Данное изобретение относится к новым изомерам дигидротетрабеназина, содержащим их фармацевтическим композициям, способам их получения и их терапевтическому применению.
Уровень техники
Тетрабеназин (химическое название: 1,3,4,6,7,11b-гексагидро-9,10-диметокси-3-(2-метилпропил)-2H-бензо(a)хинолизин-2-он) использовался в качестве фармацевтического лекарственного средства с конца 1950-х. Сначала используемый в качестве антипсихотического средства, в настоящее время тетрабеназин используется для лечения гиперкинетических двигательных расстройств, таких как болезнь Хантингтона, гемибаллизм, сенильная хорея, тик, поздняя дискинезия и синдром Туретта, см., например, Jankovic et al., Аm. J. Psychiatry. (1999) Aug; 156(8): 1279-81 и Jankovic et al., Neurology (1997) Feb; 48 (2): 358-62.
Первичным фармакологическим действием тетрабеназина является снижение поступления моноаминов (например дофамина, серотонина и норэпинефрина) в центральную нервную систему путем ингибирования везикулярного транспортера моноаминов человека изоформы 2 (hVMAT2). Данное лекарственное средство также блокирует постсинаптические дофаминовые рецепторы.
Тетрабеназин представляет собой эффективное и безопасное лекарственное средство для лечения различных гиперкинетических двигательных расстройств и в отличие от типичных нейролептиков не вызывает поздней дискинезии. Тем не менее тетрабеназин проявляет ряд дозозависимых побочных эффектов, в том числе вызывающих депрессию, паркинсонизм, сонливость, нервозность или тревогу, бессонницу и, в редких случаях, злокачественный нейролептический синдром.
Центральные эффекты тетрабеназина сильно напоминают эффекты резерпина, но он отличается от резерпина тем, что проявляет недостаточную активность в отношении VMAT1 транспортера. Недостаточная активность в отношении VMAT1 транспортера означает, что тетрабеназин обладает меньшей периферической активностью, чем резерпин, и следовательно, не дает побочных эффектов, связанных с VMAT1, таких как гипотензия.
Химическая структура тетрабеназина представляет собой структуру, показанную на Фигуре 1, приведенной ниже.
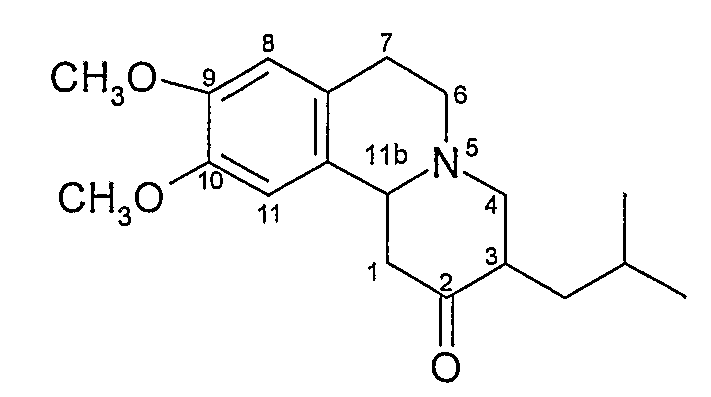
Фигура 1 - структура тетрабеназина
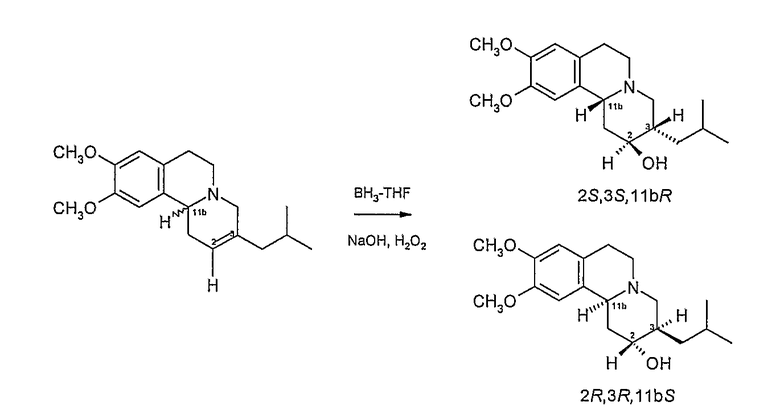
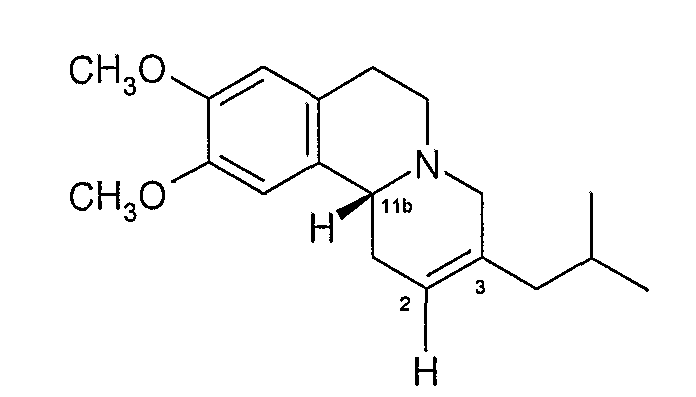
Данное соединение имеет хиральные центры при 3 и 11b атомах углерода и, следовательно, может, теоретически, существовать всего в четырех изомерных формах, как показано на Фигуре 2.
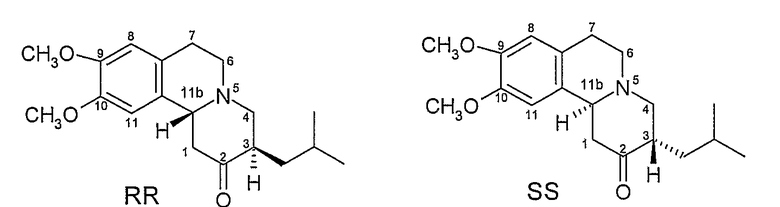
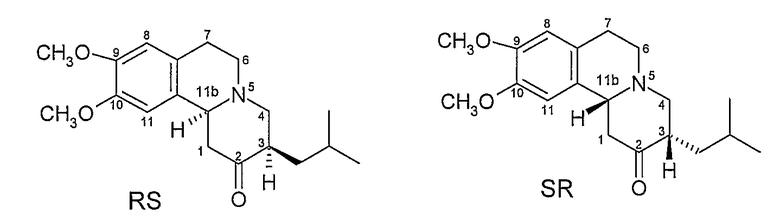
Фигура 2 - Возможные изомеры тетрабеназина
На Фигуре 2, стереохимию каждого изомера определяют, используя «R и S» номенклатуру, разработанную Cahn, Ingold и Prelog, см. Advanced Organic Chemistry by Jerry March, 4th Edition, John Wiley & Sons, New York, 1992, страницы 109-114. На Фигуре 2 и в других местах в этой патентной заявке, обозначения «R» или «S» даны в порядке номеров позиций атомов углерода. Таким образом, например, RS представляет собой сокращенное обозначение для 3R,11bS. Подобным образом, когда присутствуют три хиральных центра, как у дигидротетрабеназинов, описанных ниже, обозначения «R» или «S» перечислены в порядке атомов углерода 2, 3 и 11b. Таким образом, 2S,3R,11bR изомер в кратком виде обозначается SRR и так далее.
Коммерчески доступный тетрабеназин представляет собой рацемическую смесь RR и SS изомеров и получается, что RR и SS изомеры (здесь и далее называемые по отдельности или в совокупности транс-тетрабензином, поскольку атомы водорода в положениях 3 и 11b имеют транс взаимную ориентацию) представляют собой наиболее термодинамически стабильные изомеры.
Тетрабеназин имеет довольно низкую и изменчивую биодоступность. Он интенсивно метаболизируется посредством пресистемного метаболизма, и небольшое количество или неизмененный тетрабеназин обычно определяется в моче. Основным метаболитом является дигидротетрабеназин (химическое название 2-гидрокси-3-(2-метилпропил)-1,3,4,6,7,11b-гексагидро-9,10-диметокси-бензо(a)хинолизин), который образуется восстановлением 2-кетогруппы в тетрабеназине, и считается, что он в первую очередь отвечает за активность лекарственного средства (см. Mehvar et al., Drug Metab. Disp, 15, 250-255 (1987) и J. Pharm. Sci., 76, No. 6, 461-465 (1987)).
Ранее были идентифицированы и охарактеризованы четыре изомера дигидротетрабеназина, все они являются производными более стабильных изомеров RR и SS исходного тетрабеназина и имеют транс взаимную ориентацию между атомами водорода в положениях 3 и 11b) (см. Kilbourn et al., Chirality, 9: 59-62 (1997) и Brossi et al., Helv. Chim. Acta., vol. XLI, No. 193, pp l793-1806 (1958).
Этими четырьмя изомерами являются (+)-α-дигидротетрабеназин, (-)-α-дигидротетрабеназин, (+)-β-дигидротетрабеназин и (-)-β-дигидротетрабеназин. Рассматриваемые структуры четырех известных изомеров дигидротетрабеназина показаны на Фигуре 3.
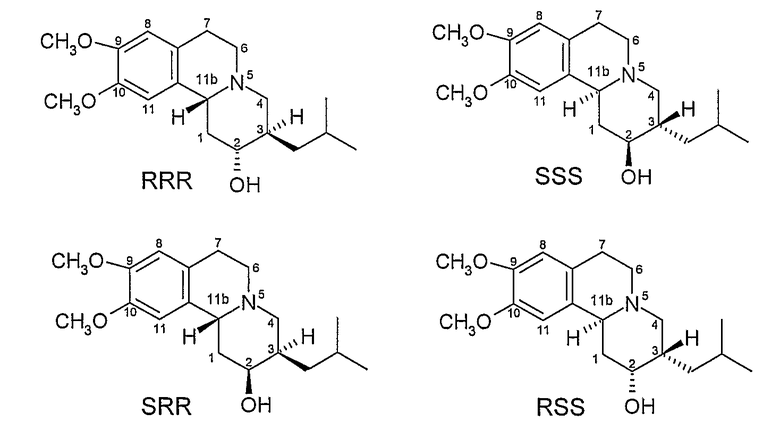
Фигура 3 - Структуры известных изомеров дигидротетрабеназина
Kilbourn et al. (см. Eur. J. Pharmacol., 278: 249-252 (1995) и Med. Chem. Res., 5: 113-126 (1994)) изучали специфическое связывание отдельных радиоактивно-меченных изомеров дигидротетрабеназина, в головном мозге крыс, находящихся в сознании. Они обнаружили, что (2R,3R,11bR)изомер (+)-α-[11C]дигидротетрабеназина накапливался в участках головного мозга, связанных с более высокими концентрациями нейронального мембранного транспортера дофамина (DAT) и везикулярного транспортера моноаминов (VMAT2). Однако по существу неактивный изомер (-)-α-[11C]дигидротетрабеназина был почти равномерно распределен в головном мозге, давая возможность предположить, что специфического связывания с DAT и VMAT2 не происходило. Исследования in vivo коррелировали с исследованиями in vitro, которые демонстрировали, что изомер (+)-α-[11C]дигидротетрабеназина показывает Ki для [3H]метокситетрабеназина >2000 раз выше, чем Ki для изомера (-)-α-[11C]дигидротетрабеназина.
К настоящему времени, насколько осведомлены заявители, изомеры дигидротетрабеназина, полученные из нестабильных RS и SR изомеров (здесь и далее названные по отдельности или в совокупности цис-тетрабеназином, поскольку атомы водорода в положениях 3 и 11b имеют цис-взаимную ориентацию) тетрабеназина ранее не были выделены и охарактеризованы, и данные о биологической активности этих соединений до настоящего времени не были опубликованы.
Сущность изобретения
В настоящее время было обнаружено, что изомеры дигидротетрабеназина, полученные из нестабильных RS и SR изомеров (««цис» изомеров») тетрабеназина являются не только стабильными, но, неожиданно, обладают хорошими биологическими свойствами. В частности, некоторые изомеры имеют профили рецепторной активности, которые предполагают ряд преимуществ по сравнению с используемым в настоящее время RR/SS тетрабеназином. Например, некоторые изомеры, хотя и имеют высокую аффинность к VMAT2, демонстрируют значительно сниженное или незначительное связывание с дофаминовыми рецепторами, следовательно, они едва ли вызывают дофаминергические побочные эффекты, встречающиеся у тетрабеназина. Ни один из этих изомеров не показывал ингибирования переносчика дофамина (DAT). Кроме того, исследования некоторых изомеров на крысах показали, что они не имеют нежелательных седативных побочных эффектов, ассоциированных с тетрабеназином. Отсутствие седативной активности связано с очень низкой аффинностью некоторых изомеров к адренергическим рецепторам. Более того, принимая во внимание, что одним из побочных эффектов тетрабеназина является депрессия, некоторые изомеры дигидротетрабеназина демонстрируют аффинность к белку-переносчику серотонина (SERT), следовательно, они могут обладать антидепрессантным действием.
В связи с этим, в первом аспекте, данное изобретение относится к 3,11b-цис-дигидротетрабеназину.
В другом аспекте, данное изобретение относится к фармацевтической композиции, содержащей 3,11b-цис-дигидротетрабеназин и фармацевтически приемлемый носитель.
Данное изобретение также относится к 3,11b-цис-дигидротетрабеназину по существу в чистом виде, например, с изомерной чистотой более 90%, обычно более 95% и более, предпочтительно более 98%.
Термин «изомерная чистота» в контексте настоящего изобретения относится к количеству присутствующего 3,11b-цис-дигидротетрабеназина относительно общего количества или концентрации дигидротетрабеназина всех изомерных форм. Например, если 90% общего содержания дигидротетрабеназина, присутствующего в композиции, представляет собой 3,11b-цис-дигидротетрабеназин, то изомерная чистота составляет 90%.
Данное изобретение дополнительно относится к композиции, содержащей 3,11b-цис-дигидротетрабеназин, по существу свободный от 3,11b-транс-дигидротетрабеназина, предпочтительно содержащей менее 5% 3,11b-транс-дигидротетрабеназина, более предпочтительно менее 3% 3,11b-транс-дигидротетрабеназина и, наиболее предпочтительно менее 1% 3,11b-транс-дигидротетрабеназина.
В другом аспекте данное изобретение относится к 3,11b-цис-дигидротетрабеназину для применения в медицине или лечении, например, при лечении гиперкинетических двигательных расстройств, таких как болезнь Хантингтона, гемибаллизм, сенильная хорея, тик, поздняя дискинезия и синдром Туретта, или при лечении депрессии.
Еще в одном аспекте, данное изобретение относится к применению 3,11b-цис-дигидротетрабеназина для получения лекарственного средства для лечения гиперкинетических двигательных расстройств, таких как болезнь Хантингтона, гемибаллизм, сенильная хорея, тик, поздняя дискинезия и синдром Туретта, или лечения депрессии.
Еще в одном дополнительном аспекте, данное изобретение относится к способу профилактики или лечения гиперкинетических двигательных расстройств, таких как болезнь Хантингтона, гемибаллизм, сенильная хорея, тик, поздняя дискинезия и синдром Туретта или лечения депрессии у пациента, нуждающегося в такой профилактике или лечении, который включает введение профилактически или терапевтически эффективного количества 3,11b-цис-дигидротетрабеназина.
Термин «3,11b-цис-», используемый здесь, означает, что атомы водорода в 3- и 11b-положениях структуры дигидротетрабеназина находятся в цис-взаимной ориентации. Изомеры по данному изобретению, следовательно, представляют собой соединения формулы (I) и их антиподы (зеркальные отображения).
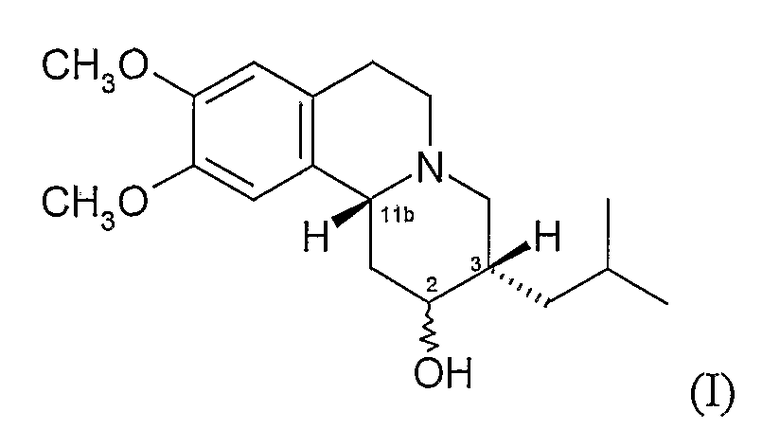
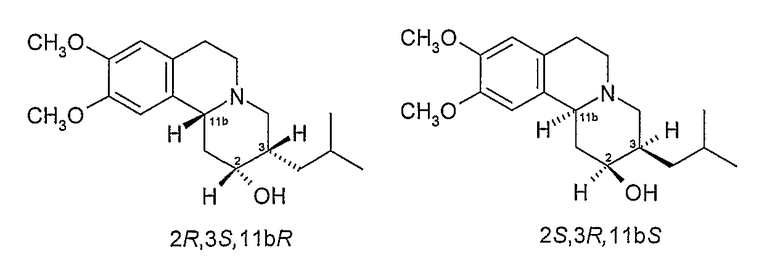
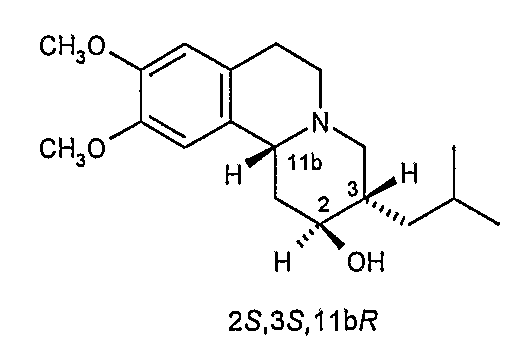
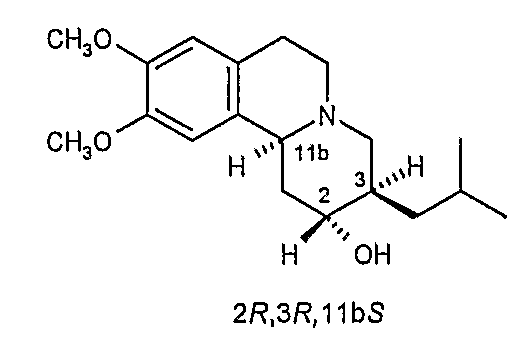
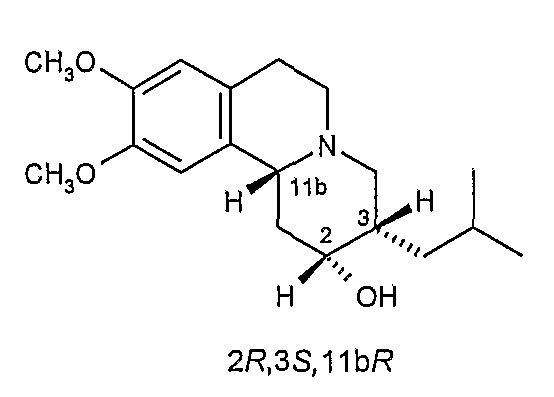
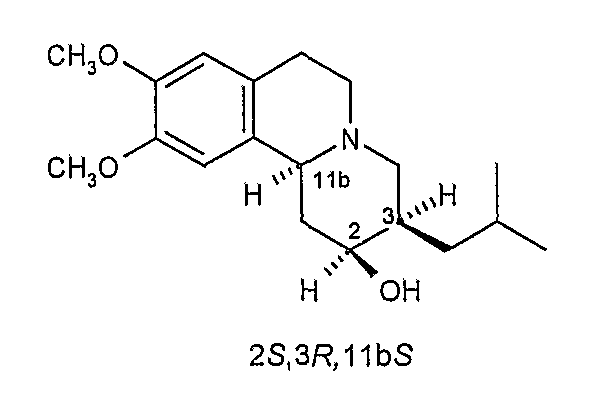
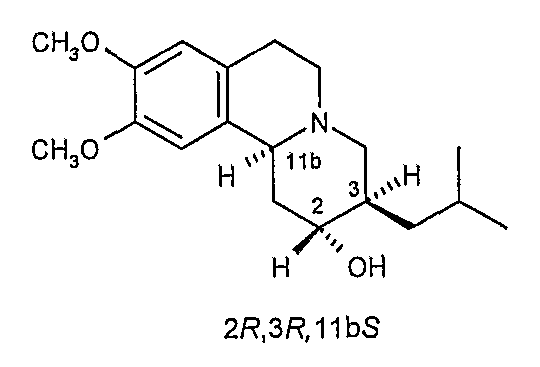
Существует четыре возможных изомера дигидротетрабеназина, имеющих 3,11b-цис конфигурацию, и они представляют собой 2S,3S,11bR изомер, 2R,3R,11bS изомер, 2R,3S,11bR изомер, и 2S,3R,11bS изомер. Эти четыре изомера были выделены и охарактеризованы и, в другом аспекте, данное изобретение относится к индивидуальным изомерам 3,11b-цис-дигидротетрабеназина. В частности, данное изобретение относится к: (a) 2S,3S,11bR изомеру 3,11b-цис-дигидротетрабеназина, имеющего формулу (Ia):
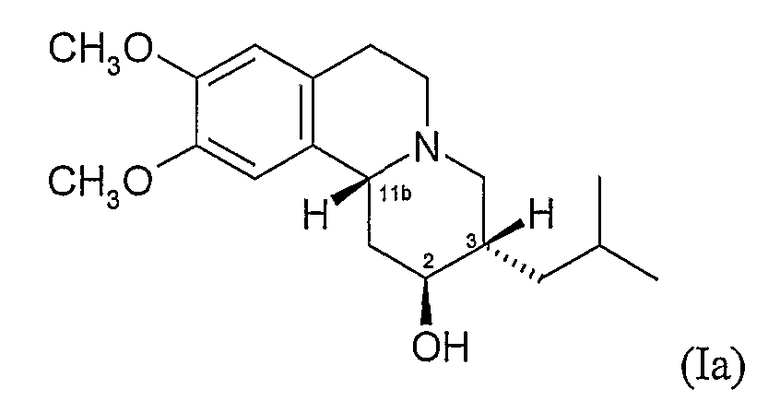
(b) 2R,3R,11bS изомеру 3,11b-цис-дигидротетрабеназина, имеющего формулу (Ib):
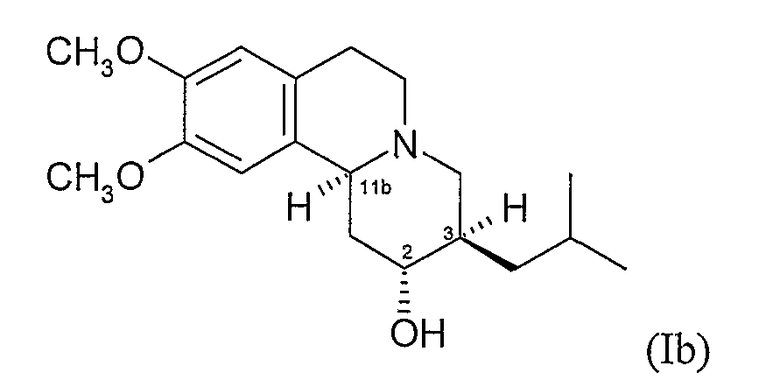
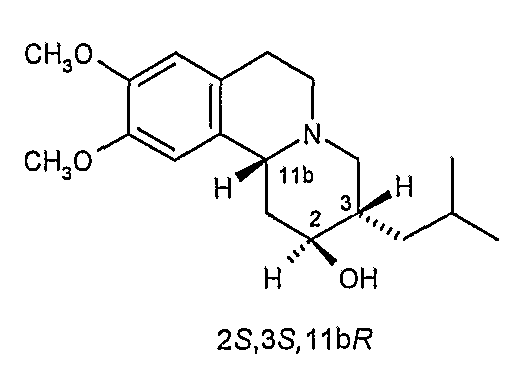
(c) 2R,3S,11bR изомеру 3,11b-цис-дигидротетрабеназина, имеющего формулу (Ic):
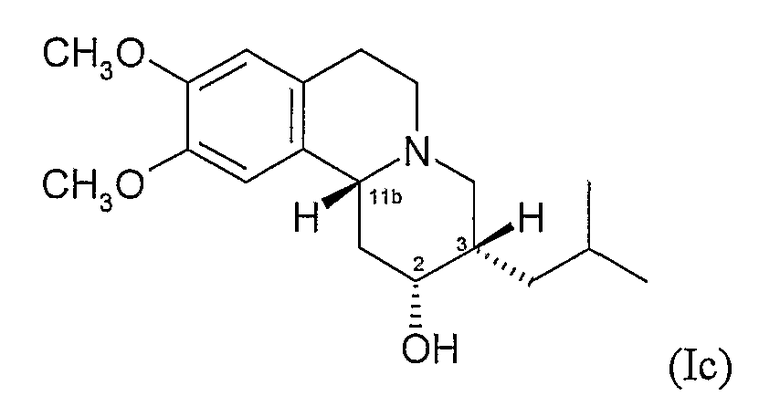
и
(d) 2S,3R,11bS изомеру 3,11b-цис-дигидротетрабеназина, имеющего формулу (Id):
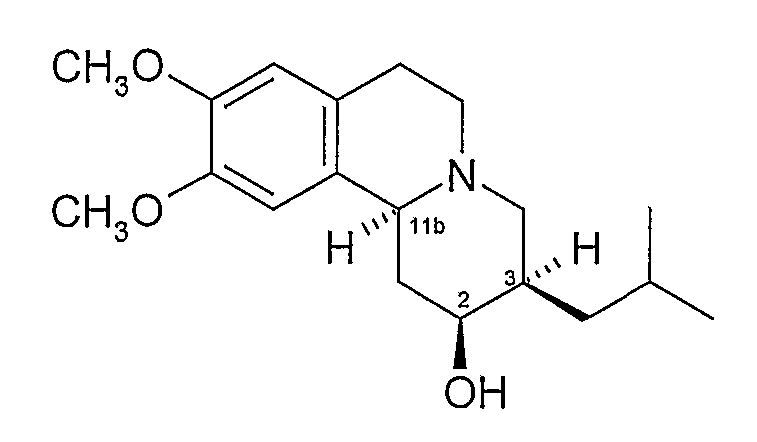
Индивидуальные новые изомеры по данному изобретению могут быть охарактеризованы по их спектроскопическим, оптическим и хроматографическим свойствам.
Предпочтительными изомерами являются правовращающие (+)изомеры.
Не предполагая конкретной абсолютной конфигурации или стереохимии, четыре новых изомера могут быть охарактеризованы следующим образом:
Изомер A
Оптическая активность, измеренная с помощью дисперсии оптического вращения (ORD) (метанол, 21°C): левовращающий (-) ИК-спектр (твердый KBr), 1H-ЯМР-спектр (CDCl3) и 13C-ЯМР-спектр (CDCl3) по существу такие, как описано в Таблице 1.
Изомер B
Оптическая активность, измеренная с помощью дисперсии оптического вращения (ORD) (метанол, 21°C): правовращающий (+)ИК-спектр (твердый KBr), 1H-ЯМР-спектр (CDCl3) и 13C-ЯМР-спектр (CDCl3) по существу такие, как описано в Таблице 1.
Изомер C
Оптическая активность, измеренная с помощью дисперсии оптического вращения (ORD) (метанол, 21°C): ИК-спектр (твердый KBr), 1H-ЯМР-спектр (CDCl3) и 13C-ЯМР-спектр (CDCl3) по существу такие, как описано в Таблице 2.
Изомер D
Оптическая активность, измеренная с помощью дисперсии оптического вращения (ORD) (метанол, 21°C): левовращающий (-) ИК спектр (твердый KBr), 1H-ЯМР спектр (CDCl3) и 13C-ЯМР спектр (CDCl3) по существу такие, как описано в Таблице 2.
Значения ORD для каждого изомера даны в примерах, приведенных ниже, но следует отметить, что такие значения даны в качестве примера и могут варьировать в зависимости от степени чистоты изомера и влияния других переменных, таких как колебания температур и действие остаточных молекул растворителя.
Каждый из энантиомеров A, B, C и D может присутствовать по существу в энантиомерно чистой форме или в виде смесей с другими энантиомерами данного изобретения.
Термины «энантиомерная чистота» и «энантиомерно чистый» в контексте настоящего изобретения относится к количеству данного энантиомера 3,11b-цис-дигидротетрабеназина, присутствующему относительно общего количества или концентрации дигидротетрабеназина всех энантиомерных и изомерных форм. Например, если 90% общего дигидротетрабеназина, присутствующего в данной композиции, находится в виде единственного энантиомера, то энантиомерная чистота составляет 90%.
В качестве примера, в каждом аспекте и варианте осуществления данного изобретения, каждый индивидуальный энантиомер, выбранный из изомеров A, B, C и D, может иметь энантиомерную чистоту по меньшей мере 55% (например, по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99%, 99,5% или 100%).
Изомеры по данному изобретению также могут находиться в форме смесей одного или нескольких изомеров A, B, C и D. Такие смеси могут быть рацемическими смесями или нерацемическими смесями. Примеры рацемических смесей включают в себя рацемическую смесь изомера А и изомера В, и рацемическую смесь изомера С и изомера D.
Фармацевтически приемлемые соли
Если только иное не требуется по контексту, ссылка в этой заявке на дигидротетрабеназин и его изомеры включает в своем объеме не только свободное основание дигидротетрабеназина, а также его соли и, в частности, кислотно-аддитивные соли.
Конкретные кислоты, из которых образуются кислотно-аддитивные соли, включают в себя кислоты, имеющие значение pKa менее 3,5 и, чаще, менее 3. Например, кислотно-аддитивные соли могут образовываться из кислоты с pKa в диапазоне от +3,5 до - 3,5.
Предпочтительные кислотно-аддитивные соли включают в себя соли, образованные с сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, камфорсульфоновая кислота и нафталинсульфоновая кислота.
Кислотой, заслуживающей особого внимания, из которой могут быть образованы кислотно-аддитивные соли, является метансульфоновая кислота.
Кислотно-аддитивные соли могут быть получены описанными здесь способами или обычными химическими методами, такими, которые описаны в публикации Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 страниц, август 2002. В основном такие соли могут быть получены путем взаимодействия соединения в форме свободного основания с соответствующим основанием или кислотой в воде или в органическом растворителе, или в смеси двух; главным образом используют безводную среду, такую как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Соли обычно представляют собой фармацевтически приемлемые соли. Однако соли, не являющиеся фармацевтически приемлемыми, также могут быть получены в виде промежутоных форм, которые затем можно преобразовать в фармацевтически приемлемые соли. Такие не-фармацевтически приемлемые солевые формы также составляют часть данного изобретения.
Способы получения изомеров дигидротетрабеназина
В дополнительном аспекте предлагается способ (Способ А) получения дигидротетрабеназина по данному изобретению, который включает в себя взаимодействие соединения формулы (II):
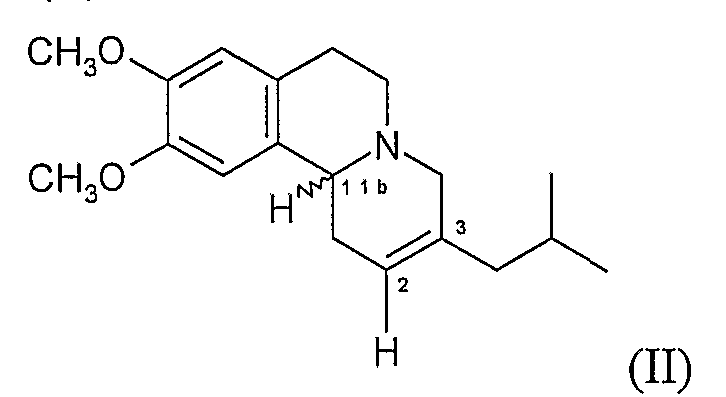
с реагентом или реагентами, подходящими для гидрирования 2,3-двойной связи в соединении формулы (II) и, в последствии, где требуется, отделение и выделение желаемой изомерной форы дигидротетрабеназина.
Гидрирование 2,3-двойной связи можно провести гидроборированием с использованием боранового реагента, такого как диборан или боран-эфир (например, боран-тетрагидрофуран (ТГФ)) с получением промежуточного алкил-боранового продукта присоединения с последующим окислением алкил-боранового продукта присоединения и гидролизом в присутствии основания. Гидроборирование обычно проводят в сухом полярном апротонном растворителе, таком как простой эфир (например, ТГФ), обычно при неповышенной температуре, например, при комнатной температуре. Боран-алкеновый продукт присоединения обычно окисляют окисляющим агентом, таким как перекись водорода, в присутствии основания, обеспечивающего источник гидроксид-ионов, как, например, гидроксид аммония или гидроксид щелочного металла, например, гидроксид калия или гидроксид натрия. Последовательность реакций Способа А гидроборирование-окисление-гидролиз обычно дает изомеры дигидротетрабеназина, в которых атомы водорода в положениях 2- и 3-транс взаимную ориентацию.
Соединения формулы (II) могут быть получены путем восстановления тетрабеназина с получением дигидротетрабеназина с последующим дегидрированием дигидротетрабеназина. Восстановление тетрабеназина можно осуществить с использованием алюмогидридного реагента, или боргидридного реагента, такого как боргидрид натрия, боргидрид калия или боргидридное производное, например, алкилборгидрид, такой как три-втор-бутилборгидрид. Альтернативно, стадию восстановления можно проводить с использованием каталитического гидрирования, например, на скелетном никелевом катализаторе гидрирования или оксидоплатиновом катализаторе. Подходящие условия для проведения стадии восстановления описаны более подробно ниже или их можно найти в US 2843591 (Hoffmann-La Roche) и Brossi et al., Helv. Chim. Acta., vol. XLI, No. 193, pp l793-1806 (1958).
Поскольку тетрабеназин, используемый как исходное вещество для реакции восстановления, обычно представляет собой смесь RR и SS изомеров (т.е. транс-тетрабеназин), дигидротетрабеназин, образованный на стадии восстановления, будет иметь такую же транс-конфигурацию в 3- и 11b-положениях и примет форму одного или нескольких известных изомеров дигидротетрабеназина, показанных на Фигуре 3, указанной выше. Таким образом, Способ А может включать в себя взятие известных изомеров дигидротетрабеназина, дегидрирование их до формы алкена (II), а затем «регидрирование» алкена (II) с использованием условий, которые дают желаемые новые цис-изомеры дигидротетрабеназина по данному изобретению.
Дегидрирование дигидротетрабеназина в алкен (II) можно проводить с использованием целого ряда стандартных условий для дегидрирования спиртов до образования алкенов, см., например, J. March (idem) страницы 389-390 и приведенные ссылки. Примеры таких условий включают в себя применение дегидрирующих агентов на основе фосфора, таких как галогениды фосфора или оксигалогениды фосфора, например POCl3 и PCl5. В качестве альтернативы для прямого дегидрирования, гидроксильная группа дигидротетрабеназина может быть преобразована в уходящую группу L, такую как галоген (например, хлорин или бромин), а затем подвергнута воздействию условий (например, присутствие основания) для удаления H-L. Преобразование гидроксильной группы в галогенид может достигаться использованием способов, хорошо известных опытному химику, например реакцией с тетрахлоридом углерода или тетрабромидом углерода в присутствии триалкил- или триарилфосфина, такого как трифенилфосфин или трибутилфосфин.
Тетрабеназин, используемый в качестве исходного вещества для восстановления, для получения дигидротетрабеназина может быть получен коммерчески или может быть синтезирован способом, описанным в US 2830993 (Hoffmann-La Roche).
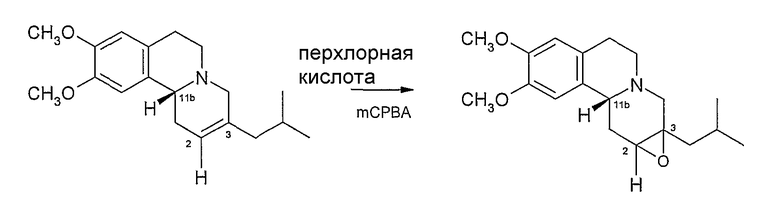
Данное изобретение также относится к способу (Способ В) получения дигидротетрабеназина по данному изобретению, который включает в себя воздействие на соединение формулы (III):
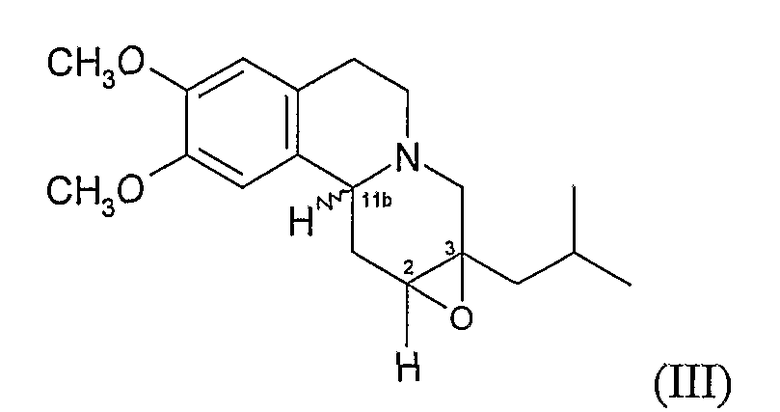
условий для раскрытия цикла 2,3-эпоксидной группы в соединении формулы (III), а в дальнейшем, где требуется, отделение и выделение желаемой изомерной формы дигидротетрабеназина.
Раскрытие цикла можно осуществить в соответствии с известными способами раскрытия эпоксидного цикла. Однако предпочтительным способом раскрытия эпоксидного цикла в настоящее время является восстановительное раскрытие цикла, которое может достигаться с использованием восстанавливающего агента, такого как боран-ТГФ. Реакцию с боран-ТГФ можно проводить в полярном апротонном растворителе, таком как простой эфир (например, тетрагидрофуран) обычно при температуре окружающей среды, образованный таким образом борановый комплекс впоследствии гидролизуется нагреванием в присутствии воды и основания при температуре кипения растворителя. В Способе B обычно образуются изомеры дигидротетрабеназина, в которых атомы водорода в положениях 2- и 3- имеют цис-взаимную ориентацию.
Эпоксидные соединения формулы (III) могут быть получены эпоксидированием алкена формулы (II), указанной выше. Реакцию эпоксидирования проводят с использованием условий и реагентов, хорошо известных опытному химику, см., например, J. March (idem), страницы 826-829 и ссылки. Обычно перкислота, такая как мета-хлорпербензойная кислота (MCPBA), или смесь перкислоты и дополнительного окисляющего агента, такого как перхлорная кислота, могут быть использованы для осуществления эпоксидирования.
В тех случаях, когда исходные вещества для способов А и В, указанных выше, представляют собой смеси энантиомеров, тогда продукты этих способов обычно будут представлять собой пары энантиомеров, например, рацемические смеси, возможно вместе с диастереоизомерными примесями. Нежелательные диастереоизомеры могут быть удалены такими способами, как хроматография (например, ВЭЖХ) и индивидуальные энантиомеры могут быть разделены рядом способов, известных опытному химику. Например, их можно разделить посредством:
(i) хиральной хроматографии (хроматография на хиральном носителе); или
(ii) образования соли с оптически чистой хиральной кислотой, отделение данных солей двух диастереоизомеров фракционной кристаллизацией, а затем выделение дигидротетрабеназина из данной соли; или
(iii) образование производного (такого как сложный эфир) с оптически чистым хиральным агентом, образующим производные (например, этерифицирующим агентом), отделение полученных в результате эпимеров (например, хроматографией), а затем превращение данного производного в дигидротетрабеназин.
Один способом разделения пар энантиомеров, полученных в результате каждого из Способов А и В, и который был признан особенно эффективным, является этерификация гидроксильной группы дигидротетрабеназина оптически активной формой кислоты Мошера, такой как R (+) изомер, показанный ниже, или его активной формой:
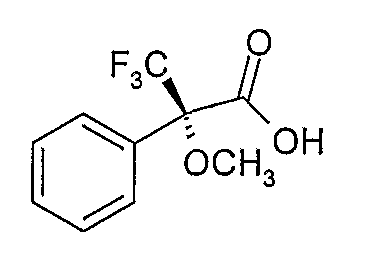
Полученные в результате сложные эфиры двух энантиомеров дигидротетрабеназина затем могут быть разделены с помощью хроматографии (например, ВЭЖХ) и разделенные сложные эфиры подвергали гидролизу с получением индивидуальных изомеров дигидробеназина, используя основание, такое как гидроксид щелочного металла (например, NaOH) в полярном растворителе, таком как метанол.
В качестве альтернативы использованию смесей энантиомеров, в качестве исходных веществ в способах А, а затем последующему проведению разделения энантиомеров, способы А и В каждый можно проводить на взятых в отдельности энантиомерах исходных веществ, что в результате приводит к продуктам, в которых преобладает отдельно взятый энантиомер. Отдельные энантиомеры алкена (II) могут быть получены путем проведения стереоселективного восстановления RR/SS тетрабеназина с использованием три-втор-бутилборгидрида лития с получением смеси SRR и RSS энантиомеров дигидротетрабеназина, разделения энантиомеров (например, фракционной кристаллизацией), а затем дегидрированием отделенного индивидуального энантиомера дигидротетрабеназина с получением преимущественно или исключительно индивидуального энантиомера соединения формулы (II).
Способы A и B более подробно проиллюстрированы ниже на схемах 1 и 2 соответственно.
Схема 1
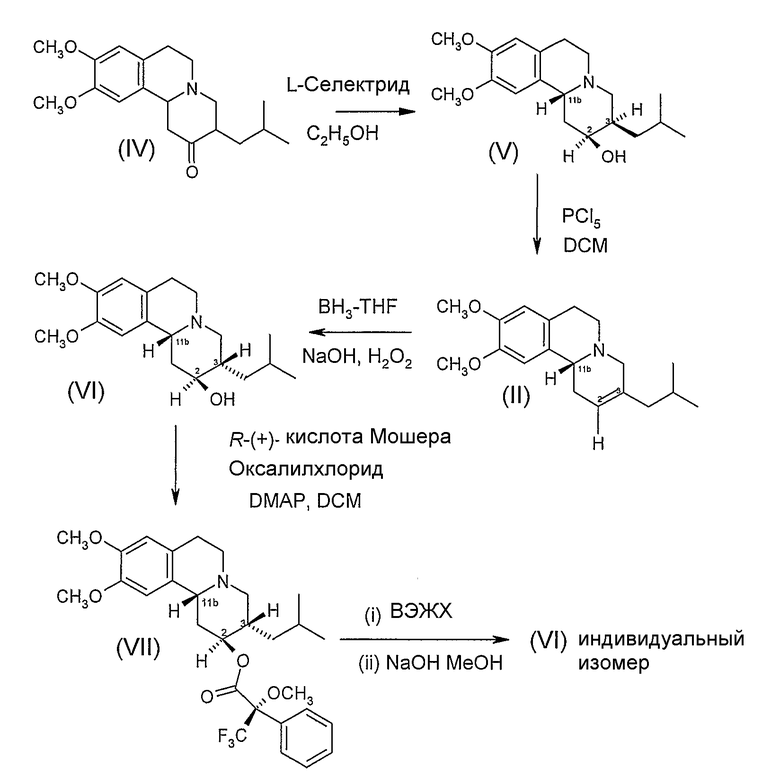
Схема 1 иллюстрирует получение индивидуальных изомеров дигидротетрабеназина, имеющих 2S,3S,11bR и 2R,3R,11bS конфигурации, в которых атомы водорода, присоединенные к положениям 2- и 3- расположены в транс взаимной ориентации. Эта схема реакции включает в себя описанный выше Способ А.
Исходной точкой последовательности реакций на Схеме 1 является коммерчески доступный тетрабеназин (IV), который представляет собой рацемическую смесь RR и SS оптических изомеров тетрабеназина. В каждых RR и SS изомерах атомы водорода в положениях 3- и 11b- расположены в транс взаимной ориентации. В качестве альтернативы использованию коммерчески доступного соединения, тетрабеназин может быть синтезирован в соответствии с процедурой, описанной в патенте США № 2830993 (см. в конкретном примере 11).
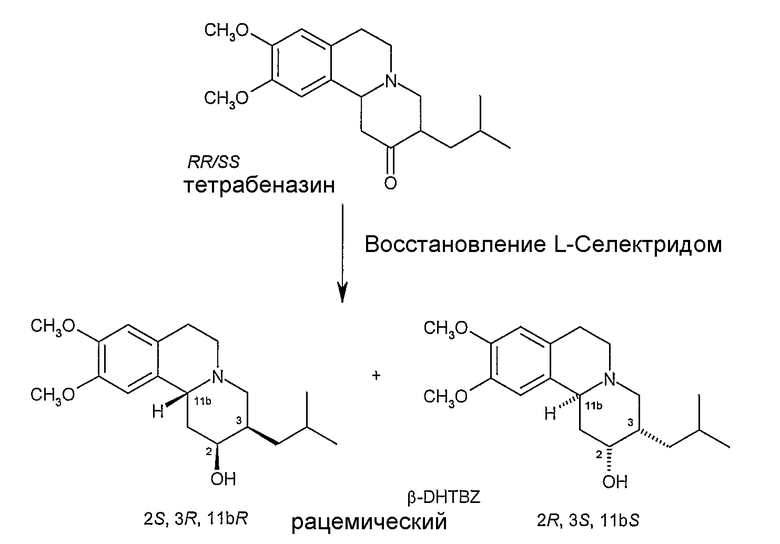
Рацемическую смесь RR и SS тетрабеназина восстанавливают с использованием боргидридного восстанавливающего агента три-втор-бутилборгидрида лития («L-Селектрид») с получением смеси известных 2S,3R,11bR и 2R,3S,11bS изомеров (V) дигидротетрабеназина, из которых для простоты показан только 2S,3R,11bR изомер. Используя более стерически затрудненный L-Селектрид в качестве боргидридного восстанавливающего агента, а не боргидрида натрия, образование RRR и SSS изомеров дигидротетрабеназина сводится к минимуму или подавляется.
Изомеры дигидротетрабеназина (V) взаимодействуют с дегидрирующим агентом, таким как пентахлорид фосфора, в апротонном растворителе, таком как хлорированный углеводород (например, хлороформ или дихлорметан, предпочтительно дихлорметан) с образованием ненасыщенного соединения (II) в виде пары энантиомеров, из которых только R-энантиомер показан на Схеме. Реакцию восстановления обычно проводят при температуре ниже комнатной температуры, например, примерно при 0-5°C.
Ненасыщенное соединение (II) затем подвергают стереоселективной регидратации с получением дигидротетрабеназина (VI) и его зеркального изображения или антипода (не показан), в котором атомы водорода в положениях 3- и 11b- расположены в цис-взаимной ориентации, а атомы водорода в положениях 2- и 3- расположены в транс-взаимной ориентации. Стереоселективную регидратацию проводят методом гидроборирования с использованием боран-ТГФ в тетрагидрофуране (ТГФ) с образованием промежуточного боранового комплекса (не показан), который затем окисляют перекисью водорода в присутствии основания, такого как гидроксид натрия.
Затем можно провести начальную стадию очистки (например, с помощью ВЭЖХ) с получением продукта (V) последовательности реакции регидратации, в виде смеси 2S,3S,11bR и 2R,3R,11bS изомеров, из которых на Схеме показан только 2S,3S,11bR изомер. Для разделения изомеров смесь обрабатывают R(+)кислотой Мошера в присутствии оксалилхлорида и диметиламинопиридина (DMAP) в дихлорметане с получением пары диастереоизомерных сложных эфиров (VII) (из которых показан только один диастереоизомер), которые затем можно разделить, используя ВЭЖХ. Индивидуальные сложные эфиры затем можно гидролизовать, используя гидроксид щелочного металла, такой как гидроксид натрия, с получением отдельного изомера (VI).
В одном варианте последовательности стадий, показанных на Схеме 1, следующих за восстановлением RR/SS тетрабеназина, результирующую смесь энантиомеров дигидротетрабеназина (V) можно разделить с получением индивидуальных энантиомеров. Разделение можно провести путем образования соли с хиральной кислотой, такой как (+) или (-)камфорсульфоновая кислота, разделения полученных в результате диастереоизомеров фракционной кристаллизицией с получением соли отдельного энантиомера, а затем высвобождения из соли свободного основания.
Отделенный энантиомер дигидротетрабеназина можно подвергнуть дегидратации с получением отдельного энантиомера алкена (II). Последующая регидратация алкена (II) затем даст преимущественно или исключительно отдельно взятый энантиомер цис-дигиротетрабеназина (VI). Преимуществом этого варианта является то, что он не включает в себя образование сложных эфиров кислоты Мошера, и, следовательно, избегает хроматографического разделения, обычно используемого для разделения сложных эфиров кислоты Мошера.
Схема 2 иллюстрирует получение индивидуальных изомеров дигидротетрабеназина, имеющих 2R,3S,11bR и 2S,3R,11bS конфигурации, в которых атомы водорода, присоединенные в положениях 2- и 3- находятся в цис взаимной ориентации. Эта схема реакции заключает в себе описанный выше Способ В.
Схема 2
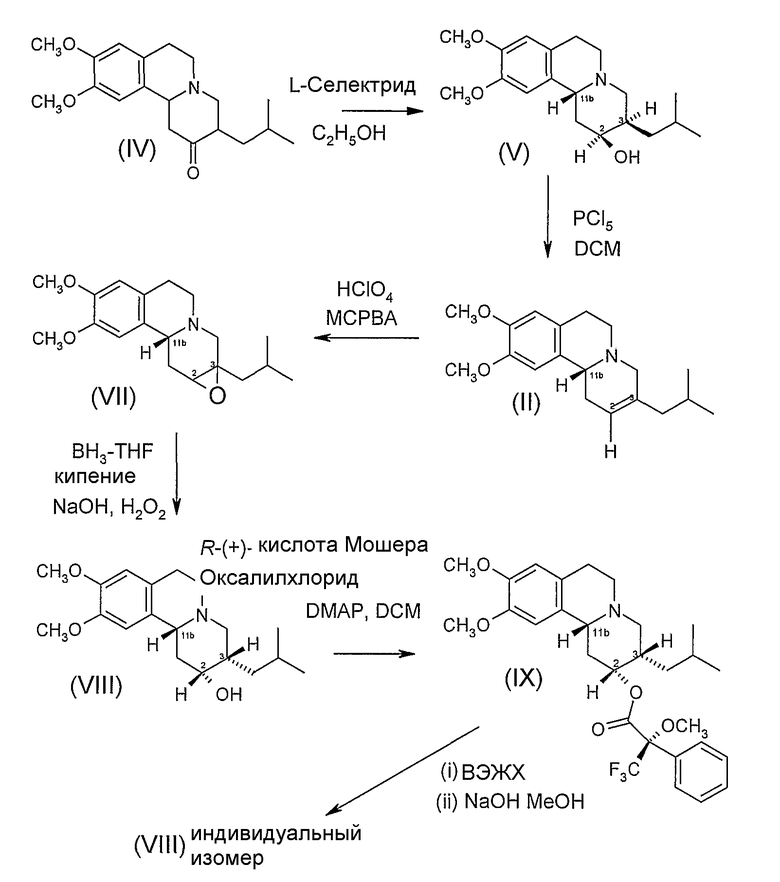
На Схеме 2, ненасыщенное соединение (II) получают восстановлением тетрабеназина с образованием 2S,3R,11bR и 2R,3S,11bS изомеров (V) дигидротетрабеназина и дегидрированием с использованием PCl5 способом, описанным выше на Схеме 1. Однако вместо проведения гидроборирования соединения (II), 2,3-двойную связь преобразовывают в эпоксидную, реакцией с мета-хлорпербензойной кислотой (MCPBA) и перхлорной кислотой. Реакцию эпоксидирования удобно проводить в спиртовом растворителе, таком как метанол, обычно примерно при комнатной температуре.
Эпоксид (VII) затем подвергают восстановительному раскрытию цикла, используя боран-ТГФ в качестве электрофильного восстанавливающего агента с получением промежуточного боранового комплекса (не показано), который затем окисляют и расщепляют перекисью водорода в присутствии щелочи, такой как гидроксид натрия, с получением дигидротетрабеназина (VIII) в виде смеси 2R,3S,11bR и 2S,3R,11bS изомеров, из которых для простоты показан только 2R,3S,11bR. Обработка смеси изомеров (VIII) R (+) кислотой Мошера в присутствии оксалилхлорида и диметиламинопиридина (DMAP) в дихлорметане дает пару эпимерных сложных эфиров (IX) (из которых показан только один эпимер), который затем можно отделить хроматографией и подвергнуть гидролизу гидроксидом натрия в метаноле, способом, описанным выше, в отношении Схемы 1.
Полагают, что химические производные (II) и (III) являются новыми и представляют дополнительный аспект данного изобретения.
Биологические свойства и терапевтическое применение
Тетрабеназины оказывают свои терапевтические эффекты путем ингибирования везикулярного транспортера моноаминов VMAT2 в головном мозге и путем ингибирования как пре-синаптических, так и пост-синаптических дофаминовых рецепторов.
Новые изомеры дигидротетрабеназина по данному изобретению также являются ингибиторами VMAT2, с изомерами C и B дающими наибольшую степень ингибирования. Подобно тетрабеназину, соединения по данному изобретению имеют только низкую аффинность к VMAT1, изоформе VMAT, обнаруженной в периферических тканях и некоторых эндокринных клетках, тем самым указывая на то, что они не должны давать побочных эффектов, связанных с резерпином. Соединения C и B также не проявляют ингибирующей активности в отношении катехол О-метилтрансферазы (COMT), моноаминоксидазы изоформ A и B и изоформ 1d и 1b 5-гидрокситриптамина.
Неожиданно изомеры C и B также демонстрируют заметное расхождение в VAMT2 и дофамин рецепторной активности в том смысле, что хотя они являются высокоактивными в связывании с VMAT2, оба соединения проявляют только слабую связывающую активность в отношении рецептора дофамина, или она не существует, и недостаточную связывающую активность в отношении переносчика дофамина (DAT). Фактически, ни один из изомеров не проявляет значительной DAT-связывающей активности. Это дает основания предположить, что данные соединения могут не иметь дофаминэргических побочных эффектов, вызываемых тетрабеназином. Изомеры С и В также являются либо слабоактивными, либо неактивными в качестве ингибиторов адренэргических рецепторов, и это дает основания предположить, что данные соединения могут не иметь адренэргических побочных эффектов, часто встречающихся у тетрабеназина. Действительно, в исследованиях двигательной функции, проводимых на крысах, тетрабеназин демонстрировал дозозависимый седативный эффект, тогда как после введения изомеров В и С по изобретению седативные эффекты не наблюдались.
Кроме того, как изомер С, так и изомер В являются мощными ингибиторами белка-переносчика серотонина SERT. Ингибирование SERT является одним механизмом, посредством которого антидепрессанты, такие как флуоксетин (Prozac®) оказывают свои терапевтические эффекты. Следовательно, способность изомеров С и В ингибировать SERT указывает на то, что эти изомеры могут действовать как антидепрессанты, в отличие от тетрабеназина, для которого депрессия является хорошо известным побочным эффектом.
На основании исследований, проведенных к настоящему времени, предусматривается, что дигидротетрабеназиновые соединения по данному изобретению будут эффективны при профилактике или лечении заболеваний и состояний, для которых в настоящее время используется или предлагается тетрабеназин. Таким образом, в качестве примера и без ограничения, дигидротетрабеназиновые соединения по данному изобретению могут быть использованы для лечения гиперкинетических расстройств, таких как болезнь Хантингтона, гемибаллизм, сенильная хорея, тик, поздняя дискинезия, дистония и синдром Туретта.
Также предполагается, что дигидротетрабеназиновые соединения по данному изобретению могут быть эффективны при лечении депрессии.
Эти соединения в основном будут назначать пациенту, нуждающемуся в таком назначении, например, пациенту - человеку или животному, предпочтительно человеку.
Эти соединения обычно будут вводить в количествах, которые являются терапевтически или профилактически эффективными и которые в основном являются нетоксичными. Однако в определенных ситуациях польза от введения дигидротетрабеназинового соединения по данному изобретению может перевешивать вред от любых токсических или побочных эффектов, при этом можно считать целесообразным введение соединений в количествах, которые связаны с некоторой степенью токсичности.
Обычная суточная доза соединения может находиться в интервале от 0,025 миллиграмм до 5 миллиграмм на килограмм массы тела, например, до 3 миллиграмм на килограмм массы тела, и чаще от 0,15 миллиграмм до 5 миллиграмм на килограмм массы тела, хотя, когда требуется, можно вводить большие или меньшие дозы.
Для примера, начальную исходную дозу 12,5 мг можно вводить 2-3 раза в день. Эту дозу можно увеличить на 12,5 мг каждые 3-5 дней до достижения максимально переносимой и эффективной дозы для индивидуума, определяемой лечащим врачом. В конечно счете, количество введенного соединения будет соответствовать природе заболевания или физиологическому состоянию, подвергаемого лечению, и терапевтическим эффектам и наличию или отсутствию побочных эффектов, вызываемых установленным режимом дозирования, и на усмотрение лечащего врача.
Фармацевтические композиции
Изобретение также относится к дигидротетрабеназиновым соединениям, определенным здесь ранее, в виде фармацевтических композиций.
Фармацевтические композиции могут быть в любой форме, подходящей для перорального, парентерального, местного, интраназального, внутрибронхиального, глазного, ушного, ректального, внутривагинального или чрескожного введения. В тех случаях, когда композиции предназначены для парентерального введения, они могут быть получены для внутривенного, внутримышечного, внутрибрюшинного, подкожного введения или для прямой доставки к целевому органу или ткани путем инъекции, инфузии или другими способами доставки.
Фармацевтические лекарственные формы, подходящие для перорального введения, включают в себя таблетки, капсулы, таблетки в форме капсулы, пилюли, лекарственные леденцы, сиропы, растворы, спреи, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, спреи, облатки или пластинки и буккальные пластинки.
Фармацевтические композиции, содержащие дигидротетрабеназиновые соединения по данному изобретению, могут быть составлены в соответствии с известными способами, см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Таким образом, таблетированная композиция может содержать единичную дозу активного соединения вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например лактозой, сахарозой, сорбитом или маннитом; и/или разбавителем несахарного происхождения, таким как карбонат натрия, фосфат кальция, тальк, карбонат кальция, или целлюлоза или ее производные, такие как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, и крахмалы, как, например, кукурузный крахмал. Таблетки также могут содержать стандартные ингредиенты в качестве связующих и гранулирующих агентов, как, например, поливинилпирролидон, дезинтегранты (например, разбухающие поперечносшитые полимеры, такие как проперечносшитая карбоксиметилцеллюлоза), смазывающие вещества (например, стеараты), консерванты (например, парабены), антиоксиданты (например BHT), буферные агенты (например, фосфатные или цитратные буферы), и шипучие вещества, такие как цитрат/бикарбонатные смеси. Такие эксципиенты хорошо известны и не нуждаются здесь в более подробном обсуждении.
Составы в виде капсул могут быть твердыми желатиновыми или мягкими желатиновыми и могут содержать активный компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы можно получать из животного желатина или его эквивалентов синтетического или растительного происхождения.
Твердые лекарственные формы (например, таблетки, капсулы и т.п.) могут быть покрытыми или не иметь покрытия, но обычно они имеют покрытие, например, защитное пленочное покрытие (например, воск или глазурь) или покрытие, контролирующее высвобождение. Это покрытие (например, полимер типа EudragitTM) может быть предназначено для высвобождения активного компонента в желаемом месте желудочно-кишечного тракта. Таким образом, это покрытие можно выбрать таким образом, чтобы оно разрушалось при определенных значениях рН в желудочно-кишечном тракте, тем самым селективно высвобождая соединение в желудке или в подвздошной или двенадцатиперстной кишке.
Вместо покрытия, или в дополнение к нему, лекарственное средство может находиться в твердой матрице, содержащей агент, контролирующий высвобождение, например агент, замедляющий высвобождение, который может быть приспособлен к селективному высвобождению соединения в условиях различной кислотности или основности в желудочно-кишечном тракте. Альтернативно, материал матрицы или покрытия, задерживающего высвобождение, может принимать форму эрозируемого полимера (например, полимер малеинового ангидрида), который по существу непрерывно эрозируется при прохождении лекарственной формы через желудочно-кишечный тракт.
Композиции для местного применения включают в себя мази, кремы, спреи, пластыри, гели, жидкие капли и вкладки (например, внутриглазные вкладки). Такие композиции могут быть получены в соответствии с известными способами.
Композиции для парентерального введения обычно находятся в виде стерильных водных или масляных растворов или тонкодисперсных суспензий, или могут быть получены в форме мелкоизмельченного стерильного порошка для приготовления состава со стерильной водой для инъекций для немедленного применения.
Примеры составов для ректального или интравагинального введения включают в себя пессарии и суппозитории, которые могут быть, например, получены из веществ, принимающих определенную форму или воскообразных веществ, содержащих активное соединение.
Композиции для введения ингаляционным путем могут быть в форме ингаляционных порошковых композиций или жидких, или порошкообразных спреев, и могут быть введены в стандартной форме с использованием порошковых ингаляционных устройств. Такие устройства хорошо известны. Для введения путем ингаляции порошкообразные составы обычно содержат активное вещество вместе с инертным твердым порошкообразным разбавителем, таким как лактоза.
Соединения по данному изобретению главным образом будут находиться в лекарственной форме для однократного приема и, по существу, как правило, будут содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, состав, предназначенный для перорального введения, может содержать от 2 миллиграмм до 200 миллиграмм активного ингредиента, чаще от 10 миллиграмм до 100 миллиграмм, например, 12,5 миллиграмм, 25 миллиграмм и 50 миллиграмм.
Активное соединение вводят пациенту, нуждающемуся в этом (например, пациенту человеку или животному), в количестве, достаточном для достижения желаемого терапевтического эффекта.
ПРИМЕРЫ
Следующие не ограничивающие объем изобретения примеры иллюстрируют синтез и свойства дигидротетрабеназиновых соединений настоящего изобретения.
ПРИМЕР 1
Получение 2S,3S,11bR и 2R,3R,11bS изомеров дигидротетрабеназина 1A. Восстановление RR/SS тетрабеназина.
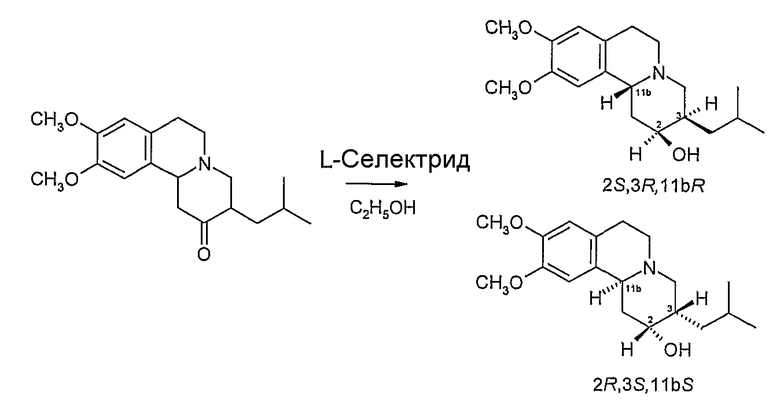
К перемешиваемому раствору рацемата RR/SS тетрабеназина (15 г, 47 ммoль) в этаноле (75 мл) и тетрагидрофуране (75 мл) при 0°С в течение 30 минут медленно добавляют 1М L-Селектрид® в тетрагидрофуране (135 мл, 135 ммол, 2,87 экв.). После окончания добавления смесь перемешивают при 0°С 30 минут и дают нагреться до комнатной температуры.
Смесь выливают на измельченный лед (300 г) и добавляют воду (100 мл). Раствор экстрагируют диэтиловым эфиром (2×200 мл) и объединенные эфирные экстракты промывают водой (100 мл) и частично сушат над безводным карбонатом калия. Окончательную сушку осуществляют с использованием безводного сульфата магния и после фильтрации удаляют при пониженном давлении растворитель (защита от света, температура бани <20°С) с получением светло-желтого твердого остатка.
Твердый остаток суспендируют в петролейном эфире (30-40°С) и отфильтровывают с получением белого порошкообразного твердого остатка (12 г, 80%).
1B. Дегидратация восстановленного тетрабеназина
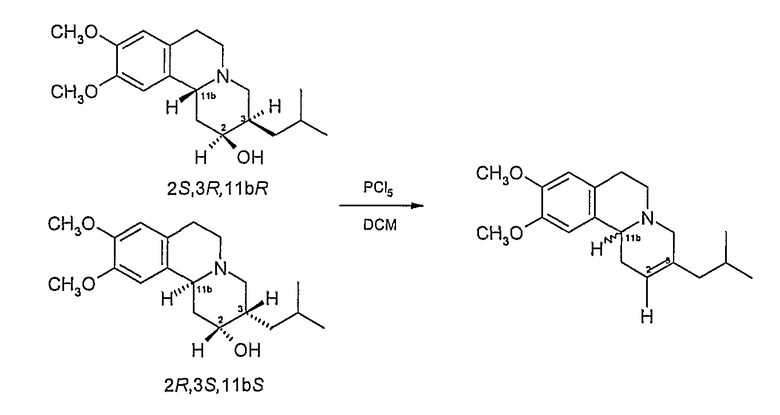
К перемешиваемому раствору восстановленного тетрабеназинового продукта примера 1А (20 г, 62,7 ммоль) в дихлорметане (200 мл) при 0°С добавляют порциями в течение 30 минут пятихлористый фосфор (32,8 г, 157,5 ммол, 2,5 экв.). По окончании добавления реакционную смесь дополнительно перемешивают при 0°С в течение 30 минут и раствор медленно вливают в 2M водный раствор карбоната натрия, содержащий измельченный лед (0°С). Начальное выделение газообразной кислоты останавливают подщелачиванием смеси (примерно pH 12), используя твердый карбонат натрия.
Щелочной раствор экстрагируют этилацетатом (800 мл) и объединенные органические экстракты сушат над безводным сульфатом магния. После фильтрации удаляют при пониженном давлении растворитель до получения коричневого масла, которое очищают с помощью колоночной хроматографии (кремнезем, этилацетат) с получением получистого алкена в виде желтого твердого остатка (10,87 г, 58%).
1C. Гидратация сырого алкена из примера 1B
Раствор сырого алкена (10,87 г, 36,11 ммоль) из примера 1B в ТГФ (52 мл) обрабатывают, добавляя при комнатной температуре по каплям 1M боран-ТГФ (155,6 мл, 155,6 ммол, 4,30 экв.). Реакционную смесь перемешивают 2 часа, добавляют воду (20 мл) и подщелачивают раствор до pH 12 30% водным раствором гидроксида натрия.
К щелочной реакционной смеси при перемешивании добавляют 30% водный раствор перекиси водорода (30 мл), раствор нагревают с обратным холодильником 1 час и дают охладиться. Добавляют воду (100 мл) и экстрагируют смесь этилацетатом (3×250 мл). Органические экстракты объединяют, сушат над безводным сульфатом магния и после фильтрации удаляют при пониженном давлении растворитель до получения желтого масла (9 г).
Полученное масло очищают с использованием препаративной ВЭЖХ (Колонка: Lichrospher Si60, 5 мкм, 250×21,20 мм, подвижная фаза:гексан:этанол:дихлорметан (85:15:5); УФ 254 нм, скорость потока: 10 мл/мин-1) при 350 мг на впрыскивание с последующим концентрированием представляющих интерес фракций под вакуумом. Затем полученное масло растворяют в эфире и еще раз концентрируют под вакуумом с получением указанного выше рацемата дигидротетрабеназина в виде желтой пены (5,76 г, 50%).
1D. Получение эфирных производных Мошера
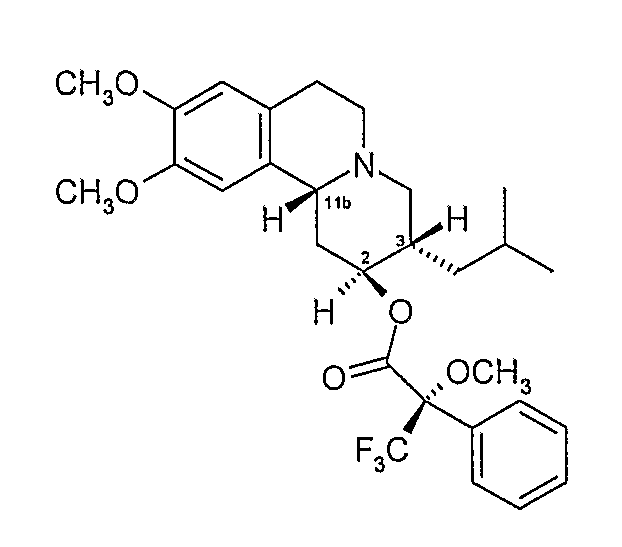 и
и 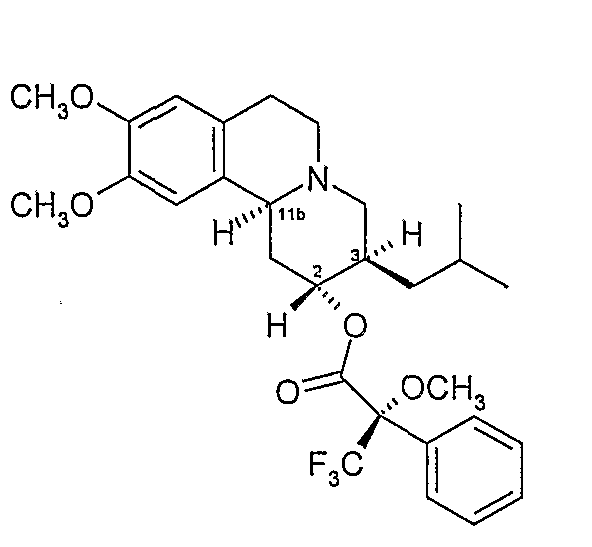
К безводному дихлорметану (50 мл) добавляют R-(+)-α-метокси-α-трифторметилфенилуксусную кислоту (5 г, 21,35 ммоль), оксалилхлорид (2,02 мл) и ДФА (0,16 мл) и раствор перемешивают при комнатной температуре 45 минут. Раствор концентрируют при пониженном давлении и остаток еще раз поглощают безводным дихлорметаном (50 мл). Полученный раствор охлаждают на водяной бане со льдом и добавляют диметиламинопиридин (3,83 г, 31,34 ммоль) с последующим добавлением предварительно высушенного раствора (сито 4Å) твердого продукта Примера 1C (5 г, 15,6 ммоль) в безводном дихлорметане. После перемешивания при комнатной температуре в течение 45 минут добавляют воду (234 мл) и смесь экстрагируют эфиром (2×200 мл). Эфирный экстракт сушат над безводным сульфатом магния, пропускают через слой кремнезема и элюируют продукт эфиром.
Собранный эфирный элюат концентрируют при пониженном давлении до получения масла, которое очищают с помощью колоночной хроматографии (кремнезем, гексан:эфир (10:1)).
Представляющие интерес собранные фракции упаривают и удаляют растворитель при пониженном давлении с получением твердого остатка, который далее очищают колоночной хроматографией (кремнезем, гексан:этилацетат(1:1)) с получением трех основных компонентов, которые частично разрешаются в виде пиков 1 и 2 эфира Мошера.
С помощью препаративной ВЭЖХ трех компонентов (Колонка: 2× Lichrospher Si60, 5 мкм, 250×21,20 мм, подвижная фаза: гексан:изопропанол (97:3), УФ 254 нм; скорость потока: 10 мл/мин-1) при загрузке 300 мг с последующим концентрированием под вакуумом представляющих интерес фракций получают чистые эфирные прозводные Мошера.
Пик 1 (3,89 г, 46,5%)
Пик 2 (2,78 г, 33%)
Соответствующие двум пикам фракции подвергают гидролизу с выделением индивидуальных изомеров дигидротетрабеназина, которые были идентифицированы и охарактеризованы как изомеры А и В. Каждый из изомеров A и B имеет одну из следующих структур
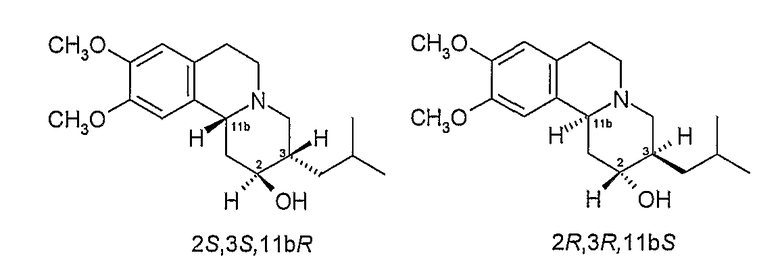
1Е. Гидролиз пика 1 с получением изомера А
20% водный раствор гидроксида натрия (87,5 мл) добавляют к раствору эфира Мошера пик 1 (3,89 г, 7,27 ммоль) в метаноле (260 мл), смесь перемешивают и нагревают с обратным холодильником 150 минут. После охлаждения до комнатной температуры добавляют воду (200 мл) и раствор экстрагируют эфиром (600 мл), сушат над безводным сульфатом магния и после фильтрации концентрируют при пониженном давлении.
Остаток растворяют в этилацетате (200 мл), раствор промывают водой (2×50 мл), органическую фазу сушат над безводным сульфатом магния и после фильтрации концентрируют при пониженном давлении с получением желтой пены. Это вещество очищают с помощью колоночной хроматографии (кремнезем, градиент элюирования этилацетат:гексан (1:1) к этилацетату). Представляющие интерес фракции объединяют и удаляют растворитель при пониженном давлении. Остаток поглощают эфиром и при пониженном давлении еще раз удаляют растворитель с получением изомера А в виде грязно-белой пены (1,1 г, 47%).
Изомер А, который предположительно имеет или 2S,3S,11bR или 2R,3R,11bS конфигурацию (абсолютная стереохимия не определялась), охарактеризован с помощью 1H-ЯМР, 13C-ЯМР, ИК, масспектрометрии (МС), хиральной ВЭЖХ и дисперсии оптического вращения (ДОВ). Данные ИК, ЯМР и МС для изомера А приведены в таблице 1 и данные хиральной ВЭЖХ и дисперсии оптического вращения приведены в таблице 3.
1F. Гидролиз пика 2 с получением изомера B.
20% водный раствор гидроксида натрия (62,5 мл) добавляют к раствору эфира Мошера пик 2 (2,78 г, 5,19 ммоль) в метаноле (185 мл), смесь перемешивают и нагревают с обратным холодильником 150 минут. После охлаждения до комнатной температуры добавляют воду (142 мл) и раствор экстрагируют эфиром (440 мл), сушат над безводным сульфатом магния и после фильтрации концентрируют при пониженном давлении.
Остаток растворяют в этилацетате (200 мл), раствор промывают водой (2×50 мл), органическую фазу сушат над безводным сульфатом магния и после фильтрации концентрируют при пониженном давлении. К остатку добавляют петролейный эфир (30-40°С)и еще раз концентрируют раствор под вакуумом с получением изомера В в виде белой пены (1,34 г, 81%).
Изомер В, который предположительно имеет или 2S,3S,11bR или 2R,3R,11bS конфигурацию (абсолютная стереохимия не определялась), охарактеризован данными 1H-ЯМР, 13C-ЯМР, ИК, масспектрометрии, хиральной ВЭЖХ и ДОВ. Данные ИК, ЯМР и MС для изомера В приведены в таблице 1 и данные хиральной ВЭЖХ и ДОВ приведены в таблице 3.
ПРИМЕР 2
Получение 2R,3S,11bR и 2S,3R,11bS изомеров дигидротетрабеназина
2A. Получение 2,3-дегидротетрабеназина
Раствор, содержащий рацемическую смесь (15 г, 47 ммоль) RR и SS энантиомеров тетрабеназина в тетрагидрофуране, подвергают восстановлению с помощью L-Селектрид® , используя метод примера 1А с получением смеси 2S,3R,11bR и 2R,3S,11bS энантимеров дигидротетрабеназина в виде белого порошкообразного твердого остатка (12 г, 80%). Затем частично очищенный дигидротетрабеназин дегидратируют с помощью PCl5 в соответствии с методом примера 1В с получением получистой смеси 11bR и 11bS изомеров 2,3-дегидротетрабеназина (11bR энантимер которого показан ниже) в виде желтого твердого остатка (12,92 г, 68%).
2B. Эпоксидирование сырого алкена из примера 2A
К перемешиваемому раствору сырого алкена из примера 2A (12,92 г, 42,9 ммоль) в метаноле (215 мл) добавляют раствор 70% перхлорной кислоты (3,70 мл, 43 ммоль) в метаноле (215 мл). К реакционной смеси добавляют 77% 3-хлорпероксибензойную кислоту (15,50 г, 65 ммоль) и полученную смесь перемешивают 18 часов при комнатной температуре, защищая от света.
Реакционную смесь вливают в насыщенный водный раствор сульфита натрия (200 мл) и добавляют воду (200 мл). К полученной эмульсии добавляют хлороформ (300 мл) и подщелачивают смесь насыщенным водным раствором бикарбоната натрия (400 мл).
Собирают органический слой и водную фазу промывают добавлением хлороформа (2×150 мл). Объединенные хлороформные слои сушат над безводным сульфатом магния и после фильтрации удалят растворитель при пониженном давлении, получая коричневое масло (14,5 г, выход >100% - возможные остатки растворителя в продукте). Вещество используют без дополнительной очистки.
2C. Восстановительное раскрытие эпоксидного кольца из 2B
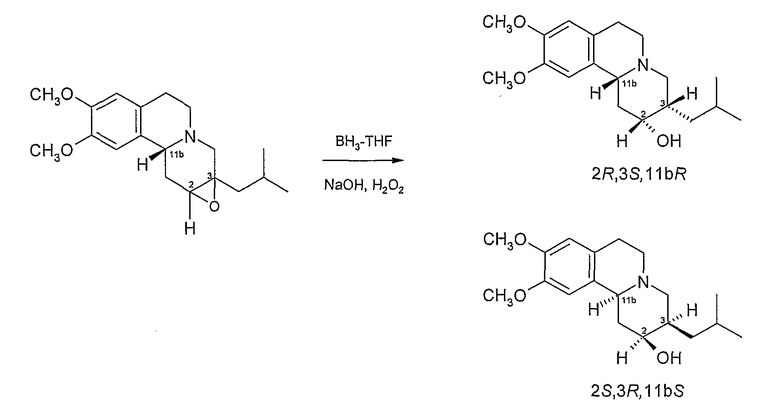
Перемешиваемый раствор сырого эпоксида из примера 2B (14,35 г, 42,9 ммол, принимая 100% выход) в сухом ТГФ (80 мл) медленно обрабатывают 1M боран/ТГФ (184,6 мл, 184,6 ммоль) в течение 15 минут. Реакционную смесь перемешивают два часа, добавляют воду (65 мл) и раствор при перемешивании нагревают с обратным холодильником 30 минут.
После охлаждения к реакционной смеси добавляют 30% раствор гидроксида натрия (97 мл) с последующим добавлением 30% раствора перекиси водорода (48,6 мл) и реакционную смесь дополнительно перемешивают и нагревают с обратным холодильником 1 час.
Охлажденную реакционную смесь экстрагируют этилацетатом (500 мл), сушат над безводным сульфатом магния и после фильтрации удаляют растворитель при пониженном давлении с получением масла. Добавляют к маслу гексан (230 мл) и раствор повторно концентрируют при пониженном давлении.
Масляный остаток очищают с помощью колоночной хроматографии (кремнезем, этилацетат). Объединяют представляющие интерес фракции и удаляют растворитель при пониженном давлении. Остаток еще раз очищают, используя колоночную хроматографию (кремнезем, градиент гексан к эфиру). Объединяют представляющие интерес фракции и при пониженном давлении упаривают растворители, получая бледно-желтый твердый остаток (5,18 г, 38%).
2D. Получение эфирных производных Мошера 2R,3S,11bR и 2S,3R,11bS изомеров дигидротетрабеназина
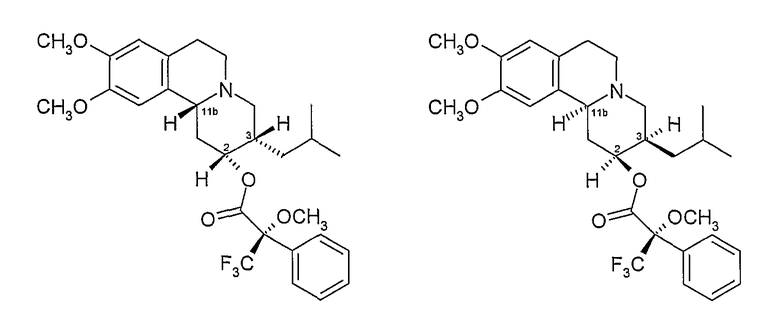
К безводному дихлорметану (46 мл) добавляют R-(+)-α-метокси-α-трифторметилфенилуксусную кислоту (4,68 г, 19,98 ммоль), оксалилхлорид (1,90 мл) и ДФА (0,13 мл) и раствор перемешивают при комнатной температуре 45 минут. Раствор концентрируют при пониженном давлении и остаток еще раз поглощают безводным дихлорметаном (40 мл). Полученный раствор охлаждают на водяной бане со льдом и добавляют диметиламинопиридин (3,65 г, 29,87 ммоль) с последующим добавлением предварительно высушенного раствора (сито 4Å) твердого продукта Примера 2C (4,68 г, 14,6 ммоль) в безводном дихлорметане (20 мл). После перемешивания при комнатной температуре в течение 45 минут добавляют воду (234 мл) и смесь экстрагируют эфиром (2×200 мл). Эфирный экстракт сушат над безводным сульфатом магния, пропускают через слой кремнезема и продукт элюируют эфиром.
Собранный эфирный элюат концентрируют при пониженном давлении до получения масла, которое очищают с помощью колоночной хроматографии (кремнезем, гексан:эфир (1:1)).
Упариванием представляющих интерес собранных фракций и удалением растворителя при пониженном давлении получают розовый твердый остаток (6,53 г).
С помощью препаративной ВЭЖХ твердого остатка (Колонка: 2× Lichrospher Si60, 5 мкм, 250×21,20 мм, подвижная фаза: гексан:изопропанол (97:3), УФ 254 нм; скорость потока: 10 мл/мин-1) при загрузке 100 мг с последующим концентрированием под вакуумом представляющих интерес фракций получают твердый остаток, который суспендируют в петролейном эфире (30-40°С) и собирают фильтрованием с получением чистых эфирных производных Мошера.
Пик 1 (2,37 г, 30%)
Пик 2 (2,42 г, 30%)
Фракции, соответствующие двум пикам, подвергают гидролизу с выделением индивидуальных изомеров дигидротетрабеназина, которые были идентифицированы и охарактеризованы как изомеры С и D. Каждый из изомеров C и D имеют одну из следующих структур
2F. Гидролиз пика 1 с получением изомера С
20% водный раствор гидроксида натрия (53 мл) добавляют к перемешиваемому раствору эфира Мошера пик 1 (2,37 г, 4,43 ммоль) в метаноле (158 мл), смесь перемешивают при нагревании с обратным холодильником 150 минут. После охлаждения к реакционной смеси добавляют воду (88 мл) и полученный раствор экстрагируют эфиром (576 мл). Органический экстракт сушат над безводным сульфатом магния и после фильтрации удаляют растворитель при пониженном давлении. К остатку добавляют этилацетат (200 мл) и раствор промывают водой (2×50 мл). Органический раствор сушат над безводным сульфатом магния и после фильтрации удаляют растворитель при пониженном давлении.
Этот остаток обрабатывают петролейным эфиром (30-40°С) и полученный суспендированный твердый остаток собирают фильтрованием. Фильтрат концентрируют при пониженном давлении и собирают фильтрованием вторую партию суспендированного твердого остатка. Объединяют оба собранных твердых остатка и сушат при пониженном давлении с получением изомера С (1,0 г, 70%).
Изомер С, который предположительно имеет или 2R,3S,11bR, или 2S,3R,11bS конфигурацию (абсолютная стереохимия не определялась), охарактеризован данными 1H-ЯМР, 13C-ЯМР, ИК, масспектрометрии, хиральной ВЭЖХ и ДОВ. Данные ИК, ЯМР и МС для изомера C приведены в таблице 2 и данные хиральной ВЭЖХ и оптической вращательной дисперсии приведены в таблице 4.
2G. Гидролиз пика 2 с получением изомера D.
20% водный раствор гидроксида натрия (53 мл) добавляют к перемешиваемому раствору эфира Мошера пик 2 (2,42 г, 4,52 ммоль) в метаноле (158 мл), смесь перемешивают при нагревании с обратным холодильником 150 минут. После охлаждения к реакционной смеси добавляют воду (88 мл) и полученный раствор экстрагируют эфиром (576 мл). Органический экстракт сушат над безводным сульфатом магния и после фильтрации удаляют растворитель при пониженном давлении. К остатку добавляют этилацетат (200 мл) и раствор промывают водой (2×50 мл). Органический раствор сушат над безводным сульфатом магния и после фильтрации удаляют растворитель при пониженном давлении.
Остаток обрабатывают петролейным эфиром (30-40°С) и полученный суспендированный твердый оранжевый остаток собирают фильтрованием. Твердый остаток растворяют в этилацетате-гексане (15:85) и очищают колоночной хроматографией (кремнезем, градиент этилацетат:гексан (15:85) к этилацетату). Объединяют представляющие интерес фракции и удаляют растворитель при пониженном давлении. Остаток суспендируют в петролейном эфире (30-40°С) и полученную суспензию собирают фильтрованием. Собранный твердый остаток сушат при пониженном давлении и получают изомер D в виде белого твердого остатка (0,93 г, 64%).
Изомер D, который предположительно имеет или 2R,3S,11bR, или 2S,3R,11bS конфигурацию (абсолютная стереохимия не определялась), охарактеризован данными 1H-ЯМР, 13C-ЯМР, ИК, масспектрометрии, хиральной ВЭЖХ и ДОВ. Данные ИК, ЯМР и МС для изомера D приведены в таблице 2 и данные хиральной ВЭЖХ и ДОВ приведены в таблице 4.
Приведенные в таблицах 1 и 2 данные инфракрасных спектров определяли с помощью KBr дискового метода. 1H ЯМР спектры снимали в растворах дейтерированного хлороформа с использованием Varian Gemini ЯМР спектрометра (200 MГц). 13C-ЯМР спектры снимали в растворах дейтерированного хлороформа с использованием Varian Gemini ЯМР спектрометра (50 MГц). Масс-спектры получены с использованием спектрометра Micromass Platform II (ES+ условия). Приведенные в таблицах 3 и 4 данные Дисперсии Оптического Вращения (ДОВ) получены с помощью прибора Optical Activity PolAAr 2001 в растворе метанола при 24°C. Определение времени удерживания при ВЭЖХ проводили с использованием HP1050 ВЭЖХ-хроматографа с УФ-детектором.
В таблицах 1 и 2 приведены данные спектроскопии.
или
6,57 δ 1H (c);
3,84 δ 6H (c);
3,55 δ 1H
(ушир., д);
3,08 δ 1H (м);
2,79 δ 2H (м);
2,55 δ 3H (м);
2,17 δ 1H (м);
1,72 δ 6H (м);
1,02 δ 1H (м);
0,88 δ 6H (т)
или
6,58 δ 1H (c);
3,92 δ 1H (м);
3,84 δ 6H (c);
3,15 δ 1H (м);
2,87 δ 3H (м);
2,43 δ 4H (м);
1,81 δ 1H (м);
1,64 δ 4H (м);
1,21 δ 1H (м);
0,94 δ 3Н (д);
0,89 δ 3H (д)
В таблицах 3 и 4 приведены данные хроматографии и ДОВ.
и
и
ПРИМЕР 3
Альтернативный способ получения изомера В и получение мезилатной соли
3A. Восстановление RR/SS тетрабеназина
1M L-Селектрид® в тетрагидрофуране (52 мл, 52 ммол, 1,1 экв.) медленно в течение 30 минут добавляют к охлаждаемому (ледяная баня), перемешиваемому раствору рацемата тетрабеназина (15 г, 47 ммоль) в тетрагидрофуране (56 мл). После окончания добавления смеси дают нагреться до комнатной температуры и дополнительно перемешивают в течение 6 часов. Тонкослойная хроматография (кремнезем, этилацетат) показывает наличие только очень небольших количеств оставшегося исходного вещества.
Смесь выливают на перемешиваемую смесь измельченного льда, (112 г), воды (56 мл) и ледяной уксусной кислоты (12,2 г). Полученный желтый раствор промывают эфиром (2×50 мл) и подщелачивают путем медленного добавления твердого карбоната натрия (примерно 13 г). К смеси добавляют при перемешивании петролейный эфир (30-40°C) (56 мл) и собирают путем фильтрования сырой β-DHTBZ (ДГТБЗ) в виде белого твердого остатка.
Сырой твердый остаток растворяют в дихлорметане (примерно l50 мл) и полученный раствор промывают водой (40 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении примерно до 40 мл. Образуется тонкая суспензия белого твердого остатка. Добавляют петролейный эфир (30-40°C) (56 мл) и суспензию перемешивают 15 минут при температуре в лаборатории. Продукт собирают фильтрованием и промывают на фильтре петролейным эфиром до снежно-белого цвета (30-40°C) (40-60 мл) перед сушкой на воздухе при комнатной температуре с получением β-DHTBZ (10,1 г, 67%) в виде белого твердого остатка. Анализ с помощью тонкослойной хроматографии (кремнезем, этилацетат) показывает наличие только одного компонента.
3B. Получение и фракционная кристаллизация соли камфорсульфоновой кислоты и рацемического β-DHTBZ
Продукт примера 3A и 1 эквивалент (S)-(+)-камфор-10-сульфоновой кислоты растворяют при нагревании в минимальном количестве метанола. Полученному раствору дают остыть и затем медленно разбавляют эфиром до полного завершения образования твердого осадка. Полученный белый кристаллический твердый остаток собирают фильтрованием и перед сушкой промывают эфиром.
Соль камфорсульфоновой кислоты (10 г) растворяют в смеси горячего абсолютного этанола (170 мл) и метанола (30 мл). Перемешивают полученный раствор и дают остыть. Через два часа образовавшийся осадок собирают фильтрованием в виде белого кристаллического твердого остатка (2,9 г). Пробу кристаллического вещества встряхивают в отдельной воронке с избытком насыщенного водного раствора карбоната натрия и дихлорметана. Органическую фазу отделяют, сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток растирают в порошок с петролейным эфиром (30-40°С) и еще раз концентрируют органический раствор. Анализ соли с помощью хиральной ВЭЖХ с использованием колонки Chirex(S)-VAL и (R)-NEA 250×4,6 мм и гексан:этанола (98:2) в качестве элюента при скорости потока 1 мл/мин показывает, что выделенный β-DHTBZ обогащен одним энантиомером (энантимерный избыток (е.е.) примерно 80%).
Обогащенную соль камфорсульфоновой кислоты (14 г) растворяют в горячем абсолютном этаноле (140 мл) и добавляют пропан-2-ол (420 мл). Перемешивают полученный раствор, и в течение одной минуты начинает образовываться осадок. Смеси дают остыть до комнатной температуры и перемешивают один час. Образовавшийся осадок собирают фильтрованием, промывают эфиром и сушат с получением белого кристаллического твердого остатка (12 г).
Кристаллическое вещество встряхивают в отдельной воронке с избытком насыщенного водного раствора карбоната натрия и дихлорметана. Органическую фазу отделяют, сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток растирают в порошок с петролейным эфиром (30-40°С) и еще раз концентрируют органический раствор с получением (после сушки в вакууме) (+)-β-DHTBZ (6,6 г, ДОВ +107,8о). Выделенный энантимер имеет е.е. примерно >97%.
3C. Получение изомера B
К перемешиваемому, охлаждаемому (водяная баня со льдом) раствору продукта примера 3В (6,6 г, 20,6 ммоль) в дихлорметане (90 мл) непрерывно добавляют в течение десяти минут раствор пятихлористого фосфора (4,5 г, 21,6 ммол, 1,05 экв.) в дихлорметане (55 мл). По окончанию добавления полученный желтый раствор дополнительно перемешивают в течение десяти минут перед выливанием на быстро перемешиваемую смесь карбоната натрия (15 г) в воде (90 мл) и измельченного льда (90 г). Смесь дополнительно перемешивают 10 минут и переносят в делительную воронку.
Разделяют фазы, удаляют коричневый дихлорметановый слой, сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении с получением сырого алкенового промежуточного соединения в виде коричневого масла (примерно 6,7 г). Анализ с помощью тонкослойной хроматографии (кремнезем, этилацетат) показывает, что в сыром продукте не осталось (+)-β-DHTBZ.
Сырой алкен поглощают (в атмосфере сухого азота) безводным тетрагидрофураном (40 мл) и добавляют при перемешивании в течение пятнадцати минут раствор борана в ТГФ (1 M раствор, 2,5 экв., 52 мл). Затем реакционную смесь перемешивают при комнатной температуре два часа. Анализ с помощью тонкослойной хроматографии (кремнезем, этилацетат) показывает, что в реакционной смеси не осталось алкенового промежуточного соединения.
К перемешиваемой реакционной смеси добавляют раствор гидроксида натрия (3,7 г) в воде (10 мл) с последующим добавлением водного раствора перекиси водорода (50%, примерно 7 мл) и образовавшуюся двухфазную смесь перемешивают при нагревании с обратным холодильником один час. Анализ с помощью тонкослойной хроматографии (кремнезем, этилацетат) показывает образование продукта с предполагаемой для изомера В величиной Rf. Наблюдается также наличие характерного неполярного компонента.
Реакционной смеси дают охладиться до комнатной температуры и выливают в делительную воронку. Удаляют верхний органический слой и концентрируют при пониженном давлении для удаления основного количества ТГФ. Остаток поглощают эфиром (стабилизированный (BHT), 75 мл), промывают водой (40 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении с получением светло-желтого масла (8,1 г).
Желтое масло очищают колоночной хроматографией (кремнезем, этилацетат:гексан (80:20), с увеличением до 100% этилацетата), желаемые фракции собирают, объединяют и концентрируют при пониженном давлении с получением светлого масла, которое обрабатывают эфиром (стабилизированный, 18 мл) и концентрируют при пониженном давлении с получением изомера В в виде светло-желтой твердой пены (2,2 г).
Хиральная ВЭЖХ в условиях, указанных в примере 3B, подтверждает образование изомера B c энантимерным избытком (e.e.) более 97%.
Оптическое вращение, измеренное на поляриметре Bellingham Stanley ADP220, составляет [αD] +123,5
3D. Получение мезилатной соли изомера B
Метансульфонатную соль изомера B получают растворением смеси 1 эквивалента изомера B из примера 3C и 1 эквивалента метансульфоновой кислоты в минимальном количестве этанола с последующим добавлением диэтилового эфира. Образующийся при этом белый осадок собирают фильтрованием и сушат в вакууме с получением мезилатной соли с выходом примерно 85% и чистотой (ВЖЭХ) примерно 96%.
ПРИМЕР 4
Скрининг VMAT-2 связывающей активности с использованием анализа связывания
[ 3 H]дигидротетрабеназина
Дигидротетрабеназин является мощным и селективным ингибитором VMAT-2, и связывается с этим везикулярным транспортером с высокой аффинностью (нМ пределы). [3H]дигидротетрабеназин успешно использовался в течение многих лет в качестве радиолиганда для мечения VMAT-2 в головном мозге людей, быков и грызунов (например, Scherman et al. J.Neurochem. 50, 1131-1136 (1988); Near et al. Mol. Pharmacol. 30, 252-257 (1986); Kilbourn et al. Eur. J. Pharmacol. 278, 249-252 (1995); и Zucker et al. Life Sci. 69, 2311-2317 (2001)).
Четыре изомера дигидротетрабеназина A, B, C и D тестировали на их способность ингибировать транспортер VMAT-2, используя описанный ниже анализ.
Методы и материалы
Мембраны переднего мозга взрослых крыс (породы Wistar) получали в основном как описано Chazot et al. (1993) Biochem. Pharmacol. 45, 605-610. Везикулярные мембраны полосатого тела взрослых крыс получали, главным образом, как описано Roland et al. (2000), JPET 293, 329-335. 10 мкг мембран инкубировали при 25°C с [3H]дигидротетрабеназином (18-20 нМ) в 50 мМ HEPES pH 8,0 (аналитический буфер), в течение 60 минут, и связанный радиолиганд собирали скорой фильтрацией под вакуумом на GF/B стекловолоконных фильтрах. Неспецифическое связывание определяли в параллельных образцах в присутствии 2 мкМ немеченого тетрабеназина. Радиоактивность подсчитывали в сцинтилляторе в β-счетчике. Трижды анализировали весь интервал концентраций (логарифмические и полулогарифмические единицы) четырех тестируемых соединений (Изомеры A, B, C и D) (интервал: 10-11-10-4 M). Тестируемые соединения и тетрабеназин растворяли в ДМСО с концентрацией исходного раствора 10 мM, а затем разведения готовили в аналитическом буфере. Для каждого соединения проводили три независимых эксперимента. Анализировали данные и строили кривую, используя пакет программ GraphPad Prism 3.2.
Результаты
Сначала получали препарат мембран переднего мозга P2 (Chazot et al., 1993) и анализировали, как описано в основном протоколе. На выходе получали очень низкий уровень специфической связывающей активности.
Затем получали везикулярный препарат полосатого тела, который давал на выходе значительный уровень стабильных специфичных [3H]дигидротетрабеназину сайтов связывания (5-6 пмоль/мг белка). Это хорошо сопоставляется с опубликованными данными (Roland et al., 2000). Этот препарат использовали для дальнейших анализов.
Параметры конкурентного связывания для тестируемых соединений
Соответствие двум сайтам
-5,98±0,33
<-4,0
% сайтов
47%
53%
Соответствие двум сайтам
-5,15±0,11
-7,13±0,26
% сайтов
74%
26%
Данные представляют собой средние значения ± SD для трех независимых экспериментов. Значения KI определяли, исходя из опубликованного значения KD 1,2 нM для полосатого тела крыс (Roland et al., 2000).
Общий фармакологический профиль на основании суммарных значений KI представляет собой Изомер C > Изомер B > Изомер D >> Изомер A.
Примечательно, и Изомер B, и Изомер A давали пологие кривые конкурентности, которые больше всего соответствовали двухсайтовой модели связывания.
Изомер A показал сайты высокой аффинности (KI=59 нM) и низкой аффинности (KI<5,9 мкМ аффинность), каждый внес вклад приблизительно в 50% всех сайтов. Это может указывать на то, что Изомер A может различать разные сайты связывания VMAT-2 в полосатом теле.
ПРИМЕР 5
Функциональные анализы VMAT
Функциональный анализ VMAT2
Синаптические везикулы полосатого тела крыс получали, главным образом, как описано в примере 3. Таким образом, мембранный препарат P2 полосатого тела крыс (Chazot et al., 1993) ресуспендировали и гомогенизировали в ледяной дистиллированной воде. Осмолярность восстанавливали добавлением 25 мM HEPES и 100 мM тартрата калия (pH 7,5, 4°C). Затем препарат центрифугировали в течение 20 минут при 20000×g (4°C). Результирующую S3 фракцию удаляли, добавляли сульфат магния (с получением конечной концентрации 1 мM, pH 7,5, 4°C), и смесь центрифугировали при 100000×g в течение 45 минут. Конечная P4 фракция содержит синаптические везикулы для анализа.
Аликвоту 100 мкл (приблизительно 2,5 мкг белка) синаптических везикул предварительно инкубировали с увеличенными концентрациями тестируемых соединений C и B (свежеприготовленных в виде исходного раствора 10-2 M в ДМСО) в течение 30 минут (интервал концентраций 10-9 M - 10-4 M), а затем в течение 3 минут в аналитическом буфере (25 мM HEPES, 100 мM тартрата калия, 1,7 мM аскорбиновой кислоты, 0,05 мM EGTA, 0,1 мM EDTA, 2 мM ATФ-Mg2+, pH 7,5), в присутствии [3H]дофамина (30 нM конечная концентрация) при 30°C. Затем реакцию прекращали добавлением ледяного аналитического буфера pH 7,5, содержащего 2 мM MgSО4 вместо 2 мM ATФ-Mg2+, и осуществляли быструю фильтрацию через Whatman фильтры, пропитанные 0,5% полиэтиленимином. Фильтры трижды промывали холодным буфером, используя коллектор Бранделя (Brandel Harvester). Задержанную на фильтрах радиоактивность подсчитывали, используя жидкостной сцинтилляционный счетчик, а неспецифическое связывание определяли измерением захвата везикулярного [3H]дофамина при 4°C. Этот способ был основан на способе, описанном Ugarte YV et al. (2003) Eur. J. Pharmacol. 472, 165-171. Селективный захват VMAT-2 определяли, используя 10 мкM тетрабеназина.
И соединение C (наблюдаемая IC50=18±2 нM), и соединение B (наблюдаемая IC50=30±3 нM) ингибировали захват [3H]дофамина в стриатные визикулы через транспортер VMAT-2 с функциональной аффинностью (профиль C>B), сходной с их соответствующей аффинностью, определенной с использованием анализа связывания
[3H]дигидротетрабеназина.
B. Функциональный анализ VMAT1
Существует ограниченное количество нативных тканей, которые обладают только VMAT1, изолировано от VMAT2. Однако тетрабеназин демонстрирует по меньше мере в 200 раз большую аффинность к VMAT2 по сравнению с VMAT1, и эта избирательность может быть использована для блокировки влияния VMAT2 в функциональном анализе (Erickson et al. (1996) PNAS (USA) 93, 5166-5171). Хромаффинные клетки надпочечников выделяли от молодых взрослых крыс SD, главным образом, как описано у Moshharov et al. (2003) J Neurosci. 23, 5835-5845. Таким образом, надпочечники препарировали в ледяном PBS, капсулу и мозговое вещество желез удаляли и оставшееся мозговое вещество измельчали. После многократного промывания с использованием PBS, ткань инкубировали с IA раствором коллагеназы, свободным от Ca2+ (250 Ед/мл) в течение 30 минут при 30°C с легким перемешиванием. Обработанную ткань промывали три раза, и диссоциированные клетки центрифугировали при 3000 об/мин до образования осадка, который ресуспендировали в PBS. Везикулярную фракцию выделяли аналогичным образом, как описано для препарата головного мозга.
100 мкл (приблизительно 2,5 мкг белка) синаптических везикул предварительно инкубировали с повышенными концентрациями тестируемого соединения (полученными как описано ранее для анализа связывания) в течение 30 минут (интервал концентраций 10-9 M - 10-4 M). Этот анализ проводили в течение 3 минут при 30°C в аналитическом буфере (25 мM HEPES, 100 мM тартрата калия, 1,7 мM аскорбиновой кислоты, 0,05 мM EGTA, 0,1 мM EDTA, 2 мM ATФ-Mg2+, pH 7,5), в присутствии [3H]дофамина (30 нM конечная концентрация). Захват [3H]дофамина измеряли в присутствии 10 мкM тетрабеназина (селективно блокирует VMAT2 при этой концентрации). Неспецифический захват определяли путем определения захвата везикулярного [3H]дофамина при 4°C. Затем реакцию останавливали добавлением ледяного аналитического буфера pH 7,5, содержащего 2 мM MgSО4 вместо 2 мM ATФ-Mg2+, осуществляли скорую фильтрацию через Whatman фильтры, пропитанные 0,5% полиэтиленимином. Фильтры трижды промывали холодным буфером, используя коллектор Бранделя (Brandel Harvester), и задержанную на фильтрах радиоактивность подсчитывали, используя жидкостной сцинтилляционный счетчик.
В присутствии 10 мкM тетрабеназина, и соединение В, и соединение С слабо ингибировали захват [3H]дофамина, значения IC50 больше 10-5 M для обоих соединений. Это указывает на то, что оба соединения имеют низкую аффинность в отношении VMAT-1. Кроме того, эти данные показывают, что оба соединения имеют по меньшей мере 2-порядка величины селективности для VMAT-2 по сравнению с VMAT-1.
ПРИМЕР 6
Исследования рецепторного связывания и связывания белка-переносчика
Четыре изомера дигидротетрабеназина A, B, C и D подвергали анализам специфического связывания для исследования их способности связываться с рецепторами и белками-переносчиками, описанными ниже. Результаты изложены в Таблице 6.
(a) α 2A адренергический рецептор:
Ссылка: S.Uhlсn et al. J. Pharmacol. Exp. Ther., 271: 1558-1565 (1994)
Источник: рекомбинантные Sf9 клетки насекомых и человека
Лиганд: 1 нM [3H] MK-912
Носитель: 1% ДМСО
Время/температура инкубации: 60 минут при 25°C
Инкубационный буфер: 75 M Трис-HCl, pH 7,4, 12,5 мM MgCl2, 2 мM EDTA
Неспецифический лиганд: 10 мкМ WB-4101
Kd: 0,6 нM
Bmax: 4,6 пмоль/мг белка
Специфическое связывание: 95%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(b) α 2B адренергический рецептор:
Ссылка: S. Uhlen et al., Eur. J. Pharmacol., 33(1): 93-1-1 (1998)
Источник: рекомбинантные клетки CHO-K1 человека
Лиганд: 2,5 нM [3H]Rauwolscine
Носитель: 1% ДМСО
Время/температура инкубации: 60 минут при 25°C
Инкубационный буфер: 50 мM Трис-HCl, 1 мM EDTA, 12,5 мM MgCl2, pH 7,4, 0,2% BSA при 25°C
Неспецифический лиганд: 10 мкM празозина
Kd: 2,1 нM
Bmax: 2,1 пмоль/мг белка
Специфическое связывание: 90%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(c) D 1 рецептор дофамина:
Ссылка: Dearry et al., Nature, 347: 72-76, (1990)
Источник: рекомбинантные клетки CHO человека
Лиганд: 1,4 нM [3H]SCH-23390
Носитель: 1% ДМСО
Время/температура инкубации: 2 часа при 37°C
Инкубационный буфер: 50 мM Трис-HCl, pH 7,4, 150 нМ NaCl, 1,4 нM аскорбиновой кислоты, 0,001% BSA
Неспецифический лиганд: 10 мкM (+)-бутакламола
Kd: 1,4 нM
Bmax: 0,63 пмоль/мг белка
Специфическое связывание: 90%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(d) D 2L рецептор дофамина:
Ссылка: Bunzo et al., Nature, 336: 783-787 (1988)
Источник: рекомбинантные клетки CHO человека
Лиганд: 0,16 нM [3H]спиперона
Носитель: 1% ДМСО
Время/температура инкубации: 2 часа при 25°C
Инкубационный буфер: 50 мM Трис-HCl, pH 7,4, 150 нM NaCl, 1,4 нM аскорбиновой кислоты, 0,001% BSA
Неспецифический лиганд: 10 мкM галоперидола
Kd: 0,08 нM
Bmax: 0,48 пмоль/мг белка
Специфическое связывание: 85%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(e) D 3 рецептор дофамина:
Ссылка: Sokoloff et al., Nature, 347: 146-151, (1990)
Источник: рекомбинантные клетки CHO человека
Лиганд: 0,7 нM [3H]спипенон
Носитель: 1% ДМСО
Время/температура инкубации: 2 часа при 37°C
Инкубационный буфер: 50 мM Трис-HCl, pH 7,4, 150 нM NaCl, 1,4 нM аскорбиновой кислоты, 0,001% BSA
Неспецифический лиганд: 25 мкM S(-)-сульпирид
Kd: 0,36 нM
Bmax: 1,1 пмоль/мг белка
Специфическое связывание: 85%
Количественный способ: Радиолигандное связывание
Критерий достоверности: >50% от максимальной стимуляции или ингибирования
(f) имидазолиновый I 2 рецептор (центральный):
Ссылка: Brown et al., Brit. J. Pharmacol., 99: 803-809, (1990)
Источник: кора головного мозга крыс Wistar
Лиганд: 2 нM [3H]идазоксана
Носитель: 1% ДМСО
Время/температура инкубации: 30 минут при 25°C
Инкубационный буфер: 50 мM Трис-HCl, 0,5 мM EDTA, pH 7,4 при 25°C
Неспецифический лиганд: 1 мкM идазоксана
Kd: 4 нМ
Bmax: 0,14 пмоль/мг белка
Специфическое связывание: 85%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(g) Сигма σ 1 рецептор:
Ссылка: Ganapathy et al., Pharmacol. Exp. Ther., 289: 251-260, (1999)
Источник: человеческие клетки jurkat
Лиганд: 8 нM [3H]галоперидола
Носитель: 1% ДМСО
Время/температура инкубации: 4 часа при 25°C
Инкубационный буфер: 5 мM K2HPО4/KH2PО4 буфер pH 7,5
Неспецифический лиганд: 10 мкM галоперидола
Kd: 5,8 нM
Bmax: 0,71 пмоль/мг белка
Специфическое связывание: 80%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(h) Сигма σ 2 рецептор:
Ссылка: Hashimoto et al., Eur. J. Pharmacol., 236: 159-163, (1993)
Источник: головной мозг крыс Wistar
Лиганд: 3 нM [3H]ифенпродил
Носитель: 1% ДМСО
Время/температура инкубации: 60 минут при 37°C
Инкубационный буфер: 50 мM Трис-HCl, pH 7,4
Неспецифический лиганд: 10 мкM ифенпродила
Kd: 4,8 нМ
Bmax: 1,3 пмоль/мг белка
Специфическое связывание: 85%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(i) переносчик серотонина (SERT):
Ссылка: Gu et al., J. Biol. Chem., 269 (10): 7124-7130, (1994)
Источник: рекомбинантные клетки HEK-293 человека
Лиганд: 0,15 нМ [125I] RTI-55
Носитель: 1% ДМСО
Время/температура инкубации: 3 часа при 4°С
Инкубационный буфер: 100 мМ NaCl, 50 мM Трис-HCl, 1 мкM лейпептина, 10 мкм PMSF, pH 7,4
Неспецифический лиганд: 10 мкM имипрамина
Kd: 0,17 нM
Bmax: 0,41 пмоль/мг белка
Специфическое связывание: 95%
Количественный способ: Радиолигандное связывание
Критерий достоверности: ≥50% от максимальной стимуляции или ингибирования
(j) переносчик дофамина (DAT):
Ссылка: Giros et al., Trends Pharmacol. Sci., 14, 43-49 (1993) Gu et al., J. Biol. Chem., 269 (10): 7124-7130 (1994)
Источник: рекомбинантные клетки CHO человека
Лиганд: 0,15 нМ [125I] RTI-55
Носитель: 1% ДМСО
Время/температура инкубации: 3 часа при 4°C
Инкубационный буфер: 100 мM NaCl, 50 мM Трис-HCl, 1 мкM лейпептина, 10 мкM PMSF, pH 7,4
Неспецифический лиганд: 10 мкM номифензина
Kd: 0,58 нM
Bmax: 0,047 пмоль/мг белка
Специфическое связывание: 90%
Количественный способ: Радиолигандное связывание
Критерий достоверности: >50% от максимальной стимуляции или ингибирования
Процент ингибирования 10 мкМ растворами изомеров дигидротетрабеназина специфического связывания с рецептором и белками-переносчиками (значение IC50, где измерено, дано в круглых скобках)
ПРИМЕР 7
Ферментативные исследования
Изомеры B и C тестировали на их активность по ингибированию ферментов, участвующих в способинге моноаминов в ЦНС, а именно Катехол О-метилтрансферазы (COMT), моноаминоксидазы A и моноаминоксидазы B. Используемые способы анализа описаны ниже, а результаты изложены в Таблице 7.
(a) ингибирование катехол О-метилтрансферазы (COMT)
Источник: печень свиньи
Субстрат: 3 мМ катехол + S-аденозил-L-[3H]метионин
Носитель: 1% ДМСО
Время/температура предварительной инкубации: Нет
Время инкубации: 60 минут при 37°C
Инкубационный буфер: 100 мМ фосфат калия, 10 мМ MgCl2, 3 мМ DTT, содержащего 12 единиц/мл аденозиндезаминазы, pH 7,4
Количественный способ: определение количества [3H]гваякола.
Критерий достоверности: >50% от максимальной стимуляции или ингибирования
(b) ингибирование моноаминоксидазы MAO-A
Источник: рекомбинантные клетки человека
Субстрат: 50 мкM кинурамин
Носитель: 1% ДМСО
Время/температура предварительной инкубации: 15 минут при 37°C
Время инкубации: 60 минут при 37°C
Инкубационный буфер: 100 мМ KH2PО4, pH 7,4
Количественный способ: Спектрофлуориметрическое определение количества 4-гидроксихинолина
Критерий достоверности: >50% от максимальной стимуляции или ингибирования
(c) ингибирование моноаминоксидазы MAO-B
Источник: рекомбинантные клетки человека
Субстрат: 50 мкM кинурамина
Носитель: 1% ДМСО
Время/температура предварительной инкубации: 15 минут при 37°C
Время инкубации: 60 минут при 37°C
Инкубационный буфер: 100 мМ КH2PO4, pH 7,4
Количественный способ: Спектрофлуориметрическое определение количества 4-гидроксихинолина
Критерий достоверности: >50% от максимальной стимуляции или ингибирования
Процент ингибирования ферментативной активности под действием 10 мкМ растворов изомеров дигидротетрабеназина
ПРИМЕР 8
Клеточные исследования
Способность изомеров B и C ингибировать захват серотонина (5-гидрокситриптамина) клетками мезонефроса человека определяли с использованием следующих условий анализа:
Мишень: Клетки HEK-293 человека
Носитель: 0,4% ДМСО
Время/температура инкубации: 10 минут при 25°C
Инкубационный буфер: 5 мM Трис-HCl, 7,5 мМ HEPES, 120 мМ NaCl, 5,4 мМ KCl, 1,2 мМ CaCl2, 1,2 мМ MgSO4, 5 мМ глюкозы, 1 мМ аскорбиновой кислоты, pH 7,1
Количественный способ: количественное определение захвата [3H]серотонина
Критерий достоверности: > 50% ингибирования захвата [3H]серотонина по сравнению с реакцией на флуксетин.
Результаты
Было показано, что соединение B является антагонистом и вызывало 56% ингибирование захвата серотонина при концентрации 10 мкM. IC50 соединения B составила 7,53 мкM.
Также было показано, что соединение C является антагонистом и вызывало 86% ингибирование захвата серотонина при концентрации 10 мкM. IC50 соединения B составила 1,29 мкM.
ПРИМЕР 9
Анализ связывания 5-НТ 1D/1B
Способность соединения B и соединения C связываться с 5-НТ1D/1B рецепторами тестировали с использованием анализа, основанного на анализе, описанного Millan, MJ et al. (2002) Pharynacol. Biochem. Behav. 71, 589-598. [N-метил3H] GR-125743 использовали в качестве радиолиганда и для рецепторов 5-HT1D, и для рецепторов
5-HT1В. Для анализа использовали мембраны переднего мозга P2 взрослых крыс SD (Chazot et al., 1993). Используемый аналитический буфер представлял собой 50 мМ Трис-HCl pH 7,7 при комнатной температуре, содержащий 4 мМ хлорида кальция, 0,1% аскорбиновой кислоты и 10 мкM паргилина. 5-HT (10 мкM) использовали для определения неспецифического связывания. Инкубацию с 1 нM [3H]GR-125743 проводили в течение 1 часа при комнатной температуре, и реакцию останавливали скорой фильтрацией, используя коллектор Бранделя, через фильтры GF/B, предварительно пропитанные 0,1% полиэтиленимином, с последующим трехкратным промыванием ледяным буфером (дополненным 0,1% BSA). Использовали диапазон доз 10-10-10-4 M. Полученные в результате конкурентные кривые анализировали, используя пакет программ GraphPad Prism 4.
Оба соединения B и C показали слабое изменение связывания [3H][N-метил]GR-125743 с мембранами переднего мозга крыс (значения IC50>10-4M), что дает возможность предположить, что и соединение B, и соединение C обладают низкой аффинностью в отношении 5-HT1D/B рецепторов.
ПРИМЕР 10
Определение кишечной проницаемости изомеров A, B, C и D с использованием асорбционного анализа Caco-2
Абсорбционный анализ Caco-2 представляет собой общепринятую систему для in vitro оценки кишечной абсорбции лекарственных средств in vivo - см. Meunier et al., Cell Biology and Toxicology, 11: 187-194, Wils et al., Cell Biology and Toxicology, 10: 393-397 и Gres et al., Pharm Sci, 15(5): 726-732.
Этот анализ основан на способности Caco-2 клеток дифференцироваться в энтероциты при культивировании на микропористом фильтре в течение периода приблизительно 21 день. Во время периода культивирования, Caco-2 клетки подвергают спонтанным морфологическим и биохимическим изменениям, которые создают поляризованный монослой с четко определенной щеточной каемкой на апикальной поверхности, а также плотные клеточные соединения. Таким образом, эти клетки могут быть использованы в качестве in vitro модели для анализа проницаемости лекарственного средства.
Абсорбцию через Caco-2 монослой можно измерить в двух направлениях: от апикального к базолатеральному или от базолатерального к апикальному, добавляя соединение к апикальному или базолатеральному отсеку, соответственно. В различные временные точки, образцы собирали из приемного отсека для анализа скорости абсорбции, измеренной с помощью коэффициента эффективной проницаемости (Papp) через монослой.
Значение Papp отражает совокупность проницаемости тестируемого образца и по трансцеллюлярному пути (через клеточные мембраны), и по парацеллюлярному пути (через плотные соединения между клетками). Относительный вклад этих путей зависит от pKa, коэффициента распределения (log D), молекулярного радиуса и заряда тестируемого образца при заданном pH. Значения Papp затем могут быть использованы для классификации соединений по их проницаемости через Caco-2 монослои.
Коэффициенты эффективной проницаемости (Papp) четырех изомеров дигидротетрабеназина A, B, C и D при 50 мкM определяли, используя абсорбционную модель Caco-2, и классифицировали относительно соединений сравнения низкой, средней и высокой проницаемости. Радиоактивно-меченые образцы сравнения маннит (низкая), салициловая кислота (средняя) и тестостерон (высокая) использовали для классификации их по проницаемости. Значение Papp каждого тестируеого образца дигидротетрабеназина оценивали путем измерения его концентрации в донорскном и принимающем отсеках через 1 час. Абсорбцию определяли при pH 7,4 через клеточные монослои в направлении от апикального к базолатеральному. Клетки Caco-2, используемые для оценки значений Papp, выращивали в течение 26 дней на мембранах «Transwell inserts» в 12-луночных планшетах. Целостность монослоя проверяли до и после анализа абсорбции методами трансэпителиальной электрической резистентности (Ohm-см2) и Lucifer Yellow.
Материалы и методы
Матричные растворы маннита, салициловой кислоты и тестостерона готовили в метаноле с концентрацией 50 мМ. В день исследования, стандартные образцы разводили в сбалансированном солевом растворе Хенкса (HBSS), pH 7,4 до конечной концентрации 50 мкM. Радиоактивно-меченный 14C-маннит, 3H-тестостерон и 14C-салициловую кислоту затем добавляли к их соответствующей немеченой среде для получения конечной специфической активности 0,35-0,65 мкКю/мл. Образцы изомера дигидротетрабеназина готовили с концентрацией 50 мМ в ДМСО. Каждый матричный раствор дополнительно разводили в HBSS буфере (pH 7,4) до конечной рабочей концентрации 50 мкM.
Суспензии клеток Caco-2 готовили следующим образом. Размораживали один флакон первичных клеток Caco-2 ID No. Caco-29-080299 количество пассажей 29 (Американская коллекция типовых культур (VA, USA) и выращивали в 150 см2 чашке, содержащей культуральную среду Caco-2. При смыкании монослоя культуральную среду удаляли и клетки промывали 5 мл PBS. Клетки отделяли после добавления и инкубации с 0,25% трипсин-EDTA (2,0 мл на чашку) приблизительно в течение 10 минут при 37°C. Отделение клеток контролировали под микроскопом и останавливали добавлением 10 мл культуральной среды Caco-2. Жизнеспособность клеток и концентрацию определяли способом исключения трепанового синего. При определении клеточной плотности и жизнеспособности клетки Caco-2 разводили в культуральной среде до конечной рабочей концетрации 2,0×105 клеток/мл. Культуральная среда Caco-2 содержала модифицированную по способу Дульбекко среду Игла, 10% эмбриональную сыворотку теленка, 100 мкM заменимых аминокислот, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина.
Поликарбонатные мембраны Costar (размер пор 0,4 мкм) предварительно уравновешивали культуральной средой Caco-2 в течение 1 часа в инкубаторе с водяной рубашкой при 37°C с 5% CO2. Содержимое апикального отсека удаляли и заменяли на 500 мкл суспензии клеток Caco-2 (200000 клеток/мл). Затем клетки подращивали в среде в течение 26 дней в инкубаторе с водяной рубашкой при 37°C с 5% CO2.
Перед анализом, все монослои дважды промывали HBSS и измеряли их трансэпителиальную электрическую резистентность (TEER) Millicell-ERS метром. Значения TEER, полученные в отсутствие клеток вычитали как фоновый сигнал. Для эксперимента по исследованию абсорбции использовали только монослой со значениями TEER более 150 Ohm-см2.
Все эксперименты по абсорбции проводили трижды при pH 7,4. Интенсивность абсорбции каждого образца сравнения и тестируемого образца дигидротетрабеназина оценивали в апикально-базолатеральном направлении. Аликвоты (100 мкл) каждого рабочего раствора (образцы сравнения и тестируемые образы при 50 мкM) оставляли в начале исследования для определения исходной концентрации (C0).
Эксперименты по абсорбции начинали заменой содержимого донорского отсека 500 мкл сбаллансрованного солевого раствора Хенкса, содержащего тестируемые образцы или образцы сравнения. Клетки возвращали в CО2 инкубатор в течение абсорбционного анализа. Образцы 100 мкл собирали из получающего и доноского отсеков через 1 час и использовали для определения процента выхода. Радиоактивно меченные образцы сравнения, манит, салициловую кислоту и тестостерон использовали в качестве количественных контролей и для сравнительной классификации тестируемых образцов дигидротетрабеназина.
В конце эксперимента по абсорбции, целостность каждого клеточного монослоя оценивали путем мониторинга утечки Lucifer Yellow от апикальной к базальной стороне. Все растворы удаляли из апикального и базолатерального отсеков. Базолатеральные отсеки восполняли 1,5 мл свежего HBSS, тогда как апикальные отсеки заполняли 0,5 мл 120 мкг/мл раствора Lucifer Yellow. Клетки возвращали в инкубатор на период 1 час, после чего образцы 100 мкл собирали и количественно оценивали спектрофотометрически с использованием планшетного ридера SpectraMax 340PC при 428 нм.
Объем 20 мкл каждого радиоактивно меченого образца добавляли к 10 мл сцинтилляционной смеси (ScintiSafe 30%) и считали в течение 5 минут с использованием жидкостного сцинтилляционного анализатора (1900CA Tri-Carb).
Инкубации клеток Caco-2 анализировали на наличие дигидротетрабеназина с использованием последовательно способов жидкостной хроматографии и масс-спектрометрии (LC-MS/MS).
Коэффициенты эффективной проницаемости рассчитывали, используя уравнение Papp=dQ/dt×1/A×I/C0 (см/сек), в котором dQ/dt представляет собой скорость диффузии соединения (мкг/сек или область интеграции/сек), A представляет собой общую площадь поверхности клеточных мембран (см2), а C0 представляет собой исходную концентрацию (мк/мл или область интеграции/сек).
Значения апикально-базолатерального Papp различных тестируемых образцов дигидротетрабеназина сравнивали со значениями Papp, определенными для образцов сравнения. Результаты показаны в таблице 8. Соединения дигидротетрабеназина C, D и A имели значения Papp соответствующие 21,14×10-6, 24,87×10-6 и 25,52×10-6 см/секc, которые сравнимы с тестостероновым образцом сравнения.
Соединение B имеет значение Papp 11,98×10-6 см/сек, которое сходно со значением образца сравнения с салициловой кислотой.
×10-6
Соединения C, D и A имеют значения Papp, близкие к значениям для тестостерона, указывая на то, что эти соединения обладают высокой проницаемостью в модели caco-2, тогда как Соединение B имеет значение Papp между значениями салициловой кислоты и тестостерона, указывая на то, что оно обладает средней проницаемостью в модели Caco-2. Эти результаты дают возможность предположить, что четыре изомера дигидротетрабеназина должны в высокой степени абсорбироваться in vivo через эпителий кишечника.
ПРИМЕР 11
Сравнение седативных свойств тетрабеназина и изомеров В и С дигидротетрабеназина
Исследование проводили у крыс для определения, обладают ли изомеры дигидротетрабеназина по данному изобретению седативными свойствами. Действия изомеров на спонтанную двигательную активность у крыс сравнивали с эффектами, вызываемыми тетрабеназином и галоперидолом с использованием изложенных ниже способов.
Способы
Для исследования использовали самцов крыс Sprague-Dawley, (Charles River Laboratories, Saint-Germain/L'Arbresle, France), весом 200-250 г в начале исследования. Крыс содержали по 2 или 3 особи в клетке, в макролоновых клетках III типа, в помещении, в котором установлены следующие окружающие условия: температура: 20±2°C, влажность: минимум 45%, проветривание: >12 в час, свето-темновой цикл 12 ч/12 ч [на 7:00 утра]. Крысам давали возможность привыкнуть к этим условиям по меньшей мере в течение пяти дней до начала исследования. Крысы получали корм (Dietex, Vigny, France, ref. 811002) и воду (водопроводная вода в бачках для воды) без ограничения.
Растворы каждого тестируемого соединения в кукурузном масле были заново приготовлены в день эксперимента. Галоперидол был приготовлен в гидроксиэтилцеллюлозе, 0,5% в деионизированной воде. Либо носитель, либо тестируемое соединение вводили в виде единичной дозы (0,3, 1, 3 и 10 мкг/кг, 2 мл/кг интраперитонеально (и.п.)). Соединение сравнения галоперидол (1 мг/кг) вводили и.п. (2 мл/кг).
Животных помещали в клетки из органического стекла под видеокамерой, в комнате с низкой интенсивностью света (максимум 50 люкс). Через сорок пять минут и 3 часа после введения, определяли двигательную активность в течение 20 минутного периода с использованием анализатора видеоизображения (Videotrack, View Point, France). Двигательную активность регистрировали в контрольной группе (галоперидол) через 1 час после введения. Определяли количество и продолжительность амбулаторных движений. В конце измерения двигательной активности (45 минут и 3 часа), в клетке из органического стекла, закрытие век и активность оценивали в баллах следующим образом:
Закрытие век:
0: (нормальное) веки широко открыты
1: веки слегка опущены
2: птоз, опущение век приблизительно на половину
3: веки полностью закрыты
Активность:
1: очень низкая, ступор, кома, слабая ответная реакция или ее отсутствие
2: низкая, некоторый ступор, вялые, некоторые движения головы или тела
3: довольно низкая, слабый ступор, некоторые разведывательные движения с периодами неподвижности
4: нормальные, проворные, разведывательные движения/медленное оцепенение
5: довольно высокая, легкое возбуждение, напряжение, внезапное метание или оцепенение
6: очень высокая, чрезмерно резвые, возбужденные, внезапные приступы бега или движений тела
Количество случаев и продолжительность (в секундах) амбулаторных (крупных) движений и продолжительность периодов неактивности (секунды) определяли в течение двух 20 минутных периодов (через 45 минут и 3 часа после введения) используя анализатор видеоизображения (Videotrack, ViewPoint, Lyon, France). Отслеживание изображения проводили с использованием видеокамеры, расположенной над клетками из органического стекла, регистрируя общую двигательную активность. Изображения, записанные с помощью видеокамеры, оцифровывали, и смещение центра тяжести точек цифрового изображения отслеживали и анализировали, используя следующий способ: измеряли скорость смещения центра тяжести точки и устанавливали два пороговых значения для определения типа движения: пороговое значение 1 (высокая скорость) и пороговое значение 2 (низкая скорость). Когда животное двигалось, и скорость смещения центра тяжести точки была выше порогового значения 1, движение считали амбулаторным движением. Когда животное оставалось неактивным, скорость была ниже порогового значения 2, это движение считали пассивностью.
Результаты выражали в виде средних значений ± SEM 12-ти отдельных значений. Статистический анализ проводили с использованием ANOVA (однофакторный анализ) и t-теста Даннета, с непараметрическим тестом Краскэла-Уоллиса (Kruskal-Wallis) с последующим U-тестом по Манн-Витней (Mann&Whitney) на определение седативного эффекта. Значение p<0,05 принимали как показатель достоверности.
Протокол
Размер группы n=12
Группа 1: Контроль, галоперидол(1 мг/кг и. п.)
Группа 2: Носитель контрольная группа (2 мл/кг и. п.)
Группа 3: тетрабеназин (0,3 мг/кг и. п.)
Группа 4: тетрабеназин(1 мг/кг и. п.)
Группа 5: тетрабеназин (3 мг/кг и. п.)
Группа 6: тетрабеназин (10 мг/кг и. п.)
Группа 7: Изомер C (0,3 мг/кг и. п.)
Группа 8: Изомер C (1 мг/кг и. п.)
Группа 9: Изомер C (3 мг/кг и. п.)
Группа 10: Изомер C (10 мг/кг и. п.)
Группа 11: Изомер B (0,3 мг/кг и. п.)
Группа 12: Изомер B (1 мг/кг и. п.)
Группа 13: Изомер B (3 мг/кг и. п.)
Группа 14: Изомер B (10 мг/кг и. п.)
Результаты
Эти результаты показывают, что тетрабеназин вызывает дозозависимый седативный эффект через 45 минут и 3 часа после введения, тогда как Изомер B и Изомер C не демонстрируют седативного эффекта ни в какое время, хотя изомер C продемонстрировал слабый и незначительный гиперлокомоторный эффект через 3 часа после введения.
ПРИМЕР 12
Фармацевтические композиции
(i) Таблетированный состав-I
Таблетированную композицию, содержащую дигидротетрабеназин по данному изобретению, получали путем смешивания 50 мг дигидротетрабеназина с 197 мг лактозы (BP) в качестве разбавителя, и 3 мг стеарата магния в качестве смазывающего вещества и прессования в форму таблетки известным способом.
(ii) Таблетированный состав-II
Таблетированную композицию, содержащую дигидротетрабеназин по данному изобретению, получали путем смешивания соединения (25 мг) с оксидом железа, лактозой, стеаратом магния, белым маисовым крахмалом и тальком, и прессования в форму таблетки известным способом.
(iii) Состав капсулы
Состав капсулы получают путем смешивания 100 мг дигидротетрабеназина по изобретению с 100 мг лактозы и заполнения полученной в результате смесью стандартных темных твердых желатиновых капсул.
Аналоги
Понятно, что многочисленные модификации и изменения могут быть выполнены в отношении отдельных вариантов осуществления данного изобретения, описанных выше, без отклонения от принципов, лежащих в основе изобретения. Все эти модификации и изменения охватываются этой заявкой.
Изобретение относится к новому 3,11b-цис-дигидротетрабеназину общей формулы (I) или к его антиподу
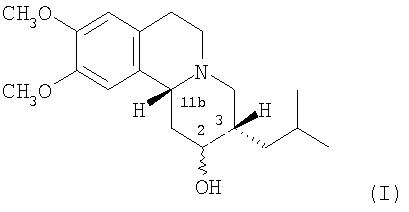 .
.
Изобретение также относится к композиции, к фармацевтической композиции, к применению 3,11b-цис-дигидротетрабеназина, к способу получения 3,11b-цис-дигидротетрабеназина по п.1, к соединениям формулы (II), (III), а также к сложному эфиру кислоты Мошера и 3,11b-цис-дигидротетрабеназина.
Технический результат - получение новых биологически активных соединений, обладающих активностью в качестве ингибитора везикулярного транспортера моноаминов VMAT2. 9 н. и 14 з.п. ф-лы, 8 табл.
1. 3,11b-цис-дигидротетрабеназин общей формулы (1) или его антипод
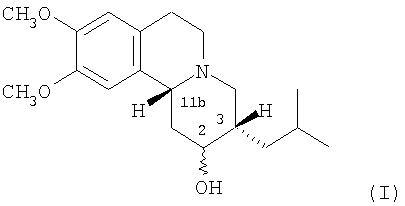 .
.
2. 3,11b-цис-дигидротетрабеназин по п.1, который находится в (+)-изомерной форме.
3. Композиция, обладающая активностью в качестве ингибитора везикулярного транспортера моноаминов VMAT2, содержащая 3,11b-цис-дигидротетрабеназин по п.1, по существу свободная от 3,11b-транс-дигидротетрабеназина.
4. Композиция по п.3, где 3,11b-цис-дигидротетрабеназин представляет собой (+)-изомер.
5. 3,11b-цис-дигидротетрабеназин по п.1, в форме 2S,3S,11bR изомера, имеющего формулу (Iа):
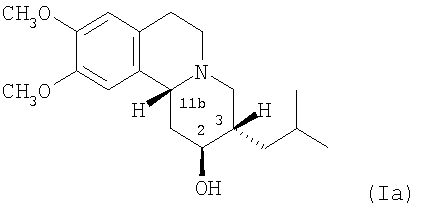
6. 3,11b-цис-дигидротетрабеназин по п.1, в форме 2R,3R,11bS изомера, имеющего формулу (Ib):
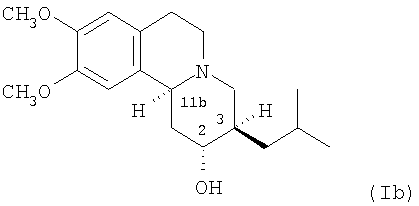
7. 3,11b-цис-дигидротетрабеназин по п.1, в форме 2R,3S,11bR изомера, имеющего формулу (Iс):
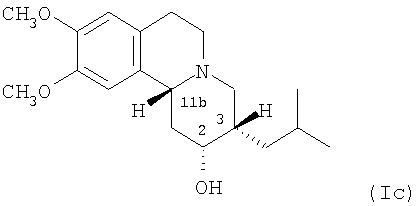
8. 3,11b-цис-дигидротетрабеназин по п.1, в форме 2S,3R,11bS изомера, имеющего формулу (Id):
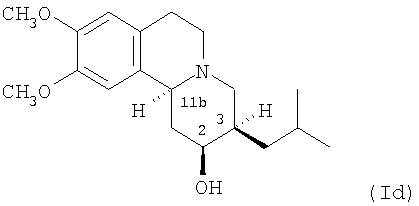
9. 3,11b-цис-дигидротетрабеназин по п.1 в форме свободного основания.
10. 3,11b-цис-дигидротетрабеназин по п.1 в форме кислотно-аддитивной соли.
11. Кислотно-аддитивная соль 3,11b-цис-дигидротетрабеназина по п.10, где соль представляет собой метансульфонат.
12. Фармацевтическая композиция, обладающая активностью в качестве ингибитора везикулярного транспортера моноаминов VMAT2, содержащая 3,11b-цис-дигидротетрабеназин по любому из пп.1-11 и фармацевтически приемлемый носитель.
13. Фармацевтическая композиция по п.12, где 3,11b-цис-дигидротетрабеназин представляет собой (+)-изомер.
14. 3,11b-цис-дигидротетрабеназин по любому из пп.1-11 для применения в медицине или терапии в качестве лекарственного средства, обладающего активностью в качестве ингибитора везикулярного транспортера моноаминов VMAT2.
15. 3,11b-цис-дигидротетрабеназин по п.14 для применения в лечении гиперкинетических двигательных расстройств или лечении депрессии.
16. 3,11b-цис-дигидротетрабеназин по п.15, где гиперкинетическое двигательное расстройство представляет собой болезнь Хантингтона, гемибаллизм, сенильную хорею, тик, позднюю дискинезию или синдром Туретта.
17. Применение 3,11b-цис-дигидротетрабеназина, охарактеризованного в любом из пп.1-11, для производства лекарственного средства для лечения гиперкинетических двигательных расстройств или лечения депрессии.
18. Применение по п.17, где гиперкинетическое двигательное расстройство представляет собой болезнь Хантингтона, гемибаллизм, сенильную хорею, тик, позднюю дискинезию или синдром Туретта.
19. Способ получения 3,11b-цис-дигидротетрабеназина по п.1, включающий взаимодействие соединения формулы (II):
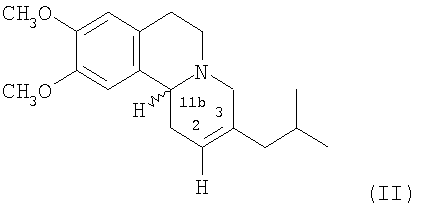
с реагентом, или реагентами, подходящими для гидрирования 2,3-двойной связи соединения формулы (II), а затем, где требуется, отделение и выделение желаемой изомерной формы 3,11b-цис-дигидротетрабеназина.
20. Способ получения 3,11b-цис-дигидротетрабеназина по п.1, включающий воздействие на соединение формулы (III):
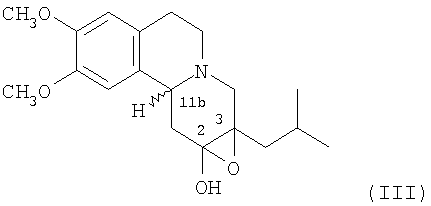
условий для раскрытия цикла 2,3-эпоксидной группы соединения формулы (III), а затем, где требуется, отделение и выделение желаемой изомерной формы 3,11b-цис-дигидротетрабеназина.
21. Соединение формулы (II):
22. Соединение формулы (III):
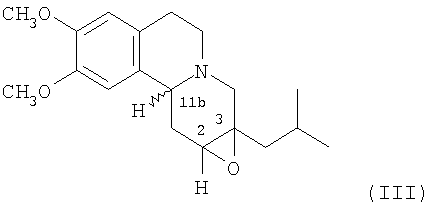
23. Сложный эфир кислоты Мошера и 3,11b-цис-дигидротетрабеназина по п.1.
| KILBOURN M.R | |||
| ЕТ AL | |||
| CHIRALITY, vol.9, no.1, 1997, p.59-62 | |||
| KILBOURN M | |||
| ЕТ AL | |||
| EUROPEAN JOURNAL OF PHARMACOLOGY, vol.278, no.3, 1995, p.249-252 | |||
| BROSSI A | |||
| ЕТ AL | |||
| HELVETICA CHIMICA ACTA, vol.41, 1958, p.1793-1806 | |||
| US 6087376 A, 11.07.2000 | |||
| US 2843591 A, 15.07.1958 | |||
| УСИЛИТЕЛЬ ЯРКОСТИ | 2024 |
|
RU2830993C1 |
| (S)-1-[(10-ХЛОР-6,7-ДИГИДРО-4-ОКСО-3-ФЕНИЛ-4H-БЕНЗО/А/ХИНОЛИЗИН-1-ИЛ)КАРБОНИЛ]-3-ЭТОКСИПИРРОЛИДИН, (S)-1-[(10-ХЛОР-6,7-ДИГИДРО-4-ОКСО-3-ФЕНИЛ-4H-БЕНЗО/А/ХИНОЛИЗИН-1-ИЛ)КАРБОНИЛ]-3-ОКСИПИРРОЛИДИН В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА, КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ГИПНОТИЧЕСКИМ НЕСЕДАТИВНЫМ ДЕЙСТВИЕМ | 1992 |
|
RU2073678C1 |

