответствии со способом связывания красителя (раствор CBB (Кумасси бриллиантового синего) для анализа белка; Nacalai Te3sque Inc.)). С использованием буфера (50 мМ Hepes (pH 7,4), 1 мМ EDTA) гомогенаты мозговой ткани крыс разбавляли так, чтобы концентрация белка в них составляла 60 мкг/мл, посредством этого получая ферментные растворы.
(2) Измерение активности FAAH
К 200 мкл ферментного раствора добавляли 50 мкл раствора субстрата (2 мкМ анандамида с флюоресцентной меткой (Арахидонил-AMC (7-амино-4-метил Кумарин)) (BIOMOL), Hepes (pH 7,4) 1 мМ EDTA и 0,5 мг/мл BSA (бычьего сывороточного альбумина)) с последующей реакцией при комнатной температуре в течение 90 минут. Измерение проводили сцинтилляционным счетчиком микропланшет (TopCountTM; Beckman).
При установке активности FAAH контрольных крыс, не получавших тестируемое вещество, установленной на 100% и активности FAAH, не содержащего тканевого гомогената буфера (50 мСМ Hepes (pH 7,4), 1 мМ EDTA), установленной на 0%, определяли относительную величину (%)активности FAAH тканевого гомогената крыс, которым вводили тестируемое вещество.
Присутствие терапевтического эффекта при нейропатической боли может быть подтверждена способами, известными специалистам в данной области, или модифицированными указанными способами. Например, используя модель перевязки спинномозговых нервов L5/L6 у крыс, которая производится в соответствии с частичной модификацией способа Kim и Chung (Pain, Vol. 50, pp. 355-363, 1992), можно оценить улучшающий эффект соединения для значимого снижения порога реакции на тактильное раздражение (аллодинию), и на основании этого можно подтвердить эффект лечения нейропатической боли.
Пример тестирования 3
Эффект против аллодинии соединения у крыс с перевязкой L5/L6 спинномозговых нервов (модель нейропатической боли)
5-6-недельного самца крысы SD подвергали операции перевязки шелковыми нитями их левосторонних спинномозговых нервов L5 и L6 под пентобарбиталовым наркозом. Для оценки анальгетического эффекта использовали тест с волосками фон Фрея. То есть регистрировали одергивание задней лапы животного при раздражении волосками, при котором минимальную силу воздействия волосков, которая вызывала реакцию одергивания, рассматривали как порог реакции на механическое раздражение (логарифм грамм). В предварительном тесте было подтверждено, что порог реакции лапы животного на оперированной стороне был заметно снижен (при аллодинии) в пределах от 7-го дня до 14-го дня после операции, эффект против аллодинии тестируемого соединения оценивали в любой день в пределах от 7-го дня до 14-го дня после операции. В день перед днем, когда предполагалось испытывать тестируемое соединение, измеряли порог реакции перед введением тестируемого соединения. Испытуемых животных группировали так, чтобы было небольшим различие средних величин порога реакции между группами и их колебания внутри групп перед введением тестируемого соединения. При оценке тестируемых соединений измеряли порог реакции после введения тестируемого соединения. 3 мг/кг тестируемого соединения перорально вводили за 60 минут до измерения порога реакции. При установке порога реакции лап с оперированной и не оперированной стороны в группе с введением растворителя соответственно на 0% и 100% рассчитывали активность тестируемого соединения в отношении его эффекта против аллодинии (частоты восстановления).
Для нескольких репрезентативных соединений по настоящему изобретению результаты тестирования в примере тестирования 1 (величины IC50) и результаты тестирования в примере тестирования 3 (частота восстановления) показаны ниже. В связи с этим, в таблице “-“ означает, что измерение не проводилось.
Сравнительное соединение А: Соединение примера 126 в патентном документе 3
В результате описанного выше теста было показано, что соединение формулы (I) обладает ингибирующей активностью в отношении FAAH и является эффективным на моделях нейропатической боли. Поэтому, соединение формулы (I) может применяться в качестве средства для предотвращения и/или лечения различных заболеваний, связанных с FAAH. Кроме того, его можно применять, наряду с другими показаниями, в качестве средства для лечения нейропатической боли.
Фармацевтическая композиция, содержащая один или более видов соединения формулы (I) или его фармацевтически приемлемой соли в качестве активного ингредиента, может быть получена в соответствии с общепринятым способом с использованием фармацевтического носителя, фармацевтического эксципиента или тому подобного, что обычно используется в данной области.
Введение может проводиться любым видом перорального введения посредством таблеток, пилюль, капсул, гранул, порошков, жидких препаратов и тому подобных, или парентерального введения посредством инъекций, таких как внутрисуставная, внутривенная, внутримышечная или другие, суппозиторий, глазных капель, глазных мазей, чрескожных жидких препаратов, мазей, трансдермальных систем, жидких препаратов для введения через слизистые оболочки, накладок для введения через слизистые оболочки, ингаляционных препаратов и тому подобных.
В качестве твердой композиции для перорального введения применяются таблетки, порошки, гранулы или тому подобные формы. В такой твердой композиции один или два или более активных ингредиентов смешиваются, по меньшей мере, с одним неактивным эксципиентом, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, магния алюминометасиликат и/или тому подобные. В соответствии с обычным способом, композиция может содержать неактивные добавки, такие как смазывающие вещества, такие как стеарат магния и тому подобные, разрыхлители, такие как натрия карбоксиметилкрахмал и тому подобные, стабилизаторы и солюбилизирующие агенты. Таблетки или пилюли могут быть покрыты сахарным покрытием или при необходимости пленкой гастро- или энтеросолюбильного вещества.
Жидкая композиция для перорального введения включает фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры или тому подобные и содержит общепринятый инертный разбавитель, такой как очищенная вода или этанол. В дополнение к инертному разбавителю, жидкая композиция может содержать адъюванты, такие как солюбилизирующие агенты, смачивающие агенты и суспендирующие агенты, подслащивающие агенты, отдушки, ароматизаторы и антисептики.
Инъекционные препараты для парентерального введения включают стерильные, водные или неводные растворы, суспензии или эмульсии. В качестве водного растворителя могут использоваться дистиллированная вода для инъекции или физиологический солевой раствор. Примеры неводного растворителя включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и тому подобные, спирты, такие как этанол и тому подобные, Полисорбат 80 (Фармакопея) и т.д. Такая композиция может, кроме того, содержать агенты тоничности, антисептики, смачивающие агенты, эмульгирующие агенты, диспергирующие агенты, стабилизаторы или солюбилизирующие агенты. Они стерилизуются, например, фильтрацией через задерживающий бактерии фильтр, смешиванием с бактерицидными средствами или облучением. Кроме того, они могут также использоваться получением стерильных твердых композиций и растворением или суспендированием их в стерильной воде или стерильном растворителе для инъекции перед их применением.
Средство для наружного применения включает мази, пластыри, кремы, желе, катаплазмы, аэрозоли, лосьоны, глазные капли, глазные мази и тому подобные. Эти средства содержат общепринятые мазевые основы, лосьонные основы, водные или неводные препараты, суспензии, эмульсии и тому подобные. Примеры мазевых основ или лосьонных основ включают полиэтиленгликоль, пропиленгликоль, белый вазелин, отбеленный пчелиный воск, полиоксиэтилен, гидрированное касторовое масло, глицерил моностеарат, стеариловый спирт, цетиловый спирт, лауромакрогол, сорбитан сесквиолеат и тому подобные.
В качестве средств для введения через слизистые оболочки, таких как ингаляционные средства, средства для интраназального введения и тому подобные, используются средства в твердом, жидком или полужидком состоянии, и они могут быть получены в соответствии с общеизвестными способами. Например, к ним могут при целесообразности добавляться известные эксципиенты, а также агенты, регулирующие рН, антисептики, ПАВ, смазывающие агенты, стабилизаторы, загустители или тому подобные ингредиенты. Для их введения могут использоваться соответствующие устройства для ингаляции или инсуффляции. Например, соединение может вводиться отдельно или в виде порошков составленной смеси, или в виде раствора или суспензии объединением его с фармацевтичски приемлемыми носителями с использованием общеизвестных устройств или аэрозольных распылителей, таких как ингаляционное устройство для введения отменной дозы и тому подобные. Ингаляторы сухого порошка или тому подобные устройства могут быть предназначены для одноразового или множественного применения, и могут применяться сухие порошки или содержащие порошок капсулы. Альтернативно, они могут быть в форме аэрозольного спрея в герметизированных баллончиках, в которых используется соответствующий вытеснитель, такой как хлорфторалкан или гидрофторалкан, или подходящий газ, такой как диоксид углерода или тому подобный.
В случае перорального введения целесообразно, чтобы суточная доза обычно составляла примерно от 0,001 до 100 мг/кг, предпочтительно, от 0,1 до 30 мг/кг, а предпочтительнее, от 0,1 до 10 мг/кг массы тела, и эта доза вводится одной порцией или дробно 2-4 порциями. Также, в случае внутривенного введения, целесообразная суточная доза составляет примерно от 0,0001 до 10 мг/кг массы тела, и введение проводится один раз в сутки или два или более раз в сутки. Кроме того, средство для введения через слизистые оболочки вводится в дозе примерно от 0,001 до 100 мг/кг массы тела один раз в сутки или два или более раз в сутки. Доза соответствующим образом определяется в каждом конкретном случае с учетом симптомов, возраста и пола и тому подобных факторов.
Соединение формулы (I) может применяться в комбинации с различными терапевтическими средствами или профилактическими средствами, применяемыми для лечения заболеваний, при которых соединение формулы (I) считается эффективным, как описано выше. Комбинированный препарат может вводиться одновременно или отдельно и непрерывно или через желательный интервал времени. Препараты, подлежащие совместному введению, могут представлять собой смесь или получены отдельно.
Примеры
Ниже способы получения соединения формулы (I) будут более детально описаны со ссылкой на примеры. В связи с этим, настоящее изобретение не ограничивается соединениями, описанными в примерах ниже. Также, способы получения соединений, представляющих исходный материал, показаны в примерах получения. Кроме того, способы получения соединения формулы (I) не ограничиваются способами получения конкретных примеров, приведенных ниже, и соединение формулы (I) может быть получено в соответствии с комбинацией таких способов получения или способами, очевидными специалистам в данной области.
Пример получения 1
При охлаждении льдом к смеси трет-бутил 4-(4-фенил-1,3-тиазол-2-ил)пиперидин-1-карбоксилата (5,79 г) и DCM (30 мл) добавляли 4М гидрохлорид/диоксан (30 мл) с последующим перемешиванием в течение 3 часов. Полученное твердое вещество собирали фильтрацией, промывали простым диизопропиловым эфиром и затем сушили при пониженном давлении с получением 4-(4-фенил-1,3-тиазол-2-ил)пиперидина гидроксихлорида (4,51 г).
Пример получения 2
К смеси трет-бутил 4-(аминокарбонтионил)пиперидин-1-карбоксилата (500 мг) и DMF (5 мл) добавляли 2-бром-1-(4-хлорфенил)этанон (573 мг) с последующим перемешиванием при комнатной температуре в течение 2 часов, и растворитель выпаривали при пониженном давлении. К остатку добавляли воду и этилацетат, и органическую фазу отделяли. Органическую фазу промывали водой и насыщенным рассолом и затем сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении, и остаток сушили при пониженном давлении. К остатку добавляли DCM (6 мл) и, при охлаждении льдом 4 М, гидрохлорид/диоксан (6 мл) с последующим нагреванием до комнатной температуры и затем перемешиванием в течение 4 часов. Растворитель выпаривали при пониженном давлении, к остатку добавляли простой диизопропиловый эфир и небольшое количество метанола, и полученное твердое вещество собирали фильтрацией. Твердое вещество промывали простым диизопропиловым эфиром и затем сушили при пониженном давлении с получением 4-[4-(4-хлорфенил)-1,3-тиазол-2-ил]пиперидина гидрохлорида (437 мг).
Пример получения 3
Смесь диизопропиламина (3,23г) и THF (20 мл) охлаждали до 0°С, и к ней медленно добавляли 1,57М н-бутиллития/гексана (20,4 мл) с последующим перемешиванием при такой же температуре в течение 1 часа. Затем смесь охлаждали до -70°С и к ней по каплям добавляли ацетофенон (3,84 г) с последующим перемешиванием при такой же температуре в течение 1 часа (реакционная жидкость 1). Тем временем к суспензии 1-[(пиридин-3-илокси)карбонил]пиперидин-4-карбоновой кислоты (2,0 г) в THF (30 мл) добавляли CDI (1,56 г) с последующим перемешиванием при комнатной температуре в течение 1 часа (реакционная жидкость 2). Реакционную жидкость 2 охлаждали до -70°С и к ней по каплям добавляли реакционную жидкость 1 с последующим перемешиванием при такой же температуре в течение 1 часа. Затем, смесь согревали до 0°С и затем согревали до комнатной температуры. К реакционной жидкости добавляли 0,1М хлористоводородную кислоту (50 мл), затем к ней добавляли воду и этилацетат, и органическую фазу отделяли. Органическую фазу промывали водой и насыщенным рассолом и затем сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10) с получением пиридин-3-ил 4-(3-оксо-3-фенилпропаноил)пиперидин-1-карбоксилата (0,93 г).
Пример получения 4
К смеси 1-[(пиридин-3-илокси)карбонил]пиперидин-4-карбоновой кислоты (1,5 г) и DCM (15 мл) добавляли 1-гидроксибензотриазол (HOBt) (0,85 г), бензгидразин (0,86 г) и 1-этил-3-(3-диметиламинопропил)карбодиимид (WSC) гидрохлорид (1,21 г) с последующим перемешиванием при комнатной температуре в течение примерно 14 часов. К реакционной жидкости добавляли хлороформ и воду, и органическую фазу отделяли. Органическую фазу промывали водой и насыщенным рассолом и сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10). К очищенному продукту добавляли простой диизопропиловый эфир и метанол, и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением пиридин-3-ил 4-[(2-бензоилгидразин)карбонил]пиперидин-1-карбоновой кислоты (1,22 г).
Пример получения 5
При охлаждении льдом к смеси этилбензолкарбоксиимидата гидрохлорида (4,58 г) и этанола (50 мл) добавляли натрий этоксид (1,68 г) с последующим перемешиванием при такой же температуре в течение примерно 20 минут. Затем при той же температуре к ней добавляли трет-бутил 4-(гидразинкарбонил)пиперидин-1-карбоксилат (5,0 г) с последующим нагреванием до комнатной температуры, затем перемешиванием в течение 1 часа и нагревания при кипячении в сосуде с обратным холодильником в течение 1 суток. После охлаждения растворитель выпаривали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10). К очищенному продукту добавляли метанол и простой диизопропиловый эфир, и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением трет-бутил 4-(3-фенил-1H-1,2,4-триазол-5-ил)пиперидин-1-карбоксилата (2,53 г).
Пример получения 6
К смеси пиридин-3-ил 4-гидроксипиперидин-1-карбоксилата (330 мг), триэтиламина (0,25 мл) и DCM (7 мл) медленно по каплям добавляли хлорид метансульфоновой кислоты (0,13 мл) при комнатной температуре. После перемешивания в течение ночи реакционную жидкость очищали непосредственно хроматографией на колонке силикагеля (гексан/этилацетат = от 50/50 до 0/100) с получением пиридин-3-ил 4-[(метилсульфонил)окси]пиперидин-1-карбоксилата (390 мг) в виде бесцветного твердого вещества.
Пример получения 7
К раствору трет-бутил 4-[амино(гидроксиимино)метил]пиперидин-1-карбоксилата (3,0 г) в THF (30 мл) добавляли 3,5-дивторбензоил хлорид (2,4 г) и триэтиламин (3,44 мл) при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли этилацетат и воду, и органическую фазу отделяли. Органическую фазу промывали водой и насыщенным рассолом, и сушили над безводным сульфатом магния, и растворитель выпаривали при пониженном давлении. К остатку добавляли THF (25 мл) и 1М раствора тетрабутиламмония фторида/THF (12,4 мл) с последующим перемешиванием при 50°С в течение 30 минут. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ). К очищенному продукту добавляли простой диизопропиловый эфир, и полученное твердое вещество собирали фильтрацией и сушили с получением трет-бутил 4-[5-(3,5-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидин-1-карбоксилата (4,22 г) в виде оранжевого твердого вещества.
Пример получения 8
Смесь трет-бутил 4-(гидразинкарбонил)пиперидин-1-карбоксилата (1,0 г), 4-хлорбензонитрила (1,7 г), калия карбоната (0,28 г)т и бутанола (8,0 мл) нагревали при 150єС в течение 2 ч с использованием микроволнового устройства. После охлаждения растворитель выпаривали при пониженном давлении, и остаток азеотропной перегонки с толуолом. Остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10). К очищенному продукту добавляли простой диизопропиловый эфир, и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением трет-бутил 4-[3-(4-хлорфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилата (0,62 г).
Пример получения 9
К раствору 3-фенил-1H-пиразола (300 мг) в толуоле (15 мл) добавляли трет-бутил 4-гидроксипиперидин-1-карбоксилат (838 мг) и (трибутилфосфоранилиден)ацетонитрил (1,0 г) с последующим перемешиванием при 100°С в течение 4 часов. Реакционную жидкость концентрировали при пониженном давлении, и затем остаток очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 100/0 до 50/50) и снова очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 100/0 до 70/30) с получением трет-бутил 4-(3-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилата (475 мг) в виде бесцветного маслянистого вещества.
Пример получения 10
К раствору 3-(диметиламино)-2-(4-фторфенил)акрилальдегида (3,0 г) в этаноле (30 мл) добавляли гидразин моногидрат (0,90 мл) с последующим нагреванием и кипячением в сосуде с обратным холодильником в течение 3 часов. После охлаждения растворитель выпаривали при пониженном давлении до тех пор, пока количество реакционной жидкости не уменьшалось примерно до половины. К ней добавляли воду (20 мл), и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением 4-(4-фторфенил)-1H-пиразола (2,44 г) в виде желтого твердого вещества.
Пример получения 11
К смеси 6-метилпиридин-3-ола (1,8 г), CDI (2,64 г) и DMSO (18 мл) по каплям добавляли изонипекотовую кислоту (4,2 г) и смесь DMSO (18 мл) и трифторуксусной кислоты (2,5 мл) с последующим перемешиванием при комнатной температуре в течение 1 суток. К реакционной жидкости добавляли насыщенный рассол и хлороформ, и органическую фазу отделяли. Органическую фазу промывали насыщенным рассолом дважды и затем сушили над безводным сульфатом натрия, и растворитель концентрировали при пониженном давлении. К остатку добавляли простой диизопропиловый эфир/метанол, и полученное твердое вещество собирали фильтрацией и сушили с получением 1-{[(6-метилпиридин-3-ил)окси]карбонил}пиперидин-4-карбоновой кислоты (3,51 г) в виде бесцветного твердого вещества.
Пример получения 12
К смеси 1-[(пиридин-3-илокси)карбонил]пиперидин-4-карбоновой кислоты (500 мг) и DCM (10 мл) добавляли HOBt (297 мг) и WSC гидрохлорид (498 мг), с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем к ней добавляли 2-амино-1-(2-фторфенил)этанон гидрохлорид (417 мг) и триэтиламин (0,31 мл) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную жидкость очищали непосредственно хроматографией на колонке силикагеля (хлороформ/метанол = от 99/1 до 95/5) с получением пиридин-3-ил 4-{[2-(2-фторфенил)-2-оксоэтил]карбамоил}пиперидин-1-карбоксилата (423 мг) в виде бесцветного твердого вещества.
Пример получения 13
Смесь 1-трет-бутил 4-этилпиперидин-1,4-дикарбоксилата (21 г), гидразина моногидрата (40 мл) и этанола (200 мл) нагревали кипячением в сосуде с обратным холодильником в течение 22 часов. После охлаждения растворитель выпаривали при пониженном давлении, к остатку добавляли насыщенный рассол и этилацетат, и органическую фазу отделяли. Органическую фазу сушили над сульфатом магния, и растворитель выпаривали при пониженном давлении. К остатку добавляли простой диизопропиловый эфир с последующим перемешиванием в течение 1 часа, и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением трет-бутил-4-(гидразинкарбонил)пиперидин-1-карбоксилат (17,8 г).
Пример получения 14
К смеси 6-метилпиридин-3-ола (5,00 г) и ацетонитрила (44 мл) добавляли CDI (7,43 г) с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем, к ней добавляли пиперидин-4-ол (4,41 г) и 4М гидрохлорид/диоксан (23 мл) с последующим перемешиванием при 50°С в течение ночи. После охлаждения к реакционной жидкости добавляли воду и хлороформ, и органическую фазу отделяли. Органическую фазу промывали насыщенным рассолом и сушили над безводным сульфатом натрия, и растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол= от 99/1 до 90/10) с получением 6-метилпиридин-3-ил 4-гидроксипиперидин-1-карбоксилата (8,65 г) в виде бесцветного твердого вещества.
Пример получения 15
К бензил 4-{[[2-(4-фторфенил)-2-оксоэтил]карбамоил}пиперидин-1-карбоксилату (5,5 г) добавляли фосфорный оксихлорид (20 мл) с последующим перемешиванием при 80°С в течение 3 часов. После охлаждения реакционную жидкость концентрировали при пониженном давлении, и остаток азеотропной перегонки с толуолом три раза. К остатку добавляли этилацетат и воду, и органическую фазу отделяли. Органическую фазу промывали насыщенным водным раствором натрия бикарбоната и насыщенным рассолом в указанном порядке и сушили над безводным сульфатом натрия, и растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 70/30 до 30/70) с получением 4-[5(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилата (1,84) в виде бесцветного маслянистого вещества.
Пример получения 16
К раствору бензил 4-[5-4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилата (1,84 г) в этаноле (40 мл) добавляли 10% палладий/углерод (влажность 54%, 200 мг) с последующим перемешиванием в течение 6 часов в атмосфере водорода. Катализатор удаляли фильтрацией через целит, и фильтрат концентрировали при пониженном давлении. К остатку добавляли этанол и 4М гидрохлорид/диоксан (1,45 мл) и концентрировали при пониженном давлении. К остатку добавляли этанол и этилацетат с последующим перемешиванием, и полученное твердое вещество собирали фильтрацией и сушили с получением 4-[5-4-фторфенил)-1,3-оксазол-2-ил]пиперидин гидрохлорида (1,32 г) в виде бесцветного твердого вещества.
Пример получения 17
Суспензию трет-бутил 4-аминопиперидин-1-карбоксилата (3,88 г), оксоуксусной кислоты (1,48 г) и карбоната калия (4,46 г) в DMF (60 мл) перемешивали при комнатной температуре в течение 3 часов. Затем к ней добавляли 1-{[изоциано(фенил)метил]сульфонил}-4-метилбензол (3,5 г) с последующим перемешиванием при комнатной температуре в течение 14 часов. Растворитель выпаривали при пониженном давлении, к остатку добавляли этилацетат и воду, и органическую фазу отделяли. Водную фазу экстрагировали этилацетатом, объединенную органическую фазу промывали водой и насыщенным рассолом и сушили над сульфатом магния, и растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10) с получением трет-бутил-4-(4-фенил-1H-имидазол-1-ил)пиперидин-1-карбоксилата (3,1 г).
Пример получения 18
К смеси трет-бутил 4-оксопиперидин-1-карбоксилата (10,0 г), бензилгидразинкарбоксилата (16,7 г), DCM (150 мл) и уксусной кислоты (5,75 мл) добавляли триацетоксиборгидрид натрия (31,9 г) с последующим перемешиванием при комнатной температуре в течение 2,5 суток. К реакционной жидкости добавляли воду, и органическую фазу отделяли. Водную фазу экстрагировали хлороформом, и объединенную органическую фазу промывали водой, насыщенным водным раствором бикарбоната натрия и насыщенным рассолом в указанном порядке и сушили над сульфатом магния, и растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10) с получением трет-бутил-4-{2-[(бензилокси)карбонил]гидразин}пиперидин-1-карбоксилат (10,0 г).
Пример получения 19
К смеси трет-бутил 4-оксопиперидин-1-карбоксилата (10,0 г) и этанола (100 мл) добавляли 5% палладий/углерод (2,0 г) с последующим перемешиванием в атмосфере водорода в течение примерно 2 часов. Катализатор удаляли фильтрацией, и растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 90/10) с получением трет-бутил-4-гидразинпиперидин-1-карбоксилата (4,1 г).
Пример получения 20
К смеси 2-метилпиридин-3-ил 4-{[2-(трет-бутоксикарбонил)гидразин]карбоксил}пиперидин-1-карбоксилата (11,98 г) и DCM (100 мл) добавляли 4 М гидрохлорид/диоксан (100 мл) с последующим перемешиванием при комнатной температуре в течение примерно 15 часов. Растворитель выпаривали при пониженном давлении, и остаток растворяли в метаноле/воде (10/1). К нему добавляли карбонат калия (8,75 г) с последующим перемешиванием в течение примерно 3 часов. Растворитель выпаривали при пониженном давлении, и к остатку добавляли хлороформ с последующей сушкой над сульфатом магния. Растворитель выпаривали под пониженным давлением, и остаток сушили при пониженном давлении с получением 2-метилпиридин-3-ил 4-(гидразинкарбонил)пиперидин-1-карбоксилата (7,49 г).
Пример получения 21
К смеси 2,3-дифторбензонитрила (5,00 г) и этанола (55 мл) по каплям добавляли ацетил хлорид (35 мл) при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 7 дней. Реакционную жидкость концентрировали при пониженном давлении, и к остатку добавляли простой диизопропиловый эфир с последующим перемешиванием в течение 1 часа. Полученное твердое вещество собирали фильтрацией и сушили с получением этил 2,3-дифторбензолкарбоксиимидата гидрохлорида (4,68 г) в виде белого твердого вещества.
Пример получения 22
К смеси трет-бутил 4-[(2-бензоилгидразин)карбонил]пиперидин-1-карбоксилата (3,00 г) и THF ((60 мл) добавляли триэтиламин (7,2 мл) и толуолсульфонил хлорид (4,94 г) с последующим перемешиванием при 50°С в течение ночи. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 95/5 до 80/20) с получением трет-бутил 4-(5-фенил-1,3,4-оксадиазол-2-ил)пиперидин-1-карбоксилата (2,84 г) в виде бесцветного маслянистого вещества.
Пример получения 23
К смеси 1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты (5,0 г) и толуола (50 мл) добавляли CDI (3,9 г) с последующим перемешиванием при комнатной температуре в течение 3 часов. Затем к ней добавляли N'-гидроксибензолкарбоксиимидат (3,3 г) с последующим перемешиванием в течение 1,5 часов и затем нагреванием и кипячением в сосуде с обратным холодильником в течение 2 часов. После охлаждения к реакционной жидкости добавляли этилацетат и воду, и органическую фазу отделяли. Органическую фазу промывали водой и насыщенным рассолом и сушили над сульфатом магния, и растворитель выпаривали при пониженном давлении. К остатку добавляли гексан и этилацетат, и полученное твердое вещество собирали фильтрацией с получением трет-бутил 4-(3-фенил-1,2,4-оксадиазол-5-ил)пиперидин-1-карбоксилата (5,46 г).
Пример получения 24
Смесь трет-бутил 4-(3-оксо-фенилпропаноил)пиперидин-1-карбоксилата (3,1 г), гидразина моногидрата (0,5 мл), этанола (30 мл) и THF (30 мл) перемешивали при комнатной температуре в течение примерно 15 часов и при 60°С в течение 1 часа. К ней далее добавляли гидразин моногидрат (0,5 мл) с последующим перемешиванием снова при 60°С в течение 3 часов. К ней снова добавляли гидразин моногидрат (4,0 мл) с последующим перемешиванием при 60°С в течение 8 часов. После охлаждения растворитель выпаривали при пониженном давлении, и к остатку добавляли простой диизопропиловый эфир и метанол с последующим перемешиванием. Полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением трет-бутил 4-(3-фенил-1H-пиразол-5-ил)пиперидин-1-карбоксилата (2,46 г).
Пример получения 25
К раствору трет-бутил 4-гидразинпиперидин-1-карбоксилата (646 мг) и этанола (15 мл) добавляли фенилмалональдегид (444 мг) с последующим перемешиванием при 75°С в течение примерно 1,5 суток. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (гексан/хлороформ = от 50/50 до 0/100) с получением трет-бутил 4-(4-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилата (226 мг).
Пример получения 26
Смесь трет-бутил 4-[(2-оксо-2-фенилэтил)карбамоил]пиперидин-1-карбоксилата (5,0 г) и трифторацетата аммония (18,9 г) перемешивали при наружной температуре 170°С в течение 30 минут. После охлаждения к ней добавляли воду и хлорформ, и водную фазу отделяли. рН водной фазы доводили примерно до 10 24% водным раствором гидроксида натрия и экстрагировали хлороформом. Объединенную органическую фазу промывали водой и насыщенным рассолом и сушили над сульфатом магния, и растворитель выпаривали при пониженном давлении. Остаток сушили при пониженном давлении и растворяли в DCM (20 мл) и метаноле (10 мл), и к нему добавляли 4М гидрохлорид/диоксан (5,3 мл). Растворитель концентрировали при пониженном давлении, к остатку добавляли простой диизопропиловый эфир/метанол, и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением 4-(4-фенил-1H-имидазол-2-ил)пиперидина дигидрохлорида (3,29 г).
Пример получения 27
К раствору 2,5-дифторбензойной кислоты (1,95 г) в THF (40 мл) добавляли оксалилхлорид (1,5 мл) и каталитическое количество DMF с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную жидкость концентрировали при пониженном давлении и к остатку добавляли THF (40 мл). При охлаждении льдом к ней добавляли трет-бутил 4-[амино(гидроксиимино)метил]пиперидин-1-карбоксилат ((2,5 г) и триэтиламин (3,0 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли этилацетат и воду, и органическую фазу отделяли. Органическую фазу промывали водой и насыщенным рассолом и сушили над безводным сульфатом магния, и растворитель выпаривали при пониженном давлении. К раствору остатка в THF (20 мл) добавляли 1М раствор тетрабутиламмония фторида/THF (10,3 мл) с последующим перемешиванием при 50°С в течение 30 минут. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ). К очищенному продукту добавляли 4М гидрохлорид/диаоксан (40 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении, затем к остатку добавляли THF, и полученное твердое вещество собирали фильтрацией. Твердое вещество промывали THF и этилацетатом в указанном порядке и сушили при пониженном давлении с получением 4-[5-(2,5-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидина гидрохлорида (2,58 г).
Пример получения 28
К суспензии 3-(4-фторфенил)-1H-1,2,4-триазола (700 мг) в толуоле (15 мл) добавляли трет-бутил 4-гидроксипиперидин-1-карбоксилат (1,3 г) и (трибутилфосфоранилиден)ацетонитрил (2,0 г) с последующим перемешиванием при 110°С в течение 15 часов. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 100/0 до 97/3). К очищенному продукту добавляли 4М гидрохлорид/диоксан (15 мл) с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционной жидкости добавляли этилацетат, и полученное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением 4-[3-(4-фторфенил)-1H-1,2,4-триазол-1-ил]пиперидина гидрохлорда (422 мг).
Пример получения 29
К смеси 1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты (5,0 г) и DCM (50 мл) добавляли HOBt (3,09 г), 4-фторбензгидразина (3,53 г) и WSC гидрохлорида (5,02 г) с последующим перемешиванием при комнатной температуре в течение ночи. К реакционной жидкости добавляли этилацетат с последующим промыванием водой/насыщенным рассолом (1:1), насыщенным водным раствором бикарбоната натрия и насыщенным рассолом в указанном порядке и сушкой над безводным сульфатом натрия, и растворитель выпаривали при пониженном давлении. К остатку добавляли THF (160 мл), хлорид п-толуолсульфонила (8,32 г) и триэтиламин (12 мл) с последующим перемешиванием при 60°С в течение ночи. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 90/10 до 50/50) с получением трет-бутил 4-[5-(4-фторфенил)-1,3,4-оксадиазол-2-ил]пиперидин-1-карбоксилата (5,83 г) в виде бледно-коричневого твердого вещества.
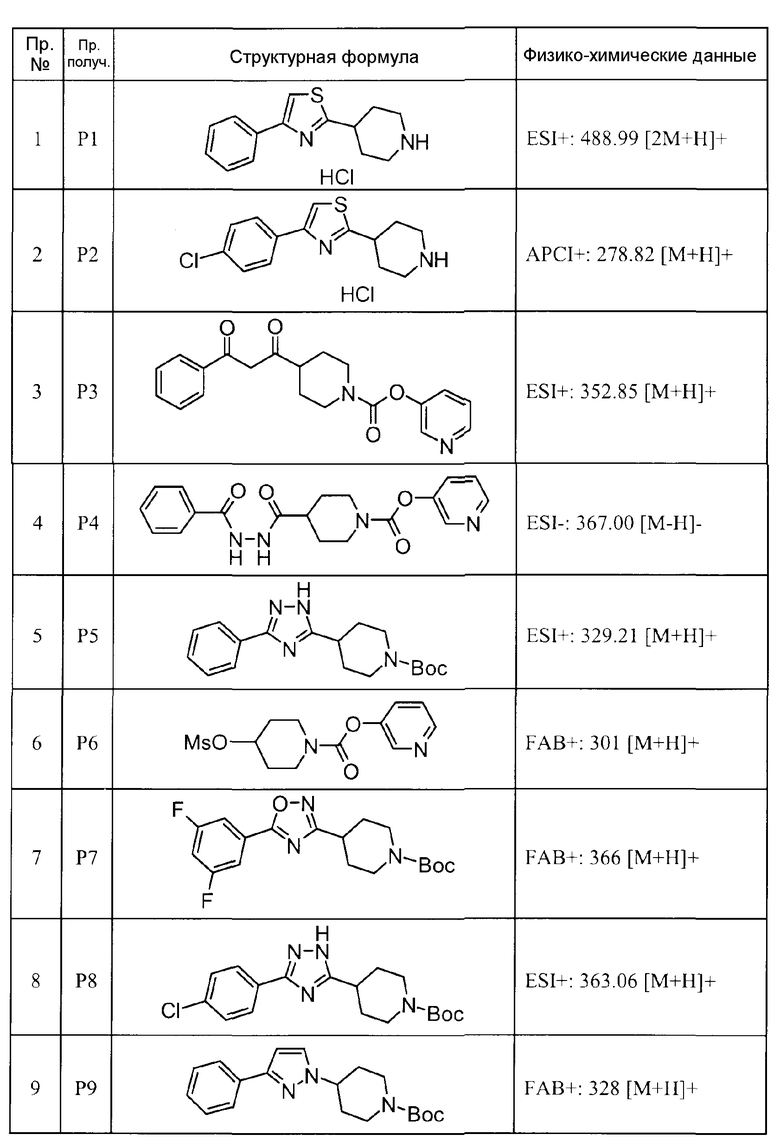
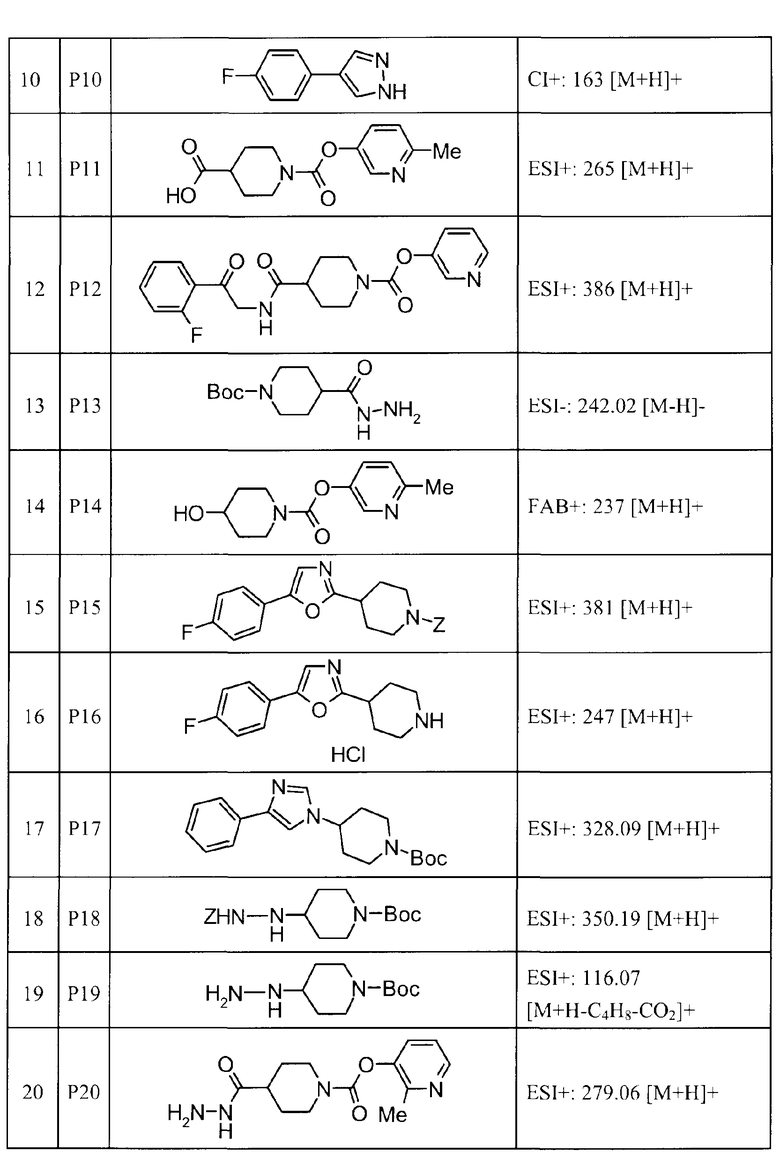
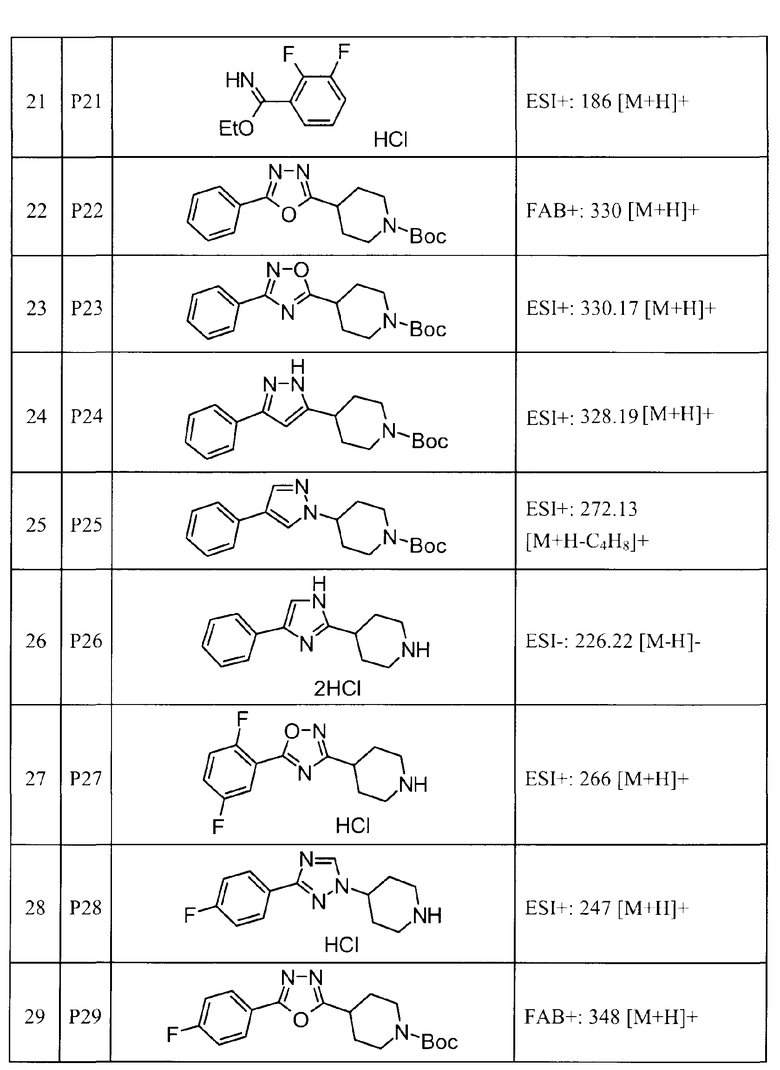
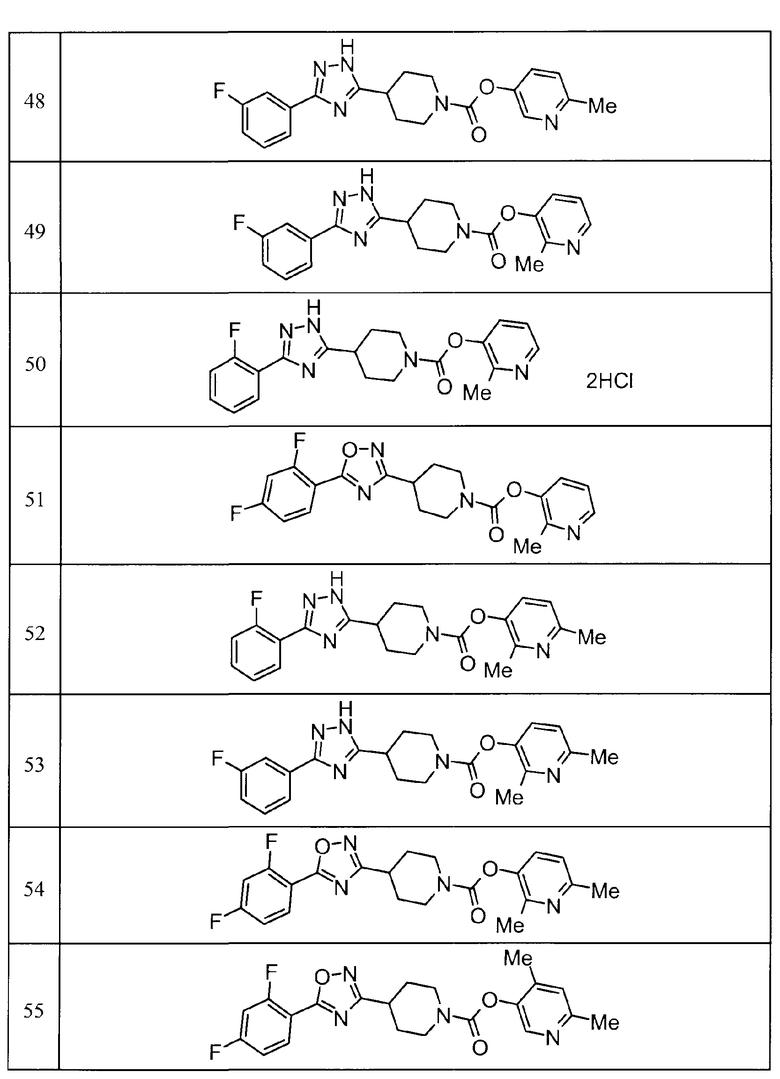
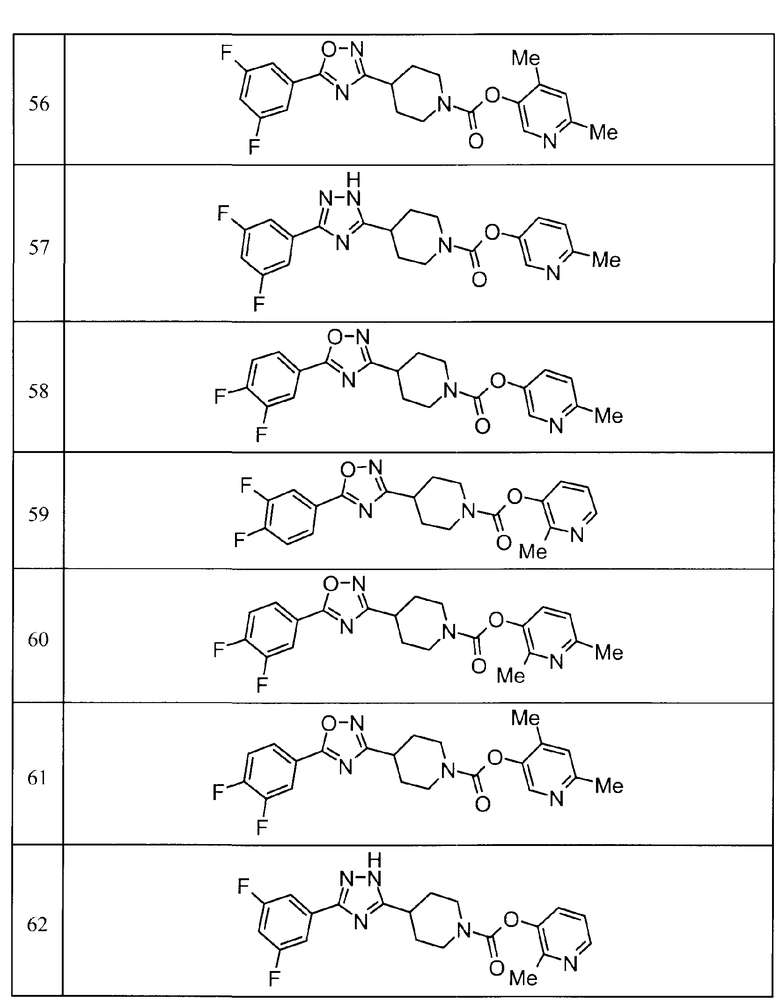
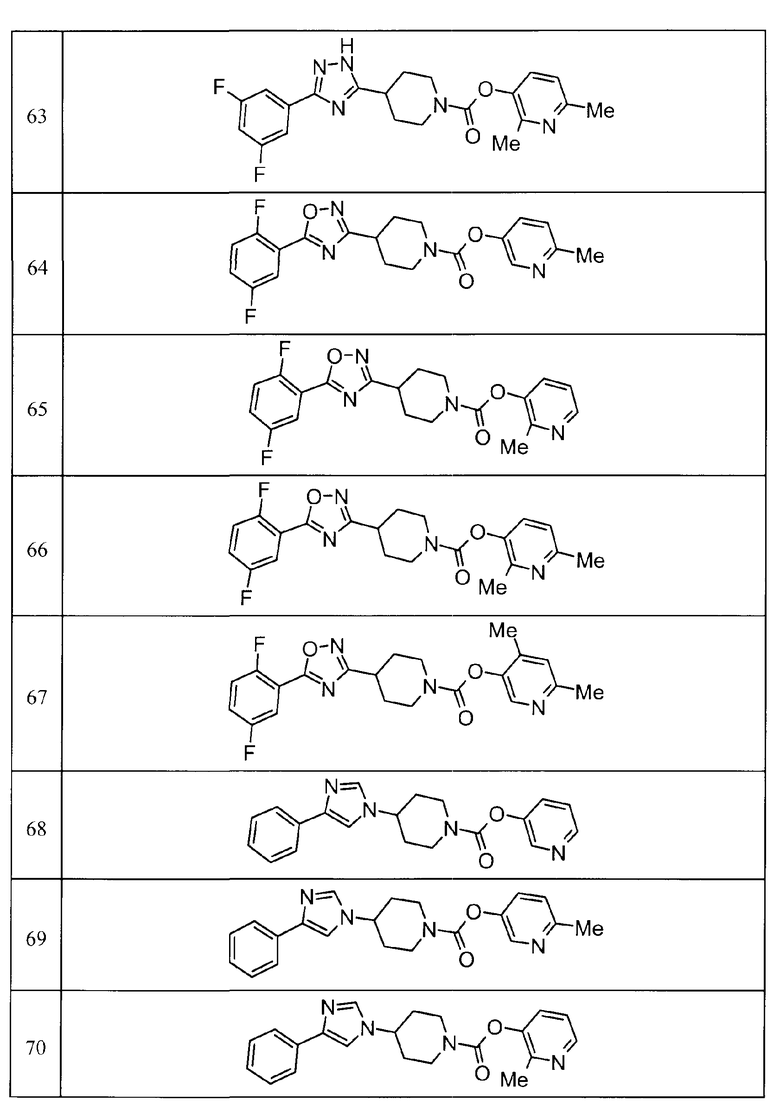
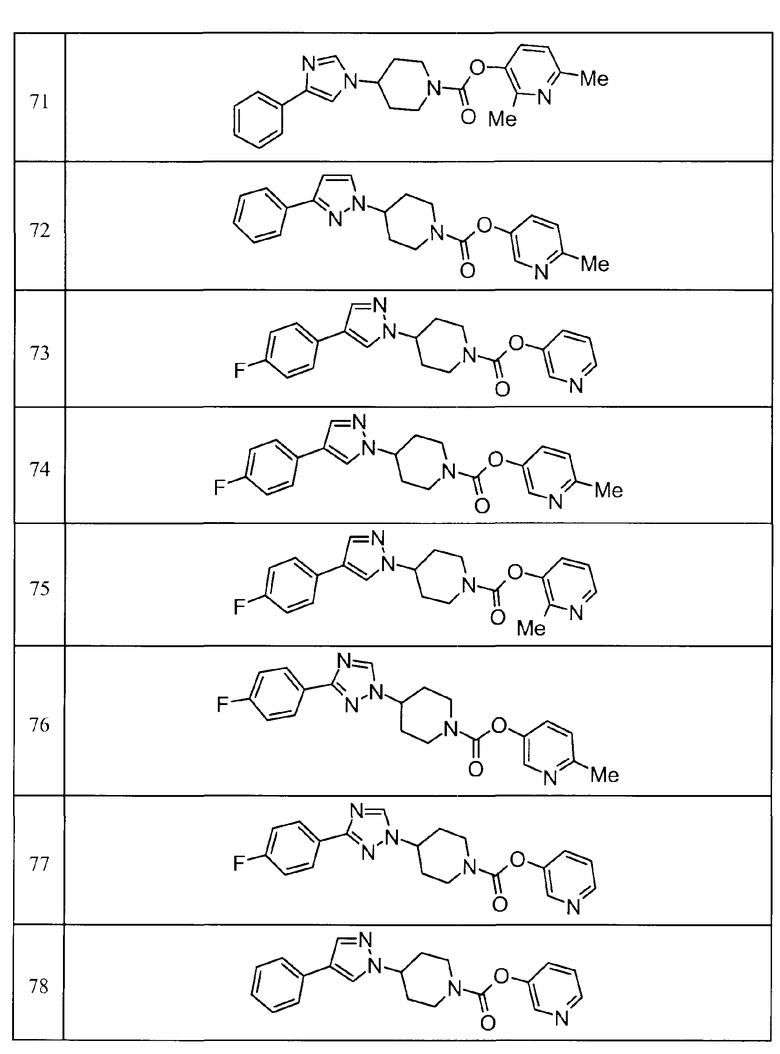
Такими же способами получения, как для соединений описанных выше примеров получения от 1 до 29, получали соединения примеров получения, как показано в таблицах, подлежащих описанию ниже, с использованием каждого из соответствующих исходных материалов. Структуры, способы получения и физико-химические данные соединений примеров получения показаны в представленных ниже таблицах 2-11.
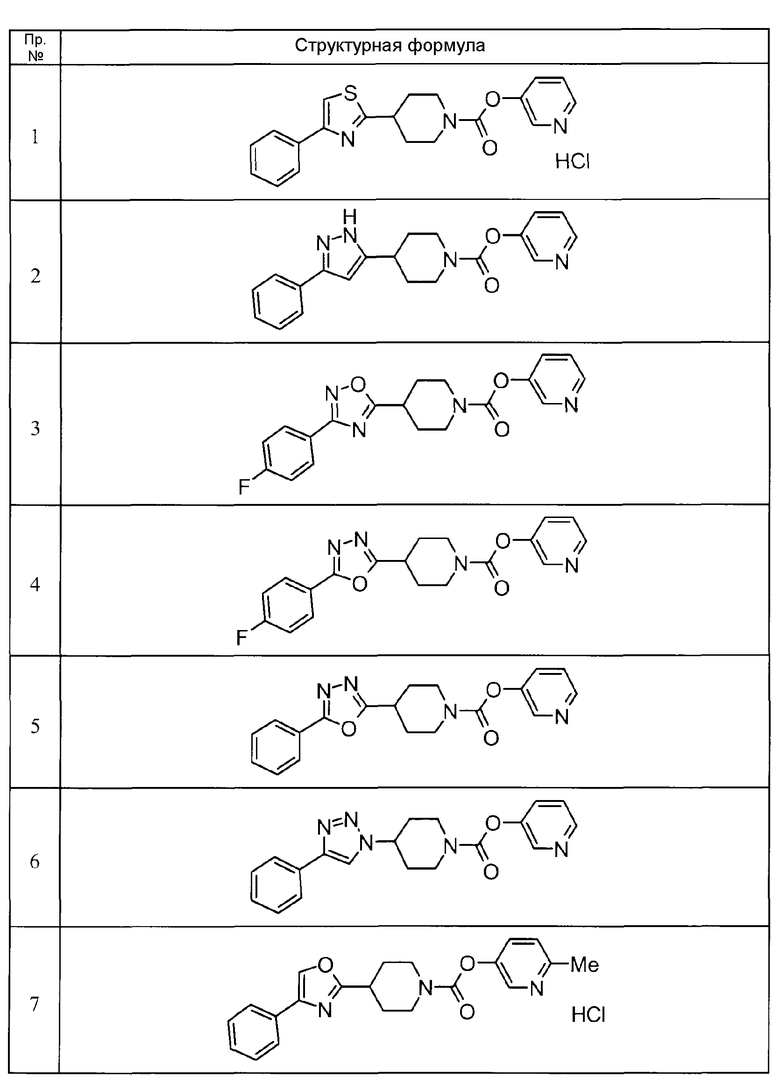
Пример 1
Такой же процедурой, как в описанном выше примере получения 14, пиридин-3-ил 4-(4-фенил-1,3-тиазол-2-ил)пиперидин-1-карбоксилат гидрохлорид получали из 4-(4-фенил-1,3-тиазол-2-ил)пиперидина гидрохлорида.
Пример 2
Такой же процедурой, как в описанном выше примере получения 24, пиридин-3-ил 4-(3-фенил-1H-пиразол-5-ил)пиперидин-1-карбоксилат получали из пиридин-3-ил 4-(3-оксо-3-фенилпропаноил)пиперидин-1-карбоксилата.
Пример 3
Такой же процедурой, как в описанном выше примере получения 22, пиридин-3-ил 4-[3-(4-фторфенил)-1,2,4-оксадиазол-5-ил]пиперидин-1-карбоксилат получали из 1-[(пиридин-3-илокси)карбонил]пиперидин-4-карбоновой кислоты.
Пример 4
К смеси 1-[(пиридин-3-илокси)карбонилпиперидин-4-карбоновой кислоты (300 мг), 4-фторбензгидразида (222 мг), HOBt (170 мг) и DCM (6 мл) добавляли WSC гидрохлорид (299 мг) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную жидкость очищали непосредственно хроматографией на колонке силикагеля (хлороформ/метанол = от 99/1 до 90/10). Остаток растворяли в THF (6 мл) и к нему добавляли толуолсульфонил хлорид (686 мг) и триэтиламин (1,0 мл) с последующим перемешиванием при 50°С в течение 8 часов. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 70/30 до 0/100). К очищенному продукту добавляли изопропанол/воду, и полученное твердое вещество собирали фильтрацией и сушили с получением пиридин-3-ил 4-[5-(4-фторфенил)-1,3,4-оксадиазол-2-ил]пиперидин-1-карбоксилата (224 мг) в виде бесцветного твердого вещества.
Пример 5
Такой же процедурой, как в описанном выше примере получения 22, пиридин-3-ил 4-(5-фенил)-1,3,4-оксадиазол-2-ил)пиперидин-1-карбоксилат получали из пиридин-3-ил 4-[(2-бензоилгидразин)карбонил]пиперидин-1-карбоксилата.
Пример 6
К смеси пиридин-3-ил 4-[(метилсульфонил)окси]пиперидин-1-карбоксилата (221 мг) и DMSO (4 мл) добавляли азид натрия (96 мг) с последующим перемешиванием при 60°С в течение 8 часов. Реакционную жидкость разбавляли этилацетатом и промывали водой и насыщенным рассолом в указанном порядке. Органическую фазу сушили над безводным сульфатом натрия, растворитель выпаривали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 70/30 до 0/100). К очищенному продукту (149 мг) и раствору этинилбензола (0,066 мл) в трет-бутаноле (10 мл) добавляли воду (2 мл), аскорбат натрия (12 мг) и сульфат меди (II) (1,5 мг) с последующим перемешиванием в течение ночи. Реакционную жидкость разбавляли этилацетатом и промывали водой и насыщенным рассолом. Органическую фазу сушили над безводным сульфатом натрия, растворитель концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 99/1 до 90/10). К очищенному продукту добавляли простой диизопропиловый эфир/этилацетат с последующим перемешиванием, и полученное твердое вещество собирали фильтрацией и сушили с получением пиридин-3-ил 4-(4-фенил)-1H-1,2,3-триазол-1-ил)пиперидин-1-карбоксилата (165 мг) в виде бесцветного твердого вещества.
Пример 7
Смесь 6-метилпиридин-3-ил 4-карбамоилпиперидин-1-карбоксилата (500 мг), 2-бром-1-фенилэтанона (453 мг) и N,N-диметилацетамида (5 мл) перемешивали при 130°С в течение 3 суток. После охлаждения к ней добавляли этилацетат и воду/насыщенный водный раствор бикарбоната натрия (1:1) с последующим перемешиванием в течение 1 часа, и реакционную жидкость фильтровали. Органическую фазу фильтрата отделяли, промывали водой/насыщенным рассолом (1:1) и насыщенным рассолом в указанном порядке и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 99/1 до 95/5). Очищенный продукт растворяли в этаноле и к нему добавляли избыточное количество 4М гидрохлорида/диоксана. Реакционную жидкость концентрировали при пониженном давлении и сушили с получением 6-метилпиридин-3-ил 4-(4-фенил-1,3-оксазол-2-ил)пиперидин-1-карбоксилата гидрохлорида (134 мг) в виде бледно-коричневого аморфного вещества.
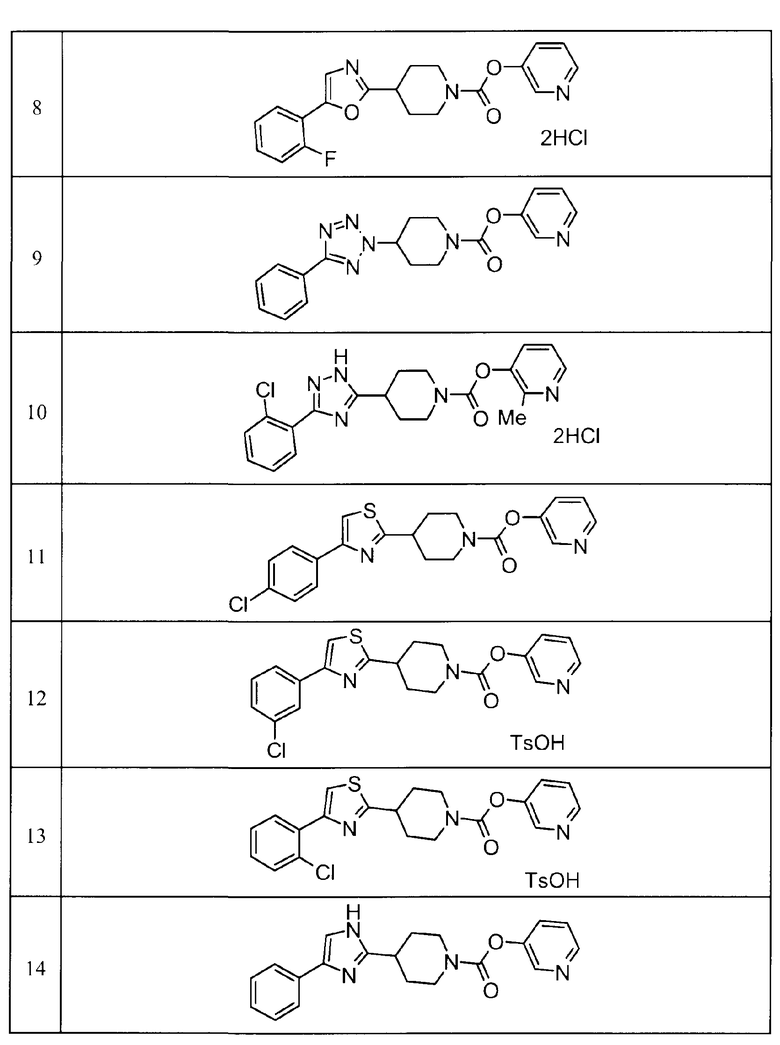
Пример 8
Такой же процедурой, как в описанном выше примере получения 15, пиридин-3-ил 4-[5-(2-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат дигидрохлорид получали из пиридин-3-ил 4-{[(2-(2-фторфенил)-2-оксоэтил]карбамоил}пиперидин-1-карбоксилата.
Пример 9
К смеси пиридин-3-ил 4-гидроксипиперидин-1-карбоксилата (300 мг), 5-фенил-1H-тетразола (217 мг), трифенилфосфина (460 мг) и THF (3 мл) добавляли по каплям 2,2 М раствор диэтилазодикарбоксилата в толуоле (0,8 мл) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную жидкость концентрировали при пониженном давлении, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 99/1 до 90/10) и снова очищали хроматографией на колонке силикагеля (гексан/этилацетат = от 70/30 до 0/100). К очищенному продукту добавляли гексан/этилацетат с последующим перемешиванием, и затем полученное твердое вещество собирали фильтрацией и сушили с получением пиридин-3-ил 4-(5-фенил-2H-тетразол-2-ил)пиперидин-1-карбоксилата (250 мг) в виде бесцветного твердого вещества.
Пример 10
К раствору этил 2-хлорбензолкарбоксиимидата гидрохлорида (435 мг) в этаноле (10 мл) добавляли метоксид натрия (107 г) с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем к нему добавляли 2-метилпиридин-3-ил 4-(гидразинкарбонил)пиперидин-1-карбоксилат (500 мг) с последующим перемешиванием при 90°С в течение 2 суток. Реакционную жидкость концентрировали при пониженным давлением, и остаток очищали хроматографией на колонке силикагеля (хлороформ/метанол = от 99/1 до 90/10). Очищенный продукт растворяли в этаноле и к нему добавляли избыточное количество 4 М гидрохлорида/диоксана с последующим перемешиванием. Реакционную жидкость концентрировали при пониженном давлении и сушили с получением 2-метилпиридин-3-ил 4-[3-(2-хлорфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилата дигидрохлорида (289 мг) в виде бледно-желтого твердого вещества.
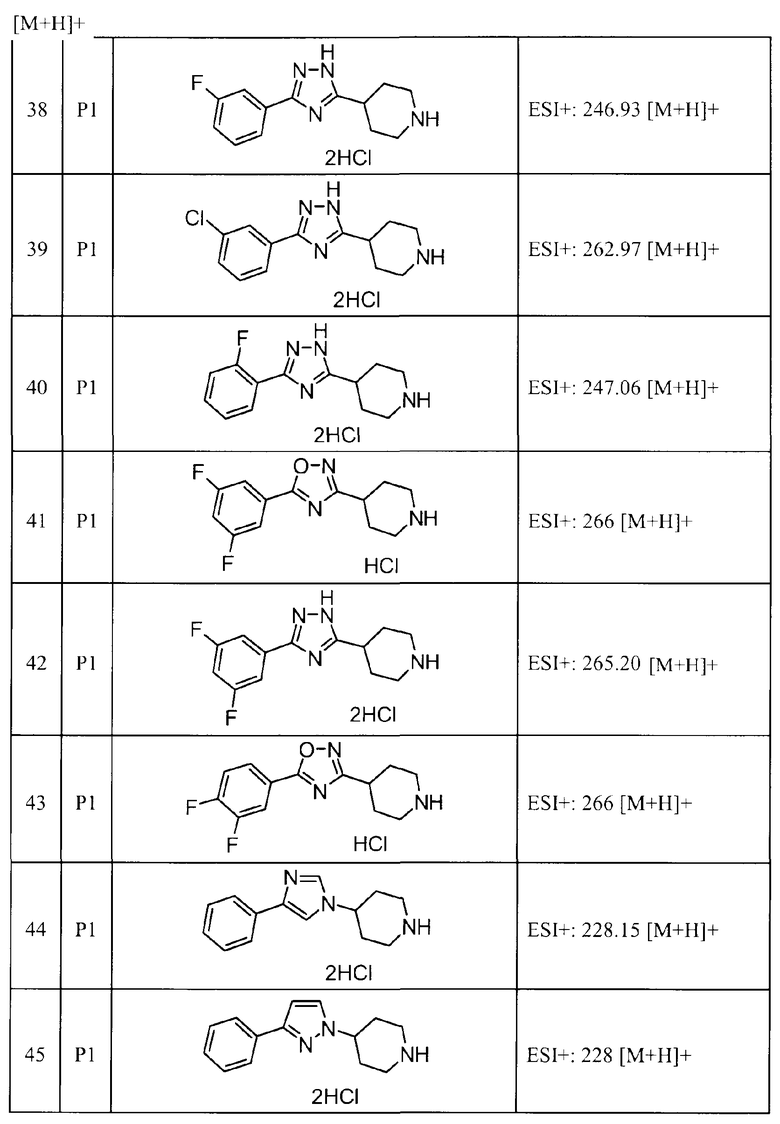
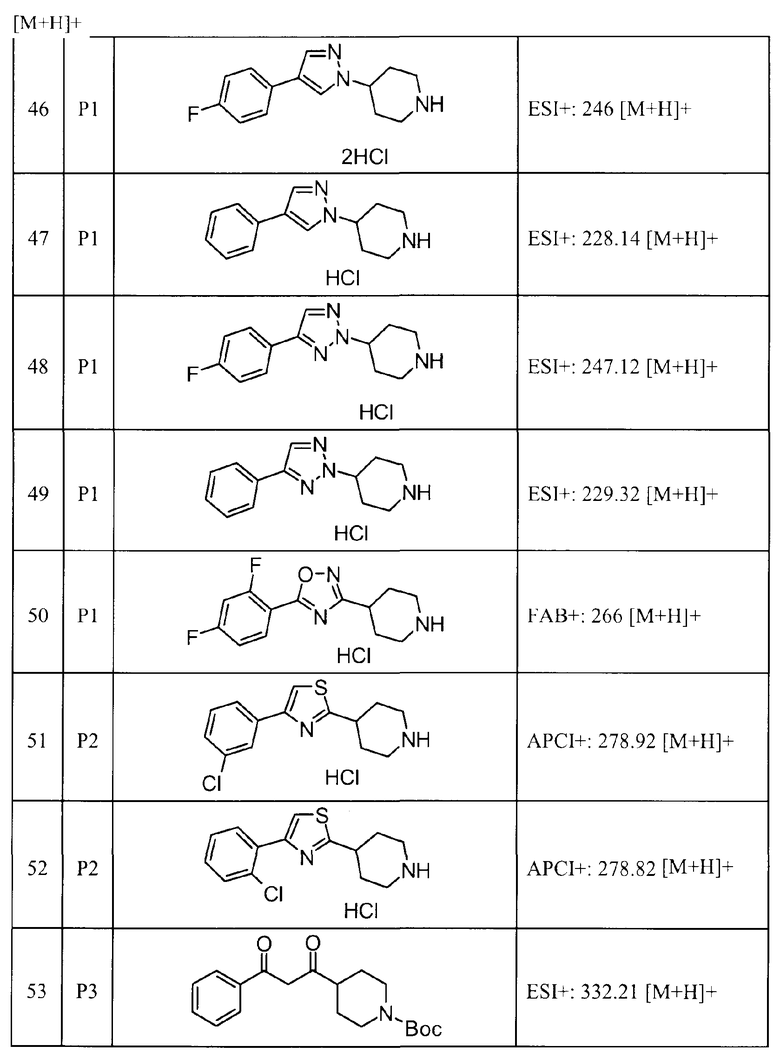
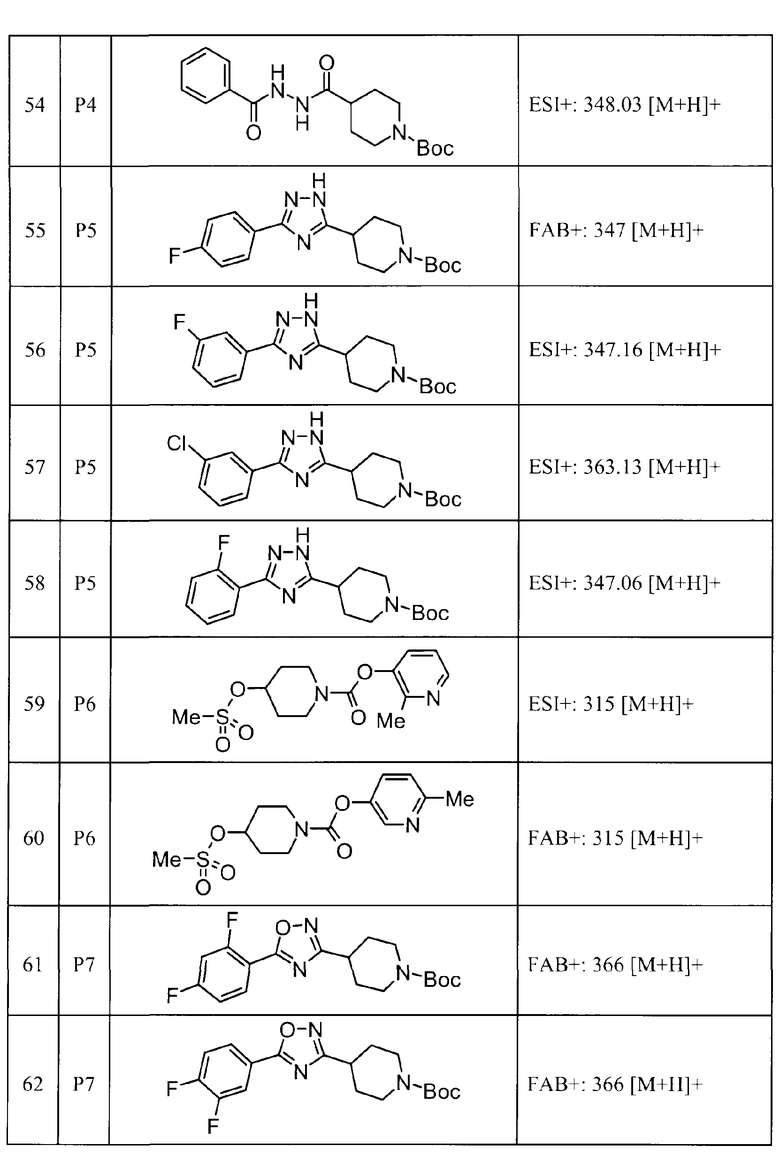
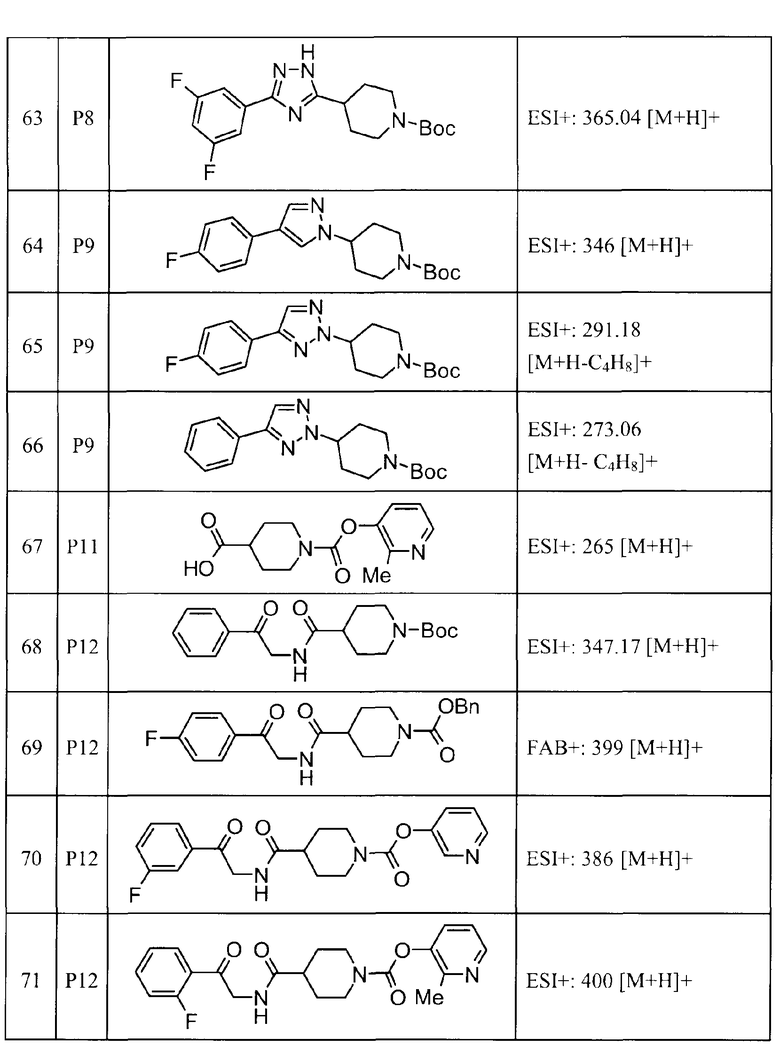
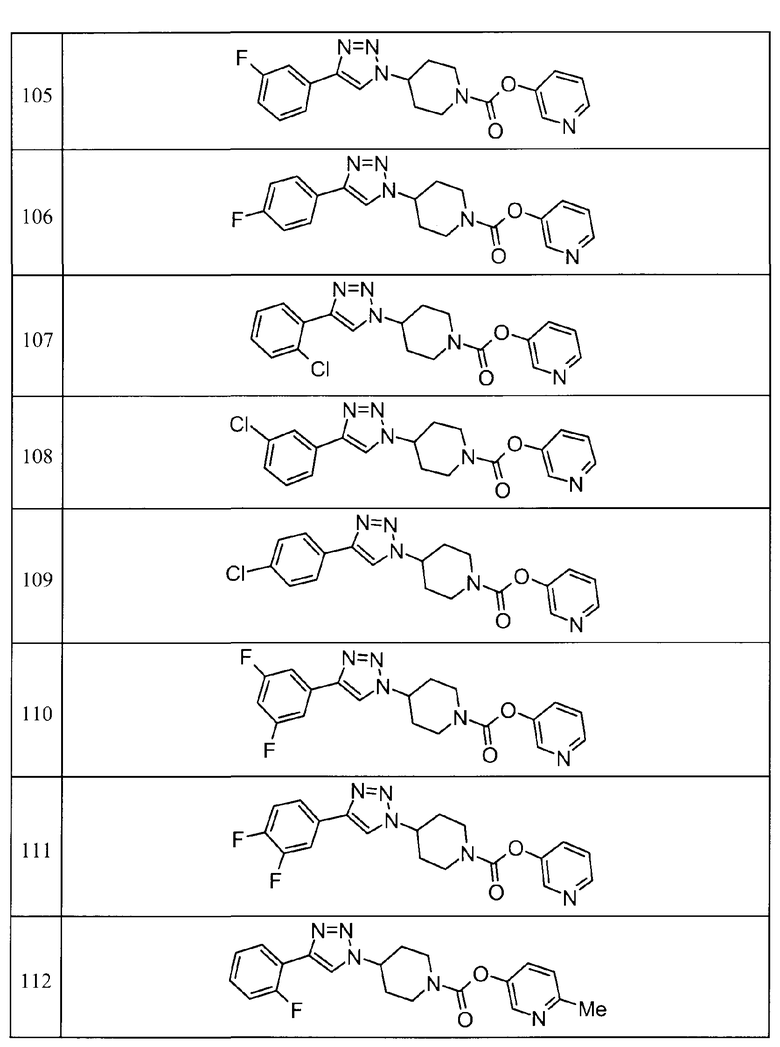
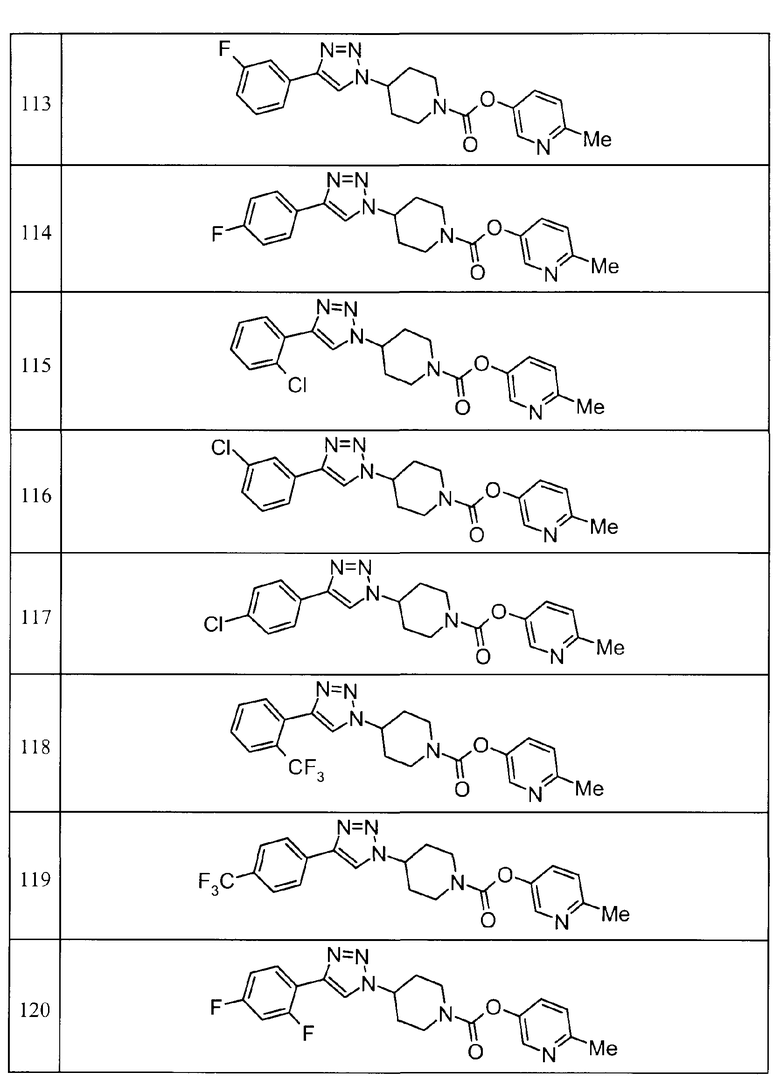
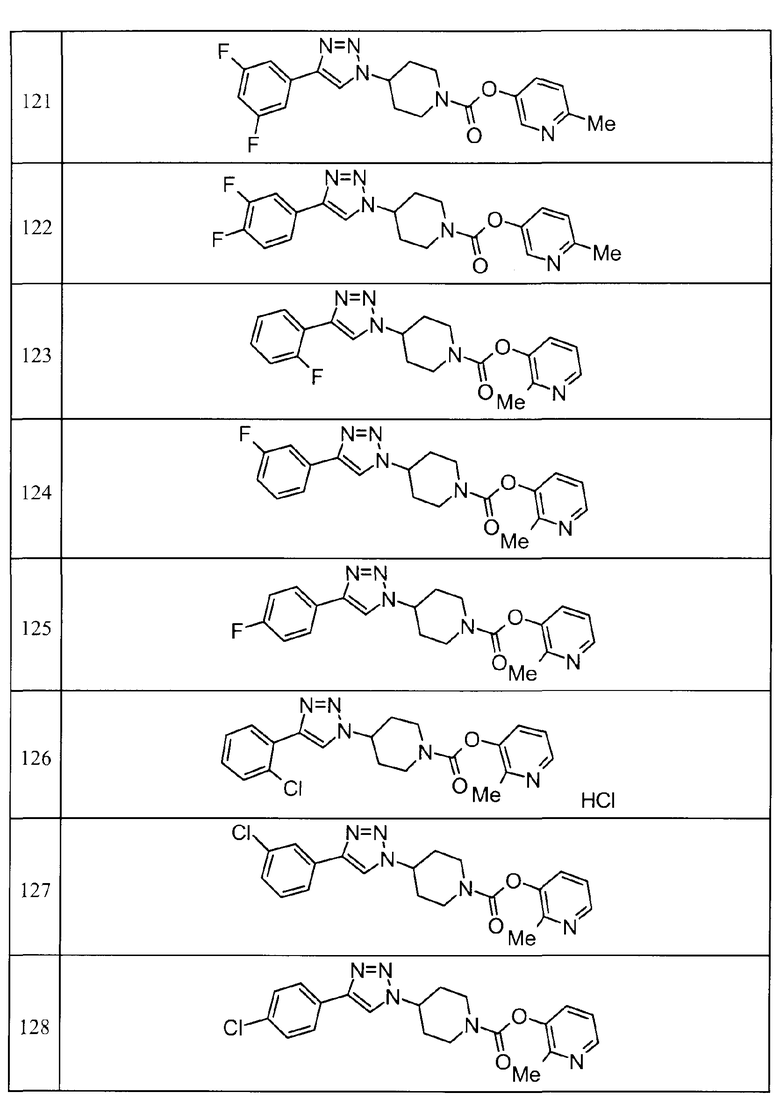
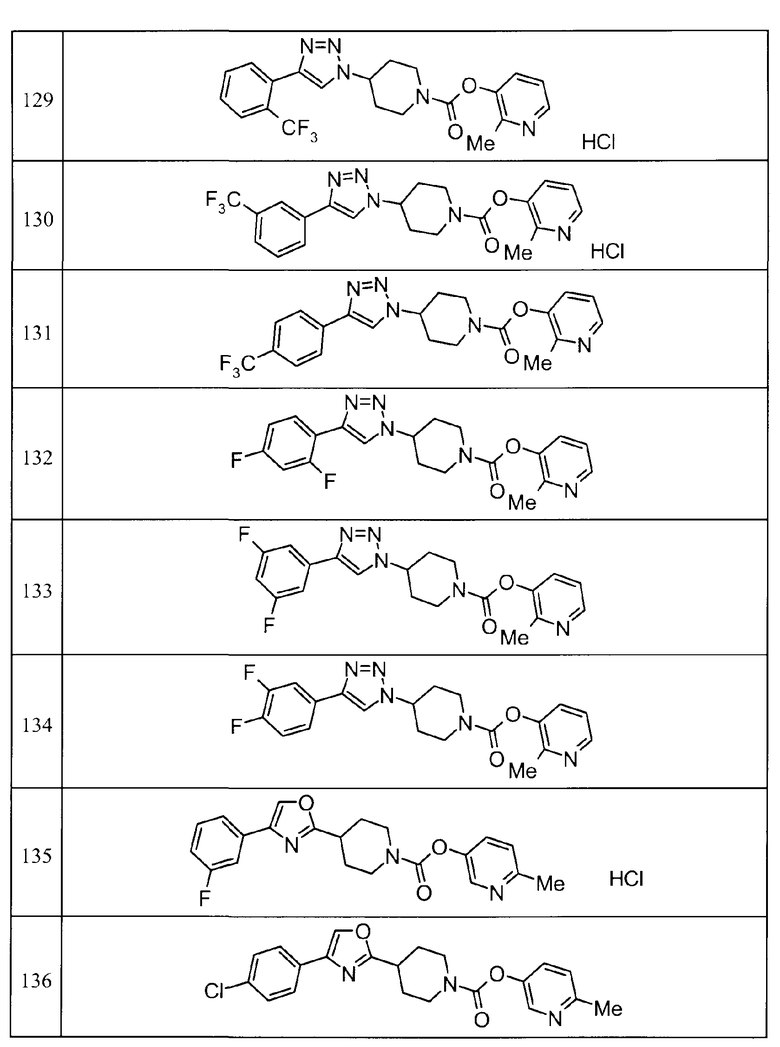
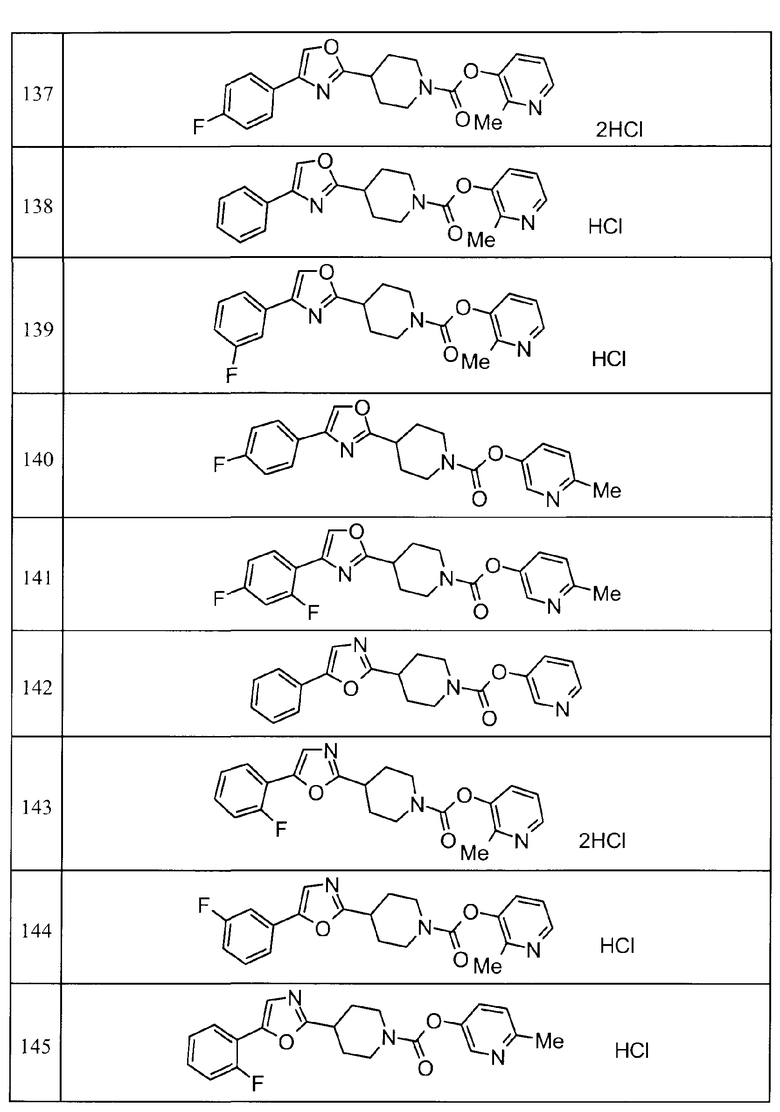
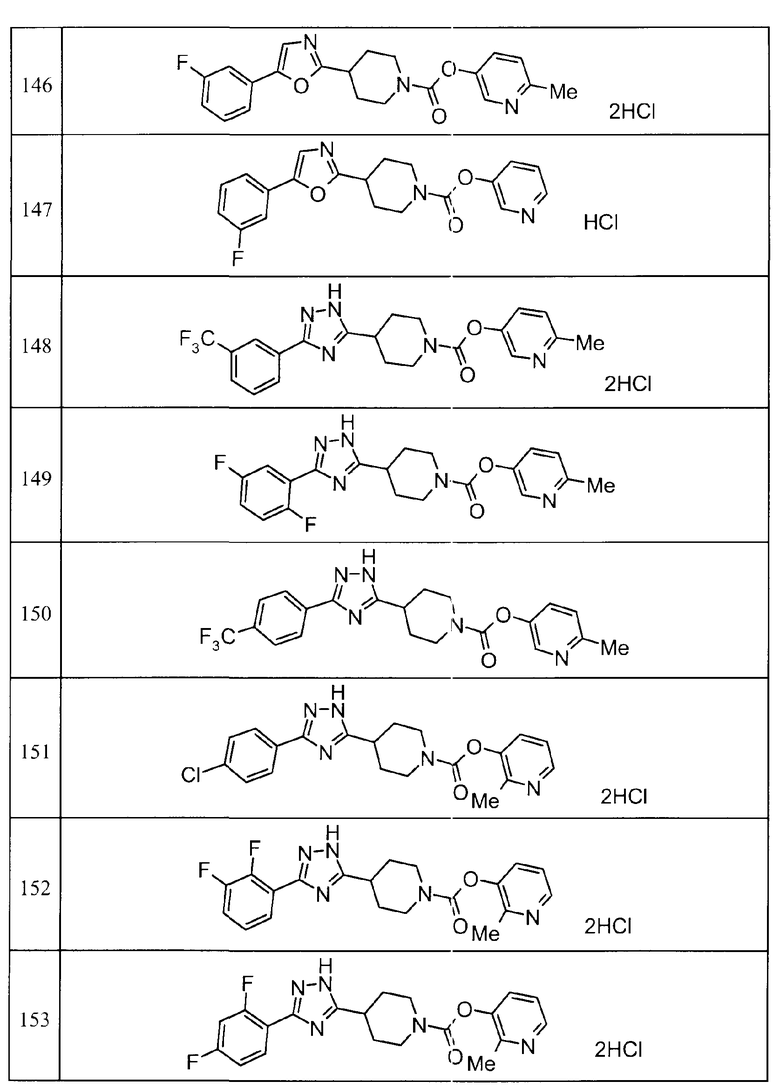
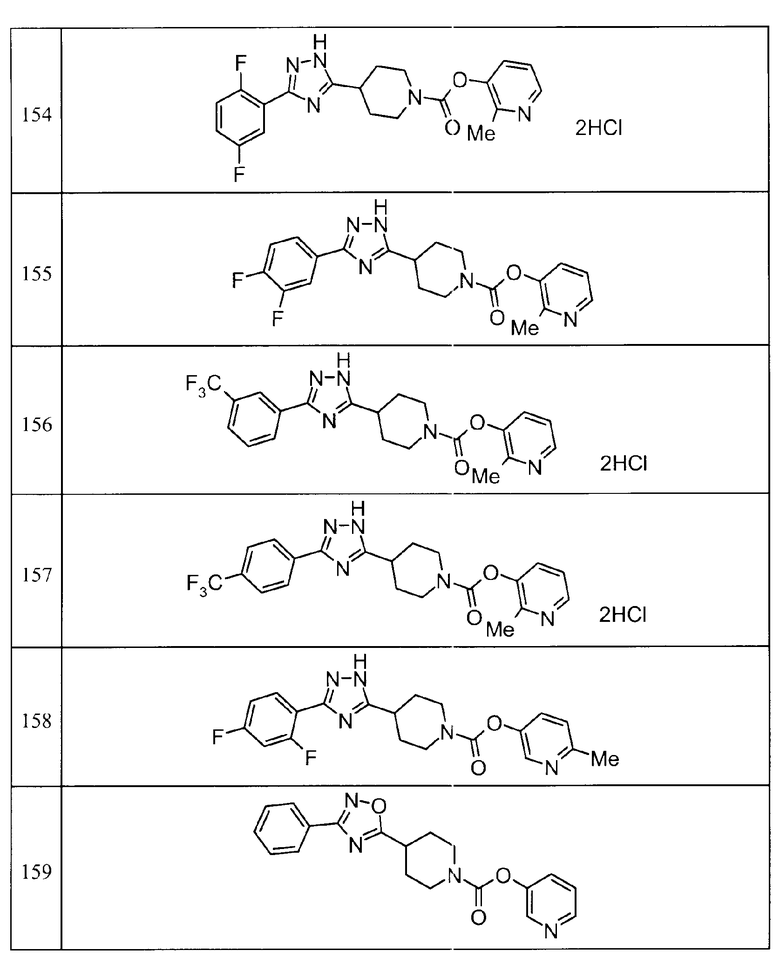
Такими же способами, как для соединений описанных выше примеров 1-10, соединения примеров, показанных в представленной ниже таблице, были получены с использованием каждого из соответствующих исходных материалов. Структуры соединений примеров 1-159 показаны в таблицах 12-31, и способы их получения и физико-химические данные показаны в таблицах 32-38.
Кроме того, следующие аббревиатуры используются в представленных ниже таблицах. Pre: Номер примера получения; Ex: Номер эксперимента; Str: Структурная формула; Syn: Способ получения (Среди представленных выше Примеров/Примеров получения, Номер примера получения и номер примера, описан такой же способ, который использовался с получением соединения. В описании P представляет Пример получения, а E представляет Пример. Например, представлено, что соединение Примера получения 30 было получено таким же образом, как соединение Примера получения 1, и соединение примера 11 было получено таким же образом, как соединение примера 1); Dat: Физико-химические данные (ЯМР: δ (м.д.) при 1Н ЯМР в DMSO-d6, FAB+ (с бомбардировкой быстрыми атомами): FAB-MS (масс-спектрометрия с бомбардировкой быстрыми атомами) (катион), FAB- (без бомбардировки быстрыми атомами): FAB-MS (анион), ESI+ (с эластораспылительной ионизацией): ESI-MS (масс-спектрометрия с эластораспылительной ионизацией) (катион), ESI- (без эластораспылительной ионизации): ESI-MS (анион), EI (электронная ионизация):EI-MS (электронная ионизация-масс-спектрометрия) (катион); CI+ (с химической ионизацией): CI-MS (химическая ионизация-масс-спектрометрия) (катион); APCI+ (с химической ионизацией при атмосферном давлении): APCI-MS (химическая ионизация при атмосферном давлении-масс-спектрометрия) (катион)); Me: метил; Et: этил; Bn: бензил; Boc: трет-бутоксикарбонил; Ms: метансульфонил; TsOH: п-толуолсульфоновая кислота; Z: бензилоксикарбонил.
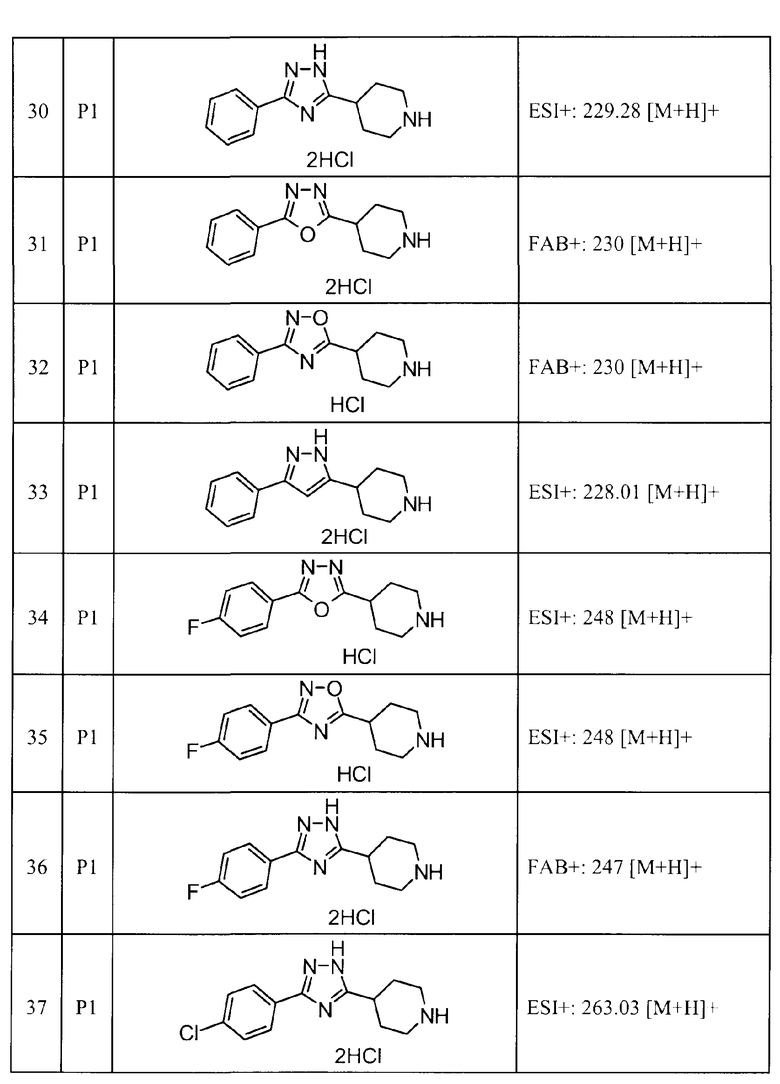
FAB+: 382,1 [M+H]+

Промышленная применимость
Соединение формулы (I) или его фармацевтически приемлемая соль обладает ингибирующей активностью в отношении FAAH и может применяться в качестве средства для профилактики и/или лечения заболеваний, связанных с FAAH. В частности, нейропатической боли.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИДИЛЬНОЕ НЕАРОМАТИЧЕСКОЕ АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛО-1-КАРБОКСИЛАТНОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2408581C2 |
| ПИРИДИЛЬНОЕ НЕАРОМАТИЧЕСКОЕ АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛО-1-КАРБОКСИЛАТНОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2408580C2 |
| ОКСАЗОЛИДИНОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВОБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2016 |
|
RU2794494C2 |
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ | 2019 |
|
RU2798336C2 |
| ТИАЗОЛИЛФЕНИЛБЕНЗОЛСУЛЬФОНАМИДОПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2012 |
|
RU2606497C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| СОЕДИНЕНИЕ ТРИАЗИНОНА И ИНГИБИТОР КАЛЬЦИЕВЫХ КАНАЛОВ Т-ТИПА | 2013 |
|
RU2645158C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗИН-3-АМИНА | 2011 |
|
RU2771819C2 |
Изобретение относится к соединениям, которые представляют собой пиридин-3-ил 4-(3-фенил-1H-1,2,4-триазол-5-ил)пиперидин-1-карбоксилат, 6-метилпиридин-3-ил 4-[3-(4-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат, 6-метилпиридин-3-ил 4-[5-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат, 2,6-диметилпиридин-3-ил 4-[5-(3,4-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидин-1-карбоксилат, 2-метилпиридин-3-ил 4-[3-(2-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат, 6-метилпиридин-3-ил 4-(3-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилат, 2-метилпиридин-3-ил 4-[5-(3-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат и 6-метилпиридин-3-ил 4-[4-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат или к их фармацевтически приемлемой соли. Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении гидролазы амидов жирных кислот (FAAH), на основе указанных соединений. Технический результат: получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения нейропатической боли. 5 н. и 8 з.п. ф-лы, 38 табл., 159 пр.
1. Соединение, которое выбрано из группы, состоящей из:
пиридин-3-ил 4-(3-фенил-1H-1,2,4-триазол-5-ил)пиперидин-1-карбоксилата,
6-метилпиридин-3-ил 4-[3-(4-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилата,
6-метилпиридин-3-ил 4-[5-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилата,
2,6-диметилпиридин-3-ил 4-[5-(3,4-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидин-1-карбоксилата,
2-метилпиридин-3-ил 4-[3-(2-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилата,
6-метилпиридин-3-ил 4-(3-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилата,
2-метилпиридин-3-ил 4-[5-(3-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилата и
6-метилпиридин-3-ил 4-[4-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилата
или их фармацевтически приемлемой соли.
2. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении гидролазы амидов жирных кислот (FAAH), содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
3. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения фармацевтической композиции для лечения нейропатической боли.
4. Применение соединения по п.1 или его фармацевтически приемлемой соли для лечения нейропатической боли.
5. Способ лечения нейропатической боли, включающий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
6. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой пиридин-3-ил 4-(3-фенил-1H-1,2,4-триазол-5-ил)пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
7. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 6-метилпиридин-3-ил 4-[3-(4-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
8. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 6-метилпиридин-3-ил 4-[5-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
9. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 2,6-диметилпиридин-3-ил 4-[5-(3,4-дифторфенил)-1,2,4-оксадиазол-3-ил]пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
10. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 2-метилпиридин-3-ил 4-[3-(2-фторфенил)-1H-1,2,4-триазол-5-ил]пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
11. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 6-метилпиридин-3-ил 4-(3-фенил-1H-пиразол-1-ил)пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
12. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 2-метилпиридин-3-ил 4-[5-(3-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
13. Соединение или его фармацевтически приемлемая соль по п.1, представляющее собой 6-метилпиридин-3-ил 4-[4-(4-фторфенил)-1,3-оксазол-2-ил]пиперидин-1-карбоксилат или его фармацевтически приемлемую соль.
| WO 2006088075 A1, 24.08.2006 | |||
| WO 2006054652 A1, 26.05.2006 | |||
| US 20070021405 A1, 25.01.2007 | |||
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2146254C1 |
| Двухвальный торфяной пресс | 1927 |
|
SU9468A1 |

