Область техники, к которой относится изобретение
Настоящее изобретение относится в основном к способу жидкофазного каталитического окисления ароматического соединения. Один аспект настоящего изобретения касается частичного окисления диалкилзамещенного ароматического соединения (в частности, пара-ксилола) с получением сырой ароматической дикарбоновой кислоты (в частности, сырой терефталевой кислоты), которое затем подвергают очистке и разделению. Другой аспект настоящего изобретения касается улучшенной барботажной колонны реакторного типа, которая обеспечивает осуществление более эффективного и экономичного способа жидкофазного окисления.
Уровень техники
Реакции жидкофазного окисления применяются в различных существующих промышленных способах. Например, жидкофазное окисление в настоящее время применяется для окисления альдегидов в кислоты (в частности, окисления пропионового альдегида в пропионовую кислоту), окисления циклогексана в адипиновую кислоту и окисления алкилзамещенных ароматических соединений в спирты, кислоты или дикислоты. Наиболее важным промышленным способом окисления в последней из указанных категорий (окисление алкилзамещенных ароматических соединений) является частичное жидкофазное каталитическое окисление пара-ксилола с образованием терефталевой кислоты. Терефталевая кислота является важным соединением, которое находит широкое применение. Основным применением терефталевой кислоты является использованием ее в качестве исходного материала для получения полиэтилентерефталата (РЕТ, ПЭТ). ПЭТ является известным пластическим материалом, который во всем мире в больших количествах применяется в производстве таких видов продукции как бутылки, волокна и упаковочные материалы.
В типичном способе жидкофазного окисления, включая частичное окисление пара-ксилола с образованием терефталевой кислоты, жидкофазный поток исходного соединения и газофазный поток окислителя вводятся в реактор и они образуют в реакторе многофазную реакционную среду. Подаваемый в реактор жидкофазный поток исходного соединения содержит, по меньшей мере, одно способное окисляться органическое соединение (в частности, пара-ксилол), а газофазный поток окислителя содержит молекулярный кислород. По крайней мере, часть молекулярного кислорода, который вводится в реактор в виде газа, растворяется в жидкой фазе реакционной среды и обеспечивает доступность кислорода для осуществления жидкофазной реакции. Если жидкая фаза многофазной реакционной среды содержит недостаточную концентрацию молекулярного кислорода (т.е. если определенные порции реакционной среды “испытывают недостачу кислорода”), то нежелательные побочные реакции могут привести к образованию примесей и/или может замедлиться протекание требуемых реакций. Если жидкая фаза реакционной среды содержит слишком мало способного окисляться соединения, то скорость реакции может оказаться нежелательно низкой. Кроме того, если жидкая фаза реакционной среды содержит избыточную концентрацию способного окисляться соединения, то дополнительные нежелательные побочные реакции могут привести к образованию примесей.
Обычные реакторы для проведения жидкофазного окисления снабжаются средствами перемешивания содержащейся в них многофазной реакционной среды. Перемешивание реакционной среды осуществляется с целью облегчить растворение молекулярного кислорода в жидкой фазе реакционной среды, поддержать относительно однородную концентрацию растворенного кислорода в жидкой фазе реакционной среды и поддержать относительно однородную концентрацию способного окисляться органического соединения в жидкой фазе реакционной среды.
Перемешивание реакционной среды, в которой проходит жидкофазное окисление, часто осуществляется с использованием механических средств перемешивания в таких реакторах, как, например, проточные реакторы смешивания (CSTR). Несмотря на то, что CSTR способны обеспечить тщательное перемешивание реакционной среды, CSTR обладают рядом недостатков. CSTR имеют относительно высокую капитальную стоимость, поскольку они, например, требуют использования дорогостоящих двигателей, изолированных от попадания жидкости подшипников и приводных валов и/или использования сложных перемешивающих устройств. Кроме того, вращающиеся и/или качающиеся механические компоненты обычных CSTR требуют регулярного технического обслуживания. Затраты труда и время простоя, связанные с проведением указанного технического обслуживания, увеличивают эксплуатационные расходы на содержание CSTR. Тем не менее, даже при проведении регулярного технического обслуживания, применяемые в CSTR механические системы перемешивания подвержены механическим отказам и могут потребовать замены через относительно небольшие промежутки времени.
Барботажные колонны реакторного типа представляют собой привлекательную альтернативу CSTR и другим снабженным механическим перемешиванием реакторам для проведения процессов окисления. Барботажные колонны реакторного типа обеспечивают перемешивание реакционной среды, не требуя использования дорогостоящего и ненадежного механического оборудования. Как правило, барботажные колонны реакторного типа включают удлиненную вертикальную зону реакции, в которой находится реакционная среда. Перемешивание реакционной среды в зоне реакции в основном обеспечивается за счет естественного всплывания пузырьков газа, которые поднимаются вверх через жидкую фазу реакционной среды. Указанное перемешивание за счет естественного всплывания, которое обеспечивается в барботажных колоннах реакторного типа, снижает капитальные и эксплуатационные затраты по сравнению с реакторами с механическим перемешиванием. Кроме того, практически полное отсутствие движущихся механических частей в барботажных колоннах реакторного типа позволяет получить систему для проведения окисления, которая в меньшей степени подвержена механическим отказам, чем реакторы с механическим перемешиванием.
В том случае, когда жидкофазное частичное окисление пара-ксилола проводится в обычном реакторе для проведения процессов окисления (в CSTR или барботажной колонне реакторного типа), то выделяемый из реактора продукт, как правило, представляет собой суспензию, содержащую сырец терефталевой кислоты (СТА) и маточный раствор. СТА содержит относительно большие уровни примесей (в частности, содержит 4-карбоксибензальдегид, паратолуиловую кислоту, флуореноны и другие окрашенные соединения), которые делают его непригодным для использования в качестве сырья при получении ПЭТ. Поэтому СТА, получаемая в обычных реакторах для проведения процессов окисления, как правило, подвергается очистке, которая превращает СТА в очищенную терефталевую кислоту (РТА), пригодную для получения ПЭТ.
Типичный способ очистки, с целью превращения СТА в РТА, включает следующие стадии: (1) замена маточного раствора СТА-содержащей суспензии на воду, (2) нагревание суспензии СТА/вода с целью растворения СТА в воде, (3) каталитическое гидрирование раствора СТА/вода с целью превращения примесей в более удобные и/или легко отделяемые соединения, (4) осаждение полученной РТА из раствора после гидрирования посредством многостадийной кристаллизации и (5) отделение кристаллов РТА от оставшихся жидкостей. Несмотря на то, что указанный обычный способ очистки является эффективным, он может оказаться очень дорогим. Индивидуальными факторами, которые вносят свой вклад в высокую стоимость обычных способов очистки СТА, являются, например, тепловая энергия, необходимая для облегчения растворения СТА в воде, катализатор, требуемый для проведения гидрирования, подача водорода, необходимого для проведения гидрирования, потери выхода продукта, вызванные гидрированием части терефталевой кислоты, и многочисленные сосуды, необходимые для проведения многостадийной кристаллизации. Таким образом, требуется получить СТА в виде такого продукта, который можно было бы очистить без использования требующего нагревания растворения в воде, гидрирования и/или многостадийной кристаллизации.
Объекты изобретения
Таким образом, объектом настоящего изобретения является более эффективный и экономичный реактор для проведения жидкофазного окисления и способ жидкофазного окисления.
Другим объектом настоящего изобретения является более эффективный и экономичный реактор и способ жидкофазного каталитического частичного окисления пара-ксилола с образованием терефталевой кислоты.
Другим объектом настоящего изобретения является барботажная колонна реакторного типа, которая обеспечивает проведение улучшенных реакций жидкофазного окисления с образованием меньшего количества примесей.
Еще одним объектом настоящего изобретения является более эффективная и экономичная система для получения очищенной терефталевой кислоты (РТА) посредством жидкофазного окисления пара-ксилола с образованием сырой терефталевой кислоты (СТА) и последующей очистки СТА с целью получения РТА.
Другим объектом настоящего изобретения является барботажная колонна реакторного типа для окисления пара-ксилола и получения СТА в виде продукта, который не требует для своей очистки использования нагрева, ускоряющего растворение СТА в воде, гидрирования растворенной СТА и/или проведения многостадийной кристаллизации РТА после гидрирования.
Следует отметить, что объем настоящего изобретения, определенный в прилагаемой формуле изобретения, не ограничивается способами и установками, которые могут реализовать все перечисленные выше объекты. Более того, в объем заявляемого изобретения могут входить различные системы, которые не позволяют получить все или какой-либо из перечисленных выше объектов. Дополнительные объекты и преимущества настоящего изобретения станут легко понятны специалисту в данной области техники после ознакомления с изложенным ниже подробным описанием изобретения и с прилагающимися к нему чертежами.
Сущность изобретения
Один из вариантов осуществления настоящего изобретения касается состава сырой терефталевой кислоты (СТА), который представляет собой множество частиц СТА, извлекаемых из реактора для проведения процессов окисления, в котором, по крайней мере, частично образуются частицы СТА, при этом типичный образец частиц СТА обладает одним или несколькими из указанных свойств: (a) содержит меньше чем приблизительно 6 м.д. мас. 4,4-дикарбоксистильбена (4,4-DCS), (b) содержит меньше чем приблизительно 400 м.д. мас. изофталевой кислоты (IPA), (c) содержит меньше чем приблизительно 25 м.д. мас. 2,6-дикарбоксифлуоренона (2,6-DCF), (d) имеет процент пропускания при 340 нм (%Т340) больше чем приблизительно 60.
Другой вариант осуществления настоящего изобретения касается состава суспензии, извлекаемой из реактора для проведения процессов окисления, при этом состав суспензии включает меточный раствор и твердые частицы сырой терефталевой кислоты (СТА), причем частицы СТА, по крайней мере, частично образуются в реакторе для проведения процессов окисления, при этом типичный образец суспензии обладает одним или несколькими из указанных свойств, которые являются обобщенными для твердых и жидких компонентов суспензии: (а) содержит меньше чем приблизительно 1500 м.д. мас. изофталевой кислоты (IPA), (b) содержит меньше чем приблизительно 500 м.д. мас. фталевой кислоты (PA), (с) содержит меньше чем приблизительно 500 м.д. мас. тримеллитовой кислоты (TMA), (d) содержит меньше чем приблизительно 2000 м.д. мас. бензойной кислоты (ВА).
Еще один вариант осуществления настоящего изобретения касается способа, который включает следующие стадии: (а) окисление пара-ксилола в жидкой фазе многофазной реакционной среды, содержащейся в зоне реакции, по меньшей мере, одного первичного реактора для проведения процессов окисления; и (b) извлечение суспензии, содержащей маточный раствор и твердые частицы сырой терефталевой кислоты (СТА), из зоны реакции, при этом типичный образец частиц СТА обладает одним или несколькими из указанных свойств: (i) содержит меньше чем приблизительно 6 м.д. мас. 4,4-дикарбоксистильбена (4,4-DCS), (ii) содержит меньше чем приблизительно 400 м.д. мас. изофталевой кислоты (IPA), (iii) содержит меньше чем приблизительно 25 м.д. мас. 2,6-дикарбоксифлуоренона (2,6-DCF), (iv) имеет процент пропускания при 340 нм (%Т340) больше чем приблизительно 60.
Другой вариант осуществления настоящего изобретения касается способа, который включает следующие стадии: (а) подача исходного рециклированного растворителя, по меньшей мере, в один реактор для проведения процессов окисления; (b) окисление способного окисляться соединения в жидкой фазе многофазной реакционной среды, содержащейся в зоне реакции реактора для проведения процессов окисления; и (с) извлечение суспензии, содержащей маточный раствор и твердые частицы сырой терефталевой кислоты (СТА), из зоны реакции, при этом типичный образец частиц СТА содержит меньше чем приблизительно 5 м.д. мас. 2,7-дикарбоксифлуоренона (2,7-DCF).
Еще один вариант осуществления настоящего изобретения касается способа, который включает следующие стадии: (а) окисление пара-ксилола в жидкой фазе многофазной реакционной среды, содержащейся в зоне реакции, по меньшей мере, одного реактора для проведения процессов окисления; и (b) извлечение суспензии, содержащей меточный раствор и твердые частицы сырой терефталевой кислоты (СТА), из зоны реакции, при этом типичный образец частиц СТА обладает одним или несколькими из указанных свойств, которые являются обобщенными для твердых и жидких компонентов суспензии: (i) содержит меньше чем приблизительно 1500 м.д. мас. изофталевой кислоты (IPA), (ii) содержит меньше чем приблизительно 500 м.д. мас. фталевой кислоты (PA), (iii) содержит меньше чем приблизительно 500 м.д. мас. тримеллитовой кислоты (TMA), (iv) содержит меньше чем приблизительно 2000 м.д. мас. бензойной кислоты (ВА).
Краткое описание чертежей
Предпочтительные варианты осуществления настоящего изобретения подробно описываются далее со ссылкой на следующие чертежи
где на фиг.1 представлен вид сбоку реактора для проведения процессов окисления, который сконструирован в соответствии с одним из вариантов осуществления настоящего изобретения, и, в частности, чертеж поясняет подачу потоков исходных веществ, окислителя и флегмы в реактор, наличие многофазной реакционной среды в реакторе и отвод газа и суспензии из верхней и нижней частей реактора, соответственно;
на фиг.2 представлен в разрезе по линии 2-2 на фиг.3 увеличенный вид сбоку днища барботажной колонны реакторного типа, и, в частности, чертеж поясняет расположение и конфигурацию барботера для ввода окислителя, который применяют для подачи потока окислителя в реактор;
на фиг.3 представлен вид сверху барботера для ввода окислителя, приведенного на фиг.2, и, в частности, чертеж поясняет отверстия для подачи окислителя, расположенные в верхней части барботера для ввода окислителя;
на фиг.4 представлен вид снизу барботера для ввода окислителя, приведенного на фиг.2, в частности чертеж поясняет отверстия для подачи окислителя, расположенные в нижней части барботера для ввода окислителя;
на фиг.5 представлен в разрезе по линии 5-5 на фиг.3 вид сбоку барботера для ввода окислителя, и, в частности, чертеж поясняет ориентацию отверстий для подачи окислителя, расположенных в верхней и нижней части барботера для ввода окислителя;
на фиг.6 представлен увеличенный вид сбоку нижней части барботажной колонны реакторного типа, и, в частности, чертеж поясняет систему подачи исходных веществ внутрь реактора в нескольких разделенных по высоте позициях;
на фиг.7 представлен вид сверху в разрезе по линии 7-7 на фиг.6, в частности чертеж поясняет, как приведенная на фиг.6 система подачи исходных веществ распределяет поток исходных веществ в предпочтительную радиальную зону подачи исходных веществ (FZ) и более чем один азимутальный квадрант (Q1, Q2, Q3, Q4);
на фиг.8 представлен в разрезе вид сверху, аналогичный фиг.7, который поясняет альтернативные устройства для подачи исходных веществ в реактор с помощью байонетных трубок, каждая их которых имеет множество небольших отверстий для подачи исходных веществ;
на фиг.9 представлена изометрическая проекция альтернативной системы для подачи исходных веществ в зону реакции в нескольких разделенных по высоте позициях, которая не требует большого количества врезок в реактор, в частности чертеж показывает, что система распределения исходных веществ может, по крайней мере, частично опираться на барботер для ввода окислителя;
на фиг.10 представлен вид сбоку системы распределения исходных веществ с одной врезкой и барботер для ввода окислителя, которые приведены на фиг.9;
на фиг.11 представлен вид сверху в разрезе по линии 11-11 на фиг.10, дополнительно поясняющий систему распределения исходных веществ с одной врезкой, которая опирается на барботер для ввода окислителя;
на фиг.12 представлена изометрическая проекция альтернативного барботера для ввода окислителя, у которого все отверстия для подачи окислителя расположены внизу кольцевого элемента;
на фиг.13 представлен вид сверху альтернативного барботера для ввода окислителя, приведенного на фиг.12;
на фиг.14 представлен вид снизу альтернативного барботера для ввода окислителя, приведенного на фиг.12, в частности чертеж поясняет расположение нижних отверстий для подачи потока окислителя в зону реакции;
на фиг.15 представлен в разрезе по линии 15-15 на фиг.13 вид сбоку барботера для ввода окислителя, в частности чертеж поясняет ориентацию нижних отверстий для ввода окислителя;
на фиг.16 представлен вид сбоку барботажной колонны реакторного типа, снабженной внутренним резервуаром для деаэрации, который располагается вблизи нижнего выпускного отверстия реактора;
на фиг.17 представлен в разрезе по линии 17-17 на фиг.18 увеличенный вид нижней части барботажной колонны реакторного типа, приведенной на фиг.16, и, в частности, чертеж поясняет конфигурацию внутреннего резервуара для деаэрации, который располагается у нижнего выпускного отверстия барботажной колонны реакторного типа;
на фиг.18 представлен вид сверху в разрезе по линии 18-18 на фиг.16, который, в частности, поясняет стабилизатор потока, размещенный в резервуаре для деаэрации;
на фиг.19 представлен вид сбоку барботажной колонны реакторного типа, снабженной внешним резервуаром для деаэрации, который поясняет, каким образом часть суспензии после деаэрации, покидающая нижнюю часть резервуара для деаэрации, может быть использована для промывки линии выгрузки продуктов реакции, присоединенной к днищу реактора;
на фиг.20 представлен вид сбоку барботажной колонны реакторного типа, снабженной гибридным внутренним/внешним резервуаром для деаэрации, предназначенным для отделения газовой фазы от реакционной среды, которая выводится сбоку реактора на определенной отметке высоты;
на фиг.21 представлен вид сбоку барботажной колонны реакторного типа, снабженной альтернативным гибридным резервуаром для деаэрации, который располагается рядом с днищем реактора;
на фиг.22 представлен в разрезе увеличенный вид сбоку нижней части барботажной колонны реакторного типа, приведенной на фиг.21, и, в частности, чертеж поясняет использование альтернативного барботера для ввода окислителя, имеющего входные патрубки, в которые поток окислителя попадает через днище реактора;
на фиг.23 представлен в разрезе увеличенный вид сбоку, аналогичный фиг.22, и, в частности, чертеж поясняет альтернативные устройства для ввода потока окислителя в реактор через множество отверстий в нижнем днище реактора и необязательное применение отражательных пластинок, предназначенных для более равномерного распределения потока окислителя в реакторе;
на фиг.24 представлен вид сбоку барботажной колонны реакторного типа, в которой применяют внутренний напорный трубопровод, предназначенный для улучшения диспергирования способного окисляться соединения за счет рециркуляции части реакционной среды из верхней части реактора в нижнюю часть реактора;
на фиг.25 представлен вид сбоку барботажной колонны реакторного типа, в которой применяют внешний напорный трубопровод, предназначенный для улучшения диспергирования способного окисляться соединения за счет рециркуляции части реакционной среды из верхней части реактора в нижнюю часть реактора;
на фиг.26 представлен в разрезе вид сбоку горизонтального эжектора, который может быть использован для улучшения диспергирования способного окисляться соединения в реакторе для проведения процессов окисления, и, в частности, чертеж поясняет эжектор, в котором исходный поток жидкости используется для ввода реакционной среды в эжектор и который с большой скоростью подает смесь исходных веществ и реакционной среды в зону реакции;
на фиг.27 представлен в разрезе вид сбоку вертикального эжектора, который может быть использован для улучшения диспергирования способного окисляться соединения в реакторе окисления, и, в частности, чертеж поясняет эжектор, который смешивает жидкое исходное вещество и подаваемый газ, использует объединенную двухфазную жидкость для введения реакционной среды в эжектор и с большой скоростью направляет смесь жидких исходных веществ, подаваемого газа и реакционной среды в зону реакции;
на фиг.28 представлен вид сбоку барботажной колонны реакторного типа, содержащей многофазную реакционную среду, и, в частности, чертеж поясняет реакционную среду, которая теоретически разделена на 30 горизонтальных слоев равного объема с тем, чтобы количественно оценить определенные градиенты в реакционной среде;
на фиг.29 представлен вид сбоку барботажной колонны реакторного типа, содержащей многофазную реакционную среду, и, в частности, чертеж поясняет первый и второй дискретные 20%-ные непрерывные объемы реакционной среды, которые имеют существенно различные концентрации кислорода и/или скорости расходования кислорода;
на фиг.30 представлен вид сбоку двух расположенных один над другим реакторов, снабженных или не снабженных необязательным механическим перемешиванием, которые содержат многофазную реакционную среду, в частности чертеж поясняет, что реакторы включают дискретные 20%-ные непрерывные объемы реакционной среды, которые имеют существенно различные концентрации кислорода и/или скорости потребления кислорода;
на фиг.31 представлен вид сбоку трех примыкающих друг к другу реакторов, снабженных или не снабженных необязательным механическим перемешиванием, которые содержат многофазную реакционную среду, и, в частности, чертеж поясняет, что реакторы включают дискретные 20%-ные непрерывные объемы реакционной среды, которые имеют существенно различные концентрации кислорода и/или скорости потребления кислорода;
на фиг.32А и 32В приведен увеличенный вид частиц сырой терефталевой кислоты (СТА), полученной в соответствии с одним из вариантов осуществления настоящего изобретения, и, в частности, фотографии показывают, что каждая частица СТА имеет низкую плотность, большую площадь поверхности частиц, образованных множеством слабо связанных друг с другом субчастиц СТА;
на фиг.33А и 33В приведен увеличенный вид частиц сырой терефталевой кислоты (СТА), полученной обычным способом, и, в частности, фотографии показывают, что полученная обычным способом частица СТА имеет больший размер частиц, меньшую плотность и меньшую площадь по сравнению с частицей СТА, полученной в соответствии с настоящим изобретением, которая приведена на фиг.32А и 32В;
на фиг.34 в упрощенном виде приведена потоковая диаграмма известного способа получения очищенной терефталевой кислоты (РТА);
на фиг.35 в упрощенном виде приведена потоковая диаграмма способа получения РТА в соответствии с одним из вариантов осуществления настоящего изобретения; и
на фиг.36 приведены результаты теста на растворение за определенный промежуток времени, который описан в примерах, и, в частности, она поясняет, что кристаллы СТА по настоящему изобретению растворяются быстрее, чем кристаллы СТА, полученные обычным способом.
Подробное описание изобретения
Один из вариантов осуществления настоящего изобретения касается жидкофазного частичного окисления способного окисляться соединения. Указанное окисление предпочтительно проводят в жидкой фазе многофазной реакционной среды, которая содержится в одном или нескольких реакторов смешения. Подходящие снабженные перемешиванием реакторы включают, например, реакторы с перемешиванием с помощью барботажа (в частности, барботажные колонны реакторного типа), реакторы с механическим перемешиванием (в частности, корпусные реакторы непрерывного действия с перемешиванием) и реакторы с перемешиванием с помощью потока (в частности, газоструйные реакторы). В одном из вариантов осуществления настоящего изобретения жидкофазное окисление проводят в барботажной колонне реакторного типа.
В данном описании термин “барботажная колонна реакторного типа” обозначает реактор для проведения химических реакций в многофазной реакционной среде, при этом перемешивание реакционной среды в основном осуществляется за счет движения пузырьков газа вверх через реакционную среду. В данном описании термин “перемешивание” обозначает мощность, которая рассеивается в реакционной среде и вызывает течение и/или смешивание жидкости. В данном описании термины “большинство”, “в основном” и “преимущественно” означает больше, чем 50%. В данном описании термин “механическое перемешивание” обозначает перемешивание реакционной среды, вызываемое физическим перемещением жесткого(их) или гибкого(их) элемента(ов) относительно реакционной среды или внутри реакционной среды. Например, механическое перемешивание можно осуществить за счет вращения, колебания и/или вибрации расположенных внутри мешалок, лопастей, вибраторов или акустических диафрагм, которые размещаются в реакционной среде. В данном описании термин “перемешивание потоком” обозначает перемешивание реакционной среды, вызываемое инжекцией с большой скоростью и/или рециркуляцией одной или нескольких жидкостей в реакционной среде. Например, перемешивание потоком можно осуществить с помощью форсунок, эжекторов и/или эжекционных устройств.
В предпочтительном варианте осуществления настоящего изобретения меньше чем приблизительно 40% перемешивания реакционной среды в барботажной колонне реакторного типа в процессе окисления осуществляется механическим перемешиванием и/или перемешиванием потоком, более предпочтительно, меньше чем приблизительно 20% перемешивания осуществляется механическим перемешиванием и/или перемешиванием потоком и, наиболее предпочтительно, меньше чем 5% перемешивания осуществляется за счет механического перемешивания и/или перемешивания потоком. Количество механического перемешивания и/или перемешивания потоком, которое осуществляется в многофазной реакционной среде в процессе окисления, предпочтительно составляет меньше чем приблизительно
3 кВт/см3 реакционной среды, более предпочтительно, составляет меньше чем приблизительно 2 кВт/см3 и, наиболее предпочтительно, составляет меньше чем 1 кВт/см3.
Если теперь обратиться к фиг.1, то предпочтительную барботажную колонну реакторного типа 20 можно описать как барботажную колонну реакторного типа 20, которая состоит из кожуха реактора 22, который включает секцию проведения реакции 24 и разделительную секцию 26. Секция проведения реакции 24 ограничивает внутреннюю зону реакции 28, в то время как секция разделения 26 ограничивает внутреннюю зону разделения 30. Преимущественно поток жидкофазных исходных веществ вводится в зону реакции 28 через входные отверстия для подачи исходных веществ 32a,b,c,d. Преимущественно газофазный поток окислителя вводится в зону реакции 28 через барботер для ввода окислителя 34, расположенный в нижней части зоны реакции 28. Поток жидкофазных исходных веществ и газофазный поток окислителя совместно образуют многофазную реакционную среду 36 внутри зоны реакции 28. Многофазная реакционная среда 36 включает жидкую фазу и газовую фазу. Более предпочтительно, многофазная реакционная среда 36 представляет собой трехфазную среду, которая содержит твердофазные, жидкофазные и газофазные компоненты. Твердофазный компонент реакционной среды 36 преимущественно осаждается в зоне реакции 28 в результате реакции окисления, которая протекает в жидкой фазе реакционной среды 36. Барботажная колонна реакторного типа 20 имеет выходное отверстие для выгрузки суспензии 38, которое расположено вблизи нижней части зоны реакции 28, и отверстие для выхода газа 40, которое расположено вблизи верхней части зоны разделения 30. Поток суспензии, содержащий жидкофазные и твердофазные компоненты реакционной среды 36, выводится из зоны реакции 28 через отверстие для выгрузки суспензии 38, в то время как преимущественно газообразный поток отходящих газов выводится из разделительной зоны 30 через выходное отверстие для газа 40.
Поток жидкофазных исходных веществ, который подается в барботажную колонну реакторного типа 20 через входные отверстия для подачи исходных веществ 32a,b,c,d, преимущественно содержит способное окисляться соединение, растворитель и каталитическую систему.
Способное окисляться соединение, которое присутствует в потоке жидкофазных исходных веществ, преимущественно содержит, по крайней мере, одну углеводородную группу. Более предпочтительно, способное окисляться соединение является ароматическим соединением. Еще более предпочтительно, способное окисляться соединение является ароматическим соединением, которое содержит в качестве заместителя, по крайней мере, одну присоединенную углеводородную группу, или, по крайней мере, одну присоединенную замещенную углеводородную группу, или, по крайней мере, один присоединенный гетероатом, или, по крайней мере, одну присоединенную карбоксильную функциональную группу (-СООН). Еще более предпочтительно, способное окисляться соединение является ароматическим соединением, которое содержит, по крайней мере, одну присоединенную углеводородную группу, или, по крайней мере, одну присоединенную замещенную углеводородную группу, при этом каждая присоединенная группа содержит от 1 до 5 атомов углерода. Наконец, еще более предпочтительно, способное окисляться соединение является ароматическим соединением, которое содержит точно две присоединенные группы, при этом каждая присоединенная группа содержит точно один атом углерода и включает метильные группы и/или замещенные метильные группы и/или не более чем одну карбоксильную группу. Еще более предпочтительно, способное окисляться соединение представляет собой пара-ксилол, мета-ксилол, пара-толуальдегид, мета-толуальдегид, пара-толуиловую кислоту, мета-толуиловую кислоту и/или ацетальдегид. Наиболее предпочтительно, способное окисляться соединение представляет собой пара-ксилол.
“Углеводородная группа” в данном описании означает, по крайней мере, один атом углерода, который соединен с атомами водорода или с другими атомами углерода. “Замещенная углеводородная группа” в данном описании означает, по крайней мере, один атом углерода, который соединен, по крайней мере, с одним гетероатомом и, по крайней мере, одним атомом водорода. “Гетероатомы” в данном описании означают все атомы, отличные от атомов углерода и атомов водорода. Ароматические соединения в данном описании включают ароматический цикл, который содержит, по крайней мере, 6 атомов углерода и, еще более предпочтительно, содержит в цикле лишь атомы углерода. Подходящие примеры подобных ароматических циклов включают, однако этим не ограничиваясь, бензол, бифенил, терфенил, нафталин и другие углеродсодержащие конденсированные ароматические циклы.
Подходящие примеры способного окисляться соединения включают алифатические углеводороды (в частности, алканы, разветвленные алканы, циклические алканы, алифатические алкены, разветвленные алкены и циклические алкены); алифатические альдегиды (в частности, ацетальдегид, пропионовый альдегид, изомасляный альдегид и н.-масляный альдегид); алифатические спирты (в частности, этанол, изопропанол, н.-пропанол, н.-бутанол и изобутанол); алифатические кетоны (в частности, диметилкетон, этилметилкетон, диэтилкетон и изопропилметилкетон); алифатические сложные эфиры (в частности, метилформиат, метилацетат, этилацетат); алифатические пероксиды, надкислоты и гидропероксиды (в частности, гидропероксид трет-бутила, надуксусную кислоту и гидропероксид ди-трет-бутила); алифатические соединения с группами, которые представляют собой комбинацию вышеприведенных алифатических соединений плюс другие гетероатомы (в частности, алифатические соединения, включающие один или несколько молекулярных сегментов углеводородов, альдегидов, спиртов, кетонов, сложных эфиров, пероксидов, надкислот и/или гидропероксидов в сочетании с натрием, бромом, кобальтом, марганцем и цирконием); различные бензольные циклы, нафталиновые циклы, бифенилы, терфенилы и другие ароматические группы, содержащие одну или несколько присоединенных углеводородных групп (в частности, толуол, этилбензол, изопропилбензол, н.-пропилбензол, неопентилбензол, пара-ксилол, мета-ксилол, орто-ксилол, все изомеры триметилбензола, все изомеры тетраметилбензола, пентаметилбензол, гексаметилбензол, все изомеры этилметилбензола, все изомеры диэтилбензола, все изомеры этилдиметилбензола, все изомеры диметилнафталина, все изомеры этилметилнафталина, все изомеры диэтилнафталина, все изомеры диметилбифенила, все изомеры этилметилбифенила, все изомеры диэтилбифенила, стильбен, а также стильбен с одной или несколькими присоединенными углеводородными группами, флуорен, а также флуорен с одной или несколькими присоединенными углеводородными группами, антрацен, а также антрацен с одной или несколькими присоединенными углеводородными группами и дифенилэтан, а также дифенилэтан с одной или несколькими присоединенными углеводородными группами); различные бензольные циклы, нафталиновые циклы, бифенилы, терфенилы и другие ароматические группы, содержащие одну или несколько присоединенных углеводородных групп и/или один или несколько присоединенных гетероатомов, которые могут быть соединены с другими атомами или группами атомов (в частности, фенол, все изомеры метилфенолов, все изомеры диметилфенолов, все изомеры нафтолов, бензилметиловый эфир, все изомеры бромфенолов, бромбензол, все изомеры бромтолуолов, включая альфа-бромтолуол, дибромбензол, нафталенат кобальта, и все изомеры бромбифенилов); различные бензольные циклы, нафталиновые циклы, бифенилы, терфенилы и другие ароматические группы, содержащие одну или несколько присоединенных углеводородных групп и/или один или несколько присоединенных гетероатомов, и/или одну или несколько присоединенных замещенных углеводородных групп (в частности, бензальдегид, все изомеры бромбензальдегидов, все изомеры бромсодержащих толуиловых альдегидов, включая изомеры альфа-бромтолуиловых альдегидов, все изомеры гидроксибензальдегидов, все изомеры бромгидроксибензальдегидов, все изомеры
бензолдикарбоксальдегидов, все изомеры
бензолтрикарбоксальдегидов, пара-толуиловый альдегид,
мета-толуиловый альдегид, орто-толуиловый альдегид, все изомеры толуолдикарбоксальдегидов, все изомеры
толуолтрикарбоксальдегидов, все изомеры
толуолтетракарбоксальдегидов, все изомеры
диметилбензолдикарбоксальдегидов, все изомеры
диметилбензолтрикарбоксальдегидов, все изомеры
диметилбензолтетракарбоксальдегидов, все изомеры
триметилбензолтрикарбоксальдегидов, все изомеры этилтолуиловых альдегидов, все изомеры триметилбензолдикарбоксальдегидов, тетраметилбензолдикарбоксальдегид, гидроксиметилбензол, все изомеры гидроксиметилтолуолов, все изомеры гидроксиметилбромтолуолов, все изомеры гидроксиметилтолуиловых альдегидов, все изомеры гидроксиметилбромтолуиловых альдегидов, гидропероксид бензила, гидропероксид бензоила, все изомеры толилметилгидроксипероксидов и все изомеры метилфенолметилгидропероксидов); различные бензольные циклы, нафталиновые циклы, бифенилы, терфенилы и другие ароматические группы, содержащие одну или несколько присоединенных избранных углеводородных групп, при этом избранные группы означают углеводородные группы, и/или присоединенных гетероатомов, и/или замещенных углеводородных групп и/или групп карбоновых кислот, и/или групп надкислот (в частности, бензойной кислоты, пара-толуиловой кислоты, мета-толуиловой кислоты, орто-толуиловой кислоты, всех изомеров этилбензойных кислот, всех изомеров пропилбензойных кислот, всех изомеров бутилбензойных кислот, всех изомеров пентилбензойных кислот, всех изомеров диметилбензойных кислот, всех изомеры этилметилбензойных кислот, всех изомеров триметилбензойных кислот, всех изомеров тетраметилбензойных кислот, пентаметилбензойной кислоты, всех изомеров диэтилбензойных кислот, всех изомеров бензолдикарбоновых кислот, всех изомеров бензолтрикарбоновых кислот, всех изомеров метилбензолдикарбоновых кислот, всех изомеров диметилбензолдикарбоновых кислот, всех изомеров метилбензолтрикарбоновых кислот, всех изомеров бромбензойных кислот, всех изомеров дибромбензойных кислот, всех изомеров бромтолуиловых кислот, включая альфа-бромтолуиловые кислоты, толилуксусной кислоты, всех изомеров гидроксибензойных кислот, всех изомеров гидроксиметилбензойных кислот, всех изомеров гидрокситолуиловых кислот, всех изомеров гидроксиметилтолуиловых кислот, всех изомеров гидроксиметилбензолдикарбоновых кислот, всех изомеров гидроксибромбензойных кислот, всех изомеров гидроксибромтолуиловых кислот, всех изомеров гидроксиметилбромбензойных кислот, всех изомеров карбоксибензальдегидов, всех изомеров дикарбоксибензальдегидов, надбензойной кислоты, всех изомеров гидропероксиметилбензойных кислот, всех изомеров гидропероксиметилгидроксибензойных кислот, всех изомеров гидропероксикарбонилбензойных кислот, всех изомеров гидропероксикарбонилтолуолов, всех изомеров метилбифенилкарбоновых кислот, всех изомеров диметилбифенилкарбоновых кислот, всех изомеров метилбифенилдикарбоновых кислот, всех изомеров бифенилтрикарбоновых кислот, всех изомеров стильбена с одной или несколькими присоединенными избранными группами, всех изомеров флуоренона с одной или несколькими присоединенными избранными группами, всех изомеров нафталина с одной или несколькими присоединенными избранными группами, бензила, всех изомеров бензила с одной или несколькими присоединенными избранными группами, бензофенона, всех изомеров бензофенона с одной или несколькими присоединенными избранными группами, антрахинона, всех изомеров антрахинона с одной или несколькими присоединенными избранными группами, всех изомеров дифенилэтана с одной или несколькими присоединенными избранными группами, бензокумарина, всех изомеров бензокумарина с одной или несколькими присоединенными избранными группами).
Если способное окисляться соединение, содержащееся в жидкофазном потоке исходных веществ, является нормально твердым соединением (т.е. представляет собой твердое вещество при стандартной температуре и давлении), то предпочтительно способное окисляться соединение при введении в зону реакции 28 практически полностью растворяют в растворителе. Температура кипения способного окисляться соединения при атмосферном давлении предпочтительно должна составлять, по меньшей мере, приблизительно 50°С. Более предпочтительно, температура кипения способного окисляться соединения составляет от приблизительно 80 до приблизительно 400°С и, наиболее предпочтительно, составляет от 125 до 155°С. Количество способного окисляться соединения в жидкофазном исходном веществе предпочтительно составляет от приблизительно 2 до приблизительно 40% мас., более предпочтительно составляет от приблизительно 4 до приблизительно 20% мас. и, наиболее предпочтительно, составляет от 6 до 15% мас.
Следует также отметить, что способное окисляться соединение, содержащееся в жидкофазном потоке исходных веществ, может включать комбинацию двух или большего количества различных способных окисляться соединений. Указанные два или больше различных химических соединений могут быть смешаны друг с другом в виде жидкофазного потока исходных веществ или же могут подаваться раздельно в виде нескольких потоков исходных веществ. Например, способное окисляться соединение, содержащее пара-ксилол, мета-ксилол, пара-толуиловый альдегид, пара-толуиловую кислоту и ацетальдегид, может подаваться в реактор через один вход или через несколько раздельных входов.
Растворитель, который присутствует в жидкофазном потоке исходных веществ, преимущественно, содержит кислотный компонент и водный компонент. Растворитель, предпочтительно, присутствует в жидкофазном потоке исходных веществ в концентрации от приблизительно 60 до приблизительно 98% мас., более предпочтительно, от приблизительно 80 до приблизительно 96% мас. и, наиболее предпочтительно, от 85 до 94% мас. Кислотный компонент растворителя преимущественно представляет собой в основном низкомолекулярную органическую монокарбоновую кислоту, содержащую 1-6 атомов углерода, и более предпочтительно, содержит 2 атома углерода. Наиболее предпочтительным компонентом растворителя в основном является уксусная кислота. Кислотный компонент, предпочтительно, составляет, по крайней мере, приблизительно 75% мас. от общего количества растворителя, более предпочтительно, составляет, по крайней мере, приблизительно 80% мас. от общего количества растворителя, и наиболее предпочтительно, составляет от 85 до 98% мас. от общего количества растворителя, а остальное количество в основном составляет вода. Растворитель, который вводят в барботажную колонну реакторного типа 20, может включать небольшие количества примесей, таких как, например, пара-толуальдегид, терефтальдегид, 4-карбоксибензальдегид (4-СВА), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или суспендированные частицы. Общее количество примесей в растворителе, который вводят в барботажную колонну реакторного типа 20, преимущественно, не превышает приблизительно 3% мас.
Каталитическая система, которая содержится в жидкофазном потоке исходных веществ, преимущественно, представляет собой гомогенную жидкофазную каталитическую систему, которая может ускорять окисление (в том числе частичное окисление) способного окисляться соединения. Более предпочтительно, каталитическая система содержит, по крайней мере, один переходный металл переменной валентности. Еще более предпочтительно, переходный металл переменной валентности представляет собой кобальт. Еще более предпочтительно, каталитическая система включает кобальт и бром. Наиболее предпочтительно, каталитическая система включает кобальт, бром и марганец.
Если кобальт присутствует в каталитической системе, то количество кобальта, содержащегося в жидкофазном потоке исходных веществ, предпочтительно, должно быть таким, чтобы концентрация кобальта в жидкой фазе реакционной среды 36 поддерживалась от приблизительно 300 до приблизительно 6000 весовых частей на миллион (м.д. мас.), более предпочтительно, от приблизительно 700 до приблизительно 4200 м.д. мас. и, наиболее предпочтительно, от 1200 до 3000 м.д. мас. Если бром присутствует в каталитической системе, то количество брома, содержащегося в жидкофазном потоке исходных веществ, предпочтительно, должно быть таким, чтобы концентрация брома в жидкой фазе реакционной среды 36 поддерживалась от приблизительно 300 до приблизительно 5000 м.д. мас., более предпочтительно, от приблизительно 600 до приблизительно 4000 м.д. мас. и, наиболее предпочтительно, от 900 до 3000 м.д. мас. Если марганец присутствует в каталитической системе, то количество марганца, содержащегося в жидкофазном потоке исходных веществ, предпочтительно, должно быть таким, чтобы концентрация марганца в жидкой фазе реакционной среды 36 поддерживалась от приблизительно 20 до приблизительно 1000 м.д. мас., более предпочтительно, от приблизительно 40 до приблизительно 500 м.д. мас. и, наиболее предпочтительно, от 50 до 200 м.д. мас.
Приведенные выше концентрации кобальта, брома и/или марганца в жидкой фазе реакционной среды 36 выражены в усредненных по времени и по объему единицах. В данном описании термин “усредненный по времени” означает среднюю величину, по крайней мере, из 10 измерений, которые одинаковым образом проводятся в течение непрерывного периода, составляющего, по крайней мере, 100 сек. В данном описании термин “усредненный по объему” означает среднюю величину, по крайней мере, из 10 измерений, которые проводятся в однородном 3-мерном пространственном окружении внутри определенного объема.
Массовое отношение кобальта к брому (Co:Br) в каталитической системе, которая вводится в зону реакции 28, предпочтительно, составляет от приблизительно 0,25:1 до приблизительно 4:1, более предпочтительно, составляет от приблизительно 0,5:1 до приблизительно 3:1 и, наиболее предпочтительно, составляет от 0,75:1 до 2:1. Массовое отношение кобальта к марганцу (Co:Mn) в каталитической системе, которая вводится в зону реакции 28, предпочтительно, составляет от приблизительно 0,3:1 до приблизительно 40:1, более предпочтительно, составляет от приблизительно 5:1 до приблизительно 30:1 и, наиболее предпочтительно, составляет от 10:1 до 25:1.
Поток жидкофазных исходных веществ, который подается в барботажную колонну реакторного типа 20, может включать небольшие количества примесей, таких как, например, толуол, этилбензол, пара-толуальдегид, терефтальдегид, 4-карбоксибензальдегид (4-СВА), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или суспендированные частицы. Если для получения терефталевой кислоты используется барботажная колонна реакторного типа 20, то примесями считаются также мета-ксилол и орто-ксилол. Общее количество примесей в жидкофазном потоке исходных веществ, который подается в барботажную колонну реакторного типа 20, преимущественно, не превышает приблизительно 3% мас.
Несмотря на то, что фиг.1 поясняет вариант осуществления настоящего изобретения, в котором способное окисляться соединение, растворитель и каталитическая система смешиваются вместе и подаются в барботажную колонну реакторного типа 20 в виде одного потока, в альтернативном варианте осуществления настоящего изобретения способное окисляться соединение, растворитель и каталитическая система могут вводиться в барботажную колонну реакторного типа 20 раздельно. Например, можно подавать поток пара-ксилола в барботажную колонну реакторного типа 20 через вход, расположенный отдельно от входа(ов) для подачи растворителя и катализатора.
Газофазный поток окислителя, который подается в барботажную колонну реакторного типа 20 через барботер для ввода окислителя 34, преимущественно содержит молекулярный кислород (О2). Поток окислителя, предпочтительно, содержит от приблизительно 5 до приблизительно 40% мол. молекулярного кислорода, более предпочтительно, содержит от приблизительно 15 до приблизительно 30% мол. молекулярного кислорода и, наиболее предпочтительно, содержит от приблизительно 18 до приблизительно 24% мол. молекулярного кислорода. Остальное количество в потоке окислителя в основном составляют газ или газы, такие как азот, которые инертны в реакции окисления. Более предпочтительно, поток окислителя состоит в основном из молекулярного кислорода и азота. Наиболее предпочтительно, поток окислителя представляет собой сухой воздух, который содержит приблизительно 21% мол. молекулярного кислорода и от приблизительно 78 до приблизительно 81% мол. азота. В альтернативном варианте осуществления настоящего изобретения поток окислителя может представлять собой практически чистый кислород.
Возвращаясь вновь к фиг.1, следует отметить, что барботажная колонна реакторного типа 20 предпочтительно снабжена распределителем флегмы 42, который расположен над верхней поверхностью 44 реакционной среды 36. Распределитель флегмы 42 предназначен для введения капель преимущественно жидкофазного потока флегмы в зону разделения 30 с помощью любого известного из области техники устройства, позволяющего генерировать капли. Более предпочтительно, распределитель флегмы 42 формирует поток капель распыляемой жидкости, подаваемый вниз по направлению к верхней поверхности 44 реакционной среды 36. Указанный направленный вниз поток капель распыляемой жидкости предпочтительно оказывает воздействие (т.е. вовлекает и охватывает), по крайней мере, на 50% от максимальной площади горизонтального сечения зоны разделения 30. Более предпочтительно, капли распыляемой жидкости оказывают действие, по крайней мере, на 75% от максимальной площади горизонтального сечения зоны разделения 30. Наиболее предпочтительно, капли распыляемой жидкости оказывают действие, по крайней мере, на 90% от максимальной площади горизонтального сечения зоны разделения 30. Указанный направленный вниз поток аэрозоля флегмы может помочь предотвратить пенообразование на поверхности или над верхней частью поверхностью 44 реакционной среды 36, а также может оказать помощь при отделении любой жидкости или капелек суспензии, которые увлекаются движущимся вверх газом, направляющимся к отверстию для вывода газа 40. Кроме того, флегма может служить для снижения количества частиц и потенциально выпадающих в осадок соединений (в частности, растворенной бензойной кислоты, пара-толуиловой кислоты, 4-СВА, терефталевой кислоты и солей металлического катализатора), которые содержатся в газах, отводимых из зоны разделения 30 через выходное отверстие 40. Кроме того, введение капель флегмы в зону разделения 30 может за счет дистиллирующего действия использоваться для регулирования состава газов, отводимых через отверстие для вывода газа 40.
Поток флегмы, который подается в барботажную колонну реакторного типа 20 через распределитель флегмы 42, преимущественно имеет тот же состав, что и компонент растворителя в жидкофазном потоке исходных веществ, который подается в барботажную колонну реакторного типа 20 через входы для подачи исходных веществ 32a, b, c, d. Таким образом, поток флегмы, предпочтительно, содержит кислотный компонент и воду. Кислотный компонент потока флегмы, преимущественно, представляет собой низкомолекулярную органическую монокарбоновую кислоту, содержащую 1-6 атомов углерода, и более предпочтительно, содержит 2 атома углерода. Кислотным компонентом флегмы, наиболее предпочтительно, является уксусная кислота. Кислотный компонент, предпочтительно, составляет, по крайней мере, приблизительно 75% мас. от общего количества потока флегмы, более предпочтительно, составляет, по крайней мере, приблизительно 80% мас. от общего количества потока флегмы, и наиболее предпочтительно, составляет от 85 до 98% мас. от общего количества потока флегмы, а остальное количество составляет вода. Поскольку поток флегмы имеет практически тот же самый состав, что и растворитель в жидкофазном потоке исходных веществ, то когда в настоящем описании говорится об “общем количестве растворителя”, который подается в реактор, то в указанное “общее количество растворителя” следует включать как поток флегмы, так и долю растворителя в потоке исходных веществ.
В процессе жидкофазного окисления в барботажной колонне реакторного типа 20, предпочтительно, потоки исходных веществ, окислителя и поток флегмы практически непрерывно вводятся в зону реакции 28, в то время как потоки отходящих газов и суспензии практически непрерывно выводятся из зоны реакции 28. В данном описании термин “практически непрерывно” означает прерываемый менее чем на 10 мин период времени, длительность которого составляет, по крайней мере, 10 час. В процессе окисления способное окисляться соединение (в частности, пара-ксилол), предпочтительно, практически непрерывно вводится в зону реакции 28 в количестве, по крайней мере, приблизительно 8000 кг/час, более предпочтительно от приблизительно 13000 кг/час до приблизительно 80000 кг/час, еще более предпочтительно от приблизительно 18000 кг/час до приблизительно 50000 кг/час и, наиболее предпочтительно, от 22000 кг/час до 30000 кг/час. Несмотря на то, что скорости подачи поступающих в реакцию потоков исходных веществ, окислителя и потока флегмы, предпочтительно, должны быть практически стабильными, в данном случае следует отметить, что в одном из вариантов осуществления настоящего изобретения предполагается использование пульсирующей подачи исходных веществ, окислителя и/или флегмы с целью улучшить процессы смешивания и массопереноса. В том случае, когда подача исходных веществ, окислителя и/или потока флегмы проводится в пульсирующем режиме, скорости потоков, предпочтительно, варьируют от приблизительно 0 до приблизительно 500% от указанных в данном описании скоростей потоков при стационарном режиме работы, более предпочтительно, от приблизительно 30 до приблизительно 200% от указанных в данном описании скоростей потоков при стационарном режиме работы и, наиболее предпочтительно, от 80 до 120% от указанных в данном описании скоростей потоков при стационарном режиме работы.
Средняя пространственно-временная скорость реакции (STR) в барботажной колонне реакторного типа 20 определяется как масса исходного способного окисляться соединения на единицу объема реакционной среды 36 в единицу времени (в частности определяется количеством килограммов исходного пара-ксилола на кубический метр в час). При обычном подходе, прежде чем проводить расчет значения STR, необходимо, как правило, вычесть количество способного окисляться соединения, которое не превратилось в продукт, из количества способного окисляться соединения, которое содержится в потоке исходных веществ. Однако степени конверсии и выходы продукта, как правило, являются высокими для многих предпочтительных в данном изобретении способных окисляться соединений (в частности, пара-ксилола), а потому в данном описании удобно определять приведенный термин, как указано выше. Учитывая среди прочих капитальные затраты и текущее незавершенное производство, обычно предпочтительно проводить реакции с большими значениями STR. Тем не менее, проведение реакции с использованием все больших значений STR может оказать влияние на качество или выход продукта при частичном окислении. Барботажная колонна реакторного типа 20 особенно пригодна в том случае, когда STR способного окисляться соединения (в частности, пара-ксилола) составляет от приблизительно 25 кг/м3·час до приблизительно 400 кг/м3·час, более предпочтительно, от приблизительно 30 кг/м3·час до приблизительно 250 кг/м3·час, еще более предпочтительно, от приблизительно 35 кг/м3·час до приблизительно 150 кг/м3·час и, наиболее предпочтительно, от 40 кг/м3·час до 100 кг/м3·час.
STR для кислорода в барботажной колонне реакторного типа 20 определяется как масса молекулярного кислорода, потребляемая на единицу объема реакционной среды 36 в единицу времени (в частности, определяется количеством килограммов молекулярного кислорода, которое потребляется на один кубический метр в час). Учитывая, среди прочих, капитальные затраты и вызванное окислением потребление растворителя, реакцию, предпочтительно, следует проводить с большими значениями STR для кислорода. Тем не менее, проведение реакции с использованием все более возрастающих значений STR для кислорода в конце концов снижает качество или выход продукта при частичном окислении. Если не связывать себя какой-либо теорией, то можно предположить, что указанное явление, вероятно, связано со скоростью переноса молекулярного кислорода из газовой фазы в жидкую фазу на границе поверхности раздела и оттуда в объем жидкости. Слишком большое значение STR для кислорода, возможно, приводит к слишком низкому содержанию растворенного кислорода в объеме жидкой фазы реакционной среды.
Итоговое среднее значение STR для кислорода в данном описании определяется как масса всего количества кислорода, поглощенного во всем объеме реакционной среды 36 в единицу времени (в частности, оно определяется количеством килограммов молекулярного кислорода, который потребляется на один кубический метр в час). Барботажная колонна реакторного типа 20 особенно пригодна в том случае, когда итоговое среднее значение STR для кислорода составляет от приблизительно 25 кг/м3·час до приблизительно 400 кг/м3·час, более предпочтительно, от приблизительно 30 кг/м3·час до приблизительно 250 кг/м3·час, еще более предпочтительно, от приблизительно 35 кг/м3·час до приблизительно 150 кг/м3·час и, наиболее предпочтительно, от 40 кг/м3·час до 100 кг/м3·час.
В процессе окисления в барботажной колонне реакторного типа 20 отношение скорости массового расхода для общего количества растворителя (как в потоке исходных соединений, так и в потоке флегмы) к массовой скорости расхода способного окисляться соединения, поступающего в зону реакции 28, предпочтительно, должно поддерживаться от приблизительно 2:1 до приблизительно 50:1, более предпочтительно, от приблизительно 5:1 до приблизительно 40:1 и, наиболее предпочтительно, от 7,5:1 до 25:1. Отношение массовой скорости расхода растворителя, который поступает в виде части потока исходных веществ, к массовой скорости расхода растворителя, который поступает в виде части потока флегмы, поддерживается от приблизительно 0,5:1 до отсутствия какого бы то ни было потока флегмы, более предпочтительно, от приблизительно 0,5:1 до приблизительно 4:1, еще более предпочтительно, от приблизительно 1:1 до приблизительно 2:1 и, наиболее предпочтительно, от 1,25:1 до 1,5:1.
В процессе жидкофазного окисления в барботажной колонне реакторного типа 20 поток окислителя, предпочтительно, должен вводиться в барботажную колонну реакторного типа 20 в количестве, которое обеспечивает количество молекулярного кислорода, несколько превышающее количество кислорода, которое требуется по стехиометрии. Избыточное количество молекулярного кислорода, необходимое для получения наилучших результатов для конкретного способного окисляться соединения, влияет на общую экономику жидкофазного окисления. В процессе жидкофазного окисления в барботажной колонне реакторного типа 20 отношение массовой скорости потока окислителя к массовой скорости потока способного окисляться органического соединения (в частности, пара-ксилола), поступающего в реактор 20, предпочтительно, поддерживается от приблизительно 0,5:1 до приблизительно 20:1, более предпочтительно, от приблизительно 1:1 до приблизительно 10:1 и, наиболее предпочтительно, от 2:1 до 6:1.
Если вновь обратиться к фиг.1, то потоки исходных веществ, окислителя и флегмы, которые подаются в барботажную колонну реакторного типа 20, совместно образуют, по крайней мере, часть многофазной реакционной среды 36. Реакционная среда 36, преимущественно, представляет собой трехфазную среду, которая включает твердую фазу, жидкую фазу и газовую фазу. Как указано выше, окисление способного окисляться соединения (в частности, пара-ксилола) в основном протекает в жидкой фазе реакционной среды 36. Таким образом, жидкая фаза реакционной среды 36 включает растворенный кислород и способное окисляться соединение. Экзотермический характер реакции окисления, которая протекает в барботажной колонне реакторного типа 20, вызывает кипение/испарение части растворителя (в частности, уксусной кислоты и воды), который вводится через отверстия для подачи исходных веществ 32a, b, c, d. Таким образом, газовая фаза реакционной среды 36 в реакторе 20 образована в основном испарившимся растворителем и не растворившейся, не прореагировавшей порцией исходного окислителя. В некоторых известных из области техники реакторах для проведения окисления используют теплообменные трубки/ребра для нагревания или охлаждения реакционной среды. Однако подобная конструкция теплообменника может оказаться нежелательной для реактора по настоящему изобретению и приведенного в данном описании способа. Таким образом, барботажная колонна реакторного типа 20 предпочтительно практически не имеет поверхностей, которые контактируют с реакционной средой 36, и выделяет усредненный по времени тепловой поток больше чем 30000 Вт/м2.
Концентрация растворенного кислорода в жидкой фазе реакционной среды 36 находится в динамическом равновесии между скоростью массопереноса из газовой фазы и скоростью расходования в процессе реакции внутри жидкой фазы (т.е. она не определяется просто парциальным давлением молекулярного кислорода в поступающей газовой фазе, хотя парциально давление является одним из факторов, влияющих на скорость поступления растворенного кислорода, и оно действительно оказывает влияние на верхнюю граничную концентрацию растворенного кислорода). Количество растворенного кислорода имеет локальные вариации, при этом оно выше вблизи границ раздела пузырьков. В целом, количество растворенного кислорода зависит от баланса подачи и факторов, оказывающих влияние на потребление, в различных частях реакционной среды 36. В зависимости от времени количество растворенного кислорода зависит от однородности смешивания газа и жидкости по отношению к скоростям потребления в процессе химической реакции. Чтобы добиться согласованной подачи и расходования растворенного кислорода в жидкой фазе реакционной среды 36, усредненная во времени и по объему концентрация кислорода в жидкой фазе реакционной среды 36, предпочтительно, должна поддерживаться на уровне больше чем приблизительно 1 м.д. мольных, более предпочтительно, от приблизительно 4 до приблизительно 1000 м.д. мол., еще более предпочтительно, от приблизительно 8 до приблизительно 500 м.д. мол., и наиболее предпочтительно, от 12 до 120 м.д. мол.
Реакция жидкофазного окисления, которая проводится в барботажной колонне реакторного типа 20, преимущественно, представляет собой реакцию, которая сопровождается осаждением образовавшихся твердых веществ. Более предпочтительно реакция жидкофазного окисления, которая проводится в барботажной колонне реакторного типа 20, приводит к тому, что, по меньшей мере, приблизительно 10% мас. способного окисляться соединения (в частности, пара-ксилола), которое поступает в зону реакции 28, образует твердое соединение (в частности, частицы сырой терефталевой кислоты) в реакционной среде 36. Еще более предпочтительно, реакция жидкофазного окисления приводит к тому, что, по меньшей мере, приблизительно 50% мас. способного окисляться соединения образует твердое соединение в реакционной среде 36. Наиболее предпочтительно, реакция жидкофазного окисления приводит к тому, что, по меньшей мере, приблизительно 90% мас. способного окисляться соединения образует твердое соединение в реакционной среде 36. Общее количество твердых веществ в реакционной среде 36, предпочтительно, должно быть больше чем приблизительно 3% мас. в пересчете на усредненное по времени и объему значение. Более предпочтительно, общее количество твердых веществ в реакционной среде 36 поддерживается от приблизительно 5 до приблизительно 40% мас., еще более предпочтительно, от приблизительно 10 до приблизительно 35% мас. и, наиболее предпочтительно, от 15 до 30% мас. Значительная часть продукта реакции окисления (в частности, терефталевой кислоты), образующегося в барботажной колонне реакторного типа 20, преимущественно находится в реакционной среде 36 в виде твердых веществ, в отличие от других соединений, которые остаются растворенными в жидкой фазе реакционной среды 36. Количество твердофазного продукта реакции окисления, присутствующего в реакционной среде 36, предпочтительно, составляет, по крайней мере, 25% мас. от общей массы продукта реакции окисления (в твердой и жидкой фазе), содержащегося в реакционной среде 36, более предпочтительно, составляет, по крайней мере, 75% мас. от общей массы продукта реакции окисления, содержащегося в реакционной среде 36, и, наиболее предпочтительно, составляет, по крайней мере, 95% мас. от общей массы продукта реакции окисления, содержащегося в реакционной среде 36. Приведенные выше численные границы для количества твердых веществ в реакционной среде 36 относятся к практически стационарному режиму работы барботажной колонны реакторного типа 20 в течение практически непрерывного периода времени и не относятся к стадиям пуска, остановки или к неоптимальному режиму работы барботажной колонны реакторного типа 20. Количество твердых веществ в реакционной среде 36 определяется гравиметрическим методом. При проведении анализа гравиметрическим методом отдельная часть суспензии выделяется из реакционной среды и взвешивается. В условиях, при которых эффективно обеспечивается разделение всех твердых и жидких фаз внутри реакционной среды, свободная жидкость отделяется от порции твердых веществ седиментацией или фильтрацией таким образом, чтобы избежать потерь выпавших в осадок твердых веществ, при этом в порции твердых веществ остается меньше чем 10% от исходной массы жидкости. Оставшуюся в твердых веществах жидкость выпаривают досуха таким образом, чтобы не происходила сублимация твердых веществ. Оставшуюся порцию твердых веществ взвешивают. Отношение массы порции твердых веществ к массе исходной порции суспензии представляет собой величину фракцию твердых веществ, которая обычно выражается в процентах.
Сопровождающаяся осаждением твердых веществ реакция, которая протекает в барботажной колонне реакторного типа 20, может вызвать засорение (т.е. образование осадка) поверхности некоторых жестких конструкций, контактирующих с реакционной средой 36. Так, в одном из вариантов осуществления настоящего изобретения барботажная колонна реакторного типа 20, предпочтительно, практически не имеет в зоне реакции 28 внутренних конструктивных элементов для осуществления теплообмена, перемешивания или отклонения потока, поскольку указанные конструктивные элементы будут подвержены засорению. Если в зоне реакции 28 присутствуют внутренние конструктивные элементы, то желательно избегать применения внутренних конструктивных элементов, значительную долю внешней поверхности которых составляет обращенная вверх плоская поверхность, поскольку подобные обращенные вверх плоские поверхности в значительной степени подвержены засорению. Таким образом, если в зоне реакции 28 присутствуют какие-либо внутренние конструкции, то предпочтительно, не более чем приблизительно 20% от общего количества обращенной вверх и подверженной воздействию внешней поверхности подобных внутренних конструкций должно представлять собой практически плоские поверхности, отклонение которых от горизонтали составляет меньше приблизительно 15°.
Если вернуться вновь к фиг.1, то физическая конфигурация барботажной колонны реакторного типа 20 способствует проведению оптимизированного процесса окисления способного окисляться соединения (в частности, пара-ксилола), который приводит к образованию минимального количества примесей. Удлиненная реакторная секция 24 внутри кожуха реактора 22, предпочтительно, включает практически цилиндрический корпус 46 и днище 48. Верхняя граница зоны реакции 28 очерчивается горизонтальной плоскостью 50, которая пересекает верх цилиндрического корпуса 46. Нижняя граница 52 зоны реакции 28 очерчивается наиболее низко расположенной внутренней поверхностью днища 48. Как правило, нижняя граница 52 зоны реакции 28 располагается непосредственно рядом с отверстием для выгрузки суспензии 38. Таким образом, удлиненная зона реакции 28, очерченная внутри барботажной колонны реакторного типа 20, имеет максимальную длину “L”, измеренную от верхней границы 50 до нижней границы 52 зоны реакции 28 вдоль длинной оси цилиндрического корпуса 46. Длина L зоны реакции 28, предпочтительно, составляет от приблизительно 10 до приблизительно 100 м, более предпочтительно, составляет от приблизительно 20 до приблизительно 75 м, и наиболее предпочтительно, составляет от 25 до 50 м. Зона реакции 28 имеет максимальный диаметр (ширину) D, которая обычно равна максимальному внутреннему диаметру цилиндрического корпуса 46. Максимальный диаметр D зоны реакции 28, предпочтительно, составляет от приблизительно 1 до приблизительно 12 м, более предпочтительно, составляет от приблизительно 2 до приблизительно 10 м, еще более предпочтительно, составляет от приблизительно 3,1 до приблизительно 9 м и, наиболее предпочтительно, составляет от 4 до 8 м. В предпочтительном варианте осуществления настоящего изобретения зона реакции 28 имеет отношение длины к диаметру L:D от приблизительно 6:1 до приблизительно 30:1. Еще более предпочтительно, зона реакции 28 имеет отношение L:D от приблизительно 8:1 до приблизительно 20:1. Наиболее предпочтительно, зона реакции 28 имеет отношение L:D от 9:1 до 15:1.
Как указано выше, в зоне реакции 28 барботажной колонны реакторного типа 20 формируется многофазная реакционная среда 36. Реакционная среда 36 имеет нижнюю границу, совпадающую с нижней границей 52 зоны реакции 28, и верхнюю границу, которая располагается на верхней поверхности 44. Верхняя поверхность 44 реакционной среды 36 очерчивается горизонтальной плоскостью, которая отсекает часть верхнего уровня зоны реакции 28, где содержимое зоны реакции 28 переходит из непрерывного газофазного состояния в непрерывное жидкофазное состояние. Верхняя поверхность 44, предпочтительно, располагается на той отметке высоты, где усредненная по времени способность удерживать газ в тонком горизонтальном срезе содержимого зоны реакции 28 составляет 0,9.
Реакционная среда 36 имеет максимальную высоту H, измеренную между ее верхним и нижним концами. Максимальная ширина W реакционной среды 36 обычно равна максимальному диаметру D цилиндрического корпуса 46. При проведении жидкофазного окисления в барботажной колонне реакторного типа 20 значение Н, предпочтительно, поддерживается на уровне от приблизительно 60 до приблизительно 120% от величины L, более предпочтительно, на уровне от приблизительно 80 до приблизительно 110% от величины L и, наиболее предпочтительно, на уровне от 85 до 100% от величины L. В предпочтительном варианте осуществления настоящего изобретения реакционная среда 36 имеет отношение высоты к ширине H:W, превышающее приблизительно 3:1. Более предпочтительно, реакционная среда 36 имеет отношение H:W от приблизительно 7:1 до приблизительно 25:1. Еще более предпочтительно, реакционная среда 36 имеет отношение H:W от приблизительно 8:1 до приблизительно 20:1. Наиболее предпочтительно, реакционная среда 36 имеет отношение H:W от 9:1 до 15:1. В одном из вариантов осуществления настоящего изобретения L=H и D=W, так что различные размеры или отношения, приведенные в данном описании для L и D, применимы также и для H и W, и наоборот.
Относительно большие значения отношений L:D и H:W, приведенные в соответствии с одним из вариантом осуществления настоящего изобретения, могут обеспечить системе по настоящему изобретению несколько важных преимуществ. Как подробно рассматривается ниже, было обнаружено, что более высокие значения отношений L:D и H:W, а также некоторые другие рассматриваемые ниже особенности, способствуют формированию благоприятных градиентов концентрации молекулярного кислорода и/или способного окисляться соединения (в частности, пара-ксилола) по высоте в реакционной среде 36. Вопреки обычной практике, согласно которой предпочтение следует отдать хорошо перемешанной реакционной среде, концентрации реагентов в которой распределены более или менее равномерно, было обнаружено, что вертикальное ступенчатое распределение концентраций кислорода и/или способного окисляться соединения способствует протеканию более эффективной и экономичной реакции окисления. Минимизация концентраций кислорода и способного окисляться соединения вблизи верхней части реакционной среды 36 может помочь избежать потерь не прореагировавшего кислорода и не прореагировавшего способного окисляться соединения через верхнее отверстие для отвода газа 40. Однако если концентрации способного окисляться соединения и не прореагировавшего кислорода внутри реакционной среды 36 являются низкими, то скорость и/или селективность окисления снижаются. Таким образом, концентрации молекулярного кислорода и/или способного окисляться соединения, предпочтительно, должны быть выше вблизи нижней границы реакционной среды 36, чем вблизи верхней части реакционной среды 36.
Кроме того, большие значения отношений L:D и H:W приводят к тому, что в нижней части реакционной среды 36 устанавливается значительно большее давление, чем давление в верхней части реакционной среды 36. Указанный градиент давления по высоте является результатом высоты и плотности реакционной среды 36. Одним из преимуществ подобного градиента давления по высоте является то, что повышенное давление внизу реактора обеспечивает лучшую растворимость кислорода и лучший массоперенос, в отличие от растворимости кислорода и массопереноса, которые установились бы при значениях температуры и давления, сравнимых со значениями в верхней части более мелких реакторов. В итоге реакцию окисления можно проводить при более низких температурах, чем температуры, которые потребовались бы в более мелком реакторе. В том случае, когда барботажная колонна реакторного типа 20 используется для частичного окисления пара-ксилола с образованием сырой терефталевой кислоты (СТА), возможность осуществлять процесс при более низких температурах реакции с теми же самыми или с более хорошими скоростями массопереноса кислорода обеспечивает ряд преимуществ. Например, низкая температура окисления пара-ксилола снижает количество растворителя, который выгорает в процессе реакции. Как подробнее рассматривается ниже, низкая температура окисления способствует также формированию мелких, обладающих большой площадью поверхности, слабо связанных друг с другом и легко растворяющихся частиц СТА, которые можно подвергнуть очистке с помощью более экономичных способов, по сравнению с большими, обладающими небольшой площадью поверхности и плотными частицами СТА, которые получаются в обычных процессах окисления, осуществляемых при высокой температуре.
В процессе окисления в реакторе 20 усредненная по времени и объему температура реакционной среды 36, предпочтительно, поддерживается от приблизительно 125 до приблизительно 200°С, более предпочтительно, от приблизительно 140 до приблизительно 180°С и, наиболее предпочтительно, от 150 до 170°С. Верхнее давление в реакционной среде 36 предпочтительно поддерживается от приблизительно 1 до приблизительно 20 бар избыточного давления (бар), более предпочтительно, от приблизительно 2 до приблизительно 12 бар и, наиболее предпочтительно, от 4 до 8 бар. Разница в давлении между верхней частью реакционной среды 36 и нижней частью реакционной среды 36, предпочтительно, составляет от приблизительно 0,4 бар до приблизительно 5 бар, более предпочтительно, разница в давлении составляет от приблизительно 0,7 бар до приблизительно 3 бар и, наиболее предпочтительно, разница в давлении составляет от 1 бар до 2 бар. Несмотря на то, что обычно давление в верхней части реакционной среды 36 предпочтительно поддерживается на относительно постоянном уровне, один из вариантов осуществления настоящего изобретения предполагает использование пульсирующего верхнего давления с целью обеспечения более тщательного смешивания и/или лучшего массопереноса в реакционной среде 36. В том случае, когда верхнее давление является пульсирующим, то величина пульсирующего давления, предпочтительно, составляет от приблизительно 60 до приблизительно 140% от приведенного выше значения давления при стационарных условиях, более предпочтительно, составляет от приблизительно 85 до приблизительно 115% от приведенного выше значения давления при стационарных условиях и, наиболее предпочтительно, составляет от 95 до 105% от приведенного выше значения давления при стационарных условиях.
Еще одно преимущество большого значения отношения L:D в зоне реакции 28 заключается в том, что оно может способствовать повышению средней приведенной скорости в реакционной среде 36. Термин “приведенная скорость” и “приведенная скорость газа” в данном описании применительно для реакционной среды 36 означает объемную скорость потока газовой фазы в реакционной среде 36 на определенной отметке высоты в реакторе, деленную на площадь горизонтального сечения реактора на этой отметке высоты. Повышенное значение приведенной скорости, которую обеспечивает большое значение отношения L:D в зоне реакции 28, может способствовать локальному смешиванию и повышению способности удерживать газ в реакционной среде 36. Усредненные по времени приведенные скорости в реакционной среде 36 на одной четверти высоты, на половине высоты и/или на трех четвертях высоты реакционной среды 36, предпочтительно, превышают приблизительно 0,3 м/сек, более предпочтительно, составляют от приблизительно 0,8 до приблизительно 5 м/сек, еще более предпочтительно, составляют от приблизительно 0,9 до приблизительно 4 м/сек и, наиболее предпочтительно, составляют от 1 до 3 м/сек.
Возвращаясь вновь к фиг.1, следует отметить, что разделительная секция 26 барботажной колонны реакторного типа 20 представляет собой просто расширенную часть кожуха реактора 22, которая располагается сразу же над реакторной секцией 24. Разделительная секция 26 снижает скорость восходящего потока газовой фазы в барботажной колонне реакторного типа 20 по мере того, как газовая фаза поднимается над верхней поверхностью 44 реакционной среды 36 и приближается к выходному отверстию для газа 40. Указанное снижение скорости обращенного вверх потока газовой фазы помогает удалить жидкости и/или твердые вещества, захватываемые восходящим потоком газовой фазы, и тем самым снижает нежелательную потерю определенных компонентов, содержащихся в жидкой фазе реакционной среды 36.
Разделительная секция 26 предпочтительно включает переходную стенку 54, обычно имеющую форму усеченного конуса, широкую боковую стенку 56, обычно имеющую цилиндрическую форму, и крышку 58. Узкий нижний конец переходной стенки 54 соединен с верхней частью цилиндрического корпуса 46 реакторной секции 24. Более широкий верхний конец переходной стенки 54 соединен с нижней частью широкой боковой стенки 56. Переходная стенка 54 простирается вверх и в стороны от ее узкого нижнего конца под углом, предпочтительно, составляющим от приблизительно 10 до приблизительно 70° от вертикали, более предпочтительно, составляющим от приблизительно 15 до приблизительно 50° от вертикали и, наиболее предпочтительно, составляющим от 15 до 45° от вертикали. Широкая боковая стенка 56 имеет максимальный диаметр X, который обычно превышает максимальный диаметр D реакторной секции 24, однако в том случае, когда верхняя часть реакторной секции 24 имеет меньший диаметр, чем общий максимальный диаметр реакторной секции 24, то тогда Х в действительности может быть меньше, чем D. В предпочтительном варианте осуществления настоящего изобретения отношение X:D диаметра широкой боковой стенки 56 к максимальному диаметру реакторной секции 24 составляет от приблизительно 0,8:1 до приблизительно 4:1 и, наиболее предпочтительно, составляет от 1:1 до 2:1. Крышка 58 соединена с верхней частью широкой боковой стенки 56. Крышка 58, предпочтительно, представляет собой элемент, обычно имеющий эллиптическую форму, в центре которого находится отверстие, позволяющее газу выходить из зоны разделения 30 через отверстие для вывода газа 40. В качестве альтернативы, крышка 58 может быть любой формы, включая коническую. Зона разделения 30 имеет максимальную высоту Y, которую измеряют от верхней части 50 зоны реакции 28 до наиболее верхней части зоны разделения 30. Отношение L:Y длины зоны реакции 28 к высоте зоны разделения 30, предпочтительно, составляет от приблизительно 2:1 до приблизительно 24:1, более предпочтительно, составляет от приблизительно 3:1 до приблизительно 20:1 и, наиболее предпочтительно, составляет от 4:1 до 16:1.
Обратимся теперь к фиг.1-5 и более подробно обсудим расположение и конфигурацию барботера 34 для ввода окислителя. На фиг.2 и 3 показано, что барботер 34 для ввода окислителя может включать кольцевой элемент 60, поперечный элемент 62 и пару трубопроводов 64a, b для подачи окислителя. Указанные трубопроводы 64a, b для подачи окислителя, преимущественно, могут входить в реактор выше той отметки высоты, на которой расположен кольцевой элемент 60, а затем направлены вниз, как показано на фиг.2 и 3. В качестве альтернативы, трубопровод 64a, b для подачи окислителя может входить в реактор ниже той отметки высоты, на которой расположен кольцевой элемент 60, или же лежать в той же горизонтальной плоскости, что и кольцевой элемент 60. Каждый из трубопроводов 64a, b для подачи окислителя включает первый конец, соединенный с соответствующим входным отверстием 66a, b для подачи окислителя, которое сделано в кожухе реактора 22, и второй конец, который плавно соединен с кольцевым элементом 60. Кольцевой элемент 60, предпочтительно, образован трубками, более предпочтительно, образован множеством секций из прямых трубок и, наиболее предпочтительно, множеством секций из прямых трубок, жестко соединенных друг с другом с образованием многоугольного трубчатого кольца. Кольцевой элемент 60, предпочтительно, образован, по меньшей мере, тремя секциями из прямых трубок, более предпочтительно, образован 6-10 секциями из трубок и, наиболее предпочтительно, образован 8 секциями из трубок. Таким образом, в том случае, когда кольцевой элемент 60 образован 8 секциями трубок, он в общем случае имеет восьмиугольную конфигурацию. Поперечный элемент 62 преимущественно образован практически прямыми секциями трубок, которые плавно соединены с противоположными секциями трубок кольцевого элемента 60 и продолжаются в диагональном направлении между секциями трубок кольцевого элемента 60. Секции трубок, которые используются для поперечного элемента 62, предпочтительно, имеют практически тот же самый диаметр, что и секции трубок, которые используются для изготовления кольцевого элемента 60. Секции трубок, которые образуют трубопроводы 64a, b для подачи окислителя, кольцевой элемент 60 и поперечный элемент 62, предпочтительно, имеют номинальный диаметр, превышающий приблизительно 0,1 м, более предпочтительно, имеют номинальный диаметр, составляющий от приблизительно 0,2 до приблизительно 2 м и, наиболее предпочтительно, имеют диаметр от 0,25 до 1 м. Как, видимо, наилучшим образом показано на фиг.3, и кольцевой элемент 60, и поперечный элемент 62 содержат множество расположенных сверху отверстий 68 для подачи окислителя с целью направить поток окислителя вверх в зону реакции 28. Как, видимо, наилучшим образом показано на фиг.4, кольцевой элемент 60 и/или поперечный элемент 62 могут иметь одно или несколько расположенных внизу отверстий 70 для подачи окислителя с целью направить поток окислителя вниз в зону реакции 28. Расположенные внизу отверстия 70 для подачи окислителя могут также использоваться для загрузки жидкостей и/или твердых веществ, которые могут входить через кольцевой элемент 60 и/или поперечный элемент 62. С целью предотвращения образования твердых веществ внутри барботера 34 для ввода окислителя, поток жидкости может постоянно или периодически направляться через барботер 34 для ввода окислителя с тем, чтобы смыть любые накопившиеся твердые вещества.
Если вновь обратиться к фиг.1-4, то в процессе проведения окисления в барботажной колонне реакторного типа 20 потоки окислителя под давлением направляются через входы 66a, b для окислителя во входные трубопроводы 64a, b для окислителя, соответственно. Затем потоки окислителя направляются через трубопроводы 64a, b для подачи окислителя в кольцевой элемент 60. Как только поток окислителя входит в кольцевой элемент 60, поток окислителя распределяется по внутренним объемам кольцевого элемента 60 и поперечного элемента 62. Затем поток окислителя выталкивается из барботера 34 для ввода окислителя и попадает в зону реакции 28 через верхние и нижние отверстия 68, 70 для подачи окислителя в кольцевом элементе 60 и поперечном элементе 62.
Выходы верхних отверстий 68 для подачи окислителя пространственно отделены друг от друга в горизонтальном направлении и располагаются практически на одной и той же высоте в зоне реакции 28. Таким образом, выходы верхних отверстий 68 для подачи окислителя в общем случае располагаются вдоль практически горизонтальной плоскости, которая определяется верхней частью барботера 34 для ввода окислителя. Выходы нижних отверстий 70 для подачи окислителя пространственно разделены друг от друга в горизонтальном направлении и располагаются практически на одной и той же высоте в зоне реакции 28. Таким образом, выходы нижних отверстий 70 для ввода окислителя в общем случае располагаются вдоль практически горизонтальной плоскости, которая определяется нижней частью барботера 34 для ввода окислителя.
В одном из вариантов осуществления настоящего изобретения барботер 34 для ввода окислителя имеет, по меньшей мере, приблизительно 20 сформированных в нем верхних отверстий 68 для подачи окислителя. Более предпочтительно, барботер 34 для ввода окислителя имеет от приблизительно 40 до приблизительно 800 образованных в нем верхних отверстий для подачи окислителя. Наиболее предпочтительно, барботер 34 для ввода окислителя имеет от 60 до 400 образованных в нем верхних отверстий 68 для подачи окислителя. Барботер 34 для ввода окислителя, предпочтительно, имеет, по меньшей мере, приблизительно 1 образованное в нем нижнее отверстие 70 для ввода окислителя. Более предпочтительно, барботер 34 для ввода окислителя имеет от приблизительно 2 до приблизительно 40 образованных в нем нижних отверстий 70 для подачи окислителя. Наиболее предпочтительно, барботер 34 для ввода окислителя имеет от 8 до приблизительно 20 образованных в нем нижних отверстий 70 для подачи окислителя. Отношение количества верхних отверстий 68 для подачи окислителя к количеству нижних отверстий 70 для подачи окислителя в барботере 34 для ввода окислителя, предпочтительно, составляет от приблизительно 2:1 до приблизительно 100:1, более предпочтительно, составляет от приблизительно 5:1 до приблизительно 25:1 и, наиболее предпочтительно, составляет от 8:1 до 15:1. Диаметры практически всех верхних и нижних отверстий 68, 70 для подачи кислорода, предпочтительно, практически те же самые, так что отношение объемной скорости потока окислителя из верхних и нижних отверстий 68, 70 практически совпадают с приведенными выше отношениями для относительного количества верхних и нижних отверстий 68, 70 для ввода кислорода.
На фиг.5 показано направление потока окислителя из верхних и нижних отверстий 68, 70 для подачи окислителя. Что касается верхних отверстий 68 для подачи окислителя, то, по крайней мере, часть верхних отверстий 68 для подачи кислорода, предпочтительно, направляет поток окислителя под углом A, который отклонен от вертикали. Процентное отношение верхних отверстий 68 для подачи кислорода, которые отклонены от вертикали на угол A, предпочтительно, составляет от приблизительно 30 до приблизительно 90%, более предпочтительно, составляет от приблизительно 35 до приблизительно 80%, еще более предпочтительно, составляет от приблизительно 60 до приблизительно 75% и, наиболее предпочтительно, составляет приблизительно 67%. Величина угла A, предпочтительно, находится от приблизительно 5 до приблизительно 60°, более предпочтительно, составляет от приблизительно 10 до приблизительно 45° и, наиболее предпочтительно, составляет от 15 до 30°. Что касается нижних отверстий 70 для подачи окислителя, то, предпочтительно, практически все нижние отверстия 70 для подачи окислителя располагаются вблизи наиболее близкой к днищу части кольцевого элемента 60 и/или поперечного элемента 62. Таким образом, любая жидкость и/или любые твердые вещества, которые случайно попадают в барботер 34 для ввода окислителя, могут быть легко удалены из барботера 34 для ввода окислителя через нижние отверстия 70 для подачи окислителя. Нижние отверстия 70 для подачи окислителя, предпочтительно, направляют поток окислителя вниз практически под вертикальным углом. В данном описании верхнее отверстие для ввода окислителя может быть любым отверстием, которое направляет поток окислителя в основном вверх (т.е. под углом больше горизонтального), а нижнее отверстие для ввода окислителя может быть любым отверстием, которое направляет поток окислителя в основном вниз (т.е. под углом ниже горизонтального).
Во многих обычных барботажных колоннах реакторного типа, содержащих многофазную среду, практически вся реакционная среда, располагающаяся ниже барботера для ввода окислителя (или другого устройства для подачи потока окислителя в зону реакции), обладает очень низкой способностью удерживать газ. Как известно из области техники, “способность удерживать газ” представляет собой просто объемную фракцию многофазной среды, которая находится в газообразном состоянии. Зоны, где способность удерживать газ в среде низка, можно также обозначить как “неаэрированные” зоны. Во многих обычных содержащих суспензии барботажных колоннах реакторного типа значительная часть общего объема реакционной среды располагается ниже барботера для ввода окислителя (или другого устройства для подачи потока окислителя в зону реакции). Таким образом, значительная часть реакционной среды, которая находится на дне обычных барботажных колонн реакторного типа, неаэрирована.
Было обнаружено, что минимизация количества неаэрированных зон в подвергаемой окислению реакционной среде в барботажной колонне реакторного типа может свести к минимуму образование определенного типа нежелательных примесей. Неаэрированные зоны реакционной среды содержат относительно немного пузырьков окислителя. Указанный низкий объем пузырьков окислителя снижает количество молекулярного кислорода, доступного для растворения в жидкой фазе реакционной среды. Таким образом, жидкая фаза в неаэрированной зоне реакционной среды имеет относительно небольшую концентрацию молекулярного кислорода. Указанные обедненные кислородом неаэрированные зоны реакционной среды проявляют тенденцию к ускорению нежелательных побочных реакций, а не требуемой реакции окисления. Например, в том случае, когда пара-ксилол частично окисляется с образованием терефталевой кислоты, недостаточная доступность кислорода в жидкой фазе реакционной среды может привести к образованию нежелательно больших количеств бензойной кислоты и конденсированных ароматических циклов, в том числе, в частности, крайне нежелательных окрашенных молекул, известных как флуореноны и антрахиноны.
В соответствии с одним из вариантов осуществления настоящего изобретения жидкофазное окисление проводят в барботажной колонне реакторного типа, которая сконфигурирована и функционирует таким образом, что объемная фракция реакционной среды с низкими удерживаемыми объемами газа, сведена к минимуму. Указанную минимизацию неаэрированных зон можно количественно оценить, теоретически разбив весь объем реакционной среды на 2000 дискретных горизонтальных слоев постоянного объема. За исключением самого верхнего и самого нижнего из горизонтальных слоев, каждый горизонтальный слой имеет дискретный объем, который с боков ограничен стенкой реактора, а сверху и снизу ограничен воображаемыми горизонтальными плоскостями. Самый верхний горизонтальный слой ограничен снизу воображаемой горизонтальной плоскостью, а сверху ограничен верхней поверхностью реакционной среды. Самый нижний горизонтальный слой ограничен сверху воображаемой горизонтальной плоскостью, а снизу ограничен нижним концом реактора. После того как реакционная среда теоретически разделена на 2000 дискретных горизонтальных слоев равного объема, можно определить усредненную по времени и по объему способность удерживать газ для каждого горизонтального слоя. При использовании указанного способа для количественной оценки количества неаэрированных зон количество горизонтальных слоев, в которых усредненная по времени и объему способность удерживать газ составляет менее 0,1, предпочтительно, должно составлять меньше чем 30, более предпочтительно, должно составлять меньше чем 15, еще более предпочтительно, должно быть меньше чем 6, еще более предпочтительно, должно быть меньше чем 4 и, наиболее предпочтительно, должно быть меньше чем 2. Количество горизонтальных слоев, в которых способность удерживать газ составляет менее 0,2, предпочтительно, должно быть меньше чем 80, более предпочтительно, должно быть меньше чем 40, еще более предпочтительно, должно быть меньше чем 20, еще более предпочтительно, должно быть меньше чем 12 и, наиболее предпочтительно, должно быть меньше чем 5. Количество горизонтальных слоев, в которых величина удерживания газа составляет менее 0,3, предпочтительно, должно быть меньше чем 120, более предпочтительно, должно быть меньше чем 80, еще более предпочтительно, должно быть меньше чем 40, еще более предпочтительно, должно быть меньше чем 20 и, наиболее предпочтительно, должно быть меньше чем 15.
Возвращаясь вновь к фиг.2, следует отметить, что авторами было установлено, что более низкое размещение барботера 34 для ввода окислителя в зоне реакции 28 обеспечивает несколько преимуществ, включая сокращение количества неаэрированных зон в реакционной среде 36. При заданных значениях H высоты реакционной среды 36, длины L зоны реакции 28, максимального диаметра D зоны реакции 28 большая часть (т.е. >50 мас.%) потока окислителя, предпочтительно, должна вводиться в зону реакции 28 в пределах приблизительно 0,025Н, 0,022L и/или 0,25D от нижнего уровня 52 зоны реакции 28. Более предпочтительно, большая часть потока окислителя вводится в зону реакции 28 в пределах приблизительно 0,02Н, 0,018L и/или 0,2D от нижнего уровня 52 зоны реакции 28. Наиболее предпочтительно, большая часть потока окислителя вводится в зону реакции 28 в пределах 0,015Н, 0,013L
и/или 0,15D от нижнего уровня 52 зоны реакции 28.
В варианте конструкции, приведенном на фиг.2, расстояние по высоте Y1 между нижним уровнем 52 зоны реакции 28 и выходом верхних отверстий 68 для подачи окислителя в барботере 34 для ввода окислителя составляет меньше приблизительно 0,025Н, 0,022L и/или 0,25D, так что практически весь поток окислителя входит в зону реакции в пределах приблизительно 0,025Н, 0,022L и/или 0,25D от нижнего уровня 52 зоны реакции 28. Более предпочтительно, Y1 составляет меньше чем приблизительно 0,02Н, 0,018L и/или 0,2D. Наиболее предпочтительно, Y1 составляет меньше чем 0,015Н, 0,013L и/или 0,15D, но больше чем 0,005Н, 0,004L и/или 0,06D. На фиг.2 показана тангенциальная линия 72 в том месте, где нижний край цилиндрического корпуса 46 кожуха реактора 22 соединяется с верхним концом эллиптического днища 48 кожуха реактора 22. В качестве альтернативы, днище 48 может быть любой формы, в том числе конической формы, и тангенциальная линия, по-прежнему, определяется нижним краем цилиндрического корпуса 46. Расстояние по высоте Y2 между тангенциальной линией 72 и верхом барботера 34 для ввода окислителя, предпочтительно, составляет, по меньшей мере, приблизительно 0,0012Н, 0,001L и/или 0,01D; более предпочтительно, составляет, по меньшей мере, приблизительно 0,005Н, 0,004L и/или 0,05D; и наиболее предпочтительно, составляет, по меньшей мере, 0,01Н, 0,008L и/или 0,1D. Расстояние по высоте Y3 между нижним уровнем 52 зоны реакции 28 и выходом нижних отверстий подачи окислителя 70 барботера 34 для ввода окислителя, предпочтительно, составляет меньше чем приблизительно 0,015Н, 0,013L и/или 0,15D; более предпочтительно, составляет меньше чем приблизительно 0,012Н, 0,01L и/или 0,1D; и, наиболее предпочтительно, составляет меньше чем 0,01Н, 0,008L и/или 0,075D, но больше чем 0,003Н, 0,002L и/или 0,025D.
В предпочтительном варианте осуществления настоящего изобретения отверстия, через которые поток окислителя и поток исходных веществ направляется в зону реакции, конфигурируют таким образом, чтобы количество (по массе) окислителя или исходных веществ, подаваемых через отверстие, было прямо пропорционально открытой площади отверстия. Так, например, если 50% общей открытой площади, определяемой всеми отверстиями для подачи окислителя, располагается в пределах 0,15D от нижнего уровня зоны реакции, то 50% мас. потока окислителя входит в зону реакции в пределах 0,15D от нижнего уровня зоны реакции, и наоборот.
В дополнение к преимуществам, предоставляемым минимизацией неаэрированных зон (т.е. зон с низкой способностью удерживать газ) в реакционной среде, было показано, что окисление можно ускорить путем максимального увеличения способности удерживать газ во всей реакционной среде 36. Усредненная по времени и объему способность реакционной среды 36 удерживать газ, предпочтительно, составляет, по меньшей мере, 0,4, более предпочтительно, составляет от приблизительно 0,6 до приблизительно 0,9 и, наиболее предпочтительно, составляет от 0,65 до 0,85. Указанным выше значениям способности удерживать газ способствует несколько физических и функциональных характеристик барботажной колонны реакторного типа 20. Например, для заданного размера реактора и потока окислителя большое отношение L:D в зоне реакции 28 приводит к меньшему значению диаметра, что повышает приведенную скорость в реакционной среде 36, которая, в свою очередь, увеличивает способность удерживать газ. Кроме того, известно, что действительный диаметр барботажной колонны реакторного типа и отношение L:D влияют на среднее значение способности удерживать газ даже для заданной постоянной приведенной скорости. Кроме того, минимизация неаэрированных зон, в особенности в нижней части зоны реакции 28, способствует увеличению способности удерживать газ. Кроме того, давление в верхней части и конфигурация барботажной колонны реакторного типа могут оказать влияние на устойчивость ее работы при указанных в данном описании больших приведенных скоростях и больших значениях способности удерживать газ.
Далее, авторы настоящего изобретения установили, что для достижения высоких значений способности удерживать газ и больших значений массопереноса важно проводить процесс с оптимизированным значением давлением в верхней части реактора. Можно было бы ожидать, что проведение процесса с низким значением верхнего давления, которое в соответствии с законом Генри снижает растворимость молекулярного кислорода, должно приводить к снижению скорости массопереноса молекулярного кислорода из газовой фазы в жидкую фазу. В реакторе с механическим перемешиванием именно это и происходит, поскольку уровни аэрации и скорости массопереноса определяются конструкцией перемешивающего устройства и величиной верхнего давления. Однако для барботажной колонны реакторного типа в соответствии с предпочтительным вариантом осуществления настоящего изобретения авторы настоящего изобретения определили, каким образом можно, используя меньшую величину верхнего давления, заставить данную массу исходного газофазного окислителя занять больший объем и тем самым повысить приведенную скорость в реакционной среде 36, что, в свою очередь, приведет к повышению способности удерживать газ и повышению скорости массопереноса молекулярного кислорода.
Баланс между слиянием и разрушением капель представляет собой чрезвычайно сложное явление, которое, с одной стороны, может вызывать пенообразование, которое снижает внутренние скорости циркулирования в жидкой фазе и может потребовать применения очень и очень больших разделительных зон, а с другой стороны, может приводить к образованию небольшого количества очень больших капель, что снижает способность удерживать газ и уменьшает скорость массопереноса из потока окислителя в жидкую фазу. Что касается жидкой фазы, то известно, что ее состав, плотность, вязкость и поверхностное натяжение, среди прочих факторов, находятся в очень сложном взаимоотношении друг с другом и дают весьма сложные результаты даже в отсутствие твердой фазы. Например, лабораторные исследования показывают, что при подготовке сообщений и при оценке наблюдений даже для простых содержащих воду и воздух барботажных колонн необходимо указывать, является ли “вода” водопроводной водой, дистиллированной водой или деионизованной водой. Для сложных смесей в жидкой фазе и при добавлении твердой фазы степень сложности еще более возрастает. Поверхностные неоднородности индивидуальных частиц твердых веществ, средний размер твердых веществ, распределение размеров частиц, количество твердых веществ по отношению к жидкой фазе и способность жидкости смачивать поверхность твердого вещества, среди прочих, важны для их взаимодействия с жидкой фазой и потоком окислителя и должны учитываться при установлении итогового поведения при барботаже и при установлении картины распределения конвекционных потоков.
Таким образом, способность барботажной колонны реакторного типа правильно функционировать с указанными в данном описании высокими приведенными скоростями и высокими значениями способности удерживать газ зависит, например, от подходящего выбора: (1) состава жидкой фазы реакционной среды; (2) количества и вида осаждаемых твердых веществ, при этом оба указанных параметра можно регулировать за счет условий проведения реакции; (3) подаваемого в реактор количества исходного окислителя; (4) верхнего давления, которое оказывает влияние на объемный поток окислителя, стабильность пузырьков и посредством баланса энергии - на температуру реакции; (5) самой температуры реакции, которая влияет на свойства жидкости, свойства осаждаемых твердых веществ и удельный объем потока окислителя; и (6) конструкции и механических узлов реактора, включая отношение L:D.
Если вернуться вновь к фиг.1, то следует отметить, что было показано, что лучшее распределение способного окисляться соединения (в частности, пара-ксилола) в реакционной среде 36 может быть обеспечено за счет введения потока жидкофазных исходных веществ в зону реакции 28 в нескольких разделенных по высоте позициях. Поток жидкофазных исходных веществ, предпочтительно, вводят в зону реакции 28, по меньшей мере, через 3 отверстия для подачи исходных веществ, более предпочтительно, через 4 отверстия для подачи исходных веществ. В данном описании термин “отверстие для подачи исходных веществ” обозначает отверстия, через которые жидкофазные исходные вещества поступают в зону реакции 28 для смешивания с реакционной средой 36. Предпочтительно, по крайней мере, 2 отверстия для ввода исходных веществ должны быть отделены друг от друга по высоте расстоянием, равным, по меньшей мере, приблизительно 0,5D, более предпочтительно, равным, по меньшей мере, приблизительно 1,5D и, наиболее предпочтительно, равным, по меньшей мере, 3D. Тем не менее, наиболее высоко расположенное отверстие для подачи исходных веществ, предпочтительно, должно быть отделено по высоте от наиболее низко расположенного отверстия для подачи окислителя расстоянием, не превышающим приблизительно 0,75H, 0,65L и/или 8D; более предпочтительно, не превышающим приблизительно 0,5H, 0,4L и/или 5D; и, наиболее предпочтительно, не превышающим 0,4H, 0,35L и/или 4D.
Несмотря на то, что желательно вводить поток жидкофазных исходных веществ в нескольких местах, расположенных на разных отметках высоты, было также установлено, что лучшее распределение способного окисляться соединения в реакционной среде 36 обеспечивается в том случае, если большая часть исходных жидкофазных веществ вводится в нижнюю половину реакционной среды 36 и/или зоны реакции 28. Предпочтительно, по крайней мере, приблизительно 75% мас. исходных жидкофазных веществ подается в нижнюю половину реакционной среды 36 и/или зоны реакции 28. Наиболее предпочтительно, по крайней мере, 90% мас. исходных жидкофазных веществ подается в нижнюю половину реакционной среды 36 и/или зоны реакции 28. Кроме того, по крайней мере, приблизительно 30% мас. исходных жидкофазных веществ должно вводиться в зону реакции 28 в пределах приблизительно 1,5D от наиболее низко расположенного по высоте места, где в зону реакции 28 вводится поток окислителя. Указанное наиболее низко расположенное место, где в зону реакции 28 вводится поток окислителя, обычно находится в нижней части барботера для ввода окислителя; тем не менее, в предпочтительном варианте конструкции по настоящему изобретению рассматриваются различные альтернативные конфигурации для подачи исходного окислителя в зону реакции 28. По крайней мере, приблизительно 50% мас. жидкофазных исходных веществ, предпочтительно, вводится в пределах приблизительно 2,5D от того наиболее низко расположенного по высоте места, где поток окислителя вводится в зону реакции 28. По крайней мере, приблизительно 75% мас. потока жидкофазных исходных веществ, предпочтительно, вводится в пределах приблизительно 5D от того наиболее низко расположенного по высоте места, где поток окислителя вводится в зону реакции 28.
Каждое из отверстий для подачи исходных веществ определяет открытую поверхность, через которую осуществляется подача исходных веществ. По крайней мере, приблизительно 30% общей открытой поверхности всех отверстий для подачи исходных веществ, предпочтительно, располагается в пределах 1,5D от того наиболее низко расположенного по высоте места, где поток окислителя вводится в зону реакции 28. По крайней мере, приблизительно 50% общей открытой поверхности всех отверстий для подачи исходных веществ, предпочтительно, вводится в пределах приблизительно 2,5D от того наиболее низко расположенного по высоте места, где поток окислителя вводится в зону реакции 28. По крайней мере, приблизительно 75% общей открытой поверхности всех отверстий для подачи исходных веществ, предпочтительно, вводится в пределах приблизительно 5D от того наиболее низко расположенного по высоте места, где поток окислителя вводится в зону реакции 28.
Если вновь вернуться к фиг.1, то в одном из вариантов осуществления настоящего изобретения входные отверстия 32a, b, c, d для подачи исходных веществ представляют собой просто серию вертикально расположенных отверстий вдоль одной из сторон кожуха реактора 22. Указанные отверстия для подачи исходных веществ, предпочтительно, имеют практически одинаковые диаметры, которые составляют меньше приблизительно 7 см, более предпочтительно составляют от приблизительно 0,25 до приблизительно 5 см и, наиболее предпочтительно, составляют от 0,4 до 2 см. Барботажная колонна реакторного типа 20, предпочтительно, снабжена системой для контролирования скорости потока жидкофазных исходных веществ, который выходит из каждого отверстия. Подобная система контролирования потока, предпочтительно, включает индивидуальные вентили для контролирования потока 74a, b, c, d для каждого соответствующего входа 32a, b, c, d для подачи исходных веществ. Кроме того, барботажная колонна реакторного типа 20, предпочтительно, снабжена системой контроля потока, которая позволяет подавать, по крайней мере, часть потока жидкофазных исходных веществ в зону реакции 28 с увеличенной приведенной скоростью на входе, составляющей, по меньшей мере, приблизительно 2 метра в секунду, более предпочтительно, составляющей, по меньшей мере, приблизительно 5 м/сек, еще более предпочтительно, составляющей, по меньшей мере, приблизительно 6 м/сек и наиболее предпочтительно, составляющей от 8 до 20 м/сек. В данном описании термин “приведенная скорость на входе” означает усредненную по времени объемную скорость потока жидкофазных исходных веществ, выходящих из отверстия для подачи исходных веществ, деленную на площадь отверстия для подачи исходных веществ. По меньшей мере, приблизительно 50% мас. потока исходных веществ, предпочтительно, подается в зону реакции 28 с повышенной приведенной скоростью на входе. Наиболее предпочтительно, весь поток исходных веществ подается в зону реакции 28 с повышенной приведенной скоростью на входе.
Обратимся теперь к фиг.6-7, на которых представлена альтернативная система для подачи потока жидкофазных исходных веществ в зону реакции 28. В указанном варианте осуществления настоящего изобретения поток исходных веществ поступает в зону реакции 28 на четырех различных уровнях. Каждый уровень снабжен соответствующей системой распределения исходных веществ 76a, b, c, d. Каждая из систем распределения исходных веществ 76 содержит основной трубопровод 78 для подачи исходных веществ и патрубок 80. Каждый патрубок 80 имеет, по меньшей мере, два выхода 82, 84, соединенных с соответствующими входными трубопроводами 86, 88, которые проникают в зону реакции 28 через кожух реактора 22. Каждый из входных трубопроводов 86, 88 содержит соответствующие отверстия 87, 89 для подачи исходных веществ, предназначенные для ввода потока исходных веществ в зону реакции 28. Отверстия 87, 89 для подачи исходных веществ, предпочтительно, имеют одинаковые диаметры, которые составляют меньше приблизительно 7 см, более предпочтительно, составляют от 0,25 до приблизительно 5 см и, наиболее предпочтительно, составляют от 0,4 до 2 см. Отверстия 87, 89 для подачи исходных веществ каждой системы распределения 76a, b, c, d исходных веществ, предпочтительно, располагаются диаметрально противоположно таким образом, чтобы можно было вводить поток исходных веществ в зону реакции 28 в противоположных направлениях. Кроме того, расположенные диаметрально противоположно входы 86, 88 для подачи исходных веществ соседних систем распределения исходных веществ 76, предпочтительно, ориентированы с разворотом на 90° друг относительно друга. При проведении процесса поток жидкофазных исходных веществ подается в главный трубопровод 78 для подачи исходных веществ и затем попадает в патрубок 80. Патрубок 80 распределяет поток исходных веществ равномерно с одновременной подачей к противоположным сторонам реактора 20 через отверстия 87, 89 для подачи исходных веществ.
Фиг.8 поясняет альтернативную конструкцию, где каждая система распределения исходных веществ 76 снабжена байонетными трубками 90, 92, а не входными трубопроводами 86, 88 (приведены на фиг.7). Байонетные трубки 90, 92 вдаются в зону реакции 28 и содержат множество небольших питающих отверстий 94, 96 для подачи исходных веществ, предназначенных для ввода жидкофазных исходных веществ зону реакции 28. Небольшие отверстия 94, 96 для подачи исходных веществ в байонетных трубках 90, 92, предпочтительно, имеет тот же самый диаметр, который составляет меньше приблизительно 50 мм, более предпочтительно, составляет приблизительно от 2 до приблизительно 25 мм и, наиболее предпочтительно, составляет от 4 до 15 мм.
Фиг.9-11 поясняют альтернативную систему распределения 100 исходных веществ. Система распределения 100 исходных веществ вводит поток жидкофазных исходных веществ во множество разделенных по высоте и разделенных по горизонтали позициях, не требуя многочисленных врезок в боковой стенке барботажной колонны реакторного типа 20. Система распределения 100 исходных веществ обычно включает единственный входной трубопровод 102, коллектор 104, множество вертикальных распределительных трубок 106, горизонтальное опорное устройство 108 и вертикальное опорное устройство 110. Входной трубопровод 102 проникает через боковую стенку корпуса 46 кожуха реактора 22. Входной трубопровод 102 плавно соединен с коллектором 104. Коллектор 104 равномерно распределяет поток исходных веществ, поступающий из входного трубопровода 102, по вертикальным распределительным трубкам 106. Каждая из вертикальных распределительных трубок 106 имеет множество разделенных по высоте отверстий 112a, b, c, d для подачи исходных веществ, предназначенных для ввода потока исходных веществ в зону реакции 28. Горизонтальное опорное устройство 108 соединено с каждой из распределительных трубок 106 и препятствует относительному латеральному перемещению распределительных трубок 106. Вертикальное опорное устройство 110 преимущественно присоединено к горизонтальному опорному устройству 108 и к верхней части барботера 34 для ввода окислителя. Вертикальное опорное устройство 110 в значительной степени препятствует относительному вертикальному перемещению распределительных трубок 106 в зоне реакции 28. Отверстия 112 для подачи исходных веществ, предпочтительно, имеют практически одинаковый диаметр, который составляет меньше чем приблизительно 50 мм, более предпочтительно, составляет от приблизительно 2 до приблизительно 25 мм и, наиболее предпочтительно, составляет от 4 до 15 мм. Расстояние по высоте между отверстиями 112 для подачи исходных веществ системы распределения 100 исходных веществ, которое поясняют фиг.9-11, может быть практически таким же, как и устройство, описанное выше при рассмотрении системы распределения исходных веществ, которая приведена на фиг.1.
Было установлено, что распределение потоков в реакционной среде многих барботажных колонн реакторного типа может позволить создать неравномерное азимутальное распределение способного окисляться соединения в реакционной среде, особенно в том случае, когда способное окисляться соединение в основном подается с одной стороны реакционной среды. В настоящем описании термин “азимутальный” обозначает угол или расстояние вокруг обращенной вверх оси, вдоль которой простирается зона реакции. В настоящем описании термин “обращенный вверх” означает имеющий отклонение от вертикали в пределах 45°. В одном из вариантов осуществления настоящего изобретения поток исходных веществ, содержащий способное окисляться соединение (в частности, пара-ксилол), вводят в зону реакции через множество азимутально разделенных отверстий для подачи исходных веществ. Указанные азимутально разделенные отверстия для подачи исходных веществ могут помочь в устранении областей в реакционной среде с избыточно высокой и избыточно низкой концентрациями способного окисляться соединения. Различные системы для подачи исходных веществ, приведенные на фиг.6-11, являются примерами систем, которые обеспечивают подходящее азимутальное разделение отверстий подачи исходных веществ.
Если вновь вернуться к фиг.7, то с целью количественной оценки азимутально раздельного введения потока жидкофазных исходных веществ в реакционную среду, реакционную среду можно теоретически разделить на четыре обращенных вверх азимутальных квадранта “Q1, Q2, Q3, Q4”, имеющих приблизительно равные объемы. Указанные азимутальные квадранты “Q1, Q2, Q3, Q4“ определяются парой воображаемых пересекающихся перпендикулярных вертикальных плоскостей “P1, P2”, которые простираются за максимальные вертикальные размеры и максимальные размеры реакционной среды. В том случае, когда реакционная среда содержится в цилиндрическом реакторе, линия пересечения воображаемых пересекающихся вертикальных плоскостей P1, P2 приблизительно совпадает с вертикальной осевой линией цилиндра, а каждый из азимутальных квадрантов Q1, Q2, Q3, Q4 в общем случае представляет собой клинообразный вертикальный объем, высота которого равна высоте реакционной среды. Значительная часть способного окисляться соединения, предпочтительно, должна поступать в реакционную среду через отверстия для подачи исходных соединений, которые расположены, по крайней мере, в двух различных азимутальных квадрантах.
В предпочтительном варианте осуществления настоящего изобретения не более чем приблизительно 80% мас. способного окисляться соединения поступает в реакционную среду через отверстия для подачи исходных веществ, которые могут располагаться в одном единственном азимутальном квадранте. Более предпочтительно, не более чем приблизительно 60% мас. способного окисляться соединения поступает в реакционную среду через отверстия для подачи исходных веществ, которые могут располагаться в одном единственном азимутальном квадранте. Наиболее предпочтительно, не более чем приблизительно 40% мас. способного окисляться соединения поступает в реакционную среду через отверстия для подачи исходных веществ, которые могут располагаться в одном единственном азимутальном квадранте. Указанные параметры азимутального распределения для способного окисляться соединения определяют тогда, когда азимутальные квадранты азимутально ориентированы таким образом, что максимально возможное количество способного окисляться соединения поступает в один из азимутальных квадрантов. Например, если весь поток исходных веществ поступает в реакционную среду через два отверстия для подачи исходных веществ, которые азимутально разделены друг от друга на 89°, то при определении азимутального распределения в четырех азимутальных квадрантах, 100% мас. потока исходных веществ поступает в реакционную среду в один единственный азимутальный квадрант, поскольку азимутальные квадранты могут быть азимутально ориентированы таким образом, что оба отверстия для подачи исходных веществ располагаются в одном и том же азимутальном квадранте.
В дополнение к преимуществам, связанным с правильным азимутальным разделением отверстий для подачи исходных веществ, было также показано, что важным может оказаться подходящее радиальное разделение отверстий для подачи исходных веществ в барботажной колонне реакторного типа. Значительная часть способного окисляться соединения, которое подается в реакционную среду, предпочтительно, должна поступать через отверстия для подачи исходных веществ, радиально отделенных внутри от боковой стенки реактора. Так, в одном из вариантов осуществления настоящего изобретения значительная часть способного окисляться соединения поступает в зону реакции через отверстия для подачи исходных веществ, расположенные в “предпочтительной радиальной зоне подачи исходных веществ”, которая отделена внутри от обращенной вверх боковой стенки, ограничивающей зону реакции.
Возвращаясь вновь к фиг.7, следует отметить, что предпочтительная радиальная зона подачи исходных веществ FZ может принимать форму теоретического обращенного вверх цилиндра, размещенного по центру зоны реакции 28, внешний диаметр которого Do составляет 0,9D, где D означает диаметр зоны реакции 28. Таким образом, между предпочтительной радиальной зоной подачи исходных веществ FZ и внутренней стенкой, очерчивающей зону реакции 28, образуется внешний кольцевой зазор OA, толщина которого составляет 0,05D. Предпочтительно, лишь малая часть способного окисляться соединения может попадать или же вообще не попадает в зону реакции 28 через отверстия для подачи исходных веществ, которые расположены в указанном внешнем кольцевом зазоре OA.
В другом варианте осуществления настоящего изобретения лишь малая часть способного окисляться соединения может попадать или же вообще не попадает в центральную зону реакции 28. Таким образом, как показано на фиг.8, предпочтительная радиальная зона подачи исходных веществ FZ может принимать форму теоретического обращенного вверх кольца с центром в зоне реакции 28, внешний диаметр которого Do составляет 0,9D, а внутренний диаметр DI составляет 0,2D. Таким образом, в этом варианте осуществления настоящего изобретения из центра предпочтительной радиальной зоной подачи исходных веществ FZ “вырезается” внутренний цилиндр IC, диаметр которого составляет 0,2D. Предпочтительно, лишь малая часть способного окисляться соединения может попадать или же вообще не попадает в зону реакции 28 через отверстия для подачи исходных веществ, которые расположены в указанном внутреннем цилиндре IC.
В предпочтительном варианте осуществления настоящего изобретения значительная часть способного окисляться соединения поступает в реакционную среду 36 через отверстия для подачи исходных веществ, которые расположены в предпочтительной радиальной зоне подачи исходных веществ, независимо от того, имеет ли предпочтительная радиальная зона подачи исходных веществ описанную выше цилиндрическую или кольцеобразную форму. Более предпочтительно, по меньшей мере, приблизительно 25% мас. способного окисляться соединения поступает в реакционную среду 36 через отверстия для подачи исходных веществ, которые расположены в предпочтительной радиальной зоне подачи исходных веществ. Еще более предпочтительно, по меньшей мере, приблизительно 50% мас. способного окисляться соединения поступает в реакционную среду 36 через отверстия для подачи исходных веществ, которые расположены в предпочтительной радиальной зоне подачи исходных веществ. Наиболее предпочтительно, по меньшей мере, 75% мас. способного окисляться соединения поступает в реакционную среду 36 через отверстия для подачи исходных веществ, которые расположены в предпочтительной радиальной зоне подачи исходных веществ.
Несмотря на то, что теоретические азимутальные квадранты и теоретическая предпочтительная радиальная зона подачи исходных веществ, приведенные на фиг.7 и 8, описываются со ссылкой на распределение потока жидкофазных исходных веществ, было показано, что определенные преимущества предоставляет соответствующее азимутальное и радиальное распределение потока газофазного окислителя. Так, в одном из вариантов осуществления настоящего изобретения приведенное выше описание азимутального и радиального распределения потока жидкофазных исходных веществ может быть также использовано для описания того, каким образом поток газофазного окислителя поступает в реакционную среду 36.
Обратимся теперь к фиг.12-15, где поясняется альтернативный барботер 200 для ввода окислителя, который включает кольцевой элемент 202 и пару трубопроводов 204, 206 для подачи окислителя. Приведенный на фиг.12-15 барботер 200 для ввода окислителя похож на барботер 34 для ввода окислителя, который показан на фиг.1-11, однако имеет три следующие основные отличия: (1) барботер 200 для ввода окислителя не содержит диагональный поперечный элемент; (2) верхняя часть кольцевого элемента 202 не имеет отверстий для поступления окислителя в направлении наверх; и (3) барботер 200 для ввода окислителя имеет много дополнительных отверстий в нижней части кольцевого элемента 202.
Как, вероятно, наилучшим образом показано на фиг.14 и 15, нижняя часть кольца 202 барботера для ввода окислителя содержит множество отверстий 208 для подачи окислителя. Отверстия 208 для подачи окислителя предпочтительно сконфигурированы таким образом, чтобы, по меньшей мере, приблизительно 1% общей открытой поверхности, определяемой отверстиями 208 для подачи окислителя, располагается ниже осевой линии 210 (фиг.15) кольцевого элемента 202, при этом осевая линия 210 находится на уровне объемного центроида кольцевого элемента 202. Более предпочтительно, по меньшей мере, приблизительно 5% общей открытой поверхности, определяемой всеми отверстиями 208 для подачи окислителя, располагается ниже осевой линии 210, при этом, по меньшей мере, приблизительно 2% общей открытой поверхности определяют отверстия 208, через которые поток окислителя поступает в основном в направлении вниз под углом, составляющим в пределах приблизительно 30° от вертикали. Еще более предпочтительно, по меньшей мере, приблизительно 20% общей открытой поверхности, определяемой всеми отверстиями 208 для подачи окислителя, располагается ниже осевой линии 210, при этом, по меньшей мере, приблизительно 10% общей открытой поверхности определяют отверстия 208, через которые поток окислителя поступает в основном в направлении вниз под углом, составляющим в пределах приблизительно 30° от вертикали. Наиболее предпочтительно, по меньшей мере, приблизительно 75% общей открытой поверхности, определяемой всеми отверстиями 208 для ввода окислителя, располагается ниже осевой линии 210, при этом, по меньшей мере, приблизительно 40% общей открытой поверхности определяют отверстия 208, через которые поток окислителя поступает в основном в направлении вниз под углом, составляющим в пределах приблизительно 30° от вертикали. Фракция общей поверхности, определяемой всеми отверстиями 208 для ввода окислителя, которые располагаются выше осевой линии 210, предпочтительно, составляет меньше чем приблизительно 75%, более предпочтительно, меньше чем приблизительно 50%, еще более предпочтительно, меньше чем приблизительно 25% и, наиболее предпочтительно, меньше чем 5%.
Как показано на фиг.14 и 15, отверстия 208 для ввода окислителя включают обращенные вниз отверстия 208a и скошенные отверстия 208b. Обращенные вниз отверстия 208a сконфигурированы таким образом, что поток окислителя через них поступает в основном в направлении вниз под углом, составляющим в пределах приблизительно 30° от вертикали, более предпочтительно, составляющим в пределах приблизительно 15° от вертикали и, наиболее предпочтительно, составляющим в пределах 5° от вертикали. Скошенные отверстия 208b сконфигурированы таким образом, что поток окислителя через них поступает в основном в сторону и в направлении вниз под углом A, величина которого составляет от приблизительно 15 до приблизительно 75° от вертикали, более предпочтительно угол А составляет от приблизительно 30 до приблизительно 60° от вертикали и, наиболее предпочтительно, угол А составляет от 40 до 50° от вертикали.
Практически все отверстия 208 для подачи окислителя, предпочтительно, имеют приблизительно одинаковый диаметр. Диаметр отверстий 208 для подачи окислителя, предпочтительно, составляет от приблизительно 2 до приблизительно 300 мм, более предпочтительно, составляет от приблизительно 4 до приблизительно 120 мм и, наиболее предпочтительно, составляет от 8 до 60 мм. Общее количество отверстий 208 для подачи окислителя в кольцевом элементе 202 выбирается таким образом, чтобы удовлетворялись критерии по низкому перепаду давления, которые детально рассмотрены ниже. Общее количество отверстий 208 для подачи окислителя, сформированных в кольцевом элементе 202, предпочтительно, составляет, по меньшей мере, приблизительно 10, более предпочтительно, общее количество отверстий 208 для подачи окислителя составляет от приблизительно 20 до приблизительно 200 и, наиболее предпочтительно, общее количество отверстий 208 для подачи окислителя составляет от 40 до 100.
Несмотря на то, что на фиг.12-15 приведена весьма конкретная форма барботера 200 для ввода окислителя, следует отметить, что для достижения приведенных в данном описании преимуществ может быть использовано множество форм барботеров для ввода окислителя. Например, барботер для ввода окислителя необязательно должен иметь форму восьмиугольного кольцевого элемента, приведенного на фиг.12-13. Более того, барботер для ввода окислителя может быть образован любой конфигурацией напорного(ых) трубопровода(ов), имеющего(их) множество пространственно разделенных отверстий, через которые поступает поток окислителя. Размер, количество и направленность отверстий для подачи окислителя в напорном трубопроводе, предпочтительно, находится в указанных выше пределах. Кроме того, форма барботера для ввода окислителя, предпочтительно, такова, чтобы обеспечивалось указанное выше азимутальное и радиальное распределение молекулярного кислорода.
Независимо от конкретной формы барботера для ввода окислителя барботер для ввода окислителя, предпочтительно, имеет такую конфигурацию и функционирует таким образом, чтобы свести к минимуму перепад давления, связанный с поступлением потока окислителя через отверстия напорного(ых) трубопровода(ов). Подобный перепад давления рассчитывают как усредненное по времени статическое давление потока окислителя внутри напорного трубопровода на выходе из отверстий 66a, b барботера для ввода окислителя минус усредненное по времени статическое давление в зоне реакции на той высоте, где половина потока окислителя вводится выше заданной отметки высоты, а половина потока окислителя вводится ниже заданной отметки высоты. В предпочтительном варианте осуществления настоящего изобретения усредненный по времени перепад давления, связанный с поступлением потока окислителя из барботера для ввода окислителя, составляет меньше чем приблизительно 0,3 мегапаскалей (МПа), более предпочтительно составляет меньше чем приблизительно 0,2 МПа, еще более предпочтительно составляет меньше чем приблизительно 0,1 МПа и, наиболее предпочтительно, составляет меньше чем 0,05 МПа. В предпочтительных условиях эксплуатации приведенной в данном описании барботажной колонны реакторного типа давление потока окислителя внутри напорного(ых) трубопровода(ов) барботера для ввода окислителя, предпочтительно, составляет от приблизительно 0,35 до приблизительно 1 МПа, более предпочтительно, составляет от приблизительно 0,45 до приблизительно 0,85 МПа и, наиболее предпочтительно, составляет от 0,5 до 0,7 МПа.
Если вернуться к ранее рассмотренной форме барботера для ввода окислителя, приведенного на фиг.2-5, то желательно постоянно или периодически промывать барботер для ввода окислителя сильным напором жидкости (в частности, уксусной кислотой, водой и/или пара-ксилолом) с тем, чтобы предотвратить засорение барботера для ввода окислителя твердыми веществами. Если используется подобный сильный напор жидкости, то эффективное количество жидкости (т.е. не просто небольшое количество капель жидкости, которые естественным образом могут находиться в потоке окислителя), предпочтительно, пропускают через барботер для ввода окислителя и через отверстия для подачи окислителя в течение более чем одной минуты ежедневно. В том случае если жидкость постоянно или периодически подается через барботер для ввода окислителя, то усредненное по времени отношение массы потока жидкости, проходящей через барботер для ввода окислителя, к массе потока молекулярного кислорода, проходящего через барботер для ввода окислителя, предпочтительно, должно составлять от приблизительно 0,05:1 до приблизительно 30:1, или от приблизительно 0,1:1 до приблизительно 2:1 или даже 0,2:1 до 1:1.
В одном из вариантов осуществления настоящего изобретения значительная часть способного окисляться соединения (в частности, пара-ксилола) может подаваться в зону реакции через барботер для ввода окислителя. При использовании подобной формы барботера для ввода окислителя способное окисляться соединение и молекулярный кислород, предпочтительно, поступают из барботера для ввода окислителя через те же самые отверстия в барботере для ввода окислителя. Как указано ранее, способное окисляться соединение обычно представляет собой жидкость при заданном значении STP. Таким образом, в данном варианте конструкции из барботера для ввода окислителя может поступать двухфазный поток, при этом жидкая фаза содержит способное окисляться соединение, а газовая фаза содержит молекулярный кислород. Тем не менее, следует понимать, что при поступлении через барботер для ввода окислителя, по крайней мере, часть способного окисляться соединения может находиться в газообразном состоянии. В одном из вариантов осуществления настоящего изобретения жидкая фаза, поступающая через барботер для ввода окислителя, образована в основном способным окисляться соединением. В другом варианте осуществления настоящего изобретения жидкая фаза, поступающая через барботер для ввода окислителя, имеет практически тот же самый состав, что и поток исходных веществ, приведенный выше. В том случае, когда жидкая фаза, поступающая через барботер для ввода окислителя, имеет практически тот же самый состав, что и поток исходных веществ, то подобная жидкость может содержать растворитель и/или каталитическую систему в количествах и соотношениях, которые были описаны выше при рассмотрении состава потока исходных веществ.
В одном из вариантов осуществления настоящего изобретения, предпочтительно, по крайней мере, приблизительно 10% мас. способного окисляться соединения, вводимого в зону реакции, поступает через барботер для ввода окислителя, более предпочтительно, по крайней мере, приблизительно 40% мас. способного окисляться соединения подается в зону реакции через барботер для ввода окислителя и, наиболее предпочтительно, по крайней мере, 80% мас. способного окисляться соединения подается в зону реакции через барботер для ввода окислителя. В том случае, если часть или все количество способного окисляться соединения подается в зону реакции через барботер для ввода окислителя, то, предпочтительно, по меньшей мере, приблизительно 10% мас. всего количества молекулярного кислорода, подаваемого в зону реакции, поступает через тот же барботер для ввода окислителя, более предпочтительно, по меньшей мере, приблизительно 40% мас. способного окисляться соединения подается в зону реакции через тот же барботер для ввода окислителя и, наиболее предпочтительно, по меньшей мере, 80% мас.способного окисляться соединения подается в зону реакции через тот же барботер для ввода окислителя. В том случае, если значительная часть способного окисляться соединения подается в зону реакции через барботер для ввода окислителя, то в барботере для ввода окислителя, предпочтительно, устанавливается один или несколько термочувствительных датчиков (в частности, термопар). Указанные датчики температуры могут применяться для того, чтобы обеспечить такие условия, при которых температура в барботере для ввода окислителя не становится опасно высокой.
Если теперь обратиться к фиг.16-18, то на них приведена барботажная колонна реакторного типа 20, которая включает внутренний резервуар для деаэрации 300, который размещается внизу зоны реакции 28 вблизи выпускного отверстия 38. Было установлено, что побочные реакции, приводящие к образованию примесей, протекают с относительно высокой скоростью в процессе деаэрации реакционной среды 36. В данном описании “деаэрация” обозначает выделение газовой фазы из многофазной реакционной среды. В том случае, если реакционная среда 36 сильно аэрирована (способность удерживать газ составляет >0,3), то образование примесей минимально. В случае, когда реакционная среда 36 в значительной степени неаэрирована (способность удерживать газ составляет <0,01), то образование примесей также минимально. Однако в том случае, если реакционная среда частично аэрирована (способность удерживать газ составляет 0,01-0,3), то активируются нежелательные побочные реакции, и образуется повышенное количество примесей. Резервуар для деаэрации 300 решает указанную проблему, а также другие проблемы путем минимизации объема реакционной среды 36, которая находится в частично аэрированном состоянии, а также путем минимизации времени, которое необходимо для осуществления деаэрации реакционной среды 36. В значительной степени деаэрированная суспензия образуется в нижней части резервуара для деаэрации 300 и покидает реактор 20 через отверстие для выгрузки суспензии 38. В значительной степени деаэрированная суспензия, предпочтительно, содержит меньше чем приблизительно 5 об.% газовой фазы, более предпочтительно, содержит меньше чем приблизительно 2 об.% газовой фазы и, наиболее предпочтительно, содержит меньше чем 1 об.% газовой фазы.
На фиг.16 приведена барботажная колонна реакторного типа 20, которая включает регулятор уровня 302 и вентиль управления потоком 304. Регулятор уровня 302 и вентиль управления потоком 304 работают согласованно таким образом, чтобы поддерживать практически постоянный уровень реакционной среды 36 в зоне реакции 28. Регулятор уровня 302 способен определять (в частности, по принципу определения уровня по перепаду давления или по принципу определения уровня с помощью радиоизотопной метки) уровень верхней поверхности 44 реакционной среды 36 и генерировать управляющий сигнал 306, который соответствует уровню реакционной среды 36. Вентиль управления потоком 304 получает управляющий сигнал 306 и регулирует скорость потока суспензии через трубопровод 308 для выгрузки суспензии. Таким образом, скорость отвода суспензии через отверстие для выгрузки суспензии 38 может варьировать между максимальной объемной скоростью потока (Fmax), когда уровень реакционной среды 36 слишком высок, и минимальной объемной скоростью потока (Fmin), когда уровень реакционной среды 36 слишком низок.
Для удаления твердофазного продукта окисления из зоны реакции 28 часть его вначале должна пройти через резервуар для деаэрации 300. Резервуар для деаэрации 300 создает внутренний объем с низкой степенью турбулентности, в котором газовая фаза реакционной среды 36 может естественным путем выделяться из жидкой и твердой фаз реакционной среды 36 по мере того, как жидкость и твердые вещества продвигаются вниз по направлению к отверстию для выгрузки суспензии 38. Выделение газовой фазы из жидкой и твердой фаз вызывается естественным всплыванием газовой фазы в жидкой и твердой фазах. При использовании резервуара для деаэрации 300 переход реакционной среды 36 из полностью аэрированной трехфазной среды к полностью деаэрированной двухфазной среде осуществляется быстро и эффективно.
Что касается фиг.17 и 18, то следует отметить, что резервуар для деаэрации 300 включает обычно обращенную вверх боковую стенку 308, которая определяет зону деаэрации 312. Боковая стенка 308 простирается вверх, и отклонение ее от вертикали, предпочтительно, составляет в пределах приблизительно 30°, более предпочтительно, составляет в пределах приблизительно 10° от вертикали. Наиболее предпочтительно, боковая стенка 308 практически вертикальна. Зона деаэрации 312 отделена от зоны реакции 28 и имеет высоту h и диаметр d. Верхний конец 310 боковой стенки 308 открыт с тем, чтобы реакционная среда могла попадать из зоны реакции 28 во внутренний объем 312. Нижний конец боковой стенки 308 плавно соединен с отверстием для выгрузки суспензии 38 посредством переходной секции 314. В некоторых случаях, например, когда размер отверстия для выгрузки суспензии 38 велик или диаметр d боковой стенки 308 мал, переходную секцию 314 можно удалить. Как, видимо, наилучшим образом показано на фиг.18, резервуар для деаэрации 300 может включать также стабилизатор потока 316, который размещается в зоне деаэрации 312. Стабилизатор потока 316 может иметь любую конструкцию, способную предотвратить образование вихрей по мере того, как твердая и жидкая фазы продвигаются вниз по направлению к отверстию для выгрузки суспензии 38.
Для того чтобы обеспечить надлежащее отделение газовой фазы от твердой и жидкой фаз в резервуаре для деаэрации 300, тщательно подбираются высота h и площадь горизонтального сечения внутренней зоны деаэрации 312. Высота h и площадь горизонтального сечения внутренней зоны деаэрации 312 должны обеспечивать достаточное расстояние и время, чтобы даже в том случае, когда извлекается максимальное количество суспензии (т.е. когда суспензия извлекается со скоростью Fmax), практически весь объем пузырьков газа мог выделиться из твердой и жидкой фаз до того, как пузырьки газа достигнут выходного отверстия, расположенного внизу резервуара для деаэрации 300. Таким образом, площадь сечения зоны деаэрации 312, предпочтительно, должна быть такой, чтобы максимальная скорость перемещения вниз (Vdmax) жидкой и твердой фаз через зону деаэрации 312 была значительно меньше, чем естественная скорость подъема (Vu) пузырьков газовой фазы через жидкую и твердую фазы. Максимальная скорость перемещения вниз (Vdmax) жидкой и твердой фаз через зону деаэрации 312 достигается при максимальной объемной скорости суспензии (Fmax), которая уже была рассмотрена выше. Естественная скорость подъема (Vu) пузырьков газовой фазы через жидкую и твердую фазы варьирует в зависимости от размера пузырьков; тем не менее, естественная скорость подъема (Vu0,5) пузырьков газа с диаметром 0,5 см через жидкую и твердую фазы может быть использована в качестве величины отсечки, поскольку первоначально диаметр практически всех пузырьков в объеме в реакционной среде 36 превышает 0,5 см. Площадь сечения зоны деаэрации 312, предпочтительно, такова, что величина Vdmax составляет меньше чем приблизительно 75% от значения Vu0,5, более предпочтительно, величина Vdmax составляет меньше чем приблизительно 40% от значения Vu0,5 и, наиболее предпочтительно, величина Vdmax составляет меньше чем 20% от значения Vu0,5.
Скорость перемещения вниз жидкой и твердой фаз в зоне деаэрации 312 резервуара для деаэрации 300 рассчитывается как скорость объемного потока деаэрированной суспензии через отверстие для выгрузки суспензии 38, деленная на минимальное значение площади сечения зоны деаэрации 312. Скорость перемещения вниз жидкой и твердой фаз в зоне деаэрации 312 резервуара для деаэрации 300, предпочтительно, составляет меньше чем приблизительно 50 см/сек, более предпочтительно, составляет меньше чем приблизительно 30 см/сек и, наиболее предпочтительно, составляет меньше чем 10 см/сек.
Следует отметить, что несмотря на то, что обращенная вверх боковая стенка 308 резервуара для деаэрации 300, как указывалось, имеет цилиндрическую форму, боковая стенка 308 может состоять из множества боковых стенок, которые образуют множество форм (в частности, могут иметь треугольную, квадратную и овальную форму), при условии, что стенки ограничивают внутренний объем, который имеет соответствующий объем, площадь сечения, ширину d и высоту h. В предпочтительном варианте осуществления настоящего изобретения величина d составляет от приблизительно 0,2 до приблизительно 2 м, более предпочтительно составляет от приблизительно 0,3 до приблизительно 1,5 м и, наиболее предпочтительно, составляет от 0,4 до 1,2 м. В предпочтительном варианте осуществления настоящего изобретения величина h составляет от приблизительно 0,3 до приблизительно 5 м, более предпочтительно, составляет от приблизительно 0,5 до приблизительно 3 м и, наиболее предпочтительно, составляет от 0,75 до 2 м.
В предпочтительном варианте осуществления настоящего изобретения боковая стенка 308 практически вертикальна, так что площадь горизонтального сечения зоны деаэрации 312 практически постоянна вдоль всей высоты h зоны деаэрации 312. Максимальная площадь горизонтального сечения зоны деаэрации 312 составляет меньше приблизительно 25% от максимальной площади горизонтального сечения зоны реакции 28. Более предпочтительно, максимальная площадь горизонтального сечения зоны деаэрации 312 составляет от приблизительно 0,1 до приблизительно 10% от максимальной площади горизонтального сечения зоны реакции 28. Наиболее предпочтительно, максимальная площадь горизонтального сечения зоны деаэрации 312 составляет от 0,25 до 4% от максимальной площади горизонтального сечения зоны реакции 28. Максимальная площадь горизонтального сечения зоны деаэрации 312, предпочтительно, находится от приблизительно 0,02 до приблизительно 3 м2, более предпочтительно, от приблизительно 0,05 до приблизительно 2 м2 и, наиболее предпочтительно составляет от 0,1 до 1,2 м2. Объем зоны деаэрации 312, предпочтительно, составляет меньше чем приблизительно 5% от общего объема реакционной среды 36 или зоны реакции 28. Более предпочтительно, объем зоны деаэрации 312 составляет от приблизительно 0,01 до приблизительно 2% от общего объема реакционной среды 36 или зоны реакции 28. Наиболее предпочтительно, объем зоны деаэрации 312 составляет от 0,05 до приблизительно 1% от общего объема реакционной среды 36 или зоны реакции 28. Объем зоны деаэрации 312, предпочтительно, составляет меньше чем приблизительно 2 м3, более предпочтительно, составляет от приблизительно 0,01 до приблизительно 1 м3 и, наиболее предпочтительно, составляет от 0,05 до 0,5 м3.
Что касается фиг.19, то на ней представлена барботажная колонна реакторного типа 20, которая включает внешний резервуар для деаэрации 400. В этой конструкции аэрированная реакционная среда 36 выводится из зоны реакции 28 через расположенное на определенной высоте отверстие в боковой стенке кожуха реактора 22. С целью отделения газовой фазы от твердой и жидкой фаз выгружаемая аэрированная среда поступает во внешний резервуар для проведения деаэрации 400 через разгрузочный трубопровод 402. Отделенная газовая фаза покидает резервуар для деаэрации 400 по трубопроводу 404, в то время как практически деаэрированная суспензия выводится из резервуара для деаэрации 400 по трубопроводу 406.
На фиг.19 разгрузочный трубопровод 402 показан как приблизительно прямой горизонтальный трубопровод, который ортогонален по отношению к кожуху реактора 22. Это просто одна из удобных конфигураций; разгрузочный трубопровод 402 может быть любым другим во всех отношениях, при условии, что он приемлемым образом соединяет барботажную колонну реакторного типа 20 с внешним резервуаром для деаэрации 400. Что касается трубопровода 404, то он преимущественно присоединяется к верхней части или вблизи верхней части резервуара для деаэрации 400 с тем, чтобы можно было надежно решить проблемы безопасности, связанные с образованием застойных карманов, содержащих способное окисляться соединение и окислитель. Кроме того, для удобства трубопроводы 402 и 404 могут включать запорные устройства, такие как вентили.
В том случае, когда реакционная среда 36 выводится из реактора 20 через выход, расположенный на определенной отметке высоты, как показано на фиг.19, то барботажная колонна реакторного типа 20, предпочтительно, снабжается нижним выходным отверстием 408, расположенным у нижнего уровня 52 зоны реакции 28. Нижнее выходное отверстие 408 и связанный с ним нижний трубопровод 410 могут применяться для выгрузки (т.е. для опорожнения) реактора 20 на время остановок процесса. Один или несколько выходных отверстий 408 располагаются, предпочтительно, в нижней части, соответствующей одной трети от высоты реакционной среды 36, более предпочтительно, располагаются в нижней части, соответствующей одной четверти от высоты реакционной среды 36, и, наиболее предпочтительно, располагаются в самой низкой точке зоны реакции 28.
В случае извлечения суспензии на определенной отметке высоты и при использовании системы деаэрации, показанной на фиг.19, нижний трубопровод 410 и нижнее выходное отверстие 408 не используются для выгрузки суспензии из зоны реакции в процессе окисления. Из области техники известно, что твердые вещества имеют тенденцию оседать под действием сил тяжести из неаэрированных и по каким-либо другим причинам не перемешиваемых частях суспензии, включая трубопроводы с застойным потоком. Кроме того, высадившиеся твердые вещества (в частности, терефталевая кислота) могут затвердевать с образованием больших конгломератов за счет протекания непрерывного процесса осаждения и/или реорганизации кристаллической структуры. Таким образом, с целью предотвращения закупоривания нижнего трубопровода 410, часть деаэрированной суспензии из нижней части резервуара для деаэрации 400 может использоваться для постоянного или периодического промывания нижнего трубопровода 410 в процессе нормальной работы реактора 20. Предпочтительно, подобное промывание трубопровода 410 напором суспензии осуществляется путем периодического открывания вентиля 412 в трубопроводе 410 с тем, чтобы часть деаэрированной суспензии могла попасть по трубопроводу 410 в зону реакции 28 через нижнее выходное отверстие 408. Даже в том случае, когда вентиль 412 полностью или частично открыт, лишь часть деаэрированной суспензии протекает через нижний трубопровод 410 и вновь попадает в зону реакции 28. Оставшаяся часть деаэрированной суспензии, которая не используется для промывки нижнего трубопровода 410, выводится через трубопровод 414 из реактора 20 и поступает на последующую обработку (в частности, очистку).
В процессе нормальной работы барботажной колонны реакторного типа 20 в течение значительного периода времени (в частности, более 100 час) количество деаэрированной суспензии, которое используется для промывки нижнего трубопровода 410, предпочтительно, составляет меньше чем 50% мас. от общего количества деаэрированной суспензии, полученного в нижней части резервуар для деаэрации 400, более предпочтительно, составляет меньше чем приблизительно 20% мас. и, наиболее предпочтительно, составляет меньше чем 5% мас. Кроме того, в течение значительного периода времени средняя скорость массопереноса деаэрированной суспензии, которая используется для промывки нижнего трубопровода 410, предпочтительно, менее чем в 4 раза превышает среднюю скорость массопереноса способного окисляться соединения в зоне реакции 28, более предпочтительно, менее чем в 2 раза превышает среднюю скорость массопереноса способного окисляться соединения в зоне реакции 28, еще более предпочтительно, она меньше, чем средняя скорость массопереноса способного окисляться соединения в зоне реакции 28 и, наиболее предпочтительно, она менее чем в 0,5 раза превышает среднюю скорость массопереноса способного окисляться соединения в зоне реакции 28.
Если вновь вернуться к фиг.19, то резервуар для деаэрации 400 включает направленную практически вертикально, преимущественно цилиндрическую боковую стенку 416, которая определяет зону деаэрации 418. Зона деаэрации 418 имеет диаметр d и высоту h. Высота h измеряется расстоянием по высоте между уровнем, где аэрированная реакционная среда входит в резервуар для деаэрации 400, и нижней частью боковой стенки 416. Высота h, диаметр d, площадь и объем зоны деаэрации 418, преимущественно такие же, как и описанные выше для зоны деаэрации 312 резервуара для деаэрации 300, который приведен на фиг.16-18. Кроме того, резервуар для деаэрации 400 включает верхнюю секцию 420, образованную продолжением боковой стенки 416 за пределы зоны деаэрации 418. Верхняя секция 420 резервуара для деаэрации 400 может иметь любую высоту, однако, предпочтительно, она простирается вверх до уровня или выше уровня реакционной среды 36 в зоне реакции 28. Верхняя секция 420 обеспечивает газовой фазе достаточное пространство, чтобы она могла отделиться от жидкой и твердой фаз прежде чем газовая фаза покинет резервуар для деаэрации 400 через трубопровод 404. Следует отметить, что хотя показано, что трубопровод 404 возвращает отделившуюся газовую фазу в разделительную зону реактора 20, в качестве альтернативы, трубопровод 404 может быть соединен с кожухом реактора 20 на любой отметке высоты, превышающей уровень, на котором расположен выходной трубопровод 402. Трубопровод 404 необязательно может быть соединен с трубопроводом для отвода газа 40 таким образом, что отделившаяся газовая фаза из резервуара для деаэрации 400 объединяется с выводимым из верхней части потоком паров в трубопроводе 40 и направляется на дальнейшую переработку.
Если теперь обратиться к фиг.20, то здесь приведена барботажная колонна реакторного типа 20, которая включает гибридный внешне-внутренний резервуар для деаэрации 500. В такой конфигурации часть реакционной среды 36 выводится из зоны реакции 28 через расположенное на достаточно большой высоте отверстие 502 в боковой стенке кожуха реактора 22. Извлекаемая реакционная среда 36 затем поступает в коленчатый патрубок 504 с относительно большим диаметром и попадает в верхнюю часть резервуара для деаэрации 500. На фиг.20 показано, что коленчатый патрубок 504 соединен ортогонально с боковой стенкой кожуха реактора 22 и представляет собой плавный поворот под углом приблизительно 90°. Это всего лишь одна из удобных конфигураций, и коленчатый патрубок 504 может быть любым другим во всех отношениях, при условии, что он приемлемым образом соединяет барботажную колонну реакторного типа 20 с внешним резервуаром для деаэрации 500. Кроме того, для удобства коленчатый патрубок 504 может включать запорные устройства, такие как вентили.
В резервуаре для деаэрации 500 газовая фаза движется вверх, в то время как твердая и жидкая фазы движутся вниз. Движущаяся вверх газовая фаза может вновь попасть в коленчатый патрубок 504 и вернуться через отверстие 502 в зону реакции 28. Таким образом, в отверстии 502 может возникнуть противоток поступающей реакционной среды 36 и выходящего отделившегося газа. Деаэрированная суспензия покидает резервуар для деаэрации 500 через трубопровод 506. Резервуар для деаэрации 500 включает направленную практически вертикально, преимущественно цилиндрическую боковую стенку 508, которая ограничивает зону деаэрации 510. Зона деаэрации 510 имеет высоту h и диаметр d. Расположенное на определенной отметке высоты отверстие 502 и коленчатый патрубок 504, предпочтительно, имеют тот же диаметр, что и диаметр d зоны деаэрации 510. Высота h, диаметр d, площадь и объем зоны деаэрации 510, преимущественно такие же, как и описанные выше для зоны деаэрации 312 резервуара для деаэрации 300, который приведен на фиг.16-18.
Фиг.19 и 20 поясняют вариант конструкции барботажной колонны реакторного типа 20, где твердый продукт (в частности, сырец терефталевой кислоты), полученный в зоне реакции 28, выводится из зоны реакции 28 через расположенное на определенной отметке высоты выходное отверстие. Выгрузка аэрированной реакционной среды 36 через расположенный на определенной высоте уровень, который находится выше днища барботажной колонны реакторного типа 20, может помочь избежать накопления и застоя слабо аэрированной реакционной среды 36 в нижней части 52 зоны реакции 28. В соответствии с другими аспектами настоящего изобретения, концентрация кислорода и способного окисляться соединения (в частности, пара-ксилола) в реакционной среде 36 вблизи верхней части реакционной среды 36, преимущественно, ниже, чем вблизи днища. Таким образом, выгрузка реакционной среды 36 через находящийся на определенной высоте уровень может повысить выход за счет снижения количества не прореагировавших исходных реагентов, которые выводятся из реактора 20. Кроме того, температура реакционной среды 36 значительно варьирует в вертикальном направлении в том случае, когда барботажная колонна реакторного типа 20 работает с большими значениями STR и градиентами химического состава, как указано в настоящем описании. В указанных условиях температура реакционной среды 36, как правило, имеет локальные минимумы вблизи верха и низа зоны реакции 28. Внизу зоны образование минимума связано с испарением растворителя вблизи того места, где поступает все количество окислителя или часть окислителя. Вверху зоны образование минимума связано вновь с испарением растворителя, однако в этом случае оно вызвано снижением давления внутри реакционной среды. Кроме того, могут образоваться дополнительные локальные минимумы между верхом и низом в том месте, где в зону реакции поступают дополнительные количества исходных веществ и окислителя. Между верхом и низом зоны реакции 28 также существует один или несколько температурных максимумов, вызванных выделением тепла при протекании экзотермической реакции окисления. Выгрузка реакционной среды 36 через находящийся на определенной высоте уровень с более высокой температурой может обеспечить особые преимущества в том случае, когда последующие стадии обработки протекают при более высокой температуре, поскольку снижаются потери энергии, связанные с нагреванием выгружаемой реакционной среды, которая направляется для проведения процессов последующей обработки.
Таким образом, в предпочтительном варианте осуществления настоящего изобретения и особенно в том случае, когда последующие процессы обработки протекают при более высоких температурах, реакционную среду 36 выгружают из барботажной колонны реакторного типа 20 через расположенное(ые) на определенной высоте выходное(ые) отверстие(ия), которое(ые) находится(ятся) выше уровня(ей), где в зону реакции 28 входит, по крайней мере, 50% мас. потока жидкофазных исходных веществ и/или потока газофазного окислителя. Более предпочтительно, реакционную среду 36 выводят из барботажной колонны реакторного типа 20 через расположенное(ые) на определенной высоте выходное(ые) отверстие(ия), которое(ые) находится(ятся) выше уровня(ей), где в зону реакции 28 входит практически все количество потока жидкофазных исходных веществ и/или потока газофазного окислителя. Предпочтительно, по меньшей мере, 50% мас. компонентов твердой фазы и жидкой фазы, извлекаемых из барботажной колонны реакторного типа 20, выводится из барботажной колонны реакторного типа 20 через расположенное(ые) на определенной высоте выходное(ые) отверстие(ия). Более предпочтительно, практически все компоненты твердой фазы и жидкой фазы, извлекаемых из барботажной колонны реакторного типа 20, выводятся из барботажной колонны реакторного типа 20 через расположенное(ые) на определенной высоте выходное(ые) отверстие(ия). Предпочтительно, расположенное(ые) на определенной высоте выходное(ые) отверстие(ия) располагается(ются), по крайней мере, на расстоянии приблизительно 1D выше нижнего уровня 52 зоны реакции 28. Более предпочтительно, расположенное(ые) на определенной высоте выходное(ые) отверстие(ия) располагается(ются), по крайней мере, на расстоянии приблизительно 2D выше нижнего уровня 52 зоны реакции 28. Наиболее предпочтительно, расположенное(ые) на определенной высоте выходное(ые) отверстие(ия) располагается(ются), по крайней мере, на расстоянии 3D выше нижнего уровня 52 зоны реакции 28. При заданной высоте H реакционной среды 36 расположенное(ые) на определенной высоте выходное(ые) отверстие(я) размещается(ются), предпочтительно, высоты от приблизительно 0,2Н до приблизительно 0,8Н, более предпочтительно, высоты от приблизительно 0,3Н до приблизительно 0,7Н и, наиболее предпочтительно, высоты от 0,4Н и до 0,6Н. Кроме того, температура реакционной среды 36 на уровне расположенного на определенной высоте выходного отверстия из зоны реакции 28, предпочтительно, по крайней мере, на 1°С выше, чем температура реакционной среды 36 у нижнего уровня 52 зоны реакции 28. Более предпочтительно, температура реакционной среды 36 на уровне выходного отверстия из зоны реакции 28, расположенного на определенной высоте, от приблизительно 1,5 до приблизительно 16°С выше, чем температура реакционной среды 36 у нижнего уровня 52 зоны реакции 28. Наиболее предпочтительно, температура реакционной среды 36 на уровне выходного отверстия из зоны реакции 28, расположенного на определенной высоте, в диапазоне от 2 до 12°С выше, чем температура реакционной среды 36 у нижнего уровня 52 зоны реакции 28.
Что касается фиг.21, то здесь представлена барботажная колонна реакторного типа 20, которая включает альтернативный гибридный резервуар для деаэрации 600, располагающийся у днища реактора 20. В данной конфигурации аэрированная реакционная среда 36 выгружается из зоны реакции 28 через относительно большое отверстие 602 у нижнего уровня 52 кожуха реактора 22. Отверстие 602 обозначает открытый верхний конец резервуара для деаэрации 600. В резервуаре для деаэрации 600 газовая фаза движется вверх, в то время как твердая и жидкая фазы движутся вниз. Поднимающаяся вверх газовая фаза может вновь попасть в зону реакции 28 через отверстие 602. Таким образом, в отверстии 602 может возникнуть противоток поступающей реакционной среды 36 и выходящего отделившегося газа. Деаэрированная суспензия покидает резервуар для деаэрации 600 через трубопровод 604. Резервуар для деаэрации 600 включает направленную практически вертикально, преимущественно цилиндрическую боковую стенку 606, которая обозначает зону деаэрации 608. Зона деаэрации 608 имеет высоту h и диаметр d. Отверстие 602, предпочтительно, имеет диаметр, который совпадает с диаметром или превосходит диаметр d зоны деаэрации 608. Высота h, диаметр d, площадь и объем зоны деаэрации 608, преимущественно такие же, как и описанные выше при рассмотрении зоны деаэрации 312 резервуара для деаэрации 300, который приведен на фиг.16-18.
Что касается фиг.22, то здесь показано, что барботажная колонна реакторного типа 20, представленная на фиг.21, включает альтернативный барботер 602 для ввода окислителя. Барботер 602 для ввода окислителя включает кольцевой элемент 622 и пару входных трубопроводов 624, 626. Кольцевой элемент 622 имеет практически ту же самую форму, что кольцевой элемент 202, рассмотренный выше со ссылкой на фиг.12-15. Входные трубопроводы 624,626 направлены вверх через отверстия в днище 48 кожуха реактора 22 и подают поток окислителя в кольцевой элемент 622.
Что касается фиг.23, то здесь показано, что барботажная колонна реакторного типа 20, представленная на фиг.21, включает не содержащее барботер устройство для подачи потока окислителя в зону реакции 28. В конфигурации на фиг.23 поток окислителя подается в реактор 20 через трубопроводы 630, 632 для ввода окислителя. Трубопроводы 630, 632 для ввода окислителя соединены с соответствующими отверстиями 634, 636 для ввода окислителя в днище 48 кожуха реактора 22. Поток окислителя непосредственно направляется в зону реакции 28 через отверстия 634, 636 для ввода окислителя. Могут быть установлены необязательные отражательные пластины 638, 640 с целью отклонить поток окислителя, когда он впервые попадает в зону реакции 28.
Как указано ранее, реактор для проведения процессов окисления, предпочтительно, должен иметь такую конфигурацию и функционировать таким образом, чтобы в нем в реакционной среде не образовывались зоны с высокой концентрацией способного окисляться соединения, поскольку подобные зоны могут привести к образованию примесей. Один из способов улучшения первоначального диспергирования способного окисляться соединения (в частности, пара-ксилола) в реакционной среде, заключается в разбавлении способного окисляться соединения жидкостью. Жидкость, которую применяют для разбавления способного окисляться соединения, может выделяться из части реакционной среды, которая расположена на значительном удалении от отметки высоты, где способное окисляться соединение вводится в зону реакции. Указанная жидкость из отдаленной части реакционной среды может циркулировать до отметки высоты, которая находится рядом с отметкой высоты, где способное окисляться соединение вводится через трубопровод, который располагается внутри и/или снаружи основного реактора.
На фиг.24 и 25 показаны два предпочтительных способа поступления жидкости из отдаленной части реакционной среды до отметки высоты рядом с входом для подачи способного окисляться соединения с использованием внутреннего (фиг.24) и внешнего (фиг.25) трубопровода. Длина напорного трубопровода от его входа (т.е. отверстия(й), где жидкость попадает в трубопровод) до его выхода (т.е. отверстия(й), где жидкость выходит из трубопровода) превышает, приблизительно 1 м, более предпочтительно, превышает, приблизительно 3 м, еще более предпочтительно, превышает, приблизительно 6 м и, и наиболее предпочтительно, превышает, 9 м. Однако действительная длина реактора становится несущественной в том случае, когда жидкость поступает из отдельного резервуара, который может располагаться непосредственно над реактором или рядом с реактором, в который первоначально попадает способное окисляться соединение. Жидкость из любого отдельного резервуара, которая содержит, по меньшей мере, некоторое количество реакционной среды, является предпочтительным источником для осуществления первоначального разбавления способного окисляться соединения.
Независимо от его источника, поток жидкости через трубопровод, предпочтительно, имеет более низкую постоянную концентрацию способного окисляться соединения, чем реакционная среда, которая непосредственно примыкает, по крайней мере, к одному из выходов трубопровода. Кроме того, жидкость, протекающая по трубопроводу, предпочтительно, имеет в жидкой фазе концентрацию способного окисляться соединения, которая составляет меньше чем приблизительно 100000 м.д. мас., более предпочтительно, составляет меньше чем приблизительно 10000 м.д. мас., еще более предпочтительно, составляет меньше чем приблизительно 1000 м.д. мас. и, наиболее предпочтительно, составляет меньше чем 100 м.д. мас., при этом концентрацию измеряют перед добавлением в трубопровод инкремента исходного способного окисляться соединения и какого-либо необязательного отдельного исходного растворителя. Когда измерение проводят после добавления инкремента исходного способного окисляться соединения и необязательного исходного растворителя, то объединенный поток жидкости, попадающий в реакционную среду, предпочтительно, имеет в жидкой фазе концентрацию способного окисляться соединения, которая составляет меньше чем приблизительно 300000 м.д. мас., более предпочтительно, составляет меньше чем приблизительно 50000 м.д. мас. и, наиболее предпочтительно, составляет меньше чем 10000 м.д. мас.
Желательно поддерживать достаточно низкую скорость потока через трубопровод так, чтобы циркулирующая жидкость действительно ограничивала требуемый предельный градиент для способного окисляться соединения в реакционной среде. В связи с этим отношение массы жидкой фазы в зоне реакции, куда изначально добавляется инкремент способного окисляться соединения, к скорости массопереноса жидкости, которая проходит через трубопровод, предпочтительно, должно составлять больше чем приблизительно 0,3 мин, более предпочтительно, должно составлять больше чем приблизительно 1 мин, еще более предпочтительно, должно составлять от приблизительно 2 мин до приблизительно 120 мин и, наиболее предпочтительно, должно составлять от 3 мин до 60 мин.
Существует множество способов заставить жидкость проходить через трубопровод. Предпочтительными средствами являются сила тяжести, эжекторы всех типов, в которых в качестве жидкого носителя применяют газ или жидкость или же их сочетание, а также механические насосы всех типов. В том случае, когда используется эжектор, то в одном из вариантов осуществления настоящего изобретения в качестве жидкого носителя используется, по крайней мере, одна жидкость, выбранная из группы, которая включает способное окисляться исходное соединение (жидкость или газ), исходный окислитель (газ), исходный растворитель (жидкость) и подаваемую с помощью насоса реакционную среду (суспензию). В другом варианте осуществления настоящего изобретения в качестве жидкого носителя используется, по меньшей мере, две жидкости, выбранные из группы, которая включает способное окисляться исходное соединение, исходный окислитель и исходный растворитель. В еще одном варианте осуществления настоящего изобретения в качестве жидкого носителя используется комбинация исходного способного окисляться соединения, исходного окислителя и исходного растворителя.
Подходящий диаметр или диаметры трубопровода, через который осуществляется циркуляция, могут варьировать в зависимости от количества и свойств подаваемого вещества, энергии, которая способна вызвать движение потока жидкости, и величины капитальных затрат. Минимальный диаметр подобного трубопровода, предпочтительно, составляет больше чем приблизительно 0,02 м, более предпочтительно, составляет от приблизительно 0,06 м до приблизительно 2 м и, наиболее предпочтительно, составляет от 0,12 м до 0,8 м.
Как указано ранее, желательно контролировать поток через трубопровод в определенных заданных пределах. Из области техники известно множество средств для осуществления подобного контроля путем выбора подходящей заданной конфигурации в процессе конструирования напорного трубопровода. Другим предпочтительным вариантом конструкции является использование структурных элементов, которые могут меняться в процессе проведения процесса, а именно включают вентили всех типов и с любыми характеристиками, в том числе вентили, управляемые не только вручную, но и приводом от сервомотора, с использованием различных устройств, включая петли с обратным контролем потока с помощью датчиков или без помощи датчиков. Другими предпочтительными средствами контролирования потока жидкости-разбавителя являются варьирование подводимой энергии между входным и выходным отверстиями трубопровода. Предпочтительные средства включают изменение скорости подачи одного или нескольких жидких носителей, направляемых в эжектор, изменение энергии, подводимой к приводу насоса, изменение разницы в плотности или изменение разницы в уровнях, если используется сила тяжести. Может также применяться любое сочетание указанных предпочтительных средств.
Трубопровод, который используется для осуществления циркуляции жидкости из реакционной среды, может представлять собой трубопровод любого известного из области техники типа. В одном из вариантов осуществления настоящего изобретения трубопровод конструируют целиком или по частям, используя обычные материалы для прокладки трубопроводов. В другом варианте осуществления настоящего изобретения трубопровод конструируют целиком или по частям, используя стенку реактора в качестве одной из частей трубопровода. Трубопровод может быть сконструирован таким образом, что он целиком находится в границах реактора (фиг.24), или же трубопровод может быть сконструирован таким образом, что он целиком находится за пределами реактора (фиг.25), или же трубопровод может содержать секции, которые располагаются как внутри, так и снаружи реактора.
Авторы настоящего изобретения полагают, что в особенности в больших реакторах желательно иметь множество трубопроводов и использовать различные схемы движения жидкости через трубопровод. Кроме того, может оказаться желательным проделать множество выходных отверстий в разных местах одного из трубопроводов или всех трубопроводов. Особенности конструкции, в соответствии с другими аспектами настоящего изобретения, обеспечат баланс требуемого общего градиента между стационарными концентрациями способного окисляться соединения с помощью необходимого первоначального разбавления способного окисляться исходного соединения.
Обе фиг.24 и 25 поясняют конструкции, в которых применяется резервуар для деаэрации, соединенный с трубопроводом. Указанный резервуар для деаэрации гарантирует, что часть реакционной среды, которая используется для разбавления поступающего способного окисляться соединения, будет представляет собой практически деаэрированную суспензию. Следует однако отметить, что жидкость или суспензия, которая используется для разбавления поступающего способного окисляться соединения, может быть в аэрированной форме, а также деаэрированной форме.
Использование проходящей через трубопровод жидкости для осуществления разбавления исходного способного окисляться соединения наиболее удобно в барботажных колоннах реакторного типа. Более того, в барботажных колоннах реакторного типа преимущества, связанные с первоначальным разбавлением исходного способного окисляться соединения, могут быть достигнуты и без добавления исходного способного окисляться соединения непосредственно в трубопровод, при условии, что выходное отверстие трубопровода располагается достаточно близко к тому месту, где осуществляется подача способного окисляться соединения. В таком варианте осуществления настоящего изобретения выходное отверстие трубопровода, предпочтительно, располагается на расстоянии, которое составляет в пределах приблизительно 27 диаметров выходного отверстия трубопровода, от ближайшего места, куда подается способное окисляться соединения, более предпочтительно, располагается на расстоянии в пределах приблизительно 9 диаметров выходного отверстия трубопровода, еще более предпочтительно, располагается на расстоянии в пределах приблизительно 3 диаметра выходного отверстия трубопровода и, наиболее предпочтительно, располагается на расстоянии в пределах 1 диаметра выходного отверстия трубопровода.
Было также обнаружено, что для первоначального разбавления исходного способного окисляться соединения в барботажных колоннах реакторного типа, в соответствии с одним из вариантов осуществления настоящего изобретения, могут использоваться эжекторы потока, даже без применения трубопроводов, предназначенных для получения используемой для разбавления жидкости из отдаленной части реакционной среды. В таких случаях эжектор располагается внутри реакционной среды и имеет открытый канал, направленный из реакционной среды в горловину эжекционного устройства, куда низкое давление увлекает соседствующую реакционную среду. Примеры двух возможных форм эжекторов приведены на фиг.26 и 27. В предпочтительном варианте конструкции указанных эжекторов ближайшее место подачи способного окисляться соединения находится на расстоянии в пределах приблизительно 4 м, более предпочтительно, находится на расстоянии в пределах приблизительно 1 м и, наиболее предпочтительно, находится на расстоянии 0,3 м от горловины эжектора. В другом варианте осуществления настоящего изобретения способное окисляться соединение подается под давлением в качестве жидкого носителя. В еще одном варианте осуществления настоящего изобретения либо растворитель, либо окислитель подается под давлением в качестве дополнительного жидкого носителя вместе со способным окисляться соединением. Наконец, в еще одном варианте осуществления настоящего изобретения и растворитель, и окислитель одновременно подаются под давлением в качестве дополнительного жидкого носителя вместе со способным окисляться соединением.
Авторы настоящего изобретения полагают, что в особенности в больших реакторах желательным может оказаться использование множества эжекторов различной формы, которые размещаются в различных местах внутри реакционной среды. Особенности конструкции, в соответствии с другими аспектами настоящего изобретения, обеспечат баланс требуемого общего градиента между стационарными концентрациями способного окисляться соединения с помощью необходимого первоначального разбавления способного окисляться исходного соединения. Кроме того, авторы настоящего изобретения полагают, что струи на выходе из эжектора могут быть ориентированы в любом направлении. Если используется множество эжекторов, то каждый из них может быть ориентирован индивидуально и в любом направлении.
Как указано выше, определенные физические и эксплуатационные характеристики барботажной колонны реакторного типа 20, которая была описана выше со ссылкой на фиг.1-27, обеспечивают образование вертикальных градиентов давления, температуры и концентраций реагентов (т.е. кислорода и способного окисляться соединения) в реакционной среде 36. Как уже обсуждалось выше, указанные вертикальные градиенты способствуют протеканию более эффективного и экономичного процесса окисления, по сравнению с обычными процессами окисления, в которых образуется хорошо перемешиваемая реакционная среда с относительно однородным давлением, температурой и концентрациями реагентов во всем объеме реакционной среды. Образование градиентов для кислорода, способного окисляться соединения (в частности, пара-ксилола) и температуры по высоте позволяет использовать окислительную систему, в соответствии с вариантом осуществления настоящего изобретения, который будет далее рассмотрен более подробно.
Если обратиться теперь к фиг.28, то для количественной оценки градиентов концентраций реагентов, которые существуют в реакционной среде 36 при проведении окисления в барботажной колонне реакторного типа 20, весь объем реакционной среды 36 можно теоретически разбить на 30 дискретных горизонтальных слоев равного объема. Фиг.28 поясняет концепцию деления реакционной среды 36 на 30 дискретных горизонтальных слоев равного объема. За исключением самого верхнего и самого нижнего горизонтального слоев, каждый горизонтальный слой представляет собой дискретный объем, который ограничен сверху и снизу воображаемыми горизонтальными плоскостями, а с боков ограничен стенкой реактора 20. Самый верхний горизонтальный слой ограничен снизу воображаемой горизонтальной плоскостью, а сверху ограничен верхней поверхностью реакционной среды 36. Самый нижний горизонтальный слой ограничен сверху воображаемой горизонтальной плоскостью, а снизу ограничен днищем кожуха сосуда. После того, как реакционная среда 36 теоретически разбита на 30 дискретных горизонтальных слоев равного объема, можно определить усредненную по времени и по объему концентрацию в каждом горизонтальном слое. Индивидуальный горизонтальный слой, имеющий максимальную концентрацию из всех 30 горизонтальных слоев, можно обозначить как “C-max горизонтальный слой”. Индивидуальный горизонтальный слой, расположенный над C-max горизонтальным слоем и имеющий наименьшую концентрацию из всех горизонтальных слоев, расположенных над C-max горизонтальным слоем, можно обозначить как “C-min горизонтальный слой”. Теперь градиент концентрации по высоте можно рассчитать как отношение концентрации C-max горизонтального слоя к концентрации C-min горизонтального слоя.
Что касается количественной оценки градиента концентрации кислорода, то после того, как реакционная среда 36 теоретически разделена на 30 дискретных горизонтальных слоев равного объема, О2-max горизонтальный слой идентифицируют как слой, имеющий максимальную концентрацию кислорода из всех 30 горизонтальных слоев, а О2-min горизонтальный слой идентифицируют как слой, имеющий минимальную концентрацию кислорода из горизонтальных слоев, расположенных над О2-max горизонтальным слоем. Концентрацию кислорода в горизонтальных слоях измеряют в газовой фазе реакционной среды 36 и выражают в виде усредненного по времени и объему молярного значения относительно жидкой фазы. Отношение концентрации кислорода О2-max горизонтального слоя к концентрации кислорода О2-min горизонтального слоя, предпочтительно, составляет от приблизительно 2:1 до приблизительно 25:1, более предпочтительно составляет от приблизительно 3:1 до приблизительно 15:1 и, наиболее предпочтительно составляет от 4:1 до 10:1.
Как правило, О2-max горизонтальный слой располагается вблизи нижней части реакционной среды 36, в то время как О2-min горизонтальный слой располагается вблизи верхней части реакционной среды 36. Предпочтительно, О2-min горизонтальный слой является одним из 5 наиболее высоко расположенных горизонтальных слоев из всех 30 дискретных горизонтальных слоев. Наиболее предпочтительно, О2-min горизонтальный слой представляет собой самый верхний из 30 дискретных горизонтальных слоев, как показано на фиг.28. Предпочтительно, О2-max горизонтальный слой является одним из 10 наиболее низко расположенных горизонтальных слоев из всех 30 дискретных горизонтальных слоев. Наиболее предпочтительно, О2-max горизонтальный слой представляет собой один из 5 наиболее низко расположенных 30 дискретных горизонтальных слоев. Например, на фиг.28 О2-max горизонтальный слой обозначен как третий горизонтальный слой от днища реактора 20. Расстояние по высоте между О2-min и О2-max горизонтальными слоями, предпочтительно, составляет, по меньшей мере, приблизительно 2W, более предпочтительно, составляет, по меньшей мере, приблизительно 4W и, наиболее предпочтительно, составляет, по меньшей мере, 6W. Расстояние по высоте между О2-min и О2-max горизонтальными слоями, предпочтительно, составляет, по меньшей мере, приблизительно 0,2Н, более предпочтительно, составляет, по меньшей мере, приблизительно 0,4Н и, наиболее предпочтительно, составляет, по меньшей мере, 0,6Н.
Усредненная по времени и объему концентрация кислорода относительно жидкой фазы в О2-min горизонтальном слое, предпочтительно, составляет от приблизительно 0,1 до приблизительно 3% мол., более предпочтительно, составляет от приблизительно 0,3 до приблизительно 2% мол. и, наиболее предпочтительно, составляет от 0,5 до 1,5% мол. Усредненная по времени и объему концентрация кислорода в О2-max горизонтальном слое, предпочтительно, составляет от приблизительно 4 до приблизительно 20% мол., более предпочтительно, составляет от приблизительно 5 до приблизительно 15% мол. и, наиболее предпочтительно, составляет от 6 до 12% мол. Усредненная по времени концентрация кислорода относительно сухого продукта в отходящем газовом потоке, который отводится из реактора 20 через выходное отверстие 40, предпочтительно, составляет от приблизительно 0,5 до приблизительно 9% мол., более предпочтительно, составляет от приблизительно 1 до приблизительно 7% мол. и, наиболее предпочтительно, составляет от 1,5 до 5% мол.
Поскольку концентрация кислорода столь быстро снижается по направлению к верхней части реакционной среды 36, то желательно, чтобы потребность в кислороде снижалась в верхней части реакционной среды 36. Указанного сокращения потребности в кислороде вблизи верхней границы реакционной среды 36 можно добиться за счет создания вертикального градиента концентрации способного окисляться соединения (в частности, пара-ксилола), при этом минимальная концентрация способного окисляться соединения находится вблизи верхней части реакционной среды 36.
Что касается количественной оценки градиента концентрации способного окисляться соединения (в частности, пара-ксилола), то после того, как реакционная среда 36 теоретически разбита на 30 дискретных горизонтальных слоев равного объема, идентифицируют ОС-max горизонтальный слой как слой, в котором концентрация способного окисляться соединения максимальна из всех 30 горизонтальных слоев, и идентифицируют ОС-min горизонтальный слой как слой, в котором концентрация способного окисляться соединения минимальна из горизонтальных слоев, которые расположены над ОС-max горизонтальным слоем. Концентрацию способного окисляться соединения в горизонтальных слоях определяют в жидкой фазе и выражают в виде усредненного по времени и объему значения массовой доли. Отношение концентрации способного окисляться соединения в ОС-max горизонтальном слое к концентрации способного окисляться соединения в ОС-min горизонтальном слое, предпочтительно, составляет больше чем приблизительно 5:1, более предпочтительно, составляет больше чем приблизительно 10:1, еще более предпочтительно, составляет больше чем приблизительно 20:1 и, наиболее предпочтительно, составляет от 40:1 до 1000:1.
Как правило, ОС-max горизонтальный слой располагается вблизи нижней части реакционной среды 36, в то время как ОС-min горизонтальный слой располагается вблизи верхней части реакционной среды 36. Предпочтительно, ОС-min горизонтальный слой представляет собой один из 5 самых верхних горизонтальных слоев из всех 30 дискретных горизонтальных слоев. Наиболее предпочтительно, ОС-min горизонтальный слой является самым верхним из 30 дискретных горизонтальных слоев, как показано на фиг.28. Предпочтительно, ОС-max горизонтальный слой представляет собой один из 10 самых нижних горизонтальных слоев из всех 30 дискретных горизонтальных слоев. Наиболее предпочтительно, ОС-max горизонтальный слой представляет собой один из 5 самых нижних горизонтальных слоев из всех 30 дискретных горизонтальных слоев. Например, на фиг.28 показано, что ОС-max горизонтальный слой является пятым горизонтальным слоем от днища реактора 20. Расстояние по высоте между ОС-min и ОС-max горизонтальными слоями, предпочтительно, составляет, по меньшей мере, приблизительно 2W, где W обозначает максимальную ширину реакционной среды 36. Более предпочтительно, расстояние по высоте между ОС-min и ОС-max горизонтальными слоями составляет, по меньшей мере, приблизительно 4W и, наиболее предпочтительно, составляет, по меньшей мере, 6W. При заданном значении высоты H реакционной среды 36 расстояние по высоте между ОС-min и ОС-max горизонтальными слоями, предпочтительно, составляет, по меньшей мере, приблизительно 0,2Н, более предпочтительно, составляет, по меньшей мере, приблизительно 0,4Н и, наиболее предпочтительно, составляет, по меньшей мере, 0,6Н.
Усредненная по времени и объему концентрация способного окисляться соединения (в частности, пара-ксилола) в жидкой фазе ОС-min горизонтального слоя, предпочтительно, составляет меньше чем приблизительно 5000 м.д. мас., более предпочтительно, составляет меньше чем приблизительно 2000 м.д. мас., еще более предпочтительно, составляет меньше чем приблизительно 400 м.д. мас. и, наиболее предпочтительно, составляет от 1 м.д. мас. до 100 м.д. мас. Усредненная по времени и объему концентрация способного окисляться соединения в жидкой фазе ОС-max горизонтального слоя, предпочтительно, составляет от приблизительно 100 м.д. мас. до приблизительно 10000 м.д. мас., более предпочтительно, составляет от приблизительно 200 м.д. мас. до приблизительно 5000 м.д. мас., и, наиболее предпочтительно, составляет от 500 м.д. мас. до 3000 м.д. мас.
Несмотря на то, что в барботажной колонне реакторного типа 20 предпочтительно создаются градиенты концентрации способного окисляться соединения по высоте, объемный процент реакционной среды 36, содержащей концентрацию способного окисляться соединения в жидкой фазе больше чем 1000 м.д. мас., предпочтительно, должен быть минимальным. Усредненный по времени объемный процент реакционной среды 36, содержащей концентрацию способного окисляться соединения в жидкой фазе больше чем 1000 м.д. мас., предпочтительно, составляет меньше чем приблизительно 9%, более предпочтительно, составляет меньше чем приблизительно 6% и, наиболее предпочтительно, составляет меньше чем 3%. Усредненный по времени объемный процент реакционной среды 36, концентрация способного окисляться соединения в жидкой фазе которой превышает 2500 м.д. мас., предпочтительно, составляет меньше чем приблизительно 1,5%, более предпочтительно, составляет меньше чем приблизительно 1% и, наиболее предпочтительно, составляет меньше чем 0,5%. Усредненный по времени объемный процент реакционной среды 36, концентрация способного окисляться соединения в жидкой фазе которой превышает 10000 м.д. мас., предпочтительно, составляет меньше чем приблизительно 0,3%, более предпочтительно, составляет меньше чем приблизительно 0,1% и, наиболее предпочтительно, составляет меньше чем 0,03%. Усредненный по времени объемный процент реакционной среды 36, концентрация способного окисляться соединения в жидкой фазе которой превышает 25000 м.д. мас., предпочтительно, составляет меньше чем приблизительно 0,03%, более предпочтительно, составляет меньше чем приблизительно 0,015% и, наиболее предпочтительно, составляет меньше чем 0,007%. Авторы настоящего изобретения отмечают, что объем реакционной среды 36, содержащий повышенные уровни способного окисляться соединения, не обязательно должны находиться в одном непрерывном объеме. Во многих случаях хаотические картины потоков в барботажной колонне реакторного типа образуют две или больше непрерывных, но изолированные друг от друга порций реакционной среды 36, которые имеют повышенные уровни способного окисляться соединения. В каждый момент времени, когда проводят усреднение по времени, все подобные непрерывные, но изолированные друг от друга объемы, превышающие 0,0001 об.% от общего количества реакционной среды, добавляют друг к другу, чтобы определить общий объем, который содержит повышенные уровни концентраций способного окисляться соединения в жидкой фазе.
Помимо концентрационных градиентов кислорода и способного окисляться соединения, которые рассмотрены выше, в реакционной среде 36, предпочтительно, существует температурный градиент. Если вновь обратиться к фиг.28, то указанный температурный градиент можно количественно оценить так же, как и концентрационные градиенты, путем теоретического разбиения реакционной среды на 30 дискретных горизонтальных слоев равного объема и определения усредненной по времени и усредненной по объему температуры в каждом слое. Горизонтальный слой с наименьшей температурой из 15 самых нижних слоев может быть идентифицирован как T-min горизонтальный слой, а горизонтальный слой, расположенный над T-min горизонтальным слоем и имеющий максимальную температуру из всех слоев, расположенных над T-min горизонтальным слоем, может быть идентифицирован как “T-max горизонтальный слой”. Температура T-max горизонтального слоя, предпочтительно, по крайней мере, приблизительно на 1°С выше, чем температура T-min горизонтального слоя. Более предпочтительно, температура T-max горизонтального слоя в диапазоне приблизительно от 1,25 до приблизительно 12°С выше, чем температура T-min горизонтального слоя. Наиболее предпочтительно, температура T-max горизонтального слоя в диапазоне от 2 до 8°С выше, чем температура T-min горизонтального слоя. Температура T-max горизонтального слоя, предпочтительно, составляет от приблизительно 125 до приблизительно 200°С, более предпочтительно, составляет от приблизительно 140 до приблизительно 180°С и, наиболее предпочтительно, составляет от 150 до 170°С.
Как правило, T-max горизонтальный слой будет располагаться вблизи центральной части реакционной среды 36, в то время как T-min горизонтальный слой будет располагаться вблизи нижней части реакционной среды 36. T-min горизонтальным слоем, предпочтительно, является один из 10 самых нижних горизонтальных слоев из 15 наиболее низко расположенных горизонтальных слоев. Более предпочтительно, T-min горизонтальным слоем является один из 5 самых нижних горизонтальных слоев из 15 наиболее низко расположенных горизонтальных слоев. Например, на фиг.28 T-min горизонтальный слой показан как второй горизонтальный слой от дна реактора 20. T-max горизонтальным слоем, предпочтительно, является один из 20 срединных горизонтальных слоев из всех 30 горизонтальных слоев. Более предпочтительно, T-max горизонтальным слоем, является один из 14 срединных горизонтальных слоев из всех 30 горизонтальных слоев. Например, на фиг.28 T-max горизонтальный слой изображен как двадцатый слой от дна реактора 20 (т.е. один из 10 срединных горизонтальных слоев). Расстояние по высоте между T-min и T-max горизонтальными слоями, предпочтительно, составляет, по меньшей мере, приблизительно 2W, более предпочтительно, составляет, по меньшей мере, приблизительно 4W и, наиболее предпочтительно, составляет, по меньшей мере, 6W. Расстояние по высоте между T-min и T-max горизонтальными слоями, предпочтительно, составляет, по меньшей мере, приблизительно 0,2Н, более предпочтительно, составляет, по меньшей мере, приблизительно 0,4Н и, наиболее предпочтительно, составляет, по меньшей мере, 0,6Н.
Как указано выше, в том случае, когда в реакционной среде 36 существует температурный градиент по высоте, то реакционная среда 36 преимущественно выгружается на определенной отметке высоты, где температура реакционной среды является наибольшей, особенно в том случае, когда извлекаемый продукт на следующих этапах подвергается переработке при более высоких температурах. Таким образом, в том случае, когда реакционная среда 36 выгружается из зоны реакции 28 через одно или несколько расположенных на определенной отметке высоты выходных отверстий, как показано на фиг.19 и 20, то расположенное(ые) на определенной отметке высоты выходное(ые) отверстие(ия) должно(ы) располагаться вблизи T-max горизонтального слоя. Расположенное на определенной отметке высоты выходное отверстие, предпочтительно, находится в пределах 10 горизонтальных слоев от T-max горизонтального слоя, более предпочтительно, находится в пределах 5 горизонтальных слоев от T-max горизонтального слоя и, наиболее предпочтительно, находится в пределах 2 горизонтальных слоев от T-max горизонтального слоя.
Далее следует отметить, что многие из рассмотренных в данном описании особенностей настоящего изобретения могут быть использованы в различных реакторных системах для проведения окислительных процессов, а не только в системах, в которых применяют единственный реактор для проведения процессов окисления. Кроме того, некоторые из особенностей настоящего изобретения могут быть использованы в реакторах с механическим перемешиванием и/или реакторах с проточным перемешиванием, а не только в барботажных реакторах (т.е. барботажных колоннах реакторного типа). Например, авторы настоящего изобретения обнаружили ряд преимуществ, связанных со ступенчатым распределением/изменением концентрации кислорода и/или скорости потребления кислорода внутри реакционной среды. Преимущества, предоставляемые ступенчатым распределением концентрации/потреблением кислорода в реакционной среде, могут быть реализованы независимо от того, содержится ли полный объем реакционной среды в единственном реакторе или же в нескольких реакторах. Кроме того, преимущества, предоставляемые ступенчатым распределением концентрации/потреблением кислорода в реакционной среде, могут быть реализованы независимо от того, является(ются) ли реактор(ы) реактором(ами) с механическим перемешиванием, с проточным перемешиванием и/или с барботажным перемешиванием.
Один из способов количественной оценки формирования ступенчатого распределения концентрации кислорода и/или скорости потребления кислорода в реакционной среде заключается в сравнении двух или нескольких отдельных 20%-х непрерывных объемов реакционной среды. Указанные 20%-е непрерывные объемы необязательно должны иметь определенную форму. Тем не менее, каждый из 20%-х непрерывных объемов должен быть образован смежным объемом реакционной среды (т.е. каждый объем должен быть “непрерывным”) и 20%-е непрерывные объемы не должны перекрывать друг друга (т.е. объемы являются “отдельными”). На фиг.29-31 показано, что указанные отдельные 20%-е непрерывные объемы могут располагаться в одном и том же реакторе (фиг.29) или в нескольких реакторах (фиг.30 и 31). Следует отметить, что ректоры, приведенные на фиг.29-31, могут быть реакторами с механическим перемешиванием, с проточным перемешиванием и/или с барботажным перемешиванием. В одном из вариантов осуществления настоящего изобретения реакторы, приведенные на фиг.29-31, предпочтительно являются барботажными реакторами (т.е. барботажными реакторными колоннами).
Что касается фиг.29, то здесь представлен реактор 20, который содержит реакционную среду 36. Реакционная среда 36 включает первый отдельный 20%-й непрерывный объем 37 и второй отдельный 20%-й непрерывный объем 39.
Что касается фиг.30, то здесь представлена многореакторная система, которая включает первый реактор 720а и второй реактор 720b. Реакторы 720а,b в совокупности содержат полный объем реакционной среды 736. Первый реактор 720а содержит первую порцию реакционной среды 736а, в то время как второй реактор 720b содержит вторую порцию реакционной среды 736b. Первый отдельный 20%-й непрерывный объем 737 реакционной среды 736 изображен как объем, заключенный в первом реакторе 720а, в то время как второй отдельный 20%-й непрерывный объем 739 реакционной среды 736 изображен как объем, заключенный во втором реакторе 720b.
Что касается фиг.31, то здесь представлена многореакторная система, которая включает первый реактор 820а, второй реактор 820b и третий реактор 820с. Реакторы 820а, b, с в совокупности содержат полный объем реакционной среды 836. Первый реактор 820а содержит первую порцию реакционной среды 836а; второй реактор 820b содержит вторую порцию реакционной среды 836b; и третий реактор 820 с содержит третью порцию реакционной среды 836 с. Первый отдельный 20%-й непрерывный объем 837 реакционной среды 836 изображен как объем, заключенный в первом реакторе 820а, второй отдельный 20%-й непрерывный объем 839 реакционной среды 836 изображен как объем, заключенный во втором реакторе 820b; а третий отдельный 20%-й непрерывный объем 841 реакционной среды 836 изображен как объем, заключенный в третьем реакторе 820с.
Ступенчатое распределение доступного кислорода в реакционной среде можно количественно оценить, рассматривая 20%-й непрерывный объем реакционной среды, который содержит наиболее богатую фракцию кислорода в газовой фазе, а также рассматривая 20%-й непрерывный объем реакционной среды, который содержит наиболее обедненную мольную фракцию кислорода в газовой фазе. В газовой фазе отдельного 20%-го непрерывного объема реакционной среды, содержащего наибольшую концентрацию кислорода в газовой фазе, усредненная по времени и объему концентрация кислорода относительно жидкой фазы, предпочтительно, составляет от приблизительно 3 до приблизительно 18% мол., более предпочтительно, составляет от приблизительно 3,5 до приблизительно 14% мол. и, наиболее предпочтительно, составляет от 4 до 10% мол. В газовой фазе отдельного 20%-го непрерывного объема реакционной среды, содержащего наименьшую концентрацию кислорода в газовой фазе, усредненная по времени и объему концентрация кислорода относительно жидкой фазы, предпочтительно, составляет от приблизительно 0,3 до приблизительно 5% мол., более предпочтительно, составляет от приблизительно 0,6 до приблизительно 4% мол. и, наиболее предпочтительно, составляет от 0,9 до 3% мол. Более того, отношение усредненной по времени и по объему концентрации кислорода относительно жидкой фазы наиболее обогащенного 20%-го непрерывного объема реакционной среды к наиболее обедненному 20%-му непрерывному объему реакционной среды, предпочтительно, составляет от приблизительно 1,5:1 до приблизительно 20:1, более предпочтительно, составляет от приблизительно 2:1 до приблизительно 12:1 и, наиболее предпочтительно, составляет от 3:1 до 9:1.
Ступенчатое распределение скорости потребления кислорода в реакционной среде можно количественно оценить в терминах STR для кислорода, которые уже рассмотрены выше. STR для кислорода была ранее рассмотрена в общем смысле (т.е. с точки зрения среднего значения STR для кислорода во всей реакционной среде); однако STR для кислорода можно также рассмотреть в локальном смысле (т.е. для порции реакционной среды) с тем, чтобы количественно оценить ступенчатое распределение скорости расходования кислорода по всей реакционной среде.
Авторы настоящего изобретения обнаружили, что целесообразно заставлять STR для кислорода согласованно варьировать по всей реакционной среде с рассмотренными в данном описании градиентами, касающимися давления в реакционной среде и мольной фракции молекулярного кислорода в газовой фазе реакционной среды. Так, отношение STR для кислорода в первом отдельном 20%-м непрерывном объеме реакционной среды к отношению STR для кислорода во втором отдельном 20%-м непрерывном объеме реакционной среды, предпочтительно, составляет от приблизительно 1,5:1 до приблизительно 20:1, более предпочтительно, составляет от приблизительно 2:1 до приблизительно 12:1 и, наиболее предпочтительно, составляет от 3:1 до 9:1. В одном из вариантов осуществления настоящего изобретения “первый отдельный 20%-й непрерывный объем” расположен ближе, чем “второй отдельный 20%-й непрерывный объем” к тому месту, откуда молекулярный кислород первоначально подается в реакционную среду. Указанные большие градиенты STR для кислорода желательны, если среда для проведения частичной реакции окисления содержится в барботажной окислительной реакторной колонне или в реакторе любого другого типа, в котором создаются градиенты давления и/или мольной фракции молекулярного кислорода в газовой фазе реакционной среды (в частности, в реакторе с механическим перемешиванием образуется несколько вертикально расположенных зон перемешивания, что достигается путем использования множества лопастей, формирующих сильный радиальный поток, который может усиливаться в основном с помощью системы горизонтальных разделительных перегородок, при этом поток окислителя обычно поднимается вверх из точки подачи, расположенной в нижней части реактора, независимо от того, что внутри каждой вертикальной перемешиваемой зоны может происходить противоточное смешивание окислителя, а некоторое обратное смешивание потока окислителя может происходить между соседними вертикальными перемешиваемыми зонами). Таким образом, авторы настоящего изобретения обнаружили, что если существует градиент давления и/или мольной фракции молекулярного кислорода в газовой фазе реакционной среды, то желательно создавать похожие градиенты в потребности растворенного кислорода для осуществления химической реакции с помощью приведенных в данном описании способов.
Предпочтительными способами вызвать локальные вариации STR для кислорода являются контролирование мест подачи способного окисляться соединения и контролирования смешивания жидкой фазы в реакционной среде, с целью управления градиентами концентрации способного окисляться соединения в соответствии с описанием настоящего изобретения. Другие пригодные способы вызвать локальные вариации STR для кислорода включают принудительное изменение активности реакции путем варьирования локальной температуры и путем изменения локальной смеси катализатора и компонентов растворителя (в частности, путем подачи газа, вызывающего охлаждение за счет испарения определенной части реакционной среды, и путем подачи потока растворителя, содержащего большее количество воды, с тем, чтобы снизить активность определенной части реакционной среды).
Как уже обсуждалось выше со ссылкой на фиг.30 и 31, может оказаться целесообразным проводить реакции частичного окисления в нескольких реакторах, при этом, по крайней мере, часть, предпочтительно, по меньшей мере, 25%, более предпочтительно, по меньшей мере, 50% и, наиболее предпочтительно, по меньшей мере, 75% молекулярного кислорода, выходящего из первого реактора, направляется в один или несколько последующих реакторов с целью потребления дополнительного количества, предпочтительно, составляющего более 10%, более предпочтительно, составляющего более 20% и, наиболее предпочтительно, составляющего более 40% от количества молекулярного кислорода, покидающего первый/расположенный выше по потоку реактор. При использовании подобного последовательного потока молекулярного кислорода от одного реактора к другим, желательно чтобы реакция в первом реакторе проводилась с большей интенсивностью, чем, по крайней мере, в одном из последующих реакторов, при этом отношение усредненного по реактору значения STR для кислорода в первом реакторе к усредненному по реактору значению STR для кислорода в последующем реакторе, предпочтительно, составляет от приблизительно 1,5:1 до приблизительно 20:1, более предпочтительно, составляет от приблизительно 2:1 до приблизительно 12:1 и, наиболее предпочтительно, составляет от 3:1 до 9:1.
Как уже указывалось выше, для создания последовательного потока молекулярного кислорода в последующие реакторы, в соответствии с настоящим изобретением, пригодны все типы используемых на первом этапе реакторов (в частности, ими могут быть барботажная колонна, реактор с механическим перемешиванием, реактор с обратным смешиванием, реактор с внутренним перераспределением, реактор плунжерного типа и т.д.) и все типы последующих реакторов, которые могут отличаться или не отличаться от типа реактора, который используют на первом этапе процесса. Средствами, позволяющими осуществлять снижение усредненного по всему реактору значения STR для кислорода в последующих реакторах обычно являются уменьшение температуры, снижение концентрации способного окисляться соединения и снижение активности конкретной смеси каталитических компонентов и растворителя (в частности, уменьшение концентрации кобальта, повышение содержания воды и введение ингибиторов катализатора в виде небольших добавок ионов меди).
В процессе перемещения от первого реактора к последующему реактору поток окислителя может быть подвергнут любому известному из области техники воздействию, такому как компрессия или снижение давления, охлаждение или нагрев, удаление массы или добавление массы в любом количестве или любого типа. Тем не менее, снижение усредненного по всему реактору значения STR для кислорода в последующих реакторах особенно полезно в том случае, когда абсолютное давление в верхней части первого реактора составляет меньше чем приблизительно 2,0 МПа, более предпочтительно, составляет меньше чем приблизительно 1,6 МПа и, наиболее предпочтительно, составляет меньше чем 1,2 МПа. Кроме того, снижение усредненного по всему реактору значения STR для кислорода в последующих реакторах особенно полезно в том случае, когда отношение абсолютного значения давления в верхней части первого реактора к абсолютному значению давлению в верхней части, по меньшей мере, одного последующего реактора составляет от приблизительно 0,5:1 до 6:1, более предпочтительно, составляет от приблизительно 0,6:1 до приблизительно 4:1 и, наиболее предпочтительно, составляет от 0,7:1 до 2:1. Снижение давления в последующих реакторах ниже указанных граничных значений приводит к чрезмерному снижению доступности молекулярного кислорода, а повышение давления сверх указанных граничных значений ведет к дополнительным затратам, по сравнению с использованием подачи свежей порции окислителя.
В том случае, когда применяется последовательный поток молекулярного кислорода в последующие реакторы с меньшей величиной усредненного значения STR для кислорода, свежие порции исходного способного окисляться соединения, растворителя и окислителя могут подаваться в последующий реактор и/или в первый реактор. Потоки жидкой фазы и твердой фазы, если они присутствуют в реакционной среде, могут двигаться в любом направлении между реакторами. Вся газовая фаза или часть газовой фазы, покидающая первый реактор и поступающая в последующий реактор, может перемещаться отдельно или же в виде смеси с порциями жидкой фазы и твердой фазы, если они присутствуют в реакционной среде, поступающей из первого реактора. Поток продуктов, состоящий из жидкой фазы и твердой фазы, если они присутствуют, может быть извлечен из реакционной среды в любом реакторе системы.
Если вновь вернуться к фиг.1-29, то окисление, предпочтительно, проводят в барботажной колонне реакторного типа 20 в условиях, которые заметно отличаются, в соответствии с приведенным в данном описании предпочтительным вариантом осуществления настоящего изобретения, от условий в обычных реакторах для проведения процессов окисления. В том случае, когда для проведения жидкофазного частичного окисления пара-ксилола в сырец терефталевой кислоты (СТА), в соответствии с приведенным в данном описании предпочтительным вариантом осуществления настоящего изобретения, используют барботажную колонну реакторного типа 20, то пространственные профили локальной интенсивности реакции, локальной интенсивности испарения и локальной температуры в сочетании с режимами потоков жидких веществ в реакционной среде и предпочтительными, относительно низкими температурами окисления способствуют образованию частиц СТА, которые обладают уникальными и предпочтительными свойствами.
Фиг.32А и 32В иллюстрируют базовые частицы СТА, полученные в соответствии с одним из вариантов осуществления настоящего изобретения. На фиг.32А показаны базовые частицы СТА при 500-кратном увеличении, а при получении изображения фиг.32 В камера “наезжает” внутрь одной из базовых частиц и показывает эту частицу при 2000-кратном увеличении. Как, видимо, наилучшим образом иллюстрирует фиг.32 В, каждая базовая частица СТА, как правило, образована большим количеством маленьких агломерированных субчастиц СТА, вследствие чего базовые частицы СТА имеют относительно большую площадь поверхности, высокую пористость, низкую плотность и хорошую растворимость. Если не указано иное, различные приведенные ниже свойства СТА по настоящему изобретению измеряют для типичного образца СТА, при этом масса типичного образца составляет, по меньшей мере, 1 г и/или он включает, по меньшей мере, 10000 индивидуальных частиц СТА. Базовые частицы СТА обычно имеют средний размер частиц от приблизительно 20 до приблизительно 150 мкм, более предпочтительно, от приблизительно 30 до приблизительно 120 мкм и, наиболее предпочтительно, от 40 до 90 мкм. Субчастицы СТА обычно имеют средний размер частиц от приблизительно 0,5 до приблизительно 30 мкм, более предпочтительно, от приблизительно 1 до приблизительно 15 мкм и, наиболее предпочтительно, от 2 до 5 мкм. Относительно большую площадь поверхности базовых частиц СТА, которую иллюстрируют фиг.32А и 32В, можно количественно оценить измерением площади поверхности по методу Браунауэра-Эмметта-Теллера (ВЕТ). Базовые частицы СТА, предпочтительно, имеют среднюю площадь поверхности БЭТ, равную, по меньшей мере, 0,6 м2/г. Более предпочтительно, базовые частицы СТА имеют среднюю площадь поверхность БЭТ, которая составляет от приблизительно 0,8 до приблизительно 4 м2/г. Наиболее предпочтительно, базовые частицы СТА имеют среднюю площадь поверхность БЭТ, которая составляет от 0,9 до 2 м2/г. Физические свойства (в частности, размер частиц, площадь поверхность БЭТ, пористость и растворимость) базовых частиц СТА, полученных по оптимизированному способу окисления, в соответствии с предпочтительным вариантом осуществления настоящего изобретения, позволяют проводить очистку частиц СТА с использованием более эффективных и/или экономичные методов, как более подробно указано на фиг.35.
Приведенные выше значения средних размеров частиц определяют методом микроскопии в поляризованном свете с последующим анализом изображения. При анализе размеров частиц применяют оптический микроскоп Nikon Е800 с объективом 4× Plan Flour N.A. 0.13, цифровой фотоаппарат Spot RT™ и персональный компьютер с программный обеспечением Image Pro Plus™ V4.5.0.19 для анализа изображения. Метод анализа размера частиц включает следующие основные стадии: (1) диспергирование порошков СТА в минеральном масле; (2) размещение образца дисперсии между предметным стеклом микроскопа и покровным стеклом; (3) исследование предметного стекла с помощью микроскопии в поляризованном свете (при скрещенных поляроидах - частицы видны как яркие объекты на черном фоне); (4) получение различных изображений для каждого образца препарата (размер поля = 3×2,25 мм; размер пикселя = 1,84 мкм/пиксель); (5) проведение анализа изображения с помощью программного обеспечения Pro Plus™; (6) составление сводной таблицы из параметров измерения частиц; и (7) проведение статистической обработки данных сводной таблицы. Стадия (5) “проведения анализа изображения с помощью программного обеспечения Pro Plus™” включает следующие подстадии: (а) определение порогового значения обнаружения белых частиц на темном фоне; (b) получение двухуровневого изображения; (с) однократное применение открытого фильтра с целью отфильтровать искаженные пиксели; (d) измерение всех частиц на изображении; и (е) регистрация среднего измеренного диаметра для каждой частицы. Программное обеспечение Pro Plus™ определяет средний диаметр индивидуальных частиц как среднечисловую длину диаметров частиц, которые измеряют с интервалами в 2 градуса и которые проходят через центроид частицы. Стадия (7) “проведения статистической обработки данных сводной таблицы” включает расчет объемно-весового среднего размера частиц следующим образом. Рассчитывают объем каждой из n частиц в образце как если бы она имела сферическую форму по формуле π/6*di 3; умножают объем каждой частицы на ее диаметр, чтобы получить значение π/6*di 4; суммируют значения π/6*di 4 для всех частиц в образце; суммируют объемы всей частиц в образце; рассчитывают объемно-весовой диаметр частицы как частное от деления суммы (π/6*di 4) для всех частиц в образце на сумму (π/6*di 3) для всех частиц в образце. В данном описании “средний размер частиц” обозначает объемно-весовой средний размер частиц, который определяют вышеуказанным способом; его также обозначают как D(4,3).
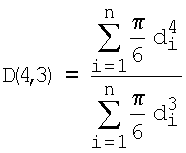
Кроме того, стадия 7 включает определение размеров частиц, для которых различные фракции общего объема образца имеют меньший размер. Например, D(v, 0,1) обозначает размер частиц, для которого 10% от общего объема образца содержит частицы меньшего размера, а 90% содержит частицы большего размера; D(v, 0,5) обозначает размер частиц, для которого половина объема образца содержит частицы меньшего размера, а половина объема образца содержит частицы большего размера; D(v, 0,9) обозначает размер частиц, для которого 90% от общего объема содержит частицы меньшего размера; и т.д. Кроме того, стадия 7 включает расчет величины D(v, 0,9) минус D(v, 0,1), которая в данном описании определяется как “распределение размеров частиц”; а также стадия 7 включает расчет значения распределения размеров частиц, деленного на D(4, 3), которое в данном описании обозначают как “относительное распределение размеров частиц”.
Далее, значение D(v, 0,1) для частиц СТА, метод измерения которого приведен выше, предпочтительно, составляет от приблизительно 5 до приблизительно 65 мкм, более предпочтительно, составляет от приблизительно 15 до приблизительно 55 мкм и, наиболее предпочтительно, составляет от 25 до 45 мкм. Значение D(v, 0,5) для частиц СТА, метод измерения которого приведен выше, предпочтительно, составляет от приблизительно 10 до приблизительно 90 мкм, более предпочтительно, составляет от приблизительно 20 до приблизительно 80 мкм и, наиболее предпочтительно, составляет от 30 до 70 мкм. Значение D(v, 0,9) для частиц СТА, метод измерения которого приведен выше, предпочтительно, составляет от приблизительно 30 до приблизительно 150 мкм, более предпочтительно, составляет от приблизительно 40 до приблизительно 130 мкм и, наиболее предпочтительно, составляет от 50 до 110 мкм. Относительное распределение размеров частиц, предпочтительно, составляет от приблизительно 0,5 до приблизительно 2,0, более предпочтительно, составляет от приблизительно 0,6 до приблизительно 1,5 и, наиболее предпочтительно, составляет от 0,7 до 1,3.
Указанные выше значения площади поверхности БЭТ измеряют на приборе Micromeritics ASAP2000 (выпускается компанией Micromeritics Instrument Corporation of Norcross, Джорджия). На первой стадии проведения измерений взвешивают от 2 до 4 г образца частиц и сушат в вакууме при температуре 50°С. Затем образец помещают в газовую систему аналитического прибора и охлаждают до температуры 77К. Измеряют изотермы адсорбции азота, как минимум, при 5 равновесных значениях давления, помещая образец в известные объемы газообразного азота и измеряя уменьшение давления. Равновесные значения давления приблизительно находятся в диапазоне Р/Р0=0,01-0,20, где Р обозначает равновесное давление, а Р0 обозначает давление паров жидкого азота при 77К. Затем строят график полученной изотермы в соответствии со следующим уравнением БЭТ:
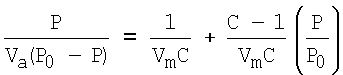
где Va обозначает объем газа, абсорбированного образцом при значении Р, Vm обозначает объем газа, необходимого, чтобы покрыть всю поверхность образца монослоем газа, а С - константа. Из полученного графика определяют значения Vm и С. Затем величину Vm переводят в площадь поверхности, используя значение площади сечения азота при 77К по уравнению:
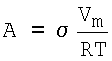
где σ - площадь сечения азота при 77К, значение Т соответствует 77К, а R - газовая постоянная.
Как указано выше, СТА, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, обладает лучшей способностью растворяться по сравнению с обычной СТА, полученной другими способами. Указанная большая скорость растворения позволяет более эффективно провести очистку СТА и/или использовать более эффективные способы очистки. Далее в описании рассматриваются способы количественной оценки скорости растворения СТА.
Скорость растворения известного количества твердых веществ в известном количестве растворителя в перемешиваемой смеси можно измерить, используя различные методики. Используемый в данном описании способ, который называют “тестом на растворение за определенный промежуток времени”, заключается в следующем. При проведении теста используется обычное окружающее давление, составляющем приблизительно 0,1 МПа. Температура окружающей среды, которую используют при проведении теста на растворение за определенный промежуток времени, составляет 22°С. Кроме того, перед проведением тестирования твердые вещества, растворитель и вся используемая для растворения оснастка полностью термически уравновешиваются при указанной температуре, а потому существенного нагревания или охлаждения химического стакана или его содержимого в течение заданного периода растворения не происходит. Порцию свежего растворителя, который представляет собой тетрагидрофуран, пригодный для проведения ВЭЖХ (его чистота составляет 99,9%), далее обозначаемый как ТГФ, в количестве 250 г помещают в чистый высокий химический стакан из KIMAX емкостью 400 мл (номер 14020 по каталогу Kimble®, Kimble/Kontes, Вайнлэнд, Нью-Джерси) без крышки, который имеет гладкие стенки и обычно цилиндрическую форму. В стакан помещают покрытый тефлоном магнитный размешиватель (номер 58948-230 по каталогу VWR, длина его составляет приблизительно 1 дюйм, диаметр 3/8 дюйма, он имеет восьмиугольное сечение; VWR International, West Chester, PA 19380), где он оседает на дно. Образец перемешивают с помощью многопозиционной магнитной мешалки Variomag® 15 (H&P Labortechnik AG, Обершлисхайм, Германия), установленной на режим вращения 800 об/мин. Перемешивание начинают не более чем за 5 мин до добавления твердых веществ и продолжают без перерыва в течение, по меньшей мере, 30 мин после добавления твердых веществ. Твердый образец частиц сырой или очищенной ТРА общей массой до 250 мг отвешивают в чашечку для взвешивания образцов, к стенкам которой частицы не прилипают. В начальное время, обозначенное как t=0, отвешенные твердые вещества разом высыпают в перемешиваемый ТГФ и одновременно включают таймер. Если все сделано правильно, ТГФ очень быстро смачивает твердые вещества и в течение 5 сек образует разбавленную, хорошо перемешиваемую суспензию. Далее получают образцы указанной смеси в следующие периоды времени, измеренные в минутах начиная от t=0: 0,08; 0,25; 0,50; 0,75; 1,00; 1,50; 2,00; 2,50; 3,00; 4,00; 5,00; 6,00; 8,00; 10,00; 15,00 и 30,00. Каждый небольшой образец берут из разбавленной, хорошо перемешиваемой суспензии с помощью нового одноразового шприца (Becton, Dickinson and Co., 5 мл, REF 30163, Franklin Lakes, NJ 07417). Сразу же после извлечения из химического стакана приблизительно 2 мл чистой жидкости образца быстро пропускают через новый неиспользованный шприцевый фильтр (диаметр 25 мм, 0,45 мкм, Gelman GHP Acrodisc FG®, Pall Corporation, Easter Hills, NY 11548) в новую снабженную биркой стеклянную пробирку. Продолжительность периода времени для каждого операции заполнения шприца, переноса на фильтр и переноса в пробирку должна составлять меньше чем приблизительно 5 сек, и указанный интервал, соответственно, начинается и заканчивается в пределах приблизительно ±3 сек от времени подготовки каждого требуемого образца. В течение приблизительно пяти минут после каждого заполнения пробирки закрывают пробками и выдерживают при приблизительно постоянной температуре до проведения следующего химического анализа. После того, как берут последний образец через 30 мин после времени t=0, все 16 образцов анализируют на содержание растворенной ТСА с помощью метода HPLC-DAD (ВЭЖХ с детектором на диодной матрице) как в общих чертах указывается в настоящем описании. В данном тесте калибровочные стандарты и полученные результаты приведены в количестве миллиграммов растворенной ТРА на грамм растворителя ТГФ (далее обозначают как “м.д. в ТГФ”). Например, если все 250 мг твердых веществ представляют собой очень чистую ТРА и все указанное количество полностью растворяется в 250 г ТГФ, который используют в качестве растворителя, прежде чем будет отобран конкретный образец, то правильно измеренная концентрация составит приблизительно 1000 м.д. в ТГФ.
Когда СТА, полученную по настоящему изобретению, испытывают с помощью описанного выше теста на растворение за определенный промежуток времени, то образец, который отбирают через одну минуту после t=0, предпочтительно, растворяется до концентрации, равной, по крайней мере, приблизительно 500 м.д. в ТГФ, более предпочтительно, до концентрации, равной, по крайней мере, приблизительно 600 м.д. в ТГФ. В образце, который отбирают через две минуты после t=0, СТА по настоящему изобретению, предпочтительно, будет растворяться до концентрации, равной, по крайней мере, приблизительно 700 м.д. в ТГФ, более предпочтительно, до концентрации, равной, по крайней мере, приблизительно 750 м.д. в ТГФ. В образце, который отбирают через четыре минуты после t=0, СТА по настоящему изобретению, предпочтительно, будет растворяться до концентрации, равной, по крайней мере, приблизительно 840 м.д. в ТГФ, более предпочтительно, до концентрации, равной, по крайней мере, приблизительно 880 м.д. в ТГФ.
Авторы настоящего изобретения обнаружили, что независимо от сложности состава образцов частиц и процесса растворения, для описания временной зависимости полного набора данных, полученного при использовании теста на растворение за определенный промежуток времени, может быть применена относительно простая отрицательная экспоненциальная модель роста. Вид уравнения, который далее обозначают как “модель растворения за определенный промежуток времени”, можно представить следующим образом:
S=A+B*(1-exp(-C*t)),
где t = время, измеренное в минутах;
S = растворимость, выраженная в м.д. в ТГФ, в момент времени t;
ехр = экспоненциальная функция по основанию натурального логарифма 2;
А, В = константы регрессии, выраженные в м.д. в ТГФ, при этом А относится в основном к быстрому растворению более мелких частиц за очень короткие промежутки времени, а сумма А+В в основном относится к общему растворению за время, близкое к окончанию заданного периода тестирования; и
С = константа времени регрессии, выраженная в обратных минутах (мин-1).
Константы регрессии подбирают таким образом, чтобы минимизировать сумму квадратов отклонений реально полученных данных от соответствующих значений, полученных при моделировании, и данный способ называют методом “наименьших квадратов”. Предпочтительным пакетом программного обеспечения для проведения указанного регрессионного анализа полученных данных является JMP Release 5.1.2 (SAS Institute Inc., JMP Software, SAS Campus Drive, Cary, NC 27513).
Когда СТА, полученную в соответствии с настоящим изобретением, исследуют с помощью теста на растворение за определенный промежуток времени, а затем проводят анализ на соответствие модели растворения за определенный промежуток времени, которая описана выше, то СТА, предпочтительно, имеет временную константу “С” больше чем приблизительно 0,5 мин-1, более предпочтительно, больше чем приблизительно 0,6 мин-1 и, наиболее предпочтительно, больше чем 0,7 мин-1.
Фиг.33А и 33В иллюстрируют обычные частицы СТА, полученные в обычном высокотемпературном процессе окисления в корпусном реакторе непрерывного действия с перемешиванием (CSTR). На фиг.33А показаны обычные частицы СТА при 500-кратном увеличении, а при получении изображения на фиг.33В камера “наезжает” внутрь частицы и показывает обычную частицу СТА при 2000-кратном увеличении. Визуальное сравнение частиц СТА по настоящему изобретению, приведенных на фиг.32А и 32В, и обычных частиц СТА, приведенных на фиг.33А и 33В, показывает, что обычные частицы имеют большую плотность, меньшую площадь поверхности, меньшую пористость и больший размер частиц, чем частицы СТА по настоящему изобретению. Действительно, обычная СТА, приведенная на фиг.33А и 33В, имеет средний размер частиц приблизительно 205 мкм, а площадь поверхности БЭТ для них составляет приблизительно 0,57 м2/г.
На фиг.34 поясняется обычный способ получения очищенной терефталевой кислоты (РТА). В обычном способе получения РТА пара-ксилол частично окисляется в высокотемпературном реакторе 700, снабженном перемешиванием. Суспензия, содержащая СТА, выгружается из реактора 700 и затем очищается в системе очистки 702. Продукт из системы очистки 702 в виде РТА вводится в разделительную систему 706 для отделения и сушки частиц РТА. Система очистки 702 вносит основной вклад в затраты на получение частиц РТА в обычных способах. Система очистки 702 обычно включает систему добавления/обмена воды 708, систему растворения 710, систему гидрирования 712 и три отдельных реактора для проведения кристаллизации 704a, b, c. В системе добавления/обмена воды 708 большая часть маточного раствора замещается водой. После добавления воды, вода/суспензия СТА подается в систему растворения 710, где смесь СТА с водой нагревается до тех пор, пока частицы СТА полностью не растворятся в воде. После растворения СТА, раствор СТА в воде подвергается гидрированию в системе гидрирования 712. Поток гидрированного продукта после системы гидрирования 712 далее подвергается кристаллизации в три стадии в реакторах для проведения кристаллизации 704a,b, а затем разделяется в разделительной системе 706.
На фиг.35 поясняется улучшенный способ получения РТА с использованием барботажной колонны реакторного типа 800, сконструированной в соответствии с вариантом осуществления настоящего изобретения. Исходная суспензия, содержащая твердые частицы СТА и маточный раствор, выгружается из реактора 800. Как правило, исходная суспензия может содержать твердые частицы СТА от приблизительно 10 до приблизительно 50% мас., а остальное составляет маточный раствор. Твердые частицы СТА, которые содержатся в исходной суспензии, как правило, содержат, по меньшей мере, приблизительно 400 м.д. мас.4-карбоксибензальдегида (4-СВА), более типично, содержат, по меньшей мере, приблизительно 800 м.д. мас.4-СВА и наиболее типично содержат 4-СВА от 1000 до 15000 м.д. мас. Исходная суспензия, которая выводится из реактора 800, подается в систему очистки 802 с целью снижения концентрации 4-СВА и других примесей, содержащихся в СТА. Более чистая/очищенная суспензия, полученная в системе очистки 802, подвергается разделению и сушке в разделительной системе 804, и таким образом получаются более чистые твердые частицы терефталевой кислоты, которые содержат меньше чем приблизительно 400 м.д. мас.4-СВА, более предпочтительно, меньше чем приблизительно 250 м.д. мас.4-СВА и, наиболее предпочтительно, от 10 до 200 м.д. мас.4-СВА.
Система очистки 802 системы получения РТА, приведенной на фиг.35, обеспечивает ряд преимуществ по сравнению с системой очистки 802, известной из области техники системы, которая приведена на фиг.34. Система очистки 802, предпочтительно, как правило, содержит систему замены маточного раствора 806, выварочный котел 808 и один кристаллизатор 810. В системе замены маточного раствора 806, по меньшей мере, 50% мас. маточного раствора, содержащегося в исходной суспензии, замещается используемым для замены свежим замещающим растворителем и таким образом образуется суспензия в замещающем растворителе, которая содержит частицы СТА и используемый для замены растворитель. Суспензия в замещающем растворителе, которая покидает систему замены маточного раствора 806, поступает в выварочный котел (или во второй реактор окисления) 808. В выварочном котле 808 вторичная реакция окисления проводится при несколько более высоких температурах, чем те, которые использовались в процессе исходной/первичной реакции окисления, которая проводится в барботажной колонне реакторного типа 800. Как указано выше, большая площадь поверхности, меньший размер частиц и низкая плотность частиц СТА, полученных в реакторе 800, приводит к тому, что определенные примеси, захваченные частицами СТА, становятся доступны для окисления в выварочном котле 808, не требуя полного растворения частиц СТА в выварочном котле 808. Таким образом, температура в выварочном котле 808 может быть ниже, чем во многих аналогичных процессах, известных из области техники. В процессе вторичного окисления, который проводится в выварочном котле 808, содержание 4-СВА в СТА, предпочтительно, снижается, по крайней мере, на 200 м.д. мас., более предпочтительно, снижается, по крайней мере, на 400 м.д. мас. и, наиболее предпочтительно, снижается, в диапазоне от 600 до 6000 м.д. мас. Температура вторичной реакции окисления в выварочном котле 808, предпочтительно, приблизительно на 10°С выше чем температура первичной реакции окисления в барботажной колонне реакторного типа 800, более предпочтительно, от приблизительно 20°С до приблизительно 80°С выше чем температура первичной реакции окисления в барботажной колонне реакторного типа 800 и, наиболее предпочтительно, от 30°С до 50°С выше чем температура первичной реакции окисления в барботажной колонне реакторного типа 800. Температура вторичной реакции окисления, предпочтительно, составляет от приблизительно 160 до приблизительно 240°С, более предпочтительно, составляет от приблизительно 180 до приблизительно 220°С и, наиболее предпочтительно, составляет от 190 до 210°С. Очищенный продукт после выварочного котла 808 требует лишь одной стадии в кристаллизаторе 810 перед разделением в разделительной системе 804. Подходящие способы проведения вторичного окисления/выварки подробно обсуждаются в патентной заявке США №2005/0065373, полное содержание которой специально включено в настоящее описание посредством ссылки.
Терефталевая кислота (в частности, РТА), получаемая в системе, приведенной на фиг.35, преимущественно, образована частицами, средний размер которых, по меньшей мере, составляет приблизительно 40 мкм, более предпочтительно, составляет от приблизительно 50 до приблизительно 2000 мкм и, наиболее предпочтительно, составляет от 60 до 200 мкм. Частицы РТА, предпочтительно, имеют среднюю площадь поверхности БЭТ меньше чем приблизительно 0,25 м2/г, более предпочтительно, указанная величина составляет от приблизительно 0,005 до приблизительно 0,2 м2/г и, наиболее предпочтительно, составляет от 0,01 до 0,18 м2/г. РТА, получаемая в системе, приведенной на фиг.35, пригодна в качестве исходного сырья для получения ПЭТ. Как правило, ПЭТ получают этерификацией терефталевой кислоты этиленгликолем с последующей поликонденсацией. Терефталевая кислота, получаемая в соответствии с вариантом осуществления настоящего изобретения, предпочтительно, используется в качестве сырья в способе получения ПЭТ в трубчатом реакторе, который описан в патентной заявке США №10/013318, поданной 7 декабря 2001, полное содержание которой включено в настоящее описание посредством ссылки.
Приведенные в настоящем описании частицы СТА с предпочтительной морфологией особенно пригодны для использования в приведенном выше окислительном выварочном процессе с целью снижения содержания 4-СВА. Кроме того, указанные предпочтительные частицы СТА предоставляют преимущества при проведении широкого спектра последующих обработок, включающих растворение и/или химические реакции с участием указанных частиц. Указанные дополнительные последующие обработки, включают, однако этим не ограничиваясь, взаимодействие, по крайней мере, с одним гидроксилсодержащим соединением с образованием сложных эфиров, в частности взаимодействие СТА с метанолом с образованием диметилтерефталата и примесных сложных эфиров; взаимодействие, по крайней мере, с одним диолом с образованием сложноэфирных мономерных и/или полимерных соединений, в частности взаимодействие СТА с этиленгликолем с образованием полиэтилентерефталата (ПЭТ); и полное или частичное растворение в растворителе, включая, однако этим не ограничиваясь, воду, уксусную кислоту и N-метил-2-пирролидон, которое может включать дальнейшую обработку, в том числе, однако этим не ограничиваясь, переосаждение более чистой терефталевой кислоты и/или селективное химическое восстановление карбонильных групп, отличных от карбоксильных групп. Особо следует отметить практически полное растворение СТА в растворителе, содержащем воду, в сочетании с частичным гидрированием, которое снижает количество альдегидов, в особенности 4-СВА, флуоренонов, фенонов и/или антрахинонов.
Авторы настоящего изобретения полагают также, что частицы СТА, обладающие раскрытыми в данном описании предпочтительными свойствами, могут быть получены из частиц СТА, которые не соответствуют раскрытым в данном описании предпочтительным свойствам (неконформные частицы СТА) с помощью способов, включающих, однако этим не ограничиваясь, механическое измельчение неконформных частиц СТА и полное или частичное растворение неконформных частиц СТА с последующим полным или частичным переосаждением.
В соответствии с одним из вариантов осуществления настоящего изобретения, предлагается способ частичного окисления способного окисляться ароматического соединения с получением одного или нескольких типов ароматических карбоновых кислот, в котором чистоту растворителя как составной части исходных веществ (т.е. “исходного растворителя”) и чистоту способного окисляться соединения как составной части исходных веществ (т.е. “исходного способного окисляться соединения”) контролируют в определенных ниже пределах. Вместе с другими вариантами осуществления настоящего изобретения это позволяет контролировать чистоту жидкой фазы и твердой фазы, если она присутствует, и объединенной фазы суспензии (т.е. твердой фазы плюс жидкой фазы) реакционной среды в определенных предпочтительных пределах, которые указаны ниже.
Что касается исходного растворителя, то известен способ окисления способного(ых) окисляться ароматического(их) соединения(ий) с образованием ароматической карбоновой кислоты, где исходным растворителем, который подают в реакционную среду, является смесь аналитически чистой уксусной кислоты и воды, которую часто применяют как в лабораторных масштабах, так и в опытном производстве. Известен также способ осуществления окисления способного окисляться ароматического соединения с образованием ароматической карбоновой кислоты, где растворитель, покидающий реакционную среду, отделяется от полученной ароматической карбоновой кислоты и затем рециклируется в реакционную среду в виде исходного растворителя, главным образом с целью снижения производственных затрат. Указанное рециклирование растворителя приводит к тому, что некоторые примеси, присутствующие в исходных веществах, и образующиеся в процессе побочные продукты с течением времени аккумулируются в рециклированном растворителе. Из области техники известно большое количество способов, позволяющих очистить рециклированный растворитель прежде, чем он будет вновь введен в реакционную среду. В общем случае более высокая степень очистки рециклированного растворителя приводит к значительно большим производственным затратам, чем меньшая степень очистки при использовании аналогичных способов очистки. Один из вариантов осуществления настоящего изобретения касается определения и установления предпочтительных пределов содержания большого числа примесей в исходном растворителе, многие из которых до настоящего времени считались практически безвредными, с целью найти оптимальный баланс между общими производственными затратами и общей чистотой продукта.
“Рециклированный исходный растворитель” обозначается в данном описании как исходный растворитель, содержащий, по меньшей мере, приблизительно 5% мас. веществ, которые уже проходили через реакционную среду, содержащую одно или несколько способных окисляться ароматических соединений, которые подвергаются частичному окислению. С учетом создаваемого запаса растворителя и продолжительности рабочего цикла в промышленной установке, порции рециклированного растворителя, предпочтительно, должны проходить через реакционную среду, по меньшей мере, один раз в течение дня проведения процесса, более предпочтительно, по меньшей мере, один раз в день в течение, по крайней мере, семи последовательных дней проведения процесса и, наиболее предпочтительно, по меньшей мере, один раз в день в течение, по крайней мере, 30 последовательных дней проведения процесса. По экономическим соображениям, предпочтительно, по крайней мере, 20% мас. исходного растворителя в реакционной среде по настоящему изобретению является рециклированным растворителем, более предпочтительно, по крайней мере, 40% мас., еще более предпочтительно, по крайней мере, 80% мас. и, наиболее предпочтительно, по крайней мере, 90% мас. исходного растворителя является рециклированным растворителем.
Авторы настоящего изобретения обнаружили, что с учетом активности реакции и металлических примесей, которые остаются в продукте окисления, концентрации выбранных металлов переменной валентности в потоке рециклированного исходного растворителя, предпочтительно, находятся в указанных далее пределах. Концентрация железа в рециклированном растворителе, предпочтительно, составляет меньше приблизительно 150 м.д. мас., более предпочтительно, составляет меньше приблизительно 40 м.д. мас. и, наиболее предпочтительно, составляет от 0 до 8 м.д. мас. Концентрация никеля в рециклированном растворителе, предпочтительно, составляет меньше приблизительно 150 м.д. мас., более предпочтительно, составляет меньше приблизительно 40 м.д. мас. и, наиболее предпочтительно, составляет от 0 до 8 м.д. мас. Концентрация хрома в рециклированном растворителе, предпочтительно, составляет меньше приблизительно 150 м.д. мас., более предпочтительно, составляет меньше приблизительно 40 м.д. мас. и, наиболее предпочтительно, составляет от 0 до 8 м.д. мас. Концентрация молибдена в рециклированном растворителе, предпочтительно, составляет меньше приблизительно 75 м.д. мас., более предпочтительно, составляет меньше приблизительно 20 м.д. мас. и, наиболее предпочтительно, составляет от 0 до 4 м.д. мас. Концентрация титана в рециклированном растворителе, предпочтительно, составляет меньше приблизительно 75 м.д. мас., более предпочтительно, составляет меньше приблизительно 20 м.д. мас. и, наиболее предпочтительно, составляет от 0 до 4 м.д. мас. Концентрация меди в рециклированном растворителе, предпочтительно, составляет меньше приблизительно 20 м.д. мас., более предпочтительно, составляет меньше приблизительно 4 м.д. мас. и, наиболее предпочтительно, составляет от 0 до 1 м.д. мас. В рециклированном растворителе, как правило, присутствуют другие металлические примеси, содержание которых обычно варьирует в низких пропорциях по отношению к одному или нескольким вышеуказанным металлам. Поддержание концентрации приведенных выше металлов в указанных предпочтительных границах позволит поддержать на соответствующем уровне и другие металлические примеси.
Указанные металлы могут появиться как примеси в любом из поступающих в процесс исходных соединений (т.е. способного окисляться соединения, растворителя, окислителя и компонентов катализатора). Кроме того, металлы могут появиться как продукты коррозии любого из узлов установки, которые контактируют с реакционной средой и/или контактируют с рециклированным растворителем. Средства контроля металлов в раскрытых выше пределах концентрации включают соответствующие технические требования и мониторинг чистоты различных исходных веществ и соответствующий выбор материалов конструкций, включая, однако этим не ограничиваясь, многие промышленные сорта титана и нержавеющих сталей, в том числе такие сорта, как сталь, выплавленная дуплекс-процессом, и стали с большим содержанием молибдена.
Авторы настоящего изобретения также установили предпочтительные границы для выбранных ароматических соединений в рециклированном растворителе. Они включают как осажденные, так и растворенные ароматические соединения, содержащиеся в рециклированном растворителе.
Неожиданно оказалось, что и сам осажденный продукт (в частности, ТРА) частичного окисления пара-ксилола является загрязняющим веществом, с которым также необходимо бороться в рециклированном растворителе. Поскольку неожиданно существуют предпочтительные границы содержания твердых веществ в реакционной среде, то любой осажденный продукт, содержащийся в исходном растворителе, прямым образом сокращает количество способного окисляться соединения, которое может быть согласованно введено в реакцию. Кроме того, было обнаружено, что попадание больших количеств осажденного твердого ТРА в рециклированный растворитель оказывает негативное влияние на свойства частиц, которые образуются в процессе осаждения из окислительной среды, что приводит к проявлению нежелательных свойств при проведении последующих операций (в частности, при фильтровании продукта, промывке растворителем, окислительном вываривании сырого продукта, растворении сырого продукта, с целью его дальнейшей обработки, и т.д.). Другая нежелательная особенность осажденных веществ в подаваемом в процесс рециклированном растворителе заключается в том, что они часто содержат очень большое количество осажденных примесей по сравнению с концентрацией примесей в объеме твердых веществ в суспензиях ТРА, из которых выделяют большую часть рециклированного растворителя. Повышенное содержание примесей, которое наблюдается в твердых веществах, суспендированных в рециклированном фильтрате, вероятно, может быть связано с временами нуклеации при осаждении определенных примесей из рециклированного растворителя и/или с охлаждением рециклированного растворителя как преднамеренного, так и вызванного потерями в окружающей среде. Например, значительно более высокие уровни концентраций сильно окрашенного и нежелательного 2,6-дикарбоксифлуоренона обнаруживаются в твердых веществах, присутствующих в рециклированном растворителе при температуре 80°С, чем в твердой ТРА, выделенной из рециклированного растворителя при температуре 160°С. Аналогично, значительно большие уровни концентрации изофталевой кислоты наблюдаются в твердых веществах, присутствующих в рециклированном растворителе, по сравнению с уровнями, которые наблюдаются в твердой ТРА из реакционной среды. Конкретное поведение специфических осажденных примесей, захваченных рециклированным растворителем, при их повторном введении в реакционную среду, видимо, может меняться. Оно, возможно, зависит от относительной растворимости примеси в жидкой фазе реакционной среды, возможно, от того, как осажденная примесь распределяется в осажденных веществах, и, возможно, от локальной скорости осаждения ТРА в том месте, где твердое вещество впервые вновь попадает в реакционную среду. Таким образом, авторы настоящего изобретения обнаружили, что важно контролировать уровни определенных примесей в рециклированном растворителе, как указано ниже, независимо от того, содержатся ли эти примеси в рециклированном растворителе в растворенной форме или же представляют собой частицы, которые захвачены рециклированным растворителем.
Количество осажденных твердых веществ, которые содержатся в рециклированном фильтрате, определяют гравиметрическим способом по следующей методике. Берут отдельный образец подаваемого в реакционную смесь растворителя по мере того, как растворитель направляется по трубопроводу в реакционную среду. Приемлемый размер образца составляет приблизительно 100 г, которые помещают в стеклянный контейнер, внутренний объемом которого составляет приблизительно 250 мл. Прежде чем давление рециклированного фильтрата сравнится с атмосферным и прежде чем образец фильтрата будет помещен в контейнер, рециклированный фильтрат охлаждают до температуры меньше 100°С; охлаждение необходимо для того, чтобы свести к минимуму испарение растворителя в течение короткого интервала времени, пока образец не будет герметично закрыт в контейнере. После того как образец отобран при атмосферном давлении, стеклянный контейнер немедленно плотно закрывают. Затем образцу дают остыть приблизительно до 20°С при температуре окружающего воздуха приблизительно 20°С и без принудительной конвекции. После того, как температура образца достигнет приблизительно 20°С, его выдерживают при указанных условиях, по меньшей мере, в течение 2 час. Затем запечатанный контейнер энергично встряхивают до тех пор, пока не будет получено визуально однородное распределение твердых частиц. Сразу же после этого в контейнер с образцом помещают магнитный перемешиватель и вращают его со скоростью, достаточной для того, чтобы поддерживать однородное распределение твердых частиц. Аликвоту объемом 10 мл смеси жидкостей с суспендированными в ней твердыми веществами отбирают с помощью пипетки и взвешивают. Затем большую часть жидкой фазы из указанной аликвоты, все еще при температуре 20°С, удаляют вакуумной фильтрацией таким образом, чтобы избежать потерь твердых веществ. Выделенные из указанной аликвоты влажные твердые вещества затем сушат таким образом, чтобы не происходила сублимация твердых веществ, и полученные сухие твердые вещества взвешивают. Отношение массы высушенных твердых веществ к массе исходной аликвоты суспензии представляет собой фракцию твердых веществ, которую обычно выражают в процентах и обозначают в данном описании как содержание “твердых веществ, осажденных при температуре 20°С” из исходного растворителя.
Авторы настоящего изобретения обнаружили, что ароматические соединения, растворенные в жидкой фазе реакционной среды, которые представляют собой ароматические карбоновые кислоты, не содержащие неароматические углеводородные группы (в частности, изофталевая кислота, бензойная кислота, фталевая кислота, 2,5,4'-трикарбоксибифенил), неожиданно оказались вредными компонентами. Несмотря на то, что химическая активность указанных соединений значительно снижается в рассматриваемой реакционной среде по сравнению со способным окисляться соединением, содержащим неароматические углеводородные группы, авторы настоящего изобретения обнаружили, что указанные соединения, тем не менее, претерпевают ряд нежелательных превращений. Таким образом, необходимо контролировать содержание указанных веществ в жидкой фазе реакционной среды в допустимых пределах. Таким образом, существуют предпочтительные границы для выбранных соединений в подаваемом в процесс рециклированном растворителе, а также предпочтительные границы для выбранных предшественников в исходном способном окисляться ароматическом соединении.
Например, авторы настоящего изобретения установили, что при проведении жидкофазного частичного окисления пара-ксилола с образованием терефталевой кислоты (ТРА) сильно окрашенную и нежелательную примесь 2,7-дикарбоксифлуоренона (2,7-DCF) практически невозможно обнаружить в реакционной среде и выгружаемом продукте, если содержание мета-замещенных ароматических соединений в реакционной среде находится на очень низких уровнях. Авторы настоящего изобретения установили, что в том случае, когда примесь изофталевой кислоты присутствует в возрастающих количествах в исходном растворителе, то образование 2,7-DCF увеличивается практически прямо пропорционально. Авторы настоящего изобретения также установили, что когда в исходном пара-ксилоле присутствует примесь мета-ксилола, то образование 2,7-DCF вновь увеличивается практически прямо пропорционально. Более того, авторы настоящего изобретения обнаружили, что даже в том случае, когда исходный растворитель и исходное способное окисляться соединение не содержат мета-замещенных ароматических соединений, некоторое количество изофталевой кислоты образуется в процессе типичного частичного окисления очень чистого пара-ксилола, особенно в том случае, когда в жидкой фазе реакционной среды присутствует бензойная кислота. Указанная самостоятельно образовавшаяся изофталевая кислота, вследствие ее большей, чем для ТРА, растворимости в растворителе, состоящем из уксусной кислоты и воды, способна накапливаться в узлах промышленных установок, в которых используется рециклированный растворитель. Таким образом, количество изофталевой кислоты в растворителе, количество мета-ксилола в исходном способном окисляться ароматическом соединении и скорость самостоятельного образования изофталевой кислоты в реакционной среде соответствующим образом следует рассматривать в балансе друг с другом и в балансе с любыми реакциями, в которых потребляется изофталевая кислота. Было установлено, что изофталевая кислота, помимо указанного выше образования 2,7-DCF, вступает в дополнительные реакции, которые рассматриваются ниже. Кроме того, авторы настоящего изобретения обнаружили, что при установлении подходящих границ для мета-замещенных ароматических соединений при частичном окислении пара-ксилола с образованием ТРА следует учитывать и другие проблемы. Оказалось, что другие сильно окрашенные и нежелательные соединения, такие как 2,6-дикарбоксифлуоренон (2,6-DCF), в значительной степени относятся к растворенным пара-замещеным ароматическим соединениям, которые всегда присутствуют в исходном пара-ксилоле, используемом в реакции жидкофазного окисления. Таким образом, подавление образования 2,7-DCF следует рассматривать в перспективе с уровнем других образующихся окрашенных примесей.
Например, авторы настоящего изобретения обнаружили, что при жидкофазном частичном окислении пара-ксилола с получением ТРА образование тримеллитовой кислоты увеличивается по мере возрастания уровней изофталевой кислоты и фталевой кислоты в реакционной среде. Тримеллитовая кислота представляет собой трехосновную карбоновую кислоту, которая вызывает разветвление цепи полимера в процессе получения ПЭТ из ТРА. Во многих применениях ПЭТ уровни разветвления цепи полимера должны быть контролируемо низкими и, таким образом в очищенной ТРА уровни тримеллитовой кислоты должны быть контролируемо низкими. Помимо образования тримеллитовой кислоты, присутствие мета-замещенных и орто-замещенных соединений в реакционной среде приводит также к образованию других трехосновных кислот (в частности, 1,3,5-трикарбоксибензола). Кроме того, увеличение содержания трехосновных кислот в реакционной среде приводит к повышению количества образуемой тетракарбоновой кислоты (в частности, 1,2,4,5-тетракарбоксибензола). В соответствии с настоящим изобретением, контролирование суммарного образования ароматических кислот, содержащих более двух карбоксильных групп, является одним из факторов при установлении предпочтительных уровней мета-замещенных и орто-замещенных соединений в подаваемом в процесс рециклированном растворителе, в исходном способном окисляться соединении и в реакционной среде.
Например, авторы настоящего изобретения обнаружили, что при жидкофазном частичном окислении пара-ксилола с получением ТРА повышенные уровни в жидкой фазе реакционной среды нескольких растворенных ароматических карбоновых кислот, не содержащих неароматические углеводородные группы, непосредственно приводят к усилению образования монооксида углерода и диоксида углерода. Указанное повышенное образование оксидов углерода приводит к снижению выхода продукта как в пересчете на окислитель, так и в пересчете на способное окисляться соединение, в последнем случае потому, что многие из совместно образующихся ароматических карбоновых кислот, которые, с одной стороны, могут рассматриваться как примеси, с другой стороны, также обладают коммерческой ценностью. Таким образом, надлежащее удаление относительно хорошо растворимых карбоновых кислот, не содержащих неароматические углеводородные группы, из рециклированного растворителя имеет экономическое значение с точки зрения предотвращения потери способного окисляться ароматического соединения и окислителя, помимо подавления образования крайне нежелательных соединений, таких как различные флуореноны и тримеллитовая кислота.
Например, авторы настоящего изобретения обнаружили, что при жидкофазном частичном окислении пара-ксилола с получением ТРА, вероятно, не удается избежать образования 2,5,4'-трикарбоксибифенила. 2,5,4'-Трикарбоксибифенил представляет собой трехосновную ароматическую кислоту, которая образуется при конденсации двух ароматических колец, вероятно, за счет конденсации растворенных пара-замещенных ароматических соединений с арильным радикалом, при этом арильный радикал, видимо, образуется при декарбоксилировании или декарбонилировании пара-замещенных ароматических соединений. К счастью, 2,5,4'-трикарбоксибифенил, как правило, образуется в меньших количествах, чем тримеллитовая кислота и обычно не приводит к значительному увеличению разветвлений молекул полимеров в процессе получения ПЭТ. Тем не менее, авторы настоящего изобретения обнаружили, что повышенные уровни 2,5,4'-трикарбоксибифенила в реакционной среде при окислении алкилзамещенных ароматических соединений, в соответствии с предпочтительными вариантами осуществления настоящего изобретения, приводят к повышенным уровням сильно окрашенного и нежелательного 2,6-DCF. Повышенное количество 2,6-DCF, вероятно, образуется из 2,5,4'-трикарбоксибифенила путем смыкания цикла с потерей молекулы воды, однако точный механизм реакции не известен. Если 2,5,4'-трикарбоксибифенилу, который лучше, чем ТРА, растворяется в растворителе, содержащем уксусную кислоту и воду, дают накапливаться в рециклированном растворителе в слишком больших количествах, то скорость конверсии 2,6-DCF может оказаться неприемлемо высокой.
Например, авторы настоящего изобретения обнаружили, что при жидкофазном частичном окислении пара-ксилола с получением ТРА ароматические карбоновые кислоты, не содержащие неароматические углеводородные группы (в частности, изофталевая кислота), если они присутствуют в жидкой фазе в достаточной концентрации, обычно приводят к умеренному подавлению химической активности реакционной среды.
Например, авторы настоящего изобретения обнаружили, что при жидкофазном частичном окислении пара-ксилола с получением ТРА осаждение часто протекает неидеально (т.е. неравновесно) по отношению к относительным концентрациям различных химических соединений в твердой фазе и в жидкой фазе. Вероятно, это вызвано тем, что скорость осаждения слишком велика для предпочтительных пространственно-временных скоростей по настоящему изобретению, что приводит к неидеальному соосаждению примесей и даже к закупориванию. Таким образом, в том случае, когда желательно ограничить концентрацию некоторых примесей (в частности, тримеллитовой кислоты и 2,6-DCF) в сырой ТРА за счет организации проведения операций на последующих стадиях процесса, то желательно контролировать концентрацию примесей в исходном растворителе, а также скорости их образования в реакционной среде.
Например, авторы настоящего изобретения обнаружили, что производные соединения бензофенона (в частности, 4,4'-дикарбоксибензофенон и 2,5,4'-трикарбоксибензофенон), которые образуются при жидкофазном частичном окислении пара-ксилола, оказывают нежелательное действие на реакционную среду, в которой получают ПЭТ, даже несмотря на то, что производные бензофенона сами по себе не являются такими же сильно окрашивающими ТРА соединениями, как флуореноны и антрахиноны. Таким образом, желательно свести к минимуму присутствие бензофенона и соответствующих предшественников в рециклированном растворителе и исходном, способном окисляться соединении. Кроме того, авторы настоящего изобретения обнаружили, что присутствие повышенных количеств бензойной кислоты, независимо от того, поступили ли они вместе с рециклированным растворителем или образовались в реакционной среде, приводит к повышенным скоростям образования 4,4'-дикарбоксибензофенона.
Если обобщить сказанное, то авторы настоящего изобретения обнаружили и провели эффективную количественную оценку неожиданной цепи реакций, характерных для ароматических соединений, не содержащих неароматические углеводородные группы, которые присутствуют при проведении жидкофазного частичного окисления пара-ксилола с образованием ТРА. Резюмируя сказанное для одного лишь случая бензойной кислоты, можно отметить, что авторы настоящего изобретения обнаружили, что повышенные уровни бензойной кислоты в реакционной среде в некоторых вариантах осуществления настоящего изобретения приводят к значительному повышению образования сильно окрашенной и нежелательной 9-флуоренон-2-карбоновой кислоты, к значительному повышению уровней 4,4'-дикарбоксибифенила, к повышенным уровням 4,4'-дикарбоксибензофенона, к умеренному подавлению химической активности пара-ксилола при его окислении, к повышенным уровням оксидов углерода и к связанным с ними потерям выхода продукта. Авторы настоящего изобретения обнаружили, что повышенные уровни бензойной кислоты в реакционной среде приводят также к образованию изофталевой кислоты и фталевой кислоты, уровни которых, в соответствии с другими аспектами настоящего изобретения, желательно контролировать в узком диапазоне. Количество и важность реакций, в которых принимает участие бензойная кислота, кажется еще более удивительным, поскольку некоторые исследователи предлагают применять бензойную кислоту вместо уксусной кислоты в качестве компонента растворителя (см., в частности, патент США №6562997). Кроме того, авторы настоящего изобретения наблюдали, что бензойная кислота образуется самостоятельно при окислении пара-ксилола со скоростями, которые оказываются весьма важными, в сравнении с ее образованием из примесей, таких как толуол и этилбензол, которые обычно содержатся в исходном способном окисляться соединении, представляющем собой коммерчески чистый пара-ксилол.
С другой стороны, авторы настоящего изобретения обнаружили незначительную пользу от дополнительного регулирования состава рециклированного растворителя в отношении присутствия способного окисляться ароматического соединения и в отношении промежуточных ароматических соединений, которые сохраняют неароматические углеводородные группы и одновременно обладают относительно высокой растворимостью в рециклированном растворителе. В общем случае, указанные соединения либо поступают в реакционную среду, либо образуются в реакционной среде со скоростями, которые приводят к тому, что их количество заметно превосходит их присутствие в рециклированном растворителе; скорость же потребления указанных соединений в реакционной среде достаточно высока, если они сохраняют одну или несколько неароматических углеводородных групп, что, соответственно, ограничивает их накопление в рециклированном растворителе. Например, при частичном окислении пара-ксилола в многофазной реакционной среде пара-ксилол испаряется в ограниченной степени по сравнению с большими количествами растворителя. Когда указанный испарившийся растворитель покидает реактор в виде части отходящего газа и конденсируется с целью извлечения для использования в качестве рециклированного растворителя, значительная часть испарившегося пара-ксилола также конденсируется. Нет необходимости ограничивать концентрацию указанного пара-ксилола в рециклированном растворителе. Например, если растворитель отделяется от твердых веществ, находящихся в суспензии, которая выводится из реакционной среды, где проводится окисление пара-ксилола, то концентрация пара-толуиловой кислоты в указанном извлекаемом растворителе будет сравнима с концентрацией пара-толуиловой кислоты в точке отвода суспензии из реакционной среды. Несмотря на то, что может оказаться важным, как указано ниже, ограничить устоявшуюся концентрацию пара-толуиловой кислоты в жидкой фазе реакционной среды, нет необходимости отдельно регулировать содержание пара-толуиловой кислоты в указанной порции рециклированного растворителя вследствие относительно хорошей растворимости и относительно низкой скорости массопереноса пара-толуиловой кислоты по сравнению с образованием пара-толуиловой кислоты в реакционной среде. По аналогии, авторы настоящего изобретения установили, что в соответствии с предпочтительными вариантами осуществления настоящего изобретения нет необходимости ограничивать в рециклированном растворителе концентрацию ароматических соединений с метильными группами (в частности, толуиловых кислот), ароматических альдегидов (в частности, терефталевого альдегида), ароматических соединений с гидроксиметильными заместителями (в частности, 4-гидроксиметилбензойной кислоты) и бромзамещенных ароматических соединений, сохраняющих, по крайней мере, одну неароматическую углеводородную группу (в частности, альфа-бром-пара-толуиловой кислоты) ниже той, которая естественным образом присутствует в жидкой фазе, отводимой из реакционной среды, образующейся при частичном окислении ксилола. Авторы настоящего изобретения неожиданно обнаружили, что также нет необходимости регулировать в рециклированном растворителе концентрации отдельных фенолов, которые обычно образуются в процессе частичного окисления ксилола, поскольку скорости образования и распада указанных соединений в реакционной среде имеют большее значение, чем их содержание в рециклированном растворителе. Например, авторы настоящего изобретения установили, что 4-гидроксибензойная кислота оказывает относительное небольшое влияние на химическую активность в предпочтительных вариантах осуществления настоящего изобретения, когда она вводится в реактор в количестве порядка 2 г 4-гидроксибензойной кислоты на 1 кг пара-ксилола, что значительно превышает ее естественное присутствие в рециклированном растворителе, несмотря на то, что, как сообщают другие авторы, она оказывает значительное отравляющее действие в подобных реакционных средах (см., в частности, W.Partenheimer, Catalysis Today 23 (1995) p.81).
Таким образом, как раскрывается в данном описании, существует множество реакций, и при установлении предпочтительных границ для различных ароматических соединений необходимо принять во внимание множество факторов. Установленные закономерности выражаются в терминах общего усредненного по весу состава всех потоков растворителя, которые поступают в реакционную среду за определенный промежуток времени, предпочтительно, в течение одного дня, более предпочтительно, в течение одного часа и, наиболее предпочтительно, в течение одной минуты. Например, если один поток растворителя поступает практически непрерывно со скоростью 7 кг в минуту, при этом содержание изофталевой кислоты в нем составляет 40 м.д. мас., а второй поток растворителя поступает практически непрерывно со скоростью 10 кг в минуту, при этом содержание изофталевой кислоты в нем составляет 2000 м.д. мас., а других потоков растворителя в реакционную среду больше не поступает, то общий усредненный по весу состав поступающего растворителя рассчитывают как (40*7+2000*10)/(7+10)=1193 м.д. мас. изофталевой кислоты. Следует отметить, что масса любого исходного способного окисляться соединения или любого исходного окислителя, которые могут смешиваться с исходным растворителем прежде, чем они попадут в реакционную среду, не учитывается при расчете общего усредненного по весу состава исходного растворителя.
Далее в таблице 1 приведены предпочтительные значения для некоторых компонентов в исходном растворителе, который подается в реакционную среду. В таблице 1 представлены следующие компоненты исходного растворителя: 4-карбоксибензальдегид (4-СВА), 4,4'-дикарбоксистильбен (4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-DCF), 2,7-дикарбоксифлуоренон (2,7-DCF), 3,5-дикарбоксифлуоренон (3,5-DCF), 9-флуоренон-2-карбоновая кислота (9F-2СА), 9-флуоренон-4-карбоновая кислота (9F-4СА), общее количество флуоренонов, включая другие флуореноны, которые не входят в приведенный список индивидуальных флуоренонов (общее содержание флуоренонов), 4,4'-дикарбоксибифенил (4,4'-DCB), 2,5,4'-трикарбоксибифенил (2,5,4'-TCB), фталевая кислота (РА), изофталевая кислота (IPA), бензойная кислота (ВА), тримеллитовая кислота (ТМА), 2,6-дикарбоксибензокумарин (2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-DCBP), 2,5,4'-трикарбоксибензофенон (2,5,4'-ТСВР), терефталевая кислота (ТРА), осажденные твердые вещества при температуре 20°С и общее содержание ароматических карбоновых кислот, у которых отсутствуют неароматические углеводородные группы. В приведенной ниже таблице 1 представлены предпочтительнее количества указанных примесей в СТА, получаемой в соответствии с вариантом осуществления настоящего изобретения.
Многие другие ароматические примеси, как правило, также присутствуют в рециклированном растворителе, и их уровни обычно варьируют в значительно меньших пропорциях по отношению к одному или нескольким из раскрываемых ароматических соединений. Способы контролирования раскрываемых ароматических соединений в предпочтительных границах, как правило, позволяют поддерживать и другие ароматические примеси на приемлемых уровнях.
Известно, что в том случае, когда в реакционной среде применяется бром, в динамическом равновесии может существовать множество ионных и органических форм брома. После того как они покинут реакционную среду и попадут в различные узлы установки, связанные с рециклированием растворителя, эти различные формы брома показывают различную устойчивость. Например, альфа-бром-пара-толуиловая кислота может в определенных условиях сохраняться в виде молекулы, а в других условиях может быстро гидролизоваться с образованием 4-гидроксиметилбензойной кислоты и бромида водорода. В настоящем изобретении, предпочтительно, по меньшей мере, приблизительно 40% мас., более предпочтительно, по меньшей мере, приблизительно 60% мас. и, наиболее предпочтительно, по меньшей мере, приблизительно 80% мас. от общего количества брома, присутствующего в общем исходном растворителе, поступающем в реакционную среду, содержится в виде одной или нескольких из следующих форм: ионы брома, альфа-бром-пара-толуиловая кислота и бромуксусная кислота.
Хотя важность и значение контролирования общей усредненной по весу чистоты исходного растворителя в раскрытых в настоящем изобретении необходимых пределах ранее не было обнаружено и/или раскрыто, подходящие способы контролирования чистоты исходного растворителя можно сгруппировать из различных методов, которые уже известны из области техники. Во-первых, любой растворитель, испарившийся из реакционной среды, как правило, обладает подходящей чистотой, при условии, что жидкие или твердые вещества из реакционной среды не захватываются испарившимся растворителем. Подача капелек растворителя с целью орошения находящегося над реакционной средой пространства, предназначенного для разделения отходящего газа, как раскрывается в настоящем изобретении, позволяет, соответственно, сократить подобное захватывание; из указанного отходящего газа можно сконденсировать рециклированный растворитель, который имеет подходящую чистоту относительно ароматических соединений. Во-вторых, более сложная и дорогостоящая очистка поступающего в реактор рециклированного растворителя, как правило, касается растворителя, который отводится из реакционной среды в жидкой форме, и к растворителю, который в значительной степени контактирует с жидкой и/или твердой фазами реакционной среды, извлекаемыми из реакционной среды (в частности, это относится к рециклированному растворителю, получаемому после фильтра, где твердые вещества концентрируются и/или промываются, к рециклированному растворителю, получаемому после центрифуги, где твердые вещества концентрируются и/или промываются, к рециклированному растворителю, отделяемому после проведения операций кристаллизации, и т.п.). Тем не менее, из области техники известны средства для осуществления необходимой очистки указанных потоков рециклированного растворителя с использованием одного или нескольких ранее раскрытых способов. Подходящими средствами контролирования осажденных веществ в рециклированном растворителе в указанных пределах являются, однако этим не ограничиваясь, гравиметрическая седиментация, механическая фильтрация с использованием фильтровальной ткани на ротационных ленточных фильтрах или ротационных барабанных фильтрах, механическая фильтрация с использованием стационарной фильтрующей среды в сосудах под давлением, гидроциклонах и центрифугах. Средствами контролирования растворенных ароматических соединений в рециклированном растворителе в указанных пределах являются, однако этим не ограничиваясь, средства, раскрываемые в патенте США №4939297, и заявка на патент США №2005-0038288, которые включены в настоящее описание посредством ссылки. Тем не менее, ни в одном из указанных известных изобретений не найдены и не раскрываются предпочтительные уровни чистоты общего исходного растворителя, которые раскрываются в настоящем изобретении. В указанных известных изобретениях лишь предлагаются способы очистки выбранных и частичных потоков рециклированного растворителя, из которых невозможно вывести приведенные в настоящем описании оптимальные значения для общего усредненного по весу состава растворителя, подаваемого в реакционную среду.
Что касается чистоты исходного способного окисляться соединения, то известно, что в нем содержится определенное количество изофталевой кислоты, фталевой кислоты и бензойной кислоты, низкие уровни которых допустимы в очищенной ТРА, используемой в производстве полимеров. Более того, известно, что указанные вещества сравнительно лучше растворяются во многих растворителях, а потому легко могут быть удалены из очищенной ТРА с помощью процессов кристаллизации. Однако из раскрываемого в данном описании предпочтительного варианта осуществления настоящего изобретения теперь известно, что контролирование уровня нескольких относительно легко растворимых ароматических веществ, а именно включающих изофталевую кислоту, фталевую кислоту и бензойную кислоту, в жидкой фазе реакционной среды неожиданно оказалось важным для контролирования уровня полициклических и окрашенных ароматических соединений, образующихся в реакционной среде, для контролирования соединений, содержащих более двух карбоксильных групп в молекуле, для контролирования реакционной способности в среде, где проводится частичное окисление, и для контролирования влияющих на выход продукта потерь окислителя и ароматического соединения.
Из области техники известно, что изофталевая кислота, фталевая кислота и бензойная кислота образуются в реакционной среде следующим образом. Примесь мета-ксилола в исходном сырье с хорошим выходом окисляется с образованием IPA. Примесь орто-ксилола в исходном сырье с хорошим выходом окисляется с образованием фталевой кислоты. Примеси этилбензола и толуола в исходном сырье с хорошим выходом окисляются с образованием бензойной кислоты. Однако авторы настоящего изобретения обнаружили, что значительные количества изофталевой кислоты, фталевой кислоты и бензойной кислоты образуются также в реакционной среде, содержащей пара-ксилол, в результате процессов, отличных от окисления мета-ксилола, орто-ксилола, этилбензола и толуола. Указанные специфичные химические реакции, возможно, включают декарбонилирование, декарбоксилирование, преобразование переходных состояний и включение метильных и карбонильных радикалов в ароматические циклы.
При определении предпочтительных границ для примесей в исходном способном окисляться соединении необходимо учитывать множество факторов. Любая примесь в исходном сырье приведет к прямым потерям в выходе продукта и затратам на очистку продукта, если требования к чистоте продукта окисления являются достаточно жесткими (в частности, в реакционной среде, где проводится частичное окисление пара-ксилола, толуол и этилбензол, которые, как правило, содержатся в коммерчески чистом пара-ксилоле, приводят к образованию бензойной кислоты, а указанная бензойная кислота в значительной степени удаляется в большинстве случаев из получаемой в промышленных масштабах ТРА). В том случае, когда продукт частичного окисления примеси, поступившей вместе с исходным сырьем, принимает участие в дополнительных реакциях, то при определении затрат на очистку исходного сырья следует принимать во внимание не только потери выхода продукта, но и учитывать и устранять другие факторы (в частности, в реакционной среде, где проводится частичное окисление пара-ксилола, этилбензол приводит, по мимо прочего, к образованию бензойной кислоты, а бензойная кислота, в свою очередь, приводит к сильно окрашенной 9-флуоренон-2-карбоновой кислоте, к изофталевой кислоте, к фталевой кислоте и к повышенным количествам оксидов углерода). В том случае, когда сама реакционная среда образует дополнительные количества примесей вследствие протекания химических реакций, напрямую не связанных с имеющимися в исходном сырье примесями, то анализ становится еще более сложным (в частности, в реакционной среде, где проводится частичное окисление пара-ксилола, бензойная кислота образуется также из пара-ксилола). Кроме того, на требования к предпочтительной чистоте исходного сырья может оказать влияние и дальнейшая обработка сырого продукта окисления. Например, затраты на удаление до приемлемых уровней прямой примеси (бензойной кислоты) и производных примесей (изофталевой кислоты, фталевой кислоты, 9-флуоренон-2-карбоновой кислоты и т.п.) могут быть одинаковыми, могут отличаться друг от друга и могут быть отличны от требований, предъявляемых к удалению в основном не относящейся к данной категории примеси (в частности, 4-СВА - продукту неполного окисления при окислении пара-ксилола с образованием ТРА).
Раскрываемые ниже диапазоны чистоты исходного пара-ксилола являются предпочтительными в том случае, когда пара-ксилол вместе с растворителем и окислителем подается в реакционную среду для частичного окисления с образованием ТРА. Указанные диапазоны являются более предпочтительными для процесса получения ТРА, который предполагает проведение послеокислительных стадий, направленных на удаление примесей, отличных от окислителя и растворителя (в частности, металлов катализатора). Указанные диапазоны еще более предпочтительны для способов получения ТРА, в которых проводится удаление дополнительного количества 4-СВА из СТА (в частности, с помощью превращения СТА в диметилтерефталат плюс примесные эфиры и последующего отделения метилового эфира 4-СВА дистилляцией, с помощью методов окислительного дегирирования, с целью превращения 4-СВА в ТРА, с помощью методов гидрирования, с целью превращения 4-СВА в пара-толуиловую кислоту, которую затем отделяют методами дробной кристаллизации). Указанные диапазоны наиболее предпочтительны для процессов получения ТРА, в которых проводится удаление дополнительного количества 4-СВА из СТА с помощью методов окислительного дегирирования с целью превращения 4-СВА в ТРА.
На основании новых данных о предпочтительных диапазонах рециклирования ароматических соединений и относительных количествах ароматических соединений, которые непосредственно образуются из содержащихся в исходном сырье примесей, в сравнении с возможными другими путями протекания химических превращений, установлены улучшенные диапазоны для примесей в неочищенном пара-ксилоле, который применяется для проведения процесса частичного окисления при получении ТРА. Ниже в таблице 2 представлены предпочтительные значения для количества мета-ксилола, орто-ксилола и этилбензола + толуола в исходном пара-ксилоле.
Специалистам должно быть понятно, что приведенные выше примеси в неочищенном пара-ксилоле могут оказать максимальное воздействие на реакционную среду после того, как продукты их частичного окисления аккумулируются в рециклированном растворителе. Например, поступление мета-ксилола в количестве, соответствующем верхнему уровню из наиболее предпочтительного диапазона, т.е. 400 м.д. мас., немедленно приводит к образованию приблизительно 200 м.д. мас.изофталевой кислоты в жидкой фазе реакционной среды в том случае, когда содержание твердых веществ в реакционной среде составляет приблизительно 33% мас. Это сопоставимо с количеством, соответствующим верхней границе, составляющей 400 м.д. мас., наиболее предпочтительного диапазона для изофталевой кислоты в рециклированном растворителе, которое после типичного испарения растворителя, проводимого с целью охлаждения реакционной среды, приводит к образованию приблизительно 1200 м.д. мас. изофталевой кислоты в жидкой фазе реакционной среды. Таким образом, наибольшее вероятное воздействие мета-ксилола, орто-ксилола, этилбензола и толуола, содержащихся в исходном неочищенном пара-ксилоле, заключается именно в накоплении с течением времени продуктов частичного окисления в рециклированном растворителе. Таким образом, приведенные выше диапазоны для примесей в исходном неочищенном пара-ксилоле, предпочтительно, следует поддерживать в течение, по крайней мере, половины каждого из дней проведения процесса в любой реакционной среде для проведения частичного окисления в конкретной промышленной установке, более предпочтительно, по крайней мере, в течение трех четвертей каждого дня, по меньшей мере, для семи последовательных дней проведения процесса и, наиболее предпочтительно, в том случае, когда среднемассовые значения композиции неочищенного пара-ксилола находятся в приведенных предпочтительных пределах, в течение, по меньшей мере, 30 дней непрерывного проведения процесса.
Способы получения содержащего примеси пара-ксилола предпочтительной степени чистоты известны из области техники и включают, однако этим не ограничиваясь, дистилляцию, методы дробной кристаллизации при температурах ниже комнатной температуры и способы с использованием молекулярных сит, включающих селективную адсорбцию в зависимости от размера пор. Тем не менее, максимальные требования, касающиеся приведенных в данном описании предпочтительных диапазонов чистоты, являются слишком завышенными и дорогостоящими по сравнению с теми, что обычно обеспечиваются коммерческими поставщиками пара-ксилола; в то же время минимальные требования, касающиеся предпочтительных диапазонов чистоты, позволяют избежать излишней дорогостоящей очистки пара-ксилола, подаваемого в реакционную среду для проведения частичного окисления, поскольку установлено и раскрыто, где объединенные эффекты самостоятельного образования примеси из самого пара-ксилола и реакций потребления примеси в реакционной среде становятся более важными, чем уровни примесей, привносимых вместе с неочищенным пара-ксилолом.
В том случае, когда содержащий пара-ксилол поток исходных веществ включает выбранные примеси, такие как этилбензол и/или толуол, окисление указанных примесей может привести к образованию бензойной кислоты. В данном описании термин “образовавшаяся из примеси бензойная кислота” обозначает бензойную кислоту, полученную из источника, отличного от ксилола в процессе окисления ксилола.
Как раскрывается в настоящем изобретении, часть бензойной кислоты, получаемая при окислении ксилола, образуется из самого ксилола. Эта бензойная кислота образуется из ксилола явно дополнительно по отношению к любой фракции образовавшейся бензойной кислоты, которая может представлять собой бензойную кислоту, полученную из примеси. Не связывая себя какой-либо теорией, авторы настоящего изобретения полагают, что бензойная кислота образуется из ксилола в реакционной среде в том случае, когда различные промежуточные продукты окисления ксилола претерпевают спонтанное декарбонилирование (потеря монооксида углерода) или декарбоксилирование (потеря диоксида углерода) с образованием арильных радикалов. Указанные арильные радикалы могут отнимать атомы водорода из одного или нескольких источников, доступных в реакционной среде, и приводить к самостоятельно образовавшейся бензойной кислоте. Независимо от механизма протекания химической реакции термин “самостоятельно образовавшаяся бензойная кислота” обозначает бензойную кислоту, которая получена из ксилола в процессе окисления.
Как также раскрывается в настоящем изобретении, в том случае, когда пара-ксилол окисляется с образованием терефталевой кислоты (ТРА), накопление самостоятельно образовавшейся бензойной кислоты приводит к уменьшению выхода продукта в пересчете на пара-ксилол и в пересчете на окислитель. Кроме того, присутствие самостоятельно образовавшейся бензойной кислоты в жидкой фазе реакционной среды коррелирует с возрастанием многих нежелательных побочных реакций, включающих, в частности, образование сильно окрашенных соединений, называемых монокарбоксифлуоренонами. Самостоятельно образовавшаяся бензойная кислота приводит также к нежелательному накоплению бензойной кислоты в рециклированном фильтрате, что приводит к дальнейшему повышению концентрации бензойной кислоты в жидкой фазе реакционной среды. Таким образом, формирование самостоятельно образовавшейся бензойной кислоты желательно свести к минимуму, однако при этом необходимо одновременно учитывать, соответственно, образующуюся из примеси бензойную кислоту, факторы, влияющие на потребление бензойной кислоты, факторы, которые связаны с другими проблемами селективности реакций, общие вопросы экономики.
Авторы настоящего изобретения обнаружили, что самостоятельное образование бензойной кислоты можно контролировать на низких уровнях за счет соответствующего выбора, например, температуры, подачи ксилола и доступности кислорода в реакционной среде в процессе окисления. Не связывая себя какой-либо теорией, авторы настоящего изобретения полагают, что низкие температуры и улучшенная доступность кислорода, вероятно, подавляют декарбонилирование и/или декарбоксилирование и тем самым позволяют избежать проблем, связанных с самостоятельным образованием бензойной кислоты. Достаточная доступность кислорода направляет арильные радикалы в сторону образования менее вредных продуктов, в частности гидроксибензойных кислот. Распределение ксилола в реакционной среде также может повлиять на равновесие между превращением арильных радикалов в бензойную кислоту или в гидроксибензойные кислоты. Независимо от механизма протекания химической реакции авторы настоящего изобретения определили условия проведения реакции, которые, несмотря на то, что они являются достаточно мягкими, чтобы снизить образование бензойной кислоты, в то же время являются достаточно жесткими, чтобы окислить значительную фракцию образовавшихся гидроксибензойных кислот с получением монооксида и/или диоксида углерода, которые легко удаляются из продукта реакции.
В предпочтительном варианте осуществления настоящего изобретения реактор окисления сконфигурирован и эксплуатируется таким образом, чтобы формирование самостоятельно образовавшейся бензойной кислоты было сведено к минимуму, а окисление гидроксибензойных кислот с образованием монооксида и/или диоксида углерода было максимальным. В том случае, когда для окисления пара-ксилола с образованием терефталевой кислоты используют реактор для проведения процессов окисления, пара-ксилол, предпочтительно, должен составлять, по меньшей мере, приблизительно 50% мас. от общего количества ксилола в потоке исходных соединений, подаваемых в реактор. Более предпочтительно, пара-ксилол составляет, по меньшей мере, приблизительно 75% мас. от общего количества ксилола в потоке исходных соединений. Еще более предпочтительно, пара-ксилол составляет, по меньшей мере, приблизительно 95% мас. от общего количества ксилола в потоке исходных соединений. Наиболее предпочтительно, пара-ксилол составляет практически все количество ксилола в потоке исходных соединений.
Если в реакторе проводится окисление пара-ксилола с образованием терефталевой кислоты, то скорость получения терефталевой кислоты, предпочтительно, должна быть максимальной, а скорость получения самостоятельно образовавшейся бензойной кислоты должна быть минимальной. Отношение скорости получения (по массе) терефталевой кислоты к скорости получения (по массе) самостоятельно образовавшейся бензойной кислоты, предпочтительно, составляет, по крайней мере, приблизительно, 500:1, более предпочтительно, составляет, по крайней мере, приблизительно, 1000:1 и, наиболее предпочтительно, составляет, по крайней мере, 1500:1. Как будет показано ниже, скорость получения самостоятельно образовавшейся бензойной кислоты, преимущественно, измеряют тогда, когда концентрация бензойной кислоты в жидкой фазе реакционной среды составляет меньше 2000 м.д. мас., более предпочтительно, составляет меньше 1000 м.д. мас. и, наиболее предпочтительно, составляет меньше 500 м.д. мас., поскольку указанные низкие концентрации позволяют снизить до приемлемо низких уровней скорости реакций, в которых бензойная кислота превращается в другие соединения.
Если объединить самостоятельно образовавшуюся бензойную кислоту и образовавшуюся из примеси бензойную кислоту, то отношение скорости получения (по массе) терефталевой кислоты к скорости получения (по массе) общей бензойной кислоты, предпочтительно, составляет, по крайней мере, приблизительно, 400:1, более предпочтительно, составляет, по крайней мере, приблизительно, 700:1 и, наиболее предпочтительно, составляет, по крайней мере, 1100:1. Как будет показано ниже, суммарная скорость получения самостоятельно образовавшейся бензойной кислоты плюс образовавшейся из примеси бензойной кислоты, преимущественно, измеряют тогда, когда концентрация бензойной кислоты в жидкой фазе реакционной среды составляет меньше 500 м.д. мас., поскольку указанные низкие концентрации позволяют снизить до приемлемо низких уровней скорости реакций, в которых бензойная кислота превращается в другие соединения.
Как раскрывается в настоящем изобретении, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к повышению образования многих других ароматических соединений, из которых несколько являются вредными примесями в ТРА; и, как раскрывается в настоящем изобретении, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к повышению образования газообразных оксидов углерода, а их образование приводит к потере выхода продукта в пересчете на окислитель или ароматические соединения и/или к потере растворителя. Кроме того, как раскрывается в настоящем изобретении, авторы настоящего изобретения обнаружили, что значительная часть из указанного повышенного образования других ароматических соединений и оксидов углерода вызвана протеканием реакций, которые превращают некоторое количество самих молекул бензойной кислоты, в отличие от бензойной кислоты, катализирующей протекание других реакций, в которых она сама не потребляется. Таким образом, “суммарное образование бензойной кислоты” определяется в данном описании как усредненная по времени масса бензойной кислоты, которая покидает реакционную среду, минус усредненная по времени масса всей бензойной кислоты, которая попадает в реакционную среду за тот же период времени. Указанное суммарное образование бензойной кислоты часто имеет положительную величину, что определяется скоростью получения образовавшейся из примеси бензойной кислоты и самостоятельно образовавшейся бензойной кислоты. Однако авторы настоящего изобретения обнаружили, что скорость превращения бензойной кислоты в оксиды углерода и в некоторые другие соединения практически линейно увеличивается по мере возрастания концентрации бензойной кислоты в жидкой фазе реакционной среды, если измерения проводятся, когда другие условия проведения реакции, включая температуру, доступность кислорода, STR и активность реакции, поддерживаются приблизительно постоянными. Так, в том случае, когда концентрация бензойной кислоты в жидкой фазе реакционной среды достаточно велика, возможно, за счет повышенной концентрации бензойной кислоты в рециклированном растворителе, то превращение молекул бензойной кислоты в другие соединения, включая оксиды углерода, может сравниться или даже превзойти химическое образование новых молекул бензойной кислоты. В этом случае величина суммарного образования бензойной кислоты может балансировать вблизи нуля или даже стать отрицательной. Авторы настоящего изобретения обнаружили, что в том случае, когда суммарное образование бензойной кислоты имеет положительную величину, то отношение скорости образования (по массе) терефталевой кислоты в реакционной среде к скорости суммарного образования бензойной кислоты в реакционной среде, предпочтительно, составляет больше чем приблизительно 700:1, более предпочтительно, составляет больше чем приблизительно 1100:1 и, наиболее предпочтительно, составляет больше чем 4000:1. Авторы настоящего изобретения обнаружили, что в том случае, когда величина суммарного образования бензойной кислоты имеет отрицательную величину, то отношение скорости образования (по массе) терефталевой кислоты в реакционной среде к скорости суммарного образования бензойной кислоты в реакционной среде, предпочтительно, составляет больше чем приблизительно 200:(-1), более предпочтительно, составляет больше чем приблизительно 100:(-1) и, наиболее предпочтительно, составляет больше чем 5000:(-1).
Авторы настоящего изобретения обнаружили предпочтительные диапазоны для состава суспензии (жидкость + твердое вещество), которая выгружается из реакционной среды, и состава порции СТА в суспензии. Неожиданно оказалось, что предпочтительный состав суспензии и предпочтительный состав СТА имеют важное практическое значение. Например, очищенная ТРА, получаемая из указанной предпочтительной СТА путем окислительного дегирирования, содержит достаточно низкий уровень всех примесей и окрашенных примесей, так что очищенная ТРА пригодна без дополнительного гидрирования 4-СВА и/или окрашенных примесей для широкого круга применений ПЭТ при изготовления волокон и упаковочных материалов. Например, предпочтительный состав суспензии, как указывается в данном описании, позволяет получить жидкую фазу реакционной среды, которая содержит относительно низкое количество важных примесей и тем самым значительно уменьшает образование других, еще более нежелательных примесей, как указано в данном описании. Кроме того, предпочтительный состав суспензии, в соответствии с предпочтительным вариантом осуществления настоящего изобретения, значительно облегчает последующую обработку жидкости, содержащейся в суспензии, с целью получения достаточно чистого рециклированного растворителя.
СТА, получаемая в соответствии с вариантом осуществления настоящего изобретения, содержит меньше примесей указанного типа, чем СТА, получаемая с использованием обычных способов и установок, в частности тех, где используют рециклированный растворитель. Примеси, которые могут присутствовать в СТА, включают следующие: 4-карбоксибензальдегид (4-СВА), 4,4'-дикарбоксистильбен (4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-DCF), 2,7-дикарбоксифлуоренон (2,7-DCF), 3,5-дикарбоксифлуоренон (3,5-DCF), 9-флуоренон-2-карбоновая кислота (9F-2СА), 9-флуоренон-4-карбоновая кислота (9F-4СА), 4,4'-дикарбоксибифенил (4,4'-DCB), 2,5,4'-трикарбоксибифенил (2,5,4'-TCB), фталевая кислота (РА), изофталевая кислота (IPA), бензойная кислота (ВА), тримеллитовая кислота (ТМА), пара-толуиловая кислота (РТАС), 2,6-дикарбоксибензокумарин (2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-DCBP), 2,5,4'-трикарбоксибензофенон (2,5,4'-ТСВР). В приведенной ниже таблице 3 представлены предпочтительные количества указанных примесей в СТА, получаемой в соответствии с вариантом осуществления настоящего изобретения.
Кроме того, СТА, полученная в соответствии с вариантом осуществления настоящего изобретения, предпочтительно, менее окрашена по сравнению с СТА, полученной с помощью обычных способов и установок, в частности тех, где используют рециклированный растворитель. Так, СТА, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, предпочтительно имеет процент пропускания при 340 нм, равный, по крайней мере, приблизительно, 25%, более предпочтительно, по крайней мере, приблизительно, 50% и, наиболее предпочтительно, по крайней мере, 60%. Далее, СТА, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, предпочтительно, имеет процент пропускания при 400 нм, равный, по крайней мере, приблизительно, 88%, более предпочтительно, по крайней мере, приблизительно, 90% и, наиболее предпочтительно, по крайней мере, 92%.
Тест на процент пропускания является мерой содержаний окрашенных, поглощающих свет примесей в ТРА или СТА. В данном описании тест заключается в измерениях, которые проводятся для порции раствора, полученного путем растворения 2,00 г сухой твердой ТРА или СТА в 20,0 мл диметилсульфоксида (ДМСО), имеющего квалификацию “чистый для анализа” или лучше. Порцию указанного раствора затем помещают в полумикропроточную ячейку Hellma, PN 176700, которая изготовлена из кварца и имеет световой путь 1,0 см и объем 0,39 мл (Hellma USA, 80 Skyline Drive, Plainview, NY 11803). Для определения пропускания через указанную заполненную проточную ячейку при разной длине волны света применяют спектрофотометр с диодной матрицей Agilent 8453 Diode Array Spectrophotometer (Agilent Technologies, 395 Page Mill Road, Palo Alto, CA 94303). После проведения соответствующей коррекции на фоновое поглощение, в том числе поглощения указанной ячейки и используемого растворителя, данные по проценту пропускания, характеризующие фракцию падающего света, которая прошла через раствор, непосредственно указываются прибором. Данные по проценту пропускания света на длинах волн 340 и 400 нм наиболее пригодны для отделения чистого ТРА от многих примесей, которые, как правило, находятся в ней.
В приведенной ниже таблице 4 представлены предпочтительнее границы для различных ароматических примесей в фазе суспензии (твердое вещество + жидкость) реакционной среды.
Указанные предпочтительные составы суспензии включают предпочтительный состав жидкой фазы реакционной среды и позволяют избежать экспериментальных трудностей, вызванных осаждением компонентов дополнительной жидкой фазы из реакционной среды на компоненты твердой фазы в процессе отбора образца из реакционной среды, разделения жидкостей и твердых веществ и изменения режимов проведения анализа.
Многие другие ароматические примеси, как правило, также присутствуют в фазе суспензии в реакционной среде и содержащейся в реакционной среде СТА и обычно варьируют с еще меньшими уровнями и/или пропорциями по отношению к одному или нескольким раскрываемым ароматическим соединениям. Контролирование раскрываемых ароматических соединений в предпочтительных границах позволяет поддерживать на приемлемых уровнях и другие ароматические примеси. Указанные предпочтительные составы для фазы суспензии в реакционной среде и для твердой СТА, которые определяют непосредственно в суспензии, достигаются благодаря осуществлению приведенных в данном описании вариантов осуществления настоящего изобретения для частичного окисления пара-ксилола с образованием ТРА.
Измерение концентрации компонентов, присутствующих с низкими уровнями, в растворителе, рециклированном растворителе, СТА, суспензии из реакционной среды и РТА проводят, используя методы жидкостной хроматографии. Далее описываются два равноценных варианта осуществления настоящего изобретения.
Способ, который в данном описании обозначают как HPLC-DAD, включает жидкостную хроматографию высокого разрешения (ВЭЖХ) совместно с детектированием на диодной матрице (DAD), с целью проведения раздельной количественной оценки молекул различных веществ в отобранном образце. Для проведения указанных измерений используют модель 1100 HPLC, оснащенную детектором на диодной матрице, который изготавливает фирма Agilent Technologies (Пало-Альто, Калифорния), хотя подходящие аналитические приборы коммерчески доступны и от других изготовителей. Как известно из области техники, как время элюирования, так и выходной сигнал детектора калибруют с помощью известных соединений, которые присутствуют в известных количествах, при этом соединения и их количества соответствуют тем, которые содержатся в реальных неизвестных образцах.
Способ, который в данном описании обозначают как ВЭЖХ-МС, включает жидкостную хроматографию высокого разрешения (ВЭЖХ), сопряженную с масс-спектрометрией (МС), с целью разделения, идентификации и количественной оценки молекул различных веществ в отобранном образце. Для проведения измерений используют аналитические приборы Alliance HPLC и ZQ MS, которые изготавливает фирма Waters Corp. (Милфорд, Массачусетс), хотя подходящие аналитические приборы коммерчески доступны и от других изготовителей. Как известно из области техники, как время элюирования, так и выходной сигнал масс-спектрометра калибруют с помощью известных соединений, которые присутствуют в известных количествах, при этом соединения и их количества соответствуют тем, которые содержатся в реальных неизвестных образцах.
Другой вариант осуществления настоящего изобретения относится к частичному окислению способного окисляться ароматического соединения при соответствующем поддержании баланса между подавлением вредных ароматических примесей, с одной стороны, и между образованием диоксида углерода и монооксида углерода, которые обозначают общим термином оксиды углерода (СОх), - с другой стороны. Как правило, указанные оксиды углерода покидают реактор вместе с отходящими газами, и они соответствуют деструктивным потерям растворителя и способного окисляться соединения, включая, в конечном счете, и предпочтительные окисленные производные (в частности, уксусную кислоту, пара-ксилол и ТРА). Авторы настоящего изобретения установили нижние границы для образования оксидов углерода, ниже которых, по-видимому, значительное образование вредных ароматических примесей, как указано ниже, и общий низкий уровень конверсии неизбежно становятся слишком неблагоприятными, чтобы можно было считать проведение процесса экономически оправданным. Авторы настоящего изобретения установили также верхние границы для образования оксидов углерода, выше которых образование оксидов углерода продолжает увеличиваться, а дальнейших улучшений, связанных с сокращением образования вредных ароматических примесей, больше не наблюдается.
Авторы настоящего изобретения обнаружили, что снижение в жидкой фазе реакционной среды концентраций способного окисляться исходного ароматического соединения и промежуточных ароматических соединений приводит к снижению скоростей образования вредных примесей в процессе частичного окисления способного окисляться ароматического соединения. Указанные вредные примеси включают конденсированные ароматические циклы и/или ароматические молекулы, содержащие больше чем нужно карбоксильных групп (в частности, при окислении пара-ксилола вредные примеси включают 2,6-дикарбоксиантрахинон, 2,6-дикарбоксифлуоренон, тримеллитовую кислоту, 2,5,4'-трикарбоксибифенил и 2,5,4'-бензофенон). Промежуточные ароматические вещества включают ароматические соединения, которые приходят вместе со способным окисляться исходным ароматическим соединением и все еще сохраняют неароматические углеводородные группы (в частности, при окислении пара-ксилола промежуточные ароматические вещества включают пара-толуальдегид, терефтальдегид, пара-толуиловую кислоту, 4-СВА, 4-гидроксиметилбензойную кислоту и альфа-бром-пара-толуиловую кислоту). Способное окисляться исходное ароматическое соединение и промежуточные ароматические вещества, сохраняющие неароматические углеводородные группы, если они присутствуют в жидкой фазе реакционной среды, видимо, приводят к вредным примесям аналогично тому, как уже указывалось в данном описании для растворенных ароматических веществ, не содержащих неароматические углеводородные группы (в частности, для изофталевой кислоты).
Решая проблему повышения активности реакции с целью подавить образование вредных ароматических примесей в процессе частичного окисления способного окисляться ароматического соединения, авторы настоящего изобретения обнаружили, что нежелательным сопутствующим результатом является увеличение образования оксидов углерода. Важно понимать, что указанные оксиды углерода являются потерей не только окислителя, но и способного окисляться соединения и окислителя. Очевидно, что существенная и иногда основная фракция оксидов углерода образуется из способного окисляться соединения, его производных, а не растворителя; а стоимость способного окисляться соединения, в пересчете на углеродную единицу, часто значительно больше, чем стоимость растворителя. Кроме того, важно понимать, что карбоновая кислота, которая является целевым продуктом (в частности, ТРА), также подвергается избыточному окислению в оксиды углерода в том случае, когда она присутствует в жидкой фазе реакционной среды.
Важно также понимать, что настоящее изобретение относится к реакциям, протекающим в жидкой фазе реакционной среды и к концентрации реагентов в ней. Этим оно отличается от некоторых известных изобретений, которые относятся непосредственно к реакциям в осажденной твердой форме ароматического соединения, сохраняющего неароматические углеводородные группы. В частности, в случае частичного окисления пара-ксилола с образованием ТРА, некоторые известные изобретения касаются количества 4-СВА, осаждаемой в твердой фазе СТА. Однако авторы настоящего изобретения обнаружили, что существует расхождение больше чем два к одному для отношения количества 4-СВА в твердой фазе к количеству 4-СВА в жидкой фазе при прочих равных параметрах для температуры, давления, катализа, состава растворителя и пространственно-временной скорости пара-ксилола, в зависимости от того, проводится ли частичное окисление в хорошо перемешиваемом автоклаве или в реакционной среде, в которой кислород и пара-ксилол распределяются в соответствии с настоящим изобретением. Кроме того, авторы настоящего изобретения обнаружили, что отношение количества 4-СВА в твердой фазе к количеству 4-СВА в жидкой фазе также может варьировать больше чем два к одному как в хорошо перемешиваемой реакционной среде, так и в реакционной среде, где формируется определенное распределение реагентов, в зависимости от скорости пространственно-временной реакции пара-ксилола, при прочих равных параметрах для температуры, давления, катализа и состава растворителя. Кроме того, 4-СВА в твердой фазе СТА, видимо, не приводит к образованию вредных примесей, и 4-СВА можно выделить из твердой фазы и легко с большим выходом доокислить в ТРА (в частности, путем окислительного дегирирования суспензии СТА, как указывается в данном описании); в то время как удаление вредных примесей является значительно более трудной и дорогостоящей процедурой, чем удаление твердой фазы 4-СВА, а образование оксидов углерода ведет к постоянным потерям в выходе продукта. Таким образом, следует различать, что данный аспект настоящего изобретения касается составов жидкой фазы реакционной среды.
Авторы настоящего изобретения обнаружили, что независимо от того, что является источником - растворитель или способное окисляться соединение - при осуществлении превращений в промышленных масштабах образование оксидов углерода непосредственно определяется уровнем общей активности реакции, независимо от варьирования в широких пределах конкретных комбинаций температуры, металлов, галогенов, температуры, кислотности реакционной среды, определяемой значением pH, концентрации воды, применяемой для достижения уровня общей активности реакции. Авторы настоящего изобретения обнаружили, что при частичном окислении ксилола следует оценивать уровень общей активности реакции с помощью концентрации толуиловых кислот в жидкой фазе на половине высоты реакционной среды, в нижней части реакционной среды и в верхней части реакционной среды.
Таким образом, важно одновременно поддерживать баланс между минимизацией формирования вредных примесей за счет повышения активности реакции и одновременно минимизацией образования оксидов углерода за счет понижения активности реакции. Таким образом, если общее образование оксидов углерода подавляется до слишком низких значений, то в этом случае образуются значительные уровни примесей, и наоборот.
Кроме того, авторы настоящего изобретения обнаружили, что растворимость и относительная активность требуемой карбоновой кислоты (в частности, ТРА) и присутствие других растворенных ароматических веществ, у которых отсутствуют неароматические углеводородные группы, является важным средством поддержания равновесия между количеством оксидов углерода и вредных примесей. Карбоновая кислота, являющаяся целевым продуктом, как правило, хорошо растворяется в жидкой фазе реакционной среды, даже когда она присутствует в твердой форме. Например, при температурах, попадающих в предпочтительные диапазоны, ТРА растворима в реакционной среде, содержащей уксусную кислоту и воду, на уровне, составляющем от приблизительно 1000 м.д. мас. до более чем 1% мас., при этом растворимость увеличивается с увеличением температуры. Независимо от того, что скорости реакций образования разных вредных примесей различны для способного окисляться исходного ароматического соединения (в частности, пара-ксилола), для промежуточных ароматических соединений (в частности, пара-толуиловой кислоты), для требуемого продукта, которым является ароматическая карбоновая кислота (в частности, ТРА), и для ароматических веществ, не содержащих неароматические углеводородные группы (в частности, изофталевой кислоты), присутствие и реакционная способность последних двух групп определяет область уменьшения выхода в зависимости от дальнейшего подавления предыдущих двух групп, способного окисляться исходного ароматического соединения и промежуточных ароматических соединений. Например, при частичном окислении пара-толуола с образованием ТРА в том случае, когда при заданных условиях содержание растворенной ТРА в жидкой фазе реакционной среды достигает 7000 м.д. мас., содержание растворенной бензойной кислоты достигает 8000 м.д. мас., содержание растворенной изофталевой кислоты достигает 6000 м.д. мас., а содержание растворенной фталевой кислоты достигает 2000 м.д. мас., то способность процесса обеспечить дальнейшее сокращение общего количества вредных примесей начинает снижаться по мере того, как активность реакции повышается и сокращается концентрация пара-толуиловой кислоты и 4-СВА в жидкой фазе до значений, меньших чем указанные уровни. Таким образом, присутствие и концентрация в жидкой фазе реакционной среды ароматических веществ, не содержащих неароматические углеводородные группы, незначительно меняется при увеличении активности реакции, и их присутствие позволяет расширить верхние границы потерь от снижения концентрации промежуточных соединений при подавлении образования вредных примесей.
Так, в одном из вариантов осуществления настоящего изобретения указываются предпочтительные диапазоны для оксидов углерода, которые по нижнему пределу определяются низкой активностью реакции и избыточным образованием вредных примесей, а по верхнему пределу определяются избыточными потерями углерода, однако указанные уровни меньше, чем те, которые ранее обнаружены и раскрыты как коммерчески приемлемые. Таким образом, образование оксидов углерода предпочтительно контролируют следующим образом. Отношение молей образовавшихся суммарных оксидов углерода к молям поступившего в реакцию способного окисляться ароматического соединения, предпочтительно, составляет больше чем приблизительно 0,02:1, более предпочтительно, составляет больше чем приблизительно 0,04:1, еще более предпочтительно, составляет больше чем приблизительно 0,05:1 и, наиболее предпочтительно, составляет больше чем 0,06:1. В то же время отношение молей образовавшихся суммарных оксидов углерода к молям поступившего в реакцию способного окисляться ароматического соединения, предпочтительно, составляет меньше чем приблизительно 0,24:1, более предпочтительно, составляет меньше чем приблизительно 0,22:1, еще более предпочтительно, составляет меньше чем приблизительно 0,19:1 и, наиболее предпочтительно, составляет меньше чем 0,15:1. Отношение молей образовавшегося диоксида углерода к молям поступившего в реакцию способного окисляться ароматического соединения, предпочтительно, составляет больше чем приблизительно 0,01:1, более предпочтительно, составляет больше чем приблизительно 0,03:1, еще более предпочтительно, составляет больше чем приблизительно 0,04:1 и, наиболее предпочтительно, составляет больше чем 0,05:1. В то же время отношение молей образовавшегося диоксида углерода к молям поданного в реакцию способного окисляться ароматического соединения, предпочтительно, составляет меньше чем приблизительно 0,21:1, более предпочтительно, составляет меньше чем приблизительно 0,19:1, еще более предпочтительно, составляет меньше чем приблизительно 0,16:1 и, наиболее предпочтительно, составляет меньше чем 0,11:1. Отношение молей образовавшегося монооксида углерода к молям поступившего в реакцию способного окисляться ароматического соединения, предпочтительно, составляет больше чем приблизительно 0,005:1, более предпочтительно, составляет больше чем приблизительно 0,010:1, еще более предпочтительно, составляет больше чем приблизительно 0,015:1 и, наиболее предпочтительно, составляет больше чем 0,020:1. В то же время отношение молей образовавшегося монооксида углерода к молям поступившего в реакцию способного окисляться ароматического соединения, предпочтительно, составляет меньше чем приблизительно 0,09:1, более предпочтительно, составляет меньше чем приблизительно 0,07:1, еще более предпочтительно, составляет меньше чем приблизительно 0,05:1 и, наиболее предпочтительно, составляет меньше чем 0,04:1.
Содержание диоксида углерода в сухом газе, который выводится из реактора для проведения процессов окисления, предпочтительно, составляет больше чем приблизительно 0,10% мол., более предпочтительно, составляет больше чем приблизительно 0,20% мол., еще более предпочтительно, составляет больше чем приблизительно 0,25% мол. и, наиболее предпочтительно, составляет больше чем 0,30%
мол. В то же время содержание диоксида углерода в сухом газе, который выводится из реактора для проведения процессов окисления, предпочтительно, составляет меньше чем приблизительно 1,5% мол., более предпочтительно, составляет меньше чем приблизительно 1,2% мол., еще более предпочтительно, составляет меньше чем приблизительно 0,9% мол. и, наиболее предпочтительно, составляет меньше чем 0,8% мол. Содержание монооксида углерода в сухом газе, который выводится из реактора для проведения процессов окисления, предпочтительно, составляет больше чем приблизительно 0,05% мол., более предпочтительно, составляет больше чем приблизительно 0,10% мол., еще более предпочтительно, составляет больше чем приблизительно 0,15% мол. и, наиболее предпочтительно, составляет больше чем 0,18% мол. В то же время содержание монооксида углерода в сухом газе, который выводится из реактора для проведения процессов окисления, предпочтительно, составляет меньше чем приблизительно 0,60% мол., более предпочтительно, составляет меньше чем приблизительно 0,50% мол., еще более предпочтительно, составляет меньше чем приблизительно 0,35% мол. и, наиболее предпочтительно, составляет меньше чем 0,28% мол.
Авторы изобретения установили, что, согласно описанию настоящего изобретения, важным фактором снижения образования оксидов углерода до указанных предпочтительных диапазонов является повышение чистоты рециклированного фильтрата и способного окисляться исходного соединения, с целью снижения концентрации ароматических соединений, которые не содержат неароматических углеводородных групп, при этом одновременно снижается образование оксидов углерода и вредных примесей. Другим фактором, согласно описанию настоящего изобретения является улучшение распределения пара-ксилола и окислителя внутри реактора. Другим фактором, позволяющим поддерживать приведенные выше предпочтительные уровни оксидов углерода, является образование в реакционной среде указанных в настоящем описании градиентов давления, температуры, концентрации способного окисляться соединения в жидкой фазе и концентрации окислителя в газовой фазе. Другим фактором, позволяющим поддерживать приведенные выше предпочтительные уровни оксидов углерода, является использование указанных в настоящем описании предпочтительных значений пространственно-временной скорости, давления, температуры, состава растворителя, состава катализатора и механической конфигурации реактора.
Важным преимуществом работы в предпочтительных границах образования оксидов углерода является то, что можно снизить потребление молекулярного кислорода, хотя и не до стехиометрического значения. Независимо от того, насколько хорошо, в соответствии с настоящим изобретением, распределяются окислитель и способное окисляться соединение, как показывает расчет, проведенный для одного лишь способного окисляться исходного соединения, должен сохраняться избыток кислорода над стехиометрическим значением с тем, чтобы могли происходить некоторые потери в виде оксидов углерода, и с тем, чтобы обеспечить за счет избытка кислорода контроль над образованием вредных примесей. Конкретно в том случае, когда способным окисляться исходным соединением является ксилол, отношение в исходных веществах массы молекулярного кислорода к массе ксилола, предпочтительно, составляет больше чем приблизительно 0,91:1,00, более предпочтительно, составляет больше чем приблизительно 0,95:1,00 и, наиболее предпочтительно, составляет больше чем 0,99:1,00. В то же время отношение в исходных веществах массы молекулярного кислорода к массе ксилола, предпочтительно, составляет меньше чем приблизительно 1,20:1,00, более предпочтительно, составляет меньше чем приблизительно 1,12:1,00 и, наиболее предпочтительно, составляет меньше чем 1,06:1,00. Конкретно в том случае, когда исходным соединением является ксилол, усредненное по времени содержание молекулярного кислорода в сухом газе, который отводится из реактора для проведения процессов окисления, предпочтительно, составляет больше чем приблизительно 0,1% мол., более предпочтительно, составляет больше чем приблизительно 1% мол. и, наиболее предпочтительно, составляет больше чем 1,5% мол. В то же время усредненное по времени содержание молекулярного кислорода в сухом газе, который отводится из реактора для проведения процессов окисления, предпочтительно, составляет меньше чем приблизительно 6% мол., более предпочтительно, составляет меньше чем приблизительно 4% мол. и, наиболее предпочтительно, составляет меньше чем 3% мол.
Еще одним важным преимуществом работы в предпочтительных границах образования оксидов углерода является то, что меньшее количество ароматического соединения превращается в оксиды углерода и другие малоценные формы. Это преимущество оценивается по сумме молей ароматического соединения, который покидает реакционную среду, деленной на сумму молей ароматического соединения, поступающего в реакционную среду в течение непрерывного периода времени, предпочтительно, в течение одного часа, более предпочтительно, в течение одного дня и, наиболее предпочтительно, в течение 30 последовательных дней. Это отношение далее обозначают как “индекс молярного отбора” для ароматических соединений в реакционной среде и выражают в числовых процентах. Если все поступающие в реакцию ароматические соединения покидают реакционную среду в виде ароматических соединений, несмотря на то, что они находятся в основном в окисленных формах поступивших в реакцию ароматических соединений, то в таком случае индекс молярного отбора имеет максимальное значение, равное 100%. Если строго 1 из каждых 100 вводимых ароматических молекул, проходя через реакционную среду, превращается в оксиды углерода и/или другие неароматические молекулы (в частности, уксусную кислоту), то индекс молярного отбора составляет 99%. Конкретно для случая, когда ксилол является основным способным окисляться исходным ароматическим соединением, индекс молярного отбора для ароматических соединений в реакционной среде, предпочтительно, составляет больше чем приблизительно 98%, более предпочтительно, составляет больше чем приблизительно 98,5% и, наиболее предпочтительно, составляет меньше чем 99,0%. В то же время, с целью сохранения достаточной общей активности реакции, индекс молярного отбора для ароматических соединений в реакционной среде, предпочтительно, составляет меньше чем приблизительно 99,9%, более предпочтительно, составляет меньше чем приблизительно 99,8% и, наиболее предпочтительно, составляет меньше чем 99,7% в том случае, когда ксилол является основным способным окисляться исходным ароматическим соединением.
Другой аспект настоящего изобретения включает образование метилацетата в реакционной среде, содержащей уксусную кислоту и одно или несколько способных окисляться ароматических соединений. Указанный метилацетат является относительно летучим соединением, по сравнению с водой и уксусной кислотой и таким образом проявляет тенденцию уноситься отходящим газом, если не применяется охлаждение или не используется блок операций, с целью извлечения метилацетата и/или его разрушения прежде, чем отходящий газ попадет в окружающую среду. Таким образом, образование метилацетата приводит к производственным потерям и капитальным затратам. Метилацетат, видимо, образуется, на первой стадии, за счет соединения метильного радикала, вероятно возникающего при разложении уксусной кислоты, с кислородом с образованием метилгидропероксида с последующим его разложением с образованием метанола и, наконец, за счет взаимодействия образовавшегося метанола с остальной уксусной кислотой, с образованием метилацетата. Независимо от того, как протекает реакция, авторы настоящего изобретения обнаружили, что если скорость образования метилацетата слишком мала, то оксидов углерода также образуется слишком мало, а вредных ароматических примесей образуется слишком много. Если скорость образования слишком высока, то тогда оксидов углерода также образуется излишне много, что приводит к потерям растворителя, способного окисляться соединения и окислителя. При использовании приведенных в настоящем описании предпочтительных вариантов осуществления изобретения мольное отношение образовавшегося метилацетата к способному окисляться исходному ароматическому соединению, предпочтительно, составляет больше чем приблизительно 0,005:1, более предпочтительно, составляет больше чем приблизительно 0,010:1 и, наиболее предпочтительно, составляет больше чем 0,020:1. В то же время мольное отношение образовавшегося метилацетата к исходному способному окисляться ароматическому соединению, предпочтительно, составляет меньше чем приблизительно 0,09:1, более предпочтительно, составляет меньше чем приблизительно 0,07:1, еще более предпочтительно, составляет меньше чем приблизительно 0,05:1 и, наиболее предпочтительно, составляет меньше чем 0,04:1.
Авторы настоящего изобретения отмечают, что для всех приведенных численных значений диапазонов верхние и нижние границы диапазонов могут быть независимыми друг от друга. Например, численные значения диапазона от 10 до 100 означают больше чем 10 и/или меньше чем 100. Таким образом, диапазон от 10 до 100 составляет основу для заявляемого ограничения больше чем 10 (без верхней границы), заявленного ограничения меньше чем 100 (без нижней границы), а также для полного диапазона от 10 до 100 (как с нижней, так и с верхней границами).
Некоторые варианты осуществления настоящего изобретения могут быть далее проиллюстрированы приведенным ниже примером, однако следует понимать, что указанный пример приведен лишь для пояснения настоящего изобретения и, если специально не указано, не ограничивает объем настоящего изобретения.
ПРИМЕР
В данном примере оценивают свойства частиц новой СТА (представлены на фиг.32А и 32В) и образцов СТА для сравнения (представлены на фиг.33А и 33В). В частности, для частиц СТА определяют размер частиц, площадь поверхности БЭТ и скорость растворения. Тестирование частиц проводят в соответствии с методиками, приведенными в разделе «Подробное описание изобретения».
Частицы новой СТА получают по способу частичного окисления коммерчески чистого пара-ксилола с использованием рециклированного фильтрата в барботажной колонне реакторного типа, в соответствии с многими вариантами осуществления настоящего изобретения, в том числе с использованием растворителя, который содержит уксусную кислоту и воду, с использованием компонентов катализатора, содержащих кобальт, марганец и бром, с использованием температуры, приблизительно составляющей 160°С на половине высоты реакционной среды с использованием предпочтительных градиентов состава газовой фазы, состава жидкой фазы и температуры внутри реакционной среды, и с использованием предпочтительного общего значения STR и предпочтительного значения STR для кислорода. Частицы СТА для сравнения получают в хорошо перемешиваемом реакторе смешения также по способу частичного окисления коммерчески чистого пара-ксилола с использованием рециклированного фильтрата растворителя, который содержит уксусную кислоту и воду, и с использованием компонентов катализатора, содержащих кобальт, марганец и бром, однако процесс предусматривает более равномерное пространственное распределение скоростей реакций, скоростей испарения растворителя, состава жидкой фазы и состава газовой фазы и, кроме того, процесс проводят при большем давлении и при температуре приблизительно 200°С.Большинство полученных результатов сведено в таблицу 5, а полный набор данных для теста на растворение за определенный промежуток времени представлен на фиг.36.
Сравнивая два типа СТА, можно отметить сходные количества наиболее мелких частиц, на что указывают значения для D(v, 0,1); однако новая СТА имеет значительно меньший средний размер частиц (D(4, 3)) и значение медианы для размера частиц (D(v, 0,5)). Новая СТА имеет также заметно более узкое относительное распределение частиц по размерам и содержит меньше очень больших частиц (D(v, 0,9)), которые, как правило, наиболее трудно растворяются и/или вступают во взаимодействия при проведении последующих операций процесса. Кроме того, несмотря на сходство в количестве очень маленьких частиц, обладающих очень большой площадью поверхности, новая СТА имеет значительно большую общую площадь поверхности, что подтверждается данными БЭТ и что можно было бы ожидать на основании визуальной инспекции микрофотографий. Комбинация всех физических факторов привела к значительно большим полезным скоростям растворения для новой СТА, несмотря на то, что конечные величины растворимости при достижении равновесия одинаковы. Было неожиданно обнаружено, что новая СТА пригодна для использования во многих последующих процессах переработки, включая растворение и/или химические реакции, как указано в данном описании, несмотря на то, что новая СТА одновременно представляет ряд трудностей и менее благоприятна для таких процессов как фильтрация и жидкостная промывка с целью удаления остатков кобальта, марганца и брома.
Настоящее изобретение подробно описано со ссылкой на предпочтительные варианты его осуществления, однако следует понимать, что могут быть предложены различные вариации и модификации, которые соответствуют сущности изобретения и входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2381212C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388738C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382759C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382758C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ В БАРБОТАЖНОЙ КОЛОННЕ РЕАКТОРНОГО ТИПА | 2005 |
|
RU2363534C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2363535C2 |
| СИСТЕМА ОКИСЛЕНИЯ С ВНУТРЕННИМ ВТОРИЧНЫМ РЕАКТОРОМ | 2006 |
|
RU2448766C2 |
| СИСТЕМА ОКИСЛЕНИЯ, ИСПОЛЬЗУЮЩАЯ ВНУТРЕННЮЮ КОНСТРУКЦИЮ ДЛЯ УЛУЧШЕНИЯ ГИДРОДИНАМИКИ | 2006 |
|
RU2418629C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388745C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2006 |
|
RU2435753C2 |
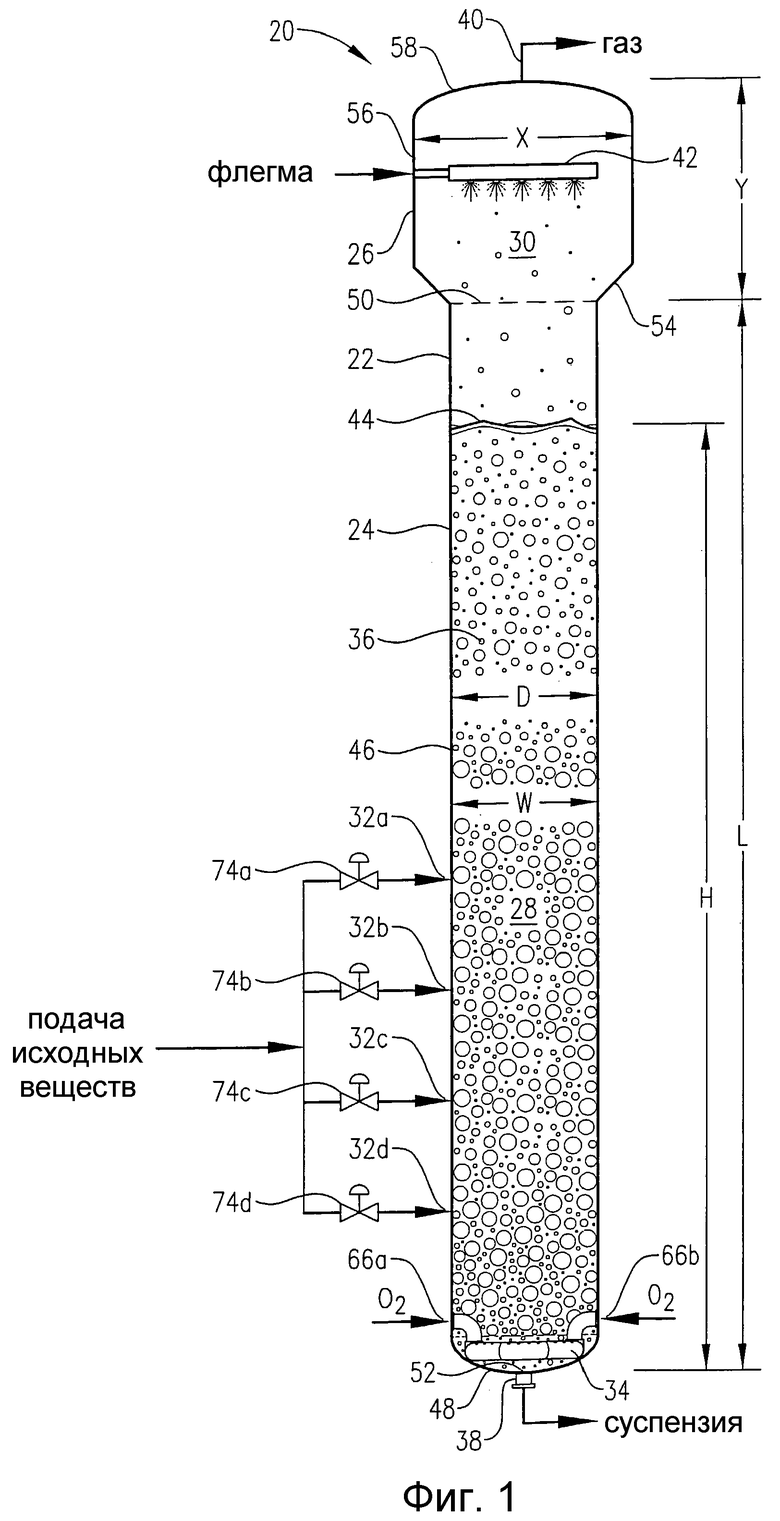
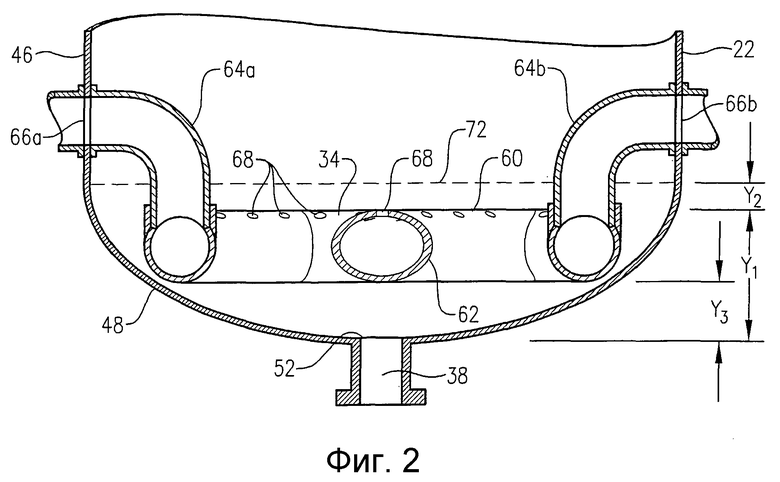
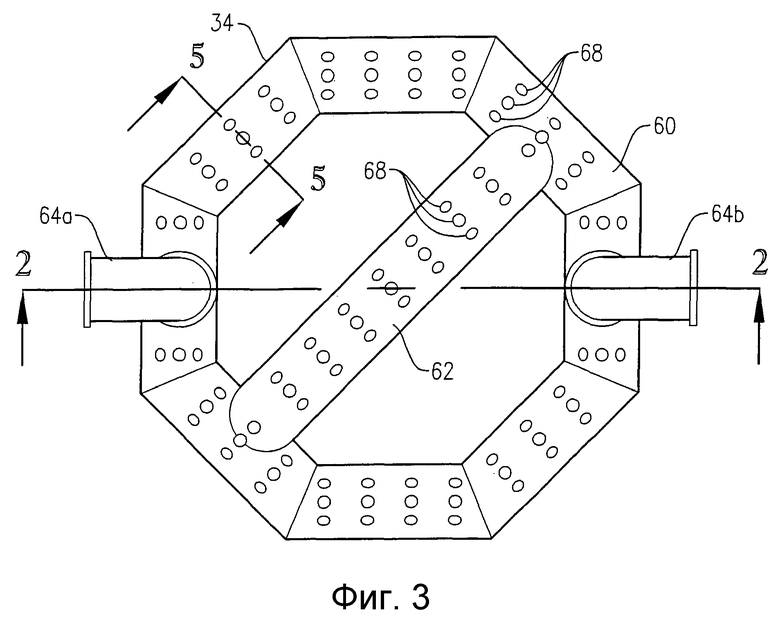
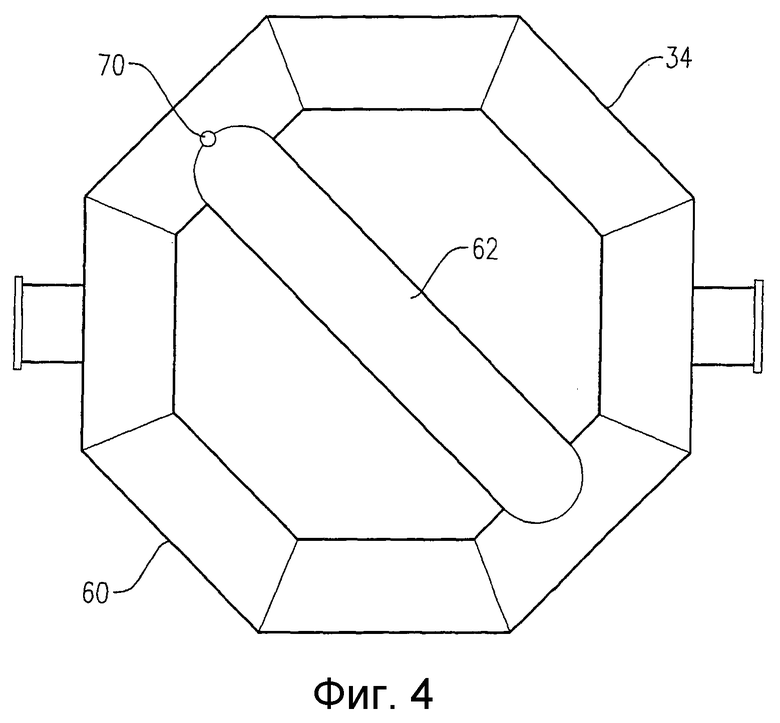
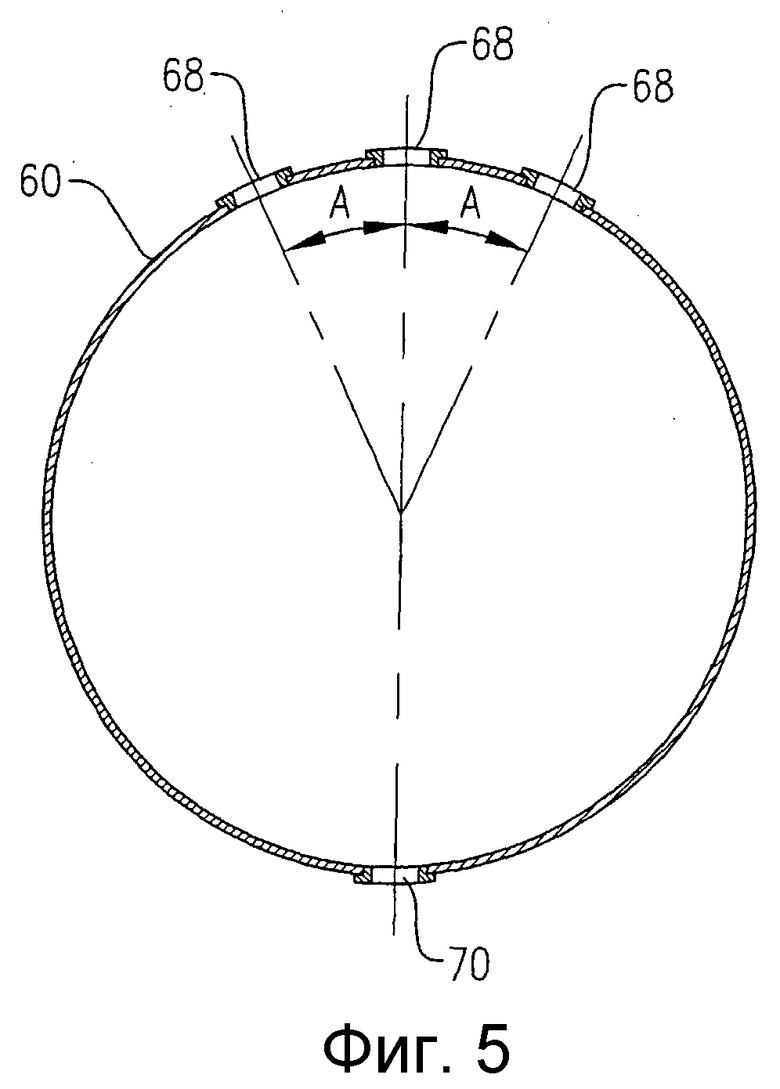
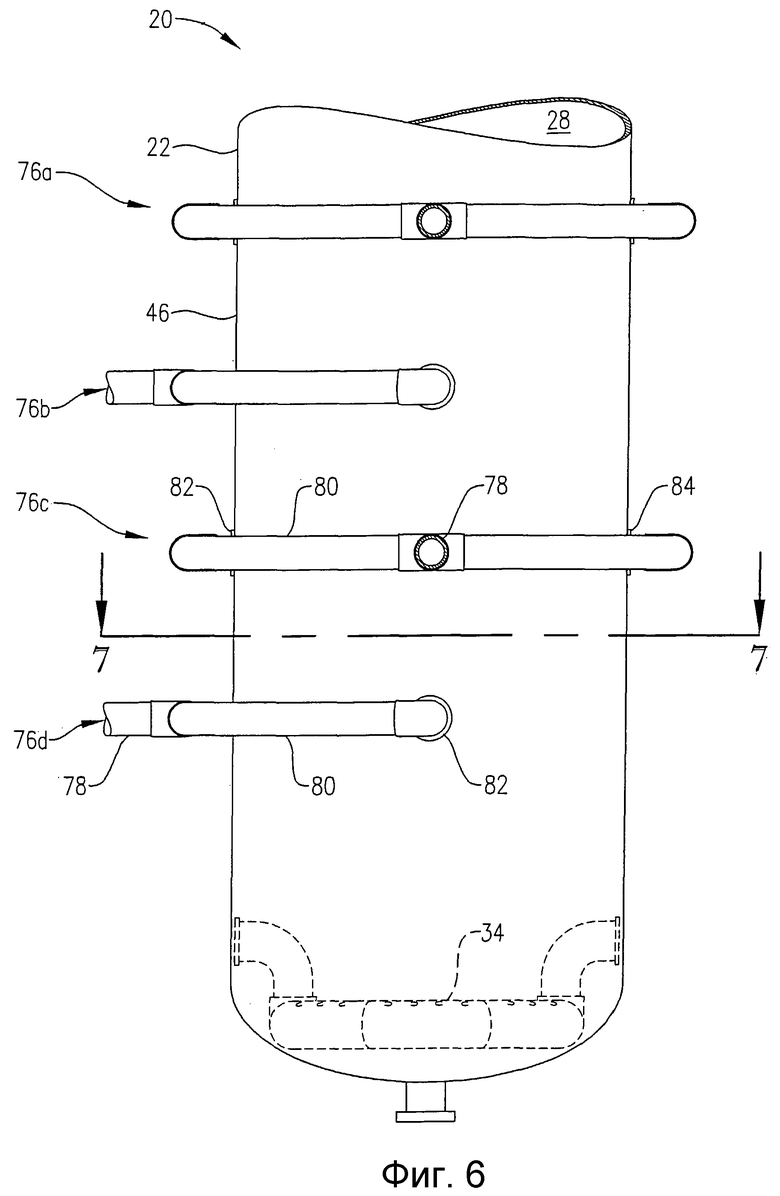
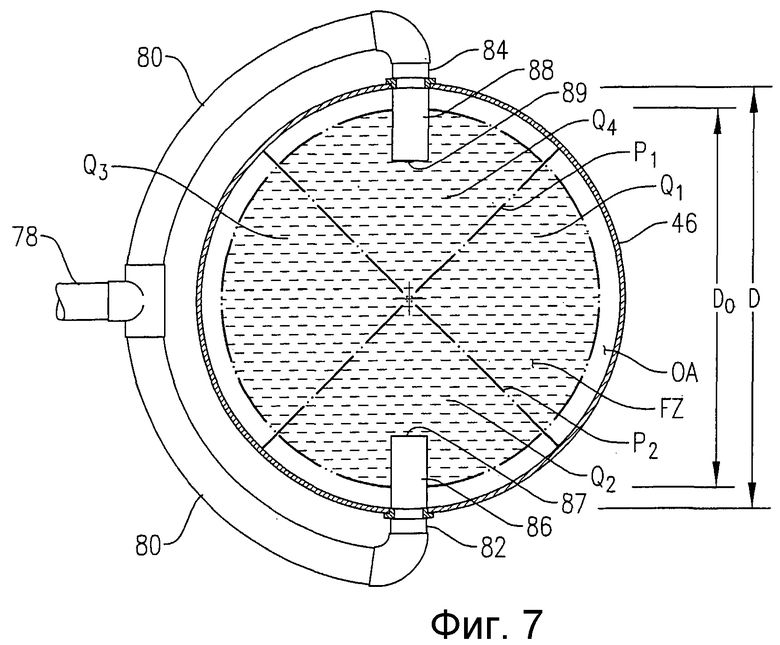
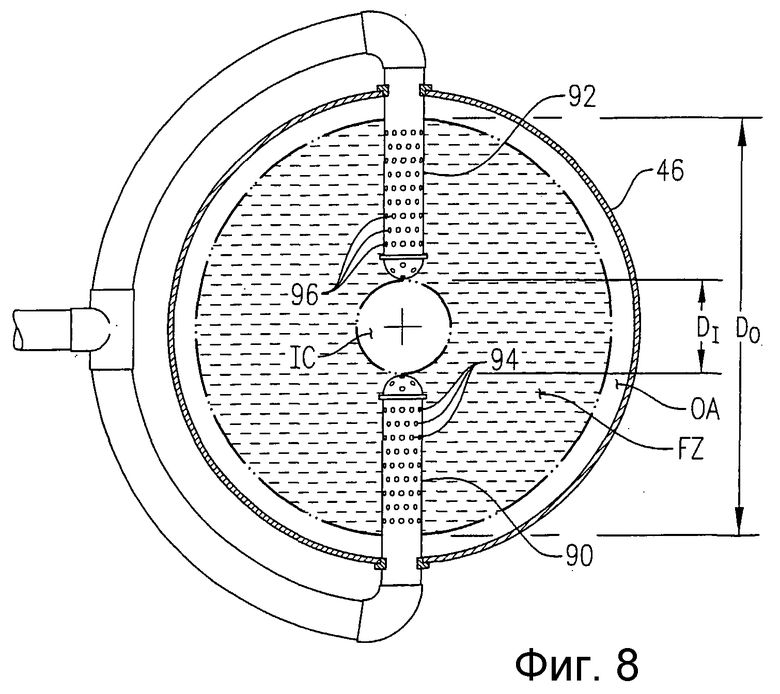
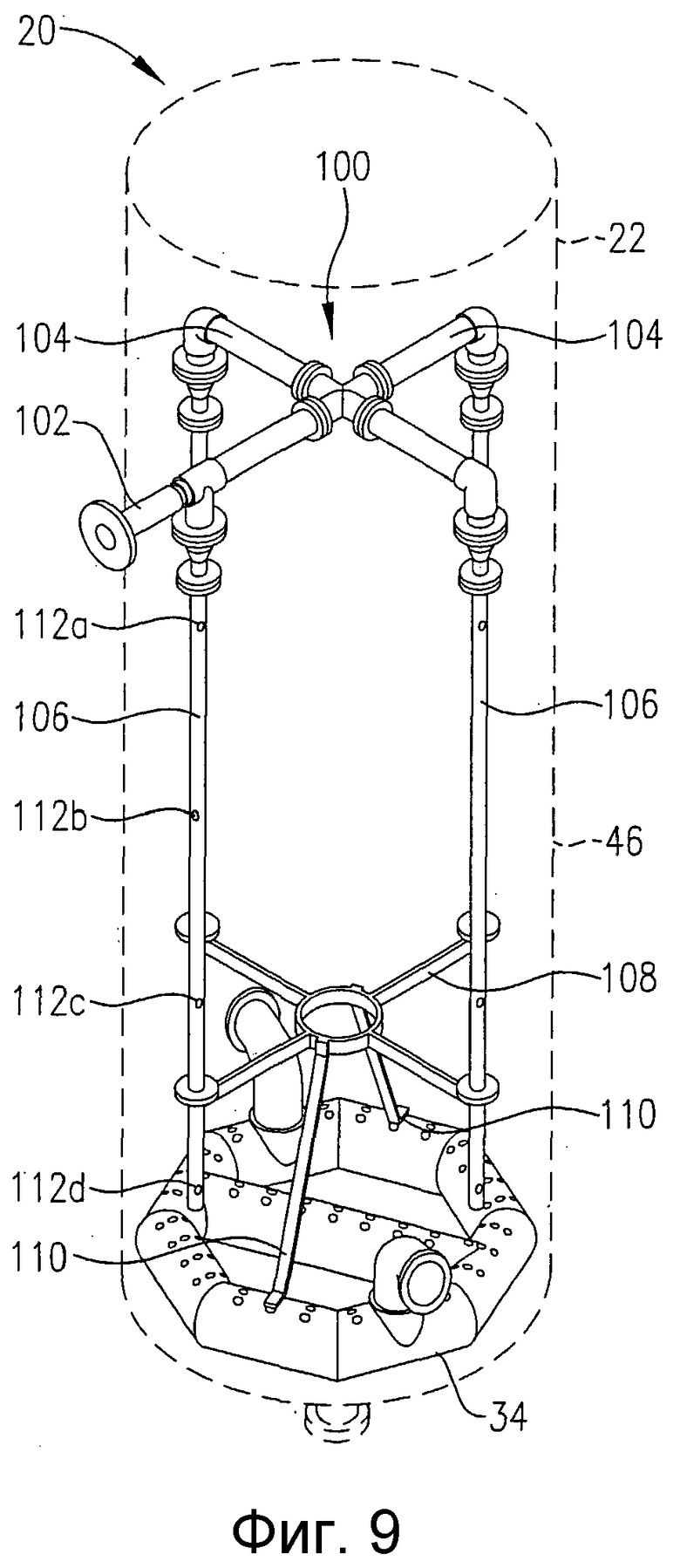
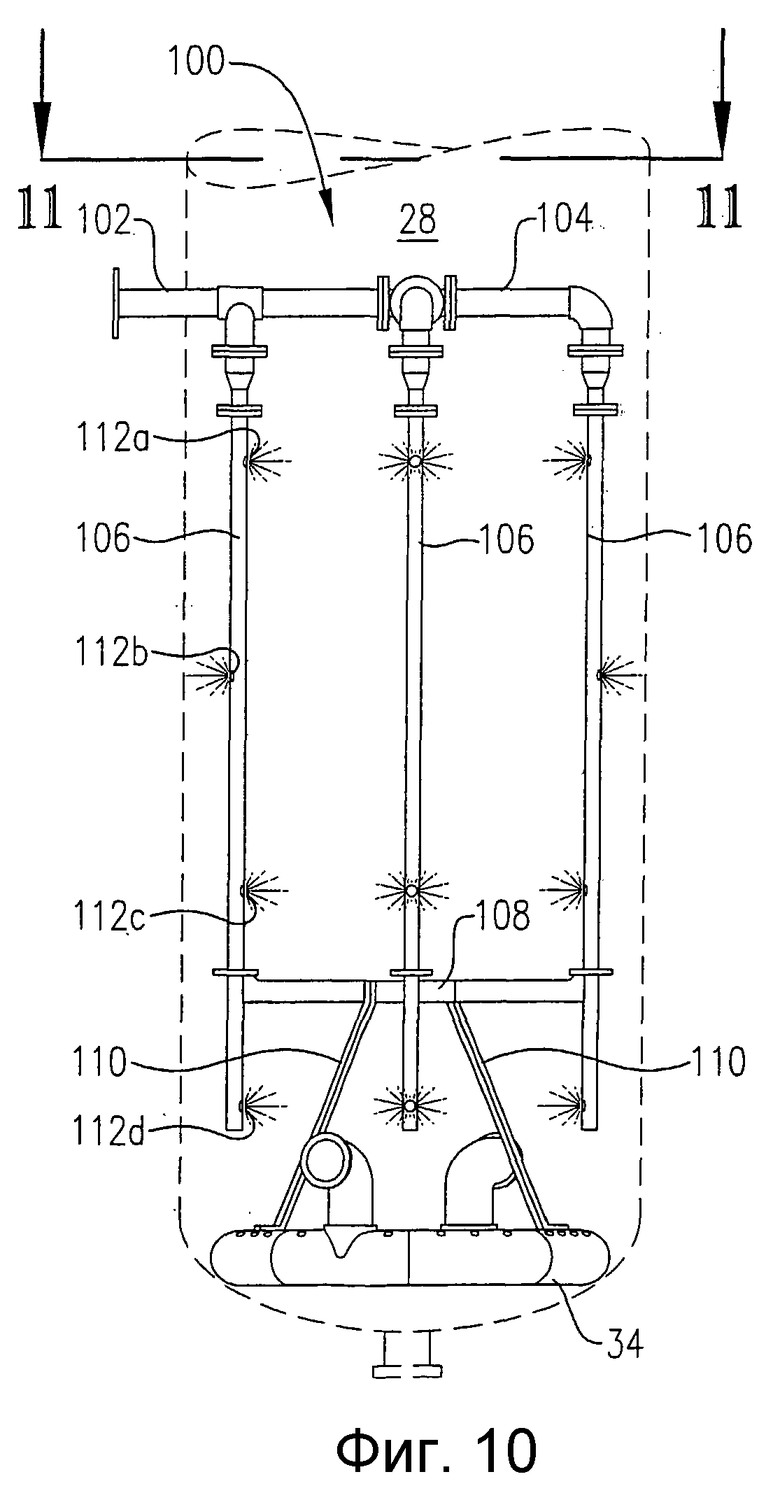
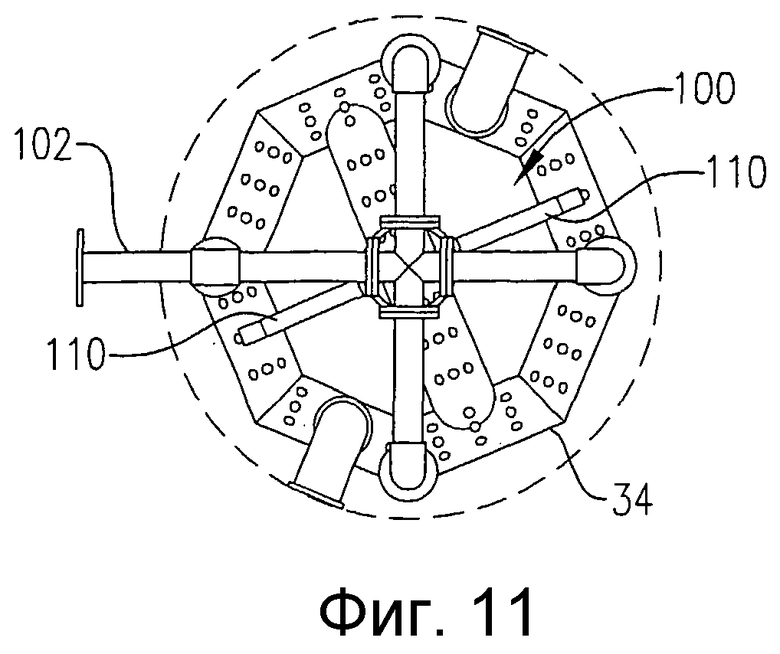
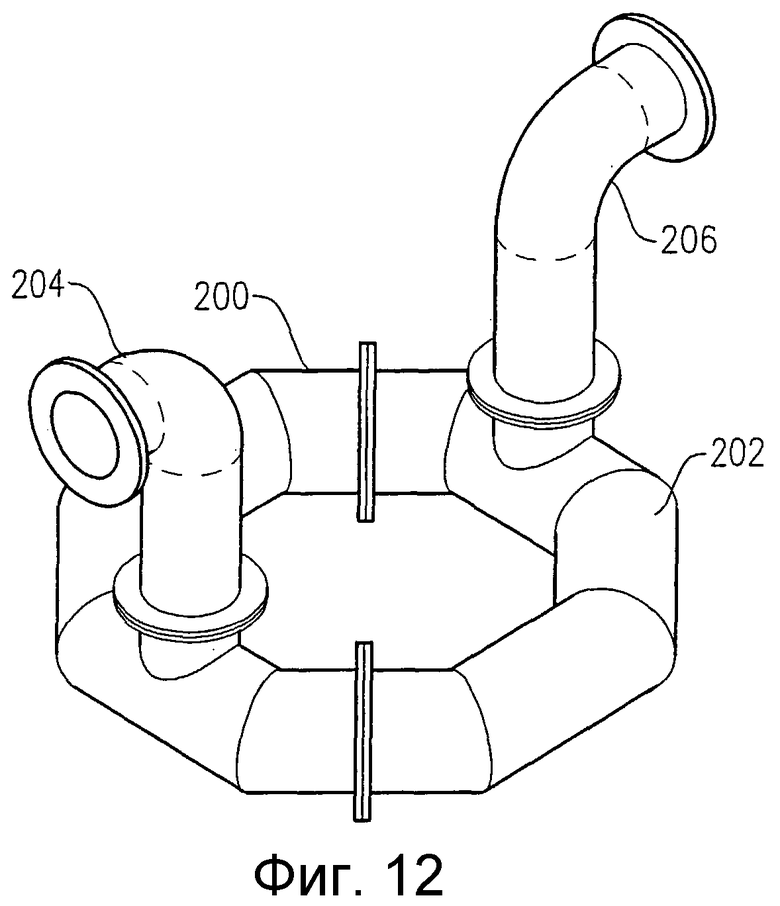
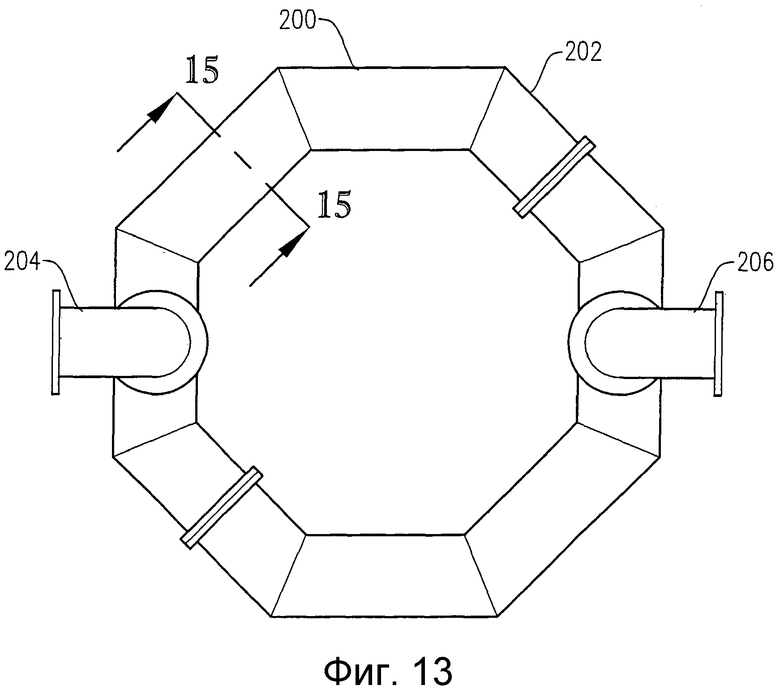
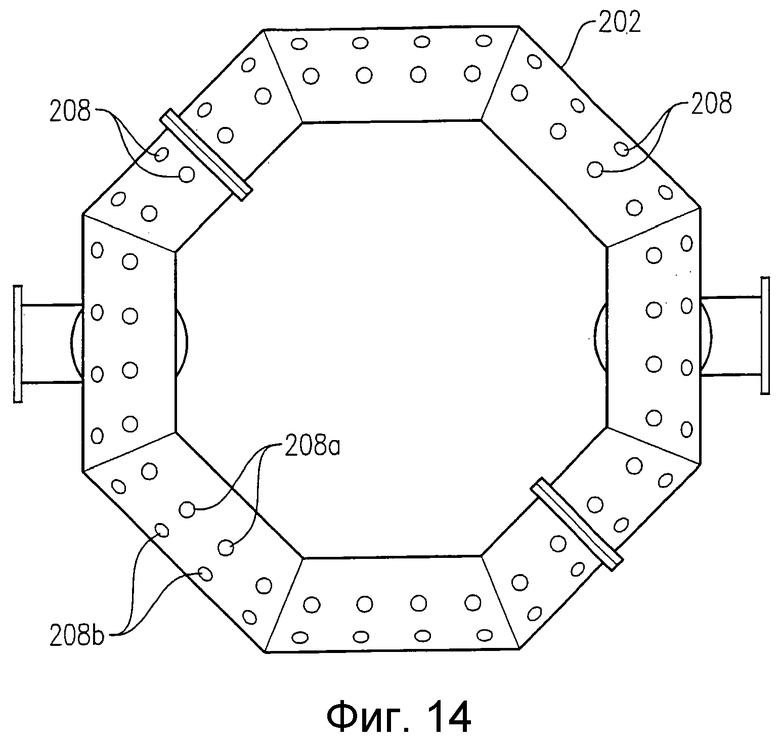
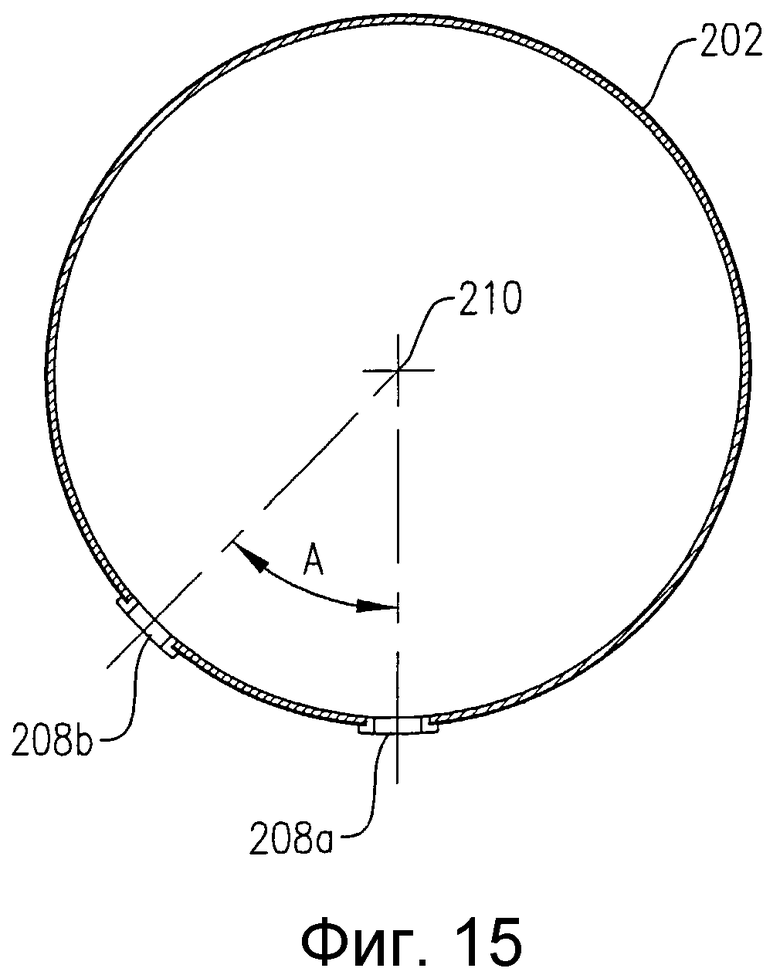
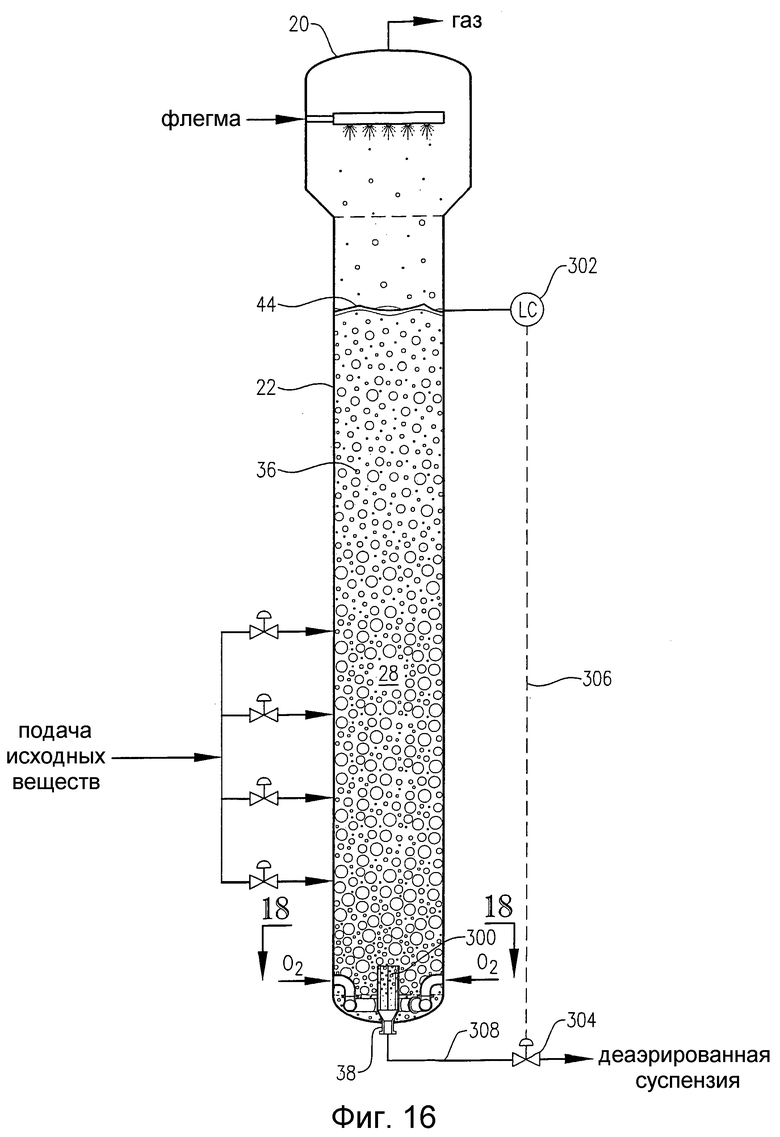
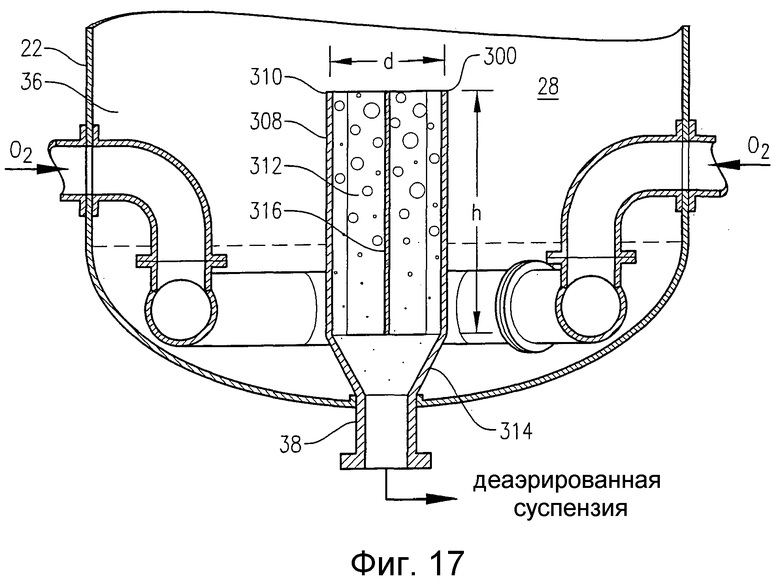
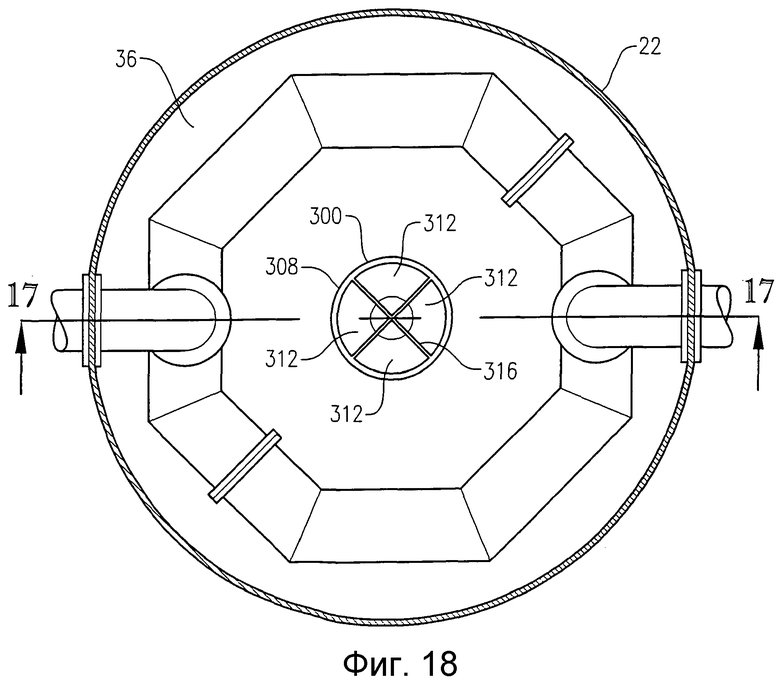
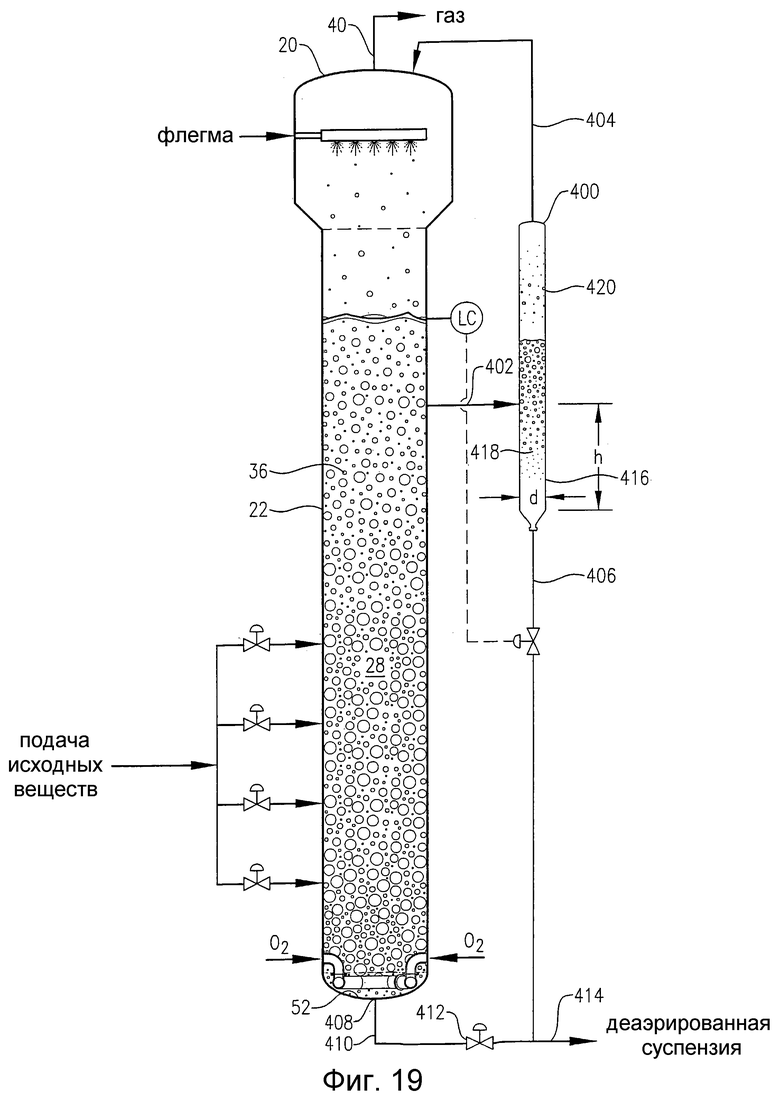
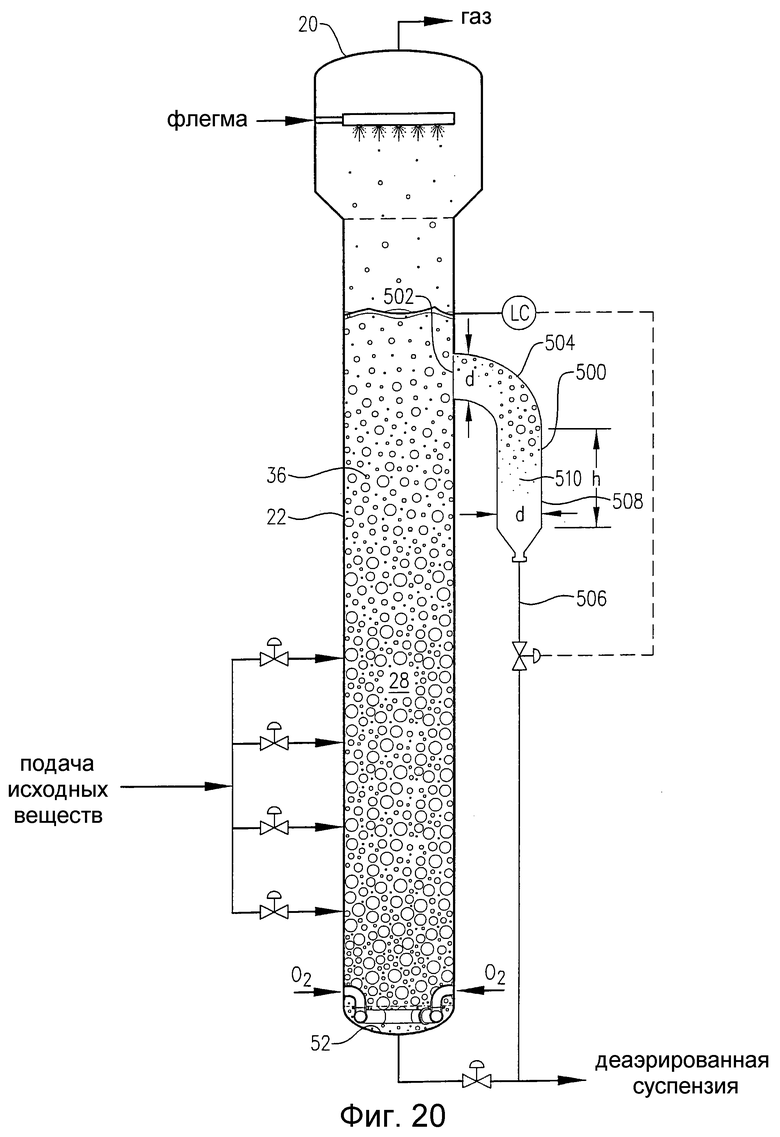
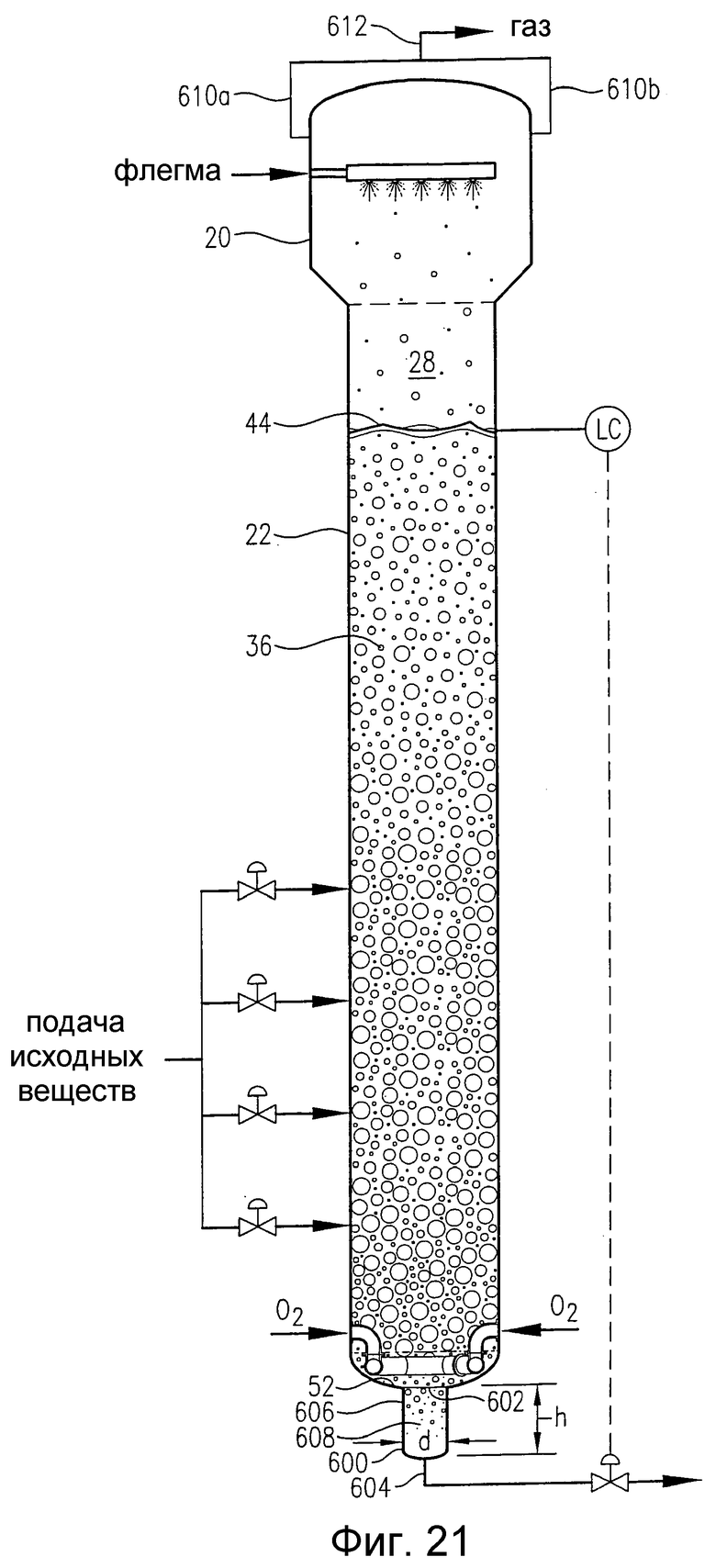
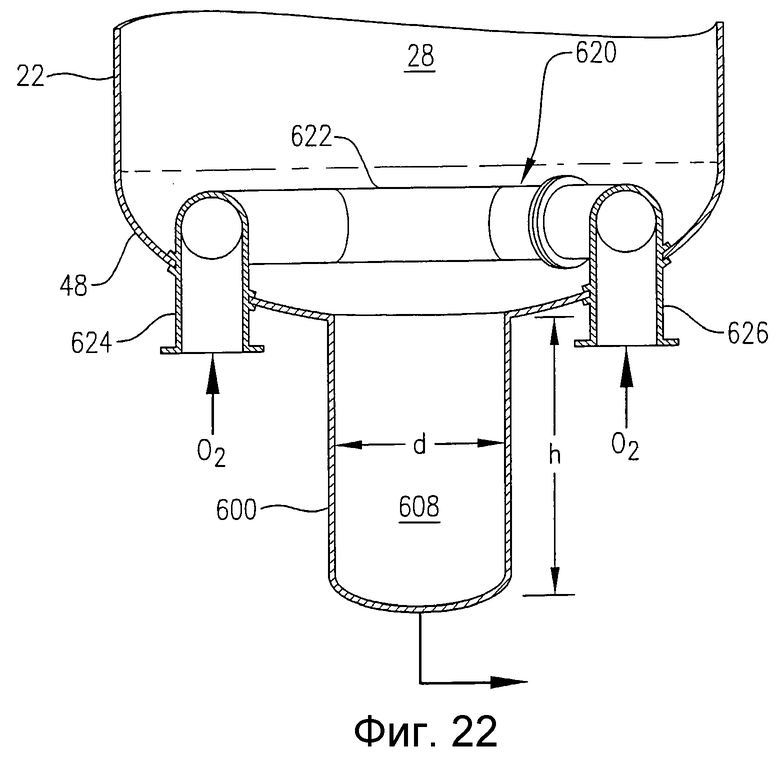
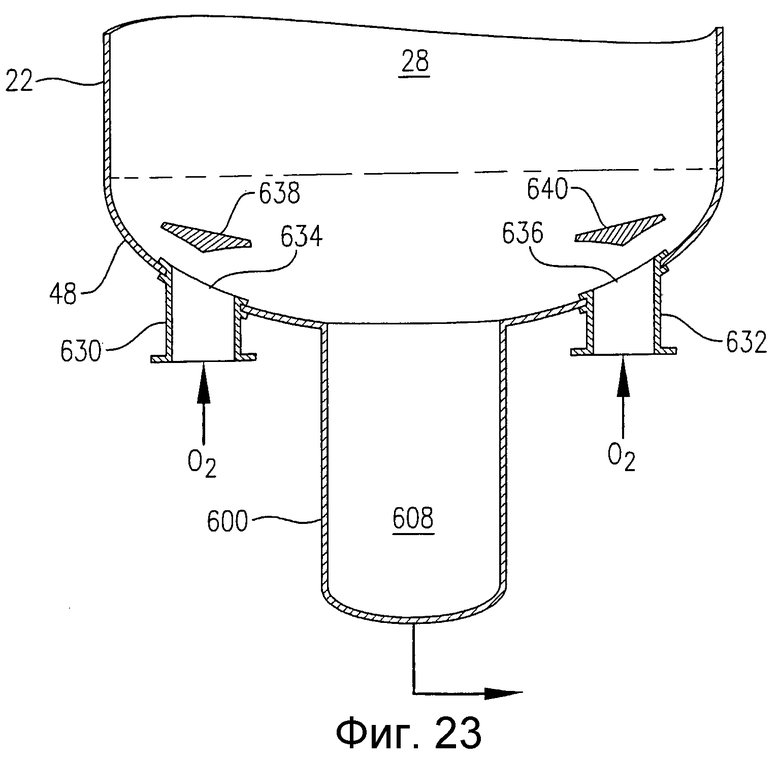
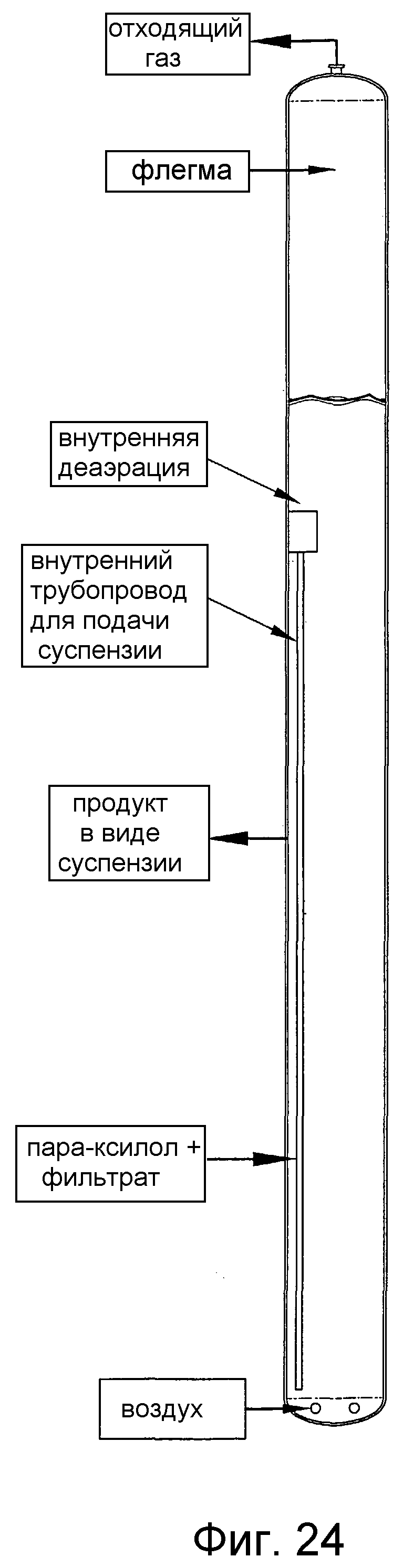
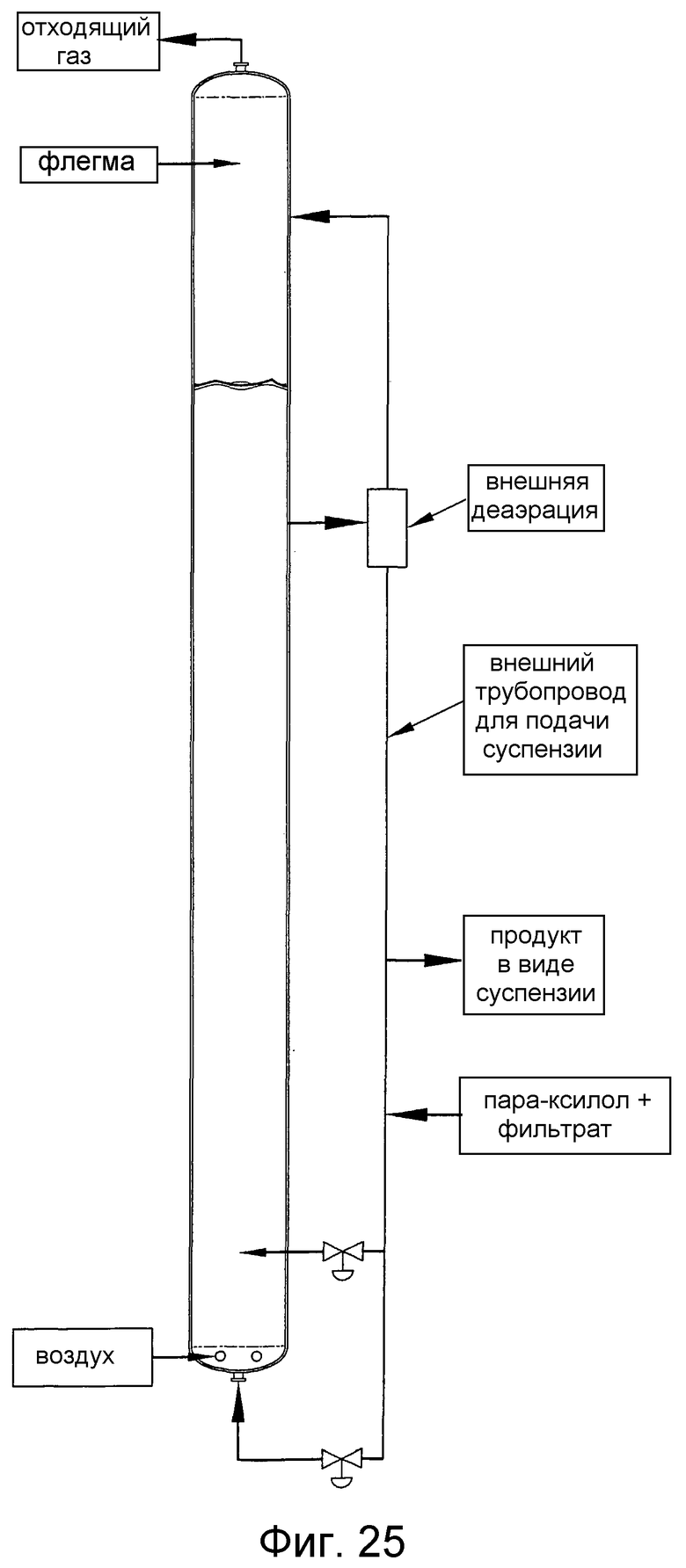
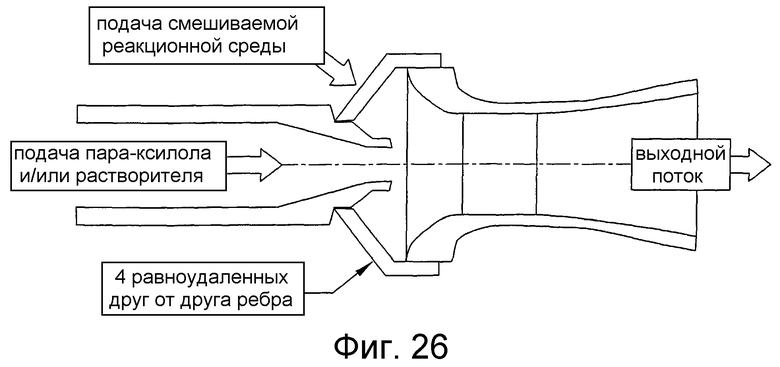
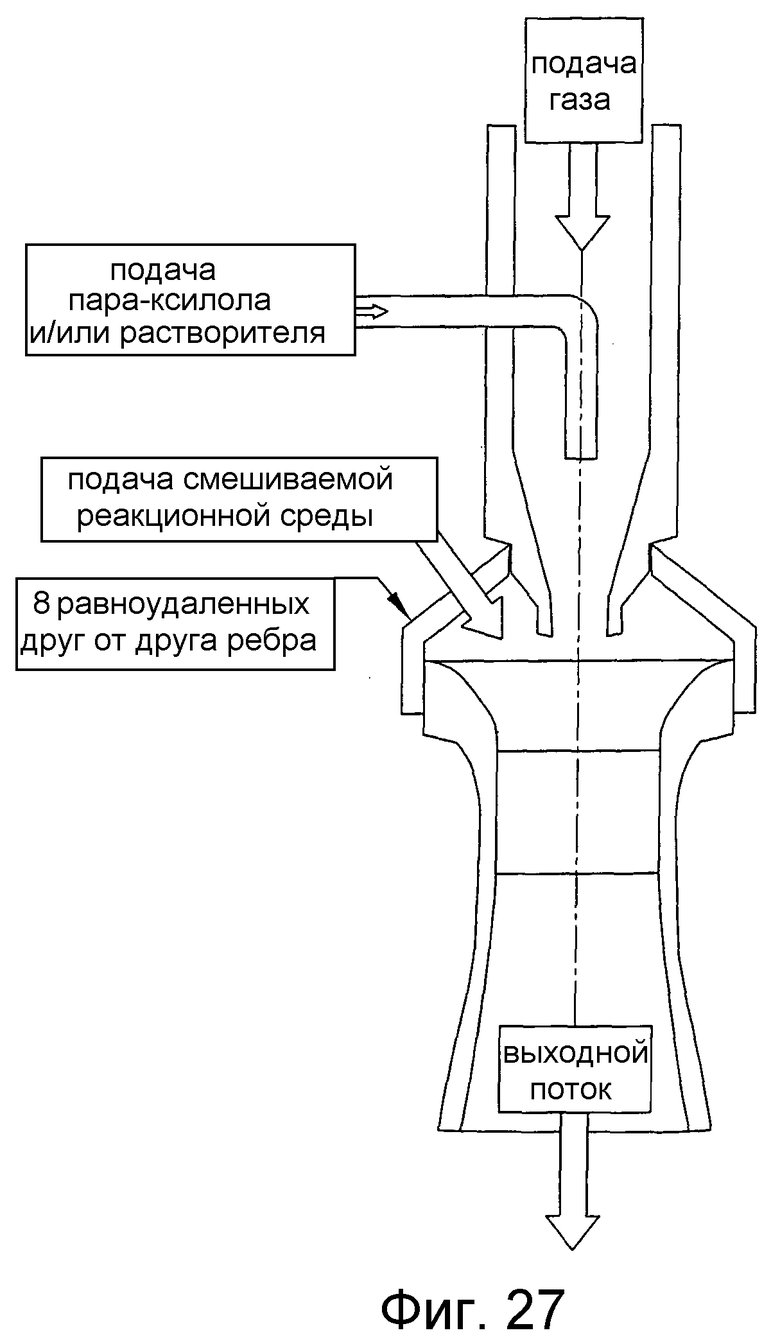
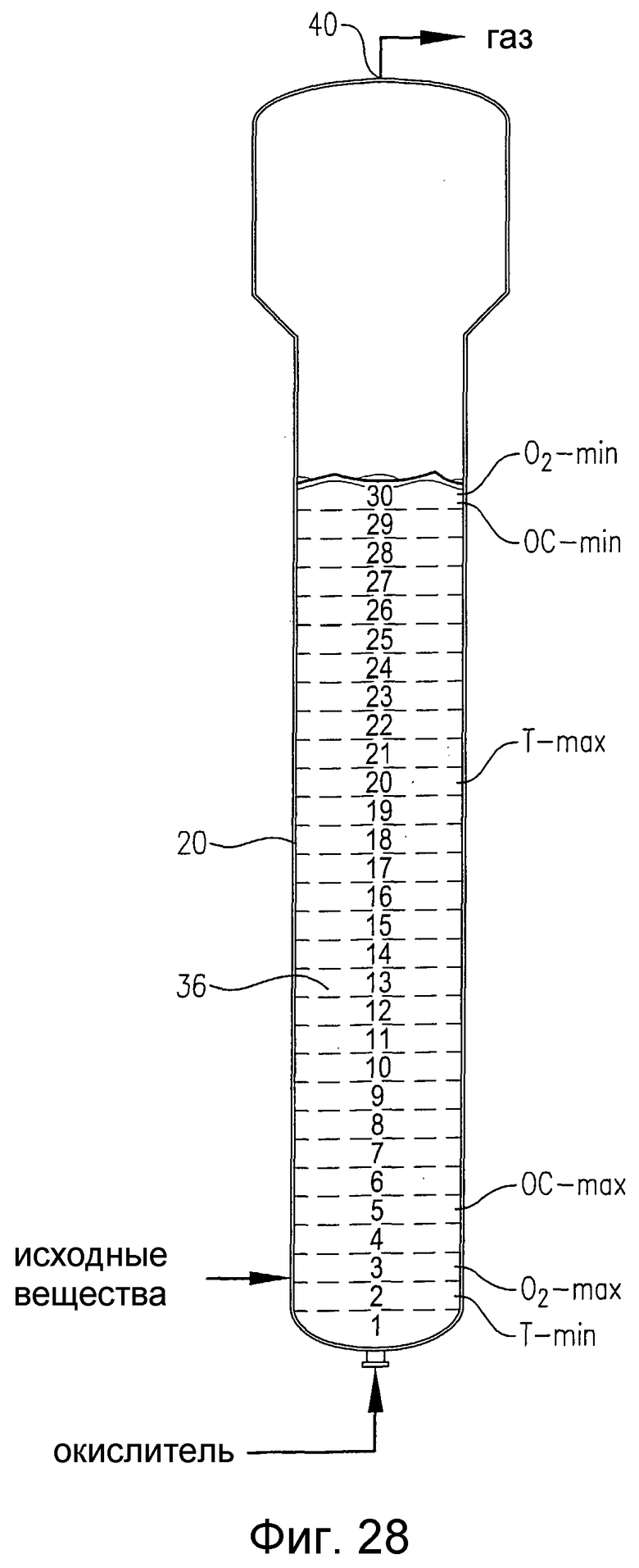
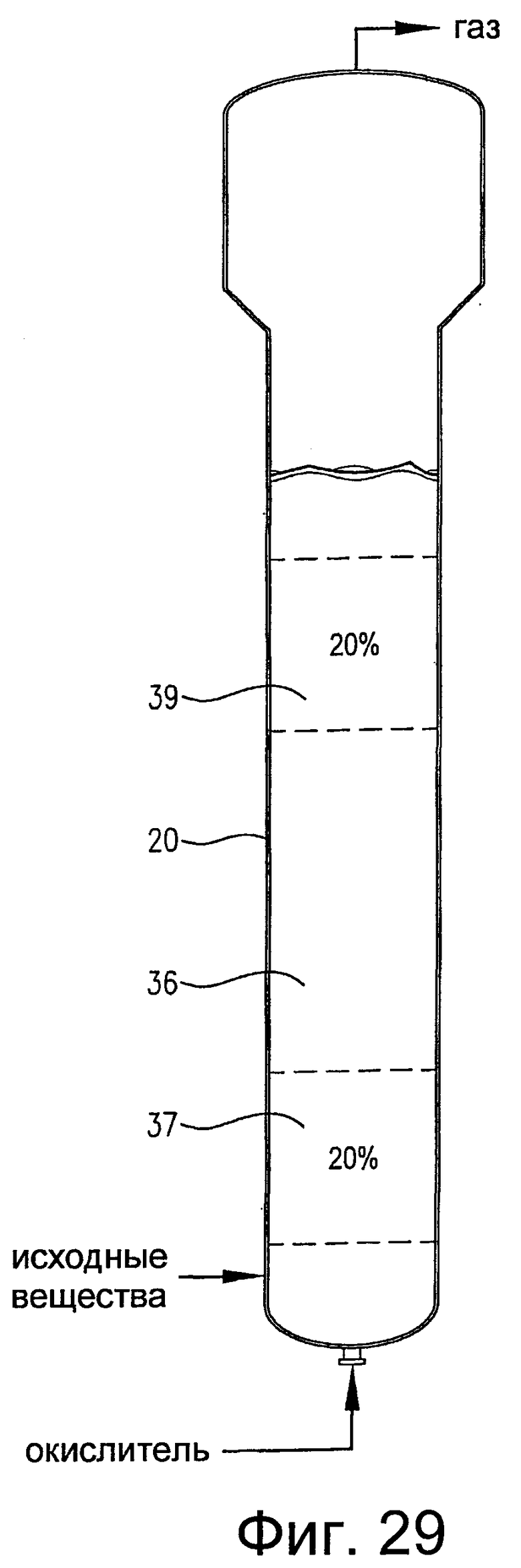
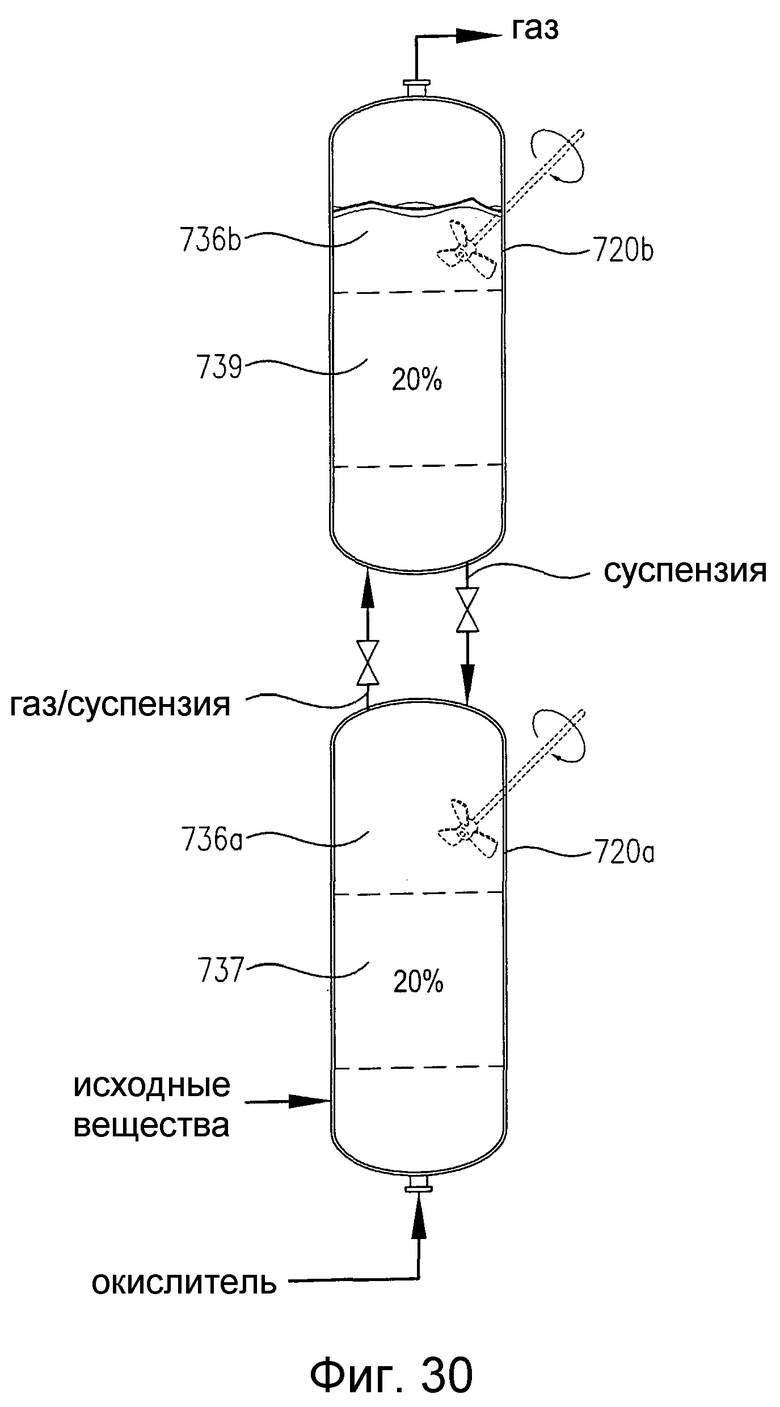
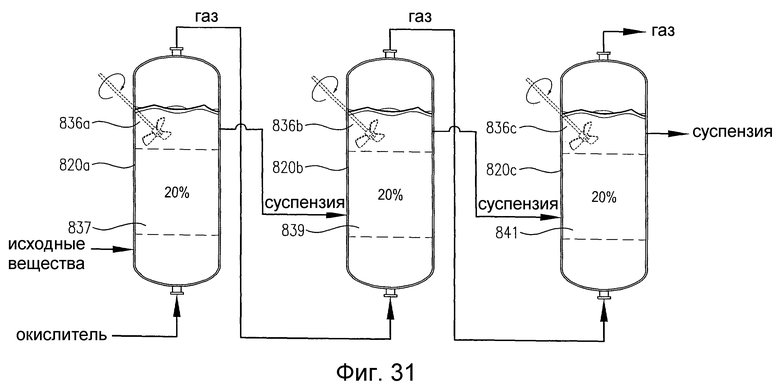
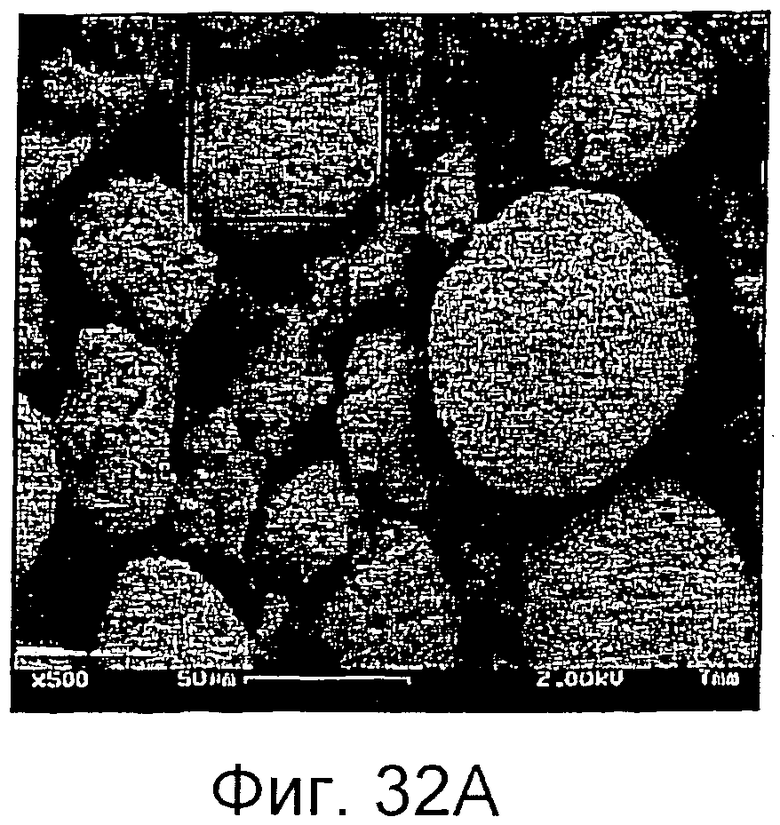



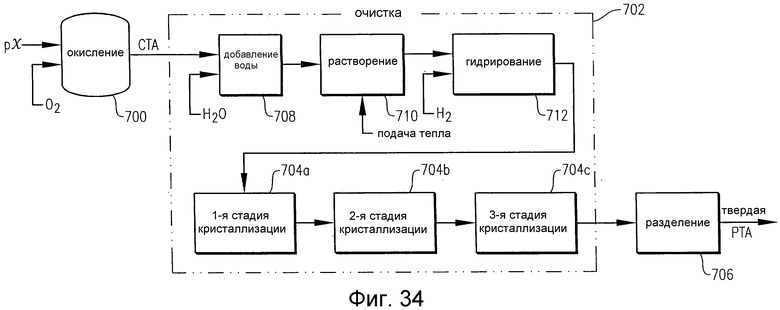
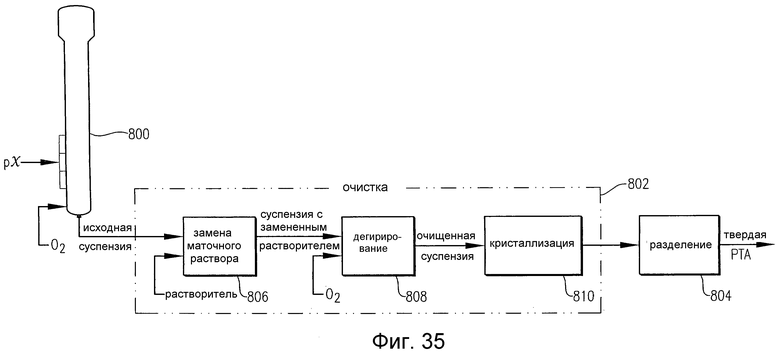
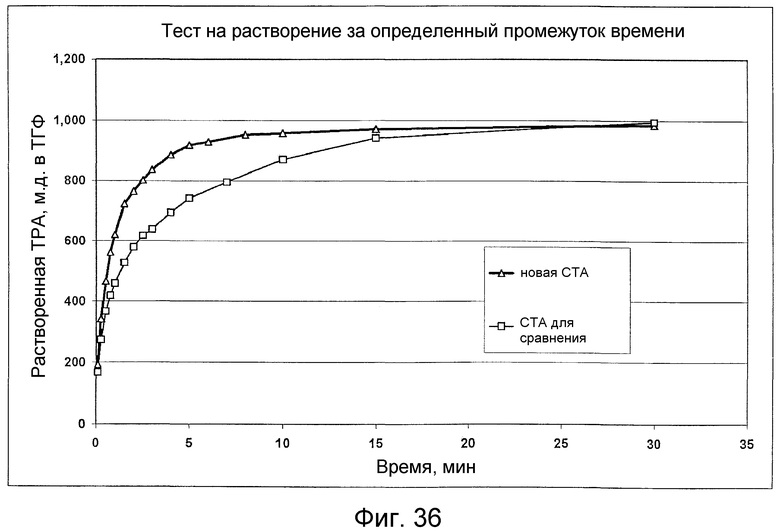
Изобретение относится к жидкофазному каталитическому окислению ароматического соединения и к получаемой сырой терефталевой кислоте. Окисление осуществляют в барботажной колонне реакторного типа, которая обеспечивает проведение высокоэффективного процесса при сравнительно низких температурах. Частица полученной сырой терефталевой кислоты, содержащая меньше чем приблизительно 100 ч./млн массовых 2,6-дикарбоксифлуоренона, имеет коэффициент пропускания на 340 нм (%Т340) больше чем приблизительно 25%, дополнительно содержит меньше чем приблизительно 12 ч./млн массовых 4,4-дикарбоксистильбена и/или содержит меньше чем приблизительно 400 ч./млн массовых изофталевой кислоты. Частица полученной сырой терефталевой кислоты, характеризующаяся средним размером в диапазоне от приблизительно 20 до приблизительно 150 мкм, растворяется в тетрагидрофуране в течение одной минуты до концентрации, по меньшей мере, приблизительно 500 ч./млн и/или характеризуется средней площадью поверхности по БЕТ больше чем приблизительно 0,6 м2/г. В том случае, когда окисляемым является параксилол, а продуктом окисления является техническая терефталевая кислота, указанный продукт может быть выделен и очищен с применением более экономически выгодных методов, чем те, которые можно использовать, если кислота получена обычным высокотемпературным способом окисления. 3 н. и 34 з.п. ф-лы, 36 ил., 5 табл.
1. Сырая терефталевая кислота, состоящая из множества твердых частиц сырой терефталевой кислоты, извлекаемых из реактора для проведения окисления, в котором указанные частицы образуются, при этом частица сырой терефталевой кислоты содержит меньше чем приблизительно 100 ч/млн. массовых 2,6-дикарбоксифлуоренона, имеет коэффициент пропускания на 340 нм (%Т340) больше чем приблизительно 25%, отличающаяся тем, что частица сырой терефталевой кислоты дополнительно содержит меньше чем приблизительно 12 ч/млн. массовых 4,4-дикарбоксистильбена и/или содержит меньше чем приблизительно 400 ч/млн. массовых изофталевой кислоты.
2. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты содержит меньше чем приблизительно 6 ч/млн. массовых 4,4-дикарбоксистильбена.
3. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты содержит меньше чем приблизительно 200 ч/млн. массовых изофталевой кислоты.
4. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты содержит меньше чем приблизительно 30 ч/млн. массовых 2,7-дикарбоксифлуоренона.
5. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты растворяется в тетрагидрофуране в течение одной минуты до концентрации, по меньшей мере, приблизительно 500 ч/млн.
6. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты имеет константу времени регрессии "С" больше чем приблизительно 0,5 мин-1, определяемую растворением за определенный промежуток времени.
7. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты растворяется в тетрагидрофуране в течение двух минут до концентрации, по меньшей мере, приблизительно 700 ч/млн. и имеет константу времени регрессии "С" больше чем приблизительно 0,6 мин-1.
8. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты имеет среднюю площадь поверхности по БЕТ больше чем приблизительно 0,6 м2/г.
9. Кислота по п.1, отличающаяся тем, что частица сырой терефталевой кислоты имеет средний размер частиц в диапазоне от приблизительно 20 до приблизительно 150 мкм.
10. Кислота по п.1, отличающаяся тем, что 90% от общего объема частиц сырой терефталевой кислоты имеют размер от приблизительно 30 до приблизительно 150 мкм.
11. Сырая терефталевая кислота, состоящая из множества твердых частиц сырой терефталевой кислоты, извлекаемых из реактора для проведения окисления, в котором указанные частицы образуются, при этом частица сырой терефталевой кислоты характеризуется средним размером в диапазоне от приблизительно 20 до приблизительно 150 мкм, отличающаяся тем, что частица сырой терефталевой кислоты растворяется в тетрагидрофуране в течение одной минуты до концентрации, по меньшей мере, приблизительно 500 ч/млн. и/или характеризуется средней площадью поверхности по БЕТ больше чем приблизительно 0,6 м2/г.
12. Кислота по п.11, отличающаяся тем, что частица сырой терефталевой кислоты имеет константу времени регрессии "С" больше чем приблизительно 0,5 мин-1, определяемую растворением за определенный промежуток времени.
13. Кислота по п.11, отличающаяся тем, что частица сырой терефталевой кислоты растворяется в тетрагидрофуране в течение двух минут до концентрации, по меньшей мере, приблизительно 700 ч/млн. и имеет константу времени регрессии "С" больше чем приблизительно 0,6 мин-1.
14. Кислота по п.11, отличающаяся тем, что частица сырой терефталевой кислоты обладает одним или несколькими из указанных свойств:
(i) содержит меньше чем приблизительно 12 ч/млн. массовых 4,4-дикарбоксистильбена,
(ii) содержит меньше чем приблизительно 400 ч/млн. массовых изофталевой кислоты,
(iii) содержит меньше чем приблизительно 100 ч/млн. массовых
2,6-дикарбоксифлуоренона,
(iv) имеет коэффициент пропускания на 340 нм (%Т340) больше чем приблизительно 25%.
15. Кислота по п.11, отличающаяся тем, что частица сырой терефталевой кислоты содержит меньше чем приблизительно 30 ч/млн. массовых 2,7-дикарбоксифлуоренона.
16. Кислота по п.11, отличающаяся тем, что 90% от общего объема частиц сырой терефталевой кислоты имеют размер от приблизительно 30 до приблизительно 150 мкм.
17. Способ получения сырой терефталевой кислоты, включающий окисление параксилола в жидкой фазе многофазной реакционной среды в зоне реакции, по меньшей мере, одного первичного реактора для проведения процесса окисления и извлечение из зоны реакции суспензии, содержащей маточный раствор и твердые частицы сырой терефталевой кислоты, при этом частица сырой терефталевой кислоты содержит меньше чем 100 ч/млн. массовых 2,6-дикарбоксифлуоренона и имеет коэффициент пропускания на 340 нм (%Т340) больше чем приблизительно 25%, отличающийся тем, что частица сырой терефталевой кислоты дополнительно содержит меньше чем приблизительно 12 ч/млн. массовых 4,4-дикарбоксистильбена и/или меньше чем приблизительно 400 ч/млн. массовых изофталевой кислоты.
18. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты обладает одним или несколькими из указанных свойств:
(v) содержит меньше чем приблизительно 9000 ч/млн. массовых изофталевой кислоты,
(vi) содержит меньше чем приблизительно 3000 ч/млн. массовых фталевой кислоты,
(vii) содержит меньше чем приблизительно 3000 ч/млн. массовых тримеллитовой кислоты,
(viii) содержит меньше чем приблизительно 15000 ч/млн. массовых бензойной кислоты.
19. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты растворяется в тетрагидрофуране в течение одной минуты до концентрации, по меньшей мере, приблизительно 500 ч/млн.
20. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты имеет константу времени регрессии "С" больше чем приблизительно 0,5 мин-1, определяемую растворением за определенный промежуток времени.
21. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты растворяется в тетрагидрофуране в течение двух минут до концентрации, по меньшей мере, приблизительно 700 ч/млн. и имеет константу времени регрессии "С" больше чем приблизительно 0,6 мин-1.
22. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты содержит меньше чем приблизительно 30 ч/млн. массовых 2,7-дикарбоксифлуоренона.
23. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты содержит меньше чем 6 ч/млн. массовых 4,4-дикарбоксистильбена.
24. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты содержит меньше чем 200 ч/млн. массовых изофталевой кислоты.
25. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты содержит меньше чем приблизительно 50 ч/млн. массовых 2,6-дикарбоксифлуоренона.
26. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты имеет коэффициент пропускания на 340 нм (%Т340) больше чем приблизительно 50%.
27. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты имеет среднюю площадь поверхности по БЕТ больше чем приблизительно 0,6 м2/г.
28. Способ по п.17, отличающийся тем, что частица сырой терефталевой кислоты имеет средний размер частиц в диапазоне от приблизительно 20 до приблизительно 150 мкм.
29. Способ по п.17, отличающийся тем, что 90% от общего объема частиц сырой терефталевой кислоты имеют размер от приблизительно 30 до приблизительно 150 мкм.
30. Способ по п.17, отличающийся тем, что в качестве первичного реактора для проведения окисления используют барботажную колонну реакторного типа.
31. Способ по п.17, отличающийся тем, что параксилол подают в зону реакции таким образом, что при теоретическом разделении зоны реакции парой пересекающихся вертикальных плоскостей на 4 вертикальных квадранта, имеющих равные объемы, не более чем приблизительно 80 мас.% параксилола поступает в зону реакции лишь в один из вертикальных квадрантов.
32. Способ по п.17, отличающийся тем, что, по крайней мере, часть зоны реакции ограничена одной или несколькими вертикальными боковыми стенками реактора, при этом, по меньшей мере, 25 мас.% параксилола поступает в зону реакции в одном или нескольких местах, расположенных внутрь от вертикальных боковых стенок на расстоянии, по крайней мере, 0,05D при максимальном диаметре зоны реакции D.
33. Способ по п.17, отличающийся тем, что окисление проводят в присутствии каталитической системы, содержащей кобальт.
34. Способ по п.33, отличающийся тем, что каталитическая система дополнительно содержит бром и марганец.
35. Способ по п.17, отличающийся тем, что, по крайней мере, часть частиц сырой терефталевой кислоты окисляют во вторичном реакторе окисления.
36. Способ по п.35, отличающийся тем, что окисление во вторичном реакторе окисления осуществляют при средней температуре, которая, по крайней мере, на 10°С выше температуры окисления в первичном реакторе окисления.
37. Способ по п.35, отличающийся тем, что окисление во вторичном реакторе окисления осуществляют при средней температуре на приблизительно 20-80°С выше, чем средняя температура в первичном реакторе окисления, при этом окисление в первичном реакторе окисления осуществляют при средней температуре от приблизительно 140 до приблизительно 180°С, а окисление во вторичном реакторе окисления осуществляют при средней температуре от приблизительно 180 до приблизительно 220°С.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 3996271 A1, 07.12.1976 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| АППАРАТ И СПОСОБ ОСУЩЕСТВЛЕНИЯ ВЗАИМОДЕЙСТВИЯ ФАЗ В СИСТЕМАХ ГАЗ-ЖИДКОСТЬ И ЖИДКОСТЬ-ЖИДКОСТЬ | 2000 |
|
RU2186614C2 |

