Изобретение относится к химии производных нитрилов, в частности к способу получения 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов общей формулы
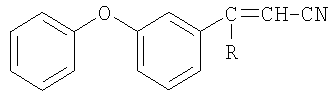
где R=H, СН3
которые являются новыми по структуре 3-феноксифенилсодержащими соединениями и могут представлять интерес в качестве полупродуктов в синтезе биологически активных веществ. Многие соединения, содержащие дифенилоксидный фрагмент, проявляют различные виды биологической активности. Так перметрин [(3-феноксифенил)метиловый эфир 3-(2,2-дихлорэтенил)-2,2-диметилциклопропанкарбоновой кислоты; смесь цис- и транс-изомеров (3:1)] и фенотрин [2,2-диметил-3-(2-метил-1-пропенил)циклопропанкарбоновой кислоты (3-феноксифенил)метиловый эфир] используются как лекарственные препараты, обладающие противопаразитарным, противопедикулезным, инсектицидным, овоцидным фармакологическим действием. Имеются примеры использования феноксифенилацетиленов, полученных на основе 1-(2-метил-4-феноксифенил)этанона и 1-(3-феноксифенил)этанона, в качестве противотромботических, противовоспалительных, жаропонижающих агентов и анальгетиков [Химическая энциклопедия: В 5 т.: Т.5. / Ред. кол.: Кнунянц И.Л. и др. - М.: Большая Российская энцикл., 1992. - 639 с].
Известен способ получения замещенных фенилакрилонитрилов, содержащих в качестве заместителей атомы хлора, водорода, нитрильную или винильную группы, заключающийся во взаимодействии стирена с цианистым водородом в присутствии кислорода, хлористого водорода, азота. В качестве катализаторов в данном процессе были использованы различные соли палладия, стронция, родия, индия, меди и кобальта [Patent US 3524874. С07С 121/70, С07С 121/72. - 1970].
Недостатками данного метода являются низкий выход фенилакрилонитрилов (7,6-28%), высокая токсичность одного из исходных соединений, а именно цианистого водорода, и использование сложной системы катализаторов.
Данный метод не приводит к получению веществ заявляемой структурной формулы.
Известен также способ получения циннамонитрила, состоящий во взаимодействии бензальдегида с ацетонитрилом в присутствии гидроокиси калия при мольном соотношении бензальдегида и гидроокиси калия, равном 1:1. Конечный продукт выделяют перегонкой с водяным паром, выход 31-45%. При вакуумной перегонке конечного продукта выход составляет 22% [Stephen A. DiBiase, James R. Beadle, George W.Gokel. Organic Syntheses, CV 7, 108].
Недостатком данного метода является достаточно трудоемкая стадия выделения продукта, большой перерасход диэтилового эфира, применяемого в качестве экстрагента и низкий выход целевого продукта.
Данный метод также не приводит к получению веществ заявляемой структурной формулы.
Задачей предлагаемого изобретения является разработка технологичного малостадийного метода синтеза 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов с хорошим выходом и высокой степенью чистоты.
Техническим результатом является расширение ассортимента химических соединений, в частности получение новых 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов с хорошим выходом, высокой степенью чистоты и упрощение стадии выделения конечного продукта.
Технический результат достигается в способе получения 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов общей формулы
где R=H, СН3
заключающийся во взаимодействии карбонильных соединений, например 3-феноксибензальдегида или 3-феноксифенилметилкетона с ацетонитрилом в присутствии гидроокиси калия, и выделении конечного продукта путем экстрагирования диэтиловым эфиром с последующей отгонкой экстрагента и очисткой целевого продукта вакуумной перегонкой. При этом процесс проводят при мольном соотношении карбонильного соединения, ацетонитрила и гидроокиси калия, равном 1:(20-22):1 соответственно в среде исходного ацетонитрила при его температуре кипения 80-82°С.
Сущностью метода является реакция присоединения ацетонитрила к карбонильным соединениям, например 3-феноксибензальдегиду или 3-феноксифенилметилкетону в присутствии гидроокиси калия в среде ацетонитрила
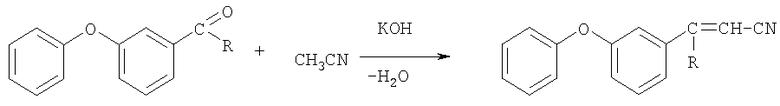
где R=H, СН3.
Способ осуществляется следующим образом.
В реакторе, оборудованном механической мешалкой, обратным холодильником с хлоркальциевой трубкой, термометром и капельной воронкой растворяют гидроокись калия в ацетонитриле при кипячении и перемешивании в атмосфере азота. Затем из капельной воронки добавляют карбонильное соединение, например 3-феноксибензальдегид или 3-феноксифенилметилкетон в ацетонитриле. Время прикапывания и синтеза зависит от реакционной способности карбонильного соединения. Далее горячий раствор выливают в стакан с дробленым льдом, после чего образуется двухфазная смесь, которую разделяют с помощью делительной воронки. Органический слой отделяют, а водный экстрагируют дважды диэтиловым эфиром. Соединяют органический слой и эфирные вытяжки, промывают дистиллированной водой. Органическую фазу сушат над сульфатом натрия. Экстрагент отгоняют, полученный 3-замещенный-3-(3-феноксифенил)-2-пропененитрил очищают вакуумной перегонкой с добавлением гидрохинона.
Выход данных продуктов после выделения составляет до 51%.
Как показали проведенные исследования, оптимальным и технологичным условием проведения реакции является ее осуществление в среде ацетонитрила при мольном соотношении карбонильное соединение: ацетонитрил: гидроокись калия 1:(20-22):1. Меньший избыток ацетонитрила приводил к некоторому снижению выхода целевого продукта за счет неполной конверсии карбонильного соединения и неполного растворения гидроокиси калия в ацетонитриле. Дальнейшее увеличение избытка ацетонитрила не влияло на выход 3-замещенных-(3-феноксифенил)-2-пропененитрилов и являлось нецелесообразным.
В прототипе циннамонитрил очищают перегонкой с водяным паром и последующей отгонкой растворителя, что значительно осложняет стадию выделения, при этом выход составляет 31-45%, степень чистоты конечного продукта авторами статьи не обсуждается. При вакуумной перегонке выход конечного продукта не превышает 22%. Предложенная нами схема выделения целевого продукта позволяет избежать трудоемкие операции и, что самое важное, достичь 48-51% выхода после перегонки 3-замещенных-(3-феноксифенил)-2-пропененитрилов на вакуумном насосе с высокой степенью чистоты 98,5%.
Оптимальной температурой реакции является 80-82°С. Снижение температуры до комнатной приводит к сильному увеличению продолжительности данного взаимодействия и снижению выхода целевого продукта, в то время как ее повышение ограничено температурой кипения ацетонитрила.
Пример 1. 3-(3-феноксифенил)-2-пропененитрил
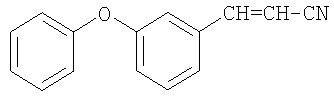
В реактор, оборудованный механической мешалкой, обратным холодильником с хлоркальциевой трубкой, термометром и капельной воронкой помещают 3 г (0,05 моль) гидроокиси калия и 40 мл ацетонитрила. Смесь кипятят при перемешивании в атмосфере азота до полного растворения шариков гидроокиси калия в ацетонитриле. Затем из капельной воронки добавляют 10 г (0,05 моль) 3-феноксибензальдегида в 20 мл ацетонитрила тонкой струей в течение 1-2 минут. После добавления всего 3-феноксибензальдегида перемешивание продолжают в течение 10 минут и горячий раствор выливают на 100 г дробленого льда в стакане. После охлаждения образуется двухфазная смесь, которую разделяют с помощью делительной воронки. Органический слой отделяют, а водный экстрагируют 2 раза по 50 мл диэтиловым эфиром. Соединяют органический слой и эфирные вытяжки, промывают дистиллированной водой. Органическую фазу сушат над сульфатом натрия. После отгонки растворителя (диэтиловый эфир), 3-(3-феноксифенил)-2-пропененитрил перегоняют в вакууме с добавлением гидрохинона. Выход - 5,3 г (0,024 моль, 48%). Т.кип. 194-200°С/4 мм рт.ст. Чистота - 98,5 % (по данным ГЖХ). ИК-спектр, υ, см-1: 1600-700 (Ar); 2218 (C≡N); 1696 (С=С); 3064 (С-Н). Спектр ЯМР 1Н, δ, м.д.: 6,84-7,2 м (9Н, С6Н5ОС6Н4); 7,44-7,46 д (1Н, Ar-СН); 5,62-5,67 д (1Н, CH-CN).
Пример 2. 3-метил-3-(3-феноксифенил)-2-пропененитрил
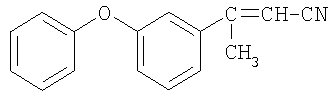
В реактор, оборудованный механической мешалкой, обратным холодильником с хлоркальциевой трубкой, термометром и капельной воронкой помещают 3 г (0,05 моль) гидроокиси калия и 40 мл ацетонитрила. Гидроокись калия должна быть как можно более свежей. Смесь кипятят при перемешивании в атмосфере азота до полного растворения шариков гидроокиси калия в ацетонитриле. Затем из капельной воронки добавляют 10 г (0,05 моль) 3-феноксифенилметилкетона в 20 мл ацетонитрила по каплям в течение 30-40 минут. После добавления всего 3-феноксифенилметилкетона перемешивание продолжают в течение 3 часов, и горячий раствор выливают на 100 г дробленого льда в стакане. После охлаждения образуется двухфазная смесь, которую разделяют с помощью делительной воронки. Органический слой отделяют, а водный экстрагируют 2 раза диэтиловым эфиром. Соединяют органический слой и эфирные вытяжки, промывают дистиллированной водой. Органическую фазу сушат над сульфатом натрия. После отгонки растворителя 3-метил-3-(3-феноксифенил)-2-пропененитрил перегоняют в вакууме с добавлением гидрохинона. Выход - 6 г (0,0255 моль, 51%). Т.кип. 215-218°С / 4 мм рт.ст. Чистота - 97,2 % (по данным ГЖХ). ИК-спектр, υ, см-1: 1600-700 (Ar); 2218 (C≡N); 1672 (С=С); 2926-3034 (СН3 ). Спектр ЯМР 1Н, δ, м.д.: 6,84-7,2 м (9Н, С6Н5ОС6Н4); 1,1-1,3 с
(3Н, СН3); 5,34-5,37 с (1Н, CH-CN).
Выводы
Предлагаемый способ позволяет получить 3-замещенные-3-(3-феноксифенил)-2-пропененитрилы, в одну стадию с хорошими выходами. К его достоинствам можно отнести препаративную простоту синтеза и легкость выделения целевых продуктов с высокой степенью чистоты. Структура синтезированных соединений подтверждена ИК-, ЯМР 1Н-спектроскопией.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3-ЗАМЕЩЕННЫХ 2-(3-ФЕНОКСИФЕНИЛ)АКРИЛОНИТРИЛОВ | 2014 |
|
RU2560178C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ФЕНОКСИФЕНИЛАЦЕТОНИТРИЛА | 2011 |
|
RU2451669C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 3-М-ФЕНОКСИФЕНИЛ-1-ФЕНИЛ-2-ПРОПЕН-1- ОНОВ | 2004 |
|
RU2266895C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛ 5-МЕТИЛ-3-(3-ФЕНОКСИФЕНИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛИЛ-4-КАРБОКСИЛАТА | 2015 |
|
RU2582127C1 |
| 3-ФЕНОКСИФЕНИЛСОДЕРЖАЩИЕ 1,3-ДИКЕТОНЫ В КАЧЕСТВЕ ИСХОДНЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ ИХ ХЕЛАТНЫХ КОМПЛЕКСОВ С ИОНАМИ МЕДИ (II) И СПОСОБ ПОЛУЧЕНИЯ 3-ФЕНОКСИФЕНИЛСОДЕРЖАЩИХ 1,3-ДИКЕТОНОВ | 2012 |
|
RU2475473C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3-ФЕНОКСИФЕНИЛ)БУТАН-1,3-ДИОНА | 2013 |
|
RU2529029C1 |
| Способ получения 2-(3-феноксифенил)-пропионовой кислоты или ее кальциевой соли | 1979 |
|
SU1039439A3 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ФЕНОКСИФЕНИЛЦИАНГИДРИНА | 2007 |
|
RU2331634C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТИЛ-2-(3-ФЕНОКСИБЕНЗОАТ)ПРОПИОНИТРИЛА | 2009 |
|
RU2398762C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛ 5-МЕТИЛ-3-(3-ФЕНОКСИФЕНИЛ)ИЗОКСАЗОЛ-4-КАРБОКСИЛАТА | 2015 |
|
RU2592281C1 |
Изобретение относится к получению новых промежуточных продуктов - 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов общей формулы 1, которые могут быть использованы в производстве биологически активных веществ, таких как инсектициды или в производстве фармакологически активных соединений. Способ получения 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов общей формулы 1
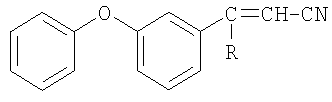
где R=H, СН3,
заключается во взаимодействии карбонильного соединения, выбранного из 3-феноксибензальдегида или 3-феноксифенилметилкетона с ацетонитрилом в присутствии гидроокиси калия с последующим выделением целевого продукта. Предпочтительно, процесс проводят при мольном соотношении карбонильного соединения, ацетонитрила и гидроокиси калия, равном 1:(20-22):1 соответственно в среде исходного ацетонитрила при его температуре кипения 80-82°С. Выделение целевого продукта, как правило, осуществляют путем экстрагирования диэтиловым эфиром с последующей отгонкой экстрагента и очисткой целевого продукта вакуумной перегонкой. Продукты получают с хорошим выходом, высокой степенью чистоты. 1 з.п. ф-лы.
1. Способ получения 3-замещенных-3-(3-феноксифенил)-2-пропененитрилов общей формулы
где R=H, СН3,
взаимодействием карбонильного соединения, выбранного из 3-феноксибензальдегида или 3-феноксифенилметилкетона с ацетонитрилом в присутствии гидроокиси калия с последующим выделением целевого продукта.
2. Способ по п.1, отличающийся тем, что процесс проводят при мольном соотношении карбонильного соединения, ацетонитрила и гидроокиси калия, равном 1:(20-22):1 соответственно, в среде исходного ацетонитрила при его температуре кипения 80-82°С, и выделение целевого продукта осуществляют путем экстрагирования диэтиловым эфиром с последующей отгонкой экстрагента и очисткой целевого продукта вакуумной перегонкой.
| S.A DIBIASE et al | |||
| "Synthesis of α,β-unsaturated nitriles from acetonitrile: cyclohexylideneacetonitrile and cinnamonitrile" Organic syntheses, 1984, v.62, p.179 | |||
| УСТРОЙСТВО ДЛЯ НАГРЕВА ЖИДКОСТИ | 2000 |
|
RU2162571C1 |
| S.A.), 20.07.1972 | |||
| FRANKE ALBRECHT et al | |||
| "The p-substituted β-methylcinnamic acid derivatives" Helvetica Chimica Acta, 1975, 58(1), p.268-278 | |||
| ПОПОВ | |||

