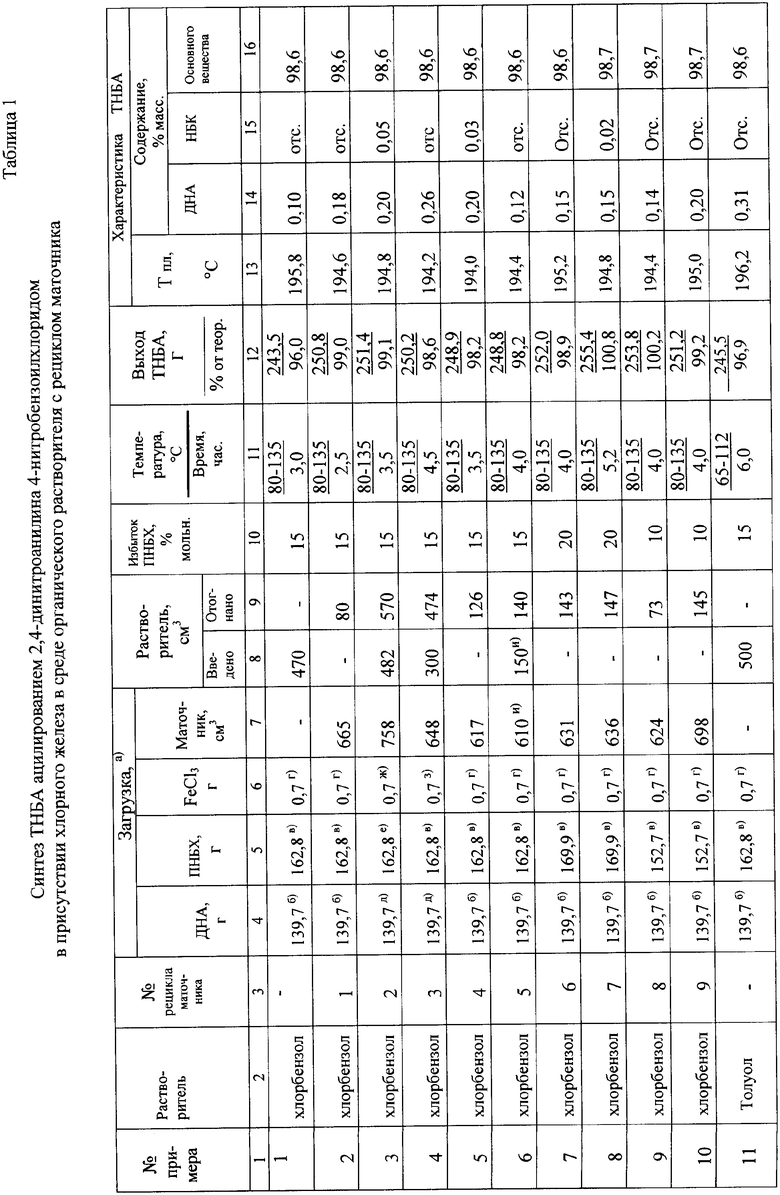
Настоящее изобретение относится к области органической химии и касается способа получения 2',4,4'-тринитробензанилида - полупродукта синтеза 5(6)-амино-2-(4-аминофенил)бензимидазола, используемого в качестве мономера в производстве термостойких и высокопрочных волокон.
Известен способ получения 2',4,4'-тринитробензанилида (в дальнейшем ТНБА) нитрованием N-(4'-нитробензоил)анилина азотной кислотой в среде концентрированной серной кислоты (Патент США 4109093, НКИ США 548/310.7, опубл. 22.08.1978). Основной недостаток данного способа - большое количество отходов - смеси концентрированной серной и азотной кислот, загрязненных примесями органических веществ.
Наиболее близким аналогом предложенного способа получения ТНБА (прототип) является способ, основанный на ацилировании 2,4-динитроанилина 4-нитробензойной кислотой в присутствии активатора - полихлорциклофосфазена и катализатора - N-окисей пиридина или β- и γ-пиколина в среде хлорбензола с последующим выделением целевого продукта путем отгонки хлорбензола с водяным паром (А.С. СССР 1025093, С07С 233/66, опубл. 20.02.1999). При температуре 130-133°С процесс заканчивается в течение 1 часа, выход 2',4,4'-тринитробензанилида составляет 93-96%. Полихлорциклофосфазен, например гексахлорциклофосфазен, синтезируют из хлорида аммония и пятихлористого фосфора. В известном способе на 1,1 г 2,4-динитроанилина используется до 0,7 г гексахлорциклофосфазена, таким образом, полихлорциклофосфазен является фактически сореагентом. Процесс в целом сопровождается образованием большого количества побочного хлорида водорода, возникает проблема утилизации фосфорсодержащих отходов производства. Пятихлористый фосфор крайне гигроскопичен, токсичен, коррозионноактивен. Поскольку пятихлористый фосфор является кристаллическим продуктом, работа с ним обусловливает ряд проблем по аппаратурному оформлению процесса. В синтезе используются дорогие труднодоступные катализаторы. По-видимому, реакция ацилирования протекает через стадию образования 4-нитробензоилхлорида in situ из 4-нитробензойной кислоты и полихлорциклофосфазена. По этим причинам синтез ТНБА по известному способу не нашел практического применения. Если использовать в синтезе хлориды фосфора, проще было бы получить 4-нитробензоилхлорид взаимодействием 4-нитробензойной кислоты с хлорокисью фосфора.
Таким образом, недостатками способа согласно прототипу являются использование труднодоступных и дорогостоящих активатора и катализаторов, их большой расход, образование значительного количества отходов производства.
Задачей настоящего изобретения является разработка способа получения ТНБА, лишенного недостатков, присущих прототипу.
Поставленная задача решается предлагаемым способом получения ТНБА ацилированием 2,4-динитроанилина (ДНА) 4-нитробензоилхлоридом (ПНБХ) в присутствии катализатора при нагревании в среде органического растворителя.
Предлагаемый способ отличается тем, что ацилирование 4-нитробензоилхлоридом, взятом в избытке более 5 мол.% по отношению к 2,4-динитроанилину, проводят при постепенном повышении температуры до кипения при атмосферном давлении, в присутствии хлорного железа в качестве катализатора, 2',4,4'-тринитробензанилид выделяют последовательными кристаллизацией, фильтрацией и нейтрализацией аммиаком в водной суспензии, реакционный маточник, образующийся после кристаллизации и фильтрации, возвращают в рецикл на следующую операцию ацилирования. Рецикл может быть осуществлен неограниченное число раз, при этом весь маточник или часть его могут быть возвращены в рецикл без какой-либо обработки. При необходимости маточник может быть очищен обработкой адсорбентом, например активированным углем.
В качестве органического растворителя при осуществлении процесса получения ТНБА могут быть использованы растворители, образующие с водой азеотроп: хлорбензол, п-ксилол, о-ксилол, техническая смесь ксилолов, этилбензол или толуол.
Реакция ацилирования 2,4-динитроанилина (ДНА) 4-нитробензоилхлоридом (ПНБХ) в присутствии хлорного железа в среде органического растворителя протекает с заметной скоростью, начиная с температуры 65-85°С. С дальнейшим повышением температуры скорость реакции возрастает и при 100-120°С процесс ацилирования в начальный период идет интенсивно с выделением хлористого водорода (см. чертеж). Для предотвращения слишком интенсивного выделения хлорида водорода и выброса реакционной массы процесс ведут при постепенном повышении температуры до температуры кипения при атмосферном давлении. С этой целью целесообразно также использовать дозированную подачу ПНБХ в зону реакции в твердом виде или в виде расплава, или в виде раствора в соответствующем растворителе при температуре, при которой реакция протекает с высокой скоростью, например при температуре кипения реакционной массы.
Предпочтительным избытком ПНБХ по отношению к ДНА является избыток 10-15 мол.%. При избытке ПНБХ менее 10 мол.% снижается выход ТНБА, а при избытке более 15 мол.% затрудняется отмывка ТНБА от избыточного ПНБХ.
Для предотвращения накопления в реакционной массе избыточного количества растворителя, который поступает с рециркулируемым маточником, с раствором ПНБХ в соответствующем растворителе или с раствором, суспензией или пастой ДНА, в процессе реакции ведут отгонку части растворителя непосредственно из реактора ацилирования, совмещая процессы ацилирования и отгонки растворителя во времени. В результате отпадает необходимость использования для отгонки растворителя дополнительного оборудования.
Нейтрализацию ТНБА в водной суспензии аммиаком для удаления остатков ПНБХ и примеси НБК целесообразно вести в более мягких условиях, чем в прототипе, а именно при 20-60°С при постепенной дозированной подаче водного раствора аммиака с целью предотвращения гидролиза ТНБА.
ДНА может быть введен в процесс в виде сухого вещества или в виде пасты с соответствующим растворителем, в котором проводится ацилирование, или в виде водной пасты. В последнем случае перед добавлением маточника и ПНБХ необходимо провести обезвоживание ДНА. Обезвоживание ведут азеотропной отгонкой воды с растворителем.
ПНБХ может быть введен в процесс в виде твердого вещества или в виде раствора в соответствующем растворителе, в котором ведут ацилирование.
Хлорное железо может быть введено в процесс в виде сухого безводного хлорного железа FeCl3 путем загрузки его непосредственно в реактор ацилирования или путем добавления его к раствору ПНБХ. Хлорное железо может быть введено в процесс также в виде кристаллогидрата FeCl3·6Н2О или в виде водного раствора путем добавления его к ДНА с последующим обезвоживанием азеотропной отгонкой с растворителем.
В соответствии с изобретением установлено, что в ходе рециклов маточника примеси не накапливаются в ТНБА в количествах, препятствующих его дальнейшей переработке в 5(6)-амино-2-(4-аминофенил)бензимидазол. Это иллюстрируется данными примеров, приведенных ниже. Температура плавления ТНБА, полученного с использованием рециклов маточника, не опускалась ниже требуемой величины не менее 194°С (см. таблицу).
В результате осуществления процесса по предлагаемому способу сокращаются расходные нормы сырья, выход ТНБА приближается к количественному, сокращается количество отходов производства, достигается значительная экономия энергоресурсов и трудозатрат, предотвращается загрязнение окружающей среды.
На чертеже представлена зависимость степени превращения ДНА от времени в реакции ацилирования 4-нитробензоилхлоридом в хлорбензоле при 120°С.
Изобретение иллюстрируется нижеследующими примерами.
Пример 1.
В трехгорлую колбу емкостью 2 дм3, снабженную мешалкой, обратным холодильником, подключенным к системе улавливания хлорида водорода, и термометром, загружают 139,7 г (0,763 г-моля) 2,4-динитроанилина (ДНА), 162,8 г (0,8776 г-моля) 4-нитробензоилхлорида (ПНБХ), 0,68 г безводного хлорного железа и 472 см3 хлорбензола. Избыток ПНБХ составляет 15 мол.% по отношению к ДНА.
Реакционную массу нагревают при перемешивании. При температуре 85°С начинается интенсивное выделение хлорида водорода. Массу нагревают, постепенно в течение 1 часа повышая температуру до 134-135°С, и дают при этой температуре в режиме кипения выдержку в течение 2 часов до завершения выделения хлорида водорода.
Далее колбу охлаждают водой, а затем в холодильнике до температуры 5-10°С и образовавшуюся суспензию фильтруют на воронке Бюхнера с отсасыванием с помощью вакуума. Осадок ТНБА промывают 210 см3 хлорбензола. Основной и промывной фильтраты объединяют. Получают 665 см3 хлорбензольного маточника, содержащего, г/дм3: ДНА 0,71; ПНБХ 20,0; ТНБА 9,87; 4-нитробензойной кислоты (НБК) 2,63; FeCl3 0.50.
Из пасты ТНБА с водяным паром отгоняют хлорбензол, суспензию охлаждают до 30-40°С и нейтрализуют 0,25%-ным раствором аммиака до рН 8. Водный аммиак добавляют постепенно, порциями дважды по 50 мл и на конечной стадии нейтрализации 20 мл. При этом последующую порцию водного аммиака добавляют, когда прореагирует аммиак, содержащийся в предыдущей порции. Далее водную суспензию ТНБА отфильтровывают, промывают водой для удаления аммониевой соли НБК. Получают 243,5 г ТНБА (96% от теоретического). Температура плавления 195,8°С, содержание, %: ДНА 0,1; НБК отс.; рН водной вытяжки 0,68.
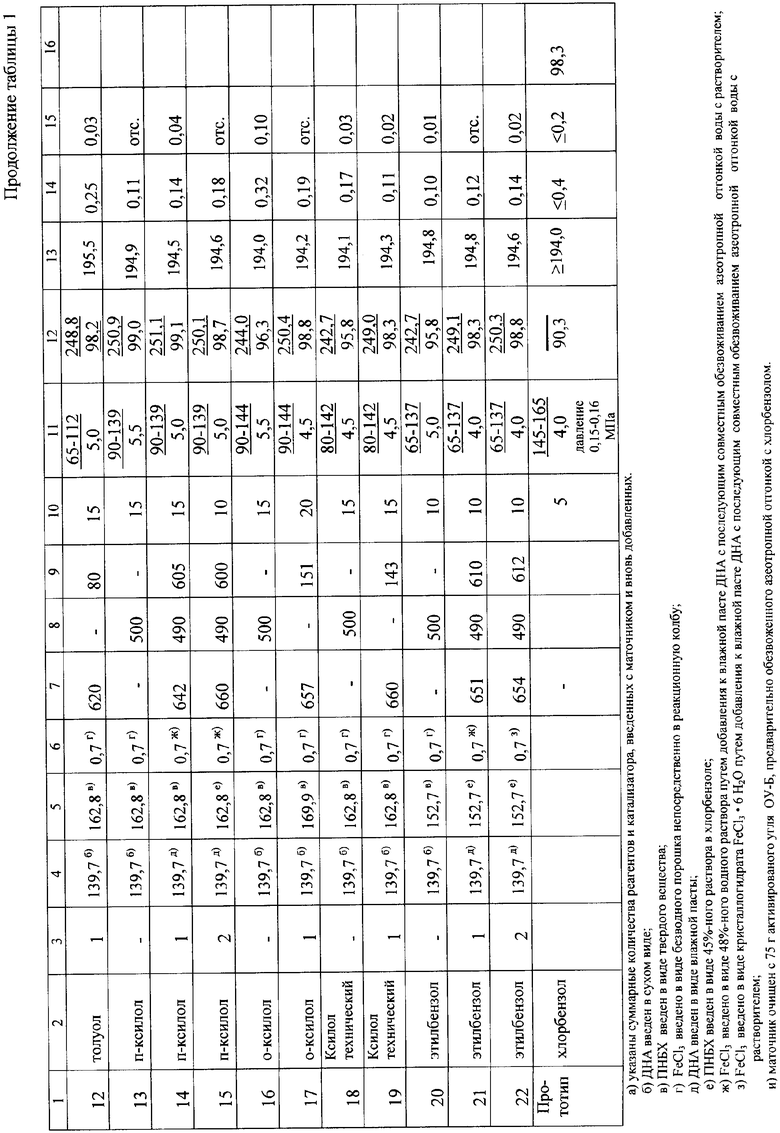
Пример 2.
В колбу, описанную в примере 1, загружают 665 см3 маточника, полученного в примере 1, 139,2 г ДНА, 149,77 г ПНБХ и 0,48 г FeCl3. Общее количество реагентов и мольный избыток ПНБХ по отношению к ДНА такие же, как в примере 1. Реакционную массу постепенно нагревают при перемешивании. Температуру в течение 0,5 часа постепенно повышают до 132°С (кипение реакционной массы) и выдерживают в течение 2 часов. В процессе выдержки отгоняют 80 см3 хлорбензола, используя прямой холодильник и совмещая процессы отгонки растворителя и ацилирования. При этом температура в реакционной массе повышается до 134-135°С. Далее реакционную массу обрабатывают, как описано в примере 1. Получают 780 см3 хлорбензольного маточника, имеющего состав, г/дм3; ДНА 0,67; ПНБХ 18,40; ТНБА 10,20; НБК 3,01; FeCl3 0,52. Этот маточник используют в следующей операции рецикла. Хлорбензольную пасту ТНБА помещают в 1200 мл воды и отгоняют хлорбензол в виде азеотропа с водой. ТНБА в водной суспензии нейтрализуют путем дозированного прибавления 280 см3 0,25%-ного водного раствора аммиака при температуре 50-35°С, доводя реакцию среды до устойчивого значения рН 8. Нейтрализованную суспензию ТНБА фильтруют и промывают водой. Выход ТНБА 250,8 г (99,0% от теоретического). Температура плавления 194,6°С; содержание, %: ДНА 0,18; НБК отс.; рН водной вытяжки 6,8.
Пример 3.
В трехгорлую колбу емкостью 2 дм3, снабженную мешалкой, термометром, трехрогой насадкой с капельной воронкой, дефлегматором с прямым холодильником и приемником, загружают 154,4 г влажной пасты ДНА, содержащей 139,15 г основного вещества, 1 г водного раствора FeCl3, содержащего 0,48 г хлорного железа и 300 см3 хлорбензола.
Содержимое колбы нагревают при перемешивании и отгоняют воду в виде аэеотропа до начала отгонки безводного хлорбензола. Затем смесь ДНА с хлорбензолом в колбе охлаждают до 70°С при перемешивании, в трехгорлую насадку устанавливают обратный холодильник, загружают 758 см3 хлорбензольного маточника, полученного в примере 2 и содержащего 0,5 г ДНА, 14,0 г ПНБХ, 7,7 г ТНБА, 2,3 г НБК, 0,4 г FeCl3 и 751 см3 хлорбензола.
Реакционную массу постепенно нагревают при перемешивании в течение 1 часа до кипения (132°С). Затем обратный холодильник убирают и при кипении реакционной массы и перемешивании из капельной воронки в течение 1,5 часов прибавляют 331 г 45%-ного раствора ПНБХ в хлорбензоле, содержащего 148,9 г ПНБХ. Общий избыток ПНБХ по отношению к ДНА в синтезе составляет 15 мол.%.
В процессе прибавления раствора ПНБХ через дефлегматор и прямой холодильник отгоняют 570 см3 хлорбензола. Далее реакционную массу обрабатывают, как примере 1. Получают 670 см3 хлорбензольного маточника, имеющего состав, г/дм3: ДНА 0,90; ПНБХ 17,30; ТНБА 13,72; НБК 2,0; FeCl3 0,54. Этот маточник направляют в следующий опыт рецикла. Хлорбензольную пасту ТНБА помещают в 1200 мл воды, отгоняют хлорбензол и нейтрализуют водным аммиаком. Расход 0,25%-ной аммиачной воды 240 см3. После фильтрации водной суспензии и промывки водой получают 251,4 г ТНБА (99,1% от теоретического). Температура плавления 194,8°С; содержание, мас.%: ДНА 0,20; НБК 0,05; рН водной вытяжки 6,9.
Примеры 4 и 5 представлены в таблице.
Пример 6.
610 см3 маточника, выделенного в примере 5, обрабатывают перед вводом в синтез ТНБА активированным углем ОУ-Б. Для этого в трехгорлую колбу емкостью 1 дм3, снабженную мешалкой, прямым холодильником и термометром, загружают 140 г водной пасты, содержащей 75 г активированного угля ОУ-Б в пересчете на 100%-ный, добавляют 255 см3 хлорбензола и отгоняют 65 мл воды в виде азеотропа с хлорбензолом до достижения температуры в парах 131°С. Далее в колбу вносят 610 см3 маточника, полученного после синтеза ТНБА в примере 5. Массу нагревают при перемешивании до 80°С, выдерживают в течение 0,5 часа, охлаждают до комнатной температуры, далее маточник отфильтровывают от угля на воронке Бюхнера с отсасыванием.
Получают 660 см3 хлорбензольного маточника, имеющего состав, г/дм3: ДНА 0,86; ПНБХ 17,2; ТНБА 7,1; НБК 2,2; FeCl3 0,44. Этот маточник направляют в следующую операцию рецикла. В реакционную колбу, как в примерах 1 и 2, загружают 634 см3 очищенного маточника, содержащего 0,55 г ДНА, 10,9 г ПНБХ, 4,5 г ТНБА и 1,4 г НБК. Добавляют 139,1 г ДНА, 151,9 г ПНБХ и 0,5 г хлорного железа. Мольный избыток ПНБХ 15%. Ацилирование ведут, как описано в примерах 1 и 2. В ходе выдержки при кипении отгоняют 180 см3 хлорбензола. ТНБА выделяют и нейтрализуют в водной суспензии аммиаком при температуре 50-30°С, постепенно добавляя 300 см3 0,25%-ного водного раствора аммиака до устойчивого значения рН 8. После фильтрации и промывки водой получают 248,8 г ТНБА (98,2% от теоретического). Температура плавления 194,4°С; содержание, мас.%: ДНА 0,12; НБК 0,01; рН водной вытяжки 6,8.
Хлорбензольный маточник в объеме 650 см3, содержащий, г/дм3: ДНА 1,1; ПНБХ 19,8; НБК 2,2; ТНБА 7,8, используют в следующей операции синтеза ТНБА.
В таблице приведены дополнительные примеры, иллюстрирующие предлагаемый способ получения ТНБА.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2', 4, 4'-ТРИНИТРОБЕНЗАНИЛИДА | 2007 |
|
RU2394810C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРЗАМЕЩЕННЫХ АМИНОАНИЛИДОВ АРОМАТИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ | 2006 |
|
RU2323207C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2',4,4'-ТРИНИТРОБЕНЗАНИЛИДА ИЗ 2,4-ДИНИТРОАНИЛИНА И 4-НИТРОБЕНЗОИЛХЛОРИДА | 2013 |
|
RU2547262C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5(6)-АМИНО-2-(4-АМИНОФЕНИЛ)БЕНЗИМИДАЗОЛА | 2006 |
|
RU2345988C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРЗАМЕЩЕННЫХ 4,4-ДИАМИНОБЕНЗАНИЛИДОВ | 2007 |
|
RU2385861C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5(6)-АМИНО-2(4'-АМИНОФЕНИЛ)-БЕНЗИМИДАЗОЛА | 2005 |
|
RU2283307C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2',4,4'-ТРИНИТРОБЕНЗАНИЛИДА ИЗ АНИЛИНА И 4-НИТРОБЕНЗОЙНОЙ КИСЛОТЫ | 2014 |
|
RU2560881C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5(6)-АМИНО-2-(4-АМИНОФЕНИЛ)БЕНЗИМИДАЗОЛА | 2013 |
|
RU2547210C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРЗАМЕЩЕННЫХ 4,4'-ДИАМИНОБЕНЗАНИЛИДОВ | 2013 |
|
RU2547268C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5(6)-АМИНО-2-(4-АМИНОФЕНИЛ)БЕНЗИМИДАЗОЛА ИЗ 2',4,4'-ТРИНИТРОБЕНЗАНИЛИДА | 2013 |
|
RU2547261C2 |
Изобретение относится к области органической химии и касается способа получения 2',4,4'-тринитробензанилида (ТНБА) - полупродукта синтеза 5(6)-амино-2-(4-аминофенил)бензимидазола, используемого в качестве мономера в производстве термостойких и высокопрочных волокон. Способ состоит в том, что проводят ацилирование 2,4-динитроанилина 4-нитробензоилхлоридом, взятом в избытке более 5 мол.% по отношению к 2,4-динитроанилину, при постепенном повышении температуры до кипения при атмосферном давлении, в присутствии хлорного железа в качестве катализатора, 2',4,4'-тринитробензанилид выделяют последовательными кристаллизацией, фильтрацией и нейтрализацией аммиаком в водной суспензии, реакционный маточник, образующийся после кристаллизации и фильтрации, возвращают в рецикл на следующую операцию ацилирования и нейтрализацию ведут при температуре не выше 60°С при дозированной подаче водного раствора аммиака. Технический результат - разработка нового эффективного способа получения 2',4,4'-тринитробензанилида. 1 н. и 5 з.п. ф-лы, 1 ил., 1 табл.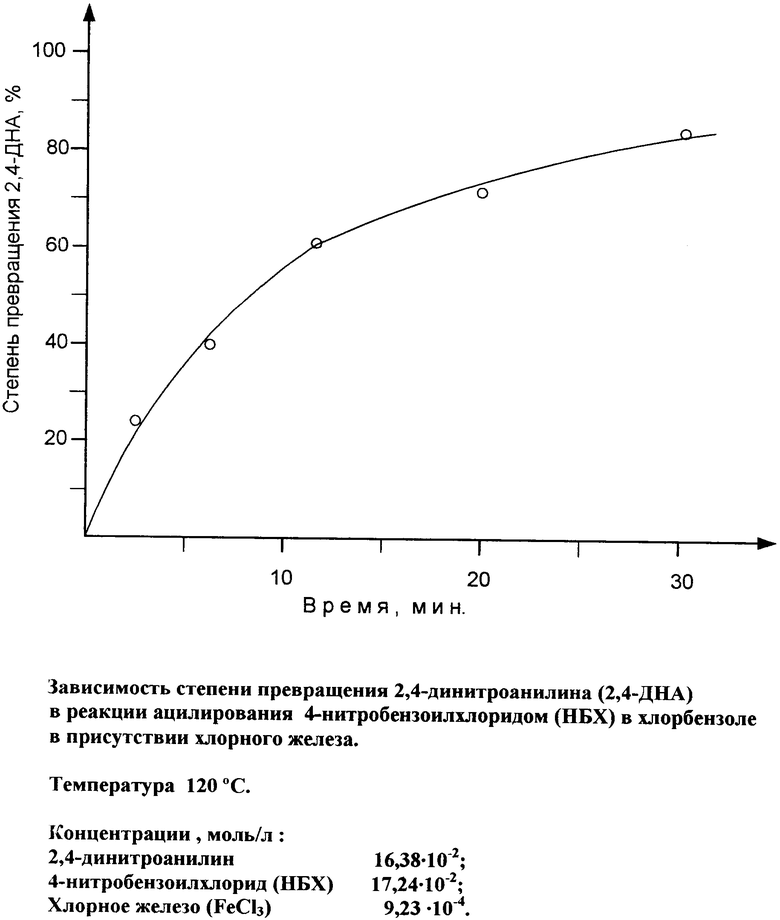
| SU 1025093, 20.02.1999 | |||
| US 4109093, 22.08.1978 | |||
| W.B.VAN HORSSEN, 1936, 55, 245-262 | |||
| Способ определения пригодности доменосодержащей пленки феррит-граната для магнитооптического управляемого транспаранта | 1988 |
|
SU1531161A1 |
| US 4061791, 06.12.1977. | |||

