Данное изобретение относится к новому химическому способу и, более конкретно, к новому химическому способу получения трет-бутил(Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}(4R,6S)-2,2-диметил[1,3]диоксан-4-ил)ацетата формулы I:
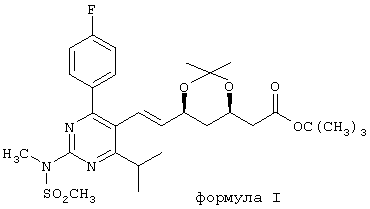
(в дальнейшем называемому ВЕМ), который полезен, например, в качестве химического полупродукта в производстве фармацевтических средств, полезных для лечения в числе прочих гиперхолестеринемии, гиперлипопротеинемии и атеросклероза. Изобретение далее включает новый исходный материал, используемый в указанном процессе, и использование процесса в производстве ингибиторов HMG СоА-редуктазы.
В Европейской патентной заявке, публикация изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновая кислота и ее натриевая соль и кальциевая соль (изображена ниже):№(ЕРА) 0521471 описывается (Е)-7-[4-(4-фторфенил)-6-
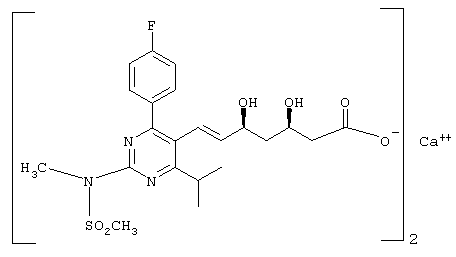
(называемые далее общим названием "Агент") в качестве ингибиторов HMG СоА-редуктазы. Агент получают восстановлением метилового эфира 7-[4-(4-фторфенил)-6-изопропил-2-[N-метил-N-метилсульфонил)амино]пиримидин-5-ил-(3R)-3-гидрокси-5-оксо-(Е)-гептеновой кислоты с последующей обработкой. Однако, Агент может быть получен из ВЕМ с помощью обработки кислотой (для отщепления ацетонидной защитной группы) с последующей обработкой основанием (для расщепления сложноэфирной группы) и (как описано в ЕРА 0521471) превращения образовавшейся вначале соли в свободную кислоту или кальциевую соль.
В настоящее время изобретателями открыт полезный и обладающий преимуществами способ получения ВЕМ.
Согласно изобретению предлагается способ получения ВЕМ (формула I), включающий взаимодействие дифенил[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-илметил]-фосфиноксида формулы III:
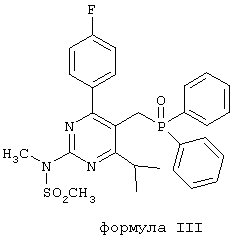
(называемого далее DPPO) с трет-бутил 2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетатом формулы II:
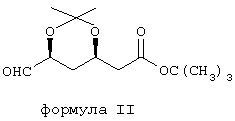
(называемого далее BFA) в присутствии сильного основания.
Процесс осуществляют в подходящем растворителе или смеси растворителей, например в простых эфирных или ароматических растворителях или их смесях. Особенно подходящими растворителями являются, например, тетрагидрофуран (ТГФ), диметоксиэтан и толуол или их смеси. Из них особенно предпочтительны ТГФ и ТГФ с толуолом.
Подходящие основания для использования в способе включают, например, амидные основания, алкилметаллы и гидриды металлов. Конкретными примерами оснований являются бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, бис(триметилсилил)амид лития, бутиллитий и гидрид натрия. Особенно предпочтительным основанием является, например, бис(триметилсилил)амид натрия (NaHMDS).
Реакция может осуществляться при температуре в пределах, например, от -20 до -90°С, такой как -40 - -90°С, например, от -40 до -80°С. Удобной температурой для проведения реакции является, например, температура смеси ацетона с твердой двуокисью углерода (около -75°С).
Процесс преимущественно осуществляют с 1,0-1,2 эквивалентами основания (на эквивалент DPPO), например, 1,05-1,2 эквивалентами и, предпочтительно, 1,05-1,12 эквивалентами. Хотя BFA может присутствовать в большом избытке, удобнее использовать 1,0-1,35 эквивалента (на эквивалент DPPO) и, предпочтительно, 1,05-1,3 эквивалента, особенно 1,05-1,15 эквивалентов.
Способ изобретения позволяет получать значительно улучшенные выходы и качество продукта по сравнению со способом, когда в качестве исходного материала вместо DPPO используют соответствующий диалкилфосфонат (-РO(Oалкил)2).
Исходный материал DPPO, представляющий собой следующий аспект настоящего изобретения, может быть получен, как описано в приведенных ниже примерах, исходя их алкилового эфира 2-амино-4-(4-фторфенил)-6-изопропилпиримидин-5-карбоновой кислоты, например метилового эфира, который может быть получен по методу, описанному в японской патентной заявке №06-256318, или этилового эфира, который может быть получен как описано в ЕРА 0521471. BFA может быть получен, как описано в ЕРА 0319847 (пример 6).
Еще одним аспектом настоящего изобретения является способ получения соединения формулы IV:
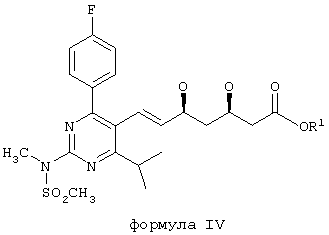
в которой R1 обозначает водород или фармацевтически приемлемый катион, который включает:
(1) взаимодействие DPPO с BFA в присутствии сильного основания (описанного выше) с образованием ВЕМ;
(2) отщепление дигидрокси-защитной группы (ацетонидной) (например, с помощью кислотного гидролиза, такого как с использованием HCl в ТГФ или ацетонитриле); и
(3) отщепление трет-бутильной сложноэфирной группы в условиях основной среды с образованием соединения формулы IV, в которой R1 является фармацевтически приемлемым катионом (например, с использованием раствора гидроксида металла в полярном растворителе, например, с использованием водного гидроксида натрия в этаноле или ацетонитриле с образованием натриевой соли);
с необязательной последующей нейтрализацией с образованием соединения формулы IV, в которой R1 обозначает водород;
и/или с последующим превращением в другое соединение формулы IV, в которой R1 обозначает фармацевтически приемлемый катион (например, превращением натриевой соли в кальциевую соль обработкой водорастворимой кальциевой солью (такой как хлорид кальция) в водных условиях).
Подходящие условия для стадий (2), (3) и последующих необязательных стадий аналогичны или являются такими же, как условия, приведенные в ЕРА 0521471 и/или ЕРА 0319847, которые включены в настоящее изобретение в качестве ссылочного материала. Для получения кальциевой соли соединения формулы IV, как это проиллюстрировано на стр.1, предпочтительно стадии (2), (3) и превращение в кальциевую соль через метиламиновую соль преимущественно осуществляют как описано в примере 7, причем названные стадии образуют следующий аспект изобретения.
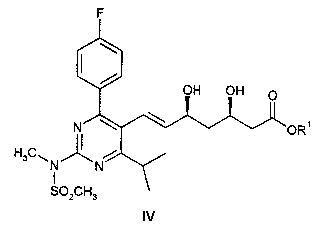
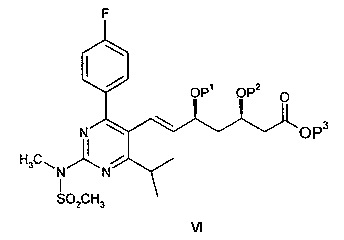
Очевидно, понятно, что в описанных выше процессах BFA можно заменить соединением общей формулы V:
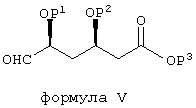
в которой Р1 и Р2 обозначают защитные группы спирта, такие как описано в ЕРА 0319847 и GB 2244705, которые включены в настоящую работу в качестве ссылочного материала, а Р3 обозначает защитную группу карбоновой кислоты, такую как C1-С8-алкил (такой, как C1-С4-алкил), с образованием соединения формулы VI:
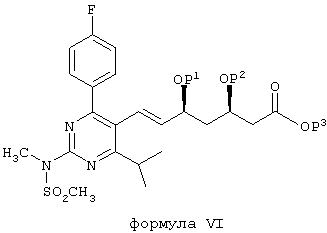
Соединение формулы VI может быть превращено в Агент путем отщепления защитных групп спирта или диола и превращения группы СООР3 в группу СООН или в ее фармацевтически приемлемую соль. Такие общие процессы составляют дополнительные признаки настоящего изобретения.
Изобретение далее иллюстрируется, но не ограничивается, следующими примерами.
Получение 1
Получение DPPO
Перемешиваемую смесь метилового эфира 4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-карбоновой кислоты (12,0 г) в толуоле (55 мл) охлаждают до -10°С и в течение 2 часов добавляют диизобутилалюминийгидрид (50 мл 1,5 М раствор в толуоле), поддерживая температуру ниже 0°С. После добавления смесь перемешивают в течение 30 мин при 0°С. К смеси добавляют метанол (0,64 мл) при поддержании температуры при 0°С. Смесь затем добавляют на протяжении 2 час к перемешиваемой смеси концентрированной соляной кислоты (23,3 мл), воды (40,5 мл) и ацетонитрила (24 мл) при 40°С, поддерживая температуру смеси при 40°С. После добавления смесь перемешивают еще в течение 30 мин при 40°С и затем продувают азотом (для удаления возможно присутствующего изобутана). Охлаждают смесь до 20°С и оставляют стоять в течение 20 мин. Органическую фазу отделяют и промывают смесью концентрированной соляной кислоты (0,7 мл) и воды (30 мл). К органической фазе добавляют ацетонитрил (24 мл) и смесь промывают раствором бикарбоната натрия (0,038 г) в воде (120 мл).
Органическую фазу нагревают до 40°С и затем от 40 до 80°С, применяя продувку азотом. Смесь концентрируют перегонкой при атмосферном давлении, собирая 54 мл дистиллята. К концентрированному раствору добавляют ацетонитрил (24 мл) и затем при перемешивании трехбромистый фосфор (1,2 мл) при поддержании температуры смеси при 20°С. После добавления смесь перемешивают в течение 30 мин при 20°С. Смесь добавляют в течение 30 мин к воде (36 мл), поддерживая температуру при 20°С. Смесь перемешивают в течение 5 мин и отделяют органическую фазу. Органическую фазу промывают раствором бикарбоната натрия (0,027 г) в воде (36 мл) и затем водой (36 мл). Органическую фазу перегоняют при пониженном давлении до тех пор, пока не будет собрано 29 мл дистиллята. Смесь охлаждают до 60°С и добавляют этилдифенилфосфинит (7,47 мл). Смесь перемешивают 3 часа при 60°С, затем нагревают до кипения с обратным холодильником. Добавляют толуол (40 мл) и смесь охлаждают до 0°С в течение 2 час. Продукт собирают фильтрованием, промывают холодным толуолом (10 мл) и сушат в вакууме при 50°С, получая DPPO (14,66 г); 1Н ЯМР (СDСl3, 270 МГц): 7,42 [м, 10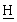 , Р(С6
, Р(С6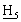 )2], 7,12 [м, 2Н, Аr-Н], 6,92 [м, 2Н, Аr-Н], 3,92 [д, 2Н, С
)2], 7,12 [м, 2Н, Аr-Н], 6,92 [м, 2Н, Аr-Н], 3,92 [д, 2Н, С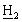 P], 3,51, 3,46 [2×с, 6Н, NС
P], 3,51, 3,46 [2×с, 6Н, NС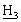 SO2С
SO2С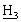 ], 3,43 [гепт., 1Н, С
], 3,43 [гепт., 1Н, С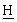 (СН3)2], 1,25 [д, 6Н, СН(С
(СН3)2], 1,25 [д, 6Н, СН(С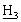 )2].
)2].
Метиловый эфир 4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-карбоновой кислоты получают следующим образом.
Смесь метилового эфира 2-амино-4-(4-фторфенил)-6-изопропилпиримидин-5-карбоновой кислоты (19,0 г), трет-пентоксида натрия (22,95 г) и диметоксиэтана (190 мл) перемешивают 30 мин при 25°С. Перемешиваемую смесь охлаждают до -10°С и добавляют по каплям метансульфонилхлорид (8,4 мл), поддерживая температуру смеси при -5°С. Через 20 мин добавляют диметилсульфат (8,1 мл) и дают смеси подогреться до 25°С. Смесь перемешивают 1 час при 25°С и добавляют раствор трет-пентоксида натрия (1,91 г) в диметоксиэтане (10 мл). Смесь перемешивают в течение 1 часа при 25°С. Добавляют раствор хлорида натрия (13,3 г) в воде (133 мл), и смесь перемешивают в течение 10 мин при 25°С. Смесь оставляют отстаиваться в течение 15 мин, и нижнюю водную фазу отделяют и отбрасывают. К оставшейся смеси добавляют воду (38 мл) и перемешивают смесь 30 мин при 25°С. Смесь затем нагревают до полного растворения. Смесь медленно охлаждают до 25°С на протяжении 1 часа. Смесь охлаждают до 0°С, перемешивают в течение 1 часа и суспендированный твердый материал собирают фильтрованием. Твердое вещество промывают холодным (0°С) раствором смеси (50/50) вода/диметоксиэтан (20 мл). Твердое вещество сушат в вакууме при 60°С, получая метиловый эфир 4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-карбоновой кислоты (19,35 г); 1Н ЯМР (270 МГц, СDСl3): 7,69 (м, 2Н), 7,14 (м, 2Н), 3,71, 3,60, 3,51 (3×с, 9Н), 3,20 (м, 1Н), 1,32 (д, 6Н).
Пример 1
Смесь DPPO (19,17 г) и ТГФ (227 мл) подогревают в течение короткого времени до 40°С до тех пор, пока не образуется прозрачный раствор, затем среду делают инертной попеременным действием вакуума и азота (5 циклов). Смесь погружают в ацетон/СO2 баню, охлаждая содержимое до -75°С. К реакционной смеси на протяжении 10 мин добавляют из капельной воронки с выравниваемым давлением бис(триметилсилил)амид натрия (37,4 мл 1,0 М раствора в ТГФ), поддерживая температуру ниже -74°С и образуя красный раствор аниона. В смесь через капельную воронку пропускают тетрагидрофуран (10 мл) для ополаскивания и смесь перемешивают еще в течение 1 часа при -76°С, получая красную суспензию. К суспензии в течение 20 мин порциями добавляют BFA (80 мл ~13,5% объем/объем раствора в толуоле) из капельной воронки с выравниваемым давлением, поддерживая температуру ниже -73°С. Ополаскивают капельную воронку над смесью толуолом (20 мл) и перемешивают смесь еще 15 мин при -76°С. Охлаждающую баню удаляют и дают суспензии подогреваться в течение 1,5 часов до 10°С. Добавляют одной порцией ледяную уксусную кислоту (3,21 г) в воде (15 г), поднимая температуру до 18°С и растворяя все твердые вещества, и смесь перемешивают еще 5 мин.
Смесь концентрируют перегонкой при атмосферном давлении (110°С в рубашке) до температуры 94°С, собирая всего 274 мл дистиллята. Концентрированную смесь охлаждают до 40°С, добавляют воду (40 мл), и смесь перемешивают 5 мин, а затем оставляют отстаиваться в течение 15 мин. Нижнюю водную фазу отбрасывают. Добавляют бикарбонат натрия (2,99 г) в воде (40 мл), и смесь перемешивают в течение 5 мин, а затем оставляют осаждаться в течение 15 мин. Нижнюю водную фазу отбрасывают. Добавляют воду (30 мл), и перемешивают смесь в течение 5 мин и дают ей осесть в течение 15 мин. Нижнюю водную фазу удаляют.
Органическую фазу переносят в перегонный аппарат с толуолом (20 мл) и концентрируют перегонкой при атмосферном давлении (125-130°С в рубашке) до температуры 116°С, собирая 85 мл дистиллята. Создают вакуум (400-500 мбар) и собирают еще 16,5 мл дистиллята до температуры 111°С. Сбрасывают вакуум и оставляют концентрированную смесь охлаждаться до 80°С. Добавляют при быстром перемешивании теплый МеОН (140 мл, 50°С) и оставляют смесь самоохлаждаться до 20°С на протяжении 30 мин, в течение которых выпадает в осадок твердое вещество. Суспензию дополнительно охлаждают до 2°С в течение 30 мин, затем твердое вещество собирают фильтрованием на пористом фильтре, применяя отсасывание до максимально сухого состояния. Твердое вещество промывают холодным МеОН (60 мл, 2°С) и снова отсасывают до максимально сухого состояния, а затем переносят в вакуумный сушильный шкаф и сушат в течение ночи (50°С, 200 мбар), получая ВЕМ (14,01 г, 67,7%). 1Н ЯМР (СDСl3, 270 МГц) 7,65 [м, 2Н, Аr-Н], 7,09 [м, 2Н, Аr- Н], 6,52 [дд, 1Н, Аr-СН=С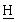 ], 5,47 (дд, 1Н, Аr-С
], 5,47 (дд, 1Н, Аr-С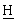 =СН], 3,57, 3,50 [2×с, 6Н, NС
=СН], 3,57, 3,50 [2×с, 6Н, NС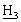 , SО2С
, SО2С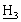 ], 3,38 [гепт., 1Н, Аr-С
], 3,38 [гепт., 1Н, Аr-С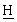 Ме2], 2,45, 2,30 [2×дд, 2Н, C
Ме2], 2,45, 2,30 [2×дд, 2Н, C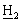 CO2-t-Bu], 1,55, 1,13 [дт, дд, 2Н, С
CO2-t-Bu], 1,55, 1,13 [дт, дд, 2Н, С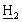 ацетонида], 1,50, 1,40 [2с, 6Н, С(С
ацетонида], 1,50, 1,40 [2с, 6Н, С(С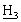 )2 ацетонида], 1,45 [с, 9Н, СO2С(С
)2 ацетонида], 1,45 [с, 9Н, СO2С(С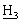 )2], 1,27 [дд, 6Н, АrСН(С
)2], 1,27 [дд, 6Н, АrСН(С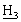 )2].
)2].
Примеры 2-6
Описанную в примере 1 процедуру осуществляют, используя приведенные в таблице 1 соотношения реагентов и температуры. Приведены также выходы получаемого ВЕМ.
Пример 7
Смесь ВЕМ (5,0 г) и ацетонитрила (35 мл) перемешивают в инертной атмосфере при 40°С. На протяжении 30 мин к полученному раствору добавляют 0,02 М соляную кислоту (9,5 мл), поддерживая температуру при 35-42°С. Смесь перемешивают 3 часа при 40°С и затем охлаждают до 25°С. Добавляют при перемешивании при 25°С 1,0 М раствор гидроксида натрия (9,5 мл), и смесь перемешивают при 25°С еще один час. Добавляют хлорид натрия (4,7 г), и смесь охлаждают до -5°С в течение одного часа. Добавляют при -5°С достаточное количество 1 М раствора соляной кислоты (9,5 мл) и хлорида натрия (2,4 г) до достижения рН 3,4-4,0, и смесь перемешивают при данной температуре в течение 5 мин. Смесь оставляют отстаиваться в течение 10 мин при -5°С, в результате чего образуются два слоя. Нижний слой отделяют и удаляют. К оставшемуся раствору добавляют при -5°С ацетонитрил (65 мл), и смесь фильтруют через фильтрующий материал. Добавляют при -5°С 40%-ный раствор метиламина в воде (1,1 мл), и смесь подогревают до 30°С в течение 40 мин и выдерживают при данной температуре в течение 90 мин. Затем смесь охлаждают до 0°С на протяжении 40 мин и поддерживают при данной температуре в течение 90 мин. Образовавшееся твердое вещество собирают фильтрованием и промывают ацетонитрилом (2×12 мл). Твердое вещество, представляющее собой метиламиновую соль соединения формулы IV (R1=МеNН
Изобретение относится к промежуточному продукту - трет-бутил(Е)-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)-амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетату, который может быть использован в синтезе соединения формулы IV, обладающего действием ингибитора HMG CoA-редуктазы, а следовательно, может быть использовано для получения фармацевтических средств для лечения, например, гиперхолестеринемии, гиперпротеинемии и атеросклероза. Изобретение также относится к способу получения указанного промежуточного продукта реакцией нового исходного соединения - дифенил[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)-амино]пиримидин-5-илметил]фосфиноксида с трет-бутил 2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетатом в присутствии сильного основания в простых эфирных или ароматических растворителях или их смесях при температуре в пределах от –200С до -900С. Изобретение также относится к способу получения соединения формулы IV:
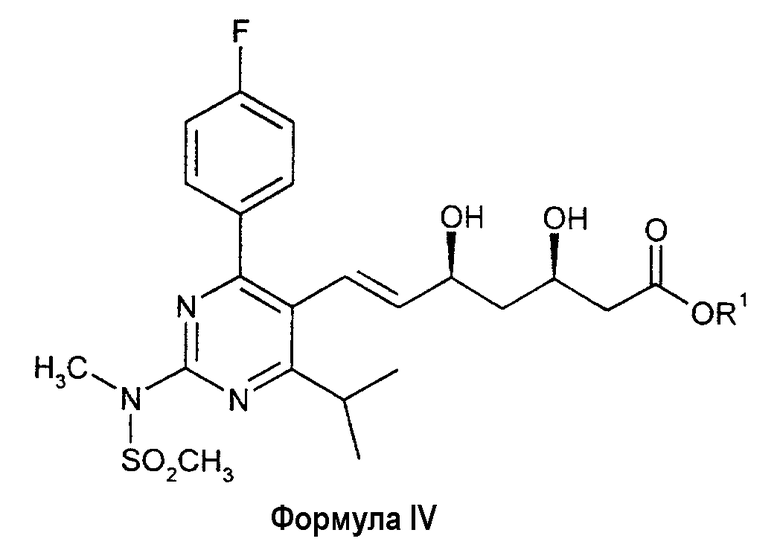
в которой R1 обозначает водород или фармацевтически приемлемый катион, и к способу получения промежуточных соединений формулы VI
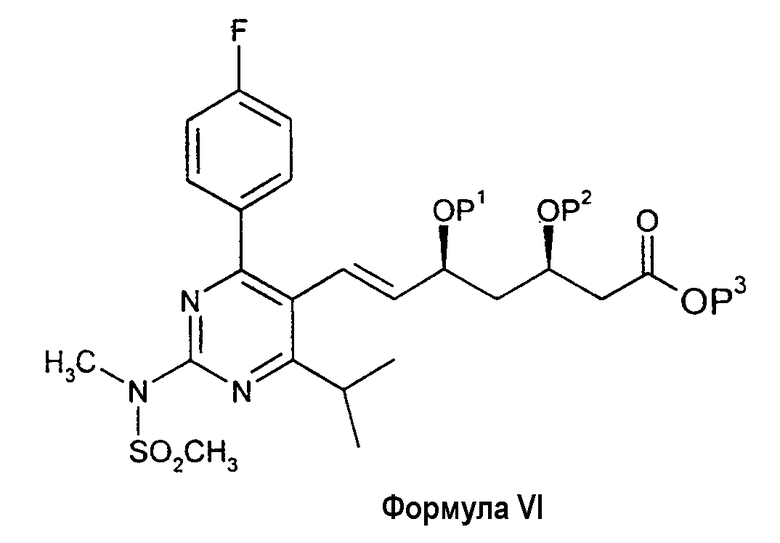
где Р1 и Р2 каждый независимо представляет (1-4С)алкил или группу  и где Р3 представляет собой (1-8С)алкил. Использование новых промежуточных соединений и предлагаемые способы позволяют повысить качество и выход продуктов 5 н. и 4 з.п. ф-лы, 1 табл.
и где Р3 представляет собой (1-8С)алкил. Использование новых промежуточных соединений и предлагаемые способы позволяют повысить качество и выход продуктов 5 н. и 4 з.п. ф-лы, 1 табл.
 -(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетата, включающий реакцию дифенил[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)-амино]пиримидин-5-илметил]фосфиноксида с трет-бутил 2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетатом в присутствии сильного основания, при этом процесс осуществляют в простых эфирных или ароматических растворителях или их смесях при температуре в пределах от –20 до -900С.
-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетата, включающий реакцию дифенил[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)-амино]пиримидин-5-илметил]фосфиноксида с трет-бутил 2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетатом в присутствии сильного основания, при этом процесс осуществляют в простых эфирных или ароматических растворителях или их смесях при температуре в пределах от –20 до -900С.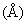 -(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетат.
-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетат.
в которой R1 обозначает водород или фармацевтически приемлемый катион, который включает:
(1) реакцию дифенил [4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-илметил]фосфиноксида с трет-бутил-2-[(4R,6S)-6-формил-2,2-диметил-1,3-диоксан-4-ил]ацетатом в присутствии сильного основания, при этом процесс осуществляют в простых эфирных или ароматических растворителях или их смесях при температуре в пределах от -20 до -900С с получением трет-бутил -(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетата формулы I;
-(6-{2-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил]винил}-(4R,6S)-2,2-диметил[1,3]диоксан-4-ил]ацетата формулы I;
(2) отщепление от продукта стадии (1) дигидроксизащитной группы с помощью кислотного гидролиза;
(3) отщепление от продукта стадии (2) трет-бутильной сложноэфирной группы в условиях основной среды с образованием соединения формулы IV, в которой R1 является фармацевтически приемлемым катионом; с последующей необязательной нейтрализацией с образованием соединения формулы IV, в которой R1 обозначает водород;
и/или с последующим необязательным превращением полученного соединения формулы IV в другую его соль, когда R1 обозначает фармацевтически приемлемый катион.
включающий реакцию дифенил[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-илметил]фосфиноксида с соединением формулы V
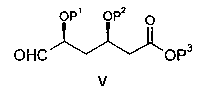
в присутствии сильного основания, где Р1 и Р2 каждый независимо представляет группу
 ,
,
где Р3 представляет собой (1-8С)алкил.
| US 6260440A, 09.11.1993.WO 94/17079A1,04.08.1994.BD CA of STN 124:29477: SOLLADIE GUI et al | |||
| “Chiral Sulfoxides en Asymmettric synthesis of Lactonic Moiety of (+)-Compactin and (+)-Mevinolin.Application to a Compactin Analog” Journal of Organic Chemistry(English),1995,60(24),p.774-7.BD CA of STN 151:211777:PRASAD KARA et al | |||
| “A novel diastereoselective synthesis of the lactone moiety of compactin” Tetrahedron Letters (English),1984,25(23),p.2435-8 | |||
| Способ обработки грубых шерстей на различных аппаратах для мериносовой шерсти | 1920 |
|
SU113A1 |
| “Stereoselective synthesis of HR-780, a now highly potent HMG-CoA reductase inhibitor ” Tetrahedron Letters (English),1990,31(18),p.2545-8.BD CA of STN 118:255237 MINAMI TATSUYA et al | |||
| Счетная таблица | 1919 |
|
SU104A1 |

